Note: Descriptions are shown in the official language in which they were submitted.
CA 02238633 2004-09-O1
21489-9410
- 1 -
ALPHA-SUBSTITUTED ARYLSULPHONAMIDO HYDROXAMIC ACIDS AS
TNF-ALPHA AND MATRIX METALLOPROTEINASE INHIBITORS
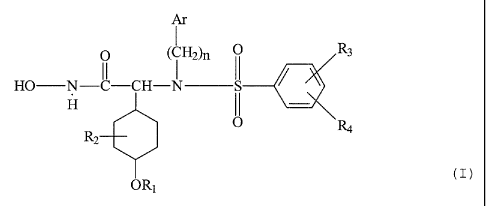
The present invention relates to the etherified cyclohexyl-
and arylsulfonamido-substituted hydroxamic acids of
formula I
Ar
H2)n ~ ~ n3
HO N-C-CH-N S
H
O
R2
OR1 ( I )
wherein
Ar represents carbocyclic aryl, heterocyclic aryl or biaryl;
R1 represents lower alkyl, cycloalkyl, (carbocyclic or
heterocyclic aryl)-lower alkyl, lower alkoxy-lower alkyl,
carbocyclic aryl, heterocyclic aryl, cycloalkyl-lower alkyl
or halogen-lower alkyl;
RZ represents hydrogen or lower alkyl;
R3 and R4 represent independently hydrogen, lower alkyl,
lower alkoxy, halogen, hydroxy, acyloxy, lower alkoxy-lower
alkoxy, trifluoromethyl or cyano; or R3 and R4 together on
adjacent carbon atoms represent lower alkylenedioxy;
n represents an integer from 1 to 5;
pharmaceutically acceptable prodrug derivatives thereof; and
pharmaceutically acceptable salts thereof;
CA 02238633 2005-05-04
21489-9410
- 1a -
further to a process for the preparation o:f these compounds,
to pharmaceutical compositions comprising -these compounds,
to the use of these compounds for the therapeutic treatment
of the human or animal body or for the manufacture of a
pharmaceutical composition.
According to one aspect of the present invention, there is
provided a compound of the formula I
Ar
O (C1
HO N-C-CH-N
R2
ORl (I)
wherein
Ar represents pyridyl;
Rl represents lower alkyl, cycloalkyl, carbocyclic-lower
alkyl, lower alkoxy-lower alkyl, or halogen-lower alkyl;
R2 represents hydrogen or lower alkyl;
R3 and R4 represent independently hydrogen, lower alkyl,
lower alkoxy, halogen, hydroxy, acyloxy, lower alkoxy-lower
alkoxy, trifluoromethyl or cyano; or R3 and R4 together on
adjacent carbon atoms represent lower alky:Lenedioxy;
n represents an integer from 1 to 5;
wherein cycloalkyl represents a saturated cyclic
hydrocarbon, unsubstituted or substituted by lower alkyl,
which contains 3 to 10 ring carbons;
CA 02238633 2005-05-04
21489-9410
- 1b -
a pharmaceutically acceptable prodrug derivative thereof in
which the CONHOH group is derivatised in the form of an
0-aryl wherein aryl is derived from an organic carboxylic
acid, carbonic acid or carbamic acid; or a substituted or
unsubstituted 0-benzyl derivative; or a pharmaceutically
acceptable salt thereof.
The compounds of the invention depending on the nature of
the substituents, possess one or more asymmetric carbon
atoms. Also the cyclohexane substituents <~re either cis or
trans to each other. The resulting diaste:reoisomers,
enantiomers and geometric isomers are encompassed by the
instant invention.
CA 02238633 1998-OS-26
WO 97J22587 PCTlEP96/05362
-2-
Preferred are the compounds of the invention wherein the configuration of the
asymmetric
carbon atom of the a,-aminohydroxamic acid moiety to which is attached the
cyclohexane
ring corresponds to that of a D-amino acid precursor and is assigned the (R)-
configuration.
Pharmaceutically acceptable prodrug derivatives are those that may be
convertible by
solvolysis or under physiological conditions to the free hydroxamic acids of
the invention
and represent such hydroxamic acids in which the CONHOH group is derivatized
in form
of an O-acyl or an optionally substituted O-benzyl derivative. Preferred are
the optionally
substituted O-benzyl derivatives.
Prodrug acyl derivatives are preferably those derived from an organic carbonic
acid, an
organic carboxylic acid or a carbamic acid.
An acyl derivative which is derived from an organic carboxylic acid is, fox
example, lower
alkanoyl, phenyl-lower alkanoyl or unsubstituted or substituted aroyl, such as
benzoyl.
An acyl derivative which is derived from an organic carbonic acid is, for
example,
allcoxycarbonyl, especially lower alkoxycarbonyl, which is unsubstituted or
substituted by
carbocyclic or heterocycIic aryl or is cycloalkoxycarbonyl, especially
C3-C7-cycloalkyloxycarbonyl, which is unsubstituted or substituted by lower
alkyl.
An acyl derivative which is derived from a carbamic acid is, for example,
amino-carbonyl
which is substituted by lower alkyl, carbocyclic or heterocyclic aryl-lower
allcyl,
carbocyclic or heterocyclic aryl, lower alkylene or lower alkylene interrupted
by O or S.
Prodrug optionally substituted O-benzyl derivatives are preferably benzyl or
benzyl
mono-, di-, or tri-substituted by e.g. lower alkyl, lower alkoxy, amino,
nitro, halogen
and/or trifluoromethyl.
Pharmaceutically acceptable salts of the acidic compounds of the invention are
salts
formed with bases, namely cationic salts such as alkali and alkaline earth
metal salts, such
as sodium, lithium, potassium, calcium, magnesium, as well as ammonium salts,
such as
ammonium, trimethyl-ammonium, diethylammonium, and tris-(hydroxymethyl)-methyl-
ammonium salts.
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96105362
-3-
Similarly acid addition salts, such as of mineral acids, organic carboxylic
and organic
sulfonic acids e.g. hydrochloric acid, methanesuifonic acid, malefic acid, are
also possible
provided a basic group, such as pyridyl, constitutes part of the structure.
The general definitions used herein have the following meaning within the
scope of the
present invention, unless otherwise specified.
The term "lower" referred to above and hereinafter in connection with organic
radicals or
compounds respectively defines such as branched or unbranched with up to and
including
7, preferably up to and including 4, and advantageously one or two carbon
atoms.
A lower alkyl group is branched or unbranched and contains 1 to 7 carbon
atoms,
preferably 1-4 carbon atoms, and represents for example methyl, ethyl, propyl,
butyl,
isopropyl or isobutyi.
A lower alkoxy (or alkyloxy) group preferably contains 1-4 carbon atoms, and
represents
for example methoxy, ethoxy, propoxy, isopropoxy, butoxy or isobutoxy.
Halogen preferably represents chloro or fluoro but may also be bromo or iodo.
Aryl represents carbocyclic or heterocyclic aryl.
Carbocyclic aryl represents monocyclic or bicyclic aryl, for example phenyl or
phenyl
mono-, di- or tri-substituted by one, two or three radicals selected from
lower alkyl, lower
alkoxy, hydroxy, halogen, cyano, trifluoromethyl, Iower alkylenedioxy and
oxy-C2-C3-alkylene; or 1- or 2-naphthyl. Lower alkylenedioxy is a divalent
substituent
attached to two adjacent carbon atoms of phenyl, e.g. methylenedioxy or
ethylenedioxy.
Oxy-C2-C3-alkylene is also a divalent substituent attached to two adjacent
carbon atoms
of phenyl, e.g. oxyethylene or oxypropylene. An example for oxy-C2-C3-alkylene-
phenyl
is 2,3-dihydrobenzofuran-5-yl.
Preferred as carbocyclic aryl is phenyl or phenyl monosubstituted by lower
alkoxy,
halogen, lower alkyl or trifluoromethyl, especially phenyl or phenyl
monosubstituted by
lower alkoxy, halogen or trifluoromethyl, and in particular phenyl.
Heterocyclic aryl represents monocyclic or bicyclic heteroaryl, for example
pyridyl,
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
-4-
quinolinyl> isoquinolinyl, benzothienyl, benzofuranyl, benzopyranyl,
benzothiopyranyl,
furanyl, pyrrolyl> thiazolyl, oxazolyl, isoxazolyl, triazolyl, tetrazolyl>
pyrazolyl,
imidazolyl, thienyl, or any said radical substituted, especially mono- or di-
substituted, by
e.g. lower alkyl or halogen. Pyridyl represents 2-, 3- or 4-pyridyl,
advantageously 3- or
4-pyridyl. Thienyl represents 2- or 3-thienyl, advantageously 2-thienyl.
Quinolinyl
represents preferably 2-, 3- or 4-quinolinyl, advantageously 2-quinolinyl.
Isoquinolinyl
represents preferably 1-, 3- or 4-isoquinolinyl. Benzopyranyl,
benzothiopyranyl represent
preferably 3-benzopyranyl or 3-benzothiopyranyl, respectively. Thiazolyl
represents
preferably 2- or 4-thiazolyl, advantageously 4-thiazolyl. Triazolyl is
preferably 1-, 2- or
S-(1,2,4-triazolyl). Tetrazolyl is preferably 5-tetrazolyl. Imidazolyl is
preferably
4-imidazolyl.
Preferably, heterocyclic aryl is pyridyl, quinolinyl, pyrrolyl, thiazolyl,
isoxazolyl,
triazolyl, tetrazolyl, pyrazolyl, imidazolyl, thienyl, or any said radical
substituted,
especially mono- or di-substituted, by Lower alkyl or halogen; and in
particular pyridyl.
Biaryl is preferably carbocyclic biaryl, e.g. biphenyl, namely 2-, 3- or 4-
biphenyl,
advantageously 4-biphenyl, each optionally substituted by e.g. lower alkyl,
lower alkoxy,
halogen, trifluoromethyl or cyano.
G~cloalkyl represents a saturated cyclic hydrocarbon optionally substituted by
lower alkyl
which contains 3 to 1 Q ring carbons and is advantageously cyclopentyl,
cyclohexyl,
cycloheptyl or cyclooctyl optionally substituted by lower alkyl.
Carbocyclic aryl-lower alkyl represents preferably straight,chain or branched
aryl-C1-C4-alkyl in which carbocyciic aryl has meaning as defined above, e.g.
benzyl or
phenyl-(ethyl, propyl or butyl), each unsubstituted or substituted on phenyl
ring as defined
under carbocyclic aryl above, advantageously optionally substituted benzyl.
Heterocyclic aryl-lower alkyl represents preferably straight chain or branched
heterocyclic
aryl-C 1-C4-alkyl in which heterocyclic aryl has meaning as defined above,
e.g. 2-, 3- or
4-pyridylmethyl or (2-> 3- or 4-pyridyl}-(ethyl, propyl or butyl); or 2- or 3-
thienylmethyl or
(2- or 3-thienyl)-(ethyl, propyl or butyl); 2-, 3- or 4-quinolinylmethyl or (2-
, 3- or
4-quinolinyl)-(ethyl, propyl or butyl); or 2- or 4-thiazolylmethyl or (2- or
4-thiazolyl)-(ethyl, propyl or butyl).
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
-S-
Cycloalkyl-lower a.tkyl represents e.g. (cyclopentyl- or cyclohexyl)-(methyl
or ethyl).
Acyl is derived from an organic carboxylic acid, carbonic acid or carbamic
acid.
Acyl represents e.g. Iower alkanoyl, carbocyclic aryl-lower alkanoyl, Iower
alkoxycarbonyl, aroyl, di-lower alkyiaminocarbonyl or di-lower alkylamino-
lower
alkanayl. Preferably, acyl is lower alkanoyl.
Lower alkanoyl represents e.g. Ct-C~-alkanoyl including formyl, and is
preferably
C2-C4-alkanoyl such as acetyl or propionyl.
Aroyl represents e.g. benzoyl or benzoyl mono- or di-substituted by one or two
radicals
selected from lower alkyl, lower alkoxy, halogen, cyano and trifluoromethyl;
or I- or
2-naphthoyl; and also e.g. pyridylcarbonyl.
Lower alkoxycarbonyl represents preferably Cl-C4-alkoxycarbonyl, e.g.
ethoxycarbonyl.
Lower a3kylene represents either straight chain or branched alkylene of 1 to 7
carbon
atoms and represents preferably straight chain alkylene of 1 to 4 carbon
atoms, e.g. a
methylene, ethylene, propylene or butylene chain, or said methylene> ethylene,
propylene
or butylene chain mono-substituted by C1-C3-alkyl {advantageously methyl) or
disubstituted on the same or different carbon atoms by C1-C3-alkyl
(advantageously
methyl), the total number of carbon atoms being up to and including 7.
Lower alkylenedioxy is preferably ethylenedioxy or methylenedioxy.
Esterified carboxyl is for example lower alkoxycarbonyl or benzyloxycarbonyl.
Amidated carboxyl is for example aminocarbonyl, mono- or di-lower
alkylaminocarbonyl.
A particular embodiment of the invention consists of the compounds of formula
I in which
the asymmetric carbon of the a.-aminohydroxamic acid moiety is of the (R)-
configuration,
namely compounds of formula II
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
Ar
I R
I ~ ( I HZ)n ( I
F-10-N-C-CH-N S ~ (R)
H II
O R
4
R2
ORZ
wherein Ar, R1, R2, R3 and R4 have meaning as defined above, pharmaceutically
acceptable prodrug derivatives thereof and pharmaceutically acceptable salts
thereof.
A further embodiment represents the above compounds having the trans
configuration
with respect to the 1,4-substituents on the cyclohexane ring, particularly
those of formula
III
O (CH2)o O R3
II I fl
HO-N-C-CH-N S (1~)
H I
O R4
R2
r
OR1
wherein
Ar represents carbocyclic or heteroeyclic aryl;
Rl represents lower alkyl, cycloalkyl, (carbocyclic or heterocyclic aryl)-
lower alkyl or
lower alkoxy-lower alkyl;
RZ represents hydrogen or lower alkyl;
R3 is hydrogen, lower alkoxy or halogen;
R4 is hydrogen or lower alkoxy; or
R3 and Ra together on adjacent carbon atoms represent methylenedioxy; and
n is 1-4;
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
pharmaceutically acceptable prodrug derivatives thereof;
and pharmaceutically acceptable salts thereof.
Also preferred are said compounds of formula III wherein Ar represents
heterocyclic aryl
as defined above; RI represents lower alkyl, cycloalkyl as defined above or
Iower
alkoxy-lower alkyl; R2 represents hydrogen or lower alkyl; R3 and R4 are
hydrogen or
lower alkoxy; and n is I-4; pharmaceutically acceptable prodrug derivatives
thereof; and
pharmaceutically acceptable salts thereof.
Preferred are said compounds wherein R3 is at the para position and R4 is at
the meta
position.
Further preferred are the said compounds of formula III wherein Ar is
heterocyclic aryl as
defined above; RF is lower alkyl; R2 is hydrogen; R3 is para-lower alkoxy; R4
is hydrogen;
and n is 1 or 2; and pharmaceutically acceptable salts thereof.
Particularly preferred are compounds of formula III wherein Ar is pyridyl,
especially 3- or
4-pyridyl; R1 is Iower alkyl, especially straight chain C2-C~-alkyl; RZ and R4
are
hydrogen, R3 is para-lower alkoxy; and n is 1; and pharmaceutically acceptable
salts
thereof.
Further preferred are said compounds wherein Ar is 4-pyridyl; Ri is C2-
C4alkyl; R2 and
R4 are hydrogen; R3 is para-ethoxy; and n is 1; and pharmaceutically
acceptable salts
thereof.
Special mention should be made of the following sub-group of a group of
compounds of
the invention: compounds (of formula I, II or III respectively) wherein R1 is
C2-C~alkyl.
The invention relates especially to the specific compounds described in the
examples,
pharmaceutically acceptable prodrug derivatives thereof and pharmaceutically
acceptable
salts thereof, and in particular to the specific compounds described in the
examples and
pharmaceutically acceptable salts thereof.
The compounds of the invention exhibit valuable pharmacological properties in
mammals
including man.
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
_g_
Firstly, they are inhibitors of TNF-alpha converting enzyme {TNF-alpha
convertase) and
thus inhibit TNF-alpha activity, e.g. suppress the production andlor release
of TNF alpha,
an important mediator of inflammation and tissue growth. Such properties
render the
compounds of the invention primarily useful for the treatment of tumors
(malignant and
non-malignant neoplasms) as well as of inflammatory conditions in mammals,
e.g. for the
treatment of arthritis (such as rheumatoid arthritis), septic shock,
inflammatory bowel
disease, Crohn's disease and the like.
Further, the compounds of the invention also inhibit matrix degrading
metalloproteinase
enzymes such as gelatinase, stromelysin, collagenase, and macrophage
metalloelastase.
Thus the compounds of the invention inhibit matrix degradation and are also
useful for the
treatment of gelatinase-, stromelysin-, collagenase- and macrophage
metalloeiastase-dependent pathological conditions in mammals. Such conditions
include
tumors {by inhibiting tumor growth, tumor metastasis, tumor progression or
invasion
and/or tumor angiogenesis), such tumors being e.g. breast, lung, bladder,
colon, ovarian
and skin cancer. Other conditions to be treated with the compounds of the
invention
include osteoarthritis, bronchial disorders (such as asthma by inhibiting the
degradation of
elastin), atherosclerotic conditions (by e.g. inhibiting rupture of
atherosclerotic plaques),
as well as acute coronary syndrome, heart attacks (cardiac ischemia), strokes
(cerebral
ischemias), and restenosis after angioplasty.
Further conditions to be treated with the compounds of the invention are
inflammatory
demyelinating disorders of the nervous system in which myelin destruction or
loss is
involved {such as multiple sclerosis), optic neuritis, neuromyelitis optica
(Devic's
disease), diffuse and transitional sclerosis (Schilder's disease) and acute
disseminated
encephalomyelitis, also demyelinating peripheral neuropathies such as
Landry-Guillain-Barre-Strohl syndrome for motor defects; also tissue
ulceration (e.g.
epidermal and gastric ulceration), abnormal wound healing, periodontal
disease, bone
disease (e.g. Paget's disease and osteoporosis).
Ocular applications of the compounds of the invention include the treatment of
ocular
inflammation, corneal ulcerations, pterygium, keratitis, keratoconus, open
angle
glaucoma, retinopathies, and also their use in conjunction with refractive
surgery (laser or
incisional) to minimize adverse effects.
The compounds are particularly useful for the treatment of intZammatory
conditions, such
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
-9-
as rheumatoid arthritis, and of tumors.
Beneficial effects are evaluated in pharmacological tests generally known in
the art, and as
illustrated herein.
The above-cited properties are demonstrable in in vitro and in vivo tests,
using
advantageously mammals, e.g. rats, guinea pigs, dogs, rabbits, or isolated
organs and
tissues, as well as mammalian enzyme preparations. Said compounds can be
applied in
vitro in the form of solutions, e.g. preferably aqueous solutions, and in vivo
either
enterally or parenterally, advantageously orally, e.g. as a suspension or in
aqueous
solution. The dosage in vitro may range between about 10-5 molar and 10'1
molar
concentrations. The dosage in vivo may range, depending on the route of
administration,
between about 0.1 and 100 mg/kg.
The inhibition of the production and secretion of TNF-alpha (by inhibition of
TNF-oc
convertase) can be determined e.g. as described in Nature 370> 555, 558
(1994).
The effect on the production of soluble TNF-alpha by LPS-stimulated THP-1
cells can be
determined as follows:
Tissue culture medium used is RPM 1640 (Gibco cat #11875-036) containing 10%
fetal
calf serum, I% penicillin and streptomycin. THP-I cells (ATCC #202-TIB) at 1 x
10+5
cells/well are added to 100 ~.l medium or test compound. Cells are pre-
incubated with
compound for 30 minutes in a 37°C humidified chamber with 5% C02 and
then stimulated
with i00 ng/ml of LPS (Sigma cat #L-4391) for 4 hours. Plates are then
centrigued and
100 p.1 of conditioned medium for TNF analysis is harvested. The amount of TNF-
alpha
in control and test cultures is determined by ELISA using recombinant TNF-
alpha for the
standard curve, using TNF ELISA plates (Genzyme) for TNF analysis. Absorbance
readings and data calculations are performed on a Molecular Devices plate
reader. Results
are expressed in ICSO's of test compound.
The effect on the plasma concentration of TNF-alpha in the mouse following
intravenous
injection of endotoxin can be determined as follows:
Female Balb-CbyJ mice are dosed by gavage with test compound in 0.1 mI
cornstarch
vehicle/ 10 grams body weight. One to four hours after administration of test
compound,
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96105362
-10-
0.1 mg/kg Lipopolysaccharide from E. coli 0127:B8 (Difco #3880-25-0) in saline
is
injected i.v. One hour after i.v. injection of LPS, blood is collected for
determination of
plasma TNF-alpha using mouse TNF-alpha ELISA kit (Genzyme). Eight mice are
used
per treatment group. Results are expressed as % inhibition of mean TNF-alpha
concentration in control mice.
The effect on the synovial fluid concentration of TNF-alpha in an inflamed rat
knee can be
determined as follows:
Female Lewis rats are dosed by gavage with test compound in 0.1 ml cornstarch
vehicle.
Oiie to four hours after administration of test compound 0.1 mg
Lipopolysaccharide from
E. coli 0127:B8 (Difco #3880-25-0) is injected into both knees. Two hours
after
infra-articular LPS injection, knees are lavaged with 0.1 ml saline and 2
lavages from
same rat are pooled. TNF-alpha is measured using mouse TNF-alpha ELISA kit
(Genzyme) which crossreacts with rat TNF-alpha. Results are expressed as %
inhibition
of mean TNF-alpha concentration in synovial fluid from saline-injected knees.
Antiinflammatory activity can be determined in standard inflammation and
arthritic
animal models well-known in the art, e.g. the adjuvant arthritis model in rats
and the
collagen II induced arthritis model in mice [Mediators of Inflam. 1, 273-279
(1992)].
One test to determine the inhibition of stromelysin activity is based on its
hydrolysis of
Substance P using a modified procedure of Harrison et aI (Harnson, R.A.,
Teahan J., and
Stein R., A semicontinuous, high performance chromatography based assay for
stromelysin, Anal. Biochem. 180, 110-113 (1989)). In this assay, Substance P
is
hydrolyzed by recombinant human stromelysin to generate a fragment, Substance
P 7-11,
which can be quandtated by HPLC. In a typical assay, a 10 mM stock solution of
a
compound to be tested is diluted in the assay buffer to 50 p.M, mixed 1:l with
8 p.g
recombinant human stromelysin (mol. wt. 45-47 kDa, 2 Units; where I Unit
produces 20
rrimoles of Substance P 7-I I in 30 minutes) and incubated along with 0.5mM
Substance P
in a final volume of 0.125 ml for 30 minutes at 37°C. The reaction is
stopped by adding
mM EDTA and Substance P 7-1 I is quantified on RP-8 HPLC. The ICSO for
inhibition
of stromelysin activity and Ki are calculated from control reaction without
the inhibitor.
Stromelysin activity can also be determined using human aggrecan as a
substrate. This
assay allows the confirmation in-vitro that a compound can inhibit the action
of
CA 02238633 1998-OS-26
WO 97/22587 PCTlEP96/05362
-11-
stromelysin on its highly negatively-charged natural substrate, aggrecan
(large aggregating
prtoeoglycan}. Within the cartilage, proteoglycan exists as an aggregate bound
to
hyaluronate. Human proteoglycan aggregated to hyaluronate is used as an enzyme
substrate. The assay is set up in 96-well microtiter plates allowing rapid
evaluation of
compounds. The assay has three major steps:
1 ) Plates are coated with hyaluronate (human umbilical chord, 400 ug/ml),
blocked with
BSA (5 mg/ml), and then proteoglycan (human articular cartilage D1 -
chondroitinase
ABC digested, 2 mg/ml} is bound to the hyaluronate. Plates are washed between
each
step.
2} Buffers + inhibitor {1 to 5,000 nM) + recombinant human stromelysin (1-3
Units/well}
are added to wells. The plates are sealed with tape and incubated overnight at
37°C. The
plates are then washed.
3) A primary (3B3} antibody (mouse IgM, 1:10,000) is used to detect remaining
fragments. A secondary antibody, peroxididase-linked anti-IgM, is bound to the
primary
antibody. OPD is then added as a substrate for the peroxidase and the reaction
is stopped
with sulfuric acid. The ICSO for inhibition of stromelysin activity is
graphically derived
and Ki is calculated.
Collagenase activity is determined as follows: ninety six-well, flat-bottom
microtiter
plates are first coated with bovine type I collagen (35 ug/well) over a two-
day period at
30°C using a humidified and then dry atmosphere; plates are rinsed, air
dried fox 3-4
hours, sealed with Saran wrap and stored in a refrigerator. Human recombinant
fibroblast
collagenase and a test compound (or buffer) are added to wells (total volume =
0. I ml) and
plates are incubated for 2 hours at 35°C under humidified conditions;
the amount of
collagenase used per well is that causing approximately 80% of maximal
digestion of
collagen. The incubation media are removed from the wells, which are then
rinsed with
buffer, followed by water. Coomasie blue stain is added to the wells for 25
minutes,
removed, and wells are again rinsed with water. Sodium dodecyl sulfate (20% in
50°l0
dimethylformamide in water) is added to solubilize the remaining stained
collagen and the
optical density at 570 nM wave length is measured. The decrease in optical
density due to
collagenase (from that of collagen without enzyme} is compared to the decrease
in optical
density due to the enzyme in the presence of test compound, and percent
inhibition of
enzyme activity is calculated. ICso's are determined from a range of
concentrations of
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
- i2 -
inhibitors (4-5 concentrations, each tested in triplicate), and Ki values are
calculated.
The effect of compounds of the invention in-vivo can be determined in rabbits.
Typically,
four rabbits are dosed orally with a compound up to four hours before being
injected
infra-articularly in both knees (N=8) with 40 Units of recombinant human
stromelysin
dissolved in 20 mM Tris, 10 mM CaCl2, and 0. IS M NaCI at pH 7.5. Two hours
later the
rabbits are sacrificed, synoviai lavage is collected, and keratan sulfate (KS)
and sulfated
glycosaminoglycan (S-GAG) fragments released into the joint are quantitated.
Keratan
sulfate is measured by an inhibition ELISA using the method of Thonar (Thonar,
E.J.-M.A., Lenz, M.E., Klinsworth, G.K., Caterson, B., Pachman, L.M.,
Glickman, P.,
Katz, R., Huff, J., Keuttner, K.E. Quantitation of keratan sulfate in blood as
a marker of
cartilage catabolism, Arthr. Rheum. 28, 1367-1376 (1985)). Sulfated
glycosaminoglycans
are measured by first digesting the synovial lavage with streptomyces
hyaluronidase and
then measuring DMB dye binding using the method of Goldberg (Goldberg, R.L.
and
Kolibas, L. An improved method for determining proteoglycan synthesized by
chondrocytes in culture. Connect. Tiss. Res. 24" 265-275 (1990)). For an i.v.
study, a
compound is solubilized in I ml of PEG-400, and for a p.o. study, a compound
is
administered in 5 ml of fortified corn starch per kilogram of body weight.
The effect in protecting against cartilage degradation in arthritic disorders
can be
determined e.g. in a surgical model of osteoarthritis described in Arthritis
and
Rheumatism, Vol. 26, 875-886 (I983).
The effect on ulcerations, e.g. ocular ulcerations, can be determined in the
rabbit by
measuring the reduction of corneal ulceration following an alkali burn to the
cornea.
Macrophage metalloelastase (MME) inhibitory activity can be determined by
measuring
the inhibition of the degradation of [3H]-elastin by truncated recombinant
mouse
macrophage metalloelastase as follows:
About 2 ng of recombinant truncated mouse macrophage metalloeiastase (FASEB
Journal
Vol. 8, A151, 1994), purified by Q-Sepharose column chromatography is
incubated with
test compounds at the desired concentrations in the presence of 5 nM CaCl2,
400 nM
NaCI, [3H]elastin (60,000 cpm/tube), and 20 mM Tris, pH 8.0, at 37°C
overnight. The
samples are spun in a microfuge centrifuge at 12,000 rpm for 15 minutes. An
aliquot of
the supernatant is counted in a scintillation counter to quantitate degraded
[3H]elastin.
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
-I3-
IC5Q's are determined from a range of concentrations of the test compounds and
the
percent inhibition of enzyme activity obtained.
The effect of the compounds of the invention for the treatment of emphysema
can be
determined in animal models described in American Review of Respiratory
Disease 117,
1109 (1978).
The antitumor effect of the compounds of the invention can be determined e.g.
by
measuring the growth of human tumors implanted subcutaneously in Balb/c nude
treated
mice according to methodology well-known in the art in comparison to placebo
treated
mice. Illustrative tumors are e.g. estrogen dependent human breast carcinoma
BT20 and
MCF7, human bladder carcinoma T24, human colon carcinoma Colo 205, human lung
adenocarcinoma A549 and human ovarian carcinoma NIH-OVCAR3.
The effect on tumor angiogenesis can be determined e.g. in rats implanted with
Walker
256 carcinoma in pellets to stimulate angiogenesis from vessels of the limbus,
as
described by Galardy et al, Cancer Res. 54, 4715 (1994).
The effect of the compounds of the invention on atherosclerotic conditions can
be
evaluated using atherosclerotic plaques from cholesterol-fed rabbits which
contain
activated matrix metalloproteinases as described by Sukhova et al, Circulation
90, i 404
{ 1994). The inhibitory effect on matrix metalloproteinase enzyme activity in
rabbit
atherosclerotic plaques can be determined by in situ zymography, as described
by Galis et
al, J. Clin. Invest. 94, 2493 (1994), and is indicative of plaque
stabilization.
The effect on restenosis and vascular remodeling can be evaluated in the rat
ballooned
carotid artery model.
The effect on demyelinating disorders of the nervious system, such as multiple
sclerosis,
can be evaluated by measuring the reversal of experimental antioimmune
encephalo-
myelitis in mice, e.g. as described by Gijbels et al, J. Clin. Invest. 94,
2177 (1994).
As inhibitors of TNF-alpha convertase and matrix metalloproteinases the
compounds of
the invention are particularly useful in mammals as antiinflammatory agents
for the
treatment of e.g. osteoarthritis, rheumatoid arthritis, and as antitumor
agents for the
treatment and prevention of tumors growths, tumor metastasis, tumor invasion
or
CA 02238633 1998-OS-26
WO 97122587 PCT/EP96/05362
-14-
progession.
The compounds of formula I can be prepared e.g. by condensing a carboxylic
acid of
formula IV
I R
O (CH~n O 3
1I I II
HO-C-CH-N S '
I!
O R
4
R2
OR 1
or a reactive functional derivative thereof, wherein Ar, n and Rl-R4 having
meaning as
defined hereinabove, with hydroxylamine of formula V,
NH2-OH (V)
optionally in protected form, or a salt thereof;
and, if necessary, temporarily protecting any interfering reactive group(s),
and then
liberating the resulting compound of the invention; and, if required or
desired, converting
a resulting compound of the invention into another compound of the invention,
and/or, if
desired, converting a resulting free compound into a salt or a resulting salt
into a free
compound or into another salt; and/or separating a mixture of isomers or
racemates
obtained into the single isomers or racemates; and/or, if desired, resolving a
racemate into
the optical antipodes.
In starting compounds and intermediates which are converted to the compounds
of the
invention in a manner described herein, functional groups present, such as
amino,
carboxyl and hydroxy groups, are optionally protected by conventional
protecting groups
that are common in preparative organic chemistry. Protected amino, carboxyl
and
hydroxy groups are those that can be converted under mild conditions into free
amino and
hydroxy groups without the molecular framework being destroyed or other
undesired side
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/053b2
-15-
reactions taking place.
The purpose of introducing protecting groups is to protect the functional
groups from
undesired reactions with reaction components under the conditions used for
carrying out a
desired chemical transformation. The need and choice of protecting groups for
a
particular reaction is known to those skilled in the art and depends on the
nature of the
functional group to be protected {hydroxy group, amino group, etc.), the
structure and
stability of the molecule of which the substituent is a part and the reaction
conditions.
Well-known protecting groups that meet these conditions and their introduction
and
removal are described, for example, in J.F.W. McOmie, "Protective Groups in
Organic
Chemistry", Plenum Press, London, New York, 1973, T. W. Greene, "Protective
Groups in
Organic Synthesis", Wiley, New York, 1991.
In the processes cited herein, reactive functional derivatives of carboxylic
acids represent,
for example, anhydrides especially mixed anhydrides, acid halides, acid
azides, lower
alkyl esters and activated esters thereof. Mixed anhydrides are preferably
such from
pivalic acid, or a Iower alkyl {ethyl, isobutyl) hemiester of carbonic acid;
acid halides are
for example chlorides or bromides; activated esters for example succinimido,
phthalimido
or 4-nitrophenyl esters; lower alkyl esters are for example the methyl or
ethyl esters.
Also, a reactive esterified derivative of an alcohol in any of the reactions
cited herein
represents said alcohol esterified by a strong acid, especially a strong
inorganic acid, such
as a hydrohalic acid, especially hydrochloric, hydrobromic or hydroiodic acid,
or sulphuric
acid, or by a strong organic acid, especially a strong organic sulfonic acid,
such as an
aliphatic or aromatic sulfonic acid, for example methanesulfonic acid, 4-
methylbenzene-
sulfonic acid or 4-bromobenzenesulfonic acid. A said reactive esterified
derivative is
especially halogen, for example chloro, bromo or iodo, or aliphatically or
aromatically
substituted sulfonyloxy, for example methanesulfonyloxy, 4-
methylbenzenesulfonyloxy
(tosyloxy) or trifluoromethanesulfonyloxy.
The above process for the synthesis of compounds of the invention can be
carried out
according to methodology generally known in the art for the preparation of
hydroxamic
acids and derivatives thereof.
The synthesis according to the above process (involving the condensation of a
free
CA 02238633 1998-OS-26
WO 97/22587 PC'TJEP96/05362
-16-
carboxylic acid of formula IV with an optionally hydroxy protected
hydroxylamine
derivative of formula V can be carried out in the presence of a condensing
agent, e.g.
l, I'-carbonyldiimidazole, or N-(dimethylaminopropyl}-N'-ethylcarbodiimide or
dicyclohexyicarbodiimide, with or without I-hydroxybenzotriazole in an inert
polar
solvent, such as dimethylformamide or dichloromethane, preferably at room
temperature.
The synthesis involving the condensation of a reactive functional derivative
of an acid of
formula IV as defined above, e.g. an acid chloride or mixed anhydride with
optionally
hydroxy protected hydroxylamine, or a salt thereof, in presence of a base such
as
triethylamine can be carried out, at a temperature ranging preferably from
about -7$°C to
+75°C, in an inert organic solvent such as dichloromethane or toluene.
Protected forms of hydroxylamine (of formula V) in the above process are those
wherein
the hydroxy group is protected for example as a t-butyl ether, a benzyl ether,
a
triphenylmethyl ether, a tetrahydropyranyl ether, or as a trimethylsilyl
derivative.
Removal of said protecting groups is carried out according to methods well
known in the
art, e.g. hydrogenolysis or acid hydrolysis. Hydroxylamine is preferably
generated in situ
from a hydroxylamine salt, such as hydroxylamine hydrochloride.
The starting carboxylic acids of formula IV can be prepared as follows:
An amino acid of formula VI
0
HO- C - CH- NH2
(VI)
~2
OH
wherein Ra is hydrogen or lower alkyl, which is optionally este~fied e.g. with
a lower
alkanol {such as methanol) or with benzyl alcohol, is treated with a reactive
functional
derivative of the appropriate sulfonic acid of the formula VII
CA 02238633 1998-OS-26
WO 97122587 PCT/EP96/05362
-17-
R3
S03H (VII)
R4
wherein R3 and R4 have meaning as defined hereinabove, e.g. with the
corresponding
sulfonyl chloride, in the presence of a suitable base, such as triethylamine
or
dicyclohexylamine, using a polar solvent such as tetrahydrofuran, dioxane or'
acetonitrile
to obtain a compound of the formula VIIC
O O R3
RS- O - C - CH- NH- S (VIII)
O R4
R2
OH
wherein R2-R4 have meaning as defined above and RS is hydrogen or a carboxyl
protecting group, e.g. lower alkyl or benzyl.
The starting materials of formula VI, VII and XII are either known in the art,
or can be
prepared by methods well-known in the art or as described herein.
Optically active D-aminoacids of formula VI (the R-enantiomers) can be
prepared
according to methods known in the art, e.g. according to methods described in
Coll.
Czech. Comm. 49, 712-742 (1984) and Angew. Chem. int. Ed. (Engl.) 27, 1194
{1988).
The intermediates of formula VIII can be converted to the intermediates of
formula IX
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
_I8_
O O R3
i~ !!
RS-O-C-CH-NH-s
O R4
R2
OR 1
wherein RI-RS having meaning as defined above, by treatment with a reactive
esterified
derivative of the alcohol of the formula
RI-OH (X)
wherein Ri has meaning as defined in formula I, under conditions well known in
the art
for ether formation.
Alternatively, the ether intermediates of formula IX can be prepared by
reduction of
ketone compounds of formula XI
O O R3
Ii !!
RS- O - C - CH-NH- S (XI)
O R4
R2
O
wherein R2-RS have meaning as defined in formula VIII, in the presence of an
alcohol of
formula X (RI-OH). The reductive O-alkylation can be carried out essentially
as
described in J. Am. Chem. Soc. 94, 3659 (I972), using mono-, dl- or
trialkylsilanes or
mono-, dl- or triarylsilanes in acidic medium, e.g. in the presence of
trifluoroacetic acid.
The resulting cis and trans isomers can be separated by known methods, such as
chromatography on silica gel.
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
-19-
Alternatively, the ketone intermediates of formula XI wherein R2 is hydrogen
can be
converted to the tertiary alcohol intermediates of formula VIII wherein R2 is
lower alkyl
(and R2 and OR1' are located on the same carbon atom) according to
conventional
methods, and such are subsequently etherified with a reactive esterified
derivative of
Ri-OH, such as the trifluoromethanesulfonyl derivative.
The ketones of formula XI can in turn be prepared by oxidation of alcohols of
formula
VIII by treatment with e.g. sodium hypochlorite in the presence of a free
radical, e.g.
TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy free radical).
Treatment of an intermediate of formula IX with a reactive esterified
derivative (such as
the halide, e.g. the chloride, bromide or iodide derivative) of the alcohol of
the formula
XII
Ar-(CH2)nOH (XII)
wherein Ar and n have meaning as defined herein, in the presence of an
appropriate base,
such as potassium carbonate or dicyclohexylamine, in a polar solvent, such as
dimethylformamide yields an ester of a compound of formula IV. The ester can
then be
converted to the acid of formula IV, using either hydrogenolysis or standard
mild methods
of ester hydrolysis, preferably under acidic conditions, the method depending
on the
nature of the esterifying group.
The above-mentioned reactions are carried out according to standard methods,
in the
presence or absence of diluent, preferably such as are inert to the reagents
and are solvents
thereof, of catalysts, condensing or said other agents respectively and/or
inert atmo-
spheres, at low temperatures, room temperature or elevated temperatures
(preferably at or
near the boiling point of the solvents used), and at atmospheric or super-
atmospheric
pressure. The preferred solvents, catalysts and reaction conditions are set
forth in the
appended illustrative examples.
The invention further includes any variant of the present processes, in which
an inter-
mediate product obtainable at any stage thereof is used as starting material
and the
remaining steps are carried out, or the process is discontinued at any stage
thereof, or in
which the starting materials are formed in situ under the reaction conditions,
or in which
the reaction components are used in the form of their salts or optically pure
antipodes.
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
-20-
Compounds of the invention and intermediates can also be converted into each
other
according to methods generally known per se.
The invention also relates to any novel starting materials and processes for
their
manufacture.
Depending on the choice of starting materials and methods, the new compounds
may be in
the form of one of the possible isomers or mixtures thereof, for example, as
substantially
pure geometric (cis or traps) isomers, optical isomers (antipodes), racemates,
or mixtures
thereof. The aforesaid possible isomers or mixtures thereof are within the
purview of this
invention.
Any resulting mixtures of isomers can be separated on the basis of the physico-
chemical
differences of the constituents, into the pure geometric or optical isomers,
diastereoisomers, racemates, for example by chromatography anctlor fractional
crystallization.
Any resulting racemates of final products or intermediates can be resolved
into the optical
antipodes by known methods, e.g. by separation of the diastereoisomeric salts
thereof,
obtained with an optically active acid or base, and liberating the optically
active acidic or
basic compound. The hydroxamic acids or carboxylic acid intermediates can thus
be
resolved into their optical antipodes e.g. by fractional crystallization of d-
or 1-(alpha-
methylbenzylamine, cinchonidine, cinchonine, quinine, quinidine, ephedrine,
dehydro-
abietylamine, brucine or strychnine)-salts.
Finally, acidic compounds of the invention are either obtained in the free
form, or as a salt
thereof.
Acidic compounds of the invention may be converted into salts with
pharmaceutically
acceptable bases, e.g. an aqueous alkali metal hydroxide, advantageously in
the presence
of an ethereal or alcoholic solvent, such as a lower alkanol. From the
solutions of the
latter, the salts may be precipitated with ethers, e.g. diethyl ether.
Resulting salts may be
converted into the free compounds by treatment with acids. These or other
salts can also
be used for purification of the compounds obtained.
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
-21 -
Compounds of the invention having basic groups can be converted into acid
addition salts,
especially pharmaceutically acceptable salts. These are formed, for example,
with
inorganic acids, such as mineral acids, for example sulfuric acid, a
phosphoric or
hydrohalic acid, or with organic carboxylic acids, such as (C1-C4)-
aikanecarboxylic acids
which, for example, are unsubstituted or substituted by halogen, for example
acetic acid,
such as saturated or unsaturated dicarboxylic acids, for example oxalic,
succinic, malefic or
fumaric acid, such as hydroxycarboxylic acids, for example glycolic, lactic,
malic, tartaric
or citric acid, such as amino acids, for example aspartic or glutamic acid, or
with organic
sulfonic acids, such as (C t-C4)-alkane- or arylsulfonic acids which are
unsubstituted or
substituted, for example, by halogen, for example methanesulfonic acid.
Preferred are
salts formed with hydrochloric acid, methanesulfonic acid and malefic acid.
In view of the close relationship between the free compounds and the compounds
in the
form of their salts, whenever a compound is referred to in this context, a
corresponding
salt is also intended, provided such is possible or appropriate under the
circumstances.
The compounds, including their salts, can also be obtained in the form of
their hydrates, or
include other solvents used for their crystallization.
The pharmaceutical compositions according to the invention are those suitable
for enteral,
such as oral or rectal, transdermal and parenteral administration to mammals,
including
man, to inhibit TNF-alpha converting enzyme and matrix-degrading
metalloproteinases,
and for the treatment of disorders responsive thereto, comprising an effective
amount of a
pharmacologically active compound of the invention, alone or in combination,
with one or
more pharmaceutically acceptable carriers.
The pharmacologically active compounds of the invention are useful in the
manufacture of
pharmaceutical compositions comprising an effective amount thereof in
conjunction or
admixture with excipients or carriers suitable for either enteral or
parenteral application.
Preferred are tablets and gelatin capsules comprising the active ingredient
together with a)
diluents, e.g. lactose, dextrose, sucrose, mannitol, sorbitol, cellulose
and/or glycine; b)
lubricants, e.g. silica, talcum, stearic acid, its magnesium or calcium salt
and/or
polyethyleneglycol; for tablets also c) binders e.g. magnesium aluminum
silicate, starch
paste, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose and
or
polyvinylpyrrolidone; if desired d) disintegrants, e.g. starches, agar,
alginic acid or its
sodium salt, or effervescent mixtures; and/or e) absorbants, colorants,
flavors and
CA 02238633 1998-OS-26
WO 97122587 PCT/EP96/05362
-22-
sweeteners. Inlectable compositions are preferably aqueous isotonic solutions
or
suspensions, and suppositories are advantageously prepared from fatty
emulsions or
suspensions. Said compositions may be sterilized and/or contain adjuvants,
such as
preserving, stabilizing, wetting or emulsifying agents, solution promoters,
salts for
regulating the osmotic pressure and/or buffers. In addition, they may also
contain other
therapeutically valuable substances. Said compositions are prepared according
to
conventional mixing, granulating or coating methods, respectively, and contain
about
0.1 to 75 %, preferably about 1 to 5Q °k, of the active ingredient.
Suitable formulations for transdermal application include an effective amount
of a
compound of the invention with carrier. Advantageous carriers include
absorbable
pharmacologically acceptable solvents to assist passage through the skin of
the host.
Characteristically, transdermal devices are in the form of a bandage
comprising a backing
member, a reservoir containing the compound optionally with carriers,
optionally a rate
controlling barrier to deliver the compound of the skin of the host at a
controlled and
predetermined rate over a prolonged period of time, and means to secure the
device to the
skin.
Suitable formulations for topical application, e.g. to the skin and eyes, are
preferably
aqueous solutions, ointments, creams or gels well-known in the art.
The pharmaceutical formulations contain an effective TNF-alpha convertase
inhibiting
amount andlor matrix-degrading metalloproteinase inhibiting amount of a
compound of
the invention as defined above, either alone or in combination with another
therapeutic
agent, e.g. an anti-inflammatory agent with cyclooxygenase inhibiting
activity, or other
antirheumatic agents such as methotrexate, each at an effective therapeutic
dose as
reported in the art. Such therapeutic agents are well-known in the art.
Examples of antiinflammatory agents with cyclooxygenase inhibiting activity
are
diclofenac, naproxen, ibuprofen, and the like.
In conjunction with another active ingredient, a compound of the invention may
be
administered either simultaneously, before or after the other active
ingredient, either
separately by the same or different route of administration or together in the
same
pharmaceutical formulation.
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
-23-
The dosage of active compound administered is dependent on the species of warm-
blooded animal (mammal), the body weight, age and individual condition, and on
the form
of administration. A unit dosage for oral administration to a mammal of about
50 to 70 kg
may contain between about IO and 1000 mg, advantageously between about 25 and
250 mg of the active ingredient.
The present invention also relates to methods of using the compounds of the
invention and
their pharmaceutically acceptable salts, or pharmaceutical compositions
thereof, in
mammals for inhibiting TNF-alpha activity and inhibiting the matrix-degrading
metalloproteinases, e.g. stromelysin, gelatinase, collagenase and macrophage
metalloelastase, for inhibiting tissue matrix degradation, and for the
treatment of
TNF-alpha and matrix-degrading metalloproteinase dependent conditions as
described
herein, e.g. inflammation, rheumatoid arthritis, osteoarthritis, also tumors
(tumor growth,
metastasis, progression or invasion), pulmonary disorders, and the like
described herein.
Tumors {carcinomas) include mammalian breast, lung, bladder, colon, prostate
and
ovarian cancer, and skin cancer, including melanoma and Kaposi's sarcoma.
The following examples are intended to illustrate the invention and are not to
be construed
as being limitations thereon. Temperatures are given in degrees Centrigrade.
If not
mentioned otherwise, all evaporations are performed under reduced pressure,
preferably
between about 15 and 100 mm Hg (= 20-133 mbar}. The structure of final
products,
intermediates and starting materials is confirmed by standard analytical
methods, e.g.
microanalysis and spectroscopic characteristics (e.g. MS, IR, NMR).
Abbreviations used
are those conventional in the art. The concentration for [oc]D determinations
is expressed
in mg/ml.
Example 1: N-(t-Butyloxy)-2(R)-[(4-methoxybenzenesulfonyl}(4-picolyl)amino]-
-~-(trans-4-propoxycyclohexyl) acetamide (0.84 g, 1.5 mmol) is dissolved in
dichloroethane {50 mL} containing ethanol (0.1 mL, 1.5 mmol} in a round bottom
flask,
and the reaction is cooled to -i0°C. Hydrochloric acid gas (from a
lecture bottle} is
bubbled through for 10 minutes. The reaction is sealed, allowed to slowly warm
to room
temperature and stirred for 4 days. The solvent is reduced to 1/3 volume by
evaporation
and triturated with ether. The mixture is filtered, filter cake removed, and
dried in vacuo
to provide N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-
2-(trans-4-propoxycyclohexyl)-acetamide hydrochloride as a white solid, m.p.
135-140°C,
of the formula
CA 02238633 1998-05-26
WO 97/22587 PCT/EP96/05362
-24-
~N ~ HCl
l
0
HO\ ~N'
N~ ~ S ~ ~ OCHg
0/ ~~
OCH2CH2CH3
The starting material is prepared as follows:
D-4-hydroxyphenylglycine {10 g) is dissolved in 3N sodium hydroxide (20 ml).
Water
(180 ml} and then Raney nickel (27 g) are added. The reaction mixture is
hydrogenated at
about 3 atmospheric pressure and 50-80°C overnight. The reaction
mixture is filtered
through Celite and reduced in volume to about 85 ml and dioxane (85 ml) is
added. The
solution of 4-hydroxycyclohexylglycine (see Coil. Czech. Chem. Comm. 49, 712-
742
( 1984)) is cooled to 0°C and treated with triethylamine ( 11.37 mI)
and
4-methoxybenzenesulfonyl chloride (10.95 g). The reaction mixture is allowed
to warm to
room temperature and stirred over the weekend. The dioxane is removed in vacuo
and the
remaining aqueous solution is diluted with 1N hydrochloride acid. The
resulting
precipitate is collected, washed with water and ether to yield (R)-N-(4-
methoxybenzene-
stilfonyl)-4-hydroxycyclohexylglycine.
A mixture of crude (R)-N-(4-methoxybenzenesulfonyl)-4-hydroxycyclohexylglycine
(7.0
g, 20.4 mmol) in dimethylformamide (100 mL) containing N,N-dicyclohexylamine
(3.7 g,
20.4 mmol) and benzyl bromide (3.5 g, 20.4 mmol) is stirred at room
temperature for 24
hours. The mixture is diluted with water and extracted with ethyl acetate. The
combined
organic extracts are washed with brine, dired (Na2S04), filtered, and
concentrated in
vacuo to yield (R)-N-(4-methoxybenzenesulfonyl)-4-hydroxycyclohexylglycine
benzyl
ester as a mixture of diastereomers.
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
-25-
To a solution of crude (R)-N-(4-methoxybenzenesulfonyl)-4-
hydroxycyclohexylglycine
benzyl ester (8.67 g, 20 mmol) in dichloromethane (66 mL) at 0°C is
added a solution of
sodium bromide (2.06 g, 20 mmol) in water (10 mL) dropwise followed by
addition of
2,2,6,6-tetramethyl-1-piperidinyloxy free radical (TEMPO, 27 mg). To this
mixture is
added dropwise an aqueous solution of 5%o sodium hypochlorite (34.2 mL, 34.3
mmol,
Clorox brand) and water (34.2 mL) in which the pH is adjusted to 8.6 with
sodium
bicarbonate before addition. Addition time of the resulting pH adjusted
aqueous sodium
hypochlorite solution is 30 minutes and stirring is continued for another 20
minutes while
maintaining a reaction temperate of 0°C. The dichloromethane layer is
separated and
successively washed with 10% aqueous potassium hydrogen sulfate (40 mL), a
small
amount of 10%o aqueous potassium iodide (3x30 mL), 10% aqueous sodium
thiosulfate (60
mL), and brine (40 mL). The organic layer is dried (MgS04), filtered, and
concentrated in
vacuo to provide solid which could be further purified by recrystallization
from ethyl
acetate to furnish (R)-N-(4-methoxybenzenesulfonyl)-4-oxocyclohexylgiycine
benzyl
ester.
To a mixture of {R)-N-(4-methoxybenzenesulfonyl)-4-oxocyclohexylglycine benzyl
ester
(15 g, 34.6 mmol) in n-propanol (7 mL, 93.2 mmol) containing phenylsilane {5.2
mL, 43.3
mmol) is added dropwise trifluoroacetic acid and the mixture is stirred at
room
temperature overnight. The mixture is diluted with ethyl acetate and washed
with
saturated aqueous sodium bicarbonate. The organic layer is dried (MgS04),
filtered, and
concentrated in vacuo. The crude product is purified by silica gel
chromatography ( l % to
5°1o ethyl acetate/methylene chloride) to provide (R)-N-(4-
methoxybenzenesulfonyl)-
cis-4-propoxycyclohexylglycine benzyl ester and (R)-N-(4-
methoxybenzensulfonyl)-
traps-4-propoxycyclohexylglycine benzyl ester.
To a solution of (R)-N-(4-methoxybenzenesulfonyl)-traps-4-
propoxycyclohexylglycine
benzyl ester (4.0 g, 8.42 mmol) in dimethylformamide (55 mL) is added 4.-
picolyl chloride
hydrochloride ( 1.5 g, 8.95 mmol) followed by potassium carbonate ( I 1.6 g,
84.2 mmol).
The reaction mixture is stirred at room temperature overnight. The mixture is
then diluted
with water and extracted with ethyl acetate. The combined organic extracts are
washed
with brine, dried (Na2S04) and the solvent is evaporated to give benzyl
2{R)-[(4-methoxybenzenesulfonyl){4-picolyl)amino]-2-(traps-4-
propoxycyclohexyl)-
acetate as a crude product.
A solution of benzyl 2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-
(traps-4-
CA 02238633 2004-09-O1
21489-9410
-26-
propoxycyclohexyl)-acetate (3.0 g, 5 mmol) in ethanol (50 mL) containing 3N
hydrochloric acid (S mL, 15 mmol) is hydrogenated at 50 psi in the presence of
596
palladium on charcoal (200 mg) at room temperature for 4 hours. The reaction
mixture is
filtered through celite washing with ethanol and concentrated in vacuo to
provide
2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-(traps-4-
propoxycyclohexyl) acetic
acid hydrochloride as a crude product.
2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-(traps-4-
propoxycyclohexyl) acetic
acid hydrochloride (2.65 g, 4.82 mmol), 1-hydroxybenzotriazole (0.65 g, 4.81
mmol),
4-methylmorpholine (2.93 mL, 26.5 mmol), and O-t-butylhydroxylamine
hydrochloride
( 1.81 g, 14.4 mmol) are dissolved in methylene chloride ( 100 mL). N-
[dimethylamino-
propyl]-N'-ethylcarbodiimide hydrochloride (1.1 g, 5.8 mmol) is added, and the
inaction is
stirred overnight. The reaction is then diluted with water and extracted with
methylene
chloride. The combined organic extracts are washed with brine, dried (Na2S04),
and the
solvent is evaporated. The crude product is purified by silica gel
chromatography (596
methanoUmethylene chloride) to give N-(t-butyloxy)-2(R)-[(4-
methoxybenzenesulfonyl)-
(4-picolyl)amino]-2-(traps-4-propoxy-cyclohexyl)-acetamide.
Example 2: The following compounds ain pinpaind similarly to example 1:
(a) N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-(traps-4-
methoxy-
cyclohexyl)-acetamide hydrochloride, m.p. 145-155°C.
(b) N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-(traps-4-
ethoxy-
cyclohexyl)-acetamide hydrochloride, m.p. 128-135°C.
(c) N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-(traps-4-
butoxy-
cyclohexyl)-acetamide hydrochloride, m.p. 132-137°C.
(d) N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-(traps-4.-
pentoxy-
cyclohexyl)-acetamide hydrochloride, m.p. 135-145°C.
(e) N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-[traps-4-
(2-phenethyloxy)cyclohexyl]-acetamide hydrochloride, m.p. 124-130°C.
(f) hI-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
-27-
[traps-4-{2-(I-naphthyl)-ethoxy)cyclohexyl]-acetamide hydrochloride, m.p. 125-
140°C.
(g) N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-(traps-4-
isopropoxycyclohexyl)-acetamide hydrochloride, m.p. 140-145°C.
(h) N-hydroxy-2{R)-[{4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-(traps-4-
isobutoxy-
cyclohexyl)-acetamide hydrochloride, m.p. 126-134°C.
(l) N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-(traps-4-
cyclohexyloxycyclohexyl)-acetamide hydrochloride, m.p. 135-144°C.
(j) N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-[traps-4-
(2-methoxyethoxy)cyclohexyl]-acetamide hydrochloride, m.p. 108-lI7°C.
(k) N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-[traps-4-
(2-fluoroethoxy)cyclohexyl]-acetamide hydrochloride, m.p. 130-i41°C.
{I) N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-{traps-4-
neopentoxycyclohexyl)-acetamide hydrochloride, m.p. 125-134°C.
(m) N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-(cis-4-
methoxy-
cyclohexyl)-acetamide hydrochloride, m.p. 142-149°C.
(n) N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl){3-picolyl)amino]-2-(traps-4-
ethoxy-
cyclohexyl)-acetamide hydrochloride.
(o) N-hydroxy-2(R)-[(4-benzenesulfonyl)(4-picolyl)amino]-2-(traps-4-methoxy-
cyclohexyl}-acetamide trifluoroacetate, m.p. 160-165°C.
(p) N-hydroxy-2(R)-[{4-ethoxybenzenesulfonyl)(4-picolyl)amino]-2-(traps-4-
methoxy-
cyclohexyl)-acetamide hydrochloride, m.p. 131°C.
{q) N-hydroxy-2(R)-[(4-propoxybenzenesulfonyl)(4-picolyl)amino]-2-{traps-4-
propoxy-
cyclohexyl)-acetamide hydrochloride, m.p. 163-165°C.
(r) N-hydroxy-2(R)-[(4-butoxybenzenesulfonyl)(4-picolyl)amino]-2-(traps-4-
propoxy-
CA 02238633 1998-OS-26
WO 97/22587 PCT/EP96/05362
-28-
cyclohexyl)-acetamide hydrochloride, m.p. 163-165°C.
(s) N-hydroxy-2(R)-[(3,4-dimethoxybenzenesulfonyl){4-picolyl)amino] -2-{trans-
4-
methoxycyclohexyl)-acetamide hydrochloride, m.p. 164°C.
{t) N-hydroxy-2(R)-{ (4-methoxybenzenesulfonyl)[2-(4-pyridyl)ethyl]amino }-2-
(trans-4-
ethoxycyclohexyl)-acetamide.
(u) N-hydroxy-2(R)-[(4-ethoxybenzenesulfonyl)(4-picolyl)amino]-2-{trans-4-
propoxy-
cyclohexyl)-acetamide hydrochloride, m.p. I31°C.
(v) N-hydroxy-2(R)-[(4-isobutoxybenzenesulfonyl){4-picolyl}amino]-2-(trans-
propoxy-
cyclohexyl)-acetamide hydrochloride, m.p. 145-I46°C.
(w) N-hydroxy-2(R)-[(4-ethoxybenzenesulfonyl)(4-picolyl)amino]-2-(trans-4-
ethoxy-
cyclohexyl)-acetamide hydrochloride, m.p. 150-155°C.
(x) N-hydroxy-2(R)-[(4-ethoxybenzenesulfonyl)(4-picolyl)amino]-2-(trans-4-
isobutoxy-
cyclohexyl)-acetamide hydrochloride, m.p. 168-I69°C.
(y} N-hydroxy-2(S)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-2-(traps-4-
propoxy-
cyclohexyl)-acetamide trifluoroacetate, m.p. 165-174°C.
Example 3:
(a} To a solution of N-(triphenyimethoxy)-2(R)-[(4-methoxybenzenesulfonyl)(4-
picolyl)-
amino]-2-{traps-4-methoxy-4-methylcyclohexyl) acetamide (348 mg, 0.48 mmol) in
methylene chloride at 0°C containing triethylsilane (260 p.L, 1.63
mmol) is added
trifluoroacetic acid (260 p.L> 3.4 mmol) dropwise. After 20 minutes, the
reaction mixture
is directly concentrated in vacuo and diluted with methylene chloride (4 mL).
The
resulting solution is cooled to 0°C and acidified with hydrogen
chloride gas. The solvent
is again removed in vacuo and the residue redissolved with methylene chloride.
The
solution is triturated by addition of pentane to precipitate out product. The
supernatent is
removed and the process repeated until all triphenylmethane is removed. The
remaining
solid precipitate is N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-
picolyl)amino]-2-
(traps-4-methoxy-4-methylcyclohexyl)-acetamide hydrochloride, m.p.
133°C.
CA 02238633 1998-OS-26
WO 97!22587 PCT/EP96/05362
-29-
The starting material is prepared as follows:
A solution of (R)-N-(4-methoxybenzenesulfonyl)-4-oxocyclohexylglycine benzyl
ester
(see example l, 5.0 g, 11.6 mmol), in methylene chloride (35 mL) at room
temperature is
added to a solution of titanium tetrachloride ( 1.0 M in methylene chloride)
(2 L2 mL, 21.2
mmol) and dimethyl zinc (1.0 M in heptane) (23.0 mL, 23.0 mmol) at-78°C
in
dichloromethane (20 mL). The reaction mixture is stirred at -78°C for
30 minutes, then
warmed slowly to room temperature over 2.5 hours. The reaction mixture is
poured into
water (700 mL} and extracted with chloroform. The combined organic extracts
are
washed with water, dried (MgS04}, filtered, and concentrated in vacuo. The
crude
product is purified by silica gel chromatography (40%, ethyl acetate/hexanes)
to provide
(R}-N-(4-methoxybenzenesulfonyl}-trans-4-hydroxy-4-methylcyclohexylglycine
benzyl
ester and {R)-N-(4-methoxybenzenesulfonyl)-cis-4-methyl-4-
hydroxycyclohexylglycine
benzyl ester.
To a solution of (R}-N-(4-methoxybenzenesulfonyl)-trans--4-hydroxy-4-methyl-
cyclohexylglycine benzyl ester (600.0 mg, 1.34 mmol) in methylene chloride (
15 mL}
containing 2,6-di-tert-butylpyridine (755 w!, 3.36 mmol) is added methyl
trifluoromethanesulfonate (305 p.L, 2.68 mmol) dropwise at room temperature.
The
reaction mixture is stirred at room temperature overnight then quenched with a
small
amount of methanol. The mixture is diluted with chloroform, then washed with
saturated
aqueous ammonium chloride and water. The organic layer is dried (MgS04),
filtered, and
concentrated in vacuo. The crude product is purified by silica gel
chromatography (35~
ethyl acetate/hexanes) to provide (R)-N-(4-methoxybenzenesulfonyl)-trans-4-
methoxy-4-
methylcyclohexyiglycine benzyl ester.
Hydrogenolysis of the benzyl ester to the acid and treatment with O-
tritylhydroxylamine
(instead of O-t-butylhydroxylamine) as in example 1 yields
N-(triphenylmethoxy)-2(R)-[{4-methoxybenzenesulfonyl )(4-picolyl)amino]
2-(trans-4-methoxy-4-methylcyclohexyl)acetamide.
(b) Similarly prepared is N-hydroxy-2{R)-[4-methoxybenzenesulfonyl)(4-picolyl)-
amino]-2-(cis-4-methoxy-4-methyl-cyclohexyl)-acetanaide hydrochloride m.p.
128°C.
Example 4: Preparation of 3000 capsules each containin~~ 25 mg of the active
ingredient,
CA 02238633 2004-09-O1
21489-9410
-30-
for example, N-hydroxy-2(R)-[(4-methoxybenzenesulfonyl)(4-picolyl)amino]-
2-(traps-4-propoxycyclohexyl)-acetamide:
Active ingredient 75.00 g
Lactose 750.00 g
TM
Avicel PH 102 300.00 g
(microcrystalline cellulose)
TM
Polyplasdone XL 30.00 g
(polyvinylpyrrolidone)
Purified water q.s.
Magnesium stearate 9.00 g
The active ingredient is passed through a No. 30 hand scxeen.
TM TM
The active ingredient, lactose, Avicel PH 102 and Polyplasdone XL are blended
for 15
minutes in a mixer. The blend is granulated with su~cient water (about 500
mL), dried in
an oven at 35°C overnight, and passed through a No. 20 screen.
Magnesium stearate is passed through a No. 20 screen, added to the granulation
mixture,
and the mixture is blended for 5 minutes in a mixer. The blend is encapsulated
in No. 0
hard gelatin capsules each containing an amount of the blend equivalent to 25
mg of the
active ingredient~