Note: Descriptions are shown in the official language in which they were submitted.
CA 02241845 1998-06-29
WO 97!25317 PCT/LTS96/i9569
4,5-DIHYDRONAPHTH[I,2-CIISOXAZOLES AND DERIVATIVES THEREOF HAVING CNS
ACTIVITY
The present invention is directed to certain novel
compounds and their use as pharmaceutical agents having unique
central nervous system acti~rity.
This invention relates to 4,5-dihydronaphth(1,2-
c]isoxazoles and derivatives thereof, and their use as serotonin
5-FiT~ antagonists. which may be useful for the treatment of
anxiety, psychiatric disorders. schizophrenia, nausea, vomiting
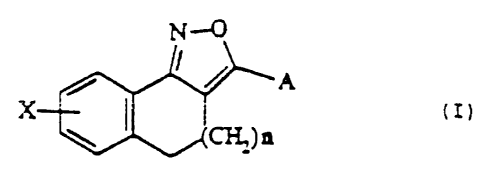
and the control of drug dependency, of general formula (I?:
N-~
t~)
X ~ )n
\ H2
wherein A is hydrogen. hydroxy,
CA 02241845 1998-06-29
2
WO 97/25317 PCT/US96/19569
_ HIV ~(C fi2~ ~ ,O
or
yR N~R
t i
U
wherein
Rt is hydrogen. an alkyl group o~ 1 to 6 carbons, optionally
substituted with hydroxy, alkoxy or amino substitution; aryl or
heteroaryl, optionally substituted with halogen, hydroxy or
alkoxy; or benzyl optionally substituted with halogen, hydroxy
or alkoxy;
n is an integer o~ 1 or 2;
Z is nitrogen, CH or CEOH);
m is an integer o~ I to 3; and
X is hydrogen, hydroxy or alkoxy:
CA 02241845 1998-06-29
3
WO 97/25317 PCT/1JS96/19569
or a pharmaceutically acceptable additional salt thereof, or
-where applicable, a geometric or optical isomer or racemic
mixture thereof.
The present invention also relates to a process for
preparing these compounds, pharmaceutically acceptable additzcn
salts thereof, as well as the pharmaceutical acceptable
compositions thereof, and a method of using the compounds as
seroton 5-HT~ antagonists.
I0
Throughout the specification and appended claims, a given
chemical formula or name shall encompass all stereo and optical
isomers where such isomers exist. Additionally, a given
chemical formula or name shall encompass the pharmaceutically
acceptable additional salts thereof.
In a preferred embodiment of the invention are compounds of
formula (I) wherein
A is
N ~(C HZ~m
y
Rt
' wherein
CA 02241845 1998-06-29
4
WO 97/25317 PCT/US96/19569
R1 is hydrogen. an alkyl group of I to 6 carbons,
optionally substituted with hydroxy, alkoxy or amino
substitution; aryl or heteroaryl. optionally substituted with
halogen, hydroxy or alkoxy; or benzyl optionally substituted
with halogen, hydroxy or alkoxy;
n is an integer of 1 or 2;
Z is nitrogen;
to
m is an integer of 1 to 3; and
X is hydrogen, hydroxy oz alkoxy.
More preferred, are compounds of formula (I) wherein
R1 is hydrogen, or an alkyl group of 1 to 3 carbons;
2Q n is 1:
Z is nitrogen;
m is 1 or 2; and
CA 02241845 1998-06-29
WO 97/25317 ~ PCT/US96/19569
X is hydrogen.
The novel compounds of the present inventicn and the
intermediates thereto may be prepared by the reaction sequence
illustrated hereinbelow. The substituents Z, m, n and X are
generally as defined above unless otherwise indicated.
N~OH
N-~
I
l
1 o X ~ --~, X ~ / H PO- ~
\ CH2~ ' ~CH~n Et3N
2 3
N-O N-O
,- ~ / t H_A / ~ / A
X \ ~ CHI base \ ~ [CH~n
4 1
According to the preparation scheme, hydroxyisoxazoles 3 are
prepared from oximes 2 in a solvent such as tetrahydrofuran
(TFiF) at a temperature of from about 25'C to about reflux
temperature of the solvent for a period of from about 0.25 to
.about 4 hours according to the methods of Griffiths and Olofson
(Jerome S. Griffiths, et al., J. Chi. Scx. C, 974 (I971) and
G.N. Barber and R.A. Olofson, J. OrQ. Chess. d3, 3015 (1978)).
The hydroxyisoxazoles 3 are converted to chloroisoxazoles 4 via
CA 02241845 1998-06-29
WO 97!253I7 ~' PCT/US96/19569
treatment with phosphorous oxychloride in the presence of a
suitable base, such as triethylamine, at a temperature of from
about 100' to about 200°C for a period of from about 0.25 to
about 4 hours in a manner similar to that utilized by Adembri e~ ,
a1. (G. Adembri and P.Tedeschi, Hull. 8ci. F'ac. Chic. Ind.
BoloQns 23, 203 (I965)). Fntermediates ~ are treated with an
appropriate nucleopbl1e H-A (wherein A is defined hereinbefore)
at a temperature of from about 100' to about 200° C with or
without added base in an appropriate solvent, such as N-
methylpyrrolidinone, to provide the novel compounds 1 of the
invention.
These compounds may be prepared by the following
representative examples. The examples are exemplary and should
not be construed as limiting the invention disclosed herein.
EXAMPLE 1
3-Chloro-4,5-dihydronaphth[I,2-c]isoxazole
N-O N-4
/ I
~ ( v ~ w
\ \
To a stirred mixture of 4,5-dihydronaphth(I,2-c]isoxazol-3-
(3aH)-one (7.258, 38.77mmol) in phosphorus oxychloride (10.84m1,
116.3mmol), triethylamine (5.40m1, 38.77mmo1) was added
CA 02241845 1998-06-29
7
WO 97/25317 PCT/US96/19569
dropwise_ After completion of addition, the mixture was heated
to reflux while stirring. After 2 hours, no starting material
remained as shown by T~C [silica, ethylacetate (EtOAc)]. The
mixture was cooled to room temperature, poured into 300 ml of
ice water, and extracted with CHZC12_ The organic extracts were
combined, dried over MgSO, and concentrated irt vacuo. The
resultant solid was filtered through silica using CHZClz eluent
to provide 6.2g of crude product. This crude product was
recrystallized from a minimum of heptane to provide a product as
needles, mp of 57-59'C, homogeneous by thin layer chromatography
(TLC) [silica, CH~CiZ, Rf=0.80] . The Infrared (IR) (CHC1~) ,
nuclear magnetic resonance (rain) (CDC1~) , and Mass Spectrum
(M'=205, EI, 70eV) were consistent with the structure. The yield
was 5.4178 (26.4mmol, 68.16$).
I5
Elemental Analysis
Calculated Found
C 64.25 64.02
H 3.92 3.86
Cl I7.24
N 6.8I 6.77
O 7.78
EXAMPLE 2
3-(4-Methyl-1-piperazinyl)-4,S-dihydronaphth[1,2-c]isoxazole
CA 02241845 1998-06-29
WO 97/25317 8 PCT/US9b/19569
N --~ L1-O
/ / ~ 1 / j / N
~N ''C H3
A stirred mixture of 3-chloro-4,5-dihydronaphth[I,2-
c]isoxazole (2.658, 12.93mmo1), N-methyl piperazine (30m1,
270.4mmo1) and KZCO~ (3.578, 25.87mmol) under NZ was lowered into
an oil bath preheated to 150'C. The mixture was heated while
stirring under N2 for 2 hours. At that time, TLC [CH~C1~] showed
no remaining starting material. The mixture was removed from
the heating bath and allowed to cool to room temperature. IC
was then partitioned between heptane/HiO. The heptane phase was
washed with water, dried over MgSO" filtered and concentrated
in vacuo to yield a solid. This crude product was
recrystallized from heptane/ether tEt20) to provide the product
as needles, mp of 92-94~C, homogeneous by TLC [silica, I:1
CH~OH:EtOAc, .Rt=0.39] . The IR (CHC1~), rain tCDCI,) and Hass
Spectrum (M'=269. EZ, 70eV) were consistent with the structure.
The yield was 1.2555g (9.67mmo1, 36.090 .
CA 02241845 1998-06-29
PCT/LTS96/19569
WO 97/25317
Elemental Analysis
Calculated Found
C 71.35 71.34
H 7.11 6.98
N 15.60 15.78
O 5.94
EXAMPLE 3
3-(4-(2-Hydroxyethyl)-I-piperazinyl)-4,5-dihydronaphth(1,2-
c]isoxazole
N ~ N --a
~ / N ~
--~ \ ~ ~ '~OH
A stirred mixture of 3-chloro-4,5-dihydronaphth[1,2-
c]isoxazole (3.0g, 14.63mmo1), 1-(2-hydroxyethyl)-piperazine
(17.95m1, 146.3mmo1) and K20~ (4.1g, 29.3mmo1) in 18m1 of N-
methlypyrrolidinone under NZ was lowered into an oil bath
2d preheated to 150'C. The mixture was heated while stirring under
Nz for Z hour. At that time, TLC (CHZClZ) showed no remaining
starting mater3.al_ The mixture was removed from the heating
bath, allowed to cool to room temperature, and diluted with HZO.
Upon the addition of heptane, a solid precipitated. The solid
was collected, washed with heptane and HZO, and dried in vacuo
CA 02241845 1998-06-29
WO 97/Z5317 1 ~ PCT/US96/19569
(O.lmm) at 85~C overnight to provide pure product, mp oL 137-
- 138~C, homogeneous by TLC (silica, 1:1 CH~OH:EtOAc, Rt=0.67).
The IR (CHC1,) , NMR (CDCI,) and Mass Spectrum (M'=299, EI, 70eV) '
were consistent with the structure. The yield was 2.6038
(8.70mmo1, 59.470 .
Elemental Analysis
Calculated Found
C 68.21 68.12
H 7.07 7.01
N 14.04 14.14
O 10.69
3-(1-Homopiperazinyl)-4,5-dihydronaphth(1,2-c)isoxazole
K
,. ~ / !
N --O N ---0
\ ~ -~ \
A stirred mixture of 3-chloro-4,5-dihydronaphth(1,2-
c~isoxazole (3.0g, 14.63 mmol) homopiperazine (I4.668, 146.3
mmol) and FC~CO~ (4.048, 29.3mmo1) in 16m1 of N-methyl-
pyrrolidinone under N~ was lowered into an oil bath preheated
to 150'C. The mixture was heated while stirring under NZ for
45 minutes. At that time, TLC (CH2C1Z) showed no remaining
CA 02241845 1998-06-29
PCT/US96l19569
WO 97/25317 11
starting material. The mixture was removed from the heating
bath, allowed to cool to room temperature, diluted with H,O and
'" extracted with EtZO. The EtZO phase was dried over MgSO"
filtered and concentrated in vacc~o. The crude solid obtained
was recrystallized from heptane/EtzO and dried in vac~o (O.Imm)
at 85°C overnight to provide pure product, mp of 79-81°C,
homogeneous by TLC [silica, 1:1 CH,OH:EtOAc, Rf=0.17j_ The IR
(CHC13) , NMR (CDC1~) and Mass Spectrum (M'=269, EI, 70eV) were
consistent with the structure. The yield was 1.9698 (7.32nnmol,
50.03~k) .
Elemental Analysis
Calculated Found
C 71.35 71.45
H 7.11 7.29
N 15.60 15.56
O 5.94
2.0 3- ( 1-Piperazinyl) -4, S-dihydronaphth [ 1, 2-cJ isoxazole
N -U N --~
~ / I / ~ / N
_N H
\ ~ ~/\
CA 02241845 1998-06-29
WO 97/25317 1 PC'T/US96/19569
A stirred mixture of 3-chloro-4,5-dihydronaphth[1,2-
- c]isoxazole (5.0g, 24.9mmo1), piperazine (34.28, 397.7mmo1) and
K2C03 (6.738, 48.7mmo1) in 40m1 of N-methylpyrrolidinone under N2 -
was lowered into an oil bath preheated to 150'C. The mixture
was heated while stirring under NZ for 45 minutes. At that time,
TLC (CHzClZ) showed no remaining starting material. The mixture
was removed from the heating bath, allowed to cool to room
temperature and extracted with Et20. This organic phase was
washed twice with H20, dried over MqSO" filtered and
concentrated in vacuo to obtain a crude solid. The solid was
collected, recrystallized from heptanelEt=O and dried in vacuo
(O.lmm) at 85'C to provide pure product, mp of 97-99'C,
homogeneous by TLC (silica, I:1 CH~OH:CHZC12, Rf=0.35). The IR
(CHC13) , NMR (CDCl3f and Mass Spectrum (M'=255, EI, 70eVy were
consistent with the structure. The yield was 3.3728 (I3.22mmo1,
54.19$) .
Elemental Analysis
Calculated Found
C 70.56 70.38
H 6.71 6.67
N 16.46 16.47
O 6.27
CA 02241845 1998-06-29
13
WO 97/25317 PCT/LTS96/19569
- 3-(4-Benzyl-1-piperazinyl)4,5-dihydronaphth(I,2-cjisoxazole
N -O N _.p
S / r ~ ) / I / N
~N
A stirred mixture of 3-chloro-4,5-dihydronaphth(1,2-
. cjisoxazole (2.0g, 9.75mmol), 1-benzylpiperazine (17m1,
IO 97.5mmo1) and KZCO~ !2.7g, 19.5mmo1) in 18m1 of N-
methylpyrrolidinone under NZ was lowered into an oil bath
preheated to 150'C. The mixture was heated while stirring under
N~ for 2 hours. At that time, TLC (CF~ZC1Z) showed no remaining
starting material. The mixture was removed from the heating
15 bath, allowed to cool to room temperature and extracted with
heptane. The organic phase was dried over MgSO" filtered and
ccncentrated_in vacuo to obtain a crude solid. The solid was
collected. titrated with Et20. recrystallized from EtZO and dried
in vacuo i0.lmm) at 85'C to provide pure product. mp of 164-
20 166'C, homogeneous by TLC (silica, 1:1 EtOAc, Rf=0.80J. The IR
(CFiCI~) , NMR (CDC1~) and Mass Spectrum (M'=345, EI, 70eV) were
consistent with the structure. The yield was 1.219g (3.53mmol,
36.240 .
CA 02241845 1998-06-29
14
WO 97/25317 PCT/US96/19569
Elemental Analysis
Calculated Found
C 76.49 76.49
H 6.71 ' 6.85
N 12.16 12.09
O 4.63
3-Hydroxy-8-methoxy-4,5-dihydronaphth[I,2-cjisoxazole
CH3 N'C~ CH; N--0
d ~ / A
l ~ ~
To a mechanically stirred mixture or 7-methoxy a-tetralone
oxime (5.0g, 26.1Bmmo1) in anhydrous THF (150m1) at 0°C under Nz
was slowly added n-butyl-lithium (n-HuLi) (23.0m1 of a 2.5M
solution in hexane, 57.60mmol). The mixture was stirred at 0°C
for 30 minutes, then CO~ gas was bubbled into the solution. (As
this addition progressed, a solid precipitate began to form).
After 15 minutes, COi addition was stopped and NZ flow was
restored. The thick mixture was stirred and warmed slowly to
room temperature for I'~ hours, then 6N HMSO, (150m1)
was slowly added which dissolved the solids. The TLC showed
traces of starting oxime and a mixture of desired product and an
CA 02241845 1998-06-29
WO 97/25317 PCT/US96/19569
intermediate which was not isolated. Stirring was continued for
- 4 hours at which time the intermediate was completely converted
to product. The mixture was extracted exhaustively with EtOAc.
The organic fractions were combined, washed once with HZO, once
5 with brine, dried over MgSO, and filtered. Concentration in
vacuo caused the precipitation of a solid which was collected,
titrated with EtOAc, and dried in vacuo to provide the product
as a solid, mp of I35-138'C, homogeneous by TLC [silica, 10:90
CH~OH:EtOAc, Rf=0.46] . The ZR (KHr) , NMR (DMSO-ds) and Mass
to Spectrum (M"=217, EZ, 70eV) were consistent with the structure.
The yield was 2.04968 tg.45mmo1, 36.080 .
Elemental Analysis
Calculated Found
15 C 66.35 66.02
H 5.10 5.03
N 6.45 6.22
O 22.10
EXAMPLE 8
3-Chloro-8-methoxy-4,5-dihydronaphth(1,2-c)isoxazole
CH3 N-~ CHI N-~
~ / H ~ / I / !
2 S ~ ( -a.
CA 02241845 1998-06-29
16
WO 97/25317 PCT/ITS96/t9569
To a stirred mixture of.3-hydroxy-8-methoxy-4,5-
dihydronaphth[1,2-cJisoxazole (lO.Og, 46.08mmo1) in phosphorus
oxychloride (I2.8m1, I37.3mmo1), triethylamine (6.42m1, ' ,
46.08mmol) was added dropwise. After completion of addition,
the mixture was heated to reflux while stirring. After 4 hours,
no starting material remained as shown by TLC [silica, EtOAc).
The mixture was cooled to room temperature, poured into 400m1
of ice water, and extracted with heptane. The organic extracts
were combined, dried over MgSO,. filtered and concentrated in
vacuo. Concentration of the filtrate in vacuo caused a solid !-o
precipitate. The solid was triturated with heptane and dried in
vacuo to provide the product as needles, mp of 55-57'C,
homogeneous by TLC [silica, CH~C12, Rf=0.45j . The IR (CHCl,) ,
NMR (CDC1~) and Mass Spectrum (M'=235, ET, 70eV) were consistent
with the structure. The yield was 7.758 t32.98mmo1, 71.57~s).
Elemental Analysis
Calculated Found
C 61.16 61.29
H 4.28 4.16
C1 15.04
N 5.94 5.90
O 13.58
CA 02241845 1998-06-29
17
WO 97/25317 PCT/US96/19569
EXAMPLE 9
- 3-(tI-Methyl-4-piperidinyl)oxyj-4,5-dihydronaphth(1,2-
c)isoxazole
. 5 ~CH3
N
N --a N --a
/ ~ / ~ /
HC 1
To a stirred solution mixture of 4-hydroxy-N-methyl
piperidine (S.OSg, 43.89mmo1) in 100m1 of N-methyipyrrolidinoae
under NZ was added NaH (1.758 of a 608 dispersion in oil,
43.89mmo1). The mixture was stirred at room temperature for 15
minutes, then a solution of 3-chloro-4,5-dihydronaphth(I,2-
c)isoxazole t3-Oq. I4.63mmo1) in 15m1 N-methylpyrrolidinone was
added in one portion. The stirred mixture was lowered into an
oil bath preheated to 150'C. After 20 minutes TLC (CFiZCI2J
showed no starting materials remaining. The mixture was removed
from the heating bath and allowed to cool to room temperature.
it was then partitioned between heptane/KzO. The heptane phase
was washed with water, dried aver MgSO" filtered and
concentrated in vacuo. This crude oil obtained was taken up in
EtZO, filtered, and the FiCl salt precipitated by the addition of
ethanoiic HC1. This salt was recrystallized from CFiiClz/EtZO to
CA 02241845 1998-06-29
18
WO 97/25317 PCT/US96/19569
provide the product as a solid, mp of 147-150'C, homogeneous by
- TLC [silica, 1:I CH,OH:EtOAc, Rfa0.02j. The IR (KBr), NHR
(CDC1~) and Mass Spectrum (M'+1=285, CI, methane) were consistent
with the structure. The yield was 1.2994g (4.05mmol, 36.090 .
Elemental Analysis
Calculated Found
C 63.65 63.55
H 6.60 6.63
C1 11.05
N 8.73 8.78
O 9.97
3-(I-Piperazinyl)-8-methoxy-4,5-dihydronaphth[1,2-cjisoxazole
CH3 N--a ~H3 ; -O
/ !
/ N
~" ~ \ _NH
\ \\
A stirred mixture of 3-chloro-8-methoxy-4,5-
dihydronaphth[1,2-cjisoxazole (2.0g, B.Slmmol), piperazine
(7.0g, 80.6mmo1) and KiCO, (2.4g, l7.lmmol) in 8.0m1 of N-
methylpyrrolidinone under NZwas lowered into an oil bath
preheated to 150'C. The mixture was heated while stirring under
NZ for 20 minutes. At that time TLC [CHZClZj showed no starting
CA 02241845 1998-06-29
19
WO 97/25317 PCT/US96/19569
material remai:~ed. The mixture was removed from the heating
- bath and allowed to cool to room temperature. Upon dilution of
the reaction mixture with HzO, a solid precipitated which was
collected and dried in vacuo to provide pure product, mp of 86-
88°C, homogeneous by TLC (silica. 1:I CH~OH:CH~Cil, Rf=0.37],
The IR (CHCl,) , NMR (CDC1,) and Mass Spectrum (M'=285, EI, 70eV~
were consistent with the structure. The yield was 1.9328
(6.78nnmol, 79.668) .
Elemental Analysis
Calculated Found
C 67.35 66.99
H 6.71 6.77
N 14.73 14.53
O 11.21
EXAMPLE 11
3-(I-Homopiperazinyl)-8-methoxy-4,5-dihydronaphth(1,2-
c]isoxazole
CH3 N-~ CH3 N--O
d , ~ ~ N~H
-~ W
A stirred mixture of 3-chloro-8-methoxy-4,5-
dihydronaphth[1,2-c]isoxazole (2.66q, II.32mmo1), homopiperazine
CA 02241845 1998-06-29
PCT/US96/19569
WO 97/25317 20
(11.40g, 113.2mo1) and KzCO~ (3.138, 22.68mmoi) in IO.Oml of N-
- methylpyrrolidinone under NZ was lowered into an oil bath
preheated to 150°C. The mixture was heated while stirring under '
Nz for 20 minutes. At that time, TLC (CHZC12) showed no starting
material remained. The mixture was removed from the heating
bath, allowed to cool to room temperature and diluted with H20,
which caused a solid to precipitate. The crude solid was dried,
recrystallized from EtZO and dried in vacvo (O,lmm) at 85°C, to
provide pure product, mp of 106-109°C, homogeneous by TLC
(silica, 1 : 1 CH~OFi:CH2ClZ, Rf=0.18] . The IR (CHC1~) . NP~iR (CDC1,)
and Mas3 Spectrum (M'=299, EI, 70e~) were consistent with the
structure. The yield was 1.79488 (6.OOmmol, 53.03$).
Elemental Analyai3
Calculated Found
C 68.21 68.24
H 7.07 7.11
N 14.04 14.00
O 10.69
~s
CA 02241845 1998-06-29
2 I.
WO 97/25317 PCT/US96119569
EXAMPLE 12
-3-(1-(4-(p-Chlarophenyl)-4-hydroxy-piperidinyl)-8-methoxy-4,5-
- dihydrvnaphth(1,2-c)isoxazole
° C H3 N --~ C H3 N -<3
~ ~ z ~ , ~ ~ N off
--- \ I
A stirred mixture of 3-chloro-8-methoxy-4,5-
dihydronaphth(1.2-c)isoxazole (2.0g, 8.51mmo1), 4-(p-
chlorophenyl)-4-hydroxy-dipiperidine (3.6g, 17.02mo1) and KZCO,
(2.358, 17.02mmo1) in 6m1 of N-methylpyrrolidinone under NZwaa
lowered into an oil bath preheated to 150'C. The mixture was
heated while stirring under NZ for 1 hour. At that time, TLC
(CHZC12] showed no remaining starting material. The mixture was
removed from the heating bath and allowed to cool to room
temperature. Upon dilution of the reaction mixture with H2o, a
solid precipitated which was recrystallized from EtOAc and dried
in vacuo (O.Imm) at 85'C to provide pure product, mp of 174-
177'C, homogeneous by TLC [silica, 2:1 heptane:EtOAc, Rf=0.263].
Z0 The IR (CFIC1~}, I~MR tCDCl~) and t~iass Spectrum (M'=410, E.I., 70eV)
were consistent with the structure. The yield was 2.37988
(5.60mmo1, 68.20%).
CA 02241845 1998-06-29
22
WO 97/25317 PCT/US96/19569
Elemental Analysis
Calculated Found
C 67.23 67.24 '
H 5.64 5.75
C1 8.63
N 6.82 8.78
O 11 . 68
3-[(endo)-8-Methyl-8-azabicyclo(3.2.I]oct-3-yl)oxy]-8-methoxy-
4,5-dihydronaphth(1,2-c]isoxazole
C H3 N --0 C H3 N ---~
/. / / I
N ~C H3
To a stirred mixture of tropine (5.418, 38.31mmo1) in i0 ml
of (THF) under Nz at 0'C was slo~rly added n-BuLi tl5.Om1 of a
2.5M solution in hexanes. 38.31mmo1?. The mixture was stirred
for I5 minutes while allowed to warm to room temperature, then a
solution of 3-chloro-8-methoxy-4,5-dihydronaphth[1,2-c]isoxazole
(3.0g, 12.76mmo1) in 30m1 N-methylpyrrolidinone was added in one
portion. The internal temperature increased to 99-100'C and was
maintained there. After 3 hours, TLC (CHZClz] showed no starting
material remaining. The mixture was removed from the heating
bath and allowed to cool to room temperature. It was then
CA 02241845 1998-06-29
23
WO 97/25317 PCT/LJS96/19569
partitioned between heptane/HZO. The heptane phase was washed
- with HiO, dried over MgSO" filtered and concentrated in vacuo,
whereupon it solidified. This crude solid was recrystallized
from a minimum of heptane and dried in vacuo to provide the
product as a solid, mp of 102-104'C,~homogeneous by TLC [silica,
1:1 CH~OH:CHZClz, Rf$0.20] . The IR (CHC1~) , HI~iR (CBCll) and Mass
Spectrum (M'+341, CI, methane) were consistent with the
structure. The yield was 1.37298 (4.038mmo1, 31.640 .
Elemental Analysis
Calculated Found
C 70.57 70.47
H 7.I1 7.25
., ,
ca a . c .7 t3 . I 4
O 14.10
EXAMPLE 14
3-C(endo-8-Methyl-8-azabicyclo[3.2.1)oct-3-yl)oxy]-4,5-
dihydronaphth[1,2-c]isoxazole hydrochloride hemihydrate
N--~ N-0
~ / I ~ / / ,
F~IC1 ~ ~N~CH3
o-s H20
To a stirred mixture of tropine (4.48, 3I.16mmo1) in IOml
CA 02241845 1998-06-29
PCT/US96/19569
WO 97/25317 24
of
THF under NZ at 0°C was slowly added n-HuLi (12.47m1 of a 2.5M
solution in hexanes, 31.16mmo1). The mixture was stirred for 15
minutes while allowed to warm to room temperature, then a
solution of 3-chloro-4.5-dihydronaphth[1,2-cJisoxazole (2.138,
10.39mmo1) in 30m1 N-methylpyrrolidinone was added in one
portion. The stirred mixture was lowered izito an oil bath
preheated to 150'C. The internal temperature increased to 85'C
and was maintained there. After 3 hours, TLC [CHZC12J showed no
remaining starting material. The mixture was removed from the
heating bath and allowed to cool to room temperature. It was.
then partitioned between heptane/H~O. The heptane phase was
washed with HZO, dried over MgSO" filtered and concentrated in
vacuo, to provide the free base as an oil, which resisted
attempts at crystallization. The oil was taken up in Et20 and
the HC1 salt was precipitated by the addition of ethanolic HC1.
This crude solid was recrystallized from EtZO/CH2Clz and dried
in vacuo at 85'C to provide the product as a solid, mp o8 167-
170'C, (darkens at ca. 150°C) homogeneous by TLC [silica, 1:1
CH30H:CHiCIZ, R~=0.14] . The IR (CHCl~), NMR (CDC13) and Mass
Spectrum (M'+1=311, CI, methane) were consistent with the
structure. Analysis and NMR confirmed the hemihydrate
structure. The yield was 1.2688 (3.563mmol, 34.29$).
CA 02241845 1998-06-29
WO 97/25317 PCTlLJS96/19569
Elemental Analysis
Calculated Found
C 64.12 64.25
H 6.80 6,77
C1
N 7.87 7.70
O 9.23
3-tl-(4-(6-Fluorobenzisoxazol-3-yl)-piperidinyl)-e-methoxy-4,5-
dihydronaphth[1,2-c)isoxazole
CHj N -0 CH3 N -O
i ;
I - I rr-o
r
/ \
A stirred mixture of 3-chloro-8-methoxy-4,5
dihydronaphth[1,2-c]isvxazole (2.0g, B.Slmmol), 4-(6
F
fluorobenzisoxazol-3-yl)-piperidine (2.8g, 12.76mmo1) and KZCO,
(2.35g, 17.02mmo1) in IOml of N-methylpyrzolidinone under NZwas
lowered into an oii bath preheated to I50'C. The mixture was
heated while stirring under N2 for 90 minutes. At that time TLC
iCH2Clz) showed no remaining starting material. The mixture was
removed from the heating bath and allowed to cool to room
CA 02241845 1998-06-29
WO 97/25317 26 PCT/LJS96/19569
temperature. Upon dilution of the reaction mixture with H,C, a .
-solid precipitated which was collected, dried, dissolved in
CHzCl2 and filtered through neutral alumina. The fractions
containing desired product were combined and concentrated, and
S the resultant solid obtained was triturated with EtzO to provide
a solid, mp of 18I-183'C, homogeneous by TLC [silica, 2:1
Heptane:EtOAc, Rf=O.15J. The IR (CHClj), NMR (CDC1~) and Mass
Spectrum (M'=419, EI, 70eV) were consistent with the structure.
The yield was 1.13188 (2.70mmol, 31.700 .
Elemental Analysis
Calculated Found
C 68.72 68.47
H 5.29 5.28
F 4.53
N 10.02 9.97
O 11.44
CA 02241845 1998-06-29
WO 97/25317 2 ~ PCT/LJS96/19569
rvwuor c l c
3-(1-(4-2-Oxo-1-benzimidazoiinyl)piperidinyl))-B-methoxy-4,5-
- dihydronaphth(1,2-cjisoxazole
CH3 N--~ CHI N-~
0
t a ~ ~ /
-" \ I N NH
A stirred mixture of 3-chloro-8-methoxy-4,5-
dihydronaphth(I.2-c]isoxazole t2.57g, 10.9mmo1), 4-t2-oxo-1-
benzimidazolinyl)piperidine t4.74g, 21.8mmo1) and KZCO~ (3.028,
21.8mmo1) in I2m1 of N-methylpyrrolidinone under Nz was lowered
into an oil bath preheated to I50'C. The mixture was heated
I5 while stirring under NZ for 4 hours. At that time, TLC tCH2C12)
showed no remaining starting material. The mixture was removed
from the heating bath and allowed to cool to room temperature.
Upon dilution of the reaction mixture with H20. a solid
precipitated which was collected, dried, dissolved in CHZC12 and
20~ filtered through neutral alumina using CHZClz and then 1:1
CH2CIz:EtzO. The fractions containing desired product were
combined and concentrated, and the resultant solid obtained was
triturated with EtOAc and dried in vacuo (O.lamn Hg, 85'C) to
provide a solid, mp of 211-214'C. homogeneous by TLC [silica,
CA 02241845 1998-06-29
WO 97!25317 2$ PCT/US96l19569
EtOAc, Rf=0.38]. The IR (CHC1,), NMR iCDCl,) and Mass Spectrum .
(M'=416, EI, 70eV) were consistent with the structure. Tr.e yield
was 1.6028 (3.85mmo1, 33.33$).
Elemental Analysis
Calculated Found
C 69.2I 68.88
H 5.8I 5.90
N 13.45 13.14
O 11.52
EXAHPLE I7
3-[(Quinuclidin-3-yl)oxy]-8-methoxy-4,5-dihydronaphth[1,2-
c)isoxazole hydrochloride
CH3 N--p CH3 N--~ L~-~l
/ i ~ , ~
1 --- ~ i
H-m
To a stirred mixture of 3-quinuclidinol (4.878, 38.28mmo1)
in IOml of THF under NZ at 0'C was slowly added n-BuLi (15.328 0~
a 2.5M solution in hexanes, 38.28mmo1). The mixture was stirred
for 10 minutes while allowing to warm to room temperature, then
a solution of 3-chloro-e-methoxy-4,5-dihydronaphth(1,2-
CA 02241845 1998-06-29
PCT/US96119569
WO 97/25317 29
c]isoxazole (3.0g, i2.76mmo1) in 30m1 N-methyipyrrolidinone was
added in one portion. The stirred mixture was lowered into an
oil bath preheated to 150°C. The internal temperature increased
to 85°C and was maintained there. After 3 hours, TLC (CHZClZ]
showed no remaining starting material. The mixture was removed
from the heating bath and allowed to cool to room temperature.
It was then partitioned between heptane/HZO. The heptane phase
was dried over MgSO" filtered and concentrated in vacuo to
provide the free base as an oil. The oil was taken up in Et20
and the HC1 salt was precipitated by the addition of ethanolic
HC1. This solid was collected and dried in vacuo (O.Imm Hg,
85°C) to provide the product as a solid, mp o~ 133-136°C,
homogeneous by TLC (silica, 1:1 CH~OH:CH2C12, Rf=0.23]. The IR
(KBr), NMR (DMSO-d,) and Mass Spectrum (M'+1=326, EI, 70eV) were
consistent with the structure. The yield was 0.965g (2.39mmol,
18.79$).
Elemental Analysis
Calculated Found
C 62.89 62.91
H 6.39 6.28
C1 9.77
N 7.72 7.51
O 13.23
CA 02241845 1998-06-29
WO 97/25317 PCT/US96/19569
r v." "., . .. . ..
_ 5,6-Dihydro-4H-benzo[6,7]cyclohept[I,2-c]isoxazol-3-01
,OH O
N
5 ' . E ~ H
To a mechanically-stirred mixture of I-benzosuberone oxime
(lO.Og, 57.1mmo1) in anhydrous THF (200m1) at 0'C under N2 was
iQ slowly added n-BuLi (50.3m1 of a 2.5M solution in hexane,
125.62mmo1). The mixture was stirred at 0'C for 30 minutes,
then COz gas was bubbled into the solution. After 15 minutes,
COZ addition was stopped and N2 flow was restored. The thick
mixture was stirred and warmed slowly to room temperature for 1'-~
15 hours, then 6N HZSO, (220m1) was slowly added, which dissolved
the solids. Stirring way continued for 18 hours, at which time
the TLC [EtOAc] showed a mixture of starting oxime and product
(starting oxime was beat visualized using 2:1 heptane:EtOAc
eluent). The mixture was poured into a separatory funnel, and
2a the organic phase drawn oft. The aqueous phase was extracted
with EtQAc, and the organic phase and the EtOAc extracts were
combined, washed with HZO, dried over MgSO, and filtered.
Concentration in vacuo caused the precipitation of a solid which
was collected and dried in vacuo to provide the product as a
CA 02241845 1998-06-29
31
WO 97/25317 PCT/US96/19569
solid, mp of 165-168'C, homogeneous by TLC (silica, EtzO,
-Rf~0.28] . The IR (KBr) , NMR (DMSO-d6) and Mass Spectrum (M'=201,
EI, 70 eV) were consistent with the structure. The yield was
3.03248 (15.09mmo1, 26.42$).
Elemental Analysis
Calculated Found
C 71.63 71.45
H 5.51 5.50
IO N 6.96 6.91
O 15.90
EXAHPLE 19
3-(1-(4-(2-Oxo-I-benzimidazolinyl)piperidinyl))-4,5-
dihydronaphth(I,2-c]isoxazole hemihydrate
N~ N-0 O
/ ~ / 1 / ~ / N
-'~' \ ~ N NH
o.s HZO
A stirred mixture of 3-chloro-4,5-dihydronaphth(I,2-
c]isoxazole (3.18, 15.12mmo1), 4-(2-oxo-I-benzimidazolinyl)-
piperidine (8.28, 37.8mmo11 and KZCO, (4.28, 30.24mmo1) in I9ml
of N-methylpyrrolidinone under N=was lowered into an oil bath
CA 02241845 1998-06-29
PCT!l1JS96/19569
WO 97/25317 32
preheated to 150'C. The mixtuze was heated while stirring under
_N2 far 90 minutes. At that time, TLC tCHzClz) showed no
remaining starting material. The mixture was removed from the -
heating bath and allowed to cool to room temperature. Upon
dilution of the reaction mixture with H20, a solid precipitated
which was collected, dried, dissolved in CHZC12 and filtered
through neutral alumina using CHZCI~ and then I:1 CHZCI~:Et20.
The fractions containing desired product were combined and
concentrated, and the solid obtained was recrystallized from
l0 EtOAc and dried in vacuo (O.lmm Hg, lI0'C) to provide a solid,
mp of 229-233°C, homogeneous by TLC [silica, EtOAc, Rf=0.54].
The IR (KHr), NMA (CDC13) and Mass Spectrum (M'=386, EI, 70eV)
were consistent with the structure. Analysis and NMR confirmed
a hemihydrate structure. The yield was 1.I03g t2.79mmol,
18.45$).
Elemental Analysis
Calculated Found
C 69.82 70.25
H 5.86 5.64
N 14.17 14.22
O 8.28
CA 02241845 1998-06-29
PCT/CTS96/19569
WO 97/25317 33
Preferred pharmaceutically acceptable addition salts
-include salts of inorganic acids such as hydrochloric,
hydrobromic, sulfuric, nitric, phosphoric and perchloric acids;
as well as organic acids such as tartaric, citric, acetic,
succinic. malefic, fumaric, and oxalic acids.
The active compounds of the present invention may be
administered orally, for example, with an inert diluent or with
an edible carrier. They may be enclosed in gelatin capsules ar
compressed into tablets. For the purpose of oral therapeutic
administration, the compounds may be incorporated with
excipients and used in the form of tablets, troches, capsules,
elixirs. suspensions, syrups, waters, chewing gums and the like.
These preparations should 'contain or form at least 0.5~ of
ZS active compound, but may be varied depending upon the particular
form and may conveniently be from about 4 to about 75~ of the
weight of the unit. The amount of compound present in such
composition is such that a suitable dosage of active compound
will be obtained_ Preferred compositions and preparations
2o according to the present invention are prepared so that an oral
dosage unit form contains from about I.0 to about 300mqs of
active compound.
The tablets, pills. capsules, troches and the like may also
25 contain the following ingredients: a binder such as
CA 02241845 1998-06-29
WO 97/25317 34 ~'CT/US96/19569
microcrystalline cellulose, gum tragacanth or gelatin; an
-excipient such as starch or lactose, a disintegrating agent such
as alginic acid. Primogel'°t, corn starch and the like; a '
lubricant such as magnesium stearate or Sterotex~; a glidant
such as colloidal silicon dioxide: and a sweetening agent such
as sucrose or saccharin or a flavoring agent such as peppermint,
methyl salicylate, or orange flavoring may be added. When the
dosage unit form is a capsule, it may contain, in addition to
materials of the above type. a liquid carrier such as fatty oil.
Other dosage unit forms may contain other various materials
which modify the physical form of the dosage unit, for example,
as coatings. Thus tablets or pills may be coated with sugar,
shellac, or other enteric coating agents. A syrup may contain,
in addition to the active compounds, sucrose as a sweetening
agent and certain preservatives, dyes and colorings an flavors.
Materials used in preparing these various compositions should be
pharmaceutically pure and non-toxic in the amounts used.
For the purpose of parenteral therapeutic administration,
the active compounds of the invention may be incorporated into a
solution or suspension. These preparations should contain at
least 0.1~ of the aforesaid compound, but may be varied from
about 0.5 to about 30~ of the weight thereof. The amount of
compound in such composition is such that a suitable dosage of '
active compound will be obtained. Preferred compositions and
CA 02241845 1998-06-29
WO 9'7/25317 35 PCT/US96119569
preparations according to tre invention are prepared so that a
-parenteral dosage unit contains from about 0.5 to about 100mgs
- of active compound.
The solutions or suspensions may'also include the following
components; a sterile diluent such as water for injection,
saline solution, fixed oils, polyethylene glycols, glycerine,
propylene glycol or other synthetic solvents; antibacterial
agents such as benzyl alcohol or methyl parabens: antioxidants
l0 such a3 ascorbic acid or sodium bisulfite; chelating agents such
as EDTA; buffers such as acetates, citrates or phosphates and
agents for the adjustment of tonicity such as sodium chloride or
dextrose. The parenteral preparation can be enclosed in
ampules, disposable syringes or multiple done vials made of
glass or plastic.
The compounds of the invention may be useful as 5-HT,
antagonists on the coronary chemoreflex for the treatment of
anxiety, psychiatric disorders, nausea and vomiting by virtue of
their ability to bind to rat entorhinal cortex membranes.
'H-GR 65630 Binding to Rat Entorhinal Cortex Membranes
Studies have been performed to determine the affinity of
the compounds of the invention for the SHT, binding site in the
brain. This study or assay may be useful for predicting the
CA 02241845 1998-06-29
WO 97/25317 36 PCT/US96/I9569
potential of compounds to exhibit antiemetic, anxiolytic or
_atypical antipsychotic profiles.
Originally, it was believed that SHT~ binding sites existed
only in the periphery. However, with the recent introduction of
potent and selective SHT, antagonist drugs such as GR65630,
Zacopride, ICS 205 930 and MDL 72222 (Bemesetron, CtSHt,C12N02) ,
data from binding studies have indicated that SHT, binding sites
are also located in selected areas of the brain. The highest
levels of SHT~ binding sites have been detected in limbic and
dopamine containing brain areas tentorhinal cortex, amygdala,
nucleus accumbens and tubezculum olfactorium) (Kilpatrick, G.J.
et a1. Identification and distribution of SHT~ receptors in rat
brain using radioliqand binding. Nstur~ 330: 746-748). Besides
possessing selective binding in dopamine rich areas, SHT,
antagonists have been reported to block behavioral effects
associated with certain drugs of abuse tnicotine and morphine?
and to be active in behavioral tests predictive of anxiolytic
activity. Based vn these selective regional binding results and
behavioral studies, SHT; antagonists may have a therapeutic
benefit in disease states believed to be associated with
excessive dopaminergic activity, i.e., schizophrenia. anxiety
and drug abuse_
CA 02241845 1998-06-29
PCT/US96119569
WO 97/25317 3 7
In accardance with the above-discussed assay, a 0.05M of
_Krebs-Hepes buffer, pH 7.4 was prepared as follows:
11.928 Hepes
10.528 NaCl
0.3738 KCl
0.2778 CaCl2~
0.2448 MgC12.6Hz0
q.s. to 1 liter with distilled HZO,
bring pH up to 7.4 (at 4~C) with 5N NaOH
['H]-GR65630 (87.OCi/mmoi) was obtained from New England
Nuclear. For .ICSa determinations: ['H]-GR65630 was made up to a
concentration of l.OnM in Krebs-Hepes buffer such that when
1001 is added to each tube. a final concentration of 0.4nM is
attained in the 250t,~1 assay.
GR38032F was obtained from Research Biochemical Inc.
GR38032F was made up to a concentration of 500E~H in Krebs-Hepes
a0 buffer. SO~cl of Krebs-Hepes were added to each of 3 tubes for
determination of nonspecific binding (yields a final
concentration of 100E,~H in the 2501.c1 assay? .
For most assays, a 50~s1 stock solution was prepared in a
suitable solvent and serially diluted with Krebs-Hepes buffer
CA 02241845 1998-06-29
38
WO 97J25317 PCT/US96/i9569
such that when 501 of drug is combined with the total 2501
_ assay, a final concentration from 10's to 10'9M was attained.
Characteristically, seven concentrations may be used for each
assay; however, higher or lower concentrations may be used,
depending on the potency of the drug.
During tissue preparation, Male Wistar rats (I5-200g) were
decapitated, the entorhinal cortex removed, weighed and
homogenized in IO volumes of ice cold O.OSM Krebs-Hepes buffer,
IO pH 7.4. The homogenate is centrifuged at 48,000q for 15 minutes
at 4'C. The resulting pellet was rehomogenized in fresh Kreba-
Hepes buffer and recentrifuged at 48,000q for I5 minutes at 4'C.
The final pellet was resuspended in the original volume of ice-
cold Krebs-Hepes buffer. This yielded a final tissue
concentration of I.2 tv l.6mg/ml with the addition of 100u1 to
the assay. Specific binding was approximately 55 to 65$ of the
total bound ligand.
In conducting the assay, the followinq volumes were utilized:
100E,c1 of Tissue suspension;
IOOul of ['HJ-GR65630; and
501 SOOM GR38032F (Vehicle for binding)
or appropriate drug concentration
i~I
CA 02241845 2001-04-20
39
Sample tubes were kept on ice for additions, then vortexed and
-incubated with continuous shaking for 30 minutes at 37a~, At the
end of the incubation period, the incubate is diluted witty S ml
of ice-cold Krebs-Hepes buffer and immediately vacuum filtered
through G~lhatmanTM GF/B filters, followed by two 5m1 washes with'
ice-cold Krebes-Hepes buffer. The filters are dried and counted
in 10 ml of liquid scintillation cocktail. Specific GR 55630
binding is defined as the difference between the total binding
and that bound in the presence of 100E.c,H GR38032F. ICsa values
14 were derived from computer-derived log-prabit analysis.
Various compounds of the invention wEare subjected to the
above-described assay and the results the affinity for 5 HT,
receptors are reported in Table I, below.
~5
TABLE I
Affinity for 5-HT~ Receptor-Displacement of 'H-GR 65630
Compound ICS, ~.
,-
Ex. 3 0.868
Ex. 4 0.083
Ex. S 0.056
CA 02241845 2001-04-20
Ondansetron 0.089
(standard)
ICS 205 930 0.039
(standard)
5
Measurement of 5 HT, Antagonist Effects in the Bezold-Jarisch
Assay
This assay evaluates the effect of these compounds as 5-HT,
to antagonists. They were examined in this aissay on the coronary
chemoreflex (Bezold-Jarisch) initiated by 5-HT~ in vivo and
characterized by leading inhibition of sympathetic outflow and
increased activity of the cardiac vagus, Leading. to profound
bradycardia and hypotension. The values «btained allow for
15 continuous monitoring of arterial pressure and heart rate
responses by these compounds over an extended period of time to
determine their effecicy for 5 HT~ antagonism.
The catheters were prepared from TygonTM tubing (45cm length,
20 0.05mm, ID) bonded to TeflonTM tubing (0.38mm, ID). The
mechanical bonding was achieved by insertion of the TeflonTM
tubing (5mm) into the dilated (ethylene dichloride, 3-4 min.)
tip of the TygonTM tubing. The junction was then sealed with
vinyl glue, the catheters were soaked in cold sterilization
25 solution (Amerse instrument germicide) an,d flushed thoroughly
CA 02241845 1998-06-29
PCT/US96/19569
WO 97/25317 41
with saline prior to implantation.
Long Evans rats were anesthetized with sodium pentobarbital
(50mq/kg, ip). The catheters filled with hepranized saline (100
U/ml) were inserted in the left femoral artery and vein and
passed into the abdominal aorta and inferior vena cava,
respectively. The catheters were then sutured to the underlying
muscle and the free ends were passed subcutaneously and
exteriorized through an incision on the top of the skull. The
l0 catheters were then secured to the skin with sutures,
nitrafurazone powder was dusted aver the area of the incision
and the incision was closed using 3-O silk sutures. The
catheters were flushed with saline and sealed with metal
obturators. Patentcy of the two catheters was maintained by
daily flushing with hepranized saline (0.2 ml of 100 U/ml). The
rata were given 48 hours recovery prior to obtaining
cardiovascular data.
In the anesthetized rat model the catheters were not
exteriorized, data was collected acutely under the influence of
general anesthesia.
The baseline data Arterial Blood Pressure(mm Hg,
' systalic/diastolic) and Heart Rate (beats/min) were recorded and
the rats were injected with 5-HT (3-7.5ug/kg, iv). The
CA 02241845 1998-06-29
42
WO 97/25317 PCTlUS96/19569
individual response to the 5-EiT intervention was determined and
the compound was then administered singlely or in an ascending
dose range. The rata were challenged with 5-HT again at '
intervals postdosing and the peak response was recorded.
Several compounds o~ the invention were tented according to
the above-described assay and the results are reported in Table
Ii, below.
CA 02241845 1998-06-29
WO 97/25317 43 PCT/US96/19569
TABLE II
-Inhibitory Potency of 5-HT~ Antagonists on Reflex Hradycardia
Induced by Intravenous 5-HT3 in the Anesthetized Long-Evans Rat
Compound Dose, mg/kg, ip ~S Inhibition of
Bezold-Jarisch
Reflex (Values are
mean SEH, 2-3
rats/dose!
Ondansetron 3.0 57.3 t 9.7
Ondansetron 10.0 94.6 t 2.7
Ex. 4 0.03 58.6 t 16.4
Ex. 4 0.05 83.3 t 8.2
Ex. 4 O.IO 93.0 t 1.0
Ex. 5 1.0 55.6 t 9.7
Ex. 5 3.0 89.3 t 2.9
In accordance with Table II, maximal reductions in heart rate
induced by SFiT, (e.g. Bezold-Jarisch reflex) occurred 15 to 60
minutes after administration.