Note: Descriptions are shown in the official language in which they were submitted.
CA 02239508 1998-06-04
W O 97/21437 PCTnB96/01496
NAPHTh'iYL-S~JBST1TUTED BENZlMlDAZOi_E DERIVATIVES AS
ANTI-COAGUI~NTS
.,
held o~ the hl.'~ liU~
The prosent invent~on is directed to naphthyl-substituted ber~ derivatives and
5 their ph~.."acet~t!c~lly accc:~LaL~le ~alts, which inhibit certain enzymes in the coagulation ~c-~acle,
such as factor Xa and factor lla ILl~ru~b;~ thereby being useful as anti-~oagu~ ts. It also
ralates to ph~ Jtio~l ~ u".l-oa;Lions cu. ~;~ld the derivatives or their phallll~ceutically
acc~Lal~le salts, and their ....:LIIo.l:, of use.
8ACKGROUND OF Th'iE INVENTION
Factor Xa is a member of the trypsin-like serine ptoLt:ase class of enzymes. A one-to-one
binding of factors Xa and Va with calcium ions and phospho::r7id forms the pruLl..u,,,bi,,ase
complex which converts ~-uLlllolllbin to factor lla (Ll,,u,,,i.:.,). Tl.lu...bill, in turn, converts
fibrinogen to fibrin which poly",t~ es to form insoluble fibrin.
In the coa~ ation cascade, the is,uLl,ru",bi~ se complex is the convergent poi~of the
15 intrinsic (surface activated ) and ~)~L,in:.ic Ivessel injury-tissue factor) pathways (8;ochemistry
(199i), Vol. 30, p. 10363; and Cell (1988), Vol. 53, pp. 505-518). The model of the
roag~lation r~c~e has been refined further with the discovery of the mode of action of tissue
factor pathway inhibitor (TFPI) (Seminars in ~Jemato/ogy (1992), Vol. 29, pp. 159-1 61 ). TFPI is a
circulating multi-domain serine protease ir;hibitor with three Kunitz-like domains which competes
20 with factor Va for free factor Xa. Once formed, the binary cu..-l~le7c of factor Xa and TFPI
becomes a potent inhibitor of the factor Vlla and tissue factor complex.
Factor Xa can be activated by two distinct complexes, by tissue factor-factor Vlla
co,..ple~c on the "Xa burst" paLll~a~r and by the factor IXa-Vll!a co~"pl~ ,c (TENase) of the
"sustained Xa" pathway in the coagulation cascade. After vessel injury, the ~Xa burst" pathway
25 is activated via tissue factor (TF~. Up regulation of the coagutation r~ ade occurs via increased
factor Xa production via the "sustained Xa" pathway. Down regulation of the coaguiation
c~cr~d~3 occurs with the f~.,..dLon of the factor Xa-TFPI complex, which not oniy removes factor
Xa but also inhibits further factor formation via the "Xa burst" pathway. Consequently, there is a
,~ natural regulation of the coagulation cascade by factor Xa.
Published data with the proteins a~ .La:,ii7 and tick anti-coagulant peptide (TAP)
~, demonstrate that factor Xa inhibitors are ~rtica- ;ous anti-roa~~ nts (Thrombosis and
Haemostasis (1992), Voi. 67, pp. 371-376; and Science (1990), Vol. 248, pp. 593-~96). The
active site of factor Xa can be blocked by either a meuban;a."-based or a tight bindir7g inhibitor la
Sl,~ 1 l l IJTE SHEET (RULE 21;~
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96tO1496
tight binding inhibitor differs from a mechanism-based inhibitor by the lack of a covalent link
between the enzyme and the inhibitor). Two types of mechanism-based inhibitors are known,
reversible and irreversible, which are distinguished by ease of hydrolysis of the enzyme-inhibitor .
link (Thrombosis Res (19~t2), Vol. 67, pp. 221-231; and Trends Pharmaco/. Sci. (1987), Vol. 8,
pp. 303-307). A series of guanidino compounds are examples of tight-binding inhibitors "
(Thrombosis Res. (1980), Vol. 19, pp. 339-349). Arylsulfonyl-arginine-piperidinecarboxylic acid
derivatives have also been shown to be tight-binding inh;L,iLors of thrombin (Biochem. ~1984),
Vol. 23, pp. 85-90), as well as a series of arylamidine-containing compounds, including
3-amidinophenylsryl derivatives (Thrombosis Res. (1983), Vol. 29, pp. 635-642) and
10 bis(amidino)benzyl cycloketones (ThrombosisRes. (1980), Vol. 17, pp. 545-548). Therapeutic
utility of these compounds, however, is limited by their poor selectivity for factor Xa.
Related Disclosures
European Published Patent Application 0 540 051 (Nagahara et a/.) describes aromatic
amidine derivatives which are stated to be capable of showing a strong anticoagulant effect
15 through reversible i,.hiLiLion of factor Xa.
The synthesis of a,o'-bis~amidinobenzylidene)cycloalkanones and o,o'-bis(amidino-
benzyl)cycloalkanones is described in Pharmazie (1977), Vol. 32, No. 3, pp. 141-145. These
compounds are disclosed as being serine protease inhibitors.
SUMMARY OF THE INVENTION
This invention is directed to compounds or their pharmaceutically acceptable salts which
are anti-coagulants by inhibiting enzymes in the coagulation cascade, such as human factor Xa
and factor lla (Ll-ro~llbi--), and are therefore useful as pha.l..acolo~;cal agents for the treatment of
disease-states .;I.~ ;L~ ed by thrombotic activity.
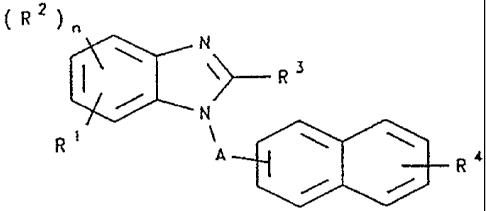
Accor~ -gly, in one aspect, this invention provides compounds of formula
( R ) n
~ ~ ( I )
R, A~ ~R 4
25 wherein:
n is 0 to 3;
A is a branched or straight chain alkylene, -C~O)- or -S(O)2-;
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
R1 is hydrogen, -oR5 or-N(R5)R6;
earh R2 is independently nitro, alkyl (optionally substituted by halo, aryl, -C(O)OR8,
-C(O)NIR8)R9,-N(R8)R9),-oR5,-NIR7)R7,-N(R7)R9,-NIR8)R9,-N(R8)C(o)R7,-C(O)OR8
-C(o)N(R7)R9, -C(O)N(R8)R9, or a heterocyclyl optionally substituted by one or more
~t 5 substituents selected from the group consisting of -C(NH)N(R8)R9, -C(NH)N(H)OR8,
-C(NH)N(H)C(O)R8, -C(NH)N(H)C(O)OR8, -C(O)ORa, -C(O)N(R8)R9, -R10-C(O)OR8,
-R10-C(O)N~R8)R9 and -S03H;
R3 is hydrogen or alkyl optionally substituted by one or more substituents selected from the group
consisting of halo, alkenyl, hydroxy, alkoxy, aryl (optionally substituted by alkyl, hydroxy,
halo, -N(R8)R9, -C(O)OR8, -C(O)N(R8)R9), aryloxy (optionally substituted by alkyl, hydroxy,
halo, -N(R8)R9, -C(O)OR8, -C(O)N(R8)R9), aralkoxy (optionally substituted by alkyl,
hydroxy, halo, -N(R8)R9, -C(O)OR8,-C(O)N(R8)R9), haloalkenyl, cycloalkyl, imidazolyl,
indolyl, adamantyl (optionally substituted by halo, alkyl, -N(R8)R9, -C(O~OR8 or-C(O)N(R8)R9), -C(O)OR8, -N(R8)R9, -C(O)N(R8)R9, -C(O)(CH2)mOR8 (where m is 1 to 4),
-N(R8)C(O)R8, -N(R8)C(O)N(R8)R9, -N(R8)C(NH)N(R8)R9, -OP03H2 and -SR8;
R4 is -C(NH)-N(R8)R9, -C(NH)N(H)OR8, -C(NH)N(H)C(O)R8 or -C(NH)N(H)C(O)OR8;
each R5 is independently:
hydrogen; or
alkyl optionally substituted by one or more substituents selected from the groupconsiaLing of halo, alkenyl, hydroxy, alkoxy, aryl (optionally substituted by alkyl,
hydroxy, halo, -N(R8)R9, -C(O)OR8 or -C(O)N(R8)R9), aryloxy (optionally
substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8 or -C(O)N(R8)R9), aralkoxy
(optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8 or
-C(O)N(R8)R9), haloalkenyl, cycloalkyl, imidazolyl, indolyl, adamantyl (optionally
substituted by halo, alkyl, -N(R8)R9, -C(O)OR8 or -C(O)N(R8)R9), -C(O)OR8,
-N(R8)R9, -C(O)N(R8)R9, -C(O)(CH2)pOR8 (where p is 1 to 4), -N(R8)C(O)R8,
-N(R8)C(O)N(R8)R9, -N(R8)C(NH)N~R8)R9, -OP03H2 and -SR8; or
aryl optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8, or -C(O)N(R8)R9; or
heterocyclyl optionally substituted by one or more substituents selected from the
group consisting of 1-iminoalkyl, -C(O)OR8, -C(O)N(R8)R9,
-C(NH)N(R8)R9, -C(NH)N(H)OR8, -C(NH)N(H)C(O)OR8, -R10-C(O)OR8,
-R10-C(O)N(R8)R9 and -SO ~H;
R6 is hydrogen, alkyl, -R10-C(O)OR8, -R10-C~O)N(R8)R9, -C(o)R7, or aralkyl (optionally substituted
by alkyl, halo, -N(R8)R9, -C(O)OR8, or -C(O)N(R8)R9);
35 R7 is a b.dl,ched or straight chain alkylene substituted by one or more substituents sele,_led from
the group consisting of halo, hydroxy, alkoxy, aryl (optionally substituted by alkyl,
hydroxy, halo, -N(R8)R9, -C(O)OR8), aryloxy, aralkoxy, alkenyl, haloalkenyl, cycloalkyl,
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/01496
imidazolyl, indolyl, adamantyl (optionally substituted by halo, alkyl, hydroxy, -C(O)OR8 or
-N(R8)R9), -C(o)OP~8~ -N(R8~R9, -C(O)N(R8)R9, -C(O)(CH2)qOR8 (where q is 1 to 4),
-N(R8)C(O)R8, -N(R8)C(o)N(R8)R9, -N(R8)C(NH)N(R8)R9, -OPO3H2 and -SR8;
each R8 and R9 is independently hydrogen, alkyl, aryl or aralkyl; and
5 each R10 is independently a branched or straight chain alkylene;
or a pharmaceutically accepLdble salt thereof.
In another aspect, this invention provides compositions useful in treating a human having
a disease-state characterized by thrombotic activity, which co.l.posiLion comprises a
therapeutically effective amount of a compound of the invention as described above, or a
10 pharmaceutically acceptsble salt thereof, and a pharmaceutically ac-~epL~l~,lc excipient.
In another aspect, this invention provides a method of treating a human having adisease-state characterized by thrombotic activity, which method comprises a.l,..i.,isL,~ring to a
human in need thereof a therapeutically effective amount of a compound of the invention as
described above.
In another aspect, this invention provides a method of treating a human having adisease-state alleviated by the inhibition of factor Xa, which method comprises administering to a
human in need thereof a therapeutically effective amount of a compound of the invention as
described above.
In another aspect, this invention provides a method of treating a human having a20 disease-state alleviated by the inhibition of factor lla (thrombin~, which method comprises
ad..-i.-;stering to a human in need thereof a therapeutically effective amount of a compound of the
inv~ntion as described above.
In another aspect, this invention provides a method of inhibiting human factor Xa ;n vitro
or in vivo by the aclr.,in,~L.~Lion of a compound of the invention.
In another aspect, this invention provides a method of inhibiting human factor lla
(thrombin) in vitro or in vivo by the admini:,L~Lion of a compound of the invention.
DETAILED DESCRIPTION OF THE INVENTION
D~ti..iliol~s
As used in the specification and appended claims, unless specified to the contrary, the
30 following terms have the meaning indicated:
"Halo" refers to bromo, chloro, fluoro or iodo.
"Alkyl" refers to a straight or brd-,c.l-ed chain monovalent radical consisting solely of
carbon and hydrogen, containing no unsaturation and having from one to six carbon atoms, e.g.,
methyl, ethyl, n-propyl, isopropyl (1-methylethyl), n-butyl, t-butyl (1,1-dimethylethyl), sec-butyl
35 (1-methylpropyl), n-pentyl, n-hexyl, and the iike.
"Alkenyl" refers to a straight or branched chain monovalent radical consisting solely of
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/01496
carbon and hydrogen, containing unsaturation and having from one to six carbon atoms, e g.,
ethenyl, n-prop-2-enyl, n-prop-1-enyl, n-but-2-enyl, n-but-3-enyl, 1-methylprop-1-enyl, and the
like.
"Alkylene" refers to a straight or branched chain divalent radical consisting solely of
carbon and hydrogen, containing no unsaturation and having from one to six carbon atoms, e.g.,
methylene, ethylene, n-propylene, isopropylene (1-methylethylene), n-butylene, t-butylene (1,1-
dimethylethylene), sec-butylene (1-methylpropylene), n-pentylene, n-hexylene, and the like.
"Haloalkenyl" refers to an alkenyl radical, as defined above, substituted by one or more
halo atoms, e.g., 1-bromoethenyl, n-1-chloroprop-2-enyl, n-3-chloroprop-1-enyl, n-3-chlorobut-2-
10 enyl, n-4-bromobut-3-enyl, 1-(chloro)methylprop-1-enyl, and the like.
"Alkoxy" refers to a radical of the formula -~i~a where Ra is alkyl as defined above, e.g.,
methoxy, ethoxy, n-propoxy, t-butoxy, and the like.
"Alkanol" refers to an alkane of one to five carbons which is substituted by a hydroxy
radical, e.g., methanol, ethanol, isopropanol, and the like.
"Aryl" refers to the phenyl or naphthyl radical.
"Aralkyl" refers to a radical of the formula -RaRb where Ra is alkyl as defined above and Rb
is sryl as defined above, e.g., benzyl.
"Aralkoxy" refers to a radical of the formula -ORC where Rc iS aralkyl as defined above,
e.g., benzyloxy, (phenyl)ethoxy, and the like.
"Amidino" refers to the radicsl -CtNH)NH2.
"Heterocyclyl" refers to a stabie 5- to 10-membered monocyclic or bicyclic ring radical
which is either saturated or unsaturated, and which consists of carbon atoms and from one to
three heteroatoms selected from the group cons; .Ling of nitrogen, oxygen and sulfur, and wherein
the nitrogen, carbon or sulfur atoms may be optionally oxidized, and the nitrogen atom may be
25 optionally qual~e~"i~ed, and including any bicyclic group in which any of the above-defined
heterocyclic ring radicals is fused to a benzene molecule. The heterocyclic ring radical may be
attached at any heteroatom or carbon atom which results in the creation of a stable structure.
Examples of such heterocyclic radicals include, but are not limited to, pi~ idinyi, piperazinyl,
2-oxopiperazinyl, 2-oxopiperidinyl, 2-oxopyrrolidinyl, 2-oxo~ I, azepinyl, pyrrolyl,
30 4-piperidonyl, pyrrolidinyl, pyrazolyl, py,azolid;nyl, i",idazoiyl, imidazolinyl, imiclA~ yl, pyridyl,
pyrazinyl, pyrimidinyl, pyridazinyl, oxazolyl, oxa~Olidi.,yl, indanyl, isoxazolyl, isoxaLolldillyl,
morpholinyl, thiazolyl, thiazolidinyl, isothiazolyl, quinuclidinyl, isothiazolidinyl, indolyl, isoindolyl,
indolinyl, isoindolinyl, octahydroindolyl, octahydroisoindolyl, quinolyl, isoQuinolyl,
decahydroisoquinolyl, benzimidazolyl, thiadiazolyl, benzopyranyl, benL~lLllid~olyl~ benzoxazolyl,
35 furyl, tetrahydrofuryl, tetrahydropyranyl, thienyl, b~llLolll;~ ,rl, thiamorpholinyl, thiamorpholinyl
sulfoxide, thiamorpholinyl sulfone, and oxadiazolyl. Preferred heterocyclic radicals for the
purposes of this invention are imidazolyl, piperidinyl, pyrrolidinyl, and indolyl.
CA 02239~08 1998-06-04
W O 97/21437 PC~B96/0~496
"Cycloalkyl" refers to a 5- to 7-membered ring radical containing solely carbon and
hydrogen atoms and no-unsaturation, i.e., cyclopentyl, cyclohexyl, and cycloheptyl.
"Factor lla" refers to thrombin.
"DEAD" refers to diethyl azodicarboxylate.
"THF" refers to tetrahydrofuran.
"HPLC" refers to high pressure liquid chromotagraphy.
"DMF" refers to dimethylformamide.
"Optional" or "optionally" means that the subsequently described event of circumstances
may or may not occur, and that the description includes instances where said event or
10 circumstance occurs and instances in which it does not. For example, "heterocyclyl optionally
substituted by one or more substituents selected from the group consisting of 1-iminoalkyl,
-C~O)OR8, -C(O)N(RB)R9, -C(NH)N~R8)R9, -C(NH)N(H)OR8, -C(NH)N(H)C(O)OR8, -R1 0-C(O)OR8,
-Rl0-C(O)N(R8)R9 and -SO31t" means that the heterocyclic radical, as defined above, may or may
not be substituted by the listed substituents and that this desc,iylion includes both substituted
15 heterocyclic radicals and heterocyclic radicals having no substitution. In addition, it is understood
that the various substitutions must be feasibly possible, within the realm of knowledge of a
chemist of ordinsry skill in the art and result in stable compounds.
"Pharmaceutically acceptable salt" includes both acid and base addition salts.
"Pharmaceutically accep~ acid addition salt" refers to those salts which retain the
20 biological effectiveness and properties of the free bases, which are not biologically or otherwise
undesirable, and which are formed with inorganic acids such as hydrochloric acid, hydrobromic
acid sulfuric acid, nitric acid, phosphoric acid and the like, and organic acids such as acetic acid,
trifluoroacetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, maleic acid, malonic
acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic
25 acid, methanesulfonic acid, ~LIIanesulfonic acid, p-toluenesulfonic acid, salicylic acid, and the like.
"Pharmaceutically acceptable base addition salt" refers to those salts which retain the
biological effectiveness and properties of the free acids, which are not biologically or otherwise
undesirable. These salts are prepared from addition of an i"u,~anic base or an organic base to the
free acid. Salts derived from inorganic bases include, but are not limited to, the sodium,
30 potassium, lithium, ammonium, calcium""a~l,esium, iron, zinc, copper, ma,.ganese, aluminum
salts and the like. Preferred inorganic salts are the ammonium, sodium, potassium, calcium, and
magnesium salts. Salts derived from orgsnic bases include, but are not limited to, salts of ,,
primary, secondary, and tertiary amines, substituted amines including naturally occurring
substituted amines, cyclic amines and basic ion exchange resins, such as isopropylamine,
35 trimethylamine, diethylamine, triethylamine, tripropylamine, ethanolamine,
2-dimethylaminoethanol, 2-diethylaminoethanol, trimethamine, dicyclohexylamine, Iysine, arginine,
histidine, caffeine, proc ,e, hydrabamine, choline, betaine, ethylenediamine, glutosd",:.,e,
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/01496
-7-
methylglucamine, theobromine, purines, piperazine, piperidine, N-ethylpiperidine, polyamine resins
and the like. Particularly preferred organic bases are isopropylamine, diethylamine, ethanolamine,
trimethamine, dicyclohexylamine, choline and caffeine.
"Therapeutically effective amount" refers to that amount of a compound of formula (I)
5 which, when administered to a human in need thereof, is sufficient to effect treatment, as defined
below, for disease-states alleviated by inhibition of factor Xa or factor lla. The amount of a
compound of formula (l) which constitutes a "therapeutically effective amount" will vary
depending on the compound, the disease-state and its severity, and the age of the human to be
treated, but can be determined routinely by one of ordinary skill in the art having regard to his
10 own knowledge and to this disclosure.
"Treating" or "treatment" as used herein cover the treatment of a disease-state in a
human, which disease-state is alleviated by inhibition of factor Xa or by factor lla; and include:
(i) preventing the disease-state from occurring in a human, in particular, when such
human is predisposed to the disease-state but has not yet been diagnosed as having it;
~ii) inhibiting the disease-state, i.e. ~ sLi~g its development; or
(iii) relieving the disease-state, i.e., causing r~y,e;,a;on of the disease-state.
The yield of each of the reactions described herein is expressed as a percentage of the
theoretical yield.
The compounds of the invention, or their pharmaceutically accepLable salts, may have
20 asymmetric carbon atoms in their structure. The compounds of the invention and their
pharmaceutically scceptable salts may Lhe~fur~: exist as single aL~ oisomers~ racemates, and as
mixtures of enantiomers and diasLel~o",ers. All such single stereoisomers, racemates and
mixtures thereof are intended to be within the scope of this invention.
In addition, the compounds of the invention may exist as individual regioisomers or
25 mixtures thereof.
The nomenclature used herein for the compounds of the invention is basically a modified
form of the l.U.P.A.C. system, wherein the compounds are named as derivatives of benzimidazole
with the following numbering system:
( R ) n
R 7 A_~ ~ R
A-,corclingly, a compound of the invention selected from formula (I) wherein A is
30 methylene, n is 1, R1 j5 piperidin-4-yloxy substituted on the nitrogen by 1-iminoethyl, R2 jS
CA 02239508 1998-06-04
W 097/21437 PCT~B96/01496
hydrogen, R3 is isopropyl and R4 is -C(NH)NH2, i.e,,
~ NX N H
[~ "~ ~ N H 2
J~
HtC NH
is named herein as 1-(4-amidinonaphth-1-yl)methyl-2-isopropyl-6-(N-(1-iminoethyl)piperidin-4-
yloxy)benzimidazoie; and wherein its regioisomer, i.e.,
~ r N H 2
n> < N H
N
H3C ~NH
Is named herein as 1-(4-amidinonaphth-1-yl)methyl-2-isopropyl-5-(N-11-iminoethyl)piperidin-4-
5 yloxy)benzi.~ 7O'Q.
Utility and f~ lr '_n
A. Utility
The compounds of the invention are inhibitors of factor Xa and factor lla and therefore
useful as anti-coagulsnts in treating disease-states characterized by thrombotic activity based on
10 factor Xa's or factor lla's role in the coagulation cascade ~see Background of the Invention
above). A primary indication for the compounds is prophylaxis for long term risk following
myocardial infarction. Additional indications are prophylaxis of deep vein thrombosis (DVT)
CA 02239~08 1998-06-04
WO 97t21437 PCT~B96/01496
following orthopedic surgery or prophylaxis of selected patients following a transient ischemic
attack. The compounds of the invention may also be useful for indications in which coumadin is
currently used, such as for DVT or other types of surgical intervention such as coronary artery
bypass graft and percutaneous transluminal coronary angioplasty. The compounds are also useful
for the treatment of thrombotic complications associated with acute promyelocytic leukemia,
.et~c~ multiple myelomas, disseminated intravascular coagulation associated with septic shock,
purpura fulminanas associated infection, adult les,~ Lory distress syndrome, unstable angina, and
thrombotic complications associated with aortic valve or vascular prosthesis. The compounds are
also useful for prophylaxis for thrombotic rliseaces, in particular in patients who have a high risk
10 of developing such disease.
In addition, the compounds of the invention are useful as in vitro diagnostic reagents for
inhibiting factor Xa or factor lla in the coagulation cascade.
B. Testing
The primary bioassays used to demonstrate the inhibitory effect of the compounds of the
15 invention on factor Xa or factor lla are simple chromogenic assays involving only serine protease,
the compound of the invention to be tested, substrate and buffer (see, e.g., Thromoosis Res.
(1979), Vol. 16, pp. 245-254). For example, four tissue human serine prc,L~ases can be used in
the primary bioassay, free factor Xa, prothrombinase, thrombin (factor lla) and tissue
plasminogen activator (tPA). The assay for tPA has been successfully used before to
20 demonstrate undesired side effects in the inhibition of the fibrinolytic process (see, e.g., J. Med.
Ch~m. (1993), Vol. 36, pp. 314-319).
Another bioassay useful in demonstrating the utility of the compounds of the invention in
inhibiting factor Xa demonstrates the potency of the compounds against free factor Xa in citrated
plasma. For example, the anticoagulant efficacy of the compounds of the invention will be tested
25 using either the prothrombin time (PT), or activated partial thromboplastin time (aPTT) while
selectivity of the compounds is checked with the thrombin clotting time (TCT) asssy. Correlation
of the K; in the primary enzyme assay with the Kj for free factor Xa in citrated plasma will screen
against compounds which interact with or are inactivated by other plasma components.
Correlation of the Kj with the t:,~Lension of the PT is a necessary in vitro demonstration that
30 potency in the free factor Xa inhibition assay translates into potency in a clinical coagulation
assay. In addition, extension of the PT in citrated plasma can be used to measure duration of
action in subsequent pharmacodynamic studies.
For further information on assays to demonstrate the activity of the compounds of the
invention, see R. Lottenberg et a/., Methods in ~nzymology (1981), Vol. 80, pp. 341-361, and
35 H. Ohno et al., Thrombosis Research (1980), Vol. 19, pp. 579-588.
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
--1 0-
C. General A~ Iiun
Administration of the compounds of the invention, or their pharmaceutically acceptable
salts, in pure form or in an appropriate pharmaceutical composition, can be carried out via any of
the accepted modes of administration or agents for serving similar utilities. Thus, adminisL,dlilJn
6 can be, for example, orally, nasally, parenterally, topically, transdermally, or rectally, in the form
of solid, semi-solid, Iyophilized powder, or liquid dosage forms, such as for example, tablets,
suppositories, pills, soft elastic and hard gelatin ~:~psulec, powders, solutions, suspensions, or
aerosols, or the like, pll rt,dbly in unit dosage forms suitable for simple administration of precise
dosages. The compositions will include a conventional pharmaceutical carrier or excipient and a
10 compound of the invention as the/an active agent, and, in addition, may include other medicinal
agents, pharmaceutical agents, carriers, adluvants, etc.
Generally, depending on the intended mode of ad.,.inial alion~ the pharmaceutically
accepldble compositions will contain about 1% to about 99% by weight of a compound(s) of the
invention, or a pharmaceutically a~ceplable salt thereof, and 99% to 1% by weight of a suitable
15 pharmaceutical excipient. Pre~erably, the co~,po~ilion will be about 5% to 75% by weight of a
compoundls) of the invention, or a pharmaceutically acce~lal)le salt thereof, with the rest being
suitable pharmaceutical e~ , lls.
The preferred route of ad.l.i..isl.dlion is oral, using a convenient daily dosage regimen
which can be adjusted accor ling to the degree of severity of the disease-state to be treated. For
20 such oral ad.";l,i2,l,dlion, a pharmaceutically acceptable composition containing a compound(s) of
the invention, or a pharmaceutically accepLdble salt thereof, is formed by the incorporation of any
of -tb~e normally employed t:, cifJienl ;, such as, for exa."ple, phâllllaceutical grades of mannitol,
lactose, starch, pregelatinized starch, magnesium stearate, sodium saccha-il~e, talcum, cell~ ose
ether derivatives, glucose, gelatin, sucrose, citrate, propyl gallate, and the like. Such
25 compositions take the form of solutions, suspensions, tablets, pills, c~rs~ les powders, sustained
release formulations and the like.
Ple:relaLly such co-,-poaiLions will take the form of capsule, caplet or tablet and therefore
will also contain a diluent such as lactose, sucrose, dicalcium phosphate, and the like; a
d;sinlt:~ldl.l such as ctuscallllellose sodium or derivatives thereof; a lubricant such as magnesium
30 stearate and the like; and a binder such as a starch, gum acacia, polyvinylpyrrolidone, gelatin,
cellulose ether derivatives, and the like.
The compounds of the invention, or their pharmaceutically accep~able salts, may also be
formulated into a suppository using, for example, about 0.5% to about 50% active ingredient
dicrosed in a carrier that slowly dissolves within the body, e.g., polyoxyethylene glycols and
35 polyethylene glycols (PEG~, e.g., PEG 1000 (96%) and PEG 4000 (4%J.
Liquid pharmacsutically admi"; .l, Is compositions can, for example, be prepared by
dissolving, dispersing, etc., a compound(s) of the invention (about 0.5% to about 20%), or a
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/01496
pharmaceutically acceptable salt thereof, and optional pharmaceutical adiuvants in a carrier, such
as, for example, water, saline, aqueous dextrose, glyceroi, ethanoi and the like, to thereby form a
solution or suspension.
If desired, a pharmaceutical composition of the invention may also contain minor amounts
of auxiliary substances such as wetting or emulsifying agents, pH buffering agents, antioxidants,
and the like, such as, for example, citric acid, sorbitan monolaurate, triethanolamine oleate,
butylated hydroxytoluene, etc.
Actual methods of preparing such dosage forms are known, or will be apparent, to those
skilled in this art; for example, see Remingron's Pharm~ceutical Sciences, 1 8th Ed., (Mack
10 Publishing Company, Easton, Pennsylvania, 1990). The composition to be adl"i";~ d will, in
any event, contain a therapeutically effective amount of a compound of the invention, or a
pharmaceutically accepldble salt thereof, for treatment of a disease-state alleviated by the
inhibition of factor Xa in accordance with the teachings of this invention.
The compounds of the invention, or their pharmaceutically acceplt,ble salts, are15 ad~"il,i5leled in a therapeutically effective amount which will vary depending upon a variety of
factors including the activity of the specific compound employed, the metabolic stability and
length of action of the compound, the age, body weight, general health, sex, diet, mode and time
of &dlllin;~laliun, rate of excretion, drug combination, the severity of the particular
disease-states, and the host undergoing therapy. Generally, a therapeutically effective daily dose
20 is from about 0.14 mg to about 14.3 mg/kg of body weight per day of a compound of the
invention, or a pharmaceutically acce~ e salt thereof; p~rt:.ably, from about 0.7 mg to about
1 0~~g/kg of body weight per day; and most preferably, from about 1.4 mg to about 7.2 mg/kg of
body weight per day. For example, for ad", ~ ,dlion to a 70 kg person, the dosage range would
be from about 10 mg to about 1.0 gram per day of a compound of the invention, or a
25 pharmaceutically acceptable salt thereof, pl~7~elaiJiy from about 50 mg to about 700 mg per day,
and most ~ rt:,ably from about 100 mg to about 500 mg per day.
r, .:f~ d Embod ru~
Of the compounds of the invention as set forth above in the Summary of the ~nvention,
several groups of compounds are plt:re"ed.
One preferred group is that group of compounds of formula ~I) wherein n is 0 or 1; A is
alkylene; Rl is -oR5 or-N(R5)R6; each R2 is independently nitro, alkyl (optionally substituted by
-C(O)OR8), -oR5, -N(R7)R9, -C(o)oR3, -C(O)N(R8)R9 or piperidinyl optionally substituted by
-C(O)OR8 or -Rl0-C(O)OR8: R3 is hydrogen or alkyl optionally substituted by one or more
substituents selected from the group consisting of halo, -C(O)OR8, or -C(O)N(R8)R9; R4 is
35 -C(NH)NH2; each R5 is independently hydrogen; or alkyl optionally substituted by one or more
substituents selected from the group consisting of -C(O)OR8, -C(O)N(R8)R9 and phenyl (optionally
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
substituted by -C(O)OR8); or piperidinyl or pyrrolidinyl, each optionally substituted by 1-iminoalkyl,
-C(NH)N(R8)R9, -R10-C(OJOR8 or -S03H; R6 is hydrogen, alkyl, benzyl (optionally substituted by
-C(O)OR8), -R10-C(O)OR8, -R10-C(O)N(R8)R9 or -C(o)R7; R7 is a branched or straight chain
alkylene substituted by one or more substituents selected from the group consisting of -C(O~OR8
5 and aryl ~optionally substituted by -C~O~OR8); each R8 and R9 is independently hydrogen or alkyl;
and each R10 is independently a branched or straight chain alkylene.
Of this group of compounds, a preferred subgroup of compounds is that subgroup wherein
n is 0 or 1; A is methylene: R1 is -oR5 or -N(R5)R6; R2 ;5 independently nitro, methyl (substituted
iby -C(O)OR8), -oR5, -N(R7)R9, -C(O)OR8, -C(O)N(R8)R9 or piperidinyl optionally substituted by
10 -C(O)OR8 or -R10-C(O)OR8; R3 is hydrogen or alkyl optionally substituted by -C(O)OR8 or
-C(O)N(R8)R9; R4 is -C(NH)NH2; each R5 is independently hydrogen; or alkyl optionally substituted
by -C(O)OR8, -C(O)N(R8)R9 or phenyl (optionally substituted by -C(O)OR8); or piperidinyl optionally
substituted by 1-iminoalkyl, -R10-C(O)OR8 or -S03H; R6 is hydrogen, alkyl, benzyl (optionally
substituted by -C(O)OR8) or -R10-C(O)OR8; R7 is a branched or straight chain alkylene substituted
15 by one or more substituents selected from the grotip consisting of -C(O)OR8 and aryl (optionally
substituted by -C(C~)OR8); each R8 and R9 is independently hydrogen, methyl or ethyl; and each
Rl~ is independently a branched or straight chain alkylene.
Of this subgroup of compounds, a pr~r~ d class of compounds is that class wherein n is
0; A is methylene: R1 is -oR5; R3 is hydrogen or alikyl optionally substituted by -C(O)OR8 or
20 -C(O)N(R8)R9; R4 is -C(NH)NH2; R5 is piperidinyl optionaliy substituted by 1-iminoalkyl; and R8 and
R9 are independently hydrogen, methyl or ethyl.
Of this class of compounds, the following compounds are more preire.,ùd:
1 -(4-amidinonaphth- 1 -yl)methyl-2-methyl-6-(N-( 1 -iminoethyl)piperidin-4-yloxybenzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(N-( 1 -iminoethyl)pi~.e~ i.li. ~-4-yloxybenzimidazole,
25 1-(4-amidinonaphth-1-yl)methyl-2-methyl-6-(N-~1-iminoethyl)p:, - id;.l-3-yloxybenzimidazole,
1-~4-amidinonaphth-1-yl)methyl-2-ethyl-6-(N-(l-iminoethyl)piperi, in-4-ylox~-,el. i...idazole,
1 -~4-amidinonaphth-1 -yl)methyl-2-ethyl-5-(N-~1 -iminoethyl)piperidin-4-yloxybenzimidazole,
1-(4-amidinonaphth-1-yl)methyl-2-isopropyl-6-(N-(1-iminoethyl)pi~ lidi.,-4-yloxybenzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-5-~N-~1 -iminoethyl)piperidin-4-yloxy)benzimidazole,
30 1 -~4-amidinonaphth- 1 -yl)methyl-2-t-butyl-6-(N-( 1 -iminoethyl)pi~.u, idin-4-yloxy) benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-t-butyl-5-(N-(1 -iminoethyl)piperidin-4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-6-(N-( 1 -iminoethyl)piperidin-4-yloxy)benzimidazole,
1-(4-amidinonaphth-1-yl)methyl-5-(N-(l-iminoethyl)piperidin-4-yloxy)b8llcill-;d~ ~18,
1 -(4-amidinonaphth-1 -yl)methyl-2-propyl-6-(N-(1 -iminoethyl)piperidin-4-yloxy)benzimidazole,
35 1-(4-amidinonaphth-1-yl)methyl-2-propyl-5-(N-(1-iminoethyl)piperidin-4-yloxy)benzimidazole,
1 -~4-amidinonaphth-1 -yl)methyl-2-propyl-6-(piperidin-4-yloxy)ber ci,--id~ Gle,1 -(4-amidinonaphth-1 -yl)methyl-2-propyl-5-(piperidin-4-yloxy)benzimidazole,
CA 02239~08 1998-06-04
W O 97/21437 PCTn~96/01496
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-6-(piperidin-4-yloxy)benzimidazole,1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-5-(piperidin-4-yloxy)benzimidazole,1 -(4-amidinonaphth-1 -yl)methyl-2-sec-butyl-6-(N-( 1 -iminoethyl)piperidin-4-yloxy)benZimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-sec-butyl-5-(N-(1 -iminoethyl)piperidin-4-yloxy~benzimidazole,
1-(4-amidinonaphth-1-yl)methyl-2-n-butyl-6-(N-(1-iminoethyl)piperidin-4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-n-butyl-5-(N-(1 -iminoethyl)piperidin-4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-(2-carboxyethyl)-6-(N-( 1 -iminoethyl)piperidin-
4-yloxy) benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-(2-carboxyethyl)-5-(N-( 1 -iminoethyl)piperidin-
4-yloxy)benzimidazole, and
1 -(4-amidinonaphth-1 -yl)methyl-2-(2-aminocsrbonylethyl)-6-(N-( 1 -iminoethyl)piperidin-
4-yloxy)benzimidazole.
Of the subgroup of compounds, another preferred class of compounds is that classwherein n is O; A is methylene; R1 is -N(R5)R6; R3 is hydrogen or methyl; R4 is -C(NH)NH2; R5 is
15 piperidinyl optionally substituted by 1-iminoalkyl; R6 is hydrogen, -R10-C(O)OR8 or -C(O)N(R8)R9;
R8 and R9 are independently hydrogen or methyl; and Rl~ is a branched or strsight chain alkylene.
Of this class of compounds, the following compounds are more pr~:ter,ed:
1-(4-amidinonaphth-1-yl~methyl-6-(N-(1-iminoethyl)piperidin-4-yl)d,l ' ~obellzimidazole,
1 -14-amidinonaphth-1 -yl)methyl-2-methyl-6-(N-(N'-( 1 -iminoethyl)piperidin-
4-yl)-N-((methoxycarbonyl)methyl)amino)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-6-(N-(N'-(1 -iminoethyl)piperidin-
4-yi)-N-((aminocarbonyl)methyl)amino)benzimidazole,
1 -(4-smidinonaphth-1 -yl)methyl-2-methyl-6-(N-(N'-(1 -iminoethyl)piperidin-
4-yl)-N-(3-carboxypropyl)amino)benzimidazole,
25 1-(4-amidinonsphth-1-yl)methyl-2-methyl-6-(N-(N'-(1-i",inoeLl,yl)piperidin-
4-yl)-N-(2-(methoxycarbonyl)propyl)amino)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-6-(N-(N'-(1 -iminoethyl)piperidin-
4-yl)-N-(2-(carboxy~propyl)amino)ben~;"~ ~a -le; and
1 -(4-amidinonaphth-1-yl)methyl-2-methyl-5-(N-(N'-(1 -il--illo~zLl,~l)p;,~e,idin-
4-yl)-N-(2-(carboxy)propyl)amino)benzimidazole.
Of the subgroup of compounds, another preferred class of compounds is that classwherein n is 1; A is methylene; R1 is -oR5; R2 is nitro, -ORs, -N(R7)R9, -C(O)OR8, or piperidinyl
(optionaliy substituted by -C(O)OR8); R3 is methyl or 1-isopropyl; R4 is -C(NH)NH2; each R5 is
independently hydrogen or alkyl optionally substituted by -C(O)OR8, -C(O)N(R8)R9, aryl (optionally
35 substituted by -C(O)OR8), or piperidinyl ~optionally substituted by -R10-C(O)OR8 or 1-iminoethyl);
R7 is a branched or straight chain alkylene substituted by -C(O)OR8 or phenyl (optionally
substituted by -C(O)OR8); and each R8 and R9 is independently hydrogen or methyl.
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/01496
-14-
Of this class of compounds, the foliowing compounds are more preferred:
1 -t4-amidinonaphth-1 -yi)methyl-2-methyl-5-hydroxy-6-~N-( 1 -iminoethyl)piperidin-
4-yloxy) benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-taminocarbonyl)methoxy-6-(N-t 1 -imino-
ethyl)piperidin-4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5~(carboxy)methoxy-6-(N-(1 -imino-
ethyl)piperidin-4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(4-(methoxycarbonyl~benzyloxy)-
6-tN-( 1 -iminoethyl)piperidin-4-yloxy)benzimidazole,
10 1-(4-amidinonaphth-1-yl)methyl-2-methyl-5-t4-(carboxy)benzyloxy)-
6-(N-( 1 -iminoethyl)piperidin-4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-carboxy-6-(N-(1 -iminoethyl)piperidin-
4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(3-(methoxycarbonyl)benzyloxy)-
6-(N-(1-iminoethyl)piperidin-4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(3-(carboxy)benzyloxy)-
6-(N-tl-iminoethyl)~ ,e,i ii,--4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(methoxycarbonyl)methoxy-6-(N-( 1 -imino-
ethyl)piperidin-4-yloxy)benzimidazole,
20 1-(4-amidinonaphth-1-yl)methyl-2-methyl-5-(1-(aminocarbonyl)ethoxy)-6-(N-tl-imino-
ethyl)piperidin-4-yloxy)benzimidazole,
,amidinonaphth-1 -yl)methyl-2-methyl-5-( 1 -(methoxycarbonyl)ethoxy)-6-(N-( 1 -imino-
ethyl)piye,i iill-4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(1 -(carboxy)ethoxy)-6-(N-(1 -imino- ethyl)pipe,idil~-4-yloxy)ben i,-lidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-4-methoxy-6-(pi~eridin-4-yloxy)benzimidazole,
1-(4-amidinonaphth-1-yl)methyl-2-isopropyl-7-methoxy-5-(~, i ii-l-4-yloxy)ben ;-,-ida ole,
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyi-4-methoxy-6-(N-(1 -iminoethyl)piperidin-
4-yloxy)ben~i" ,idazole,
301-(4-amidinonaphth-1-yl)methyl-2-isopropyl-7-methoxy-5-(N-(1-iminoethyl)pi --i ~-
4-yloxy)ben i...ids ~ la,
1 -(4-amidinonaphth- 1 -yl)methyl-2-methyl-5-(N-(4-carboxy~benzylamino~-
6-(N-(1 -iminoethyl)piperidin-4-yloxy)benzimidazole,
1 -(4-amidinonaphth- 1 -yl~ methyl-2-isopropyl-4-(4-carboxy~ " i ii"-1-
35yl)-6-(N-(1-iminoethyl~piperidin-4-yloxy)benzimidazole,
1 -(4-amidinonaphth- 1 -yl~methyl-2-isopropyl-4-(carboxy)methoxy-6-(N-( 1 -imino-
ethyl)piperidin-4-yloxy)ben i.-,id~ole,
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96101496
-15-
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-7-nitro-6-(N-(1 -imino-
ethyl)piperidin-4-yloxy)benzimidazole,
1 -(4-amidinonaphth- 1 -yl)methyl-2-methyl-7-nitro-6-(N-( 1 -imino-
ethyllpiperidin-4-yloxy~benzimidazole,
1-(~amidinonaphth-1-yl)methyl-2-isopropyl-5-(N-(2-carboxyprop-2-yl)amino)-
6-(N-( 1 -iminoethyl)piperidin-4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-5-(N-~4-carboxy)benzylamino)-
6-(N-(1 -iminoethyllpiperidin-4-yloxy)benzimidazole,
1 -(~amidinonaphth-1 -yl)methyl-2-isopropyl-5-~N-(2-carboxyethyl)amino)-
6-(N-(1-iminoethyl)pipeddin-4-yloxy)benzimidazole~ and
1 -(4-amidinonaphth- 1 -yl)methyl-2-methyl-5 -(N-(carboxymethyl)piperidin-4-yloxy) -
6-(N-(1 -iminoethyl)piperidin-4-yloxy)benzimidazole.
Of the subgroup of compounds, a preferred class of compounds is that class wherein n is
1; A is methylene; R1 is -N(R5)R6; R2is-C(O)OR8 or -C(O)N(R8)R9; R3is 1-isopropyl; R4is
15 -C(NH)N~12; R5is piperidinyl optionally substituted by 1-iminoethyl; R6 j5 hydrogen; and each R8
and R9is independently hydrogen or methyl.
Of this class of compounds, the following compounds are more prere.,~:d:
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-4-carboxy-6-(N-( 1 -iminoethyl)piperidin-
4-ylamino)benzimidazole, and
20 1-(4-amidinonaphth-1-yl)methyl-2-isopropyl-4-aminocarbonyl-
6-(N-( 1 -iminoethyl)piperidin-4-ylamino)benzimidazole.
P~.,pa..~tiun of Compounds of The invention
In the following Reaction Schemes, only one ~~:y;oison,er is shown as being prepared,
although one of ordinary skill in the art, having the full di~clc!sure of this specification, including
25 the Preparations and Examples, would realize that certain steps in the Reaction Schemes result in
mixtures of regioisomers, which can be separated and isolated by conventional techniques.
A. Prepnration of Compounds of Formulae (la) and ~Ib~.
Compounds of formulae (la) and (Ib) are compounds of the invention and are prepared as
shown below in Reaction Scheme 1 wherein A is a branched or straight chain alkylene, -C(0)- or
30 -S(0)2-; R2 ;5 alkyl (optionally substituted by halo, aryl, -C(O)OR8, -C(O)N(R8)R9, -N(R8)R9), -oR5,
-N(R7)R7,-N(R7)R9,-N(R8)R9,-N(R8)C(o)R7,-C(o)oR8~-C(o)N(R7)R9~-c(o)N(R8)R9; R3ishydrogen or alkyl optionally substituted by one or more substituents s~ected from the group
consisting of halo, alkenyl, hydroxy, alkoxy, aryl (optionally substituted by alkyl, hydroxy, halo,
-N(R8)R9, -C(O)OR8, -C(O)N(R8)R9), aryloxy (optionally substituted by alkyl, hydroxy, halo,
3~ -N(R8)R9, -C~O)OR8, -C(O~N(R8)R9), aralkoxy (optionally substituted by alkyl, hydroxy, halo,
CA 02239~08 1998-06-04
W 097/21437 PCT~B96/01496
-16-
-N~R8)R9, -C(O)OR8, -C~o)N(R8)R9), haloalkenyl, cycloalkyl, imidazolyi, indolyl, adamantyl
~optionally substituted by halo, alkyl, -N(R8)R9, -CIO)OR8 or -C(o)N(R8)R9), -C(O)OR8, -N(R8)R9,
-C(O)N(R8)R9, -C(O)(CH2)mOR8 (where m is 1 to 4), -N(R8)C(O)R8, -N(R8)C(O)N(R8)R9,
-NtR8~C(NH)N(R8)R9, -OP03H2 and -SR8; Rs j5 independently hydrogen; or alkyl optionally
5 substituted by one or more substituents selected from the group consisting of halo, alkenyl,
hydroxy, alkoxy, aryl (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8 or
-C(O)N(R8)R9), aryloxy (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8 or
-C(o)N(R8)R9), aralkoxy (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8 or
-C(o)N(R8)R9), haloalkenyl, cycloalkyl, imidazolyl, indolyl, adamantyl (optionally substituted by
10 halo, alkyl, -N(R8)R9, -C(O)OR8 or -C(O)N(R8)R9), -C(O)OR8, -N(R8)R9, -C(O)N(R8)R9,
-C(O)(CH2)pOR8 (where p is 1 to 4), -N(R8)C(O)R8, -N(R8)C(O)N(R8)R9, -N(R8)C(NH)N(R8)R9,
-OP03H2 and -SR8; or aryl optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8, or
-C(O)N(R8)R9; R7 is a branched or straight chain alkylene substituted by one or more substituents
selected from the group consisting of halo, hydroxy, alkoxy, aryl (optionally substituted by alkyl,
15 hydroxy, halo, -N(R8)R9, -C(O)OR8), aryloxy, aralkoxy, alkenyl, haloalkenyl, cycloalkyl, imidazolyl,
indolyl, adamantyl (optionally substituted by halo, alkyl, hydroxy, -C(O)OR8 or -N(R8)R9),
-C(O)OR8, -N(R8)R9, -C(O)N(R8)R9, -C~O)(CH2)qOR8 (where q is 1 to 4), -N(R8)C(O)R8,
-N(R8)C(o)N(R8)R9, -N(R8)C(NH)N(R8)R9, -OP03H2 and -SR8; each R8 and R9 is as described
above in the Summary of the Invention; R11 is hydroxy or halo; and X is halo and W is a
20 protecting group for nitrogen such as tert-butoxycarbonyl:
CA 02239508 1998-06-04
W O 97/21437 PCT~B96/01496
-17-
Re&~ S~heme 1
N H ~
(2)
R2
R 3--C ( O ) R ~ ~ ~N\~--R 3
' . Protec I N
HO W
( 3 )
R 3 ~ ~ N\? R 3
( 4 ) ~ W ~~ H
W W
(s) (6)
J fl ~CN
W (8)
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-18-
R~ac~ion Scheme 1 continued
R2
S ( ~ ">~ ~ H
~ ~ , ~/ ( I b )
HN~NH2
Compounds of formulae (1), (4) and (7) are commercially available, for example, from
Aldrich Co., or may be prepared acco.~ling to methods known to one skilled in the art.
Compounds of formula ~7) may also be prepared accordi,-g to the methods disclosed in European
5 Published Patent ~Fplic t;~n 0 540 051.
In general, compounds of formulae (la) and (Ib) are prepared by first treating a compound
of formula (1) in a r~rotic solvent, preferably, methanol, with a hydrogenating agent, for example,
with palladium on carbon in the presence of hydrogen, for 1 to 24 hours, pn~ rdbly, for 12
hours, with vigorous shaking. The compound of formula (2) is isolated from the reaction mixture
by first treating the reaction mixture with a strong acid, pr~r~.dl~l~ 4N HCI, followed by filtration
and concenL~dl;on.
The compound of formula (2) so formed is then dissolved in an aqueous acidic solvent,
such as 4N aqueous HCI, and then an excess molar amount of the compound of formula
R3-C(o)R1 1 where R1 t j5 hydroxy is added to the solution. The resulting reaction mixture is
refluxed for 2 to 16 hours, preferably for 12 hours, and then basified, preferably with potassium
bicarbonate, at ambient temperature. The product is isolated from the reaction mixture through
extraction and evaporation to yield the unprotected analog of the compound of formula (3). The
analog is dissolved in an aprotic solvent, p~er~.dbly tetrahydrofuran, and treated with di-tert-
butyldicarbonate, at ambient temperature, for 2 to 12 hours, prerelably for 3 hours. The
CA 02239~08 1998-06-04
WO 97/21437 PCTnB96/01496
19
compound of formula (3~ is isolated from the reaction mixture by aqueous extraction, followed by
column chromatography.
Alternatively, the compound of formula (2) is dissolved in an aprotic solvent, preferably
pyridine, and then treated with the compound of formula R3-C(o)R1 1 where R1 1 is chloro. The
resulting reaction mixture is stirred for 4 to 16 hours, preferably for 16 hours, at ambient
te",p~,aLure. The product is isolated from the reaction mixture by a~ueous exl,~.;lion and
recrystallization to produce an intermediate amide, which is treated with a mineral acid, preferably
1N HCI. The resulting reaction mixture is refluxed for Z to 12 hours, pl~rerably for 3 hours, and
then basified at ambient temperature. The resulting compound is dissolved in an aprotic solvent,
10 preferably tetrahydrofuran, and treated with di-tert-butyldicarbonate, at ambient temperature, for
2 to 12 hours, preferably for 3 hours. The compound of formula (33 is then isolated from the
reaction mixture in a manner similar to that which is described above.
The compound of formula (3) is then dissolved in an aprotic solvent, preferably,tetrahydrofuran, to which is added the compound of formula (4) in the presence of
15 triphenylphosphine and diethylazodicarboxylate (DLAD) in excessive molar amount at ambient
temperature. The resulting reaction mixture is stirred for 1 to 14 hours, pre~,al,ly for 12 hours.
Isolation by column chr~JlllaLography yielded the compound of formula (5), which is then dissolv.,d
in a protic solvent, pr~rerably~ methanol. The resulting solution is then treated with a base,
preferably ammonia, in a sealed flask and stirred for 2 to 16 hours, preferably, for 3 hours, at
20 45~C to 70~C, preferably, at 50~C. The compound of formula (6) is then isolated from the
reaction mixture through conventional techniques, such as conce.-l-aLion and column
chrclcnatography .
The compound of formula (6) is then dissolved in an aprotic solvent, prereraL,ly, DMF, and
treated with a strong inorganic base, such as sodium hydride. The resulting reaction mixture is
25 stirred for 30 minutes to 3 hours, preferably for 1 hour, st ambient temperature. A compound of
formula (7) is then added to the reaction mixture, and the resulting mixture is stirred for 1 to 24
hours, prt:reral,ly for 20 hours, at ambient temperature. Isolation through conventional
techniques, such as aqueous extraction and column chromatography yielded the compound of
formula (8).
The compound of formula (8) is then dissolved in an anhydrous alkanol, preferably ethanol
and the resulting solution is then treated with an anhydrous mineral acid, pr~ft:lLbly HCI, while
.,.ai~l..;ning the reaction temperatures between about -78~C and ambient temperature for
between 2 hours and 24 hours, and allowing the telllperaLure to rise to ambient te""~eral~lre
whiie monitoring for reaction completion, for example, through reverse phase HPLC. The solvent
35 is then removed and the resulting residue dissolved in fresh anhydrous alkanol, preferably ethanol.
The resulting solution is then treated with anhydrous ammonia at ambient pressure or in a sealed
flask, at temperatures from between ambient temperature and 100~C for about 1 to about ~
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-20-
hours. The compound of formula ~la) are then isolated from the reaction mixture by standard
techniques, such as concentration and reverse phase HPLC.
Alternatively, instead of treating the resulting soiution above with anhydrous ammonia,
the resulting solution is treated with a compound of the formula -NH20R8 to prepare the
corresponding compound of formula ~la) wherein R4 is -C(NH)N~H)OR8 substituent.
The compound of formula (la) may then be dissolved in a protic solvent, preferably
methanol, and then, in the presence of a base, p,ete.~bly triethylamine, be treated with the
appropriate imidate, preferably ethyl acetimidate, at ambient te",pe,tlLure, for 1 to 16 hours,
preferably for 3 hours. The product, the compound of formula (Ib), is isolated from the reaction
10 mixture by standard techniques, such as concenL~aLion and reverse phase HPLC.Compounds of formulae ~la) and (Ib) wherein R2 contains -C~O)N(R8)R9 or -C(O)OR8 where
each R8 and R9 are independently alkyl, aryl or aralkyl may be hydrolyzed under acidic conditions
to prepare compounds of formula (la) and (Ib) where R2 contains -C(O)OR8 where R8 j5 hydrogen.
Compounds of formula (la) and (Ib) where R2 contains -C~O)OR8 where R8 j5 hydrogen
15 may be amidated or esterified under standard conditions to produce compounds of formulae (la)
and (Ib) where R2 contains -C(O)OR8 where R8 j5 alkyl, aryl or aralkyl, or compounds of formulae
(la) and (Ib) where R2 conl~ina -C(O)N(R8)R9 or -C(o)N(R7)R9 where R8 and R9 are independently
hydrogen, alkyl, aryl or aralkyl and R7 is as defined above in the Summary of the Invention.
Compounds of formulae (ia) and (Ib) where R2 is nitro may be reduced under standard
20 conditions to produce compounds of formulae (la) and (Ib) where R2 is amino, which can further
be treated with the appropriate alkylating agent to produce compounds of formulae (la) and ~Ib)
wh~e R2 is -N(R7)R7, -N(R7)R9, -N(R8)R9 or -N(R8)C(o)R7 where each R8 snd R9 is hydrogen,
alkyl, aryi or aralkyl and R7 is as defined above in the Summary of the Invention.
Compounds of formula (Ib) may further be treated with the approp,i~ acid halides,
25 prefe,dlJI~ acid chlorides, or with the appropriate acid anhydrides or equivalents, to yield
compounds of the invention wherein R4 is -C(NH)NIH)C(O)R8. Alternatively, compounds of
formula (Ib) may further be treated with calLallloyl chlorides, or their equivalents, to yield
compounds of the invention where R4 is -C(NH)N(H)C(O)OR8.
B. F~e~ lion of Cornro~n~lc of Formula ~14a)
Compounds of formula (14a) are i~ r",ediates in the preparation of the compounds of the
invention and are prepared as shown below in Reaction Scheme 2 wherein R3 is hydrogen or alkyl
optionally substituted by one or more substituents selected from the group conai,,Li~g of halo,
alkenyl, hydroxy, alkoxy, aryl (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8,
-C(O)N(R8)R9), aryloxy (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8,
35 -C(O)N(R8)R9), aralkoxy (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8,
-C(O)N(R8)R9), haloalkenyl, cycloalkyl, imidazolyl, indolyl, adamantyl (optionally substituted by
CA 02239~08 1998-06-04
W O 97/21437 PCT/IB96/01496
-21-
halo, alkyl, -NtR8tR9, -C(O~OR8 or -C(O)N(R8)R9), -C(O)OR8, -NlRs)R9, -C(o)N(R8)R9,
-C(O)(CH2)mOR8 (where m is 1 to 4), -N(R3)C(o)R8, -N(R8~C(O)N(R8)R9, -N(R8)C(NH)N(R8)R9,
-OPO3Hz and -SR8; R5 is hydrogen; or R5 is alkyl optionally substituted by one or more
substituents selected from the group consisting of halo, alkenyl, hydroxy, alkoxy, aryl (optionally
5 substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8 or -C(o)N(R8)R9), aryloxy (optionally
substituted by aikyl, hydroxy, halo, -N~R8)R9, -C(O)OR8 or -C(O)N(R8)R9), aralkoxy (optionally
substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8 or -C(o)N(R8)R9), haioalkenyl, cycloalkyl,
imidazolyi, indolyl, adamantyl (optionally substituted by halo, alkyl, -N(R8)R9, -C(O)OR8 or
-C(O)N(R8)R9), -C(O)OR8, -N(R8)R9, -C(O)N(R8)R9, -C(O~(CH2)pOR8 (where p is 1 to 4),
1~ -N(R81C(O)R8, -N(R8)C(o)N(R8)R9, -N(R8)C(NH)N(R3)R9, -OPO3H2 and -SR8; R8, R9 and R10 are as
defined above in the Summary of the Invention; R11 is hydroxy or halo; Y is a protecting group for
oxygen, such as tetrabutyldimethylsilyl, and W is a protecting group for nitrogen, such as tert-
butoxycarbonyl:
_
CA 02239~08 1998-06-04
WO97/21437 PCT~B96/01496
-22-
Reaction Scheme 2
F OCH3 OH
F ~ ~ R 3 - C ( O ) R ~
( 9 ) ( ~ O ) ( I I )
OY ORS
P r o t e c t ~ >_ 3 X R S ~ >--R 3
H H
~12) (~3)
ORs ORs
3 . (1 3 ) ~ R 3 ~ R 3
W W
( I 4 ) ( ~ 40 )
Compounds of formula (9) and the acid chloride used in Step 1 are commercially available,
for example, from Lancaster Synthesis, Inc., or may be prepared accordi.lg to methods known to
those skilled in the art.
In general, the compounds of formula (14a) are pr~pa,ed by first dissolving a compound of
formula (9) in an aprotic solvent, prefersbly tetrahydrofuran, and saturating the solution with
ammonia (gas) at 0~C. After the saturation, the reaction mixture is sealed and warmed to
ambient temperature and stirred for 3 to 16 hours, preferably for 12 hours. The reaction mixture
is filtered and the filtrate is concentrated and dissolved in a protic solvent, preferably methanol,
10 and treated with an excessive molar amount of an alkaline alkoxide, preferably sodium methoxide,
and the reaction mixture is stirred at ambient temperature for 1 to 6 hours, p,~r~:rably 3 hours.
The compound of formula (10) was isolated from the reaction mixture through standard isolation
techniques, such as exLla~ on and concentration.
The compound of formula (10) is then dissolved in an aprotic basic solvent, such as
15 pyridine, and treated with a slight molar excess of the compound of formula R3-C(O)CI. The
reaction mixture is stirred for 1 to 16 hours, preferably for 1 hour, at room temperature. Isolation
through conventional isolation techniques, such as evaporation, aqueous extraction and
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-23-
recry~ ion, afforded the corresponding amide. The amide is dissolved in a protic solvent,
preferably ethanol, in the presence of a strong acid, for example, 4N HCI, and then reduced under
standard reducing conditions, such as palladium on carbon in the presence of hydrogen, to form
the corresponding amine, which is isolated from the reaction mixture through standard
techniques, such as filtration and concentration. The resulting product is then dissolved in a
mineral acid, such as 2N HCI, and refluxed for 3 to 12 hours, preferably 4 hours. The product is
isolated through concentration and dissolved in aqueous hydrogen bromide and refluxed for 1 to
16 hours, preferably 3 hours. The product is concentrated and redissolved in H20 and an
inorganic base, such as potassium bicarbonate. The compound of formula 111) is isolated from
10 the reaction mixture by conventional methods, such as aqueous extraction and concentration.
The compound of formula (11) is then dissolved in an aprotic solvent, preferably, DMF, to
which a weak base, such as imidazole, is added. To this reaction mixture is added an excessive
molar amount of a bulky silyl halide, such as tert-butyldimethylsilyl chloride. The resulting
reaction mixture is stirred for 1 to 12 hours, preferably for 1 hour, at ambient temperature. The
15 compound of formula (12) is then isolated from the reaction mixture through standard techniques,
such as extraction and concenL-dLion.
The compound of formula (12~ is then dissolved in an sprotic solvent, such as DMF, and
treated with a strong base, such as sodium hydride, at ambient temperature. The reaction is
stirred for 1 to 6 hours, prere-t-bly for 4 hours, and the compound of formula XR5 is added to the
20 reaction mixture, while stirring, over a period of 1 to 3 hours. The compound of formula (13) is
isolated from the reaction mixture by stsndard techniques, such as extraction and column
chromatography .
The benzimidazole nitrogen of the compound of formula (13) is then protected in a
manner similar to the process descrlbed above for a compound of formula ~2) to produce the
25 compound of formula (14). The compound of formula (14) is then dissolved in an aprotic solvent,
such as DMF, and cooled to 0~C. Tetra-butyl ammonium fluoride, in an aprotic solvent, is then
added to the solution, and the resulting mixture was stirred for 30 minutes to 3 hours, preferably
for 1 hour at O~C. The compound of formula (14a) is then isolated from the reaction mixture
through standard techniques, such as aqueous extraction and column chromatography.
The compound of formula (1 4a) may then be treated in a manner similar as described
above for compound of formula ~3) to produce compounds of the invention.
In addition, the dimethoxybenzimidazole compound formed in the process of making the
compound of formula (11) from the compound of formula (10) may be dissolved in aqueous
hydrogen bromide and refluxed for a shorter time period, pr~rerably, for about 3 hours, to produce
35 the corresponding mono-hydroxymethoxy benzil--;dazole of compound (13), i.e., the compound
wherein R5 is methyl and Y is hydrogen. This compound is then treated in the similar manner as
described above for the compound of formula (13) to produce compounds of the invention.
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/~1496
-24-
C. P~p~.~lion of Compounds of Formula (22)
Compounds of formula (22) are intermediates in the preparation of the compounds of the
invention and are prepared as shown below in Reaction Scheme 3 wherein R3 is hydrogen or alkyl
optionaliy substituted by one or more substituents selected from the group consisting of halo,
5 alkenyl, hydroxy, alkoxy, aryl ~optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8,
-C(O)N(R8)R9), aryloxy (optionally substituted by alkyl, hydroxy, halo, -N(R3)R9, -C(O)OR8,
-C(O)N(R8~R9), aralkoxy (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8,
-C(O)N(R8)R9), haloalkenyl, cycloalkyl, imidazolyl, indolyl, adamantyl (optionally substituted by
halo, alkyl, -N(R8)R9, -C~O)OR8 or -c(o)N(R8)R9t~ -C(O)OR8, -N(R8)R9, -C(O)N(R8)R9,
10 -C(O)(CH2)mOR8 (where m is 1 to 4~, -N(R3)C(o)R8, -N(R8)C(O)N(R8)R9, -N(R8)C(NH)N(R8)R9,
-OP03H2 and -SR8; R8 and R9 are each independently hydrogen, alkyl, aryl or aralkyl; R10 is a
branched or straight chain alkylene; R12 is alkyl; X is halo; M is an alkaline metal anion and W is a
protecting group for nitrogen, such as tert-butoxycarbonyl:
CA 02239508 1998-06-04
WO 97/21437 PCT~B96/01496
-25-
Reaction Scheme 3
R ~ ~-C ( O ) oR8
N O
(g) ( ~5) ( I 6~
~ -C( O )OR
2( I 6 ) + H 2 N ~ N N ~ 2
F N
H~3
( 1 7 )
R - C ( O ) O R
3 . ( I 7 ) + R ~ 20 M+ ~ N
~,N ~2
R t 2 o~J~N
H
( ~ 8 )
~ ~ ~ C ( O ) O R 8 ~ C ( O ) O R
4 . ( I ,3 ) + N R - C ( O ) X~ N
R 1 2 0 J~ N 11~R 1 2 ~ J~C NN\>_ R 3
( I 9 ) ( 2 0 )
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/01496
-26-
Re~-~lion Scheme 3 continued
R~0_c(o)oR8 R~~-C(O)OR~
5 ~ ( 2 0 ) N P r o I e c I
H O /~ ~ R 3 H O /~ N~ R
H W
(21 ) (~2)
Compounds of formulae (9) and (15), and the acid halide used in Step 4 are commercially
avaiiable or may be prepared according to methods known to those skilled in the art.
In general, compounds of formula (22) are prepared by first dissoiving a compound of
formula (9) in an aprotic solvent, preferably acetonitrile, and the resulting solution is chilled to
sbout -1 0~C. An equivalent molar amount of a compound of formula (15) is then added to the
solution in the presence of a base, ptt:r-:.ably diisopropylethylamine, and the resulting solution is
allowed to warm to ambient temperature. The reaction mixture is stirred for 3 to 12 hours,
preferably for 5 hours. Isolation through conventional techniques, such as evaporation of the
10 solvent, exl,d~Lion and concenL.t,lion, yields the corresponding compound of formula (16~.
The compound of formula (16) is then dissolved in an aprotic solvent, p,efelably~c~t~niL,ile and the resulting solution is chilled to about -10~C. E3enzylamine is added slowly to
the solution in the p(esence of a base, preferably cliisopropylethylamine, and the resulting reaction
mixture is allowed to warm to ambient temp,anilure. The reaction mixture is then refluxed for 24
15 to 48 hours, preferably for about Z4 hours. Conventional isolation techniques, such as removal
of the solvent, aqueous organic extraction, and concsriLIaLion yields the compound of formula
(1 7).
The compound of formula (17) is then dissolved in a protic solvent, preferably a alkanol
such as methanol. An excessive molar amount of an alkaline alkoxide corresponding to the
20 alkanol used, such as sodium methoxide, is then added to the reaction mixture under nitrogen.
The resulting mixture is refluxed for 1 to 12 hours, preferably for 6 hours, and the reaction
allowed to cool to ambient Le-,.pe.aL.Ire. The solvent is removed and the resulting residue is
dissolved in an organic solvent. Conventional isolation techniques, such as aqueous extraction,
concenL~dLion and chromatography yields the compound of formula (18).
The compound of formula (18) is then dissolved in a protic solvent and treated with a
reducing agent, such as palladium on carbon, in the presence of an acid, such as HCI. The
resulting mixture is then hydrogenated under pressure and the solids filtered out of the solution to
CA 02239~08 1998-06-04
W O g7/21437 PCT~B96/01496
-27-
yield the HCI salt of the compound of formula (19).
The salt form of the compound of formula 119) is dissolved in an organic basic solvent, .
preferably, pyridine. An excessive molsr amount of a compound of formula R3-C(o)X is added to
the solution at about 0~C. The mixture is allowed to warm to ambient temperature and then
stirred for 12-16 hours, preferably for 12 hours. The solvent is removed and conventional
isolation techniques, such as extraction an orç~anic solvent and conce"L~dLion, provides the
product which is then dissolved in a strong mineral acid, prere,dbly 4N HCI and the resulting
solution is refluxed for 8 to 16 hours, prt:rerably 16 hours. The acid ;s removed by evaporation
and the resulting residue is then neutralized to pH 7 with a mild inorganic base, such as sodium
10 bicarbonate. Standard isolation techniques, such as removal of the resulting water by
conce~L~ ion and trituration with tetrahydrofuran, provides the compound of formula (20).
The compound of formula (20) is then dealkylated in a manner similar to that described
above for compounds of formula (11) to produce a compound of formula (21) and N-protected in
a manner similar to that described above for compounds of formula (2) to produce the compound
15 of formula 122).
The resulting compound of formula (Z2) is then treated in a manner similar to that
des~(iL,ed in Reaction Scheme 1 for compounds of formula (3) to produce compounds of the
invention .
D. ~ .OI;OI~ of C~ ou., is of Formula 132~
Compounds of formula (32) are intermediates in the plepd~Lion of the compounds of the
in~rention and are ptepa,~d as shown below for Reaction Scheme 4 wherein A is a branched or
straight chain alkylene; R3 is hydrogen or alkyl optionally substituted by one or more substituents
selected from the group consisting of halo, alkenyl, hydroxy, alkoxy, aryl (optionally substituted
by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8, -C(O)N(R8)R9), aryloxy (optionally substituted by
25 alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8, -C(O)N(R8)R9), aralkoxy (opLion-~y substituted by alkyl,
hydroxy, halo, -N(R8)R9, -C(O)OR8, -C(O)N(R8)R9), haioalkenyl, cycloalkyl, imidazolyl, indolyl,
ad&lllallLyl (optionally substituted by halo, alkyl, -N(R8)R9, -ClO)OR8 or -C(o)N(R8)R9), -C(O)OR8,
-N(R8)R9, -C(O)N(R8)R9, -C(O)(CH2)mOR8 (where m is 1 to 4), -N(R8)C(O)R8, -NIR8)C(O)N(R8)R9,
-N(R8)C(NH)N(R8)R9, -OP03H2 and -SR8; R5 is hydrogen; or R5 is alkyl optionally substituted by
30 one or more substituents selected from the group consiaLi,-g of halo, alkenyl, hydroxy, alkoxy,
aryl (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8 or -C(O)N(R8)R9), aryloxy
(optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8 or -C(O)N(R8)R9), aralkoxy
(optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(o)oR5 or -C(o)N(R8)R9), haloalkenyl,
cycloalkyl, imidazolyl, indolyl, adamantyl (optionally substituted by halo, alkyl, -N(R8)R9, -C(O)OR
35 or -C(O)N(R8)R9), -C(O)OR8, -N(R8)R9, -C(O)N(R8)R9, -C(O)(CH2)pOR8 (where p is 1 to 4),
-N(R8)C(OlR8, -N(R8)C(O)N(R8)R9, -N(R8)C(NH)N(R8)R9, -OPO3H2 and -SR8; or R5 is aryl optionally
CA 02239508 1998-06-04
WO 97/21437 PCT~B96/01496
substituted by alkyl, hydroxy, halo, -N(R8)R9, -CtO)OR8, or -C(O~N(R8)R9; R8, R9 and R10 are as
defined above in the Summary of the Invention; X is halo; each Y is an oxygen protecting group,
such as tert-butyldimethylsilyl; and each W is a nitrogen protecting group such as tert-
butoxycarbonyl:
CA 02239508 l998-06-04
W O 97/21437 PCT~B96/01496
-29-
Reaction Scheme 4
HO HO
H 2 ~ R 3 P r o i e c ~ ~C \~ R 3
( 2 4 ) ( 2 5 ) r o N
[~ >--
HO
W
(26)
Y o
H O ~ ~ ~ R 3
~X~ W
( 4) ~ J
W ( 27 )
HO ro
3 . ( 2 7 ) ~ ~C ~ R 3 ~ ~ i e c ~ ~ N~ R 3
[~~ W [~0 H
W ( 28 ) W ( 29 )
4 . ( 2 9 ~ + ~_~3C N ~ 5
W ( 30 )
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-30-
Reaction Scheme 4 continued
HO
5 . ( 3 0 ) ~ ~ R 3
[~~ A ~ C N
W( 3 1 ~
RsO
. ( 3 ~ ) + X - R 5 ~ C N
W (32)
Compounds of formulae (24), (4) and m sre commercially available or may be prepared
by methods known to those skilled in the art or by the methods disclosed herein.In general, the compounds of formula (32) are prepared by first dissolvinçl a compound of
5 formula (24~ in an aprotic solvent, plerelaiJly~ tetrahydrofuran. The solution is basified to a pH of
7 to 10, pre:ierai ly, to pH 8, with a mild i"orçtanic base, p~ererably~ sodium bicarbonate, at
ambient l~lllperaLure. An excessive molar amount of a nitrogen-prute-;Li"g reagent, such as di-
tert-butyldicsrbonate, is sdded to the solution snd the resulting m;xture wss stirred at ambient
temperature for 0.5 to 24 hours, preferably for 12 hours. The solvent is removed and the
resulting residue is diluted and extracted with an organic solvent. Extraction and concentration
provides the compound of formula 125).
The compound of formula (25~ is then clissolved in an aprotic solvent, preferabiy, DMF. A
mild base, preferably, imidazole, and an oxygen-pruLt5~;Li"~ reagent, such as tert-butyldimethylsilyl
chloride, is added to the solution. The resulting mixture is stirred for 30 minutes to 5 hours,
preferably for 1 hour, at ambient temperature. The compound of formula 126) is isolated from the
reaction mixture by extraction and evaporation.
The compound of formula (26) is then dissolved in an aprotic solvent, preferably,
tetrahydrofuran, and then reacted in a manner similar to thst described above for compounds of
formula (3) to produce the compound of formuls ~27).
The compound of formula (27) is then dissolved in a protic solvent, prefersbly, methanol,
and then deproLeuLtd by ammonolysis st 0~ to 50~C, preferably at 20~C, while stirring for 5 to
48 hours, preferably for 12 hours. Conventional isolation techniques, such as concenLIc:Lion and
CA 02239~08 l998-06-04
W O 97/21437 PCTnB96/01496
-31-
drying provides the compound of formula (28).
The compound of formula ~28) is then dissolved in an aprotic solvent, preferably DMF, .
and 0-protected in a manner similar to that described above for the compound of formula (26) to
produce the compound of formula (29).
The compound of formula (29) is then treated in a similar manner as that described above
for the compounds of formula (6) to provide compounds of formula (30). The compound of
formula ~30) is then dissoived in an aprotic solvent, preferably, tetrahydrofuran, and treated with
a deprotecting agent, such as tetrabutyl ammonium fluoride, at ambient temperature. After
stirring the reaction mixture for 30 minutes to an hour, prererably for 30 minutes, the compound
10 of formula (31~is isolated from the reaction mixture through conventional techniques, such as
extraction and conce"L~nLion.
The compound of formula ~31)is then dissolved in an aprotic solvent, such as DMF. A
strong base, such as sodium hydride, is added to the solution, and the resulting mixture is
allowed to stir for 30 minutes to 2 hours, prt7re~ably for 30 minutes, at ambient temperature. The
16 compound of formula XR5 is added to the reaction mixture, and the resulting mixture is stirred for
30 minutes to 3 hours, pr~re,~Lly, for 1 hour. The compound of formula (32) is then isolated
from the reaction mixture through conventional isolation techniques, such as aqueous extraction,
concentration and column chromatography.
The compound of formula (32) is then treated in a similar manner as that described above
20 for compounds of formula (8) to provide compounds of the invention.
E. F~.",....Jtiol) of Compounds of Formuia (36~
Compounds of formula (36) are intermediates in the preparation of the compounds of the
invention and are prepared as shown below in Reaction Scheme 5 wherein A is a straight or
branched chain alkylene; R3 is hydrogen or alkyl optionally substituted by one or more
25 substituents selected from the group consisting of halo, alkenyl, hydroxy, alkoxy, aryl (optionally
substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)ORB, -C~O)N(R8)R9), aryloxy ~optionally
substituted by alkyl, hydroxy, halo, -N(R3)R9, -C(O)OR8, -C(O)N(R8)R9~, aralkoxy (optionally
substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8, -C(O)N(R8)R9), haloalkenyl, cycloalkyl,
imidazolyl, indolyl, adamantyl (optionally substituted by halo, alkyl, -N(R8)R9, -C(O)OR8 or
30 -C(O)N(R8)R9), -C(O)OR8, -N(R8)R9, -C(o)N(R8)R9, -C(O)(CH2)mOR8 (where m is 1 to 4),
-N(R8)C(O)R8, -N(R8)C(O)N(R8)R9, -N(R8)C(NH)N(R3)R9, -OP03H2 and -SR8; R8, R9 and Rl~ are as
defined above in the Summary of the Invention; X is hslo; Y is a prùLecLil,9 group for oxygen,
such as tert-butyldimethylsilyl; and w1 is a ~ruL~-;Lillg group for nitrogen, such as tert-
butoxycarbonyl and w2 is a di~rt:,el)L pruLe.;Ling group for nitrogen, such as benzyloxycarbonyl
35 (CBZ):
CA 02239508 1998-06-04
WO 97~1437 PCT~B96/01496
-32-
Re~lion Scheme 5
YO HO
[X~ \>--R 3 [~ \>--R
j ~ C N ~ j ~ C N
(30~ 1 (33
W W
~0
2 . ( 3 3 ) + ~N--W [j~ \>--R 3 ( 3 4 )
~j ~ A ~ ~} C N
H W
~0
3 ' ( 3 4 ) ' [~ R 3 ( 3 5 )
[~0 A_~CN
W
IR -C(O)OR
~0
3 5 ~ X R ~ - C ( O ) O R 8 [~ R 3 ( 3 6 )
~0 A_~CN
N
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/01496
-33-
Compounds of formula (30) are prepared according to methods disclosed herein.
Compounds of formula (4) and XR10-C(O)OR8 are commercially available, or may be prepared
accorcling to methods known to those skilled in the art.
In general, compounds of formula (36) are prepared by first dissolving a compound of
formula (30) in an aprotic solvent, such as tetrahydrofuran, and then treating the resulting
solution in a manner similar to that as described above for compounds of formula (30) to produce
a compound of formula (33).
The compound of formula (33) is then treated with a compound of formula of formula (4)
in a similar manner as described above for compounds of formula (26) to produce a compound of
10 formula (34).
The compound of formula (34) is then dissolved in a mixture of a protic and an aprotic
solvent, such as a 9:1 mixture of methylene chloride and methanol. An organic acid, such as
trifluoroacetic acid, is then added to the solution. The resulting reaction mixture is stirred for 3 to
24 hours, preferably for 6 hours, at ambient temperature. The compound of formula (35) is then
15 isolated from the reaction mixture by conventional techniques, such as concentration.
The compound of formula (35) is then dissolved in an aprotic solvent, such as
tetrahydrofuran. A compound of formula XR10-C(O)OR9 in the presence of a mild base,
pr~relably, potassium carbonate. The resulting reaction mixture is stirred at ambient temperature
for 30 minutes to 6 hours, preferably for 1 hour. Conventional isolation techniques, such as
20 exL.a.;lion with an organic solvent, dryin~, concentration and chromatography provides a
compound of formula (36).
The compound of formula (36) is then treated in a manner similar to that described above
for the compound of formula (8) to provide compounds of the invention.
F. F~~p~.~lioll of Compounds of Formula (45)
Compounds of formula (45) are i~ -edic~es in the p.~pa.~Lion of compounds of theinvention and are prepared as shown below in Reaction Scheme 6 wherein A is a branched or
straight chain alkylene; R2 is alkyl (optionslly substituted by halo, aryl, -C(O)OR8, -ClO)NtR8)R9,
-N~R8)R9), -oR5, -NtR7)R7, -N(R7)R9, -N(R8)R9, -N(R8)C(o)R7, -C(O~OR8, -C(o)N(R7)R9,
-C(O)N(R8)R9, or a heterocyclyl optionally substituted by one or more substituents selected from
30 the group consisting of -C(NH)N(R8)R9, -C(NH)N~H)OR8, -C(NH)N(H)C~O)R8, -C(NH)N(H)C(O)OR8,
-C(o)oR5, -CtO)N(R8)R9, -R10-C(O)OR8, -R10-CtO)N(R8)R9 and -S03H; R3 is hydrogen or alkyl
optionally substituted by one or more substituents selected from the group consisting of halo,
alkenyl, hydroxy, alkoxy, aryl (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8,
-C(O)N(R8)R9), aryloxy (optionally substituted by alkyl, hydroxy, halo, -NtR8)R9, -C(o)oR3,
35 -C(O)N(R8)R9), aralkoxy (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8,
-C(O)N(R8)R9), haloalkenyl, cycloalkyl, imidazolyl, indolyl, adamantyl (optionally substituted by
CA 02239508 1998-06-04
W O 97/21437 PCT~B96/01496
-~4-
halo, alkyl, -N(R8)R9, -C(O)OR8 or -C(O)N(R8)R9), -C(O)OR8, -N(R8)R9, -C(O~N(R8)R9,
-C(O)(CH2)mOR8 (where m is 1 to 4), -N(R8)C(O)R8, -N(R8)C(O)N(R8)R9, -N(R8)C(NH)N(R8)R9,
-OP03H2 and -SR8; R7, R8, R9 and R10 are 85 defined above in the Summary of the Invention; X
is halo; and W is a protecting group for nitrogen, such as tert-butoxycarbonyl:
Re~,Lion Scheme 6
H O ( R - C ) 2 ~ ~"~ o J~ R 3
( 37 ) ( 38 )
R ~<~ N o 2
2 . ( 3 8 ) ~ [~N,H
HO
o~R3
( 3 9 )
3 . ( 3 9 ) ,, ~ N N 2 R ~ NN~ R 3
HO o~R3 HO H
( 40 ) ( 4 1 )
4 ~ ( 4 1 ) P r o t e c t ~ R 3
HO W
( 42 )
CA 02239508 1998-06-04
W O 97/21437 PCT~B96/01496
-35-
Reaction Scheme 6 continued
R2
H O ~ ~ \~ R ( 4 3 )
(4) [~~ \W
W
R2
R 3 ( 4 4 )
~1~
XA R2
6 . ( 4 4 ) ~ ,~ C N ~ R 3
( 7 ) C~ t ; C N
W ( 4 5 )
Compounds of formulae (373, (4) and (7) are co,.,r"c:~ ~"y available, for example, from
Aldrich Co., or may be prepared according to methods known to those skilied in the art.
Compounds of formula (R3-C~o))2o are commercially available, for example, from Aldrich Co., or
5 may be prepared according to methods known to those skilled in the art.
In general, compounds of formula (45) are prepared by first dissolving a compound of
formula (37) in an aprotic basic solvent, such as pyridine. The appropriate anhydride is added to
the solution at ambient le""~er~L-lre. The resulting reaction mixture is stirred for 1 to 48 hours,
pr~telai,ly, for 12 hours, at ambient temperature. Removal of the solvent affords the compound
10 of formula ~38).
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-36-
The compound of formula (38) is then dissolved in protic acidic solvent, such astrifluoroacetic acid. Concentrated nitric acid is added to the solution at smbient temperature.
The resulting mixture is stirred at ambient temperature for 30 minutes to 12 hours, preferably for
2 hours. Isolation through recrystallization gives the compound of formula (39) and its nitro
regioisomer.
The compound of formula (39) is then dissolved in a protic solvent, preferably, methanol,
and treated with a reducing agent under standard conditions, such as palladium on carbon in the
presence of hydrogen under pressure at ambient temperature. The resulting mixture is filtered
and the filtrate is concentrated to give the compound of formula (40). The compound of (40) is
10 refluxed for 1 to 3 hours, preferably for 2 hours, in a mild organic acid, such as acetic acid, which
corresponds to the anhydride used in Step 1. The acidic solvent is removed and the compound of
formula (41 ) is isolated from the reaction mixture through extraction, drying and concentration.
The compound of formula (41~ is then protected in a manner similar to that described
above for the compound of formula (2) to produce the compound of formula (42), which is further
15 treated in a manner similar to that described above for the compound of formula t3) to produce
compounds of formula ~45) and compounds of the invention.
G. F~'~.p~.al;On of Compounds of Formulae (Ic) and (Id)
Compounds of formulae (Ic) and (Id) are compounds of the invention and are prepared as
shown below in Reaction Scheme .7 wherein A is a strai~ht or branched chain alkylene; R2 is
20 nitro, alkyl (optionally substituted by halo, aryl, -C(O)OR8, -C(o)N~R8)R9, -N(R8)R9), -oR5,
-N~R,.7)R7, -N(R7)R9, -N(R8)R9, -N(R8)C(o)R7, -C(O)OR8, -C(o)N(R7)R9, -C(o)N(R3)R9, or a
heterocyclyl optionally substituted by one or more substituents selected from the group consisting
of -C(NH)N(R8)R9, -C(NH)N(H)OR8, -C(NH)N(H)C(O)R8, -C(NH3N(H~C(O)OR8, -C(O)OR8,
-C(O)N(R8)R9, -R10-C(O)OR8, -R10-C(O)N(R8)R9 and -S03H; R3 is hydrogen or alkyl optionally
25 substituted by one or more substituents selected from the group consisting of halo, alkenyl,
hydroxy, alkoxy, aryl (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8,
-C(O)N(R8)R9), aryloxy (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8,
-C(O)N(R8)R9), aralkoxy (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8,
-C(O)N(R8)R9), haloalkenyl, cycloalkyl, imidazolyl, indolyl, adamantyl (optionally substituted by
30 halo, alkyl, -N(R8)R9, -C(O)OR8 or -C(O)N(R8)R9), -C(O)OR8, -N(R8)R9, -C(O)N(R8)R9,
-C(O)~CH2)mOR8 (where m is 1 to 4), -N(R8)C(O)R8, -N(R8)C(O)N(R8)R9, -N(R8)C(NH)N(R8)R9,
-OP03H2 and -SR8; R7, R8, R9 and R10 are as defined above in the Summary of the Invention; X
is halo; and W is a protecting group for nitrogen, such as tert-butoxycarbonyl:
CA 02239508 l998-06-04
W O 97/21437 PCT~B96/01496
-37-
Reaction Scheme 7
R~ R + ¦ ~ ~C N~H>--R
(46)
( 67 ) ~N
( 4 8 )
XA R2
2 ~ ~! R ~ + ~ ¦ J H>--
( 4 9 )
H ~ H
< ~ H 2
N ( I d )
H j C
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/03496
-38-
Compounds of formula 146) are prepared according to methods described below in the
Preparations. Compounds of formula (47) and (7) are commercially avaiiabie, for example, from
Aidrich Co., or may be prepared according to methods known to those skilied in the art.
In general, the compounds of formuiae (Ic) and (Id) are prepared by first dissolving a
compound of formuia (46) in a protic acidic solvent, such as a mixture of methanol and acetic
acid. An excessive molar amount of a compound of formula (47)is then added to the solution at
ambient temperature in the presence of a reducing agent, such as NaCNBH4. The reaction
mixture is stirred at ambient temperature for 30 minutes to 3 hours, preferabiy, for 1 hour, and
the solvent removed. Conventional isolation techniques, such as aqueous extraction,
10 concentration and column chromatography provides the compound of formuia (48).
The compound of formula (48)is then treated in a manner similar to that described above
for the compound of formula (6? to produce the compound of formula (49), which is then treated
in a manner similar to that described above for the compound of formula (8) to produce the
compound of formula (Ic), which is then treated in a manner simiiar to that described above for
15 the compound of formula (la) to produce the compound of formula (Id).
H. i~el~a.c.~ ) of Comrou~ds of Formuls (55)
Compounds of formula (5~) are intermediates in the preparation of compounds of the
invention and are prepared as shown below in Reaction Scheme 8 wherein R3 is hydrogen or alkyl
optionally substituted by one or more substituents selected from the group consisting of halo,
20 alkenyl, hydroxy, alkoxy, aryl (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8,
-C~ N(R8)R9), aryloxy (optionally substituted by aikyl, hydroxy, halo, -N(R8)R9, -C(O~OR8,
-C(O)N(R8)R9), aralkoxy (optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8,
-C(O)N(R8)R9), haloalkenyl, cycloalkyl, imidazolyl, indolyl, adamantyl (optionally substituted by
halo, alkyl, -N(R8)R9, -C(O)OR8 or -C(O)N(R8)R9), -C(O)OR8, -N(R8)R9, -C(O)N(R8)R9,
25 -C(O)(CH2)mOR8 (where m is 1 to 4), -N(R8)C(O)R8, -N(R8)C(O)N(R8)R9, -N(R8)C(NH)N(R8)R9,
-OP03H2 and -SR8; R6 is hydrogen, alkyl, -R10-C(O)OR8, -R10-C(O)N(R8)R9, -C(o)R7, or aralkyl
(optionally substituted by alkyl, halo, -N(R8)R9, -C(O)OR8, or -C(O)N(R8)R9); R7, R8, Rg and R10
are as defined above in the Summary of the Invention; X is halo; and W is a protecting group for
nitrogen, such as tert-butoxycarbonyl and WZ is a iirrerer.l protecting group for nitrogen, such as
30 tosyl:
CA 02239508 1998-06-04
WO 97/21437 PCT~B96/01496
-39-
Re&cliollScheme 8
H ~ N N l1 R Z~L~ N
(50) (47)
R 2 ~! ( s I )
2 . ( 5 1 ) P r o I e c I ~ \~ R 3
H--N
( 5 2 )
3 . ( 5 2 ) + R 6 _ X ~ \>--R 3
( 5 3 ) R --N W
( 5 4 )
4 . ( 5 4 ) --' ~ NN\>--R 3
R --N H
I (s5)
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-40-
Compounds of formula (50) are prepared according to methods disclosed herein.
Compounds of formula (47) and R6X sre commercially available or may be prepared according to
methods known to those of ordinary skill in the art.
In general, the compounds of formula (55) are produced by first treating a compound of
5 formula (50) with a compound of formula (47) in a manner similar to that described above for the
compound of formula (46) to produce a compound of formula (51). The compound of formula
(51 ) is then dissolved in an aprotic solvent, preferably, DMF, at about 0~C. A strong base, such
as sodium hydride, is added to the solution in an excessive molar amount. The solution is stirred
for about 30 minutes. A nitrogen-protecting producing group, such as tosyl chloride, is added to
10 the reaction mixture, and the resulting mixture is stirred for 30 minutes to 3 hours, preferably, for
1 hour, at about 0~C. Conventionai isolation techniques, such as extraction and concentration
yields the compound of formula (52).
The compound of formula (5Z) is then dissolved in an aprotic solvent, such as DMF, in the
presence of a mild base, such as potassium carbonate, and the compound of formula (53), such
15 as methyl iodide. The resulting reaction mixture is stirred for 10 to 24 hours, prere-ably, for 12
hours, at ambient temperature. Conventional isolation techniques, such as ex~ .Lion by organic
solvent and concentrat;on, yields the compound of formula ~54~.
When the compound of formula ~53) is other than a methyl halide or ethyl bromo acetate
and contains an alkylene group, an allyl halide is used to produce the compound of formula ~54)
20 and then the compound is reduced to produce the corresponding alkylene-containing R6 group.
The compound of formula (54) is then de-prc,~ d by dissolving it in a protic solvent,
such as methanol, in the presence of a mild nucleophilic agent, such as an alkoxide or a mineral
hydroxide, for example, sodium hydroxide. The reaction mixture is stirred for 1 to 3 hours,
p.ererably for 1 hour, at ambient temperature. Conventional isolation techniques, such as
25 evaporation and column chromatography yields the compound of formula (55).
The compound of formula (55) is then further treated in a manner similar to that described
above for compounds of formula (6) to produce compounds of the invention.
I. r-~pb.Jt;ol) of Compounds of Formulae (60) snd (62)
Compounds of formulae (60) and (62) are in~t:.-l.ediates in the preparation of compounds
30 of the invention and are prepared as shown below in Reaction Scheme 9 wherein A is a straight
or branched chain alkylene; R3 is hydrogen or alkyl optionally substituted by one or more
substituents selected from the group consislil1~ of halo, alkenyl, hydroxy, alkoxy, aryl (optionally
substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O~OR8, -C(O)N(R8)R9), aryloxy (optionally
substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8, -C(O~N(R8)R9), aralkoxy (optionally
35 substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8, -C(O)N(R8)R9), haloalkenyl, cycloalkyl,
imidazolyl, indolyl, adamantyl (optionally substituted by halo, alkyl, -N(R8)R9, -C(O~OR8 or
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/~1496
-41-
-C(O~N(R8lR9), -C~O)OR8, -N(R8)R9, -C(O)N(R8)R9, -C(O)(CH2~mOR8 (where m is 1 to 4),
-N(R8)C(o)R3, -N(R8)C(O)N~R8)R9, -N(R8)C(NH)N(R8)R9, -OPO3H2 and -SR8; R6 is hydrogen, alkyl,
-R10-C(O)OR8, -R10-C(O)N(R8)R9, -C(o)R7, or aralkyl (optionally substituted by alkyl, halo,
-N(R8)R9, -C(O)OR8, or -C(o)N(R8)R9); R7 is a branched or straight chain alkylene substituted by
5 one or more substituents selected from the group consisting of halo, hydroxy, alkoxy, aryl
(optionally substituted by alkyl, hydroxy, halo, -N(R8)R9, -C(O)OR8), aryloxy, aralkoxy, alkenyl,
haloalkenyl, cycloalkyl, imidazolyl, indolyl, adamantyl (optionally substituted by halo, alkyl,
hydroxy, -C(O)OR8 or -N(R8~R9), -C(O)OR8, -N(R8)R9, -C(O)N(R8)R9, -C(O)~CH2)qOR8 (where q is 1
to 4), -N(R8)C(O)R8, -N(R8)C(O)N(R8)R9, -N(R8)C(NH)N(R3)R9, -OPO3H2 and -SR8; R8, R9 and Rl~
10 are as defined above in the Summary of the Invention; X is halo; and W is a protecting group for
nitrogen, such as tert-butoxycarbonyl:
CA 02239508 1998-06-04
W 097/21437 PCT~B96/01496
-42-
Reaction Scheme 9
02N 02N
[~ \>--~ 3 [~C \>--R 3 ~C \>--R 3
H3C0 H H~CO H H0 H
( 56 ) ( 57 ) ( 58 )
02N
H O ~ ~ ~ R 3
( 4) \ H
~> (59)
N~W
3 . ( 5 9 ) + ? ~ ~ 2 N \~ R 3
~~ A ~L C N
W ( 6 0 )
H2N
4 . ( 6 0 ) ~ R 3
~~ A~CN
W ( 6 1 )
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-43-
Reaction Scheme 9 continued
R7
H--N
5. (61! + R 7 X ~ ~ R 3
A ~ C N
W (62)
Compounds of formulae (56), (4), (7) and R7X are commercially available, for example,
from Aldrich Co., or may be prepared according to methods known to those skilled in the art.
In general, the compounds of formula (62) are prepared by first dissolving a compound of
5 formula ~56) in a protic solvent, prt:re.àbly, trifluoroacetic acid, in the presence of nitric acid at
ambient te",p~-~-Lure. The resulting reaction mixture is stirred for 6 to 16 hours, prefe-ably, for
12 hours. The solvents are removed by evaporation and the residue neutrali~ed to about pH 7
with a miid base, such as sodium bicarbonate. Conventional isolation techniques, such as
ehL.~.;Lion, concentration and column chromatography yields the compound of formula (57).
The compound of formula (57) is then de-methylated in a manner similar to that des~i.il,ed
above for compound of formula (11) to produce the compound of formula (58), which is then
treated in a manner similar to that described above for the compound of formula (3) to produce
the ~;ompound of formula (59).
The compound of formula (59) is then treated in a manner similar to that described above
15 for the compound of formula (7) to produce the compound of formula (60), which is then
dissolved in a basic aprotic solvent, such ss pyridine. A mild reducing sgent, such ss tin (Ill)
chloride dihydrate, is then added to the solution. The resulting slurry is then heated to 50~ to
60~C, preferably to 50~C, for 10 to 16 hours, preferably for 14 hours. The solvent is removed
and the resulting residue dissolved in an organic solvent. Conventional isolation techniques, such
20 a filtrstion, conce~L~c~Lion snd chromstography, yields the compound of formula (61).
The compound of formula (61) is then dissolved in sn sprotic solvent, prefersbly, DMF.
An excessive molar smount of a compound of formuls R7X is then added to the solution in the
presence of a mild bsse, such as potassium bicarbonate. The resction mixture is stirred for 24 to
48 hours, prefersbly for 48 hours, at 20~C to 55~C, preferably at 20~C. Conventional isolation
25 techniques, such as t~ ;Lion and chromatoqraphy, yields the compound of formula (62).
The compound of formula (62) is then treated in a manner similar to that described above
for the compound of formula (8) to produce compounds of the invention.
In addition, the compound of formula ~60) may be treated in a manner similar to thst
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/01496
described above for the compound of formula (8~ to produce compounds of the invention.
In addition, all compounds of the invention that exist in free base form or free acid form
may be converted to their pharmaceutically acceptable saits by treatment with the appropriate
inorganic or organic acid, or by the appropriate inorganic or organic base. Salts of the
5 compounds of the invention can aiso be converted to the free base form or to the free acid form
or to another salt.
* ~I * * ~
The foilowing specific preparations and examples are provided as a guide to assist in the
practice of the invention, and are not intended as a limitation on the scope of the invention.
PREPARATION 1
Compounds of formula (2)
To a solution of 4-amino-3-nitrophenol (25.0 9, 162 mmol) and methanol (300 mL) was
added 10% Pd/C (300 mg). The reaction was placed under hydrogen and shaken for 12 hours.
Afterward, 4 N HCI (50 mL) was added. The mixture was then filtered through celite. The
15 filtrate was concentrated to give 3,4-diaminophenol.
PREPARATION 2
Compounds of formula (3)
A. A solution of 3,4-diaminophenol (5.20 9, 42.0 mmol), isobutyric acid (5.80 mL,
63.0 mmol) and 4 N HCI (50 mL) was refluxed for 16 hours. After cooling to ambient
20 temperature, the reaction was neutralized (KHCO~). Filtration afforded a dark solid. The solid
was dissolved in THF (100 mL) and H20 (20 mL). Di-tert-butyldicc,,LonaLe (4.09 9, 18.7 mmol)
was added to the solution. After stirring for 12 hours, the reaction mixture was poured into H2O
(50 mL). The resulting mixture was extracted with ethyl acetate (2x150 mL). The organic layers
were washed with brine (10 mL), dried (Na2SO4) and concentrated. The resulting oil was
25 chromatographed on SiO2 (50 9) with hexane/ethyl acetate (1:1) to afford 1-tert-butoxycarbonyl-
6-hydroxy-2-isopropylbe~ ida~c~le.
B. In a similar manner, the following compounds of formula (3) were made:
1 -tert-butoxycarbonyl-6-hydroxybenzimidazole;
1 -tert-butoxycarbonyl-6-hydroxy-2-methylben~;" ,icla~ole;
30 1-tert-butoxycarbonyl-6-hydroxy-2-trifluoromethylbenzimidazole;
1 -tert-butoxycarbonyl-6-hydroxy-2-eth~lL~"~i,.,i~iR7ole;
1 -tert-butoxycarbonyl-6-hydroxy-2-propylbenzimidazole;
1 -tert-butoxycarbonyl-6-hydroxy-2-butylben~in ,idazole;
CA 02239~08 l998-06-04
W O 97t21437 PCT~B96/01496
-45-
1-tert-butoxycarbonyl-6-hYdroxy-2-isobutylben-imidazole; and
1 -tert-butoxycarbonyl-6-hydroxy-2-tert-butylben7imidazole .
C. Alternatively, succinic anhydride (11.2 9, 113 mmol, 2 eq) and 3,4-diaminophenol
(7 g, 56 mmoL, 1 eq) were dissolved in 150 mL dry DMF and the mixture was heated to 100~C
for 3 hours. The DMF was removed in vacuo, and the residue was dissolved in 350 mL 4N HCI
and refluxed for 14 hours. The reaction was cooled, the water was removed in vacuo and the
residue was dissolved in 200 mL methanol and to this was added 20 mL concenL~aLed sulfuric
acid. This mixture was refluxed overnight. The methanol was stripped off and the residue
neutralized to pH 7 with saturated aqueous sodium bicarbonate. The water was removed ;n
10 vacuo and the residue was triturated with THF (4x100 mL). The combined THF fractions were
concentrated to about 100 mL volume and di-tert-butyldicarbonate (12 g, 56 mmol, 1 eq.) was
added. The mixture was stirred overnight at ambient temperature and then concentrated. The
crude oil was chromatographed (1:1 ethyl acetate/hexanes) to afford 1-tert-butoxycarbonyl-6-
hydroxy-2-(methoxycarbonylethyl)benzimidazole as a colorless solid, 5 g ~42%, yield).
PREPARATION 3
Compounds of formula (5)
A. DEAD (0.60 mL, 3.80 mmol) was added to a solution of 1-tert-butoxycarbonyl-6-hydroxy-2-isopropylbenzimidazole (690 mg, 2.30 mmol), 1-tert-butoxycarbonyl-4-
hydroxypiperidine (~25 mg, 4.60 mmol), triphenylphosphine (904 mg, 3.45 mmol) and THF
20 (5 mL~ at 25 ~C. After stirring for 12 hours, the solvent was removed in vacuo. Chromatography
(SiO~2, 100 9) of the resulting oil with hexanelethyl acetate (1:1) afforded 1-tert-butoxycarbonyl-
6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-2-isopropylbeu~i"~id~ole.
B. In a similar manner, the following compounds of formula (5) were made:
1-tert-butoxycarbonyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxY)ben~ 7Ole;
25 1 -tert-butoxycarbonyl-6-(N -(tert-butoxycarbonyl)~;,,e, id;n-4-yloxy)-2-methylbenzimidazole;
1 -tert-butoxycarbonyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy) -2-trifluoromethyl-
ben~i, . ,idaLole;
1 -tert-butoxycarbonyl-6-~N-(tert-butoxycarbonyl)p;,Jc:, idi..-4-yloxy)-2-ethylbel1~i--.ida~ole;
1 -tert-butoxycarbonyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-2-propylbenzimidazole;
30 1-tert-butoxycarbonyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-2-butylbenzimidazole;
1 -tert-butoxycarbonyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-2-isobutylbenzimidazole;
1 -tert-butoxycarbonyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-2-tert-butyl-
beu~i",;dazole; and
1 -tert-butoxycarbonyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-2-isopropyl- 4-methoxybenzimidazole.
CA 02239~08 l998-06-04
W O 97/21437 PCT~B96/01496
-46-
PREPARATION 4
Compounds of formuta (8)
A. A solution of 1-tert-butoxycarbonyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-2-
isopropylbenzimidazole (425 mg 0.92 mmol) and methanol ~10 mL) was cooled in a dry
5 ice/acetone bath and NH3 (9) was bubbled in. The reaction flask was sealed and heated to 50~C
for 12 hours. The solvent was removed in vacuo. The resulting oil was chromatooraphed on
SiO2 (50 9) using ethyl acetate to afford a clear oil (a compound of formula (6)). To a solution of
the oil and DMF (15 mL) was added NaH (28.0 mg 0.70 mmol). The solution was stirred for 1
hour before 7-bromomethyl-2-naphthonitrile (178 mg 0.72 mmol) was added. After stirring for
10 20 hours the reaction was poured into H20 (50 mL). The aclueous layer was extracted with
ethyl acetate (2x60 mL). The organic layers were washed with brine (50 mL), dried (Na2S04)
snd conce~LIt~Led. The resulting oil was chromatographed on SiO2 (20 9) with ethyl acetate to
afford 6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-2-isopropyl-1-(4-cyanonaphth-1-
yl)methyll)e~ "idazole and 5-~N-(tert-butoxycarbonyl)piperidin-4-yloxy)-2-isopropyl-1-(4-
15 cyanonaphth-1-yl)methyll,e,.~ 7nle as a 1:1 mixture of compounds.
B. In a similar manner the following compounds of formula (8) were made:
6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1-(4-cyanonaphLI,-1-yl)be~ idszole and
5-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1 -(4-cyanonaphth-1 -yl)ben~i, l ,idazole;
6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1 -(4-cyanonaphth-1 -yl)-2-methyl-
benzimidazole and 5-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1-(4-cyanonaphth-1-yl)-2-
methylbenzimidazole;
6-lN-(tert-butoxycarbonyl)piperidin-4-yloxy)-1 -(4-cyanonaphth-1 -yi)-2-trifluoromethyl-
benzimidazole and 5-(N-(tert-butoxycarbonyl),~ eridi~1-4-yloxy)-1-(4-cyanonaphth-1-yl)-2-
trifluoromethylbenzimidazole;
25 6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1-(4-cyanonaphth-1-yl)-2-ethylb~ i.,.idazole
and 5-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1-(4-cyanonaphth-1-yl)-2-
ethylbenzimidazole;
6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1-(4-cyanona~ l.-1-yl)-2-propyl-
benzimidazole and 5-(N-(tert-butoxycarbonyl),ui~,e.. 'i ~-4-yloxy)-1-(4-cyanonaphth-1-yl)-2-
propylbel-~i---idazole;
6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1 -(4-cyanonaphth-1 -yl)-2-but~Jlben~i".idazole
and 5-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1-(4-cyanonaphth-1-yl)-2-
butylbenzimidazole;
6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1 -(4-cyanonaphth-1 -yl)-2-isobutyi-
benzimidazole and 5-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1-(4-cyanonaphth-1-yl)-2-
isobutylbenzimidazole;
8-(N-~tert-butoxycarbonyl)piperidin-4-yloxy)-1 -(4-cyanonaphth-1 -yl)-2-tert-butyl-
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/01496
-47 -
benzimidazole and 5-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1-(4-cyanonaphth-1-yl)-2-
tert-but~11L en~il"ida~ole;
6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1 -(4-cyanonaphth-1 -yl)-2-isopropyl-
4-methoxybenzimidazole and 5-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1-(4-
cyanonaphth-1-yl)-2-isopropyl-4-methoxybenzimidazole;
6-(N-(terr-butoxycarbonyl)piperidin-4-yl)amino-1 -(4-cysnonaphth-1 -yl)benzimidazole;
6-(N-(tert-butoxycarbonyl)piperidin-4-yl)((methoxycarbonyl)methyl)amino-1 -(4-
cyanonaphth-1 -yl)-2-methylbenzimidazole;
6-(N-(tert-butoxycarbonyl)piperidin-4-yl)(3-(methoxycarbonyl)prop-1 -yl)amino-1 -(4-
cyanonaphth-1 -yl)-2-methylben~i,.,id~ole;
6-(N-(tert-butoxycarbonyl)piperidin-4-yl)~2-(methoxycarbonyl)prop-1 -yl)amino-1 -(4-
cyanonaphth-1 -yl)-2-methylbenzimidazole;
5-(N-(tert-butoxycarbonyl)piperidin-4-yl) (2-(methoxycarbonyl)prop-1 -yl)amino-1 -(4-
cyanonaphth-1 -yl)-2-methylbenzimidazole;
15 6-~N-(tert-butoxycarbonyl)piperidin-4-yl)amino-2-methyl-5-nitro-1 -(4-cyanonaphth-
1-yl)be,.Li",id le; and
6-(N-ltert-butoxycarbonyl)piperidin-4-yl)amino-2-isopropyl-5-nitro-1 -(4-cyanonaphth-
1 -yl)ben~i",idd~ole.
PREPARA~ION 5
Compounds of formula (10)
To a solution of 1,3,5-trifluoro-2-nitrobenzene (25.0 p, 141 mmol) and THF (30 mL)
cooled in a dry ice/acetone bsth was bubbled in NH3 (9). After saturation, the reaction tube was
sealed and warmed to ambient temperature. After stirring for 12 h, the reaction mixture was
filtered and the filtrate concentrated to afford a yellow oil. The yellow oil was dissolved in
25 methanol (150 mL). Over 1 hour, sodium methoxide (27.0 9, 500 mmol) was added. The
reaction was stirred for 3 hours, then H2O (500 mL) was added. The mixture washed with ethyl
acetate (3x700 mL). The organic layers were washed with brine (50 mL), dried (Na2SO2) and
concenL~dled to afford 3,5-dimethoxy-2-nitroaniline as a red solid.
PREPARATION 6
Compounds of formula (11)
A. To a solution of 3,5-dimethoxy-2-nitroaniline (55.0 9, 321 mmol) and pyridine(400 mL) was added isobutyryl chloride (41.0 mL, 391 mmol). After stirring for 16 hours, the
reaction mixture was conce"l,~lled in vacuo. The resulting oil was partitioned between H20
(200 mL) and ethyl acetate (200 mL~. The aqueous layer was ~xL,acLt:d with more ethyl acetate
35 (2x200 mL). The organic layers were washed with brine (50 mL), dried (MgSO4) and
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-48-
concentrated. The solid was recrystaliized from hexane/ethyl acetate at 5~C to afford yellow
crystais. To a slurry of the yellow crystals, 4N HCI ~200 mL) and ethanol (250 mL) was added
10% Pd/C (1.30 9). The slurry was placed under 50 psi of H2 (9) and shaken for 4 hours. The
mixture was then filtered and conce~lL~dLion of the filtrate afforded a white solid. The white solid
was dissolved in 2N HCI (200 mL) snd refiuxed for 4 hours. The reaction mixture was
concentrated. The resulting solid was dissolved in 48% HBr (100 mL) and H20 (100 mL) and
refluxed for 15 hours. The reaction was then cooied to ambient It:lllperaLLlre and concenL.~Led.
The solids were dissolved in H20 (200 mL) and neutralized ~KHC03). The aqueous layer was then
extrscted with n-butanol ~3x500 mL). The organic layer was washed with brine (50 mL), treated
10 with activated charcoal and concentrated to afford 4,6-dihydroxy-2-isopropylbenzimidazole, a
compound of formula (11) .
B. Alternatively, a solution of 4,6-dimethoxy-2-isopropylbenzimidazole (12.70 9,57.66 mmol) and 48% HBr ~100 mL) was refluxed for 3 hours, and then the solution was cooled
to ambient temperature and concentrated. The resulting solids were dissolved in H20 ~100 mL)
15 and neutralized ~NaHC03). A brown precipitate formed which was isolated by filtration. The
precipitate was dissolved in MeOH ~100 mL~, treated with charcoal and concellLlaL~d to afford 6-
hydroxy-2-isopropyl-4-methoxybenzimidazole as a brown solid.
PREPARATION 7
Compounds of formula (12) and (13)
To a mixture of 4,6-dihydroxy-2-isopropylbenzimidazole t36.0 g, 113 mmol~, imidazole
~35_g 9, 514 mmol) and DMF (200 mL) was added tert-butyldi~ Ll-ylsilyl chloride (40.0 9,
265 mmol). The dark mixture was stirred for 10 hours, then it was partitioned between H20
~500 mL) and ethyl acetate ~500 mL). The aqueous layer was exLld~Led with more ethyl acetate
~2x500 mO. The combined organic layers were washed with brine (100 mL), dried (Na2504),
25 treated with activated charcoal and con-;e~L~Lt:d to afford a dark oil, 4,6-
di~tetrabutyldimethylsilyl30xy-2-isopropyibenz;".id~Lole. The oil ~12.2 9, 29 mmol) was dissolved
in DMF ~150 mL) and treated with NaH (1.76 g, 44.0 mmol). After 4 hours, the solution was
placed in an ice bath, then methyl bromoacatate ~1.50 mL, 15.8 mmol) was added dropwise.
The reaction was stirred for 40 minutes, then a second portion of methyl bromoacetate ~1.20 mL,
30 12.6 mmol) was added. After stirring for 20 more minutes, the reaction mixture was added to
H20 ~300 mL). The aqueous layer was t~xLla~;Led with ethyl acetate ~3x300 mL). The combined
organic layers were washed with brine (50 mL), dried (Na2SO4) and conce--l-aLed to afford a oil.
Chromatography ~SiO2, 500 9) of the oil with hexane/ethyl acetate (1:1) afforded 2-isopropyl-4-
(carboxy)methyl-6-(tetrabutyld;"~Lhyl-silyl)oxybenzimidazole.
CA 02239~08 l998-06-04
WO 97~1437 PCT~B96/01496
-49-
PREPARATION 8
Compounds of formula (14a)
A. To a mixture of 2-isopropyl-4-~carboxy)methoxy-6-(tetrabutyldimethyl-
siiyl)oxybenzimidazole t9.00 9, 18.7 mmol) and THF (100 mL) was added di-tert-butyldicarbonate
~23.0 9, 105 mmol). The reaction mixture was refluxed for 7 hours. After cooling to ambient
temperature, it was conce~LI ~led. The resulting oil was purified by chromatography (500 9 SiO2)
with hexane/ethyl acetate (Z:1) to afford a clear oil. A solution (0.01 M) of the clear oil in THF
1300 mL) was placed in an ice bath. After a few minutes tetrabutyl ammonium fluoride (8.00 mL
of a 1.0 M solution, 8 mmol) was added. The reaction mixture was stirred for a few minutes,
10 then the reaction mixture was partitioned between H2O (300 mL~ and ether 1300 mL). The
aqueous layer was extracted with more ether 12x300 mL). The combined organic layers were
washed with brine (50 mL), dried (Na2SO4) and concentrated to afford a yellow oil.
Chromatography (10 9 SiO2) using ether afforded 1-tert-butoxycarbonyl-2-isopropyl-4-
(carboxy)methoxy-6-hydroxybenzimidazole.
B. Alternatively, to a mixture of 6-hydroxy-2-isopropyi-4-methoxyL,en i",idazole(3.44 9, 18 mmol), THF 150 mL) and H20 (50 mL) was added di-tert-butyl~ a-i onate (8.00 q,
37 mmol). The mixture was refluxed for 16 hours. ArLt: .r.c..ds, the reaction mixture was
extracted with ethyl acetate I3x50 mL). The organic layers were washed with brine, dried
tNa2SO4) and concentrated. Chromatogrsphy ~SiO2, 100 9) of the resulting oil with hexane/ethyl
20 acetate (1:1) afforded 1-tert-butoxycarbonyl-6-hydroxy-2-isopropyl-4-methoxybenzimidazole.
PREPARATION 9
Compounds of formula (16)
In a 1 L flask, 2,4,6-trifluoro-2-nitrobenzene (8.2 mL, 70 mmoL, 1.1 eq.) was added to
25 500 mL of dry aceLùniLlile and the solution chilled to -10~C. Diisoproi,ylethylamine (33 mL,
191 mmol, 3 eq) and ethyl isonipecotate (10 q, 64 mmoL, 1 eq.) were added slowly (approx.
20 min.) as a combined solution via addition funnel, under nitrogen. The reaction turned yellow
almost immediately, and it was warmed to 20~C slowly. After 5 hours at 20~C, thin layer
chromatography showed the reaction to be nearly complete (9515 hexanes/ethyl acetate). The
30 &ct5LuniL~ile was removed in vacuo and the resulting yeilow oil was Ji;.~olved in 1 L of ethyl
acetate. This solution was washed with water (3x100 mL), brine (1x300 mL), dried over sodium
sulfate, and concentrated to give 20 9 of a yellow oil. This material was identified as 6-(4-
(ethoxycarbonyl)piperidin-1-yl)-2,4-difluoro-1-nitrobenzene by lH NMR.
PREPARATION 10
Compounds of formula (17)
6-(4-(Ethoxycarbonyl)piperidin-1-yl)-2,4-difluoro-1-nitrobenzene (20 9, 64 mmoL, 1 eq)
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-50-
was dissolved in 300 mL of dry acetonitrile and the solution chilled to -10~C.
Diisopropylethylamine (33 mL, 191 nmol, 3 eq~ and benzyl amine ~7 mL, 64 mmol, 1 eq) were
added slowly (approx. 20 min.) as a combined solution via addition funnel, under nitrogen. The
reaction turned yellow almost immediately, and it was warmed to 20~C slowly. After 48 hours
at 20~C, thin layer chromatography showed the reaction was only about half con,Fl~te. The
reaction was heated to 50~C for 24 hours, cooled, the acetonitrile was removed in vacuo and the
resulting orange oil was dissolved in 1 L of ethyl acetate. This solution washed with water
(3x100 mL), brine (1x300 mL), dried over sodium sulfate, and concellL. ,Led to give 20 9 of an
orange oil. Chromatography (95/~ hexanes/ethyl acetate) gave 12 9 (48% yield) of 2-N-
10 benzylamino-6-(4-(ethoxycarbonyl)piperidin-1 -yl)-4-fluoro-1 -nitrobenzene.
PREPARATION 11
Compounds of formula (18)
2-/\/-Benzylamino-6-(4-(ethoxycarbonyl)piperidin-1-yl)-4-fluoro-1-nitrobenzene (11 9,
27 mmol, 1 eq) was added to 200 mL of dry methanol and to the mixture was added slowly
15 (approx. 20 min.) sodium methoxide (5.0 g) as a solution in 100 mL methanol, under nitrogen.
The reaction was heated to reflux for 6 hours, cooled, the methanol was removed jn vacuo and
the resulting red solid was dissolved in 1 L of ethyl acetate. This solution washed with water
~3x100 mL), brine ~1x300 mL), dried over sodium sulfate, and concentrated to give a dark red oil.
Chromato~raphy (10-50% ethyl acetate in hexanes~ gave 4.3 9 (38% yield) of 2-N-benzylamino-
20 6-(4-(methoxycarbonyl)piperidin-1-yl)-4-methoxy-1-nitrobenzene.
PREPARATION 12
- Compounds of formula (19)
2-N-Benzylamino-6-~4-(methoxycarbonyl)piperidin-1-yl)-4-methoxy-1-nitrobenzene (4.3 9,
10 mmol, 1 eq) was dissolved in 100 mL ll.t:Lllanol and to the mixture was added a few srams of
25 10% Pd/C (wet Degussa type) followed by 50 mL of 4N HCL. The mixture was hydrogenated at
50 psi for 2 hours, the solids filtered out with Celite; and the filtrate concel.L,aLed to give a light
tan solid. The HCI salt form of the diamine ~quant. yield) was identified as 1,2-diamino-6-(4-
(methoxycarbonyl)piperidin-1-yl)-4-methoxybenzene by 1H NMR.
PREPARATION 13
Compounds of formula (20)
1,2-Diamino-6-~4-(methoxycarbonyl~piperidin-1-yl)-4-methoxybenzene bis-hydrochloride
(3.7 9, 10 mmol, 1 eq) was dissolved in 100 mL dry pyridine and to the mixture was added
isobutyryl chloride (2.2 mL, 20 mmoL, 2 eq.) at 0~C. The mixture was allowed to slowly warm
to 20~C and stirred overnisht. The pyridine was removed in vacuo and the residue was dissolved
CA 02239~08 1998-06-04
W O 97~1437 PCT~B96/01496
-51-
in 500 mL of ethyl acetate. This solution was washed with water (3x100 mL), brine t1x300 mL~,
dried over sodium sulfate, and concentrated to give 3.5 9 (87% yield) of a dark brown residue.
This material was identified as 1,2-di((1-methylethyl)carbonyl)amino-6-(4-
~methoxycarbonyl)piperidin-1-yl)-~methoxybenzene by 1H NMR. The residue (3.5 9, 10 mmol, 1
eq) was dissolved in 100 mL 4N HCI and the mixture was refluxed overnight. The HCI was
removed in vacuo and the residue was neutralized to pH 7 with saturated aqueous sodium
bicarbonate. The water was removed in vacuo and the residue was triturated with THF
(4x100 mL). The combined THF fractions were dried over sodium sulfate and concentrated to
give 2.5 9 t69% yield) of a dark solid. This material was identified as 2-isopropyl-4-~4-
10 tcarboxy)piperidin-1-yl)-6-methoxybenzimidazole by ~H NMR.
PREPARATION 14
Compounds of formula ~21)
4-(~(Carboxy)piperidin-1-yl)-6-methoxy-2-isopropylben i, lidazole ~2.5 9, 7.4 mmol, 1 eq)
was dissolved in 60 mL 48% HBr and the mixture was refluxed for 3 hours. The HBr was
15 removed in vacuo and the residue was dissolved in 200 mL methanol and to this was added ZO
mL concerL,d~ed sulfuric acid. This mixture was rsfluxed overnight. The methanol was stripped
off and the residue neutralized to pH 7 with saturated aqueous sodium bicarbonate. The water
was removed in vacuo and the residue was triturated with THF (4x100 mL). The combined THF
fractions were dried over sodium sulfate and concen~ d to give 2 9 of a dark red solid. This
20 material was identified as 4-(4-(methoxycarbonyl)piperidin-1-yl)-6-hydroxy-2-isop~opylbenzimidazole by 1H NMR (with some methoxy product still remaining). Chromatography
(2.5:1 ethyl acetate/hexanes) gave the desired product as a colorless soiid, 800 mg (32%, yield).
PREPARATION 15
Compounds of formula (22)
4-(4-(Methoxycarbonyl)piperidin-1-yl)-6-hydroxy-2-isopropylbeu ;" ia7ole (800 mg,
2.4 mmol, 1 eq) was dissolved in 20 mL THF and to the solution was added di-
tert-butyldicarbonate t520 mg, 2.4 mmoL, 1 eq.) and the resulting mixture was refluxed. Thin
layer chromatography indicated that the reaction was sluggish, Ll-a~t:rore several 1 eq. aliquots of
di-tert-butyldicarbonate were added over a period of 3 days of reflux. The reaction was cooled,
30 the THF removed ;n vacuo, and the residue chromatographed (4:1 hexanes/ethylacetate) to give
1-tert-butoxycarbonyl-4-~4-(methoxycsrbonyl~piperidin-1-yl)-6-hydroxy-2-isopropylbe" ;",idd~ole
as a colorless solid, 590 mg (59%, yield).
_
CA 02239~08 1998-06-04
W O 97/21437 PCTnB96/01496
-52-
PREPARATION 16
Compounds of formula (25)
2-Methyl-5,6-dihydroxybenzimidazole (100 9) in THF (1 L) was basified to pH 8 with
aqueous NaHCO3, and to the resulting solution was then added di-tert-butyldicarbonate (104 ~).
After stirring for 3 hours, the THF was removed, and the aqueous residue was diluted and
exL~c.uLed with ethyl acetate. The organic layer was dried and concentrated to afford 1-tert-
butoxycarbonyl-2-methyl-5,6-dihydroxybenzimidszole as a white solid.
PREPARATION 17
Compounds of formula (26) and (27)
To 1-tert-butoxycarbonyl-2-methyl-5,6-dihydroxybenzimidazole (12 9) in DMF (100 mL)
was added imidazoie (6.5 g) and tert-butyldimethylsilyl chloride (7.5 g) with vigorous stirring.
After stirring for 1 hour, the reaction was worked up between ethyl acetate/water. The organic
layer was dried and concentrated to afford a mixture of 1-tert-butoxycarbonyl-2-methyl-5-(tert-
butyld;~lleLll~lsiiyl)oxy-6-hydroxybenzimidazole~ 1-tert-butoxycarbonyl-2-methyl-6-~tert-
15 butyldimethylsiiyl)oxy-5-hydroxybenzimidazole, and 1-tert-butoxycarbonyl-2-methyl-5,6-di~(tert-
butyldimethylsilyl)oxy)ben~i...ida~ole. To the mixture in THF (150 mL) was first added N-tert-
butoxycarbonyl-4-hydroxy-piperidine (12 9), PPh3115 g), and then DEAD (10 mL) in a dropwise
fashion. After stirring at ambient te.~pe.aLLlre for 1 hour the solvent was removed and the
residue was purified and the regioisomers sepa.dled by silica gel chromatography (hexane/ethyl
20 acetate, ylddienL) to afford 1-tert-butoxycarbonyl-2-methyl-5-(tert-butyld;.lle:Lll~lsilyl)oxy-6-(N
ltert-butoxYcarbonYl)piperidin-4-yloxy)benzimidazole, 1-tert-butoxycarbonyl-2-methyl-6-(tert-
butyldimethylsilyl)oxy-5-(N-(tert-butoxycarbonyl)piperidin-4-yioxy)ben~;,.,idazole, and 1-tert-
butoxycarbonyl-2-methyi-5,6-di((tert-butyld ,I~:Ll-ylsilyl)oxy)be~ Ic.
PREPARATION 18
Compounds of formula (28) and (29)
To 1-tert-butoxycarbonyl-2-methyl-5-(tert-butylJ;~ Ll-ylsilyl)oxy-6-(N-(tert-
butoxycarbonyl)piperidin-4-yloxy)be--~;.-.;da~ole (5 9) in Ill~LI~dnol (100 mL) was bubbled in
ammonia at 0~C. After stirring for 12 hours at ambient Lelll~JeldLIJre in a sealed vessel the
30 reaction mixture was concenL,dLl:d and dried to afford 2-methyi-5-hydroxy-6-(N-(tert-
butoxycarbonyl)piperidin-4-yloxy)ben~;.,.;dazole (a compound of formula (28)). To the resulting
product in DMF was added imidazole (0.66 g) and (tert-butyldimethylsilyl)chloride (1.5 g). After
stirring at ambient temperature for 1 hour the reaction was worked up between ethyl acetate and
H20. The organic layer was dried, conce~L~dLed and purified by silica gel chlullld~uylaphy
35 (hexane/ethyl acetate/CH2CI2/methanol, y- ~jLnL) to afford 2-methyl-5-(tert-
butyldimethylsilyl)oxy-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)benzimidazole.
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-53-
PREPARATION 19
Compounds of formula (30)
To 2-methyl-5-(tert-butyldimethylsilyl3Oxy-6-~N-(tert-butoxycarbonyl)piperidin-4-
yloxy)benzimidazole ~6.6 9) in DMF (50 mL) was added NaH (0.65 9~ at ambient temperature.
After stirring at ambient temperature for 30 minutes 7-bromomethyl-2-naphthonitrile (3.88 9) was
added. The reaction mixture was stirred at ambient temperature for 1 hour. The reaction was
worked up between ethyl acetate and ~'2~ The organic layer was dried concentrated and
purified by silica gel chromatography ~hexane/ethyt acetate/CH2CI2/methanol gradientl to afford
2-methyl-5-(tert-butyldimethylsilyl~oxy-6-(N-(tert-butoxycarbonyl~piperidin-4-yloxy)-1 -(4-
10 cyanonaphth-1-yl)methylbenzimidazole and 2-methyl-6-(tert-butyldimethylsilyl)oxy-5-(N-(tert-
butoxycarbonyl~piperidin-4-yloxy)-1 -(4-cyanonaphth-1 -yl~methylbenzimidazole.
PREPARATION 20
Compounds of formula (31)
To 2-methyl-5-(tert-butyldimethylsilyl)oxy-6-(N-(tert-butoxycarbonyl~piperidin-4-yloxy)-1-
15 (4-cyanonaphth-1 -yl)methylbenzi". ~7nle (3 9) in THF (50 mL) was added tetrabutyl ammonium
fluoride (3 mL 1 M)) at ambient temperature. After stirring at ambient temperature for 30
minutss the solvent was removed. The reaction was woriced up between ethyl acetate and H2O.
The organic layer was dried and conce~L~nl~:d to afford 2-methyl-5-hydroxy-6-(N-(tert-
butoxycarbonyl)piperidin-4-yloxy)-1-(4-cyanonaphth-1-yl)methylbe-, ;",idazole.
PREPARATION 21
Compounds of formula (32)
A. To 2-methyl-5-hydroxy-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1-(4-
cyanonaphth-1-yl)methylbenzimidazole (1.11 9) in DMF (50 mb) was added NaH (0.15 9) at
ambient temperature. After stirring at ambient for 30 minutes ethyl bromoacetate (0.4 m~) was
25 added. The reaction was stirred at ambient L.:",per ,L~3re for 1 hour and was worked up between
ethyl acetate and H20. The organic layer was dried, concenll~Le:d and purified by silica gel
chromatography (hexane/ethyl acetate/CH2CI2/methanol, ~ L) to afford 2-methyl-
5-(ethoxycarbonylmethoxy)-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1 -(4-cyanonaphth-1 -
yl)methylbenzimidazole.
B. In a similar manner, the following compounds of formula (32) were made:
2-methyl-5-(1 -(methoxycarbonyl)ethoxy)-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy) -
1 -(4-cyanonaphth-1 -yl)methylbenzimidazole;
2-methyl-5-(4-(methoxycarbonyl)benzyloxy)-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-
1-(4-cyanonaphth-1-yl)methylbenzimidazoie; and
35 2-methyl-5-(3-(methoxycarbonyl)benzyloxy)-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-
.
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/01496
-54-
1 -~4-cyanonaphth- 1 -yl)methylbenzimidazole.
PREPARATION 22
Compounds of formula (33)
To 2-methyl-6-(tert-butyldimethylsilyl)oxy-5-(N-(tert-butoxycarbonyl)piperidin~-yloxy)-1-
(4-cyanonaphth-1-yl~methylbenzimidazole (3 9) in THF (50 mL) was added Bu4NF (4 mL, 1 M)~ at
ambient temperature. After stirring at ambient temperature for 30 minutes the solvent was
removed. The reaction was worked up between ethyl acetate and H20. The organic layer was
dried, concentrsted and purified by silica gcl chromatography (hexane/ethyl
acetate/CH2CI2/methanol, gradient) to afford 2-methyl-6-hydroxy-5-(N-(tert-
10 butoxycarbonyl)piperidin-4-yloxy)-1 -~4-cyanonaphth-1 -yl)methylbenzimidazole.
PREPAP~ATION 23
Compounds of formula (34)
To 2-methyl-6-hydroxy-5-~N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1-(4-cyanonaphth-1-
yl)methylbenzimidazole (2.8 9) in THF (50 mL) was first added N-benzyloxycarbonyl-4-
15 hydroxypiperidine (1.7 9), PPh3 (1.9 9~, and then DEAD ~1.2 mL~ in a dropwise fashion. After
stirring at ambient temperature for 1 hour the solvent was removed and the residue was
separated by silica gel chromatography ~hexane/ethyl acetate, gradient) to afford 2-methyl-6-~N-
(benzyloxycarbonyl)piperidin-4-yloxy)-5-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1 -(4-
cysnonaphth-1-yl)methylben i-~-;da ole.
PREPARAT~ON 24
Compounds of formula (35) and (36)
To 2-methyl-6-(N-(benzyloxycarbonyl)piperidin-4-yloxy)-5-(N-(tert-butoxycarbonyl)piperidin-
4-yloxy)-1-(4-cyanonaphth-1-yl)methylbenzimidazole (4 9) in CH2CI2/methanol ~100 mL, 9:1, v/v)
was added trifluoroacetic acid (20 mL). After stirring for 6 hours at ambient temperature the
25 reaction was concenLraLt:d to afford the trifluoroacetic acid salt of 2-methyl-6-(piperidin-4-yloxy)-
~;-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-1-(~cyanonaphth-1-yl)methylbenzimidazole (a
compound of formula (35)). To the resulting product in THF (50 mL) was added ethyl
bromoacetate (0.72 mL) and K2CO3 (5 g). After stirring at ambient temperature for 1 hour the
reaction was worked up between ethyl acetate and H20. The organic layer was dried,
30 concenL-~Led and purified by silica gel chromatography (hexane/ethyl acetate/CH2CI2/methanol,
~J,adienL) to afford 2-methyl-6-(N-(ethoxycarbonyl-methyl)piperidin-4-yloxy)-5-(N-(tert-
butoxycarbonyl)piperidin-4-yloxy)-1 -(4-cyanonaphth-1 -yl)methylbenzimidazole.
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
PREPARATION 25
Compounds of formula (38)
A.To 5-amino-2-methoxycarbonylphenoi ~60 9) in pyridine (200 mL) was added
acetic anhydride (70 mL) at ambient temperature. After stirring at ambient temperature for 14
hours the solvent was removed to afford 5-acetylamino-2-methoxycarbonyl-1-acetoxybenzene.
B. In a similar manner, the following compound of formula (38) was prepared:
5-(2-methyl-1 -oxopropyl)amino-2-methoxycarbonyl-1 -(2-methyl-1 -oxopropoxy~benzene.
PREPARATlON 26
Compounds of formula (39)
A. To 5-acetylamino-2-methoxycarbonyl-1-acetoxybenzene (20 g) in trifluoroaceticacid (150 mL) was added HNO3 (5 mL) at ambient temperature. After stirring at ambient
temperature for 2 hours the solvent was removed. The resulting residue was recrystallized from
ethyl acetate to afford a mixture of 5-acetylamino-4-nitro-2-methoxycarbonyl-1-hydroxybenzene
and 5-acetylamino-6-nitro-2-methoxycarbonyl-1-hydroxybenzene.
B. In a similar manner, the following compounds of formula (39) were made:
5-(2-methyl-1-oxopropyl)amino-4-nitro-2-methoxycarbonyl-1-hydroxybenzene: and
5-(2-methyl-1 -oxopropyl)amino-6-nitro-2-methoxycarbonyl-1 -hydroxybenzene.
PREPARATION 27
Compounds of formulae (41)
A. To a mixture of 5-acetylamino-4-nitro-2-methoxycarbonyl-1-hydroxybenzene and
5-acetylamino-6-nitro-2-methoxycarbonyl-1-hydroxybenzene (15 9) in methanol (50 mL) was
added Pd/C (10 9). The resulting mixture was hydrogenated at 50 psi for 3 hours until H2 intake
d "inished. The catalyst was filtered off and the filtrate was concen~l~Led to afford a mixture of
5-acetylamino4-amino-2-methoxycarbonyl-1-hydroxybenzene and 5-acetylamino-6-amino-2-
25 methoxycarbonyl-1-hydroxybenzene. This mixture was then refluxed in acetic acid (200 mL) for
2 hours. After removal of acetic acid the residue was worked up between ethyl acetate and
aqueous NaHCO3. The organic layer was dried and concenLl~led to afford a mixture of 2-methyl-
6-hydroxy-5-methoxy-carbonylbenzimidazole and 2-methyl-4-hydroxy-5-methoxycarbonyl-
ben~ 701e.
B. In a similar manner, the following compound of formulae (41 ) was made:
2-isopropyl-6-hydroxy-5-methoxycarbonylbenzimidazole.
PREPARATION 28
Compounds of formula (42)
To a mixture of 2-methyl-6-hydroxy-5-methoxycarbon~lL,erl~illlidazole and 2-methyl-4-
CA 02239~08 1998-06-04
W O 97~1437 PCT~B96/01496
-56-
hydroxy-5-methoxycarbonylbenzimidazole (7.2 9) in THF (50 mL) was added di-tert-butyldicarbonate (7.6 9) and triethylamine (5 mL). After stirring at 50~C for 6 hours the solvent
was removed and the residue was triturated with ethyl acetate and filtered to afford 1-tert-
butoxycarbonyl-2-methyl~6-hydroxy-5-methoxycarbonylbenzimidazole and 1-tert-butoxycarbonyl-
5 2-methyl-4-hydroxy-5-methoxycarbonylbenzimidazole.
PREPARATlON 29
Compounds of formula (43)
To a mixture of 1-tert-butoxycarbonyl-2-methyl-5-hydroxy-6-methoxycarbonyl-
benzimidazole and 1-tert-butoxycarbonyl-2-methyl-6-hydroxy-5-methoxycarbonylbenzimidazole
10 (3.7 9) in THF (50 mL) was first added N-tert-butoxycarbonyl-4-hydroxypiperidine (3.2 9), PPh3
(4.2 9) and then DEAD (2.5 mL) in a dropwise fashion. After stirring at ambient temperature for
4 days the solvent was removed and the residue was separated by silica gel chromatography
(hexane/ethyl acetate/CH2C12/methanol, glt~.l;enL) to afford a mixture of 1-tert-butoxycarbonyl-
2-methyl-5-methoxycarbonyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)ben~i",ida~ole and 1-tert-
15 butoxycarbonyl-2-methyl-6-methoxycarbonyl-5-(N-(tert-butoxycarbonyl)piperidin-4-
yloxy) benzimidazole.
PREPARATION 30
Compounds of formulae (44) and (45)
To a mixture of 1-tert-butoxycarbonyl-2-methyl-5-methoxycarbonyl-6-(N-~tert-
20 buto~cycarbonyl)piperidin-4-yloxy)benzimidazole and 1-tert-butoxycarbonyl-2-methyl-6-
methoxycarbonyl-5-(N-(tert-butoxycarbonyl~piperidin-4-yloxy)ben~ 7Ole (2.5 9~ in methanol
~200 mL) was bubbled in ammonia at 0~C. After stirring for 2 hours at ambient temperature the
reaction mixture was concentrated. The resulting residue was purified by silica geJ
chromatography (hexane/ethyl acetate/CH2C12/methanol, ~, ~ ,L) to afford 2-methyl-5-
25 methoxycarbonyl-6-tN-(tert-butoxycarbonyl)F . i.li,l-4-yloxy)ben~i", '~701e. To this product
(0.66 9) in l:)MF (20 mL) was added NaH (0.075 9) at ambient te"",e~Lure. After stirring at
ambient temperature for 30 minutes 7-bromomethyl-2-naphthonitrile (0.5 9~ was added at 0~C.
The reaction mixture was stirred at ambient temperature for 0.5 hours. The reaction was worked
up between ethyl acetate and H2O. The organic layer was dried and concentrated to afford a
30 mixture of 1-(4-cyanonaphth-1-yl)methyl-2-methyl-5-methoxycarbonyl-6-(N-(tert-
butoxycarbonyl)piperidin-4-yloxy) benzimidazole and 1 -(4-cyanonaphth- 1 -yl)methyl-2-methyl-6-
methoxycarbonyl-5-(N-(tert-butoxycarbonyl)pi~ue,idi-,-4-yloxy)be,)~ ida~ole in a ratio of 2 to 1,
respectively.
CA 02239~08 1998-06-04
W O 97/21437 PCTnB96/01496
-57-
PREPARATION 31
Compounds of formulfl (46)
To 3,5-dinitro-1-carboxy-2-chlorobenzene (53 g) in acetonitrile I500 mL) and triethylamine
(100 mL) was added benzylamine at 0~C in a dropwise fashion. The reaction was allowed to
5 warm to ambient temperature and stirred at ambient temperature for 1 hour. The solvent was
removed under reduced pressure to afford the crude product, 3,5-dinitro-1-carboxy-2-
(benzyl~aminobenzene. The product was dissolved in methanol/12N HCI (400 mL, 3:1, v/v), and
was hydrogenated at 60 psi for 3 hours until H2 intake stopped. The solvent was removed under
reduced pressure to afford the crude product, 2,3,5-triamino-1-carboxybenzene. To the product
10 in pyridine ~400 mL) was added isobutyric anhydride (200 mL), and the reaction mixture was
stirred at ambient temperature for 14 hours. The sotvent was removed under reduced pressure to
afford the crude product, 2,3,5-tri(isopropylcarbonylamino)-1-carboxybenzene. The product was
refluxed in methanol/12N HCI (500 mL, 4:1, v/v) for 16 hours to afford the desired product, 2-
isopropyl-4-methoxycarbonyl-6-aminobenzimidazole, after removal of the solvents under reduced
15 pressure.
PREPARATION 32
Compounds of formula (48)
To 2-isopropyl-4-methoxycarbonyl-6-aminoben~i-,-ida~ole in methanol/acetic acid (300 mL,
2:1, v/v) was added N-tert-butoxycarbonyl-4-piperidone (80 9), foilowed by NaCNBH4 (9 9) at
20 ambient temperature. After stirring st ambient l~:r,-pe-dL--re for 1 hour the solvents were removed
and~the resulting residue was worked up between ethyl acetate and H20. The organic layer was
dried, concentrated, and purified by silica gel chromatography (hexane/ethyl
acetate/CH2CI2/methanol, gradient) to afford 2-isopropyl-4-methoxycarbonyl-6-(N-(tert-
butoxycarbonyl)piperidin-4-ylamino)benzi,ll ~ ~le as a foam.
PREPARATION 33
Compounds of formula (49)
To 2-isopropyl-4-methoxycarbonyl-6-(N-(tert-butoxycarbonyl)piperidin-4-
ylamino)ben~i...idazole (3.3 9) in DMF (200 mL) was added NaH (0.35 9) at ambient temperature.
After stirring at ambient temperature for 30 minutes, the reaction flask was cooled to -10~C and
30 7-bromomethyl-2-naphthonitrile (2.2 g) was added. The reaction was allowed to warm to
ambient ltemperature and stirred at ambient temperature for 15 hours. The reaction was worked
up between ethyl acetate and H20. The organic layer was dried, concenl~ d and purified by
silica gel chromatography (hexane/ethyl acetate/CH2CI2/methanol, gradient) to afford 1-(4-
cyanonaphth-1 -yl)methyl-2-isopropyl-4-methoxycarbonyl-6-(N-(tert-butoxycarbonyl)piperidin-4-
35 ylamino)benzimidazole, and 1-(4-cyanonaphth-1-yl)methyl-2-isopropyl-7-methoxycarbonyl-5-(N-
CA 02239~08 1998-06-04
WO 97~1437 PCT~B96/01496
-58-
(tert-butoxycarbonyl) -piperidin-4-ylamino) benzimidazole.
PREPARATION 34
Compounds of formula (51)
To a solution of 5-aminobenzimidazole (5.2 9, 40 mmol)"V-tert-butoxycarbonyl-4-
h piperidone ~8.0 9, 40 mmol), and acetic acid ~3 mL) in methanol (250 mL) snd methyiene chloride
(50 mL) at 0~C was added sodium cyanoborohydride (3.77 9, 60 mmol). The solution was
stirred for 20 minutes at 0~C when it was allowed to warm to ambient temperature. After 12
hours, the reaction was worked up wlth ethyl acetate and water, dried ~MgSO4), and
chrul,la~ugraphed on silica gel eluting with 7% methanol in methylene chloride to recover 9.47 9
10 (77%) of 5-((N-(tert-butoxycarbonyl)piperidin-4-yl)amino)ben~ da~ole.
PREPARATION 35
Compounds of formula (52) and (54)
A. To a solution of 5-~lN-(tert-butoxycarbonyl)piperidin-4-yl)amino)benzimidazole
11.50 9, 4.75 mmol) in DMF (30 mL) at 0~C was added NaH (210 mg, 5.2 mmol). The solution
15 was stirred for 0.5 hours when tosyl chloride (1.0 9, 5.2 mmol) was added. The reaction mixture
was stirred for 1 hour at 0~C when it was worked up with ethyl acetate and water, dried
(MgSO4), and concenLIaLt:d to an oil to afford 1-tosyl-5-((N-(tert-butoxycarbonyl)piperidin-4-
yl)amino)benzimidazole and its re~ioisoll,er, 1-tosyl-6-((N-(tert-butoxycarbonyl)piperidin-4-
yl)amino)ben~i"l;da~ . The resulting oil was taken up in DMF (30 mL) when it was charged with
20 K2CO3 (4 9) and methyl iodide (3 mL). After 12 hours at ambient temperature, the reaction
mixture was worked up with ethyl acetate and water, dried (MgSO4), and concentrated to an oil.
The resulting tosylated product, 1-tosyl-5-((N-(tert-butoxycarbonyl)piperidin-4-yl)(methyl)amino)benzimidazole and the corresponding regioi:.o."er, was taken up in methanol
(80 mL) and NaOH (0.3 9, 7.5 mmol) was added. After 1 hour at ambient temperature, the
25 methanol was removed in vacuo and the product recovered via chromatography on silica gel
sluting with 5% to 10% methanol in methylene chloride to give 473 mg (30%) 5-((N-(tert-
butoxycarbonyl) piperidin-4-yl) (methyl) amino) beu~i" ,;dazole .
B. In a similar manner, to a solution of 1-tosyl-2-methyl-5-((N-(tert-
butoxycarbonyl)piperidin-4-yl)amino)ben~i,..idazcl~ (2.7 9, 5.58 mmol) in DMF (15 mL) was added
30 K2CO3 (3.85 g, 28 mmol) and methyl 3-bromo-2-methylene-propionate (1 9, 5.58 mmol). The
solution was stirred at ambient temperature for 12 hours when it was worked up with ethyl
acetate and water, dried (MgSO4), and conce"l-aLed to recover a foam (2.~ 9) containing 1-tosyl-
2-methyl-~-(N-(tert-butoxycarbonyl)piperidin-4-yl)(~2-methoxycarbonyl)prop-2-en-1 -
yl)aminobenzimidazole and trace starting mate~ial and DMF.
35 C. To a solution of the product of Pa"l~,(aph B above, 1-tosyl-2-methyl-5-(N-(tert-
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/01496
-59-
butoxycarbonyl)piperidin-4-yl)(2-~methoxycarbonyl)prop-2-en-1-yl)aminobenzimidazole, (2.5 9) in
ethanol (100 mL) and ethyl acetate (30 mL3 was charged with Raney nickel (1 mL of a 50%
water siurry) and subjected to an atmosphere of hydrogen (20 psi). After 6 hours, the catalyst
was filtered off, the solution dried (M~SO4) and concentrated to a solid to recover 1.5 g of 1-
tosyl-2-methyl-5-(/\/-(tert-butoxycarbonyl)piperidin-4-yl)(2-(methoxycarbonyl)prop-1-
yl)aminobenzimidazole.
D. To a solution of 1-tosyl-2-methyl-5-(N-(tert-butoxycarbonyl)piperidin-4-
yl)aminoben~iml~ole (6.1 9, 12.1 mmol) and triethylamine (3.4 mL, 24 mmol) in methylene
chloride (30 mL) at 0~C was added ethyl succinyl chloride (2.1 mL, 14.5 mmol). After 2.5 hours,
10 the solution was filtered and extracted with water and 1 M NaOH to recover 1-tosyl-2-methyl-5-
(N-(tert-butoxycarbonyl)piperidin-4-yl)((2-(ethoxycarbonyl)ethyl)carbonyl)aminobenzimidazole.
E. In a manner similar to that described above in Paragraph A wherein the compound
is treated with sodium hydroxide in methanol, the following compounds of formula (55) were
made:
15 2-methyl-5-(N-(tert-butoxycarbonyl)piperidin-4-yl)(2-~methoxycarbonyl)prop-1 -
yl)aminoben~;"-idazole;
2-methyl-5-(N-(tert-butoxycarbonyl)piperidin-4-yl)((methoxycarbonyl)-
methyl)aminobenzimidazole:
2-methyl-5-(N-(tert-butoxycarbonyl)piperidin-4-yl)((2-(ethoxycarbonyl)ethyl)-
carbonyl)a",i"oben~ -idazole; and
2-methyl-5-(N-(tert-butoxycarbonyl)p;,~,eridill-4-yl)(3-tmethoxycarbonyl~-
prop-1 -yl)aminobenzimidazole.
PREPARATION 36
Compounds of formula (56)
To a solution of 1,2-diamino-4-methoxybenzene (50 9) in pyridine (400 mL) at 0~C was
added isobutyryl chloride (100 mL). After 3 hours, the solvent was removed in vacuo and the
solid partitioned between methylene chloride and wster. The orqanic layer was concentrated and
taken up in 4 N HCI, and then heated to reflux overnight. The solution was concentrated to a
solid, neutralized with sodium bicarbonate, then conce,.ll~L~d once again to a solid. The solid
30 was washed with THF to recover 2-isopropyl-5-methoxy-6-nitrobenzimidazole (29 S~)-
PREPARATION 37
Compounds of formula (57)
A. 2-lsopropyl-5-methoxyben~illl;da~ole (50 9) was dissolved in 150 mL TFA and to
the mixture was added 5 mL 90% nitric acid. The reaction was stirred overnight, and the
35 solvents removed in vacuo and the residue neutralized to pH 7 with saturated aqueous sodium
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/01496
-60-
bicarbonate. The residue was extract~d with ethyi acetate (4x100 mL). The combined ethyl
acetate fractions were dried over sodium sulfate and concentrated to give a dark residue.
Chromatography (3% methanol/methylene chloride) gave 2-isopropyl-4-nitro-5-
methoxybenzimidazole as a yellow solid, 24 9. Also obtained was 25 9 of 2-isopropyl-5-nitro-6-
5 methoxybenzimidazole.
B. In a similar manner, the following compound of formula (57) was made:
2-methyl-4-nitro-5-methoxybenzimidazole and 2-methyi-5-nitro-6-methox~,L,e,.~i".idazole.
C. Alternatively, to a solution of 2-isopropyl-5-methoxybenzimidazole (50 9~ in
trifluoroacetic acid (150 mL) at ambient temperature was added fuming HNO3 (15 mL). The
10 reaction was stirred overnight, the solvent removed in vacuo and the residue quenched with
potassium carbonate. The aqueous layer was then extracted with ethyl acetate, the organic layer
dried and concentrated to recover a mixture of the regioisomers, 2-isopropyl-6-methoxy-
5-nitrobenzimidazole and 2-isopropyl-4-nitro-5-met~oxyl,el1~i".idazole (3:1 ratio) and. The
regioisomers were separated by chromatography (3% methanol/methylene chloride) to recover 2-
15 isopropyl-6-methoxy-5-nitrobenzimidazole (24 9) .
PREPARATION 38
Compounds of formula (58)
A. A solution of 2-isopropyl-6-methoxy-5-nitrobenzimidazole (t7 9) in concentrated
HBr (300 mL) was refluxed for 7 hours when the solvent was removed in v~cuo. The residue
20 was quenched with potassium carbonate and extracted with ethyl acetate. The organic layer was
dried and conce~ Led to recover 2-isopropyl-6-hydroxy-5-nitroben~;.,-id~,~ole as well as an HBr
addition product (approx. 2:1 ratio).
B. In a similar manner, the following compound of formula ~58) was made:
2-methyl-6-hydroxy-5-nitrobenzimidazole;
25 2-methyl-5-hydroxy-4-nitroben~i",idazole; and
2-isopropyl-5-hydroxy-4-nitroben~il, ~idazole .
PREPARATION 39
Compounds of formula (59)
A. To a solution of Z-isopropyl-6-hydroxy-5-nitrobenzimidazole and the HBr addition
30 product (15 9), N-tert-butoxycarbonyl-4-hydroxypiperidine (17.7 9), PPh3 (23.1 9), and THF
(150 mL) at room temperature was added DEAD (14 mL). After 4 hours, the solvent was
removed in vacuo and the product recovered from the crude using chromatography (3%
methanol/methylene chloride) to yield 2-isopropyl-5-(/~/-(tert-butoxycarbonyl)p;~,e.idin-4-yloxy)-6-
ni~"~ben~i,--idaZole (16-5 Ç1)-
35 B. In a similar manner, the following compound of formula (59) was made:
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/Ot496
-61-
2-methyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy~-5-nitrobenzi" '~7c 1e;
2-methyl-5-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-4-nitrobenzimidazole; and
2-isopropyl-5-~N-(tert-butoxycarbonyl)piperidin-4-yloxy)-4-nitroben~ 701~.
PREPARATION 40
Compounds of formula (60)
A. To a solution of 2-isopropyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-5-
nitrobenzimidazole (16.5 9) in DMF (50 mL) at 0~C was added NaH (2.45 9). After 40 minutes,
7-bromomethyl-2-naphthonitrile (11 9) was added and the reaction stored in the freezer (-35~C)
overnight. The reaction was worked up with ethyl acetate and water and the organic layer dried
10 and concentrated to an oil. The product was isolated using 2% methanol/methylene chloride on
silica gel to give a mixture of 1-(4-cyanonaphth-1-yl)methyl-2-isopropyl-6-(N-(tert-
butoxycarbonyl)piperidin-4-yloxy)-5-nitrobenzimidazole and 1-(4-cyanonaphth-1-yl)methyl-2-
isopropyl-5-(N-(tert-butoxycarbonyl~piperidin-4-yloxy)-6-nitroben~ idazole (2.5:1 ratio, 16.5 9).
B. In a s;milar manner, the following compound of formula (60) was made:
15 1-14-cyanonaphth-1-yl)methyl-Z-methyl-6-(N-(tert-butoxycarbonyl)p;~ edd ~-4-yloxy)-
5-nitrobenzimidazole;
1 -(4-cyanonaphth-1 -yl)methyl-2-methyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-
7-nitrobenzimidazole; and
1 -(4-cyanonaphth-1 -yl)methyl-2-isopropyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-
7-nitroben~;~ "ida~ole.
PREPARATION 41
Compounds of formula (61)
A. 1-(4-Cyanonaphth-1-yl)methyl-2-isopropyl-7-nitro-8-(N-(tert-
butoxycarbonyl)piperidin-4-yloxy)ben~i,l-idazole (12 9, 21 mmoL, 1 eq.) was dissolved in 400 mL
25 dry pyridine and to this was added tin ~ chloride dihydrate (71 9, 315 mmoL, 15 eq.) which
formed a finely divided suspension. The slurry was heated to 50~C for 14 hours. The pyridine
was stripped off, and the salts triturated in 1 L ethyl acetate. The salts were removed by
filtration through Celite and the filtrate was conct:~LlaLed to give a
brown oil. Chromatography (3/2 ethyl acetate/hexanes) gave 7.6 9 (67% yield) of 1-~4-
30 cyanonaphth-1-yl)methyl-2-isopropyl-7-amino-6-(N-(tert-butoxycarbonyl)piperidin-4-
yloxy)benzimidazole as a tan foam.
B. Alternatively, to a solution 1-(4-cyanonaphth-1yl)methyl-2-isopropyl-5-(N-(tert-
butoxycarbonyl)piperidin-4-yloxy)-6-nitroben~ildida~Gle and 1-(4-cyanonaphth-1-yl)methyl-2-
isopropyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-5-nitrObell~illlidd~ole (16.5 9) in
CA 02239~08 1998-06-04
W O 97/21437 PCT/IB96/01496
-~2-
triethylamine (100 mL) and pyridine (50 mL) was added SnCi2 H2O ~32.7 9). The reaction was
warmed to 60~C for 3 hours when the solvent was removed in vacLlo. The solid was taken up in
ethyl acetate and filtered. The ethyl acetate was removed ;n vacuo and the product isolated
using siiica gel eluting with 5% methanol/methylene chloride to recover 1-(4-cyanonaphth-1-
5yl)methyl-2-isopropyl-6-(N-~tert-butoxycarbonyl)piperidin-4-yloxy)-5-aminobenzimidazote (5.7 9).
C. In a similar manner, the following compound of formula (61) was made:
1 -(4-cyanonaphth-1 -yl)methyl-2-methyi-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-
5-aminobenzi."i '~7-~1e.
PREPARATION 42
10Compounds of formula (62)
A. 1-(4-cyanonaphth-1-yl)methyl-2-isopropyl-7-amino-6-(N-(tert-
butoxycarbonyl)piperidin-4-yloxy)benzimidazole (5.5 9, 10 mmol, 1 eq.) was dissolved in 50 mL
dry DMF and to this was added methyl bromoacetate (1.2 mL, 12 mmoL, 1.2 eq.) and then
potassium carbonate (1.4 9, 1.2 eq.). The reaction was stirred at 20~C for 2 days. TLC
15 indicated the reaction to be nearly complete. The reaction mixturs was worked up between
water and ethyl acetate. The ethyl acetate was removed in v,~cuo and the product was isolated
by chromatography (3/2 hexanes/ethyl acetate) to afford 5 g of 1-(4-cyanonaphth-1-yl)methyl-
2-isopropyl-7-((methoxycarbonyl)methyl)amino-6-(N-~tert-butoxycarbonyl)piperidin-4-
yloxy)benzimidazole with a smali amount of the diaddition product.
B. Alternatively, to a suspension of 1-(4-cyanonaphth-1-yl)methyl-2-isopropyl-
6-~N-(tert-butoxycarbonyi~piperidin-4-yloxy)-5-aminobenzimidazole (500 mg), potassium carbonate
(642 mg), and DMF was added methyl 4-bromomethylbenzoate 1212 mg). After 2 days at
ambient temperature, the reaction was worked up with ethyl acetate and water. The organic
layer was dried and the product isolated on silics qel using 2% methanol/methylene chloride to
25 recover 1-(4-cyanonaphth-1-yl)methyl-2-isopropyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-5-
(N-(4-me2hoxycarbonylbenzyl)-amino)benzimidazole (440 mg).
C. In a similar manner, the following compounds of formula (62) were made:
1 -(4-cyanonaphth- 1 -yl)methyl-2-isopropyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-
5-(N-(2-methoxycarbonylethyl)amino)benzimidazole;
30 1-(4-cyanonaphth-1-yl)methyl-2-isopropyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-
5-(N-(1-methoxycarbonyl-1-methylethyl)amino)be-, i,..idd ole;
1 -(4-cyanonaphth-1 -yl)methyl-2-methyl-6-(N-(tert-butoxycarbonyl)piperidin-4-yloxy)-
5-(N-(4-methoxycarbonylbenzyl)amino)benzimidazole; and
1 -(4-cyanonaphth-1 -yl~methyl-2-isopropyl-6-(N-(tert-butoxycarbonyl)Piperidin-4-yloxy)-
5-lN-(4-methoxycarbonylbenzyl)amino)benzimidazole.
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-63-
EXAMPLE 1
Compounds of formula (la)
A. To a slurry of 1-(4-cyanonaphth-1-yl)methyl-2-isopropyi-6-(N-(tert-
butoxycarbonyl)piperidin-4-yloxy~benzimidazole and 1-(4-cyanonaphth-1-yl)methyl-2-isopropyl-5-
(N-ltert-butoxycarbonyl~Piperidin-4-yloxy)bell~inlidc- ole (320 mg, 62 mmoi) and ethanol (10 mL)
cooled in a dry ice/acetone bath was bubbled HCI (9). After the solution was saturated, the
reaction flask was sealed and the temperature maintained at 5~C for 16 hours. The solvent was
removed in vacLto. The residue was dissolved in ethanol (20 mL~. The solution was cooled in a
dry ice/acetone bath and ammonia (g) was bubbled in. The reaction flask was sealed then heated
10 at 80~C for 4 hours. The solvent was removed. The mixture was separated by HPLC on a C18
Dynamax column with a 5-20% ace~onil,ile in water gradient with 0.1 % trifluoroacetic acid to
afford 1-(4-amidinonaphth-1-Yl~methyl-2-isopropyl-6-(piperidin-4-yloxy~benzimidazole
ditrifluoroacetate salt, 1H NMR (300 MHz, DMS0) ~ 9.40 (s, 2H), 9.20 (s, 2H), 8.60 (br s, 2H),
8.40 (s, 1H), 8.18 (d, 1H), 8.14 (d, 1H), 7.90-7.75 (m, 3H), 7.60 (m, 1H~, 7.40 (s, 1H), 7.25
15 (dd, 1H), 6.05 (s, 2H), 4.70 (m, 1H~, 3.65 (septuplet, 1H), 3.30-3.20 (m, 2H), 3.15-3.00 (m,
2H), 2.18-2.00 ~m, 2H~, 1.90-1.80 (m, 2H), 1.40 (d, 6H); and 1-(4-amidinonaphth-1-yl)methyl-2-
isopropyl-5-(piperidin-4-yloxy)benzimidazole ditrifluoroacetate salt,1H NMR (300 MHz, DMS0) ~
9.40 (s, 2H), 9.20 (s, 2H), 8.60 (br s, 2H), 8.40 (s, 1H), 8.18 (d, 1H), 8.14 (d, 1H), 7.90- 7.75
(m, 3H), 7.65-7.60 (m, 2H), 7.25 (dd, 1H), 6.05 (s, 2H), 4.70 (m, 1H), 3.65 (septuplet, 1H),
20 3.30-3.20 (m, 2H), 3.15-3.00 (m, 2H), 2.18-2.00 (m, 2H), 1.85-1.70 (m, 2H), 1.35 (d, 6H).
B. In a similar manner, the following compounds were made:
1-(4,amidinonaphth-1-yl)methyl-2-propyl-6-(piperidin-4-yloxy~ben i,lli ~ole, 'H
NMR (300 MHz, DMS0~ ~ 9.40 (s, 2H~, 9.20 (s, 2H), 8.60 (br s, 1H), 8.40 (s, 1 H), 8.18
(d, 1H), 8.10 (s, 1H), 7.95 (s, 1H~, 7.85 (d, 1H), 7.75 (d, 1H~, 7.65 (d, 1H~, 7.40 (s,
1H), 7.20 (dd, 1H), 5.95 (s, 2H), 4.70 (m, 1H), 3.30-3.00 (m, 6H), 2.18-2.00 (m, 2H),
1.85-1.70 (m, 4H), 0.90 (t, 3H);
1-(4-amidinonaphth-1-yl~methyl-2-propyl-5-(piperidin-4-yloxy)benzimidazole, 1H NMR (300 MHz,
DMS0) ~ 9.40 (s, 2H), 9.20 (s, 2H), 8.60 (br s, 2H), 8.40 (s, lH~, 8.18 (d, 1H), 8.14 (d,
1H~, 7.90- 7.80 (m, 3H), 7.65 (d, 1H), 7.60 (s, 1H), 7.25 (dd, 1H), 6.00 (s, 2H), 4.70
(m, 1H), 3.30-3.00 (m, 6H), 2.18-2.00 (m, 2H), 1.85-1.70 (m, 4H~, 0.90 (t, 3H~;
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-4-methoxy-6-(piperidin-4-yloxy)benzimidazole,
tH NMR (300 MHz, DMS0) ~ 9.40 (s, 2H), 9.20 (s, 2H), 8.60 (br s, 2H), 8.40 (s, 1H~,
8.18 (d, 1H), 8.14 (d, 1H~, 7.80 (d, 1H~, 7.75 (s, 1H), 7.60 (d, 1H), 7.20 (s, 1H), 6.85
(s, 1H~, 6.00 (s, 2H), 4.70 (m, 1H), 4.05 (s, 3H), 3.60 (septuplet, 1H), 3.30-3.25 (m,
2H), 3.15-3.05 (m, 2H), 2.18-2.00 (m, 2H), 1.90-1.80 (m, ZH), 1.35 (d, 6H);
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-7-methoxy-5-(piperidin-4-yloxy)benzimidazole,
1H NMR (300 MHz, DMS0) ~ 9.40 (s, 2H), 9.20 (s, 2H), 8.60 (br s, 2H), 8.40 (s, 1H),
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-64-
8.18 ~d, 1it), 8.14 (d, 1H), 7.80 (d, 1H~, 7.75 (s, 1H), 7.60 (d, 1H), 6.97 (s, lH), 6.80
(s, 1H), 6.05 (s, 2H~, 4.80 (m, lH), 3.85 (s, 3H3, 3.65 ~septuplet, lH), 3.30-3.25 (m,
2H), 3.20-3.05 (m, 2H), 2.18-2.00 (m, 2H~, 1.90-1.80 (m, 2H), 1.40 (d, 6H);
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-6-(piperidin-4-Y'oXY)benZimidazole~ ' H
NMR (300 MHz, DMS0) ~ 9.40 (s, 2H), 9.20 (s, 2H), 8.60 (br s, 1H), 8.40 (s, lH), 8.18
(d, 1H), 8.10 (s, lH), 7.90-7.75 (m, 3H), 7.70 ~d, 1H), 7.58 (s, 1H), 7.20 (dd, lH), 5.95
(s, 2H), 4.70 (m, 1H), 3.30-3.20 lm, 2H), 3.10-3.00 (m, 2H), 2.80 (s, 3H), 2.10-2.00 (m,
2H), 1.85-1.75 (m, 2H);
1 -(4-amidinonaphth- 1 -yl)methyl-2-methyl-5-(piperidin-4-yloxy)benzimidazole,
1H NMR (300 MHz, DMS0) ~ 9.40 (s, 2H), 9.20 (s, 2H), 8.60 (br s, lH), 8.40 (s, 1H),
8.18 (d, 1H), 8.10 (s, 1H), 7.99 (s, lH), 7.95 (d, lH), 7.75 (d, lH), 7.65 (d, lH), 7.40
(s, ltl), 7.20 (dd, lH), 5.95 (s, 2H), 4.80 (m, lH), 3.30-3.20 (m, 2H), 3.10-3.00 (m,
2H), 2.90 (s, 3H), 2.10-2.00 (m, 2H), 1.95-1.85 (m, 2H);
1-(4-amidinonaphth-1-yl)methyl-6-(piperidin-4-yloxy)benzimidazolP, lH NMR (300 MHz, DMS0)
9.80 (s, 1 H), 9.40 (s, 2H), 9.20 (s, 2H), 8.60 ( br s, 2H), 8.40 (s, 1 H), 8.20-8.00 (m,
3H), 7.90-7.75 (m, 3H), 7.60 (s, 1H), 7.25 (dd, 1H), 6.00 ~s, 2H), 4.70 (m, 1H), 3.30-
3.20 (m, 2H), 3.20-3.00 (m, 2H), 2.20-2.00 (m, 2H), 1.85-1.75 (m, 2H);
1-(4-amidinonaphth-1-yl)methyl-5-(piperidin-4-yloxy)benzimidazole, 1H NMR (300 MHz, DMS0) o
9.80 (s, 1H), 9.40 (s, 2H), 9.20 (s, 2H), 8.60 (br s, 2H), 8.40 (s, 1H), 8.20-8.00 (m, 3H),
7.gO-7.75 (m, 3H), 7.45 (s, 1H), 7.20 (dd, 1H), 6.00 (s, 2H), 4.80 (m, 1H), 3.30-3.20
(m, 2H), 3.20-3.00 (m, 2H), 2.20-2.00 (m, 2H), 1.90-1.70 (m, 2H);
1 -~4~amidinonaphth-1 -yl)methyl-2-methyl-5-(aminocarbonyl)methoxy-6-(piperidin-
4-yloxy)be,~il"idazel~,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-6-(aminocarbonyl)methoxy-5-(piperidin- 4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-6-(carboxy)methoxy-5-(piperidin-
4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-6-(piperkli"-3-yloxy~benzimidazole,
1 -(4-am;dinonaphth- 1 -yl)methyl-2-(2-amino ,a, Lonylethyi)-6-(piperidin-
4-yloxy)ben i,llidacole,
i-(4-amidinonaphth-1-yl)methyl-2-(2-carboxyethyl)-6-(pi~ eridin-
4-yloxy)benzimidazole,
1-(4-amidinonaphth-1-yl)methyl-2-ethyl-6-(pi~,eridi"-4-yloxy)benzimidazole, lH
NMi~ (300 MHz, DMS0) ~ 9.40 (s, 2H), 9.20 (s, 2H), 8.50 (br s, 1 H), 8.40 (s, 1 H), 8.20-
8.05 (m, 2H), 7.90-7.80 (m, 3H), 7.65 (d, 1H), 7.60 (s, 1H) 7.25 (dd, 1H), 6.00 (s, 2H),
4.70 (m, lH), 3.30-3.00 (m, 6H), 2.10-2.00 (m, 2H), 1.90-1.70 (m, 2H), 1.35 (t, 3H);
1-(4-amidinonaphth-1-yl)methyl-2-ethyl-5-(piperidin-4-yloxY)ben~illlida~ole~ lH
CA 02239508 1998-06-04
W O 97/21437 PCT~B96101496
-65-
NMR (300 MHz, DMS0~ ~ 9.40 (s, 2H), 9.20 (s, 2H), 8.60 (br s, lH), 8.40 (s, 1H), 8.15
(d, 1H), 8.10 (d, 1H), 7.90 (s, lH), 7.85 (d, 1H), 7.75 (d, 1H), 7.60 (d, 1H), 7.40 (s, 1H),
~ 7.20 (dd, 1H), 6.00 (s, 2H), 4.70 (m, 1H), 3.30-3.00 (m, 6H), 2.10-2.00 ~m, 2H), 1.90-
1.70 (m, 2H), 1.35 (t, 3H);
1-(4-amidinonaphth-1-yl)methyl-2-methyl-5-(carboxy)methoxy-6-(piperidin-
4-yloxy)benLi~ilid - ' ~,
1 -(4-amidinonaphth-1 -yl)methyl-6-(N-(piperidin-4-yl)-N-(4-methoxycarbonylbenzyl)-
amino) benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-6-(N-(piperidin-4-yl)-N-(4-carboxybenzyl)-
amino)benzimidazole, and
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-6-(N-(piperidin-4-yl)-N-(2-(methoxy-
carbonyl)ethyi)amino)benzimidazole.
E~CAMPLE 2
Compounds of formula (Ib)
1 5 A. To a solution of 1-(4-smidinonaphth-1-yl)methyl-2-isopropyl-6-(piperidin-4-
yloxy)benzimidazole and 1-(4-amidinonaphth-1-yl)msthyl-2-isopropyi-5-(piperidin-4-
yloxy)benzimidazole as a 1 :1 mixture of compounds (288 mg, 0.65 mmol) and methanol ~5 mL)
was added triethylamine (0.54 mL, 3.90 mmol) and ethylacetimidate (320 mg, 2.60 mmol). After
stirring for 16 hours, the reaction was concentrated. The mixture was separated on HPLC on a
C18 Dynamax column with a 1-20% acetonil,ilc in water gradient with 0.1 % trifluoroacetic acid
to-~fford 1-(4-amidinonaphth-1-yl)methyl-2-isopropyl-6-(N-(1-iminoethyl~piperidin-4-
yloxy)benzimidazole ditrifluoroacetate salt; 1H NMR (300 MHz, DMS0~ ~ 9.40 (s, 2H), 9.20 (s,
2H), 9.15 (s, 1H), 8.60 (s, 1H), 8.40 (s, 1H), 8.20-8.10 (m, 2H), 7.90-7.70 (m, 3H), 7.60-7.50
(m, 2H), 7.24 (d, 1H), 6.00 (s, 2H), 4.80 (m, 1H), 3.80-3.40 (m, 5H), 2.28 (s, 3H), 2.05 (m,
2H), 1.80 (m, 2H), 1.30 (d, 6H); and 1-(4-t.,ll;d:.. onaphth-1-yl)methyl-2-isopropyl-5-(N-(1-
iminoethyl)piperidin-4-yloxy)ben~il"idazole ditrifluoroacetate salt; 1H NMR (300 MHz, DMS0)
9.40 (s, 2H), 9.20 (s, 2H), 9.10 (s, lH), 8.60 (s, lH), 8.40 (s, lH), 8.20-8.00 (m, 2H), 7.90-
7.75 (m, 3H), 7.60 (dd, lH), 7.40 (s, lH), 7.25 (dd, lH), 6.00 (s, 2H), 4.80 (m, 1 H), 3.80-3.65
(m, 3H), 3.55 (m, 2H), 2.30 (s, 3H), 2.10 (m, 2H), 1.80 (m, 2H), 1.40 (d, 6H).
B. In a similar manner, the following compounds were made:
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-6-(N-(1 -iminoethyl)piperidin-4-yloxy)benzimidazole,
1H NMR (300 MHz, DMS0) ~ 9.40 (s, 2H), 9.30 (s., 2H), 9.15 (s, 1 H), 8.60 (s, 1 H), 8.40
(s, lH), 8.20-8.05 (m, 2H), 7.90-7.75 (m, 3H), 7.65 (d, lH), 7.60 (s, lH), 7.25 (dd, lH),
6.00 (s, 2H), 4.80 (m, lH), 3.80-3.65 (m, 2H), 3.60-3.50 (m, 2H), 2.90 (s, 3H), 2.25 (s,
3H), 2.10-2.00 (m, 2H), 1.80-1.65 (m, 2H).
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(N-(1 -iminoethyl)piperidin-4-yloxy)benzimidazole,
CA 02239~08 1998-06-04
WO 97/21437 PCTnB96/01496
-66-
1H NMR (300 MHz, DMS0) ~ 9.40 (s, 2H), 9.20 (s, 2H), 9.10 (s, 1H), 8.60 (s, 1H), 8.40
(s, 1H), 8.20-8.00 (m, 2H), 7.95 (s, 1H), 7.80 (d, 1H~, 7.70 (d, 1H), 7.65 (d, 1H), 7.40
(s, 1H), 7.20 (dd, 1H), 5.95 (s, 2H), 4.80 (m, 1H), 3.85-3.65 (m, 2H), 3.60-3.50 (m,
2H), 2.90 (s, 3H~, 2.25 (s, 3H), 2.10-2.00 (m, 2H), 1.95-1.85 (m, 2H);
5 1-(4-amidinonaphth-1-yl)methyl-2-methyl-6-(N-(1-iminoethyl)piperidin-
3-yloxy)benzimidazole, 1H-NMR (300 MHz, DMS0) ~ 9.50 (s, 2H~, 9.45 (s,2H), 9.30 (d,
1H~, 8.80 (s, 1H~, 8.40 (s, 1H~, 8.20 (dd, 2H~, 7.80 (dd, 2H~, 7.60 (dd, 2H), 7.20 (d,
1H), 5.90 (s, 2H), 4.80 (m, 1H), 3.90-3.40 (m, 4H), 2.80 (s, 3H), 1.95-1.50 (m, 6H);
t-(4-amidinonaphth-1-yl)methyl-2-ethyl-6-(N-(1-iminoethyl)piperi li-l-
4-yloxy)benzimidazole, 1H NMR (300 MHz, DMS0) ~ 9.40 (s, 2H), 9.30 (s, 2H), 9.15 (s,
1H), 8.60 (s, 1H), 8.40 (s, 1H), 8.20-8.05 (m, 2H), 7.90-7.75 (m, 3H), 7.65 ~d, 1H),
7.60 (s, 1H), 7.25 ~dd, 1H), 6.00 (s, 2H), 4.80 (m, 1H), 3.80-3.65 (m, 2H), 3.60-3.50
(m, 2H), 3.20 (q, 2H), 2.25 (s, 3H), 2.10-2.00 (m, 2H), 1.80-1.65 (m, 2H), 1.25 (t, 3H);
1 -(4-amidinonaphth-1 -yl)methyl-2-ethyl-5-(N-(1 -iminoethyi)piperidin-4-yloxY)benzimidazole,
1H NMR (300 MHz, DMS0) ~9.40 (s, 2H), 9.20 (s, 2H), 9.18 (s, 1H), 8.60 (s, 1H), 8.40
(s, 1H), 8.15 (d, 1H), 8.10 (d, 1H), 7.90 (s, lH), 7.85 (d, 2H), 7.65 (d, 1H), 7.40 (s, 1H),
7.20 (dd, 1H), 6.00 (s, 2H), 4.80 (m, 1H), 3.80-3.65 (m, 2H), 3.60-3.50 (m, 2H), 3.25
(q, 2H), 2.30 (s, 3H), 2.10-2.00 (m, 2H), 1.90-1.70 (m, 2H), 1.40 (t, 3H);
1 -(4-amidinonaphth-1 -yl)methyl-2-t-butyl-6-(N-(1 -iminoethyl)~ipel i.li, .-
4-yloxy)ben ;.";da~Ole, 1H NMR (500 MHz, DMS0) ~ 9.40 (s, 2H), 9.20 (s, 2H), 9.18 (s,
1H), 8.60 (s, 1H), 8.40 (s, 1H), 8.18 (d, lH), 8.10 (d, 1H), 7.80- 7.77 (m, 2H), 7.70 (d,
1H), 7.59 (s, 1H), 7.25-7.20 (m, 2H), 6.00 (s, 2H), 4.60 (m, 1H), 3.70-3.60 (m, 2H),
3.50-3.40 (m, 2H), 2.30 (s, 3H), 2.00-1.90 (m, 2H), 1.75-1.60 (m, 2H), 1.48 (s, 9H);
1 -(4-amidinonaphth-1 -yl)methyl-2-t-butyl-5-(N-(1 -iminoethyl)piperidin-
4-yloxy)ben i", ~701e, lH NMR (500 MHz, DMS0) ~ 9.40 (s, 2H), 9.10 (m, 3H), 8.60 (s,
1H3, 8.30 (s, lH), 8.18 (d, lH), 8.10 (d, 1H), 7.80 (d, 1H), 7.70- 7.60 (m, 2H), 7.45 (d,
1H), 7.40 (s, 1H), 7.20 (d, 1H), 6.00 (s, 2H), 4.60 (m, 1H), 3.80-3.70 (m, 2H), 3.60-
3.50 (m, 2H), 2.30 (s, 3H), 2.20-2.00 (m, 2H), 1.90-1.60 (m, 2H), 1.60 (s, 9H);
1-(4-all nonapllLII-1-yl)methyl-6-(N-(1-iminoethyl)piperidin-
4-yloxy)benzi",: ~7nle, 1 H NMR (300 MHz, DMS0) ~ 9.70 (s, 1H), 9.45 (s, 2H), 9.30 (s,
2H), 9.20 (s, 1H), 8.60 (s, 1H), 8.40 (s, 1H), 8.25-8.18 (m, 2H), 8.10 (s, 1H), 7.90-7.80
(m, 3H), 7.60 (s, 1H), 7.30 (dd, 1H), 6.00 (s, 2H), 4.80 (m, 1H), 3.85-3.70 (m, 2H),
3.60-3.50 (m, 2H), 2.35 (s, 3H), 2.15-2.05 (m, 2H), 1.95-1.85 (m, 2H);
1 -(4-amidinonaphth-1 -yl)methyl-5-(N-(1 -iminoethyl)piperidin-
4-yloxy)benzimidazole, lH NMR (300 MHz, DMS0) ~ 9.80 (s, 1H), 9.40 (s, 2H), 9.Z0 (s,
2H), 9.10 (s, 1H) 8.60 (s, 1H), 8.40 (s, 1H), 8.15-8.00 (m, 3H), 7.90-7.75 (m, 3H), 7.45
(s, 1H), 7.20 (dd, 1H), 6.00 (s, 2H), 4.80 (m, 1H), 3.80-3.70 (m, 2H), 3.60-3.45 (m,
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
2H), 2.30 (s, 3H), 2.20-2.00 (m, 2H), 1.90-1.70 (m, 2H);
1 -(4-amidinonaphth-1 -yl)methyl-2-propyl-6-~N-~1 -iminoethyl)piperidin-4-yloxy)benzimidazole,
1H NMR (300 MHz, DMS0) ~9.40 (s, 2H), 9.20 (s, 2H), 9.18 (s, lH), 8.60 (s, 1H), 8.40
(s, lH), 8.18 (d, 1H), 8.14 (d, 1H), 7.90- 7.80 (m, 3H), 7.65 (d, 1H), 7.60 (s, 1H), 7.25
(dd, 1H), 6.00 ~s, 2H), 4.80 ~m, lH), 3.80-3.65 (m, 2H), 3.60-3.40 ~m, 2H), 3.20 ~t, 2H),
2.30 ~s, 3H), 2.18-2.00 ~m, 2H), 1.85-1.70 (m, 4H), 0.90 (t, 3H~;
1 -~4-amidinonaphth-1 -yl)methyl-2-propyl-5-~N-(1 -iminoethyl)piperidin-
4-yloxy)benzi,I,iddzoIe, 1H NMR ~300 MHz, DMS0) ~ 9.40 Is, 2H), 9.20 (s, 2H), 9.18 ~s,
1H), 8.60 (s, 1H), 8.40 (s, 1H), 8.15 (d, 1H), 8.10 (d, lH), 7.90 (s, lH), 7.80 (d, lH),
7.70 (d, 1H), 7.63 (s, 1H), 7.40 (s, 1H), 7.20 (dd, 1H), 6.00 (s, 2H), 4.80 (m, lH), 3.80-
3.65 ~m, 2H), 3.62-3.50 ~m, 2H), 3.23 ~t, 2H), 2.30 ~s, 3H), 2.20-2.00 ~m, 2H), 1.85-
1.70 (m, 4H), 1.00 ~t, 3H);
1-~4-amidinonaphth-1-yI)methyI-2-sec-butyI-6-~N-~1-i".inoe~ 13piperidin-4-yIoxy)benzimidazoIe,
1H NMR (300 MHz, DMS0) ~ 9.40 (s, 2H), 9.20 (s, 2H), 9.18 (s, 1H), 8.60 (s, 1 H), 8.40
(s, lH), 8.18 (d, lH), 8.10 (d, lH), 7.90- 7.77 (m, 3H), 7.65 (d, lH), 7.59 (s, 1H), 7.25
(dd, 1H~, 6.00 (s, 2H), 4.80 (m, lH), 3.80-3.70 (m, 2H), 3.60-3.50 (m, 2H), 3.10 (d,
2H), 2.30 (s, 3H), 2.18-2.00 (m, 3H), 1.85-1.70 (m, 2H), 0.90 (d, 6H);
1-(4-~ idil)ond,uhLII-1-yl)methyl-2-sec-butyl-5-(N-(1-iminoethyl)piperidin-
4-yloxy)benzimidazole, 1H NMR (300 MHz, DMS0) d9.40 (s, 2H), 9.20 (s, 2H), 9.18 (s,
lH), 8.60 (s, lH), 8.40 (s, lH), 8.18 (d, lH), 8.10 (d, lH), 7.90 (s, lH), 7.85 (d, lH),
7.70 (d, lH), 7.60 (d, 1H), 7.45 (s, 1H), 7.20 (d, lH), 6.00 (s, 2H), 4.80 (m, lH), 3.80-
3.70 (m, 2H), 3.60-3.50 (m, 2H), 3.10 (d, 2H), 2.30 (s, 3H), 2.18-2.00 (m, 3H), 1.85-
1.70 (m, 2H), 0.95 (d, 6H);
1 -(4-amidinonaphth-1 -yl)methyl-2-n-butyl-6-(N-(1 -iminoethyl)piperidin-4-yioxy)benzimidazole,
1H NMR (300 MHz, DMSO) ~ 9.40 (s, 2H), 9.20 (s, 2H), 9.18 (s, 1H), 8.60 (s, 1H), 8.40
(s, lH), 8.18 (d, lH), 8.10 (d, lH), 7.90- 7.80 (m, 3H), 7.70-7.60 (m, 2H), 7.25 (dd,
1H), 6.00 (s, 2H), 4.80 (m, lH), 3.80-3.70 (m, 2H), 3.60-3.50 (m, 2HI, 3.20 (t, 2H),
2.30 (s, 3H), 2.18-2.00 (m, 2H), 1.85-1.60 (m, 4H~, 1.40 Im, 2H), 0.90 ~t, 3H);
1-(4-t,lll: iinonaphth-l-yl)methyl-2-n-butyl-5-(N-(1-i l i"o :LI,~I)p:"e(idil,-
4-yloxy)benzimidazole, 1H NMR (300 MHz, DMS0) ~ 9.40 (s, 2H), 9.20 (s, 2H), 9.1 8 (s,
1H), 8.60 (s, 1H), 8.40 (s, 1H), 8.18 (d, lH), 8.10 (d, lH), 7.90 (s, lH), 7.83 (d, lH),
7.75 (d, lH), 7.60 (d, 1H), 7.40 (s, 1H), 7.20 (d, 1H), 6.00 (s, 2H), 4.80 (m, 1H), 3.80-
3.70 (m, 2H), 3.60-3.50 (m, 2H), 3.25 (t, 2H), 2.30 (s, 3H), 2.18-2.00 (m, 2H), 1.86-
1.65 (m, 4H), 1.40 (m, 2H), 0.95 (t, 3H);
35 1-(4-amidinonaphth-1-yl)methyl-2-(2-carboxyethyl)-6-(N-(1-iminoethyl)piperidin-
4-yIoxy)be" i"~ 7~ , 1H-NMR 9.45 (s, 2H), 9.39 (s, 2H), 9.20 (s, 1H), 8.60 (s, lH),
8.40 (s, 1H), 8.10 (dd, 2H), 7.80 ( d, 1H), 7.70 (dd, 3H), 7.45 (s, 1H), 7.10 ~d, 1H),
CA 02239~08 1998-06-04
W O 97/21437 PCTnB96/01496
-68-
5.90 (s, 2H), 4.70 (m, 1H), 3.70 (m, 2H), 3.50 (m, 2H), 3.30 ~t, 2H), 3.10 tm, 1H~, 2.90
(t, 2H), 2.30 (s, 3H), 2.10 (m, 2H), 1.75 (m, 2H);
1 -(4-amidinonaphth-1 -yl)methyl-2-(2-carboxyethyl)-5-(N-(1 -iminoethyl)piperidin-
4-yloxy)benzimidazole, 1H-NMR 9.45 (s, 2H), 9.40 (s, 2H), 9.20 (s, 1H), 8.85 ( s, 1H),
8.40 (s, 1H), 8.10 (dd, 2H), 7.80 (m, 2H), 7.65 (dd, 2H), 7.40 (s, 1H), 7.10 (d, 1H),
5.95 (s, 2H), 4.85 (m, 1H), 3.80 (m, 2H), 3.50 (m, 2H), 3.25 (m, 2H), 3.10 (m, 1H),
2.95 (m, 2H), 2.30 (s, 3H), 2.10 ( m, 2H), 1.75 (m, 2H);
1 -(4-amidinonaphth-1 -yl)methyl-2-(2-aminocarbonylethyl)-6-(N-(1 -iminoethyl)piperidin-
4-yloxy)benzimidazole, 1H-NMR 9.42 (s, 2H), 9.35 (s, 2H), 9.20 (s, 1H), 8.60 (s, 1H),
8.40 (s, 1H), 8.10 (dd, 2H), 7.85 (d, 1H), 7.70 (m, 3H), 7.50 (s, 1H), 7.45 (s, 1H), 7.10
(d, 1H), 7.00 (s, 1H), 5.90 (s, 2H), 4.70 (m, 1H), 3.70 (m, 2H), 3.50 (m, 2H), 3.20 ~m,
3H), 2.80 (t, 2H), 2.00 (m, 2H), 1.70 (m, 2H);
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-6-(N-(N'-(1 -iminoethyl)piperidin-
4-yl)-N-((methoxycarbonyl)methyl)amino)benzimidazole, lH NMR (300 MHz, DMS0) ~
1.70 (m, 4H), 2.25 (s, 3H), 2.80 (s, 3H), 3.20 (m, 2H), 3.50 (s, 3H), 3.90 (m, lH), 4.10
(m, 2H), 4.15 (s, 2H). 5.90 (s, 2H), 7.00 (s, 1H), 7.05 (d, lH), 7.B0 ~d, 1H), 7.70 ld,
1H), 7.80 (d, lH), 7.90 (s, 1H), 8.10 (d, 1H), 8.15 (d, 1H), 8.40 (s, 1H), 8.50 (s, 1H),
9.10 (s, 1H), 9.15 (s, 2H), 9.40 (s, 2H);
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-6-(N-(N'-(1 -iminoethyl)piperidin-
4-yl)-N-((a,,,;~loca.L,onyl)methyl)amino)ben~i,.,idc.~ole, 1H NMR (300 MHz, DMS0) ~ 1.80
(m, 4H), 2.25 (s, 3H), 2.80 (s, 3H), 3.25 (m, 2H), 3.80 (s, 2H), 3.95 (m, 1H), 4.15 (m,
2H), 5.90 (s, 2H), 7.00 (d, 1H), 7.10 (s, 1H), 7.40 (br s, 1H), 7.60 (d, 1H), 7.70 (d, 1H),
7.83 (d, 1H), 7.95 (s, 1H), 8.10 (d, 1H), 8.15 (d, 1H), 8.40 (s, 1H), 8.55 (s, 1H), 9.10
(s, 1H), 9.15 (s, 2H), 9.40 (s, 2H);
25 1-(4-amidinonaphth-1-yl)methyl-2-methyl-6-(N-(N'-(1-iminoethyl)piperidin-4-yl)-
N-(3-carboxypropyl)amino)benzimidazole, lH NMR (300 MHz, DMS0) ~ 1.60 (m, 2H),
1.80 (m, 4H), 2.20 (m, 2H), 2.25 (s, 3H), 2.80 (s, 3H), 3.20 (m, 4H), 3.95 (m, 1H), 4.10
(m, 2H), 5.90 (s, 2H), 7.15 (m, 2H), 7.65 (d, 1H), 7.70 (d, 1H), 7.80 (d, 1H), 7.95 (s,
1H), 8.10 (d, 1H), 8.15 (d, 1H), 8.40 (s, 1H), 8.58 ~s, 1H), 9.10 (m, 3H), 9.20 (s, 2H);
30 1 - (4-amidi no naphth- 1 -yl) methyl-2-methyl-6-(N-(N'-(1 -i mi noethyl )
4-yl)-N-(2-(methoxycarbonyl)propyl)amino)benzimidazole, 1H NMR (300 MHz, DMS0) ~0.80 (d, 3H), 1.60 (m, 4H), 2.20 (s, 3H), 2.40 (m, 1H), 2.80 (s, 3H), 3.10 (m, 3H), 3.30
(m, 1H), 3.40 (s, 3H), 3.90 (m, 2H), 4.05 (m, 1H), 5.85 (d, 1H), 5.90 (d, 1H), 7.10 (d,
1H), 7.20 (d, 1H), 7.60 (d, 1H), 7.65 (d, 1H), 7.80 (d, 1H), 7.90 (s, 1H), 8.05 (d, 1H),
8.10 (d, 1H), 8.35 (s, 1H), 8.50 (m, 1H), 9.05 (br s, 1H), 9.10 (s, 2H), 9.40 (s, 2H);
1 -(4-amidinonaphth- 1 -yl)methyl-2-methyl-6-(N-(N'-(1 -iminoethyl)piperidin-
4-yl)-N-(2-(carboxy)propyl)amino)benzimidazole, 1H NMR (300 MHz, DMS0) ~0.85 (d,
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-69-
3H), 1.70 (m, 4H), 2.20 (s, 3H), 2.30 Im, 1H), 2.85 (s, 3H), 3.10 (m, 3H), 3.40 (m, 1H),
3.90 (m, 2H), 4.10 (m, 1H), 5.85 (d, 1H), 5.90 (d, 1H), 7.15 (d, 1H), 7.15 (d, 1H), 7.60
- (d, 1H), 7.65 (d, 1H), 7.80 (d, 1H), 7.95 (s, 1H), 8.05 (d, 1H), 8.10 (d, lH), 8.40 (s, 1H),
8.50 (s, 1H), 9.05 (br s, 1H), 9.10 (s, 2H), 9.40 (s, 2H);
1-(4-amidinonaphth-1-yl)methyl-2-methyl-5-(N-(N-(1-iminoethyl)piperidin-
4-yl)-N-(2-(carboxy)propyl)aminolbenzimidazole, lH NMR (300 MHz, DMS0) ~ 1.00 (d,
3H), 1.70 (m, 4H), 2.20 (s, 3H), 2.50 (m, 1H), 2.90 (s, 3H), 3.10 (m, 3H), 3.40 (m, 1H),
3.90 (m, 2H), 4.10 (m, 1H), 5.85 (s, 2H), 7.15 (d, lH), 7.20 (d, lH), 7.55 (d, 1H), 7.60
(d, 1H), 7.80 (d, 1H), 7.95 (s, 1H), 8.00 (d, 1H), 8.05 (d, lH), 8.40 (s, lH), 8.50 (s, lH),
9.05 (br s, lH), 9.10 (s, 2H), 9.40 (s, 2H);
1 -(4-amidinonaphth- 1 -yl)methyl-2-methyl-5-hydroxy-6-(N-(1 -iminoethyl)piperidin-
4-yloxy)benzimidazole, 1H NMR (DMS0, 300 MHz) ~ 9.50 (s, 4H), 9.20 (s, 1 H), 8.65 (s,
1H), 8.40-7.20 (m, 9H), 5.92 (s, lH), 5.88 (s, 1H), 4.80 (m, 0.5HI, 4.70 (m, 0.5H),
3.80-3.45 (m, 4H), 2.90 (s, 1.5H), 2.80 (s, 1.5H), 2.30 (s, 3H), 2.10-1.70 (m, 4H) (a
mixture of two ,e~~ioiso,llers);
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(aminocarbonyl)methoxy-6-(N-(1 -imino-
ethyl)piperidin-4-yloxy)benzimidazole, 1H NMR (DMS0, 300 MHz) o 9.46 ~s, 4H), 9.21 (s,
1~, ~.,~ (s, 1t~ 7,38 ~m, 1(~1~, 5.92 !s, lH!, 6.88 !s, lH!, 4 82 !m, Or5H!~ 4 76
(m, 0.5H), 4.62 (s, 1H), 4.52 (s, 1H), 3.80-3.40 (m, 4H), 2.80 (s, 1.5H), 2.78 (s, 1.5H),
2.25 (s, 1.5H), 2.23 (s, 1.5H), 2.16-1.75 (m, 4H) (a mixture of two regioisomers);
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(carboxy)methoxy-6-(N-(1 -imino-
ethyl)piperidin-4-yloxy)benzimidazole, 1H NMR (DMS0, 300 MHz) ~ 9.60 (s, 2H), 9.50 (s,
2H), 9.20 (s, 1H), 8.70 (s, 1H), 8.40-7.40 (m, 9H), 5.96 (s, 2H), 4.90 (s, 1H), 4.80 (s,
1H), 4.70 (m, 1H), 3.80-3.50 (m, 4H), 2.80 (s, 3H), 2.30 (s, 3H), 2.15-1.70 (m, 4H) (a
mixture of re~ioi~o" ,e, :,);
1 -(4-amidinonaphth- 1 -yl)methyl-2-methyl-5-(4-(methoxycarbonyl)benzyloxy)-
6-(N-(1-iminoethyl)piperidin-4-yloxy)be" i",idazole, 1H NMR (DMS0, 300 MHz) ~ 9.42 (s,
2H), 9.30 (s, 2H), 9.18 (s, 1H), 8.60 (s, 1H), 8.40-7.40 (m, 12H), 5.90 (s, 2H), 5.35 (s,
2H), 4.70 (m, 1H), 3.90 (s, 3H), 3.80-3.40 (m 4H), 2.70 (s, 3H), 2.30 (s, 3H), 2.10-1.70
(m, 4H);
1 -(4-amidinonaphth-1 -yl~methyl-2-methyl-5-(4-(carboxy)benzyloxy)-
6-(N-(1-iminoethyl)piperidin-4-yloxy)benzimidazole, 1H NMR (DMS0, 300 MHz) ~ 9.42
(s,2H), 9.30 (s,2H), 9.18 (s,1H), 8.60 (s, 1H), 8.18-7.40 (m, 12H), 5.90 (s, 2H~, 5.30 (s,
2H), 4.70 (m, 1H), 3.80-3.40 (m, 4H), 2.70 (s, 3H), 2.30 (s, 3H), 2.10-1.70 (m, 4H);
1-(4-amidinonaphth-1-yl)methyl-2-methyl-5-carboxy-6-(N-(1-iminoethyl)piperidin-
4-yloxy)ben i"lidd ole, 1H NMR (DMS0, 300 MHz) ~ 9.40 (s, 4H), 9.20 (s, 1H), 8.63
~s,1H), 8.40-7.60 (m, 9H), 6.00 (s, 2H), 4.90 (m, 1H), 3.70 (m, 4H), 2.70 ~s, 3H), 2.30
CA 02239~08 1998-06-04
W O 97~1437 PCT~B96/~t496
-70-
~s, 3H), 2.05-1.80 (m, 4H);
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(carboxy)methoxy)-
6-(N-(1 -iminoethyl)piperidin-4-yloxy)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-((aminocarbonyl)methoxy)-
6-(N-(1-iminoethyl)piperidin-4-yloxy)benzimidazole, lH NMR ~DMS0, 300 MHz) ~ 9.60 (s,
2H), 9.30 (s, 2H), 9.12 (s, lH), 8.60 (s, 1H), 8.40-7.30 (m, 10H), 5.90 (s, 2H), 4.70 (m,
lH), 4.60 (s, 2H), 3.80-3.40 (m, 4H), 2.70 (s, 3H), 2.30 (s, 3H), 2.12-1.80 (m, 4H);
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(3-(methoxycarbonyl)benzyloxy)-
6-(N-(1-iminoethyl)piperidin-4-yloxy)benzimidazole, 1H NMR (DMS0, 300 MHz) ~ 9.65 (s,
2H), 9.30 (s, 2H), 9.16 (s, 1H), 8.60 (s, 1H), 8.40-7.40 (m, 12H), 5.90 (s, 2H), 5.30 ~s,
2H), 4.70 (m, 1H), 3.90 (s, 3H), 3.80-3.40 (m, 4H), 2.70 (s, 3H), 2.36 (s, 3H), 2.20-
1.80 (m, 4H);
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-6-(3-(carboxy)benzyloxy)-
6-(N-(1-iminoethyl)piperidin-4-yloxy)benzimidazole, 1H NMR (DMS0, 300 MHz) ~ 9.40 (s,
2H), 9.10 (s, 1H), 9.05 (s, 2H), 8.60 (s, lH), 8.40-7.40 (m, 12H), 5.90 (s, 2H), 5.35 (s,
2H), 4.70 (m, lH), 3.80-3.45 (m, 4H), 2.70 (s, 3H), 2.25 (s, 3H), 2.05-1.70 (m, 4H);
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(methoxycarbonyl)methoxy-6-(N-(1 -imino-
ethyl)pipe,idin-4-yloxy)benzimidazole, 1H NMR (DMS0, 300 MHz) ~ 9.45 (s, 2H), 9.10 (s,
3H), 8.60 (s, 1H), 8.40-7.40 (m, 8H), 5.90 (s, 2H), 5.00 (s, 2H), 4.70 (m, 1H), 3.70 (s,
3H), 3.60 (m, 4H), 2.80 (s, 3H), 2.30 (s, 3H), 2.10-1.80 (m, 4H);
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(1 -(aminocarbonyl)ethoxy)-6-(N-(1 -imino-
ethyl),~ .i.lin-4-yloxy)ben~ le, 1H NMR (DMS0, 300 MHz) ~ 9.40 (s, 2H), 9.20 (s,3H), 8.60 Is, 1H), 8.40-7.30 (m, 10H), 5.95 (s, 2H,), 4.70 (m, 2H), 3.80-3.50 (m, 4H),
2.80 (s, 3H), 2.30 (s, 3H), 2.10-1.80 (m, 4H), 1.50 (d, 3H);
25 1-(4-amidinonaphth-1-yl)methyl-2-methyl-5-(1-(methoxycarbonyl)ethoxy)-6-(N-(1-imino-ethyl)-
piperidin-4-yloxy)benzimidazole, 1H NMR (DMS0, 300 MHz) ~S 9.45 (s, 2H), 9.30 (s, 2H),
9.20 (s, 1H), 8.60 (s, lH), 8.40-7.35 (m, 8H), 5.90 (s, 2H), 5.10 (m, 1H), 4.70 (m, lH),
3.90 (s, 3H), 3.70 (m, 4H), 2.76 (s, 3HI, 2.30 ~s, 3H), 2.10-1.80 (m, 4H), 1.60 (d, 3H);
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(1 -(carboxy)ethoxy)-6-(N-(1 -imino-ethyl)piperidin-4-yloxy)benzimidazole, 1H NMR (DMS0, 300 MHz) ~ 9.45 (s, 2H), 9.35 (s,
2H), 9.18 (s, 1H), 8.60 (s, 1H), 8.40-7.30 (m, 8H), 5.96 (s, 2H), 5.00 (m, 1H), 4.70 (m,
1H), 3.60 (m, 4H), 2.78 (s, 3H), 2.30 (s, 3H), 2.10-1.80 (m, 4H), 1.60 (d, 3H);
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-4-methoxy-6-(N-(1 -iminoethyl)piperidin-
4-yloxy)ben~i"-' '~701e, 1H NMR (300 MHz, DMS0) ~ 9.40 (s, 2H), 9.20 (s, 2H~, 9.18 (s,
1H), 8.60 (s, 1H), 8.40 (s, 1H), 8.18 (d, 1H), 8.14 (d, 1H), 7.85 (d, 1H), 7.75 (s, 1H),
7.60 (d, 1H), 7.25 (s, 1H), 6.80 (s, 1H), 6.00 (s, 2H), 4.80 (m, 1H), 4.10 (s, 3H), 3.80-
3.65 (m, 2H), 3.60-3.45 (m, 3H), 2.30 (s, 3H) , 2.18-2.00 (m, 2H), 1.90-1.80 (m, 2H),
CA 02239~08 1998-06-04
W O 97/21437 ~CT~B96/01496
-71 -
1.35 (d, 6H);
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-7-methoxy-5-(N-(1 -iminoethyl)p;~e~ idi"-
- 4-yloxy)benzimidazole, 1 H NMR (300 MHz, DMS0) ~ 9.40 (s, 2H~, 9.20 (s, 3H), 8.60 (br
s, 1H), 8.40 (s, 1H), 8.20-8.10 (m, 3H), 7.90-7.80 (m, 3H), 7.ô5 (d, 1H), 7.00 (5, 1H),
6.80 (s, 1H), 6.10 (s, 2H), 4.90 (m, 1H), 3.90-3.55 (m, 6H), 2.35 (s, 3H), 2.20-2.10 (m,
2H), 2.00-1.80 (m, 2H), 1.40 (d, 6H);
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(N-(4-carboxy)benzylamino)-
6-(N-(1-iminoethyl)piperidin-4-yloxy)benzimidazole, (approx 1:1 mixture o~ rer~ioisomers)
lH NMR (300 MHz, DMSO) ~ 1.85 (m, 2H), 2.00 (m, 2H), 2.25 (s, 3H), 2.60 (s, 1.5H),
2.75 (s, 1.5H), 3.60 (m, 2H), 3.80 (m, 2H), 4.40 (s, 1H), 4.50 (s, 1H), 4.80 (m, 0.5H),
4.90 (m, 0.5H), 5.65 (s, 1H), 5.85 (s, 1H), 6.50 (s, 0.5H), 6.60 (s, 0.5H), 7.25 (d, 1H),
7.30 (s, 1H), 7.40 ~m, 1H), 7.50 (s, 0.5H), 7.60 (d, 1H), 7.70 (d, 1H), 7.75 (d, 1H), 7.85
(d, 0.5H), 7.90 (d, 1H), 8.00 (d, lH), 8.05 (d, 1H), 8.10 (d, 1H), 8.35 (d, lH), 8.60 (s,
1H), 9.10 (s, 2H), 9.15 (s, 1H), 9.35 (s, 2H);
1-(4-amidinonaphth-1-yl)methyl-2-isopropyl-4-(4-carboxypiperidin-1-yi)-
6-(N-(l-iminoethyl)piperidin-4-yloxy~benzimidazole, lH-NMR 9.38 (s, 2H), 9.05 (s, 3H),
8.50 (s, lH), 8.38 (s, lH), 8.05 (dd, 2H), 7.80 (d, lH), 7.70 (s, 1H), 7.58 (d, lH), 7.25
(s, lH), 6.70 (s, 1H), 5.95 (s, 2H), 4.70 (m, 1H), 3.65 (m, 2H), 3.50 (m, 4H), 2.85 (t,
2H), 2.40 (m, 2H), 2.20 (s, 3H), 1.95 (m, 5H), 1.70 (m, 2H), 1.30 (d, 6H);
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-4-(carboxy)methoxy-6-(N-(1 -imino-
ethyl)pi~,r,id ~-4-yloxy)benzi".ida,ole, lH-NMR 9.38 (s, 2H), 9.05 (s, 2H), 8.75 (s, 1H),
8,40 ( s, 1H), 8.05 (dd, 2H), 7,80 (d, 1H), 7.70 (s, 1H), 7.60 (d, 1H), 7.25 (s, 1H), 6.75
(s, lH), 5.95 (s, 2H), 5.00 (s, 2H), 4.70 (m, lH), 3.80 - 3.40 (m, 5H), 2.20 (s, 3H), 2.00
(m, 2H), 1.85 (m, 2H), 1.30 (d, 6H);
1-(4-amidinonaphth-1-yl)methyl-2-isopropyl-7-nitro-6-(N-(1-imino-
ethyl)piperidin-4-yloxy)ben~i,l,ir' He, lH-NMR 9.38 (s, 2H), 9.15 (s, 1H), 9.05 (s, 2H),
8.80 (s, 1H), 8.35 (s, 1H), 8.10 (d, lH), 8.05 (d, 1H), 7.95 (d, lH), 7.80 (d, 1H), 7.70
(s, lH), 7.60 (d, 1H), 7.50 (d, 1H), 5.95 (s, 2H), 5.00 ( m, 1H), 3.60-3.45 (m, 5H), 2.25
(s, 3H), 2.00 (m, 2H), 1.30 (d, 6H), 1.30 (d, 6H);
1-(4-amidinonaphth-1-yl)methyl-2-methyl-7-nitro-6-(N-(l-imino-
ethyl)piperidin-4-yloxy)benzimidazole, lH NMR (DMS0, 300 MHz) ~ 9.45 (s, 2H), 9.38 (s,
2H), 9.20 (s, 1H), 8.65 (s, 1H), 8.42-7.30 (m, 8H), 5.81 (s, 2H), 4.90 (m, 1H), 3.60 (m,
4H), 2.60 (s, 3H), 2.23 (s, 3H), 2.10 (m, 2H), 1.80 (m, 2H);
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-5-(N-(2-carboxyprop-2-yl)amino)-
6-(N-(1-iminoethyl)piperidin-4-yloxy)ben~i-,-idA~ole, lH NMR (300 MHz, DMS0) ~ 1.25 (d,
6H), 1.55 (s, 6H), 1.80 (m, 2H), 2.00 (m, 2H), 2.25 (s, 3H), 3.50 (m, 3H), 3.70 (m, 2H),
4.80 (m, 1H), 5.95 (s, 2H), 6.70 (s, 1H), 7.60 (m, 2H), 7.70 (s, lH), 7.80 (d, 1H), 8.05
CA 02239~08 1998-06-04
W 097/21437 PCT~B96/01496
(d, lH), 8.10 (d, lH), 8.40 (s, 1H), 8.60 (s, 1H), 9.10 (s, 2H), 9.15 (s, 1H), 9.40 (s, 2H);
1 -~4-amidinonaphth-1 -yl)methyl-2-isopropyl-5-(N-(4-carboxy)benzyiamino)-
6-(N-(1-iminoethyl)piperidin-4-yloxy)benzimidazole, lH NMR (300 MHz, DMS0) ~ 1.20 (d,
6H), 1.90 (m, 2H), 2.00 (m, 2H), 2.20 (s, 3H), 3.45 Isept, 1H), 3.60 (m, 2H), 3.80 (m,
2H), 4.55 (s, 2H), 4.93 (m, 1H), 5.95 (s, 2H), 6.45 (s, 1H), 7.45 ~d, 2H), 7.57 (s, 1H),
7.60 (d, 1H), 7.65 (s, 1H), 7.80 (d, 1H), 7.90 (d, 2H), 8.05 (d, 1H), 8.10 (d, 1H), 8.37
~s, 1H), 8.60 (s, 1H), 9.10 (s, 2H), 9.15 (s, 1H), 9.18 (s, 2H);
1 -(4-amidinonaphth-1 -yl)methyl-2-isopropyl-5-(N-(2-carboxyethyl)amino)-
6-(N-(1-iminoethyl)piperidin-4-yloxy)benzimidazole, lH NMR (300 MHz, DMS0) ~ 1.20 (d,
6H), 1.80 ~m, 2H), 1.95 (m, 2H), 2.20 (s, 3H), 2.55 (m, 2H), 3.40 (m, 2H), 3.50 (m, 3H),
3.70 (m, 2H), 4.80 (m, 1H), 5.95 (s, 2H), 6.85 (s, 1H), 7.50 (m, 2H), 7.65 (s, lH), 7.75
(d, 1H), 8.00 (d, 1H), 8.05 (d, 1H), 8.30 (s, 1H), 8.50 (s, 1H), 9.10 (s, 3H), 9.30 (s, 2H);
1 -(4-amidinonaphth-1 -yl)methyl-2-methyl-5-(N-(carboxymethyl)piperidin-4-yloxy)-
6-(N-(1-iminoethyl)piperidin-4-yloxy)benzimidazole, lH NMR (DMS0, 300 I\AHz) ~ 10.40 (s,
1H), 9.70 (s, 2H), 9.45 (s, 3H), 8.98 (s, 1H), 8.70-7.80 (m, 8H), 6.24 (s, 2H), 5.00 (m,
2H), 4.48 (s, 2H), 4.0-3.40 (m, 8H), 3.20 (s, 3H), 2.60 (s, 3H), 2.40-2.00 (m, 8H);
1 -(4-amidinonaphth-1 -yl)methyl-6-~N-tN'-(1 -i."inoeLI-~yl)piperidin-
4-yl)-N-((4-methoxycarbonyl)benzyl)amino)bèn~ J~
1 -(4-amidinonaphth-1 -yl)methyl-6-(N-(N'-(1 -iminoethyl)piperidin-
4-yl)-N-((4-carboxy)benzyl)amino)benzimidazole,
1 -(4-amidinonaphth-1 -yl)methyl-2-trifluoromethyl-6-(N-(1 -iminoethyl)piperidin-
4-yloxy)ben~in,ida~ole, 1H NMR (300 MHz, DMS0) ~ 9.40 (s, 2H), 9.20-9.t5 (m, 3H),
8.60 (s, 1H), 8.45 (s, 1H), 8.20-8.10 lm, 3H), 8.00 (d, lH), 7.80 (d, 2H), 7.43 (s, 1H),
7.20 (d, 1 H), 5.40 (s, 2H), 4.85-4.75 (m, 1 H), 4.20 (dd, 2H), 3.60-3.40 (dt, 2H), 2.80-
2.60 (m, 2H, obscured by DMS0 signal), 2.40 (s, 3H), 2.15-2.00 (m, 2H);
1-(4-amidinonaphth-1-yl)methyl-6-(N-(N'-(1-iminoethyl)~,- i.lin-
4-yl)-N-((2-(ethoxycarbonyl)ethyl)carbonyl)amino)be"zi",-'~701e, and
1 -(4-amidinonaphth-1 -yl)methyl-6-(N-(N'-(1 -i",:.,oe~l,yl)piperidin-
4-yl)-N-((2-(carboxy)ethyl)carbonyl)amino)ben~i",
EXAMPLE 3
Compounds of formula (Ic)
To a mixture of 1-(4-cyanonaphth-1-yl)methyl-2-isopropyl-4-methoxycarbonyl-6-(N-(tert-
butoxycarbonyl)piperi.lil,-4-ylamino)benzimidazole, 1-(4-cyanonaphth-1-yl)methyl-2-isopropyl-7-
methoxycarbonyl-5-(N-(tert-butoxycarbonyl)p;,.e.idin-4-ylaminobenzi",;dazole, and 2-isopropyl-4-
35 methoxycarbonyl-6-(N-(tert-butoxycarbonyl)pipt(idin-4-yl)((4-cyanonaphth-1-
yl)methyl)aminobenzimidazole [4 9, 50% 1-(4-cyanonaphth-1-yl~methyl-2-isopropyl-4-
CA 02239~08 l998-06-04
WO 97/21437 PCT~B96/01496
-73-
methoxycarbonyl-6-(N-(t~t-butoxycarbonyl)piperidin-4-ylamino)ben~;"lid~ le~ in ethanol (150 mL)
cooled in an ice water bath was bubbled HCI (g). After the solution was saturated, the reaction
flask was sealed and the temperature maintained at ambient temperature for 16 hours. The
solvent was removed under reduced pressure. The residue was dissolved in ethanol ~50 mL).
The solution was cooled in a dry ice/acetone bath and ammonia (g) was bubbled in. The reaction
flask was sealed and then heated at 70~C for 4 hours. The solvent was removed. The mixture
was separated by HPLC on a Ct8 Dynamax column with a 3-25% ac~Lonil-ile in water gradient
with 0.1 % trifluoroacetic acid to afford 1-(4-amidinonaphth-1-yl)methyl-2-isopropyl-4-
aminocarbonyl-6-(piperidin-4-ylamino)-benzimidazole as a white solid after concentration and
10 freeze-drying removal of the solvent.
EXAMPLE 4
Compounds of formulae (Id)
A. To 1-(4-amidinonaphth-1-yl~methyl-2-isopropyl-4-aminocarbonyl-6-(piperidin-4-ylamino)benzimidazole ~1 9) in methanol (50 mL) was added triethylamine (3 mL) and
15 ethylaceLi,-, ~late (1 g). After stirring for 3 hours, the reaction was concentrated. The residue
was purified by HPLC on a C18 Dynamax column with a 3-25% acetonitrile in water gradient
with 0.1 % trifluoroacetic acid to afford 1-(4-amidinonaphth-1-yl)methyl-2-isopropyl-4-
aminocarbonyl-6-(N-(1-iminoethyl)piperidin-4-ylamino)benzimidazole, 1H NMR (DMS0, 300 MHz)
9.70 (s, 2H), 9.58 (5, 2H), 9.40 (s, 1H), 9.35 (s, lH), 8.82 (s, 1H), 8.62-7.20 (m, 9H), 6.10 (s,
20 2H), 4.10-3.50 (m, 6H), 2.50 (s, 3H), 2.26 (m, 2H), 1.70 (m, 2H), 1.60 (d, 6H), as white solid
after freeze drying removal of the solvent.
B. In a similar manner, the following compound of formula (Id) was made:
1 -(4-amidinonaphth-1 -yl)methyl-6-(N-(1 -iminoethyl)piperidin-
4-yl)aminobenzimidazole, 1H NMR (300 MHz, DMS0) ~ 1.40 (m, 2H), 2.00 ~m, 2H), 2.25
(s, 3H), 3.30 (m, 2H), 3.60 (m, 2H), 3.80 (m, 1H), 3.95 (m, 1H), 5.90 (s, 2H), 6.90 ~s,
1tl), 7.00 (d, 1H), 7.60 (d, 1H), 7.80 (d, lH), 7.85 (d, 1H), 8.00 (s, lH), 8.10 (d, lH),
8.15 (d, 1H), 8.40 (s, 1H), 8.55 (s, 1H), 9.15 (br s, 3H), 9.40 (s, 2H), 9.55 (s, 1H).
EXAMPLE 5
1 -(4-Amidinonaphth-1 -yl)methyl-2-isopropyl-4-aminocarbonyl-6-(N-(1 -iminoethyl)piperidin-
30 4-ylamino)benzimidazole (2 9) was heated at 80~C in 12N HCI for 12 hours. The solvent was
removed under reduced pressure. To the resulting residue in methanol (80 mL) was added
triethylamine (10 mL) and ethylacetimidate (2 g). After stirring for 12 hours, the reaction mixture
was concenL,~Led. The residue was purified by HPLC on a C18 Dynamax column with a 3-25%
acetonitrile in water ~,adie"L with 0.1 % trifluoroacetic acid to afford 1 -(4-amidinonaphth-1-
35 yl)methyl-2-isopropyl-4-carboxy-6-(N-(l-iminoethyl)piperidin-4-ylamino)benzimidazole~ 1H NMR
CA 02239~08 1998-06-04
W 097/21437 PCT~B96/01496
-74-
(DMS0, 300 MHz) ~ 9.70 (s, 2H), 9.60 (s, 2H), 9.40 (s, 1H), 8.93 (s, 1H), 8.65-7.40 (m, 8H),
6.30 (s, 2H), 4.20-3.50 (m, 6H), 2.55 (s, 3H), 2.30 (m, 2H), 1.70 (m, 2H), 1.60 ~d, 6H), as a
white solid after freeze-drying removal of the solvent.
EXAMPLE 6
This example illustrates the preparation of represerLaLive pharmaceutical compositions for
oral ad~ ,aLion conLa;.l;l-g a compound of the invention, or a pharmaceutically acceptable salt
thereof, e.g.,1-(4-amidinonaphth-1-yl)methyl-2-n-butyl-5-(N-(1-iminoethyl)piperidin-4-
yloxy)benzimidazole:
A. Inqredients % wt./wt.
Compound of the invention 20.0%
Lactose 79-5 %
Mâgnesium stearate 0.5Yo
The above ingredients are mixed and dispensed into hard-shell gelatin capsuies containing
100 mg each, one capsule would approximate a total daily dosage.
15 B. InQredients % wt./wt.
Compound of the invention 20.0~h
Magnesium stearate 0.9%
Starch 8.6%
Lactose 79.6%
PVP ~polyvinylpyrroiidine) 0.9%
The above ingredients with the exception of the magnesium stearate are combined and
granulated using water as a granulating liquid. The formulation is then dried, mixed with the
magnesium stearate and formed into tablets with an appropridle tableting machine.
C. I n4, ecl;e. I l~
Compound of the invention 0.1 9
Propylene glycol 20.0 9
Polyethylene glycol 400 20.0 9
Polysorbate 80 1.0 9
Water q.s. 100 mL
The compound of the invention is dissolved in propylene glycol, polyethylene glycol 400
and polysorbate 80. A sufficient quantity of water is then added with stirring to provide 100 mL
of the solution which is filtered and bottled.
D. In4r~ enL:i % wt.lwt.
Compound of the invention 20.0%
Peanut Oil 78.0%
Span 60 2.0%
CA 02239~08 1998-06-04
W 0 97/21437 PCT~B96/01496
-75-
The above ingredients are melted, mixed and filled into soft elastic capsule
E. Inqredients % wt./wt.
Compound of the invention 1.0%
Methyl or carboxymethyl cellulose 2.0%
0.9% saline q.s. 100 mL
The compound of the invention is dissolved in the cellulose/saline solution, filtered and
bottled for use.
EXAMPLE 7
This example illustrates the preparation of a representative pharmaceutical formulation for
parenteral admin;stration containing a compound of the invention, or a pharmaceutically
accepLdble salt thereof, e.g., 1-(4-amidinonaphth-1-yl~methyl-2-t-butyl-5-~N-(1-iminoethyl)piperidin-4-yloxy)benzimidazole:
Inqredients
Compound of the invention 0.02 g
Propylene glycol 20.0 9
Polyethylene glycol 40020.0 g
Polysorbate 80 1.0 g
0.9% saline solution q.s. 100 mL
The compound of the invention is dissolved in propylene glycol, polyethylene glycol 400
and poiysorbate 80. A sufficient quantity of 0.9% saline soiution is then added with stirring to
provide 100 mL of the l.V. solution which is filtered through a 0.2 ~ membrane filter and
pacl~aged under sterile conditions.
EXAMPLE 8
This ~X&Ill, le illustrates the preparation of a representative pharmaceutical composition in
25 suppository form conlai~ 9 a compound of the invention, or a pharmaceutically ac~;dpLdi le salt
thereof, e.g., 1-(4-amidinonaphth-1-yl)methyl-2-methyl-6-(N-~N-(1-iminoethyllpiperidin-4-yl~-N-
((aminocarbonyllmethyllamino)ben~i"li~701e:
In~redients % wt./wt.
Compound of the invention 1.0%
Polyethylene glycol 1000 74 5%
Polyethylene glycol 4000 24.5%
The ingredients are melted together and mixed on a steam bath, and poured into moldscontaining 2.5 9 total weight.
CA 02239~08 1998-06-04
WO 97/21437 PCT~B96/01496
-76-
EXAMPLE 9
This example illustrates the preparation of a representative pharmaceutical formulation for
insufflation containing a compound of the invention, or a pharmaceutically acceptable salt
thereof, e.g., 1-(4-amidinonaphth-1-yl)methyl-2-propyl-5-(N-(1-iminoethyl)piperidin-4-
5 yloxy)benzimidazole:
Inqredients % wt./wt.
Micronized compound of the invention 1.0%
Micronized lactose 99.0%
The ingredients are milled, mixed, and packaged in an insufflator equipped with a dosing
1 0 pump.
EXAMPLE 10
This example illustrates the preparation of a representative pharmaceutical formulation in
nebulized form containing a compound of the invention, or a pharmaceutically acceptable salt
thereof, e.g., 1 -(4-amidinonaphth- 1 -yl)methyl-2-methyl-5-( 1 -(methoxycarbonyl)ethoxy) -6-(N-~ 1 -
1 5iminoethyl)piperidin-4-yloxy)benzimidazole:
Inu,eclienL~ % wt./wt.
Compound of the invention 0.005%
Water 89.995%
Ethanol 1 0.000%
The compound of the invention is dissolved in ethanol and blended with water. The
formulation is then packaged in a nebulizer equirped with a dosing pump.
EXAMPLE 1 1
This example illustrates the prepaltlLion of a represe,-t~Live pharmaceutical formulation in
aerosol form conLain;.~g a compound of the invention, or a pharmaceutically acc~:pLable salt
25 thereof, e.g., 1-(4-amidinonaphth-1-yi)methyl-2-isopropyl-4-carboxy-6-(N-(1-iminoethyl)piperidin-4-
ylamino)benzimidazole:
Inaredients ~h wt.lwt.
Compound of the invention 0.10%
Propellant 11/12 98.90%
Oleic acid 1.00%
The compound of the invention is dispersed in oleic acid and the propellants. The
resulting mixture is then poured into an aerosol container fitted with a metering valve.
-
CA 02239~08 1998-06-04
W O 97/21437 PCT/IB96/01496
EXAMPLE ~2
(/n vitro assay for Factor Xa and Thrombin)
This assay demonstrates the activity of the compounds of the invention towards factor
Xa, ~hrorllbill and tissue plasminogen activator. The activities were determined as an initiai rate
5 of cleavage of the peptide p-nitroaniiide by the enzyme. The cleavage product, p-niLIoa~ e~
absorbs at 405 nm with a molar extinction coefficient of 9920 M-~cm~1.
Rea~ents and Solutions:
Dimethyl sulfoxide (DMS0) (Baker analyzed grade~.
Assay buffer:
50 mM TrisHCI, 150 mM NaCI, 2.5 mM CaCI2, and 0.1% polyethylene glycol
6000, pH 7.5.
Enzymes (Enzyme Research Lab.):
1. Human factor Xa stock solution: 0.281 mg/mb in assay buffer, stored at -80~C (working
solution (2X): 106 ng/mL or 2 nM in assay buffer, prepared prior to use).
15 2. Human thrombin stock solution: Stored at -80~C (working solution (2X): 1200 ng/mL or
40 nM in assay buffer, prepare prior to use~.
3. Human tissue pla:,n-inogen activator (tPA) (Two chains, Sigma~ stock solution: 1 mç~/mL,
stored at -80~C (working solution (2X): 1361 ng/mL in assay buffer, prepare prior to use).
Chromogenic suL:,L,~Ies (Pharmacia Hepar Inc.):
20 1. S2222 (FXa assay~ stock solution: 6 mM in dH20, store at 4~C (working solution ~4X):
656,L~M in assay buffer).
2. S2302 (Thrombin assay) stock solution: 10 mM in dH20, stored at 4~C (working solution
(4X): 1200 ~M in assay buffer).
3. S2288 (tPA assay) stock solution: 10 mM in dH20, stored at 4~C (working solution (4X):
1484,uM in assay buffer).
(All substrate working solutions were prepared on assay day 5.)
Standard inhibitor compound stock solution:
5 mM in DMS0, stored at -20~C.
Test compounds (compounds of the invention) stock solutions:
10 mM in C)MS0, stored at -20~C.
Assav orocedure:
Assays were performed in 96-well ~ uLilt:r plates in a total volume of 200 ~I. Assay
conducted in final concentration of 50 mM TrisHCI, 150 mM NaCI, 2.5 mM CaC12, 0.1 %
polyethylene glycol 6000, pH 7.5, in the absence or presence of the standard inhibitor or the test
35 compounds and enzyme and substrate at followin~ concellllaLions: (1) 1 nM factor Xa and 164
,uM S2222; (2) 20 nM thrombin and 300,11M S2302; and (3) 10 nM tPA and 371 ~M S2288.
Concentrations of the standard inhibitor compound in the assay were from 5,uM to 0.021,uM in 1
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-78-
to 3 dilution. Concentration of the test compounds in the assay typically were from 10 ~M to
0.041 ~M in 1 to 3 dilution. For potent test compounds, the concentrations used in the factor Xa
assay were further diluted 100 fold (100 nM to 0.41 nM) or 1000 fold (10 nM to 0.041 nM). All
substrate concentrations used are equal to their Km values under the present assay conditions.
5 Assays were performed at ambient temperature.
The first step in the assay was the preparation of 10 mM test compound stock solutions
in DMSO (for potent test compounds, 10 mM stock solutions were further diluted to 0.1 or
0.01 mM for the factor Xa assay), followed by the preparation of test compound working
solutions (4X) by a serial dilutions of 10 mM stock solutions with Biomek 1000 (or Multiprobe
10 204) in 96 deep well plates as foliows:
la) Prepare a 40,uM working solution by diluting the 10 mM stock 1 to 250 in assay
buffer in 2 steps: 1 to 100, and 1 to 2.5.
~b) Make another five serial dilutions ~1:3) of the 40,uM solution (600 ,uL for each
concentration). A total of six diluted test compound solutions were used in the assay.
Standard inhibitor compound (5 mM stock) or DMSO ~control) went through the same dilution
steps as those described above for test compounds.
The next step in the assay was to dispense 50,LIL of the test compound working solutions
14X) (from 40 uM to 0.164 uM), in duplicate, to microtiter plates with Biomek or MP204. To this
was added 100 ~L of enzyme working solution (2X) with Biomek or MPZ04. The resulting
20 solutions were incubated at ambient temperature for 10 minutes.
To the solutions was added 50 ,LIL of substrate working solution (4X~ with Biomek or
MP204.
The enzyme kinetics were measured at 405 nm, at 10 seconds interval, for five minutes
in a THERMOmax piate reader at ambient temperature.
25 Calculation of K~ of the test comDounds:
Enzyme rates were calc~-lat.od as mOD/min based on the first two minutes readings. The
IC50 values were dt:Ler,..il.ed by fitting the data to the log-logit eciuation ~linear) or the Morrison
equation (non-linear) with an EXCEL spread-sheet. Ki values were then obtained by dividing the
IC50 by 2. Routinely, Ki~factor Xa) values lower than 3 nM were cAlc~ t~d from the Morrison
30 equation.
Compounds of the invention, when tested in this assay, demonstrated the ability to inhibit
human factor Xa and human thrombin.
EXAMPLE 1 2
(In vitro assay for Human Prothrombinase)
This assay demon~ Les the ability of the compounds of the invention to inhibit
prothrombinase. Prothrombinase (i'Tase) catalyzes the activation of prothrombin to yield
CA 02239~08 1998-06-04
W O 97/21437 PCT~B96/01496
-79-
fragment 1.2 plus thrombin with meizothrombin as the intermediate. This assay is an end point
assay. Activity of the prothrombinase is measured by activity of thrombin (one of the reaction
products) or by the amount of thrombin formed/time based on a thrombin standard curve (nM vs
mOD/min~. For determination of ICso (PTase) of the compounds of the invention, PTase activity
5 was expressed by thrombin activity (mOD/min).
Materials:
Enzymes:
1. Human factor Va (Haematologic Technologies Inc., Cat# HCVA-0110) working solution:
1.0 mg/mL in 50% glycerol, 2 mM CaC12, stored at -20~C.
10 2. Human factor Xa (Enzyme Res. Lab. cat# HFXalO11) workin~ solution: 0.281 mg/mL in
assay buffer ~without BSA), stored at-80~C.
3. Human prothrombin ~FII) ~Enzyme Res. Lab., Cat# HP1002) working solution:
Diluted Fll to 4.85 mg/mL in assay buffer (without BSA), stored at -80~C.
Phospholipid ~PCPS) vesicles:
PCPS vesicles (80%PC, 20%PS) were prepared by modification of the method reported
by Barenholz et a/., ~iochemistry (1977), Vol. 16, pp. 2806-2810.
Phosphatidyl serine (Avanti Polar Lipids, Inc., Cat#840032):
10 mg/mL in ;hlorofor.ll, purified from brain, stored -20~C under nitrooen or
argon .
Phosphatidyl Choline ~Avanti Polar Lipids, Inc., Cat# 850457):
50 mg/mL in cl,lorotc.l.ll, synthetic 16:0-18:1 Palmitoyl-Oleoyl, stored at -20~C
under nitrogen or argon.
Spectrozyme-TH ~American Diagnostica Inc., Cat# 238L, 50 ~moles, stored at room
temperature) working solution: Dissolved 50 ~moles in 10 mL dH2O.
25 BSA ~Sigma Chem Co., Cat# A-7888, FractionV, RIA grade).
Assay buffer: 50 mM TrisHCI, pH 7.5, 150 mM NaCI, 2.5 mM CaCI2, 0.1% PEG 6000
~BDH), 0.05% BSA ~Sigma, Fr.V, RIA grade).
For one plate sssav. prepare the following working solutions:
1. Prothrombinase complex:
30 ~a) 100,uM PCPS (27.5 ~L of PCPS stock ~4.36 mM) diluted to final 1200,uL with
assay buffer.
~b) 25 nM Human factor Va: 5.08 ,uL of Va stock (1 mg/mL) was diluted to final
1200 ~L with assay buffer.
(c) 5 pM Human factor Xa: Dilute Xa stock (0.281 mg/mL) 1 :1,220,000 with assay
buffer. Prepare at least 1200 ~I.
Combine equal volumeç (1100,uL) of each component in the order of PCPS, factor Va and
factor Xa. Let stand at ambient temperature for 5 to 10 minutes and use immediately, or
- CA 02239~08 1998-06-04
W O 97~1437 PCTnB96/01496
-80-
store in ice ~bring to ambient temperature before use).
2. 6,uM Human prothrombin IFII): dilute 124~L of Fll stock (4.85 mg/mL) to final 1400~L
with assay buffer.
3. ZO mM EDTA/Assay buffer: 0.8 mL of 0.5 M EDTA (pH 8.5) plus 19.2 mL assay buffer.
4. 0.2 mM Spectrozyme-TH/EDTA buffer: 0.44 mL of SPTH stock (5 mM) pius 10.56 mL of
20 mM EDTA/assay buffer.
5. Test compounds (compounds of the invention):
Prepare a working solution ~5X) from 10 mM stock (DMSO) and make a series of 1:3dilution. Compounds were assayed at 6 concenL~Lions in duplicate.
10 Assav conditions and procedure:
Prothrombinase reaction was performed in final 50 ~L of mixture containing PTase ~20 uM
PCPS, 5 nM hFVa, and 1 pM hFXa), 1.2 uM human factor ll and varied concentration of the test
compounds (5 ~M to 0.021 JIM or lower concentration range). Reaction was started by addition
of PTase and incubated for 6 minutes at room temperature. Reaction was stopped by addition of
15 EDTA/buffer to final 10 mM. Activity of thrombin Iproduct) was then measured in the presence
of 0.1 mM of Spectrozyme-TH as substrate at 405 nm for 5 minutes (10 second intervals), at
ambient temperature, in a THEROmax microplate reader. Reactions were performed in 96-well
micl oliLe~ plates.
In the first step of the assay, 10 ~L of diluted test compound t5X) or buffer was added to
20 the plates in dupliGate. Then 10 ,uL of pr.,lhor.lbin (hFII) (5X) was added to each well. Next 30
,uL PTase was added to each well, mix for about 30 seconds. The plates were then incubated at
ambient temperature for 6 minutes.
In the next step, 50 ~L of 20 mM EDTA (in assay buffer) was added to each well to stop
the reaction. The resulting solutions were then mixed for about 10 seconds. Then 100 ,uL of 0.2
25 mM spectro~yme was added to each well. The thrombin reaction rate was then measured at
405 nm for 5 minutes (at 10 second intervals) in a Molecular Devices microplate reader.
Calculations:
Thrombin reaction rate was ~ .ressed as mOD/minute using OD readings from the five
minute reaction. IC!;o values were c~lclll~t~d with the log-lo~it curve fit program.
The compounds of the invention demonstrated the ability to inhibit thrombinase when
tested in this assay.
EXAMPLE 13
(In v;vo assayJ
The following assay demonsll~L~s the ability of the compounds to act as anti-coagulants.
Male rats (250-330 9) were ane~Lll~ ed with sodium pentobarbital (90 mg/kg, i.p.) and
prepared for surgery. The left carotid artery was cannulated for the measurement of blood
CA 02239~08 1998-06-04
W O 97/21437 PCT/IB96/01496
-81-
pressure as well as for taking blood samples to monitor clotting variables (prothrombin time (PT)
and activated partial thromboplastin time (aPTT~). The tail vein was cannulated for the purpose of
administering the test compounds Ir.e., the compounds of the invention and standards~ and the
thromboplastin infusion. The abdomen was opened via a mid-line incision and the abdom;nal vena
5 cava was isolated for 2-3 cm distal to the renal vein. All venous branches in this 2-3 cm segment
of the abdominal vena cava were ligated. ~ollowing all surgery, the animals were allowed to
stabilize prior to beginning the experiment. Test compounds were ad~ -i .Le.ed as an intravenous
bolus (t=0). Three minutes later ~t=3), a 5-minute infusion of thromboplastin was begun. Two
minutes into the infusion (t=~;), the abdominal vena cava was ligated at both the proximal and
10 distal ends. The vessel was left in place for 60 minutes, after which it was excised from the
animal, slit open, the clot (if any) carefully removed, and weighed. SI~L;~LiCC,I analysis on the
results was performed using a Wilcoxin-matched-pairs signed rank test.
The compounds of the invention, when tested in this assay, demonstrated the ability to
clot the blood.
1 5
While the present invention has been described with r~:r~ nce to the specific
embodiments thereof, it should be understood by those skilled in the art that various changes
may be made and equivalents may be substituted without departing from the true spirit and scope
of the invention. In addition, many modifications may be made and equivalents may be
20 substituted without departing from the true spirit and scope of the invention. In addition, many
modifications may be made to adapt a particular situation, material, composition of matter,
process, process step or steps, to the objective, spirit and scope of the present invention. All
such modifications are intended to be within the scope of the claims appended hereto.