Note: Descriptions are shown in the official language in which they were submitted.
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
TITLE
Vitronectin Receptor Antagonists
FIELD OF THE INVENTION
This invention relates to pharm~eu~ically active compounds which inhibit
the vitronectin receptor and are useful for the treatment o~ infl~mm~tion, cancer and
cardiovascular disorders, such as atherosclerosis and restenosis, and ~ e:~e.s
wherein bone resorption is a factor, such as osteoporosis.
BACKGROUND OF TH~ INVEI~TION
Integrins are a ~upelr~lllily of cell adhesion receptors, which are
transmembrane glycoproteins expressed on a variety of cells. These cell surface
adhesion receptors include gpIIb /IIIa, the fibrinogen receptor, and o~vM3, the
15 vitronectin receptor. The fibrinogen receptor gpIIb /IIIa is expressed on the platelet
surface and it m~ tes platelet aggregation and the formation of a hemostatic clot at
the site of a bleeding wound. Philips, et al., Blood., 1988, 71, 831. The vitronectin
receptor av1~3 is expressed on a number of cells, including endothelial, smooth
muscle, osteoclast, and tumor cells, and, thus, it has a variety of functions. The aV133
20 receptor expressed on the membrane of osteoclast cells mediates the bone resportion
process and contributes to the development of osteoporosis. Ross, et al., J. Biol.
Chem., 1987, 262, 7703. The ocv~3 receptor expressed on human aortic smooth
muscle cells ~tim~ tPs their migration into neointima, which leads to the formation
of atherosclerosis and restenosis after angioplasty. Brown, et al., Cardiovascular
25 Res., 1994, 28, 1815. Additionally, a recent study has shown that a CCv~3 antagonist
is able to proIrlote turnor regression by in.lu.~ing aEioptosi~. of angiogenic l~lood
vessels. Brooks, et al., Cell, 1994, 79, 1157. Thus, agents that would block thevitronectin receptor would be useful in treating diseases m~ t~d by this receptor,
such as osteoporosis, atherosclerosis, restenosis and cancer.
~ -- 1 --
CA 02241633 1998-06-26
W O97/24119 PCT~US96/20748
The vitronectin receptor is known to bind to bone matrix proteins, such as
osteopontin, bone sialoprotein and thrombospondin, which contain the tri-peptideArg-Gly-Asp (or RGD) motif. Thus, Horton, et al., Exp. Cell Res. lg91, 195, 368,disclose that RGD-cont:lining peptides and an anti-vitronectin receptor antibody(23C6~ inhibit dentine resorption and cell spreading by osteoclasts. In addition,
Sato, et al., J. Cell Biol. 1990, 111, 1713 disclose that echicf~fin, a snake venom
peptide which contains the RGD sequence, is a potent inhibitor of bone resorption in
tissue culture, and inhibits attachment of osteoclasts to bone. Fisher, et al.,
Endocrinology 1993, 132, 1411, has further shown that echistatin inhibits bone
resorption in vivo in the rat. BertoIini etal., J. Bone Min. ~es., 6, Sup. 1, S146, 252
have shown that cylco-S,S-N~C-acetyl-cy~leillyl-N~c methyl-argininyl-glycyl-
aspartyl-penicillzlmin~ inhibits osteoclast ~tt:~hmt-nt to bone. EP 528 587 and 528
586 report substituted phenyl derivatives which inhibit osteoclast m~ tçA bone
resorption.
Alig et al., EP 0 381 033, Hartman, et al., EP 0 540,334, Blackburn, et al.,
WO 93/08174, Bondinell, et al., WO 93/00095, Blackburn, et al. WO 95/04057,
Egbertson, et al, EP 0 478 328, Sugihara, et al. EP 529,858, Porter, et al., EP 0 542
363, and Fisher, et al., EP 0 635 492 ~1icclc)se certain compounds that are useful for
inhibiting the fibrinogen receptor. It has now been discovered that certain
a~ iately substituted compounds are potent inhibitors of the vitronectin receptor.
In particular, it has been discovered that such compounds are more potent inhibitors
of the vitronectin receptor than the fibrinogen receptor and such compounds contain
a fibrinogen receptor antagonist template.
SUM[MARY OF THE INVENTION
This invention comprises compounds of the formula (I)-(V) and
(XXI)-(XXII) as described hereinafter, which have pharmacological activity for the
inhibition of the vitronection receptor and are useful in the treatment of
infl~mm:-fion, cancer and cardiovascular disorders, such as atherosclerosis and
restenosis, and ~lice~cçs wherein bone resorption is a factor, such as osteoporosis.
CA 02241633 1998-06-26
W O g7/24119 PCTrUS96120748
This invention is also a pharm~reutical composition comprising a compound
according to formula (I)-(V) (XXI)-(XXII)and a ph~rm:~reutically carrier.
This invention is also a method of treating ~ e~es which are m~ t~d by
" the vitronectin receptor. In a particular aspect, the compounds of this invention are
5 useful for treating atherosclerosis, restenosis, infl~mm~tion, cancer and diseases
wherein bone resorption is a factor, such as osteoporosis.
DETAILI~D DESCRIPTION
This invention compri~es novel compounds which are more potent inhibitors
10 of the vitronectin receptor than the fibrinogen receptor. The co~ oullds of the
instant invention comprise a fibrinogen receptor antagonist template that is linked to
a nitrogen-cont~ining five-membered ring, which is optionally fused to an aromatic
six-membered ring. The fibrinogen receptor antagonist template is substituted by an
aliphatic substituent which contains an acidic moiety. It is preferred that about
15 fourteen intervening covalent bonds via the shortest intramolecular path will exist
between the acidic group of the fibrinogen receptor antagonist template and the
nitrogen of the optionally fused five-membered ring.
As used herein, the term "fibrinogen recel~lor antagonist template" means the
core structure of a fibrinogen receptor antagonist, said core being substituted by an
20 acidic group and said core being linked to an organic group substituted with a basic
nitrogen moiety. A fibrinogen receptor antagonist is an agent that inhibits the
binding of fibrinogen to the platelet-bound fibrinogen receptor GPIIb-IIIa. It is an
object of this invention that a fibrinogen receptor antagonist is converted to avitronectin receptor antagonist by replacing the organic group substituted with a
25 basic nitrogen moiety in a fibrinogen receptor antagonist with an optionally fused
nitrogen-cont~ining five-membered ring, preferably an imidazole ring and, most
preferably, a ben~imi~ ole ring.
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
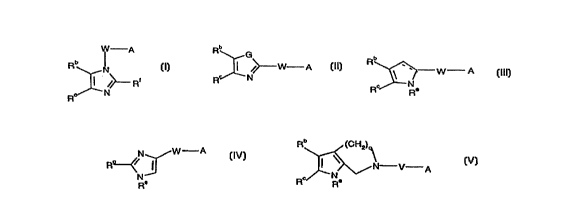
This invention conl~lises compounds of formula (I)~
W A Rb G
,~ /~ R' ~W--A
(I) or (Il) or (1ll) or
R" Rc~N--W A
(IV) or (V)
wherein:
W is - (CHRg)b-V'- or Ar;
A is a fibrinogen receptor antagonist template;
V' is CONR21 or NR2~CO;
GisNRe,SorO;
Rg is H, Cl 6alkyl, Het-CO 6alkyl, C3 7cycloalkyl-CO 6alkyl or Ar- CO 6alkyl;
R21 is Het-CO 6alkyl-U'-CI 6alkyl-~ C3 7cycloalkyl-CO 6alkyl-U'-CI 6alkyl-, or
Ar-CO 6alkyl-U'-C I 6alkyl-;
U' is CONRf or NRfCO;
Rf is H, Cl 6alkyl or Ar-CI 6alkyl;
Re is H, Cl 6alkyl, Ar-CI 6alkyl, Het-CI 6alkyl, C3 7cycloalkyl-CI 6alkyl,
(CH2)qOH or (CH2)kCO2Rg;
kisO, 1 or2;
q is 1 or 2;
bisO, 1 or2;~0 Rb and RC are independently sele.cte~l from H, Cl 6alkyl, Ar-CO 6alkyl, Het-
CO 6alkyl, or C3 6cycloalkyl-CO 6alkyl, halogen, C1~3, ORf, S(O)kRf, CORf,
NO2, N(Rf)2, Co(NRf)2, CH2N(Rf)2, or Rb and RC are joined together to
form a five or six membered aromatic or non-aromatic carbocyclic or
heterocyclic ring, optionally substituted by up to three substituents chosen
-- 4 --
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
from halogen, CF3, Cl 4alkyl, ORf~ S(O)kRf, CORf, CO2Rf OH, N02,
N(Rf)2, CO(NRf)2, and CH2N(Rf)2, or methylenedioxy;
or a pharm~-~elltically acceptable salt thereof.
This invention also comprises compounds of formula (XXI)-(XXII):
I~I~ G
~ B A ~ /~ B A
~XXI) or (XXII)
wherein:
B is a linking moiety of the form -(CHRg)a-U- (CHRg)b-V-;
A is a fibrinogen receptor antagonist template;
G is NRe, S or O;
Rg is H, Cl 6alkyl, Het-CO 6alkyl, C3 7cycloalkyl-C0 6alkyl or Ar- C0 6alkyl;
Rk is Rg, -C(O)Rg, or -C(O)ORf;
Ri is is H, Cl 6alkyl, Het-CO 6alkyl, C3 7cycloalkyl-C0 6alkyl, Ar- C0 6alkyl,
Het-CO 6aL~cyl-U-C1 6alkyl-, C3 7cycloalkyl-CO 6alkyl-U'-Cl 6alkyl-, or
Ar-CO 6alkyl-U'-CI 6alkyl- or Cl 6alkyl;
lS Rf is H, Cl 6alkyl or Ar-CI 6alkyl;
Re is H, Cl 6alkyl, Ar-CI 6alkyl, Het-CI 6alkyl, C3 7cycloalkyl-CI 6alkyl,
(CH2)qOH or (CH2)kCO2Rg;
U, U' and V independently are absent or CO, CRg2, C(=CRg2), S(O)k, O, NRg,
CRgORg, CRg(ORk)CRg2, CRg2CRg(ORk), C(O)CRg2, CRg2C(O), CON Ri
N Ri CO OC(O), C(O)O, C(S)O, OC(S), C(S)NRg, NRgC(S), S(O)2NRg,
NRgS(O)2, N=N, NRgNRg, NRgCRg2, NRgCRg2, CRg2O, OCRg2, C_C,
CRg=CRg~ Ar or Het;
kisO, 1 or2;
~ qis I or2;
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
aisO, 1 or2;
bisO, 1 or2;
Rb and RC are independently selected from H, Cl 6alkyl, Ar-CO 6alkyl, Het-
CO 6alkyl, or C3 6cycloalkyl-CO 6alkyl, halogen, CF3, oRf, S(o)kRf, CORf,
N02, N(Rf)2, Co(NRf)2, CH2N(Rf)2, or Rb and RC are joined together to
forrn a five or six membered aromatic or non-aromatic carbocyclic or
~ heterocyclic ring, optionally- substituted by up to three substituents chosen
from halogen, CF3, Cl 4alkyl, ORf, S(O)kR~, CORf, C02Rf OH, N02,
N(Rf)2, CO(NRf)2, and CH2N(Rf)2; or methylenedioxy;
10 or pharm~reuticSllly acceptable salts thereof.
Preferably, U' is CONRf or NRfCO.
Also included in this invention are pharm:3relltically acceptable addition
salts, complexes or prodrugs of the compounds of this invention. Prodrugs are
considered to be any covalently bonded carriers which release the active parent drug
lS according to formula (I) in vivo. In cases wherein the compounds of this invention
may have one or more chiral centers, unless specified, this invention includes each
unique nonracernic compound which may be synth~o~i7ç~1 and resolved by
conventional techniques. In cases in which compounds have unsaturated carbon-
carbon double bonds, both the cis (Z) and trans (E) isomers are within the scope of
20 this invention. In cases wheleill compounds may exist in tautomeric forms, such as
O OR'
keto-enol tautomers, such as ~~ and b ~, and each tautomeric form is
contemplated as being included within this invention whether existing in equilibrium
or locked in one form by a~ iate substitution with R'.
The compounds of formula (I) - (V) and (XXI) - (XXII) inhibit the binding
of vitronectin and other RGD-co..~ lg peptides to the vitronectin (ocv1~3) receptor.
Inhibition of the vitronectin receptor on osteoclasts inhibits osteoclastic boneresorption and is useful in the tre~tm~nt of ~ e~sç~ wherein bone resorption is
associated with pathology, such as osteoporosis. Additionally, since the compounds
-- 6-
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96120748
of the instant invention inhibit vitronectin receptors on a number of different types
of cells, said compounds would be useful in the tre~tm~n~ of infl~mm~tion and
cardiovascular diseases, such as atherosclerosis and restenosis, and would be useful
as anti-m~t~cta~ir and ~ntitllmt~r agents.
In a particuar embodiment, the compounds of this invention are of the
formula (II), wherein Rb and Rc are joined to form an aromatic ring ct-nt~ining up to
two nitrogen atoms. In a preferred embodiment Rb and Rc are joined to form an
optionally substituted phenyl ring according to formula (IIa):
RY ~C /~ W-A
R (IIa)
wherein G is N-R, S, CH or 0.
L N-CO
Suitably W is -(CHRg)aNRiCO- or ~~ , or, when G is CH, W is
-CH2CH2NRiCO-wherein Ri is a methylene group :lt~hl-cl to G.
Preferably W is -CHRgNRiC0-.
Suitably Ri is H, Cl 6alkyl, C3 7cycloalkyl, Ar or Cl 6alkyl substituted by
one to three groups chosen from halogen, CN, NRg2, ORg, SRg, C02Rg, and
CON(Rg)2, Ar, Het or C3 7cycloalkyl. In particular, Ri is H, methyl, butyl,
cyanomethyl, carboxymethyl, phenylethyl or ben7imi~ 701ylmethyl.
Suitably RX, RY and RZ are independently chosen from Cl 6alkyl, methoxy,
20 nitro, trifluoromethyl, fluoro, chloro, amino or Rx and RY are adjacent to one another
and are joined to form a methylenedioxy group.
Preferably G is NRe.
Suitably Re is H, Cl 4alkyl, Ar, Het or Cl 4alkyl substituted by Ar or Het.
More suitably, Re is H, methyl or ben7.imi~1~7olylmethyl.
-- 7 --
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
In another speeific embodiment, Rb and Rc form a six membered aromatic
ring eon~ining one or two nitrogen atoms according to formulas (IIb-d):
N R~ \~W A ~ \~W A
(lIb) (IIc) (IId)
5 wherein G, RX and RY are as above for formula (IIa).
Specifically, the compounds of this invention are comprised of a nitrogen-
cont~ining optionally fused five-m~mhered ring, a linking group W, and a fibrinogen
reeeptor antagonist template A. In partieular, the fibrinogen reeeptor antagonist
template A is as defmed in Bondinell, et al., WO 93/00095, publich~ocl January 7,
1993, of the sub-formula (VI):
D A'--A (VI)
Al to As form an ~reeccihle ~ub~LiLuLed seven-membered ring, which may be
saturated or unsaturated, optionally eontaining up to two heteroatoms ehosen from
15 the group of O, S and N wlle~ S and N may be optionally oxi~
Dl to D4 form an aeeessible substituted six membered ring, optionally
cont~ining up to two nitrogen atoms;
R is at least one substituent ehosen from the group of R7, or Q-Cl 4alkyl,
Q-C2 4alkenyl, Q-C24alkynyl, optionally substituted by one or more of =O, Rl 1 or
20 R7;
R* is H, Q-Cl 6alkyl, Q-C1 6oxoalkyl, Q-C2 6alkenyl, Q-C3 40xoalkenyl,
Q-C3 40xoalkynyl, Q-C24alkynyl, C3 6eyeloalkyl, Ar or Het, optionally substituted
by one or more of Rl l;
Q is H, C3 6eyeloalkyl, Het or Ar;
CA 0224l633 l998-06-26
PCTrUS96/20748
W O 97/24119
R7 is -COR8, -COCR'2R9, -C(S)R8, -S(O)mOR', -S(O)mNR'R", -PO(OR'),
-PO(OR')2, -B(OR')2, -NO2 and Tet;
R8 is -OR', -NR'R", -NR'SO2R', -NR'OR', -OCR'2C(O)OR',
-OCR'2OC(O)-R', -OCR'2C(O)NR'2, CF3 or AAl;
R9 is -OR', -CN, -S(O)rR', S(O)mNR'2, -C(O)R' C(O)NR'2 or -CO2R';
Rl I is H, halo, -OR12, -CN, -NR'RI2, -NO2, -CF3, CF3S(O)r, -CO2R',
-CONR 2, Q-C0 6alkyl-, Q-Cl 6oxoalkyl-, Q-C2 6alkenyl-, Q-C2 6alkynyl-, Q-Co
6alkyloxy-, Q-C0 6alkylamino- or Q-C0 6alkyl-S(O)~;
R12 is R', -C(O)R', -C(O)NR'2, -C(O)OR15, -S(O)mR' or S(O)mNR'2;
Rl3 is R', -CF3, -SR', or -OR';
Rl4 is R', C(O)R', CN, NO2, SO2R' or C(O)oRl5;
Rls is H, Cl 6alkyl or Ar-Co 4alkyl;
R' is H, Cl 6alkyl, C3 7cycloalkyl-Co 4alkyl or Ar-Co 4alkyl;
R" is R', -C(O)R' or-C(O)OR15;
R"' is R" or AA2;
AAl is an amino acid attached through its amino group and having its
carboxyl group optionally protected, and AA2 is an amino acid attached through its
carboxyl group, and having its amino group optionally protected;
mis 1 or2;
nisOto3;
pisOor l;and
tisOto2;or
ph~ celltic~lly acceptable salts thereof.
With reference to formula (VI), suitably,
Al is CRlRl, CRl, NRl, N, O or S(O)x;
A2 iS CR2R2~ CR2, NR2;
A3 is CR3R3, CR3, NR3, N, O or S(O)x;
g
CA 02241633 l99X-06-26
W O 97/24119 PCTrUS96120748
A4 is CR4R4, CR4, NR4, or N;
A5 is CRsRs, CRs, NR5, N, O or S(O)x;
Dl-D4 are CRIl~ CR6 orN;
Rl and Rl are R* or R, or together are =O;
R2 and R2 are R*, R or =O;
R3andR3 areR*,Ror=O;
R4 and R4 are R*, R or =O; .
Rs and R5 are R*, R or =O; and
xisOto2.
More suitably, Al is CRlRl, CR1, NRl, N, O or S; A2 is CR2R2, NR2 or
CR2; A3 is CR3R3; A4 is CR4R4, CR4, NR4, or N; AS is CR5Rs, CR5, NR5, N, O;
Dl - D4 are CH; R2 or R4 are R; R3,R3 and R5,R5 are =0 or R*,H.
Preferably, Al is CHRl, CRl, NR", N or S; A2 is CR2 or CR2R2; A3 is
CR3R3; A4 is CR4R4 or NR4; As is CR5Rs, and Dl - D4 are CH.
In one embodiment, Al is CRl, A2 is CR2, A3 is C=O, A4 is NR4 and As
are CHRs.
In another embodiment, Al is NRl, A2 is CHCR2, A3 is CR3R3, A4 is NR4,
and A5 are C=O.
In yet another embodiment, Al and A4 are C=O, A2 is NR2, A3 is CHR3 and
20 AS is NRs.
In a preferred embodiment, Al is NRI, A2 is CHR2, A3 is C--O, A4 is NR'
and A5 is CHRs.
Representative sub-form~ of (VI) are given by each of formulas (VIa)-
(VIi) below:
,R-I ~5N,FI,~R3,
R R2 N R2 R?~R '
(VIa) (VIb) (VIc)
- 10-
CA 02241633 1998-06-26
W O 97/24119 PCT~US96120748
, R~
(VId) (VIe) (VIf)
{~ ~ )~ R3
Rl R1 2 , R2 or R1 R1'
(VIg) (VIh) (VIi)
Specific embodiments of this invention wherein the fibrinogen receptor
antagonist template A is of the sub-formula (VI) are named in the Examples.
Preferred compounds of this invention are:
5-[[[(Benzimidazol-2-yl)methyl]methylamino]carbonyl]-lH-ben7.imicl:~701e-
2-aminoacetic acid;
(+)-2,3,4,5-Tetrahydro-7-[[[(ben7imi~i~7.ol-2-yl)methyl}-
methylamino~carbonyl]-4-(3 ,3-dimethylbutyl)-3-oxo- 1 H- 1 ,4-benzodiazepine-2-
acetic acid;
(S)-2,3,4,5-Tetrahydro-4-methyl-3-oxo-7-[[[(5-trifluoromethylb~n7.imi(1~7.ol-
2-yl)methyl]methylamino]carbonyl]- 1 H- 1 ,4-benzodiazepine-2-acetic acid;
(S)-2,3,4,5-Tetrahydro-7-[[[(4,7-dimethoxyben7.imi~1~7.ol-2-
yl)methyl]methylamino~carbonyl]-4-methyl-3-oxo- 1 H- 1 ,4-benzodiazepine-2-acetic
acid;
(+)-2,3,4,5-Tetrahydro-7-[[[(benzimidazol-2-yl)methyl]amino]carbonyl]-4-
(3 ,3-dimethylbutyl)-3-oxo- 1 H- 1 ,4-benzodiazepine-2-acetic acid;
~. (S)-2,3,4,5-Tetrahydro-7-[[[(4-methylb~n7.imicl~7.ol-2-
yl)methyl~methylamino]carbonyl]-4-methyl-3-oxo- 1 H- 1 ,4-benzodiazepine-2-acetic
acid;
- 11 -
CA 02241633 1998-06-26
W O 97/24119 PCTAUS96/20748
S3-2,3,4,5-Tetrahydro-7-[[N-[(ben7.imill~7nl-2-yl)methyl]-N-(4-
anninobutyl3amino]carbonyl]-4-methyl-3-oxo-lH-1,4-benzodiazepi.le-2-acetic acid;(S)-2,3,4,5-Tetrahydro-7-[[N-(ben ~.i ., ~idzl701-2-yl)methyl-N-(2-
cyanomethyl)amino]carbonyl]-4-methyl-3-oxo-lH-1,4-b~.n70~ Ppine-2-acetic
5 acid;
(S)-2,3,4,5-Tetrahydro-7-[[[(be~7.imi~1~7.ol-2-yl)methyl]amino~carbonyl]-3-
oxo-4-(4-phth~limiclobutyl)-lH-l~4-benzodiazepine-2-acetic acid;
4-[[r3-(Ren7.imitl~701-2-yl)propyl]amino]carbonyl]piperidine-1-acetic acid;
4-[[~3-(Ben7.imi~1~701-2-yl)propyl]amino]carbonyl]phenylacetic acid;
(S)-2,3,4,5-Tetrahydro-7-[[[(4-aza-5,7-dimethylben7.imi~ 7-1-2-
yl)methyl3methylamino~carbonyl]-4-methyl-3 -oxo- 1 H- 1 ,4-benzodiazepine-2-acetic
acid;
(~t)-2,3,4,5-Tetrahydro-7-[[[(bel~7.i,..i~1~7.ol-2-yl)methyl3methylamino~-
carbonyl}-4-[2-(3 ,4-methylenedioxyphenyl)ethyl]-3-oxo- 1 H- 1 ,4-benzodiazepine-2-
acetic acid;
(~)-2,3 ,4,5-Tetrahydro-7-[[[(ben7.imidz-701-2-yl)methyllamino]carbonyl]-4-
(2-methoxyethyl)-3 -oxo- 1 H- 1 ,4-benzodiazepine-2-acetic acid;
(S)-2,3,4,5-Tetrahydro-7-[[[(bt~n7.imi~1~7.ol-2-yl)methyl]methylamino]-
carbonyl]-4-methyl-3-oxo- 1H- 1 ,4-benzodiazepine-2-~f et~mi~le;
(+)-2,3,4,5-Tetrahydro-7-[[[[1-[(ben7.imi~1~7.ol-2-yl)methyl]ben7.imi(1~7.ol-2-
yl]methyl]amino]carbonyl]4-methyl-3-oxo-lH-1,4-ben7.orliz~7epine-2-acetic acid;
(S)-2,3,4,5-Tetrahydro-7-[[[(be.n7.imi-1~7.~-1-2-
yl)methyl]methylamino]carbonyl~-3 -oxo- 1 H- 1 ,4-benzodiazepine-2-acetic acid;
(S)-2,3,4,5-Tetrahydro-7-[[bis[(benzimidazol-2-yl)methyl]amino]carbonyl]-
4-methyl-3-oxo- lH-1 ,4-benzodiazepine-2-acetic acid;
(+)-2,3,4,5-Tetrahydro-7-r[[(4-azaben7.imit1~701-2-
yl)methyl~methylamino]carbonyl]-4-(3 ,3 -dimethylbutyl)-3-oxo- 1 H- 1,~
benzodiazepine-2-acetic acid;
(i~)-2,3,4,5-Tetrahydro-7-[[[(ben7.imi<1~7.Ql-2-
yl)methyl]methylamino]carbonyl]-3-oxo-4-(2,2,2-trifluoroethyl)-lH-1,4-
benzodiazepine-2-acetic acid;
- 12-
CA 0224l633 l998-06-26
W O 97/24119 PCT~US96/20748
(~)-2,3,4,5-Tetrahydro-7-t[2-(ben7.imid~7.ol-2-yl)acetyl~amino]-S-oxo-4-(2-
phenylethyl)- 1 H- 1 ,4-benzodiazepine-2-acetic acid;
, (+)-2,3,4,5-Tetrahydro-7-[[[(ben7.imi-1~7.ol-2-yl)methyl]amino]carbonyl]-3-
oxo-4-(2,2,2-trifluoroethyl)- lH- 1 ,4-benzodiazepine-2-acetic acid;
" S (S)-2,3,4,5-Tetrahydro-7-~[[(5,6-difluoroberl7.imifl:~7.ol-2-
yl)methyl]methylamino]carbonyl]-4-methyl-3-oxo- lH- 1 ,4-benzodiazepine-2-aceticacid;
(+)-2,3,4,5-Tetrahydro-7-[[bis[(ben7.imicl~7.ol-2-yl)methyl]amino]carbonyl]-
3-oxo-4-(2-phenylethyl)- lH- 1 ,4-ben70~ 7.epine-2-acetic acid;
(S)-2,3,4,5-Tetrahydro-7-[[[~4-aza-5-methylben7imid:~7.ol-2-
yl)methyl]arnino]carbonyl]-4-methyl-3-oxo- 1 H- 1 ,4-benzodiazepine-2-acetic acid;
(S)-2,3,4,5-Tetrahydro-4-methyl-7-[[[(4-nitroben7.imi(1~7.ol-2-
yl)methyl]methylaminolcarbonyl]-3-oxo- lH- 1 ,4-benzodiazepine-2-acetic acid;
(+)-2,3,4,5-Tetrahydro-7-[[[(4-aza-S-methylben7.imi~ 7.ol-2-
15 yl)methyl]amino]carbQnyl]-3-QxQ-4-(2,2,2-trifluoroethyl)- lH- 1 ,4-benzodiazepine-2-
acetic acid;
(+)-4-~4-[[L( iH-Ben7.imidi~7.oi-2-yi)melhyljmethylamino]ccubonyl]phenyl]=3=
phenylbutanoic acid;
(+)-3-[[[4-(4-Azabe.,~ 7.Ql-2-yl)butanoyl]glycyl]amino]-4-pentynoic
20 acid;
(S)-2,3,4,5-Tetrahydro-7-[[~[ 1 -(2-hydroxyethyl)ben7.imkl:~701-2-
yl3methyl]amino]carbonyl]-4-methyl-3-oxo-lH-1,4-benzodiazepine-2-acetic acid;
(+)-2,3,4,5-Tetrahydro-7-[[[(4-aza-5-methylben~.i...irl~7.ol-2-
yl)methyl]arnino]carbonyl]-4-(2-methoxyethyl)-3-oxo- 1 H- 1 ,4-benzodiazepine-2-
25 acetic acid;
(S)-2,3,4,5-Tetrahydro-7-[[[(4-aminoben7.imill~701-2-
yl)methyl]methylarnino]carbonyl~-4-methyl-3-oxo-lH-1,4-berl7.o~ 7Ppine-2-acetic
acid;
Ethyl (S)-2,3,4,5-tetrahydro-7-[[~(ben7.imid~7.nl-2-
30 yl)methyl]methylamino]carbonyl]~-methyl-3-oxo-lH-1,4-ben7.o~ 7.P.pine-2-
acetate;
- 13-
CA 02241633 1998-06-26
W O 97/24119 PCTAUS96/20748
(S~-2,3,4,5-Tetrahydro-7-~[[(benzimidazol-2-
yl)methyl]methylamino]carbonyl]-4-methyl-3-oxo- 1 H- 1 ,4-benzodiazepine-2-acetic
acid, ~(2,2-dimethyl-2-methoxyacetyl)oxy]methyl ester;
2,3,4,5-Tetrahydro-7-[[[( l R)-(b~ 701-2-
yl)ethyl]methylarnino]carbonyl]-4-methyl-3-oxo- 1 H- 1 ,4-benzodiazepine-(2S)-acetic
acid;
(_)-N-t2-(Aminomethyl)-4-[[[(4-aza-5-methylben7.imi~1~7.(31-2-
yl)methyl]methylamino]carbonyl]phenyl]aspartic acid;
(+)-2,3,4,5-Tetrahydro-4-methyl-3-oxo-7-[[[(phen~nthrimi~1~7.ol-2-
yl)methyl]amino~carbonyl]-1H-1,4-benzodiazepine-2-acetic acid;
(+)-2,3,4,5-Tetrahydro-7-[3-(benzimidazol-2-yl)phenyl]-4-methyl-3-oxo- lH-
1,4-ben70di~7~pine-2-acetic acid;
- (+)-4-[4-[[[(Ben7imid~701-2-yl)methyl]methylamino]carbonyl]phenyl]-3-
(dimethylaminocarbonyl)butanoic acid;
(S)-2,3,4,5-Tetrahydro-7-[[[(ben7.imiclz-7nl-2-yl)methyl]amino]carbonyl]-3-
oxo-4-[2-(pyrid-3-yl)ethyl]-lH-1,4-bP.n70~ 7~pine-2-acetate;
(S)-2,3,4,5-Tetrahydro-7-[[[(4-aza-5-methylben7imi~1~701-2-
yl)methyl]methylamino]carbonyl]-3-oxo- 1 H- 1 ,4-benzodiazepine-2-acetic acid;
(+)-2,3,4,5-Tetrahydro-7-[[N-[(ben7imi-1~7.- 1-2-yl)methyl]-N-[[4-(2-
carboxybenzoyl)amino]butyl]amino]carbonyl]-3-oxo-4-(2-phenylethyl)- 1~-1,4-
benzodiazepine-2-acetic acid;
(+)-2,3,4,5-Tetrahydro-7-[[N-[(ben7.imi-1~7.ol-2-yl)methyl3-N-[[4-(4-azido-2-
hydroxybenzoyl)amino]butyl]amino]carbonyl]-3-oxo-4-(2-phenylethyl)- 1 H- 1,4-
benzodiazepine-2-acetic acid;
Ethyl (S)-2,3,4,5-tetrahydro-7-[[[(4-aza-5-methylbe~7imid~701-2-
yl)methyl]amino~carbonyl]-4-methyl-3-oxo- I H- 1 ,4-benzodiazepine-2-acetate;
2,3,4,5-Tetrahydro-7-~[N-[(ben7.imil1~7rl-2-yl)methyl]-N-[[[(+)-
biotinoyl]amino]butyl]amino]carbonyl]-3-oxo4-(2-phenylethyl)- 1 H- 1,4-
benzodiazepine-(2RS)-acetic acid;
- 14-
CA 02241633 1998-06-26
W O 97/24119 PCTnUS96/20748
2,3,4,5-Tetrahydro-7-[[[(lS)-(ben,.i...i-1~7.Ql-2-
yl)ethyl]methylamino]carbonyl]-4-methyl-3-oxo-lH-1 ,4-benzodiazepine-(2S)-acetic
acld;
(S)-2,3,4,5-Tetrahydro-7-[[[(imi~1~7.o( 1 ,2a)pyrid-2-
S yl)methyl]methylamino]carbonyl] -4-methyl-3-oxo- 1 H- 1 ,4-bçn7.0rli ~7epine-2-acetic
acid;
(S)-2,3,4,5-Tetrahydro-7-rt[(benz; ., .i~7.ol-2-yl)methyl]amino~carbonyl]-3-
oxo-lH-1,4-berl7.orli:~7çpine-2-acetic acid;
(+)-5-[[2,3,4,5-Tetrahydro-7-~[[(benzimidazol-2-yl)methyl]amino]calbollyl]-
3-oxo-4-(2-phenylethyl)- 1 H- 1 ,4-benzodiazepin-2-yl]methyl]tetra_ole;
(S)-2,3,4,5-Tetrahydro-7-[[[(4-aza-5-methylbel.~,i..licl~7.ol-2-
yl)methyl]amino]carbonyl]-3-oxo- 1 H- 1 ,4-benzodiazepine-2-acetic acid;
(+)-2,3,4,5-Tetrahydro-7-[3-(ben7.imicl~7ol-2-yl)propyl]-4-methyl-3-oxo-lH-
1,4-benzodiazepine-2-acetic acid;
(+)-2,3,4,5-Tetrahydro-7-[[N-[(benzill~dazol-2-yl)methyl]-N-(4-
aminobutyl)amino]carbonyl] -3-oxo-4-(2-phenylethyl)- 1 H- 1 ,4-benzodiazepine-2-acetic acid;
(+)-2,3,4,5-Tetrahydro-7-[[[(ben7imi~ 7.ol-2-yl)methyl]amino]carbonyl]-4-
methyl-3-oxo- lH- 1 ,4-benzodiazepine-2-(N-hydroxy)aret~mide;
Ethyl (+)-3-[[[2-(Ben7.imi~7.~1-2-yl)ethyl]amino]succinoyl]amino-4-
pentynoate;
(+)-3-[[[2-(Bçn7.imi(1~7.ol-2-yl)ethyl~amino]succinoyl]amino4-pentynoic
acid;
(+)-2,3 ,4,5-Tetrahydro-7-[[N-[(benzimidazol-2-yl)methyl] -N-[[4-(4-azido-3-
iodo-2-hydroxybenzoyl)amino]butyl] amino]carbonyl] -3 -oxo-4-(2-phenylet'nyl)- 1 H-
1,4-benzodiazepine-2-acetic acid;
2,3,4,5-Tetrahyciro-7-~[[(iS)-(ben7.imi~l~7.Qi-2-yi)ethyi]amino3carbonyl]-4-
methyl-3-oxo- 1 H- 1 ,4-benzodiazepine-(2S)-acetic acid;
2,3,4,5-Tetrahydro-7-[[[( 1 R)-(benzimidazol-2-yl)ethyl]amino]carbonyl3-4-
methyl-3-oxo-lH-1,4-benzodiazepine-(2S)-acetic acid; and
.
- 15-
CA 02241633 1998-06-26
w o 97n4119 PCTrUS96/20748
(+)-7-[[[(4,5-Dimethyl-lH-imi~1~7~1-2-yl)methyl]methyla~mino]carbonyl~-
2,3,4,5-tetrahydro-4-methyl-3-oxo- lH- 1 ,4-benzodiazepine-2-acetic acid;
or pharmaceutically acceptable salts thereof.
The most preferred fibrinogen receptor antagonist template is of the sub-
S formula (VIa), wherein CR2R2 is CHCH2CO2H, CR3R3 is C=O, and CRsRs isCH2. Vitronectin fibrinogen receptor antagonism is particularly pronounced when
the A-W- substituent is attached to the 7-position of the 3-oxo-2,3,4,5-tetrahydro-
lH- 1 ,4-benzodiazepine ring system.
In the formula below the definitions for the ~.ub~.LiLuents are as defined in
10 ft rm~ (I)-(V) and (XX)-(XXI), unless specified otherwise.
Another embodiment of a preferred fibrinogen receptor template A is
represented by the 1,4-benzodiazepine 2,5-dione of sub-formula (VII);
~Y
R (VII)
1 5 wherein:
Y is H, Cl 4alkyl, Cl 4alkoxy, Cl 4alkoxycarbonyl, F, Cl, Br, I, CF3, oRf,
S(O)kRf, CORf, NO2, N(Rf)2, CO(NRf)2, CH2N(Rf)2, methylenedioxy, CN, CO2Rf,
OC(O)Rf, or NHC(O)Rf; and
Rh is (CH2)qC02R
The plepal~tion and the use of tnis sub-structure in ~l~ing fibrinogen
receptor antagonists of this sub-formula is detailed in Bondinell, et al., WO
93/OOO9S published January 7, 1993 and Blackburn, et al., WO 93/08174, publishedApril 29, 1993.
- 16-
..
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
Table I, below, sl~mm~ries other ~l~felled fibrinogen receptor templates that
are included within the scope of the present invention. Such templates are:
Table I
(VIII)
Ais~R ~R22 J~R2~ j~R22
or
/ ~R
whGreill:
R21 and R22 independently are H or -Z-CO2Rfor Z-CON~Rf)2 with the proviso that
one of Al or A2 is -ZCO2Rf or Z-CON(Rf)2;
Zis-CH2-,~O(CH2)q~~~NRf(CH2)q~~~S(CH2)q~-CH2CH2-,-CH(CH3)CH2-,
-(CH2)3-, -CH=CH-, -C(CH3)=CH-, CH2-CH=CH- or CH=CHCH2; and
Y is H, Cl 4alkyl, Cl 4alkoxy, Cl 4alkoxycarbonyl, F, Cl, Br, I, CF3, oRf, S(O)kRf,
CORf, NO2, N(Rf)2, CO(NRf)2, CH2N(Rf)2, methylenedioxy or Z-CORf,
in Alig, et al., EP 0 381 033, published August 8, 1990.
(IX)
R6SO2NH CO2R~
20 wherein:
-17-
CA 02241633 1998-06-26
W O 97124119 PCTAUS96/20748
R6 is aryl, Cl lOalkyl, C3 6cycloalkyl, C4 1oaraLkyl, Cl loalkoxyalkyl,
Cl loalkaryl, Cl loalkylthio~lkyl, Cl loaLkoxythioalkyl, Cl loalkylarnino,
C4 1 oaralkylarnino, C 1-1 oalkanoylarnino, C4_ 1 oaralkanoylarnino, C 1-1 oaL~anoyl,
C4 10aralkanoyl, or Cl locarboxyaLkyl; and
S Y is H, Cl 4alkyl, Cl 4aLlcoxy, Cl 4alkoxycarbonyl, F, Cl, Br, I, CF3, ORf,
S(o)kRf, CORf, NO2, N(Rf)2, Co(NRf)2, CH2N(Rf)2, methylenedioxy, CN, CO2Rf,
OC(o)Rf, or NHC(O)Rf,
in Egbertson, et al., EP 0 478 328, published April 1, 1992.
(X)
--M~\M2~G~ CHCO2Rf
wherein:
Ml is CH or N;
M2 is CH or N, with the proviso that when Ml is CH, M2 is N; and
G' is N or N~3R",
in Eldred, et al., EP 0542 363, published May 19, 1993.
. (XI)
--M~\M2{><0H
--/ CH2CO2R~
wherein:
Ml is CH or N; and
M2 is CH or N, with the proviso that when Ml is CH, M2 is N,
in Porter, et al., EP 0 537 380, published April 21, 1993.
I
CA 02241633 1998-06-26
W O 97t24119 PCTrUS96/20748
~XIIl
~3 . M ~ Rh
wherein:
M1 isCHorN;
S Y is H, Cl 4alkyl, Cl 4alkoxy, Cl 4alkoxycarbonyl, F, Cl, Br, I, CF3, ORf,
S(O)kRf, CORf, NO2, N(Rf)2, CO(NRf)2, CH2N(Rf)2, methylenedioxy, CN, Co2Rf,
OC(O)Rf, or NHC(o)Rf;
D3 is CH2 or C=O; and
Rh is (cH2)qco2Rf7
10in Klinnick, et al., EP 0 635,492, published January 25, l99S.
(XIII)
\B~ N
wherein:
Y is H, C~ 4alkyl, C1 4alkoxy, Cl 4alkoxycarbonyl, F, Cl, Br, I, CF3, oRf,
lS S(o)kRf, CORf, NO2, N(Rf)2, Co(NRf)2, CH2N(Rf)2, methylenedioxy, CN, CO2Rf,
OC(O)Rf, or NHC(O)Rf;
Rh is ~CH2)nCO2Rf; and
B is HsC)~ , ~N H3C ~
- in Blackburn, et al., WO 9S/04057, published February 9, 1995.
~V)
- 19-
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
,~
N~ L -C02R9
O
wherein:
L* is -C(O)NRg-(CH2)-, ~C(O)~(CH2)q~~ NRg-(CH2)q-, ~O~(CH2)q~~ or
S(O)k-(CH2)q~~
in Hartman, et al., EP 0 540 331, published May 5, 1993.
~V)
N N--CH2 Co2R9
CO~o
in Sugihara, et al., EP 0 529,858, published March 3, 1993.
(XVI~
~~
~ "'--CO R~
wherein:
Y is H, Cl 4alkyl, Cl 4alkoxy, Cl 4alkoxycarbonyl, F, Cl, Br, I, CF3, ORf,
S(o)kRf, CoRf1, N02, N(Rf)2, Co(NRf)2, CH2N(Rf)2, methylenedioxy, CN,
CO2Rf, Oc(o)Rf~ or NHC(O)Rf,
in ~imm~i~b~h, et al., EP 0 483 667, published May 6, 1992.
(XVII)
~(CH2)oco2Ro
N
in Linz, et al., LP 0 567 968, published November 3, 1993.
(XVIII)
- 20 -
CA 0224l633 l998-06-26
W ~ 97/2~119 PCTAUS96/20748
Z"' Z~
~<Co2R9
wherein:
Rd is Het-C0 6alkyl; and
Z", Z"' independently are hydrogen, Cl 4alkyl, halo, ORf, CN, S(o)kRf,
5 Co2Rf, or OH,
in Bovy, et al., EP 0 539 343, published April 28, 1993.
Compounds of this invention comprising specific fibrinogen templates are
named in the examples. These examples illustrate the invention, but do not limit the
10 scope of the invention.
The above descriptions of fibrinogen receptor templates for use in the present
invention were taken from pending published patent applications. Reference should
be made to such patent applications for their full disclosures, including the methods
of plc~ali~lg said templzlt~s and specific compounds using said templates, the entire
15 disclosure of such patent applications being incol~oldl~d herein by reference.
Table II, below, describes other fibrinogen receptor antagonists, whose core
structures would be useful in carrying out the instant invention. Reference should be
made to the patent applications and other publications for their full disclosures,
20 including the methods of preparing said tt-mpl~t~s and specific compounds using
said templates, the entire disclosure of the noted patent applications and otherpublications being incorporated herein by reference. Since it is contemplated that
any fibrinogen receptor antagonist that is linked to an optionally fused nitrogen-
cont~ining five-m.-.mhP.red ring will possess the novel utility described herein, the
25 list below does not limit the scope of the present invention.
CA 02241633 1998-06-26
WO 97124119 PCT~US96/20748
Table II
Adir et Col~r:-~nie
FR 928004, June 30, 1992, Fauchere, J. L., et al.
EP 0578535, June 29, 1993, Fauchere, J-L, et al.: Describes X-RGDW-OH analogs,
where X contains a cationic arnine.
CA 2128560, Jan. 24, 1995, Godfroid, J-J, et al., substituted pipel~ines.
Asahi Breweries, Ltd.
JP 05239030, Sep. 17, 1993, aminomethyltetrahydroisoquinolines.
Asahi Glass
WO 90/02751, Ohba, M. et al.: Sept. 8, 1989: Describes cyclic RGD-con~ining
peptides.
WO 90/115950, Mar. 22, 1990, Ohba, M., et al.
EP 0406428, 1/9/91: Describes cyclic RGD-cont:~ining peptides
WO 92/09627, Isoai, A. et al.: Nov. 29, 1991: Describes cyclic RGD-cont~ining
peptides.
~s~c~PIls AG
DE 4207254, (Der 93-289298/37) Mar. 7, 1992, Zoller, G., et al.: Describes
guanidinopropyl-4-oxo-2-thioimida_olidin-3-yl-Asp-X analogs
EP 93904010, Feb. 24, 1993, Zoller, G., 4-oxo-2-Thioxoimi~1~7Olidine Derivatives.
EP 0565896, Mar. 18, 1993, Klinger, O, et al.: Describes
guanidinoethylphenyloxyacetyl-Asp-X analogs.
EP 0566919, (Der 93-338002/43) Apr. 3, 1993, Zoller, G., et al.: Describes
guanidinopropyl-4-oxo-2-thioimidazolidin-3-yl-Asp-X analogs.
EP 580008, (Der 94-027663/04) July 6, 1993, Zoller, G., et al.: Describes S-m-
guanidinophenyl-2,4-dioxoimi(l~7.Qlidin-3yl)acetyl-Asp-Phg.
DE 224414, July 6, 1993, Zoller, G., et al.: Describes 5-m-~tl:~ni~inophenyl-2,4-
dioxoimidazolidin-3yl)acetyl-Asp-Phg.
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
EP 584694, (Der 94-067259/09) Apr. 2, 1994, Zoller, G., et al.: Describes 5-m-
guanidinophenyl-2,4-dioxoimidazolidin-3yl~acetyl-Asp-Phg .
" DE 4301747, (Der 94-235891/29) Jul. 28, 1994, Zoller, G., et al.: Describes 5-m-
guanidinophenyl-2,4-dioxoimi(1~7O1idin-3yl)acetyl-Asp-Phg analogs.
DE 4308034, (Der 94-286666/36) Sept. 15, 1994, Klinger, O. et al.: Describes 5-m-
guanidinophenyl-2,4-dioxoimi~i~7O1idin-3yl)acetyl-Asp-Phg analogs.
DE 4309867, Sept. 29, 1994, Klingler, O, et al.: Describes 5-m-guanidinophenyl-
2,4-dioxoimi~ 7.olidin-3yl)acetyl-Asp-Phg.
Chiron
WO 93/07169, ~Der 93-134382/16), Mar. 15, 1993, Devlin, J. J., et al.: Describes RGD peptides.
Ciba Geigy
EP 0452210, (Der 91-305246/42) Apr, 5, 199Q, describes aminoalkanoyl-GDF
analogs.
EP 0452257, Mar. 26, 1991, Allen, M. C., et al.: Describes aminoalkanoylAsp-Phe
analogs.
COR Ther~renti~c
WO 90/15620, June 15, 1990: Describes cyclic RGD-cont~ining peptides.
EP 0477295, Apr. 1, 1992: Scarborough, R. M. et al.
WO 92/08472, May 29, 1992, Scarborough, R. M. et al.
WO 93/223356, April 27. 1993, Swift, R. L., et al.: Describes cyclic RGD-
c--nt~inin~ peptides.
EP 0557442, Sept. 1, 1993, Scarborough, R. M., et al.
Scarborough, R. M.; Rose, J. W.; Hsu, M. A.; Phillips, D. R.; Fried, V. A.;
Campbell, A. M.; N--nni77i, L.; Charo, I. F., Barbourin, A GPIIb-IIIa-
Specific Integrin Antagonist from the Venom of Sistrurus M. Barbouri, J.
BioL Chem., 266, 9359, 1991.
- 23 -
CA 0224l633 l998-06-26
W O97/24119 PCT~US96/20748
Daiichi Pharm Co Ltd.
JP 05078344-A, ~Der 93-140339/17) Mar. 30, 1993: Describes Bis-
amidinoheter~cycles, eg. benzofurans.
S DuPont Merck
WO 93/07170, Apr. lS, 1993: Describes cyclic-RGD-con~ining peptides.
WO 94/11398, May 26, 1994: Wells, G. J. et al. Describes cyclic RGD cont~ining
peptides.
IL 109237, Ju1. 31, 1994.
WO 94/22909, (Der 94-333113/41) Oct. 13, 1994: DeGrado W. F., et al.
WO 94/22910, (Der 94-333114/41 Oct. 13, 1994: DeGrado W. F., et al. Prodrugs.
WO 94/22494, (Der 94-332838/41) Oct. 13, 1994: DeGrado W. F., et al. Cyclic
peptides
EP 625164, Nov. 23, 1994: Degrado, W. F., et al. Cyclic peptides.
Mousa, S. A.; Bozarth, J. M.; Forsythe, M. S.; Jackson, S. M.; Leamy, A.; Diemer,
M. M.; Kapil, R. P.; Knabb, R. M.; Mayo, M. C.; Pierce, S. K.; al., e.,
~ntiplzltelet and An-ilhloll-botic Efficacy of DMP 728, a Novel Platelet
GPIIb/IIIa Receptor Antagonist, Circulation, 89, 3, 1994.
~ackson, S.; DeGrado, W.; Dwivedi, A.; Parthasarathy, A.; Higley, A.; Krywko, J.;
Rockwell, A.; Markwalder, J.; Wells, G.; Wexler, R.; Mousa, S.; Harlow, R.,
Template-Constrained Cyclic Peptides: Design of High-Affinity T .ig~n~1~ for
GPIIb/IIIa, J. Amer. Chem. Soc., 116, 3220, 1994
Ellem Ind Farma Spa
GB 2207922, Aug, 3, 1988, describes linear RGD analogs.
Farmitalia Erba SRL Carlo
EP 611765 (Der 94-265375/33), Aug 24, 1994: Cozzi, P., et al. Describes 5-(2-
~y~ lylmethyl-2-imi~ 7Ql-l-yl)-1-cyclohexylethylidene)aminoxypentanoic
acid.
- 24 -
CA 02241633 1998-06-26
W O 97124119 PCTrUS96/20748
Fuji Photo Film
JP 04208296-A (Der. 92-303598/38), Nov. 30, 1990, Describes RGD peptides.
JP 04213311-A (Der. 92-305482/38), Nov. 27, 1990, Describes mllltimeric RGD
peptides.
JP 04217693-A, (Der 92-312284/38), Oct. 23, 1990, Descirbes mllltim~ric RGD
peptides.
JP 04221394-A (Der. 92-313678/38), Oct. 26, 1990, Describes mllltimf~ric RGD
peptides.
JP 04221395-A (Der. 92-313679/38), Oct. 26, 1990, Describes m-lltim~ric RGD
peptides.
JP 04221396-A (Der. 92-313680/38), Oct. 26, 1990, Describes mllltim~ric RGD
peptides.
JP 04221397-A (Der. 92-313681/38), Dec. 20, 1990, Describes mllltim~ril~ RGD
peptides.
EP 0482649 A2, April 29, 1992, Kojima, M. et al.: Describes RGD peptides.
EP 0488258A2, June 3, 1992, Komazawa, H., et al: Describes RGD peptides.
EP 503301-A2, Feb. 14, 1991, Kitaguchi, H. et al.: Describes RGD peptides.
JP 05222092, May 21, 1993, Nishikawa, N., et al.: DescribesT in~r X-RGDS.
JP 06239885, (Der 94-313705/39), Aug 30, 1993, Nishikawa, N. et al.: Describes
mllltim~ric RGD peptides.
WO 9324448, (Der 93-405663/50), Dec. 9, 1993, Nishikawa, N., et al.: Describes
ml-ltimf~ric retro-inverseo RGD peptides.
JP 06228189, (Der 94-299801/37), Aug. 16, 1994. Describes RGD peptides.
EP 619118, (Der 94-311647/39), Oct. 12, 1994, Nishikawa, N. et al.: Describes
linear RGD peptides.
Fujisawa
EP 0513675, May 8, 1992, N. Umekita, et al.: Describes
amidinophenyloxyalkanoyl-Asp-Val-OH analogs.
WO 9409030-A1, Apr. 28, 1994, T~k~ gi, H., et al.: Describes
Amidinophenoycbutanoyl-Asp-Val-OH analogs.
- 25 -
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
EP 0513675, (Der 92-383589/47): Describes Ami~lin~phenyloxybutyrl-Asp-Val
analogs.
WO 9500502, Jan, 5, 1995, Oku, T., et al.,: Describes "aminopi~e~ e
derivatives."
FR 144633: Thromb Haem. 69, 706, 1993.
Cox, D.; Aoki, T.; Seki, J.; Motoyama, Y.; Yoshida, K., Pent~mitline: A Speci~lcNonpeptide GPIIb/IIIa Antagonist, Thromb. Haem., 69, 707, 1993.
G~nPntef h
WO 90/15072 (Der 91007159): Describes RGD-cont~inin~ peptides:
WO 91/01331 (Der91058116), July 5, 1990, P. L. Barker, et al.: Describes cyclic
RGD-corltziinin~ peptides
WO 91/04247, Sept. 24, 1990, T. R. Webb: Describes (guanidinoalkyl)Pro-GD
analogs.
WO 91/11458 (Der 91252610), Jan. 28, 1991, P. L. Barker, et al.: Describes cyclic
RGD-cont~inin~ peptides
WO 92/07870, Oct. 24, 1991 J. P. Burnier, et al.: Describes cyclic RGD-
c-n~ining peptides.
WO 92/17492, Oct. 15, 1992, Burnier, J. P. et al.: Describes cyclic RGD-cont~ining
peptides.
CA 2106314, Oct. 6, 1992, Burnier, J. P. et al.
WO 93/08174, Oct. 15, 1991, B. K. Blackburn, et al.: Describes 2,5-dioxo-1,4-
ben70fliz~7~pines.
CA 2106314, Oct. 6, 1992, Burnier, J. P., et al.
EP 0555328, Aug. 18, 1993, J. P. Burnier, et al.
WO 95/04057, Feb. 9, 1995, Blackburn, B. K., et al.: Describes 1,4-benzodiazepines
con~ining a heterocyclic at positions 1,2.
Scarborough, R. M., Naughton, M. A., Teng, W., Rose, J. W., Phillips, D. R.,
N~nni77i, L, Arfsten, A., Campbell, A. M., and Charo, I. F., J. Biol. Chem.
268, 1066, 1993.
- 26 -
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/207~8
Dennis, M. S.; Henzel, W. J.; Pitti, R. M.; T., L. M.; Napier, M. A.; Deisher, T. A.;
Bunting, S.; Lazarus, R., Platelet Glycoprotein IIb-IIIa Protein Antagonists
,~ from Snake Venoms: Evidence for a Family of Platelet-Aggregation Inhibitors, Proc. Natl. Acad. Sci. USA, 87, 2471, 1989.
Barker, P. L.; Bullens, S.; Bunting, S.; Burdick, D. J.; Chan, K. S.; Deisher, T.;
Eigenbrot, C.; Gadek, T. R.; Gant7os, R.; Lipari, M. T.; Muir, C. D.; Napier,
M. A.; Pitti, R. M.; Padua, A.; Quan, C.; Stanley, M.; Struble, M.; Tom, J. Y.
K.; Burnier, J., P., ~yclic RGD Peptide Analogues as Antiplatelet
A~ lbotics, J. Med. Chem., 35, 2040, 1992.
McDowell, R. S.; Gadek, T. R., Structural Studies of Potent Constrained RGD
Peptides, J. Amer. Chem. Soc., 114, 9245, 1992.
Glaxo
EP 537980, Oct. 13, 1992, B. Porter, et al.: Describes six cis-4-~4-(4-
amidinophenyl)-l-piL,el~illyl]-l-hydroxycyclohPx~nP~eetic acid analogs.
EO 0542363, Nov. 10, 1992, Porter, B., et al.: Describes 4-~-4-arnidinophenyl-
~i~e~ yl]-piperidine-l-acetic acid analogs.
WO 93/22303, Jan. 11, 1993, Middlemiss, D., et al.: Describes amidinophenyl-
arylpiper~7inP~etic acid analogs.
WO 93/22303, Jan. 11, 1993,1\/ricl~llemi.~, D., et al.: Describes arnidinophenyl-
arylpiper~7ineiqcetic acid analogs.
WO 93/14077, Jan. 15, 1993, B. Porter, et al.: Describes arnidinophenyl-piperizinyl-
piperidine-acetic acid analogs.
EP 609282 Al, Aug. 10, 1994, Porter, B. et al.: Describes cyclohexane acetic acid
derivatives.
EP 612313, Aug. 31, 1994, Porter, B ., et al. Describes alpha-alkylpiperidineacetic
acid derivatives.
EP 93911769, Apr. 20, 1994, Midlerniss, D., et al.
EP 637304 Al, Eeb. 8, 1995, MiclrllPrni~ D., et al. Piperazine Acetic acid
- 30 Derivatives.
- 27 -
CA 0224l633 l998-06-26
W O 97/24119 PCTrUS96/20748
Hann, M. M.; Carter, B.; Kitchin, J.; Ward, P.; Pipe, A.; Broornhead, J.; Hornby, E.;
Forster, M.; Perry, C., An Investigation of the Bioactive Conformation of
ARG-GLY-ASP ContAining Cyclic Peptides and Snake Venom Peptides
Which Inhibit Human Platelet Aggregation, In Molecular Recognition:
S Chemical and Biochemical Problems 11", S. M. Roberts, Ed., The Royal
Society o~ Chemistry, Cambridge, l 9g2.
Ross, B. C. Nonpeptide Fibrinogen Receptor Antagonists", (SAR leading to the
discovery of GR 144053), In Seventh RSC-SCI Medicinal Che~
Symposium, The Royal Society of Chemistry l~ine Ch~mic~l~ and
Medicinals Group and SCI Fine Ch~qmi~ Group, Churchill College,
Carnbridge, 1993, L20.
Pike, N. B.; Foster, M. R.; Hornby, E. J.; Lumley, P., Effect of the Fibrinogen
~eceptor Antagonist GR144053 Upon Platelet Aggregation Ex Vivo
Following Intravenous and Oral ~lmini~tration to the Marmoset and
Cynomologous Monkey, Thromb. Haem., 69, 1071, 1993.
Hoechst
DE 4009506, Mar. 24, 1990, Konig, W., et al.: Describes Hydantoin-(Arg-Gly)-
Asp-X analogs.
Hoffn~nn-La Roche
AU 9344935, (Der 94-118783/15), Mar. 10, 1994,: Describes Cyclic RGD analogs.
EP 0592791, Apr. 20, 1994, Bannwarth. W. et al.: Describes Cyclic RGD analogs.
25 Kogyo Gijl~uil-
JP 06179696, June 28, 1994, Maruyama, S., et al.: Describes Gly-Pro-Arg-Pro-Pro
and analogs.
Kyowa Hakko Kogyo KK
JP 05078244-A, Mar. 30, 1993: Describes dibenzo(b,e)oxepine derivatives.
- 28 -
CA 0224l633 l998-06-26
W O97/24119 PCTrUS96/20748
Laboratoire Chauvin
WO 9401456, Jan. 20, 1994, Regnouf, D. V. J. et al.: Describes Ac-Arg-Gly-Asp-
NHBn analogs.
- La Jolla Cancer Res. Fndn
WO 9500544, Jan. 5, 1994, Pierschbacher, M. D. et al.
US 079441, Jan 5, 1994, Pierschbacher, M. D. et al.: Describes RGD Peptides.
Lilly / COR
EP 0635492, Jan. 25, 1995, Fisher, M. J., Happ, A. M., Jakubowski, J. A., Kinnick,
M. D., Kline, A. D., Morin, Jr., J. M., Sall, M. A., Vasileff, R. T.,: Describescompounds with 6,6-templates.
Medical Univ~. Dily of South Carolina
EP 587770, Mar. 23, 1994 Halushka, P. V., Spicer, K. M.
Merck
EP 0368486 (Der 90-14g427/20), Nov. 10, 1988: Describes X-R-Tyr-D-Y analogs.
EP 0382451 (Der 90248531): Descirbes RGD-cont~ining snake venom inhibitors.
EP 03~538 (Der 90248420): Descirbes RGD-cont~ining snake venom inhibitors.
EP 0410537, July 23, 1990, R. F. Nutt, et al.: Describes cyclic RGD-cont~ining
peptides.
EP 0410539, July 25, 1990, R. F. Nutt, et al.: Describes cyclic RGD-cont~inin~
peptides.
EP 0410540, July 25, 1990, R. F. Nutt, et al.: Describes cyclic RGD-cont~inin~
peptides.
EP 0410541, July 25, 1990, R. F. Nutt, et al.: Describes cyclic RGD-cont~inin~
peptides.
EP 0410767, July 26, 1990, R. F. Nutt, et al.: Describes linear RGD-cont~ining
peptides.
- 29 -
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
EP 0411833, July 26, l99Q R. F. Nutt, et al.: Describes cyclic RGD-con~ining
peptides.
EP 0422937, Oct. 11, 1990, R. F. Nutt, et al.: Describes cyclic RGD-cont~ining
peptides.
S EP 0422938, Oct. 11, 1990, R. F. Nutt, et al.: Describes cyclic RGD-co~t~ining
peptides.
EP 0487238, Octover 13, 1991, T. M. Connolly, et al.: Describes Linear RGD-
cont~ining
EP 0437367 (Der 91209968), M. Sato et al.: Describes cyclic RGD-con~ i
peptides, as inhibitors of osteoclast-m~ tl-A bone resorption.
EP 576898, Jan. S, 1994, Jonczyk, A., et al.: Describes linear RGD peptide analogs
for use in inhibition of cell adhesion.
WO 9409029, Apr. 28, 1994, Nutt, R. F. and Veber, D. F., describes
piperidinylethylpyrrolidinylacetyl-Asp-Trp(tetrazoles) .
EP 618225, (Der 94-304404/38) Oct. 5, 1994, Describes RGD peptide analogs as
antimetastatic colllpou~ds.
DE 4310643, (Der 94-311172/39), Oct. 6, 1994, Jonczyk, A.. et al.,: Describes
cyclic RGD analogs as :~ntimt-,t~t~ti~, agents.
NO 9404093, Oct. 27, 1994, Jonczyk, A.. et al.
~P 0632053, Jan. 4, l99S, Jonczyk, A.. et al.,: Describes cyclic RGD analogs as
:~ntiml~t:~tatic agents.
EP 0479481, Sept. 25, 1991, M. E. Duggan et al.: Describes X-GlyAsp-Y linear
semipeptides.
EP 0478328, Sept. 26, 1991, M. S. Egbertson, et al.: Describes tyrosine derivatives.
EP 0478362, Sept. 27, 1991 M.3~. Duggan et al.: Describes X-Gly-(3-
phenethyl),BAla analogs.
EP 0478363, Sept. 27, 1991, W. L. Laswell, et al.: Describes Tyrosine
sulfon~mil:1ec .
EP 0512829, May, 7, 1992, Duggan, M. F., et al.: Describes chiral 3-hydroxy-6-(4-
piperidinyl)heptanoyl-,B-X-,B-Ala-OH analogs, with variations on X and the
central alkanoyl chain.
- 30 -
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
EP 0512831, May, 7, 1992, Duggan, M. E., et al.: Describes chiral 2-oxo-3-
(Piperidinylethyl)piperidinylacetyl-~-X-13-Ala-OH analogs, with variations
on X and the central piperidinyl ring.
EP 0528586, August 5, 1992, M. S. Egbertson, et al.: Describes tyrosine
sulfon~mi(1~s as inhibitors of osteoclast-mediated bone resorption.
- EP 0528587, August 5, 1992, M. S. Egbertson, et al.: Describes tyrosine
sulfon~ s as inhibitors of osteo~last-m.-di~t~-l bone resorptiQn.
EP 0540334, October 29, 1992, G. D. Hartman, et al.: Describes bç~ oles.
US 5227490, Feb. 21, 1992, G. D. Hartman, et al.: Describes Tyrosine
sulfonamides.
CA 2088518, Feb. 10, 1993, Egbertson, M. S., et la. aminoalkyl-phenyl derivs. as bone resorption inhibts.
US 5206373-A, (Der 93-151790tl8) Apr. 27, 1993, Chung, J. Y. L., et al.: Describes
MK-383-type compounds.
WO 9316994, (Der 93-288324/36), Sep. 2, 1993, Chung, J. Y. L., et al.: Describes pyridinylbutyl-L-Tyrbutylsulfon~mi~le
US 5264420-A, Nov. 23, 1993, Describes piperidinylalkyl-Gly-betaAla analogs.
US 5272158, Dec. 21, 1993, Hartman, G. D. et al.,: Describes
piperidinylethylisoinole analogs.
US 5281585, Jan. 25, 1994, Ihle, N., et al.,: Describes 3-(piperidinylethyl)-
piperidinone analogs.
GB 945317 A, Mar. 17, 1994 (Priority US 34042A, Mar. 22, 1993).
GB 2271567 A, Apr. 20, 1994, Hartman, G. D. et al.: Describes compounds
replacing Tyr with beta-phenylsuccinate.
US 5294616, (Der 94-091561/11) Mar. 15, 1994, Egbertson, M. S., et al.
US 5292756, (Der 94-082364) Apr. 8, 1994, Hartrnan, G. D. et al.
WO 9408577, Apr. 28, 1994, Ha~ an, G-. D., et al.
WO 9408962, Apr. 28, 1994, Hartman, G. D., et al.
WO 9409029, (Der 94-151241/18) Apr. 28, 1994, Hartman, G. D., et al. Describes
- 30 piperidinylpyrrolinylacetyl-Asp-Trp-tetrazoles.
US 5312923, May 17, 1994, Chung, J. Y. L. et al.
- 31 -
CA 02241633 1998-06-26
W O97/24119 PCT~US96/20748
HU 9400249, May 30, 1994, Gante, J. et al.,: Describes pi~c-~ine analogs.
WO 9412181, (Der 94-199942/24), Jun. 9, 1994, Egbertson, M. S. et al.,: Describes
piperidinylethyloxyphenyl acetic acid analogs ,
US 5321034, June 14, 1994, Duggan, M. E., et al.: Describes Piperidinylalkyl-
bet~mino acids.
US 5334596, Aug. 2, 1994, Hartman, G. D. et al.
EP 0608759 A, Aug. 3, 1994, GAnte, J. P. et al.: Describes amidin~i~e~inyl
culllpolll,ds.
WO 9418981, (Der 94-293975/36) Sep. 1, 1994, Claremon, D. A.. et al.: Describes
Many different amine surrogate.
GB 2276384, (Der 94-287743/36) Sep. 28, 1994, Claremon, D. A.., Liverton, N..,:
Describes piperidinylethylquinazoline analogs.
WO 9422825, Oct. 13, 1994, Claremon, D. A.. Liverton, N. J.,: Describes
piperidinylethyl-retro-ben70~i~7epine analogs.
EP 0623615A, Nov. 9, 1994, E251~ 7., P. et al: Describes
arnidinophenyloxazolidinylmethyl-piperidine-4-carboxylic acid and analogs.
WO 9504531, Feb. 16, 1995, Hartman, G D., et al.: Describes
piperidinylalkylheterocycles .
Nutt, R. F.; Brady, S. F.; Colton, C. D.; Sisko, J. T.; Ciccarone, T. M.; Levy, M. R.;
Duggan, M. E.; Imagire, I. S.; Gould, R. J.; Anderson, P. S.; Veber, D. F.,
Development of Novel, Highly Selective Fibrinogen Receptor Antagonists as
Potentially Useful AnLi~ ulllbotic Agents, In Peptides, ~hemistry and
Biology, Proc. 12th Amer. Peptide Symp., J. A. Smith and J. E. Rivier, Ed.,
ESCOM, Leiden, 1992; 914.
Hartman, G. D.; Egbertson, M. S.; H~lc7~nko, W.; Laswell, W. L.; Duggan, M. E.;
Smith, R. L.; Naylor, A. M.; Manno, P. D.; Lynch, R. J.; Zhang, G.; Chang,
C. T. C.; Gould, R. J., Non-peptide Fibrinogen Receptor Antagonists. 1.
Discovery and Design of Exosite Inhibitors, J. Med. Chem., 35, 4640, 1992.
Gould, R. J.; Barrett, S.; Ellis, J. D.; Holahan, M. A.; Stranieri, M. T.; Theoharides,
A. D.; Lynch, J. J.; Friedman, P. A.; Duggan, M. E.; Ihle, N. C.; Anderson,
P. S.; Hartman, G. D., Characterization of L-703,014, A Novel Fibrinogen
- 32 -
.
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
Receptor Antagonist, Following Oral ~lmini~tration to Dogs, Thromb.
Haem., 69, 539, 1993.
Merrell Dow
WO 93/24520, May 14, 1993, Harbeson, S. L., et al.: Describes cyclic RGD
- peptides.
WO 9324520, Dec. 9, 1993, Harbeson, Bitonti,J., A.,: Describes cyclic RGD analogs
as antimetastatic agents.
WO 9429349, Dec. 22, 1994, Harbeson, Bitonti,J., A.,: Describes cyclic RGD
analogs as ~ntim~t~tz~tic agents.
Nippon Steel Corp
WO 9405696, Mar. 17, 1993, Sato, Y., et al,.
EP 628571, Dec. 14, 1994, Sato, Y. et al.
WO 9501371, Jan. 12, 1995, Sato, Y. et al.: Describes RWSRGDW analogs.
ONO Phar
JP 05286922 (Der 93-383035/48), Describes guanidinophenol alkylbenzoic acid
esters.
Roche
EP 038,362, Feb. 19, 1990, M. Muller, et al.: Describes X-NHCHYCO-Gly-Asp-
NHCHZCO2H analogs.
EP 0372486, June, 13, 1990, Allig, L., et al.
EP 0381033, July, 8, 1990, Allig, L., et al.
EP 0384362, August 29, 1990, Allig, L. et al.: Describes amidinophenyl-linked Gly-
Asp-X se~ lides.
EP 0445796, Sept. 11, 1991, Allig, L. et al.: Describes amidinophenyl-linked Gly-
Asp-X selllipeplides.
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
E~P 0505868, Sept. 30, 1992, Allig, L. et al.: Describes N-acyl-alphaamino acid
derivatives, ie. analogs from EP0381003 with variations in the
phenyloxyacetic acid group.
US 5273982-A, (Der94-006713/01) Dec. 28, 1993: Describes arnidinophenyl-linked
S Gly-Asp-X se~ ~Lides.
Alig, L.; 3~denhofer, A.; Hadvary, P.; Hllr7l 1er, M.; Knopp, D.; Muller, M.; Steiner,
B.; Trzeciak, A.; Weller, T., Low Molecular Weight, Non-peptide Fibrinogen
Receptor Antagonists, J. Me~ Chem., 35, 4393, 1992.
Rhone-Poulenc Rorer
US 49525G2, Sept. 29, 1989, S. I. Klein et al.: Describes X-Gly-Asp-Val-OH
analogs.
US 5064814, (Der 91-353169/48) Apr. 5, 1990: Describes Piperidinyl-azetidinyl-
Asp-X analogs.
WO 9104746, Sept. 25, 1990, S. I. Klein et al.: Describes X-Asp-Val-OH analogs.
WO 91/05562, Oct. 10, 1989, S. I. Klein et al.: Describes X-Gly-Asp-Val-OH
analogs.
WO 91/07976, (Der 91-192965) Nov. 28, 1990, S. I. Klein et al.: Describes X-
cycloAA-Asp-Val-OH analogs.
WO 91/04746, S. I. Klein et al.: Describes des-ArninoArginine RGD analogs.
WO 92/18117, Apr. 11, 1991, S. I. Klein et al.: Describes X-Asp-Val-OH analogs.
US 5086069, (Der 92-064426/08) Apr. 2, 1992,: Describes X-Gly-Asp-Val-OH
analogs.
WO 92~17196, Mar. 30, 1992, S. I. Klein et al.: Describes X-Gly-Asp-Val-OH
analogs.
US 5328900, (Der 94-221950/27) Jul. 12, 1992,: Describes X-~ ti~inyl-Asp-Val-
- OH analogs.
US 5332726, (Der 94-241043/29) Jul. 26, 1994,: Describes guanidinoalkanoyl-(N-
alkyl)Gly-Asp-Val-OH analogs.
WO 93/11759, Dec. 7, 1992, S. I. Klein et aL: Describes Bis-gn~nitlinoaklanoic acid
analogs.
- 34 -
CA 02241633 1998-06-26
W O97/24119 PCTrUS96/20748
EP 0577775, Jan 12, 1994, Klein, S. I. et al.
CA 2107088, Sept. 29, 1992, Klein, S.I. et al.
.~9~-~o7
EP 0560730, Mar. 8, 1993 G. Kottirisch and R. Metternich: Describes
amidinophenyl~lkAn~mid-S-a-acetic acid analogs.
G. Kottirisch, et al. Biorg. Med. Chem. Lett 3, 1675-1680, 1993, Describes
amidinophenylacetyl-(Gly-Asp-~-lactam mimetic)analogs.
S~L~ g AG
E 530937, Mar. 10, 1993, Noeski-Jungblut, C., et al. "Collagen rnd~lce~l Platelet
Aggregation Inbitor."
Searle / Monsanto
EP 0319506, (Der 89-3195506) Dec. 2, 1988, S. P. Adams, et al.: Describes RGD-X
analogs.
EP 0462,960, June, 19.1991, Tjoeng, F. S., et al.: Describes guanidinooctanoyl-
Asp-Phe analogs.
US 4857508, S. P. Adams, et al.: Describes RGD analogs.
EP 0502536, (Der 92-301855~ Mar. 3, 1991, R. B. ( Ts~rl~n(l, et al.: Describes
amidinophenylalkanoyl-Asp-Phe analogs.
EP 0319506, Dec. 2, 1988, S. P. Adams et al.: Describes RGDX analogs.
US 4992463, Aug. 18, 1989: Describes guallidinoalkanoyl-Asp-X analogs.
US 5037808, Apr. 23, l990: Describes guanidinoalkanoyl-Asp-X analogs.
EP 0454651 A2, Oct. 30, 1991, Tjoeng, F. S., et al.: Describes amidinoalkanoyl-
Asp-X analogs.
US 4879313, July, 20, 1988: Describes guanidinoalkanoyl-Asp-X analogs.
WO 93/12074, Nov. 19, 1991~ N. Abood, et al.: Describes amidinophenylalkanoyl-
,~-X-AlaOH analogs.
- 35 -
CA 02241633 1998-06-26
W O97/24119 PCT~US96/20748
WO 93/12103, Dec. 11, 1991, P. R.Bovy, et al.: Describes amidinophenylalkanoyl-
,B-X-lactone analogs.
US 5091396, Feb. 25, 1992, Tjoeng, F. S., et al.: Describes amidinoalkanoyl-Asp-X
analogs.
S WO 92/15607, Mar. 5, 1992, C~rl:~nll, R. B., et al.: Describes
~miflinophenylalkanoyl-Asp-X analogs.
WO 93/07867, Apr. 29, 1993, P. R. Bovy, et al.: Describes amidinophenyl-
amidopropionyl-,B-X-AlaOH analogs.
US 888686, May 22, 1992, Bovy, P. R. et al.
CA 2099994, Sept. 7, 1992, (~T~rlzln~1,R. B., et al.
US 5254573, Oct. 6, 1992, Bovy, P. R., et al.: Describes
amidinophenylamidopropionyl-,B-X-,~-Ala-OH.
(PF54C06), EP 0539343, Oct. 14, 1992, P.R. Bovy et al.: Describes
amidinophenylamidopropionyl-13-X-,B-Ala-OH.
WO 93/12074, Nov. 27, 1992, N. A. Abood, et al.: Describes
amidinophenylalkylamido-(R)-Asp-(i.e. retro-Asp)-alkyl and aryl amides and
sulfon~midec
WO 93/12103, Dec. 11, 1992, P. R. Bovy et al.: Describes amidinophenylalkanoyl-
Asp-X lactones
EP 0 539343, Apr. 28, 1993, Bovy, P. R., et al.
EP 0542708, May, 19, 1993, Bovy. P. R., et al.
WO 94/00424, June 23, 1993, Abood, N. A., et al.: Describes
amidinophenylalkanoic acid lactones related to previous compounds.
WO 93/16038, Aug. 16, 1993, Miyano. M. et al.: Describes
amidinophenylpentanoyl-~-arylsulfonarnidomethyl-,13-Ala-OH analogs.
WO 93US7975, Aug. 17, ~993, Zablocki, J. A., Tjoeng, F. S.
WO 93/18058, Sept. 16, 1993, Bovy, P. R. et al.: Describes
amidinophenylamidoproionoyl-Asp-X-OH analogs.
US 5254573, Oct. 19, 1993, Bovy, P. R., et al., Describes amidinophenylpropionyl-
amino acid dervs.
- 36 -
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
US, 5272162, Dec. 21, 1993, Tjoeng, F. S., et al.: Describes amidinophenyl-X-
NHCO-13-Y-,13-Ala-OH analogs.
EP 0574545, Dec. 22, 1993, Garland, R. B., et al.: AmidinophenylX-Asp Analogs.
WO 9401396, Jan. 20, 1994, Tjoeng, F. S., et al., Describes
amidinophenylalkyla~nido-amino acid derivatives.
- WO 9405694, (Der 94-101119/12) Mar. 17, 1994, Zablocki, et al.: Describes
amidinophellylalkylamido-amino acid derivatives.
US 5314902, May 24, 1994, Adams, S. P. et al.: Describes
amidinophenylami~loalk~noyl derivatives.
WO 9418162, Aug, 18, 1994, Adams, S. P., et al.: Describes
amidinophenylalkanoyl-amino acid derivatives.
WO 9419341, Sept. 1, 1994, Tjoeng, F. S., et al.: Describes arnidinophenylnipecotic
acid derivatives.
US 5344837, (Der 94-285503/35), Sept. 6, 1994, Zablocki, J. A., et al.
EP 614360, Sept. 14, 1994, Bovy, P. R., et al.
WO 9420457, (Der 94-302907/37) Sep. 15, 1994, Tjoeng, F. S. et al.
Amin~inophenyl compounds with central ring.
WO 9421602, (Der 94-316876/39), Sept. 29, 1994, Tjoeng, F. S., et al. Describes
guanidinoalkylaminocarbonylaminoacid derivatives.
WO 9422820, Oct. 13, 1994, Abood, N. A., et al.: Describes
amidinophenylpyrollidinonyl-,l~-Ala derivatives.
EP 630366, Dec. 28, 1994, Bovy, P. R., et al.
US 5378727, Jan. 3, 1995, Bovy, P. R. et al.
K. F. Fok, et al., Int. J. Peptide Prot. Res., 38, 124-130, 1991, SAR of RGDY
analogs.
J. A. Zablocki, et al. J. Med. Chem. 35, 4914-4917, 1992, SAR sumnary of
guanidinoalkanoyl-Asp-Phe analogs.
Tjoeng, F. S.; Fok, K. F.; Zupec, M. E.; ~T~rl~n<l, R. B.; Miyano, M.; Panzer-Knodle,
S.; King, L. W.; Taite, B. B.; Nicholson, N. S.; Feigen, L. P.; Adams, S. P.,
Peptide Mimetics of the RGD Sequence, In Peptides, Chem. and Biol. Proc.
-
- 37 -
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
12th Amer. Peptide Symp., J. A. Smith and J.E. Rivier, Fd., ESCOM,
Leiden, 1992; 752.
Nicholson, N.; Taite, B.; Panzer-Knodle, S.; Salyers, A.; Haas, N.; Szalony, J.,Zablocki, J.; Feigen, L.; Glenn, K.; Keller, B.; Broschat, K.; Herin, M.,
J~ min,P.; lesne, M., An Orally Active Glycoprotein nbmIa Antagonist -
SC-54684, Thromb. Haem., 69, 975, 1993.
S~ hKlin~ Beecham Corporation
WO 93/00095, WO 94/14776, and WO 95/18619, Bondinell, et al., Describes (6,7)
bicyclic fibrinogen templates.
WO 94/12478, Keenan, et al.,, Describes ~6,5) bicyclic fibrinogen templates.
WO 94/22440, c~ h~n~ et al., Describes (8,6) bicyclic fibrinogen templates.
WO 94/22444, ~ h~n, et al., Describes ~8,6,7) tricyclic fibrinogen templates.
WO 94/29273, Samanen, J., Describes (6,6) bicyclic fibrinogen templates.
Sumitomo Pharm. Co. Ltd.
WO 9501336, June 6, 1994, Ikeda, Y., et at., Describes piperidinyloxyacetyl-Tyr- piperidinyloxyacetic acid d~liv~liv~s.
S~ ol~o Seiyaku KK
JP 06025290, (Der 94-077374/10) Feb. 1, 1994. Describes mllltimPric RGDT.
Ta~ho Pharm. (Te~in, Ltd)
JP 05230009, (Der 93-317431/40, Feb. 24, 1992: Describes amidino-Cbz-meta-
aminol?heuylpl~,pionate.
JP 9235479, Feb. 24, 1992: Describes Amidinophenylc~l,alnates.
(PFD4C06), WO 94/17804, Aug. 18, 1994, ~i71l~him~ Y. Phatm. Comp for
Treating Cerebral Thrombosis.
(EP 634171), Jan. 18, 1995, Ni7.11~him~, M. Pharm. Comp forTreating Cerebral
Thrombosis. Pro~t~l~nfiin.~
- 38 -
CA 02241633 1998-06-26
W O97/24119 PCTrUS96120748
Takeda
EP 0529858, Apr. 3, 1993, H. Sugihara, et al.: Describes amidinobenzoyl-Gly-
Piperazinone analogs.
EP 606881, Jul. 20, 1994, Cyclic peptides with beta and gamma turns.
EP 614664, Sept. 14, 1994, Miyake, A., et al: Quinoloneearboxylic Acids as eell
- adhesion inhibitors.
Tanabe
WO 89/07609, T. J. Lobl, et al.: Deseribes RGD analogs.
WO 92JOO995, July 9, 1991, T. J. Lobl, et al.: Describes eyelic RGD analogs.
WO 93/08823, Nov. 6, 1991, T. C. MeKenzie: Deseribes guanidinoalkanoyl-Gly-
Asp-X analogs.
CA 2087021, Jan 10, 1991, Lobl, T. J., et al: Deseribes eyclic RGD analogs.
WO 92/08464, Nov. 15, 1991, T. C. McKenzie, et al.: Describes.
Telios / La Jolla Cancer Research
US. 4578079, Nov. 22, 1983, E. Ruocl~hti, and M. Pierschbacher: Describes X-
RGD-Y analogs.
US. 4614517, June 17, 1985, E. Ruoslahti, and M. Piersehbacher: Describes X-
RGD-Y analogs.
US. 4792,525, June 17, 1985, E. Ruoslahti, and M. Pierschbacher: Describes X-
RGD-Y analogs.
US 4879237, (Der 90-154405/20) May, 24, 1985, Describes X-RGD-Y analogs.
WO 91/15515, (Der 91-325173/44) Apr. 6, 1990, describes eyelic RGD analogs.
US. 5041380, 1991, E. R~lo~l~hti, and M. Piersehhaeher: Deseribes RGD-X analogs.WO 95/00544 Jan. 5, 1995, Cra;g, W. S., et. al.
Cheng, S.; Craig, W. S.; Mullen, D.; Tschopp, J. F.; Dixon, D.; Piersehbaeher, M.
F., Design and Synthesis of Novel Cyclic RGD-Cont~inin~ Peptides as
Highly Potent and Selective Integrin ocIIb,133 Antagonists, J. Medicin. Chem.,
37, 1, 1994.
c.
- 39 -
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
Collen, D., Lu, H. R.; Stassen, J.-M.; Vreys, I.; Yasuda, T.; Bunting, S.; Gold, H. K.,
A~ olllbotic Effects and Bleeding Time Prolongation with Synthetic
Platelet GPIIbMIIa Inhibitors in Animal Models of Platelet-Mediated
Thrombosis, ~hrombosis and Haemostasis, 71, 95, 1994.
Temple U.
WO 9409036, (Der 94-151248/18), Apr. 28, 1994, Describes ~ in~-gTin peptides.
Terumo KK
JP 6279389, Oct. 4, 1994, Obama, H., et al.: Describes 3-(4-
arnidinophenyloxymethyl)phenylamidopropionic acid analogs (ala Roche I-
35).
Karl Thomae / Boel~ r Ingelheim
EP 0483667, May 6, 1992, E~immPl~bach, F., et al.: Describes amidinobiphenyl-
oxymethyl-2-pyrrolidinone-acetic acid.
EP 0496378, Jan. 22, 1992, Himm~l~bArh, F., et al.: Describes amidinobiphenyl-
aminocarbonylcyclohexylcarboxylic acid anlogs.
EP 0503548, Sep. 16, 1992, Himmelsbach, F., et al.: Describes arnidinophenyl-
pyrrolidinone-phenylpropionic acid analogs.
AU A-86926/91, May 7, 1992, Hirnmelsbach, F. et al.: Describes amidinophenyl
compounds.
EP 0528369, Feb. 24, 1993, Austel, V., et al.: Describes Ami~Tinobiphenyl-
oxymethyl-2-pyrrolidinone-acetic acid.
EP 0537696, Apr. 21, 1993 Linz, G., et al.: Describes A~nidinophenyl-pyri-lA7ine analogs.
DE 4124942, Jan. 28, 1993, Hirnmelsbach, F. et al.: Describes Ami~lin---
triarylproionic acid analogs.
DE 4129603, Mar. 11, 1993, Pieper, H, et Al.: Describes amidinobiphenyl-
I~ç ~iT.~itlA7ole.
-40 -
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/2~748
EP 0547517 A1, (Der 93-198544) ~une 23, 1993, Soyka, R., et al.: Describes pyridyl
compounds.
EP 0567966, Nov. 3, 1993, Himmelsbach, F., et al.: Describes arnidinobiphenyl-
oxymethyl-2-pyrrolidinone-acetic acid.
6 5 EP 0567967, Nov. 3 1993, Weisenberger, J., et al.: Describes amidinobiphenyl-
2 oxymethyl2-pyrrolidinone-acetic acid.
EP 0567968, Nov. 3, 1993, Linz, G., et al.: Describes amidinobiphenyl-lactam-acetic
acid and amidinophenyll~f t~mphenylpropionic acid analogs.
EP 0574808, June 11, 1993, Pieper, H., et al.: Describes amidinobiphenyl-X-acetic
acid ester analogs.
Der 93-406657/51, Austel, V.., et al.: Describes amidinobiphenyl analogs.
EP 587134, (Der 94-085077/11) Mar. 16, 1994, Himmerlsbach, F. D. D., et al.,
Describes amidinophenyltriazolone analogs.
EP 589874, Apr. 6, 1994, Grell, W., et al.
~P534005), DE 4234295, Apr. 14, 1994, Pieper, H., et al., Describes heteroaryl-
azacyclohexylcarboxylic acid analogs.
EP 0592949, Apr. 20, 1994, Pieper, H. D., et al., Describes arnidinophenyl-
4piperi-1in~mi~1o-4-cyclohexylcarboxylic acid analogs.
EP 596326, May, 11, 1994, Maier, R. et al.
DE 4241632, June 15, 1994, Himmelsbach, F., et al., Describes
piperidinophenylamido-phenyl~lvL,ionyl analogs.
EP 0604800 A, Jul. 6, 1994, Himmelsbach, F. et al., Describes
piperidinophenylamido-phenyl~l~nine derivatives.
DE 4302051, (Der 94-235999/29) July, 28, 1994, describes compounds con~inin~ a
2H-pyrazol-5-one.
EP 0608858 A, Aug, 3, 1994, Linz, G. D., et al., Describes amidino-biphenyl
compounds.
DE 4304650, (Der 94-256165/32), Aug, 18, 1994, Austel, V., et al., describes
compounds with a 5,6 template.
- 30 EP 611660, Aug, 24, 1994, Austel, V., et al., Describes tricyclic template.
-41-
CA 02241633 1998-06-26
W O97/24119 PCT~US96~0748
DE 4305388, (Der 94-264904/33), Aug. 25, 1994, I~Timm~l~b~rh~ F., et al.,
Describes 6,6 and 7,6 templates.
(P5D4005), EP 612741, (Der 94-265886/33), Aug. 31, 1994, Himmelsbach, F., et
al., Describes 6,6 and 7,6 templates.
EP 0639575 A, Feb. 22, 1995, Linz, G., et al.: Describes tetrahydrothiazolo-
[5,4,c]pyridine cation replacements.
DE 4324580, Jan. 26, 1995, Linz, G. et al.
EP 0638553, Feb. 15, 1995, Himm~l~b~h, F., et al.
F. rTiumm~olsbach, V. Austel, G. Kruger, H. Pieper, H. Weisenberger, T. H. Muller,
and W. G. Eisert, in XIIth Int. Symp. on Med. Chem. Basel, Book of
Abstracts, 47, 1992.
V. Austel, W. Eisert, F. Himmelsbach, G. Kruger, G. Linz, T. Muller, H. Pieper, and
J. Weisenberger, Natl. Mtg. Amer. Chem. Soc. Book of Abstracts, Denver,
Div. Med. Chem., 1993.
Muller, T. H.; Schurer, H.; Waldmann, L.; Bauer, E.; ~immPlsbach, F.; Binder, K.,
Orally Activity of BIBU 104, a Prodrug of the Non-peptide Fibrinogen
Receptor Antagonist BIBU 52, in Mice and Monkeys, Thromb. Haem., 69,
975, 1993.
Univ. California
WO 94/14848, July, 7, 1994, Zanetti, M. RGD peptides from CDR.
Univ. New York
WO 94/00144, June 29, 1993, Ojima, I. et al.: Describes RGD peptide mllltimtors.
Yeda lRes. and Dev. Co.
WO 93/09795, (Der 93-182236/22), Lido, O. et al.: Describes Gn~ni~inopentanoic
acid analogs.
7PnP(~ -
-42 -
CA 02241633 1998-06-26
W O 97/24119 PCTAUS96/20748
WO 9422834, Oct. 13, 1994, Wayne, M. G., et al. Describes pyridinopiperazino-
phenylcarbonyl-arnino acids.
WO 9422835, Oct. 13, 1994, Wayne, M. G., et al. Describes pyridinopiperidino-
amidophenylacetic acids.
5EP 632016, Jan. 4, 1995, Brewster, A. G.. , et al. Describes
- pyridinopropionylhyd,d~i"ylbenzoyl analogs.
EO 632019, Jan. 4, 1995, Brown, G., Shute, R. E.
EO 632020, ~an. 4, 1995, Brown, G., Shute, R. E.
~n cases wherein the compounds of this invention may have one or more
chiral centers, unless specified, this invention includes each unique nonracemiccompound which may be synthesized and resolved by conventional techniques. In
cases in which compounds have unsaturated carbon-carbon double bonds, both the
cis (Z) and trans (E) isomers are within the scope of this invention. The m~z~nin~ of
15 any substituent at any one occurrence is independent of its m~ning, or any other
substituent's m~ning, at any other occurrence.
Abbreviations and symbols commonly used in the peptide and chemical arts
are used herein to describe the compounds of this invention. In general, the amino
acid abbreviations follow the IUPAC-IUB Joint Commission on Biochemical
20 Nomenclature as described in Eur. J. Biochem., 158, 9 (1984).
Cl 4alkyl as applied herein means an optionally substituted alkyl group of 1
to 4 carbon atoms, and inc.lucles methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl
and t-butyl. Cl 6alkyl additionally includes pentyl, n-pentyl, isopentyl, neopentyl
and hexyl and the simple aliphatic isomers thereof. C0 4alkyl and C0 6alkyl
25 additionally indicates that no alkyl group need be present ~e.g., that a covalent bond
is present).
Any Cl 4alkyl or Cl 6 alkyl, C2 6 alkenyl, C2 6 alkynyl or Cl 6 oxoalkyl may
be optionally substituted with the group RX, which may be on any carbon atom that
results in a stable structure and is available by conventional synthetic techniques.
30 Suitable groups for Rx are Cl 4alkyl, OR, SR, Cl 4alkyl, Cl 4alkylsulfonyl,
-43 -
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
C I 4alkylsulfoxyl, -CN, N(R )2, CH2N(R )2. -N02, -CF3, -C02R -CON(R )2~ -COR,
-NR C(O)R, OH, F, Cl, Br, I, N3, or CF3S(O)~-,wherein r is O to 2.
Ar, or aryl, as applied herein, means phenyl or naphthyl, or phenyl or
naphthyl substituted by one to three subctitllent~ sueh as those defined above for
5 alkyl, espeeially Cl~alkyl, Cl 4alkoxy, Cl 4alkthio, trifluoroalkyl, N3, OH, C02H
F, Cl, Br or I.
Het, or heterocyele, in~lic~tl~.s an optionally ~llb~liLuled five or six membered
monoeyelic ring, or a nine or ten-membered bieyelie ring cf-nt~ining one to three
heteroatoms ehosen from the group of nitrogen, oxygen and sulfur, whieh are stable
10 and available by eonventional ehPmic~l synthesis. Illustrative heterocycles are
benzofuran, ben7imicl~01e, bellzoL)yl~l, benzothiophene, biotin, furan, imicl~ole,
indoline, morpholine, piperidine, ~iL~t;ld~ille, pyrrole, pyrro}idine, pyridine,tetrahydropyridine, pyridine, thiazole, thiophene, quinoline, isoquinoline, and tetra-
and ~lhyd~ quinoline and isoquinoline. Any ~cçc.cihle combination of up to
15 three substit~entc on the Het ring, such as those defined above for alkyl that are
available by ehemieal synthesis and are stable are within the seope of this invention.
C3 7cyeloaL~yl refers to an optionally substituted carbocyelic system of three
to seven earbon atoms, whieh may eontain up to two unsaturated earbon-earbon
bonds. Typieal of C3 7eyeloalkyl are eyelopropyl, cyclobutyl, cyclopentyl,
20 cyelopentenyl, eyelohexyl, eyelohexenyl and eyeloheptyl. Any eombination of up to
three substi~uent.~, such as those defined above for alkyl, on the cycloalkyl ring that
is available by conventional chemieal synthesis and is stable, is within the seope of
this invention.
When Rb and Rc are joined together to form a five- or six-membered
25 aromatie or non-aromatic carbocyclic or heterocyclie ring fused to the ring to whieh
Rb and Rc are attached, the ring formed will generally be a five- or six-membered
heterocycle s~lecte~ from those listed above for Het, or will be a phenyl, eyclohexyl
or cyclopentyl ring. Preferably Rb and Rc will be -D1=D2-D3=D4 wherein Dl - D4
are independently CH, N or C-RX with the proviso that no more than two of D 1 - D4
--44--
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
are N. Most preferably, when Rb and Rc are joined together they form the group
-CH=CH-CH=CH-.
- Certain radical groups are abbreviated herein. t-Bu refers to the tertiary butyl
radical, Boc refers to the t-butyloxycarbonyl radical, Fmoc refers to the
5 fluorenylmethoxycarbonyl radical, Ph refers to the phenyl radical, Cbz refers to the
- benzyloxycarbonyl radical, BrZ refers to the o-bromobenzyloxycarbonyl radical,
ClZ refers to the o-chlorobenzyloxycarbonyl radical, Bzl refers to the benzyl radical,
4-MBzl refers to the 4-methyl benzyl radical, Me refers to methyl, Et refers to ethyl,
Ac refers to acetyl, Alk refers to Cl 4alkyl, Nph refers to 1- or 2-naphthyl and cHex
10 refers to cyclohexyl. Tet refers to 5-tetrazolyl.
Certain reagents are abbreviated herein. DCC refers to
dicyclohexylcarbo-liimi(~, DMAP refers to dimethylaminopyridine, DIEA refers to
diisopropylethyl amine, EDC refers to 1-(3-dimethylaminopropyl)-3-
ethylcarborliimi(le, hydrochloride. HOBt refers to l-hydroxybenzotri~7Ole, THF
15 refers to tetrahydrofuran, DIEA refers to diisopropylethylamine, DME refers to
dimethoxyethane, DMF refers to dimethylform~micle, NBS refers to N-
bromosuccinimide, Pd/C refers to a palladium on carbon catalyst, PPA refers to 1-
propanephosphonic acid cyclic anhydride, DPPA refers to diphenylphosphoryl
azide, BOP refers to benzotriazol-l-yloxy-tris(dimethyl-amino)phosphonium
20 hexafluorophosphate, HF refers to hydrofluoric acid, TEA refers to triethylamine,
TFA refers to trifluoroacetic acid, PCC refers to pyridinium chlorochromate.
Compounds of the formula ~ (V) are prepared, for example, by reacting a
compound of formula (XIX) with a compound of formula (XX), wherein Ll and L2
are groups which may react to fo;m a covalent bond in the moiety W, by methods
25 generally known in the art.
~ ~> L~ > W-A
RC N + Ll-A RC N
(XIX) (XX) (I)
Typical methods include coupling to form amide bonds, nucleophilic
displ~rçm~nt reactions and p~ m catalyzed couplings. For instance, when W
- 45 -
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
contains an ether or amine linkage, the bond may be formed by a displacement
reaction, and one of Ll and L2 will contain an amino or hydroxy group and the other
will contain a displaceable group, such as a chloro, bromo or iodo group. When Wcontains an amide bond, typically one of Ll and L2 will contain an amino group, and
5 the other will contain a carboxylic acid group. In another approach, Ll may be an
aryl or heteroaryl bromide, iodide or trifluoromethylsulfonyloxy derivative and L2
may contain an amino group and the amide linkage may be formed by p~ m-
catalyzed aminocarbonylation with carbon monoxide in a suitable solvent such as
dimethylform~micle or toluene.
It will be apparent that the precise identity of Ll and L2 will be dependent
upon the site of the linkage being }'ormed. General methods for preparing the
linkage -(CHR")~-U-(CHR")s-V- are described, for example, in EP-A 0 372 486 and
EP-A 0 381 033 and LP-A 0 478 363, which are incol~oldt~d herein by reference.
For in~t~nre, if V is CONH, Ll may be -NH2, L2 may be O~I (as in an acid)
15 or Cl (as in an acid chloride), and R6 may be W-(CRr2)q-Z-(CR'Rl0)r~U-(CRI2)
C(O), with any functional groups optionally protected. For example, R6 may be
(benzyloxycarbonyl-amidino)benzoyl- or (N~Boc,NgUan-Tos)arginyl-. When L2 is
OH, a coupling agent is used.
Similarly, if V is NHCO, Ll may be -CO2H or CO-Cl, L2 may be -NH2, and
R6 may be W~(CR12)q~Z(CRIRl~)r-u-(cRl2)s-. For example, R6 may be
(benzyloxycarbonyl-amidino)phenyl, (benzyloxycarbonylamino)methylbenzyl- or 6-
(benzyloxycarbonylamino)hexyl- .
Where V is NHSO2, Ll may be SO2Cl, L2 may be -NH2 and R6 may be as
above. Where V is SO2NH, Ll may be -NH2 and L2 may be SO2CI. Methods to
25 prepare such sulfonyl chlorides are disclosed, for in~t~n~e, in J. Org. ~he~l., 23,
1257 (1958).
If V is CH=CH, Ll may be -CHO, L2 may be CH=P-Ph3 and R6 may be W-
(CR12)q-Z-(CRlRlO)~U-(CRl2)s-- ~l~rnzlt~ly, Ll may be CH=P-Ph3, L2 may be
CHO, e.g., R6 may be W~(CR~2)q~Z~(CRlRl0)r-u-(cRl2)s-l-cHo.
-~6 -
~ .
CA 02241633 1998-06-26
W O97/24119 PCTrUS96/20748
Where V is CH2CH2 may be obtained by reduction of a suitably protected
compound wherein V is CH=C~I.
Where V is CH20, CH2N or C--C, L1 may be -OH, -NH or
-C~ C H,respectively; L2 may be -Br; and R6 may be
W~(CR~2)q~Z(CR~RI~)r~u-(cR~2)s-~ For example, R6 may be
(benzyloxycarbonylarnino)-methylbenzyl- or 2-(N-benzyl-4-piperidinyl)-ethyl.
Similarly where U or V is OCH2, NR'CH2 or C~ C, Ll may be -CH2Br and L2 may
be -OH, -NH or -C= C H, le~.~e.;lively. Alternately, when U or V is C--C, Ll maybe Br, I or CF3SO3, L2 may be C--C H and the coupling may be catalyzed by
p~ m and a base.
Compounds wherein V is CHOHCH2 may be prepared from a suitably
protected compound where V is CH=CH by the procedure disclosed in J. Org.
Chem., 54, 1354 (1989).
Compounds wherein V is CH2CHOH may be obtained from a suitably
protected compound where V is CH=CH by hydroboration and basic oxidation as
disclosed in Tet. Lett., 31, 231 (1990).
The core 6-7 fused ring system is prepared of formula (VI) by methods well
known in the art, e.g., Hynes, et al., J. Het. Chem., 1988, 25, 1173; Muller, et al.,
Helv. Chim. Acta., 1982, 65, 2118; Mori, et al., Heterocycles, 1981, 16, 1491.
Similarly, methods for preparing benzazepines, 1,4-benzofhi~7.e.pin~ s, 1,4-
benzoxazepines and 1,4-benzodiazepines are known and are disclosed, for in.~t~n~e,
in Bondinell, et al., International Patent Application WO 93/00095.
The sl~htom~s which follow detail the ~l~dLion of the compounds of the
instant invention.
- 47 -
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
S. ' - ~ I
CH30~ ~ CH~0~[~
o CO2Et
CH30~ H0 ~
3 C02Et 4 C02Et
TfO ~ HO C~
C02Et C02Et
g_--~ N r ~ ~ h
a) EtOAc/LiN(TMS)2, THF; b) Et3SiH, BF3 ~ OEtz, CH2Cl2; c) H2~ 10% Pd/C, EtOH;
d) EtSH, AICl3, CH2Cl2; e) Tf20, 2,6-h-ti~lin~., CH~Cl2; f) CO, KOAc, Pd(OAc)2,
5 dppf, DMSO; g) 2-(methylaminomethyl)l~e~ ole dihydrochloride, EDC,
EIOBt ~ H20, (i-Pr)2NEt, CH~CN; h) 1.0 N NaOH, EtOH.
- 48 -
CA 0224l633 l998-06-26
W O 97/24119 PCTAUS96/20748
An ~p,op.iately substituted deoxybenzoin, such as 2-(4-methoxyphenyl)-l-
phenylethanone (Chem. Ber. 1958, 91, 755-759), is reacted in an aldol-type reaction
with the enolate of ethyl acetate, which can be generated from ethyl acetate on
exposure to an a~pr~p.iate amide base, for in~t~nre lithium diisopropylamide (LDA)
5 or lithium bis(trimethylsilyl)amide (LiN(TMS)2), to afford I-2. Fre~uently, THF is
- the solvent of choice for an aldol reaction, although THF in the presence of various
additives, for in.~t~nre HMPA or TMEDA, is often used. Reaction of I-2 with
triethylsilane (Et3SiH) in the presence of boron trifluoride etherate (BF3 ~ OEt2)
according to the general protocol of Orphanopoulos and Smonu (Synth. Commun.
10 1988, 833) for the reduction of tertiary benzylic alcohols affords I-3, together with
the olefinic product derived from ~ limin:~tion of the alcohol. The olefinic product
can be conveniently converted to I-3 by hydrogenation over a F~ m catalyst,
such as palladium metal on activated carbon (Pd/C), in an ~pl~pliate inert solvent,
for in~t:~nre mr.th~nol, ethanol, or ethyl acetate. Removal of the methyl ether of I-3
15 to give I-4 can be accomplished by reaction with eth~nethiol (EtSH) in the presence
of a Lewis acid catalyst, preferably anhydrous ~lnminllm trichloride (AlCl3), in an
inert solvent, for in~t~nce CH2Cl2. Other useful methods for removal of a methylether are described in Greene, "Protective Groups in Organic Synthesis" (published
by Wiley-Interscience). Alcohol I-4 is converted to its trifluoromethanesulfonate
20 ester I-5 by reaction with trifluoromethanesulfonic anhydride (Tf2O) in the presence
of a suitable non-nucleophilic amine base, such as 2,6-lutidine, in an inert solvent,
generally CH2Cl2. I-~ reacts with carbon monoxide (CO) in the presence of
potassium acetate, 1, l '-bis(diphenylphosphino)ferrocene (dppf), and a F~ lillmcatalyst, for in~t~nce palladium acetate (Pd(OAc)2), in a suitable solvent, preferably
25 DMSO, according to the general method described by Cacchi and Lupi (Tet. Lett.
1992, 33, 3939) for the carboxylation of aryl trifluorometh~nt-sulfonates. The
resulting benzoic acid derivative I-6 is converted to an activated form of the
carboxylic acid using, for example, EDC and HOBt, or SOCl2, and the activated
form is subsequently reacted with an ~pl~,p.iate amine, for in~t~nre 2-
30 (methylaminomethyl)methylben7.imi-l~7.ole dihydrochlnri~le, in a suitable solvent
- 49 -
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
such as DMF, CH~Cl2, or CH3CN, to afford I-7. Df~pçn-lin~ on whether acid
neutralization is required, an added base, such as diisopropylethylamine ((i-Pr)2NEt)
or pyridine, may be used. Many additional methods for converting a carboxylic acid
to an amide are known, and can be found in standard reference books, such as
5 "Compendium of Organic Synthetic Methods", Vol. I - VI (published by Wiley-
Interscience), or Bodansky, "The Practice of Peptide Synthesis" (published by
Springer-Verlag). The ethyl ester of I-7 is hydrolyzed using aqueous base, for
example, LiOH in aqueous THF or NaOH in aqueous methanol or ethanol, and the
intermediate carboxylate salt is acidified with a suitable acid, for instance TFA or
10 HCl, to afford the carboxylic acid I-8. ~lt~rn~tively, the interm~ t.o carboxylate
salt can be isolated, if desired, or a carboxylate salt of the free carboxylic acid can be
prepared by methods well-known to those of skill in the art.
- 50 -
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
Scheme II
~ HO2C~ t-BUO2C~0~ t-Bu02C~C02CH3
~- 1 23
t-BuO2C ~ t-BuO2C ~ e
4 CO2CH3 5 CO2H
t-Bu02C ~ t-Bu02C~,CH3)
CON(CH3)2 CO2Et
H~2C~ CON(CH3)2 ~ N~N~ "CH3)2
~CO2Et (~NH CH3 CO2Et
8 9
N~ N J~ CH3)2
~ NH CH3 CO2H
a) isobutylene, TfOH, CH2C12; b) methyl acrylate, Pd(OAc)2, P(tol)3, (i-Pr)2NBt,propionitrile; c) H2, 10% Pd/C, MeOH, EtOAc; d) 1.0 N LiOH, THF, H2O; e)
dimethylamine hydrochloride, EDC, HOBt ~ H20, (i-Pr)2NEt, CH3CN; f) LiN(TMS)2,
- THF, then BrCH2CO2Et; g) TFA, CH2Cl2; h) 2-(methylaminomethyl)ben7.imi(1~7.ole
dihydrochloride, EDC, HOBt H20, (i-Pr),NEit, CH3CN; i) 1.0 N LiOH, THF, H20.
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96t20748
Commercially available 4-bromobenzoic acid (II-l) is converted to the tert-
butyl ester II-2 by reaction with isobutylene in the presence of a catalytic arnount of
an acid, such as trifluorom~thAn.osulfonic acid (TfOH) or sulfuric acid, in _n inert
solvent, generally CH2Cl2 or diethyl ether. Alternative methods for the formation of
5 tert-butyl esters are described in Greene, "Protective Groups in Organic Synthesis"
(published by Wiley-Interscience). Other esters can be employed, as long as theyare compatible with the subsequent chemistry and can be removed selectively whendesired. A Heck-type reaction between II-2 and methyl acrylate affords II-3. Thegeneral conditions for the Heck reaction have been reviewed by Heck (Org.
lO Reactions 1982, 27, 345). For ~I-2, reaction with methyl acrylate in the presence of
palladium ~II) acetate (Pd(OAc)2) and tri-ortho-tolylphosphine (P(tol)3) in an inert
solvent, such as CH3CN, propionitrile, or toluene, in the presence of an ~p.o~,iate
acid scavenger, such as diisopropylethyla~lnine ((i-Pr)2NEt), affords II-3. Reduction
of the a,l3-unsaturated ester of II-3 to afford the saturated compound II-4 occurs
15 under standard hydrogenation conditions, for instance reaction with hydrogen gas in
the presence of a suitable catalyst, preferably palladium metal on activated carbon
(Pd/C), in an inert solvent, generally m(~thAn~l, ethanol, ethyl acetate, or mixtures
thereof. The methyl ester of II-4 is hydrolyzed using aqueous base, for example,LiOH in aqueous THF or NaOH in aqueous methanol or eth_nol, and the
20 interm~-liAte carboxylate salt is acidified with a suitable acid, for in~t~n~e T~A or
HCl, to afford the carboxylic acid II-5. Tnis is converted to an activated form of the
carboxylic acid using, for example, EDC and HOBt, or SOC12, and the activated
form is subsequently reacted with an a~3p~ iate a~nine, for in~tAnce dimethylamine
hydrochloride, in a suitable solvent such as DMF, CH2CI~, or CH3CN, to afford II-6.
25 Depending on whether acid neutralization is required, an added base, such as
diisopropylethylamine ((i-Pr)2NEt) or pyridine, may be used. Many additional
methods for converting a carboxylic acid to an amide are known, and can be foundin standard reference books, such as "Compendium of Organic Synthetic Methods",
Vol. I - VI (published by Wiley-Interscience), or Bodansky, "The Practice of Peptide
30 Synthesis" (published by Springer-Verlag). Reaction of II-6 with an amide base, for
CA 0224l633 l998-06-26
W O 97/24119 PCT~US96/20748
in.~t:~nCe~ lithium bis(trimethylsilyl)amide (3_iN(TMS)2), sodium
bis(trimethylsilyl)amide (NaN(TMS)2), pot~c~ m bis(trimethylsilyl)amide
, (KN(TMS)2), or lithium diisopropylamide (LDA), in an inert solvent, generally THF
or ethylene glycol dimethyl ether (DME), affords an interm~ te amide enolate.
5 This is generally not isolated, but rather is reacted in situ with an electrophile, for
example ethyl bromoacetate, to afford the alkylated product II-7. Various additives
known to those of skill in the art, for in~t~n~e HMPA, teLlalllelhylethylen~ mine
(TMEDA), or 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU), can be
used to improve the eff1ciency of the alkylation reaction. ). The tert-butyl ester
10 group of II-7 is removed under acidic conditions, generally with TFA or HCl, in an
inert solvent, usually CH2Cl2, 1~4-dioxane, or l~ Lules thereof, to afford the acid II-
8. Other useful methods for the removal of tert-butyl esters are described in Greene,
"Protective Groups in Organic Synthesis" (published by Wiley-Interscience). II-8 is
converted to II-9 under the general conditions described in Scheme I for the
conversion of I-6 to I-7, and II-9 is converted to II-10 by the general conditions
described in Scheme I for the conversion of I-7 to I-8.
CA 02241633 1998-06-26
W O 97/24119 PCTAJS96/20748
Schem~. m
~_NH a~_NH O COzCH3
~N CO H
0 11
~ NH N ~ ~ COzEt
0 11
N ~ N ~ ~ H CO2H
g_NH O
a) 3-carbomethoxypropionyl chloride, (i-Pr)2NEt, CH2Cl2; b) 1.0 N NaOH, MeOH;
c) ethyl 3-amino-4-pentynoate, EDC, HOBt ~ H20, (i-Pr)2NEt, CH~CN, DMF; d) 1.0
N LiOH, THF, H20, CH3CN.
Readily available 2-(2-aminoethyl)be~,~.;.l.itl~7.ole is reacted with 3-
1() carbomethoxy~lopionyl chloride in the presence of an al,~r.,l,liate acid scavenger,
such as triethylamine, diisopropylethylamine, or pyridine, in a neutral solvent,generally CH2CI2, to afford III-2. The methyl ester of III-2 can be hydrolyzed to
afford III-3 under the general conditions described in Scheme 1 for the conversion
- 54 -
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
of I-7 to I-8. Alternatively, III-l can be reacted with succinic anhydride in the
presence of an a~plopliate base, such as triethylamine, diisopropylethylamine, or
,~ pyridine, in a neutral solvent, generally CH2Cl2, to afford III-3 directly. III-3 is
converted to III-4 by reaction with the known ethyl 3-amino4-pentynoate
5 (W093/07867) under the general conditions described in Scheme I for the
conversion of I-6 to I-7. Hydrolysis of the ethyl ester of III-4 to provide III-5 is
accomplished according to the general conditions conditions described in ~cheme I
for the conversion of I-7 to I-8.
CA 02241633 1998-06-26
W O 97~4119 PCTAJS96/20748
SchemP IV
NH2 N~--C02CH3
~NH2 a,b ~--NH c
N1 2
N ~ C02H
~NH
Il . 0 11
H2N ~ CO2Et d ~ Boc'N ~ H''''~,'C02Et e ~
0 11
H2N~HN~CO2E~ HX f
0 11
N ~ ~ N ~J'--H CO2Et
~NH O
~ Il
--N~ H
N 8
a) methyl 4-(chloroformyl)butyrate, Et3N, THF; b) AcOH; c) 1.0 N NaOH, MeOH;
d) Boc-Gly, EDC, H013t ~ H,O, (i-Pr)~NEt, CH3CN; e~ TFA, CH,CI2; f) 3, EDC,
HOBt ~ H20, (i-Pr)2NEt, CH3CN; g) 1.0 N LiOH, THF, H20.
The synthesis of IV-8 is acc~-mpli~h~l by the reaction of two separately
prepared interm~ tes, IV-3 and IV-6. The preparation of intP.rmP.rli~h? IV-3 begins
- ~6 -
CA 02241633 1998-06-26
W O 97/24119 PCTnUS96/20748
with colllll,er-;ially available 2,3-diaminopyridine (IV-l). Accordinf to this sch~mP,
IV-1 is acylated with methyl 4-(choloroformyl)butyrate in the presence of a suitable
acid scavenger, such as triethylamine, diisoL,loL\ylethylarnine, or pyridine, in a
neutral solvent, generally CH2Cl2 or THF, to afford an interm~ t~ monoacylated
- S derivative. This delivativ~ is then cyclized, for example using r~nu~ing acetic acid,
- to afford IV-2. Hydrolysis of the methyl ester of IV-2 under the general conditions
described in Scheme 1 for the conversion of I-7 to I-8 affords IV-3. The preparation
of the intermediate IV-6 begins with the coupling of the known ethyl 3-amino-4-
pentynoate (W093/07867) with culll.llelcially available tert-butoxycarbonylglycine
(Boc-Gly) under standard peptide bond forming conditions described in the
previously referenced Bodansky publication, and in Scheme 1 for the conve,~ion of
I-6 to I-7. The product of this reaction, IV-5, is deprotected to IV-6 under acidic
conditions which are known to effect removal of a Boc protecting group. Such
conditions are described in the previously referenced Bodansky and Greene
publications. The two interm~ t~s IV-3 and IV-6 are coupled under standard
peptide coupling conditions as previously described to afford IV-7, which is
hydrolyzed to IV-8 according to the general methods described in Scheme 1 for the
conversion of I-7 to I-8.
- 57 -
CA 02241633 1998-06-26
W O97/24119 PCT~US96/20748
Schen~ V
,CH3 ,CH3
H~ N~
CO2CH3 Boc CO2CH3
HO2C~I N'r'~
3 g_NH 4
,~ N ~--~--SnBu3
~=~ NH 5
~O2CH3
,CH3
7 CO2H
S a) (Boc)20, DMAP, CH3CN; b) isobutylchloroformate, Et3N, THF, then 1,2- phenylen~ mine, then AcOH; c) (n-Bu3Sn)2, (PPh3)2PdCl2, DMF; d) CuI,
(PPh3)2PdCI2, DMF; e) 4 M HCI/dioxane; f) 1.0 N NaOH, MeOH.
- 58 -
CA 02241633 1998-06-26
W O 97/24119 PCTAUS96/20748
The synthesis of V-7 is accomplished by the reaction of two separately
prepared interme~ tt~s, V-2 and V-5. V-2 is conveniently prepared by reaction ofthe readily available V-l with di-tert-butyl dicarbonate ((Boc)2O) in the presence of
an acylation catalyst, preferably 4-dimethylaminopyridine (DMAP) or 4-
- 5 pyrrolidinopyridine, in a neutral solvent, for example CE~3CN, THF, or CH2Cl2. The
preparation of interm~ te V-5 begins with colll,ne,l~;ially available 3-iodobenzoic
acid (V-3), which is converted to the ben7imi~1~7- 1e derivative V-4. According to
this scheme, V-3 is reacted with isobutyl chloroformate in the presence of a suitable
amine base, such as triethylamine, diisopropylethylamine, or 4-methylmorpholine,in a neutral solvent, generally CH2Cl2 or THF, to afford an interm~ te mixed
anhydride delivalivt;. Without isolation, this derivative is reacted with an
a~ pliate pheny!çne~ mine to produce a mono-N-acylated phenylen~ mine
inferm/ ~ te. This int~ rm~ t~ is then cyclized to V-4 using acetic acid. Reaction
of V-4 with bis(tributyltin) to produce V-5 occurs under palladium catalysis using,
for example bis(triphenylphosphine)p~ m (II) chloride ((PPh3)2PdCl2), in an
inert solvent, usually DMF. Stille-type coupling of V-2 with V-5 to afford V-6 is
m~ ted by a p~ m catalyst, for instance bis(triphenylphosphine)p~ lm (II)
chloride ((PPh3)2PdCl2), in the presence of copper (I) iodide (CuI), in a suitable
neutral solvent, generally DMF. To obtain V-7, the protecting groups of V-6 are
removed by well-established methods known to those of skill in the art and
described in the previously cited Greene publication. Thus, the Boc protecting
group is removed under acidic conditions, such as 4 M HCl in dioxane or TFA in
CH2Cl2, and the methyl ester is hydrolyzed as generally described in Scheme 1 for
the conversion of I-7 to I-8.
59
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
Scheme VI
CH, HO~,
Boc COzCH3 Boc C02CH3
,CH3
HO ~N
Boc CO2CH3
HO2C ~ = O
N~
Boc CO2CH3
,CH3
Boc CO2CH3
6 CO2H
S a) 3-butyne-1-ol, (PPh3)2PdCl2, PPh3, CuI, Et3N; b) H2, 10% Pd/C, EtOH; c) 2,2,6,6-
tetramethyl-oxopiperitlinil-m chloride, CH2Cl2, then NaCl02, Na2HPO37 2-rnethyl-2-
butene, H20; d) isobutyl chlc: lvfo~ ate, Et3N, then 1 ,2-phenylene~ mint-" thenAcOH; e) 1.0 N LiOH, THF, H20; f~ TFA, CH2Cl2.
- 60 -
CA 0224l633 l998-06-26
W O 97/24119 PCTrUS96/20748
Compound VI-1, the ~ al~lion of which is described in Scheme V, is
reacted with 3-butyne-1-ol in the presence of a catalytic amount of a p~ flinm salt,
usually bis(triphenylphosphine)palladium (II) chloride ((PPh3)2PdCl~), together with
a catalytic amount of copper (I) iodide (CuI), in an amine solvent, such as
S triethylamine (Et3N), to afford VI-2. A phosphine ligand, such as
triphenylphosphine (PPh3), can be added to improve the efficiency of the reaction.
Reduction of the acetylenic unit of VI-2 is accomplished under standard
hydrogenation conditions which are well-known to those of skill in the art. The
rçs-llting compound, VI-3, is oxididized to the corresponding carboxylic acid VI-4,
by the two-step method described by Wovkulich (J. Org. Chem. 1993, 58, 832-839).Many alternative m~thofl~ for the oxidation of a primary alcohol to the
corresponding carboxylic acid have been described, and can be found in such
reference volumes as "Compendium of Organic Synthetic Methods" (published by
Wiley-Interscience). Conversion of the carboxylic acid of VI-4 to the
ben7imid:~701e derivative VI-5 follows the procedures described in the above
Schemes. The methyl ester of VI-5 is removed as described in the above Schemes,
and the Boc protecting group is removed under acidic conditions, such as 4 M HClin dioxane or TFA in CH2Cl2, to afford VI-6.
- 61 -
CA 0224l633 l998-06-26
W O 97/24119 PCTrUS96/20748
~r.hPme VII
HOzC--~ ~ N ~ c
~=~ NH
N ~ ~ N ~ ~
NTs ~ NTs
\~/ 3 \~/ 4
N ~ NOH ~ ~_< N ~NOH
~=~ NTs ~ NTs 6
N--O N--O
N ~CO2tBu h N~co2H
NTs ~ NTs
~;~N~CO2Et
~ CO2H
a) l,2-pheny1enP~ mine, DCC, DMF, CH2Cl2; b) AcOH, THF; c) TsCl, NaH, THF;
S d) 03, CH2Cl2, MeOH, then DMS; e) NH20H ~ HCl, NaOAc, MeOH; f) NCS, DMF;
g) tert-butyl 3-butenoate, Et3N; h) 4 M HCI/dioxane, CH2Cl2; i) ethyl 3-
aminobutyrate, EDC, HOBt H2O, (i-Pr)2NEt, CH3CN; j) l.0 N LiOH, THF, H2O.
- 62 -
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
Commercially available 4-pentenoic acid (VII-l) is converted to the
bPn7imi~1~701e derivative VII -2 using to the general procedures described
,~ previously. Protection of one of the nitrogen atoms of the ben7.imid~7ole moiety in
VII -2 can be accomplished by reaction with a sulfonyl chloride, for in~t~n~e p-5 toluenesulfonyl chloride, in the presence of a suitable base, generally sodium- hydride or an aqueous alkali metal hydroxide, in an inert solvent, preferably THF, to
afford VII -3. ~Itern~tive protecting groups known to those of skill in the art may
be used, as long as they are co~ ible with the subsequent chemistry and can be
removed when desired. Such protecting groups are described in Greene, "Protective
Groups in Organic Synthesis" (published by Wiley-Interscience). Oxidative
cleavage of the olefin of VII -3 to afford the aldehyde VII -4 can be conveniently
accomplished by ozonolysis in an inert solvent, usually CH2Cl2 or a ll~ Ul'~ of
CH2Cl2 and MeOH, followed by in-situ reduction of the ozonide with a suitable
reducing agent, generally dimethylsulfide (DMS) or triphenylphosphine. ~ltr.rn~tive
mPthocl~ for oxidative cleavage, such as the Lemieux-Johnson reaction (J. Org.
Chem. 1956, 21, 478) can also be used. The aldehyde is converted to the aldoximeVII -5 by standard procedures known to those of skill in the art, and this aldoxime is
oxi~li7ed to the oximinoyl chloride derivative VII -6 by the methods described in
WO 95tl4682 and WO 95/14683. Reaction of VII -6 with an olefin, such as tert-
butyl 3-butenoate (Tet. Lett. 198~;, 26, 381 -384), in the presence of a suitable base,
for in.~t~n~e triethylamine or diusopropylethylarnine, in an inert solvent such as
benzene or toluene, according to the protocol described in WO 95/14682 and WO
95/14683, gives the cycloadduct I-7. The tert-butyl ester of VII -7 is removed under
standard acidic conditions, generally TFA in CH2Cl2 or HCl in dioxane, to give the
carboxylic acid VII -8. The carboxylic acid is activated using, for example, EDCand HOBt, or SOCl2, and the activated form is subsequently reacted with an
a~prupliate amine, for in.~tAn~e a suitable derivative of ~ nin~, in a neutral
solvent, such as DMF, CH2Cl2, or CH3CN, to afford VII -9. Depending on whether
acid neutralization is required, an added base, such as diisopropylethylamine ((i-
Pr)2NEt) or pyridine, may be used. Many additional methods for converting a
- 63 -
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
carboxylic acid to an amide are known, and can be found in standard reference
books, such as "Compendium of Organic Synthetic Methods", Vol. I - VI (publishedby Wiley-Interscience), or Bodansky, "The Practice of Peptide Synthesis" (published
by Springer-Verlag). Derivatives of ,13--alanine are readily available in either racemic
5 or optically pure form by a variety of methods known to those of skill in the art. A
reprçsent~tive method is described in WO 93/07867. The ethyl ester and sulfonyl
protecting groups of VII -9 are removed using aqueous base, for example, LiOH inaqueous THF or NaOH in aqueous methanol or ethanol. The interm~ tf .
carboxylate salt is acidified with a suitable acid, for in.ct~nc~.e TFA or HCl, to afford
10 the carboxylic acid VII -10. Alternatively, the int~ te~ carboxylate salt can be
isolated, if desired, or a carboxylate salt of the free carboxylic acid can be prepared
by methods well-known to those of skill in the art.
-64-
CA 02241633 1998-06-26
W O 97/24119 PCTAUS96/20748
Scheme vm
I'IH, I ~N ~0
~CO2Bn ~ ~CO2Bn
~ CO2Bn
,N ~o
5 CH3
O CO2Bn
CH~f ~C~, ~
O CO2H
~N ~ N ~f ~
S a) COCl2 in toluene, Na2CO3, H20; b) ,B-alanine benzyl ester tosylate, DMAP,
- pyridine; c) CH3I, 2,6-lllti~1inç, DMF; d) BrCH2COBr, Et3N, CH2Cl2; e) NaH, DMF,
f) CO, (Ph3P)2PdCl2, DIEA, 2-(methylaminomethyl)ben7.imi~1~701e dihydrochloride,NMP; g) H2, Pd/C, EtOH.
-65-
CA 02241633 1998-06-26
W O97/24119 PCTrUS96/20748
Following the procedures of US 5403836 and WO 9504057, except starting
from 2-amino-4-iodobenzoic acid (VIII-1) rather than 2-amino-5-iodobenzoic acid,compound VIII -5 is prepared. Reaction of VIII -5 with an ~plopliate amine, for
inct~nre 2-(methyl~minom~ thyl)benzimidazole, in a carbon monoxide atmosphere,
5 in the presence of a p~ rlil~m catalyst, preferably (Ph3P)2PdCl2, in an inert solvent,
optimally 1-methyl-2-pyrrolidinone (NMP) gives the amide VIII -6. Depending on
whether acid neutralization is required, an added base, such as
diisopropylethylamine ~DIEA) or pyridine, may be used. The benzyl ester of VIII -6
is removed to afford VIII -7 under standard hydrogenolysis conditions well-known10 to those of skill in the art. ~lt~rn:~tively, the benzyl ester can be saponified using
aqueous base, for example, LiOH in aqueous THF, or NaOH in aqueous methanol or
ethanol. The intern~ te carboxylate salt is acidified with a suitable acid, for
instance TFA or HCl, to afford the carboxylic acid. If desired, the intern~ t~-
carboxylate salt of VIII -7 can be isolated, or a suitable salt of the carboxylic acid
15 can be prepared by methods well-known to those of skill in the art.
-6~-
CA 02241633 1998-06-26
W O 97/24119 PCTAJS96120748
Scheme TX
O O
,~~ a ~ ~CO2Et
~~CO2Et ~[~CO2Et
3 4
~CO2Et ~ ~CO2Et
SMe ¦ N
H3C 6
OCO2H
7 ~
S a) ,B-alanine ethyl ester hydrochloride, DMAP, pyridine; b) BrCH2COBr, Et3N,
CH2Cl2; c) NaH, DMF; d) Lawesson's reagent, THF, 50~ C; e) CH3I, (n-Bu)4NHSO4,
NaOH, CH2Cll, H20; f~ ~loL)al~ylalI~ine, pyridine HCl, toluene; g) CO,
(Ph3P)lPdCl2, DIEA, 2-(methylaminomethyl)benzimidazole dihydrochloride, NMP;
h) LiOH, THF, H20.
-6~ -
CA 02241633 1998-06-26
W O 97124119 PCT~US96/20748
Following the procedures of US 5403836 and WO 9504057, except starting
from 4-iodoisatoic anhydride (IX-1, see Scheme I) rather than 5-iodoisatoic
anhydride, compound IX-6 is prepared. IX-6 is converted to IX-7 following the
procedures described in Scheme XIII for the conversion of XIII -5 to XIII -7.
-68-
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
SchP,rnP. X
- BocN ~
~, N b
02H l~N~CO2Et
HN~
~, N ~
~N~CO2Et
~
H OH H Cl
4 5
~N
H N ~ e
N ~
N ~ CO2Et
~N
H N
l~, N ~
7 ~ N ~ C02H
.,
S a) l-Boc-~i~e,azille, NaBH3CN, HCl, MeOH; b) 4 M HCl/dioxane, CH2Cl2; e)
SOCl2, CH2Cl2; d) 3, DIEA, DMF; e) 1.0 N NaOH, MeOH.
-69 -
CA 02241633 1998-06-26
W O97/24119 PCTrUS96/20748
Reductive amination of the readily available 1-(ethoxycarbonylmethyl)~
piperidone (~-1, EPA 0 542 363 A2) with commercially available l-Boc-pi~e~ le
and an ~ p.iate reducing agent, preferably sodium cyanoborohydride, affords
amine X-2. The reaction is generally conducted under acidic catalysis, usually with
S HCl, in a hydroxylic solvent, for in~tzln~e methanol or ethanol. The Boc protecting
group is removed under acidic conditions, preferably HCI/dioxane or TFA, in a
suitable solvent, such as CH2Cl2, to the give amine X-3. This reacts with 2-(2-
chloroethyl)ben~illlida_ole (X-~;) in the presence of an ~ liate acid scavenger,
for instance diisopropylethylamine (DIEA), in a polar solvent, preferably DMF, to
give the coupled product X-6. 2-(2-Chloroethyl)bt~.n7;n.icl~7Qle can be preparedfrom 2-(2-hydroxyethyl)b~n7imid~7- 1e by reaction with a suitable halogenating
reagent, such as thionyl chloride, or carbon tetrachloride in the presence of
triphenylphosphine, in an inert solvent, for example CH2Cl2. The ethyl ester of X-6
is removed using aqueous base, for example, LiOH in aqueous THF or NaOH in
15 aqueous methanol or ethanol. The int~rm~ te~ carboxylate salt is acidified with a
suitable acid, for in.stzlnce TFA or HCl, to afford the carboxylic acid X-7.
.~lt~rn~tively, the interTn~ te~ carboxylate salt can be isolated, if desired, or a
carboxylate salt of the free carboxylic acid can be prepared by methods well-known
to those of skill in the art.
-7~ -
CA 02241633 1998-06-26
W O 97124119 PCTAUS96/20748
Sch~me XI
HN~
~, N ~f ~
~C02tBu
OH
N~
NH ~N~
2 CO2tBu
OH
--N
NH ~N~
3 ~,CO2H
OH
S a) 2-(3-bromopropyl)bç~7.imi~ 7.ole, DIEA, DMF, b) 4 M HClldioxane, CH2CI2.
The readily available piperazine derivative Xll-l (EPA 0 537 980 Al) is
reacted with the readily available 2-(3-bromopropyl)benzimidazole (J. Org. Chem.1962, 27, 2165) in the presence of an a~ ,pliate acid scavenger, for instance
10 diisopropylethylamine (DIEA), in a polar solvent, preferably DMF, to give tnecoupled product XI-2. The tert-butyl ester protecting group is removed under
standard acidic conditions, preferably HCl/dioxane or TFA, in a suitable solvent,
such as CH2Cl2, to the carboxylic acid XI-3. If desired an a~r~,l)liate salt of the
carboxylic acid can be prepared by methods well-known to those of skill in the art.
,.
~ -7~-
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96120748
Sch~.mP.XTT
o ~ N N ~ "" ~ OC 3
b,c ~ ~N~ NH~~O-Bn
O O O
NH~N~",~NH ~OH
O O O
S a) 2-(benzirnidazolyl)propionic acid, BOP-Cl, NMM, CH2Cl2; b) LiOH, THF, H20;
c) benzyl ,13-~l~nin~tP, EDC, HOBt H20, NMM, CH2Cl2; d) H2, 10% Pd/C, AcOH,
THF, H2O.
The procedures of Beavers et. al., WO 95/25091, Exam~?le 1, were followed
to give XII-4, except (2-b~n7imicl~701yl)propionic acid was substituted for Na-Boc-
D-lys(Cbz)-OH.
CA 0224l633 l998-06-26
W O97/24119 PCTrUS96/20748
Scheme XIII
~I O
Br ~ CO2CH3 N =< C~2CHs
~3~NH
o
~ C~2H
=~N--W c
~NH
=~ N~ ~ C02CH3
~NH 4
=~ N~ N ~ CO2H
,~NH 5
a) 2-(arninomethyl)ben7.tmicl:~7.Qle, Et3N, benzene; b) 1.0 N LiOH, MeOH, H20; c)
,13-alanine ethyl ester, BOP, Et3N, CH3CN.
-73-
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
A suitably functil-n~li7P.d arnine, such as 2-(aminomethyl)ben7imi(l;~7ole~ is
reacted with dimethyl 4-bromomethylbenzene-1,3-dicarboxylate (XIII-l;
.y~ esi~ as in EP 0540334Al) under the general conditions described for the
~,c~dLion of l-H-isoindole-5-carb~-x~mitl~7 2,3-dihydro-N-(2-carboxy-ethyl)-2-[2-
S (piperidinyl)ethyl]-3-oxo (preparation 1-12, EPA 0 540 334 Al), to afford XIII -2.
The methyl ester of XIII -2 is hydrolyzed using aqueous base, for example, LiOH in
aqueous THF or NaOH in aqueous methanol or ethanol, and the int.orm~ t~
carboxylate salt is ~ci~lifi~.-l with a suitable acid, for instance TFA or HCl, to afford
the carboxylic acid XIII -3. The carboxylic acid of XIII -3 is converted to an
activated form of the carboxylic acid using, for example, EDC and HOBt, SOC12, or
BOP reagent, and the activated form is subsequently reacted with an a~ iate
amine, for in~t:~nre ,B-alanine ethyl ester, in a suitable solvent such as DMF, CH2Cl2,
or CH3CN, to afford XIII -4. Depending on whether acid neutralization is required,
an added base, such as diisopropylethylamine ((i-Pr)2NEt) or pyridine, may be used.
Many additional methods for converting a carboxylic acid to an amide are known,
~ and can be found in standard reference books, such as "Compendium of Organic
Synthetic Methods", Vol. I - VI (published by Wiley-Interscience), or Bodansky,
"The Practice of Peptide Synthesis" ~published by Springer-Verlag). Ester
hydrolysis as described above for the conversion of XIII -2 to XIII -3 then affords
XIII -S. Alternatively, the int~rm.o~ii~tf~. carboxylate salt of XIII -~ can be isolated, if
desired, or a carboxylate salt of the free carboxylic acid can be prepared by methods
well-known to those of skill in the art.
-74-
CA 0224l633 l998-06-26
PCTrUS96/20748
W O 97/2~119
Scheme XIV
NH~ ~OH N' ~3~OH
'~3'o CO~BnCH~'O CO2Bn
ICH3 0
d N ~N~
~NH O O
\G/ 5
CO2Bn
N ~ ICH~3~
~q~NH O O
\~/ 6 CO2H
a) (Boc)20, NaOH, 1,4-dioxane, HlO; b) BrCH2COlBn, K2C03, acetone; c) 4 M
HCI/dioxane; d) 2-(ben7.imi~1~7.olyl)acetic acid, EDC, DIEA, DMF; e) H2, 5 % Pd/C,
MeOH.
XIV-1 is treated with di-tert-butyl dicarbonate and sodium hydroxide in
10 aqueous dioxane to afford XIV -2, which is alkylated on oxygen with benzyl
- bromo~f.et~te and potassium carbonate in acetone to give XIV -3. The Boc group in
-7~-
CA 02241633 1998-06-26
W O 97124119 PCTAUS96/20748
XIV -3 is removed with hydrogen chloride in dioxane, and the rçsllltin~ XIV -4 is
acylated on nitrogen with 29ben7imi-1~7Qlyl)acetic acid, EDC and DIEA in DMF to
give XIV -5. The benzyl ester in XIV -5 is cleaved by tre~tml-.nt with hydrogen and
Fz~ linm-on-carbon in methanol to give XIV -6.
S
Sch.ome XV
H2N ~ NH ~O
al
NH ~ NH~ NH ~f O J3
O O
b
NH ~ NH~ NH ~OH
O O
a) 2-(benzirnidazolyl)acetic acid, EDC, DIEA, DMF; b) NaOH, H20,
CH30H.
XI-l, prepared as described in Alig et. al., EPA 0372486, is con~ n~e~ with
a suitable substituted carboxylic acid, such (2-ben7imitl~701yl)acetic acid, in the
15 presence of EDC and DIEA, and in a suitable solvent, e.g., DMF or acetonitrile.
-7~ -
CA 02241633 1998-06-26
W O97124119 PCT~US96/20748
Many additional methods for converting a carboxylic acid to an amide are known,
and can be found in standard reference books, such as "Compendium of Organic
,. Synthesis". Vol. I-VI (published by Springer-Verlag). Hydrolysis of the ester is
accomplished by saponirlcation with a suitable reagent, e.g., sodium hydroxide, in a
5 suitable solvent, e.g., aqueous methanol. AAternatively, a benzyl ester may be- converted to the acid by tre~tmfA.nt with a suitable catalyst, e.g., Pd/C, and hydrogen
in a suitable solvent, e.g., methanol, ethanol or acetic acid.
Schem~ XVI
1~
~[3~OH
H2N ~ N3 O ~<0
al 1 O-tBu
~3 0 ,<o
2 O-tBu
b l
~OH
Q--N o ~W
NH ~W--NH~N~} --COzH
a) 2-(benzill,Adazolyl)acetic acid, EDC, DIEA, DMF; b) TFA
CA 0224l633 l998-06-26
W O 97/24119 PCTAJS96/20748
XVI-l, prepared as described in Alig et. al., EPA 0505868, is condensed
with a suitable substituted carboxylic acid, such (2-ben7imi~1~7Olyl)acetic acid, in the
presence of EDC and DIEA, in a suitable solvent, e.g., DMF or acetonitrile, to give
XVI -2. Many additional methods for converting a carboxylic acid to an amide are5 known, and can be found in standard reference books, such as "Compendium of
Organic Synthesis". Vol. I-VI (published by Springer-Verlag). Hydrolysis of the
ester in XVI -2 is accomplished with trifluoroacetic acid or hydrogen chloride to
give XVI -3. Alt~rn~tively, the ester in XVI -2 may be saponified with a suitable
reagent, e.g., lN NaOH, in a suitable solvent, e.g., methanol.
Seht-mt- XVII
~IL N N ~ O-tBu
H2N 0~O t
H3CO
a l
~0
H3CO 2
b l
~ ~ A co H
H3CO
a) 2-(b~n7imi-1~701yl)acetic acid, EDC, DIEA, DMI;'; b) TFA, CH2Cl2.
-78 -
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
XVII-1, prepared as described in Sugihara, et. al., EP 0529858, is condensed
with a suitable substituted carboxylic acid, such as (2-bell7imi~1~701yl)acetic acid, to
" give XVII -2, and the tert-butyl ester is cleaved with TFA, following the general
procedure of Sugihara, et. al., Example 59, to give XVII -3. Many additional
- 5 methods for converting a carboxylic acid to an amide are known, and can be found
~ in standard reference books, such as "Compe~-lium of Organic Synthesis", Vol. I-VI
(published by Springer-Verlag).
Schem~ XVITI
Ms-o Q--N
~~~0 a ~ 2 4~ 0
0--t-Bu
O-tBu
~0
3 "~0
OH
a) 4-r2-(benzimidazolyl)methyl]phenol, Cs2CO3, DMF; b) TFA
Compound XVIII-1, prepared as described in Himmelsbach, et. al.,
Australian Patent Application AU-A-86926/91, Example VI~28), is treated with a
suitable ~ub~ uled phenol, such as 4-[2-(benzimidazolyl)methyl]phenol, prepared
-79 -
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
by the general procedure of Wahlgren and Addison, J. Heterocycl. Chem., 1989, 26,
541-3, following the general method of Himmelsbach, et. al., Example 3(51), to give
XVIII -2. The tert-butyl ester in XVIII -2 is hydrolyzed with lN NaOH in CH30H .
following the general procedure of Himmelsbach, Example 7(3) to give XVIII -3.
5 Alternatively, the tert-butyl ester may be cleaved with TFA or HCl.
-8~-
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
Scheme XIX
N ~CO2CH3
NH2 H ~ H 2
- ,¦, b
Q~ N ~ CO2CH3
C02CH3
~c,d CO2CH3
Q--~ ~~/
OCH3
'¦, e CO2H
~ ~~
OCH3
a) HO2CCH2Ph-4-CH2CH~CO2CH3, Ph2POCl, Et3N, DMAP, THF; b) NaH, DMF,
BrCH2CO2CH3; c) KOt-Bu, THF, DMF; d) KOt-Bu, CH3I. DMF; e) LiOH, THF,
H20.
The procedures of Linz, et. al., EP 0567968, are used to prepare XIX-5,
10 except (2-ben~.imi~1~7.olyl)m~th~n~mine is su~ led for 4-cyanoaniline.
_g~
CA 0224l633 l998-06-26
W O 97/24119 PCT~US96/20748
Sch~me XX
MeO ~C~ MeO ~C~ b
NH N ~ CO2Et
HO ~ N ~ co2Et
3 4
R-NHCO ~ e HO2C ~
N ~ CO2Et N ~ CO2Et
6 5
19
R-NHCO~C~ R = [~C \~
N ~ CO2Et
S a) ClCH2CO2Et, Et3N, DMF; b) BBr3; c) (CF3SO)20; d) CO, Pd(OAc)~, PPh3, DIEA,
NMP, NH4HCO3, H20; e) H2N-R, EDC / HOBt, DIEA, DMF; f) H2N-R, CO,
Pd(OAc)2, PPh3, DIEA, NMP, NH4HCO3, H20; g) lN NaOH, HOEt
Scheme XX provides a method for the preparation of 1,2,3,4-
tetrahydroisoquinoline compounds as exemplary fibrinogen receptor antagonists, as
described in M. J. Fisher et al. (EP 0635492, Jan. 25, 1995). Accordingly, a 6-
methoxy-3,4-dihydroisoquinoline, such as compound XX-~I is prepared by the
method described by D. J. Sall and G. L. Grunewald (J. Med. Chem. 1987, 30,
- 8~ -
.
CA 0224l633 l998-06-26
W O 97/24119 PCT~US96/20748
2208-2216). The isoquinoline is treated with a haloacetic acid ester in the presence
of a tertiary amine to afford the 2-acetic acid ester, as exemplified by compound
XX-2. The 6-methoxy compound is converted into the corresponding 6-hydroxy
compound by methods known in the art, for example with BBr3, which is converted
5 into the triflate with trifluorosulfonic acid anhydride. p~ m catalyzed
carbonylation affords the 6-carboxy compound, such as compound XX-5, which is
then condensed with an amine, as exemplified by (2-ben7.imi~1~7.olyl)acetic acid,
employing a standard amide bond forming reagent to give the desired amide, such as
compound XX-6. Saponification affords XX-7. Alternatively, the palladium
10 catalyzed carbonylation reaction with the triflate, exemplified by compound XX-4,
may be trapped with said aminomethyl compound to provide, after saponification,
the corresponding 6-(2-bçn7.imi~ 7.Qlyl)methylaminocarbonyl compound, XX-7.
Sch~otne XXI
HO ~ CF3SO3~
~N~CO2Et ~N~CO2Et
3 O ~ 4 ~
R-NHCO~ e HO2C~
~N~CO2Et ~N ~CO2Et
6 ~ 5 ~
lg
R-NHCO~,~ R = ~N~
N ~ CO2Ft ~J--NH
7 o
a) 1. LiN(TMS)2, 2. ClCH2CO2Et, DMF; b) BBr3; c) (CF3SO2)20; d) CO, Pd(OAc)2,
PPh3, DIEA, NMP, NH4HC03, H20; e) H2N-R, EDC / HOBt, DIEA, DMF; f) H2N-R
CO, Pd(OAc)2, PPh2, DIEA, NMP, NH4HCO3, H20; g) lN NaOH, HOEt
-8~-
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
Scheme XXI provides a method for the pl~al~lion of 3,4-
dihydroisoquinolin- l-one compounds as exemplary fibrinogen receptor antagonists,
as described M. J. Fisher et al. (~P 0635492, Jan. 25, 1995). Accordingly, the 1-oxo
compound XXI-1, prepared by the method described by D. J. Sall and G. L.
Grunewald (J. Med. Chem. 1g87, 30, 2208-2216), is treated with a base, such as
LiN(TMS)~, and a haloacetic acid eser to give a 2-acetic acid ester, as exemplified by
compound XXI-2. The 1-oxo compound is then employed in the analogous series of
reactions deployed in Scheme XX, sub~ g the corresponding 1-oxo analog, as
shown in Scheme XXI, to provide XXI-7. As in Scheme XX, alternatively, the
p~ m catalyzed carbonylation reaction with the triflate, exemplified by
compound XXI-4, may be trapped with an amine to provide, after saponification, the
amide, XXI-7.
Sch~mf XXII
H2N~C~ a, b R-CONH~
N ~ CO2tBu N ~ CO2H
~N
~ ~ NH
a) RCO-X; b) TFA / CH2C12
Scheme XXII provides a method for the pr~alaLion of 6-acylaminotetraline
compounds as exemplary fibrinogen receptor antagonists, as described M. J. Fisher
et al. (;EP 0635492, Jan. 25, 1995). Accordingly, a 6-arnino-2-tert-
butyloxyca.1.onyl-tetral-1-one, exemplified by compound XXII-l, which is prepared
according to the methods described in M. J. Fisher et al. (EP 0635492, Jan. 25,
1995), is con-l~.n.~ed with an activated derivative of a carboxylic acid, such as the
-84-
CA 0224l633 l998-06-26
W O 97/24119 PCTrUS96/20748
activated derivative of (2-bPn7imi-~7olyl3acetic acid, to provide, after
deesterification, the amide, XXII-2.
Sch~rnP XXlII
HO ~[~G CF3SO3 ~0~ CO2Et
R-NHCO~ c HO2C~
COzEt ~ CO2Et
4 3
1 e
R-NHCO = N~
a) CF3SO2O; b) CO, Pd(OAc)2, PPh3, DIEA, NMP, NH4HCO3, H2O; c) H2N-R, EDC
/ HOBt, DIEA, DMF; d) H2N-R, CO, Pd(OAc)2, PPh3, DIEA, NMP, NH4HCO3,
10 H2O; e) lN NaOH, HOEt
Scheme XXIII provides a method for the plc;paldlion of 6-aminoacyltetraline
compounds as exemplary fibrinogen receptor antagonists, as described M. J. Fisher
et al. (EP 0635492, Jan. 25, 1995). Accordingly, an ethylo~ycall,onylmethyl-6-
15 hydroxy-tetral-l-one, exemplified by compound XXIII-l, which is prepared
according to the methods described in M. J. Fisher et al. (EP 0635492, Jan. 25,
-85 -
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96120748
1995), is treated with triffic anhydride to provide the triflate, as exem~ ed bycompound XXIII -2, which is employed in a p~ m catalyzed carbonylation
reaction to afford a carboxylic acid, such as compound XXIII -3, which is then
condensed with an amine to provide, after deesterifi-~tion, the 6-aminoacyl
S compound, XXIII -5. ~AltPrn~tively~ the palladium catalyzed carbonylation reaction
with the triflate exemplified by compound XXIII -2, may be trapped with said
amine compound to provide, after saponification, the corresponding 6-aminoacyl
compound, XXII~ -5.
Scheme XXIV
02N~ EtO IP~o S CO2Et
4 ,/
e
H2N ~ + ~ CO2Et
6 7
1 f~9 1 f'9
R-CONH~ RCONH~ ~ CO2H
8 9
.~ N \
a) BrCH2CO2Et, KzCO3~ NaI; b) 1. DBU, EtOH, 2. HCl, EtOH; c) DiBAL, -78 ~ C; d)
15 NaH, THF; e)Hz, 10% Pd-C; f) R2CO-X; g)lN NaOH, MeOH
-8~-
CA 02241633 1998-06-26
W O97/24119 PCTrUS96120748
Scheme XXIV provides a method for the preparation of 5-
acylaminobenzofuran and S-acylaminodihydrobenzofuran compounds as exemplary
r fibrinogen receptor antagonists, as described in M. L. Denney, et al. (~P 0655439,
31/5/95). Accordingly, a 4-nitrosalicylaldehyde, exemplified by compound XXIV-1
5 , is treated with a haloacetic acid ester to give the phenoxyacetic acid ester,
exemplified by compound XXIV -2. A 2-alkoxycarbonylfuran, exemplified by
compound XXIV -3, is obtained by treating the aldehyde with base, for example
with DBU. The 2-alkoxycarbonyl group is reduced to the aldehyde, for example
with DiBAB. Wittig reaction affords the 2-acrylate ester, exemplified by compound
10 XXIV -4, which is reduced to the benzofuran-2-propionic acid ester, exemplified by
compound XXIV -5 and the dihydrobenzofuran-2-propionic acid ester, exemplified
by co~ )o-llld XXIV -6. The amine XXIV -~ is then condensed with an activated
derivative of a carboxylic acid to provide, after deesterification, the amide 5, XXIV -
8. ~lt~rnzltively, the amine XXIV -6 is condensed with an activated derivative of a
15 carboxylic acid to provide, after dççstçrifiç~tion, the amide, XXIV -7.
-8~-
CA 02241633 1998-06-26
W O97/24119 PCTrUS96/20748
Schem~ XXVa
HO ~ CO2Et ~ CO2Et b,c
a-l a-2
TBDMSO ~ CHO TBDMSO ~
EtO2P ~ a-~ ~ CO2Et
~e,f
HO ~ CO2Et ~ CO2Et
a-6 a-7
a)l.TBDMS-Cl, imid~ole; b)DiBAl-H, -78 ~C, d) NaH, THF; e) H2, 5% Pd-C; f)
Et4N+ F -
CA 0224l633 l998-06-26
W O 97/24119 PCTrUS96/20748
Scheme XXVb
HO ~ CF3SO3 ~ CO2Et
a-6 b-1
d/
~' I b
R-NHCO~ CO~EtCO2Et
b-3 b-2
l e
R-NHCO~co2H R- [~NH
b-4
a) ~CF3SO2)20; b) CO,Pd(OAc)2, PPh3, DTF.A, NMP, NH4HCO3, H20; c) H2N-R,
EDC / HOBt, DIEA, DMF; d) H2N-R, CO, Pd(OAc)2, PPh2, DIEA, NMP, NH4HCO3,
H20 e) lN NaOH, EtOH
-89-
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
SchemP XXVc
CO2Et
a-7 c-1
~ 1
R-NHCO ~ CO2Et CO2Et
C-3 C-2
l e
R-NHCO~co2H R =[~NH
S C-4
a) (CF3SO2)20; b) CO, Pd(OAc)2, PPh3, DlF.A, NMP, NH4HC03, H20; c) H~N-R,
EDC / HOBt, DIEA, DMF; d) H2N-R, CO, Pd(OAc)2, PPh2, DIEA, NMP, NH4HCO3,
H20 e) lN NaOH, BtOH
Scheme XXV provides a method for the ~rt;p~dtion of 5-
aminoacylbenzofuran and 5-aminoacyldihydrobenzofuran compounds as exemplary
fibrinogen receptor antagonists, as described in M. L. Denney, et al. (EP 0655439,
31/5/95). Accordingly, a 5-hyd~ ybellzofuran-2-carboxylic acid ester, such as
compound XXVn-l, prepared in the manner of M. L. Denney, et al. (~P 0655439,
31/5/95), is treated with TBDMS-Cl to provide the TBDMS delivalive of the ester,XXVa-2. The ester is reduced to an aldehyde, such as co,llpolJ.ld XXVa-3. Wittig
9~
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/2074.~.
reaction affords the acrylic acid ester, XXVa-4. Catalytic reduction affords thebenzofuran-2-acetic acid ester and the dihydl. bell of uran-2-acetic acid ester.r Cleavage of the silyl ether group of each ester, by methods known to the art, affords
either the benzofuran-2-acetic acid ester, XXVa-~, or the dihydrobenzofuran-2-
S acetic acid ester, XXVa-6.
~ As seen in Scheme XXVb and XXVc, each alcohol in turn may be converted
to a carboxylic acid via p~ m catalyzed carbonylation, such as compound
XXVb-2 or XXVc-2, which is then condensed with an amine to provide, after
~lees~.; rication, the amide XXVb-4 or XXVc-4. Alternatively, the p~ lm
10 catalyzed carbonylation reaction with the triflate exemplified by compound XXVb-
1, or XXVc-1, may be trapped with said aminomethyl compound to provide, after
deesterification, the corresponding 6-aminoacyl compound, XXVb-4 or XXVc-4.
Amide coupling reagents as used herein denote reagents which may be used
15 to form peptide bonds. Typical coupling methods employ carbo-liimicles, activated
anhydrides and esters and acyl halides. Reagents such as EDC, DCC, DPPA, PPA,
BOP reagent, HOBt, N-hydroxysuccinimi(le and oxalyl chloride are typical.
Coupling methods to form peptide bonds are generally well known to the art.
The methods of peptide synthesis generally set forth by Bodansky et al., THE
20 PRACTICE OF ~ SYNTHESIS, Springer-Verlag, Berlin, 1984, Ali et al. in
J. Med. Chem., 29, 984 (1986) and J. Med. Chem., 30, 2291 (1987) are generally
illustrative of the technique and are incorporated herein by reference.
Typically, the amine or aniline is coupled via its free arnino group to an
a~L.,~,iate carboxylic acid substrate using a suitable carbodiimide coupling agent,
25 such as N,N' dicyclohexyl carbodiimide (DCC), optionally in the presence of
catalysts such as l-hydroxybenzotriazole (HOBt) and dimethylamino pyridine
(DMAP). Other methods, such as the formation of activated esters, anhydrides or
acid h~licles, of the free carboxyl of a suitably protected acid substrate, and
subsequent reaction with the free amine of a suitably protected amine, optionally in
30 the presence of a base, are also suitable. For example, a protected Boc-amino acid or
_9~
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
Cbz-amidino benzoic acid is treated in an anhydrous solvent, such as methylene
chloride or tetrahydrofuran(THF), in the presence of a base, such as N-methyl
morpholine, DMAP or a trialkylamine, with isobutyl chlc,loroll,late to form the
"activated anhydride", which is subsequently reacted with the free amine of a second
5 protected amino acid or aniline.
The compounds of formula (XIX) and (XX) are commercially available or
are prepared by methods known in the art such as illustrated herein disclosed insta~ldard reference books, like the COMPENDIUM OF ORGANIC S~rHETIC METHODS,
Vol. I-VI (Wiley-Interscience). A generally applicable route to ben7imifl~7oles is
disclosed in Nestor et al, J. Med. Chem. 1984, 27, 320. Representative methods for
preparing compounds of formula (XX) are also common to the art and may be
found, for instance, in EP-A 0 381 033.
Acid addition salts of the compounds are prepared in a standard manner in a
suitable solvent from the parent compound and an excess of an acid, such as
15 hydrochloric, hydl~lvll~ic, hydrofluoric, sulfuric, phosphoric, acetic, trifluoroacetic,
maleic, succinic or methanesulfonic. Certain of the compounds form inner salts or
zwitterions which may be acceptable. Cationic salts are prepared by treating theparent compound with an excess of an ~lk~lint? reagent, such as a hydroxide,
carbonate or alkoxide, containing the ~lupliate cation; or with an appl.,p~iate
20 organic amine. Cations such as Li+, Na+, K+, Ca++, Mg++ and NH4+ are specific
examples of cations present in ph~rms~reutir~lly acceptable salts.
- This invention also provides a pharm~-~entical composition which comprises
a compound according to formula (I)-(V) and (XXI)-(XXII) and a ph~rm~reutically
acceptable carrier. Accordingly, the compounds of formula (I)-(V) and (XXI)-
25 (XXII) may be used in the m~nuf~tllre of a m~-lic~m~nt. Pharmaceutical
compositions of the compounds of formula (I)-(V) and (XXI)-(XXII) prepared as
hereinbefore described may be form~ te~l as solutions or lyophilized powders forparenteral ~rlmini~tration~ Powders may be recon.ctitnte~1 by addition of a suitable
diluent or other ph~rm~entic~lly acceptable carrier prior to use. The liquid
30 fi~rm~ tion may be a buffered, isotonic, aqueous solution. Examples of suitable
diluents are normal isotonic saline solution, standard 5% dextrose in water or
_ g
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96t20748
buffered sodium or ammonium acetate solution. Such formulation is especially
suitable for parenteral ~rlmini~tration, but may also be used for oral ~rlmini~tration or
cont~inP~l in a metered dose inhaler or nebulizer for in~llffl~tion It may be desirable
to add excipients such as polyvinylpyrrolidone, gelatin, hydroxy cellulose, acacia,
polyethylene glycol, m~nnitol, sodium chloride or sodium citrate.
Alternately, these compounds may be encapsulated, tableted or prepared in a
emulsion or syrup for oral ~lmini~tration Ph~rm~eeutiç~lly acceptable solid or
liquid carriers may be added to enh~nre or stabilize the composition, or to facilitate
plG~ald~ion of the composition. Solid carriers include starch, lactose, calcium
sulfate dihydrate, terra alba, m~gn~ m stearate or stearic acid, talc, pectin, acacia,
agar or gelatin. Liquid carriers include syrup, peanut oil, olive oil, saline and water.
The carrier may also include a sllct~inP~l release m~teri:~l such as glyceryl
monostearate or glyceryl distearate, alone or with a wax. The amount of solid
carrier varies but, preferably, will be between about 20 mg to about 1 g per dosage
unit. The pharmaceutical pl~l,aldLions are made following the conventional
techniques of pharmacy involving milling, mixin~, gr:~n~ ting, and compressing,
when necessary, for tablet forms; or milling, mixing and filling for hard gelatin
capsule forms. When a liquid carrier is used, the ~lepaldlion will be in the form of a
syrup, elixir, emulsion or an aqueous or non-aqueous suspension. Such a liquid
fnrmnl~tion may be ~flmini.~tered directly p.o. or filled into a soft gelatin capsule.
For rectal ~lmini~tration, the compounds of this invention may also be
combined with excipients such as cocoa butter, glycerin, gelatin or polyethyleneglycols and molded into a suppository.
The compounds described herein are antagonists of the vitronectin receptor,
and are useful for treating diseases whe~ the underlying pathology is attributable
to ligand or cell which interacts with the viLIoile~;lin receptor. For instance, these
compounds are useful for the treatment of diseases wherein loss of the bone matrix
creates pathology. Thus, the instant compounds are useful for the tre~tment of
ostoeporosis, hyperparathyroidism, Paget's disease, hypercalcemia of mz~ n~3ncy,- 30 osteolytic lesions produced by bone m~t~t~ , bone loss due to immobilization or
sex hormone deficiency. The compounds of this invention are also believed to have
9~
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
utility as ~ntitl~mor, anti-angiogenic, ~ntiinfl~mm~tory and anti-mPt~t~tic agents,
and be useful in the treatment of atherosclerosis and restenosis.
The compound is ~lmini~tered either orally or L)a-ellLe,dlly to the patient, in a
manner such that the concentration of drug is sufficient to inhibit bone resorption, or
S other such indication. The ph:lrm~re~lti(~l composition cont~ining the peptide is
~lmini~tered at an oral dose of between about 0.1 to about 50 mg/kg in a manner
conci~tent with the condition of the patient. Preferably the oral dose would be about
0.5 to about 20 mg/kg. For acute therapy, parenteral ~r~ . alion is prefeilcd. An
intravenous infusion of the peptide in 5% dextrose in water or normal saline, or a
10 similar formulation with suitable excipients, is most effective, although an
cc~ r bolus injection is also useful. Typically, the parenteral dose will be
about 0.01 to about 100 mg/kg; preferably between 0.1 and 20 mg/l~g. The
compounds are ~lmini~tered one to four times daily at a level to achieve a total daily
dose of about 0.4 to about 400 mg/kg/day. The precise level and method by which
15 the compounds are ~lmini.~tf red is readily fletermin~d by one routinely skilled in the
art by comparing the blood level of the agent to the concentration required to have a
therapeutic effect.
The compounds may be tested in one of several biological assays to
determine the concentration of compound which is required to have a given
20 I)h~ ological effect.
Tnhi'~it JI~ of vilrv~ binding
Solid-Phase [3H7-sK&F-107260 Binding to av~3: Human placenta or human
platelet av~B3 (0.1-0.3 mg/mL) in buffer T (cont~ining 2 mM CaC12 and I %
octylglucoside) was diluted with buffer T collt;1i.. i.. g 1 mM CaC12, 1 mM MnC12,
1 mM MgC12 (buffer A) and 0.05% NaN3, and then imm~ t~ly added to 96-well
ELISA plates (Corning, New York, NY) at 0.1 rnL per weIl. 0.1 - 0.2 ~g of ocv,133
was added per well. The plates were incubated overnight at 4~C. At the time of the
experiment, the wells were washed once with buffer A and were incubated with
0.1 mL of 3.5% bovine serum alburnin in the same buffer for 1 hr at room
-94-
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
telllpeldLuie. Following in~nb:~tion the wells were aspirated completely and washed
twice with 0.2 mL buffer A.
Compounds were dissolved in 100% DMSO to give a 2 mM stock solution,
which was diluted with binding buffer (15 mM Tris-HCl (pH 7.4), 100 mM NaCl,
1 mM CaC12, 1 mM MnC12, 1 mM MgC12) to a final compound concentration of
100,uM. This solution is then diluted to the required final compound concentration.
Various concentrations of unlabeled antagonists (0.001 - 100 ~M) were added to the
wells in triplicates, followed by the addition of 5.0 nM of [3H]-SK&F-107260 (65 -
86 Ci/mmol).
The plates were incubated for 1 hr at room lelllpeldLul~. Following
incubation the wells were aspirated completely and washed once with 0.2 mL of ice
cold buffer A in a well-to-well fashion. The receptors were solubilized with 0.1 mL
of 1% SDS and the bound [3H]-SK&F-107260 was determin~-1 by liquid
scin~ tion counting with the addition of 3 mL Ready Safe in a Beckman LS Liquid
Scintill~tic~n Counter, with 40% efficiency. Nonspecific binding of [3H3-SK~F-
107260 was ~et~rmined in the presence of 2 ,uM SK&F-107260 and was consistently
less than 1 % of total radioligand input. The ICso (concentration of the antagonist to
inhibit 50% binding of [3H]-SK&F-107260) was determined by a nonlinear, least
squares curve-fitting routine, which was modified from the LUNDON-2 program.
The Ki (dissociation constant of the antagonist) was calculated according to theequation: Ki = ICso/( l + L/Kd), where L and Kd were the concentration and the
dissociation constant of [3H]-SK&F-107260, respectively.
Compounds of the present invention inhibit vitronectin binding to SK&F
107260 in the concentration range of about 0.001 to 50 micromolar.
Compounds of this invention are also tested for in vitro and in vivo bone
resorption in assays standard in the art for ev~ ting inhibition of bone formation,
such as the pit formation assay disclosed in EP 528 587, which may also be
performed using human osteoclasts in place of rat osteoclasts, and the ovarectomi7ed
- rat model. described by Wronski et al., Cells and Materials 1991, Sup. 1, 69-74.
_95
CA 02241633 1998-06-26
W O 97124119 PCT~US96/20748
Vascular smooth linuscle cell ~ ..lion assay
Rat or human aortic smooth muscle cells were used. The cell migration was
monitored in a Transwell cell culture chamber by using a polycarbonate membrane
with pores of 8 um (Costar). The lower surface of the filter was coated with
vitronectin. Cells were suspended in DMEM supplemented with 0.2% bovine serum
albumin at a concentration of 2.5 - 5.0 x 106 cells/mL, and were pretreated with test
compound at various concentrations for 20 min at 20~C. The solvent alone was used
as control. 0.2 mL of the cell suspension was placed in the upper c~ nlent of
the chamber. The lower co~ lllent contained 0.6 rnL of DMEM supplemented
with 0.2% bovine serum albumin. lncubation was carried out at 37~C in an
atmosphere of 95% air/5% CO2 for 24 hr. After incubation, the non-migrated cellson the upper surface of the f1lter were removed by gentle scraping. The filter was
then fixed in methanol and stained with 10% Giemsa stain. Migration was measuredeither by a) counting the number of cells that had rnigrated to the lower surface of
the filter or by b) extracting the stained cells with 10% acetic acid followed by
~terrninin~ the absorbance at 600 nM.
PARATHYROIDE~TOMIZED RAT MODEL
Each experimental group consists of 5-6 male Sprague-Dawley rats. The rats are
parathyroidectomized (by the vendor, Taconic Farms) 7 days prior to use. Twenty four
hours prior to use, circ~ tin~ ionized calcium levels are measured in whole blood
immediately after it has been withdrawn by tail venipuncture into hep~rini7f r1 tubes. Rats
are included if ionized Ca level (measured with a Ciba-Corning model 634 calcium pH
analyzer) is _1.2 mM/L. The rats are then put on a diet of calcium-free chow anddeionized water. At the start of the experiment the rats weigh approximately 100g.
Baseline Ca levels are measured and the rats are ~irnini~tered control vehicle (saline) or
compound (dissolved in saline) as a single intravenous (tail vein) bolus injection followed r
im mf.~ t~ly by a single ~ubc~lt:~nPous injection of either human paldlhyl~id hormone 1-34
peptide (hPI'Hl-34, dose 0.2mg/kg in saline/0.1% bovine serum albumen, Bachem, Ca)
_9~_
CA 02241633 1998-06-26
W O97/24119 PCT~US~6120748
or the PTH vehicle. The calcemic response to PTH (and any effect of compound on this
response) is measured 2h after co~ oulld/PTH allmini~tration.
.
RAT ULNA DRII~T MODEL
Each experimental group consists of 8-10 male Sprague-Dawley or Wistar rats of
aL~plo~illlately 30-40g body weight at the start of the e~elilllent. The agent being tested is
~rlmini~trred by an a~prop.iate route as single or multiple daily doses for a period of
seven days. Prior to ~lmini~tration of the first dose, the rats are given a single dose of a
10 fluorescent marker (tetracycline 25mg/lcg, or calcein lOmg/lcg) that labels the position of
bone forming surfaces at that point in time. After dosing of compound has been
completed, the rats are killed and both forelimbs are removed at the elbow, the foot is
removed at the ankle and the skin removed. The sample is frozen and mounted vertically
on a microtome chuck. Cross sections of the mi(l~h~ft region of the ulna are cut in the
15 cryostat. The rate of bone resorption is measured morphom~-tric~lly in the medial-dorsal
portion of the cortical bone. The mea~ule.llellt is done as follows: the amount of bone
resorbed at the periosteal surface is equal to the t1ict~nre by which the periosteal surface
has advanced towards the fluorescent label which had been incorporated at the endosteal
bone formation surface on day zero; this fli~t~nre is calculated by subtracting the width of
20 bone between the label and the periosteal surface on day 7 from the width on day zero; the
resorption rate in microns per day is calculated by dividing the result by 7.
HUMAN OSTEOCLAST RESORPTION ASSAY ("PIT ASSAY")
25 ~ Aliquots of osteoclastoma-derived cell suspensions are removed from liquid
nitrogen strorage, warmed rapidly at 37~C and washed x l in RPMI- 1640 m~ m by
centrifugation (lOOOrpm, 5 mins at 4~C).
~ Aspirate the medium and replace it with murine anti-HLA-DR antibody, diluted
1:3 in RPMI-1640 ml rlillm Incubate for 30 mins on ice and mix the cell suspension
.~
frequently.
_
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
~ The cells are washed x2 with cold RPMI-1640 by c~ L irugation (1000 rpm, 5 rnins
at 4~C) and the cells are transferred to a sterile 15 rnl centrifuge tube. The number of
mononuclear cells are enumerated in an il~l~roved Nellh~ r counting chamber.
S
~ Sufficient m~n~tic beads (5 / mononuclear cell), coated with goat anti-mouse IgG,
are removed from their stock bottle and placed into 5 ml of fresh mP~ lm (this
washes away the toxic azide preservative). The m~ lm is removed by immobilizing
the beads on a magnet and is replaced with fresh medium.
~ The beads are mixed with the cells and the suspension is incub~t~cl for 30 mins on
ice. The suspension is mixed frequently.
~ The bead-coated cells are immobilized on a magnet and the rem~ining cells
15 (osteoclast-rich fraction) are dec~nt~d into a sterile 50 ml centrifuge tube.
~ Fresh mto-lillm is added to the bead-coated cells to dislodge any trapped osteoclasts.
This wash process is repeated xlO. The bead-coated cells are discarded.
20 ~ The osteoclast~s are enullleldt~d in a counting chamber, using a large-bore disposable plastic pasteur to charge the chamber with the sample.
~ The cells are pelleted by centrifugation and the density of osteoclasts ~dj-lste~l to
1.5xlO4/ml in EMEM m~ lm, supplemented with 10% fetal calf serum and
25 1.7gllitre of sodium bicarbonate.
~ 3ml aliquots of the cell suspension ( per tre~tmP~t) are ~ieC~ntl ~i into 15mlcenkifuge tubes. The cells are pelleted by centrifugation.
30 ~ To each tube 3m1 of the a~p~ .;ate treatment are added (diluted to 50 uM in the
EMEM medium). Also included are ~pl~,pliate vehicle controls, a posiLive conkol
gg
CA 02241633 1998-06-26
W O 97/24119 ~T~US96/20748
(87MEM1 diluted to 100 ug/ml) and an isotype control (IgG2a diluted to 100
ug/ml). Incubate at 37~C for 30 mins.
~,
~ 0.5ml aliquots of the cells are seeded onto sterile dentine slices in a 48-well plate
- 5 and incubated at 37~C for 2 hours. Each treatment is screened in quadruplicate.
.,
~ The slices are washed in six changes of warm PBS (lO ml / well in a 6-well plate)
and then placed into fresh tre:~tmf~nt or control. Incubate at 37~C for 48 hours.
10 Tartrate l~- ~;C~nt acid Ph~3s~ ce (TRAP) Pl~ce-lul~ (selective stain for cells
of the osteoclast lineage).
~ The slices are washed in phosphate buffered saline and fixed in 2% gluteraldehyde
(in 0.2M sodium cacodylate) for 5 mins.
~ They are washed in water and incubated in TRAP buffer for 5 mins at 37~C.
~ Following a wash in cold water they are incubated in cold acetate buffer / fast red
garnet for 5 mins at 4~C.
~ Excess buffer is aspirated, and the slices are air dried following a wash in water.
~ The TRAP positive osteoclasts are enumerated by bright-field microscopy and are
then removed from the surface of the dentine by sonication.
~ Pit volumes are dele~ ed using the Nikon/Lasertec ILM2 1 W confocal
microscope.
Human o~leo~lr--l le3~ ~io.. and adhesion assavs
30 Pit resorption and adhesion assays have been developed and standardized usingnormal human osteoclasts derived from osteoclastoma tissue. The osteoclast
_99_
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
population is negatively selected from the osteoclastoma cell suspensions using
magnetic beads (Dynal Inc, NY). These beads are coated with a murine monoclonal
antibody that recognizes a hurnan class II major histocompatihility antigen that is
present on a large number of mononuclear cells in the cell suspensions. The cells
5 that express this antigen, and consequently bind the beads, are removed from the
mixture of cells using a m~n~t The osteoclast-rich suspension is then ready to use
in the assays det~ below.
Resor~lio.- sssay (with ELISA readout)
10 Enriched preparations of osteoclasts are preincubated for 30 minllfes at 37~C with
test compound (4 doses) or controls. They are then seeded onto bovine cortical bone
slices in wells of a 48-well tissue culture plate and are in~ubat-~-l for a further 2 hours
at 37nC. The bone slices are washed in six changes of warm phosphate buffered
saline (PBS), to remove non-adherent cells, and are then returned to wells of a 48
15 well plate con~ining fresh compound or controls. The tissue culture plate is then
incubated for 48 hours at 37"C. The supern~t~n~c from each well are aspirated into
individual tubes and are screened in a cornpetitive ELISA that detects a collagen
peptide that is released during the resorption process. This is a commercially
available ELISA (Osteometer, Denmark) that contains a rabbit antibody that
20 specifically reacts with an 8-amino acid se~uence (Glu-Lys-Ala-His- Asp-Gly-Gly-
Arg) that is present in the carboxy-tçrmin~l telopeptide of the al-chain of type I
collagen. The results are expressed as % inhibition of resorption colllpal~;d to a
vehicle control.
25 Adhesion assay
Osteoclastoma-derived osteoclasts are preincubated with compound (4 doses) or
controls at 37~C for 30 minutes. The cells are then seeded onto osteopontin-coated
slides (human or rat osteopontin, 2.5ug/ml) and incubated for 2 hours at 37~C. Non
adherent cells are removed by washing the slides vigorously in P~S and the cells30 r~m~ining on the slides are fixed in acetone. The osteoclasts are stained for tartrate-
resistant acid phosphatase (TRAP), a selective marker for cells of this phenotype,
and are enumerated by light microscopy. The results are expressed as % inhibition
of adhesion colll~alcd to a vehicle control.
-10~
.
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96120748
Inhibition of RGD-m~ t.-d GPIIb-IIIa binding
pllrific~tion of GPIIb-ma
Ten units of ollt~l~tP-l, washed human platelets (obtained from Red Cross)
were lyzed by gentle stirring in 3% octylglucoside, 20 mM Tris-HCl, pH 7.4, 140
mM NaCl, 2 mM CaC12 at 4~C for 2 h. The lysate was centrifuged at lOO,OOOg for 1
h. The sUpprn~t~nt obtained was applied to a 5 mL lentil lectin sepharose 4B
column (E.Y. Labs~ preequilibrated with 20 mM Tris-HCl, pH 7.4, 100 mM NaCl, 2
mM CaC12, 1 % octylglucoside (buffer A). After 2 h incubation, the column was
washed with 50 rnL cold buffer A. The lectin-retained GPIIb-IIIa was eluted withbuffer A containing 10% dextrose. All procedures were performed at 4~C. The
GPIIb-IIIa obtained was >95% pure as shown by SDS polyacrylamide gel
electrophoresis .
Incorporation of GPIIb-IIIa in Liposomes.
A mixture of phosphatidylserine (70%) and phosphatidylcholine (30%)
(Avanti Polar Lipids) were dried to the walls of a glass tube under a stream of
nitrogen. Purified GPIIb-IIIa was diluted to a final concentration of 0.5 mg/rnL and
mixed with the phospholipids in a protein:phospholipid ratio of 1:3 (w:w). The
mixture was resuspended and sonicated in a bath sonicator for 5 min. The mixturewas then dialyzed overnight using 12,000- 14,000 molecular weight cutoff dialysis
tubing against a 1000-fold excess of 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 2 mM
CaC12 (with 2 changes). The GPIIb-IIIa-cont~ining liposomes wee centrifuged at
12,000g for 1~ min and resuspended in the dialysis buffer at a final protein
concentration of approximately 1 mg/mL. The liposomes were stored at -70C until
needed.
Competitive Binding to GPIIb-IIIa
The binding to the fibrinogen receptor (GPIIb-ma) was assayed by an
indirect competitive binding method using [3H]-SK&F-107260 as an RGD-type
ligand. The binding assay was performed in a 96-well filtration plate assembly
(Millipore Corporation, Bedford, MA) using 0.22 um hydrophilic durapore
membranes. The wells were precoated with 0.2 mL of 10 ,ug/mL polylysine (Sigma
Chemical Co., St. Louis, MO.) at room temperature for 1 h to block nonspecific
35 binding. Various concentrations of unlabeled ben7~ 7~rines were added to the
-1~1-
CA 0224l633 l998-06-26
W O 97/24119 PCT~US96/20748
wells in quadruplicate. [3H]-SK&F-IQ7260 was applied to each well at a final
concentration of 4.5 nM, followed by the addition of 1,ug of the purified platelet
GPIIb-IIIa-cont~ining liposomes. The mi~ulcs were incubated for 1 h at room
tel,,pcldture. The GPIIb-ma-bound [3H]-SK&F-107260 was seperated from the
S unbound by filtration using a Millipore filtration manifold, followed by washing
with ice-cold buffer (2 times, each 0.2 rnL). Bound radioactivity rem:~ining on the
filters was counted in 1.5 mL Ready Solve (Beckm~n Instr lm~nt~, Fullerton, CA) in
a Beckman Liquid Scintillation Counter (Model LS6800), with 40% efficiency.
Nonspecific binding was ~et~ in.o~t in the presence of 2 ,uM unlabeled SK&F-
107260 and was con~i~te-ntly less than 0.14% of the total radioactivity added to the
samples. All data points are the mean of quadruplicate ~leterrnin~tions.
Competition binding data were analyzed by a nonlinear least-
squares curve fitting procedure. This method provides the IC50 of the antagonists
(concentration of the antagonist which inhibits specific binding of [3H]-SK&F-
107260 by 50% at equilibrium). The IC50 is related to the equilibrium dissociation
constant (Ki) of the antagonist based on the Cheng and Prusoff equation: Ki =
IC50/(l~L/Kd), where L is the concentration of t3H~-SK&F-107260 used in the
co~llpeLiLive binding assay (4.5 nM), and Kd is the dissociation constant of r3H]-
SK&P-107260 which is 4.5 nM as ~ rmin~l by Scatchard analysis
Compounds of the present invention inhibit the vitronectin binding to SK&P
007260 with a Ki at the vitronectin receptor that is about ten-fold greater than that
for the fibrinogen receptor. Preferred compounds have a Ki at the vitronectin
receptor that is thirty-fold greater than that at the fibrinogen receptor. The most
preferred compounds have a Ki at the vitronectin receptor that is a hundred-foldgreater than that at the fibrinogen receptor.
The examples which follow are intended in no way to limit the scope of this
invention, but are provided to illustrate how to make and use the compounds of this
invention. Many other embodiments will be readily a~cnt to those skilled in the
art.
CA 02241633 1998-06-26
W O 97/2411g PCTrUS96/20748
G~o,n~ral
Nuclear magnetîc resonance spectra were recorded at either 250 or 400 MHz
using, respectively, a Bruker AM 250 or Bruker AC 400 spectrometer. CDC13 is
deuteriochloroform, DMSO-d6 is hPxAfle~ riodimethylsulfoxide, and CD30D is
tetradeuteriomethanol. Chemical shifts are reported in parts per million (o)
downfield from the internal standard tetramethylsilane. Abbreviations for NMR
data are as follows: s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet,
dd=doublet of doublets, dt=doublet of triplets, ~ arent, br-broad. J inclic~tes
the NMR coupling constant measured in Hertz. Continuous wave infrared (IR)
spectra were recorded on a Perkin-Elmer 683 infrared spectrometer, and Fourier
transform infrared (FTIR) spectra were recorded on a Nicolet Impact 400 D infrared
spectrometer. IR and FTIR spectra were recorded in trAn~mi~ion mode, and band
positions are reported in inverse wavenumbers (cm-l). Mass spectra were taken oneither VG 70 FE, PE Syx API III, or VG ZAB HF instruments, using fast atom
bombardment (FAB) or ele~ v~L~ldy (ES) ionization techniques. Elemental analyseswere obtained using a Perkin-Elmer 240C elemental analyzer. Melting points were
taken on a Thomas-Hoover melting point apparatus and are uncorrected. All
te~ ela~ules are reported in degrees Celsius.
~nAltt~ch Silica Gel GF and E. Merck Silica Gel 60 F-254 thin layer plates
were used for thin layer chromatography. Both flash and gravity chromatography
were carried out on E. Me~ck Kieselgel 60 (230-400 mesh) silica gel. Analytical and
preparative HPLC were carried out on Rainin or Berkm~n chromatographs. ODS
refers to an octadecylsilyl derivatized silica gel chromatographic support. 5 ,u Apex-
ODS in~lic~ s an octadecylsilyl derivatized silica gel chromatographic support
having a nominal particle size of 5 ,u, made by Jones Chromatography, Littleton,Colorado. YMC ODS-AQ~ is an ODS chromatographic support and is a registered
tra(1~mArk of YMC Co. Ltd., Kyoto, Japan. PRP-l~) is a polymeric (styrene-
divinylbenzene) chromatographic support, and is a registered trademark of T~milton
Co., Reno, Nevada) Celite(~) is a filter aid composed of acid-washed diatomaceous
silica, and is a registered trAtl~m~rk of Manville Corp., Denver, Colorado.
Methyl (+)-7-carboxy4-methyl-3-oxo-2,3,4,5-tetrahydro- lH- 1,4-
- 35 benzodi~epine-2-acetate, methyl (2S)-7-carboxy-4-methyl-3-oxo-2,3,4,5-
tetrahydro- 1 H- 1,4-benzodiazepine-2-acetate, methyl (2R)-7-carboxy-~methyl-3-
oxo-2,3,4,5-tetrahydro-lH-1,4-ben7o~ 7~pine-2-acetate, methyl (+)-7-carboxy-4-
103
CA 02241633 1998-06-26
W O 97/24119 PCTAUS96/20748
isopropyl-3-oxo-2,3,4,5-tetrahydro- 1 H- 1,4-benzodiazepine-2-acetate,
methyl (+~-7-carboxy-3-oxo-2-(2-phenylethyl)-2,3,4,5-tetrahydro- 1 H- 1,4-
benzodiazepine-2-acetate, methyl (+)-8-carboxy-2-methyl-3-oxo-2,3,4,5-tetrahydro-
lH-2-benzazepine-4-acetate, methyl (+) 7-amino-5-oxo-~(2-phenylethyl)-lH-1,4-
5 benzodiazepine-2-acetic acid, and tert-butyl 4-fluoro-3-methylbenzoate were
prepared by the method of Bondinell, et al., WO 93/00095. Methyl (~)-7-carboxy-4-
(2-methoxyethyl)-3-oxo-2,3,4,5-tetrahydro- 1 H- 1,4-benzodiazepine-2-acetate, methyl
(~)-7-carboxy-4-[2-(3,4-methylenedioxyphenyl)ethyl] -3-oxo-2,3,4,5-tetrahydro- 1 H-
1,4-benzodiazepine-2-acet~tf- m~thyl (+)-7-carboxy-3-oxo-2,3,4,5-tetrahydro-1~I-
1,4-benzodiazepine-2-acetate, methyl (+)-2,3,4,5-tetrahydro-7-[[[(ben7imi~l~7Ql-2-
yl)methyl~amino]carbonyl]-4-methyl-3-oxo-lH-1,4-benzodiazepine-2-acetate, (S)-
2,3,4,5-tetrahydro-7-t[[(bf~n7:imi-1~7.ol-2-yl)methyl]methylamino]carbonyl]~
methyl-3-oxo-lH-1,4-benzodiazepine-2-acetic acid, 2-
(methylaminomethyl)ben~imic~7ole dihydrochloride, and 4-aza-5-rnethyl-2-
(methylamino)methylben7imif~70le were prepared according to P50256-1.
Preparation 1
Preparation of 2-(aminomethyl)-4-~a-5-methylben7.imi-1~7--1e dihydrochloride
a) 2,3-Diamino-6-methylpyridine
10% Pd/C (3.2 g, 3 mmole) was added to a solution of 2-Amino-6-methyl-3-
nitropyridine (2.30 g, 15 mmole) in absolute EtOH (150 mL), and the mixture was
shaken at RT under H2 (50 psi). After 1.5 hr, the mixture was filtered through
celite~), and the filtrate was concentrated under vacuum to afford the title compound
as a yellow oil. This was used without further purification: 'H NMR (250 MHz,
CD30D) ~ 6.82 (d, lH), 6.36 (d, IH), 2.25 (s, 3H).
b) 2-Amino-3-[(benzyloxycarbonyl)glycyl~amino-6-nlethylL)ylidine
DCC (3.09 g, 15 mmole) was added to a solution of 2,3-diamino-6-
methylpyridine (15 mmole) and Cbz-glycine (3.14 g, 15 mmole) in DMF (19 mL)
and CH2Cl2 (19 mL) at 0~C under argon. When the DCC had dissolved, the slightly
cloudy solution was warrned to RT. After 18.5 hr, the rnixture was filtered through
celite(~, and the filtrate was concentrated to dryness on the rotavap. The residue was
reconcentrated from xylenes (to remove DM~) to leave a yellow solid. Silica gel
chromatography (10% MeOH/CHCl3) gave the title compound (2.24 g, 48%) as a
yellow solid: TLC Rf (10% MeOH/CHCl3) 0.57; 'H NMR (250 MHz, DMSO - d6) o
104
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
9.11 (br s, 1 H), 7.48 - 7.60 (br t, 1 H), 7.20 - 7.48 (m, 6 H), 6.40 (d, 1 H),
5.69 (br s, 2 H), 5.06 (s, 2 H), 3.82 (d, 2 H), 2.23 (s, 3 H).
c) 4-Aza-2-(benzyloxycarbonyl)aminomethyl-5-methylbenzimidazole
S A solution of 2-amino-3-[(benzyloxycarbonyl)glycyl]amino-6-
melhyl~ylidine (2.24 g, 7.13 mmole) in glacial AcOH (70 mL) was heated to refluxunder argon. After 17 hr, the solution was concentrated (rotavap, high vacuum), and
the residue was reconcentrated from toluene (to remove AcOH). The r~slllting
yellow oil was treated with hot EtOAc (20 mL) and the ll~i~lUlG was cooled to RT.
The solid was collected by suction filtration and washed with EeOAc to afford the
title compound (1.72 g, 81%) as an off-white solid: TLC R, (15% MeOH/CHCl3)
0.63; MS (ES) m/e 297.4 (M + H)+.
d) 2-(Arninomethyl)-4-aza-5-methylben7imi-l~7ole dihydrochloride
10% Pd/C (153 mg, 0.14 mmole) was added to a solution of 4-aza-2-
(benzyloxycarbonyl)aminomethyl-5-methylben7imi(1~7Ole (213.4 mg, 0.72 mmole)
and 1.0 N HCl (1.44 mL, 1.44 mmole) in absolute EtOH (7.2 mL). The llli~lult; was
purged with H2, then was stirred briskly at RT under H2 (balloon). After 2 hr, the
reaction was filtered through celite, and the filtrate was concentrated on the rotavap
to leave the title compound as an off-white solid: MS (ES) m/e 163.2 (M + H)+.
rr~l)aidlion 2
Preparation of methyl (+)-2.3~4.5-tetrahydro-7-carboxy-4-~3.3-dimethylbutyl)-3-
oxo- lH- 1.4-ben7.odiazepine-2-acetate
a) tert-Butyl 3-[(3,3-dimethylbutyl)amino]methyl-4-nitrobenzoate
A ~ u~e of tert-butyl 3-methyl-4-nitrobenzoate (WO 93/00095; 17.7 g,
74.7 mmol), NBS (19.9 g, 112.0 mmol), benzoyl peroxide (1.81 g, 7.47 mmol), and
CC14 (370 mL) was heated at reflux. After 17.5 h, the reaction was cooled
thoroughly in ice and filtered to remove the pleci~ilaled succinimicle. The filtrate
was concentrated to leave a brownish-yellow oil.
This oil ~4.2 g, 13.29 mmol), was dissolved in dry THF (50 mL), and 3,3-
dimethylbutylamine (3.0 g, 29.64 mmol) was added all at once. The orangish-
yellow solution was stirred at RT for 80 min, then was concentrated to remove the
THF. The residue was diluted with Et20 (150 mL) and washed sequentially with
1.0 N NaOH (25 mL) and H2O (25 mL). The combined aqueous layers were back-
105
CA 02241633 1998-06-26
W O 97/24119 PCT~US96120748
extracted with Et2O (50 mL~, and the combined organic layers were
washed with brine (25 mL) and dried (MgS04). Concentration gave the crude title
compound (4.19 g, 94%~ as a light-brown oil: MS (ES) rn/e 337.2 (M+H)+.
S b) tert-Butyl 3-[[N-(3,3-dimethylbutyl)-N-(tert-butoxycarbonyl)]amino]methyl-4-
nitrobenzoate
Di-tert-butyl dicarbonate (4.0 g, 18.39 mmol) was added all at once to a
solution of tert-butyl 3-[(3,3-dimethylbutyl)amino]methyl-4-nitrobenzoate (4.12 g,
12.26 mmol) in CHCl3 (80 mL) at RT. After 18 h, the reaction was concen~lated
and reconcentrated from hexanes (to remove CHCl3). Silica gel chromatography
(10-25% EtOAc/hexanes) gave the title compound (5.0 g, 93%) as a yellow oil: MS
(ES) m/e 437.2 (M+H)+, 459.2 (M+Na)+.
c) tert-Butyl 4-amino-3-[~N-(3,3-dimethylbutyl)-N-(tert-
butoxycarbonyl)]amino]methyl benzoate
10% Pd/C (1.0 g, 0.94 mmol) was added to a solution of tert-butyl 3-[[N-
(3,3-dimethylbutyl)-N-(tert-butoxycarbonyl)]amino]methyl~-nitrobenzoate (4.95 g,11.35 mmol) in EtOAc (50 mL), and the mixture was shaken on a Parr apparatus at
RT under H2 (55 psi). After 4 h, the reaction was filtered through celite~, and the
filtrate was concentrated to afford the title compound (4.3 g, 93%) as a reddish-
brown oil: MS (ES) m/e 407.4 (M+H)+.
d) tert-Butyl (~t)-4-[2-(1,4-dimethoxy-1,4-dioxobutyl)amino~-3-[[N-(3,3-
dimethylbutyl)-N-(tert-butoxycarbonyl)]amino]methylbenzoate
A solution of tert-butyl 4-amino-3-[[N-(3,3-dimethylbutyl)-N-(tert-
butoxycarbonyl)]arnino]methylbenzoate (5.6 g, 13.79 mmol) and dimethylacetylene
dicarboxylate (1.86 mL, 15.17 mmol) in MeOH (28 mL) was heated at reflux for 1
h, then was cooled to RT. The rçslllting solution was combined with MeOH (80
mL) and 10% Pd/C (2.9 g, 2.76 mmol), and the mixture was shaken on a Parr
apparatus at RT under H2 (50 psi). After 22 h, the reaction was filtered throughcelite~), and the filtrate was concentrated on the rotavap. The residue was
reconcentrated from CHC13 (to remove MeOH), then was chromatographed on silica
gel (25% EtOAc/hexanes). The title compound (2.64 g, 42%) was obtained as a
faintly yellow oil: MS (ES) m/e 551.2 (M+H)+.
e) Methyl (+)-2,3,4,5-tetrahydro-7-carboxy-4-(3,3-dimethylbutyl)-3-oxo-lH-1,4-
benzodiazepine-2-acetic acid
106
CA 02241633 1998-06-26
PCTrUS96120748
W 097/24119
TFA (25 mL) was added all at once to a solution of tert-butyl (+)4-
~2-(1 ,4-~iimPth~xy-1 ,4-dioxobutyl)amino]-3-~[N-(3,3-dimethylbutyl)-N-(tert-
butoxycarbonyl)]amino]methylbenzoate (12.64 g, 4.8 mmol) in anhydrous CH2Cl2
(25 mL) at 0~C, and the faintly yellow solution was warmed to RT. After 1 h, thesolution was concenLlaLed on the rotavap, and the residue was reconcentrated from
toluene (to remove residual TFA). The resulting oil was combined with toluene (50
mL) and Et3N (3.34 mL, 24 mmol), and the mixture was heated to reflux. A light
yellow, homogeneous solution was produced. After 16 h, the reaction was
concentrated on the rotavap to leave a solid residue. This was dissolved in a
minimllm of MeOH (ca. 10 mL), diluted with H2O (10 mL), and acidif1ed with
glacial AcOH to pH 4.5. The ~ lulc was filtered, and the precipitate was washed
sequentially with MeOH and E~t20, then was dried in high vacuum to afford the title
compound (1.88 g, 93%) as a nearly colorless powder: MS (ES) m/e 363.2 (M+H)+.
~lGI)aldlion 3
Preparation of Bisr(ben7imi-1~7ol-2-yl)methyllamine tris(trifluoroacetate)
a) Bis[[1-N-(tert-butoxycarbonyl)bçn7imi(1~7Ol-2-yl]methyl]-N-(tert-
butoxycarbonyi)amine
To a stirred solution of 2-aminomethylbenzimidazole dihydrochloride
hydrate (6.26 g, 28.4 mmol) and triethylamine (4.0 mL, 28.4 mmol) in dry THF (50mL) was added l-(tert-butoxycarbonyl)-2-(bromomethyl)bçn7imit1~7c 1e (P50256-1;
2.00 g, 9.48 mmol) in THF (30 mL),. After 8 h, a solution of di-tert-butyl
dicarbonate (10.0 g, 45.84 mmol~ in CHC13 (50 mL) was added slowly. The
resulting mixture was stirred at RT overnight then was concentrated. The residuewas taken up in CH2C12 (150 mT.), and washed sequentially with water (60 rnL), 5%
NaHCO3 (60 mL), and brine (60 mL). Drying (MgSO4), concentration, and silica
gel chromatography (6% MeOH/CH2C12) gave the title compound (0.46 g, 8%) as a
faint yellow oil: MS (ES) m/e 578.4 (M+H)+.
b) Bis[(bPn7imic1:~701-2-yl)methyl]amine tris(trifluoroacetate)
A solution of TFA (3 mL) and CH2C12 (9 mL) at RT was added all at once to
bis[[1 -N-(tert-butoxycarbonyl)benzimidazol-2-yl]methyl}-N-(tert-
butoxycarbonyl)amine (0.23 g, 0.4 mmol). After 35 min, the solution was
concentrated on the rotavap, and the residue was reconcentrated from toluene (to 107
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96~0748
remove residual TFA) to afford the title compound (0.17 g, 68%) as an off-
white powder: MS (ES) m~e 278.0 ~M+H)+.
Preparation 4
Preparation of 2-rrl-r(benzimidazol-2-yl)methyllben7imi(1~7ole~methyllamine
bi~(Jri lluorQacetate)
a) [[l-N-(tert-Butoxycarbonyl)be~7imi-1~701-2-yl3methyl]-N-(tert-
butoxycarbonyl)amine
To a stirred solution of 2-aminomethylben7imidzl701e dihydrochloride
hydrate (3.0 g, 13.63 mmol) and triethylamine (8.44 rnL, 61.3 mmol) in dry CH~C12
(50 mL) a solution of di-tert-butyl dicarbonate (6.54 g, 30.0 mmol) in CH2Cl2 (50
rnL) was added at 0~C. The reaction was stirred at RT for 1 h, then more of
triethylamine (1.9 mL, 13.8 mmol) and di-tert-butyI dicarbonate (2.97 g, 13.63
mmol) were added. The reslllting mixture was stirred at RT for 24 h, then was
concenkated. The residue was taken up in CH2Clz (50 mL) and washed sequentially
with 0.5 N HCl (2x40 mL), 5% NaHC03 (50 mL), and brine (50 mL). The crude
product was recrys~lli7ecl from CH2Cl~/ether to give the title compound (2.8 g, 59%)
as a wnite powder: MS (ES) m/e 348.2 (M~H)+.
b) 2-[[1-[[( 1-(tert-Butoxycarbonyl)ben7imirl~701-2-
yl3methyl]ben7.imitl~7.01e]methyl]-N,N-di-(teLt-butoxycarbonyl)amine
To a stirred solution of [[l-N-(tert-butoxycarbonyl)bPn7imirl~7Ql-2-
yl]methyl]-N-(di-tert-butoxycarbonyl)amine (0.6 g, 1.73 mmol) and NaH (0.1 g,
4.17 mmol) in dry THF (12 mL) and DMF (4 mL) was added 1-(tert-
butoxycarbonyl)-2-(bromomethyl)ben7imi~ 701e ~0.6 g, 1.93 nimol). Theresulting
Lul~ was stirred at RT for 1 h, then was concenLIdL~d. The residue was taken up
in CH2C12 (100 mL) and washed sequentially with water (50 mL), 5% NaHC03 (30
mT ), and brine (30 mL). Drying (MgS04), concentration, and silica gel
chromatography (2:3 EtOAc/hexanes) gave the title compound (0.27 g, 27%) as a
faint yellow oil: MS (ES) m/e 578.2 (M~H)+.
c) 2-~[l-[(Ben7imi~1~7-)l-2-yl)methyl]bçn7imitl~7~1e~methyl]amine
bis(trifluoro~et~1r)
A solution of TFA/CH2C12 (30 ml, 25%) at RT was added all at once to 2-[[1-
[[( 1 -N-tert-butoxycarbonyl)bel-7 i ~ 701-2-yl]methyl]ben7imi~1~701e]methyl]-N,N-
108
CA 0224l633 l998-06-26
W O 97/24119 PCT~US96/20748
di-(tert-butoxycarbonyl)amine (0.25 g, 0.43 mmol). After 25 min, the
solution was concentrated on the rotavap, and the crude product was recryst~ d
from CH2Cll/ether to give the title co~ oulld (0.17 g, 63%) as an off-white powder:
MS (ES) m/e 278.0 ~M+H)+.
~xamI)le 1
a~dLion of (+)-2.3~4.5-tetrahydro-7-rrr(4-~a-5-methylbellzi.ll~dazol-2-
yl)methyllaminolcarbonyll -4-(2-methoxyethyl)-3-oxo- 1 H- 1.4-benzodiazepine-2-
a~tic acid
a) Methyl (+)-2,3,4,5-tetrahydro-7-[[[(4-aza-5-methylben7imiclzl7Ql-2-
yl)methyl]amino]carbonyl]~(2-methoxyethyl)-3 -oxo- 1 H- 1,4-benzodiazepine-2-
acetate
EDC (138 mg, 0.72 mrnole) was added to a solution of methyl (+)-7-
carboxy-4-(2-methoxyethyl)-3-oxo-2,3,4,5-tetrahydro- lH- 1,4-benzodiazepine-2-
acetate (202 mg, 0.60 mmole), 2-(arninomethyl)-4-aza-5-methylben7imicl~7Ole
dihydrochloride (0.72 mmole), HOBt ~ H2O (97 mg, 0.72 mmole), and
diisopropylethylamine (0.84 mL, 4.8 mmole) in anhydrous CH3CN (3 mL) at RT.
After 16 hr, the reaction was concentrated, and the residue was reconcellllaL~d from
xylenes/CHCl3. Silica gel chromatography (15% MeOH/CHCI3) gave the title
compound (impure): TLC Rf (15% MeOH/CHCI3) 0.55; MS (ES) m/e 481.5 (M +
H)t. This was used without further purification.
b) (+)-2,3,4,5-Tetrahydro-7-[tt(4-aza-S-methylben7imi~1~7ol-2-
yl)methyl]amino~carbonyll-4-(2-methoxyethyl)-3-oxo- 1 H- 1,4-benzodiazepine-2-
acetic acid
A two-phase mixture of methyl ( )-2,3,4,5-tetrahydro-7-[[[(4-aza-5-
methylbel.~.i...ic1sl7.Ql-2-yl)methyl]amino]carbonyl]-4-(2-methoxyethyl)-3-oxo-lH-
1,4-benzodiazepine-2-acetate (0.60 mrnole), 1.0 N LiOH (1.8 mL, 1.8 mmole), and
THF (4.2 mL) was stirred at RT for 45 min, then was concentrated to remove the
THF. The aqueous layer was washed with Et2O (2 x 2 mL), and the Et2O layers werediscarded. The aqueous layer was diluted with CH3CN (2 mL) and acidified with
TFA (0.23 mL). The res-llting solution was concentrated to dryness on the rotavap,
and the residue was purified by ODS chromatography (12~o CH3CN/H20 contS~ini
0.1% TFA (250 mL), then 15% CH3CN/H20 cont:~ining 0.1% TFA). Concentration
and lyophilization gave the title compound (199.5 mg, 50% for two steps) as a light
109
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
yellow powder: HPLC (PRP~ , 15% CH3CN/H20 cont~ining 0.1%
TFA) K' = 1.4; MS (ES) rn/e 467 (M + H)'. Anal. Calcd for C23El26N6Os ~ 1.5
CF3COzH 1.33 H2O: C, 47.21; H, 4.60; N, 12.70. Found: C, 47.20; H, 4.73; N,
12.79.
S .
Examl~le 2
Pl~a~tion of (_)-2~3~4~s-tetr~ydro-7-rrr(ben7im~ 7~ol-2-
yl)methyllaminolcarbonyll-4-(2-methoxyetl~ -3-oxo-lH-1.4-ben7.o-lJa7epine-2-
acetic acid
a) Methyl (_)-2,3,4,5-tetrahydro-7-[[t(bel ,7i . l . if 1~7f~l-2-yl)methyl]arnino]carbonyl]
4-(2-methoxyethyl)-3-oxo- lH- 1 ,4-benzodiazepine-2-acetate
EDC (230 mg, 1.2 mmole) was added to a solution of methyl (+)-7-carboxy-
4-(2-methoxyethyl)-3-oxo-2,3,4,5-tetrahydro- lH- 1 ,4-benzodiazepine-2-acetate
(336.4 mg, 1.0 mmole), 2-(aminomethyl)be~7imif1~7f-1e dihydrochloride hydrate
(264 mg, 1.2 mmole), HOBt ~ H2O (162 mg, 1.2 mmole), and diisopropylethylamine
(0.70 mL, 4.0 mmole) in anhydrous DMF (5 mL) at RT. After 17 hr, the reaction
was concentrated, and the residue was reconcentrated from xylenes (2 x) to remove
DMF. The residue was diluted with H2O (3 mL) and extracted with CHCl3 (3 x 5
mL). The combined extracts were treated with MeOH (2 rnL) to dissolve a
precipitate, then were dried (MgSO,) and concentrated. Reconcentration from
xylenes (to remove residual DMF) left a light yellow solid. This was dissolved in
MeOH/CHCI3, and the solution was concentrated to leave an oil. Silica gel
chromatography (10% MeOH/CHCl3) gave an off-white solid, which was triturated
with EtOAc (3 mL) to afford the title compound (397.1 mg, 85%) as a colorless
solid: TLC R, (10% MeOH/CHCl3) 0.46; MS (ES) m/e 466.2 (M + H)~.
b) (_)-2,3,4,5-Tetrahydro-7-[~[(ben7imi(1~7Ol-2-yl)methyl]amino]carbonyl]-4-(2-
methoxyethyl)-3-oxo- lH- 1 ,4-benzodiazepine-2-acetic acid
1.0 N LiOH ( 1.0 mL, 1.0 mmole) was added to a suspension of methyl (+)-
2,3,4,5-tetrahydro-7-[[[~benzimidazol-2-yl)methyl3amino]carbonyl]~(2-
methoxyethyl)-3-oxo-lH-1,4-ben7o~ 7f-pine-2-acetate (397 mg, 0.85 mmole) in
THF (4.3 rnL) and H20 (3.3 mr ) at RT. The light yellow mixture was stirred at 40 -
50~C for 1 hr, and the reslllting homogeneous solution was then stirred at RT for
17.5 hr. The reaction was concentrated, and the rçslllting oil was dissolved in H20
(4 mL). The solution was filtered to remove particnl~tes, and the filtrate was
110
CA 0224l633 l998-06-26
W O97/24119 PCTrUS96/20748
neutralized with 1.0 N HCl (1.0 rnL). The yellowish solid was collected
and triturated with good stirring with hot 1: 1 CH3CN/H2O. The resulting solid was
collected, washed with plenty of 1: 1 CH3CN/H2O, and dried in high vacuum (40~C)to afford the title compound (327.9 mg, 85%) as a colorless powder: HPLC (PRP-
S l(E~), 15% CH3CN/H20 cont:~inin~ 0.1% TFA) K' = 4.6; MS (ES) m/e 452.2 (M +
H)~. Anah Calcd for C23H25N5O5: C, 61.19; H, 5.58; N, 15.51. Found: C, 61.18; H,5.58; N, 15.39.
~ExamE)le 3
Preparation of (+)-4-~4-~rr(lH-~en7~imicla7Ql-2-
yl)methyllmethylaminolcarbonyllphenyll-3-phenylbutanoic acid
a) Ethyl 3-hydroxy-4-(4-methoxyphenyl)-3-phenylbutanoate
Anhydrous EtOAc (4.3 mL, 44 mmole) was added dropwise over 5 - 6 rnin
to a solution of lithium bis(trimethylsilyl)amide (1.0 M in THF, 40 mL, 40 mmole)
in dry THF (60 mL) in a flame-dried flask at -78~C under argon. The yellow
solution was stirred at -78~C for 0.5 hr, then a solution of 2-(4-methoxyphenyl)-1-
phenyleth~n<)nP (Chem. Ber. 195S, 91, 755-759; 4.53 g, 20 mmole) in dry THF (20
ml) was added dropwise over 12 rnin. Additional THF (2 mL) was used in transfer.After 0.5 hr, The reaction was quenched with satd NH4Cl (120 mL) and warmed to
RT. EtOAc extraction, drying (MgSO4), concentration, and silica gel
chromatography (20% EtOAc/hexanes) gave the title compound (6.13 g, 96%) as a
light yellow oil: TLC Rf (20% EtOAc/hexanes) 0.34; MS (ES) m/e 315.2 (M + H)~.
b) Ethyl 4-(4-methoxyphenyl)-3-phenylbutanoate
Boron trifluoride etherate (4.8 mL, 39 mmole) was added dropwise over 3
rnin to a solution of ethyl 3-hydroxy-4-(4-methoxyphenyl)-3-phenylbutanoate (6.13
g, 19.5 mmole) and triethylsilane (6.2 mT, 39 mmole) in anhydrous CH2C12 (49 rnL)
at 0~C under argon. The reaction was stirred at RT overnight, then was quenched
with 5% NaHCO3 (100 mL). The ll~i~lul~ was stirred briskly for 10 min, then was
separated. The aqueous layer was extracted with CH2Cl2 (100 mL), and the
combined organic layers were dried (Na2SO4) and concentrated. The residue was
reconcentrated from hexanes (to remove CH2Cl2) to leave a yellow oil. This was
- 35 dissolved in absolute EtOH (100 mT.), and 10% Pd/C (775 mg, 1.95 mmole) was
added. The mixture was shaken on a Parr apparatus at RT under H2 (50 psi) for 2 hr,
then was filtered through celite~3). The filtrate was concentrated, and the residue was
111
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
chromatographed on silica gel (15 % EtOAc/hf~x~n~s). The title compound
(5.27 g, 91%) was obtained as a colorless oil: TLC Rr (15% EtOAc/hexanes) 0.40;
MS (ES) m/e 299.2 (M + H)+.
c) Ethyl 4-(4-hydroxyphenyl)-3-phenylbutanoate
Anhydrous ~ minllm trichloride (4.49 g, 33.7 mmole) was added all at once
to solution of ethyl 4-(4-methoxyphenyl)-3-phenylbutanoate (2.01 g, 6.74 mmole)
and eth~nethiol (2.5 mT., 33.7 mrnole) in anhydrous CH2Cl2 (67 mL) at 0~C under
argon. The yellow solution was warmed to RT and stirred for 3 hr, then was
recooled to 0~C and ~luen~hl cl with cold 3 N HCl (67 mL). The mixture was stirred
for 5 rnin, then was separated. The aqueous layer was extracted with CH2Cl2 (2 x100 mL), and the combined organic layers were dried (Na2SO4) and concentrated.
Silica gel chromatography (25% EtOAc/hexanes) gave the title compound (1.84 g,
96%) as a colorless oil: TLC Rf (30% EtOAc/hexanes) 0.47; MS (ES) m/e 285.2 (M
+ H)'.
d) Ethyl 3-phenyl-4-[4-(trifluoromethanesulfonyloxy)phenyl]butanoate
Trifluorom~th~n.oslllfonic anhydride (1.4 mL, 8.4 mrnole) was added rapidly -
dropwise to a solution of ethyl 4-(4-hydroxyphenyl)-3-phenylbutanoate (1.84 g, 6.47
rnmole) and 2,6-lutidine (1.5 mL, 12.9 mmole) in anhydrous CH2Cl2 (32 mL) at -
78~C under argon. After 0.5 hr, the yellow solution was warmed to RT and stirredfor 1 hr. The reaction was diluted with Et2O (150 mL) and washed sequentially with
1.0 N HCl (15 mL), 5% NaHCO3 (15 mL), and saturated brine(15 mL). Drying
(MgS04), concentration, and silica gel chromatography (15% EtOAc/hexanes) gave
the title compound (2.62 g, 97%) as a nearly colorless oil: TLC Rf (20%
EtOAc/hexanes) 0.55; MS (ES) m/e 417.0 (M + H)~.
e) Ethyl 4-(4-carboxyphenyl)-3-phenylbllt:~n~-~t~
A mixture of ethyl 3-phenyl~-[4-
~trifluornm~th:-nesulfonyloxy)phenyl]butanoate (2.62 g, 6.29 mmole), anhydrous
KOAc (2.47 g, 25.16 mmole), Pd(OAc)2 (70.6 mg, 0.31 mrnole), dppf (697.4 mg,
1.26 mmole), and anhydrous DMSO (31 mL) was purged with carbon monoxide
(three evacuation/ carbon monoxide purge cycles, followed by bubbling carbon
monoxide through the mixture for 5 min), then was heated at 70~C under a balloonof carbon monoxide. After 3.5 hr, the reaction was diluted with H2O (31 mL),
cooled in ice, and acidified with 1.0 N HCl (25 mL). CH2C12 extraction (2 x 100
mL), drying (MgSO4), concentration, and reconcentration from toluene left a
112
:
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
reddish-orange liquid. Silica gel chromatography (1% AcOH in 7:3
toluene/EtOAc) gave the title compound (1.78 g, 91 %) as a cream-colored solid:
TLC Rf (1% AcOH in 7:3 toluene/EtOAc) 0.47; MS (ES) m/e 313.2 (M + H)+.
S f) Ethyl (+)-4-[4-[~[(lH-bell~hllidazol-2-yl)methyl]methylamino~carbonyllphenyl]-
3 -phenylbutanoate
- EDC (230 mg, 1.2 mmole) was added to a solution of ethyl 4-(4-
carboxyphenyl)-3-phenylbutanoate (312.4 mg, 1.0 rnrnole), 2-
(methylaminomethyl)ben7imi(1z-7ole~dihydrochloride (281 mg, 1.2 mmole), HOBt
lQ H2O (162 mg, 1.2 rnmole), and diisopropylethylamine (0.70 mL, 4.0 mmole) in
anhydrous CH3CN (5 mL) at RT. After 18 hr, the reaction was concentrated, and the
brown residue was chromatographed on silica gel (5% MeOH in 1: 1 EtOAc/CHCl3).
The title compound (439.2 mg, 96%) was obtained as a light orange foam: TLC Rf
(5% MeOH in 1: 1 EtOAc/CHCl3) 0.50; MS (ES) m/e 456.2 (M + H)+.
g) (~)~[4-[[[(}H-Benzimidazol-2-yl)methyl]methylamino]carbonyl]phenyl]-3-
phenylbutanoic acid
A solution of ethyl (_)-4-[4-[[[(lH-ben7imitl~7ol-2-
yl)methyl]methylamino]carbonyl]phenyl~-3-phenylbutanoate (439.2 mg, 0.96
20 mmole) and 1.0 N NaOH (1.2 mL, 1.2 mmole) in EtOH (8.4 mL) was stirred at
50~C. After 24 hr, the reaction was concentrated to dryness and the residue was
purified by ODS chromatography (35% MeOH/H20). Concentration and
lyophilization gave the title compound (412.2 mg, 86%) as a colorless powder:
HPLC (PRP-l(~), 35% CH3CN/H20 cont~inin~ 0.1% TFA) K' = 1.4; MS (ES) 428
25 (M + H)', 450 (M + Na)~. Anal. Calcd for C26H2JN3O3Na 2.75 H2O: C, 62.58; H,
5.96; N, 8.42. Found: C, 62.34; H, 5.84; N, 8.44.
Rx~m,l?le 4
30 ~le~)a dlion of (+~-4-14-rrr(ben7imi~1~7~-1-2-
yl)methyllmethylaminolcarbonyllphenyll-3-(dimethylaminocarbonyl)butanoic acid
a) tert-Butyl 4-bromobenzoate
Trifluoromethanesulfonic acid (0.18 rnL, 2 rnmole) was added dropwise to a
- 35 mixture of 4-bromobenzoic acid (20.10 g, 100 rnrnole), anhydrous CH2Cl2 (100 mL),
and condensed isobutylene (-78~C; 100 mL), and the rf s~ in~ mixture was allowedto reflux under a dry ice/acetone condenser. After 40 min, more isobutylene (30
113
CA 0224l633 l998-06-26
W O 97/24119 rcTrusg6/2o748
mL) was added, and reflux was continued for an additional 20 min. The
reaction was poured into Et2O (500 mL) and washed sequentially with 1.0 N KOH (2x 50 rnL), H2O (50 mL), and satd brine (50 mL). Drying (MgSO4), concentration,
and silica gel chromatography (5% EtOAc/hexanes) gave the title compound (15.28
g, 59%) as a light yellow oil: TLC Rf (5% EtOAc/hexanes) 0.59; MS (ES) m/e
259/257 (M + H)+.
b) Methyl 3-[4-(tert-buto~y~;all~onyl)phenyl]propenoate
A solution of tert-butyl 4-hromobenzoate (5.14 g, 20 mmole), methyl
- 10 acrylate (9.1 rnL, 100 mrnole), Pd(OAc)2 (224.5 mg, 1 rnrnole), tri-o-tolylphosphine
(608.8 mg, 2 rnmole), and diisopropylethylamine (7.0 mL, 40 mmole) in
propionitrile (100 rnL) was heated at reflux for 3 hr, then was concentrated on the
rotavap. The residue was diluted with Et2O (200 mL) and washed sequentially with1.0 N HCl (2 x 50 mT), 5% NaHCO3 (50 rnL), and satd brine (50 mL). Drying
(MgSO4), concentration, and silica gel chromatography (15% EtOAc/hexanes) gave
the title compound (3.34 g, 64%) as a light yellow solid: TLC Rf (20%
EtOAc/hexanes) 0.51; MS (ES) rn/e 263.0 (M + H)+.
c) Methyl 3-[4-(tert-butoxycarbonyl)phenyl]propanoate
10% Pd/C (2.71 g, 2.55 mrnole) was added to a solution of methyl 3-[4-(tert-
butoxycarbonyl)phenyl]propenoate (3.34 g, 12.73 mmole) in EtOAc (65 rnL) and
MeOH (65 rnL), and the mixture was shaken on a Parr a~paldLus at RT under H2 (50psi). After 3 hr, the reaction was filtered through celite~, and the rlltrate was
concentrated to dryness on the rotavap. Reconcentration from hexanes left the title
compound (3.27 g, 97%) as a cloudy, grayish oil: TLC Rf (20% EtOAc/hexanes)
0.63; MS (ES) m/e 265.0 (M + H)+.
d) 3-t4-(tert-Butoxycarbonyl)phenyl]propanoic acid
A rnixture of methyl 3-[4-(tert-butoxycarbonyl)phenyl]propanoate (3.27 g,
12.37 rnrnole), 1.0 N LiOH (14.8 mL, 14.8 mmole), THF (31 mL), and H2O (16 mL)
was stirred at RT for 1.5 hr, then was concentrated on the rotavap to remove theTHF. The aqueous solution was washed with Et20 ~2 x 30 rnL), and the Et,O layerswere discarded. The aqueous layer was acidified with 1.0 N HCl (ca. 17 mL), and
the mixture was extracted with CHCl3 (3 x 50 mL). Drying (Na2SO4) and
concent;ation gave the title compound (3.04 g, 98%) as a colorless powder: mp 88.5
- 89.5~C; MS (DCVNH3) m/e 268.0 (M ~ NH4)+.
114
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
e) N,N-Dimethyl 3-[4-(tert-butoxycarbonyl)phenyl]prop~n~mi~l
EDC (2.09 g, 10.88 mmole) was added to a solution of 3-[4-(tert-
butoxycarbonyl)phenyl]propanoic acid (2.27 g, 9.07 mmole), di~llelllylamine
hydrochloride (0.88 g, 10.88 mmole), HOBt ~ H2O (1.47 g, 10.88 mmole), and
diisopropylethylamine (3.2 rnL, 18.14 mmole) in anhydrous CH3CN (45 mL) at RT.
After 19.5 hr, the reaction was concentrated, and the residue was chromatographed
on silica gel (EtOAc). The title compound (2.46 g, 98%) was obtained as a colorless
oil: TLC Rf (EtOAc) 0.52; MS (ES) mte 278.4 (M + H)~.
f) Ethyl 4-[4-(tert-butoxycarbonyl)phenyl]-3-(dimethylaminocarbonyl)butanoate
A solution of lithium bis(trimethylsilyl)amide in THF (1.0 M, 5.8 mL, 5.8
mmole) was added dropwise over 2.5 min to a solution of N,N-dimethyl 3-[4-(tert-butoxycarbonyl)phenyl]prop~n~mi~lf~ (1.34 g, 4.83 mmole) in dry THF (48 mL) at -78~C under argon. The yellow solution was stirred at -78~C for 0.5 hr, then ethyl
bromoacetate (2.7 mL, 24.15 mmole) was added over 15 sec down the walls of the
flask (to precool). After 0.5 hr, the reaction was poured into satd NH4Cl (50 rnL),
and the mixture was extracted with EtOAc (2 x 100 mL). Drying (MgSO4),
concentration, and reconcentration from xylenes left a light yellow oil. Silica gel
chromatography (1: 1 EtOAc/hexanes) gave the title compound (453.5 mg, 26%) as alight yellow oil: TLC Rf (1: 1 EtOAc/hexanes) 0.44, MS (ES) m/e 364.2 (M + H)+.
g) Ethyl 4-(4-carboxyphenyl)-3-(dimethylaminocarbonyl)b~lt:~noz-tf~
TFA (2.3 mL) was added all at once to a solution ethyl 4-[4-(tert-
butoxycarbonyl)phenyl] 3-(dimethylaminocarbonyl)butanoate (168.6 mg, 0.46
mmole) in anhydrous CH2Cl2 (2.3 rnL) at 0C. The solution was stirred at RT for 0.5
hr, then was concentrated to dryness on the rotavap. The residue was reconcentrated
from toluene to afford the title colllpoulld as a light yellow oil: MS (ES) mle 308.0
(M + H)1.
h) Ethyl (+)-4-[4-t[[(ben7 i ~ I .ifl~7ol-2-yl)methyl]methylamino~call,onyl]phenyl}-3-
(dimethylaminocarbonyl)bl~t~ o~t~
EDC (105.8 mg, 0.55 mmole) was added to a solution of ethyl 4-(4-
carboxyphenyl)-3-(dimethylaminocarbonyl)butanoate (0.46 mmole), 2-
(methylaminomethyl)ben7imicl~7ole dihydrochloride (129.2 mg, 0.55 mmole),
- 35 HOBt ~ H2O (74.6 mg, 0.55 mmole), and diisopropylethylamine (0.32 mL, 1.84
mmole) in anhydrous CH3CN (2.3 mL) at RT. After 22 hr, the reaction was
concentrated, and the yellow residue was chromatographed on silica gel (10%
115
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
MeOH in 1:1 EtOAc/CHCl3). The title compound (191.5 mg, 92%) was
obtained as a light yellow oil: TLC Rf (10% MeOH in 1:1 EtOAc/CHCl3) 0.44; MS
(ES) m/e 451 (M + H)+.
S i) (V-4-[4-[r[(l~en7.imicl~7.ol-2-yl)methyl]methylamino]carbonyl]phenyl]-3-
~dimethylaminocarbonyl)butanoic acid
A solution of ethyl (i)-4-[4-[[[(benzirnidazol-2-
yl)methyl]methylamino]carbonyl]phenyl]-3-(dimethylaminocarbonyl)butanoate
(191.5 mg, 0.43 mmole) and 1.0 N LiOH (0.52 mL, 0.52 mmole) in T~IF (2.2 mL)
and H~O (1.6 mL) was stirred at RT for 17 hr, then was acidified with TFA (0.10
rnL, 1.29 mmole). Concentration left an aqueous residue which was purified by
ODS chromatography (17~o CH3CN/H2O cont:~inin~ 0.1% TFA; chromatographed
againusingl5%CH3CN/H2Ocont~ining0.1%TFA). Concentrationand
lyophilization gave the title compound (133.4 mg, 4791'o) as a colorless powder:HPLC (PRP-1(~), 20% CH3CN/H20 cont:lining 0.1% TFA) K' = 1.3; MS (ES) m/e
423.2 (M + H)+. Anal. Calcd for C23H26N,O4 ~ 2 CF3CO2H ~ 0.5 H2O: C, 49.17; H,
4.43; N, 8.49. Found: C, 49.13; H, 4.62; N, 8.52.
T~x~ ?le S
Preparation of (S)-2.3.4.5-tetrahydro-7-rrr(ben7imi~7ol-2-
yl)mPthyllmethylaminolcarbonyll-4-mPthyl-3-oxo-lH-1 .~ben70diazepine-2-acetic
:~rid. r(2.2--limethyl-2-methoxyacetyl)oxy~methyl ester
a) (S)-2,3,4,5-Tetrahydro-7-[r[[1-(tert-butoxycarbonyl)bc~ 7Q1-2-
yl]methyl]methylamino]carbonyl]-4-methyl-3 -oxo- 1 H- 1 ,4-benzodiazepine-2-acetic
acid
To a ~ Lulc of (S)-2,3,4,5-tetrahydro-7-r[[(ben7imi~ 7ol-2-
yl)methyl~methylamino]carbonyl3-4-methyl-3-oxo- I H- l ,4-benzodiazepine-2-acetic
acid (444 mg, 1.0 mmol), triethylamine (0.1464 mT.,l.OS mmol) in DMF (8 mL),
was added dropwise di-tert-butyl dicarbonate (230 mg, 1.05 mmol) in DMF (2 mL).
The reaction ll~i~lule was stirred at RT for 18 h. An aliquot was assayed to inrii~ ~te.
only 50% conversion. Another quantity of triethylamine and di-tert-butyl
dicarbonate were added and stirring was continued for another 18 h. An aliquot still
in-liC~tefl some unreacted material, so a third quantity of reagents was added and the
reaction was stirred for another 18 h. The reaction mixture was concentrated to
dryness and the residual oil was triturated with water, filtered and vacuum dried at
116
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
40-50~C, to give a white solid of the title compound ( 0.442 g, 85%). MS
fES) m/e 522.4 ~M+H]+.
b) (S)-2,3,4,5-Tetrahydro-7-[[l[l-(tert-butoxycarbonyl)ben7imi~l~7ol-2-
yl]methyl]methylamino]carbonyl]-4-methyl-3-oxo- lH- 1,4-benzodiazepine-2-acetic
acid, ~(2,2-dimethyl-2-methoxyacetyl)oxy]methyl ester
To a solution of the compound of Example Sa, (0.209 g, 0.4 mmol) in dIy
acetone (10 mL) was added anhydrous polassiulll carbonate (0.25 g, 1.8 mmol). The
reaction mixture was stirred at RT and under argon for 1 h. 2-methoxy-2-
methylpropanoic acid chloromethyl ester, US Patent 4,602,012, July 22, 1986,
(0.334 g, 2.0 mmol) was then added, followed by tetrabutylammonium iodide (0.03
g, 0.08 mmol). The reaction was stirred at RT under argon for 48 h. It was then
filtered and the filtrate was concentrated to a yellow oily residue of the titlecompound (0.67g, q~nti~ative yield). TLC RF 0.48 (silica gel, 6% methanol in
methylene chloride). MS ~ES) m~e 652.2 [M+H]+.
c) (S)-2,3,4,5-Tetrahydro-7-[[[(ben7imicl~7ol-2-yl)methyl]methylamino]carbonyl]-4-
methyl-3 -oxo- 1 H- 1,4-benzodiazepine-2-acetic acid, [(2,2-dimethyl-2-
methoxyacetyl)oxy]methyl ester
To a solution of the compound in Example Sb (0.67 g, 1 mmol) in methylene
chloride (5 mL) was added TFA (1 mL). The reaction was stirred at RT under argonfor 4 h. It was concentrated to dryness, and the residue was evaporated with
methylene chloride three times to remove TFA traces, to give the title compound
(0.4 g, 73%). This was purified on a flash silica column (step gradient, 2-3%
methanol in methylene chloride). The fractions cont:linin~ the pure compound were
collected, concentrated to yield the title compound (65 mg ) as an off-white solid.
MS (ES) m/e 552.2 [M+H]+. 'H NMR (400 MHz, (CDCl3) ~ 7.6 (br s, lH), 7.22 (m,
6H), 6.5 ( d, lH), 5.85 ( d, lH), 5.8 (d, lH), 5.4 (d, lH), 5.05 (m, lH), 4.79 (q, 2H),
4.3 (d, lH), 3.7 (d, lH), 3.25 ( s, 3H), 3.15 (s, 3H), 3.05 ( s, 3H), 3.02 ( dd, lH), 2.7
(dd, lH), 1.4 (s, 6H). Anal. Calcd for C~8H33N507 1.25 H20: C, 58.58; H, 6.23; N,
12.20. Found: C, 58.60, H, 5.94, N, 12.00.
Fxam~?le 6
P.~paldlion of fi)-2.3.4.5-tetrahydro-7-rrrfben~imi~ 7Ql-2-
yl)methyl~ aminolcarbonyll-4-methyl-3-oxo- 1 H- 1.4-benzodiazepine-2-fN-
hydroxy~ fzlmi~l~
117
CA 02241633 1998-06-26
W O 97124119 PCT~US96/20748
a) (~)-2,3,4,5-Tetrahydl o-7-[[~(benzimidazole-2-yl)methylamino]call,ollyl]-4-
methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-(N-hydroxy)~cetz~
NaOMe (Aldrich, 25 wt. % solution in MeOH, 2.2 mL, 9.7 mmole) was
added to a solution of hydroxylamine hydrochloride (0.67 g, 9.7 mmole) in MeOH
(40 mT-) at 45~C, and the ~ Lul~e was stirred for 5 min. Methyl (~-2,3,4,5-
tekahydro-7-[[[(kpn7imi~ Ql-2-yl)methyl]amino]carbonyl]-4-methyl-3-oxo-lH-
1,4-benzodiazepine-2-acetate (0.82 g, 1.9 mmole) was s-lcpçn~lP-l in MeOH (2 mL)and THF (15 mL) and added dropwise to the above solution. The reaction was then
stirred at 45~C for 24 h. The ll~ lul~ was concentrated in vacuo, and was then
treated with 10% CH3CN/H20 conl~inin~ 0.1% TFA (5 mL). All material dissolved,
and then a solid precipitated out. Half of this material was dissolved in the mobile
phase by addition of excess TFA and was purified by. prepd.d~ e HPLC (YMC
ODS-AQ, 50 x 250 mm, flow rate = 80 mL/min, 10% CH3CN/H20 con~z~ining 0.1%
T~A; tR = 57 min) to yield the title compound (91 mg, 22%) as a white solid. MS
(ES) m/e 423.1 [M+H] '. 'H NMR (400 MHz, DMSO-d6) ~ 9.06 (bt, J = 4 Hz, lH),
7.77 (m, 2H), 7.58 (m, 2H), 7.50 (m, 2H), 6.60 (d, J = 10 Hz, lH), 6.40 (bs, lH),
5.52(d,J= l9Hz, IH),5.18(bt,J=9Hz, lH),4.85(d,J=6Hz, lH),3.83(d,J=
19 Hz, lH), 2.95 (s, 3H), 2.60 (dd, J = 17, 9 Hz, lH), 2.28 (dd, J = 15, 7 Hz, IH).
Anal. Calcd for C2,H22N6O" ~ 1.5 C2HF3O2 1.0 H2O): C, 47.14; H, 4.20; N, 13.74.
Found: C, 46.95; H, 4.24; N, 13.37.
F.x~nu?le 7
Preparation of (+)-2.3.4~5-tetrahydro-7-r3-(benzimidazol-2-yl~pher~vll-4-methyl-3-
oxo- 1 H- 1 ~4-benzodiazepine-2-acetic acid
a) 2-(3-Iodophenyl)benzilllidazole
To a cold solution of 3-iodobenzoic acid (5~0 g, 20 mmol) and Et3N (3~7 mL,
26 mmol) in THF (50 mL) was added isobutylchloroformate (2~9 mL, 21 mmol3.
The solution was stirred for 1 h at 10~C All of the solution was added slowly to a
solution of 1,2-diaminobenzene (2~2 g, 20 mmol) in THF (50 mL)~ After 18 hr, thereaction was concentrated and the residue was partitioned between EtOAc and 5%
Na2CO3~ The layers were separated and the EtOAc layer was washed with water.
Concentration of the organic layer gave a residue which was treated with EtOAc and
allowed to stand for 1~ min. Filtration gave a solid which was treated with AcOH(50 rnL) and heated to 110~C After 18 hr, the solution was concentrated. The
- 118
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
residue was treated with EtOAc and the solution was filtered to give the
title cc,~ oulld (3.14 g, 50%): MS (ES) m/e 321.2 (M+H)~.
b) 2-[(3-Tributylstannyl)phenyl]l,en,;...icl~7Ole
S A solution of 2-(3-iodophenyl)ben7imitl~7Ole (1.0 g, 3.1 mmol),
bistributyltin (3.9 mL, 6.2 mmol) and PdCl~(PPh3)2 (100 mg, 0.14 rnmol) in DMF
(10 mL) was heated to 90~C under argon. After 2 hr, the solution was concentrated.
The residue was treated with hexane and filtered. EtOAc was added and the solution
was filtered. The filtrate was concentrated to give the title compound (812 mg,
54%): MS (ES) m/e 485.4 (M+H)'.
c) Methyl (i)-2,3,4,5-tetrahydro-1-(tert-butoxycarbonyl)-7-iodo-4-methyl-3-oxo-
1 H- 1,4-benzodiazepine-2-acetate
To a solution of methyl-7-iodo-4-methyl-3-oxo-lH-1,4-benzodiazepine-2-
acetate (1.6 g, 4.3 mmol) and DMAP (10 mg, 0.08 mmol) in CH3CN (10 mL) was
added di-tert-butyl dicarbonate (2.0 g, 8.6 mmol), and the solution was stirred at RT.
Additional di-tert-butyl dicarbonate (a total of 8 g, 34.4 mmol) was added
periodically until the reaction went to completion. Concentration and silica gelch~ lugraphy gave the title compound (1.8 g, 90%): MS (ES) mle 497.2 (M+Na)'
d) Methyl (i)-2,3,4,5-tetrahydro-7-[3-(ben7imi~1~7ol-2-yl)phenyl]- 1 -(tert-
butoxycarbonyl)-4-methyl-3-oxo- lH- 1,4-benzodiazepine-2-acetate
A mixture of 2-[(3-tributylstannyl)phenyl3ben7imicl~7ole (0.24 g, 0.5 mmol),
methyl (+)- 1 -(tert-butoxycarbonyl)-7-iodo-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-
2-acetate (0.223 g, 0.7 mmol), CuI (10 mg, 0.05 mmol), and PdCl2(PPh3)z (40 mg,
0.05 mmol) in DMF (10 mL) was heated to 100~C under argon. After 18 hr, the
solution was concentrated. Silica gel chromatography gave the' title compound (0.06
g, 22%): MS (ES) m/e 541.5 (M+H)t.
e) Methyl (_)-2,3,4,5-tetrahydro-7-[3-(be~.~i.. ifl~7ol-2-yl)phenyl]-4-methyl-3-oxo-
lH- 1,4-benzodiazepine-2-acetate
A solution of methyl (+)-2,3,4,5-tetrahydro-7-[3-(ben7imi~1~7~ 1-2-yl)phenyl]-
1 -(tert-butoxycarbonyl)-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetate (0.06 g,
0.11 mmol) in 4 M HCl/dioxane (3 mL) was stirred for 1 hr at RT. The solution was
concentrated to give the title compound (0.05 g, 100%): MS (ES) m/e 441.4 (M+H)~.
119
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
f) (_)-2,3,4,5-Tekahydro-7-[3-(be~ idazol-2-yl)phenyl]-4-methyl-3-
oxo-lH-1,4-ben7O~ 7~pine-2-acetic acid
1.0 N NaOH (0.22 mL, 0.22 mmol) was added dropwise to a solution of
methyl (+)-2,3,4,5-tetrahydro-7-[3-(b~n7imill~7ol-2-yl)phenyl]-4-methyl-3-oxo-lH-
1,4-benzodiazepine-2-acetate (0.05 g, 0.11 mmol) in 1: 1 MeOH/H2O (2 mL) at RT,
and the resulting solution was stirred for 18 hr, then was concentrated. The residue
was dissolved in H,O and the solution was acidified with AcOH to pH 4 (Litmus
paper). Filtration gave the title compound (0.005 g, 10%): 'H NMR (250 MHz,
DMSO-d6) o 2.4-2.9 (m, 2H), 3.0-3.1 (s, 3H), 3.8-4.0 (d, lH), 5.0-5.1 (m, lH), 5.5-
5.6 (d, lH), 6.7-6.8 (d, lM), 7.5-8.5 (m, 1 lH); MS (ES) m/e 427.5 (M+H)f. Anal.Calcd for C25H"N4O3 1.5 HCl - 1.0 AcOH 0.5 H20: C, 58.94; H, 5.22; N, 10.18.
Found: C, 59.00; H, 5.15; N, 9.92.
Fx~rr~le 8
P1~AI ~lion of (+)-2.3.4.5-tetrahydro-4-methyl-3-oxo-7-r~rtph~ n~ l ., hnidazol-2-
yl)m~ thyll"minolcarbonyll- 1 H- 1.4-benzodiazepine-2-acetic acid
a) 2-[[(N-Benzyloxycarbonyl)amino]methyl]phen~nthrimi~1~7Ole
Following the general procedure of Exarnple 7(a), except substituting N-Cbz-
glycine for the 3-iodobenzoic acid, and 9,10-tliz~minophen~nthrene for the 1,2-
diaminoben_ene, the title compound (0.41 g, 45%) was prepared: MS (ES) m/e
382.4 (M+H)i.
b) 2-(Aminomethyl)phen"nthrimi(1~7Ole
A solution of 2-t[(N-benzyloxycarbonyl)amino~methyl]ph~ ~nthrimidazole
(0.2 g, 0.52 mmol) in 30% HBr in acetic acid (0.8 mL) was stirred at RT for 1 hr.
The solution was concentrated and the residue was treated with Et2O. Filtration gave
the title compound (0.138 g, 80%) as an oily residue: MS (ES) m/e 248.3 (M+H)~.
c) Methyl (_)-2,3,4,5-tetrahydro-4-methyl-3-oxo-7-[[~(phen~nthrimi~z~7ol-2-
yl)methyl]amino]carbonyl]- lH- 1,4-benzodiazepine-2-acetate
EDC (0.08 g, 0.42 mmol) was added to a solution of 2-
(aminomethyl)ph~n:~nthrimi~ le (0.138 g, 0.42 rnmol), methyl ( )-7-carboxy~
methyl-3-oxo-2,3,4,5-tetrahydro-lH-1,4-benzodiazepine-2-acetate (0.123 g, 0.42
mmol), HOBt H~O (0.063 g, 0.42 mmol) and Et,N (0.14 mL, l mmol) in anhydrous
~20
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
DMF (5 rnL~ at RT. After 18 hr, the reaction was concentrated, and the
residue was partitioned between EtOAc and 5% NaHCO3. The layers were separated
and the organic layer was washed with H2O. Drying (NazSO4) and concentration
gave the title compound (0.2 g, 90%): MS (ES) m/e 522.4 (M+H)+.
d) (+)-2~3~4~5-Tetrahydro-4-methyl-3-oxo-7-[[[(phpn~nthrim~ 7ol-2
- yl)methyl]arnino]carbonyl]-lH-1,4-bçn7o~ 7~pine-2-acetic acid
Following the procedure of Example 7(f), methyl (+)-2,3,4,5-tetrahydro-4-
methyl-3-oxo-7-[[[(phen~nthrimi(lzl71>1-2-yl)methyl]amino}carbonyl]-lH-1,4-
benzodiazepine-2-acetate (0.2 g, 0.38 mmol) was saponified and purified to give the
title compound (0.014 g, 10%): MS (ES) m/e 508.5 (M+H)+. Anal. Calcd for
C29H25N5O4 1.0 TFA 3.0 H2O: C, 55.11; H, 4.77; N, 10.37. Found: C, 55.38; H,
5.13; N, 10.74.
F.x~nl~l?le 9
Ple~)a dlion of methyl (+)-7-carboxy-4-(2.2~2-trifluoroethyl)-3-oxo-2.3.4.5-
tetr:~ydro- 1 H- 1.4-benzodiazepine-2-acetate
a) tert-Butyl 3-[(2,2,2-trifluoroethyl)arnino]methyl-4-nitroben7O~t~
tert-Butyl 3-bromomethyl-4-nitrobenzoate (2.4 g, 8 mmol)was dissolved in
dry THF (50 mL), and 2,2,2-trifluoroethylamine (3 rnL, 38 mmol) was added all atonce. The orangish-yellow solution was stirred at RT for 40 min, then was
concentrated to remove the TH~. The residue was diluted with Et20 (100 rnL) and
washed twice with 10 % aqueous Na2CO3 (50 mL) and brine (50 rnL). The organic
layer was dried (MgSO4). Concentration and silica gel chromatography (2.5% -
10% EtOAc/hexanes) gave the title compound (1.6 g, 63%) as a yellow oil: IH
NMR (250 MHz, CDC13) o 8.21 (d, J = 1.3 Hz, lH), 8.03 (dd, J = 8.4, 1.3 Hz, lH),7.96 (d, J = 8.4 Hz, lH), 4.20 (s, 2H), 3.24( q, J = 9.3 E~z, 2H), 1.62 (s, 9H).
b) tert-Butyl 3-[[N-(2,2,2-trifluoroethyl)-N-(tert-buto~yc~l~onyl)3amino]methyl-4-
nitro~en7O~te
Di-tert-butyl dicarbonate (2.15 g, 10 mmol) was added all at once to a
solution of tert-butyl 3-[(2,2,2-trifluoroethyl)amino]methyl-4-nitrobenzoate (1.6 g, 5
mmol) in CH2Cl2 (25 mT.) at RT. The reaction was concentrated and heated to 50~Cunder vacuum for 18 hours. Silica gel chromatography (2% - 5% EtOAc/h~-x~n~s)
gave the title compound (2 g, 96%) as a yellow oil: ~H NMR (400 MH_, CDCl3)
121
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
7.85-8.15 (m, 3H), 4.75-5.05 (m, 2H), 3.80-4.10 (m, 2H), 1.60 (s, 9H),
1.15-1.80 (m, 9H).
c) tert-Butyl 4-amino-3-[[N-(2,2,2-trifluoroethyl)-N-(tert-
butoxycarbonyl)]amino~methylben70~tl ~
10% Pd/C (.4 g, .4 mmol) was added to a solution of tert-butyl 3-[[N-(2,2,2-
trifluoroethyl)-N-(tert-butoxycarbonyl)3amino~methyl-4-nitrobenzoate (2.0 g, 5
mmol) in EtOAc (20 mL), and the ~ Lul~ was shaken on a Parr apparatus at RT
under H2 (55 psi). After 4 h, the reaction was filtered through celite(~), and the
filtrate was concentrated to afford the title compound (1.9 g, 99%) as a colorless oil:
lH NMR (400 MHz, CDC13) ~ 7.76 (dd, J = 8.5 Hz, 1.8 Hz, lH), 7.68 (d, J = 1.8 Hz,
lH), 6.62 (d, J = 8.4 Hz, lH), 4.53 (s, 2H), 3.69 (m, 2H), 1.58 (s, 9H), 1.51(m, 9H).
d) tert-Butyl (+)-4-[2-(1,4-dimethoxy- 1,4-dioxobutyl)anuno]-3-[[N-(2,2,2-
trifluoroethyl~-N-(tert-butoxycarbonyl)]amino]methylbenzoate
A solution of tert-butyl 4-amino-3-[[N-(2,2,2-trifluoroethyl)-N-(tert-
butoxycarbonyl)]amino]methylbenzoate (1.9 g, S mmol) and dimethylacetylene
dicarboxylate (0.58 mL, S.S mmol) in MeOH (10 mL) was heated at reflux for 60
rnin, then was cooled to RT. The resulting solution was combined with MeOE~ (20
mL) and 10% Pd/C (0.5 g, .S mrnol), and the mixture was shaken on a Parr apparatus
at RT under H2 (50 psi). After 3 h, the reaction was filtered through celite~, and the
filtrate was concentrated on the rotavap. The title compound (1.6 g, 62%) was
obtained as a faintly yellow oil: IH NMR (40V MHz, CDC13) ~ 7.85 (dd, J = 8.4, 2.0
Hz, lH), 7.68 (d, J = 2.0 Hz, lH), 6.65 (d, J = 8.4 Hz, lH), 6.15 (br s, lH), 4.55~.70
(m, 2H), 4.40 (1/2 AB, J = 15.3 Hz, lH), 3.71 (s, 3H), 3.70 (s, 3H), 3.35-3.50 (m,
2H), 2.95 (dd, J = 16.9, 6.8 Hz, lH), 2.84 (dd, J = 16.9, 6.9 Hz, lH), 1.56 (s, 18H).
e) Methyl (+)-7-carboxy-4-(2,2,2-trifluoroethyl)-3-oxo-2,3,4,5-tetrahydro-lH-1,4-
benzodiazepine-2-acetate
TFA (7 mL) was added all at once to a solution of tert-butyl (+)-4-[2-(1,4-
~lim~thoxy- 1,4-dioxobutyl)amino}-3-[[N-(2,2,2-trifluoroethyl)-N-(tert-
butoxycarbonyl)]amino]methylbenzoate (1.6 g, 3 mmol) in anhydrous CH2C12 (20
rnL) at 0~C, and the faintly yellow solution was warmed to RT. After 2 h, the
solution was concentrated on the rotavap, and the residue was reconcentrated from
toluene (to remove residual TFA). The reSlllting oil was combined with toluene (10
mL) and Et3N (2 mL, lS mmol), and the mixture was heated to reflux under argon.
After 18 hr the solution was allowed to cool and was concenll~ted to dryness under
122
CA 02241633 1998-06-26
W O 97/24119 PCT~US96120748
vacuum. The residue was dissolved in a miniml~m of MeOH (ca. 15 mL)
at reflux, diluted with H2O ~10 ml), and acidified with glacial AcOH (4 drops). The
mixture was kept in the refrigerator overnight then was filtered. The solid was dried
under high vacuum to afford the title compound (0.80 g, 76%) as a tan powder: lHNMR (400 MHz, DMSO-d6) o 7.61 (2, lH), 7.57 ~dd, J = 8.5, 2 Hz, lH), 6.63 (d, J =
2 Hz, lH), 6.59 (d, J = 8.5 Hz, lH), 5.59 (d, ~ = 16.7 Hz, lH), 5.25 (m, lH), 4.28 (m,
2H), 4.15 (d, J = 16.7 Hz, lH), 3.61 (s, 3H), 2.86 (dd, J = 16.8, 8.7 Hz, lH), 2.74
(dd, J = 16.8, 5.4 Hz, lH).
f) Methyl (+)-2,3,4,5-tetrahydro-7-[[~(benzimidazol-2-yl)methyl]amino]carbonyl]-3-oxo-4-(2,2,2-trifluoroethyl)- 1 H- 1 ,4-bçn70~ 7epine-2-acetate
EDC (0.16 g, 0.86 mmol) was added at RT to a solution of methyl (+)-7-
carboxy4-(2,2,2-trifluoroethyl)-3-oxo-2,3 ,4,5-tetrahydro- 1 H- 1 ,4-benzodiazepine-2-
acetate (0.20 g, 0.71 mmol), HOBt ~ H2O (0.12 g, 0.86 mmol), 2-
aminomethylb~n7imill~701e dihydrochloride (0.19 g, 0.86 mmol), DIEA (0.5 mL,
2.8 mmol), and acetonitrile (5 mL) under argon. The rçslllting solution was stirred
at RT overnight, then was concentrated. The residue was partitioned between ethyl
acetate and water, and the layers were separated. The organic phase was washed
with brine, dried (MgSO4), and concentrated. Silica gel chromatography ~1% - 10%CH30H in CH2Cl~) gave the title compound (0.12 g, 44%) as a tan solid: NMR (400
MHz, DMSO-d6) o 8.59 (t, J = 5 Hz, lH), 7.61 (m, 2H), 7.50 (m, 2H), 7.16 (m, 2H),
6.57 (d, J = 11.1 Hz, lH), 6.17 (d, J = 5 Hz, lH), 5.53 (d, J = 16.7 Hz, lH), 5.13 (m,
lH), 4.75 (m, 2H), 4.10 (m, 2H), 3.62 (s, 3H), 2.94 (dd, J = 16.8, 8.5 Hz, lH), 2.69
(dd, J = 16.8, 5.4 Hz, lH).
g) (+)-2,3,4,5-Tetrahydro-7-[[[(bçn7imi~1~7ol-2-yl)methyl]amino~carbonyl]-3-oxo-4-(2,2,2-trifluoroethyl)- 1 H- 1 ,4-benzodiazepine-2-acetic acid
A solution f methyl(f)-2,3,4,5-tetrahydro-7-[[[(bçn7imi~l~7ol-2-
yl)methyl]amino]carbonyl]-3-oxo4-(2,2,2-trifluoroethyl)- lH- 1 ,4-ben7o~ 7~pine-2-
acetate (0.12 g, 0.25 mmol) and lithium hydroxide monohydrate (0.017 g, 0.4 mmol)
in THF (10 mL), CH30H (2 mL), and H2O (2 mL) was stirred at RT overnight. It
was then concentrated and the residue was dissolved in water. The solution was
brought to pH 4 with 3 N HCI, then was refrigerated for 1 hour. The resulting solid
was collected by filtration and dried to give the title co~ oul-d (0.11 g, 90%) as a
~ 35 white solid: Ms (ES) m/e 476 ~M+H]~. Anal. Calcd for C22H2oN5F304 1.25 H20: C,
53.07; H, 4.55; N, 14.06. Found: C, 52.85; H, 4.36; N, 13.98.
123
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
Example 10
Preparation of f+)-2.3.4.5-tetr~ dro-7-rrrbenzimidazol-2-
yl)methyllmethylaminolcarbon,yll-3-oxo-4-(2.2.2-trifluoroethyl)- 1 H- 1.4-
S berl7~ 7Ppine-2-acetic acid
a) Methyl (+)-2,3,4,5-tetrahydro-7-[[[ben7imi(1:-7ol-2-
yl)methyl]methylamino]carbonyl~-3-oxo-4-(2,2,2-trifluoroethyl)- I H- 1,4-
benzodiazepine-2-acetate
Using the procedures of Example 9(f), except substituting 2-
(methylaminomethyl)bçn7imic~7ole dihydrochloride for the 2-
aminomethylbenzimidazole dihydrochloride, the title compound was prepared: 'H
NMR (400 MHz, CDCl3) ~ 7.67 f~m, 2 H), 7.37 (m, 2 H), 7.25 (m, 2 H), 6.54 (d, J =
8 Hz, 1 H), 5.46 (d, J = 16.7 Hz, 1 H), 5.20 (m, 1 H), 5.04 (s, 2 H), 4.71 (m, 1 H),
4.17(m,1H),3.94(m,1H),3.92(d,J=16.7,1H),3.74(s,3H),3.23(s,3H),2.98
(m, 1 H), 2.74 (m, 1 H).
b) (i)-2,3,4,5-Tetrahydro-7-[trbçn7imiti~7ol-2-yl)methyl]methylamino]carbonyl]- -
3-oxo-4-(2,2,2-trifluoroethyl)- 1 H- 1,4-benzodiazepine-2-acetic acid
Using the procedure of Example 9(g), methyl (+)-2,3,4,5-tetrahydro-7-
[[rbenzimidazol-2-yl)methyl]methylamino]carbonyl] -3-oxo-4-(2,2,2-trifluoroethyl)-
lH-1,4-benzodiazepine-2-acetate was saponified to afford the title compound: MS
f~ES) mte 490.2 [M+H~+. Anal. Calcd for C23H22N5F30" 2.25 H20: C, 52.12; H,
5.04; N,13.21. Found: C, 52.00; H, 5.12; N, 13.09.
Fx~n~l?le 11
Prep ~ration of (+)-2.3.4.5-tetrahydro-7-rrr(4-aza-S-methylben7.imicl~7ol-2-
yl~rnf~tllyllaminolcarbonyll-3-oxo-4-f2.2.2-trifluoroethyl)-lH-1.4-benzodiazepin~-2-
zlf~Ptic acid
a) Methyl (+)-2,3,4,5-tetrahydro-7-~[r(4-aza-5-methylben7imirl:~7OI-2-
yl)methyl]amino]carbonyl]-3-oxo-4-(2,2,2-trifluoroethyl)- 1 H- 1,4-benzodiazepine-2-
acetate
Using the procedures of Fx:~rnpl~ 9(f~, except ~ubsLiLuLing 2-(aminomethyl)-
4-aza-5-methylben7imi~ 7O1e dihydrochloride for the 2-~minom( thylben7imi-l~7oledihydrochloride, the title colllp-)ul~d was prepared: MS (ES) rnte 505.2 ~M + H)+.
124
CA 02241633 1998-06-26
W O97124119 PCTrUS96/20748
b) (~ 2,3,4,5-Tetrahydro-7-[[[(4-aza-5-methylben7imi~1~7ol-2-
yl)methyl]amino}carbonyl]-3-oxo-4-(2,2,2-trifluoroethyl)- lH- 1,4-b~4n7.o-1iA7epine-2-
acetic acid
Using the procedure of Example 9(g), methyl (~)-2,3,4,5-tetrahydro-7-[[[(4-
aza-5-methylbenzimidazol-2-yl)methyl]amino]carbonyl]-3-oxo-4-(2,2,2-
~ trifluoroethyl)-lH-1,4-benzodia~epine-2-acetate was saponified to afford the title
compound: MS (ES) m/e 491.2 (M+H)+. Anal. Calcd for C~H2lN6F3O4 - 2 7/8 H2O:
C, 48.73; H,4.97; N, 15.50. Found: C, 48.50; H, 4.59; N, 15.33.
F.x~n~le 12
al~lion of (+)-2.3.4.5-tetrahydro-7-rrr(ber~7imi(1~7ol-2-
yl)methyllmethylamino]carbonyll-4-r2-(3.4-methylenedioxyphenyl)ethyl]-3-oxo-
lH-1.4-benzotli~7~-pine-2-acetic acid
a) Methyl (+)-2,3,4,5-tetrahydro-7-[[[(ben7imi~ 7~-1-2-
yl)methyl3methylamino]carbonyl]-4-[2-(3,4-methylenedioxyphenyl)ethyl]-3-oxo-
lH- 1,4-benzodiazepine-2-acetate
EDC (0.10 g, 0.55 mmol) was added at RT to a solution of methyl 7-
carboxy-4-[2-(3,4-methylenedioxyphenyl)ethyl]-3-oxo- 1 H- 1,4-benzodi~epine-2-
acetate (0.14 g, 0.3 mmol), 2-(methylaminomethyl)ben7imicl:~701e dihydrochloride(0.12 g, 0.51 mmole), HOBt ~ H20 (0.072 g, 0.55 mmol), and DIEA ~0.32 mL, 1.84
mmole), in DMF (5 mL) under argon. The resulting solution was stirred at RT
overnight, then was concentrated. The residue was partitioned between ethyl acetate
and water, and the layers were separated. The aqueous phase was extracted with
ethyl acetate, and the combined organic phases were washed with brine, dried
(MgSO4), and concentrated. Silica gel chromatography gave the title compound
(0.11 g, 59%) as a colorless foam: 'H NMR (CDCl 3) o 7.62 (m, 2H), 7.31 (m, 2H),7.20 (d, J = 8.1 Hz, lH), 7.07 (s, lH), 6.65 (d, J = 7.9 Hz, lH), 6.60 (s, lH), 6.55 (d,
J=7.9Hz, lH),6.46(d,J=8.1 Hz, lH),5.90(d,J=5.4Hz,2H),5.26(d,J= 16.5
Hz, lH), 5.02 (m, lH), 4.93, (d, J = 14.6, lH), 4.83 (d, J = 14.6 Hz, lH), 4.51 (d, J =
5 Hz, lH), 3.74 (s, 3H), 3.71 (m, lH), 3.60 (m, lH), 3.58 (d, J = 16.5 Hz, lH), 3.18
(s, 3H), 2.99 (dd, J = 16, 6.8 Hz, lH), 2.70 (m, lH).
b) (+)-2,3,4,5-Tetrahydro-7-[[[(ben7imi~1~7Ol-2-yl)methyl]methylamino]carbonyl]-4-~2-(3,4-methylenedioxyphenyl)ethyl] -3-oxo- 1 H- 1,4-benzodiazepine-2-acetic acid
125
CA 02241633 1998-06-26
W O97/24119 PCT~US96/20748
To a solution stirred at RT of methyl (_)-2,3,4,5-tetrahydro-7-
[[[(benzirnidazol-2-yl)methyl]methylamino]carbonyl]-4-[2-(3,4-
methylenedioxyphenyl)ethyl]-3-oxo-lH-1,4-benzodiazepine-2-acetate (0.11 g, 0.19
rnmol) in THF (1 mL) was added a solution of lithium hydroxide monohydrate (0.01g, 0.23 rnrnol) in H20 (1 mL). The r~s~ ing solution was stirred at RT overnight,
then was concentrated to dryness. The residue was dissolved in H2O and the
solution was washed with ethyl acetate, then was brought to pH 4 with 3 N HCl.
The res~ ing precipitate was collected by ~lltration and dried to give the titlecompound (0.055 g, 51%) as a white solid. MS (ES) m/e 556.2 [M+H]+. Anal.
Calcd for C3nH29NsO6 H2O: C, 62.82; H, 5.45; N, 12.21. Found: C, 62.69; H, 5.26;N, 12.15.
Fx~ ?le 13
~lel)a-~lion of (+)-2.3.4.5-tetrahydro-7-rl2-(benzimidazol-2-yl)acetyll~mino]-5-oxo-
4-(2-phenylethyl)- 1 H- 1.4-benzodi~epine-2-acetic acid
a) Methyl (+)-2,3,4,5-tetrahydro-7-[t2-(be~ idazol-2-yl)acetyl]amino]-5-oxo-4-
(2-phenylethyl)- 1 H- 1,4-benzodiazepine-2-acetate
EDC (0.27 g, 1.4 rnmole) was added at RT to a solution of (_)-2,3,4,5-
tetrahydro-7-amino-5-oxo4-(2-phenylethyl)- 1 H- 1,4-benzodiazepine-2-acetate (0.40
g, 1.1 mmol), ben7imi~1~7Qle-2-acetic acid (Archiv. Der Pharmazie 1960, 293, 758;
0.25 g, 1.4 rnmol), HOBt ~ H[2O (0.20 g, 1.5 mmol), and DIEA (0.35 mL, 2 mmol) in
acetonitrile (10 rnL). The res~ ing solution was stirred for 2 days, then was
concentrated to dryness. The residue was partitioned between ethyl acetate and
water, and the layers were separated. The organic phase was washed with brine,
dried (MgSO4), and concentrated. Silica gel chromatography (1% - 10% CH30H in
CH2Cll) gave the title compound (0.21 g, 36%) as an amber foam: MS (ES) m/e
512.2 [M+H]+.
b) (+)-2,3,4,5-Tetrahydro-7-t[2-(benzimidazol-2-yl)acetyl]amino]-5-oxo4-(2-
phenylethyl)- 1 H- 1,4-benzodiazepine-2-acetic acid
A solution of methyl (_)-2,3,4,5-tetrahydro-7-[~2-(ben7.imi~ 1-2-
yl)acetyl]amino]-5-oxo4-(2-phenylethyl)- 1 H- 1,4-benzodiazepine-2-acetate (0.21 g,
0.41 mmole) and lithium hydroxide monohydrate (0.022 g, 0.52 mmol) in THF (10
mL) and H20 (2 mL) was stirred at RT overnight, then was then concentrated. The
residue was dissolved in water and the solution was washed with ethyl acetate, then
126
CA 02241633 1998-06-26
W O 97124119 PCT~US96/20748
was brought to pH 4 with 3 N HCl. The resulting precipitate was collected
by filtration and dried to give the title compound (0.12 g, 59%) as an off-white solid:
MS (ES) m/e 498.2 [M+H]+. Anal. Calcd for C28H27N5O4 ~ 1.5 H2O: C, 64.11; H,
5.76; N, 13.35. Found: C, 64.36; H, 5.57; N, 13.21.
Exarnple 14
Preparation of (S)-2.3~4.5-tetrahydro-7-rrr(benzimidazol-2-
yl)methyllmethylaminolcarbonyl1 -4-methyl-3-oxo- 1 H- 1.4-benzodiazepine-2-
acet~mi~l~
a) (S)-2,3,4,5-tetrahydro-7-t[[(bçn7imi~1~7Ol-2-yl)methyl]methylamino]carbonyl]-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-~et~mi~le
Methyl (+)-2,3,4,5-tetrahydro-7-[[[(ben ~ 7Ol-2-
yl)methyl]amino]callJonyl]-4-methyl-3-oxo-lH-1,4-benzodiazepine-2-acetate (330
mg, 0.76 mmole) in dry MeOH (10 mL) was cooled in an ice bath as ammonia was
bubbled into the solution for 0.5 hr. The reaction was then allowed to sit stoppered
at RT for 18 hr. After concentration, the residue was purified by silica gel flash
chromatography (90: 10 CH2Cl2/MeOH) to give the title compound (52%) as a white
solid: MS (ES) m/e 421.2 ~M+H~'. Anal. Calcd for C2~H24N603 1.5 H~O: C, 59.05;
H, 6.08; N, 18.78. Found: C, 58.90; H, 6.04; N, 18.45.
Fx~n~le 15
~ aldtion of (+)-S-rr2.3.4.5-tetrahydro-7-rrr(benzimidazol-2-
yl)methyllamino~carbonyll-3-oxo4-(2-phenylethyl)- 1 H- 1.4-benzodiazepin-2-
yllm~thylltetrazole
a) Methyl (+)-2,3,4,5-tetrahydro-7-(tert-butoxycarbonyl)-3-oxo-4-(2-phenylethyl)-
lH-l,~benzodiazepine-2-acetate
Methyl (+)-2,3,4,5-tetrahydro-7-carboxy-3-oxo~-(2-phenylethyl)- 1 H- 1,4-
b~n70~ 7~pine-2-acetate (1.0 g, 2.6 mmole) was suspended in toluene (10 mL), andN,N-dimethylformz-micle-di-tert-butyl acetal (5 mL, 20.8 mmole) was added
dropwise. The reaction mixture was heated at 80~C for 1.5 hr, then was cooled toRT and poured into 5% Na~CO3 solution. The layers were separated, and the
aqueous was extracted with toluene (2 x). The combined organic layers were
127
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
washed with brine, dried over MgS04, filtered and evaporated to give the
title compound (0.9lg, 82%). MS (ES) m/e 439.2 [M+H]+.
b) (i)-2,3,4,5-Tetrahydro-7-(tert-butoxycarbonyl)-3-oxo-4-(2-phenylethyl)- lH- 1,4-
benzodiazepine-2-acetic acid
A solution of methyl (i)-2,3,4,5-tetrahydro-7-(tert-butoxycarbonyl)-3-oxo-4-
(2-phenylethyl)-lH-1,4-benzodiazepine-2-acetate (1.5 g, 3.4 mmole) in ethylene
glycol dimethyl ether (160 mL) was treated with H2O (20 mT ) and 0.91 N NaOH (5
mT.). The reaction was stirred under argon at RT for 24 hr, then was acidified to pH
3 with glacial AcOH, concentrated to a small volume (10 mL), and poured into iceH2O. The precipitated solid was collected and dried giving the title compound inqll~ntit~tive yield. MS (ES) m/e 425.2 ~M+H]+.
c) (+)-2,3,4,5-Tetrahydro-7-(tert-butoxycarbonyl)-3-oxo-4-(2-phenylethyl)- lH- 1,4-
1 5 benzodiazepine-2-[N-(2-cyanoethyl)~cet~mide]
A solution of (i)-2,3,4,5-tetrahydro-7-(tert-butoxycarbonyl)-3-oxo-4-(2-
phenylethyl)-lH-1,4-benzodi~epine-2-acetic acid (1.2 g, 2.6 mmole) in dry DMF
(12 mL) under argon was treated with diisopropylethylamine (0.65 g, 5 mrnole),
EDC (0.764 g, 4 rnmole) and HOBt ~ H2O (0.54g, 4 mmole). The resulting solution
was stirred for 10 min, then was treated with a solution of 3-aminopropionitrilefumarate in dry DMF (2 mL) cont~inin~ diisopropylethylamine (0.85 g, 6.6 lnrnole).
The reaction was stirred under argon ~or 18 hr, then was concentrated to dryness.
The residue was partitioned between H,O and EtOAc, and the layers were separated.
The organic layer was washed with brine, dried over MgSO4, filtered and
concentrated. The oily residue was purified by silica gel flash chromatography (98:2
CH2Cl~/MeOH) to give the title colllpoulld (750 mg, 50%): MS (ES) 477.2 [~+H]+.
d) (i)-1-(2-Cyanoethyl)-5-[[2,3,4,5-tetrahydro-7-(tert-butoxycarbonyl)-3-oxo-4-(2-
phenylethyl)- lH- 1 ,4-benzodiazepin-2-yl]methyl]tetrazole
A solution of ~+)-2,3,4,5-tetrahydro-7-(tert-butoxycalbollyl)-3-oxo~-(2-
phenylethyl)-lH- 1 ,4-benzodi~epine-2-[N-(2-cyanoethyl)acet~mide] (750 mg, 1.3
mmole) in dry THF (15 mL) under argon was treated with triphenylphosphine (1.14
g, 4.6 mrnole), trimethylsilyl~ide (0.52 g, 4.6 mmole), and diethyl~odicarboxylate
(0.8 mL, 4.6 mmole) at RT under argon. After 50 hr, the reaction was concentrated
to dryness, and the residue was purified by silica gel flash chromatography (98.5:1.5
CH2Cl2/MeOH) to give the tit~e co~ ou,ld (0.56g, 86%): MS (ES) m/e 502.2
[M+H]+.
128
CA 0224l633 l998-06-26
W O97/24119 PCTrUS96/20748
e) (+)-1-(2-Cyanoethyl)-5-[[2,3,4,5-tetrahydro-7-carboxy-3-oxo-4-(2-phenylethyl)-
lH-1,4-benzodiazepin-2-yl]methyl]tetrazole
A solution of (+)-1-(2-cyanoethyl)-5-[[2,3,4,5-tetrahydro-7-(tert-
butoxycarbonyl)-3-oxo-4-(2-phenylethyl)- 1 H- 1,4-benzodi~epin-2-
yl~methyl]tetrazole (0.5 g, 1 mmole) in CH2Cl2 (20 mL) at RT under argon was
treated with 4 M HCl in dioxane (10 mL). After 20 hr, the reaction was
concentrated to dryness, and the residue was diluted with 5% Na2CO3. The solution
was extracted with EtOAc, and the ELOAc layer was discarded. The aqueous layer
was acidified with dil HCl and extracted with EtOAc (3x). The combined EtOAc
extracts were washed with brine, dried over MgSO4 and evaporated to yield the title
compound (0.36g, 81%): MS(ES) m/e 445.4 [M+H]+.
f) (+)-1-(2-Cyanoethyl)-5-[[2,3,4,5-tetrahydro-7-[[[(b~n7imi~1~7ol-2-
yl)methyl]amino]carbonyl] -3 -oxo-4-(2-phenylethyl)- 1 H- 1,4-benzodiazepin-2-
yl]methyl]tetrazole
A solution of (+)-1-(2-cyanoethyl)-5-[[2,3,4,5-tetrahydro-7-carboxy-3-oxo-4-
(2-phenylethyl)-lH-l,~ben7ofli~7epin-2-yl]methyl]tetrazole) (360 mg, 0.8 rnmole)dry DMF (5 mL) was treated under argon with DIEA (129 mg, 1 ~nmole), EDC (172
mg, 0.9 mmole) and HOBt ~ H2O (122 mg, 0.9 mmole). The reaction was stirred at
RT for 10 min, then a solution of 2-aminomethylben7imit~701e dihydrochloride
hydrate (352 mg, 1.6 mmole) in DMF (2 mL) cont:lining DIEA (413 mg, 3.2
mmole) was added. After 20 hr, the reaction was concentrated to dryness and the
residue was partitioned between }~2O and EtOAc. The layers were separated, and the
aqueous layer was extracted with EtOAc. The organic layers were combined, dried
over MgSOt, filtered and concentrated. The residue was purified by silica gel flash
chromatography (95:5 CH2Cl2/MeOH) to give the title compound (120 mg, 21%):
MS (ES) m/e 575.2 [M+H]+.
g) (+)-S-[r2,3,4,5-Tetrahydro-7-[[[(b~ 7-)1-2-yl)methyl]amino]carbonyl]-3-
oxo~-(2-phenylethyl)- l H- 1,4-benzodiazepin-2-yl]methyl]tetrazole
A solution of (+)-1-(2-cyanoethyl)-5-[[2,3,4,5-tetrahydro-7-[[[(ben7imi~ 7Ol-
2-yl)methyl]amino]carbonyl]-3-oxo-4-(2-phenylethyl)- I H- 1,4-benzodiazepin-2-
yl]methyl]tetrazole (100 mg, 0.2 mmole) in MeOH (1 mL) was treated with
~ 35 thiophenol (0.02 mL) followed by 1 N NaOH solution (2.2 mL). After 3 hr, the
reaction was concentrated to dryness, and the residue was purified by silica gel~pa,a~ive TLC (85: 15 CH2Cl2/MeOH). The isolated product was dissolved in HzO,
129
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
and the solution was filtered to remove insoluble m~t~ri~l~ The filtrate
was treated with 2 drops of glacial AcOH. The precipitated solid was collected and
dried to give the title compound (45 mg, 41%): MS (ES) m/e 522.2 ~M+H]'. Anal.
Calcd for C3~ Ng04 ~ 2.25 H2O: C, 56.15; H, 5.52; N,19.65. Found: C, 56.51; H,
5.05; N, 19.72.
~x~rn,ple 16
Preparation of (S)-2.3.4.5-tetrahydro-7-rrrfbe~ .idazol-2-
10 yl~methyllaminolcarbonyll-3-oxo4-r4-r(2-carboxybenzoyl)aminolbut-1-yll-lH-1.4-
ben70~ pine-2-acetic acid
a) N-[[2-(N-4-hydroxybut- 1-yl)aminomethyl-4-tert-butoxycarbonylJphenyl~-L-
aspartic acid ,l3-methyl ester
A mixture of N-[[2-formyl-4-tert-butoxycarbonyl]phenyl]-L-aspartic acid ,13-
methyl ester (WO 95/18619; 2.55g, 7.26 mmol), 4A molecular sieves, and 4-
hydroxybutylamine (0.64 g, 7.26 mmol) in MeOH (35 mL) was stirred under argon
at RT for 30 min, then sodium cyanoborohydride (0.49 g, 0.79 mmol) and acetic
acid (0.3 mL) were added. The reaction mixture was kept at RT overnight and then20 the solvent was elimin~te~l in vacuo. The residue was dissolved in H20 and the
solution was acidified to pH 4 with dil HCl. EtOAc extraction, drying (MgSO4),
filtration, and conce~ dLion gave the title compound ( 1.75 g, 57%) as a pale yellow
solid: TLC Rf (4:20:20:56 MeOH/EtOAc/hexane/Cl2CH~) 0.22; 1~ NMR (CDCl3) o
1.55 (s, 9H), 1.56 (m, 2H), 1.80 (m, 2H), 3.01 (m, 4H), 3.55 (m, 2H), 3.70 (s, 3H),
25 4.05 (m, lH), 4.40 (m, lH), 4.55 (m, lH), 6.81 (d, J = 8.4 Hz, lH), 7.70 (s, lH), 7.89
(d, J = 8.4 Hz, lH).
b) Methyl (S)-7-(tert-butoxycarbonyl)-2,3,4,5-tetrahydro-3-oxo-4-(4-hydroxybut- 1 -
yl)- 1 H- 1,4-benzodi~epine-2-acetate
To a solution of N-[[2-(N-4-hydroxybut-1-yl)aminomethyl-4-tert-
butoxycarbonyl]phenyl]-L-aspartic acid ,B-methyl ester (1.75 g, 4.1 mmol) and
triethylamine (1.15 mL, 8.2 mmol) in dichloromethane (150 mL) under argon at RT
was added benzotriazol- 1 -yloxy-tris(dimethylamino)phosphonium
hexafluorophosphz-~ (2.08 g, 14.7 mmol). The reaction mixture was stirred
35 overnight at RT, then was washed sequentially with ice-cold dil HCl, water, 5%
sodium bicarbonate, saturated brine, and then dried (MgS04). Filtration and
concentration left a residue which was purified by silica gel flash column
130
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
chromatography (5% mPth~nol:ethyl acetate) to give the title compound
(0.631 g, 38%): TLC Rf (4% MeOH/EtOAc) 0.26; IH NMR (CDC13) o 1.46-1.61
(m, 4H), 1.57 (s, 9H), 2.64 (d, J = 6.9 Hz, lH), 2.66 (dd, J = 15.9, 6.3 Hz, lH), 2.99
(dd, J = 15.6, 6.9 Hz, lH), 3.56-3.54 (m, 4H), 3.74 (s, 3H), 3.84 (d, J = 16.2 Hz, lH),
4.54 (m, lH), 5.10 (m, lH), 5.41 (d, J = 16.2 Hz, lH), 6.49 (d, J = 8.3 Hz, lH), 7.59
(d, J = 1.8 Hz, lH), 7.67 (dd, J = 8.3, 1.8 Hz, lH); MS (ES) m/e 407.2 [M+H]+; ~CC~D
= - 185.4~ (c = 1, CH30H).
c) Methyl (S)-7-(tert-butoxycarbonyl)-2,3,4,5-tetrahydro-3-oxo-4-(4-
phth~limic~obut-1-yl)-lH-1,4-benzodiazepine-2-acetate
To a solution of methyl (S)-7-(tert-butoxycarbonyl)-2,3,4,5-tetrahydro-3-
oxo-4-(4-hydroxybut-1-yl)-lH-1,4-benzodiazepine-2-acetate (437 mg, 1.07 mmol)
and triphenylphosphine (308 mg, 1.17 mmol) in THF (20 mL) at RT under argon
was sequentially added phth~limi~ (173 mg, 1.17 mmol) and diethyl
azodicarboxylate (205 mg, 1.17 mmol). The reaction ll~ib~luie was stirred overnight
at RT, the solvent was removed, and the residue was purified by silica gel flashcolumn chromatography (4:20:20:56 m~ths~n~ll/ethyl acetatethexane/methylene
chloride) to give the tit}e compound (0.430 g, 75%): TLC Rf (4:20:20:56
MeOH/EtOAc/hexane/Cl2CH2) 0.32; IH NMR (CDCl3) o 1.55 (s, 9H), 1.55-1.61 (m,
4H), 2.68 (dd, J = 14.0, 5.7 Hz, 2H), 2.98 (dd, J = 14.0, 6.6 Hz, lH), 3.46-3.64 (m,
4H), 3.71 (s, 3H), 3.85 (d, J = 16.5 Hz, lH), 4.63 (d, J = 4.4 Hz, lH), 5.08 (dd, J =
5.7, 6.6 Hz, lH), 5.37 (d, J = 16.5 Hz, lH), 6.48 (d, J = 8.3 Hz, lH), 7.67 (s, lH),
7.69 (d, J = 8.3, 1.8 Hz, lH), 7.72-7.76 (m, 2H), 7.81-7.86 (m, 2H).
d) Methyl (S)-2,3,4,5-tetrahydro-7-carboxy-3-oxo-4-(4-phth:~limidobutyl)-lH-1,4-benzodiazepine-2-acetate
To a solution of methyl (S)-7-(tert-butoxycarbonyl)-2,3,4,5-tetrahydro-3-
oxo-4-(4-phth~limidobutyl)-lH-1,4-benzodiazepine-2-acetate (660 mg, 0.89 m~nol)
in dichloromethane (20 mL) was added 4 N HCI/dioxane (5 mT, 20 mmol) at RT
under argon. The reaction mixture was stirred for 18 hr. The s~lspen~ion was
concentrated to give the title compound as an off-white solid (425 mg, 98%): IH
NMR (CDCl3) ~ 1.55-1.61 (m, 4H), 2.71 (dd, J = 14.1, 6.0 Hz, 2H), 3.01 (dd, J =
14.1, 6.3 Hz, lH), 3.50-3.65 ~m, 4H), 3.75 (s, 3H), 3.89 (d, J = 16.5 Hz, lH), 4.68
- (d, J = 4.5 Hz, lH), 5.12 (dd,3 = 6.0, 6.3 Hz, lH), 5.40 (d, J = 16.6 Hz, lH), 6.41
- 35 (bs, lH), 6.53 (d, J = 8.4 Hz, lH), 7.69-7.75 (m, 4H), 7.82-7.85 (m, 2H); MS (l~S)
m~e 480.2 [M+H]+.
131
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
e~ Methyl (S)-2,3,4,5-tetrahydro-7-[[[(b~n7imi-1~7Ol-2-
yl)methyl3amino]carbonyl]-3-oxo~-(4-phth:~limiclobut-1-yl)-lH-1,4-
benzodiazepine-2-acetate
EDC (240 mg, 1.25 mmol) was added to a stirred solution of methyl (S)-
2,3,4,5-tetrahydro-7-carboxy-3-oxo-4-(4-phth~limi~lQbut-l-yl)-lH-1,4-
benzodiazepine-2-acetate (0.85 g, 0.88 mmol), 2-(aminomethyl)'oe~.7i.,~ 7Ole
dihydrochloride (230 mg, 1.04 mmol), HOBt -H2O (169 mg, 1.25 mmol), and
diisopropylethylamine (0.78 mL, 4.5 mmol) in anhydrous acetonitrile (10 rnL) at
RT. After 19 h, the reaction was concentrated on the rotavap (high vacuum), and the
residue was partitioned between H20 (5 mL) and EtOAc (20 mL). The layers were
separated and the organic layer was washed with H2O (5 rnL). Drying (MgSO4),
concentration, and silica gel chromatography (5% MeOH/CH2Cl2), gave the title
compound (230 mg, 43%) as an off-white solid: TLC Rf (5% MeOH/Cl2CH2) 0.30;
lH NMR (CD30D) o 1.42-1.56 (m, SH), 2.63 (dd, J = 6.4, 16.2 Hz, lH), 2.95 (dd, J= 6.7, 16.2 Hz, lH), 3.33-3.40 (m, 2H), 3.48-3.55 (m, 2H), 3.57 (d, J = 16.5 Hz, lH),
3.67 (s, 3H), 4.72~.80 (m, 3H), 5.03 (dd, J = 6.4, 6.7 Hz, lH), 5.20 (d, J = 16.5 Hz,
lH), 6.44 (d, J = 8.4 Hz, lH), 7.18-7.21 (m, 2H), 7.52-7.63 (m, 6H); 7.74-7.76 (m,
2H), 9.08 (br s, lH).
f) (S)-2,3,4,5-Tetranydro-7-[t[(b~n7imicl~7ol-2-yl)methyl]amino]carbonyl]-3-oxo-4-[4-[(2-carboxybenzoyl)amino]but-1-yl]- lH-1,4-bt-.n7oflizl7~pine-2-acetic acidLiOH (30 mg, 0.71 mmole) was added at RT to a solution of methyl (S)-
2,3,4,5-tetrahydro-7-[[[(benzimidazol-2-yl)methyl]amino]carbonyl]-3-oxo-4-(4-
phth:~limi~lobut-l-yl)-lH-1,4-benzodiazepine-2-acetate (223 mg, 0.33 mmol) in
MeOH (2 mL) and H20(3 mL). The reaction mixture was sti;red at RT for 19 hr.
Acidification with dil HCl to pH 4 and concentration produced a solid. Filtration
gave the title compound (145 mg, 66%) as a white solid: ~a]D - -100.4~ (c = 1,
CH30H); IH NMR (CD30D) ~ 1.32-1.65 (m, 5EI), 2.58 (dd, J = 16.4, 6.7 Hz, lH),
2.90 (dd, J = 16.4, 7.9 Hz, lH), 3.06 (m, lH), 3.69 (m, lH), 4.00 (d, J = 16.7 Hz,
lH),4.69(brs,2H),5.11 (dd,J=7.9,6.7Hz, lH~, 5.36(d,J= 16.7Hz, lH),6.50
(d, J = 8.4 Hz, lH), 7.29 (m, 2H), 7.37 (m, 4H); 7.51 (m, 2H), 7.664 (s, lH), 7.74 (d,
J = 6.8 Hz, lH); MS (ES) m/e 613.2 [M+H]+. Anal. Calcd for C32H32N607 -1.5
H2O: C, 60.0g; H, 5.51; N, 113.14. Found: C, 59.77; H, 5.46; N, 12.98.
E~ ?le 17
132
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/207~8
F'lc~aldlion of (+)-7-r3-(benzill,idazol-2-yl)propyll-4-methyl-3-oxo-
2.3.4.5-tetrahydro- 1 H- 1.4-benzodiazepine-2-acetic acid
a) Methyl (_)- 1 -(tert-butoxycarbonyl)-7-(4-hydroxy- 1 -butyn- 1 -yl)-4-methyl-3-oxo-
2,3,4,5-tetrahydro- 1,4-benzodiazepine-2-acetate
3-Butyn-l-ol (65 mg, 0.93 mmol), bis(triphenylphosphine)pS~ m (II~
chloride (5 mg, 0.007 mmol), triphenylphosphine (10 mg, 0.038 mmol), and
copper(I) iodide (10 mg, 0.052 mmol) were added under an Ar atmosphere to a
solution of methyl (+)-2,3,4,5 -tetrahydro- 1 -(tert-butoxycarbonyl)-7-iodo-4-methyl-
3-oxo-lH-1,4-benzodiazepine-2-acetate (obtained as in example 7c; 440 mg, 0.94
mmol) in triethylamine (34 m~ ). The reaction mixture was heated to reflux for 4 hr,
then was filtered through celite(~), and the filtrate was concentrated. The residue was
purified by silica gel flash column chromatography (4:20:20:56
MeOH/EtOAc/hexane/CI2CH2) to give the title compound (390 mg, g4%) as a pale
yellow liquid: TLC Rf (5% MeOH: Cl2CH2) 0.37; IH NMR (400 MHz, CDCl3) o
7.34 (dd, J = 1.8, 8.1 Hz, lH), 7.31 (d, J = 1.8 Hz, lH), 7.13 (d, J = 8.1 Hz, lH),
5.15-5.23 (m, lH), 4.75 (d, J = 14.4 Hz, lH), 3.68 (d, J = 14.4 Hz, lH), 3.63-3.67
(m, 2H), 3.59 (s, 3H), 3.04, (s, 3H), 2.88 (dd, J = 5.5, 15.2 Hz, lH), 2.45 (t, J = 6.4
Hz, 2H), 2.26 (dd, J = 9.5, 15.2 Hz, lH), 2.04 (br s, lH), 1.34 (br s, 9H); MS (ES)
m/e 417 [M+H]+.
b) Methyl ( )- 1 -(tert-butoxycarbonyl)-7-(4-hydroxybut- 1 -yl)4-methyl-3-oxo-
2,3,4,5-tetrahydro- 1,4-benzodiazepine-2-acetate
10% Pd/C (40 mg) was added to a solution of methyl (+)-l-(tert-
butoxycarbonyl)-7-(4-(hydroxy- 1 -butyn- 1 -yl)-4-methyl-3-oxo-2,3,4,5-tetrahydro-
1,4-benzodiazepine-2-acetate (370 mg, 0.89 mmol) in EtOH (20 rnL), and the
mixture was shaken on a Parr apparatus at RT under H2 (50 psi). After 12 h, the
reaction was filtered through celite~, and the filtrate was concentrated to afford the
title compound (350 mg, 94%) as a pale yellow liquid: TLC Rf (4:20:20:56
MeOH/EtOAc/hexane/CI2CH2) 0.55; IH NMR (400 MHz, CDCl3) o 7.10-7.19 (m,
3H), 5.59-5.77 (m, lH), 4.85 (d, J = 15.0 Hz, lH), 3.68-3.60 (m, SH), 3.12 (s, 3H),
2.85 (dd, J = 5.4, 15.3 Hz, lH), 2.65 (t, J = 6.4 Hz, 2H), 2.34 (dd, J = 10.0, 15.3 Hz,
lH), 1.30-1.78 (m, 13H); MS (ES) rn/e 421 LM+H]+.
-
35 c) Methyl (_)-l-(tert-butoxycarbonyl)-7-(4-carboxybut-1-yl)-4-methyl-3-oxo-
2,3,4,5-tetrahydro- 1,4-benzodiazepine-2-acetate
~33
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
To a solution of methyl (+)-l-(tert-butoxycarbonyl)-7-(4-
hydroxybut- I -yl)4-methyl-3-oxo-2,3,4,5-tetrahydro- l ,4-ben7orli~7epine-2-acetate
(350 mg, 0.82 mmol) in CH2Cl2 at 0~C was added 2,2,6,6-tetrarnethyl-
oxopiperi~linillm chloride (J. Org. C*em. 1985, 50, 3930-3931; 220 mg, 1.1 rnmol).
S The mixture was stirred for 2 hr at 0~C under Ar atmosphere. 2-Methyl-2-butene (1
mL) was added, followed by the addition of a freshly prepared solution of NaClO2~0.76 g, G.7 mmol), NaH2PO, ~ H20 (0.78 g, 5.68 mrnol) and H20 (25 mL). The
cooling bath was removed, and the rnixture was taken up in EtOAc and washed
successively with 0.05 M HCl and brine. Drying (MgSO4), concentration, and silica
gel chromatography (5% AcOH in 4:20:20:56 MeOH/EtOAc/hexane/Cl2CH2), gave
the title compound (350 mg, 98%): TLC Rf (5% AcOH in 4:20:20:56
MeOH/EtOAc/hexane/Cl~CH2) 0.32; IH NMR (400 MHz, CDC13) o 7.13-7.18 (m,
3H), 5.60-5.69 (m, lH), 4.83 (d, J = 14.2 Hz, lH), 3.76 (d, J = 14.2 Hz, lH), 3.66 (s,
3H), 3.12 (s, 3H), 2.93 (dd, J = 4.5, 15.3 Hz, lH), 2.67 (t, J = 7.3 Hz, 2H), 2.36 (t, J
= 7.3 Hz, 2H), 2.30-2.34 (m, lH), 1.95 (q, J = 7.3 Hz, 2H), 1.34 (s, 9H); MS (ES)
m/e 435 [M+H]+.
d) Methyl (+)-l-(tert-butoxycarbonyl)-7-t3-(~ 7.ol-2-yl)propyl]-4-methyl-3-
oxo-2,3,4,5-tetrahydro- 1,4-benzodiazepine-2-acetate
To a stirred and cooled (-10~C) rnixture of methyl (+)-l-(tert-
butoxycarbonyl)-7-(4-carboxybut- 1 -yl)-4-methyl-3-oxo-2,3,4,5-tetrahydro- 1,4-
benzodiazepine-2-acetate (350 mg, 0.8 mmol) and Et3N (81 mg, 0.8 rnrnol) in
anhydrous THF (8 mL) was added isobutylchloroformate (97 mg, 0.8 mrnol). After
10 min, a solution of 1,2-phenylenefli:~min~ (1.43 g, 0.9 mmol) in THF (2 rnL) was
added. Stirring was continued at RT overnight, then the solvents were evaporated.
The residue was dissolved in EtOAc, and the solution was washed seqllentiz3lly with
aqueous NaHCO3 and brine. Drying (MgSO4) and concentration produced a pale
yellow solid. This was dissolved in glacial AcOH (5 rnL), and the reaction was
heated to 60~C. After 3 hr, the mixture was cooled, concentrated, neutralized with
2.5 N NaOH, and extracted with CH2C12. Drying (MgSO4), concentration, and
silica gel chromatography (gradient 1-5% MeOH/CH2C12) gave the title compound
(200 mg, 50%): TLC Rf (4:20:20:56 MeOH/EtOAc/hexane/Cl2CH2) 0.18; lH NMR
(400 MHz, CDC13) o 7.57-7.61 (m, 2H), 7.15-7.28 (m, 4H), 7.08 (s, lH), 5.60-5.55(m, lH), 4.75-4.88 (m, lH), 3.71 (d, J = 14.2 Hz, lH), 3.70 (s, 3H), 3.10 (s, 3H),
2.94 (dd, J = 4.2, 15.3 Hz, lH), 2.85-2.90 (m, 2H), 2.73-2.79 (m, 2H), 2.32-2.36 (m,
lH), 2.15-2.23 (m, lH), 1.34 and 1.55 (br s, rotamers, 9H); MS (ES) m/e 507
[M+H]+.
134
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
e) (+)-7-[3-(Ben7imi(i~7ol-2-yl)propyl~-4-methyl-3-oxo-2~3~4~s-tetrahydro- lH- 1,4-
benzodiazepine-2-acetic acid
LiOH (17 mg, 0.71 mmole) was added at RT to a solution of methyl (+)- 1-
(tert-butoxycarbonyl)-7-[3-(ben7imi~1~7.ol-2-yl)propyl]-4-methyl-3-oxo-2,3,4,5-
tetrahydro-1,4-benzodiazepine-2-acetate (200 mg, 0.395 mmol) in MeOH (2 mL)
and H20 (3 mL). The reaction mixture was stirred at RT for 4 hr. Aci~lifi~tion
with dil HCl to pH 4 and concentration produced a white solid. This was dissolved
in a mixture of methylene chloride (10 mL) and trifluoroacetic acid (5 mL) at 0~C,
and the reaction was kept at 0~C for 30 min. The solvents were ~v~olat~d and theresidue was triturated with ether and purified by ODS flash chromatography
(gradient 10 to 18% CH3CN/H2O cont~ining 0.1% TFA). Concentration and
lyophilization gave the title compound (75 mg, 48%) as a colorless powder: IH
NMR ((400 MHz, CDC13) â 7.58-7.61 (m, 2H), 7.37-7.39 (m, 2H), 6.79 (d, J = 8.2
Hz, lH), 6.68 (s, lH), 6.39 (d, J = 8.2 Hz, lH), 5.18(d, J = 16.8 Hz, lH~, 4.77 (dd, J
= 7.0, 7.5 Hz, lH), 3.63 (d, J = 16.8 Hz, lH), 3.03-3.19 (m, 3H), 2.97 (s, 3H), 2.83
(dd, J = 7.0, 16.4 Hz, lH), 2.52-2.56 (m, 2H), 2.08-2.12 (m, 2H); MS (ES) m/e
393.0 tM+H]~- Anal. Calcd for C22H24N4O3 ~-C2HF3O2 ~ 0.5 H2O: C, 55.92; H,
5.08; N, 10.89. Found: C, 56.16; H, 4.92; N, 10.88.
Example 18
Plc~paldtion of (S)-2.3.4.5-tetrahydro-7-rrN-r(ben7imi~1~7-)1-2-yl)mPtllyl~-N-(4-
aminobutyl)aminolcarbonyll-4-methyl-3-oxo- 1 H- 1.4-benzodiazepine-2-acetic acid
a) 4-[(B~n7imid~7OI-2-yl)methylJaminobutyronitrile
To a stirred mixture of 2-aminomethylben7imi~i~701e dihydrochloride (0.5 g,
2.2717 mmol) and NaHCO3 (0.67 g, 7.951 mmol) in dry DMF (10 mL) was added 4-
bromobutyronitrile (0.37 g, 2.4989 mmol). After stirring at RT for 24 hr, the
mixture was concentrated. The residue was taken up in H2O and extracted with
CH2Cl2. The organic extracts were dried over MgS04, concentrated, and purified by
silica gel flash column chromatography (5% MeOH/CH2Cl2)to give the title
compound (0.15 g, 35%) as a brown oil: 'H NMR (250 MHz, DMSO-d6) ~ 1.82 (m,
- 2H), 2.45 (t, J = 4 Hz, 2H~, 2.85 (t, J = 4 Hz, 2H), 4.11 (s, 2H), 7.14 (m, 2H), 7.50
35 (m, 2H).
-
135
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
b) Methyl (S)-2,3,4,5-tetrahydro-7-[[N-[(benzimidazol-2-yl)methyl]-N-
(4-cyanopropyl)amino]carbonyl]-4-methyl-3-oxo- lH- 1,4-benzodiazepine-2-acetate
To a stirred mixture of 4-~(b~n7im;~ 7Ol-2-yl)methyl]aminobutyronitrile
(0.159 g, 0.7422 mmol), methyl 2,3,4,5-tetrahydro-7-carboxy-4-methyl-3-oxo-lH-
1,4-benzodiazepine-2-acetate ( 0.217 g, 0.7422 mmol), HOBt H,O (0.120 g, 0.8906
mmol), and i-Pr2NEt (0.192 g, 1.4844 mmol) in dry CH~CN (7 mL) was added EDC
(0.265 g, 0.8906 mmol). After stirring at RT for 48 hr, the ~ lule was
concentrated. The residue was taken up in H2O and extracted with CH2Cl2. The
organic layer was washed se~uentially with saturated NaHCO3 and brine, dried over
MgSO4, and concentrated to give a brown oil. Silica gel flash column
chromatography (3% MeOH/CH2CI2) gave the title compound (0.261 g, 74%) as an
off white foam: 'H NMR (250 MHz, DMSO-d6): o 1.95 (m, 2H), 2.66 (dd, J = 16.4,
3.5 Hz, lH), 2.78 (dd, J = 16.4, 3.5 Hz, lH), 2.85 (t, J = 8.7 Hz, 2H), 3.45 (t, J = 8.7
Hz, 2H), 3.60 (s, 3H), 3.80 (d, J = 16 Hz, lH), 4.52 (s, 2H), 4.84 (d, J = 2.9 Hz, 2H),
5.15 (m, lH), 5.48 ~d, J = 16 Hz, lH), 6.40 (d, J = 3.5 Hz, IH), 6.54 (d, J = 8.3 Hz,
lH), 7.25 (m, 4H);,7.50 (m, lH), 7.62 (m, lH).
c) (S)-2,3,4,5-Tetrahydro-7-[[N-[(ben7imi~1~7.ol-2-yl)methyl~-N-(4-
cyanopropyl)amino]carbonyl~-4-methyl-3-oxo- lH- 1,4-benzodiazepine-2-acetic acidTo a stirred solution of methyl (S)- 2t3,4,5-tetrahydro-7-[~N-[(benzimidazol-
2-yl)methyl]-N-(4-cyanopropyl)amino]carbonyl]-4-methyl-3-oxo- lH- 1,4-
benzodiazepine-2-acetate (0.261 g, 0.5478 mmol) in MeOH (5 mL) was added 2.5 N
NaOH (0.7 mL, 1.6433 mmol). After stirring at RT overnight, the mixture was
concentrated. The residue was taken up in H2O, and the solution was acidified with
6 N HC1 to pH = 4. The white solid was filtered and dried to afford the title
compound ~0.21 g, 81%): 'H NMR (250 MHz, DMSO-d6): ;~ 1.95 (m, 2H), 2.66
(dd, J = 16.4, 3.5 Hz, lH), 2.78 (dd, ~ = 16.4, 3.5 Hz, lH), 2.85 (t, J = 8.7 Hz, 2H),
3.45(t,J=8.7Hz,2H),3.80(d,J= 16Hz, lH),4.52(s,2H),4.84(d,J=2.9Hz,
2H), 5.15 (m, lH), 5.48 (d, J = 16 Hz, lH), 6.40 (d, J = 3.5 Hz, lH), 6.54 (d, J = 8.3
Hz, lH), 7.25 (m, 4H), 7.50 (m, lH), 7.62 (m, lH~.
d) (S)-2,3,4,5-Tetrahydro-7-[~N-L(benzimidazol-2-yl)methyl]-N-(4-
aminobutyl)amino]carbonyl]-4-methyl-3-oxo- lH- 1,4-benzodiazepine-2-acetic acid
A mixture of (S)-2,3,4,5-tetrahydro-7-[[N-[(ben~i...i-1~7- 1-2-yl)methyl]-N-(4-
cyanopropyl)amino]carbonyll-4-methyl-3-oxo- lH- 1,4-benzodiazepine-2-acetic acid(0.200 g, 0.4325 mmol) and NH40H (1 mL, 30% solution) in MeOH (5 mL) was
hydrogenated over Ra/Ni at RT for 24 hr. The catalyst was ~lltered off, and the
136
.
CA 0224l633 1998-06-26
W O97/24119 PCT~US96/20748
filtrate was concentrated and purified by reverse phase chromatography
(10% CH3CN/H10 contzlining 0.1% TFA) to give the title compound (0.100 g, 33%)
as an off white solid: 'H NMR ( 400 MHz, DMSO-d6) ~ 1.45 (m, 2H), 1.72 (m, 2H),
2.54 (dd, J = 16.4, 3.5 Hz, lH), 2.70 (m, 2H), 2.75 (dd, J = 16.4, 3.5 Hz, lH), 2.95
(s, 3H), 3.65 (t, J = 8.7 Hz, 2H), 3.85 (d, J = 16 Hz, lH), 5.05 (s, 2H), 5.15 (m, lH),
5.48 (d, J = 16 Hz, lH), 6.65 (d, J = 8.3 Hz, lH), 7.20 (m, 2H), 7.61 (m, 2H), 7.75 (s,
2H), 7.85 (m, 2H); IR (KBr) 3425, 3000, 3100, 1728, 1675, 1630, 1625, 1613 cm~';MS (ES) m/e 479 (M+H). Anal. Calcd for C25H3"N604 ~ 2 CF3CO2H: C, 49.30; H,
4.56; N,11.89. Found: C, 49.22; H, 4;89; N, 11.84.
Fx:-ml?le 19
Preparation of ~S)-2.3.4.5-tetrahydro-7-rrN-r(benzimidazol-2-yl)methyl-N-(2-
cyanomethyl)amino~carbonyll-4-methyl-3-oxo- lH- 1.4-benzodiazepine-2-acetic acid
a) t(Ben7imi(1~7Ol-2-yl)methyl]~minn~retonitrile
Following the procedure of Example in 18(a), except substituting
bromoacetonitrile for the 4-bromobutyronitrile, the title compound was prepared as
an off white solid (0.15 g, 35%): lH NMR (250 MHz, DMSO-d6): o 3.71 (s, 2H),
3.98 (s, 2H), 7.14 (m, 2H), 7.50 (m, 2H).
b) Methyl (S)-2,3,4,5-tetrahydro-7-[[N-[(ben7imicl~7(-1-2-yl)methyl-N-(2-
cyanomethyl)amino]carbonyl]-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetate
Following the procedure of Example 18(b), except substituting
[(ben7imi~701-2-yl)methyl]~mino~retQnitrile for 4-l (bçn7imi~1~7ol-2-
yl)methyl]aminobutyronitrile, the title compound was prepared as an of ~ white foam
(0.487 g, 66%): 'H NMR (250 MHz, DMSO-d6) ~ 2.66 (dd, J--16.4, 3.5 Hz, lH),
2.78 (dd, J = 16.4, 3.5 Hz, lH), 2.92 (s, 2H), 3.60 (s, 3H), 3.80 (d, J = 16 Hz, lH),
4.52 (s, 2H), 4.84 (d, J = 2.9 Hz, 2H), 5.15 (m, lH), 5.48 (d, J = 16 Hz, lH), 6.40 (d,
J = 3.5 Hz, lH), 6.54 (d, J = 8.3 Hz, lH), 7.25 (m, 4H), 7.50 (m, 2H), 7.62 (m, 2H).
c) (S)-2,3,4,5-Tetrahydro-7-[[N-[(b~n,; . . ,i~1~7ol-2-yl)methyl-N-(2-
cyanomethyl)amino]cal L o~yl]-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetic acid
Following the procedure in Example 18(c), methyl (S)-2,3,4,5-tetrahydro-7-
~ 35 [[N-[(bell~i.. id~7ol-2-yl)methyl-N-(2-cyanomethyl)amino]carbonyl]-4-methyl-3-
- oxo-lH-1,4-benzodiazepine-2-acetate was saponified, and the product was
y~ ~;.lli7e~l from EtOH, to give the title compound as a white solid (0.420 g,
137
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
89%): 'H NMR ( 400 MHz, DMSO-d6) ~i 2.66 (dd, J = 16.4, 3.5 Hz, lH),
2.78 (dd, J = 16.4, 3.5 Hz, lH~, 2.92 (s, 2H), 3.80 (d, J = 16 Hz, lH), 4.52 (s, 2H),
4.84 (d, J = 2.9 Hz, 2H), 5.15 (m, lH), 5.48 (d, J = 16 Hz, lH), 6.40 (d, J = 3.5 Hz,
lH), 6.54 (d, J = 8.3 Hz, lH), 7.25 (m, 4H), 7.50 (m, lH), 7.62 (m, lH); MS (ES)m/e 465 (M+H)~. Anal. Calcd for C23H22N6O4 ~ 2 HCl: C, 53.19; EI, 4.66; N, 16.18;
Found: C, 52.98; H, 4.43; N, 16.53.
Fx~ ?le 20
Preparation of (S)-2.3.4.5-tetrahydro-7-rr~ben~ ol-2-
yl)methyl~methyiamino~carbonyll-3-oxo-lH-l~4-benzo~ n~-2-acetic açid
a) Dimethyl (R)-2-trifluoromethanesulfonylsuccinate
To a stirred, cooled (0~C) mixture of dimethyl D-malate (5.5 g, 33.9213
mmol), and dry pyridine (2.82 g, 35.7164 mmol) in dry CH2Cl2 (55 mL) was added
trifluoro..,t~lh~neslllfionic anhydride (10.0 g, 35.7164 mmol) dropwise. After stirring
at 0~C for 4 hr, the ~ ule was cluenched with H~O and the layers were separated.The organic layer was washed sequentially with dil HCI and brine, dried over
MgSO4, and concentrated to give title compound (8.50 g, 96%) as a white solid: 'H
NMR( 250 MHz, CDCI3: ~ 3.10 (d, J = 5.8 Hz, 2H), 3.74 (s, 3H), 3.78 (s, 3H), 5.52
(t, J = 5.8 Hz, lH).
b) Dimethyl D-(2-cyanophenyl)malate
A mixture of 2-arninobenzonitrile (0.5 g, 4.2323 mmol), 2,6-di-tert-
bu~ylpylidine (0.85 g, 4.4439 mmol), and dimethyl (R)-2-
trifluorometh~n~slllfonylsuccinate in 2:1 hexane/chloroform (25 mL) was stirred at
RT for 76 hr. The mixture was concentrated, and the residue was taken up in H2O
and extracted with EtOAc. The organic extracts were washed sequentially with 10%HCI and brine, dried over MgSO4, concentrated, and purified by silica gel flash
column chromatography (10% EtOAc/hexane) to give the title compound (0.886 g,
80%) as a yellow solid: 'H NMR (250 MHz, CDCl3) ~ 2.95 (d, J = 5.8 Hz, lH), 3.74(s, 3H), 3.78 (s, 3H), 4.60 (m, lH), 5.28 (d, J = 5.8 Hz, lH), 6.73 (d, J = 8.5 Hz, lH),
6.80 (t, J = 8.5 Hz, lH), 7.47 (m, 2H).
c) Methyl (S)-2,3,4,5-tetrahydro-3-oxo-lH-1,4-benzodiazepine-2-acetate
A solution of dimethyl D-(2-cyanophenyl)malate (10.75 g, 41.0006 mmol) in
MeOH (pre-saturated with NH3 (g) for 10 min, 100 mL) was hydrogenated over
138
CA 0224l633 l998-06-26
W O 97/24119 PCTrUS96/20748
Ra/Ni at 55 psi for 48 hr. The catalyst was filtered off, and the filtrate was
concentrated and purified by silica gel flash colurnn chromatography (40%
EtOAc/hexane) to give the title compound (5.03 g, 53%) as an off white solid: 'HNMR (250 MHz, CDCl3) o 2.65 (dd, J = 16.3, 7.6 Hz, lH), 2.99 (dd, 16.3, 5.9 Hz,
lH), 3.74 (s, 3H), 3.95 (dd, J = 16, 6.9 Hz, lH), 4.79 (m, lH), 4.95 (dd, J = 16, 5.3
Hz, lH), 6.55 (t, J = 5.3 Hz, lH), 6.65 (d, J = 7.6 Hz, lH), 6.78 (d, J = 7.6 Hz, lH),
6.97(d,J=7.6Hz, lH),7.15(d,J=7.6Hz, lH).
d) Methyl (S)-2,3 ,4,5-tetrahydro-7-bromo-3-oxo- 1 H- 1 ,4-benzodiazepine-2-acetate
A mixture o~ methyl (S)-2,3,4,5-tetrahydro-3-oxo-lH-1,4-benzodiazepine-2-
acetate (5.03 g, 21.4746 mmol) and n-Bu4NBr3 (10.35 g, 21.4746 mmol) in CHC13
(100 mL) was stirred at RT for 3 hr, then the ~ lule was concentrated. The residue
was taken up in H20, stirred, and filtered to afford the title compound (5.61 g, 83~o)
as an off white solid: 'H NMR (250 MHz, CDCl3) o 2.74 (dd, J = 16.3, 7.6 Hz, lH),
3.05 (dd, J = 16.3, 5.9 Hz, lH), 3.75 (s, 3H), 4.05 (dd, J = 16, 6.9 Hz, lH), 4.73 (t, J
=5.9Hz, lH),4.86(dd,J= 16,5.3Hz, lH),6.68(d,J=7.6Hz, lH),6.75 (t,J=5.3
Hz, lH), 7.14 (s, lH), 7.25 (d, J = 7.6 Hz, lH).
e) Methyl (S)-2,3,4,5-tetrahydro-7-[[N-[(ben7imi~1~71 1-2-yl)methyl]-N-
methylamino]carbonyl]-3-oxo- 1 H- 1 ,4-benzodiazepine-2-acetate
A mixture of methyl (S)-2,3,4,5-tetrahydro-7-bromo-3-oxo-lH-1,4-
benzodiazepine-2-acetate (1.5 g, 4.77905 mmol), 2-
(methylaminomethyl)ben~imi-1~7Qle dihydrochloride (2.24 g, 9.5809 mrnol),
triphenylphosphine (1.26 g, 4.7905 mmol), n-Bu3N ( 6.21 g, 33.5333 mmol), and
(Ph3P)4Pd (1.10 g, 0.9581 mmol) in N-methyl 2-pyrrolidinone (20 rnL) was flushedwith argon and carbon monoxide for 10 min. The mixture was then heated at 100-
105~C under a carbon monoxide balloon for 8 hr. The mixture was cooled and
acidified with 6 N HCl to pH = 2. The solution was extracted with EtOAc, and theEtOAc layer was discarded. The aqueous layer was neutralized with 30% NaOH
and extracted with CH2Cl2. The organic extracts were dried over MgSO4,
concentrated, and purifled by silica gel flash column chromatography (5%
MeOH/CH2Cl2) to give the title compound (1.62 g, 80%) as an off white solid: 'H
NMR (250 MHz, DMSO-d6): ~; 2.65 (dd, J = 16.3, 7.6 Hz, lH), 2.81 (dd, J = 16.3,
5.9 Hz, lH), 3.05 (s, 3H), 3.60(s, 3H), 3.75 (dd, J = 16.3, 6.9 Hz, lH), 4.78 (s, 2H),
4.95 (m, lH), 5.05 (dd, J = 16, 5.3 Hz, lH), 6.20 (d, J = 5.9 Hz, lH), 6.55 (d, J = 7.6
Hz, lH), 7.25 (m, 4H), 7.55 (m, 2H), 8.21 (t, J = 5.3 Hz, lH).
139
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
f) (S)-2,3,4,5-Tetrahydro-7-[[[berl7imi(1:l7Ol-2-
yl)methyl]methylamino]ccubollyl]-3-oxo- 1 H- I ,4-ben70~ 7epine-2-acetic acid
Following the procedure of Example 18(c), methyl (S)-2,3,4,5-tetrahydro-7-
[~N-t(ben7.imi(1~7.ol-2-yl)methyl]-N-methylamino]carbonyl]-3-oxo-lH-1,4-
benzodiazepine-2-acetate was saponified to afford the title compound (0.060 g,
57%) as an off white solid: 'H NMR ( 400 MHz, DMSO-d6) ~ 2.52 (dd, J = 16.3, 7.6Hz, lH), 2.84 (dd, J = 16.3, 5.9 Hz, lH), 3.20 (s, 3H), 3.75 (dd, J = 16.3, 6.9 Hz,
lH), 4.95 (t, J = 5.9 Hz, lH), 5.05 (dd~ J = 16, 5.3 Hz, lH), 5.10 (s, 2H), 6.59 (d, J =
7.6 Hz, lH), 7.12 (s, lH), 7.20 (d, J - 7.6 Hz, lH), 7.48 (m, 2H), 7.69 (m, 2H), 7.90
(d, J = 5.3 Hz, lH); IR ( KBr) 3600-3100, 3100-2800, 1681, 1613, 1601, 1485,
1445, 1314, 830, 764, 742 cm l; MS (ES) m/e 422 (M+H)~. Anal. Calcd for
C2,H21N5O4: C, 61.91; H, 5.20; N, 17.19. Found: C, 61.57; H, 5.32; N, 17.29.
Fx~ml?le 21
Preparation of (S~-2.3.4.5-tetrahydro-7-rrrrl-(2-hydroxyethyl)ben7imi~1~7ol-2-
yllmetllyllaminolcarbonyll4-methyl-3-oxo- lH- 1.4-benzodi~epine-2-acetic açid
a) Ethyl 2-[[(ben7imi-1~7O1-2-yl)methyl]amino]acetate
A mixture of 2-aminomethyll~e~ 701e dihydrochloride hydrate (4.0 g,
18.1736 mmol), NaHCO3 (7.63 g~ 90.868 mmol), and ethyl 2-bromoacetate (4.55 g,
27.2603 mmol) in dry DMF (60 rnL) was stirred at RT for 24 hr, then was
concentrated. The residue was taken up in H~O and extracted with CH2CI2. The
organic extracts were dried over MgSO4, concentrated, and purified by silica gelflLash column chromatography (5% MeOH/CH2Cl2) to give the title compound (0.50
g, 12%) as a brown oil: 'H NMR (250 MHz, CDCI3) ~ 1.95 (s, 3H), 3.48 (s, 2H),
4.50 (m, 4H), 7.25 (m, 2H), 7.35 (m, lH), 7.73 (m, lH).
b) Methyl (S)-2,3,4,5-tetrahydro-7-[[~[1 -(2-acetyloxyethyl)ben7imi~1~7ol-2-
yl]methyl]amino]~l,onyl]4-methyl-3-oxo-lH-1,4-benzodiazepine-2-acetate
Following the procedure of Example 18(b), except substituting ethyl 2-
[[(benzimidazol-2-yl)methyl]amino]acetate for the 4-[(ben7imid~7ol-2-
yl)methyl]aminobutyronitrile, the title compound (0.251 g, 78.5%) was prepared as a
white solid: ;H NMR (250 MHz, CDCl3) ~ 1.95 (s, 3H~, 2.66 (dd, J = 16.4,3.5 Hz,
lH), 2.95 (s, 3H), 3.05 (dd, J = 16.4, 3.5 Hz, lH), 3.60 (d, J = 16 Hz, lH), 3.75 (s,
3H), 4.45(d, J = 5.9 Hz, 2H), 4.62 (s, 2H), 4.92 (t, J = 5.9 Hz, 2H), 5.10 ( m, lH),
140
.~
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
5.40(d,J= 16Hz, lH),6.49(d,J=8.3Hz, lH),7.32(m,3H),7.60(m,
2H), 7.71 (m, lH), 8.15 (t, J = 5.3 Hz, lH).
c) (S)-2.3,4,5-Tetrahydro-7-[[[[1-(2-hydroxyethyl)bçn7imi-1~7ol-2-
yl]methyl]amino]carbonyl]-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetic acid
To a stirred, partial suspension of methyl (S)-2.3,4,5-tetrahydro-7-[[[[1-(2-
acetyloxyethyl)ben7imi~ 7ol-2-yl]methyl]amino]carbonyl]-4-methyl-3-oxo-lH-1,4-
benzodiazepine-2-acetate (0.241 g, 0.475 mmol) in THF (5 mL) was added 1.0 N
LiOH (1.4 mL, 1.425 mmol). After stirring at RT overnight, the mixture was
concentrated. The residue was taken up in H2O and acidified with AcOH to pH = 4.The off-white solid was filtered and lliluldt~d with acetone to afford the titlecompound (0.16 g, 75%) as a white solid: 'H NMR (400 MHz, DMSO-d6):o 2.54
(dd, J = 16.4, 3.5 Hz, lH), 2.95 (s, 3H), 3.05 (dd, J = 16.4, 3.5 Hz, lH), 3.60 (d, J =
16 Hz, lH), 4.45 (d, J = 5.9 Hz, 2H), 4.62 (s, 2H), 4.92 (t, J = 5.9 Hz, 2H), 5.10 (m,
lH),5.40(d,J= 16Hz, lH),6.49(d,J=8.3Hz, lH),7.32(m,3H),7.60(m,2H),
7.71 (m, lH), 8.15 (t, J = 5.3 Hz, lH); MS (ES) m/e 452 (M+H)+. Anal. Calcd for
C23H25N5O5 - 0.75 H2O: C, 59.71; H, 5.72; N, 15.14. Found: C, 59.65, H, 5.70; N,14.88.
Exarnple 22
Preparation of (+)-2.3~4.5-tetrahydro-7-rrN-(b~n7imicl~7ol-2-yl)methyl-N-rr4-(2-carboxybenzoyl)amino~butyllaminolcarbonyll-3-oxo-4-(2-phenylethyl)- 1 H- 1.4-
benzot1i~7~-pine-2-acetic acid
a) 4-~[(Benzimidazol-2-yl)methyllamino]butylphthz~limide
A mixture of 2-aminomethylbenzimidazole dihydrochloride hydrate (22.10
g, 100.7269 mmol), NaHCO3 (42.40 g, 503.6347 mmol), and 4-
bromobutylphth~limicle (34.10 g, 120.8723 mmol) in dry DMF (250 mL) was heated
at 100-110~C for 6 hr, then was cooled and concenllatt;d. The residue was taken up
in H2O and extracted with CH2Cl2. The organic extracts were dried over MgSO4,
concentrated, and purified by silica gel flash column chromatography (5%
MeOH/CH2Cl2) to give the title compound (10.8 g, 31%) as a brown foam: IH NMR
(250 MHz, CDCl3) ~ 1.65 (m, 2H), 1.85 (m, 2H), 2.75 (t, J = 8.9 Hz, 2H), 3.78 (t, J =
8.9 Hz, 2H), 4.17 (s, 2H), 7.20 (m, 2H), 7.60 (m, 2H), 7.72 (m, 2H), 7.88 (m, 2H).
-
141
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
b) Methyl ~+)-2,3,4,5-tetrahydro-7-[[N-(ben~i.l.if1z-7Ql-2-yl)methyl-N-[~4-
(phth~limido)butyl]amino]carbonyl]-3-oxo-4-(2-phenylethyl)-lH-1,4-
benzodiazepine-2-acetate
To a stirred mixture of 4-[[(~çn7imi~l~7Ql-2-
yl3methyl]amino]butylphth:~1imi~ (1.75 g, 5.0525 mmol), methyl (+)-2,3,4,5-
tetrahydro-7-carboxy-3-oxo-4-(2-phenylethyl)- 1 H- 1,4-benzodiazepine-2-acetate
(1.61 g, 4.2104 mmol), HOBt H2O (0.69 g, 5.0525 mmol), and i-PrlNEt (1.10 g,
8.4209 mmol) in dry CH3CN (30 mL) was added EDC (1.50 g, 5.0525 mmol). After
stirring at RT for 24 hr, the mixture was concentrated. The residue was taken up in
H2O and extracted with CH2Cl2. The organic extracts were washed sequentially with
saturated NaHCO3 and brine, dried over MgSO4, concentrated, and purified by silica
gel ~lash column chromatography (5% MeOH/CH2Cl2) to give the title compound
(2.85 g, 95%) as a yellow foam: 'H NM~ (250 MHz, DMSO-d6) o 1.60 (m, 2H),
2.65 (m, 2H), 2.85 (dd, J = 16.4, 3.5 Hz, lH), 3.55 (m, 4H), 3.65 (s, 3H), 4.00 (d, J=
16.0 Hz, lH), 4.18 (q, J = 8.9 Hz, 2H), 4.75 (s, 2H), 5.15 (m, lH), 5.45 (d, J = 16.0
Hz, lH), 6.23 (d, J = 5.3 Hz, lH), 6.57 (d, J = 7.6 Hz, lH), 7.20 (m, 7H), 7.55 (m,
4H), 7.90 (m, 4H).
c) Methyl (+)-tetrahydro-7-[[N-[ber-~mi~1~7Ol-2-yl)methyl]-N-~4-
aminobutyl)amino]carbonyl~-3-oxo~(2-phenylethyl)- 1 H- 1,4-benzodiazepine-2-
acetate
A mixture of methyl (+)-2~3~4~5-tetrahydro-7-[[N-(ben7imi~ 7Ql-2-
yl)methyl-N-[[4-(phth~limido)butyl]amino]carbonyl]-3-oxo-4-(2-phenylethyl)-lH-
1,4-benzodiazepine-2-acetate (0 50 g, 0.7015 mmol) and hydrazine (0.07 g, 2.1045mmol) in MeOH (5 mL) was refluxed for 6 hr, then was cooled and concentrated.
The residue was taken up in H2O, acidified to pH = 2 with 6N HCl, and filtered to
remove a white solid. This solid was discarded. The aqueous filtrate was extracted
with EtOAc, and the EtOAc layer was discarded. The aqueous layer was basified topH = 9 with Na2CO3 and extracted with CHCl3. The organic layer was dried over
MgSO~ and concentrated to give the title compound (0.41 g, 89%) as an off white
solid: 'H NMR (250 MHz, DMSO-d6) ~ 1.47 (m, 2H), 1.75 (m, 2H), 2.65 (m, SH),
2.85 (dd, J = 16.3, 5.9 Hz, lH~, 3.65 (s, 7H), 4.05 (d, J = 16.0 Hz, lH), 5.05 (s, 2H),
5.15 (m, lH), 5.45 (d, J = 16, lH), 6.65 (d, J = 7.6 Hz, lH), 7.25 (m, 6H), 7.41 (s,
lH), 7.60 ~m, 2H), 7.85 (m, 2H).
142
CA 02241633 1998-06-26
WO 97124119 rCT/US96/20748
d) (+)-2,3,4,5-Tetrahydro-7-[[N-(ben~i...icl~7Ql-2-yl)methyl-N-[[4-(2-
carboxybenzoyl)amino]butyl]amino]carbonyl]-3-oxo-4-(2-phenylethyl)- 1 H- 1,4-
benzodiazepine-2-acetic acid
To a stirred solution of methyl (+)-tetrahydro-7-[[N-[ben7imi-1~7Ol-2-
yl)methyl]-N-(4-aminobutyl)amino]carbonyl]-3-oxo-4-(2-phenylethyl)- 1 H- 1,4-
benzodiazepine-2-acetate (0.34 g, 0.47 mmol) in THF was added 1.0 N LiOH (1.2
mL). After stirring at RT overnight, the mixture was concentrated, and the residue
was acidified with AcOH to pH = 4. The solid was filtered and lliLuldt~d with
acetone/ether to give the title compound (0.130 g, 39%) as a white solid: 'H
NMR(400 MHz, DMSO-d6) ~ 1.47 (m, 2H), 1.75 (m, 2H), 2.54 (dd, J = 16.3, 3.5 Hz,
lH), 2.65 (m, 2H), 2.85 (dd, J = 16.3, 5.9 Hz, lH~, 3.20 (m, 2H), 3.75 (m, 4H), 4.05
(d, J = 16.0 Hz, lH), 5.05 (s, 2H), 5.15 (m, lH), 5.45 (d, J = 16, lH), 6.65 (d, J = 7.6
Hz, lH), 7.25 (m, 6H), 7.41 (s, lH), 7.60 (m, 2H), 7.85 (m, 2H); IR (KBr) 3400,
3326, 3100-3000, 1721, 1637, 1626, 1616, 1607,1300, 750, 694 cm~'; MS (ES) m/e
717 (M+H)~. Anal. Calcd for C40H38N6O8 - 3 H2O: C, 63.56; H, 5.87; N, 11.12.
Found: C, 63.56; H, 5.83; N, 11.04.
Fx~ml?le 23
Preparation of (+)-2.3.4.5-tetrahydro-7-rrN-(ben7imid~7ol-2-yl)methyl~-N-rr4-(4-azido-2-hydroxybenzoyl)aminolcarbonyll -3-oxo-4-(2-phenylethyl)- 1 H- 1.4-
bell7odiazepine-2-acetic acid
a) Methyl (+)-2,3,4,5-tetrahydro-7-~[N-(be~7imi~1~7Ol-2-yl)methyl]-N-[[4-(4-azido-
2-hydroxybenzoyl)amino~carbonyl]-3-oxo-4-(2-phenylethyl)- 1 H- 1,4-
benzodiazepine-2-acetate
To a mixture of methyl (+)-2~3~4~s-tetrahydro-7-[[N-(brn7imi~l~7ol-2-
yl)methyl-N-[[4-(phth~limido)butyl]amino]carbonyl]-3-oxo-4-(2-phenylethyl)-lH-
1,4-benzodiazepine-2-acetate (0.409 g, 0.702 mmol), 4-~.ido.c~licylic acid-N-
hydroxysuccinimide ester (0.194 g, 0.702 mmol), and i-Pr2NEt (0.272 g, 2.106
mmol) in dry 2: 1 CH3CN/DMF (10 mL) was added EDC (0.25 g, 0.8424 mmol).
After stirring at RT for 24 hr, the mixture was concentrated. The residue was taken
up in H2O, stirred, and ~lltered to afford, after drying, the title compound (0.204 g,
39%) as an off white solid: 'H NMR (250 MHz, DM~O-d6) ~ 1.40 (m, 2H), 1.75 (m,
2H), 2.65 (m, 3H), 2.75 (dd, J = 16.3, 5.9 Hz, lH), 3.20 (m, 2H), 3.51 (m, 4H), 3.65
(m, 3H), 3.95 (d, J = 16 Hz, lH), 4.79 (s, 2H), 5.12 (m, lH), 5.37 (d, J = 16 Hz, lH),
143
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
6.20 (s, lH), 6.55 (m, 3H), 7.20 (m, 8H), 7.55 (m, 3H), 7.~5 (d, J = 7.6 Hz,
lH), 7.98 (s, lH), 8.75 (s, lH).
b) (+)-2,3,4,5-Tetrahydro-7-[[N-(be~ 7nl-2-yl)methyl3-N-[[4-(4-azido-2-
hydroxybenzoyl)amino]carbonyl]-3-oxo4-(2-phenylethyl)- 1 H- 1,4-benzodiazepine-
2-acetic acid
Following the procedure of Example 22(d), methyl (+)-2,3,4,5-tetrahydro-7-
[[N-(benzimidazol-2-yl)methyl]-N-[[4-(4-azido-2-hydroxybenzoyl~arninoJcarbonyl]-3-oxo-4-(2-phenylethyl)-lH-1,4-benzodiazepine-2-acetate was saponified to affordthe title compound (0.100 g, 50%) as a white solid: 'H NMR (400 MHz, DMSO-d6)
~ 1.40 (m, 2H), 1.75 (m, 2H), 2.65 (m, 3H), 2.75 (dd, J = 16.3, 5.9 Hz, lH), 3.20 (m,
2H), 3.51 (m, 4H), 3.95 (d, J = 16 Hz, IH), 4.79 (s, 2H), 5.12 (m, lH), 5.37 (d, J =
16 Hz, lH), 6.20 (s, lH), 6.55 (m, 3H), 7.20 (m, 8H), 7.55 (m, 3H), 7.85 (d, J = 7.6
Hz, lH), 7.98 (s, lH), 8.75 (s, lH); MS (ES) m/e 730 (M~H)+. Anal. Calcd for
C39H39N9O6 ~ 2.~ ~20: C, 60.46; H, 5.72; N, 16.27. Found: C, 60.46; H, 5.43; N,
15.90.
F~ nple 24
rl~palalion of 2.3.4.5-tetrahydro-7-rrN-r(ben7imi(1~7ol-~-yl)methyl-N-rrr(+)-
biotinoyllaminolbutyll~minolcarbonyll-3-oxo-4-(2-phenylethyl)-lH-1.4-
benzo~ pine-(2RS)-acetic acid
a) Methyl 2,3,4,5-tetrahydro-7-[~N-[(benzimidazol-2-yl)methyl-N-[[[(+)-
biotinoyl]amino]butyl]amino~carbonyl~-3-oxo-4-(2-phenylethyl)- l H- 1,4-
benzodiazepine-(2RS)-acetate
To a mixture of methyl (+)-tetrahydro-7-[[N-[ben7imirl~7ol-2-yl)methyl]-N-
(~aminobutyl)arnino~carbonyl]-3-oxo-4-(2-phenylethyl)- 1 H- 1,4-benzodiazepine-2-
acetate (0.40 g, 0.6865 mmol), (+)-biotin (0.17 g, 0.6865 mmol), HOBt ~ H2O (0.11
g, 0.8239 mmol), and i-Pr2NEt (0.18 g, 1.3730 mmol) in 1:2 DMF/CH3CN (12 rnL)
was added EDC (0.25 g, 0.8239 mmol). After stirring at RT for 24 hr, the mixturewas concentrated. The residue was taken up in H2O and extracted with CHCl3. The
organic extracts were washed sequentially with saturated NaHCO3 and brine, driedover MgSO4, concentrated, and purified by silica gel flash column chromatography(10% MeOH/CH2CI2~ to give the title compound (0.24 g, 44%) as a yellow foam: lH
NMR (250 MHz, DMSO-d6) o 1.30 (m, 2H), 1.60 (m, 4H), 2.05 (t, J = 8.9 Hz,
2H);,2.60 (m, 3H), 2.68 (dd, J = 16.3, 5.9 Hz, lH), 3.10 (m, 4H), 3.45 (m, 2H), 3.60
144
CA 0224l633 l998-06-26
W O 97/24119 PCT~US96/20748
(m, 2H), 3.65 (s, 3H), 4.01 (d, J = 16 Hz, lH), 4.12 (t, J = 8.9 Hz, lH),
4.30 (t, J = 8.9 Hz, lH), 4.78 (s, 2H), 5.10 (m, lH), 5.45 (d, J = 16 Hz, lH), 6.20 (d,
J = 5.3 Hz, lH), 6.40 (d, J = 8.9 Hz, 2H), 6.55 (d, J = 7.6 Hz, lH), 7.25 (m, 9H),
7.45 (m, lH), 7.55 (m, lH), 7.70 (t, J = 8.6 Hz, lH).
b) 2,3,4,5-Tetrahydro-7-[[N-[(bçn7imicl~7ol-2-yl)methyl-N-r[[(+)-
biotinoyl]amino]butyl]amino]carbonyl]-3-oxo-4-(2-phenylethyl)-lH-1,4-
benzodiazepine-(2RS)-acetic acid
To a stirred solution of methyl 2,3,4,5-tetrahydro-7-[[N-[(benzimidazol-2-
yl)methyl-N-[[[(+)-biotinoyl]amino]butyl]amino]carbonyl]-3-oxo-4-(2-phenylethyl)-
lH-1,4-benzodiazepine-(2RS)-acetate (0.24 g, 0.2967 mmol) in 1:2 THF/MeOH (6
mL) was added 1.0 N LiOH (0.44 mL). After stirring at RT overnight, the mixture
was concentrated. The residue was taken up in H2O and acidified with AcOH to pH
= 4. The off-white solid was filtered, and triturated with hot acetone to give the title
compound (0.160 g, 68%) as a white solid: 'H NMR (400 MHz, DMSO-d6) d 1.30
(m, 2H), 1.60 (m, 4H), 2.05 (t, J = 8.9 Hz, 2H), 2.60 (m, 3H), 2.68 (dd, J = 16.3, 5.9
Hz, lH), 3.10 (m, 4H), 3.45 (m, 2H), 3.60 (m, 2H), 4.01 (d, J = 16 Hz, lH), 4.12 (t, J
= 8.9 Hz, lH), 4.30 (t, J = 8.9 Hz, lH), 4.78 (s, 2H), 5.10 (m, lH), 5.45 (d, J = 16
Hz, lH), 6.20 (d, J = 5.3 Hz, lH), 6.40 (d, J = 8.9 Hz, 2H), 6.55 (d, J = 7.6 Hz, lH),
7.25 (m, 9H), 7.45 (m, lH), 7.55 (m, lH), 7.70 (t, J--8.6 Hz, lH); MS (l~S) m/e 795
(M+H)~. Anal. Calcd for C42H5"N806S 1.75 H20: C, 61.04; H, 6.52; N, 13.56.
Found: C, 60.89; H, 6.24; N, 13.31.
F.x~ml?le 25
P.epa~a~ion of (+)-2.3~4~5-tetrahydro-7-rrN-r(benzimidazol-2-yl)methyl~-N-(4-
aminobutyl)~minolcarbonyll-3-oxo-4-(2-phenylethyl)-lH-1.4-benzodiazepine-2-
acetic acid
a) (+)-2,3,4,5-Tetrahydro-7-[[N-[(ben7imid~7ol-2-yl)methyl]-N-(4-
aminobutyl)amino]carbonyl]-3-oxo-4-(2-phenylethyl)- lH- 1,4-benzodiazepine-2-
acetic acid
Following the procedure in Example 24(b), methyl (+)-2,3,4,5-tetrahydro-7-
[[N-[ben7imifl~701-2-yl)methyl]-N-(4-aminobutyl)amino]carbonyl]-3-oxo-4-(2-
phenylethyl)-lH-1,4-benzodiazepine-2-acetate was saponi~led to give the title
compound (0.250 g, 80%) as an off white solid: 'H NM~ (400 MHz, DMSO-d6)
1.37 (m, 2H), 1.62 (m, 2H), 2.52 (dd, J = 3.5 Hz, lH), 2.64 (m, 2H), 2.75 (dd, J =
145
CA 0224l633 l998-06-26
W O 97/24119 PCT~US96/Z0748
16.3, 5.9 Hz, lH), 3.51 (m, 4H), 3.91 (d, J = 16 Hz, lH), 4.98 (s, 2H), 5.05
(m, lH),5.37(d,J= 16Hz, lH),6.53(d,J=7.9Hz,2H),7.17(m,7H),7.52(m,
lH), 7.62 (s, lH), 7.78 (m, lH); IR ( KBr): 3386, 3100-3000, 1647, 1613, 1403, 740,
699 cm '; MS (ES) m/e 569 (M+H)+. Anal. Calcd for C32H36N604 ~ 2.75 H20: C,
62.18; H, 6.77; N, 13.60. Found: C, 62.11; H, 6.68; N, 13.57.
Fxzlrnple 26
Preparation of (+)-2~3.4~5-tetrahydrb-7-rfN-r(benzimidazol-2-yl)methyll-N-rr4-(4-
azido-3-iodo-2-hydroxyben7oyl~aminolbutyllaminolc~1Jon~111-3-oxo-4-(2-
ph~r~ylethyl)- 1 H- 1.4-ben~otli~7~pine-2-acetic acid
a) 3-Iodo-4-~7i~1os~1icylic acid-N-hydroxysllccinimifl. ester
To a stirred mixture of 4-azidosalicylic acid N-hydroxysuccinimide ester
(0.500 g, 1.8103 mmol) and silver trifluoroacetate (0.44 g, 1.9913 mmol) in CHCI3
(10 mL) was added iodine (0.510 g, 1.9913 mmol). After stirring at RT overnight,the reaction was filtered to remove a solid precipitate. The filtrate was washedsequentially with H2O, saturated NaHCO3 and brine, then was dried over MgSO~.
Concentration gave the title compound (0.703 g, 97%) as a light purple solid: 'HNMR (250 MHz, CDCl3) ~ 2.98 (s, 4H), 6.83 (d, J = 7.6 Hz, lH), 8.05 (d, J = 7.6 Hz,
lH).
b) Methyl (+)-tetrahydro-7-[[N-[(ben7imic1~7O1-2-yl)methyl]-N-[[4-(4-azido-3-
iodo-2-hydroxybenzoyl)amino]butyl]arnino]carbonyl]-3-oxo-4-(2-phenylethyl)-lH-
1.4-benzodiazepine-2-acetate
Following the procedure in Example 22(b), except substituting 3-iodo-4-
azidosalicylic acid-N-hydroxysuccinimide ester for the 4-[[(~n7imi~1~7ol-2-
yl)methyl]amino~butylph~h~limicl~, the title compound (0.312 g, 56%) was prepared
as a yellow foam: 'H NMR (250 MHz, DMSO-d6) ~ 1.42 (m, 2H), 1.60 (m, 2H),
2.52 (dd, J = 16.3, 3.5 Hz, lH), 2.63 (m, 2H), 2.79 (dd, J = 16.3, 5.9 Hz, lH), 3.25
(s, 2H), 3.55 (m, 6H), 3.65 (s, 3H), 3.95 ~d, J = 16 Hz, lH), 4.75 (s, 2H), 5.02 (m,
lH), 5.35 (d, J = 16 Hz, lH), 6.14 (d, J = 5.3 Hz, lH), 6.52 (d, J = 7.9 Hz, lH), 6.86
(d, J = 7.9 Hz, lH~, 7.25 (m, lOH), 7.51 (s, 2H), 7.90 (d, J = 7.9 Hz, lH), 9.01 (s,
lH).
146
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
c) (+)-Tetrahydro-7-[[N-[(bçl ~ i . I ,i(1~7Ol-2-yl)methyl~-N-[~4-(4-a_ido-3-
iodo-2-hydroxybenzoyl)amino]butyl] amino]carbonyl] -3 -oxo-4-(2-phenylethyl)- 1 H-
1.4-ben7.o~ 7~pine-2-acetic acid
Following the procedure in Example 22(c), methyl (+)-tetrahydro-7-[[N-
[(bç~ 7Ol-2-yl)methyl]-N-[[4-(4-a7ido-3-iodo-2-
hydroxybenzoyl)amino]butyl]amino]carbonyl] -3-oxo4-(2-phenylethyl)- 1 H- 1.4-
- benzodiazepine-2-acetate was saponified. Purification by silica gel flash colurnn
chromatography (0.5, 0.5, 9.5 AcOH/MeOH/CH2Cl2) gave the title compound (0.170
g, 58%) as an off white solid: 'H NMR (400 MHz, DMSO-d6) ~ 1.42 (m, 2H), 1.60
~m, 2H), 2.52 (dd, J = 16.3, 3.5 Hz, lH), 2.63 (m, 2H), 2.79 (dd, J = 16.3, 5.9 Hz,
lH), 3.25 (s, 2H), 3.55 (m, 6H), 3.95 (d, 16, lH), 4.75 (s, 2H), 5.02 (m, lH), 5.35 (d,
J= 16Hz, lH),6.14(d,J=5.3Hz, lH),6.52(d,J=7.9Hz, lH),6.86(d,J=7.9
Hz, lH), 7.25 (m, 10H), 7.51 (s, 2H), 7.90 (d, J = 7.9 Hz, lH), 9.01 (s, lH); MS (ES)
mle 856 (M+H)+; IR (KBr): 3360, 3100-3000, 2116, 1704, 1643, 1610, 1586, 1477,
1305, 1274, 766, 700 cm~'. Anal. Calcd for C39H38IN906 ~ 4.5 H20: C, 50.01; H, 5.06;
N, 13.46. Found: C, 50.19; H, 5.01; N, 13.12.
Fx~ml?le 27
Preparation of 5-rrr(ben7imid~7ol-2-yl)methyllmethylaminolcarbonyll-lH-
be~ 7Ole-2-aminoacetic acid
a) Methyl 5-[~[(benzimida_ol-2-yl)methyl]methylamino]carbonyl]-lH-
benzimidazole-2-;~mino~cetate
Diisopropylethylamine (1.1 mL, 6.48 mmol) was added to a stirred solution
of methyl 5-carboxy-b~n7imi~1~7Ole-2-~mino~et~t~ (0.24 g, 0.96 mmol), 2-
(methylaminomethyl)bellzil,.idazole bis-trifluoroacetate (0.56 g, 1.44 mmol), HOBt
H2O (0.19 g, 1.44 mmol), and EDC (0.28 g, 1.44 mmol) in anhydrous DMF (8 mL)
at RT. After 23 h, the reaction mixture was diluted with CH2Cll (100 mL) and
washed sequentially with 5% NaHCO3 (30 mL) and brine (30 mL). Drying
(MgSO4), concentration, and silica gel chromatography (10% MeOH/CH2Cl2) gave
the title compound (0.16 g, 42%~ as an off-white solid: MS (ES) m/e 393.0
(M+H)+.
A
35 b) 5-[[[(Ben7imicl~701-2-yl)methyl]methylatnino~carbonyl]-lH-bçn7imic~7Ole-2- aminoacetic acid
147
CA 02241633 1998-06-26
W O97/24119 PCT~US96~0748
1.0 N LiOH (1.0 rnL, 1.0 mmol) was added dropwise at RT to a
mixture of methyl 5-[[r(ben7imid~7-~1-2-yl)methyl]methylamino]carbonyl]-lH-
ben7.imi~1~701e-2-aminoacetate (0.16 g, 0.41 mmol) in THF (10 rnL) and H20 (10
mL). After 1 h, the reaction ~ Lule was concentrated to a small volume on the
5 rotavap and cooled in an ice bath before neutralizing with 1.0 N AcOH (1.0 mL).
The solid was collected, washed with cold H20, and air dried to give the title
compound (0.15 g, 100%) as an off white solid: MS (ES) rn/e 379.2 (M+H)+.
~F~m,J?le 28
I~e~aldlion of (_)-2.3~4~s-Tetrakydro 7-rrrfbenzimidazol-2-
yl)methyllmethy!;lmino~carbonyll-4-(3.3-~lim~thylbutyl)-3-oxo-lH-1.4-
ben7n~ 7epine-2-acetic acid
a) Methyl (V-2,3,4,5-tetrahydro 7-[[[(bc~7imi-1~7ol-2-
yl)methyl]methylarnino]carbonyl]-4-(3,3-dimethylbutyl)-3-oxo- lH- 1,4-
benzodiazepine-2-acetate
Diisopropylethylamine (0.94 rnL, 5.4 mmol) was added to a stirred solution
of methyl (_)-7-carboxy-4-(3,3 -dimethylbutyl)-3-oxo-2,3,4,5-tetrahydro- 1 H- l ,4-
benzodia_epine-2-acetic acid (0.39 g, 1.08 mmol), 2-
(methylamin~lllet}lyl)ben7imi~1~701e bis(trifluoroacetate) (0.42 g, 1.08 rnmol), ),
EIOBt H2O (0.22 g, 1.62 mmol), and EDC (0.31 g, 1.62 mmol) in anhydrous DMF
(8 mL) at RT. After 23 h, the reaction mlixture was diluted with CH2Cl~ (100 mL)and washed sequentially with 5% NaHCO3 (2 x 25 ml) and brine (25 mL). Drying
(MgSO4), concentration, and silica gel chromatography (7% MeOH/CH2Cl2) gave
the title compound (0.39 g, 71%) as a white solid: MS (ES) m/e 506.4 (M+H)+.
b) (i)-2,3,4,5-Tetrahydro 7-~[[(bç~ 7Ol-2-yl)methyl]methylamino]carbonyl]-
4-(3,3-dimethylbutyl) -3 -oxo- 1 H- 1,4-benzodia_epine-2-acetic acid
1.0 N LiOH (1.Q rnL, 1.0 mrnol) was added dropwise at RT to a mixture of
methyl (i)-2,3,4,5-tetrahydro 7-~[(b~imi~7(~l-2-
yI)methyl]methylamino]carbonyl]-4-(3,3-dimethylbutyl)-3-oxo- lH- 1,4-
benzodiazepine-2-acetate (0.38 g, 0.75 mmol) in THF (10 mL) and H2O (10 mL).
After 50 min, the reaction mixture was concentrated to a small volume on the
rotavap and cooled in an ice bath before neutralizing with 1.0 N AcOH (2.5 mL).
The solid was collected, washed with a cold H20, and air dried to give the titlecompound (0.27 g, 73%) as a white solid: MS (ES) rn/e 492.2 (M~H)+.
148
CA 02241633 1998-06-26
W O 97124119 PCTrUS96/20748
F.xample 29
Preparation (_)-2.3.4.5-Tetrahydro 7-rrr(ben7imi{~7ol-2-
yl)methyl7aminolcarbonyll-4-(3 .3-dimethylbutyl)-3-oxo- 1 H- 1 .4-benzodiazepine-2-
acetic acid
-
a) Methyl (+)-2,3,4,5-tetrahydro 7-[l[~be.n7imi~1~7~.1-2-yl)methyl]amino]carbonyl]-
4-(3,3-dimethylbutyl)-3-oxo- lH-1,4-benzodiazepine-2-acetate
Diisopropylethylamine (0.79 rnL, 4.56 mmol) was added to a stirred solution
of methyl (+)-7-carboxy-4-(3 ,3-dimethylbutyl)-3-oxo-2,3,4,5-tetrahydro- lH- 1,4-
benzodiazepine-2-acetic acid (0.33 g, 0.91 mmol), 2-(aminomethyl)ben7imi~1~7Ole
dihydrochloride hydrate (0.3 g, 1.36 mmol), HOBt H2O (0.18 g, 1.36 mmol), and
ED(~ (0.26 g, 1.36 mmol) in anhydrous DMF (8 mL) at RT. After 20 h, the reaction15 rnixture was diluted with CH Cl2 (70 mL) and washed sequentially with 5% NaHCO3
(2 x 20 mL) and brine (20 mL). Drying (MgSO4), concentration, and silica gel
chromatography (7% MeOH/CH2Cl2) gave the title compound (0.25 g, 56%) as a
white solid: MS (ES) m/e 492.4 (M+H)+.
20 b) (_)-2,3,4,5-Tetrahydro 7-[[[(be~ 7Ol-2-yl)methyllamino]carbonyl]4-(3,3-
dimethylbutyl)-3-oxo-lH-1,4-benzotli:l7epin~-2-acetic acid
1.0 N LiOH (1.0 mL, 1.0 mmol) was added dropwise at RT to a mixture of
methyl ( )-2,3,4,5-tetrahydro 7-[[[(benzimidazol-2-yl)methyl~amino]carbonyl]-4-
(3,3-dimethylbutyl)-3-oxo-lH-1,4-benzodiazepine-2-acetate (0.24 g, 0.49 mmol) in25 THF (8 mL) and H2O (8 mL). After 2.5 h, the reaction mixture was concentrated to
a small volume on the rotavap and cooled in an ice bath before neutralizing with 1.0
N AcOH (1.2 mL). The solid was collected, washed with cold H20 ,and air dried togive the title compound (0.25 g, 109%) as a white solid: MS (ES) m/e 478.2
(M+H)+.
l~xample 30
Plc;palation of (+)-2.3.4~5-Tetrahydro 7-rrr(4-azaben7.i~ 7ol-2-
yl)methyllmethylaminolcarbonyl~-4-(3 .3-dimethylbutyl)-3-oxo- 1 H- 1.4-
35 benzodiazepine-2-acetic acid
149
CA 0224l633 l998-06-26
W O 97t24119 PCTrUS96/20748
a) Methyl (+)-2,3,4,5-tetrahydro 7-[[[(4-~7~hen7imi~l~7~ 2-
yl)methyl]methylamino]carbonyl]-4-(3,3-dimethylbutyl)-3-oxo- 1 H- 1,4-
benzodiazepine-2-acetate
Diisopropylethylamine (0.53 mL, 3.0 mmol) was added to a stirred solution
of methyl (+)-7-carboxy-4-(3,3-dimethylbutyl)-3-oxo-2,3,4,5-tetrahydro- IH- 1,4-benzodiazepine-2-acetic acid (0.22 g, 0.61 mmol), 2-(methylamino)methyl-4-
azaben7imi~1~701e diacetate (0.29 g, 1.0 mmol), ~IOBt ~ H2O (0.12 g, 0.91 mmol),and EDC (0.17 g, 0.91 mmol) in anhydrous CH3CN (12 mL) at RT. After 21 h, the
reaction mixture was conce~ dl~d~ diluted with CEI~Cl2 (100 mL), and washed
sequentially with 5% NaHCO3 (2 x 20 mL) and brine (20 mL). Drying (MgSO4),
concentration, and silica gel chromatography (7% MeOH/CH2Cl2) gave the title
compound (0.147 g, 48%) as a white solid: MS (ES) m/e 507.4 (M+H)+.
b) (+)-2,3,4,5-Tetrahydro 7-[[[(4-~7~h~ 7ol-2-
yl)methyl]methylamino]carbonyl]-4-(3,3-dimethylbutyl)-3-oxo-lH-1,4-
benzodiazepine-2-acetic acid
1.0 N LiOH (0.69 mL, 0.69 mmol) was added dropwise at RT to a rnixture of
methyl (+)-2,3,4,5-tetrahydro 7-[[[(4-azaben7i,..il1~7Ol-2-
yl)methyl]methylamino]carbonyl]-4-~3,3-dimethylbutyl)-3-oxo-lH- 1,4-
benzodiazepine-2-acetate (0.14 g, 0.276 mmol) in THF (8 mL) and H2O (8 mL).
After 2 h, the reaction n~ib~ ; was concentrated to a small volume on the rotavap
and cooled in an ice bath before neutralizing with 1.0 N AcOH (0.69 mL). The solid
was collected, washed with cold H20, and air dried to give the title compound
(0.074 g, 54%) as a white solid: MS (ES) m/e 493.2 (M+H)+.
F;.x~ml?le 31
Preparation of (Sl-2~3.4.5-tetrahydro-7-rrrl l-r(ben7imicl~7Ql-2-
yl~m~tl~yllben7imicl~7ol-2-yllmetllyllarnino~cal1,o,l~1l-4-methyl-3-oxo-lH-l ~4-benzodiazepine-2-acetic acid
- a) Methy} (S)-2,3,4,5-tetrahydro-7-[[[[l-[(benzimid~ol-2-yl)methyl]bçn7imi~ ol-
2-yl]methyl]amino~carbonyl]-4-methyl-3-oxo-lH-1,4-ben7O~ 7epine-2-acetate
Diisopropylethylamine (0.27 mL, 1.53 mmol) was added to a stirred solution
of methyl (S)-7-carboxy-4-methyl-3-oxo-2,3,4,5-tetrahydro- 1 H- 1,4-benzodi~epine-
2-acetate trifluoroacetate (0.14 g, 0.34 mmol), 2-[[l-[(ben7imi(i~7Ol-2-
yl)methyl]be~7imitl~7c~e]methyl~amine bis(trifluoroacetate) (0.17 g, 0.34 mmol), 150
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
HOBt H2O (0.064 g, 0.48 mmol), and ~DC (0.091 g, 0.48 mmol) in
anhydrous DMF (10 mL) at RT. After 22 h, the reaction mixture was concentrated,
diluted with CH2Cl2 (70 mL), and washed sequentially with 5% NaHCO3 (2x30 mL)
and brine (20 mL). Drying (MgSO4), concentration, and silica gel chromatography
(7% MeOH/CH2Cl2) gave the title compound (0.080 g, 43%) as a white solid: MS
~ (ES) m/e 552.2 (M+H)+.
b) (S)-2,3,4,5-Tetrahydro-7-[[~rl-[(ben7imid~7-~1-2-yl)methyl]ben7imi~ 7ol-2-
yl]methyl]amino]carbonyl]-4-methyl-3-oxo-lH-1,4-benzodiazepine-2-acetic acid
1.0 N LiOH (0.36 mL, 0.36 mmol) was added dropwise at RT to a rnixture of
methyl (S)-2,3,4,5-tetrahydro-7-[[[[l-[(ben7i~ 7ol-2-yl)methyl]ben7imi~t~7Ql-2-
yl]methyl]amino]carbonyl]-4-methyl-3-oxo- lH- 1,4-benzodiazepine-2-acetate (0.08g, 0.14 mmol) in THF (5 mL) and H2O (4 mL). After 2 h, the reaction mixture was
concentrated to a small volume on the rotavap and cooled in an ice bath before
neutralizing with 1.0 N AcOH (0.69 mL). The solution was lyophilized to a crude
product (0.086 g) as a white powder. Purification on C-18 Bond Elute (0%-20%
CH3CN/H2O cont:~ining 0.1 % TFA) afforded the title compound as a white powder:
MS (ES) m/e 538.2 (M+H)+.
Example 32
Prep~r~tion of (S)-2.3.4.5-Tetrahydro-7-rrbisr(ben7imicl~7ol-2-
yl)methyllarninolcarbonyll-4-methyl-3-oxo-lH-1.4-benzodiazepine-2-acetic acid
a) Methyl (S)-2,3,4,5-tetrahydro-7-[rbis[(ben7imi~1~7ol-2-
yl)methyl]amino~carbonyl]-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetate
Diisopropylethylamine (0.22 mL, 1.29 mmol) was added to a stirred solution
of methyl (S)-7-carboxy-4-methyl-3-oxo-2,3,4,5-tetrahydro- 1 H- 1,4-benzodiazepine-
2-acetate trifluoroz-~-et~te (0.075 g, 0.26 mmol), bis[(ben7imi<1~7Ol-2-
yl)methyl]amine tris(trifluoro~cet~te) (0.16 g, 0.26 mmol), HOBt ~ H2O (0.05 g, 0.36
mmol), and EDC (0.069 g, 0.36 mmol) in anhydrous CH3CN (10 mL) at RT. After
17 h, the reaction rnixture was concentldted, diluted with CH2Cl2 (80 mL), and
washed sequentially with 5% NaHCO3 (2 x 20 mL) and brine (20 mL). Drying
- (MgSO4), concentration, and silica gel chromatography (7% MeOH/CH2Cl2) gave
the title compound (0.05 g, 36%) as a white solid: MS (ES) m/e 552.2 (M+H~+.
-
151
:
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
b) (S)-2,3,4,5-Tetrahydro-7-[[bist(ben7irni~ 7nl-2-
yl)methyl]amino]carbonyl]-4-methyl-3-oxo-lH-1,4-benzodia_epine-2-acetic acid
1.0 N LiOH (0.23 mL, 0.23 mmol) was added dropwise at RT to a mixture of
methyl ~S)-2,3,4,5-tetrahydro-7-[[bis[(ben7imi~7ol-2-yl)methyl]amino]carbonyl]-4-
methyl-3-oxo-lH-1,4-benzodiazepine-2-acetate (0.05 g, 0.09 mmol) in THF (6 mL)
and H20 (4 rnL). After 1 h, the reaction ~l~ixAluie was concentrated to a small
volume on the rotavap and cooled in an ice bath before neutralizing with 1.0 N
AcOH (0.3 mT.). The solid was collected, washed with cold H~O, and air dried to
give the title compound (0.048 g, 98%) as a white solid: MS (ES) m/e 538.2
(M+H)+.
Fx~mple 33
P~pdld~ion of f_)-2.3.4.5-tetrahydro-7-rrbisr~ben7imi~7ol-2-
yl)methyllamino~carbonyll-3-oxo-4-(2-phenethyl)-lH-1 ~4-benzofli~7~ 1~ine-2-acetic
B~
a) Methyl (+)-2,3,4,5-tetrahydro-7-[[bis[(ben7imi<1~7Ol-2-
yl)methyl]amino]carbonyl~-3-oxo-4-(2-phenethyl)-lH-1,4-berl7o~ 7Ppine-2-acetate
Diisopropylethylamine (0.3 mT, 1.74 mmol) was added to a stirred solution
of methyl (+)-7-carboxy-3-oxo-2-(2-phenylethyl)-2,3,4,5-tetrahydro- lH- 1,4-
benzodiazepine-2-acetate (0.11 g, 0.29 rnmol), bis[(ben7imi-1~7Ol-2-yl)methyl]amine
tris(trifluoroacetate) (0.18 g, 0.29 mmol), HOBt ~ H2O (0.058 g, 0.43 mmol), andEDC (0.083 g, 0.43 mmol) in anhydrous CH3CN (12 mL) at RT. After 21 h, the
reaction mixture was concelltld~d, diluted with CH2Cl2 (100 rnL), and washed
sequentially with 5% NaHCO3 (2 x 20 mL) and brine (20 mL). Drying (MgSO4),
concentration, and silica gel chromatography (7% MeOH/CH2CI2) gave the title
compound (0.13 g, 70%) as a white solid: MS (ES) m~e 642.2 (M+H)+.
b) (+)-2,3,4,5-Tetrahydro-7-[[bis[(ben7imi-1~7nl-2-yl~methyl]amino]carbonyl]-3-
oxo~-(2-phenethyl)- 1 H- l ,4-benzodiazepine-2-acetic
1.0 N LiOH (0.6 mL, 0.6 rnmol) was added dropwise at RT to a mixture of
methyl (~)-2~3~4~5-tetrahydro-7-[[bis~(be~7~ 7nl-2-yl)methyl]amino]carbonyl]-3
oxo-4-(2-phenethyl)- 1 H- 1,4-benzodiazepine-2-acetate (0.13 g, 0.20 mmol) in THF
(5 mL) and H20 (5 rnL). After 18 h, the reaction mixture was concentrated to a
small volume on the rotavap and cooled in an ice bath before neutralizing with 1.0 N
AcOH (0.6 mT.). The solution was Iyophilized to a crude product (0.092 g, 77~o) as .
152
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
a white powder. ODS chromatography (step gradient, 5%-30%
CH3CN/H2O cont~inin?~ 0.1 % TFA) afforded the title compound as a white powder:
MS (ES) mle 628.2 (M+H)+.
Ex:~ml?le 34
Preparation of ethyl (~)-3-rrr2-(ben7imidazol-2-yl~ethyl~aminolsuccinoyllamino-4-
pentynoate
a) (+)-4-Ethynyl-2-azetidinone
4-Acetoxy-2-~7~ti(1inone (9.0 g, 69.7 mmol) was added slowly to a solution
of ethynylm~gne~iumchloride (31 mL of a 0.5 M THF solution, 0.55 mmol) at 0~C.
After 1.5 h, 1.0 N HCl (100 mL) was added, and the mixture was taken up in EtOAc(300 mL) and washed sequentially with 1.0 N HCl (100 mL), saturated NaHCO3
(100 mL), and brine (100 mT.). After drying (MgSO4) and concentration, the titlecompound (4.57 g, 69%) was obtained as a light brownish solid: MS (ES) mJe 96.0
(M+H)+.
b) Ethyl (+)-3-amino-4-pentynoate
A mixture of (_)-4-ethynyl-2-:~7~ tirlinnne (1.3 g, 13.68 mmol), EtOH (54
ml), and concentrated HCl (6 mL) was heated to reflux for 18 h. The reaction wascooled to RT before adjusting the pH to 8.0 using saturated NaHCO3. The reactionwas extracted with EtOAc (3 x 70 mL), and the combined EtOAc layers were
washed with brine (50 ml). Drying (MgSO4) and concentration gave the title
compound (1.06 g, 55%) as a brownish liquid: MS (ES) m/e 141.9 (M+H)+.
c) Methyl-[[2-(benzimid~ol-2-yl)ethyl]amino]succinate
3-Carbomethoxypropionylchloride(0.6 g, 4.0 mmol) was added at 0~C to a
stirred solution of 2-aminoethylben7imi~ 701e ~ eet~te (1.13 g, 4.0 mmol) and
diisol~lu~ylethylamine (2.59 g, 20 mmol) in dry CHzClz (45 rnL). After stirring for
1.5 h at RT, the reaction mixture was diluted with CH2Cl2 (50 mr ) and washed
sequentially with H2O (30 mL), 5% NaHCO3 (30 ml), and brine (30 mL). Drying
(MgSO4), concentration, and silica gel chromatography (8% MeOH/CH2C12) gave
the title compound (0.2 g, 18%) as a yellow solid: MS (E~) m/e 276.4 (M+H)+.
. d) [[2-(Ben7imi~1~7-)1-2-yl)ethyl]amino]succinic acid
153
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
A mixture of methyl-[[2-(ben7imi~ 7Ol-2-
yl)ethyl]annino]succinate(0.2 g, 0.73 m~nol), 1.0 N NaOH (1.82 ml, 1.82 mmol) and
MeOH (10 rnL) was stirred at RT for 24 h, then was concentrated to dryness. H2O
(5 mL) was added, the solution was neutralized with 1.0 N HCl (1.82 mL), and theresnlt;ng solution was lyophilized to give the crude title compound (0.23 g) as an
off-white powder: MS (ES) m/e 261.9 (M+H)+.
e) Ethyl (_)-3-[~[2-(ben7.imi-1~7~ 2-yl)ethyl]amino]succinoyl]amino-4-pentynoateDiisopropylethylamine (0.32~mL, 1.83 mrnol) was added to a stirred solution
of ethyl (+)-3-amino-4-pentynoate (0.12 g, 0.88 mmol), [[2-(be~ 7Ql-2-
yl)ethyl]arnino]succinic acid (0.19 g, 0.73 mmol), HOBt H20 ~0.15 g, 1.1 mmol),
and EDC (0.21 g, 1.1 mmol) in anhydrous CH3CN (15 ml) and DMF (3 mL) at RT.
After 23 h, the reaction mixture was concentrated, diluted with CH2Cl2 (100 mL),and washed sequentially with 5% NaHCO3 (2 x 25 mL) and brine (25 mL). Drying
(MgSO4), concentration, and silica gel chromatography (7% MeOH/CH2Cl2) gave
the title compound (0.07 g, 25%) as a white solid: MS (ES) mle 385.4 (M+H)+.
F.~mRle 35
P~ ~dlion of (_1-3-rrr2-(Ben7imitl~7ol-2-yl)ethyllaminolsuccinoyllamino-4
pentynoic acid
a) (+)-3-r[r2-(Ben7imifi~7- 1-2-yl)ethyl]amino]succinoyl]amino-4-pentynoic acid
1.0 N LiOH (0.78 mL, 0.78 rnmol) was added dropwise at RT to a rnixture of
ethyl (+)-3-[r[2-(bçn7imi~ 7ol-2-yl)ethyl]amino]succinoyl]amino-4-pentynoate
(0.12 g, 0.31 mmol) in THF (5 mL), H2O (5 ml) and CH3CN (1 mL). After 3 h, the
reaction mixture was concentrated to a small volume on the rotavap and cooled in an
ice bath before neutralizing with 1.0 N AcOH (0.78 mL). The solution was
lyophili7~l1 to a crude product (0.167 g) as a white powder. ODS chromatography
(10% CH3CN/H2O cont~lining 0.1% TFA) afforded the title compound as a white
powder: MS (ES) m/e 357.1 (M+H)+.
~;xample 36
Prep:lration of (+)-3-rrr4-(4-Azal~el.7i.. id~zol-2-yl)butanoyll~lycyllaminol-4-
pentynoic acid (SB-237554)
154
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
a) Methyl (4-azabenzimidazol-2-yl)butyrate
Triethylamine (319 mL, 22.9 mmol) was added to a mixture of 2,3-
diaminopyridine (2.5 g, 22.9 mmol) and methyl 4-(chloroformyl)butyrate (3.77g,
22.9 mmol) in dry THF (50 mL) at 0~C. After stirring for 16 h at RT, the reaction
was concentrated to dryness under vacuum. The residue was dissolved in glacial
AcOH (25 mL) and was heated at 110~C. After 93 h, the reaction was allowed to
- cool to RT and was conct;ntldL~;d under vacuum. The dark brown residue was
diluted with H2O (40 mL) and CH2Cl2 (40 mL), and the mixture was neutralized to
pH 7 using 5 N NaOH. The layers were separated and the aqueous layer was furtherextracted with CH2C12 (2x100 mL). The combined organic layers were washed
sequentially with 5% NaHCO3 (2 x 30 mL)and brine (30 mL). Drying (MgSO4),
concentration, and silica gel chromatography (7% MeOH/CH2Cl2) gave the title
compound (0.47, 9%): MS (ES) m/e 220.0 (M+H)+.
~5 b) (4-Azabçn7imi(1zl7Ql-2-yl)butyric acid
A lllii'~Ul'~ of methyl (4-:~7zlhen7imidazol-2-yl)butyrate (0.47 g, 2.13 mmol),
1.0 N NaOH (6 mL, 6.0 mmol) and MeOH (10 mL) was stirred at RT for 5.5 h, then
was concentrated to dryness. The residue was diluted with H2O (2 mL) and
neutralized with 1.0 N HCl (0.73 mL). The resulting solid was collected and air
dried to give the title compound (0.32 g, 73%) as a yellow powder: MS (ES) m/e
206.0 (M+H)+.
c) Ethyl (_3-3-[[(N-tert-butoxycarbonyl)glycyl]amino]-4-pentanoate
Diisopropylethylamine (0.92 mL, 5.32 mmol) was added to a stirred solution
of ethyl I_)-3-amino-4-pentynoate (0.3 g, 2.13 mmol), Boc-Gly (0.56 g, 3.19 mmol),
HOBt H2O (0.43 g, 3.19 mmol), and EDC (0.61 g, 3.19 mmol) in anhydrous
CH3CN (15 mL) at RT. After 34 h, the reaction ll~Ult; was concentrated, diluted
with CH2Cl2 (70 mL), and washed sequentially with 5% NaHCO3 (2x15 mL) and
brine (15 mL). Drying (MgSO4), concentration, and silica gel chromatography (1:1EtOAc/Hexane) gave the title compound (0.5 g, 79%) as a colorless oil: MS (ES)
m/e 299.2 (M+H)+.
d) Ethyl (:!~)-3-~(glycyl)amino}-4-pPnt~no~tl~ trifluoroacetate
A solution of TFA (5 mL) and CH2Cl2 (15 mL) at RT was added all at once
to ethyl (_)-3-~[(N-tert-butoxycarbonyl)glycyl]amino]-4-pe~t~no~te (0.5 g, 1.68
mmol). After 30 min, the solution was concentrated on the rotavap, and the residue
155
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
was reconcentrated from toluene (to remove residual TFA) to afford the
title compound (0.55g, 106%) as a light yellow syrup: MS (ES) m/e 199.2 (M+H)+.
e) Ethyl (+)-3-~[[4-(4-azaben7imi~ 2-yl)butanoyl]glycyl]amino}-4-pentynoate
Diisopropylethylamine (0.94 mL, 5.43 mmol) was added to a stirred solution
of ethyl (+)-3-[(glycyl)amino]-4-pçnt~n-)~tlq trifluoro~l~et~te (0.55 g, 1.76 mmol), (4-
azabenzimidazol-2-yl)butyric acid (0.32 g, 1.55 mmol), HOBt ~ H2O (0.31 g, 2.33
mmol), and EDC (0.45 g, 2.33 mmol) in anhydrous CH3CN (15 mL) at RT. After 64
h, the reaction mixture was concentrated, diluted with CH2Cl2 (100 mL), and washed
sequentially with 5% NaHCO3 (2 x 25 mL) and brine (25 rnL). Drying (MgSO4),
concentration, and silica gel chromatography (7% MeOH/CH2CI2) gave the title
compound (0.11 g, 18%) as a white solid: MS (ES) m/e 386.4 (M+H)+.
f) (_)-3-[~[4-(4-Azaben7imi~1~7ol-2-yl)butanoyl]glycyl]amino]-4-pentynoic acid
1.0 N LiOH (0.71 mL, 0.71 mmol) was added dropwise at RT to a mixture of
ethyl (+)-3-[[[4-(4-~abt n7imi~ 7ol-2-yl)butanoyl]glycyl~amino]-4-pentynoate (0.11
g, 0.285 mmol) in THF (5 mL), H2O (5 mL) and CH3CN (1 mL). After 2 h, the
reaction mixture was concentrated to a small volume on the rotavap and cooled in an
ice bath before neutralizing with 1.0 N AcOH (0.70 mL). The solution was
lyophilized to a crude product (0.1 g, 100%) as a white powder. ODS
chromatography (5% CH3CN/H20 cont~ining 0.1% TFA) afforded the title
compound as a white powder MS (ES) m/e 358.4 (M+H)+.
Fxz~m~?le 37
Preparation of (S)-2.3.4.5-tetrahydro-7-~rr(4-aza-$-methylben7irr i~ ol-2-
yl)methyllmethylaminolcarbonyll-3-oxo- lH- 1.4-benzodiazepine-2-acetic acid
a) Dimethyl D-malate-o-trifluorom~th~npslllfonate
A solution of dimethyl-D-malate (12.96 g, 80 mmol) and pyridine (6.8 mT.,
84 mmol) in CH2Cl~ dry (50 mL) was added dropwise under argon at 0~C to a
solution of trifluo~ nesulfonic anhydride (14.2 rnL, 84 mmol) in dry CH2Cl2
(40 rnL) in a flame dried flask. The resulting yellowish-orange mixture was stirred
at 0~C for 30 rnin, and then at RT for 4 hr. The reaction was qllen~h~l by adding
H20 (50 rnL), and the layers were separated. The organic layer was washed
sequentially with H2O (3 x) and brine. Drying (MgS04) and concentration gave thetitle compound (22.45 g, 95%) as an off white solid: MS(ES) m/e 295 (M + H)+.
156
CA 0224l633 l998-06-26
W O 97/24119 PCTrUS96/20748
b) Dimethyl-N-(2-cyanophenyl)-D-aspartate
A solution of dimethyl D-malate-O-trifluo~ n--sulfonate (22.4 g, 76.2
mmol) in CHCl3 (40 mL) and hexanes (40 mL) was added at to a solution of 2-
aminobenzonitrile (9.0 g, 76.2 mmol) and 2,6-di-tert-butylpyridine in CHCl3 (50
mT) and hexanes (50 mL) in a flame dried flask at 0~C under argon. The resulting- mixture was stirred at 0~C for 30 min, then at RT for 3 days. The solvent was
removed in vacuo and the residue was taken into EtOAc and washed sequentially
with 5% HCI (10 x) and brine. Drying (MgSO4), concentration and silica gel flashchromatography (12% EtOAc/hexanes) gave the title compound (12.3 g, 62%) as a
clear oil: MS(ES) mte 263.3 (M + H)+.
c) Methyl (S)-2,3,4,5-tetrahydro-3-oxo- lH- 1 ,4-benzodiazepine-2-acetate
A mixture of dimethyl-N-(2-cyanophenyl)-D-aspartate ~12 g, 45.7 mmol),
Et3N (7.64 mL, 54.84 mmol) and Raney-Ni (46 g, wet, prewashed with CH30H) in
CH30H (200 mL) was stirred at RT under H2 (balloon) for 2 days. The catalyst wasremoved by filtration and washed with CH30H (3 x). Concentration and silica gel
flash chromatography (0-5% CH3OH/CH2Cl2) gave the title compound (7.93 g, 74
%) as a white solid: MS(ES) m/e 235.3 (M + H)+.
d) Methyl (S)-2,3,4,5-tetrahydro-7-bromo-3-oxo- 1 H- 1 ,4-benzodiazepine-2-acetate
Tetrabutylammonium tribromide (5.16 g, 10.7 mmol) was added portionwise
to a solution of methyl (S)-2,3,4,5-tetrahydro-3-oxo-lH-1,4-benzodiazepine-2-
acetate (2.5 g, 10.7 mmol) in CHCl3 (50 mL), and the llliY~tUl'e was stirred at RT for 2
days. H2O (30 mL) was then added, and the organic layer was separated and washedsequentially with H20 and brine. Drying (MgSO~), concentration, and silica gel
flash chromatography (0-5% CH3OH/CH2Cl2) gave the title compound (1.99 g,
60%)) as a white solid: MS(ES) m/e 313.0 (M + H)+.
e) Methyl (S)-2,3,4,5-tetrahydro-7-[t[(4-aza-5-methylbeu7i.l.i~l~7Ol-2-
yl)methyl]methylamino]carbonyl]-3-oxo- 1 H- 1 ,4-benzodiazepine-2-acetate
A ~ tule cont~inin~ methyl (S)-2,3,4,5-tetrahydro-7-bromo-3-oxo-lH-1,4-
benzodiazepine-2-acetate (624 mg, 2 mmol), 2-(aminomethyl)4-a_a-5-
methylben7imicl~7ole dihydrochloride (695 mg, 2.8 mmol), DIEA (1.8 mL, 10
mmol), and (Ph3P)2PdCl2 (126 mg, 0.18 mrnol) in NMP (22 mL) was heated to
110~C under a CO balloon for 48 hr. The solvent was removed on the rotavap (highvacuum) and the residue was puri~led by silica gel flash chromatography (0.5 - 5%
157
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96120748
CH3OEI/CH2Cl2) to give the title compound (170 mg, 19.5%) as a pale
yellow solid: MS (ES) m/e 437.5 (M + H)'.
f~ (S)-2,3,4,5-Tetrahydro-7-[~[(4-aza-5-methylben7imitlz-~ol-2-
5 yl)methyl]methylamino]carbonyl]-3-oxo- 1 H- 1,4-benzodiazepine-2-acetic acid
1.0 M LiOH (0.6 mL, 0.6 mmol) was added dropwise to a solution of methyl
(S)-2,3,4,5-tetrahydro-7-[[[(4-aza-5-methylbçn~.imi~1~7.ol-2- -
yl)methyl}methylamino]carbonyl]-3-oxo-lH- 1,4-benzodiazepine-2-acetate (170 mg,
0.39 rnmol) in CH30H (5 mL) and THF (5 mL)at RT. The resulting rnixture was
10 stirred for 20 hr and then was concenlldted. The residue was dissolved in H20,
acidified with 30% TFA, and purified by ODS chromatography (5% CH3CN/H20
contz-ining 0.1% TFA). Concentration and lyophilization gave the title compound as
an off white powder: [a],,25 -74.5o (c = 1, CH30H); MS (ES) m/e 423.2 (M + H)~.
Anal. Calcd for C2,H22N604 ~ 2 TFA ~ 1.75 H20: C, 44.03; H, 4.06; N, 12.32. Found:
C, 44.33; H, 4.04; N, 12.28.
Fx~nlple 38
Plep~dlion of (S)-2.3~4.5-tetrahydro-7-rrr(ben7imitl~zo1-2-
yl)metbyl~minolcarbonyll-3-oxo- 1 H- 1 ~4-benzodiazepine-2-acetic acid
a) Methyl (S)-2,3,4,5-tetrahydro-7-iodo-3-oxo- l H- 1,4-benzodiazepine-2-acetatePyridine-ICI complex: Iodine monochloride (100 mL, 1 M solution in
CH2Cl2) was added slowly to a solution of pyridine (8.5 mL, 105 mmol) in dry
CH2Cl2 (20 mL) at 5~C under argon so that the temperature was m~int~in~d between10 - 15~C. The mixture was stirred at 5 - 10~C for 20 min, then hexanes (50 mL)
was added, and the rnixture was stirred in the cold bath for another 30 min. Thesolid was collected by suction filtration, washed sequentially with hexanes and
petroleum ether, and dried to afford the reagent (22.5 g) as a yellow, crystalline
solid.
Pyridine-ICl complex (1.27 g, 5.28 mmol) was added portionwise to a
solution of methyl (S)-2,3,4,5-tetrahydro-3-oxo-lH-1.4-benzodiazepine-2-acetate
(1.18 g, 4.8 mmol) in CH2Cl2 (20 rnL) and CH30H ~20 mT). The resulting mixture
was stirred at RT for 40 rnin, then l M NaHSO3 (20 mL) was added. The solid was
collected by suction filtration and washed with Et2O. Drying yielded the title
compound (1.72 g, qu~ntit~tive) as an off-white solid: MS (ES) m/e 361.2 (M + H)~.
158
CA 02241633 1998-06-26
W O 97/24119 PCTAUS96/20748
b) Methyl (S)-2,3,4,5-tetrahydro-7-r[[(ben7imi~ 7OI-2-
yl)methyl]amino]carbonyl]-3-oxo- l H- 1,4-benzo~ 7~pine-2-acetate
A mixture of methyl (S)-2,3,4,5-tetrahydro-7-iodo-3-oxo-lH-1,4-
benzodia_epine-2-acetate (1.08 g, 3 mmol), 2-aminomethylben7imi~1~7Ole
S dihydrochloride hydrate (924 mg, 4.2 mmol), DIEA (2.6 mL, 15 mmol), and
(Ph3P)2PdCl2 (211 mg, 0.3 mmol) in NMP (30 mL) was heated to 110~C under a CO
- balloon for 3 hr. The solvent was removed on the rotavap (high vacuum) and the
residue was purified by silica gel flash chromatography (0-7% CH30H/CH2Cl2) to
afford the title compound (530 mg, ~44%) as an off white solid: MS(ES) m/e 408.1
(M+H)t.
c) (S)-2,3,4,5-Tetrahydro-7-{[[(ben7imi~ 7ol-2-yl)methyl]amino]carbonyl]-3-oxo-
1 H- 1,4-benzodiazepine-2-acetic acid
Following the procedure of Example 37(f), except substituting methyl (S)-
2,3,4,5-tetrahydro-7-[[[(ben7imi(1z~7Ol-2-yl)methyl]amino]carbonyl]-3-oxo-lH-1,4-
benzodiazepine-2-acetate for methyl (S)-2,3,4,5-tetrahydro-7-[[[(4-aza-5-
methylbenzimidazol-2-yl)methyl]methylamino3carbonyl]-3-oxo- lH- 1,4-
benzodiazepine-2-acetate, the title compound (66%) was ~ al~,d as a white
powder: [a]n25 -145.3~ (c = 1, CH30H); MS (ES) m/e 394.2 (M + H)t. Anal. Calcd
for C2nH,gN5O4 - 2 TFA 0.125 H2O: C, 46.22; H, 3.43; N, 11.23. Found: C, 46.13;
H, 3.78; N, 11.49.
E~am~l?le 39
~lGpaldlion of (+)-N-r2-(aminomethyl~-4-rrr~4-~7~-S-methylbe~7,imi-1~7ol-2-
yl)methyl~methylaminolcarbonyllphenyl~aspartic acid
a) Methyl (+)-2,3,4,5-tetrahydro-7-[[[(4-a_a-5-methylben7imi~1~7ol-2-
yl)methyl]methylamino]carbonyl3 -3-oxo- 1 H- 1,4-benzodia_epine-2-acetate
EDC (130 mg, 0.75 mmol) was added to a solution of methyl (+)-2,3,4,5-
tetrahydro-7-carboxy-3-oxo-lH-1,4-benzodiazepine-2-acetate (190 mg, 0.68 mmol),
2-(aminomethyl)-4-aza-S-methylben7imi~l~7ole dihydrochloride (169 mg, 0.68
mmol), HOBt H2O (101 mg, 0. 75 mmol), and DIEA (0.39 mL. 2.24 mmol) in
anhydrous DMF at RT. After 20 hr, the reaction was concentrated on the rotavap
(high vacuum), and the residue was cl~ atographed on silica gel (1 - 6.5%
CH3OH/CH2Cl2) to afford the title compound (260 mg, 88%) as a white solid: MS
(ES) m/e 437.5 (M ~ H)'.
159
CA 02241633 1998-06-26
W O 97/24119 PCTAUS96120748
b) (_)-N-[2-(Aminomethyl)-4-[[[(4-aza-5-methylben7imitl~7O1-2-
yl)methyl]methylamino}carbonyl]phenyl]aspartic acid
Following the procedure of Example 37(f), except substitl-ting methyl (+)-
2,3,4,5-tetrahydro-7-[[[(4-~a-5-methylben7imi~1~7O1-2-
yl)methyl]methylamino]carbonyl]-3-oxo-lH-1,4-ben7O~ 7~pine-2-acetate for
methyl (S)-2,3,4,5-tetrahydro-7-[[[(4-aza-5-methylben7imi~1~701-2-
yl)methyl]methylamino]carbonyl]-3-oxo- 1 H- l ,4-bçn70~ 7epine-2-acetate, the title
compound (66%) was prepared as a white powder: MS (ES) m/e 441.2 (M + H)+.
Anal. Calcd for C2,H2"N6O5 - 2 TFA 2.25 H2O: C, 42.08; H, 4.38; N, 11.78. Found:C, 42.01; H, 4.18; N, 11.55.
Fx~rru?le 40
Plepaldlion of (S~-2~3.4.5-tetrahydro-7-lrr(4-aza-5-methylbell7i.. i~zl7ol-2-
yl)mt~thyll~minolcarbQ~.~yll-3-oxo-lH-1~4-benzodiazepine-2-acetic acid
a) Methyl (S)-2,3,4,5-tetrahydro-7-[[[(4-aza-5-methylberl 7i mi~l ,.7 ol-2-
yl)methyl]amino]carbonyl]-3-oxo- I H- 1,4-benzodiazepine-2-acetate
Following the procedure of Example 38(b), except ~7u1)~7LiLu~il.g 2-
(aminomethyl)-4-aza-5-methylbell,i..,i-lz~7Ole dihydrochl(-ride for 2-
aminomethylbe~7imicl~7ole dihydrochloride hydrate, the title compound (63%) was
prepared as an amber solid: MS ~ES) m/e 423 (M + H)'.
b) (S)-2,3,4,5-Tetrahydro-7-[[[(4-aza-5-methylben7imicl~7O1-2-
yl)methyl]amino]carbonyl~-3-oxo- 1 H- 1,4-benzodiazepine-2-acetic acid
Following the procedure of Example 37(f), except substituting methyl (S)-
2,3,4,5-tetrahydro-7-[[[(4-aza-5-methylb~n7imi~ 7ol-2-yl)methyl]amino]carbonyl]-3-oxo- lH- 1,4-benzodiazepine-2-acetate for methyl (S)-2,3,4,5-tetrahydro-7-t[[(4-
aza-5-methylbe~-7i.. i-l~7~-1-2-yl)methyl]methylamino]~ul,o.,yl]-3-oxo-lH-l,~benzodiazepine-2-acetate, the title colll~oulld (50%) was prepared as a white
powder: MS (~S) m/e 409.2 (M + H)~. Anal. Calcd for C2"H2"N6O4 ~ 1.75 TFA -
H~O: C, 45.09; H, 3.82; N,3 .45. Found: C, 45.18; H, 4.10; N, 13.58.
E~mple 41
160
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
Preparation of (S)-2.3.4.5-Tetrahydro-7-rrr(bPnzimitl:~7ol-2-
yl)methyllaminolcarbonyll-3-oxo-4-r2-(pyrid-3-yl)ethyll-lH- 1.4-ben7-1diazepine-2-
acetate
5 a) tert-Butyl 4-fluoro-3-[[2-(pyrid-3-yl)ethyl~amino]benzoate
A mixture of tert-butyl 4-fluoro-3-methylbenzoate (3.83 g, 18.22 mmole),
- NBS (3.57 g, 20.24 mmole), benzoyl peroxide (0.22 g, 0.91 mmole), and CC14 (90
mL) was heated at reilux. After 16 hr, the reaction was cooled in ice/H20 and
filtered, and the filtrate was concentrated. The residue was passed through a short
10 pad of silica gel (20 % EtOAc/hexane) to remove baseline m~t~.ri~ls~ and the filtrate
was concentrated. The residue was dissolved in THF ~90 mI,), and 3-(2-
aminoethyl)pyridine (6.97 g, 57 mmol).) was added rapidly. The addition a~eal ,dto be mildly endothermic. The reaction was stirred overnight then was concentrated.
The residue was diluted with Et2O (100 mL) and washed sequentially with 1.0 N
15 NaOH (30 mL), H2O (30 mT.), and brine (30 mL). Drying (MgSO4), concentration,and silica gel chromatography (10 % MeOH/CH2Cl2) gave the title compound (2.58
g, 59 %) as a yellow oil: MS (ES) m/e 331 (M + H)+.
b) tert-Butyl (S)-4-fluoro-3-[2-aza-4-(benzyloxycarbonyl)amino-3,6-dioxo-6-
20 methoxy-2-[2-(pyrid-3-yl)ethyl]hexyl]benzoate
DCC (1.86 g, 9 mmol)was added to a solution of tert-butyl 4-fluoro-3-[[2-
(pyrid-3-yl)ethyl]amino]benzoate (2.7 g,.8.18 mmol), N-Cbz-L-aspartic acid ~-
methyl ester (J. Am. Chem. Soc. l9S7, 79, 5697; 2.53 g, 9 mmol.), and HOBt H2O
(1.2 g, 9 mmol) in anhydrous DMF (10 mL) at RT. After 24 hr, the mixture was
25 diluted with Et20 (25 rnL) and filtered. The filtrate was concentrated to dryness,
and the residue was diluted with Et2O (50 rnL) and washed with H2O (2 x 10 mL)
and brine (10 mL). Drying (MgSO4), concentration, and silica gel chromatography
(CH2Cl2) gave the title compound (2.4 g, 49%)as a colorless oil: MS (ES) m/e 594(M +H)+.
c) tert-Butyl (S)-4-fluoro-3-[4-amino-2-aza-3,6-dioxo-6-methoxy-2-[2-(pyrid-3-
yl)ethyl]hexyl]benzoate
A mixture of tert-butyl (S)-4-fluoro-3-[2-aza-4-(benzyloxycarbonyl)arnino-
3,6-dioxo-6-methoxy-2-[2-(pyrid-3-yl)ethyl]hexyl]benzoate (2.4 g, 4 mmol.), 10 %35 Pd/C (184 mg, 0.17 mmole), and MeOH (17 mL) was shaken at RT under H2 (50
psi). After 1.5 hr, the reaction was filtered through celite(~3) and concentrated. Silica
161
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
gel chromatography (10 % MeOH in 1:1 EtOAc/CHC13) gave the title
compound (1.1 g, 59 %) as a colorless oil: MS (ES) 460 (M + H)+.
d) Methyl (S)-2,3,4,5-tetrahydro-7-(tert-butoxycarbonyl)-4-[2-(pyrid-3-yl)ethyl]-3-
5 oxo- lH- 1,4-benzodiazepine-2-acetate
A solution of tert-butyl (S)-4-fluoro-3-t4-amino-2-aza-3,6-dioxo-6-methoxy-
2-[2-(pyrid-3-yl)ethyl]hexyl]ben70~te (0.64 g 1.39 mrnol) in anhydrous DMSO (5.7mL) was heated under argon in an oil bath set at 120-125~C. After 17.5 hr, the
reaction was cooled in ice/H20 and~diluted with H20 (12 mL). The mixture was
10 extracted with EtOAc (3 x 20 mL), and the combined EtOAc layers were washed
with H2O (10 mL) and brine (10 mL). Drying (MgSO4), concentration, and silica
gel chromatography (9: 1 CH2Cl2/MeOH) gave the title compound (0.15 g, 33%) as anearly colorless solid: MS (ES) m/e 440 (M +H)+.
e) Methyl (S)-2,3,4,5-tetrahydro-7-carboxy-4-[2-(pyrid-3-yl)ethyl]-3-oxo-lH-1,4-benzodiazepine-2-acetate
4 M HCI/dioxane (0.5 mL) was added to a solution of methyl (~)-2,3,4,5-
tetrahydro-7-(tert-butoxycarbonyl)-4- [2-(pyrid-3-yl)ethyl]-3 -oxo- 1 H- 1,4- -
benzodiazepine-2-acetate(0.18 g, 4.1 mrnol) in anhydrous CH2Cl2 (5 mL) and the
reaction was stirred at RT overnight. Concentration in vacuo followed by
reconcentration from toluene (3 x 10 mL) gave the title compound (0.12 g, 65%):
MS (ES) m/e 384 (M + H)+.
f) Methyl (S)-2,3,4,5-tetrahydro-7-[t[(ber 7imitl~7ol-2-yl)methyl]amino]carbonyl]-
3-oxo-4-[2-(pyrid-3-yl)ethyl]- 1 H- 1,4-benzodiazepine-2-acetate
A mixture of methyl (S)-2,3,4,5-tetrahydro-7-carboxy-4-[2-(pyrid-3-
yl)ethyl]-3-oxo-lH-1,4-benzo~ 7epine-2-acetate (0.12 g, 0.26 mmol) and thionyl
chloride (15 mL) was refluxed for 1 h. The re~slllting orange solution was
concentrated to dryness to leave a yellow-orange foam. This was dissolved in
CH2C12 (10 mL) and added dropwise to a solution cont~inin~ 2-
aminomethylbe..~ilni~l~701e dihydrochloride hydrate (0.058 g, 0.26 mmol), pyridine
(0.72 g, 9.1 mmol), and triethylamine (0.55 g, 5.46 mmol) in CH2C12 (15 mL) at
0~C under argon. The reaction mixture was then stirred in RT under argon. After
25.5 h, CH2C12 (200 mL) and 5% NaHCO3 (50 rnL) were added to the reaction
35 mixture to give a light yellow precipitate which was filtered and air-dried to give the
title compound (0.030g.,~% yield): MS (ES) m/e 513 (M+H)+.
162
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
g) (s)-2~3~4~5-Tekahydro-7-~[[(ben~ 7ol-2-
yl)methyl]amino]carbonyl] -3-oxo-4-[2-(pyrid-3-yl)ethyl]- 1 H- 1,4-benzodiazepine-2-
acetate
1.0 N LiOH (0.57 mL, 0.57 mmol) was added dropwise at RT to a mixture of
S methyl (S)-2,3,4,5-tetrahydro-7-[[[(ben7imi(1~7~-1-2-yl)methyl3amino]carbonyl]-3-
oxo-4-[2-(pyrid-3-yl)ethyl]-lH-1,4-benzodia_epine-2-acetate (0.030 g, 0.059 mmol)
- in THF (4 mL) and H2O (5 mL). The resulting light brownish-yellow solution was
stirred for 21.5 h, then was concenkated on the rotavap. The resulting residue was
lyophilized to give the crude product as a yellowish powder. ~ liv~ HPLC
(PRP-l~ column, step gradient, 10-20% CH3CN/H2O contzlinin~ 0.1% TFA)
afforded the title compound (0.OlOg 34% yield): MS (ES) m/e 499(M+H)+. Anal.
Calcd .for C27H26N6O4 - 3 C2HF3O2 ~ 3 HCI - 3 H2O: C, 37.41; H, 3.41; N,7.52.
Found: C, 37.6; H, 3.52; N, 7.52.
Example 42
Preparation of Ethyl (S)-2.3.4.5-tetrahydro-7-rrr~ben7imid~7ol-2-
yl)methyl~methylaminolcarbonyll-4-methyl-3-oxo- 1 H- 1.4-benzodiazepine-2-acetate
a) Ethyl (s)-2~3~4~s-tekahydro-7-~[[(be~ 7~l-2-
yl)methyl]methylamino]carbonyl]-4-methyl-3-oxo- lH- 1,4-benzodia_epine-2-acetateHCl gas was bubbled into EtOH (200 mL) for 10 min, then (S)-2,3,4,5-
tetrahydro-7-[[[(ben7imi~ 7ol-2-yl)methyl]methylamino]carbonyl]~-methyl-3-oxo-
lH-1,4-benzodia7epine-2-acetic acid (2.00 g, 4.5 mmol) was added. The reaction
was stirred at RT for 24 hr, then was concentrated to dryness on the rotavap. The
residue was reconcenkated from toluene (2 x) to remove residual EtOH, then was
chromatographed on silica gel (gradient: 7% MeOH/CH2Cl2 (1 L) then 10%
MeOH/CH2Cl2). The r~sllltin~ residue was dissolved in EtOH, and Et2O was added
to precipitate a solid. This was collected and washed with Et2O to afford the title
compound as a white solid: MS (ES) m/e 450.2 (M + H)+. Anal. Calcd for
C24H27NsO4 ~ 1.5 H2O: C, 60.49; H, 6.35; N, 14.70. Found: C, 60.41; H, 6.27; N,
14.38.
Fx~mple 43
Preparation of 4-~rr3-(lH-ben7imi~1~7ol-2-yl)propyllaminolcarbonyllpiperidine-l-acetic acid
163
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
a~ 2-[3-~N-tert-Butoxycarbonyl)aminopropyl3benzirnidazole
A solution of isobutylchloroformate ( 10.2 rnL, 79 mmol) in THF (25 rnL)
was added to a solution of 4-(tert-butoxycarbonyl)arninobutyric acid (Organic
S ~;ynthesis 1984, 63, 160; 13.5 g, 0.066 mole) and triethylamine (11 mL, 80 mmol) in
THF ( 50 mL) at 0~C under argon. After 0.5 h, a solution of 1,2-phenylen~ mine
(7 g, 64.8 rnmole) in THF (50 rnL) was added dropwise to the res-ltting white
suspension. The reaction was stirred for 18 h, then was ffltered, and the filtrate was
concentrated to give a serni-solid. This was dissolved in AcOH (100 rnL), and the
solution was heated at 70~C for 18 h. The reaction mixture was concentrated, andthe residue was reconcentrated several times from toluene. Silica gel flash
chromatography gave the title compound (6.0 g, 33%): MS (ES) m/e 276 [M+H]t.
b) 2-(3-Aminopropyl)bc~.~i...itl~7ole dihydrochloride
A solution of 2-[3-(N-tert-butoxycarbonyl)aminopropyl3ben7imirls-7.nle (1.2 g,
4.3 mmol) and 4 M HCl in dioxane (20 mL) in CH2C12 (25 ml) was stirred at RT
for 18 h. The resulting white suspension was filtered to give the title compound(1.07 g, 97%).
20 c) Ethyl ~[[[3-(lH-ben,.iid~7.ol-2-yl)propyl]arnino]carbonyl]piperidine-l-acetate
A ~ lule of ethyl 4-carb-~y~ 3elidine-1-acetate hydrochloride (Yellin's SB
223913 CIP)(0.76 g, 3 mmol) in thionyl chloride (10 mL) was heated to reflux for15 min, then was concentrated to dryness. After evaporation several times from
toluene, the residue was dissolved with 2-(3-aminopropyl)ben7imi~1~7Ole
dihydrochloride (0.77 g, 3 mmol) and DIEA (3 mL) in DMF (25 mL). After 18 h,
the reaction mixture was partitioned between EtOAc (50 mL) and 5% NaHCO3 (100
mL), and extracted with EtOAc (2 x 50 mL). The combined organic extracts were
washed seq~lenti:~ily with H20 and saturated NaCl solution, then were dried overMgSO4. Concentration and silica gel flash chromatography (6% MeOH/CH2C12)
gave the title compound (40 mg, 3%).
b) 4-[[~3-(Rçn7.imida_ol-2-yl)propyl]amino~carbonyl]piperidine-1-acetic acid
lN NaOH solution (0.4 mL, 0.4 mmol) was added to a stirred solution of
ethyl 4-[[3-~lH-ben7imi~l~7ol-2-yl)propylamino]carbonyl]piperidine-l-acetate (40mg, 0.1 mmol) in MeOH (10 mL)at RT. After 18 h, the mixture was neutralized
with AcOH (1 mL) and concentrated to remove the MeOH. The aqueous solution
was loaded onto an XAD-2 column, and eluted with H20 (500 mL), then with 20%
164
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/2074P
CH3CNt H20. The fractions con,~zlining the product were pooled and
lyophilized to give the title compound (9 mg, 25%). MS (ES~ m/e 345.2 [M+H]+.
Anal. Calcd for C,8H24N4O3 0.75 H2O: C, 60.40; H, 7.18; N, 15.65. Found: C,
60.48; H, 7.16; N, 15.40.
Lxample 44
Plc~aldliQn of 4-rrr3-(ben7imi~l~7f 1-2-yl)propyllaminolcarbonyllphenylacetic acid
a) Ethyl 4-[[[3-~ben7imiclz~ 1-2-yl)propyl]amino]carbonyl]-l-phenylacetate
A mixture of ethyl 2-(4-carboxyphenyl)acetate (Yellin's SB 223913 CIP)(0.5
g, 2.4 mmol), 2-(3-aminopropyl)ben7imi~7ole dihydrochloride (0.7 g, 2.8 mmol),
HOBt ~ H20 ~0.36 g, 2.6 mmol), EDC (0.5 g, 2.6 mmol), and DIEA (1.5 mL, 8.8
mmol) in DMF (15 mL) was warmed briefly and stirred at RT for 18 h. The reactionmixture was partitioned between EtOAc ~50 mL) and 5% NaHCO3 (100 mL), and
extracted with EtOAc (2 x 50 mL). The combined organic extracts were washed
sequentially with H2O and saturated NaCI solution, then were dried over MgSO4.
The evaporated residual solid was triturated with Et2O to give the title compound
(0.56 g, 66%): MS (ES) m/e 366.0[M+H~+. Anal. Calcd for C2lH23N3O3 0.25 H2O:
C, 68.18; H,6.40; N, 11.36. Found: C, 68.16; H,6.26; N,11.36.
b) 4-[[[3-(Ben7imi~1~7(-1-2-yl)propyl]amino]carbonyl]phenylacetic acid
1 N NaOH solution (6 rnL, 6 mmol) was added to a stirred solution of ethyl 4-
[[[3-(lH-bPn7imicl~7n-2-~71)propyl]arnino]carbonyl]-l-phenylacetate (0.38 g, 1
25 mmol) in MeOH (15 mL)at RT. After 4 h, the mixture was neutralized with AcOH
(6 mL) and the resulting solid was filtered to give the title compound (77 mg, 22%):
MP 108-110~C; MS (ES) rn/e 345.2 [M+H]+. Anal. Calcd for Cl9HlgN3O3 0.6 H2O:
C, 65.54; H, 5.85; N, 12.07. Found: C, 65.63; H, 5.65; N, 11.95.
Fx~m~le 45
Preparation of (S)-2.3.4.5-tetrahydro-4-m~t~yl-3-oxo-7-rrr(S-
trifluoromethylben~ 7ol-2-y~)methyl~methylaminolcarbonyll - l H- l .4-
benzodiazepine-2-acetic acid
a) 3,4-Diaminobenzotrifluoride
165
CA 02241633 1998-06-26
W O 97~4119 PCT~US96/20748
4-Amino-3-nitroben~oLlinuoride (3.7070 g, 17.98 mmol) was
dissolved in MeOH, and a catalytic amount of 10% Pd/C was added. The reaction
was purged with H2, and stirred at RT under H2 (balloon). After 24 hr, the reaction
was filtered through a bed of celite~, and the filtrate was evaporated under vacuum
to yield the title compound (3.0878 g, 98%). The material was used without
characlefizdLion.
b) 2-[N-(Benzyloxycarbonyl)-N-methyl]aminomethyl-5-
trifluoromethylben7imi~ 1e
Cbz-sarcosine (3.9950 g, 17.13 mmol) was dissolved in dry THF, and
isobutylchloroformate (2.5 mL, 19.27 mmol) was added, followed by triethylamine
(5.0 mL, 35.95 mmol). The mixed anhydride was allowed to form at RT for 30 min,
then was added to a solution of 3,4-diaminobenzotrifluoride (3.0818 g, 7.53 mmol)
in dry THF. After 20 hours at RT, the reaction was ~vapoldted under vacuum. The
residue was partitioned between EtOAc and 1.0 N NaHCO3, and the layers were
separated. The aqueous layer was extracted with EtOAc, and the combined organic
layers were dried (MgSO4), filtered and e-v~oldled under vacuum. The residue wasreconcentrated from toluene, then was dissolved in glacial AcOH (125 mL). The
solution was heated at 110~C for 24 hr, then the AcOH was evaporated under
vacuum. The residue was reconcentrated from toluene, then was adsorbed onto
silica gel and loaded onto a dry silica gel flash chromatography column. The
column was eluted with 1:1 CHCl3/Et,0 to afford the title compound (2.9397 g,
47.2%): TLC R, (1:1 CH2Cl~/Et2O) 0.57; MS (ES) rn/e 364.2 (M+H)+; 'H NMR (250
MHz, CDC13) ~ 8.0 - 7.2 (m, 9H), 5.05 (s, 2H), 4.84 (s, 2H), 3.07 (s, 3H).
c) 2-(Methylaminomethyl)-5-trifluoromethylben7.imicl~701e
2-[N-(Benzyloxycarbonyl)-N-methyl]aminomethyl-S-
tri~uoromethylben7imi(1~7ole (2.9397 g, 8.09 mmol) was dissolved in MeOH, and a
catalytic amount of 10% Pd/C was added. The reaction was purged with H2, then
was stirred at RT under H2. After 5 hr, the reaction ~ lure was filtered through a
bed of celite(~), and the filtrate was evaporated under vacuum to leave a tan colored
oil. Analysis by 400 MHz NMR showed the Cbz protecting group was still present,
so the residue was resubmitted to the reaction conditions. After 18 hr, the catalyst
was removed by filtration through a bed of celite(~) and the filtrate was ~;v~oldted
under vacuum to yield the title compound (1.7809 g, 96%): lH NMR (250 MHz,
CDC13) o 7.76 - 7.32 (m, 4H), 4.32 (s, 2H), 2.59 (s, 3H).
166
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
d) Methyl (S)-2,3,4,5-tetrahydro4-methyl-3-oxo-7-[[[(5-
trifluoromethylben7im~ 7Ol-2-yl)methyl]methylamino]carbonyl]- lH- 1,4-
benzodiazepine-2-acetate
Methyl (S)-7-carboxy-4-methyl-3-oxo-2,3 ,4,5-tetrahydro- l~I- 1,4-
S benzodiazepine-2-acetate (179.2 mg, 0.61 mmol) was weighed into a 100 mL round
bottomed flask. CH3CN (10 mL) was added, followed by HOBt ~ H2O (97.9 mg,
0.72 mmol) and EDC (149.3 mg, 0.78 mmol). After all the solids had dissolved, a
solution of 2-(methylarninomethyl)-5-trifluoromethylben7imi~1~701e (186.1 mg, 0.81
mmol) in CH3CN was added with diisopropylethylamine (0.25 rnL, 1.44 mmol).
After 24 hr at RT, the reaction was evaporated under vacuum, and the residue waschromatographed on silica gel (3% MeOH/CHCl3) to afford the title compound
(308.1 mg, 100%): TLC Rf (5% MeOH/CHCl3) 0.21; lH NMR (250 MHz, CDC13)
7.83-7.16 (m, 7H), 5.37 (d, 1~), 5.05-4.70 (m, 3H), 2.96 (m, 3H), 3.72 (s, 3H),
3.16 (s, 2H), 2.11 (s, 3H); MS (ES) m/e 504.0 (M+H)~.
e) (s)-2~3~4~5-Tetrahydro-4-methyl-3-oxo-7-[[[(s-trifluoromethylben~ 7Q1-2
yl)methyl]methylamino]carbonyl] -1 H- 1 ,4-benzodiazepine-2-acetic acid
Methyl (S)-2,3,4,5-tetrahydro-4-methyl-3-oxo-7-[[[(5-
trifluoromethylb~n7imi~1~7ol-2-yl)methyl]methylamino~carbonyl]-lH-1,4-
benzodiazepine-2-acetate (308.1 mg, 0.61 mmol) was dissolved in MeOH (S mL).
H20 (5 mL) was added, followed by 1.0 N NaOH (2.0 mL, 2.0 mmol). After 24 hr
at RT, the reaction was neutralized with 1.0 N HCl (2.0 mL). The milky white
mixture was stirred at RT for 15 min, then was diluted with H2O, and the precipitate
was collecte~l on a sintered glass funnel. The white powder was dried in a vacuum
de~ c~tQr overnight to yield the title compound (268.0 mg, 90%): M~ (ES) m/e
490.2 (M+H)'. Anal. Calcd for C23H~N5O4F3 2.25 H2O ~ 0.25 HCl: C, 51.24; H,
5.00; N, 12.99. Found: C, 51.44; H, 4.96; N, 12.45.
F.x:~ml?le 46
Ple~ alio l of (S)-2.3.4.5-tetrahydro-7-rrr(4.7-dimethoxyben7imicl~7.ol-2-
yl)methyllmethylamino~carbonyll-4-methyl-3-oxo- lH- 1 .4-benzodiazepine-2-aceticacid
~'
a) 2-[N-(Benzyloxycarbonyl)-N-methyl]~n~innmPthyl-4,7--1im~thoxybenzimidazole
~ Following the procedure of Example 45(b), except sul)~lilulillg 1,2-~ mino-
3,6-dimethoxybenzene for the 3,4-diaminobenzotrifluoride, the title compound was 167
CA 02241633 1998-06-26
W O 97~4119 PCT~US96~0748
prepared: MS (ES) m/e 356.2 (M+H)'; lH NMR (250 MHz, CDC13) o
7.34 (s, SH), 6.54 (d, 2H), 5.18 (s, 2H), 4.65 (s, 2H), 3.95 (s, 3H), 3.86 (s, 3H), 3.03
(s,3H).
S b) 4,7-Dimethoxy-2-(methylaminomethyl)ben7imi(1~7ole
2-[N-(Benzyloxycarbonyl)-N-methyl]aminomethyl-4,7-
dimethoxybenzimidazole (186.5 mg, 0.53 mmol) was dissolved in MeOH, and a
catalytic amount of 10% Pd/C was added. The reaction was purged with Hz, then
was stirred at RT under H~ (balloon). ~After 20 hr, the reaction was filtered through
celite(~, and the filtrate was eva~?oraLed under vacuum to yield the title compound
(96.9 mg, 83%): 'H NMR (250 MHz, CDC13) ~ 6.52 ~s, 2H), 3.94-3.86 (m, 6H),
2.36 (s, 3H).
c) Methyl (S)-2,3,4,5-tetrahydro-7-[[[(4,7-~lim~thQxyben7imifl~7ol-2-
yl)methyl]methylamino]carbonyl]4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetate Methyl (S)-7-carboxy~-methyl-3-oxo-2,3,4,5-tetrahydro-lH-1,4-
benzodiazepine-2-acetate (112.3 mg, 0.38 rnmol) was weighed into a 100 mL round
bottomed flask. CH3CN was added, followed by HOBt ~ H~O (62.3 mg, 0.46 mmol~
and EDC (120.0 mg, 0.63 mmol). When the solids had all dissolved,
diisopropylethylamine (0.1 mL, 0.57 mmol) was added, followed by a suspension of4,7-~lim~thoxy-2-(methylaminomethyl)be~-7i~ 7ole (96.8 mg, 0.44 mmol) in
CH3CN cont5.inin~ diisopropylethylamine (0.1 mL, 0.57 mmol). After 2.5 days at
RT, the reaction was evaporated under vacuum. The residue was evaporated once
with toluene, then was chromatographed on silica gel (CHCl3, then 5%
MeOH/CHCl3) to afford the title compound (152.0 mg, 80.0%): TLC R~ (5%
MeOH/CHCl3) 0.35; MS (ES) m/e 496.2 (M+H)t; H NMR (250 MHz, CDC13) o
7.25 (m, 2H), 6.56 (s, 2H), 5.36 (d, lH), 3.91 (s, 6H), 3.70 (s,3H), 3.08 (s, 3H).
d) (S)-2,3,4,5-Tetrahydro-7-[[[(4,7-dimetho~ybel-7il..i~1~7~ 2-
yl)methyl]methylarnino]carbonyl]-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetic
acid
Following the procedure of Example 45(e), methyl (S)-2,3,4,5-tetrahydro-7-
t[[~4,7-dimethoxybçn7.imic~7.(-1-2-yl)methyl]methylamino]carbonyl]-4-methyl-3-
oxo-lH-1,4-benzodiazepine-2-acetate was saponified to afford the title compound
(110.0 mg, 74%): MS (ES) m/e 482.2 (M+H)~. Anal Calcd for C~,~H27N5O6 ~ 0.75
H2O: C, 58.23; H, 5.80; N, 14.15. Found: C, 58.26; H, 5.59; N, 13.90.
168
CA 02241633 1998-06-26
W O97/24119 PCTAUS96120748
Fxample 47
~l~dlion of (S)-2.3.4.5-tetrahydro-7-rrr(4-methylbenzimidazol-2-
yllmethyllmethylaminolcarbonyll-4-methyl-3-oxo-lH-1.4-benzodiazepine-2-acetic
5 a~id
a) 1,2-Diamino-3-methylbenzene
Following the procedure of Example 45(a), except ~ub~ li..g 2-methyl-6-
nitro~nilin.- (3.0204 g, l9.g8 mmol) for the 4-amino-3-nitrobenzotrifluoride, the title
compound (2.4815 g) was prepared. This was used without char~( le, i7~1 ;on.
b) 2-[N-(Benzyloxycarbonyl)-N-methyl]aminomethyl-4-methylben7imitl:~7ole
C bz-sarcosine (4.6466 g, 19.92 mmol) was dissolved in dry THF in a 100 mL
round-bottomed flask. Triethylamine (3.0 mL, 21.57 mmol) was added, followed by
isobutylchlolufol-llate (2.8 mL, 21.59 mmol). The white reaction mixture was stirred at
RT for Q.5 hr, then was added to a solution of 1,2-diamino-3-methylbenzene (2.4815 g)
in dry THF at -20 to -30~C. After 20 min, the reaction was warmed to RT and stirred
there for 16 hr. The reaction was evaporated under vacuum and the residue was
partitioned between EtOAc and 1.0 N NaHCO3. The layers were separated, and the
aqueous layer was extracted with EtOAc. The combined organics were dried (MgSO4),
filtered, and evaporated under vacuum. The residue was reconcentrated from toluene and
the dried solid was dissolved in glacial AcOH (150 mL). The solution was heated at
110~C for 18 hr, then was concentrated under high vacuum. The residue was
reconcentrated from toluene, then was adsorbed onto silica gel and loaded onto a dry
silica ge} flash chromatography column. The column was eluted with 1: 1 CH2Cl2/Et2O to
afford the title compound (3.1586 g, 51%): MS (ES) m/e 310.2 (M+H)~; 'H NMR (250MHz, CDCl3) ~ 7.35-7.01 (m, 10H), 5.00 ~s, 2H), 4.72 (s, 2H), 2.99 (s, 3H), 2.55 (s, 3H).
c) 4-Methyl-2-(methylaminomethyl)ben7imi<1~7Ole
30 Following the procedure of Example 45(c), except subslil~li.. g 2-[N-
(ben_yloxycarbonyl)-N-methyl]aminomethyl-4-methylben7imit1~37.01e for the 2-[N-
(benzyloxycarbonyl)-N-methyl]aminomethyl-5-trifluoromethylb~on7imi(1~7nle, the title
compound (2.9916 g, qn~ v~) was prepared: 'H NMR (250 MHz, CDCl3) o 7.36-
7.01 (m, 4H), 4.01 (s, 2H), 2.52 (s, 3H), 2.41 (s, 3H).
d) Methyl (S)-2,3,4,5-tetrahydro-7-[[[(4-methylben7imi~1~7ol-2-
, yl)methyl]methylamino]carbonyl]-4-methyl-3-oxo- 1 H- 1,4-benzodi~epine-2-acetate
169
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96120748
Methyl (S)-7-carboxy-4-methyl-3-oxo-2,3,4,5-tetrahydro-lH-1,4-
benzodiazepine-2-acetate (188.5 mg, 0.64 mmol) was weighed into a 100 mL round
bottomed flask. CH3CN was added, followed, sequentially, by HOBt ~ H2O (103.5 mg,
0.77 mmol), EDC (149.3 mg, 0.78 mmol), and diisopropylethylamine (0.15 ml, 0.86
S mmol). After 15 min, a solution of 4-methyl-2-(methylaminomethyl)ben7imi~1~7ole
(273.8 mg, 1.56 mmol) in CH3CN was added. CH2Cl2 (5 mL) was added to dissolve
some material. After 18 hr at RT, the reaction was concentrated, and the residue was
chromatographed on silica gel (5% MeOH/CHCl3) to afford the title compound (307.3
mg, q~l~ntifz~tive): MS (ES) rn/e 450.2 (M+H)t; 'H NMR (250 MHz, CDCl3) ~ 7.23-7.03
(m, 7H), 6.41 (br s, lH), 5.33 (d, J = 16.3 Hz, lH), 3.69 (s, 3H), 3.46 (s, 3H), 3.10 (s,
3H).
e) (S)-2,3,4,5-Tetrahydro-7-[[L(4-methylbçn7imidz-~ol-2-
yl)methyl]methylamino]carbonyl3-4-methyl-3-oxo- lH- 1,4-benzodiazepine-2-acetic
acid
Following the procedure of Example 45(e), methyl (S)-2,3,4,5-tetrahydro-7-[[[(4-methylbçn7imicls-7ol-2-yl)methyl]methylamino]carbonyl]-4-methyl-3-oxo-lH-1,4-
benzodiazepine-2-acetate ~307.3 mg, 0.68 mmol) was saponified to afford the title
compound (243.9 mg, 82%): MS (ES) rn/e 436.2 (M+H)+. Anal Calcd for C23H25N504 -2.75 H2O: C, 56.96; H, 6.34; N, 14.44: Found: C, 56.72; H, 6.27; N, 14.26.
F.x~m~G~le 48
~dLion of (S)-2.3.4.5-tetrahydro-7-rrr(4-aza-5.7-dim~tllylbe~ idazol-2-
yl)methyllme~llylaminolcarbonyll-4-methyl-3-oxo- 1 H- 1.4-benzodiazepine-2-acetic
a) 2-Amino-4,6-dimethyl-3-nitropyridine
2-Arnino-4,6-dimelhyl~ylidine (5.55 g, 45.43 mmol) was weighed into a 500
mL round bottomed flask. The flask was cooled to -78~C. Concentrated HzS04 (25
mL, 450 mmol) was added, followed by concentrated HNO3 (3.5 mL, 56.0 mmol).
The ~ui~Lul~ became a solid frozen mass. The cooling bath was removed and the
reaction was allowed to warm to RT. After about 15 min there was an exothermic
reaction with the release of some nitrous oxide gas, and the reaction became a very
dark red color. The reaction was heated at 85 - 90~C for 3 hr, then was cooled to
RT, diluted with ice, and neutralized with 6 N NaOH (160 mL). The aqueous
solution was extracted with EtOAc (3 x), and the combined EtOAc layers were dried
170
.
CA 02241633 1998-06-26
W O 97/24119 PCTrJS96/20748
(MgSO4), filtered, and evaporated under vacuum. The resulting yellowish-
orange solid was adsorbed onto silica gel and flash chromatographed on a dry silica
gel colurnn. The column was eluted with 1: 1 CHCI3/Et2O to afford the title
compound (1.0650 g, 14%): MS (ES) m/e 168.0 (M+H)'.
s
b) 2,3-Diamino-4,6-dim~;lhyl~ylidine
- Following the procedure of Example 45(a), except substituting 2-amino-4,6-
dimethyl-3-nitl~ylidine (1.0650 g, 6.37 mmol) for the 4-amino-3-
niLluben~otrifluoride, the title compound (836.1 mg, 95.7%) was prepared. This was
used without char~ct~ri7~tion.
c) 4-Aza-2-[N-(benzyloxycarbonyl)-N-methyl]aminomethyl-5,7-
dimethylbenzimidazole
Following the procedure of Example 45(b), except sub~ 2,3-diamino-
4,6-dhllt;lllylpyridine (836.1 mg, 6.09 mmol) for the 3,4-diaminobenzotrifluoride,
the title compound (1.2273 g, 62%) following silica gel chromatography (3%
MeOH/CHCl3): MS (ES) m/e 325.0 (M~H)+.
d) 4-Aza-2-(methylaminomethyl)-5,7-dimethylben7imitl:~7ole
Following the procedure of Example 45(c), except substituting 4-aza-2-[N-
(benzyloxycarbonyl)-N-methyllarninomethyl-5,7-dimethylben7imi~1~7.ole (1.2273 g,3.78 mmol) for the 2-[N-(benzyloxycarbonyl)-N-methyl~aminomethyl-5-
trifluoromethylbenzimidazole, the title compound was obtained as a white powder
following trituration with Et2O. This material was used without characterization.
e) Methyl (S)-2,3,4,5-tetrahydro-7-[[[(4-aza-5,7-dimethylbe7imi~1~7c!1-2-
yl)methyl]methylamino~carbonyl~-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetate
Methyl (S)-7-carboxy-4-methyl-3-oxo-2,3,4,5-tetrahydro- lH- 1,4-
benzodiazepine-2-acetate (175.0 mg, 0.60 mmol) was weighed into a 100 mL round
bottomed flask. CH3CN (10 mL) was added, followed sequentially, by HOBt ~ H~O
(115.9 mg, 0.86 mmol), EDC (124.9 mg, 0.65 mmol), and diisopropylethylamine
(0.13 mL, 0.75 mmol). A ~uspel sion of 4-aza-2-(methylaminomethyl)-5,7-
dimethylben7imicl~7~-1e (144.5 mg, 0.76 mmol) and diisopropylethylamine (0.13
mL, 0.75 mmol) in CH3CN was added, and the reaction was stirred at RT. After 22
- 35 hr, the reaction was evaporated under V~l~;UUlll, and the residue was co-evaporated
with toluene. Silica gel chromatography (3% MeOH/CHCl3 (1 L) then 5%
171
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96120748
MeOH/CHCI3) gave the title compound (76.9 mg, 28%): MS (ES) m/e
465.2 (M+H)+.
f) (S)-2,3,4,5-Tetrahydro-7-[[[(4-aza-5,7-dimethylben7imi~ 7OI-2-
yl)methyl]methylarnino]carbonyl-4-methyl-3-oxo-lH-1,4-benzodiazepine-2-acetic
acid
Methyl (S)-2,3,4,5-tetrahydro-7-[[[(4-a_a-5,7-dimethylbç7imi~7ol-2-
yl)methyl]methylamino]carbonyl]-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetate
(76.9 mg, 0.17 mmol) was dissolved in MeOH (5 mL) and HlO (5 rnL), and 1.0 N
- 10 NaOH (0.5 mL, 0.5 mmol) was added. After 24 hours at RT, the reaction was
neutralized with 1.0 N HCl (0.5 mL), and the solvents were evaporated under
vacuum. ODS chromatography (gradient: 5% CH3CN/H20 cont"inin~ 0.1% TFA
(500 mL), then 10% % CH3CN/H20 cont"ining 0.1 % TFA (500 mT~), then 15% %
CH3CN/H~0 cont"ining 0.1% TFA (500 mL), then 30% % CH3CN/EI~0 cont~ining
0.1 % TFA (500 mL)) gave a residue which was co-evaporated once with toluene anddried under high vacuum. The resulting residue was dissolved in MeOH (5 mL) and
precipitated with Et,O. The white solid was collected on a sintered glass funnel and
dried in a vacuum desiccator overnight to yield the title compound (52.0 mg, 68%):
HPLC (ODS column, 1.5 mL/rnin; gradient 5-50% CH3CN/H20 cont~ining 0.1%
TFA) tR 12.38 min; MS (ES) m/e 451.2 (M+H) ' . Anal Calcd for Cr,H~6N604 1 H20
1 CF3CO2H: C, 51.55; H, 5.02; N, 14.43: Found: C, 51.34; H, 5.00; N, 14.41.
Fx~ml?le 4g
~l~p~dtion of (S)-2.3.4.5-tetrahydro-7-rrr(5.6-difluoroben7imi~1~7ol-2-
yl)methyllmethylaminolcarbonyll-4-methyl-3-oxo- 1 H- 1.4-benzodiazepine-2-açeticacid
a) 1,2-Diarnino-4,5-difluorobenzene
Following the procedure of Example 45(a), except substit-lting 4,5-difluoro-
2-nitroaniline (2.0 g, 11.49 mmol) for the 4-amino-3-nitTobellzol~ ori~ , the title
compound was prepared. This was used without characterization.
b) 2-[N-(Benzyl-~yca~bonyl)-N-methyl~aminomethyl-5,6-difluorobel-7i . . . i~ 7ole
Following the procedure of Example 47(b), except substituting 1,2-~ mino-
4,:~-difluoro~en~ne. for the 1,2~ mino-3-methylbenzene, and running the AcOH
cyclization step at 80~C instead of at 110~C, the title compound (1.3767 g, 36%) was
172
CA 02241633 1998-06-26
W O 97/2411g PCTrUS96/20748
prepared: TLC Rf (1: 1 CH2C12/Et2O) 0.42; MS (ES) m/e 332.0 (M+H)+; IH
NMR (250 MHz, CDC13) ~ 7.50-7.14 (m, 8H), 5.13 (s, 2H), 4.61 (s, 2H), 3.06 (s,
3H).
S c) 5,6-Difluoro-2-(methylaminomethyl)ben7imid~7Ole
Following the procedure of Example 46(b), except substituting 2-[N-
- (benzyloxycarbonyl)-N-methyl]aminomethyl-5,6-difluorobçn7imi-1~7ole (1.3767 g,
4.16 mmol) for the 2-[N-(benzyloxycarbonyl)-N-methyl]~minom~thyl-4,7-
~lim~thoxybe~ mdazole, the title compound (875.6 mg, qll~ntit~tive) was prepared:
10 MS (ES) m/e 198.0 (M+H)'.
d) Methyl-(S)-2,3,4,5-tetrahydro-7-[[[5,6-difluoroben7imill~7ol-2-
yl)methyl]methylamino]carbonyl]-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetate
Methyl (S)-7-carboxy-4-methyl-3-oxo-2,3,4,5-tetrahydro-lH-1,4-
15 benzodiazepine-2-acetate (415.7 mg, 1.42 mmol) was taken up in CH3CN, and HOBt
~ H2O (209.3 mg, 1.55 mmol) and EDC (314.9 mg, 1.64 mmol) were added. After 5
min, and diisopropylethylamine (0.25 mL, 1.64 mmol) was added, which produced a
clear, colorless solution. A solution of 5,6-difluoro-2-
(methylaminomethyl)ben7imicl~701e (284.5 mg, 1.44 rnmol) in CH3CN was added.
20 After 30 minutes, the reaction became slightly turbid, so more
diisopropylethylamine (0.25 mL) was added, which made the reaction again clear
and colorless. After 24 hr, the reaction was evaporated under vacuum. The residue
was co-evaporated once with toluene, then was chromatographed on silica gel
(CHCl3 (0.25 L), then 2% MeOH/CHCl3 (1.5 L), then 5% MeOH/CHCl3) to afford
25 the title compound (456.8 mg, 68%): MS (ES) m/e 472.2 (M+H)t; 'H NMR (250
MHz, CDCl3) o 7.34-7.08 (m, 6H), 6.44 (br s, lH), 5.39 (d, J = 16.2 Hz, lH), 3.70 (s,
3H), 3.14 (s, 3H), 2.96 (s, 3H).
e) (S)-2,3,4,5-Tetrahydro-7-[[[(5,6-difluorobçr 7imi~ 7ol-2-
30 yl)methyl]methylamino]carbonyl]-4-methyl-3-oxo- lH- 1,4-benzodiazepine-2-acetic
acid
Methyl (S)-2,3,4,5-tetrahydro-7-[[[5,6-difluoroben7imi~1~7-1-2-
yl)methyl]methylamino]carbonyl] -4-methyl-3-oxo- l H- 1,4-ben70~ 7~pine-2-acetate
(456.8 mg, 0.97 mmol) was dissolved in MeOH (10 mL) and H2O (10 ml). 1.0 N
35 NaOH (3.0 mL, 3.0 mmol) was added and the reaction was stirred at RT. After 18
hr, the reaction was neutralized with 1.0 N HCl (3.0 mT .). A white precipitate
formed which was collected on a sintered glass filter and dried in a vacuum
173
CA 02241633 1998-06-26
W O 97~4119 PCTrUS96/20748
desiccator. ODS chromatography (gradient: 10% CH3CN/H20 Cont~ining
0.1% TFA (500 rnL), then 18% % CH3CN/H20 cont~inin~ 0.1% TFA (500 mL), then
25% % CH3CN/H20 cont~inin~ 0.1% TFA (500 mL)) gave a residue which was co-
evaporated once with toluene. The rçslllting residue was dissolved in a small
5 amount of MeOH and precipitated with Et2O to give the title co~ oulld (330.9 mg)
as a white powder: HPLC (ODS column; 1.5 mL/min; gradient 5-50% CH3CN/H~O
contzlining 0.1% TFA) tR = 14.12 min; MS (ES) m/e 458.2 ~M+H)~. Anal Calcd for
C22H2,N504F2 ~ 2.5 H2O: C, 52.57; H, 5.22; N, 13.94: Found: C, 52.76; H, 5.15; N,
67.
F.x~m~ple 50
Preparation of (S)-2.3.4.5-tetrahydro-7-rr~(4-:~7~-5-methylbe1l7i..lid~7.ol-2-
yl)methylJamirlolcarbonyll-4-m~o-tllyl-3-oxo-lH-l~4-benzodiazepine-2-acetic acid
a) Methyl (S)-2,3,4,5-tetrahydro-7-[[~(4-aza-5-methylben7imi-1~7nl-2-
yl)methyl]amino]carbonylJ-4-methyl-3-oxo- lH- 1,4-benzodiazepine-2-acetate
Methyl (S)-7-carboxy-4-methyl-3-oxo-2,3,4,5-tetrahydro- 1 H- 1,4- -
benzodiazepine-2-acetate (228.8 mg, 0.78 mmol) was taken up in CH3CN, and HOBt
~ H~O (154.2 mg, 1.14 mmol), EDC (179.4 mg, 0.94 mmol), and
diisopropylethylamine (0.50 ml~ ,, 0.94 rnmol) were added sequentially. A solution
of 2-(aminomethyl)-4-aza-5-methylben7imi(1~7(1e dihydrochloride (125.4 mg, 0.77
mmol) in CH3CN /DMF was added, and the reaction was stirred at RT. After 24 hr,
the reaction was evaporated under vacuum~ and the residue was co-evaporated oncewith toluene. Silica gel chromatography (CHCl3 (0.25 L) then 3% MeOH/CHCl3
(0.5 L), then 5% MeOH/CHCl3) gave the title compound (159.9 mg, 48%): MS (ES)
m/e 437.2 (M+H)+.
b) (S)-2,3,4~5-Tetrahydro-7-[~[(4-aza-5-methylbenzimidazol-2-
yl)methyl]amino]carbonyl]-4-methyl-3-oxo- lH- 1,4-benzodia_epine-2-acetic acid
Following the procedure of Fx~mrle 48(f), methyl (S)-2,3,4,5-tetrahydro-7-
[[[(4-aza-5-methyl~en7imi~ 7Ol-2-yl)methyl]aminoJcarbonyl]-4-methyl-3-oxo-lH-
1,4-benzodiazepine-2-acetate (159.9 mg, 0.37 mmol) was saponified and purified to
afford the title compound: MS (ES) m/e 423.39 (M+H)~. Anal. Calcd for
C2,H22N6O4 - 0.5 H2O 1.25 TFA: C, 52.64; H, 5.02; N, 16.74. Found: C, 52.65; H,
5.02; N, 16.74.
174
CA 02241633 1998-06-26
W O 97/24119 PCTAUS96/20748
Fx~m,ple 51
Preparation of (S)-2.3.4.5-tetrahydro-4-m~tllyl-7-rrr(4-nitrobçn7imicla7ol-2-
yl)methyl~methylaminolcarbonyll-3-oxo-lH-1.4-benzo~ 7~pine-2-acetic acid
- 5
a) 2-[N-(tert-Butoxycarbonyl)-N-methyl]aminomethyl-4-nitroben7imi<1~7ole
e Boc-sarcosine (2.0320 g, 10.74 mmol) was dissolved in dry THF and cooled
in a dry-ice/acetone bath to -15~C. Triethylamine (5.0 mL, 3.6375 mmol) was
added, followed by isobutylchlor~fullllate (1.5 mL, 11.56 mmol). After 0.5 hr, the
mixture was added to a solution of 1,2-diamino-3-nitrobenzene (1.3047 g, 10.77
mmol) in dry THF at -20~C, and the reaction was allowed to warm to RT. After 24
hr, the reaction was evaporated under vacuum and the residue was partitioned
between EtOAc and 1.0 N NaHCO3. The layers were separated, and the aqueous
layer was extracted with EtOAc. The combined organics were dried (MgSO4),
filtered, and evaporated under vacuum. The residue was dissolved in glacial AcOH(100 mL), and the solution was heated at 75~C. After 24 hr, the reaction was
evaporated under vacuum, and the residue was co-evaporated with toluene (2 x).
The m~t~ri~l was adsorbed onto silica gel and flash chromatographed on a dry silica
gel column (gradient: CHC13 (0.5 L), then 2% MeOH/CHCl3 (1 L), then 5%
MeOH/CHCl3) to afford the title compound (2.2089 g, 75%): 'H NMR (250 MHz,
CDCl3) ~ 8.10 (dd, 2H), 7.40-7.32 (m, lH), 4.69 (s, 2H), 3.02 (s, 3H), 1.54 ~s, 9H).
b) 2-(Methylaminomethyl)-4-niliuben,i.l.i(1~7Ole
2-[N-(tert-Butoxycarbonyl)-N-methyl3aminomethyl-4-nitroben7.imi~ 7.ole
(2.2089 g, 8.05 mmol) was treated with 4 N HCl in dioxane at RT. After the
addition, there was an imm~ te precipitation of a white solid. After 4 hr, the
reaction was evaporated under vacuum and the residue was triturated with diethylether to give the title compound (1.639 g) as a white solid. This was used without
chara~ ation.
c) Methyl-(S)-2,3,4,5-tetrahydro-7-1[[(4-nitrobe~7.imi~1~7.ol-2-
yl)methyl]methylamino~carbonyl-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetateFollowing the procedure of Example 49(d), except substitllting 2-
(methylaminomethyl)4-nitroben7.imi~1~7c-1e for 5,6-difluoro-2-
(methylaminomethyl)ben7imi-1~7ole, the title compound (292.9 mg, qn~ntit:~tive)
was plep~d: MS(ES) m/e 481.2 (M~H)+.
175
CA 0224l633 l998-06-26
W O 97/24119 PCTrUS96/20748
d) (S)-2,3,4,5-Tetrahydro-7-t[[(4-nitro~çn7imitl~7ol-2-
yl)methyl]methylamino]carbonyl-4-methyl-3-oxo- l H- l ,~bP,n7.o~ .pine-2-acetic
acid
Following the procedure of Example 45(e), methyl ~S)-2,3,4,5-tetrahydro-7-
[[[(4-nitrobenzimidazol-2-yl)methyl]methylamino]carbonyl-4-methyl-3-oxo- 1 H- 1,4-
benzodiazepine-2-acetate (292.9 mg, 0.61 mmol) was saponified to afford the title
compound (211.0 mg, 68%): MS (ES) m/e 467.4 (M+H)+. Anal. Calcd for
C22H22N6O6 2.5 H20: C, 52.12; H, 5.27; N, 16.58: Found: C, 52.07; H, 4.97; N,
16.40.
F.xzln~?le 52
.,.tion of (S)-2.3.4.5-tetrahydro-7-rrr(4-arninoben7imi-l~7ol-2-
yl)m~thyllmethylaminolcarbonyl-4-methyl-3-oxo-lH- 1 ~4-benzodiazepine-2-acetic
~i~
a) (s)-2~3~4~5-tetrahydro-7-[[[(4-amino~rn7imi~ 7ol-2-
yl)methyl] methylamino}carbonyl-4-methyl-3 -oxo- 1 H- 1,4-benzodiazepine-2-acetic
acid
(S)-2,3,4,5-Tetrahydro-7-[[[(4-nitroben7imir1~ol-2-
yl)methyl]methylamino]carbonyl-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetic
acid (108.7 mg, 0.21 mmol) was dissolved in MeOH, and a catalytic amount of 10%
PdlC was added. The reaction was purged with Hz, then was stirred at RT under H2(balloon). After 20 hr, the catalyst was removed by filtration through celite(g), and
the filtrate was evaporated under vacuum. The resulting solid was dissolved in
MeOH, reprecipitated with Et20, dried in a vacuum rlecicc;~tor, and purified by ODS
chromatography (gradient: H2O cont~inin~ 0.1% TFA (500 mL), then 5%
CH3CN/H20 cont~ining 0.1% TFA (500 mL), then 10% % CH3CN/H70 Cont~inin~
0.1% TFA (500 mL), then 15% % CH3CN/H20 confz-inin~ 0.1% TFA (500 rnL), then
20% % CH3CN/H20 cont~inin~ 0.1% TFA (500 mL), then 25% % CE~3CN/Hz0
contz~ining 0.1% TFA (500 rnL), then 30% % CH3CNIH20 cont:~lininp: 0.1% TFA
(500 mL)). The rçsnltin~ m~tPri~l was co-evaporated once with toluene then was
triturated with diethyl ether to afford the title compound (34.7 mg): MS (ES) m/e
437.5 (M+H)~; Anal. Calcd for C~24N6O4 1.5 H2O 1.5 TFA: C, 47.32; H, 4.53; N.
13.24: Found: C, 47.35, H, 4.86; N. 13.61.
Fx~m;l?le 53
176
CA 02241633 1998-06-26
W O 97/24119 PCTnUS96120748
?ari.lion of 23.4~5-tetrahydro-7-rrr(lR~-fb~n7imi(1~7ol-2-
yl)ethyllmethylarninolcarbonyll-4-m~thyl-3-oxo-lH-1.4-benzodia_epine-(2S~-aceticacid
-
a) 2-[1 (R)-~N-(Benzyloxycarbonyl)-N-methyl]aminoethyl~ben7.imifl~7.ole
Following the procedure of Example 47(b), except ~ub~liluLillg Cbz-N-
methyl-D-alanine for the Cbz-sarcosine, and substituting 1,2-phenylen~ mine for
the 1,2-diamino-3-methylbenzene, and running the AcOH cyclization step at 80~C
10 instead of at 110~C, the title compound was prepared: MS (ES) m/e 310.2 (M+H)+.
b) 2-[1(R)-(Methylaminoethyl)]ben7.imi~ 7.01e
Following the procedure of Example 46(b), except sub~ ulillg 2-[l(R)-tN-
~benzyloxycarbonyl)-N-methyl]aminoethyl]ben7.imi(1~7.01e for the 2-[N-
15 (benzyloxycarbonyl)-N-methyl]aminomethyl-4,7-dimethoxyben7imic~:~7.ole, the title
compound (276.0 mg, 48%) was prepared: MS(ES) m/e 176.2 (M+H)+.
c) Methyl 2,3,4,5-tetrahydro-7-[[[(lR)-(ben7.imi~1~7.c!1-2-
yl)ethyl]methylamino]ca~bollyl]-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetate
Following the procedure of Example 49(d), except substituting 2-[l(R)-
(methylaminoethyl)]benzimidazole for the 5,6-difluoro-2-
(methylaminomethyl)1)~ll7.il"i(1~7.ole, the title compound (203.5 mg, 90%) was
prepared: MS (ES) m/e 450.5 (M+H)~.
25 d) 2,3,4,5-Tetrahydro-7-[~[(lR)-(bçn7.imi-1~7.ol-2-yl)ethyl]methylamino]carbonyl]-
4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetic acid
Following the procedure of Example 49(e), methyl 2,3,4,5-tetrahydro-7-
[[[(lR)-(b~7imi~1~7.ol-2-yl)ethyl]methylamino~ca~bonyll-4-methyl-3-oxo-lH-1,4-
benzodiazepine-2-acetate was saponified to afford the title compound (179.3 mg,
30 75%) following ODS chromatography: MS (ES) m/e 436 5 (M~H)+. Anal. Calcd
for C23H25N504 0.75 H20 0.75 TFA: C, 55.01; H, 5.14; N, 13.10; Found: C, 54.98;
H, 5.42; N. 12.75.
F.x~n~le 54
177
CA 0224l633 l998-06-26
W O 97/24119 PCTrUS96/20748
Pl~alion of ethyl fS)-2.3~4.$-tetrahydro-7-rrr(4-Aza-5-
met~lylben7.imid~ol-2-yl)methyllaminolcarbonyll4-metlly1-3-oxo- 1 H- 1.4-
'~n70~ 7~pine-2-acetate
S a) Ethyl (S)-2,3,4,5-tetrahydro-7-[[[(4-Aza-5-methylben7imi~1~701-2-
yl)methyl]amino]carbonyl]4-methyl-3-oxo- lH- 1 .4-bt~n70tli~7~pine-2-acetate
(S)-2,3,4,5-Tetrahydro-7-[[[(4-aza-S-methylbçn7.imi~7.oI-2-
yl)methyl]arnino]carbonyl]-4-methyl-3-oxo- 1 H- 1 ,4-benzodiazepine-2-acetic acid
~0.5 g) was dissolved in EtOH, and the solution was cooled in an }ce bath to 0~C.
Gaseous HCI was bubbled into the solution until the solution was saturated, then the
flask was sealed with a rubber septum and the cooling bath was removed. The
reaction was stirred at RT for 20 hr, then the solvents were evaporated under
vacuum. The residue was co-evaporated three times with toluene (3 x), then was
dissolved in EtOH and precipitated with Et20. The solid was collected on a sintered
glass funnel and dried in a vacuum ~iesiccatQr overnight to afford the title compound
(483.9 mg): MS (ES) m/e 451.4 (M+H)~. Anal. Calcd for C23H26N6O, ~ HCl ~ 1.375
H20: C, 53.98; H, 5.86; N, 16.42. Found: C, 54.00; H, 5.82; N, 16.42.
F.xz-m~?le S5
PIG~al~Lion of 2.3.4.5-tetrahydro-7-rrr(lS)-(ben7imi-1~7.ol-2-
yl)etl~yllmethyl~minolcarbonyll-4-methyl-3-oxo-lH-1 .4-benzodi~epine-~2S)-aceticacid
a) 2-[l(S)-[N-(tert-Butoxycarbonyl~-N-methyl~aminoethyl]ben7imi~1~701e
Following the procedure of Example 51(a) except substituting Boc-N-
methyl-L-alanine for the Boc-sarcosine, and sub..liLuLillg 1,2-phenylene~ min~ for
the 1,2-diamino-3-nitroben7~n~, the title compound (1.7792 g, 65%) was prepared
following recryet:llli7~tion from CHCI3/hexanes: MS (ES) m/e 276.4 (M+H)~.
b) 2-[l(S)-(Methylaminoethyl)]ben7imi~1~7.ole
Following the procedure of Example 51(b), except substituting 2-[l(S)-~N-
(tert-butoxycal'L,ollyl)-N-methyl]aminoethyl]bel-7i . . .i-i~7Qle for the 2-[N-(tert-
butoxycarbonyl)-N-methyl]aminomethyl4-nitroben7imi~ 701e, the title compound
35 was prepared. This was used without charactt-ri7~tion.
178
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
c) Methyl (S)-2,3,4,5-tetrahydro-7-[[t(1 S)-(ben7.imi~1~701-2-
yl)ethyl]methylamino]carbonyl]-4-methyl-3-oxo- 1 H- 1,4-bçn7.o~ 7P-pine-2-acetate
Following the procedure of Example 49(d), except substihlting 2-[l(S)-
(methylaminoethyl)]ben7.imitl~7.(1e for the 5,6-difluoro-2-
S (methylaminomethyl) ben7.imi~1~7.01e, the title compound (414.7 mg, 88%) was prepared: MS (ES) m/e 450.2 (M+H)+.
d) 2,3,4,5-Tetrahydro-7-t[[(lS)-(ben7.imi~ 7.~1-2-yl)ethyl]methylamino]carbonyl]-
4-methyl-3-oxo- lH- 1,4-benzodiazepine-2-acetic acid
1(~ Following the procedure of Example 45(e), methyl (S)-2,3,4,5-tetrahydro-7-
[[[(lS)-(ben7.imi~1~7.ol-2-yl)ethyl~methylamino]carbonyl]-4-methyl-3-oxo-lH-1,4-benzodiazepine-2-acetate was saponified to afford the title compound (117.2 mg):MS (ES) m/e 436.2 (M+H)+. Anal Calcd for C23H~5N504 0.75 H20 0.75 TFA: C,
55.05; H, 5.14; N, 13.10; Found: C, 55.14; H, 5.38; N, 13.04.
Ex~mple S6
P~ io~ of 2.3.4.5-tetrahydro-7-rrr(lS)-(b~n7imi~1~7.ol-2-
yl)ethyllaminolcarbonyll-4-methyl-3-oxo-lH-1.4-ben7.o~;~7.~pine-(2S)-acetic acid
a) 2-r 1 (S)-(tert-Butoxycarbonyl)aminoethyl]ben7imi~1~7.ole
Following the procedure of Example 51 (a) except substituting Boc- L-
alanine for the Boc-sarcosine, and sul)~LiLuLil~g 1,2-phenylene~ mine for the 1,2-
diamino-3-nitroben7ene7 the title compound (714.7 mg, 25%) was prepared: MS
(ES) m/e 262.4 (M+H)+.
b) 2-[1 (S)-(Aminoethyl)]ben7imi(1~7.ole
Following the procedure of Example 51(b), except subsliluli~lg 2-[l(S)-(tert-
butoxycarbonyl)aminoethyl]ben7imi~1~701e for the 2-[N-(tert-butoxycarbonyl)-N-
methyl]aminomethyl-4-nitroben7.imi~1~7.01e, the title compound was prepared. This
was used without characterization.
c) Methyl 2,3,4,5-tetrahydro-7-[t[(1S)-(ben7imi~ 7.ol-2-yl)ethyl)]amino]carbonyl]-
4-methyl-3 -oxo- l H- 1,4-ben 70~ 7epine-2-acetate
Following the procedure of Example 49(d), except substit~ting 2-[l(S)-
(aminoethyl)]ben7.imi~1~7.r 1e for 5,6-difluoro-2-(methylaminomethyl)be~- ~.i " ,icl~7.~1e,
the title compound (270.7 mg, 80%) was p~ l.,d: MS (ES) m/e 436.0 (M+H~+.
179
CA 02241633 1998-06-26
W O 97/24119 PCT~US96120748
d) 2,3,4,5-Tetrahydro-7-t[[(lS)-(be~7imid~7ol-2-yl)ethyl)]amino]carbonyl]-4-
methyl-3-oxo- lH-1,4-benzodiazepine-2-acetic acid
Following the procedure of Example 45(e), methyl 2,3,4,5-tetrahydro-7-
[[[(1 S)-(benzimidazol-2-yl)ethyl)]amino]carbonyl]-4-methyl-3-oxo- lH- 1,4-
benzodia_epine-2-acetate was saponified to afford the title compound (158.1 mg,
61%): MS (ES) m/e 422.0 (M+H)+. Anal. Calcd for C2lH23N5O4 ~ 1.75 H20: C,
58.37; H, 5.90; N, 15.46; Found: C 58.17; H, 5.77; N, 15.08.
F.~z~ml?le 57
P~ lion of 2.3.4~5-tetr~l~ydro-7-rrr(lR~-(ben~ 7ol-2-
yl)ethyllarninolcarbonyll-4-methyl-3-oxo-lH-1.4-benzodiazepine-(2S~-acetic acid
a) 2-[1 (R)-(Benzyloxycarbonyl)aminoethyl]ben7imi~1~701e
Following the procedure of Example 47(b~, except substituting Cbz-D-
alanine for the Cbz-sarcosine, and substituting 1,2-phenylenedi~minc for the 1,2-
diarnino-3-methylbenzene, and rurming the AcOH cyclization step at 80~C instead
of at 110~C, the title compound (1.1455 g, 43%) was prèpared: MS (ES) m/e 296.4
(M+H) i .
b) 2-(l(R)-Aminoethyl)ben~imi-1~7O1e
Following the procedure of Fx~mple 46(b), except sub~iLu~ g 2-[l(R)-
(benzyloxycarbonyl)aminaethyl]ben7imi~ 701e for the 2-[N-(benzyloxycarbonyl)-N-
methyl]aminomethyl-4,7-dimethoxyben~ 7Qle, the title compound (258.1 mg,
93%) was ~l~palt;d: MS(ES) m/e 161.9 (M+H)t.
c) Methyl 2,3,4,5-tetrahydro-7-[[[( lR)-(ben7imi~1~7~-1-2-yl)ethyl~amino]carbonyl]-
4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-(2S)-acetate
Following the procedure of Example 49(d), except substituting 2-(l(R)-
aminoethyl)ben7imi~701e for the 5,6-difluoro-2-
(methylarninomethyl)benzimidazole, the title compound (263.6 mg, 84%) was
prepared: MS (ES) m/e 436.3 (M+H)+.
d) 2,3,4,5-Tetrahydro-7-[[[(lR)-(be~7imi(i~7ol-2-yl)ethyl]amino]carbonyl]-4
methyl-3-oxo- lH- 1,4-ben7o~ 7topine-2-acetic acid
180
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
Following the procedure of F.xz-mple 49(e), methyl 2,3,4,5-
tetrahydro-7-[[[(lR)-(b~n7imi~1~7ol-2-yl)ethyl]amino]carbonyl]~-methyl-3-oxo-lH-1,4-benzodiazepine-(2S)-acetate was saponified to afford the title compound (125.0
mg, 49%): MS (ES) m~e 422.0 (M+H)t. Anal. Calcd for C22H23N5O4 ~ 0.5 H20 ~ 1.25
HCl: C, 55.51; H, 5.35; N, 14.71. Found: C, 55.70; H, 5.47; N, 14.53.
- Example $8
Pr~a,dLion of (S)-2.3~4.5-tetrahydro-7-rrrCjmifl:~7o( 1 .2a)pyrid-2-
yl)methyllmethylaminolcarbonyl~-4-methyl-3-oxo-lH-1.4-benzodiazepine-2-acetic
acid
a) 2-Carboethoxyirnidazo[ 1 ,2a]pyridine
2-Aminopyridine (4 g, 42.50 mmol) was dissolved in MeOH (50 mL). Ethyl
bromopyruvate (8.3 g, 42.50 mmol) was added and the reaction was stirred at 70~Cfor 2 hr. The solvent was then elimin~te-l and the solution was neutralized with 1 M
NaOH. The reaction was extracted with EtOAc, and the combined EtOAc layers
were washed with brine. Drying (MgSO4), filtration, concentration, and silica gel
ilash column chromatography (2% MeOH/Cl2CH2) gave the title compound (4.5 g,
56%) as a pale yellow solid: 1H NMR (400 MHz, CDC13) o 1.43 (t, J = 7 Hz, 3H),
4.45(q,J=7Hz,2H),6.86(t,J=6.6Hz, lH),7.24(t,J=6.6Hz, lH).
b) 2-Hydroxymethylimicl~7O[1,2a]pyridine
2-Carboethoxyimidazo[1,2a]pyridine (0.5 g, 2.81 mmol) was dissolved in
dry THF at 0~C, and then a solution of lithium ~ minl~m hydride (0.5 mL of a 1.0M in THF) was added. The reaction was allowed to warm to RT and stirred for 1 hr.
H2O (0.2 rnL) was added, followed by 15% NaOH (0.2 mL), and finally H2O (0.6
mL). The solids were removed by filtration and washed with hot THF (2 x 100 mL)
and hot CHCl3 (4 x 100 mL). The filtrate and washings were combined and dried
(MgSO4). Following filtration the solvents were removed under reduce pressure the
residue was purified by silica gel flash column chromatography (5%
MeOH/Cl2CH2~ to obtain the title co~ u-ld (0.1 g, 25~o) as pale yellow liquid: 'H
NMR (400 MHz, CDCl3) o 4.85 (s, 2H), 6.76 (t, J = 6.8 Hz, lH), 7.15 (t, J = 6.8 Hz,
lH), 7.53 (s, lH), 7.54 (d, J = 6.7 Hz, lH), 8.1 (d, J = 6.7 Hz, lH). MS (ES) m/e
149 (M + H) +.
,.
c) 2-Chloromethylimi~1~7o[ 1 ,2a]pyridine
181
CA 0224l633 l998-06-26
W O 97/24119 PCTnjS96nO748
Thionyl chloride (0.4 mL, 3.2 mmol) was added to a solution of 2-
hydroxymethylimi(l~70[1,2a]pyridine (0.4 g, 2.7 mmol) in CHC13 (30 mL) at 0~C
After stirring at RT for 1 hr, the suspension was poured into a mixture of ice, 10%
NaHCO3, and CHCl3. The layers were separated, and the aqueous phase was
extracted with CHCI3. The organic extracts were combined and dried (MgS04).
Following filtration, the solvents were removed under reduce pressure to obtain the
title compound (0.4 g, 89%). lH NMR (400 MHz, CDCl3) o 4.7 (s, 2H), 6.7 (t, lH~, -
7.2 (t, lH), 7.5 (d, lH), 7.6 (s, lH), 8.0 (d, lH); MS (ES) m/e 167 (M + H)+.
d) 2-(Methylaminomethyl)imidazo[1,2a]pyridine
Freshly condensed methylamine (15 mL) was added to a solution of 2-
chloromethylimifl~70~1,2a]pyridine (317 mg, 2 mmol) in EtOH (5 mL) at 0~C, and
the reaction mixture was allowed to stir at 0~C for 2 h. The solvent was then
~limin~tto~l and the residue was purified by reverse-phase column chromatography(C-18 silica gel, H20 cont~ining 0.1 % TFA). Lyophilization gave the title
compound (461 mg, 87%) as a white solid: IH NMR (250 MHz, CDC13) o 2.5 (s,
3H), 4.0 (s, 2H), 6.7 (t, lH), 7.2 (t, lH), 7.5 (d, l~I), 7.6 (s, lH), 8.1 (d, lH). MS
(ES) m/e 162 (M + H)'.
- 20 e) Methyl (S)-2,3,4,5-tetrahydro-7-[[[(imidazo(1,2a)pyrid-2-
yl)methyl]methylamino~carbonyl]-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetate
Methyl (S)-7-carboxy4-methyl-3-oxo-2,3,4,5-tetrahydro- lH- I ,~
benzodiazepine-2-acetate (828.2 mg, 2.83 mmol) was taken up in CH3CN, and HOBt
~ H20 (398.1 mg, 2.92 mmol), EDC (562.9 mg, 2.94 mmol), and
diisopropylethylamine (1 mL, 5.74 mmol) were added sequentially. When the
solids had dissolved, a solution of 2-(methylaminomethyl)imi~ 70[1,2a]pyridine
(460.7 mg, 2.86 mmol) and diisopropylethylamine (1.5 mL, 8.61 mmol) in CH3CN
was added, and the reaction was stirred at RT. After 24 hr, the reaction was
concentrated, and the residue was co-~v~olat~d with toluene (2 x). The rçsl-lting
residue was chromatographed on silica gel (5% MeOH/CHCl3) to give the title
compound (694.6 mg, 56%): MS (ES) m/e 436.4 (M~H)~.
f) (S)-2,3,4,5-Tetrahydro-7-[[[(imi-1~7.o(1,2a)pyrid-2-
yl)methyl]methylamino~carbonyl]-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-aceticacid
Methyl (S)-2,3,4,5-tetrahydro-7-~[[(imidazo(1,2a)pyrid-2-
yl)methyl]methylamino]carbonyl]-4-methyl-3-oxo- lH- 1,4-benzodiazepine-2-acetate 182
"
CA 0224l633 l998-06-26
W O 97/24119 PCT~US96/20748
(663.4 mg, 1.52 mmol) was dissolved in MeOH (10 mL). H2O (10 mL)
was added, followed by 1.0 N NaOH (5 mL, 5.0 mmol), and the reaction was stirredat RT. After 20 hr, the reaction was neutralized with 1.0 N HCl (5 mL), and the
solution was evaporated under vacuum. The residue was purified by ODS
chromatography (gradient: H2O containing 0.1% TFA (500 mL), then 5%
CH3CN/H20 con~aining 0.1% TFA (500 mL), then 10% CH3CN/H20 co~
- 0.1% TFA (500 mL), then 15% CH3CN/H2O cont~ining 0.1% TFA (500 mL), then
20% CH3CN/H2O cont~ining 0.1% TFA (500 rnL)) followed by rechromatography
on ODS (gradient: H20 cont~inin~ 0.1~TFA (250 mL), then 10% CH3CN/H2O
cont~ining 0.1% TFA (1.5 L), then 20% CH3CN/H20 c~-nt~ininp 0.1% TFA (1 L).
Fractions containing pure m~t~ri~l were combined and concentrated. The residue
was co-evaporated with toluene, then was dissolved in MeOH and reprecipitated
with l~t20 to afford the title compound (96.4 mg): MS (ES) m/e 421.9 (M+H)~.
Anal. Calcd for C22H23N5O4 ~ 0.25 H2O ~ TFA: C, 53.38; H, 4.57; N, 12.97. Found:C, 53.68; H, 4.97; N, 12.94.
E~xample 59
Preparation of (+)-7-rrr(4~5-Dimethyl-lH-;mi~ ol-2-
yl)methyllmethylaminolcarbonyll-2~3.4.5-tetrahydro-4-methyl-3-oxo-lH-1~4-
benzodiazepine-2-acetic acid
a) N-(Carbobenzyloxy)-N-(methyl)acetonitrile
Cbz chloride (7.40 rnL, 49.3 mmol) was added slowly at RT to a solution of
N methylaminoacetonitrile hydrochloride (5.0 g, 46.92 mmol) and triethylamine
(13.4 mL, 96.2 mmol) in dichloromethane (200 mL). The reaction was stirred at RT18 h, and the mixture was washed with lN HCl, water and brine. The organic layerwas dried (MgSO4~ and concentrated to yield the title compound (6.97 g, 73%) as a
clear oil.
b) N-(Carbobenzyloxy)-N-(methyl)aminothio~ret~mi~le
Hydrogen sulfide was bubbled through a solution of N-(carbobenzyloxy)-N-
(methyl)acetonitrile (15 g, 73.5 mmol) and triethylarnine (30.75 mL, 220.6 mmol) in
DMF (250 mL). After 20 min, the flask was closed and the reaction was stirred atRT for 18 h. The reaction was then poured into 2 N NaHCO3 (1 L ) and extracted
with dichloromPth~ne. The combined organic phase was washed with 1: 1
.
183
CA 02241633 1998-06-26
W O 97/24119 PCT~US96t20748
water/brine (5 x), dried (MgSO4), and concentrated to give a yellow oil
which was purified by silica gel llash chromatography (step gradient, 40-50% ethyl
acetate/hexane) to yield the title compound (12.26 g, 70%) as a white solid: MS
(ES) rn/e 239.0 [M+H]+.
s
c) N-(Carbobenzyloxy)-N-methyl-S-(methyl)acetothioimidate
Iodomt-.th~nP (17.66 mL, 283.6 mmol) was added at RT to a solution of N-
(carbobenzyloxy)-N-(methyl)aminothio~et:lmide(6.75 g, 28.36 mmol) in acetone
100 mL). The solution was stirred at RT in the dark for 3 h, and the reslllting
precipitate was filtered to yield the title compound (9.63 g, 89%) as a white solid:
MS (ES) m~e 253.4 [M+H]+.
d) (+)-2-Amino-3,3-dimethoxybutane
Sodium cyanoborohydride was added to a solution of 3,3-dimethoxy-2-
butanone (1.32 g, 10 mmol) and ammonium acetate (1.1 g, 100 mmol) in methanol
(30 rnL). The pH was adjusted to 6 with methanolic HCl, and the reaction was
stirred at RT overnight and concentrated. The residue was dissolved in water, and
the pH was adjusted to 5 with aqueous HCl. The res-lltin~ solution was extractedwit_ ether (3 x), and the aqueous phase was basified to pH 10 with Na2CO3, and
extracted with ether. The organic layer was dried (MgSO4) and concentrated to yield
the title compound (1.1 g, 83%) as a clear oil: MS (ES) m/el34.2 [M+Hl+.
e) N-(Carbobenzyloxy)-N-methyl-(4,5-dimethyl- lH-imidazol-2-yl)meth~n~rnine
A solution of (~)-2-amino-3,3-~l;m~tho~y~uLalle (1.05 g, 7.89 mmol) and N-
(carbobenzyloxy)-N-methyl-S-(methyl)~< etofhi~ imidate (2.0 g, 5.26 mmol) in
methanol (30 mL) was warmed at 60~C for 2 h, and concentrated to give a yellow
oil. The crude oil was dissolved in 6 N HCl (30 mL) and stirred at RT 1 h. The
solution was basified to pH 12 wieh aqueous NaOH and then extracted with
dichlor~)m~th~ne. The combined organic phase was dried (MgSO4) and
concentrated to give a brown oil which was purified by silica gel flash
chromatography (4% methanol/dichloromethane) to yield the title compound (0.590
g, 41%) as a clear oil: MS (ES) m/e 274.0 [M+H~+.
f~ N-Methyl-(4,5-dimethyl-lH-imi~ 7,t~1-2-yl)~ nzlrninr.
A solution of N-(carbobenzyloxy)-N-methyl-(4,5-dimethyl-lH-imidazol-2-
yl)mto.thz-nz~mine (0.35 g, 1.28 mmol) in methanol (15 mL) and glacial acetic acid (5
184
CA 0224l633 l998-06-26
W O 97/24119 PCTrUS96/20748
mL), cont~ining 10% Pd/C (0.035 g), was shaken in a H2 atmosphere (45
Psi) for 6 h. The reaction was filtered and filtrate conce~ dl~;d to give the title
compound (0.22 g, 86%) as a brown oil which was used in the next step without
further purification: MS (ES) m/e 140 [M+H]+.
g) Methyl (_)-7-[[[(4,5-dimethyl-lH-imi-1~7ol-2-yl)methyl]methylamino]carbonyl]-- 2,3,4,5-tetrahydro-4-methyl-3-oxo- 1 H- 1,4-benzodiazepine-2-acetate
A solution of N-methyl-(4,5-dimethyl-lH-imi(1~7ol-2-yl)meth~n~minP (0.20
g, 1.1 mmol) and methyl (_)-7-carboxy-2,3,4,5-tetrahydro-4-methyl-3-oxo-1~-1,4-
benzodia_epine-2-acetate (0.320 g, 1.1 mmol) in the presence of DIEA (0.287 ml,
1.65 mmol) was stirred at RT. EDC (0.316 g, 1.65 mmol) was then added, followed
by DMAP (0.013 g, 0.11 mmol). The mixture was stirred at RT for 18 h, and
concentrated to give an oil which was purified by silica gel flash chromatography
(step gradient, 0.5-2% methanol/dichloromPthzlne) to yield the title compound (0.060
g, 9%) as a clear oil: MS (ES) m/e 414.2 [M+H]+.
h) (+)-7-[[[(4,5-Dimethyl-lH-imidazol-2-yl)methyl]methylamino]carbonyl]-
2,3,4,5-tetrahydro-4-methyl-3-oxo- lH- 1,4-benzodiazepine-2-acetic acid
1 N Sodium hydroxide (3 eq) was added to a solution of methyl (+)-7-[[[(4,5-
dimethyl-lH-imidazol-2-yl)methyl]methylamino]carbonyl]-2,3,4,5-tetrahydro-4-
methyl-3-oxo-lH-1,4-benzodiazepine-2-acetate (0.060g, 0.145 mmol), the mixture
was stirred at RT for 5 h, and concentrated. The residue was dissolved in water and
the pH was adjusted to S with 50% acetic acid. The solution was concentrated andpurified by MPLC (ODS-AQ, 10% acetonitrile/water cont~inin~ 0.1% TFA, UV
detection at 220 nm) to yield the title compound (0.050 g, 86%) as a white solid:
MS (ES) m/e 400.2 [M+H]+.
Example 60
Preparation of 3-rr3-r2-(ben7imill~7- 1-2-yl)ethyllisoxazolin-SfR.S)-yllacetyllamino-
3(R.S)-meth~ upalloic acid
a) 4-(Ben7imi-1~7Ol-2-yl)- 1 -butene
According to the general procedures of Preparation 4 in P50256-1, except
~ul~Li~ulillg 4-pentenoic acid for the Boc-sarcosine, the title compound is prepared.
-
b) 4-( l -Toluenesulfonylben7imi~l~7Ql-2-yl~- l -butene
185
CA 02241633 1998-06-26
W O 97124119 PCTrUS96/20748
Sodium hydride is added carefully to a solution of 4-(bP.n7.imi(1~7.01-
2-yl)-1-butene (50 mmole) and 4-toluenesulfonyl chloride (55 mmole) in dry THF
(2~)0 mr .). The reaction is stirred at RT until complete, then is quenched withsaturated NH,,CI (200 mL~, and the mixture is extracted with EtOAc. The combined5 organic extracts are dried (MgSO4) and concentrated, and the residue is purified by
silica gel chromatography to give the title compound.
-
c) 4-(l-Toluenesulfonylben7.imi-1~7.ol-2-yl)-l-butanal
Ozone is bubbled into a solutiolI of 4-(1-toluenesulfonylben7imi~l~7.Ql-2-yl)-
l-butene (40 mmole) in CH2Cl~ (160 mL) and MeOH ~40 mL) at -78~C until the blue
color persists, then the excess ozone is removed by bubbling argon through the
solution. Dry dimethylsulfide (excess) is added, and the reaction is warmed to RT.
The reaction is stirred at RT until complete, then is concentrated, and the residue is
chromatographed on silica gel to afford the title c~ oulld.
d) 4-(l-Toluenesulfonylben7.imi~1~7.(-1-2-yl)-l-butanal oxime
Hydroxylamine hydrochloride (33 rnmole) is added to a solution of 4-(1-
toluenesulfonylben7imi~1~7.ol-2-yl)-1-butanal (30 mmole) and anhydrous sodium
acetate ~66 mmole) in MeOH (150 mL) at 0~C. The reaction is stirred at 0~C until20 complete, then is concentrated, and the residue is partitioned between H20 and
EtOAc. The layers are separated, and the aqueous layer is extracted with EtOAc.
The combined organic layers are washed sequentially with 5% NaHCO3 and
saturated brine, dried (MgSO4~, and concentrated to afford the title compound.
2~ e) 4-(l-Toluenesulfonylbenzimi~ 7.ol-2-yl)-l-butanoxirninoyl chloride
According to the procedure of Example l(b) in WO 95/14682, except
substituting 4-(1-toluenesulfonylbe~7.imifl~7.01-2-yl)-l-butanal oxime for the 4-
cyanobenzoxime, the title compound is prepared.
f) tert-Butyl ~3-[2-(1 -toll-~neslllfonylbPn7imi(1~7ol-2-yl)ethyl]isoxazolin-5(R~S)-
yl~acetate
According to the procedure of Example 1 (d) of WO 95/14682, except
sub.~ uling 4-(1-toluenesu~nylben7.imi<~ Ql-2-yl)-1-butanoximinoyl chloride for
the 4-cyanobenzoximinoy~ chloride, and ~ub~liluling tert-butyl 3-butenoate for the
methyl 3-butenoate, the title compound is ~l~al~d.
186
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
g) [3-[2-(1-Tnllleneslllfonylbenzimidazol-2-yl)ethyl]isoxazolin-S(R,S)-
yl]acetic acid
4 M HCl in dioxane (10 mL) is added to a solution of tert-butyl [3-[2-(1-
toluenesulfonylben7imi(1~7nl-2-yl)ethyl]isoxazolin-5~R,S)-yl]acetate (5 mmole) in
C~I2CH2 (40 mL) at 0~C. The reaction is stirred at RT until complete, then is
concentrated to afford the title compound.
h) Ethyl 3-[[3-[2-(1-toluenesulfonylben7imi-1~7ol-2-yl)ethyl]isoxazolin-5(R,S)-
yl]acetyl]amino-3(R,S)-methylpropanoate
EDC (1.2 mmole) is added to a solution of [3-[2-(1-
toluenesulfonylbenzimidazol-2-yl)ethyl]isoxazolin-5(R,S)-yl]acetic acid (1 mmole),
ethyl 3(R,S)-aminobutyrate (1.2 mmole), HOBt H2O (1.2 mmole), and
diisopropylethylamine (4 mmole) in anhydrous CH3CN (5 rnL) at RT. The reaction
is stirred at RT until complete, then is concentrated, and the residue is purified by
silica gel chromatography to afford the title compound.
i) 3-[[3-[2-(Ben7imi-1~7ol-2-yl)ethyl]isoxazolin-5(R,S)-yl]acetyl]amino-3(R,S)-
methylpropanoic acid
1.0 N LiOH (2.5 mmole) is added to a solution of ethyl 3-[3-[2-(1-
toluenesulfonylb~ n7imi~1~7ol-2-yl)ethyl]isoxazolin-5(R,S)-yl]acetyl]amino-3(R,S)-
methylpropanoate (0.5 mmole) in THF (2.5 ml). The reaction is stirred at RT until
complete, then is neutralized with l.0 N HCl. The solution is concentrated and the
residue is purified by reverse-phase chromatography to afford the title compound.
Fxample 61
Preparation of 3-{3.4-dihydro-8-rrr(ben7imi(1~7nl-2-
yl)methyl~methy}aminolcarbonyll-1-methyl-2~5-dioxo-lH-1.4-benzodiazepine}-4-
propanoic acid
a) 2-Amino-4-iodobenzoic acid
The title compound is prepared from the oxidation of 4-iodo-2-nitrotoluene
to give 4-iodo-2-niLI~bell~oic acid according to the method of Sasson, et al., J. Org.
Chem. 1986, 51, 2880-2883, followed by reduction of the nitro group using iron and
acetic acid.
187
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
b) 4-Iodoisatoic anhydride
To a mechanically stirred ice-cold solution of 2-amino-4-iodobenzoic acid
(26.3 g, 0.1 mol), sodium carbonate ~10.6 g, 0.1 mol) and water (250 mL), is slowly
added, via an addition funnel, a solution of phosgene in toluene (80 mL of 1.93 M
S solution). After 2 h, the precipitated product is i.~ol~te~l by filtration and the solid is
washed cuccessively with water (200 mL), a 1: 1 mixture of ethanol/ether (300
mL),and ether (200 mL). Drying under vacuum yields the title compound.
c) N-(2-Amino-4-iodobenzoyl)-,13-alanine benzyl ester
A magnetically stirred solution of 4-iodoisatoic anhydride (5.0 g, 0.0173
mol)"1~ alanine benzyl ester tosylate (5.85 g, 0.0173 mol), and
dimethylaminopyridine (0.5 g, 0.0041 mol) in pyridine (35 rnL) is heated for 2 h at
80~C. The reaction mixture is allowed to cool to RT and concentrated in vacuo.
The resulting residue is dissolved in ethyl acetate (100 mL), and washed
successively with 10% cupric sulfate (2 x 50 rnL), saturated sodium bicarbonate (I x
50 mL) and brine (1 x 50 mL). Drying (Na2SO4), filtration, concentration, and silica
gel chromatography (1:1 EtOAc/hex~n~ s) gives the title compound.
d) N-(4-Iodo-2-methylaminobenzoyl)-,13-alar~ine benzyl ester
A m~gnç~ lly stirred solution of N-(2-amino-4-iodobenzoyl)-13-alanine
benzyl ester (2.0 mmol), 2,6-lutidine (0.35 mL, 3.0 mmol) and methyl iodide (0.19
mL, 3.0 mmol) in DMF (15 mL) is heated at 50~C for 15 h. The reaction mixture isallowed to cool to RT and concentrated in vacuo. The r~s-llting residue is dissolved
in ethyl acetate (75 rnL), and washed succl ccively with 10% citric acid (1 x 50 mL),
saturated sodium bicarbonate (1 x 50 mL) and brine (I x 50 mL). Drying (Na2SO4),filtration, concentration, and silica gel chromatography (gradient 35-65 %
EtOAc/hexanes) gives the title compound.
e) Benzyl 3-~3,4-dihyro-8-iodo- 1-methyl-2,5-dioxo- lH- 1,4-benzodiazepinel-4-
propanoate
To a cold (-30~C) m~gn~ti~:~lly stirred solution of N-(4-iodo-2-
methylarninobenzoyl)-,13-alanine benzyl ester (0.305 g, 0.69 mrnol) and triethylamine
(0.144 g, 1.04 mmol) in methylene chloride (3 mL) is added slowly a solution of a-
bromoacetyl bromide (0.09 mL, 1.04 mmol) in methylene chloride (2 mL) under
argon. The reaction mixture is allowed to warm to RT and stir for 2 h. The llli~lulG
is diluted with methylene chloride (40 ml ) and washed s-lcce~ivGly with 10% citric
acid (1 x 50 rnL) and saturated sodium bicar~onate (1 x 50 mL), dried (Na2SO4),
188
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
filtered and concentrated in vacuo. The resultin~ residue is dissolved in
DMF (3 ml) and added via an addition funnel to a slurry of sodium hydride (25 mg,
1.04 mrnol) in DMF (2 mL) which is cooled to 0~C. After 2 h of stirring, the
mixture is poured over an ice cooled solution of 10% citric acid (50 mL) and
5 extracted with ethyl acetate (3 x 40 ml). The combined extracts are washed with
saturated sodium bicarbonate (1 x 50 mL), dried (Na2SO4), filtered and concentrated.
Silica gel chromatography (gradient 40-70% EtOAc/hexanes) gives the title
compound.
f) Benzyl-3-[3,4-dihyro-8-t[[(ben7imi(1~7Ol-2-yl)methyl]methylamino]carbonyl]
methyl-2,5-dioxo- 1 H- 1,4-benzodiazepine]-4-propanoate
A mixture of benzyl 3-[3,4-dihyro-8-iodo- 1-methyl-2,5-dioxo- lH-1,4-
benzodiazepine]-4-propanoate(2 mmol), 2-(methylaminomethyl)ben~ill,idazole
dihydrochloride (3 mmol), DIEA (1.8 mL, 10 mmol), and (Ph3P)2PdC12 (140 mg,
0.2 mrnol) in N-methyl-2-pyrrolidinone (20 mL) is heated to 110~C under CO
balloon for 3 h. The mixture is then concentrated and the residue is purified bysilica gel flash chromatography to give the title compound.
g) 3-[3,4-Dihydro-8-[[[(ber-7imi~1~7Ol-2-yl)methyl]methylamino]ca.bollyl]-l-
methyl-2,5-dioxo- 1 H- 1,4-benzodiazepine]-4-propanoic acid
A mixture of benzyl-3-[3,4-dihyro-8-[[[(bel,,.i...ifl~7Ql-2-
yl)methyl]methylamino]carbonyl3 - 1 -methyl-2,5-dioxo- 1 H- 1,4-benzodiazepine3-4-
propanoate (2 mmol) and 10% Pd/C (0.02 g) in ethanol (100 mL) is hydrogenated inan atmosphere of H2 (50 psi3 for 6 h. The catalyst is removed by filtration, and the
filtrate is concentrated under vacuo to afford the title compound.
Fx~m~le 62
~re~aldlion of 3-{4H-imi-l~7Q~1~2-al~1.41benzQdiazepine-5(6H)-l-methyl-6-oxQ-9-
rrr(ber~7imi~ 701-2-yl)methyllmethylaminolcarbonyl~ }-4-propanoic acid
a) Ethyl N-(2-amino-4-iodobenzoyl)-,B-alanine
A m~gneti~lly stirred solution of 4-iodoisatoic anhydride (0.0173 mol)"l3
alanine ethyl ester hydrochloride (0.0173 mol), and dimethylaminopyridine (0.5 g,
- 35 0.0041 mol) in pyridine (35 mL) is heated for 2 h at 80~C. The reaction mixture is
allowed to cool to RT and concentrated in vacuo. The resulting residue is dissolved
in ethyl acetate (100 mL), and washed successively with 10% cupric sulfate (2 x 50
189
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/2~748
mL), saturated sodium bicarbonate (1 x 50 rr~ ) and brine (1 x 50 rnL).
Drying (Na2SO4), filtration, concentration, and silica gel chromatography (1:1
EtOAc/hexanes) gives the title compound.
b) Ethyl-3-[3 ,4-dihyro-8-iodo-2,5-dioxo- 1 H- 1 ,4-benzodiazepine]-4-propanoateTo a cold (-30~C) m:~gnPti~z~lly stirred solution of ethyl N-(2-amino-4-
iodobenzoyl)-,B-alanine (0.69 mmol), and triethylamine (0.144 g, 1.04 mmol) in
methylene chloride (3 rnL) is added slowly a solution of a~-bromoacetyl bromide
(0.09 mL, 1.04 mmol) in methylene chloride (2 mL) under argon. The reaction
mixture is allowed to warm to RT and stir for 2 h. The mixture is diluted with
methylene chloride (40 mL) and wash snccç~ively with 10% citric acid (1 x 50 mT.)
and saturated sodium bicarbonate (1 x 50 mL), dried (Na2SO;), filtered, and
concentrated in vacuo. The rçsllltin~ residue is dissolved in DMF (3 mL) and added
via an addition funnel to a slurry of sodium hydride (25 mg, 1.04 mmol) in DMF (2
mL) which is cooled to 0~C. After 2 h of stirring, the rnixture is poured over an ice
cooled solution of 10% citric acid (50 mL) and extracted with ethyl acetate (3 x 40
rnL). The combined extracts are washed with saturated sodium bicarbonate ( 1 x 50
rnL), dried (Na2SO4), filtered, concentrated, and chromatographed on silica gel to
afford the title compound.
c) Ethyl-3-[3 ,4-dihyro-8-iodo-2-thione-5-oxo- 1 H- 1 ,4-benzodiazepine~-4-propanoate
To a solution of ethyl-3-[3,4-dihyro-8-iodo-2,5-dioxo- 1 H- 1,4-
benzodiazepine]-4-propanoate ( 1.0 g, 2.49 mmol) in THF ( 10 mL) at RT and underan atmosphere of nitrogen is added Lawesson's reagent ( 1.0 g), and the reaction is
heated at 50~C for 2 h. The reaction mixture is allowed to cool to RT and is
concentrated in vacuo. ~ilica gel chromatography (gradient 40-60%
EtOAc/hexanes) gives the title compound.
d) Ethyl-3-[4H-imi-1~7o~1,2-a][1,4]benzodiazepine-5(6H)-1-methyl-6-oxo-9-iodo]-
4-propanoate
To a vigorously stirred biphasic solution of ethyl-3-[3,4-dihyro-8-iodo-2-
thione-S-oxo-lH-1,4-benzodiazepine]-4-propanoate (0.95 g, 2.27 mmol), methyl
iodide (0.2 g) and a catalytic amount of tetrabutylammonium hydrogen sulfate in
CH2CI2 (10 rnL) and water (10 mL) is added 2 N NaOH (1.2 mL) at RT. After 2 h,
the layers are separated and the aqueous layer is washed with CH2Cl2 (2 x 25 rnL).
The combined organic extracts are dried (Na2SO4), filtered and concentrated in
vacuo. The resulting residue is dissolved in toluene (10 rnL) and treated with
190
CA 02241633 1998-06-26
W O 97/24119 PCTr~S96/20748
pl~pa.~yl amine (0.64 mL, 4-fold excess) and pyridine hydrogen chloride
(0.23 g, 1 mol eq). The reaction is heated to reflux for 6 h, then was allowed to cool
to RT. Concentration and silica gel chromatography (EtOAc) gives the title
compound.
s
e) Ethyl-3-[4H-imi~l~7~[1,2-a][1,4]benzodiazepine-5(6H)-l-methyl-6-oxo-9-
[[[(benzimidazol-2-yl)methyl]methylamino]carbonyl] }-4-propanoate
A rnixture of ethyl-3-[4H-imi~1~7O[ l ,2-a] ~ 1,4]benzodiazepine-5(6H)- 1 -
methyl-6-oxo-9-iodo]-4-propanoate(2 mmol), 2-(methylaminomethyl)ben7imi(1z~7c-1e(3 mmol), DIEA (1.8 mL, 10 mmol), and (Ph3P)2PdCl2 (140 mg, 0.2 mmol) in N-
methyl-2-pyrrolidinone (20 mL) is heated to 110~C under a CO balloon for 3 h. The
mixture is then concentrated and the residue is purified by silica gel flash
chromatography to give the title compound.
f) 3-[4H-Imidazo[1,2-a][1,4]benzodiazepine-5(6H)-l-methyl-6-oxo-9-
[[[(ben7imi~i~7Ql-2-yl)methyl]methylamino]carbonyl]]4-propanoic acid
A solution of ethyl-3-[4H-imi~l~7o[l~2-a][l~4]benzodiazepine-5(6H)
methyl-6-oxo-9-[[t(ben7imi~1~7ol-2-yl)methyl]methylamino]carbonyl] }-4-
propanoate (54 mmol), LiOH H20 (0.79 mmol), THF (5 mL), and water (2 mL) is
stirred at RT overnight. The mixture is concentrated, the residue is dissolved in
water, and the resulting solution is neutralized with 3 N HCl. The precipitate is
collected and dried in vacuo to afford the title compound.
E~xample 63
P~ lion of 4-r4-r2-(lH-ben7~imi~l~7~ol-2-yl)ethyll-l-piperazin
piperi~1inP~çetiç acid
a) Ethyl 4-[4-[(tert-butoxycarbonyl)]-1-pipe~ lyl]-1-piperi~1inP~et~t~
The title compound is prepared from tert-butyl l-piperazinecarboxylate
(Aldrich) and ethyl 4-oxo-1-piperitlinP~ret:ltP (Porter et al. EPA 0 542 363 A2) by
NaCNBH3 reductive amination according to the method of Porter et al., EPA 0 542
363 A2.
- 35
b) Ethyl 4-(1-~ipe~dzinyl)- 1-piperi(1in~P;Icet~tP
191
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
A solution of ethyl 4-[4-t(tert-butoxycarbonyl)]-1-pi~e~ yl]-l-
piperitlin~a~et~t~ and 4 M HCl/dioxane in CH2Cl2 is stirred at RT for 18 h. The
reaction mixture is concentrated to give the title compound as the hydrochloride salt.
5 c) 2-[2-Chloroethyl)]be.n7.imirl~7.ole
A solution of 2-ben7.imici:~7.Qleethanol and thionyl chloride in CH2Cl~ is
heated at reflux for 2 h. The mixture is evaporated to give the title compound.
d) Ethyl of 4-[4-[2-(lH-bPn7imi<1~7.Ql-2-yl)ethyl]-l-pipeld~ yl]-l-
10 piperitlinf.~e~slt~
A solution of ethyl 4-(1-pi~ela~ yl)-1-pipericlinP~ret~te, 2-[2-
chloroethyl)]ben7imi/i~7.Qle, and Dl~EA in DMF is stirred at RT for 18 h. The
mixture is concentrated and purified by chromatography to give the title compound.
e) 4-[4-[2-(lH-Ben7imi(1~7- 1-2-yl)ethyl]-1-~ .dzinyl]-1-piperi~1inP.~reti~ acidA solution of ethyl of 4-[4-[2-(lH-ken7.imiti~7.--1-2-yl)ethyl]-l-pipera_inyl]-l-
piperi~linea~et~te. and 1.0 N NaOH in MeOH is stirred at RT. After 18 h, the mixture
is neutralized with AcOH, cle.s~ltP.cl through an XAD-2 column, and lyophiii7P.-l to
give the title compound.
F,x~mrle. 64
Prep~ration of 1 -hydroxy-4-r4-r3-( 1 H-be~7imi~1~7.ol-2-yl)propyll- 1 -piperazinyll-
eyclohex:~neacetic acid
a) tert-Butyl l-hydroxy4-[4-[2-(lH-bçn7.imi-1~7.ol-2-yl)propyl~ ;ldzinyl]-
cyclohexaneacetate
A solution of tert-butyl l-hydroxy-4-(1-pipel~inyl)-cyclohexs~nP~-~et~tP
(EPA O 537 980 Al~, 2-(3-bromopropyl)ben7imi(1~7.ole (J. Org. Chem. 1962, 27,
3U 2165), and DIEA in DMF is stirred at RT for 18 h. The l~ Lul~iS concentrated and
purified by chromatography to give the title compound.
b) 1 -Hydroxy-4-[4-[2-~ lH-ben7.imi~l~701-2-yl)propyl]- l-pipera_inyl]-
cyclohex:~n~ etic acid
A solution of tert-butyl l-hydroxy~-[4-[2-(lH-ben7imi~1~7.ol-2-yl~propyl]-l-
yip~,.dzinyl]-cyclohex~nP.~cet~te and 4 M HCl/dioxane in CH~Cl2 is stirred at RT.
After 18 h, the mixture is evaporated to give the title compound.
192
CA 0224l633 l998-06-26
W O97/24119 PCTrUS96/20748
Ex~mple 6S
Preparation of N-r3-~l-rr2-(2-Ben7imi(1~7olvl)ethyllcarbonyllpiperidinyllcarbonyll-
5 ,13-alanine
Following the procedures of Beavers et. al., WO 95/25091, Example 1,
except ~ubstitl-tin~ (2-ben7imi~701yl)propionic acid for N~'-Boc-D-lys(Cbz)-OH,
gives the title compound.
Example 66
Preparation of 2-r~Ben7imi(1~7ol-2-yl)methyll- 5-r2-~carboxy-ethyl)amino~call,ollyll
-2.3-dihydro-3-oxo- 1 H-isoindole
Following the procedures of rl~Jaldlion 1-12 in Hartman, et al., EP 0 540
334 Al, for the prepa-dlion of l-H-isoindole-5-carboxamide, 2,3-dihydro-N-(2-
carboxy-ethyl)-2-[2-(piperidinyl)ethyl]-3-oxo, except substituting 2-
(aminomethyl)be.l~ lidazole (Aldrich) for Boc-4-piperidine-2-ethylamine, the title
compound is pl~pal~ed.
Bxample 67
Ple~ lion of r3(R)-r2-(be~ 7ol-2-yl)ethyll-2-oxopiperidinyllacetyl-3(R)-
methyl -~-alanine
a) Methyl 4-(ben7imifl~7Ol-2-yl) butanoate
Following the procedure of Example 36(a), except sub~LiLuLillg 1,2-
diaminobenzene for the 2,3-diaminopyridine, the title compound is prepared.
b) 4-(Ben7imi~ 7OI-2-yl) butanoic acid
Following the procedure of Example 36(b), methyl 4-(benzimidazol-2-yl)
butanoate is saponified to afford the title compoud.
c) [3(R)-r2-~Ben7.imi~ ol-2-yl)ethyl]-2-oxopiperidinyl]acetyl-3(R)-methyl-,13-
- 35 alanine
.
193
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
Following the procedure of Duggan et al (J. Med. Chem. 1995, 38,
3332), except using 4-(ben7imid~7Ol-2-yl) butanoic acid instead of (N-Boc-
piperidin-4-yl3butanoic acid, the title compound is prepared.
E~ rr~le68
.
Preparation of 4-rrrr2-(benzimidazolyl)methyllcall,unyllmethylaminol-
acetyllphenoxyacetic acida) 4-[2-(Boc-methylamino)acetyl]phenol
A solution of di-tert-butyl dicarbonate (5.96 g, 27.3 mmol) in 1,4-dioxane
(25 mL) was added dropwise at 0~C to a mixture of 4-t2-
(methylamino)acetyl]phenol hydrochloride (5.0 g, 24.8 mmol), 1,4-dioxane (30 mL),
H2O (25 rnL) and 1.0 N NaOH (25 mL, 25 mmol). After 24 h, the reaction was
warmed to RT and stirred for 1.5 h. More 1.0 N NaOH (25 mL, 25 mmol) was
added, and the reaction was stirred for an additional 0.5 h at RT, and concentrated.
The residue was diluted with EtOAc (80 mL), and the ll~ix.Lule was acidified to pH 2
using 1.0 M NaHSO4. The res-llsin~ mixture was extracted with EtOAc, and the
combined organic layers were washed with H20 and dried (Na2S04). Filtration and
concentration gave the title compound (6.49 g, 99%): lH NMR (250 MHz, CDCl3)
~ 6.70-8.05 (m, 4 H), 4.53 (s, 2H), 2.98 (s, 3H), 1.50 (s, 9H).
b) Benzyl 4-[2-(Boc-methylamino)acetyl]phenoxyacetate
A mixture of the compound of Example 68(a) (5.04 g, 19.0 mmol) and
K2CO3 (2.63 g, 19.0 mmol) in acetone (100 mL) was stirred at reflux under argon
for lh. The mixture was cooled to RT and benzyl bromo~etS-~ (5.23 g, 22.8 mmol)
was added. The reaction was heated at reflux for 18 h, then was cooled and filtered.
The filter cake was washed with acetone, and the filtrate was concentrated. The
residue was dissolved in CH2Cl2 (300 mL) and washed sequentially with H2O (50
mL) and brine (50 mL). Drying ~Na2SO4), concentration, and flash
chromatography ( silica gel, 1 :3 EtOAc/hexanes) yielded the title compound (7.28 g,
93%): 1H NMR (250 MHz, CDCl3) o 6.85-7.95 (m, 9 H), 5.23 (s, 2H), 4.71 (s, 2H),
- 4.55 (d, 2H), 2.95 (d, 3H), 1.45 (d, 9H).
c) Benzyl 4-r2-(methylamino)acetyl]phenoxyacetate hydrochloride
- A mixture of the compound of Example 68(b) (7.26 g, 17.57 mmol) and 4 M
HCl in 1,4-dioxane (150 mL) was stirred for 1 h at RT. Concentration and
trituration with Et2O afforded the title compound as a white powder (5.93 g, 97%):
194
CA 0224l633 l998-06-26
W O 97/24119 PCTrUS96/20748
1H NMR (250 MHz, CD30D) ~ 7.05-8.00 (m, 9 H), 5.23 (s, 2H), 4.88 (s,
2H), 4.65 (s, 2H), 2.80 (s, 3H).
d) Benzyl 4-~[1:[2-
S (bçn7imicl~701yl)methyl3carbonyl]methylalI~ino]acetyl~phenoxyacetate
A mi~lule of the compound of Example 68(c)(1 mmol), 2-
(ben7imicl~7Olyl)acetic acid (1 mmol), EDC (1.5 mmol), and D~ 3 mrnol) in
DMF (25 mL) is stirred at RT. The mixture is poured in to 5% NaHCO3 and
extracted with EtOAc. The organic phase is washed with H2O, dried (MgSO4) and
concentrated. The residue is chromatographed (silica gel) to give the title
compound.
e) 4-[[[[2-(Ben7imit1~7Olyl)methyl]carbonyl]methylamino]acetyl]phenoxyacetate
The compound of Example 68(d)(1 mmol) and lN NaOH (1.5 mL) in
CH30H (20 mL) is stirred and concentrated. The residue is dissolved in H2O,
extracted with CH2C12, and the aqueous phase is adjusted to pH 5 with dilute HCl to
give the title compound.
Fx~mI?le 69
Pl~;yaldlion of 4-rrrr2-(Benzimidazolyl)methyllcarbonyllmethylaminolacetyll-1.2-l?henylenedioxvdiacetiç acid
a) 4-[2-(Boc-methylamino)acetyl~- 1,2-dihydroxybenzene
Following the procedure of Example 68(a), except substituting adrenalone
hydrochloride (5.0 g, 23.0 mmol) for 4-[2-(methylamino)acetyl]phenol
hydrochloride, the title compound (1.2 g, 19%) was prepared following flash
chromatography (silica gel, 1: I EtOAc/hexanes): MS (ES) m/e 282.2 [M+H]+.
b) Dimethyl 4-[2-(Boc-methylamino)acetyl]-1,2-phenylenedioxy~ ret~te
Following the procedure of Example 68(b), except substituting the
compound of Example 69(a) (0.9 g, 3.2 mmol) for the compound of Example 68(a)
and methyl bromoacetate (1.23 g, 8.0 mmol) for ~enzyl bromo~cet~e, the title
co.llpoul~d (1.11 g, 81%) was prepared: MS (ES) m/e 426.2 [M+H]+.
- 35
c) Dimethyl 4-~2-(methylamino)acetyl]- 1,2-phenylenedioxydiacetate hydrochloride 195
CA 0224l633 l998-06-26
W O 97/24119 PCTrUS96~0748
Following the procedure of Example 68(c), except substituting the
compound of Example 69(b) (1.1 1 g, 2.6 mmol) for the compound of Example
68(b), the title colllpoulld was l~r~al~;d (1.1 g, qu~..l ;l~tive): MS (ES) m/e 326.0
[M+H]+.
d) Dimethyl 4-[[[[2-(ben7imi-1A7QIyl)methyl]carbonyl]methylarnino]acetyl]-1,2-
phenylenedioxyrliAcetAtP
Following the procedure of procedure of Exarnple 68(d), except sub~ ; ugthe ~;ul~ o~-nd of Example 69(c) for the compound of Example 68(c), gives the title
10 compound.
e) 4-[[[[2-(Benzimidazolyl)methyl]carbonyl]methylamino]acetyl]- 1,2-
phenylenedioxydiacetic acid
Following the procedure of procedure of l~xarnple 68(e), except substituting
15 the compound of Example 69(d) for the compound of Example68(d), gives the title
compound.
Fx~mple 70
Preparatisn of N-r3-rrrt2-Ben7imi-1A7olyl)metllyllçarbonyllamirlolbenzoyll-¦3-
alar~ine
a) Benzyl N-[3-[[t(2-bçn7imi(1:~7olyl)methyl]carbonyl]arl~ino]benzoyl]-,B-AI~ninAte
A ~ Y.lul~ of benzyl N-(3-aminobenzoyl)-,B-~IAninAt~ (Alig, et. al., EPA
372486)(1 mmol), (2-b~7imi~l~7Q}yl)acetic acid (1 mmol), EDC (1.5 mmol), and
DIEA (3 rnmol) in DMF (25 mL) is stirred at RT. The mixture is poured into 5%
NaHCO3 and extracted with EtOAc. The combined organic phase is washed with
H2O, dried (MgSO4) and concentrated. The residue is chromatographed (silica gel)to give the title compound.
b) N-[3-[[[(2-Bçn7imi~1A7olyl)methyl]carbonyl]amino]benzoylJ-~3-alanine
A mixture of the compound of Example 70(a)( 1 mmol) and lN NaOH ( 1.5
mL) in CH30H (20 mL) is stirred and coneell~ldled. The residue is dissolved in
H2O, extracted with CH2Cl2, and the aqueous phase is adjusted to pH 5 with dilute
HCl to give the title compound.
F.~ 7 1
196
CA 0224l633 l998-06-26
W O 97/24119 PCTrJS96/20748
Pl~al~tion ofrrl-rN-rr(2-Ben7.imi~7.olyl)methyl]carbonyl~tyrosyll-4-
piperidinylloxylacetic acid
=
S a) tert-Butyl [[l-[N-[[(2-ben7imi~lA7olyl)methyl]c~ln,llyl]tyrosyl]-4
piperidinyl]oxy]acetate
A mixture of tert-butyl [( l-tyrosyl-4-piperidinyl)oxy]acetate (Alig, et. al.,
EPA 372486)(1 mmol), (2-ben7imi~1A7olyl)acetic acid (1 mmol), EDC (1.5 mmol),
and DIEA (3 mmol) in DMP (25 mL) is stirred at RT. The mixture is poured into
10 5% NaHCO3 and extracted with EtOAc. The combined organic phase is washed
with H2O, dried (MgSO4) and concentrated. The residue is chromatographed (silicagel) to give the title compound.
b) [[l-[N-[[(2-Benzimida_olyl)methyl]call~ollyl]tyrosyl]~-piperidinyl]oxy]acetic15 acid
A mixture of the compound of Example 71(a)(1 mmol) and CF3CO2H in
CH2CI2 is stirred and concentrated to give the title compound.
F.x~ ?le 72
P-~pal~lion of fS)-4-rrr(2-Benzimidazolyl)methyllcarbonyllglycyll-3-
m~thoxycarbonylmethyl-2-oxopipe-d~ine-l-acetic acid
Following the procedure of Sugihara, et. al., EP 0529858, Example 59,
except substit~-ting (2-ben7imitlA701yl)acetic -acid for 4-amidinobenzoic acid
25 hydrochloride, gives the title compound.
PxArnple 73
Preparation of (3S.5S)-5-rr4-[(2-Ben71midA7olyl)methyllphenylloxymethyl~-3-
30 carboxymethyl-2-pyrrolidinone
a) 4-[(2-Ben7imi-1A7l-1yl)methyl]phenol
Pollowing the general procedure of Wahlgren and Addison, J. Heterocycl.
- Chem., 1989, 26, 541-543, except substitl-ting 4-(hydroxy)phenylacetic acid for 2-
35 (hydroxy)phenylacetic acid, gives the title compound.
..
-
197
CA 02241633 1998-06-W O97/24119 PCT~US96/20748
b) (3S,5S)-5-t[4-[~2-Benzimidazolyl)methyl]phenyl3O~y~ hyl]-3-t(tert-
butoxycarbonyl)methyl] -2-pyrrolidinone
Following the procedure of Hirnmelsbach, et.al., Australian Patent
Application AU-A-86926/91, Example 51, substituting the compound of Example
5 73(a) for 4'-cyano-3'-fluoro-4-hydroxy)biphenyl, gives the title compound.
c) (3S,SS)-5-[~4-[(2-Ben7imi-1~7olyl)methyl~phenyl]oxymethyl]-3-carboxymethyl-2-pyrrolidinone
The compound of Example 73(b) is treated with CF3CO2H in CH2CI2 to give
the title compound.
F~ le 74
Prçparatioll of 1-r~2-Ben7imi~ m~tllyll-3-r4-(2-carboxyethyl)phenyl~-
1 5 m~,thoxy-3-pyrrolin-2-one
Following the procedures of Linz, et. al., EP 0567968, except sub~ (2-
ben7imi-1~701yl)methsln:~min/ for 4-cyano~nilin~, gives the title colllpo~l,ld.
Fx~ ple 75
Plti~,a,~tion of 2-r6-(ben7imi~1~7ol-2-yl) metllylaminocarbonyl)-12.3
tetrahydroisoquinolinyllacetic acid
a) 6-Methoxy- 1,2,3 ,4-tetrahydroisoquinoline
6-Methoxy-1,2,3,~tetrahydroisoquinoline is prepared according to the
method of D. J. Sall and G. L. Grunewald (J. Med. C~hem. 1987, 30, 2208-2216).
b) Ethyl 2-[6-Methoxy-1,2,3,~tetrahydroisoquinolinyl]acetate
A solution of the compound of Example 75(a) ( 1.1 mmol), ethyl
chloroacetate (1.17 mmol), and pot~ssillm carbonate (1.17 mrnol) in acetonitrile (10
mL) is stirred for 18 hr. The mixture is then partitioned in a mixture of EtOAc and
H20. The organic phase is rotary evaporated to an oil, which is purified by silica gel
chromatography to afford the title co~ oulld.
198
CA 02241633 1998-06-26
W O 97/2411g PCTrUS96/20748
c) Ethyl 2-[6-Hydroxy-1,2,3,4-tetrahydroisoquinolinyl]acetate
A solution of the compound of Example 75(b3 (0.249 g, 1.0 mmol) and
boron tribromide ( lM in CH2Cl2, 1.0 mL, 1.0mmol, mL) is stirred at -70 ~C for 2 hr
and then stirred at RT for 12 hr. The solution is rotary evaporated to an oil. The
- S residue is taken up in EtOAc. EtOAc is washed with water (lX), 5% NaHCO3(2X),
water (lX). EtOAc is dried over Mg2SO4, filtered and rotary evaporated to afford the
title compound.
d) Ethyl 2-[6-trifluoromethylsulfonyloxy-1,2,3,4-tetrahydroisoquinolinyl]acetateA solution of the compound of Example 75(c) (0.235 g, 1.0 mmol),
trifluorosulfonic acid anyhdride (0.23 mL, l.lmmol,) and Et3N (0.32 mL, 1.5 mmol)
in CH2Cl2 (S ml)is stirred for 8 hr The solution is rotary evaporated to an oil. The
residue is taken up in EtOAc. EtOAc is washed with 5% NaHCO3 (2X), water (lX).
EtOAc is dried over Na2SO4, filtered and rotary evaporated to afford the title
lS compound.
e) Ethyl 2-[6-carboxy-1,2,3,4-tetrahydroisoquinolinyl~acetate
A solution of the compound of Example 75(d) (0.367 g, 1.0mmol),
pall~ m(II)bis-acetate (0.022 g, 0.1mmol,), triphenylphosphine (0.262 g,
1.0mmol), diisopropylamine (0.34 mL, 2.5 mmol), NMP (S mL), in aqueous
~mmonillm carbonate (10%) is stirred for 8 hr under an atmosphere of carbon
monoxide. The solution is rotary evaporated to an oil. The residue is purified by
silica gel chromatography to afford the title compound.
f) Ethyl -[6-(ben7imicl~7Ol-2-yl)methylaminocarbonyl)-1,2,3,4-
tetrahydroisoquinolinyl]acetic acid
A solution of the compound of Example 75(e) (0.263 g, 1.0 mmol), the
compound of (2-ben~ 7Olyl)acetic acid (0.34g, 1.0 mmol), EDC (0.19lg, 1.0
- mmol), HOBt (0.151 g, 1.0 mmol) and triethylamine (0.235 mL, 2.0 mmol) in
DMF(7 mL) is stirred for 8 hr. The solution is rotary evaporated to an oil. The
}esidue is purified by silica gel column to afford the title compound.
199
CA 0224l633 l998-06-26
W O97/24119 PCTrUS96/20748
g) 2-[6-(ben7imi~1~7ol-2-yl)methylarninocarbonyl)-1,2,3,4-
tetrahydroisoquinolinyl]acetic acid
A solution of the compound of Example 75(f) ( 0.42 g, 1.0 mmol) in aqueous
1 N sodium hydroxide (1.5 mL, 1.5 mmol) and ethanol ( 5 mL) is stirred for 8 hr.The solution is rotary evaL,ol~led to an oil. The residue is purified by silica gel
column to afford the title compound.
Example 76
Pre~?~ration of 2-r6-fbell~i...id~7ol-2-yl)methylaminoca~l,ollyl)-l-oxo-1.2.3~4-tetrahydroisoquinolir~ acetic acid
a) 6-Methoxy- 1 -oxo- 1,2,3,4-tetrahydroisoquinoline
6-Methoxy-l-oxo-1,2,3,4-tetrahydroisoquinoline is p~ d according to the
method of D. J. Sall and G. L. Grunewald, J. Med. Chem. (1987), 30, 2208-2216.
b) Ethyl 2-[6-Methoxy-l-oxo-1,2,3,4-tetrahydroisoquinolinyl]acetate
A mixture of the compound of Example 76(a) (0.39 mmol) and NaH (0.17 g,
0.43 mmol, 60% oil dispersion) in THF (5 rnL) is heated to reflux for 1 hr and then
allowed to cool to room ~e~ el~ture. Ethyl chloroacetate (0.43 mmol) is added tothe mixture, and the mixture is allowed to stir for 1 hr. The mixture is quenched
with water (10 rnL) and washed with EtOAc (2X 15 rnL). The organic layers are
combined, washed with water (10 rnL) and rotary evaporated to an oil. The residue
is purified by silica gel chromatography to afford the title compound.
c) Ethyl 2-[6-Hydroxy-l-oxo-1,2,3,4-tetrahydroisoquinolinyl~acetate
A solution of the compound of Example 76(b) (0.263 g, 1.0 mmol) and
boron tribromide (lM solution in CH~Cll, 1.1 mL) is stirred at -70 ~C for 2 hr and
then at RT for 4 hr. The solution is rotary evaporated to an oil. The residueis taken
up in EtOAc. EtOAc is washed with water (lX), 5% NaHCO3 (2X), water (lX).
200
CA 02241633 1998-06-26
W O97/24119 PCTrUS96/20748
EtOAc is dried over MgSO4, filtered and rotary evaporated to afford the
title compound.
~. d) Ethyl 2-[6-trifluoromethylsulfonyloxy-1-oxo-1,2,3,4-
- 5 tetrahydroisoquinolinyl]acetate
A solution of the compound of F.~mple 76(c) (3.4 mmol) and
trifluorosulfonic acid anyhdride (3.4 mmol, mL) in pyridine (5 mL) is chilled at 0~
and allowed to warm to room temperature for 1 hr. The mixture is quenched with
water (5 mL~ and washed with EtOAc (2X 7 mL). The organic layers are combined,
washed with water (7 rnL) and rotary evaporated to an oil. The residue is purified by
silica gel chromatography to afford the title compound.
e) Ethyl 2-[6-carboxy-1-oxo-1,2,3,4-tetrahydroisoquinolinyl~acetate
A solution of the compound of Example 76(d) (0.23 g, 1.0 mmol),
palladium(II)bis-acetate (0.026 g, 0.1 mmol), triphenylphosphine (0.262 g, 1.0
mmol), diisopropyla~nine (0.23 mL, 2.0 mmol), NMP (7 mL), in aqueous
ammonium carbonate "(10 %) is stirred for 8 hr under an atmosphere of carbon
monoxide. The solution is rotary evaporated to an oil. The residue is purified by
silica gel chromatography to afford the title compound.
f) Ethyl -[6-(bc~ 7Ol-2-yl)methylaminocarbonyl)- 1,2,3,4-
tetrahydroisoquinolinyl]acetic acid
A solution of the compound of Example 76(e) (0.34 g, 1.0 mmol), the
compound of (2-ben~ idazolyl)acetic acid (0.43 g, 1.0 mmol), EDC (0.191 g, 1.0
mmol), HOBt (0.15 g, 1.0 mmol) and triethylamine (0.234 mL, 2.3 mmol) in DMF(8
mL) is stirred for 8 hr. The solution is concentratred. The residue is purified by
silica gel chromatography to afford the title compound.
~lt~ tively, a solution of the compound of Example 76(d) (0.23 g, 1.0
mmol), p~ m(II)bis-acetate (0.026 g, 0.1 mmol), triphenylphosphine (0.262 g,
- 1.0 mmol), diisopropylarnine (0.25 mL, 2.1 mmol), NMP (7 mL), and the compound
201
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
of Interm~ t~ A (0.31 g, 1.0 mmol) in aqueous ammonium carbonate
~10%) is stirred for 8 hr under an atmosphere of carbon monoxide. The solution is
rotary evaporated to an oil. The residue is purified by silica gel chromatography to
afford the title compound.
g) 2-[6-((ben7imi~l~7ol-2-yl)methylaminocarbonyl)-1,2,3,4-
tetrahydroisoquinolinyl]acetic acid
A The solution of the compound of Example 76(f) (0.25 g, 1.0 mmol) in
aqueous 1 N sodium hydroxide (1.5 mL, 1.5 mmol) and ethanol (8 mL) is stirred for
10 8 hr. Solution is rotary evaporated to an oil. The residue is purified by silica gel to
afford the title compound.
Rx~mple 77
Preparation of 2-r6-((be~ 7~1-2-yl)methylcarbonyl:~mino)tetralinlacetic acid
a ) tert-butyl-5-amino-tetraline-2-acetate
Tert-butyl-5-amino-tetraline-2-acetate is prepared according to the methods
described in M. J. Fisher, et al. (Scheme 12 and Example 28, parts A-D, EO
0635492, Jan. 25, 1995).
b) 2-[6-((benzimidazol-2-yl)methylcarbonylamino)tetralin]acetate
A solution of the compound of Example 77 (a) (0.261 g, 1.0 mmol), 2-
(aminomethyl)ben7imid~7O1e(0.256g, l.Ommol),EDC(0.19lg, l.Ommol) 1-
hydroxybenzotriazole hydrate (0.152 g, 1.0 mmol), and triethylamine (0.234 mL, 2.1
mmol) in DMF (5 mL) is allowed to stir for ~ hr. The solution is rotary evaporated
to an oil. The residue is purified by silica gel chromatography to afford the title
compound.
A solution of the crude ester amide (0.32 g, 1.0 mmol) and trifluoroacetic
acid (5 mL) in methylene chloride (~ mL) is allowed to stir for 1 hr. The solution is
202
CA 0224l633 l998-06-26
W O97/24119 PCT~US96/20748
rotary evaporated to an oil. The residue is treated with Et2O. Filtration and
drying in vacuo afforded the title compound.
;,
F.x~mple 78
~l~;palalion of 2-r6-((benzimidazol-2-yl)methylaminocarbonyl)tetr~linJacetic acid
a) Ethyl-5-hydroxy-tetraline-2-acetate
The compound Ethyl-5-hydroxy-tetraline-2-acetic acid is prepared according
to the method of M. J. Fisher et al. (EP 0635492, Scheme 6 and Example 20, parts A-D, p. 71).
b) Ethyl-5-trifluoromethylsulfonyloxy-tetraline-2-acetate
A solution of the compound of Example 78(b) (0.321g, 1.0 mmol) in CH2Cl~
(10 mL) is cooled to O~ C.Trifluoromethylsulfonic acid anhydride ( 0.125 mL, 1.1mmol) is added. The solution is stirred for 2 hr. The solution is rotary evaporated to
an oil. The residue is taken up in EtOAc. EtOAc is washed with water (lX), 5%
NaHCO3, water (lX). EtOAc is dried over MgSO4, filtered. Filtrate is rotary
evaporated to afford the title compound.
c) Ethyl 6-carboxy-tetraline-2-acetate.
A solution of the compound of Example 78(c) (0.26 g, 1.0 mmol),
palladium(II)bis-acetate (0.023g, 0.1 mmol), triphenylphosphine ~0.262g, 1.0
mmol), diisopropylamine (0.245 mL, 2.1 mmol), NMP (10 mL ), in aqueous
25 ammonium carbonate (10%) is stirred for 8 hr under an ~tmosphere of carbon
monoxide. The solution is rotary evaporated to an oil. The residue is purified by
silica gel chromatography to afford the title compound.
- d) Ethyl -[6-((ben7imi~l~7ol-2-yl)methylaminocarbonyl)-tetraline-l-acetate
A solution of the compound of Example 78(c) (0.34 g, 1.0 mmol), the
compound of (2-ben7imicl~olyl)acetic acid (0.32g, 1.0 mmol), EDC (0.191 g, 1.0
203
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96120748
mmol), HOBt ~0.152 g, 1.0 mmol) and triethylamine (0.23 mL, 2.1 mrnol3
in DMF(6 mL) is stirred for 8 hr. The solution is rotary evaporated to an oil. The
residue is purified by silica gel chromatography to afford the title compound.
~It.orll~tively, a solution of the compound of Example 78(b) ~0.34 g, 1.0
mmol), palladium(II)bis-acetate (0.023g, 0.1 mmol), triphenylphosphine (0.262g,
1.0 mmol), diisopropylarnine (0.23 mL, 2.1 mmol), NMP (10 mL), and the
compound of IntermPrli~te A (0.32 g, 1.0 mmol), in aqueous ammonium carbonate
(10%) is stirred for 8 hr under an zitm~spht~re of carbon monoxide. The solution is
rotary evaporated to an oil. The residue is purified by silica gel chromatography to
afford the title compound.
e) 2-~6-((be.~i."~ 7Ol-2-yl)methylaminocarbonyl)-tetraline-2-acetic acid.
A solution of the compound of Example 78(d) (0.31 g, 1.0 mmol) in aqueous
1 N sodium hydroxide (1.5 mL, 1.5 mmol) and ethanol ( 5 mL) is stirred for 8 hr.The solution is rotary evaporated to an oil. The residue is purified by silica gel
chromatography to afford the title compound.
Fx~mple 79
Preparation of 2-r5-((be~7,i l ll;dzl~ol-2-yl)methylcarbonylamino)-benzofuranl-
.
proplomc acld
a) Ethyl 1-carboxymethyloxy~-nitrosalicylaldehyde
A solution of 1-hydroxy~-nitrosalicylaldehyde (Aldrich) ( 0.167 g, 1.0
mmol), ethyl bromoacetate (0.166 g, 1.0rnmol) potSlc~inm carbonate ( 0.276 g,
2.0mmol) and sodium iodide (0.015 g, 0.1 mmol) in THF ( 10mL) is heated to 80 ~Cfor 24 hr. The solution is rotary evaporated to an oil and the residue is purified by
silica gel chromatography to afford the title compound.
b) Ethyl 2-carboxy-5-nitrobenzofuran
204
CA 02241633 1998-06-26
W O 97/24119 PCTrUS96/20748
A solution of the compound of Example 79(a) (0.229 g, 1.0 mmol),
DBU (0.152 g, 1.0 mmol) in ethyl alcohol ( 10 mL) is allowed to stir at RT for 18 hr.
The solution is rotary evaporated to an oil and the residue is treated with EtOH (10
mL). The solution is bubbled with HCl.gas for 2 minllt~s and refluxed for 5 hr. The
- 5 solution is rotary evaporated to an oil. EtOAc is added and washed with water (2X),
5% citric acid (2X), water (1~), 5% NaHCO3.(2X) and water (lX). EtOAc is rotary
evaporated to afford the title compound.
c) Ethyl 2-(2-carboxy)ethylene-5-nitrobenzofuran
A cold solution (-78 ~C) of the compound of Example 79(b) ( 0.235 g, 1.0
mmol)in THF (5 mL) is treated with DiBAL ( 1.0 M in THF, 1.0 mL, 1.0 mmol).
The solution is stirred at -78 ~C for 30 min~ltes and RT for 3 hr. The solution is
treated with CH3COOH (3 mL) followed by water (2 rnL). The solution is rotary
evaporated to an oil and treated with toluene to azeotrope off the acetic acid. Drying
in vacuo afforded the crude aldehyde.
A solution of the phosphonate ester (0.224 g, 1.0 mmol),in THF (5 mL) is
treated with sodium hydride (60% suspension in mineral oil, 0.04 g, 1.0 rnmol)at 0~
C for 1 hr. To the solution is added the aldehyde (0.235 g, 1.0 mmol). The solution
is stirred at RT for 18 hr. The solution is rotary evaporated to an oil and the residue
is purified by silica gel chromatography to afford the title compound.
d) tert-butyl 2-[5-amino-benzofuranyl]propionate
A solution of the compound of Example 79(c) ( 0.261 g, 1.0 rnrnol) in
ethanol ( 5 mL) containing 10% palladium-on-carbon(0.026 g, 10% wt) is
hydrogenated at 45 psi for 1 hr. The solution is filtered through Celite and the- filtrate is rotary evaporated to an oil. Silica gel cl~ t~ graphy affords the title
compound.
e) 2-~5-((bçn7imicl~7Ql-2-yl)methylcarbonylamino)-benzofuran]-propionic acid
205
CA 02241633 1998-06-26
W O 97/24119 ~CT~US96/20748
A solution of the compound of Example 79(d) ( 0.263 g, 1.0
mmol), EDC ( 0.191 g, 1.0 mmol), 1-hydroxybenzotriazole ( 0.150 g, 1.0 mmol), the
compound of 2-(aminomethyl)benzimidazole ( 0.234 g, 1.0 mmol) and triethylamine
(0.288 mL, 2.0 mmol) in DMF (5.0 mL) is allowed to stir for 18 hr. The solution is
rotary evaporated to an oil and the residue is purified by silica gel chromatography
to afford the title compound.
A solution of the crude ester ( 0.263 g, 1.0 mmol) in MeOH (3.0 mL) is
treated with 1 N NaOH ( l .S mL, l .S mmol) and water (2 ml). The solution is
stirred at RT for 18 hr. The solution is rotary evaporated to an oil and purified by
reversed phase chromatography to afford the title compound.
F.x~?le 80
Preparation of 2-rS-((benzirnidazol-2-yl)methylcarbonylamino)-2.3-dihydro-
ben7Ofilranl-propionic acid
a) Ethyl-2-[S-amino-2,3-dihydro-benzofuranyl3propionate
In the chromatographic purification of the compound of Example 79(d) the
-20 title compound is also obtained.
b) 2-[5-(6-arninopyridinyl-2-methylcarbonylamino)-benzofuran]-propionic acid
A solution of the compound of Example 80(a) (0.263 g, 1.0 mmol), EDC
(0.19lg, 1.0 rnmol), 1-hydroxybenzotriazole (0.lSg, 1.0 m~nol), the compound of 2-
(aminomethyl)ben7imi.1~701e (0.32 g, 1.0 mmol) and triethylamine (0.288 mL, 2.0
mmol) in DMF (5.0 mL) is allowed to stir for 18 hr. The solution is rotary
evaporated to an oil and the residue is purified by silica gel chromatography toafford the title compound.
A solution of the crude ester ( 0.290g, 1.0 rnmol) in MeOH (5.0 mL) is
treated with lN NaOH (1.2 mL, 1.2 mmol) for 18 hr. The solution is rotary
206
.
CA 02241633 1998-06-26
W O 97/24119 PCT~US9~/20748
evaporated to an oil and purified by reversed-phase chromatography to
afford the title compound.
-
Fxample 81
S
Preparation of 2-r5-(6-aminopyridinyl-2-nlethylaminocarbonyl)-benzofuranl-
propionic acid
a) 2-Ethyloxycarbonyl-5-(tert-butyl-dimethylsilyloxy)-benzofuran
A solution of 2-Ethyloxycarbonyl-5-(hydroxy)-benzofuran is prepared via
the procedure described in M. L. Denny, et al. (EP 0655439, 31,5,95) (0.206 g, 1.0
mmol), tert-butyl-dimethylsilylchloride (0.23 mL, 1.0 mmol) and imidazole ( 0.34 g,
1.0 mmol) in THF is allowed to stir for 4 hr. The solution is rotary evaporated to an
oil. EtOAc is added and washed with water. EtOAc is rotary evaporated to afford
the title compound.
b) 2-Hydrocarbonyl -5-(tert-butyl-dimethylsilyloxy)-benzofuran
A cold solution (-78 ~ C) of the compound of Example 81 (a) ( 0.35 g, 1.0
mmol)in THF (5 mL) is treated with DiBAL ( 1.0 M in THF, 1.0 mL, 1.0 mmol).
The solution is stirred at -78 ~ C for 30 minutes and RT for 3 hr. The solution is
treated with CH~COOH (3 mL) followed by water (2 mL). The solution is rotary
evaporated to an oil and treated with toluene to azeotrope off the acetic acid. Drying
in vacuo afforded the aldehyde.
c) Ethyl 2-[5-(tert-butyl-dimethylsilyloxy)-benzofuran]-acrylate
A solution of the phosphonate ester (0.224 g, 1.0 mmol),in THF (5 rnL) is
treated with sodium hydride (60% s-lcpen~ion in mineral oil, 0.04 g, 1.0 mmol)at 0"
C for 1 hr. To the solution is added the above aldehyde(0.235 g, 1.0 mmol). The
solution is stirred at RT for 18 hr. The solution is rotary evaporated to an oil and the
residue is purified by silica gel chromatography to afford the title compound.
207
CA 02241633 1998-06-26
W O 97/24119 PCT~US96J20748
d) 2-Ethyl 2-~-5-(hydroxy)-benzofuran]-propionate
A mixture of the compound of Example 81(c) (0.234g, 1.2 mmol) and 10%
p~ m-on-carbon (0.023 g, 10% wt) in EtOH( S mL).is hydrogenated at 50 psi
5 for 1 hr. Filtration through Celite and concentration afforded.the ester (0.169 g,
56%).
A solution of the crude ester (0.34 g, 1.0 mmol) and tetraethylammonium
fluoride (0.149 g, 1.0 mmol) in THF (10 mL) is allowed to stir at RT for 18 hr. The
10 solution is rotary evaporated to an oil and purified by silica gel ch~ atography.
e) 2-Ethyl 2-[-5-(trifluoromethylsulfonyloxy)-benzofuran]-propionate
A solution of the compound of Example 81(d) (0.366 g, 1.0 mmol) and
Et3N(0.23 mL, 1.5 mmol) in CH2Cl2 (10 mL) at 0 "C is treated with
trifluoromethylsulfonic acid anhydride (0.21 mL, 1.1 mmol). After 2 hr solution is
rotary evaporated to an oil. The residue is taken up in EtOAc. EtOAc is washed
successively with water(lX), 5% Na~ICO3 (2X), water (lX). EtOAc is dried over
MgSO4, and filtered. Filtrate is rotary evaporated to afford the title compound.
20 f) 2-Ethyl 2-[-5-(carboxy)-benzofuran3-propionate
A solution of the compound of Example 81(e) (0.366g, 1.0 mmol),
p~ m(II) bis-acetate (0.023g, 0.1 mmol), triphenylphospine (0.262g, 1.0 mmol),
diisopropylethylamine (0.23 mL, 2.1 mmol), NMP (7 mL) in aqueous sodium
bicarbonate (10%, 6 mL) is allowed to stir. The solution is rotary evaporated to an
25 oil. The residue is purified by silica gel chromatography to afford the title compound.
g) 2-Ethyl 2-~-5-(6-(6-aminopyridinyl-2-methylaminocarbonyl)-benzofuran]-
propionate
208
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/20748
A solution of the compound of Example 81(f) (0.366 g, 1.0 mmol), the
compound of (2-ben7imid~701yl)acetic acid (0.23g, 1.0 mmol), EDC (0.191 g,
1.0mmol), HOBt (0.152g, 1.0mmol) and triethylamine (0.235 mL, 2.1mmol) in
DMF(8 mL) is stirred for 8 hr. The solution is rotary ev~poldled to an oil. The
5 residue is purified by silica gel chromatography to afford the title compound.
Alt~-rn~tively, a solution of the compound of Example 81(e) (0.366g,
l .Ommol), palladium(II)bis-acetate (0.023g, 0.1 mmol), triphenylphosphine (0.262g,
1.0 mmol), diisopropylamine (0.23 mL, 2.1 mmol), NMP (10 mL ), and the
compound of Intern~ t(~ A (0.32g, 1.0 mmol) in aqueous ammonium carbonate
(10%, 10 mL) is stirred for 8 hr under an atmosphere of carbon monoxide. The
solution is rotary evaporated to an oil. The residue is purified by silica gel
chromatography to afford the title compound.
h) 2-[-5-(6-(6-aminopyridinyl-2-methylaminocarbonyl)-benzofuran]-propionic acid
A solution of the col"pou~d of Example 81(g) (0.366g, 1.0 mmol) in
aqueous 1 N sodium hydroxide (1.5 mL, 1.5 mmol) and ethanol ( 8 rnL) is stirred for
8 hr. The solution is rotary eva~o,~l~d to an oil. The residue is purified by silica gel
chromatography to afford the title compound.
Ex~n~le 8~
Preparation of 2-rS-((benzi",idazol-2-yl)methylaminocarbonyl)-2.3-dihydro-
ben7:r~furan~-propionic acid
a) 2-tert-Butyl 2-[-5-(hydroxy)-2,3-dihydro-benzofuran]-propionate
The chromatographic purification in Example 81 (d) also provides the title
compound.
b) 2-tert-Butyl 2-[-5-(trifluoromethylsulfonyloxy)-2,3-dihydro-benzofuran]-
propionate
209
CA 02241633 1998-06-26
W O 97/24119 PCT~US96/207~.
A cold solution of the compound of Example 82(a) (0.28 g, 1.0
mmol) and Et3N (0.23 mL, 2.1 mmol) in CH2Cl2 (5 mT.)is treated with
trifluoromethylsulfonic acid anhydride (0.15 mL, 1.1 mmol) for 2 hr. The solution
is rotary evaporated to an oil. The residue is taken up in EtOAc. EtOAc is washed
5 with water (lX), 5% NaHCO3(2X), water (lX). EtOAc is dried over MgSO4,
filtered, and filtrate is rotary evaporated to afford the title compound.
c) 2-tert-Butyl 2-[-5-(carboxy)-2,3-dihydro-benzofuran]-propionate
A solution of the compound of Example 82(b) (0.24 g, 1.0 mmol),
10 p~ um(II) bis-acetate (0.023g, 0.1 mmol), triphenylphospine (0.262g, 1.0 mmol),
diisopropylethylamine (0.23 rnL, 2.1 mmol), NMP (8 mL ) in aqueous sodium
bicarbonate (10 mL, 10%) is allowed to stir at RT for 8 hr. The solution is rotary
evaporated to an oil. The residue is purified by silica gel chromatography to afford
the title compound.
d) 2-tert-Butyl 2-[-5-(6-(6-aminopyridinyl-2-methylaminocarbonyl)-2,3-dihydro-
benzofuran] -propionate
A solution of the compound of Example 82(c) (0.366g, 1.0 mmol), the
compound of (2-be~ 7olyl)acetic acid ( 0.23g, 1.0 mmol), EDC (0.19lg, 1.0
20 mmol), HOBt (0.152g, 1.0 ~nol) and triethylamine (0.23 mL, 2.1 mmol) in DMF(8mL) is stirred for 8 hr. The solution is rotary evaporated to an oil. The residue is
purified by silica gel chromatography to afford the title compound.
~It~ tively, a solution of the compound of Example 82(b) (0.366g, 1.0
25 mmol), p~ m(II)bis-acetate (0.023 g, 0.1 mmol), triphenylphosphine (0.2~2 g,
1.0 rnmol), diisopropylamine (0.23 mL, 2.1 mmol), NMP (10 mL), and the
compound of Interm~ te A (0.23 g, 1.0 mmol), in aqueous ammonium carbonate
(10%, 10 mL) is stirred for 8 hr under an atmosphere of carbon monoxide for 8 hr.
The solution is rotary evaporated to an oil. The residue is purified by silica gel
30 chromatography to afford the title compound.
210
CA 02241633 1998-06-26
W O97/24119 PCT~US96/20748
e) 2-[-5-(6-((ben7imit1~7Ql-2-yl)methylaminocarbonyl)-2,3-dihydro-
benzofuran]-propionic acid
. A solution of the compound of Example 82(d) (0.37 g, 1.0 mmol) in aqueous
1 N sodium hydroxide ( 1.5 mL, 1.5 mmol) and ethanol (8 mL) is stirred for 8 hr.- 5 The solution is rotary eva~oldted to an oil. The residue is purified by silica gel
chromatography to afford the title compound.
.
Example 83
lQ ~lcpald~ion of 1 -r( lH-ben7imi(1~7ol-2-yl)methyll-3-{4r(2-
ethoxycarbonyl)ethyl)lphenyl }-3-oxo-imidazolidine
a) 2-Aminomethyl-l-p-toll-enl-sl-lfonyl-benzimidazole
A solution of 2-Methylben7imi~701e (1 mmol), p-toluenesulfonyl chloride
(1.05 mmol) and Et3N (1.05 mmol) in water (2 mT ) and THF (1 mL) is allowed to
stir for 4 hr. The ~ L~nc is concentrated by rotary evaporation and diluted withwater (10 mL). The solution is washed with EtOAc (2X 15 mL). The organic layers
are combined, washed with water (S mL) and rotary evaporated to an oil.
A solution of the aforementioned oil (2-Methyl- 1 -p-toluenesulfonyl-
benzimidazole) (1 mmol) and N-bromosuccinimide (1.05 mrnol) in CCl4 (10 mL) is
allowed to reflux for 18 hr. Upon cooling a white solid precipitates from solution.
The solid is filtered, triturated with CCl4 and dried in vacuo to a solid.
A solution of Boc2NH (1 mmol) and pot~sil-m hydroxide ~1 mmol~ in
ethanol (S mL) is allowed to stir for 1 hr. Anhydrous ethyl ether (15 mL) is added
and the mixture is filtered to afford the salt as a white solid. A solution of the
aforementioned solid 2-bromomethyl-l-p-toluenesulfony!bl~n7i..~ 701e (1 mmol)
and (Boc)2N~ Kt (mmol) in THF (10 mT.)is to stir at 60~ for 18 hr. The mixture is
30 rotary evaporated to an oil.
A solution of the 2-[bis-(tert-butyloxycarbonyl)aminomethyl]- l-p-
toluenesulfonylben7im~ 701e (1 mmol) in TFA (1.1 mmol) and CH2Cl2 is allowed
to stir for 1 hr. The solution is rotary ev~o-dled to an oil, which is purified by
35 chromatography to afford the title compound.
211
CA 02241633 1998-06-26
W O97/24119 PCT~US96/20748
b) Ethyl 2-[4-(2-hydroxyethylamino)phenyl]propionate
This compound is prepared following the procedure of F. ~imm~l~bach et al.
~Example V, p. 44, EP 0587134, Sept. 8, 1993), in which glycolaldehyde dimer
(Aldrich) (mmol) is added to a solution of methyl 2-(4-aminophenyl)propionate 1 (1
mmol) in aqueous acetonitrile (pH 6-7) (mL), followed by sodiumcyanoborohydride
(1.1 mmol), and the mixture is allowed to stir for 1 hr. The lluhLIul~ is rotaryevaporated to an oil, and the residue is dissolved in a mixture of ice water and ethyl
acetate. The water layer is neutralized with 4 N sodium hydroxide and washed with
ethyl acetate. The organic phase is rotary evaporated to an oil. A solution of the oil
in ethyl acetate is purified on a silica gel column to give the title compound.
c) N-[(l-p-toluenesulfonyl-lH-be~ 7~l-2-yl)methyl]-N~-hydroxyethyl-N~
{4[(2-ethoxycarbonyl)ethyl)]phenyl } -urea
This compound is Lnc~cd following the procedures of F. Himmelsbach et
al. (EP 0587134, Sept. 8, 1993 and EP 0612741, Feb.21, 1994), in which a solution
of the compound of Example 83(a) (1 mmol) and phosgene (1.1 mmol) in THF (20
mL) is allowed to stir at -20~ for 20 minutes The compound of Example 83(b) (1.0mmol) is added to the solution and the r~cllltin~ mixture is allowed to stir for 18 hr.
The resulting solution is rotary evaporated. A solution of the residue in ethyl acetate
is washed with 5% citric acid, followed by water. The organic phase is rotary
evaporated to an oil. . A solution of the oil in ethyl acetate is purified on a silica gel
column to give the title compound.
d) N'-[(l-p-toluenesulfonyl-lH-ben7imifl~7ol-2-yl)methyl]-N3-{4t(2-
ethoxycarbonyl)ethyl)]phenyl } -2-oxo-imidazolidine
This compound is prepared following the procedures of F. Himmelsbach et
al. (Example III, EP 0587134, Sept. 8, 1993 and EP 0612741, Feb.21, 1994), in
which a solution of the compound of Example 83(c) (1 mmol),
mf-th~n~sulfonylchloride (1.2 mmol) and triethylamine (1.2 mrnol) in methylene
chloride (5 ml) is allowed to stir at 0~ for 1 hour. The ~ lul~; is partitioned in a
mixture of water and methylene chloride. The organic phases are combined and
dried over anhydrous sodium sulfate and rotary evaporated.
A solution of the residue and sodium iodide (1.1 mmol) in acetone (5 mL) is
heated to reflux for 3 hr and then rotary evaporated to an oil. Potassium-
bis(trimethylsilyl)azide (1.2 mmol) is added to a solution of the residue in DMF (5
mL), cooled to 0~. The solution is allowed to warrn to room t~ln~el~lul~ over 30 212
,
CA 02241633 1998-06-26
PCT~US96120748
W O 97/24119
min. and the rotary evaporated to an oil. The residue is partitioned in a
ul~ of water and methylene chloride. The organic phases are combined and
dried over anhydrous sodium sulfate and rotary evaporated. A solution of the oil in
ethyl acetate is purified on a silica gel column to give the title compound.
e) N~ H-bçn7im~ 7ol-2-yl)methyl]-N3-{4[(2-carboxyl)ethyl)]phenyl}-2
imicl~olidine
A solution of the compound of Example 83(d) ( 1 mmol) in THF (S mL) and
1 N sodium hydroxide (1.2 mL, 1.2 mmol) is allowed to stir for 18 hr. The mixture
10 is neutralized with conc. hydrochloric acid and purified on a silica gel column to
give the title compound.
F,~ m,ple 84
Parenteral Dosage Unit Composition
A preparation which contains 20 mg of the compound of Example 1 as a
sterile dry powder is prepared as follows: 20 mg of the compound is dissolved in 15
mL of distilled water. The solution is filtered under sterile conditions into a 25 ml
multi-dose ampoule and lyophilized. The powder is reconctit-lte~l by addition of 20
mT of 5% dextrose in water (DSW) for intravenous or intr~mllccul~r injection. The
dosage is thereby det~,rmin~l by the injection volume. Subsequent dilution may be
made by addition of a metered volume of this dosage unit to another volume of
DSW for injection, or a metered dose may be added to another m~c,h~nicm for
en~ g the drug, as in a bottle or bag for IV drip infusion or other injection-
infusion system.
Exarnple 85
C)ral Dosa~e Unit Composition
A capsule for oral ~f1mini,ctration is prepared by mixing and rnilling 50 mg of
the compound of Example 1 with 75 mg of lactose and 5 mg of m~gnl~,citlm stearate.
The rçs--ltin~ powder is screened and filled into a hard gelatin capsule.
F,x:~mple 86
Oral r~osa~e Unit Com,~osition
A tablet for oral a~1minictration is prepared by mixing and gr~n~ ting 20 mg
of sucrose, 150 mg of calcium sulfate dihydrate and 50 mg of the compound of
J
213
CA 0224l633 l998-06-26
W O 97/24119 PCTAUS96/20748
Example 1 with a 10% gelatin solution. The wet granules are screened,
dried, mixed with 10 mg starch, 5 mg talc and 3 mg stearic acid; and co~ r~ssed
into a tablet.
The above description fully discloses how to make and use the present
- invention. However, the present invention is not limited to the particular
embodiments described hereinabove, but includes all modifications thereof withinthe scope of the following claims. The various references to journals, patents and
other publications which are cited herein comprises the state of the art and areincorporated herein by reference as though fully set forth.
214
r