Note: Descriptions are shown in the official language in which they were submitted.
CA 0224011~ 1998-06-09
WO 97t21435 PCT/US96/20004
TITLE~ OF THE INVENTION
~ AN7rAGONISTS ~F GONADOTROPIN RELEASING HORMONE
BACKGROUN~ OF THE INVENTION
The gonadotropin-releasing hormone (GnRH), also
referred to as luteinizing hormone-releasing hormone (LHRH), is a
decapeptide that plays a key role in hllm~n reproduction. The hormone
is released from the hypoth~l~mus and acts on the pituitary gland to
stimulate the biosynthesis and secretion of luteinizing hormone (LH) and
follicle-stimulating hormone (FSH). LH released from the pituitary
gland is primarily responsible for the regulation of gonadal steroid
production in both sexes, whereas FSH regulates spermatogenesis in
males and follicular development in females. GnRH agonists and
antagonists have proven effective in the treatment of certain conditions
which require inhibition of LH/FSH release. In particular, GnRH-based
therapies have proven effective in the treatment of endometriosis,
uterine fibroids, polycystic ovarian disease, precocious puberty and
several gonadal steroid-dependent neoplasia, most notably cancers of the
prostate, breast and ovary. GnRH agonists and antagonists have also
been utili~ed in various assisted fertilization techniques and have been
investigated as a potential contraceptive in both men and women. They
have also shown possible utility in the treatment of pituitary
gonadotrophe adenomas, sleep disorders such as sleep apnea, irritable
bowel syndrome, premenstrual syndrome, benign prostatic hyperplasia,
hirsutism, as an adjunct to growth hormone therapy in growth hormone
deficient children, and in murine models of lupu.s. The compounds of
the invention may also be used in combination with bisphosphonates
(bisphosphonic acids) and other agents, such as growth hormone
secretagogues, e.g. MK-0677, for the treatment and the prevention of
disturbances of calcium, phosphate and bone metabolism, in particular,
for the prevention of bone loss during therapy with the GnRH
antagonist, and in combination with estrogens, progesterones,
antiestrogens, antiprogestins and/or androgens for the prevention or
CA 02240115 1998-06-09
WO 97121435 PCT/US96/20004
treatment of bone loss or hypogonadal symptoms such as hot flashes
during therapy with the GnRH antagonist.
Additionally, a compound of the present invention may be
co-aclminixtered with a Soc-reductase 2 inhibitor, such as finasteride or
epristeride; a So~-reductase 1 inhibitor such as 4,7,B-dimethyl-4-aza-5Oc-
cholestan-3-one, 3-oxo-4-a~;a-4,7,B-dimethyl- 16,~-(4-chlorophenoxy)-
5Oc-androstane, and 3-oxo-4-aza-4,7,B-dimethyl-16,B-(phenoxy)-5~-
androstane as disclosed in WO 93/23420 and WO 95/11254; dual
inhibitors of So~-reductase 1 and Soc-reductase 2 such as 3-oxo-4-aza-
17,13-(2,5-trifluoromethylphenyl-carbamoyl)-Sa-androstane as disclosed
in WO 95/07927; antiandrogens such as flutamide, casodex and
cyproterone acetate, and alpha-l blockers such as prazosin, terazosin,
doxazosin, tamsulosin, and alfuzosin.
Further, a compound of the present invention may be u.sed
in combination with growth hormone, growth ho~none releasing
horrnone or growth hormone secretagogues, to delay puberty in growth
hormone de~lcient children, which will allow thern to continue to gain
height before fusion of the epiphyses and cessation of growth at puberty.
Current GnRH antagonists are GnRH-like decapeptides
which are generally ?~flministered intravenously or subcutaneously
presumably because o~ negligible oral activity. These have amino acid
substitutions usually at positions one, two, three, six and ten.
Non-peptide GnRH an~agonists offer the possible advantage
of oral ~lm;n.~tration. Non-peptide GnRH antagonists have been
described in European Application 0 219 292 and in De, B. et al., J.
~ed. Chem., 32, 2036-2038 (1989), in WO 95/28405, WO 95/29900
and EP 0679642 all to Takeda Chemical Industries, Ltd.
Substituted indoles known in the art include those described
in the ~ollowing patents and patent applications. US Patent No.
5,030,640 discloses alpha-heterocyclic ethanol aminoalkyl indoles which
are potent r3-agonists. US Patent No. 4,544,663 discloses indolamine
derivatives which are allegedly useful as male anti-i~ertility agents. WO
90/05721 discloses alpha-amino-indole-3-acetic acids useful as anti-
diabetic, anti-obesity and anti-atherosclerotic agents. French patent
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
2,181,559 discloses indole derivatives with .sedative, neuroleptic,
analgesic, hypotensive, an~iserotonin and adrenolytic activity. Belgian
patent 8793~1 discloses 3-aminoalkyl-lH-indole-5-thioamide and
carboxamide derivatives as cardiovascular agents used to treat
hypertension, Raynaud's disease and migraine.
SUMMARY OF THE INVENTION
The present invention relates to compounds which are non-
peptide antagonists of GnRH which can be used to treat a variety of sex-
hormone related conditions in men and women, to methods for their
preparation, and to methods and pharmaceutical compositions cont~ining
said compounds for use in m~mm~
Because of their activity as antagonists of the hormone
GnRH, the compounds o~ the present invention are useful to treat a
variety of se~-hormone related conditions in both men and women.
These conditions include endometriosis, uterine fibroids, polycystic
ovarian disease, hirsutism, precocious puberty, gonadal steroid-
dependent neoplasia.s such as cancers of the prostate, breast and ovaly,
gonadotrophe pituitary adenomas, sleep apnea, irritable bowel
syndrome, premenstrual syndrome and benign prostatic hypertophy.
They are also useful as an adjunct to treatment of growth horrnone
deficiency and short stature, and for the treatment of systemic lupus
erythematosis. Further, the compounds of the invention may be useful
in in vitro fertilization and as contraceptives. The compounds may also
be useful in combination with androgens, estrogens, progesterones,
antiestrogens and antiprogestogens for the treatment of endometriosis,
fibroids and in contraception. They may also be useful in combination
with testosterone or other androgens or antiprogestogens in men as a
contraceptive. The compounds may also be used in combination with an
angiotensin-converting enzyme inhibitor such as Fn~l~pril or Captopril,
an angiotensin II-receptor antagonist such as Losartan or a renin
inhibitor for the treatment of uterine fibroids. Additionally, the
compounds of the invention may also be used in combination with
bisphosphonates (bisphosphonic acids) and other agents, for ~e
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
treatment and the prevention of dis~urbances of calcium, phosphate and
bone metabolism, in particular, for the prevention of bone loss during
therapy with the GnRH antagonist, and in combination with estrogens,
progesterones and/or androgens for the prevention or treatment of bone
loss or hypogonadal symptoms such as hot flashes during therapy with
the GnRH antagonist.
Additionally, a compound of the present invention may be
co~ mini~tered with a So~-reductase 2 inhibitor, such as finasteride or
epristeride; a So~-reductase 1 inhibitor such as 4,7,3-dimethyl-4-aza-50~-
cholestan-3-one, 3-oxo-4-aza-4,7,B-dimethyl-16,13-(4-chlorophenoxy)-
Sa-androstane, and 3-oxo-4-aza-4,7~-dimethyl-1613-(phenoxy)-Soc-
androstane as disclosed in WO 93/23420 and WO 9S/1 1254; dual
inhibitors of Sa-reductase 1 and Sa-reductase 2 such as 3-oxo-4-aza-
17,3-(2,5-trifluoromethylphenyl-carbamoyl)-50c-androstane as disclosed
in WO 95/07927; antiandrogens such as flutamide, casodex and
cyproterone acetate, and alpha-1 blockers such as prazosin, terazo.sin,
doxazosin, tamsulosin, and alfuzosin.
Further, a compound of the present invention may be used
in combination with growth horrnone, growth hormone releasing
hormone or growth hormone secretagogues, to delay puberty in growth
honnone deficient children, which will allow them to continue to gain
height be~ore fusion of the epiphyses and cessation of growth at puberty.
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
DETAILED DESCRIPTION OF THE l~VENTION
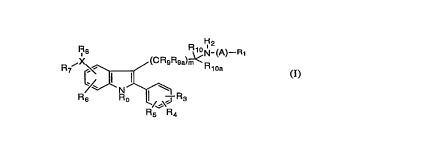
The present invention relates to compounds of the general
formula
~8 R~N- (A)--
R6 Ro /~\'J R3
R~; R4 (1)
wherem
A is Cl-C6 alkyl, substituted C1-C6 alkyl, C3-C7 cycloalkyl,
substituted C3-C7 cycloaLkyl, C3-C6 alkenyl, substituted C3-
C6 alkenyl, C3-c6 alkynyl, substituted C3-c6 alkynyl, Cl-C6
alkoxy, or Co-C~ aLkyl-S(O)n-Co-cs alkyl, Co-Cs alkyl-O-
Co-Cs alkyl, Co-Cs alkyl-NR1g-Co-Cs alkyl where R1g and
the Co-Cs alkyl can be joined to form a ring,
~ ,~N-(cH2)p-
R16 ' or a single bond;
Ro is hydrogen, C1-C6 alkyl, substituted C1-C6 alkyl, wherein the
substituents are as defined below; aryl, substituted aryl,
araLkyl or substituted aralkyl, wherein the substituents are as
defined for R3, R4 and Rs;
Rl is
~/'~ ~R-~
wherein:
CA 02240115 1998-06-09
WO 97/2143S PCT/US96/20004
Y is B, C or a bond;
B is 0, S(O)n, C(O), NRls or C(R1 lRl2)p
C is B(CH2)p-;
R2 is hydrogen, Cl-C6 alkyl, substituted C1-C6 alkyl, aralkyl,
substituted araLkyl, aryl, substituted aryl, alkyl -ORl 1,
Cl-C6(NRl lRl2), Cl -c6(coNR l lRl2) or C(NRl lRl2)NH;
R2 and A taken together form a ring of 5-7 atoms;
R3, R4 and Rs are independently hydrogen, C1-C6 alkyl, substituted
Cl-C6 alkyl, C2-C6 alkenyl, substituted C2-C6 alkenyl, CN,
nitro, C1-C3 perfluoroalkyl, Cl-C3 perfluoroalkoxy, aryl,
substituted aryl, aralkyl, substituted aralkyl, Rl IO(CH2)p-
~R1 1C(O)O(CH2)p-, Rl lOC(O)(CH2)p-, -(cH2)ps(o)nRl7~
-(CH2)pC(O)NRI lR12 or halogen; wherein R17 is hydrogen,
Cl-C6 alkyl, C1-C3 perfluoroalkyl, aryl or substituted aryl;
R3 and R4 taken together form a carbocyclic ring of 3-7 carbon atoms
or a heterocyclic ring con~ining 1-3 heteroatoms selected
from N, O and S;
R6 is hydrogen, C1-C6 alkyl, substituted Cl-C6 aLkyl, aryl,
substituted aryl, Cl-C3 perfluoroalkyl, CN, N02, halogen,
lR 1 lO{CH2)p-, NR 12c(o)Rl 1, NR 12c(o)NR 1 l R 12 or SOnRl l;
R7 is hydrogen, Cl-C6 alkyl, or substituted Cl-C6 alkyl, unless X
is hydrogen or halogen, then R7 is absent;
R8 is hydrogen, C~O)ORg, C(O)NRl lRl2, NR1 lRl2~ C(O)Rl 1,
NR12C(O)Rl 1, NR12C(O)NR1 13? l2, NR12S(0)2Rl 1,
NR12S(0)2NR1 lRl2, OC(O)Rl 1, OC(O)NR1 lR12, ORl 1,
SOnRl 1, S(O)nNR 1 IR12, Cl-c6 aLlcyl or substituted Cl-C6
alkyl, unless X is hydrogen or halogen, then Rx is absent; or
R7 and R8 taken together form a carbocyclic ring of 3-7 atoms;
Rg and Rga are independently hydrogen, C1-C6 alkyl, substituted Cl-C6
alkyl; aryl or substituted aryl, aralkyl or substituted araLkyl
when m~O; or
Rg and R~a taken together form a carbocyclic ring of 3-7 atoms or
when m~O;
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
~g and A taken toget~er form a heterocyclic ring cont~ining 3-7 carbon
atoms and one or more heteroatoms when m~0; or
Rlo and R10a are independently hydrogen, C1-C6 alkyl, substituted Cl-
C6 alkyl, aryl, substituted aryl, aralkyl or substituted
aralkyl; or
Rlo and Rloa taken together form a carbocyclic ring of 3-7 atoms or
ll
Rg and Rlo taken together folm a carbocyclic ring of 3-7 carbon atoms
or a heterocyclic ring containing one or more heteroatoms
when m~O; or
Rg and R2 taken together form a heterocyclic ring cont~ining 3-7
carbon atoms and one or more heteroatoms when m~0; or
Rlo and R2 taken together form a heterocyclic ring con1zlining 3-7
carbon atoms and one or more heteroatoms;
Rlo and A taken together fo~n a heterocyclic ring cont~ining 3-7
carbon atoms and one or more heteroatoms; or
R 1 1 and R 1 2 are independently hydrogen, C 1 -C6 alkyl, substituted C 1 -
C6 alkyl, aryl, substituted aryl, aralkyl, substituted aralkyl, a
carbocyclic ring of 3-7 atoms or a substituted carbocyclic
ring containing 3-7 atoms;
Rl 1 and Rl2 taken together can fonn an optionally substituted ring of 3-
7 atoms;
R 13 is hydrogen, OH, NR7R~, NR 1 1 SO2(C 1 -C6 alkyl),
NR1 1SO2(substituted Cl -C6 alkyl), NRl ISO2(aryl),
NRl 1SO2(substituted aryl), NRI 1SO2(Cl-C3
perfluoroalkyl); S02NR 1 1 (C 1 -C6 alkyl),
SO2NR 1 1 (substituted C 1 -C6 alkyl), SO2NR 1 1 (aryl),
SO2NR 1 1 (substituted aryl), SO2NR 1 1 (C 1 -C3
perfluoroalkyl); SO2NR 1 1 (C(O)C 1 -C6 aLkyl);
SO2NR 1 1 (C(O)-substituted C l -c6 alkyl); SO2NR ~ l (C(O)-
aryl); SO2NRIl(C(O)-substituted aryl); S(O)n(C1-C6 alkyl);
S(O)n
CA 02240115 1998-06-09
WO 97/21435 PCTIUS96/20004
(substituted Cl-C6 alkyl), S(O)n(aryl), S(O)n(substituted
aryl), Cl-C3 perfluoroalkyl, Cl-C3 perfluoroalkoxy, Cl-C6
alkoxy, substituted C1-C6 alkoxy, COOH, halogen, NO2 or
CN;
R14 and Rls are independently hydrogen, Cl-C6 aLkyl, substituted Cl-C6
alkyl, C~2-C6 alkenyl, substituted C2-C6 alkenyl, CN, nitro,
Cl-C3 perfluoroalkyl, Cl-C3 perfluoroalkoxy, aryl,
substituted aryl, aralkyl, substituted aralkyl, R1 IO(CH2)p-,
R 11 C(O)O(CH2)p-, R 1 l OC(O)(CH2)p-, -(CH2)pS(O)nR 17,
-(CH2)pC(O)NR 1 1 R 1 2 or halogen; wherein R 17 is hydrogen,
C~ 6 alkyl, Cl-C3 perfluoroalkyl, aryl or substituted aryl;
R16 is hydrogen, Cl-C6 alkyl, substituted Cl-C6 alkyl, or
N(R1 1R12);
R1g is hydrogen, C1-(~6 alkyl, substituted Cl-C6 aLkyl, C(O)ORg,
C(O)NRl lRl2, C(O)R~ (O)nRl l;
X is hydrogen, halogen, N, O, S(O)n~ C(O), (CR1 lRl2)p; C2-c6
alkenyl, substituted C2-C6 alkenyl, C2-C6 aLkynyl, or
substituted C2-C6 aLkynyl; when X is hydrogen or halogen,
R7 and R8 are absent; when X is O, S(O)n, C(O), or
CR 1 I R 12 only R7 or R8 is possible;
m is 0-3;
n is 0-2;
p is 0-4; and
the aLkyl, cycloalkyl, alkenyl and aLkynyl substituents are
selected from C1-C6 alkyl, C3-C7 cycloalkyl, aryl,
substituted aryl, aralkyl, substituted araLkyl, hydroxy, oxo,
cyano, (~ 6 alkoxy, fluoro, C~O)ORI 1, aryl Cl-C3 alkoxy,
substituted aryl C~ 3 alkoxy, and the aryl substituents are
as defined for R3, R4 and Rs,
or a pharmaceutically acceptable addition salt and/or hydrate thereof, or
where applicable, a geometric or optical isomer or racemic mixture
thereof.
Unless otherwise stated or indicated, the following
definitions shall apply throughout the specification and claims.
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
When any variable (e.g., aryl, heterocycle, Rl, etc.) occurs
more than one time in any constituent or in formula I, its definition on
each occurrence is independent of its definition at every other
occurrence. Also, combinations of substituents and/or variables are
permissible only if such combinations result in stable compounds.
The tersn "alkyl" is intended to include both branched- and
straight-chain saturated aliphatic hydrocarbon groups having the
specified number of carbon atoms, e.g., methyl (Me), ethyl (Et), propyl,
butyl, pentyl, hexyl, heptyl, octyl, nonanyl, decyl, undecyl, dodecyl, and
the isomers thereof such as isopropyl (i-Pr), isobutyl (i-Bu), sec-butyl
(s-Bu), tert-butyl (t-Bu), isopentane, isohexane, etc.
The term "aryl" includes phenyl and naphthyl. Preferably,
aryl is phenyl.
The term "halogen" or "halo" is intended to include
fluorine, chlorine, bromine and iodine.
The term "heterocycle" or "heterocyclic ring" is defined by
all non-aromatic, heterocyclic rings of 3-7 atoms con~ining 1-3
heteroatoms selected from N, O, and S, such as oxirane, oxetane,
tetrahydrofuran, tetrahydropyran, pyrrolidine, piperidine,
tetrahydropyridine, tetrahydropyrimidine, tetrahydrothiophene,
tetrahydrothiopyran, morpholine, hydantoin, valerolactam,
pyrrolidinone, and the like.
As used herein, the term "composition" is intended to
encompass a product comprising the specified ingredients in the
specified amounts, as well as any product which results, directly or
indirectly, from combination of the specified ingredients in the specified
amounts
In addition, it is well known to those skilled in the art that
many of the foregoing heterocyclic groups can exist in more than one
tautomeric form. It is intended that all such tautomers be included
within the ambit of this invention.
The optical isomeric forms, that is mixtures of
enantiomers, e.g., racemates, or diastereomers as well as individual
enantiomers or diastereomers of the instant compound are included.
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
- 10 -
These individual enantiomers are commonly designated according to the
optical rotation they effect by the symbols (+) and (-), (L) and (D), (1)
and (d) or combinations thereof. These isomers may also be designated
according to their absolute spatial configuration by (S) and (R), which
stands for sinister and rectus, respectively.
The individual optical isomers may be prepared using
conventional resolution procedures, e.g., treatment with an appropriate
optically active acid, separating the diastereomers and then recovering
~e desired isomer. In addition, the individual optical isomers may be
prepared by asymmetric synthesis.
Additionally, a given chemical formula or name shall
encompass pharmaceutically acceptable addition salts thereof and
solvates thereof, such as hydrates.
The compounds of the present invention, while effective
themselves, may be formulated and administered in the for~n of their
pharmaceutically acceptable addition salts for purposes o~ stability,
convenience of crys~ tion, increased solubility and other desirable
properties.
The compounds of the present invention may be
~mini~tered in the form of pharmaceutically acceptable salts. The telm
"pharmaceutically acceptable salt" is intended to include all acceptable
salts. Examples of acid salts are hydrochloric, nitric, sulfuric,
phosphoric, formic, acetiG, trifluoroacetic, propionic, maleic, succinic,
malonic, methane sulfonic and the like which can be used as a dosage
folln for modifying the solubility or hydrolysis characteristics or can be
used in sustained release or prodrug form~ ions. Depending on the
particular functionality of the compound of the present invention,
pharmaceutically acceptable salts of the compounds of this invention
include those formed from cations such as sodium, potassium,
aluminum, calcium, lithium, magnesium, zinc, and from bases such as
ammonia, çthylenediamine, N-methyl-glllt~mine, lysine, arginine,
ornithine, choline, N,N'-dibenzylethylenedi~mine, chloroprocaine,
diethanolamine, procaine, N-benzylphenethylamine, diethylamine,
piperazine, tris(hydroxymethyl)aminomethane, and
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
tetramethylammonium hydroxide. These salts may be prepared by
standard procedures, e.g. by reacting a free acid with a suitable organic
or inorganic base, or alternatively by reacting a free base with a suitable
~ organic or inorganic acid.
Also, in the case of an acid (-COOH) or alcohol group
being present, pharmaceutically acceptable esters can be employed, e.g.
methyl, ethyl, butyl, acetate, maleate, pivaloyloxymethyl, and the like,
and those esters known in the art for modifying solubility or hydrolysis
characteristics for use as sustained release or prodrug formulations.
The compounds of the present invention may have chiral
centers other than those centers whose stereochemistry is depicted in
formula I, and therefore may occur as racemates, racemic mixtures
and as individual enantiomers or diastereomers, with all such
isomeric forms being included in the present invention as well as
mixtures thereof. Furthermore, some of the crystalline forms for
compounds of the present invention may exist as polymorphs and as
such are intended to be included in the present invention. In
addition, some of the compounds of the instant invention may form
solvates with water or common organic solvents. Such solvates are
encompassed within the scope of this invention.
The compounds of the invention are prepared by the
following reaction schemes. All substituents are as defined above unless
indicated other~,vise.
CA 02240115 1998-06-09
WO 97/21435 PCT/US96120004
- 12 -
Scheme A
I ~ ~ NH2
Ro
OEt R ~ ~ ~ 3
pyridine HBr;Br2 R--
THF/CHCI3 R6 R10 R10aO
B(~H)2 3
R¢6~R--4R3~ ~
Na2CO3, LiCI R6 ll R
Pd(PPh3)4 ~ N ~1 0aO
toluene/EtOH Ro 11/ \,J R3
Rs R4
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 13 -
H2NNH2 ~ R ~,X~NH2
THF/EtOH R6 5
Reaction Scheme A
As shown in reaction Scheme ~, treatment of tryptamine
(1) with N-carboxyphth~limide in an inert organic solvent such as
tetrahydrofuran at a temperature of 20-65~C, preferably 65~C, for a
period of 12-48 hours gives the corresponding N-phthalimidotryptamine
derivative (2). The N-phthalimidotryptamine (2) could be further
modified by treatment with a bromin~ting agent such as pyridinium
hydrobromide perbromide, pyrrolidone hydrotribromide, or the like in
an inert organic solvent such as tetrahydrofuran, methylene chloride,
chloroform, or mixtures thereof at 0-25~C for a period of 30 minutes to
4 hours to provide the 2-bromotryptamine (3). Bromide (3) may be
reacted with an arylboronic acid (prepared essentially as described in:
Gronowitz, S.; Hornfeldt, A.-B.; Yang, Y.-H. Chem. Scr. 1986, 26,
311-314.) with palladium (0) catalysis, a weak base such as aqueous
sodium carbonate or the like, and a chloride source such as lithium
chloride in an inert solvent like toluene, benzene, ethanol, propanol or
mixtures thereof at a temperature of 25~-100~C, preferably 80~C, for a
period of 1-6 hours to give the 2-aryltryptamine derivative (4~. Finally,
the phth~limido group may be removed by treatment of (4) with
aqueous hydrazine in an inert solvent such as methanol or ethanol at a
temperature of 0~-25~C for a period of 4-24 hours to give tryptamine
(~)- .
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 14 -
Scheme B
HO~(A~ R
0 6
EDC, HO~
~8 NMM, CH2C12
R ~X~NHR2 R8 Rg Rg R2
S ~/~ J R~A) R
X~(A)R / R~ R4
0 8
triethylamine
CH2CI2
Reaction Scheme B
As shown in reaction Scheme B, the 2-aryltryptamine may
be condensed with a carboxylic acid of type (6) using the coupling
reagent 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
(EDC), 1,3-dicyclohexylcarbodiimide (DCC) or the like with or without
l-hydroxybenzotriazole (HOBt) and a tertiary amine base such as N-
methylmorphol~ne (NMM), triethylamine or the like in an inert organic
solvent such as methylene chloride, chloroform, dimethylformamide, or
mixtures thereof at or near room temperature for a period of 3-24
hours to provide the corresponding amide derivative (7). Alternatively,
2-aryltrypt~mine (5) can be treated with an active ester or acid chloride
of type (8) in an inert organic solvent such as methylene chloride,
chloroform, tetrahydrofuran, diethyl ether, or the like and a tertiary
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 15 -
amine base such as triethylamine, diisopropylethylamine, pyridine or the
like at a temperature of 0~-25~C for 30 minutes to 4 hours to give (7).
Scheme C
R ,~ _
7 Ro /~\~J 3
R5 R4
R8
or R ,X~ Rs~N~(A) R
LiAlH4, TH F g ~/~\J
Rs R4
Reaction ~cheme C
As shown in reaction Scheme C, the amide carbonyl of (7)
can be reduced by treatment with borane, lithium aluminllm hydride, or
equivalent hydride sources in an inert organic solvent such as
tetrahydrofuran, diethyl ether, 1,4-dioxane or the like at 25~-100~C,
preferably 65~C, for a period of 1-8 hours to give the corresponding
amine compound (9).
CA 02240115 1998-06-09
WO 97/2143S PCT/US96/20004
- 16 -
Scheme D
P'8
R9a~ NHR2
s~ ~\'RJ1~R3
Rs R4
R8
TFA 3 A s, ~es~ Rs~20~A)--R1
NaCNBH3 MeOH11 Ro /~\'J R3
R~ R4
Reaction Scheme D
As shown in reaction Scheme D, the 2-aryltryptamine (5)
can be modi~led by treatment with an aldehyde or ketone of type (10) in
the presence of a weak acid such as trifluorfoacetic acid (TFA~, acetic
acid or the like, with or without a dessicant such as 3A molecular sieves
or magnesium sulfate, and a hydride source such as sodium borohydride
or sodium cyanoborohydride, in an inert organic solvent such as
methanol, ethanol, isoprop~nol, tetrahydrofuran, dichloromethane,
chloroform, or mixtures thereof at a temperature of 0~-25~C for a
period of 1-12 hours to give the corresponding secondary or tertiary
amine derivative ( 1 1).
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
Scheme E
~,
o
~HCI R ~3~,9R9 13 R8
NHNH2 R5 R10 RlOa R7~X Rg Rg
n-butanol ~ ~NH2
R6 or- R6 ~--NJ~l Oa
12 R4~0 RlOa 5 H ~ J R3
R5 14
methanol/t-butanol
Reaction Scheme E
As shown in reaction Scheme E, treatment of an
arylhydrazine or arylhydrazine hydrochloride (12) with an
arylcyclopropyLketone of type (13) in a polar organic solvent such als
methanol, ethanol, n-propanol, isopropanol, n-butanol, t-butanol,
preferably n-butanol, at a temperature of 70~-120~C for a period of 8-
24 hours gives 2-aryltryptamine (5). Alternatively, when an
arylhydrazine or arylhydrazine hydrochloride (12) is treated with an
arylbutyl ketone of type (14) con~:~ining a leaving group (chloride,
bromide, iodide, O-methansulfonate, O-trifluoromethansulfonate, or the
like) at the 4-position in a polar solvent such as methanol, ethanol, n-
propanol, isopropanol, n-butanol, t-butanol, or mixtures thereof at
room temperature for a period of 30 minutes to 2 hours followed by
heating to a temperature of 65~-100~C for 4-24 hours, 2-aryltrypt~mine
(5) is produced.
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 18 -
Scheme F
1~ ~ R
6 ~ '~ R6 R6--~, PdCI2, CH3CN
Pd(PPh3)4
~ ~X~R CuBr, Et3N 16 R7 R8
R ,R8
7 ~ 3
R5 R4
Reaction Scheme F
As shown in reaction Scheme F, iodoanilines of type (15)
may be reacted with aryl acetylenes, an appropriate palladium (û)
catalyst such as tetrakis(triphenylphosphine)palladium, a copper (I)
halide such as cuprous bromide in an inert organic solvent such as
triethyl~min~, at a temperature of 50~-88~C for a period of 30 minlltes to
5 hours to provide the diarylacetylene (16). Acetylene (16) may be
further modified by treatment with a palladium (II) catalyst such as
palladium (II~ chloride or pall~ m (II) acetate in an inert organic
solvent such as acetonitrile at a temperature of 50~- 82~C for a period of
30 minutes to 6 hours to give 2-arylindole (17).
CA 02240ll5 l998-06-09
WO 97/21435 PCT/US96/20004
- 19 -
Scheme G
- ' n ~
P'8
HN (A) R ~ N- (A)--Rl
THF R6 ~RJ~--R -or-
/~\J LiAlH4, TH F
R6 R4
R8 R
R;~ R1
21 D D
r~5 ,~4
Reaction Scheme G
As shown in reaction Scheme G, treatment of 2-arylindole
(17) with oxalyl chloride neat or in an inert organic solvent such as
methylene chloride, chloroform, dichloroethane, tetrahydrofuran or the
like at a temperature of 25~-65~C for a period of 3-24 hours gives the
acylchloride adduct (18). The crude product (18) may be reacted with
an amine of type (19) in an inert organic solvent such as diethylether,
tetrahydrofuran, methylene chloride, chloroform or the like and an
amine base such as triethylamine, diisopropylethylamine or pyridine at a
temperature of 0~C-25~C for a period of 30 minutes to 4 hours to
provide the amide derivative (20). Amide (20) may be further modified
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 20 -
by treatment with a reducing agent such as borane or lithium aluminum
hydride in an inert organic solvent such as tetrahydrofuran at elevated
temperatures, preferably reflux, for a period of 1-5 hours to give
compound (21).
Scheme H
R8 fAr
R ~X~N- (A)--
R6~ 'J H2
22a Rs R4Pd(OH)2/C
or
R, 8 O~O~Ar
R ,X~b ~.N- (A)--
R ~ J~10a
H ~/ \,J R3
22b R5 R4
R,8
R
Reaction Scheme H
As shown in reaction Scheme H, N-benzyl derivatives of
type (22a) or N-benzyloxycarbonyl derivatives of type (22b) may be
reduced to provide the secondary almine analogs (7) by treatrnent with
hydrogen (1 atm) and an appropriate catalyst such as palladium on
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/200û4
carbon, palladium hydroxide on carbon, or the like in an inert organic
solvent such as tetrahydrofuran, ethyl acetate, methanol, ethanol, or
mixtures thereof to which has been added a weak acid such as 30%
aqueous acet~c acid for a period of 10 minlltes to 3 hours or until the
aryl group has been removed to give the secondary amine.
Scheme I
02N~ Rg~N-(A)--R1
R6 ~ ~ Oa H2, RtahnaenyO(~) Ni
R~)--
25R~ / \~ R3
Rs R4
Reaction Scheme I
As shown in reaction Scheme I, treatment of a nitroindole
of type (24) with hydrogen (1 atm) and an appropriate catalyst such as
Raney(~) Nickel in an inert organic solvent such as ethanol, methanol, or
the like at room temperature for a period of 2-12 hours gives the
corresponding aminoindole derivative (25).
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 22 -
Scheme J
H~ 11 or EDC, HOBt,
Ro /,\,J 3 diisopropyl- NMM, CH2Ci2
RR ethyl amine
~; 4 cH2Cl2
R7 ~/
R ~ (A)--
26 R/~\J
Reaction Scheme J
As shown in reaction Scheme J, amino- or hydroxyindole
(25) may be modified by acy~ation under a variety of conditions. For
example, treatment of (25) with an acid chloride, acid anhydride or
active ester and an amine base such as triethylamine,
diisopropylethyl~mine, pyridine, or the like in an inert organic solvent
such as methylene chloride, chloroform, tetrahydrofuran, or mixtures
thereof at 0~C to room temperature for a period of 1 to 12 hours gives
the corresponding amide or ester derivatives (26). Alternatively (25)
may be coupled with a carboxylic acid by one of the many dehydrating
agents commonly employed. For instance, treatment of aminoindole
(2~) with an appropriate carboxylic acid and 1-(3-
dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), 1,3-
dicyclohexylcarbodiimide (DCC) or the like with or without 1-
hydroxyben~otriazole (HOBt) and a tertiary amine base such as N-
methylmorpholine (NMM), triethylamine or the like in an inert organic
CA 02240115 1998-06-09
WO 97t21435 PCT/US96/20004
- 23 -
solvent such as methylene chloride, chloroform, dimethylformamide, or
mixtures thereof at or near room temperature for a period of 3-24
hours provides the corresponding amide or ester derivative (26).
Sch~me K
R6 I~'(A) RR11~NJ~CI R11~ 27b
N ~ J~ diisopropyl- ;or diisopropyl- ; or
Ro i --R3 ethyl amine ethyl amine
~/ \~ Ctl2C12 CH2CI2
R5 R4
R11 R7 R
i. triphosgene R12 ~ ~N-(A~--R
ii. R12R11NH 2B J R~1oa
5 R4
Reaction Scheme K
As shown in reaction Scheme K, urea or carbamate
derivatives of (25) can be prepared by treatment with a carbamoyl
chloride of type (27a), or alternatively with an isocyanate reagent of
type (27b), and an amine base such as pyridine, triethylamine,
diisopropylethyl~mine, N-methylmorpholine or the like in an inert
organic solvent such as methylene chloride, chloroform,
dimethylformamide, tetrahydrofuran or mixture.s thereof at a
temperature of 0~-65~C for a period of 1-72 hours to give (28~.
Compound (25) can also be modified by treatment with a
- bis(electrophilic) reagent such as phosgene, triphosgene, 1,1'-
carbonyldiimidazole, N,N'-disuccinimidyl carbonate, or the like with or
without the addition of an amine base such as pyridine, triethyl~min~,
CA 02240115 1998-06-09
WO 97/21435 PCT/US96120004
- 24 -
diisopropylethyl~min~, N-methylrnorpholine in an inert solvent such as
me~ylene chloride, chloroform, or the like at a temperature of -20~-
0~C ~or a period of 20 minutes to 2 hours. After this time, the reaction
mixture is treated with an appropriate mono- or disubstituted amine at
-20~-25~C for a period of 1-5 hours to give the urea or carbamate
~nalog (28).
Scheme L
R 7 ~;
25/~\'J
O~S"O ~ R~
diisopropyl- 31 /~\R
ethyl amine Rs 4
CH2CI2
R N-S-CI 30 R 'N'S~N ~)--R~
diisopropyl- R6 NR --R
ethyl amine ~~/ \,J 3
cH2Cl2 32 Rs R4
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 25 -
Reaction Scheme L
As shown in reaction Scheme L, amine (25) can be
modified by treatment with an appropriate sulfonyl chloride of type
(29) or sulfamyl chloride of type (30) with an amine base such as
pyridine, triethylamine, diisopropylethylamine, N-methylmorpholine in
an inert solvent such as methylene chloride, chloroform, dichloroethane
or the like at a temperature of -20~-25~(~ for a period of 20 minutes to 2
hours to give the corresponding N-sulfonamide (31) or N-
sulfamylamide (32) derivatives, respectively.
Scherne M
R ~ MeOH
R~; R4
R~ ~ ~ rA) R
R~ R4
Reaction Scheme M
As shown in reaction Scheme M, the 2-aryltryptamine (33)
can be modified by treatment with an epoxide such as (34) in an inert
organic solvent such as methanol, ethanol, isopropanol, butanol, tert-
butanol, or mixtures thereof at a temperature of 65~-110~C for a period
of 8-20 hours to give the corresponding amino-alcohol derivative (35).
CA 02240ll5 l998-06-09
WO 97/21435 PCT/US96/20004
- 26 -
Scheme N
tlO~(R l2R1 1 C~ Rga~,N~ (A)--Rl Rl2R1 1 NH~
R6~/~ oRa3 PyBOP
36 R5 R4
R p(R12R11~ ~; )--
R5 4
~eaction Scheme N
As shown in reaction Scheme N, amide derivatives of an
acid-cont~inin~ indole derivative such as (36) can be prepared by
treatment with an appropriate amine (Rl2R1 1 NH) and a suitable
coupling agent such as benzotriazol-l-yloxy-
tris(pyrrolidino)phosphonium hexafluorophosphate (PyBOP),
benzotriazol- l -yloxy-tris(dimethylamino)phosphonium
hexafluorophosphate (BOP), 1-(3-dimethylaminopropyl)-3-
ethylcarbodiimide hydrochloride (EDC), 1,3-dicyclohexylcarbodiimide
(DCC) or the like with or wi~out l-hydroxybenzotriazole (HOBt) and a
tertiary amine base such as N-methylrnorpholine (NMM), triethylamine
or the like in an imert organic solvent such as methylene chloride,
chloroform, tetrahydrofuran, dimethylformamide, or mixtures thereof
at or near room temperature for a period of 3-24 hours provides the
corresponding amide derivative (37).
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
- 27 -
The compounds of the present invention are useful in the
treatment of various sex-hormone related conditions in men and women.
This utility is manifested in their ability to act as antagonists of the
neuropeptide hormone GnRH as demonstrated by activity in the
following in vitro assays.
Rat pituitary GnR~ receptor bindin~ assay:
Crude plasma membranes prepared from rat pituitary tissues were
incubated in a Tris.HCI buffer (50 mM, PH. 7.5) cont~ining bovine
serum albumin (.1%), [I-125]D-t-Bu-Ser6-Pro9-ethyl amide-GnRH, and
the desired concentration of a test compound. The assay mixtures were
incubated at 4~C~ for 90-120 minutes followed by rapid filtration and
repeated w~hing~ through a glass fiber filter. The radioactivity of
membrane bound radioligands was determined in a gamma-counter.
From this data, the ICso of the radioligand binding to GnRH receptors
in the presence of test compound was estimated.
Inhibition of LH release assay:
Active compounds from the GnRH receptor binding assay were further
evaluated with an in vitro LH release assay to confirm their antagonist
activity (blocking GnRH-induced LH release).
1. Sample Preparation
The compounds to be assayed were dissolved and diluted in DMSO. The
final concentration of DMSO in the incubation medium was 0.5%.
2. Assay
The Wistar male rats (150-200 grams) were obtained from Charles
River Laboratories (Wilmington, MA). Rats were m~int~ined at a
constant temperature (25~~) on a 12-hr light, 12-hr dark cycle. Rat
chow and water were available ad libitum. The ~nim~ls were sacrificed
by decapitation and pituitary glands were aseptically removed and
placed in Hank's Balanced Salt Solution (HBSS~ in a 50-ml
- polypropylene centrifuge tube. The collection tube was centrifuged for
5 min at 250 x g, and HBSS was removed by aspiration. Pituitary
glands were transferred to a disposable petri plate and minced with a
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 2~ -
scalpel. The minced tissue was then transferred to a 50-mL disposable
centrifuge tube by suspending the tissue fragments in three successive
10-mL aliquots of HBSS containing 0.2% collagenase and 0.2%
hyaluronidase. The cell dispersion was carried out in a water bath at
37~C with gentle stirring for 30 min. At the end of ~e incubation, the
cells were aspirated 20 to 30 times with a pipet and the undigested
pituitary fragments were allowed to settle for 3 to 5 min. The
suspended cells were removed by aspiration, and then subjected to a
1200 x g centrifugation for 5 min. The cells were then resuspended in
C~ulture medium. The undigested pituitary fragments were treated with
30 mL aliquots of the digestion enzymes as above for a total of 3
digestions with the collagenase/hyaluronidase mixture. The resulting
cell suspensions were pooled, counted and diluted to a concentration of 3
x 105 cells/ml, and 1.0 ml of this suspension was placed in each well of
a 24-well tray (Costar, Cambridge, MA). Cells were m~int~ined in a
humidified 5% C02-95% air atmosphere at 37~C for 3 to 4 days. The
culture medium consisted of DMEM cont~inin~ 0.37% NaHCO3, 10%
horse serum, 2.5% fetal bovine serum, 1% non-essential amino acids,
1% gl~ mine, and 0.1% gentamycin. On the day of an experiment,
cells were washed three times 1 1/2 hrs prior to and two more tirnes
immediately before the start of the experiment with DMEM cont~ining
0.37% NaHCO3, 10% horse serum, 2.5% fetal bovine serum, 1% non-
essential amino acids(lOOX), 1% glllt~mine(lOOX), 1%
Penic;llin/Streptomycin(10,000 Units of Penicillin and 10,000
micrograms of Streptomycin per ml), and 25 mM HEPES, pH 7.4. LH
release was initiated by adding 1 ml of fresh medium containing test
compounds in the presence of 2 nM GnRH to each well in duplicate.
Incubation was carried out at 37~C for 3 hr. After incubation, medium
was removed and centrifuged at 2,000 x g for 15 min to remove any
cellular material. The supernatant fluid was removed and assayed for
LH content with a double antibody RIA procedure using materials
obtained from Dr. A. F. Parlow (Harbor-UCLA Medical Center,
Torrance, CA).
CA 0224011~ 1998-06-09
WO 97/21435 PCTIUS96/2000
- 29 -
The compounds of formula I are useful in a number of
areas affected by GnRH. They may be useful in sex-hormone related
conditions, ,sex-hormone dependent cancers, benign prostatic
hypertrophy or myoma of the uterus. Sex-hormone dependent cancers
which may bene~lt from the ~lministration of the compounds of this
invention include prostatic cancer, uterine cancer, breast cancer and
pituitary gonadotrophe adenomas. Other sex-hormone dependent
conditions which may benefit from the ~lmini~tration of the compounds
of this invention include endometriosis, polycystic ovarian disease,
uterine fibroids and precocious puberty. The compounds may also be
used in combination with an angiotensin-converting enzyme inhibitor
such as Fn~l~pril or Captopril, an angiotensin II-receptor antagonist
such as Losartan or a renin inhibitor for the treatment of uterine
fibroids.
The compounds of the invention may also be useful for
controlling pregnancy, as a contraceptive in both men and women, for
in vitro fertilization, in the treatment of premenstrual syndrome, in the
treatment of lupus erythematosis, in the treatment of hirsutism, in the
treatment of irritable bowel syndrome and for the treatment of sleep
disorders such as sleep apnea.
A ~urther use of the compounds of this invention is as an
adjunct to growth hormone therapy in growth hormone deficient
children. The compounds may be ~lmini~tered with growth hormone
or a compound which increases the endogenous production or release of
growth hormone. Certain compounds have been developed which
stimulate the release of endogenous growth hormone. Peptides which
are known to stimulate the release of endogenous growth hormone
include growth hormone releasing hormone, the growth hormone
releasing peptides GHRP-6 and GHRP-l (described in U.S. Patent No.
4,411,890, PCT Patent Pub. No. WO 89/07110, and PCT Patent Pub.
No. WO 89/07111) and GHRP-2
- (described in PCT Patent Pub. No. WO 93/04081), as well as hexarelin
(J. Endocrinol Invest., 15(Suppl 4), 45 (1992)). Other compounds
which stimulate the release of endogenous growth hormone are
CA 02240115 1998-06-09
WO 97/2143!; PCT/US96/20004
- 30 -
disclosed, for example, in the following: U.S. Patent No. 3,239,345;
U.S. Patent No. 4,036,979; U.S. Patent No. 4,411,890; U.S. Patent No.
5,206,235; IJ.S. Patent No. 5,283,241; U.S. Patent No. 5,284,841; U.S.
Patent No. 5,310,737; U.S. Patent No. 5,317,017; U.S. Patent No.
5,374,721; U.S. Patent No. 5,430,144; U.S. Paten~ No. 5,434,261; U.S.
Patent No. 5,438,136; EPO Patent Pub. No. 0,14~,230; EPO Patent Pub.
No. 0,5I3,974; PCT Patent Pub. No. WO 94/07486; PCT Patent Pub.
No. WO 94/08583; PCT Patent Pub. No. WO 94/11012; PCT Patent
Pub. No. WO 94/13696; PCT Patent Pub. No. WO 94/19367; PCT
Patent Pub. No. WO 95/03289; PCT Patent Pub. No. WO 95/03290;
PCT Patent Pub. No. WO 95/09633; PCT Patent Pub. No. WO
95/11029, PCT Patent Pub. No. WO 95/1259g; PCT Patent Pub. No.
WO 95/13069; PCT Patent Pub. No. WO 95/14666; PCT Patent Pub.
No. WO 95/16675; PCT Patent Pub. No. WO 95/16692; PCT Patent
Pub. No. WO 95/17422; PCT Patent Pub. No. WO 95/17423; Science.
260, 1640- 1643 (June 11, 1993); Ann. Rep. Med. Chem., 2~, 177- 186
(1993); Bioor~. Med. Chem. Ltrs.,_(22), 2709-2714 (1994); and Proc.
Natl. Acad. Sci. USA 92, 7001-7005 (July 1995).
Representative preferred growth ho~none secretagoues
employed in ~e present combination include the following:
1) N-[l(R)-[(1,2-Dihydro-l-methanesulfonylspiro[3H-indole-3,4'-
piperidin] - 1 '-yl)carbonyl] -2-(1 H-indol-3 -yl)ethyl] -2-amino-2-methyl-
propanamide;
2) N-[l(R)-[(1,2-Dihydro-l-methanecarbonylspiro[3H-indole-3,4'-
piperidin]- 1 '-yl)carbonyl~ -2-(1 H-indol-3-yl)ethyl]-2-amino-2-methyl-
propanamide;
3) N- [1 (R)-~(1,2-Dihydro- 1 -benzenesulfonylspiro~3H-indole-3,4'-
piperidin]- 1 '-yl)carbonyl]-2-(1 H-indol-3 -yl)ethyl]-2-amino-2-methyl-
propanamide;
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
4) N-[l(R)-[(3,4-Dihydro-spiro[2H-1-benzopyran-2,4'-piperidin]-1'-yl)
carbonyl]-2-( 1 H-indol-3-yl)ethyl]-2-amino-2-methylpropanamide;
S) N-[ 1 (R)-[(2-~cetyl- 1,2,3 ,4-tetrahydrospiro[isoquinolin-4,4'-
piperidin l- 1 '-yl)carbonyl] -2-(indol-3-yl)ethyl] -2-amino-2-methyl-
propanamide;
6) N-[l(R)-[(1,2-Dihydro-1-methanesulfonylspiro[3H-indole-3,4'-
piperidin]- 1 '-yl)carbonyl]-2-(phenylmethyloxy)ethyl]-2-amino-2-
methylpropanamide;
7) N-~ 1 (R)-[( 1 ,2-Dihydro- 1 -methanesulfonylspiro[3H-indole-3,4'-
piperidin]- 1 '-yl)carbonyl]-2-(phenylmethyloxy)ethyl]-2-amino-2-
methylpropanamide methanesulfonate;
8) N-[l(R)-[~1,2-Dihydro-1-methanesulfonylspirol3H-~ndole-3,4;-
piperidin] -1 '-yl)carbonyl] -2-(2' ,6'-difluorophenylmethyloxy)ethyl] -2-
amino-2-methylpropanamide;
9) N-[ 1 (R)-~( 1 ,2-Dihydro- l -methanesulfonyl-S-fluorospiro[3H-indole-
3 ,4'-piperidin] -1 '-yl)carbonyl] -2-(phenylmethyloxy)ethyl]-2-amino-2-
methylpropanamide;
10) N-[ l (S)-[(l ,2-Dihydro- 1 -methanesulfonylspiro[3H-indole-3,4'-
piperidin]- 1 '-yl) carbonyl ~-2-(phenylrnethylthio)ethyl]-2-amino-2-
methylpropanamide;
1 1 ) N-~ 1 (R)-[( l ,2-Dihydro- I -methanesulfonylspiro[3H-indole-3 ,4'-
piperidinl- l '-yl)carbonyl]-3-phenylpropyl]-2-amino-2-methyl-
propanamide,
12) N-[ 1 (E~ ( l ,2-Dihydro- 1 -methanesulfonylspiro[3~I-indole-3,4'-
piperidin~- l '-yl)carbonyll-3-cyclohexylpropyl]-2-amino-2-methyl-
propanamide;
_ _ _ _ _ _
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 32 -
13) N-[l(R)-[(1,2-Dihydro-l-methanesulfonylspiro[3H-indole-3,4'-
piperidin]- 1 '-yl)carbonyl]-4-phenylbutyl]-2-amino-2-methyl-
propanamide;
14) N-~l(R)-[(1,2-Dihydro-l-methanesulfonylspiro~3H-indole-3,4'-
piperidin]-1 '-yl)carbonyl]-2-(S-fluoro- 1 H-indol-3-yl)ethyl]-2-amino-2-
methylpropanamide;
l S) N-[ 1 (R)-[( 1 ,2-Dihydro- 1 -methanesulfonyl-5-fluorospiro~3H-indole-
3 ,4'-piperidin]- 1 '-yl)carbonyl~-2-(S-fluoro- 1 H-indol-3-yl)ethyl]-2-
amino-2-methylpropanamide;
16) N-[ 1 (R)-[(l ,2-Dihydro- 1 -(2-ethoxycarbonyl)methylsulfonylspiro-
1 3H-indole-3,4'-piperidin]-1 '-yl)carbonyl]-2-(1 H-indol-3-yl)ethyl~ -2-
amino-2-methylpropanamide;
17) N-[ 1 (R)-[( 1 ,2-Dihydro- 1,1 -dioxospiror3H-benzothiophene-3,4'-
piperidin]- 1 '-yl)carbonyl]-2-(phenylmethyloxy)ethyl] -2-amino-2-
methylpropanamide;
and pharmaceutically acceptable salts thereof.
The compounds of the invention may also be used in
combination with bisphosphonates (bisphosphonic acids) and other
agents, such as growth hormone secretagogues, e.g. MK-0677, for the
treatment and the prevention of disturbances of calcium, phosphate and
bone metabolism, in particular, for the prevention of bone loss during
therapy with the GnRH antagonist, and in combination with estrogens,
progesterones and or androgens for the prevention or treatment of bone
loss or hypogonadal symptoms such as hot flashes during therapy with
the Gn~H antagonist.
Bisphosphonates (bisphosphonic acids) are known to inhibit
bone resorption and are useful for the treatment of bone li~iasis as
disclosed in U.S. Patent 4,621,077 to Rosini, et al.
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
The literature discloses a variety of bisphosphonic acids
which are useful in the treatment and prevention of diseases involving
bone resorption. Representative examples may be found in the
following: U.S. Patent No. 3,251,907; U.S. Patent N o. 3,422,137; U.S.
Patent No. 3,5~4,125; U.S. Patent No. 3,940,436; U.S. Patent No.
3,944,599; U.S. Patent No. 3,962,432; U.S. Patent No. 4,054,598; U.S.
Patent No. 4,267,108; U.S. Patent N o. 4,327,039; U.S. Patent N o.
4,407,761; U.S. Patent No. 4,578,376; U.S. Patent N o. 4,621,077; U.S.
Patent N o. 4,624,947; U.S. Patent N o. 4,746,654; U.S. Patent No.
4,761,406; U.S. Patent No. 4,922,007; U.S. Patent N o. 4,942,157; U.S.
Patent No. 5,227,506; U.S. Patent No. 5,270,365; EPO Patent Pub. N o.
0,252,504; and J. Or~. Chem., 36, 3~43 (1971).
The preparation of bisphosphonic acids and halo-
bisphosphonic acids is well known in the art. Representative examples
may be found in the above mentioned references which disclose the
compounds as being useful for the treatment of disturbances of calcium
or phosphate metabolism, in particular, as inhibitors of bone resorption
Preferred bisphosphonates are selected from the group of
the following compounds: alendronic acid, etidrononic acid, clodronic
acid, pamidronic acid, tiludronic acid, risedronic acid,
6-amino-1-hydroxy-hexylidene-bi.sphosphonic acid, and 1-hydroxy-
3(methylpentylamino)-propylidene-bisphosphonic acid;
or any pharmaceutically acceptable salt thereof. A particularly
preferred bisphosphonate is alendronic acid (alendronate), or a
pharmaceutically acceptable salt thereof. An especially preferred
bisphosphonate is alendronate sodium, including alendronate sodium
trihydrate. Alendronate sodium has received regulatory approval for
marketing in the United States under the trademark FOSAMAX(~).
Additionally, a compound of the present invention may be
co-~-1mini~tered with a 5O~-reductase 2 inhibitor, such as finasteride or
epristeride; a 5Oc-reductase I inhibitor such as 4,7~ dimethyl-4-aza-5Oc-
cholestan-3-one, 3-oxo-4-aza-4,7,~3-dimethyl- 16~-(4-chlorophenoxy)-
So~-androstane, and 3-oxo-4-aza-4,7,13-dimethyl-16,(3-(phenoxy)-So~-
androstane as disclosed in WO 93/23420 and WO 95/11254; dual
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
- 34 -
inhibitors of ~(x-reductase 1 and 5cc-reductase 2 such as 3-oxo-4-aza-
17,1~-(2,5-trifluoromethylphenyl-carbamoyl)-50~-androstane as disclosed
in WO 95/07927; antiandrogens such as flutamide, casodex and
cyproterone acetate, and alpha-1 blockers such as prazosin, terazosin,
doxazosin, tamsulosin, and alfuzosin.
Further, a compound of the present invention may be used
in combination with growth hormone, growth hormone releasing
ho~none or growth hormone secretagogues, to delay puberty in growth
hormone deficient children, which will allow them to continue to gain
height before fusion of the epiphyses and cessation of growth at puberty.
For combination treatment with more than one active agent,
where ~e active agents are in separate dosage formulations, the active
agents may be zl~lmini~tered separately or in conjunction. In addition,
the ~lmini.~tration of one element may be prior to, concurrent to, or
subsequent to the ~flmini~tration of the other agent.
The pharmaceutical cornpositions cont~ining the active
ingredient may be in a form suitable for oral use, for example, as
tablets, troches, lozenges, aqueous or oily suspensions, dispersible
powders or granules, emulsions, hard or soft capsules, or syrups or
elixirs. Compositions intended for oral use may be prepared according
to any method known to the art for the manufacture of pha~naceutical
compositions and such compositions may contain one or more agents
selected from the group consisting of sweetening agents, flavoring
agents, coloring agents and preserving agents in order to provide
pharmaceutically elegant and palatable preparations. Tablets contain the
active ingredient in admixture with non-toxic pharmaceutically
acceptable excipients which are suitable for the manufacture of tablets.
These excipients may be for example, inert diluents, such as calcium
carbonate, sodium carbonate, lactose, calcium phosphate or sodium
phosphate; granulating and disintegrating agents, for example, corn
starch, or alginic acid; bindin~ agents, for example starch, gelatin or
acacia, and lubricating agents, for example, magnesium stearate, stearic
acid or talc. The tablet,s may be uncoated or they may be coated by
known techniques to delay disintegration and absorption in the
CA 0224011~ 1998-06-09
WO 97/21435 PCT/[JS96/20004
- 35 -
gastrointestinal tract and thereby provide a sustained action over a
longer period. For example, a time delay material such as glyceryl
monostearate or glyceryl dis$earate may be employed. They may also
be coated by the technique described in the U.S. Patent 4,256,108;
4,166,452; and 4,~65,874 to forrn osmotic therapeutic tablets for
control release.
Formulations for oral use may also be presented as hard
gelatin capsules wherein the active ingredient is mixed with an inert
solid diluent, for example, calcium carbonate, calcium phosphate or
kaolin, or as soft gelatin capsules wherein the active ingredient is mixed
with water or an oil medium, for example peanut oil, liquid paraffin, or
olive oil.
Aqueous suspensions contain the active material in
admixture with excipients suitable for the manufacture of aqueous
suspensions. Such excipients are suspending agents, for example sodium
carboxymethylcellulose, methylcellulose, hydroxypropylmethyl-
cellulose, sodium alginate, polyvinyl-pyrrolidone, gum tragacanth and
gum acacia; dispersing or wetting agents may be a naturally-occurring
phosphatide, for example lecithin, or condensation products of an
alkylene oxide with fatty acids, for example polyoxyethylene stearate, or
condensation products of ethylene oxide with long chain aliphatic
alcohols, for example heptadecaethylene-oxycetanol, or condensation
products of ethylene oxide with partial esters derived from fatty acids
and a hexitol such as polyoxyethylene sorbitol monooleate, or
condensation products of ethylene oxide with partial esters derived from
fatty acids and hexitol anhydrides, for example polyethylene sorbitan
monooleate. The aqueous suspensions may also contain one or more
preservatives, for example ethyl, or n-propyl, p-hydroxybenzoate, one
or more coloring agents, one or more flavoring agents, and one or
more sweetening agents, such as sucrose, saccharin or aspartame.
Oily suspensions may be formulated by suspending the
active ingredient in a vegetable oil, for example arachis oil, olive oil,
sesame oil or coconut oil, or in mineral oil such as liquid paraffin. The
oily suspensions may contain a thickening agent, for example beeswax,
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
- 36 -
hard paraffin or cetyl alcohol. Sweetening agents such as those set forth
above, and flavoring agents may be added to provide a palatable oral
preparation. These compositions may be preserved by the addition of
an anti-oxidant such as ascorbic acid.
Dispersible powders and granules suitable for preparation
of an aqueous suspension by the addition of water provide the active
ingredient in admixture with a dispersing or wetting agent, suspending
agent and one or more preservatives. Suitable dispersing or wetting
agents and suspending agents are exemplified by those already
mentioned above. Additional excipients, for exarnple sweetening,
flavoring and coloring agents, may also be present.
The pharmaceutical compositions of the invention may also
be in the form of an oil-in-water emulsions. The oily phase may be a
vegetable oil, for example olive oil or arachis oil, or a mineral oil, for
example liquid paraffin or mixtures of these. Suitable emulsifying
agents may be naturally-occurring phosphatides, for example soy beans,
lecithin, and esters or partial esters derived from fatty acids and hexitol
anhydrides, for example sorbitan mnnooleate, and condensation
products of the said partial esters with ethylene oxide, for example
polyoxyethylene sorbitan monooleate. The emulsions may also contain
sweetening and flavouring agents.
Syrups and elixirs may be form~ ted with sweetening
agents, for exarnple glycerol, propylene glycol, sorbitol or sucrose.
Such formulations may also contain a demulcent, a preservative and
flavoring and coloring agents.
The pharmaceutical compositions may be in ~e forrn of a
sterile injectable aqueous or o~eagenous suspension. This suspension
may be forrn~ ted according to the known art using those suitable
dispersing or wetting agents and suspending agents which have been
mentioned above. The sterile injectable preparation may also be a
sterile injectable solution or suspension in a non-toxic parenterally-
acceptable diluent or solvent, for example as a solution in 1,3-butane
diol. Among the acceptable vehicles and solvents that may be employed
are water, Ringer's solution and isotonic sodium chloride solution. In
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/200û4
addition, sterile, fixed oils are conventionally employed as a solvent or
suspending medium. For thi,s purpose any bland fixed oil may be
employed including synthetic mono- or diglycerides. In addition, fatty
acids such as oleic acid find use in the preparation of injectables.
Compounds of Formula I may also be ~clmini~tered in the
form of a suppositories for rectal ~1mini~tration of the drug. These
compositions can be prepared by mixing the drug with a suitable non-
irritating excipient which is solid at ordinary temperatures but liquid at
the rectal temperature and will therefore melt in the rectum to release
the drug. Such materials are cocoa butter and polyethylene glycols.
For topical use, creams, ointments, jellies, solutions or
suspensions, etc., containing the compound of Formula I are employed.
(For purposes of this application, topical application shall include mouth
washes and gargles.)
The compounds for the present invention can be
~lmini~tered in intranasal form via topical use of suitable intranasal
vehicles, or via transdermal routes, using those forms of transdermal
skin patches well known to those of ordinary skill in the art. To be
~lmini.~tered in the form of a transdermal delivery system, the dosage
~lmini.~tration will, of course, be continuous rather than intermittent
throughout the dosage regimen. Compounds of the present invention
may al,so be delivered as a suppository employing bases such as cocoa
butter, glycerinated gelatin, hydrogenated vegetable oils, mixtures of
polyethylene glycols of various molecular weights and fatty acid esters
of polyethylene glycol.
The dosage regimen utilizing the compounds of the
present invention is selected in accordance with a variety of factors
including type, species, age, weight, sex and medical condition of the
patient; the severity of the condition to be treated; the route of
administration; the renal and hepatic function of the patient; and the
particular compound thereof employed. A physician or veterinarian
of ordinary skill can readily determine and prescribe the effective
amount of the drug required to prevent, counter, arrest or reverse
the progress of the condition. Optimal precision in achieving
CA 0224011~ 1998-06-09
WO 97/;~1435 PCT/US96/20004
- 38 -
concentration of drug within the range that yields efficacy without
toxicity requires a regimen based on the kinetics of the drug's
availability to target sites. This involves a consideration of the
distribution, equilibrium, and elimin~tion of a drug. Preferably,
doses of the compound of structural formula ~ useful in the method
of ~e present invention range from 0.01 to I000 mg per adult
h~lm~n per day. Most preferably, dosages range from 0.1 to S00
mg/day. For oral ~(1mini~tration, the compositions are preferably
provided in the form of tablets cont:~ining 0.01 to 1000 milligrams of
the active ingredient, particularly 0.01, 0.05, 0.1, 0.5, 1.0, 2.5, 5.0,
10.0, 15.0, 25.0, 50.0, 100 and 500 milligrams of the active
ingredient for the symptomatic adjustment of the dosage to the
patient to be treated. An effective amount of the drug is ordinarily
supplied at a dosage level of from about 0.0002 mg/kg to about 50
mg/kg of body weight per day. The range is more particularly from
about 0.001 mg/lcg to 1 mg/kg of body weight per day.
Advantageously, the active agent of the present invention
may be ~lmini~tered in a single daily dose, or the total daily dosage
may be ~1mini.stered in dividend doses of two, three or four times
daily.
The amount of active ingredient that may be combined with
the carrier materials to produce a single dosage forrn will vary
depending upon the host treated and the particular mode of
:~lmini .~tration.
It will be understood, however, that the specific dose level
for any particular patient will depend upon a variety of ~actors
including the age, body weight, general health, sex, diet, time of
7l~1mini~tration, route of ~1ministration, rate of excretion, drug
combination and the severity of the particular disease undergoing
therapy.
The following examples illustrate the preparation of some
of the compounds of the invention and are not to be construed as
limiting the invention disclosed herein.
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 39 -
EXAMPLE 1
,~,OH
Ol~Ae
OMe
N- r2-r2-~3 .4-dimethoxyphenyl)- 1 H-indol-3 -yllethyll -3 -(4-
hydroxyphenyl)propionamide
To a stirred ,solution of 3-(4-hydroxyphenyl)propionic acid
(80 mg in 4 rnL N,N-dimethylformamide) was added 1-
hydroxybenzotriazole (78 mg) and the mixture cooled to 0~(~. After 10
minutes, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
(130 mg) was added. The mixture was warmed to room temperature
and 2-[2-(3,4-dimethoxyphenyl)-lH-indol-3-yl]ethylamine (272 mg) was
added. After 17 hours the reaction was quenched by the addition of
water and extracted with ethyl acetate. The organic portion was washed
with water, 0.5M sodium bisulfate and brine, dried over sodium sulfate
and concentrated in vacuo. Purification by flash chromatography on
silica gel (methylene chloride:methanol, 95:~) gave the title compound
(227 mg). m/e = 444 (M)
Following a procedure similar to that described in Example
1, the following interrnediates were prepared:
~,~N~f(A) R
H / \,J R3
Rs R4
CA 02240115 1998-06-09
WO 97/21435 PCT/US96120004
- 40 -
Example # R1 R3,R4,R5 (CH2)n--~ m/e
lA Ph-4-0-CH2-Ph 3,4-OMe 3 505 (M + H)
lB Ph-4-OH 3,4-OMe 3 459 (M + H)
lC Ph-4-OH 3,4-OMe l 431 (M + H)
lD Ph-3,4-Cl,Cl 3,4-OMe 1 4g3 (M)
lE Ph-4-F 3,4-OMe 1 433 (1\~ + H)
1 F Ph-4-No2 3,4-OMe 3 --
lG Ph-4-NH2 3,4-OMe 3 458 (M + H)
lH Ph-4-No2 3,4-OMe 1 --
l I Ph-4-NH2 3,4-OMe 1 430 (M + H)
lJ Ph-4-OH 3,5-OMe 3 --
lK Ph-4-OH 3-Ph 3 475 (M + H)
1 L Ph-4-NH- 3,5-Me 3 526 (M + H)
COO-tBu
lM Ph-4-NH2 3,5-Me 3 426 (M + H)
lN Ph-4-N02 3,5-Me 3 4~6 (M + H)
Ph-4-OH 3-SCH3, 3 459 (M + H)
S-CH3
lP Ph-4-S02NH2 3,5-Me 0 448 (M + H)
EX~MPLE 2
H OH
r ~ ~~~
Me
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96t20004
- 41 -
3-r3-r2-r2-(3.5-dimethylphenyl)- lH-indol-3-yllethylaminol-2-
hydroxypropoxylphenol
Step 2A 2-12-( lH-indol-3-yl)-ethyll-isoindole- 1 .3-dione
To a stirred suspension of 2-(lH-indol-3-yl)ethylamine (2.0
g in 20 mL of dry tetrahydrofuran) was added N-carbethoxyphthalimide
(2.85g) and the mixture heated to reflux on an oil bath. After 48 hours
the reaction was cooled to room temperature, filtered and the filtrate
concentrated in vacllo. The resulting solid was suspended in a mixture
of hexane/methylene chloride (2.5:1) and filtered. Purification of the
collected solids by flash chromatography on silica gel (methylene
chloride:methanol, 97:3) gave the title compound (3.1 g).
Step 2B 2-r2-(2-bromo- lH-indol-3-yl)-ethyll -isoindole- 1 .3-dione
To a solution of 2-[2-(lH-indol-3-yl)-ethyl]-isoindole-1,3-
dione (1.0 g in a mixture of 10 mL dry tetrahydrofuran and 10 mL dry
chloroform) at 0 ~C was added pyridinium bromide perbromide (1.14
g) and the reaction stirred at 0 ~C. After 50 minutes, the reaction was
quenched by the addition of saturated sodium bicarbonate and extracted
with ethyl acetate. The organic portion was washed with saturated
sodium bicarbonate (3x) and ~).3M sodium bisulfate (3x) then dried over
magnesium sulfate. Purification of the concentrate by flash
chromatography on silica gel (hexane:ethyl acetate, 3:1) gave the title
compound (1.2 g).
Step 2C 2- ~ 2-r2-(3 .5-dimethylphenyl)- 1 H-indol-3 -yll -ethyl ~ - isoindole- 1 .3-dione
To a solution of 2-[2-(2-bromo-lH-indol-3-yl)-ethyl]-
isoindole-1,3-dione (150 mg in a mixture of 5 mL toluene and 5 mL
ethanol) was added 3,5-dimethylphenyl boronic acid (85 mg) followed
by 1.0 mL of 1~ sodium carbonate. To the stirred solution was added
lithium chloride (60 mg) followed by tetrakis(triphenylphosphine)
palladium (28 mg) and the mixture heated to reflux on an oil bath.
After 4 hours the mixture was cooled to room temperature and
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 42 -
concentrated in vacuo. Purification by flash chromatography on silica
gel (hexane:ethyl acetate, 5:I) gave the title compound (146 mg).
Step 2D 2-1 2-(3.5-dimethylphenyl)- lH-indol-3-yll-ethylamine
To a solution of 2-{2-[2-(3,5-dimethylphenyl)-lH-indol-3-
yl}-ethyl}-isoindole-1,3-dione (~7 mg in a mi~ture of 4 rnL
tetrahydrofuran and 4 mL ethanol) was added 0.6 mL of 95% aqueous
hydrazine and the reaction stirred at room temperature. After 18 hours
the mixture was concentrated in vacuo and purified by flash
chromatography on silica gel (methylene chloride:methanol:ammonium
hydroxide, 9:6:1) to provide the title compound (54 mg).
Step 2E 3-r3-r2-r2-(3.5-dimethylphenyl)- 1 H-indol-3-yllethylaminol- 2-hydro~ypropoxylphenol (benzyl ether)
To a solution of 2-[2-(3,5-dimethylphenyl)-lH-indol-3-yl~-
ethylamine (25.5 mg in 5 mL dry methanol) was added 12.1 mg of 1-[3-
(benzyloxy)phenoxy]-2,3-epoxypropane and the mixture heated to
reflux on an oil bath. After 7 hours the reaction mixture was cooled to
room temperature, concentrated in vacuo and the product prified by
flash chromatography on silica gel (methylene chloride:methanol, 95:5)
to give ~e ti~le compound (13.3 mg).
Step 2F 3-r3-r2-12-(3.5-dimethylphenyl)-lH-indol-3-yllethylaminol-
2-hydroxypropoxylphenol
To a solution of 3-[3-[2-[2-(3,5-dimethylphenyl)-lH-indol-
3-yl]ethylamino]-2-hydroxypropoxy]phenol (benzyl ether) (11 mg in l
mL ethanol) was added 10 mg of 10% palladium hydroxide on carbon
catalyst. The reaction flask was fitted with a hydrogen balloon,
evacuated and recharged with hydrogen (3 times) and stirred at room
temperature. After 2 hours the reaction was flushed with nitrogen,
filtered over diatomaceous earth, concentrated in vacuo and purified by
flash chromatography on silica gel ~methylene chloride:methanol, 95:5)
to provide the title compound (2.3 mg~. m/e = 431 (M+H)
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 43 -
Following a procedure similar to that described in Exarnple
2, the following compounds were prepared:
H
~N~J~(A) R
R5 R4
Example#Rl R3,R4,R5 A m/e
2A Ph-4-OH 3,4-OMe (S) CH2-0
2B Ph-4-OH 4-OMe (S) CH2-0
2C Ph 3,4-OMe (S) CH2-0
2D Ph-4-OH 3,4-OMe(S) CH2-CH2
2E Ph-4-OH 3,4-OMe (R) CH2
2FPh-3-F, 4-NH2 3,4-OMe (S) CH2-0
2G Ph-4-NHAc 3,4-OMe (S) CH2-0
2H Ph-4-NH2 3,4-OMe (S) CH2-0
21 Ph-4-OH 3,4-OMe (R) CH2-0
2J Ph-4-F 3,4-OMe (S) CH2-0 465 (M +H)
2KPh-4-Cl, 3- 3,~0Me (S) CH2-0 496 (M +H)
NH2
2L Ph-4-0-CH2- 3,4-OMe (S) CH2-0 ~~
Ph, 3-NH-
COCH3
2MPh-4-OH, 3- 3,4-OMe (S) CH2-0 520 (M + H)
NHCOCH3
2N Ph-3-CN 3,4-OMe (S) CH2-0 472 (M + H)
20Ph-3-CH20H 3,4-OMe (S) CH2-0 477 (M + H)
2P Ph-3-F 3,4-OMe (S) CH2-0 465 (M +H)
2QPh-3-CH2NH2 3,4-OMe (S) CH2-0 476 (M + H)
2R Ph-2-F 3.4-OMe (S) CH2-0 465 (M +H)
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 44 -
2S Ph-3-OCH2-Ph 3,5-Me (R,S) CH2-O 521 (M +H)
2T Ph-4-OH 3,5-Me(R,S) CH2-O431 (M + H)
2U Ph-3-C1 3,5-Me(S) CH2-O449 (M +H)
2V Ph-3-CN 3,5-Me(S) CH2-0440 (M +H)
2WPh-3-CH20H 3,5-Me(S) CH2-~445 (M +H)
EXAMPLE 3
H OH
~; ~ ~~
Me OMe
OMe
(S)-4-r3-r2-r2-(3.5-dimethylphenyl)- l -methyl-lH-indol-3-
yllethylaminol -2-hydroxypropoxylphenol
Step 3A r2-r2-(3 .4-dimethoxyphenyl)- 1 H-indol-3-yllethyll carbamic
acid benzyl ester
To a su~pension of 2-[2-(3,4-dimethoxyphenyl)-lH-indol-3-
yl]ethylamine (150 mg in 1.5 mL methylene chloride) at -78~C was
added benzyl chloroformate (O.OP~ mL) and diisopropylethyl amine
(0.093 mL) and the mixture walmed to 0 ~C. After 40 minutes, the
reaction was quenched by the addition of saturated ammonium chloride,
extracted with ethyl acetate and the organic portion dried over sodiurn
sulfate. Purification by flash chromatography on silica gel
(hexane:ethyl acetate, 2:1) gave the title compound (220 mg).
Step 3B 2-r2-(3.4-dimetho~yphenyl)- l -methyl- 1 H-indol-3-
yllethylamine
To a solution of [2-[2-(3,4-dimethoxyphenyl)-lH-indol-3-
yl]ethyl]carbamic acid benzyl ester ~lO0 mg in 1.5 mL N,N-
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 45 -
dimethylformamide) at 0~ C was added sodium hydride (9 mg) and the
mixture allowed to s~ir at 0~ C. After 10 mimltes, iodomethane (0.016
mL) was added followed by warming to room temperature for 20
minlltes and quenching by the addition of water. The mixture was
extracted with ethyl acetate, washed with water and the organics dried
over sodium sulfate to give the crude N-methylated product.
Hydrogenolysis of the crude product by a method similar to that
described in EXAMPLE 7.1 Step J gave the title compound.
Step 3C ~S)-4-r3-~2-r2-(3 ~S-dimethylphenyl)- 1 -methyl- 1 H-indol-3-
yllethylaminol -2-hydroxypropoxylphenol
The title compound was prepared following a procedure
similar to that described in Example 2 Step F. m/e - 477 (M + H~
Following a procedure similar to that described above, the
following compounds were prepared:
OH
R2
0~ ~N~ A)' R 1
Me / \,J R3
R5 R4
Example # R~ R3,R4,R5 R2 m/e
3A Ph-4-0CH2-Ph 3,4-OMe Me
3 B Ph-4-OCH2-Ph 3,4-OMe H
3C Ph-OH 3,4-OMe Me 491 (M + H)
CA 02240ll5 l998-06-09
WO 97/21435 PCT/US96/20004
- 46 -
EXAMPLE3 4.1
H J[ ~ NO2
OMe
OMe
r2- r2-(3 ~4-dimethoxyphenyl)- I H-indol-3 -yllethyll -
r2-(4-nitrophenyl )ethyll arnine
~tep 4.1 A /V-r2-r2-(3 ~4-dimethoxyphenyl)- 1 H-indol-3-yllethyll-2-(4-
nitrophenyl)acetamide
To a stirred solution of 4-nitrophenylacetic acid (lû0 mg in
2.5 mL N,N-dimethylformamide) was added 1-hydroxybenzotriazole
(90 mg) and the mixture cooled to 0~C. After 10 minutes, 1-(3-
dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (148 mg) was
added. The mixture was warmed to room temperature and 3-(2-
aminoethyl)-2-(3,4-dimethoxyphenyl)indole (316 mg) was added. After
17 hours the reaction was quenched by the addition of water and
extracted with ethyl acetate. The organic portion was washed with
water, 0.5M sodium bisulfate and brine, dried over sodium sulfate and
concentrated in vacuo. Purification by flash chromatography on silica
gel (hexane:ethyl acetate, 1:2) gave the title compound ~116 mg).
~tep 4.1 B 1 2-r2-(3 .4-dimethoxyphenyl)- I H-indol-3-yllethyl 1 -r2-(4-
nitrophenyl)ethyllamine
To a stirred solution of N-[2-[2-~3,4-dimethoxy-phenyl)-
lH-indol-3-yl]ethyl~-2-(4-nitrophenyl)acetamide (90 mg in 3 mL dry
tetrahydrofuran) was added 0.79 mL of a lM solution of borane in
tetrahydrofuran and the mixture heated slowly to reflux on an oil bath.
A~ter 2 hours the mixture was cooled to room temperature and the
excess borane quenched by the careful addition of methanol. The
mixture was concentrated to half-volume, treated with N,N-
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 47 -
dimethylethanolamine (0.60 mL) and heated to reflux on an oil bath.
After 3 hours the mixture was cooled to room temperature and
concentrated in vacuo. Purification by flash chromatography on silica
gel (methylene chloride:methanol, 96:4) gave the title compound (79
mg).
EXAMPLE 4.2
C~, ~ NH2
OMe
OMe
1 2-r2-(3 .4-dimethoxyphenyl)- 1 H-indol-3-yllethyll -r2-(4-
aminophenyl)ethyll amine
To a stirred solution of [2-[2-(3,4-dimethoxyphenyl)-l~I-
indol-3-yl]ethyl~-[2-(4-nitrophenyl)ethyl]amine (45 mg in 4 mL
methanol) was added 2N hydrochloric acid (0.020 mL) and 18 mg of
10% pal}adium hydroxide on carbon catalyst. The reaction flask was
fitted with a hydrogen balloon, evacuated and recharged with hydrogen
(3 times) and stirred at room temperature. After 40 minutes the
reaction was flushed with nitrogen, filtered over diatomaceous earth,
concentrated in vacuo and purified by flash chromatography on silica
gel (methylene chloride:methanol, 96:4) to provide the title compound
(32 mg). m/e = 416 (M + H)
l~ollowing a procedure similar to that described in
EXAMPLES 4.1 and/or 4.2, the following compounds were prepared:
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 48 -
H
O~ ~ N ~ R
N~
H ~ ~J R3
R5 R4
Example # R I R3,R4,R5 m/e
4A Ph-3-F,4-OH 3,4-OMe 435 ~M + H)
4B Ph-4-OH 3,4-OMe 417 (M ~ H)
4C Ph-3,4-C1 3,4-OMe 469 (M + H)
4D Ph-4-F 3,4-OMe 419 (M + H)
4E Ph-4-C~1 3,4-OMe 435 (M + H)
4F Ph-4-OH 3,5-Me 385 (M + H)
EXAMPLE S.l
~ Br
~;
OMe
OMe
r3-(4-bromophenyl)allyll-r2-r2-(3~4-dimethoxyphenyl)- 1~1-indol-3-
yllethyllamine
To a stirred solution of 2-[2-(3,4-dimethoxyphenyl)-lH-
indol-3-yl]ethyl~mine (261 mg in a mixture of 3 mL N,N-
dimethylformamide and 8 mL methylene chloride) at 0~ C was added a
solution of 81 mg of 4-bromocinnamyl bromide in 2 mL methylene
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 49 -
chloride and the mixture allowed to warm to room temperature. After
27 hours the reaction was quenched by the addition of water followed
by extraction with ethyl acetate. The organic portion was dried over
over sodium sulfate and purified by flash chromatography on silica gel
(methylene chloride:methanol, 95:5) to give the title compound (93 mg).
m/e = 491 (M)
EXAMPLE 5.2
OMe
OMe
4-r3-r2-rr2-(3.4-dimethoxyphenyl)-lH-indol-3-
yllethvll aminolpropyl)phenol
To a stirred solution of N-12-[2-(3,4-dimethoxyphenyl)-lH-
indol-3-yl]ethyl]-3-(4-hydroxyphenyl)propionamide (50 mg in 1.5 mL
dry tetrahydrofuran) was added 0.45 mL of a lM solution of borane in
tetrahydrofuran and the mi~ture heated slowly to reflux on an oil bath.
After 2 hours the mixture was cooled to room temperature and the
excess borane quenched by the careful addition of methanol. The
mixture was concentrated to half-volume, treated with N,N-
dimethylethanolamine (0.34 mL) and heated to reflux on an oil bath.
After 4 hours the mixture wa.s cooled to room temperature and
concentrated in vacuo. Purification by flash chromatography on silica
gel (methylene chloride:methanol, 90:10) gave the title compound (47
mg). m/e = 431 (M ~ H)
Following a procedure similar to that described in
EXAMPLES 5.1 and 5.2, the following compounds were prepared:
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 50 -
, N ~., R 1
,~ R3
R5 R4
Example ~ R 1 R3,R4,R5 rn/e
5A Ph-3-NH2~4-OH 3,4-OMe 446 (M + H)
SB Ph-4-OH 3~5-Me 399 (M + H)
~C Ph-4-so2NH2 3,5-Me 462 (M + H)
5D Ph-4-CH20H 3,5-Me 413 (M + H)
5E Ph-4-COOMe 3,5-Me 441 (M + H)
5F Ph-4-NHSO2Me 3~5-Me 476 (M + H)
EX~MPLE 6.1
~NH2
' ~ --~OH
Me
2-rr2-r2-(3 ~S-dimethylphenyl)- 1H-indol-3-vllethyll-14-(4-
hydroxyphenyl)-butyll aminol acetamide
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96t20004
To a solution of 4-[4-[[2-[2-(3,5-dimethylphenyl)-lH-indol-
3-yl]ethyl]amino]butyl]phenol (15 mg in a mixture of 0.7 mL
acetonitrile and 0.2 mL N,N-dimethylformamide) was added 0.015 mL
of diisopropylethyl amine followed by 8 mg of iodoacetamide and the
mixture stirred at room temperature. After 4.5 hours the crude
reaction mixture was applied to a silica gel preparative TLC plate and
eluted with (methylene chloride:methanol, 93:7). Isolation of the
desired band was followed by extraction with methylene
chloride:methanol (95:5) and further purification of this material by
flash chromatography on silica gel (hexane:ethyl acetate, 2:5) to give the
title compound (16 mg). m/e = 470 (M + H)
EXAMPLE 6.2
r~~ NH2
~; ~OH
Me
4-r4-r (2-aminoethyl)-1 2-r2-(3 .5-dimethylphenyl)- l H-indol-3 -
yllethyllaminolbutyllphenol
Following a procedure similar to that in Example 5.2, the
title compound was prepared from 2-[[2-[2-(3,5-dimethylphenyl)-lH-
indol-3 -yl] ethyl] -[4-(4-hydroxyphenyl)-butyl] amino] acetamide . m/e =
456 (M + H)
CA 02240115 1998-06-09
WO 9712143~; PCT/US96/20004
EXAMPLE 6.3
HNq~ NH2
~, Mo --~OH
H
Me
and
EXAMPLE 6.4
H
HNq~N~NH
OH
Me
N-r2-r2-(3~5-dimethylphenyl)- lH-indol-3-yllethyll-N-1 4-(4-
hydroxyphenvl)butyll~uanidine
and
N-r2-r2-(3.5-dimethylphenyl)- 1~-indol-3-yllethyll-N-1 4-(4-
hydroxyphenyl)butyllguanidino-~uanidine
To a solution of 4-[4-[[2-[2-(3,5-dimethylphenyl)-lH-indol-
3-yl]ethyl]amino]butyl]phenol (15 mg in 0.50 mL ethanol) in a thick-
walled vial was added 50 mg of cyanamide followed by 0.30 mL of
triethylamine. The vessel was flushed with nitrogen, sealed and heated
to 70~ C on an oil bath. After 17.5 hours the mixture was cooled to
room temperature, concentrated in vacuo and purified by ~ash
chromatography on silica gel
- (methanol:chloroform:water:trifluoroacetic acid, 20:100:3:0.3; then
repurified with methylene chloride:methanol:ammollium hydroxide,
CA 02240115 1998-06-09
WO 97/21435 PCT/US96120004
83:17:1) to give title compounds (12 mg and 5 mg, respectively). m/e =
455 (M + H), 497 (M + H)
EXAMPLE 6.5
Me
~, M~OH
H
Me
4-r4-rr2-r2-(3~5-dimethylphenyl)- lH-indol-3-
yllethyllmethylaminolbutyllphenol
To a solution of 4-[4-[~2-[2-(3,5-dimethylphenyl)-lH-indol-
3-yl]ethyl]amino]butyl]phenol (19 mg in 1 mL methanol) was added
0.020 mL of a 37% solution of formaldehyde in water followed by
0.010 mL acetic acid and 16 mg of sodium cyanoborohydride and the
mixture stirred at room temperature. After 26 hours the reaction was
quenched by the addition of acetic acid, concentrated in vacuo and the
excess acetic acid removed by toluene azeotrope. Purification of the
concentrate by flash chromatography on silica gel (methylene
chloride:methanol, 93.5:6.5; ~en methylene
chloride:methanol:ammonium hydroxide, 90:10:1) gave the title
compound (20 mg). m/e = 427 (M + H)
CA 02240115 1998-06-09
WO 97~21435 PCT/US96/20004
- 54 -
~XAMPLE 6.6
~'--OH
~M- '--~OH
H
Me
4-14-rr2-r2-(3.5-dimethylphenyl)-1~-indol-3-yllethyll-(4-
hydroxybutyl)aminolbutvllphenol
To a solution of 4-~4-[[2-[2-(3,5-dimethylphenyl)-lH-indol-
3-yl]ethyl3amino]butyllphenol (18 mg in 2 mL tetrahydrofuran) was
added 0.050 mL of 2-ethoxytetrahydrofuran followed by 0.25 mL of
30% aqueous acetic acid and the mixture stirred for 30 minutes. At this
time 0.35 mL triethylamine was added followed by 10% palladium
hydroxide on carbon catalyst. The reaction flask was fitted with a
hydrogen balloon, evacuated and recharged with hydrogen (3 times) and
stirred at room temperature. After 23 hours ~e reaction was flushed
with nitrogen, filtered over diatomaceous earth and concentrated in
vacuo and partially purified by flash chromatography on silica gel
(methylene chloride:methanol, 92:8). Repurification by HPLC (C8
methanol:water, 55:45 = 0.1% trifluoroacetic acid) gave the title
compound (2.8 mg). m/e = 485 ~M + H)
lFollowing a procedure similar to that described in
EXAMPLES 6.5 or 6.6, the following compounds were prepared:
R2
OH
R3
R5 R4
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
_ 55 _
Example # R2 R3-Rs m/e
..
6A c~3 3,4-OMe459 (M + H)
6B ((:~H2)4-Ph(4-oH) 3,5-Me561 (M + H)
EXAMPLE 7.1
H H
M ~ N~O ~ ~l3~OH
Me
Propylcarbamic acid 2-(3~5-dimethylphenyl)-3-r2-r4-(4-
hydroxyphenyl)butylaminolethyll-1H-indol-5-yl ester
Step 7.1 A 2-r2-(5-benzyloxy- 1 H-indol-3-yl)ethyllisoindole- 1 .3-dione
To a stirred suspension of 5-benzyloxytryptamine
hydrochloride (1.0 g in 10 mL of dry tetrahydrofuran) was added
triethylamine (O.SO mL) followed by N-carbethoxyphth~limide (750
mg) and the mixture heated to reflux on an oil bath. After 48 hours the
reaction was cooled to room temperature, filtered and the filtrate
concentrated in vacuo. The resulting solid was suspended in a mixture
of hexane/methylene chloride (2.5:1, 50 mL) and filtered to give the
title compound (1.3 g).
Step 7.1B 2-r2-(5-benzyloxy-2-bromo-lH-indol-3-vl)ethyllisoindole-
1 ,3-dione
To a solution of 2-[2-(5-benzyloxy-lH-indol-3-
yl)ethyl]isoindole-1,3-dione (~00 mg in a mixture of dry 25 mL
tetrahydrofuran and 25 mL dry chloroform) at 0~ C was added
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 56 -
pyridinium hydrobromide perbromide (666 mg) and the mixture stirred
at 0~ C. After 23 minutes the reaction was quenched by the addition of
saturated sodium bicarbonate and extracted with ethyl acetate. The
organic portion was washed with saturated sodium bicarbonate (3x) and
0.3M sodium bisulfate (3x~ then dried over magnesium sulfate.
Purification of the concentrate by flash chromatography on silica gel
(hexane:e~yl acetate, 7:2) followed by repurification by flash
chromatography on silica gel (methylene chloride) gave the title
compound (632 mg).
~tep 7.1 C 2-r2-r5-benzyloxy-2-(3~5-dimethylphenyl)- lH-indol-3-
yllethyllisoindole- 1 .3 -dione
To a solution of 2-[2-(5-benzyloxy-2-bromo-lH-indol-3-
yl)ethyl]isoindole-1,3-dione (500 mg in a mixture of 6 mL ethanol and
16 mL toluene) was added 3,5-dimethylphenyl boronic acid (205 mg)
followed by 2.7 mL of l!M sodium carbonate. To the stirred solution
was added lithi~n chloride (156 mg) followed ~y
tetrakis(triphenylphosphine)palladium (78 mg) and the mixture heated
to reflux on an oil bath. After 2 hours the mixture was cooled to room
temperature and concentrated in vacuo. Purification by flash
chromatography on silica gel (he~ane:methylene chloride:ethyl acetate,
15:8:1 then 12:8:1) gave the title compound (479 mg).
~tep 7.1 D 2-r2-r2-(3 .S-dimethylphenyl)-S-hydroxy- 1 H-indol-3-
yllethyllisoindole- 1 .3-dione
To a stirred solution of 2-[2-r5-benzyloxy-2-(3,~-
dimethylphenyl)-lH-indol-3-yl]ethyl]isoindole-1,3-dione (S10 mg in 20
mL dry ethyl acetate was added 197 mg o~ 10% palladium on carbon
catalyst. The reaction fla,sk was fitted with a hydrogen balloon,
evacuated and recharged with hydrogen (3 times) and stirred at room
temperature. After 37 hours the reaction was flushed with nitrogen,
filtered over diatomaceous earth and concentrated in vacuo to provide
the crude title compound (41~ mg).
Step 7.1 E 3-(2-aminoçthyl)-2-(3.5-dimethylphenyl)- 1 H-indol-5-ol
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
- 57 -
To a solution of 2-[2-[2-(3,5-dimethylphenyl)-S-hydroxy-
lH-indol-3-yl]ethyl]isoindole-1,3-dione (41~ mg in a mixture of 7 mL
ethanol and 7 mL tetrahydrofuran) was added 2.5 mL of 95% aqueous
hydrazine and the reaction stirred at room temperature. After 12 hours
the mixture was concentrated in vacuo and purified by flash
chromatography on silica gel (methylene chloride:methanol:arnmonium
hydroxide, ~9: 11: 1) to provide the title compound (228 mg).
Step 7.1 F 4-(4-benzyloxyphenyl)-N- f 2-r2-(3.5-dimethylphenyl~-5-
kydroxy-lH-indol-3-yllethyllbutyramide
To a stirred solution of 4-benzyloxyphenylbutyric acid (159
mg in a mixture of 2 mL methylene chloride and 0.5 mL N,N-
dimethylformamide) was added l-hydroxybenzotriazole (110 mg) and
1 -(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (113
mg) and the reagents allowed to mix for 30 mimltes. At this time a
solution of 3-(2-aminoethyl)-2-(3,5-dimethylphenyl)-lH-indol-5-ol (144
mg in 4 mL N,N-dimethylformamide) was added and the reaction
stirred at room temperature. After 6 hours, the mixture was
concentrated in vacuo and purified by flash chromatography on silica
gel (hexane:ethyl acetate, 4:5) to give the title compound (241 mg).
Step 7.1 G 3-12-r4-(4-benzyloxyphenyl)butylaminolethyll -2-(3.5-
dimethylphenyl)- lH-indol-5-ol
To a stirred solution of 4-(4-benzyloxyphenyl)-N-{2-[2-
(3,5-dimethylphenyl)-S-hydroxy- 1 H-indol-3 -yl]ethyl]butyramide(241
mg in 10 mL dry tetrahydrofuran) was added 4 mL of a 1~ solution of
borane in tetrahydrofuran and the mixture heated slowly to reflux on an
oil bath. After 2 hours the mixture was cooled to room temperature
and the excess borane ~uenched by the careful addition of methanol.
The mixture was concentrated to half-volume, treated with N,N-
dimethylethanolamine (1.4 mL) and heated to reflux on an oil bath.
- After 3 hours the mixture wa~ cooled to room temperature and
concentrated in vacuo. Purification by flash chromatography on silica
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 58 -
gel (methylene chloride:methanol, 92:~S) gave the title compound (234
mg)
Step 7.1H r4-(4-benzyloxyphenyl)-butyl~-r2-r2-(3.5-dimethylphenyl)- -
5-hydroxy-lH-indol-3-yllethyllcarbamic acid benzyl ester
To a solution of 3-[2-[4-(4-benzyloxyphenyl)butylamino3
ethyl~-2-(3,5-dimethylphenyl)-lH-indol-5-ol (234 mg in 5 mL of dry
methylene chloride) at -7~~ C was added benzyl chloroformate (0.082
mL) and diisopropylethylamine (0.104 mL) and the mixture stirred at
room temperature. After 1 hour the reaction was quenched by the
addition of saturated sodium bicarbonate and extracted wi~ ethyl
acetate. The organic portion was washed with saturated ammonium
chloride, dried over magnesium sulfate and concentrated in vacuo.
Purification by flash chromatography on silica gel (hexane:ethyl acetate,
3:1 then 2:1) gave the title compound (lL55 rng).
~tep 7.1I Propylcarbamic acid 3-(2-rbenzyloxycarbonyl-~4-(4-
benzyloxyphenyl)butyllaminol-ethyl~-2-(3~5-
dimethylphenyl)- I H-indol -S-yl ester
To a stirred solution of [4-(4-benzyloxyphenyl)-butyl]-[2-
[2-(3,5-dimethylphenyl)-5-hydroxy-lH-indol-3-yl]ethyl]carbamic acid
benzyl ester (20 mg in 3 mL dry methylene chloride) at 0~ C was added
triphosgene (4.9 mg) and pyridine (0.037 mL of a 10% solution in
methylene chloride) and the reagents allowed to mix for 30 minutes. At
this time, propylamine (0.040 mL) was added and the mixture allowed
to walm to room temperature. After 30 minutes, the reaction was
quenched by the addition of 0.3M sodium bisulfate and extracted with
ethyl acetate. The organic portion was washed with 0.3M sodium
bisulfate (3x) and brine, then dried over magnesium sulfate and
concentrated in vacuo. Purification by flash chromatography on silica
gel (~exane:methylene chloride:ethyl acetate, 4:5:1) gave the title
compound (17 mg).
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 59 -
Step 7.1J Propylcarbamic acid 2-(3.5-dimethylphenyl)-3-r2-r4-(4-
hydroxyphenyl)-butylaminolethyll - l H-indol-5-yl ester
t To a stirred solution of propylcarbamic acid 3-(2-
~benzyloxycarbonyl-~4-(4-benzyloxyphenyl)butyl]amino] -ethyl)-2-(3 ,5-
dimethylphenyl)-lH-indol-5-yl ester (17 mg in a mixture of 1.5 mL
tetrahydrofuran and 0.5 mL methanol) was added 16 mg of 10%
palladium on carbon catalyst followed by acetic acid (0.010 mL of a
30% solution in water). The reaction flask was fitted with a hydrogen
balloon, evacuated and recharged with hydrogen (3 times) and stirred at
room temperature. After 1.5 hours the reaction was flushed with
nitrogen, filtered over diatomaceous earth and concentrated in vacuo.
Purification by flash chromatography (methylene
chloride:methanol:ammonium hydroxide, 90:6:1) gave the title
compound (1 1 mg). m/e--514 (M + H)
PREPARATION OF SYNTHETIC INTERMEDIATES
4-(4-benzyloxyphenyl)butyric acid
Step A: 4-(4-benzyloxyphenyl)butyric acid benzyl ester
To a stirred solution of ~-hydroxyphenylbutyric acid (810
mg in 8 mL N,lV-dimethylformamide) at 0~ C was added sodium
hydride (290 mg of an 80% dispersion in mineral oil) and the mixture
allowed to warm to room temperature. Benzyl bromide (1.2 mL) was
added after 20 minutes and the mixture stirred at room temperature.
After 13 hours the reaction was quenched by the addition of saturated
ammonium chloride and extracted with ethyl acetate. The organic
portion was washed with water (4x), dried over sodium sulfate and
concentrated in vacuo. Purification by flash chromatography on silica
gel (hexane:ethyl acetate, 13:1) gave the title compound (1.45 g)
Step B: 4-(4-benzvloxyphenyl)butyric acid
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 60 -
To a stirred solution Or 4-(4-benzyloxyphenyl)butyric acid
benzyl ester (277 mg in a mixture of 3 mL methanol and 1 mL
methylene chloride) at 0~ C was added 1.5 mL of 5M sodium hydroxide
and the mixture warmed to room temperature. After 2 hours the
mixture was acidified to pH 2 by the addition of aqueous hydrochloric
acid, the aqueous portion extracted with ethyl acetate (5x) and the
resulting organics concentrated in vacuo. Purification by flash
chromatography on silica gel (methylene chloride:methanol, 94:6 then
96:4 + 0.25% TFA) gave the title compound (196 mg).
3~5-dimethylphenylboronic acid
To a solution of S-bromo-m-xylene (1.~ g in lS mL of dry
tetrahydrofuran) at -78~ C was added 6.4 mL of a 1.4M solution of
butyllithium in hexane and the mixture stirred for 20 minutes. At this
time triisopropyl borate (2.8 mL) was added and the mixture allowed to
warm to room temperature. After 1.5 hours the reaction was
concentrated in vacuo to 1/3 volume then cooled to 0~ C and treated
with 2N hydrochloric acid (9 mL) followed by warming to room
temperature. After 4 hours the mixture was made basic by the addition
of 2.5M sodium hydroxide and partitioned between ethyl ether (75 mL)
and 1.25M sodium hydroxide. The organic layer was extracted with
1.25M sodium hydroxide (2x) and the aqueous portion then cooled to 0~
C and acidified to pH 3 by the dropwise addition of conc. hydrochloric
acid. The white slurry was dissolved in methylene chloride, the organic
portion dried over magnesium sulfate and concentrated in vacuo to
provide the title compound (960 mg).
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
- 61 -
EXA~IPLE 7.2
Me~N~O~ ~,N ~~~3~
,~Me OH
H
Me
Ethylcarbamic acid 2-(3.5-dimethylphenyl)-3-r2-r4-(4-
hydroxyphenyl)butylaminolethyll-lH-indol-5-yl ester
~tep 7.2A Ethylcarbamic acid 3-(2-rbenzyloxycarbonyl-r4-(4-
benzyloxyphenyl)butyll aminol -ethyl )-2-(3 .5 -
dimethylphenyl)- I H-indol-5-yl ester
To a solution of [4-(4-benzyloxyphenyl)-butyl]-r2-[2-(3,5-
dimethylphenyl)-5-hydroxy-lH-indol-3-yl]ethyl]carbamic acid benzyl
ester (50 mg in 0.5 mL dry tetrahydrofuran) was added 0.035 mL of
ethyl isocyanate and the mixture stirred at room temperature. Over the
course of 2 weeks, additional ethyl isocyanate was added in portions and
the mixture heated to reflux for several days after which time it was
cooled to room temperature, concentrated in vacuo and purified by flash
chromatography on silica ~el (hexane:ethyl acetate, 2:1) to give the title
compound (20 mg).
~tep 7.2B Ethylcarbamic acid 2-(3.5-dimethylphenyl)-3-r2-r4-(4-
hydroxyphenyl)butvlaminolethyll-1H-indol-5-yl ester
To a stirred solution of ethylcarbamic acid 3-(2-
rbenzyloxycarbonyl-r4-(4-benzyloxyphenyl)butyl~amino] -ethyl)-2-(3 ,5-
dimethylphenyl)-lH-indol-5-yl ester (12 mg in a mixture of ~.5 mL
tetrahydrofuran and 0.5 mL methanol) was added 12 mg of 10%
palladium hydroxide on carbon catalyst followed by acetic acid (0.010
mL of a 30% solution in water~. The reaction flask was fitted with a
hydrogen balloon, evacuated and recharged with hydrogen (3 times) and
stirred at room temperature. After 1.5 hours the reaction was flushed
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 62 -
with nitrogen, filtered over diatomaceous earth and concentrated in
vacuo. Purification by flash chromatography (methylene
chloride:rnethanol:arnmonium hydroxide, 90:7:1) gave the title
compound (8.2 mg). m/e = 500 (M + H)
Following a procedure similar to that described in
EXAMPLES 7.1 and 7.2 above, the following compounds were
prepared:
~'~ ~ ~OH
Me
Example R2 R8 m/e
7A (CH2)4OH -CO-NHEt 572 (M + H)
7B (cH2)4oH-CO-N(CH2CH3)-CO-NCH2CH3 643 (M + H)
7C H-co-N(cH2cH3)-co-NHcH2cH3 571 (M + H)
7D H -CO-OCH2CH3 501 (M + H)
7E H -CO-NH-CH3 4~6 (M + H)
7F H -CO-N-(CH3)2 500 (M + H)
7G H -CO-NH-Ph 548 (M + H)
CA 02240115 1998-06-09
WO 97/Z1435 PCT/US96/20~04
- 63 -
FXAMPLE
Me H H
M ,N~N~ ,~
~N~Me ~N,S~M
Me
N-r4-(4- f 2-12-(3 .5-dimethylphenyl)-S-(3 ,3 -dimethylureido)- 1 H-indol-3-
yllethylamino ~butyl)phenyllmethanesulfonamide
Step 8A 2-(3.5-dimethylphenylethynyl)-4-nitrophenylamine
To a solution of 3,5-dimethylphenylethyne (156 mg in 7
mL of dry, nitrogen saturated triethylamine) was added 2-iodo-4-
nitroaniline (264 mg, prepared essentially as described in: Toth, I.
Helv. Chim. Acta, 1971, 54, 1486.) followed by
tetrakis(triphenylphosphine)palladium (23 mg) and cuprous bromide (10
mg) and the mixture heated to reflux on an oil bath. After 2 hours the
mixture was cooled to room temperature, concentrated in vacuo and
purified by flash chromatography on silica gel (hexane:methylene
chloride:ethyl acetate, 15:8:1) to give the title compound (256 mg).
Step 8B 2-~3.5-dimethylphenyl)-5-nitro-lH-indole
To a stirred solution of 2-(3,5-dimethylphenylethynyl)-4-
nitrophenylamine (50 mg in 3 mL of dry, nitrogen saturated
acetonitrile) was added 5 mg of palladium (rl) chloride and the mixture
heated to reflux on an oil bath. After 3 hours the mixture was cooled to
room temperature, concentrated in v~cuo and purified by flash
chromatography on silica gel (hexane:methylene chloride:ethyl acetate,
15:8:1) to provide the title compound (46 mg).
CA 02240115 1998-06-09
WO 97/21435 PCT/lJS96/20004
- 64 -
Step 8C lV-benzyl-2-r2-(3 5-dimethylphenyl)-5-nitro- lH-indol-3-yll -
N-~4-(4-methanesulfonylaminophenyl)butyll -2-oxo-
acet~mide
To a stirred suspension of 2-(3,5-dirnethylphenyl)-5-nitro-
lH-indole (59 mg in 6 mL dry dichloroethane) was added oxalyl
chloride (0.025 mL) and heated to reflux on an oil bath. After 15 hours
the mixture was cooled to room temperature, diluted with benzene and
the volatiles removed in vacuo. The resulting solid was dissolved in 3
mL dry tetrahydrofuran and cooled to 0~ C. To ~is a solution of N-[4-
(4-benzylaminobutyl)-phenyl]methanesulfonamlde (74 mg in 2 mL dry
methylene chloride) was added simlllt~neously with triethylamine (0.047
mL) and the mixture stirred for 20 minutes at 0~ C then warmed to
room temperature. After 10 minutes the reaction was quenched by the
addition of saturated sodium bicarbonate, extracted with ethyl acetate.
The organic portion was washed with saturated ammonium chloride
(2x), dried over magnesium sulfate and concentrated in vacuo.
Purification by flash chromatography on silica gel (hexane:ethyl acetate,
4:5) gave the title compound (127 mg).
Step 8D IV-~4-r4-(benzyl-f2-r2-(3.5-dimethylphenyl)-5-nitro-lH-
indol-3-yllethyl ~ amino)butyllphenyl ~ -methanesulfonamide
To a stirred solution of N-benzyl-2-[2-(3,5-
dimethylphenyl)-5-nitro- lH-indol-3-yl]-N-[4-(4-
methanesulfonylaminophenyl)butyl]-2-oxo-acetamide (73 mg in 4 mL
dry tetrahydrofuran) was added 1 mL of a lM solution of borane in
tetrahydrofuran and the mixture heated slowly to reflux on an oil bath.
After 2 hours the mixture was cooled to room temperature and the
excess borane quenched by the careful addition of methanol. The
mixture was concentrated to half-volume, treated with N,N-
dimethylethanolamine ~0.35 mL) and heated to reflux on an oil bath.
After 2.5 hours the mixture was cooled to room temperature and
concentrated in vacuo. Purification by flash chromatography on silica
gel (hexane:ethyl acetate, 3,2) gave the title compound (59 mg).
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/211004
- 65 -
Step 8E N- ~ 4-r4-( ~ 2-r5-amino-2-(3 ~-dimethylphenyl)- 1~-indol-3-
yllethvl ~benzylamino)butyllphenyl 1 methanesulfon~mi~le
To a stirred .solution of N- { 4-[4-(benzyl- { 2-[2-(3,5-
dimethylphenyl)-5 -nitro- 1 H-indol-3-yl]ethyl } amino)butyl]phenyl } -
methanesulfonamide (S9 mg in 4 mL absolute ethanol) was added ca. 15
mg of Raney(~) nickel. The reaction flask was fitted with a hydrogen
balloon, evacuated and recharged with hydrogen (3 times) and stirred at
room temperature. After 3 hours the reaction was flushed with
nitrogen, ~lltered over diatomaceous earth and concentrated in vacuo.
Purification by flash chromatography on silica gel (hexane:ethyl acetate,
1:2) gave the title compound (42 mg).
Step 8F N- ~4-r4-(benzyl- ~ 2-r2-(3.5-dimethylphenyl)-5-(3.3-
dimethylureido)- lH-indol-3-yllethvl ~-
amino)butyllphenyl ~ methanesulfonamide
To a stirred solution of N-{4-~4-({2-[5-amino-2-(3,5-
dimethylphenyl)- 1 H-indol-3 -yl]ethyl } benzylamino)butyl]phenyl } -
methanesulfonamide (15 mg in 1.5 mL of dry methylene chloride) at 0~
C was added dimethylcarbamyl chloride (0.03 mL of a 10% v/v solution
in methylene chloride) and diisopropylethylamine (0.053 mL of a 10%
v/v solution in methylene chloride) and the mixture warmed to room
temperature. After 3 days the reaction was concentrated in vacuo and
purified by flash chromatography on silica gel (methylene
chloride:methanol:ammonium hydroxide, 96:4:1) to give the title
compound (17 mg).
Step 8G N-r4-(4- ~ 2-r2-(3.5-dimethylphenyl)-5-(3.3-
dimethylureido)- 1 H-indol-3-
yllethylamino ~butyl)phenvllmethanesulfonamide
To a stirred solution of N- { 4-[4-(benzyl- ~ 2-[2-(3,5-
dimethylphenyl)-5 -(3 ,3-dimethylureido)- 1 H-indol-3 -yl ~ethyl } -
amino)butyl]phenyl}methanesulfonamide (17 mg in a mixture of 4 mL
tetrahydrofuran and 1.5 mL methanol) was added 7 mg of 10%
palladium hydroxide on carbon catalyst followed by acetic acid (0.020
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 66 -
mL of a 30% solution m water). The reaction flask was fitted with a
hydrogen balloon, evacuated and recharged with hydrogen (3 times) and
stiITed at room temperature. After 45 minutes the reaction was flushed
with nitrogen, filtered over diatomaceous earth and concentrated in
vacuo. Purification by flash chromatography (methylene
chloride:methanol:amrnonium hydroxide, 91:9:1) gave the title
compound (145 mg). m/e = 576 (M + H)
Following a procedure similar to that described above, the
following compounds were prepared:
R~ R2
~~ NH "S~~Me
Me
Example R6 R2 X-R7R8 m/e
8A H HNH-COOCH2Ph
8B HCH2P NH-COO-Et 677 (M + H)
h
8C H H NH-COO-Et 577 (M + H)
8D H HNH-co-N(cH2cH3)2 604 (M + H)
8E H HN(CH2CH3)CO-N(CH2CH3)2 632 (M + H)
8F H HNH-CO-Cyclopropyl 573 (M + H)
8G H H NH-CO-Ph 609 (M + H)
8H H H Me 637 (M + H)
H ~h
, N ~ Me
o
SU~ lUTE SHEET(RULE 26)
CA 02240115 1998-06-09
WO 97/21435 PCT/US96120004
- 67 -
81 H H CF3 745 (M + H)
~N~CF3
8J H H NH-CO-Me ~47 (M + H)
8K }I H F 645 (M + H)
H ~
'N~~'F
o
8L H HNH-CO-CH(Me)-NH-CO-Me 618 (M + H)
8M H H N(Me)-CO-Ph 623 (M + H)
8N H H N(Me)-CO-Me 561 (M + H)
H H NH-S02Me 583 (M + H)
8P H H ,N~J~OCH3 639 (M + H)
8Q H H H 1~1 639 (M + H)
~ N ~OCH3
o
8R H HNH-CO-NH(CH2CH3) 576 (M + H)
8S H H NH-CO-CH2CH3 561 (M + H)
8T _ H H NH-CO-NHMe 562 (M + H)
8U H HNH-CO-NH(CH~CH2C1~3) 590(M + H)
8V H HNH-CO-CH(CH3)2 575 (M + H)
~sW H 1~NH-CO-NH-CH(CH3)2 5~0 (M + H)
8X H HNH-CO-NH-(cyclopropyl) 5~" (M + H)
~Y H HNH-S02-(CH2CH2CH3) 61 1 (M + H)
8Z H HNH-S02-NH-(CH2CH3) 612 (M + H)
8AA H H SCH3 536 (M + H)
8BB H H S(O)CH3 552 (M + H)
8CC H H S(0)2CH3 568 (M + H)
8DD H H S~0)2NH2 569 (M + H)
8EE 6-CI H * 569 (M + H)
8F~ 6-CI HNH-CO-NH-(cyclopropyl) 622 (M + H)
Sl.~d ~ ~1 1 UTE SHE~T (RULE 26)
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 68 -
* = N02
;~XAMPLE 9
Following a procedure similar to that desc~ibed in
EXAMPLES 4.1 and 12.1 the following compounds were prepared:
H
R6~ (A)
R~ R4
EX. #Rl R3-R5 R6(CH2)n=(rn/e
A)
9APh-4-0-CH2 Ph 3,4-OMe 4 ~~
9B Ph-4-OH 3,4-OMe 4 445 (M +H)
9C Ph-4-OH 3,4-OMe I 403 (M + H)
9D Ph-4-N02 3,4-OMe 4 474 (M + H)
9E Ph-4-NH2 3,4-OMe 4 444 (M + H)
9F Ph-4-OCH3 3-OCH2(Ph- 4 535 (M + H)
3-OMe)
9G Ph-4-OH 3,4-OMe 5 549 (M + H)
9H Ph-4-OH 3,5-CF3 4 521 (M + H)
91 Ph-4-OH 3,4-OMe 6 473 (M + H)
9J Ph-4-OCH3 3,4-OMe 4 459 (M + H)
9K Ph~OH 2-Me 4 399 (M + H)
9L Ph~OH 2,4-C1 4 453 (M)
9M Ph~OH 4-F 4 403 (M + H)
9N Ph-4-OH 4-Me 4 399 (M + H)
Ph-4-OH 3-Cl,4-F 4 437 (M+ H)
CA 0224011~ l998-06-os
wo 97/21435 PCT/US96/20004
- 69 -
9P Ph-4-OH 3,5-C1 4 453 (M)
9Q Ph-4-OH - 4385 (M + H)
9R Ph-4-OH 3,5-Me 4413 (M +H)
9S Ph-4-OH 3-Me 4399 (M + H)
9T Ph-4-OH 2,6-Me 4413 (M + H)
9U Ph-4-OH 3-OMe 4415 (M + H)
9V Ph-4-OH 3,5-OMe 4445 (M + H)
9W Ph-4-OCH3 3,5-Me 4427 (M + H)
9X Ph-4-OH 3,5-Me 5-C1 4447 (M + H)
9Y Ph-4-OH 3,5-Me S-Me 4427 (M + H)
9Z Ph-4-OH 3,5-Me 5427 (M +H)
9AA Ph-4-OH 3,5-Me S-OBn 4519 (M + H)
gBB Ph-4-OH 2,3-Me 4413 (M + H)
9CC Ph-4-OH 3-N(Me)2 4428 (M + H)
9DD Ph~OH 3,5-Me 6441 (M + H)
9EE Ph-4-OH 2,5-Me 4413 (M + H)
9FF Ph-4-OH 3,5-Me 7-Me 4427 (M + H)
9GG Ph-4-OH 3,5-Me 1371 (M + H)
9HH Ph-4-OH 3,5-Me S-OMe 4443 (M + H)
9II Ph-4-OH 3-OCH2-Ph 4491 (M+H)
9JJ Ph-4-OH 3-CH(Me) 4519 (M + H)
OBn
9KK Ph-4-OH 3-Et 4413 (M + H)
9LL Ph-4-No2 3,5-Me 4442 (M + H)
9MM Ph-4-OH 3-CH(Me) 4429 (M + H)
OH
9NN Ph-4-OH 3,5-Me 6-NH- 4
C(O)CH3
9Oo Ph-4-OCH3 3-O-CH2Ph 4505 (M + H)
gpp Ph-4-NH2 3,5-Me 4412 (M + H)
9QQ Ph-4-NH- 3,5-Me 4454 (M + H)
COCH3
CA 02240115 l998-06-os
Wo 97/21435 PCT/US96/20004
- 70 -
9RRPh-4-NHS02Ph 3,5-Me 4 552 (M + H)
9SS Ph-4-NHSO2Me 3,5-Me 4490 (M + H)
9'rT Ph-4-OMe 3-OCH2(Ph- 4535 (M + H)
3-OMe)
9UU Ph-4-OH 3-SMe 4431 (M + H)
9W Ph-4-OH 3-SMe, 5-Me 4445 (M + H)
9VVW Ph-4-OH 3,5-Me 6-C1 4475 (M)
9XX Ph-4-SO2NH2 3,5-Me
9YY Ph-4-OH 3,5-Me 4-C1 4475 (M)
9Z Ph~OH 3-S~O)Me 4447 (M + H)
9AAA Ph-4-OH 3-S(O)Me, 4461 (M + H)
5-Me
9BBB Ph-4-OH 3-SO2Me 4463 (M + H)
9CCC Ph-4-OH 3-SO2Me, 5- 4477 (M + H)
Me
9DDD Ph-NHS02CF3 3,5-Me 4544 (M + H)
9EEE Ph-NHSO2Et 3,5-Me 4504 (M + H)
~ 471 ~M + H)
9FFF ~ ~ 3,4-OMe 0
H
H ~CH 471 (M + H~
9GGG,~ 3,4-OMe 0
H
Me~ 517 (M + H)
9HHHPh-4-OH 3,5-Me 6- ~ 4
Me
9IIl Ph-4-OH3-Me, 5-i-Bu 4
9JJJ Ph-4-OH3-Me, 5-Pr 4
CA 02240115 1998-06-og
wo 97/21435 PCT/US96/20004
- 71 -
9KKK Ph-4-NH23,5-Me 5- 4
NHC(O)-
NHEt
9LLL Ph-4-NHS02- 3,5-Me 4532 (M + H)
iPr
9MMM Ph-4-OH 3,5-Me5-NO2 4 --
9NNN Ph-3,4-OMe 3,5-Me 44~7 (M + H)
9OOO Ph-3,4-OH 3,5-Me 4429 (M + H)
9PPP Ph-4-OH 3,5-Me 5-Br 4492 (M + H)
9QQQ 2-naphthyl 3,5-Me 4447 (M +H)
9RRR Ph-4- 3,5-Me 4505 (M +H)
NHS02NHMe
9SSS Ph-4-CN 3,5-Me 4422 (M + H)
91~ Ph-4-F 3,S-Me 4415 (M + H)
9UUU Ph-4-OH 3,5-Me 5-Ph 4489 (M + H)
9VVV Ph-3-Br, 4- 3,5-Me 4570 (M ~ H)
NHS02-Me
9WWW Ph-4- 3,5-Me 4469 (M + H)
NHCONHMe
9XXX Ph-4-OH 3,5-Me 5- 4455 (M +H)
CH(Me)2
gyyy Ph-4-S02NH2 3,5-Me 4476 (M + H)
9ZZ l-naphthyl-4- 3,5-Me 4477 (M + H)
OMe
9AAAA l-naphthyl-4- 3,5-Me 4463 (M + H)
OH
9BBBB Ph-3-F, ~OMe 3,5-Me 4445 (M + H)
9CCCC Ph-3-F, 4-OH 3,5-Me 44310 (M +
H)
9DDDD Ph-4- 3,5-Me 4519 (M + H)
- NHS02NHEt
9EEEE Ph-4- 3,5-Me 4483 (M + H)
NHCON~HEt
CA 02240115 1998-06-09
WO g7/21435 PCT/US96/20004
- 72 -
9FFFF Ph~NHS02Me 3,5-Me 5-S02Me 5 582 (M + H)
9GGGG Ph-4-NHSO~Me 3,5-C1 5- 4
N(Et)CO-
N(Et)2
9HHHH Ph-4-OH 3,5-Me 5-F 4431 (M + H)
EXAMPLE 10
Following a procedure similar to thrat described in
EXAMPLE 9, the following compounds were prepared:
R6~ ~/\~'OH
Rs R4
Example # R3-R5 R6 m/e
10A 2-(CH)4-3 H 435 (M + H)
10B 3-(CH)4-4 H 435 (M + H)
10C 3-(CH-CH-N(Me))-4 H 438 (M+ H)
10D 2-(CH)4-3 5-OBn 541 (M+ EI)
lOE 2-(CH)4-3 5-OH 451 (M + H)
lOF 2-(CH)4-3 6-F 453 (M + H)
10G 2-(CH)4-3, 5-Me H 449 (M + H)
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 73 -
EXAMPLE 1 1
Me H
OH
H
Me
4-(4-r2-r2-(3 .5-dimethylphenyl)- 1 H-indol-3-yll-
propvlaminolbutyl)phenol
~tep 1 1 A 2-methylcyclopropanecarboxylic acid N-methoxy-N-
methyl-amide
To a solution of 2-methylcyclopropanecarboxylic acid (10 g
in a mixture of 200 mL benzene and 2 mL N,N-dimethylformamide) at
0~ C was added 10.5 mL of oxalyl chloride and the mixture stirred at 0~
C for 30 minutes then warmed to room temperature for 30 minutes. At
this time, 14.6 g of N,O-dimethylhydroxylamine hydrochloride was
added followed by 41 mL of triethylamine. The mixture was stirred at
room temperature for one hour then quenched by the addition of
saturated sodium bicarbonate. The aqueous portion was extracted with
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 74 -
ethyl acetate and the combined organics washed with brine, dried over
sodium sulfate and concentrated in vacuo. The product was purified by
distillatic~n under reduced pressure to give P,.9 g as an oil.
Step 1 1 B (3 .5-dimethylphenyl)-(2-methylcyclopropyl)methanone
To a solution of 5-bromo-mcta-xylene (5.7 mL in 120 mL
of dry tetrahydrofuran) at -7~~ C was added 30.6 mL of a 1.4M solution
of n-butyllithium in hexane and the mixture stirred at low temperature.
After 15 minutes, a solution of 2-methylcyclopropanecarboxylic acid N-
methoxy-N-methyl-arnide (5.0 g in 50 mL tetrahydrofuran) was added
dropwise over 5 minutes and the mixture then allowed to warm slowly
to room temperature. After 1 hour, the reaction was quenched by the
addition of 20 mL 2N hydrochloric acid and 40 mL water. Thi.s was
extracted with ethyl acetate washed with saturated sodium bicarbonate
and brine then dried over sodium sulfate to give 6.95 g of the crude title
compound.
Step 1 I C 2-r2-(3 .5-dimethylphenyl)- 1 H-indol-3-yllpropylamine
To a solution of phenylhydrazine hydrochloride (1.42 g in
24 mL n-butanol) at 95~ C was added a solution of (3,5-
dimethylphenyl)-(2-methylcyclopropyl)methanone 2.0 g in 16 mL of n-
butanol) and heat at 1 I0~ C for 4 hours. At this tirne the reaction was
cooled to room temperature, 25 mL of 1 N sodium hydroxide and the
mixture extracted 3x with methylene chloride. The organics were
washed with brine and dried over sodium sulfate. Purification of the
concentrate by flash chromatography on silica gel (methylene
chloride:methanol, 95:5) gave the title compound (307 mg).
~teps llD, 11E 4-(4-r2-r2-(3.5-dimethylphenyl)-lH-indol-3-yll-
propylaminolbutyl)phenol
The title compound was prepared essentially as described in
EXAMPLES 1 and 5.2 from 2-[2-(3,5-dimethylphenyl)-lH-indol-3-
yl]propyl~mine.
CA 02240ll5 l998-06-09
WO 97/21435 PCT/US96/20004
- 75 -
Following a procedure similar to that described above, the
following compounds were prepared:
R
Me
Ex R1 R2 Rg R~a R1o RlOa A
1 lA Ph-4-OH H H H CH3 H 4
I lB Ph-4-OH H Ph H H H 4
I lC Ph-4-OH -CH2CH2- H H H 4
1 lD Ph-4-OH H CH3 H H H 2
1 lE Ph-4- H CH3 H H H 4
NHS02Me
I lF Ph-4-OH H H H CH3 H 2
1 lG Ph-4-OH H H H CH3 CH3 4
I lH Ph-4- H CH3 H H H 2
NHS02Me
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 76 -
EXAMPLE 12.1
H {~OH
N ~CH3
H 1¦ l
CH3
4-(4- ~ 2-r2-(3,5-dimethylphenyl)- lH-indol-3-yllethylamino ~cyclohexyl)-
phenol
A mixture of 2-[2-(3,5-dimethylphenyl)-lH-indol-3-yl]-
ethylamine (EXAMPLE 2, Step D, 345 mg) and 4-(4-
hydroxyphenyl)cyclohexanone (62 mg) were solvated in 8 rnL dry
methanol to which ca. 2 g powdered 3A molecular sieves were added.
The pH of this mixture was adjusted to 6 by the addition of 0.65 mL of
a 10% ,solution of trifluoroacetic acid in methanol and then 90 mg
sodium cyanoborohydride was added and the mixture atirred at room
temperature. After 20 hours, the mixture was filtered through
diatomaceous earth, concentrated in vacuo and purified by flash
chromatography on silica gel I (methylene chloride:methanol:, g2:8) then
again (chloroform:methanol, 90:10) to separate the diastereomers~ to
give the title compound (isorner A 40 mg, isomer B 36 mg). m/e = 439
(M + H)
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
EXAMPLE 12.2
¦ I ~ CH30 --\~OH
CH3
1 - ~ 2-r2-(3 .5-dimethylphenyl)- 1 H-indol-3 -yllethyl ~ -3-r2-(4-
hydroxyphenyl)ethyllurea
~tep 1 2.2A 1 -r2-(4-benzyloxyphenyl)ethyll-3- f 2-r2-(3~5-
dimethylphenyl)- lH-indol-3-yllethyl ~urea
To a solution of 2-12-(3,5-dimethylphenyl)-lH-indol-3-yl]-
ethylamine (EXAMPLE 2 Step D, 39 mg in 1 mL dry methanol) was
added 64 mg [2-(4-benzyloxyphenyl)-ethyl]-carbamic acid 4-nitrophenyl
ester and the mixture stirred at room temperature. After 24 hours, the
mixture was concentrated in vacuo and the residue re-solvated in ethyl
acetate. This was washed with saturated aqueous potassium carbonate
(3x) and brine, dried over sodium sulfate and purified by flash
chromatography on silica gel (hexane:ethyl acetate, 5:4; then 1:1) to
give the title compound (73 mg).
~tep 1 2.2B 1- ~ 2-r2-(3~5-dimethylphenyl)- 1 H-indol-3 -yll ethyl ~ -3 -r2-(4-
hydroxyphenyl)ethyllurea
The title compound was prepared essentially as described in
EXAMPLE 2 Step B starting from 1-[2-(4-benzyloxyphenyl)ethyl]-3-
{2-[2-(3,5-dimethylphenyl)-lH-indol-3-yl]ethyl}urea to give the title
compound. m/e = 42~s (M + H)
-
Following a procedure similar to those described above and
in EXAMPLE 4.1, the following compounds were prepared:
CA 02240115 1998-06-09
PCT/US96/20004
WO 97/21435
- 78 -
~ C H
CH3
Example R 1 (A) m/e
12A Ph-4-0-tBu -CH2-CH2-O-CH2-
12B Ph-4-OH -(CH2)3-C(cH3)2-Ph-4-oH
1 2C Ph-4-OH -CH2-CH2-CHMe-CH2-
12D Ph-4-OH -CH2-CH(CH3)2- 427 (M + H)
12F Ph-4-OH -CNH-NH-(CH2)2-
1 2F Ph-4-OH -(CH2)3-CH(O-CH2-CH2-OH)-
1 2G Ph-4-OH -(C~2)3-C(O-CH2-CH2-O)-
12H l-(naphthyl- -CH2-C(MC)2- 463 (M + H)
4-OH)
EXAMPLE 13.1
Me
Me~N~ ,N ~ 3~
tl ~Me N ,S~ M
Me
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96120004
- 79 -
2-(2-(3 ~5-dimethylphenyl)-3- ~ 2-l 4-~4-methanesulfonylaminophenyl)-
butylaminol-ethyl ~-1H-indol-5-yl)-N.N-diethylacetamide
~ Step 13.1A r3-(2-aminoethyl)-2-(3~5-dimethylphenyl)-lH-indol-5-yll-
acetic acid ethyl ester
A mixture of 6.34 g (approximately 29.5 mmol) of ethyl 2-
(4-hydrazinophenyl)acetate hydrochloride/2-(4-hydrazinophenyl)acetic
acid hydrochloride, 6.22g (29.5 mmol) of 3-chloropropyl 3,5-
dimethylphenyl ketone, and 120 mL of absolute ethanol was stirred at
reflux under nitrogen for 12 hours. The cooled solution was
concentrated in vacuo, and the residue was partitioned between 200 mL
of ethyl acetate and 50 mL of saturated aqueous sodium carbonate
solution. The organic phase was washed with 25 mL of brine, then
dried over sodium sulfate, and filtered. The residue from concentration
of the filtrate in vacuo was purified by flash chromatography on silica
gel (elution with 95:5 CH2C12-MeOH and then 95:5:0.5 CH2C12-MeOH-
concentrated NH40H). Concentration of the product fractions gave
1.13 g (11%) of a stiff foam; nearly homogeneous by TLC in 95:5:0.5
CH2C12-MeOH-concentrated NH40H. 400 MHz lH NMR (CDC13) was
consistent with the assigned structure. Mass spectrum (PB-NH3/CI):
m/e = 351 (M + H).
Stepl3.1B (2-(3.5-dimethylphenyl)-3-~2-r4-(4-methanesulfonylamino-
phenyl)butylaminolethyl~-lH-indol-S-yl)acetic acid ethyl
ester
The reductive ~min~tion reaction of [3-(2-aminoethyl)-2-
(3,5-dimethylphenyl)-lH-indol-S-yl]-acetic acid ethyl ester and 4-~4-
(methanesulfonamido)phenyl]butyraldehyde was accomplished according
to the procedure of Example 14.1, Step 14.1B to give the titled
compound in 29% yield as a stiff foam; homogenous by TLC in 92.5:7.5
CH2Cl2-MeOH. 500 MHz 1 H NMR (CDCl3) was consistent with the
assigned structure. Mass spectrum (ESI): m/e = 576 (M + H).
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- ~0 -
S~ep 13.1 C r3-(2- ~ benzyloxycarbonyl-r4-f4-methanesulfonylamino-
phenyl)butyllamino ~ -ethy~)-2-(3.5-dimethylphenyl)- 1 H-
indol-5-yll-acetic acid ethyl ester
The reaction of (2-(3,5-dimethylphenyl)-3-{2-[4-(4-
methanesulfonylaminophenyl)butylamino]ethyl } -1 H-indol-5-yl)acetic
acid ethyl ester with benzyl chlorofo~nate was carried out according to
the procedure of Example 14.1, Step 14.1C, to give the titled compound
in 73% yield as a stiff foam; homogeneous by TL(~ in 95:5 CH2C12-
MeOH. 500 MHz lH NMR was complex, owing to the existence of
rotamers, but was consistent with the assigned structure. Mass spectrum
(ESI): m/e = 710 (M + H).
~tep 13.1 D r3-(2- f benzyloxycarbonyl-14-(4-methanesulfonylamino-
phenyI)butyll amino ~ ethyl)-2-(3 ~5 -dimethylphenyl) - I H-
indol-5-yllacetic acid
To a solution of 227 mg (0.32 mmol) of [3-(2-
{ benzyloxycarbonyl-[4-(4-methanesulfonylamino-phenyl)butyl lamino } -
ethyl~-2-(3,5-dimethylphenyl)-lH-indol-5-yl~-acetic acid ethyl ester in
4.0 mL (2.0 mmol) of 0.50 N potassium hydro7~ide in methanol was
stirred under nitrogen at 60-65 ~C as 1.0 mL of water was added
gradually, resulting in slight cloudines.s. After 3 hours, the
homogeneous solution was cooled and concentrated to small volume in
vacuo. The residue was partitioned between 10 mL of ethyl acetate and
10 mL of 0.5 N hydrochloric acid. The organic phase was dried
(m~gnesium sulfate), filtered, and concentrated in vacuo at room
temperature. Trituration of the residue with diethyl ether resulted in
solidification of the product. This material was collected on a filter and
washed with small volumes of ether. The evaporation residue from the
mother liquor was also triturated with some ether to give a solid. After
decantation of the ether, the trituration-decantation cycle was repeated
twice more. The solids were combined and dried in vacuo to give 205
mg (94%) of a powder, mp 123-125 ~C; virtually homogeneous by TLC
(92.5:7.5 CH2Cl2-MeOH). 500 MHz lH NMR (DMSO-d6) was
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
- 81 -
complex, owing to rotamers, but was consistent with the assigned
structure. Mass spectrum (ESI): m/e = 682 (M + H).
~tep 13.1 E ~ 2-15-diethylcarbamoylmethyl-2-(3 5-dimethylphenyl)- 1 H-
indol-3-yllethyl ~-r4-(4-methanesulfonylaminophenyl)butyll-
carbamic acid benzyl ester
The reaction of 34.1 mg (0.05 mrnol) of r3-(2-
{ benzyloxycarbonyl-l 4-(4-methanesulfonylaminophenyl)butyl] -
amino}ethyl)-2-(3,5-dimethylphenyl)-lH-indol-5-yl]acetic acid with
diethylamine in the presence of PyBOP reagent was accomplished
according to the procedure of Bxample 14.1, Step 14.1L. The crude
product was purified by preparative TLC on 2 1000-micron silica gel
GF plates (20 x 20 cm), which were developed in 92.5:7.5 CH2C12-
MeOH. The product bands were isolated and extracted with the same
solvent to afford 30.9 mg (84%) of nearly colorless residual glass;
virtually homogeneous by TLC in 92.5:7.5 CH2C12-MeOH. 500 MHz
1H NMR (CDC13) was complex, owing to rotamers, but was consistent
with the assigned structure. Mass spectrum (E~I): m/e = 737 (M + H).
~tep 13.1 F 2-(2-(3.5-dimethylphenyl)-3- ~ 2-r4-(4-
methanesulfonylaminophenyl)-butylaminol-ethyl ~-1~-
indol -5 -yl)-N.N-diethylacetamide
A mixture of 28.7 mg (0.039 mmol) of {2-[5-
diethylcarbamoylmethyl-2-(3,5-dimethylphenyl)- lH-indol-3-yl]ethyl } -
[4-(4-methanesulfonylaminophenyl)butyl~-carbamic acid benzyl ester, 20
mg of 20% palladium hydroxide on carbon, and 5 mL of glacial acetic
acid was shal~en with hydrogen (50 psig) in a pressure vessel. After 1
day, an additional 20 mg of catalyst was added, and .~h~kin~ with
hydrogen was continued for 3 hours more. The catalyst was removed
by filtration through Celite under nitrogen, and the filtrate was
concentrated in vacuo. The residue was reconcentrated twice from
toluene and then purified by preparative TLC on 2 1000-micron silica
gel GF plates (20 x 20 cm), which were developed in 92.5:7.5:0.75
CH2C12-MeOH-concentrated NH40H. The product band from each
CA 02240ll5 l998-06-09
WO 97/21435 PCT/US96/20004
- 82 -
plate was isolated, combined, and extracted with the same solvent.
Concentration of the extracts in vacuo afforded 15.7 mg (67%) of a
glass; virtually homogeneous by TLC in 92.5:7.5:0.75 CH2Cl2-MeOH-
concentrated NH40H. 500 MHz lH NMlR (CDC13) was consistent with
the assigned structure. Mass spectrum (ESI): m/e = 603 ~M + H).
EXAMP~}~ 13.2
Me
~ Me Me tl
Me ~N~"N~ O
~N~Me ~N,S~Me
H 1¦ I H
Me
2-(2-(3.5-dimethylphenyl)-3- ~ 2-1 4-(4-methanesulfonylaminophenyl)-
butylaminolethyl ~-1H-indol-5-yl)-N.N-diethylisobutyramide
Step 13.2A 2-r3-(2-aminoethyl)-2-(3.5-dimethylphenyl)-lH-indol-5-yll-
2-methylpropionic acid ethyl ester
By the procedure of Example 14.1 ~tep A, ethyl 2-(4-
hydrazinophenyl)-2-methylpropionate was reacted with 3-chloropropyl
3,5-dimethylphenyl ketone to afford the titled compound in 16% yield
as a stiff foam; virtually homogeneous by TLC in 95:5:0.5 CH2cl2
MeOH-concentrated NH40H. 500 MHz 1 H NMR (CDCl3) was
consistent with the assigned structure. Mass spectrum (PB-NH3/CI):
m/e=379 (M+H).
~tep 13.2B 2-f2-f3.5-dimethylphenyl)-3- ~ 2-r4-(4-
methanesulfonylamino-phenyl)butylaminolethyl ~ - l ~-indol-
5-yl)-2-methylpropionic acid ethyl ester
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
- 83 -
The reductive amination reaction of 2-~3-(2-arninoethyl)-2-
(3,5-dimethylphenyl)-lH-indol-5-yl~-2-methylpropionic acid ethyl ester
and 4-~4-(methanesulfonamido)phenyl]butyraldehyde was carried out
according to the procedure of Example 14.1 Step B to give the titled
compound in 43% yield as a stiff foam; virtually homogenous by TLC
in 92.5:7.5 CH2Cl2-MeOH. 500 MHz l H NMR (CDCl3) was consistent
with the assigned structure. Mass spectrum (PB-NH3/CI): mJe = 604
(M ~ H).
~tep 13.2C 2-r3-(2- ~ benzyloxycarbonyl -14-(4-methanesulfonylamino-
phenyl)butyllamino ~ethyl)-2-(3~5-dimethyl-phenyl)-l~-
indol-5-yll-2-methylpropionic acid ethyl ester
The reaction of 2-(2-(3,5-dimethylphenyl)-3-{2-[4-(4-
methanesulfonylarninophenyl)butylamino]ethyl }- 1~-indol-5-yl)-2-
methylpropionic acid ethyl ester with benzyl chloroformate was carried
out according to the procedure of Example 14.1 Step C, to give the
titled compound in 72% yield as a stiff foam; homogeneous by TLC in
95:5 CH2Cl2-MeOH. 500 MHz 1H NMR was complex, owing to the
existence of rotamers, but was consistent with the assigned structure.
Mass spectrum (ESI): m/e = 738 (M + H).
~tep 13.2D 2-13-(2- f benzyloxycarbonyl-14-(4-methanesulfonylamino-
phenyl~butyll amino ~ ethyl )-2-(3.5 -dimethylphenyl)- 1 H-
indol-5-yll-2-methylpropionic acid
The saponification of 2-[3-(2-{benzyloxycarbonyl-[4-(4-
methanesulfonylamino-phenyl)butyl3amino } ethyl)-2-(3,5-dimethyl-
phenyl)-lH-indol-5-yl]-2-methylpropionic acid ethyl ester was achieved
according to the procedure of Example 14.1 Step D, except that the
reaction time was increased to 30 hours, providing a qll~ntit~tive yield
of the titled compound as a powder, mp >102 ~C (gradual; paltial
decomposition); homogeneous by TLC in 92.5:7.5 C~I2C12-MeOH. 500
MHz lH NMR (DMSO-d6) was complex, owing to rotamers, but was
consistent with the assigned structure. Mass spectrum (ESI): m/e = 710
(M + H).
CA 02240115 1998-06-09
WO 97/2143S PCT/US96/20004
- 84 -
Step 13 .2E ~ 2-r5-( 1 -diethylcarbamoyl- 1 -methylethyl)-2-(3 .5-
(limethylphenyl)- 1 H-indol-3-yllethyl ~ - r4-(4-
methanesulfonylaminophenyl)butyllcarbamic acid benzyl
ester
A solution of of 71.0 mg (0.1 mmol) of 2-[3-(2-
{ benzyloxycarbonyl-[4-(4-methanesulfonylamino-
phenyl)butyl]amino}ethyl)-2-(3,5-dimethylphenyl)-lH-indol-S-yl]-2-
methylpropionic acid, 53.0 mg (0.102 mmol) of PyBOP reagent, and
14.2 lmL (10.3 mg; 0.102 mmol) of triethylamine in 400 mL of dry
methylene chloride was stirred at room temperature in a stoppered
flask. After 25 minutes, 15.5 mL (11.0 mg; 0.15 mmol) of
diethylamine was added, followed after 4 hours by an additional 36.2
mL (25.6 mg; 0.35 mmol) of diethylamine. After 1 day, the solution
was partitioned between 10 mL of 0.5 N hydrochloric acid. The
organic phase was washed with 10 mL of saturated aqueous ~odium
bicarbonate solution and then with S mL of saturated aqueous sodium
chloride solution. The ethyl acetate phase was then dried (magnesium
sulfate), filtered, and concentrated in vacuo at room temperature. The
residue was purified by preparative T~C on 4 Analtech tapered silica
gel plates (20 x 20 cm), which were developed in 94:6 CH2C12-MeOH.
The product band from each plate was isolated, combined, and extracted
with 94:6 CH2C12-MeOH. Concentration of the extracts in vacuo
yielded 66.2 mg (87%) of a nearTy colorless glass; virtually
homogeneous by TLC ~n 95:5 CH2C12-MeOH. 500 MHz lH NMR
(CDC13) was complex, owing to rotamers, but was consistent with the
assigned structure. Mass spectrum (ESI): m/e = 765 (M + H).
~tep 1 3.2F 2-(2-(3.5-dimethylphenyl)-3- ~ 2-r4-(4-
methane.sulfonylaminophenyl)-butylaminolethyl ~ -1 H-indol-
S-yl)-N.N-diethylisobutyramide
A mixture of 62.7 mg (0.082 mmol) of ~2-~S-(1-
diethylcarbarnoyl- 1 -methylethyl)-2-(3 ,5 -dimethylphenyl)- 1 H-indol-3-
yl]ethyl~-[4-(4-methanesulfonylaminophenyl)butyl]carbamic acid benzyl
CA 0224011~ 1998-06-09
WO 97/21435 PCTtlJS96/21)004
- 85 -
ester, 30 mg of 20% palladium on carbon, 10 mL of glacial acetic acid,
and 5 mL of absolute ethanol wa,s shaken with hydrogen (48 psig) in a
pressure vessel for 2 hours. The catalyst was removed by filtration
through Celite under nitrogen, and the filtrate was concentrated in
vacuo at room temperature. The residue was purified by preparative
TLC on 4 Analtech tapered silica gel plates (20 x 20 cm), which were
developed in 92.5:7.5:0.75 CH2C12-MeOH-concentrated NH40H. The
product band from each plate was isolated, combined, and extracted
with 92.5:7.5:0.75 CH2C12-MeOH-concentrated NE~40H. Concentration
of the extracts in vacuo yielded 47.2 mg (91 %) of a glass; homogeneous
by TLC in 92.5:7.5:0.75 ~H2C12-MeOH-concentrated NH40H. 500
MHz lH NMR (CDC13) was consistent with the assigned structure. Mass
spectrum (ESI): m/e = 631 (M + H).
PREPARATION OF SYNTHETIC INTERMEDIATES
Step A 4-chloro-N-methoxy-N-methylbutyramide
To a solution of 4-chlorobutyryl chloride (10.0 g in 200
mL of dry methylene chloride) was added 10.4 g of N,O-
dimethylhydroxylamine hydrochloride. The mixture was stirred under
nitrogen and maintained below 25 ~C by cooling in an ice bath as
necessary while triethylamine (29.1 mL)was added dropwise over about
20 minutes, resulting in precipitation. After 1.5 hours at room
temperature, the mixture was concentrated in vacuo. The residue was
partitioned between 100 mL of diethyl ether and 100 mL of saturated
aqueous sodium bicarbonate solution. The organic layer was washed
with an additiona~ 100 mL of saturated sodium bicarbonate, and the
ac~ueous fractions were back-extracted with ether. The combined
organic phases were dried over sodium sulfate, filtered, and
concentrated in vacllo to give 10.5 g ~90%) of an oil, which had
satisfactory purity by ~H NMR (CDC13). Mass spectrum (PB-NH3/CI):
m/e= 166(M+H).
CA 02240115 1998-06-09
WO 97/21"35 PCT/IJS96/20004
- 86 -
~tep B 3-chloropropyl 3.5-dimethylphenvl ketone
A solution of 10.2 mL ~13.9 g; 72 mmol) S-bromo-m-
xylene in 200 mL of anhydrous tetrahydrofuran was stirred under
nitrogen at -78 ~C as 35.8 mL (84 mmol) of 2.5 M n-butyllithium in
tetrahydrofuran was added dropwise. After lS minutes at -78 ~C, a
solution of 10.0 g ~60 mmol) of 4-chloro-N-methoxy-N-
methylbutyramide (from Step A) in 30 ml of anhydrous
tetrahydrofuran was added dropwise over 25-30 minlltes. The resulting
solution was maintained at -78 ~C for 45 minutes and then warmed
briefly to room temperature. The reaction was quenched by addition of
40 ml of 2 N hydrochloric acid and then partitioned between ethyl
acetate and water. The organic phase was washed with saturated
aqueous sodium bicarbonate solution and then saturated aqueous sodium
chloride solution. The organic solution was dried over sodium sulfate,
filtered, and concentrated in vacuo. Flash chromatography of the
residue afforded
8.91 g (70%) of an oil, which had satisfactory purity by 1H NMR
(CDC13).
Step AA 4-(4-nitrophenyl)butyric acid. N-methoxy-N-methylamide
A stirred solution of 6.29 g (30 mmol) of 4-(4-
nitrophenyl)butyrIc acid in 90 mL of dry methylene chloride (
m~int~med under nitrogen and cooled in a water bath) was treated with
4.17 rnL (3.03 g; 30 mmol) of triethylamine, followed by 13.26 g (30
mmol) of BOP reagent. After a few minutes, 3.22 g (33 mmol) of N,O-
dime~ylhydroxylamine hydrocholoride was added, followed by an
additional 4.59 mL (3.33 g, 33 mmol) of triethylamine. After 2.25
hours, the solution was diluted with 200 rnL of diethyl ether and washed
successively with 3 x 100 mL of 2 N hydrochloric acid, 1 x 100 mL
and 2 x 50 mL of saturated aqueous sodium bicarbonate solution, and 1
x 50 mL of saturated aqueous sodium chloride solution. The organic
phase was dried over magnesium sulfate, filtered, and concentrated in
vac~;o. Flash chromatography of the residue on silica gel (elution with
2:1 and then 3:2 hexane-EtOAc) afforded 6.27 g (~3%) of crystals, mp
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
- 87 -
39.5-41.5 ~C; homogeneous by TLC in 1:1 hexane-EtOAc. 500 MHz lH
NMR (CDCl3) was consistent with the assigned structure. Mass
spectrum (PB-NH3/C~): m/e = 253 (M + H).
Step BB 4-(4-aminophenyl)butyric acid. N-methoxy-N-methylamide
A mixture of 6.05 g (24 mmol) of 4-(4-nitrophenyl)butyric
acid, N-methoxy-N-methylamide, 50 mg of 10% palladium on carbon,
and 200 mL of ethanol was shaken with hydrogen (initial hydrogen
pressure 53 psig) for 1.5 hours, by which time hydrogen uptake had
ceased and TLC indicated complete reaction. The mixture was filtered
through Celite under nitrogen, and the filtrate was concentrated in
vacuo to yield 5.29 g of an oil; homogeneous by TLC in 95:5 CH2C12-
MeOH. 400 MHz 1 H NMR (CDC13) was consistent with the assigned
structure. Mass spectrum (PB-NH3/CI): m/e = 223 (M + H).
Step CC 4-r4-(methanesulfonamido)phenyllbutyric acid. N-methoxy-
N-methylamide
A solution of 5.33 g (24 mmol) of 4-(4-
aminophenyl)butyric acid, N-methoxy-N-methylamide in 48 mL of dry
pyridine was stirred under nitrogen with cooling in an ice bath as 1.86
mL (2.75 g; 24 mmol) of methanesulfonyl chloride was added dropwise
over about 15 minutes. After completion of the addition, the solution
was allowed to warm to room temperature. After 1.5 hours, the
solution was concentrated in vacuo at room temperature. The residue
was diluted with 10 mL of methylene chloride and partitioned between a
mixture of 100 mL of ethyl acetate + 100 mL of tetrahydrofuran and
100 mL of 2 N hydrochloric acid. The organic layer was washed with
an additional 4 x 100 mL of 2 N hydrochloric acid, then with 50 mL of
saturated aqueous sodium bicarbonate solution, and finally with 20 mL
of saturated aqueous sodium chloride solution. The organic phase was
diluted with some tetrahydrofuran, dried over magnesium sulfate, and
~ treated with charcoal. The mixture was filtered through Celite, and the
filter cake was washed with additional tetrahydrofuran. C oncentration
of the filtrate in vacuo gave 4.39 g (61%) of crystals, mp 115-117 ~(~;
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 88 -
homogeneous by TLC in 95:5 CH2C12-MeOH. 400 MHz ~H NM~
(DMSO-d6) was consistent with the assigned structure. Mass spectrum
(PB-NH3/CI): m/e = 301 (M + H).
Step DD 4-r4-(methanesulfonamido)phenyllbutyraldehyde
A mixture of 4.20 g (14 mmol) of 4-[4-(methane-
sulfonamido)phenyl~butyric acid, N-methoxy-N-methylamide and 100
mL of anhydrous tetrahydrofuran was stirred under nitogen with
cooling in an ice bath as 17.5 mL (17.5 mmol) of 1 M lithium aluminum
hydride in tetrahydrofuran was added gradually by syringe. After 0.75
hours, 70 mL of 5% potassium hydrogen sulfate solution (aqueous) was
added cautiously by syringe. The mixture was then removed from the
ice bath, diluted with 150 mL of water, and shaken with 150 mL of
ethyl acetate. The milky aqueous phase was extracted with aIl additional
50 mL of ethyl acetate. The combined organic fractions were washed
successive~y with 2 x 100 mL of 1 N hydrochloric acid, then 50 mL of
saturated aqueous sodium bicarbonate solution, and finally 50 mL of
saturated aqueous sodium chloride solution. The organic phase was
dried over magnesium sulfate, filtered, and concentrated in vacuo.
Flash chromatography of the residue on silica gel (elution with 3:2
hexane-EtOAc) yielded 2.47 g (73%) of an oil; homogeneous by TLC in
1:1 hexane-EtOAc). Upon storage in the freezer, solidification
occurred (mp 41-44 ~C). 400 MHz lH NMR (CDCl3) was consistent
with the assigned structure. Mass spectrum (PB-NH3/C~): m/e = 259
(M + NH4).
Step AAA Ethyl 2-(4-hydrazinophenyl)acetate hydrochloride and 2-
(4-hydrazinophenyl)acetic acid hydrochloride
This compound (a mixture of the ethyl ester and the
carboxylic acid) was prepared from 13.4 g (75 mmol) of ethyl 2-(4-
aminophenyl)acetate, by diazotization and stannous chloride reduction of
the diazonium salt, according to the method of L. J. ~treet, et al., J.
Med. Chem.. 36, 1529 (1993). The material was obtained in two crops.
The first crop consisted of 6.40 g of powder, mp >200 ~C. By 400 MHz
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
- 89 -
lH NMR (DMSO-d6), this material consisted of a mixture of carboxylic
acid and ethyl ester in approximately a 4:3 molar ratio. Mass spectrum
(PB-NH3/CI): 19~ (arylhydrazonium cation for the ethyl ester). The
second crop consisted of 4.60 g of powder, mp >lP~0 ~C. By 400 MHz
1H NMR (DMSO-d6), this material consisted of a mixture of carboxylic
acid and ethyl ester in approximately a 7:1 molar ratio. After
adiustment for the mixture composition of the two crops, the estimated
total yield was 69%. Because esterification of any carboxylic acid
occurs in the next step, both the ester and the acid react to give the same
product.
Step AAAA Ethyl (+1-)-2-(4-nitrophenyl)propionate
To a solution of 9.76 g (50 mmol) of (+/-)-2-(4-
nitrophenyl)propionic acid in lS0 mL of absolute ethanol was added 3.0
mL, of concentrated sulfuric acid. The resulting solution was stirred at
reflux under nitrogen. After 6 hours, the solution was cooled and
stirred vigorously as 250 mL of saturated aqueous sodium bicarbonate
solution was added gradually (Caution: foaming). The mixture was
then partitioned between 750 mL of ethyl acetate and S00 mL of water.
The organic layer was washed with 100 mL of saturated aqueous sodium
bicarbonate solution and then with 100 mL of saturated aqueous sodium
chloride solution. The organic phase was dried over magnesium sulfate,
filtered, and concentrated in vacuo to give 10.86 g (97%) of an oil;
homogeneous by TLC in 9: l hexane-ethyl acetate. 400 MHz 1 H NMR
(CDCl3) was consistent with the assigned structure.
Step BBBB Ethyl 2-methyl-2-(4-nitrophenyl)propionate
A suspension of 924 (23 mmol) of sodium hydride (60% in
oil) in 21 mL of dry N,N-dimethylformamide was stirred under
nitrogen in an ice bath as a solution of 4.6~ g (21 mmol) of ethyl (+/-)-
2-(4-nitrophenyl)propionate in 20.5 mL of dry N,N-dimethylformamide
was added gradually over about lO minutes. An intense violet color
developed during the addition. The mixture was then allowed to warm
to room temperature. After about 1 hour, the mixture was again cooled
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 90 -
in an ice bath as a solution of 1.44 mL ~3.28 g; 23 mmol) of methyl
iodide in 5 mL of dry N,N-dimethylformamide was added dropwise by
syringe over about 10 minutes, while m~in~ining the internal
temperature at 10-15 ~C. The mi~ture was allowed to warm to room
temperature, and the color changed to brown. After 1 hour, an
additional 187 mL (426 mg, 3 mmol) of iodomethane was added. By
the next day, the mixture consisted of a suspension of some grayish solid
in a golden liquid. It was stirred vigorously and quenched by gradual
addition of 10 mL of 5% aqueous potassium bisulfate solution. The
mixture was partitioned between 400 mL of diethyl ether and 400 mL
of water. The organic layer was washed with an additonal 3 x 400 mL
of water and then with 50 mL of saturated aqueous sodium chloride
solution. The organic phase was then dried over magnesium sulfate,
filtered, and concentrated in vacuo. Flash chromatography of the
residue on silica gel (elution with 19:1 hexane-ethyl acetete) yielded
4.31 g (87%) of an oil; homogeneous by TL(: in 9:1 hexane-ethyl
acetete. 400 MHz 1 H NMR (CDC13) was consistent with the assigned
structure.
Step CCCC Ethyl 2-(4-aminophenyl)-2-methylpropionate
A mixture of 4.27 g (18 mmol) of ethyl 2-methyl-2-(4-
nitrophenyl)propionate, 200 mg of 10% palladium on carbon, and 120
mL of absolute ethanol was shaken with hydrogen (initial hydrogen
pressure 47 psig) in a pressure vessel for 2 hours. The catalyst was
removed by filtration through Celite under nitrogen, and the filter cake
was washed with additional ethanol. Concentration of the filtrate in
vacuo at up to 50 ~C gave 3.74 g (100%) of an oil; homogeneous by
TLC in 4:1 hexane-EtOAc. 400 MHz lH NMR (CDC13) was consistent
with the assigned structure. Mass spectrum (ESI): m/e = 208 (M + H).
Step DDDD Ethyl 2-(4-hydrazinophenyl)-2-methylpropionate
A solution of 3.725 g (18 mmol) of ethyl 2-(4-
aminophenyl)-2-methylpropionate in 18 mL of concentrated
hydrochloric acid was stirred at -10 to -5 ~C in an ice-acetone bath as a
. =
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
- 91 -
solution of 1.29 g (18.7 mmol) of sodium nitrite in 7.5 mL of water was
added dropwise over about 15 minutes. Stirring was continued at this
temperature for an additional 30 minutes. Next, a small amount of
7 insoluble solid was removed by filtration into a cold receiving flask.
The filtrate was then added dropwise over 10-15 minutes to a solution
of 20.3 g (90 mmol) of stannous chloride dihydrate in 14.5 mL of
concentrated hydrochloric acid stirred under nitrogen in an ice-acetone
bath. The addition was carried out at such a rate that the internal
temperature remained at about -5 ~C. A gummy material separated
during the addition. After completion of the addition, stirring was
continued at -10 to -5 ~C for 1 hour. The aqueous phase was decanted,
and the residual gum was dissolved in 250 mL of ethyl acetate. The
ethyl acetate solution was treated cautiously with 250 mL of saturated
aqueous sodium bicarbonate solution and shaken in a separatory funnel.
The ethyl acetate layer was washed with 50 mL of saturated aqueous
sodium chloride solution. The entire mixture was filtered before
separation of the phases. The ethyl acetate phase was dried over
magnesium sulfate, filtered, and concentrated i7~ vacuo at room
temperature to yield 2.59 g (65%) of an oil. 500 MHz ~H NMR
(CDCl3) was consistent with the assigned structure and indicated that
only minor impurities were present.
Following a procedure similar to that described in
EXAMPLES 13.1 and 13.2, the following compounds were prepared:
R8 H
'N' Me
Me
CA 02240115 1998-06-09
W O 97/21435 PCTAUS96/20004
- 92 -
Exam,~le X-R7R8 rn/e
13A CH2COOEt 576 (M + H)
13B CH2CON(Me)2 575 (M + H)
13C C~(Me)COOEt 59() (M + H)
13D C(Me)2COOEt 604 (M t H)
13E CH(Me)CON(~t)2 617 (M + ~)
13F C(Me)2CON(Me)2 603 (M ~ H)
13G C(Me)2CON(Pr)2 659 (M + H)
FXAMPLE 14. 1
Me~N~N ~
MeJ ~J--N~ ~Me ~ ,S~M
Me
2-(3 ,5-dimethylphenyl)-3- f 2-1 4-(4-methanesulfonylaminophenyl)-
butylaminolethyl~-lH-indole-S-carboxylic acid diethylamide
Step 14.1 A 3-(2-aminoethyl)-2-(3 .5-dimethylphenyl)- 1 H-indole-5-
carboxylic acid ethyl ester
A mixture of 7.60 g (50 mmol) of 4-hydrazinobenzoic acid,
10.55 g (S0 mrnol) of 3-chloropropyl 3,5-dimethylphenyl ketone, and
200 mL of absolute ethanol was stirred under nitrogen and heated to
reflux. After 12 hours, the mixture was cooled and filtered. The solid
on the filter was washed with additional small volumes of ethanol. The
filtrate was treated with 4 mL of concentrated sulfuric acid and stirred
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
- 93 -
at reflux under nitrogen ~or 4 days. The cooled mixture was stirred in
an ice bath as a solution of sodium ethoxide (21 % w/w in ethanol) was
added dropwise under nitrogen until the mixture was basic by pH paper.
The mixture was filtered and concentrated in vacuo at 30 ~C. The
residue was partitioned between diethyl ether and water, with some
saturated aqueous sodium chloride solution added to assist in separation
of the layers. The aqueous phase was washed with an additional 100 mL
of ether. The combined organic extracts were dried over sodium
sulfate, filtered, and concentrated in vacuo. The residual gum was flash
chromatographed on silica gel (elution with 97:3:0.3 and then 95:5:0.5
CH2C12-~eOH-concentrated NH40H). Concentration of the product
fractions yielded 4.03 g of pure product as a stiff foam (virtually
homogeneous by TLC in 95:5:0.5 CH2Cl2-MeOH-concentrated
NH40H). Concentration of mixed fractions yielded an additional 0.93
g, which was rechromatographed to provide an additional 0.77 g of
pure material, for a total yield of 4.80 g (29%). 400 MHz 1 H NMR
(CDCl3) was consistent with the assigned structure. ~ass spectrum
(PB-N~3/CI): m/e = 337 (M + H).
~tep 14.1 B ~-~3.5-dimethylphenyl)-3- ~ 2-r4-(4-methanesulfonylamino-
phenyl)butylaminolethyl~-lH-indole-5-carboxy}ic acid ethyl
ester
To a dry flask were added 672 mg (2.0 mmol) of 3-(2-
aminoethyl)-2-(3,5-dimethylphenyl)-lH-indole-5-carboxylic acid ethyl
ester, 530 mg (2.2 mmol) of 4-[4-(methanesulfonamido)phenyl]-
butyraldehyde, 1.20 g (10 mmol) of magnesium sulfate, and a magnetic
stirring bar. The flask wa,s purged with nitrogen, cooled to -10 to -5 ~C
in an ice-methanol bath, and stirred as 4 mL of dry CDC13 was
introduced gradually by syringe. The mixture was stirred under
nitrogen for 15 mimltes. Next, the septum was removed, and 100 mg
(2.6 mmol) of sodium borohydride was added rapidly. The septum was
immediately replaced, and the system was again purged with nitrogen.
The mixture was stirred under nitrogen at about -5 ~C as 4 mL of dry
methanol was added gradually by syringe. After 20 minutes at this
CA 02240115 1998-06-09
WO 97/2143S PCT/US96/20004
- 94 -
temperature, the reaction was quenched by gradual syringe addition of 1
mL of acetone to destroy excess sodium borohydride. After a few more
minutes, the mixture was removed from the cooling bath and partitioned
between 25 mL of ethyl acetate and 25 mL of water. The organic layer
was washed with 10 mL of saturated aqueous sodium chloride solution,
~en dried over sodium sulfate, filtered, and concentrated in vacuo. The
residue was flash chromatographed on silica gel (elution with 97:3 and
~hen 95:5 CH2C12-MeOH). Concentration of the pooled product
fractions in vacuo yielded 663 mg (59%) of a foam; virtually
homogeneous by TLC (92.5:7.5 CH2C12-MeOH). 400 MHz lH NMR
(CDC13 + small arnount of DMSO-d6) was consistent with the assigned
structure. Mass spectrum (E~SI): m/e = 562 (M + H).
~tep 14.1C 3-(2-~benzyloxycarbonyl-r4-(4-methanesulfonvlamino-
phenyl)butyllamino ~ethyl)-2-(3.5-dimethylphenyl)- lH-
indole-5-carboxylic acid ethyl ester
A solution of 646 mg (1.15 mmol) of 2-(3,5-
dimethylphenyl)-3- { 2-[4-(4-methanesulfonylamino-
phenyl)butylamino]ethyl }-1 H-indole-5-carboxylic acid ethyl ester in 5
mL of dry methylene chloride and 5 mL of anhydrous tetrahydrofuran
was stirred under nitrogen and coolecl to -78 ~C in a dry ice-acetone
bath as 200 mL of N,N-diisopropylethylamine was added, followed by
gradual addition of 173 rnL (207 mg; 1.15 mmol, based on 95% purity)
of benzyl chloroformate was added gradually by syringe. After 30
minutes, the solution was removed from the cooling bath and allowed to
warm to room temperature. It was then partitioned be~ween 25 mL of
ethyl acetate and 25 mL of 5% potassium bisulfate aqueous solution.
The organic layer was washed with an additional 25 mL of 5%
potassium bisulfate and then with lO mL of saturated aqueous sodium
chloride solution. The organic phase was dried (magnesium sulfate).
filtered, and concentrated in vacuo. Flash chromatography of the
residue on silica gel (elution with 9~:2 CH2cl2-MeoH) afforded 611 mg
(76%) of a foam; homogeneous by TLC in 95:5 CH2cl2-MeoH. 500
MHz lH NMR was complex, owing to the existence of rotamers, but was
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
_ 95 _
consistent with the assigned structure. Mass spectrum (ESI): m/e = 696
~ (M~H).
Step 14.1 D 3-(2- ~ benzyloxycarbonyl-r4-(4-methanesulfonylamino-
phenyl)butyll amino ~ ethyl)-2-(3 ~5 -dimethylphenyl)- 1 H-
indole-5-carboxylic acid
A solution of 600 mg (0.862 mmol) of 3-(2-
~ benzyloxycarbonyl- [4-(4-methanesulfonylamino-
phenyl)butyl]amino }ethyl)-2-(3,5-dimethylphenyl)-lH-indole-5-
carboxylic acid ethyl ester in 18 mL (9 mmol) of 0.50 N potassium
hydroxide in methanol was stirred at about 60 ~C as 2.0 mL of water
was added gradually. Stirring was continued at 60-65 ~C under nitrogen
for 10 hours. The cooled mixture, which contained a white precipitate,
was concentrated to small volume in vacuo. The residual suspension
was partitioned between 25 mL of ethyl acetate and 25 mL of 0.5 N
hydrochloric acid. After the aqueous layer was separated, precipitation
began in the ethyl acetate phase. ~ilution with 25 mL of
tetrahydrofuran redissolved the precipitate. The aqueous phase was
back-extracted with 10 mL of ethyl acetate + 10 mL of tetrahydrofuran.
The combined organic phases were dried over magnesium sulfate,
filtered, and concentrated in vacuo. The residual solid was triturated
with diethyl ether, collected on a filter, and washed with some additional
ether to give (after drying) 573 mg (100%) of a powder, mp 211.5-213
~C; virtually homogeneous by TLC(92.5:7.5 CH2C12-MeOH). 500 MHz
lH NMR (DMSO-d6) was consistent with the assigned structure. Mass
spectrum (ESI): m/e = 668 (M + H).
Step 14.1E f2-rS-diethylcarbamoyl-2-(3~5-dimethylphenyl)-lH-indol-
3-yllethyl ~ -r4-(4-methanesulfonylaminophenyl)-
butyllcarbamic acid benzyl ester
To a suspension of 668 mg (0.703 mmol) of 3-(2-
{ benzyloxycarbonyl-~4-(4-methanesulfonylamino-
phenyl)butyl]amino } ethyl)-2-(3,5-dimethylphenyl)- 1 h-indole-5-
carboxylic acid in 2.7 mL of dry methylene chloride and 2.7 mL of
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 96 -
anhydrous tetrahydrofuran were added 366 mg (0.703 mmol) of PyBOP
reagent and 98 mL (71.0 mg; 0.703 mmol) of triethylamine. The
resulting solution was stirred under nitrogen at room temperature for
20 minutes. Next, 109 mL (77.1 mg; 1.05 mmol) of diethylamine was
added, and stirring was continued for 4 hours. The solution was then
partitioned between ethyl acetate and saturated aqueous sodium
bicarbonate solution. The organic layer was dried over sodium sulfate,
filtered, and concentrated in vacuo. Flash chromatography of the
residue on silica gel (gradient elution with 1-4% MeOH in CH2C12)
afforded 440 mg (87%) of a stiff foam; homogeneous by TLC in 95:5
CH2C12-MeOH. 500 MHz I H NMR (CDC13) was complex, owing to
rotamers, but was consistent with the assigned structure. Mass spectrum
(ESI): m/e = 723 (M + H).
~tep 14.1F 2-(3~5-dimethylphenyl)-3-~2-r4-(4-methanesulfonyl-
aminophenyl)butylaminolethyl ~ -1 H-indole-5 -carboxylic
acid diethylamide
A mixture of 435 mg (0.602 mmol) of {2-[5-
diethylcarbamoyl-2-(3,5-dimethylphenyl)- lH-indol-3-yl]ethyl } -[4-(4-
methanesulfonylaminophenyl)butyl]carbamic acid benzyl ester, 100 mg
of 20% palladium hydroxide on carbon, and 50 mL of 2-
methoxyethanol was shaken with hydrogen (42 psig) in a pressure vessel
for 2.25 hours. The catalyst was removed by filtration through Celite,
and the filtrate was concentrated in vacuo. Purification of the residue
by flash chromatography on silica gel (gradient elution with 5-10%
MeOH in CH2Cl2) yielded 353 mg (100%) of a stiff foam;
homogeneous by TLC in 95:5:0.5 CH2Cl2-MeOH-concentrated NH40H.
500 MHz lH NMR (CDCl3) was consistent with the assigned structure.
Mass spectrum (ESI): m/e = 589 (M + H).
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
- 97 -
EXAMPLE~ 14.2
Me O H
Me N~N ~
MelMe ~N ~Me ~ 'S'M
Me
2-(3 .5-dimethylphenyl)-3-r2-~4-14-(methanesulfonylamino)phenyll-
butylaminolethyll-~H-indole-5-carboxylic acid diisopropylamide
Step 14.2A N~N-diisopropyl-4-nitrobenzamide
A solution of 3.51 mL (2.53 g, 25 mmol) of
diisopropyl~mine and 3.62 mL (2.63 g, 26 mmol) of triethylamine in 50
mL of anhydrous tetrahydrofuran was stirred under nitrogen and
maintained at -5 ~C as a solution of 4.1 l g (22.1 mmol) in 10 mL of
anhydrous tetrahydrofuran was added dropwise over 15 minutes. The
mixture was allowed to warm gradually to room temperature. After 2
hours, the mixture was filtered, and the filtrate was partitioned between
diethyl ether and 1 N hydrochloric acid. The organic phase was then
washed with saturated sodium carbonate3 solution, then dried over
sodium sulfate and ~lltered. The filtrate was concentrated in vacuo, and
the res;due was flash-chromatographed on silica gel (gradient elution
with 2-5% MeOH in CH2Cl2) to yield 4.77 g (86%) of yellowish
crystals, mp 141.5-142 ~C; homogeneous by TLC 2:1 hexane-EtOAc.
500 MHz lH NMR (CDCl3) was consistent with the assigned structure.
.c
Step 14.2B 4-amino-N.N-diisopropylbenzamide
A mixture of 4.70 g (18.8 mmol) of N,N-diisopropyl-4-
nitrobenzamide, 200 mg of 10% palladium on carbon, and 200 mL of 2-
methoxyethanol was shaken with hydrogen at approx. 50 psig for 6.5
hours. The catalyst was removed by filtration through diatomaceous
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 98 -
earth under nitrogen. Concentration of the filtrate in vacuo afforded a
quantitative yield of a yellow solid, mp 169.5-170 ~C; homogeneous by
TLC in 95:5 CH2Cl2-MeOH. 500 MHz IH NMR (CDC13) was consistent
with the assigned structure. Mass spectrum (PB-NH3/~ m/e = 221
(M + H).
Step 14.2C 4-hydrazino-N.N-diisopropylbenzamide
Treatment of 4.2 g (19 mmol) of 4-amino-N,N-
diisopropylbenzamide with 15 mL of concentrated hydrochloric acid
and 10 mL of water was followed by agitation. The resulting solution
was m~int~ined at approx. -3 ~C as a solution of 1.32 g (19.1 mmol) of
sodium nitrite in 9 mL of water was added dropwise. After being
stirred for an additional 30 minutes at this temperature, this solution
was added portionwise to a vigorously stirred solution of 15.1 g (66.7
mmol) of stannous chloride dihydrate in 15 mL of concentrated
hydrochloric acid, which was m~int~ined at about -10 ~C. After
completion of the addition, the mixture was stirred at this temperature
for 5 minutes and then allowed to warm to room temperature. At thi.s
point, it was again cooled and basified by gradual addition of 25 mL of
50% sodium hydroxide. The resulting precipitate was collected on a
filter and partitioned between tetrahydrofuran and S N sodium
hydroxide in a 2:1 ratio. The aqueous layer was extracted 3 times with
tetrahydrofuran. The combined organic fractions were concentrated in
vacuo. The residue was taken up in CH2CI2-EtOAc, dried over sodium
sulfate, filtered, and reconcentrated to give 3.55 g (~0%) of semisolid;
homogeneous by TLC in 95:5 CH2C12-MeOH. 500 MHz lH NMR
(CDC13) was consistent with the assigned structure. Mass spectrum (PB-
NH3/CI): m/e = 236 (M ~ H).
Step 1 4.2D 3-(2-aminoethyl)-2-(3.5-dimethylphenyl)- 1 H-indole-5- h
carboxylic acid diisopropylamide
A solution of 3.51 g (14.9 mmol) 4-hydrazino-N,N-
diisopropylbenzamide (from Step 3) in 18 mL of 2-methoxyethanol was
stirred at 100 ~C under nitrogen as 3.77 g (17.P~ mrnol) of 3-
CA 0224011~ 1998-06-09
WO 97/21435 PCT/US96/20004
_ 99 _
chloropropyl 3,5-dimethylphenyl ketone in 7 mL of 2-methoxyethanol
was added dropwise over 20 minlltes. The solution was stirred at this
temperature for 5 hours, then cooled and filtered to remove a solid (a
tetrahydropyridazine by-product). The filtrate was concentrated in
vacuo, and the residue was purified by flash chromatography on silica
gel ~elution with 95:5 CH2C12-MeOH followed by a gradient of 98:2:0.2
to 92:8:0.8 CH2Cl2-MeOH-concd. NH40H) gave 1.78 g (31%) of a
brownish, stiff foam, satisfactory purity by TLC in 95:5:0.5 CH2Cl2-
MeOH-concd. NH40H. 500 MHz IH NMR (CDC13) was consistent with
the assigned structure. Mass spectrum (PB-NH3/CI): m/e = 392.2 (M +
H).
Step 14.2E 2-(3~5-dimethylphenyl)-3-r2-14-r~-(methanesulfonylamino)-
phenyllbutylaminolethyll-lH-indole-5-carboxylic acid
diisopropylamide
A mixture of 100 mg (0.25~ mmol) 3-(2-aminoethyl)-2-
(3,5-dimethylphenyl)-lH-indole-5-carboxylic acid diisopropylamide,
67.7 mg (0.281 mmol) of 4-[4-
(methanesulfonamido)phenyl]butyraldehyde, and 153 mg (1.28 mmol)
of anhydrous magnesium sulfate was purged with nitrogen and cooled in
an ice-methanol bath at about -10 to -5 ~C as 0.60 mL of dry CDCl3 was
added gradually by syringe. The mixture was stirred under nitrogen at
this temperature for 35 minlltes. The septum was removed just long
enough to add 12.5 mg (0.332 mmol) of sodium borohydride, and the
solution was repurged with nitrogen. The mixture was stirred at -10 to
-5 ~C as 0.40 mL of dry methanol was added gradually, and stirring was
continued at this temperature for several minutes. The mixture wa.s
partitioned between ethyl acetate and dilute sodium hydroxide (pH 10).
The ethyl acetate phase was dried over sodium sulfate, filtered, and
t concentrated in vacuo. The residue was purified by flash
chromatography on silica gel (gradient elution with 99:1:0.1 to 90:10:1
CH2Cl2-MeOH-concd. NH40H) to give 16.0 mg of a yellow, stiff foam,
and an additional 13.8 mg was obtained by preparative TLC of mixed
fractions (developed in 95:5:0.5 CH2Cl2-MeOH-concd. NH40H),
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 100-
affording a total of 29.8 mg (19%) of the title compound, homogeneous
by TLC in 90:10:1 CH2Cl2-MeOH-concd. NH40H. 500 MHz lH NMR
(CDC13) was consistent with ~he assigned structure. Mass spectrum
(ESI): m/e = 617.5 (M + H).
Following a procedure similar to that described above, the
following compounds were prepared:
R8 H
R7'~/ ,N~
H ~Me ~ N' Me
Me
Example X-R7R8 rn/e
14A COOEt 562 (M + H)
14B CO-N(CH2CH20H) 621 (M + H)
14C CO-NHEt 562 ~M + H)
14D CO-NH-cyclopropyl 573 (M + H)
14E Me'~~ ~~~ 617 (M + H)
Me
14F Me o 603 (M + H)
Me'~ N
Me
14G [~ ~ 643 (M + H~
N
Me
CA 02240115 1998-06-09
PCT/US96/20004
WO 97/21435
- 101 -
14H ~ 591(M+H)
HO~ IN~
Me
14I M Me ~ ~ 603 (M + H)
Me
14J O 617(M+~)
Me ~ NJ~
Me ~
14K O 605(M+H)
HO ~' N~
Me
14L ~ o 623(M +H)
Me
14M ~ 561 (M+ H)
Me~N
Me
14N ~ 605 (M + H)
MeO~
Me
140 ~ 633 (M + H)
Me~~
Me 1 Me
14P 11 ~~
MeO~
Me ~ Me
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 102-
14Q O 617(M+H)
Me~N~
Me~ Me
Me
14R Me~MeO 645 (M + H)
N J~\
Me~J
Me
14S 11 697 (M + H)
F C~N~
F3C
EXAMPLE 15
Following a procedure similar to that described in
EXAMPLES 8 and 11, the following compounds were prepared:
R7~ ~ ~; Me N--
Me
Bxample X-R7R8 Rg, Rga:Rlo, Rloa (CH2)n t = A
15A NH-coN(Et)2 Me, H:H, H 4
15B NH-CO-Ph Me, H:H, H 4
CA 02240ll5 l998-06-09
WO 97/21435 PCT/US96/20004
- 103-
lSC NH-CO-N(Me)2 Me, H:H, H 2
lSD NH-CO-N(Me)2 Me, H:H, H 4
15E N(CH2Ph)2 Me, H:Me, H 4
lSF NHC(O)Ph CH2CH20H, H:H, H 4
lSG SO2Me Me, H:H, H 4
lSH NHC(O)Ph CH2CH20Me, H:H, H 4
lSI O-cH2ph H, H:Me, Me 4
EXAMPLE 16
Following a procedure similar to that described in
EXAMPLE 14.1, the following compounds were prepared:
R8 H
R ,X,~ N ~
~Me ~OH
Me
Example X-R7R8 m/e
16A COO-CH2CH3 485 (M + H)
16B COOH 457 (M + H)
16C CO-N(CH2CH3)2 512 (M + H)
16D CO-NH-CH2Ph 546 (M + H)
16E CO-N(CH3)2 484 (M + H)
16F CO-N(iBu)2 568 (M + H)
CA 02240115 1998-06-09
WO 97121435 PCT/IJS96/ZO004
- 104-
EXAMPLl~ 17
Following a procedure similar to that described in
EXAMPLES 9 and 14.1, the ~ollowing compounds were prepared:
R8 R2
R ~ N~
~N~CH3
t, ,~
CH3
Exarnple Rl X-R7R8 R2 m/e
17A Ph-4- NHCOEt 617 (M + H)
NH(S02Me)-
COEt
17B Ph-4-S02CH2C(O)Me 652 (M + H)
NHS02CH2-
C(O)Me
1 7C Ph-4- S02Me
NHS02CH2- 610 (M + H)
C(O)Me
17D Ph-4- COOEt 562 (M + H)
S02NE~e
17E Ph-4-N02 COOEt 514 (M + H)
17F Ph-4- CON(Et)2 Et617 (M + H)
NHS02Me
17G Ph-4- CON(Et)2 589 (M + H)
S02NHMe "
17H Ph-4- CON(iBU)2 645 (M + H)
S02NHMe
CA 0224011~ 1998-06-09
PCT/US96/20004
WO 97/21435
- 105 -
17I Ph-4- CON(cyclohexyl)Et 643 (M + H)
S02NHMe
17J Ph-4-NO2 CON(iBu)2 597 (M + H)
17K Ph-4-NH2 CON(iBu)2 567 (M + H)
17L Ph-4-SMe COOEt 515 (M + H)
17M Ph-4-SMe CON(Et)2 542 (M + H)
17N Ph-4-S(O)Me CON(:~t)2 558 (M + H)
170 Ph-4- CON(Et)2 574 (M + H)
S(~)2Me
17P Ph-4-SMe CON(iBu)2 598 (M + H)
17Q Ph-4-S(O)Me CON(iBu)2 614 (M + H)
171~ Ph-4- CON(iBU)2 630 (M + H)
S(0)2Me
17S Ph-4- CON(iBu)2 666 (M + H)
NH[C=N(CO
NH2)]NHMe
17T Ph-4- CON(iBu)2 648 (M + H)
NH[C=N(CN)
]NHMe
17U Ph-4-F CON(iBu)2
17V Ph-4- COOEt 576 (M + H)
SO2N(Me)2
17W Ph-4- CON(iBU)2 659 (M + H)
SO2N(Me)2
17X Ph-4- CON(Et)2 603 (M + H)
SO2N(Me)2
17Y Ph-4- CON(Et)cyclohexyl 657 (M + H)
SO2N(Me)2
CA 02240115 1998-06-09
WO 97/21435 PCT/US96120004
- 106 -
EXAMPLE 18
Following a procedure similar to that described
EXAMPLES 4.1 and 5.1, the following compounds were prepared:
R7 H
R ,X~ N (A)--R
N ~¢~ Me
Me
Example R7-X-R8 -(A)-R 1 M/E
1 8A H O2N
,~
18B H
18C H
~OH
18D NHCO- ~OH
N(Et)2
18E SO2Me OH 569 (M + H)
~,
,Me
o 'o
CA 02240ll5 l998-06-09
WO 97/21435 PCT/US96/20004
- 107-
EXAMPLl~ 19
M~
Me
4-(4- ~ 12-(3 .5 -dimethylphenyl)- I H-indol-3-ylmethyllamino ~butyl)phenol
Step l9A r2-(3.5-dimethylphenyl)-lH-indol-3-
ylmethylldimethylamine
To 2.53 g glacial acetic acid was added 2.0 g dimethyl
amine (40% aqueous solution) followed by 1.37 g fomalin (37%
solution) then 4.0 g 2-(3,5-dimethylphenyl)-lH-indole and the mixture
atirred at 0 ~C. After 15 minutes, 40 mL ethanol was added and the
mixture allowed to warm to room temperature. After 1 hour at room
temperature, the reaction was quenched by pouring into 50 mL of lN
sodium hydroxide. The resulting mixture was extracted with methylene
chloride (4 x 10 mL) and the combined organics dried over potassium
carbonate. Concentration in vacuo gave the crude title compound (4.15
g)-
Step 19B 12-(3.5-dimethylphenyll-lH-indol-3-
ylmethylltrimethylammonium iodide
To a solution of [2-(3,5-dimethylphenyl)-lH-indol-3-
ylmethyl~dimethylamine (350 mg in 4 mL diethyl ether) was added O.S
mL iodomethane and the mixture stirred at room temperature. After 3
hours, the mixture was filtered and the solids dried in vacuo to provide
the crude title compound. (414 mg).
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 108 -
Step 19C 4-(4-~r2-(3~5-dimethylphenyl)-lH-indol-3-
ylmethyllamino ~butyl)phenol
To a solution of 4-(4-{r2-(3,5-dimethylphenyl)-lH-indol-3-
ylmethyl]amino}butyl)phenol (20 mg in l.S mL dry methanol) was
added 47 mg 4-(4-aminobutyl)phenol and the mixture stirred at room
temperature. After 32 hours, the mixture was concentrated in vacuo
and the residue purified by preaparative TLC on silica gel (methylene
chloride:methanol, 96:4) to give the title compound (12.3 mg). m/e =
234 (base)
Followirlg a procedure similar to that described above, the
~ollowing compounds were prepared:
N J~ S Me
Me
Example R1 (CH2)n = A m/e
l9A Ph-4-OMe 4 234 (base)
1913 Ph-4-OH 3 234 (base)
l9C Ph-4-OH 2 234 (base)
. .
CA 02240115 1998-06-09
WO 97/21435 rCT/US96/:;!0004
- 109-
~XAMPLE~ 20
~ '--N~
N~,J
~J!~N~Me
H
Me
2-(3 .5-dimethylphenyl)-3-1 2-(4-phenylpiperazin- 1 -yl)ethyll - I H-indole
Step 20A 1 -r2-(3.5-dimethylphenyl)- 1 H-indol-3-yll -2-(4-
phenylpiperazin- l -yl)ethane- 1 .2-dione
To a solution of 2-(3,5-dimethylphenyl)-lH-indole (75 mg
in 4 mL dry diethyl ether) was added dropwise 0.032 mL oxalyl
chloride and the mi~ture stirred at room temperature. After 45
minute~, the mixture was concentrated in ~acuo and re-solvated in 3 mL
dry tetrahydrofuran then 0.104 mL of l-phenylpiperazine was added
dropwise. After 20 minutes, the reaction was quenched by the addition
of water and the resulting mixture extracted with ethyl acetate. The
organic portion was washed with brine, dried over magnesium sulfate
and the concentrate purified by flash chromatography on silica gel
(ethyl acetate:hexane, 1:1 + 1% methanol) to give the title compound
~143 mg).
Step 20B 2-(3.5-dimethylphenyl)-3-r2-(4-phenylpiperazin-l-
yl)ethyll - l H-indole
To a solution of 1-[2-(3,5-dimethylphenyl)-lH-indol-3-yl]-
2-(4-phenylpiperazin-1-yl)ethane-1,2-dione ~57 mg in 2 mL dry
tetrahydrofuran) was added 30 mg of lithium aluminum hydrideand the
mixture heated to reflux on an oil bath. After 1 hour the mixture was
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
- 110-
cooled and quenched by the sequential addition of 1 mL water and 4 mL
ammonium hydroxide and 5 mL ethyl acetate. ~e mixture was filtered
to remove the solids. The organic portion was washed with brine, dried
over magnesium ,sulfate and the concentrate purified by flash
chromatography on silica gel (ethyl acetate:hexane, 1:5) to give the title
compound (38 mg).
m/e = 410 (M+1)
Following a procedure similar to that described above, the
following compounds were prepared:
R,8 R2
R ~X~ N (A) R1
N J\ ,~ Me
Me
Example F~2 m/e
R7-X-R8 N (A)- R1
20A H / \ /=\ 440 (M + H)
--N~N~OMe
2013 H --N~ N H --
,~,
20C H ~~S"0 514 (M + H)
\ /--N' Me
--N~
~ Y
CA 02240115 1998-06-09
WO 97/21435 PCT/US96/20004
20D H ~ 465 (M + H)
--N/~N NH
\ \
20E H / \ H 439 (M + H)
--N~ N N H2
20F NHC(O)- / \ /=\
N(Et)2 --N~
COOEt
20G NHC(O)- / \ /=\
N(Et)2 --N~
COOH
20H H--N~ N--CH2~ 635 (M + H~
kl-s=o
tBu o