Note: Descriptions are shown in the official language in which they were submitted.
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
ASP~RTYTl PROTF.~.~F. INHIBITO~S
T~CHNIC ~ FI~T.n OF TH~ IN~r~TION
The present invention relates to a novel
class of compounds which are aspartyl protease
inhibitors. In one embodiment, this invention relates
to a novel class of HIV aspartyl protease inhibitors
characterized by specific structural and
physicochemical features. This invention also relates
to pharmaceutical compositions comprising these
compounds. The compounds and pharmaceutical
compositions of this invention are particularly well
suited for inhibiting HIV-1 and HIV-2 protease activity
and consequently, may be advantageously used as anti-
viral agents against the HIV-1 and HIV-2 viruses. This
invention also relates to methods for inhibiting
aspartyl protease activity, methods for treating viral
infections using the compounds and compositions of this
invention, and methods for making intermediates and
compounds of this invention.
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
R~CKGROlnND OF TH~ IN~r~TIO~
- The human immunodeficiency virus ~"HIV") is
the causative agent for acquired immunodeficiency
syndrome ("AIDS") -- a disease characterized by the
destruction of the immune system, particularly of CD4
T-cells, wit~ attendant susceptibility to opportunistic
infections -- and its precursor AIDS-related complex
(I'ARC") -- a syndrome characterized by symptoms such as
persistent generalized lymphadenopathy, fever and
weight loss.
As in the case of several other retroviruses,
HIV encodes the production of a protease which carries
out post-translational cleavage of precursor
polypeptides in a process necessary for the formation
of infectious virions (S. Crawford et al., "A Deletion
Mutation in the 5' Part of the pol Gene of Moloney
Murine Leukemia Virus Blocks Proteolytic Processing of
the gag and pol Polyproteins", J. Virol., 53, p. 899
(1985)). These gene products include ~Ql, which
encodes the virion RNA-dependent DNA polymerase
(reverse transcriptase), an endonuclease, HIV protease,
and ~, which encodes the core-proteins of the virion
(H. Toh et al., "Close Structural Resemblance Between
Putative Polymerase of a Drosophila Transposable
Genetic Element 17.6 and pol gene product of Moloney
Murine Leukemia Virus", F.MRO J., 4, p. 1267 (1985);
L.H. Pearl et al., "A Structural Model for the
Retroviral Proteases", N~tl1re, pp. 329-351 (1987); M.D.
Power et al., "Nucleotide Sequence of SRV-1, a Type D
Simian Acquired Immune Deficiency Syndrome Retrovirus",
Science, 231, p. 1567 (1986)).
A number of synthetic anti-viral agents have
been designed to target various stages in the
CA 02243121 1998-07-14
W O 97/27180 PCT~US97101610
-- 3
repllcation cycle of HIV. These agents include
compounds which block viral binding to C~4 T-
lymphocytes (for example, soluble CD4), and compounds
which interfere with viral replication by inhibiting
viral reverse transcriptase ~for example, didanosine
and zidovudine (AZT)) and inhibit integration of viral
DNA into cellular DNA (M.S. Hirsh and R.T. D'Aqulia,
"Therapy for Human Tm~lln~de~iciency Virus Infection",
N.~ng.J.Med., 328, p. 1686 (1993)). However, such
agents, which are directed primarily to early stages of
viral replication, do not prevent the production of
~infectious virions in chronically infected cells.
Furthermore, administration of some of these agents in
effective amounts has led to cell-toxicity and unwanted
side effects, such as anemia and bone marrow
suppression.
More recently, drug design efforts have been
directed toward creating compounds which inhibit the
formation of infectious virions by interfering with the
processing of viral polyprotein precursors. Processing
of these precursor proteins requires the action of
virus-encoded proteases which are essential for
replication (Kohl, N.E. et al. "Active HIV Protease is
Required for Viral Infectivity" Proc. ~atl. Acad. Sci.
~, 85, p. 4686 (1988)). The anti-viral potential of
HIV protease inhibition has been demonstrated using
peptidal inhibitors. Such peptidal compounds, however,
are typically large and complex molecules that tend to
exhibit poor bioavailability and are not generally
consistent with oral administration. Accordingly, the
need still exists for compounds that can effectively
inhibit the action of viral proteases, for use as
agents for preventing and treating chronic and acute
viral infections. Such agents would be expected to act
CA 02243121 1998-07-14
PCT/US97/01610 ~O~IUS ~ PA~T~.-R
Vertex Pharmaceuticals Inc., et alFAT_~lA~ E
Our Ref.: B 2555 PCT ~I~BE~T~TR.~ . .
81675 ~N~H~
- 4 ~ ~ 9g8
as effective therapeutic agents in their own right. In
addition, since they act at a separate stage in the
virus life cycle from previously described
antiretroviral agents, the administration of a
combination of agents would be expected to result in
increased therapeutic efficacy.
International Publication WO 94/19329
discloses cyclic carbonyls and derivatives thereof as
protease inhibitors. International Publication Wo
95/24385 discloses sulfonamide protease inhibitors.
SUMMARY OF TH~ ~NVENTION
The present invention provides a novel class
of compounds, and pharmaceutically acceptable
derivatives thereof, that are useful as inhibitors of
aspartyl proteases, and in particular, HIV aspartyl
protease. The compounds of this invention can be used
alone or in combination with other therapeutic or
prophylactic agents, such as anti-virals, antibiotics,
immunomodulators or vaccines, for the treatment or
prophylaxis of viral infection.
According to a preferred embodiment, the
compounds of this invention are capable of inhibiting
HIV viral replication in human CD4 cells including T-
cells, monocytic lines including macrophages and
dendrocytes and other permissive cells. These
compounds are useful as therapeutic and prophylactic
agents to treat or prevent infection by HIV-l and
related viruses which may result in asymptomatic
infection, AIDS-related complex ('rARC"), acquired
immunodeficlency syndrome ("AIDS"), or similar disease
of the immune system.
I~ is a principal object of this invention to
provide a novel class of compounds that are aspartyl
~MENDED SHE~
CA 02243121 1998-07-14
~ ,; .;;, ' '
-- 5
protease lnhibitors, and particularly, HIV aspartyl
protease inhibitors. This novel class of compounds is
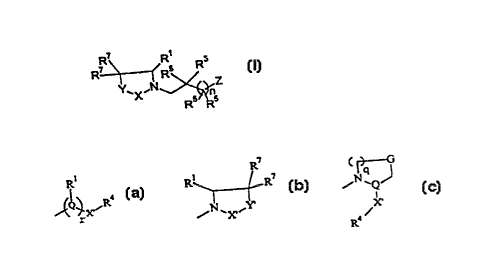
represented by formula I:
Y'X-~
( I )
wherein
each Z is
~Q~ ,R or ~ ~ or ~ N~Q J
wherein any Z may be optionally fused with R6;
each X and X' is independently selected from the
group consisting of -C(O)-, -C(O)C(O)-, -S(O)- and
-S(O)2;
eac .- and Y' is independently selected from the
group consisting of -(C(R )2)p-~ -NR2-, -(C(R )2)p-M-,
>C=C(R2)2, and -N(R2)-CH2-;
each R is independently selected from the group
consisting of hydrogen; R6; C1-C6 alkyl; C2-C6 alkenyl;
C2-C6 alkynyl; C3-C6 cycloalkyl optionally fused with
R6; C5-C6 cycloalkenyl optionally fused with R6; and
where R1's are attached to adjacent atoms, the R1's
together with their attached adjacent atoms form a
carbocyclic or heterocyclic ring system which may be
optionally fused with R6; where any member of R1 may be
optionally substituted by one or more R ;
~MEND~D SHE~T
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
each R2 is independently selected from hydrogen;
~ R ; C1-C6 alkyl; C2-C6 alkenyl; C2-C6 alkynyl; C3-C6
cycloalkyl optionally fused with R6; C5-C6 cycloalkenyl
optionally fused with R6; and where two R 's are s
attached to the same geminal atom, the R 's together
with their attached geminal atom may form a
spirocarbocycllc or spiroheterocyclic ring system;
where any member of R may be optionally substituted by
one or more R3;
each R is independently selected from oxo, OR9,.
N(R )2~ N(R )-X-R ,N(R )-X-OR , N(R )-X-N(R )2~ SR , X-
R , O-X-N(R )2~ C(O)N(R )2~ halogen, NO2, CN, COOR and
R6;
each R is independently selected from from the
group consisting of OR9; N(R9)2; X-R9; C(O)N(R9)2; R ;
C1-C6 alkyl; C2-C4 alkenyl; C3-C6 cycloalkyl optionally
fused with R6; C5-C~ cycloalkenyl optionally fused with
R ; where any member of R4 may be optionally
substituted by one or more groups independently
selected from the group consisting of R and R ;
each R is independently selected from the group
consisting of H, OH, O and R ;
each R is independently selected from the group
consisting of aryl, carbocyclyl and heterocyclyl,
wherein said aryl, carbocyclyl or heterocyclyl may be
optionally substituted with one or more groups selected
from the group consisting of oxo, -OR9, -R9, -N(R9)(R9),
-N(R )-X-R , SR , -X-R , -O-X-N(R )2~ -R -OR , -CN,
-CO2R , -X-N(R9)(R ), halogen, -NO2, and -CF3;
each R is independently selected from the group
consisting of hydrogen, OH and O;
each R is independently selected from the group
con~sisting of hydrogen, alkyl, alkenyl, alkynyl, aryl,
carbocyclyl, and heterocyclyl;
CA 02243121 1998-07-14
W O 97/27180 PCTnUS97/01610
each R is independently selected from the group
consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl,
carbocyclyl, heterocyclyl, aralkyl, carbocyclylalkyl
and heterocyclylalkyl wherein any aryl, carbocyclyl or
heterocyclyl may be optionally fused with R and
wherein any member of R may be optionally substituted
by one or more groups independently selected from the
group consisting of -OR , -N(R )2~ -CN, -NOz, -X-R , -X-
N(R8)2 -C(O~OR8, -N(R8)-XNR8, and halogen;
each Q is independently selected from CH and N;
each M is independently selected from the group
consisting of NH, -NR -, -O-, -S-, -S(O)- and -S(O)2-;
each n is 1 or 2;
each r is 0,1 or 2;
each p is independently 1 or 2;
each q is independently 1, 2 or 3; and
each G is independently selected from the group
consisting of -NH-, -NR -, -O-, -S-, -S(O)-, S(O)2,
-C(o)-, and -C(R2)2-.
An alternate object of this invention is a
novel class of compounds represented by formula IV:
R7 R
R7 ~ ~ ~ H
Y'X~N ~ N~ ,R
~IV)
wherein:
X and X' are independently -C(O)- or -S(O)2-;
Y is -(C(R )2)-M-, -(C(R )2)p-~ -N(R )- or -N(R2)-
CH2-i and
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
each Rl, R2, R7, R4, p and M is independently as defined
for formula I.
Another object of this invention is a novel class
of compounds represented by formula V:
~'x~l~z
(V)
wherein:
X is -C(O)- or -S(O)2-;
Y is -(C(R2)2)-M-, -(C(R2)2)p-, -N(R2)- or -N(R2)-
CH2-;
10 ~ R is O or H2;
each R1l is independently H, OH or O, wherein both
Rll are not simultaneously hydrogen;
Z is a structure of formula VI:
N~,~R
~4 /
(VI)
wherein any structure of formula VI is optionally fused
with an aryl, carbocyclic or heterocyclic ring and is
optionally substituted with 1-3 substituents
independently selected from R2; and
h R1 R2 R7 R4 R8, p, q, G, M, Q and X is
independently as defined for formula I.
CA 02243121 1998-07-14
W O 97127180 PCTAUS97/01610
It is also an object of this invention to
provide pharmaceutical compositions comprising the
compounds of formulas I, IV and V and methods for their
use as inhibitors of aspartyl protease, and
particularly, HIV aspartyl protease.
It is a further object of this invention to
provide methods for treating viral diseases, and in
particular HIV-related diseases, using the compounds
and compositions of this invention.
CA 02243121 1998-07-14
W ~ 97/27180 PCTrUS97/01610
-- 10 --
D~TAIT.F.D D~SCRIPTIO~ OF THE I~V~TIO~
In order that the invention herein described
may be more fully understood, the following detailed
description is set forth. In the description, the
5 following a~breviations are used:
~esian~tion Reaaent or Fra~ment
Ac acetyl
Me methyl
Et ethyl
Bn benzyl
Trityl triphenylmethyl
Asn D- or L-asparagine
Ile D- or L-isoleucine
Phe D- or L-phenylalanine
Val D- or L-valine
Boc tert-butoxycarbonyl
Cbz benzyloxycarbonyl (carbobenzyloxy)
Fmoc 9-fluorenylmethoxycarbonyl
DCC dicyclohexylcarbodiimide
DIC diisopropylcarbodiimide
EDC 1-(3-dimethylaminopropyl)-3-
ethylcarbodiimide hydrochloride
HOBt 1-hydroxybenzotriazole
HOSu 1-hydroxysuccinimide
TFA trifluoroacetic acid
DIEA diisopropylethylamine
DBU 1,8-diazabicyclo(5.4.0)undec-7-ene
EtOAc ethyl acetate
. t-Bu tert-butyl
iBu iso-butyl
DMF dimethylformamide
THP tertrahydropyran
~ THF tetrahydrofuran
DMSO dimethylsulfoxide
CA 02243121 1998-07-14
W O97/27180 PCT~US97/01610
The following terms are employed herein:
~ Unless expressly stated to the contrary, the
terms "-SO2-" and "-S(O)2-" as used herein refer to
sulfone or sulfone derivative ~i.e., both appended
groups linked to the S), and not a sulfinate ester.
The term "alkoxy" refers to an alkyl ether
radical, wherein the term "alkyl" is as defined above.
Examples of suitable alkyl ether radicals include, but
are not limited to, methoxy, ethoxy, n-propoxy,
isopropoxy, n-butoxy, isobutoxy, sec-butoxy, tert-
butoxy and the like.
The term "alkyl", alone or in combination
with any other term, refers to a straight-chain or
branch-chain saturated aliphatic hydrocarbon radical
containing the specified number of carbon atoms, or
where no number is specified, preferably from 1-10 and
more preferably from 1-5 carbon atoms. Examples of
alkyl radicals include, but are not limited to, methyl,
ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-
butyl, tert-butyl, pentyl, isoamyl, n-hexyl and the
like.
The term "alkenyl", alone or in combination
with any other term, refers to a straight-chain or
branched-chain mono- or poly-unsaturated aliphatic
hydrocarbon radical containing the specified number of
carbon atoms, or where no number is specified,
preferably from 2-10 carbon atoms and more preferably,
~rom 2-6 carbon atoms. Examples of alkenyl radicals
include, but are not limited to, ethenyl, E- and
Z-propenyl, isopropenyl, E- and Z-butenyl, E- and
Z-lsobutenyl, E- and Z-pentenyl, E- and Z-hexenyl,
E,E-, E,Z-, Z,E- and Z,Z-hexadienyl and the like.
The term "anti-viral agent" or "anti-
re_roviral agent" refers to a compound or drug which
-
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 12 -
possesses viral inhibitory activity. Such agents
include reverse transcriptase inhibitors (including
nucleoside and non-nucleoside analogs) and protease
inhibitors. Preferably the protease inhibitor is an
HIV protease inhibitor. Examples of nucleoside analog
reverse transcriptase inhibitors include, but are not
limited to, zidovudine (AZT), dideoxycytidine (ddC),
didanosine (ddI), stavudine (d4T), 3~C, 935U83, 1592U89
and 524W91. Examples of non-nucleoside analog reverse
transcriptase inhibitor include, but are not limited to
TIBO, delavirdine (U90) and nevirapine. Examples of
~IV protease inhibitors include, but are not limited to
VX-478 (Vertex, also known as 141W94 (Glaxo-Wellcome)
and KV~-478 (Kissei)), saquinavir (Ro 31-8959, Roche),
indinavir (L-735,524, Merck)), ritonavir (ABT 538,
Abbott), nelfinavir (AG 1343, Agouron), palinavir (Bila
2011 BS), U-103017 (Upjohn), XM 412 (DuPont Merck), XM
450 (DuPont Merck), BMS 186318 (Bristol-Meyers Squibb),
CPG 53,437 (Ciba Gelgy), CPG 61,755 (Ciba Geigy), CPG
70,726 (Ciba Geigy), ABT 378 (Abbott), GS 3333 (Gilead
Sciences), GS 3403 (Gilead Sciences), GS 4023 (Gilead
Sciences), GS 4035 (Gilead Sciences), GS 4145 (Gilead
Sciences), GS 4234 (Gilead Sciences), and GS 4263
(Gilead Sciences).
The term "aryl", alone or in combination with
any other term, refers to a carbocyclic aromatic
radical (such as phenyl or naphthyl) containing the
specified number of carbon atoms, preferably from 6-14
carbon atoms, and more preferably from 6-10 carbon
atoms. Examples of aryl radicals include, but are not
limited to phenyl, naphthyl, indenyl, indanyl,
azulenyl, fluorenyl, anthracenyl and the like.
~ The term "carbocycle" and "carbocyclyl"
radical, refers to a non-aromatic stable 3- to 8-
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97101610
membered carbon ring which may be saturated, mono-
- unsaturated or poly-unsaturated. The carbocycle may be
attached at any endocyclic carbon atom which resul~s in
a stable structure. Preferred carbocycles have 5-6
carbons.
The term 'Iheterocycle'' and "heterocyclyl"
radical, unless otherwise defined herein, refers to a
stable 3-7 membered monocyclic heterocyclic ring or 8-
11 membered bicyclic heterocyclic ring which is either
saturated or unsaturated, and which may be optionally
benzofused if monocyclic. Each heterocycle consists of
one or more carbon atoms and from one to four
heteroatoms selected from the group consisting of
nitrogen, oxygen and sulfur. As used herein, the terms
"nitrogen and sulfur heteroatoms" include any oxidized
form of nitrogen and sulfur, and the quaternized form
of any basic nitrogen. In addition, any ring nitrogen
may be optionally substituted with a substituent R2, as
defined herein for compounds of formula I. A
heterocyclyl radical may be attached at any endocyclic
carbon or heteroatom which results in the creation of a
stable structure. Preferred heterocycles include 5-7
membered monocyclic heterocycles and 8-10 memebered
bicyclic heterocycles. Preferred heterocycles defined
above include, for example, benzimidazolyl, imidazolyl,
imidazolinoyl, imidazolidinyl, quinolyl, isoquinolyl,
indolyl, indazolyl, indazolinolyl, perhydropyridazyl,
pyridazyl, pyridyl, pyrrolyl, pyrrolinyl, pyrrolidinyl,
pyrazolyl, pyrazinyl, quinoxolyl, piperidinyl, pyranyl,
pyrazolinyl, piperazinyl, pyrimidinyl, pyridazinyl,
morpholinyl, thiamorpholinyl, furyl, thienyl,
triazolyl, thiazolyl, ~-carbolinyl, tetrazolyl, thiazo-
li~inyl, benzofuranoyl, thiamorpholinyl sulfone,
oxazolyl, benzoxazolyl, oxopiperidinyl, oxopyrroldinyl,
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 14 -
- oxoazepinyl, azepinyl, isoxazolyl, isothiazolyl,
~urazanyl, tetrahydropyranyl, tetrahydrofuranyl,
thiazolyl, thiadiazoyl, dioxolyl, dioxinyl, oxathioly ,
benzodioxolyl, dithiolyl, thiophenyl,
tetrahydrothiophenyl and sul~olanyl, dioxanyl,
dioxolanyl, tetrahydrofurodihydrofuranyl,
tetrahydropyranodihydrofuranyl, dihydropyranyl,
tetrahydrofurofuranyl and tetrahydropyranofuranyl.
The term "halogen" refers to a radical of
fluorine, chlorine, bromine or iodine.
The terms 'IHIV protease" and "HIV aspartyl
protease" are used interchangeably and refer to the
aspartyl protease encoded by the human immunodeficiency
virus type 1 or 2. In a preferred embodiment of this
invention, these terms refer to the human
immunodeficiency virus type 1 aspartyl protease.
The term "inert solvent" refers to a solvent
reaction medium which allows the reagents to react
together at a substantially increased rate relative to
any reagent reacting with the designated solvent.
The term "leaving group" or "LG" re~ers to
groups readily displaceable by a nucleophile, such as
an amine, alcohol, phosphorous or thiol nucleophile or
their respective anions. Such leaving groups are well
known and include carboxylates, N-hydroxysuccinimide,
N-hydroxybenzotriazole, halogen (halides), tri~lates,
tosylates, mesylates, alkoxy, thioalkoxy, phosphinates,
phosphonates and the like. Other potential
nucleophiles include organometallic reagents ~nown to
those skilled in the art.
The term "protecting group" re~ers to a
suitable chemical group which may be attached to a
functional group and removed at a later stage to reveal
the intact functional group. Examples o~ suitable
protecting groups ~or various ~unctional groups are
CA 02243l2l l998-07-l4
W O 97/27180 PCT~US97/01610
- 15 -
described in T.W. Greene and P.G.M. Wuts, Protective
Groups ' n Org~nic Synthesis. 2~. F~ ., John Wiley and
Sons (1991); L. Fieser and M. Fieser, F;eser and
F;eser's Reag~nts for Org~n;c Synthes;s, John Wiley and
Sons (1994); L. Paquette, ed. ~ncycl ope~;a of Reagents
for Ora~n;c Synthes;s, John Wiley and Sons (1995).
The term "fused" whether preceded by the term
"optionally" or not, refers to a structure wherein two
distinct ring systems are joined together such that
both rings share at least two common atoms. This can
be envisioned as the replacement of a carbon-hydrogen
or nitrogen-hydrogen bond on a ring atom with a carbon-
carbon (from a second ring) or nitrogen-carbon (from a
second ring) bond. For example, a cyclohexyl ring
fused to a second cyclohexyl ring results in a
decahydronaphthalene, a cyclohexyl ring fused to a
piperidine ring results in a decahydroquinoline or
decahydroisoquinoline, or a phenyl ring fused to a
thiazole ring results in a benzothiazole.
The term "substituted", whether preceded by
the term "optionally" or not, and substitutions
contained in formulas of this invention, refer to the
replacement of one or more hydrogen radicals in a given
structure with the radical of a specified substituent.
When more than one position in a given structure may be
substituted with more than one substituent selected
from a specified group, the substituents may be either
the same or different at every position (for example,
the moiety -N(R2)(R2)). Typically, when a structure
may be optionally substituted, 0-3 substitutions are
'~ preferred, and 0-1 substitutions is more preferred.
Most preferred substituents are those which enhance
protease inhibitory activity or intracellular antiviral
activity in permissive mammalian cells or immortalized
mammalian cell lines, or which enhance deliverability
CA 02243121 1998-07-14
W O 97/27180
PCT~US97/~1610
- 16 -
by enhancing solubility characteristics or enhancing
pharmacokinetic or pharmacodynamic profiles as compared
to the unsubstituted compound. Other more preferrea
substituents include those used in the compounds shown
in Tables 1-5.
The term "pharmaceutically ef~ective amoun~"
refers to an amount effective in treating HIV infec'ion
in a patient either as monotherapy or in combination
with other agents. The term "treating" as used herein
refers to the alleviation of symptoms of a particular
disorder in a patient or the improvement of an
ascertainable measurement associated with a particular
disorder. Specifically, with respect to ~IV, effec~ive
treatment using the compounds and compositions of this
invention would result in an improvement in an HIV
associated ascertainable measurement. The term
"prophylactically effective amount" refers to an amount
ef~ective in preventing HIV infection in a patient. As
used herein, the term "patient" refers to a mammal,
including a human.
The term "pharmaceutically acceptable carrier
or adjuvant" refers to a carrier or adjuvant that ~ay
be administered to a patient, together with a compound
of this invention, and which does not destroy the
2~ pharmacological activity thereof and is nontoxic when
administered in doses sufficient to deliver a
therapeutic amount of the antiretroviral agent.
As used herein, the compounds of this
invention, including the compounds of formula I are
defined to include pharmaceutically acceptable
derivatives or prodrugs thereof. A "pharmaceutically
acceptable derivative or prodrug" means any
pharmaceutically acceptable salt, ester, salt of an
ester, or other derivative of a compound of this
-
-
CA 02243l2l l998-07-l4
W O 97/27180 PCT~US97/01610
- 17 -
invention which, upon administration to a recipient, is
capable of providing (directly or indirectly) a
compound of this invention or an inhibitorily ac;ive
metabolite or residue thereof. Particularly favored
derivatives and prodrugs are those that increase the
bioavailability of the compounds of this invention when
such compounds are administered to a mammal (e.s., by
allowing an orally administered compound to be more
readily absorbed into the blood) or which enhance
delivery of the parent compound to a biological
compartment (e.g., the brain or lymphatic system)
relative to the parent species.
Pharmaceutically acceptable salts of the
compounds of this invention include those derived from
pharmaceutically acceptable inorganic and organic acids
and bases. Examples of suitable acids include
hydrochloric, hydrobromic, sulfuric, nitric,
perchloric, fumaric, maleic, phosphoric, glycollic,
lactic, salicylic, succinic, toluene-p-sulfonic,
tartaric, acetic, citric, methanesulfonic,
ethanesulfonic, formic, benzoic, malonic, naphthalene-
2-sulfonic and benzenesulfonic acids. Other acids,
such as oxalic, while not in themselves
pharmaceutically acceptable, may be employed in the
preparation of salts useful as intermediates in
obtaining the compounds of the invention and their
pharmaceutically acceptable acid addition salts.
Salts derived from appropriate bases include
alkali metal (e.g., sodium), alkaline earth metal
(e.g., magnesium), ammonium and N-~C1_4 alkyl)4 salts.
The term "thiocarbamates" refers to compounds
containing the functional group N-SO2-O.
- The compounds of this invention contain one
or more asymmetric carbon atoms and thus occur as
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
.
- 18 -
racemates and racemic mixtures, single enantiomers,
diastereomeric mixtures and individual diastereomers.
All such isomeric forms of these compounds are
expressly included in the present invention. Each
stereogenic carbon may be of the R or S configuration.
Although the specific compounds exemplified in this
application may be depicted in a particular
stereochemical configuration, compounds having either
the opposite stereochemistry at any given chiral center
or mixtures thereof are also envisioned.
Combinations of substituents and variables
~envisioned by this invention are only those that result
in the formation of stable compounds. The term
"stable", as used herein, refers to compounds which
possess stability sufficient to allow manufacture and
which maintains the integrity of the compound for a
sufficient period of time to be useful for the purposes
detailed herein (e.g., therapeutic or prophylactic
administration to a mammal or for use in affinity
2Q chroma~ography applications). Typically, such
compounds are stable at a temperature of 40~C or less,
in the absence of moisture or other chemically reactive
conditions, for at least a week.
The compounds of the present invention may be
used in the form of salts derived from inorganic or
organic acids. Included among such acid salts, for
example, are the following: acetate, adipate,
alginate, aspartate, benzoate, benzenesulfonate,
bisulfate, butyrate, citrate, camphorate, camph-
orsulfonate, cyclopentanepropionate, digluconate,dodecylsulfate, ethanesulfonate, fumarate, glucohepta-
noate, glycerophosphate, hemisulfate, heptanoate, hexa-
no'ate, hydrochloride, hydrobromide, hydroiodide, 2-
hydroxyethanesulfonate, lactate, maleate,
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
-- 19 --
~methanesulfonate, 2-naphthalenesulfonate, nicotinate,
oxalate, pamoate, pectinate, persulfate, 3-
phenylpropionate, picrate, pivalate, propionate,
succinate, tartrate, thiocyanate, tosylate and
undecanoate.
This invention also envisions the
quaternization of any basic nitrogen-containing groups
of the compounds disclosed herein. The basic nitrogen
can be quaternized with any agents known to those of
ordinary skill in the art including, for example, lower
alkyl halides, such as methyl, ethyl, propyl and butyl
chloride, bromides and iodides; dialkyl sulfates
including dimethyl, diethyl, dibutyl and diamyl
sulfates; long chain halides such as decyl, lauryl,
myristyl.and stearyl chlorides, bromides and iodides;
and aralkyl halides including benzyl and phenethyl
bromides. Water or oil-soluble or dispersible products
may be obtained by such quaternization.
The compounds of this invention are those of
formula I:
R7
( I )
wherein
each Z is
..
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 20 -
r ~ '~r~ / X
wherein any Z may be optionally fused with R6;
each X and X' is independently selected from the
group consisting of -C(O)-, -C(O)C(O)-, -S~O)- and
-S(O)2;
each Y and Y' is independently selected from the
group consisting of -(C(R2)2)p-, -NR2-, -(C(R2)2)p-M-,
>C=C(R2)2, and -N(R2)-CH2-;
each Rl is independently selected from the group
consisting of hydrogeni R ; C1-C6 alkyl; C2-C6 alkenyl;
C2-C6 alkynyl; C3-C6 cycloalkyl optionally fused with
R6; C5-C6 cycloalkenyl optionally fused with R ; and
where R1's are attached to adjacent atoms, the R1's
together with their attached adjacent atoms form a
carbocyclic or heterocyclic ring system which may be
optionally fused with R ; where any member of R1 may be
optionally substituted by one or more R2;
each R2 is independently selected from hydrogen;
R ; C1-C6 alkyl; C2-C6 alkenyl; C2-C6 alkynyl; C3-C6
cycloalkyl optionally fused with R ; C5-C6 cycloalkenyl
optionally fused with R ; and where two R 's are
attached to the same geminal atom, the R2's together
with their attached geminal atom may form a
spirocarbocyclic or spiroheterocyclic ring system; v
where any member of R2 may be optionally substituted by
one or more R3;
CA 02243121 1998-07-14
W O 97/27180 PCT~US97101610
each R3 is independently selected from oxo, OR9,
- N(R )2~ N(R )-X-R , N(R )-X-OR , N(R )-X-N(R )2~ SR , X-
R9, O-X-N(R9)2, C(O)N(R9)2, halogen, NO2, CN, COOR9 and
R6;
each R4 is independently selected from from the
group consisting of OR ; N(R )2; X.R ; C(O)N(R )2; R i
C1-C6 alkyl; C2-C4 alkenyl; C3-C6 cycloalkyl optionally
fused with R ; C5-C6 cycloalkenyl optionally fused with
R ; where any member of R may be optionally
substituted by one or more groups independently
selected from the group consisting of R9 and R3;
~ each R is independently selected from the group
consisting of H, OH, O and Rl;
each R is independently selected from the group
consisting of aryl, carbocyclyl and heterocyclyl,
wherein said aryl, carbocyclyl or heterocyclyl may be
optionally substituted with one or more groups selected
from the group consisting of oxo, -OR9, -R9, -N(R )(R ),
-N(R )-X-R , SR , -X-R , -O-X-N(R )2~ -R -OR , -CN,
-CO2R , -X-N(R )(R ), halogen, -NO2, and -CF3;
each R is independently selected from the group
consisting of hydrogen, OH and O;
each R is independently selected from the group
consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl,
carbocyclyl, and heterocyclyl;
each R is independently selected from the group
consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl,
carbocyclyl, heterocyclyl, aralkyl, carbocyclylalkyl
and heterocyclylalkyl wherein any aryl, carbocyclyl or
heterocyclyl may be optionally fused with R and
wherein any member of R8 may be optionally substituted
by one or more groups independently selected from the
group consisting of -OR , -N(R )2~ -CN, -NO2, -X-R , -X-
N(R )2 -C(O)OR8, -N(R8)-XNR8, and halogen;
CA 02243121 1998-07-14
W O 97/27180 PCTnUS97/01610
each Q is independently selected from CH and N;
each M is independently selected from the group
consisting of NH, -NR -, -O-, -S-, -S(O)- and -S(O~2-;
each n is 1 or 2;
each r is 0,1 or 2;
each p is independently 1 or 2;
each q is independently 1, 2 or 3; and
each G is independently selected from the group
consisting of -NH-, -NR -, -O-, -S-, -S(O)-, S(O)2,
-C(O)-, and -C(R2)2-.
Except where expressly noted to the contrary,
the term ~t [variable] as defined for formula I" refers
to the definitions shown directly above. In addition,
where no reference is made to a particular definition
for a given variable, the definition is to be taken as
that defined for formula I shown directly above.
Preferred compounds of formula I are those
wherein
each Y and Y' is independently selected from the
group consisting of -(C(R )2)p-~ -NR -, -(C(R )2)p-M-,
and -N~R )-CH2-; and
each R is independently selected from oxo, OR ,
N(R9)2, N(R )-X-R , N(R )-X-OR9, SR9, X-R9, O-X-N(R )2
C(O)N(R )2~ halogen, NO2, CN, COOR and R .
Alternate preferred compounds of formula I
are those having the structure of formula IA:
Y~X,N~Z
(IA)
wherein
~ each Rl is independently selected from the group
consisting of R ; C1-C6 alkyl optionally substituted
CA 02243121 1998-07-14
W O97/27180 PCT~US97/01610
- 23 -
with R6; C2-C6 alkenyli C2-C6 alkynyl; C3-C6 cycloalkyi
optionally fused with R ; C5-C6 cycloalkenyl optionally
fused with R ; where any member of R12 may be optionally
substituted by one or more R2.
Preferred compounds of formula I are those
wherein n is equal to 1; those having the structure of
formula II:
~X--S~Z
(II)
and those having the structure of formula III:
R7 R
~7 \ ~ fH
y~x,N ~ ~ Z
(III)
Also preferred are compounds according to
formula I wherein X is -C(O)- or -S(0)2- and
Y is -lC(R )2)p-M-; those wherein X is -C~O)- or -S(0)2-
and Y is (-C(R2)2-)p; those
wherein X is -C(O)-, -C(O)C(O)- or -S(0)2-; and
Y~is -N(R )- or -N(R )-CH2-.
CA 02243121 1998-07-14
WO 97/27180 PCTrUS97/01610
- 24 -
An alternate object of this invention is a
novel class of compounds represented by formula IV:
~ ' R
R ~ ~ H 4
(IV)
wherein:
~ X and X' are independently -C(O)- or -S(O)2-;
Y is -(C(R )2)-M-, -(C(R )2)p-~ -N(R )- or -N(R )-
CH2-; and
each R1, R2, R7, R4, p and M is independently as defined
for formula I.
Another object of this invention is a novel
class of compounds represented by formula V:
R7 N
'X'
~10
(V)
wherein:
X is -C(O)- or -S(O)2-;
Y is -(C(R2)2)-M-, -(C(R2)2)p-, -N(R2)- or -N(R2)-
CH2-; 1
R is O or H2;
each R is independently H, OH or ~, wherein both
R11 are not simultaneously hydrogen;
Z is a structure of formula VI:
CA 02243l2l l998-07-l4
W O 97127180 P~TrUS97/01610
. - 25 -
~G R8
'Q ~8
R4/
(VI)
wherein any structure of formula VI is optionally fused
with an aryl, carbocyclic or heterocyclic ring and is
optionally substituted with 1-3 substituents
independently selected from R2 (where in formula V, if
R1 is H2, a methylene is implied); and
R1 R2 R7 R4 R8, p, q, G, M, Q and X is
independently as defined for formula I.
Also preferred are those compounds having the
structure of formula V, wherein
R and R are O;
compounds having the structure of formula V, wherein
R and Rll are O;
q is 1;
G is S; and
X' is -C~O)-;
compounds having the structure of formula V, wherein
R10 and R11 are O
q is 1;
G is S;
X' is -C(O)-; and
R is t-butylamino;
compounds having the structure of formula V, wherein
R and R are O;
X is -C(O)-;
Y is -(C(R )2)p-; and
R is H;
compounds having the structure of formula V wherein
CA 02243121 1998-07-14
W O 97/2718~ PCTAUS97/01610
- 26 -
Y is -~C(R )2)-;
R is H;
Rl is H2; and
one R is H and one R is OH;
Also preferred are those compounds of formula V
~wherein
X and X' is -C(O)-;
Y is -(C(R )2)-;
R is H;
R10 is H2;
one R11 is H and one R11 is OH; and
R within the de~inition of Y is selected from
hydrogen, R3 or C1-C6 alkyl optionally substituted with
1~ R ;
those compounds of formula V wherein
X and X' is -C(O)-;
Y is -(C(R2)2)-;
R is H;
R is H2;
one R11 is H and one R is OH; and
R2 within the definition of Y is selected from
hydrogen, -N (R )2 or heterocyclyl, which may be
optionally benzofused, and wherein said heterocyclyl
25 may be optionally substituted with one or more groups
selected from the group consisting of oxo, -OR , -R ,
-N(R9)(R9), --N(R9)--X-R , SR9, --X--R9,-O-X-N(R )2~ -R -
OR9, -CN, -CO2R9, -X-N(R9)(R9), halogen, -NO2, and -C~3;
those compounds of formula V wherein
X and X' is -C(O)-;
Y is -(C(R ) 2)-;
R is H;
R is H2;
CA 02243l2l l998-07-l4
W O 97/27180 PCT~US97/01610
- 27 -
one R is H and one R is OH; and
R within the definition of Y is selected rom the
group consisting of:
HO~cn ~ HO~
/ ~ ~C 3 ~C
3~ ~ N~\N~
e~N~ ~N~ <~
H H
CA 02243121 1998-07-14
W O 97~7180 PCT~US97/01610
- 28 -
~H ~N~ Ct~ Ne CH3 Me
Me
N~l2
S~N
OH
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 29 -
OH
F~F
~F ~F F~F
those compounds according to formula V wherein:
X and X' is -C(O)-;
Y is -(C(R )2)-;
R is H;
R10 is H2;
one R is H and one Rll is OH; and
at least one R2 within the definition of Y is aryl
optionally substituted with one or more groups selected
from the group consisting of oxo, -OR9, -R , -N(R )(R9),
10 -N(R )-X-R , SR , -X-R , -O-X-N(R )2~ -R -OR , -CN,
-CO2R , -X-N(R )(R9), halogen, -NO2, and -CF3;
those compounds according to formula V wherein:
X and X' is -C(O)-;
Y is -(C(R2)2)-;
R is H;
R is H2;
~ one Rll is H and one Rll is OH; and
; at least one R2 within the definition of Y is Cl-C6
alkyl optionally substituted with R ;
20 those compounds according to formula V wherein:
X and X' is -C(O)-;
Y is -(C(R2)2)-;
CA 02243121 1998-07-14
WO 97~7180 PCT~US97101610
- 30 -
R is H;
R is H2i
one R1l is H and one R1 is OH;
at least one R2 within the definition of Y is Cl-C6
alkyl optionally substituted with R3; and
at least one R3.within the definition of Y is
pyridyl, triazolyl, oxazolyl, isoxazolyl, pyrimidyl,
pyrazolyl, pyridazinyl, thiazolyl, imidazolyl, thienyl
thiadiazolyl, oxadiazolyl, tria~inyl or pyrazinyl
wherein said R3 may be optionally substituted with 1.-3
substituents selected from -OR9, -R9, -N(R9)(R9),
-N(R )-X-R , SR , -X-R , -O-X-N(R )2~ -R -OR , -CN,
-CO2R , -X-N(R9)(R9), halogen, -NO2, and -CF3.
those compounds according to formula V wherein:
X and X' is -C(O)-;
Y is -(C(R )2)-;
R is H;
R is H2;
one R is H and one R is O~;
at least one R2 withir. the definition of Y ls C1-C6
alkyl optionally substituted with R3; and
R within the definition of Y is aryl optionally
substituted with 1-3 substituents selected from -OR9,
-R , -N(R )~R ), -N(R )-X-R , SR , -X-R , -O-X-N(R )2~
-R -OR , -CN, -CO2R , -X-N(R )(R ), halogen, -NO2, and
-CF3.
Also preferred are those compounds according to
any of the aforementioned preferred co~pounds of
formula V wherein:
30. Rl is benzyl; and Z is
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
those compounds according to any of the aforementioned
preferred compounds of formula V wherein:
R1 is benzyl optionally substituted with 1-3
substituents selected from -OR9, -N~R9)(R9), SR , -X-R ,
-R9-OR , -CN, halogen, -NO2, and -CF3;
those compounds according to any of the aforementioned
preferred compounds of formula V wherein:
Rl is benzyl optionally substituted with 1-3
substituents selected from -OR , -N(R )(R ), SR , -X-R ,
-R -OR , -CN, halogen, -NO2, and -CF3; and
z is
H
those compounds according to any of the aforementioned
preferred compounds of formula V wherein R1 is benzyl
optionally substituted with 1-3 substituents selected
fro~ the group consisting of OCH3, OH and NH2;
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 32 -
~ those compounds according to any of the aforement oneà
preferred compounds of formula V wherein R is benzyl
optionally substituted with 1-3 substituents selected
from the group consisting of OCH3, OH and NH2 and
wherein Z is
~Ç
~N~J
o~NX
H
An alternate embodiment of this invention is
compounds according to formula V, wherein:
R7' R
~X~
(V)
each R is independently selected from the group
consisting of aryl, carbocyclyl and heterocyclyl,
wherein said aryl, carbocyclyl or heterocyclyl is
optionally substituted with one or more groups selected
from the group consisting of oxo, -OR , -R9, -N(R9)(R9),
-N(R )-X-R , SR , -X-R , -O-X-N(R )2~ -R -OR , -CN,
-CO2R9, -X-N(R9)(R9), halogen, -NO2, -CF3, -O-(CH2)q~R
-O-(CH2)q~OR , 2,3-methylenedioxy and 3,4-
methylenedioxy; and
ch X X' y y~ z R1 R2, R3, R4, R~, R , R , R , Q,
M, n, r, p, ~ and G is independently as defined for
formula I; and
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 33 -
those compounds according to formula V, wherein:
~ each R6 is independently selected ~rom the grouc
consisting of aryl, carbocyclyl and heterocyclyl,
wherein said aryl, carbocyclyl or heterocyclyl is
optionally substituted with one or more groups selected
from the group consisting of oxo, -OR , -R , -N(R )~R ),
-N(R )-X-R , SR , -X-R , -O-X-N~R )2~ -R -OR , -CN,
-CO2R , -X-N(R )(R ), halogen, -NO2, -CF3, ~O~(CH2)q~R
~O~(CH2)q~OR , 2,3-methylenedioxy and 3,4-
methylenedioxy;
R within the definition of Y is selected from
hydrogen, R or C1-C6 alkyl optionally substituted with
R ; and
h X X' Y Y' Z R1, R3, R9, R5, R , R , R , Q, M,
n, r, p, q and G is independently as defined for
formula I.
those compounds of formula V wherein
2 ( ) ;
Y is -N(R )-;
R is H;
R is H2; and
one R is H and one R is OH; and
those compounds of formula V wherein
Y is -(C(R )2)-M-;
M is O;
R is H;
R1 is H2; and
one R11 is H and one R11 is OH.
Also preferred is the compound of formula I
having the structure of formula IX:
., ,
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 34 -
R7
R7 \ ~ ( )H Rl
'X' ~ ~~~~
~ IX )
wherein
X is -C(O)- or -S(0)2-; and the compounds of
formula IX wherein
X is -C(O)-;
Y is -(C(R2)2)-M-; and
R is H; and those compounds of formula IX wherein
X is -C(O)-;
Y is -N(R )-; and
R is H; and those compounds of formula IX wherein
X is -C(O)-; Y is -(C(R )2)-; and R is H.
Also preferred are those compounds of formula
I having the structure of formula XII:
R7 Rl
R \~ ~ ~H R
'X' ~X~R4
(XII)
wherein
X and X' are independently -C(O)- or -S(0)2-;
those compounds of formula I having the structure of r
formula XII, wherein
X and X' are independently -C(O)- or -S(0)2-; and
CA 02243121 1998-07-14
W O 97127180 PCTAUS97101610
- 35 -
R4 is 1-amino-2-hydroxyindanyl; and
- compounds of formula I havi.ng the structure of formula
XII, wherein R is l(S)-amino-2(R)-hydroxyindany'
Also preferred are the compounds according to
formula I, having the structure of formula XIII:
R7 N~Rl
'X' ~ ~' 'R9
(XIII)
wherein
X and X' are independently -C(O)- or -S(O)2-i
compounds according formula I having the structure of
formula XIII, wherein
X is -C(O)- or -S(O)2-;
Xl is -C.(O)-;
Y is -(C(R )2)- or -N(R )-; and
R is Hi
compounds of formula I having the structure of formula
XIII, wherein
X is -C(O)-;
X' is -C(O)-;
Y is -(C(R2)2)-; and
R is H;
those compounds of formulz XIII wherein
X is -C(O)-;
Xl is -C(O)-,
. Y is -(C(R )2)-;
7 25 R is H; and
CA 02243121 1998-07-14
W O97/27180 PCT~US97/01610
- 36 -
R2 within the definition of Y is seiected from
hydrogen, R3, or C1-C~ alkyl optionally substituted with
those compounds according to formula XIII wherein:
X is -C(O)-;
X' is -C~O)-;
Y is -(C(R )2)-;
R is H; and
R within the definition of Y is selected from
hydrogen, -N(R9)2 or heterocyclyl, which may be
optionally benzofused, and wherein said heterocyclyl
~may be optionally substituted with 1-3 groups se'ected
from the group consisting of oxo, -oR9, -R , -N(R9)(R ),
-N(R9)-X-R9, SR9, -X-R9, -O-X-N(R9)2, -R -OR , -CN,
-CO2R9, -X-N(R9)(R9), halogen, -NO2, and -CF3;
those compounds according to formula XIII wherein:
X is --C (O)--;
X' is -C(O)-;
Y is -(C(R )2)-;
R is H; and
at least one R2 within the definition o~ Y is
selected from the group consisting of:
Me ~ S~
HO~CN
H ~N ~N~
H H3C Me
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 37 -
N~\N
~N~ ~N e,~ ~N~
HH
~N~ ~N~ ~XN~ [~\
H CH3 H
~H ~N ~ N~
Me CH3 Me CH3 Me
~N
Me
~,
CA 02243121 1998-07-14
WO 97127180 PCTrUS97/01610
~SVN--s~N
OH NH2 OH
S/~ ~N orV~N V
H
OH
N ~ N NF'~3
~\F ~F ~F
those compounds according to ~ormula XIII wherein:
X is -C(O)-;
X' is -C(O)-;
Y is -(C(R )2)-;
~ R7 is H; and
-
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97tO1610
at least one R2 within the definition of Y is a~yl
optionally substituted with.one or more groups selec~ed
from the group consisting of oxo, -OR, -R, -N (R ! (R !,
-N (R9) -X-R, SR9, -X-R, -O-X-N (R ) 2~ -R -OR, -CN,
5 -CO2R, -X-N (R ) (R ), halogen, -NO2, and -CF3 ;
those compounds according to formula XIII wherein:
X is -C(O)-;
X' is -C(O)-;
Y is -(C(R2)2)-;
R is H; and
at least one R within the definition of Y is C -C6
alkyl optionally substituted with R3;
those compounds according to formula XIII wherein:
X is -C(O)-;
X ' is -C (O) -;
Y is - (C (R ) 2) -;
R is H; and
at least one R within the definition of Y is
pyridyl, triazolyl, oxazolyl, isoxazolyl, pyrimidyl,
pyrazolyl, pyridazinyl, thiazolyl, imidazolyl, thienyl
thiadiazolyl, oxadiazolyl, triazinyl or pyrazinyl
wherein said R3 may be optionally substituted with 1-3
substituents selected from -OR, - R, -N (R ) (R ),
-N (R ) -X-R , SR , -X-R , -O-X-N (R ) 2, --R --OR , -CN,
-CO2R, -X-N (R ) (R ), halogen, -NO2, or -CF3;
those compounds according to formula XIII wherein:
X is -C(O)-;
X' is -C~O)-;
Y is - (C (R ) 2)
R is H; and
R3 within the definition of Y is aryl optionally
substituted with 1-3 substituents selected from -OR,
-R, -N (R ) (R ), -N (R ) -X-R, SR9, -X-R, -O-X-N (R9) 2
CA 02243121 1998-07-14
W O 97/Z7180 PCTrUS97/01610
- 40 -
-R9-OR9, -CN, -CO2R9, -X-N(R9)(R9), halogen, -NO2, or
-CF3;
those compounds according to any of the aforementioned
preferred compounds of formula XIII wherein:
each R1 is benzyl; and
each R not within the definition of Y is 2-
hydroxyindanyl.
.those compounds according to any of the aforementioned
preferred compounds of formula XIII wherein:
each R is independently selected from benzyl
optionally substituted with 1-3 substituents selected
from -OR , -N~R )(R ), SR , -X-R , -R -OR , -CN,
halogen, -NO2, and -CF3;
those compounds according to any of the aforementioned
preferred compounds of formula XIII wherein:
each R1 is independently selected from benzyl
optionally substituted with 1-3 substituents selected
from -OR , -N(R )(R ), SR , -X-R , -R -OR , -CN,
halogen, -NO2, and -CF3; and
each R not within the definition of Y is 2-
hydroxyindanyl;
those compounds according to any of the aforementioned
preferred compounds wherein:
each R1 is independently selected from benzyl
optionally substituted with 1-3 substituents selected
from the group consisting of OCH3, OH and NH2; and
those compounds according to any of the aforementioned
preferred compounds wherein:
each R1 is independently selected from benzyl
optionally substituted with 1-3 substituents selected
from the group consisting of OCH3, OH and NH~;
each R not within the definition of Y is 2- r
hydroxyindanyl.
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 41 -
Another embodiment is compounds according ~o
- formula XIII, wherein:
R7 Rl
Y'X~N ~ ,N~R9
(~II)
each R is independently selected from the group
consisting of aryl, carbocyclyl and heterocyclyl,
wherein said aryl, carbocyclyl or heterocyclyl is
optionally substituted with one or more groups selected
from the group consisting of oxo, -OR , -R , -N(R )(R ),
-N(R9)-X-R9, SR9, -X-R9, -O-X-N(R9)2, -R9-OR9, -CN,
-C02R , -X-N(R )(R ), halogen, -NO2, -CF3, ~O~(CH2)q~R ,
~O-(CH2)q~OR9 , 2,3-methylenedioxy and 3,4-
methylenedioxy; and
ach X, X , Y, Y', Z, Rl, R2, R3 R4 R5 R7 8 9
M, n, r, p, q and G is independently as defined for
formula XIII.
Another embodiment is compounds according to
formula XIII, wherein:
R7 R
R7 ~ ~ ~H Rl H
'X' ~ ,N~ 9
~XIII)
wherein R2 within the definition of Y is selected from
hydrogen, R3 or Cl-C6 alkyl optionally substituted with
R3;
CA 02243121 1998-07-14
W O 97t27180 PCT~US97/01610
- 42 -
each R6 is independently selected from the sroup
consisting o~ aryl, carbocyclyl and heterocyclyl,
wherein said aryl, carbocyclyl or heterocyclyl is
optionally substituted with one or more groups selec~ed
from the group consisting of oxo, -OR , -R , -N(R )(R ),
-N(R9)-X-R , SR , -X-R , -O-X-N(R )2~ -R -OR , -CN,
-C02R , -X-N(R )(R9), halogen, -NO2, -CF3, ~O~(CH2)q~R ,
-O-(CH2)~-OR , 2,3-methylenedioxy and 3,4-
methylenedioxy; and
, X , Y, Y', Z, R1, R3 R4 R5 R7 8 9
n, r, p, q and G is independently as defined for
formula XIII.
Another embodiment is compounds of formula I
having the structure of formula XIII, wherein
X is -C(O)-;
X' is -C(O)-;
Y is -N(R )-; and
R is H;
compounds of formula I having the structure of formula
XIII, wherein
X is -SO2-;
X' is -C(O)-i
Y is (C(R2) ) ; and
R is H; and
compounds of formula I having the structure of formula
XIII, wherein
X is -SO2-;
X' is -C~O)-;
Y is N(R2) ; and
R is H.
In an alternate embodiment, preferred
compounds are those of formula V wherein
~ R10 is H2; and
one R1l i5 H and one Rl is OH; and
CA 02243l2l l998-07-l4
W O 97/27180 PCTrUS97/01610
- 43 -
Z is selected from the group consisting of:
and ~ ~
O NH~u O ~ NH~u
and R2 is as defined in formula I; and those of formula
V wherein Z is selected from the group consisting of
N ~ and ~ N ~
O ~ NH~u O ~ NH~u
R is H2i and
one R is H and one R is OH.
Also preferred are those compounds of formula
V wherein X and X' is -C(O)-;
Y is -(C(R )2)-;
R is H;
R is H2; and
one R is H and one R is OH; and
those compounds of formula V wherein
X and X' is -C(O)-;
Y is -N(R2~-;
~ R7 i H
R10 is H2; and
CA 02243121 1998-07-14
W O 97127180 PCT~US97/01610
- 44 -
one R11 is H and one R is OH, and
those compounds of formula V, wherein
X and X' is -C(O)-;
Y is -(C(R )2)-M-;
M is O;
R is H;
R is H2; and
one Rl is H and one R 1 is OH, and the
aforementioned compounds of formula V wherein Z is
10 selected from the group consisting of:
and ~ N
O NH~u O NH~u
and R is as defined in claim 1.
Also preferred are those compounds of formula
V wherein X and X' is -C(O)-;
y is -(C(R )2)-;
R is ~;
R10 is H2i and
one R11 is H and one Rl1 is OH; and
those compounds of formula V wherein
X and X' is -C(O)-;
Y is -N(R )-;
R is H;
R10 is H2; and
one R1 is H and one R11 is OH, and
25 those compounds of formula V, wherein
X and X' is -C(O)-;
Y is -(C(R )2)-M-;
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 45 -
- R is Hi
R10 is H2; and
one Rll is H and one R is OH, and the
aforementioned compounds of formula V wherein Z is
selected from the group consisting of:
H ~ H ~ S
N and ~ N
O ~ NH~u O ~ NH~u
Also preferred are compounds of formula
I wherein:
Y'X'
( I )
Z is selected from the group consisting of -X'R4,
-N(R13-X~-R4~ -N(R1)-N(R1)-X'-R , and formula VI;
N~,,~R
X'
- R4
(VI)
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
4~ -
wherein any structure of formula VI is optionally used
with an aryl, carbocyclic or heterocyclic ring and ~s
optionally substituted with 1-3 members indepenàen,ly
selected from R2; and
each X~ X', Y, Y' R1! R2, R3, R4 R5 R6 R7 R8 9
M, n, r, p, q and G is independently as defined ln for
formula I.
- Another embodiment of this invention relates to
the process for preparing a compound of formula XIV:
R1
R6 ~ _~NH
XIV
wherein R1 and R6 are.defined as in formula I,
comprising the steps of:
(1) reacting a compound of formula XV:
R1
~NBoc
o
XV
wherein R1 is defined as in formula I,
in an inert solvent, preferably an ethereal solvent
such as diethyl ether or T~F, with a base, preferably
an alkali metal amide such as lithiumdiisopropylamlde
at a temperature between about -78 ~C to about 25 ~C;
(2) reacting the product of step (1) with an
a~dehyde R6CHO followed by an optional treatment with a
dehyrating agent, preferably Martinls sulfurane
CA 02243l2l l998-07-l4
W O 97/27180 ~CTrUS97/01610
- 47 -
dehydrating agent, wherein R6 is defined as in formula
4 I to give a compound of formula XVI:
R1
~o
XVI
wherein R and R are defined as in formula I;
- (3) reacting the product of step (2) in an ine-t
solvent, preferably methanol, with hydrogen gas in the
presence of an hydrogenation catalyst, preferably 10
palladium on carbon, followed by treatment with an
anhydrous acid, preferably trifluoroacetic acid or 4N
HCl in dioxane to give a product of formula XIV.
Another embodiment of this invention relates to
the process for preparing a compound of formula XVII:
R2$~,Nor ~NH~<
XVII
f wherein Rl and R2 are defined as in formula I,
comprising the steps of:
(1) reacting a compound of formula XVIII:
_
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 48 -
o
XVIII
wherein R and R2 are as defined in formula I,
in an inert solvent, preferably DMF or THF, with a base
preferably sodium hydride, then bromomethylacrylic acid
at a temperature between about -78 ~C to about 25 ~C;
(2) reacting the product of step (1) with an
oxidizing agent, preferably ozone and if necessary a
reductive work-up with a reducing agent such as
dimethylsulfide;
(3) reacting the-product of step (2) in an inert
solvent, such as DMF, with thioproline t-butylamide and
suitable amide-bond coupling reagents, preferably EDC,
HOBT and N-methylmorpholine, to give a product of
formula XVII.
Another embodiment of this inventlon relates to
1~ the process for preparing a compound of formula XIX:
,NH
(~ O .. '
XIX
wherein R1 and r are defined as in formula I,
comprising the steps of:
CA 02243l2l l998-07-l4
W O 97/27180 PCTnUS97/01610
~ 49 -
r (1) reacting a compound of formula XX
.~ ~
~NPG
X~C
wherein R1 is defined as in formula I and PG is a N-
protecting group, such as those described in Greene and
Wuts (infra), preferably p-methoxybenzyl, an iner'_
'solvent, preferably THF, with a base, preferably
lithiumdiisopropylamide at between about -78 ~C to
about 25 ~C, then a bis-leaving group alkane of formula
XX T:
~LG
XXi
wherein LG is selected from halo, preferably chloro or
ioào, arylsulfonate esters, preferably tosyl, and
aikylsulfonate esters, preferably mesyl, and r is
defined as in formula I, to give a product of formula
XXII:
~R1
LG~NPG
:~ 1
wnerein R and PG are defined as in formula XX and LG
and r are defined as in formula XXI;
(2) reacting the product of step (1) in an inert
sclvent, preferably THF, with a base, preferably
CA 02243121 1998-07-14
W O 97/27180 ~CT~US97/01610
- 50 -
lithiumdiisopropylamide, at between about -7~ ~C to
about 25 ~C to give a product of ~ormula XXIII:
R1
O ~
~NPG
(~ ~
X><lll
wherein R is defined as in formula I and PG is a N-
protecting group;
(3) reacting the product of step (2) in an inert
solvent with a reagent suitable for removal of the N-
protecting group PG, such as those described in Greene
and Wuts (infr~), to give a compound of formula XIX.
In another embodiment, compounds of formula I
with structures VII, VIII, IX, and X are preferred:
R~R 1 ,R~, H R 1
'X' ~N~N,X~R~ 'X ~N:s~R4
(Vll)(IXI
R~,R 'R ~ OH \--~R7
Y'X~N N'X'R 'X' ~N-X'Y'
(Vlll) (X)
where all definitions of variables for formula I apply.
Preferred R2 groups for formula I include:
Cl-C6 alkyl and alkenyl optionally substituted with R6;
where two R2 taken together form a spriocyclic ring and
Ci-C6 cycloalkyl or cycloal~enyl optionally fused with
R6 ~
CA 02243121 1998-07-14 VOSS~US & PA~TN~R
PCT/US97/01610 PArE~Jr~ LTE
Vertex Pharmaceuticals Inc., et al . ~.IE8F~T~ 4
Our Ref.: B 2555 PCT 31~75 ~'~C~EN
~. F,~. i998
-- 51
Preferred compounds of this invention of
formula I include the specific compounds contained in
Tables 1-5.
T~Rr.~ 1
~H
A ~ Z
Cmpd .
NC. A Z
~--Ph
/~ O O OM
2 ~--Ph H~in
N~N~ ~N~
O NHtBu
0 3 ~Nb,N~ o~JNH~Po
4 HN--r~' ~Ph O
N ~ ~ ,'.S
O 0
~"' ~Ph ,H~H
O NHtE~u
~MENDED Slt~T
CA 02243121 1998-07-14
- 52 -
6 ~ ~Ph f~
O NHtB u
7 --P h
~~ N~
8 ~~ H
O NHtBu
g _Ph f~
o O NHtBu
H3C ~5~ ~ ~ .S~OM~
O '0
~P h ~
o,,S ~oN ~ ~ o.. S,~OM e
12 ~ --Ph ~
~,N b,N ~ r¢~
13 3 ~ ~N ~S J~OM e
O ''O
4 ¢ ~ ~ '"~P h N~
AMENDED S~
CA 02243121 1998-07-14
.
, .
1 5 P h ~>
~N 'S
16 Ph
H C ~N _ 'P,~
O 0' '0
17 Ph
3 $ '
O 0' '0
18 Ph
E! ~N' I '
O 0' 0
19 Ph
HO I
~N _ ~/~
O O ''O
-Ph
P h ~ NIP~¢~
O 0' 0
21 Ph ~
H 2N ~N _ ~N ~S J~OM e
O 0' 0
22H3C ~N ~ N_
H O O O
23 Ph
HO~N_ ~ ',S'~
O O 0
A~hEN~o s~lEÇT
CA 02243121 1998-07-14
- 54 -
24 Ph
Ph ~
AcO~o N~ l ~ O .S.J~OMe
26 o o N~P~ h N(P,¢~
27 Plh
H N~o N ~ ~ .'.S.
28 Ph
O~N ~
29 Ph P
H3C ~O NIP~
HN~r ~ ~OMe
H3C ~ ~N ~ --No'.S. .O
31 Plh ~?
H3C~o ~ o,,S.J~OM e
3 2 Ph
N ? 'J ~J~
~E~ S~
CA 02243121 1998-07-14
33 p~
H3CO2C ~N ~ ~ O, ,S.J~OM e
3 4~P'7 ~
H~C~o ~ O''S-J~OM O
Fjh ~
~''' ~ ',S.
3 6 Plh
~N ~ ~ ~5,J~
3 7 P~
O HN--~' r~OMe
H3C O J~N ~ ~ O. .S. O~J
38 lh ~
[~' r~r
3 9 Plh ~>
H HN~ ~OMe
H C N ~N ~ ~ O S.
P h~o N -~ ~N 'S J~
41 PJh
D~o N-~ ~,~¢~
AMENDEO S~.E~T
CA 02243121 1998-07-14
- 56 -
42 .- Ph
C~N ~ ~,/¢~
O'- ''O
43 .- Ph
HO ~o N ~ ~N 'S J~
O-' ~'0
44 ~- Ph
H3CO~;( ~ ~ .'.S.--
.- Ph
H2N ~ ~ 'P,~
46 Ph
¢~ N 'S ~N ~
0''--0 O' ~'0
47 ~h
C~ ~ S
48 Ph
0'---0 O' ~-0
49 ~ ~H
O NHteu
--Ph H"f~
~ ~N HtB u
AMENDEû S~.rL~T
.
CA 02243121 1998-07-14
51 .-Ph
~ N~ o NHtBu
52 ~ ~Ph ,H~,~H
O NHtllu
2 ~ O~JNHtn ~
54 _~n ,HN ~ H
O NHtau
~ H~
J~ O NHt8u
56 ~ ,HN ~ H
~J'_~ O NHt8 u
57 ~ H~
o~ O NHt8u
~IIEND~ ~
CA 02243l2l l998-07-l4
- 58 -
58 ~ ,NH ~ H
H~~ o NHt8u
5 9 ~ ¦~H
o NHtBu
~NH~au
61 ~ u
u
~~ ~U~U
A~ E~ S~
CA 02243121 1998-07-14
_ 59
64 ~ O~NA(Bo
NH2
~N_ O~NHtBu
66 ~ ~H
[3~N_ ~ N Ht8 u
67 ~ ~H
HN~N_ O NHtBu
68 ~ ~H
¢~f H~7N_ O NHtBu
~~ O~NHtBu
A~ENDED S~.~ET
CA 02243121 1998-07-14
' . ~ ' '; ,~'_ '
-- 60
7 0 ~ t~H
EtOJ~-- O NHtEU
71 13 I~H
~NJ~-- O NHt8u
72 ~3 Of ~NH)t~ u
73 ~ ~H
0 ~
O~NHt8u
~_ ¢N~
74 ~ H~
0 '~
~;,N~ O NHt8u
~ H~
~N~ O N Ht8 u
AMENDED S~.~ET
CA 02243l2l l998-07-l4
76 ~ ~NHtBU
~ O~N HllB u
123 o~NHIB.
124 ~ H~
3,~_ O~JNHtBu
O~NH2B]
M~
6 o~NHtBu
AMEND~l~ S~
CA 022431.l l99X-07-14
, . -- ~ .. .. .. ..
-- 62
12 7 ~ H~
NC ~I_ o NHtBu
12 8 0~ ¦~H
~ 0 NHtBu
12 9 0~ ¦~H
O NHt8u
3 0 0~ ~H
O NHtBu
51 31 0~ ,HN~H
O NHt8u
13 2 0~ ,HN~H
~ O NHtBu
AMENDED SHET
CA 02243l2l 1998-07-14
- 63 -
133 ~ o ~NHtBL
134 ~ ~H
~h O NHtBu
135 ~ ~H
O NHtBu
136 ~ ~H
~ N_ o NHtBu
137 ~ ~H
~ ?~ O NHtBu
138 H~
f~titNDc~ S~
CA 02243l2l l998-07-l4
- 64 -
3 9 ~NHsB=
140 ~ ~H
~_ O~NHtBu
141 O~NH~n u
14:~ o~NH~
5 143 ~ ~H
O NHtBU
144 ~ ~ ~H
~ O~NHtBu
~?'.,lENDED SHEET
CA 02243121 1998-07-14
. . ~ . ' . .. . . .. -
, . . , . . . ..... .. -
.... .. .. .... .. ..
145 ,N~5
146 ~
~q ~N 'S'[~NH2
N
147 -N's ~ N~
148 ~ -N'S ~ NH2
N~
149 ~, ~N'S~NH~
15 0 ~N 'S~NH2
~ .NDED S~!E~T
CA 02243l2l l998-07-l4
- 66 -
151
r~N
52 ,N.SJ[~NH2
53 , ,,~ ,N~5J~NH2
54 ~N'SJ~NH2
15 5 --~ 9P~¢~NHZ
56 ~N'SJ~NH2
i;icD S~ :E~T
CA 02243l2l l998-07-l4
- 67 -
157 ~
~7;_ o. ,S, '~ N H2
15 a ~N S ~NA2
59 0'~ ~P
H2N '~?;-- ~ 'S NH2
l60 ~ ,NIl~
5 161 ~ NH~
16 2 15,P<~,N~
AMENDED S,~
CA 02243121 1998-07-14
- 68 -
163 ~ ,N ~ ~ N~
,~
~J
164 ~N~
165 ~N~
166 ~ ~ ~N~
N~N_
167 ~ ,NH~
68 ,~ N~
Ai~lE~!DED S~'.E~T
CA 02243121 1998-07-14
- 69 -
16 9 ,N~P<[~NI~
170
171 ~ ~ ~N~
17Z ~ ~ ~N~
173 ,N ~ ~ N~
174 ~ ~ N~
~N ~ ,N
Ai~iEl~DED Sl-~
-
CA 02243l2l l998-07-l4
- 70 -
175 ~N~ 'NHZ
176 ~ N~
N~ N_
7 7 0~ 'N(~5<b¢~NH2
NH2
s 17 9 ,~
1 8 0
AMENDED SHEET
CA 02243121 199X-07-14
-- 71
1 81 0,~
~0
182 ~
la3 ~N~<D[~NHz
la4 --~'N~
s las ,~
a6 ,~
Ai'viENDcD S~tEET
CA 02243121 1998-07-14
187
188 ~ ~ ~ ,N ~ ~ N~
189 ~ N~
19 0 ~NI~<~NHz
191 0~
~ ~N_
1 92 0~
A,tlEI'Jl)ED S~ET
CA 02243121 1998-07-14
- 73
19 3 0
N~
194 ~ ,NH2
195 ,NI~<~NH2
19 60~
19 7 ~ ,NH2
~N_
H ~ ~5b~NH2
,p~ Sn~
CA 02243121 1998-07-14
- 74 -
HO~ ~ NH~
200 ~ ~NHz
201 ,N~NH2
202 ~ ~ NH2
5 203 ~ ~ H2
204 H N~? _ ~~<~NH2
CA 02243121 1998-07-14
205
~_ ~''''O
206
O O ''O
N_
207 ~ ~ ~N~
~N_
2 5 9 0~ ~H
~_ O NH28u
260 ~ ,HN ~ H
O NHtB u
299 ~ ,HN ~ H
O NHtl3u
AMENDED SHEET
CA 0224312l l998-07-l4
- 76 -
~_ O~NHtnll
301 ~ ~H
O NHtBu
302 ~ ,HN ~ H
~_ O NHtl~u
303 ~ H~
O NHtE~u
O~NHt~u
Of ~NH~ U
AMENDED S, ~1 T
CA 02243121 1998-07-14
- 77 -
306 ~ H~
~ ~ O~JNHtBu
307 ~ H~
~ ~_ O~NHt8u
308 s
O N Ht8 u
3 0 9 0~ ~Hl
,p~ O NHt8u
310 0~ ~H
~_ O NHt8u
3 11H25~ O~N Ht8 u
Ai~ NDc~ SY.E'T
CA 02243l2l l998-07-l4
- 78 -
312 ~ ~H
H2~1~jJ - 0 NHtBU
313 ~ ~H
~ O~NHtBu
314 ~ ~ ,H~H
O NHt8u
Me
315 OMe 0~ ,HN~H
~_ O NHt8u
316 ~ a~HJ~llu
317 H ~ ,HN~H
~ O NHtBu
!D,~ S~E~T
CA 02243121 1998-07-14
~ . .~ .. .. ..
- 79 -
318 H ~ ,H~H
N~ O NHt8u
319 ~ ~H
~o--~h O NHt8u
320 ~ ~H
S--~ O NHt8u
321 ~ o~NHtDu
322 ~ ~H
~\5~ O NHt8u
323 H~_0~ ,H~H
~N_ O NHt8u
.'J .~ S~ F t
CA 02243121 1998-07- 14
-- 80
3 2 48nO ~ ,N~H
~N_ O NHtBu
3 2 5HO o--~ H~
O NHt8u
3 2 6MeO 0~ ~
~ N_ O NHtBu
3 2 7o~ H ., ~
~ O~NHtBu
}~
AMENDEI~ S~'EJ
CA 02243l2l l998-07-l4
- 81 -
TARr.R ~.
OH Rl
A lz
Cmpd. A Rl z
No.
78 ~.- P h H ~H
~ N"N~ Bn ~n,N 8
79 HN~ ~Ph H OH
~N~ Bn ~N ;8
8 0 ~_ Ph H OH
D~N ~ Bn ~,N 8
81 Ph Bn H CH
O~o~ ~N~I ~n,N ~8
82 ~ ' ~Ph Bn H OH
N ~N ~ ~N '~8
83 ~--Ph Bn H ~H
o ~l,N",8
8 4 Ph Bn H OH
¢~o... s,,\O ~N'~8
~E~)~ S'r,~r
CA 02243121 1998-07-14
8 5 ~, 9n H OH
8 6 ~ ,, ~ Eln n,H 8
87 ,~ 9n I~H ~ ~8
8 8 ~ Bn H OH
8 9 ~ 8 n H OH
g o ~,~_,~, Bn H OH
o~D S~
CA 02243121 1998-07-14
- 83 -
91 ~ Bn H CH
0~N~ ,N_ 8
o o
9 2 ~ Bn H OH
~ ~ 8
93 /==\ Bn H OH
~ ~n,N ""~
9 4 ~ B n~N "" OH
,~ ~ 8
<~
~,~ N~8
N_
96 ~ Bn H QH
N "'8
NC~;~
~ D S~
CA 02243121 1998-07-14
- 84 -
2 0 8 Bn H
MeO~
2 0 9 ~ Bn ~rN. 8
Me2N pN_
210 H B n H
~S'~
o~~ 11
211 ~ Bn H CH
~J~ ~n,N ",8
212 ~.~ Bn H OH
Me oJ~ N ~8
~ ~ Bn ~N 8
N~¢N_
A~llENûED S~
CA 02243l2l l998-07-l4
r
- 85 -
214 ~'~ Bn H 9H
~ ,N. ~>
~ ~ ~
215 Bn H 9H
N ""~
~0~ ~ ~3
216 Bn H 9H
rN 8
,~
ol b
~ 217 ~ Bn H OH
bJI~ ~N 8
218 ~ Bn H 9H
~ N",8
219 ~ Bn H 9H
~ ~N~8
o~N_
~ cO S~
CA 02243121 1998-07-14
- 86 -
220 ~ Bn H qH
~ "N"'8
221 ~ Bn H CH
~N'~8
222 ~ Bn ~N,~
~$ o 8
223 ~ Bn H qH
_~ ~N 8
224 ,~ Bn H qH
N'~8
225 Bn H QH
0~ ~ 8
A~El'll~ED Slt0
CA 02243121 1998-07-14
. - 87 -
226 ~ Bn H ~H
b,N ".8
H
227 ~ Bn H OH
,~ ~ 8
2 z a ~N 8
229 ~ Bn ~N 8
NC~N_
N_N .- Bn b,N ~8
N~
231 ~ Bn H OH
~'~ ~'~N~o
~ O ~
AM~ND~ SH~T
CA 02243121 1998-07-14
- 88 --
232 ~ Bn H O,H
M~ ~ ~N "8
233 ~ Bn H OH
~ ,N"'8
2 3 4 ~ Bn H
N,~8
2 3 5 ~ Bn H O,H
~ ,N,~8
Meol~N~N~
236 ~ Bn H O,H
~J~ ~n,N ~8
2 3 7 ~ Bn H O,H
N'~8
Me
Al~,1END~O St~E~
CA 02243121 1998-07-14
- 89 -
238 ~ Bn H OH
~ ~ 8
2 3 9 ~ Bn H OH
N~8
240 Bn H OH
'N'8
241 ~ Bn H OH
H--~ ~n,N "8
242 Bn H ~H
~ ,N,~8
HN~ _
2 4 3 0~ Bn HOH
~N~ - 8
N~N_
ANlEN~ED st~
CA 02243121 1998-07-14
-- 90
2 4 4 0~ Bn ~N -" OH
~h ~ 8
245 ~ Bn 8
246 ~ Bn ~N 8
2 4 7 0~ Bn H OH
~'?;- ~ 8
2 4 8 [~ Bn ~N '8
N~--
2 4 9 0~ Bn N
~' '? ~ 8
AMENDED S~
CA 02243l2l l998-07-l4
- 91 -
2 S 0 0~ Bn H OH
~ ~ 8
2 51 0~ Bn H OH
~$ ~ 8
2 5 20~ Bn H O,H
~ ~ 8
2 5 3 ~ Bn H O,H
~ ~ 8
2 5 4 ~ Bn H O,H
N ~8
255 ~ Bn H O,H
~ ~n,N,~8
AME~ID~D S. ~
CA 02243121 1998-07-14
- 92 -
2 5 60~ Bn H OH
~ ~ 8
o~J
2 61 ~ Bn~rN 8
~:$
2 62 ~ Bn H OH
~I~N ",8
Q ~Nl~N
2 6 3/=\ Bn H qH
~ ~N"'8
~0
2 64 ~ Bn H qH
~ ~N ~ o
:~_ ~ ~3
2 6 5 ~ N ON
~NI:)0 Slt~T
CA 02243121 1998-07-14
- 93 -
266 ~ Bn H OH
~n~N 8
267 ~ Bn H OH
~'?;-- "N "8
2 6 8 0~ Bn H OH
M~;_ 8
269 Bn H OH
FC~ N 8
270 Bn o
271 ~ ~N 8
AMENDED S~
CA 02243121 1998-07-14
, ; , . '
-- 94
2 7 2 ~ B n H qH
~ N"8
2 7 3~3~ B n ~N ~8
2 7 4 Bn H OH
275 ~ Bn H OH
~ "N"'8
~ 7 5 Brl H OH
277 0~ Bn H OH
~ ~ 8
AM'NDED S~E~T
CA 02243121 1998-07-14
278 ,~ Bn H OH
~N"~8
Me
279 ~ Bn H qH
MeO~_ b,N "8
280 ~ Bn H OH
~ ~N ",8
281 ,~ Bn H OH
~ N"~8
2 8 2 ~ Bn H OH
~ ~8
2 8 3 ~ Bn H OH
~ N~'8
~ ENoE~ S~E~
CA 02243121 1998-07-14
-- 96
2 84 ~ Bn H O,,H
~, ~ ~ ~N "'8
S~
285 Bn H ~H
~, ~, "N';8
Isl~h
286 ~ Bn H qH
N "'8
287 HO Bn H O,H
~ ~n~N~g
Z88 ~ Bn ~N 8
2 8 9 H~ ~~0~ Bn H
P~A, N~ O S~
CA 02243121 1998-07-14
- 97 -
290 MeO ~ Bn H qH
~ N"'8
291 o ~ Bn H qH
~ N o "N 8
292 H cH
293 ~ H -
~p_ ElrlO~0> ~rN~,8
294 ~ H qH
N';8
295 ~ H q
M~ f N 8
A~ D~D Sr~
CA 02243121 1998-07-14
- 98 -
296 2
~ MM~ N 8
298 H OH
T~Rr.r2 3
OH
A~l~,Z
Cmpd ~o. A Z
97 .--Ph rS
N N ,N,~
~ O NHt8u
98 HN--I''' ~Ph rS
~N ~,N~
~ O NHtBu
CA 02243l2l l998-07-l4
_ 99 _
g g~--Ph rS
~N ~ ~N,~
o O NHt9u
00 Ph rS
H N--I ~J ,N,~
~N ~ O NHtBu
101¢~ I ~Ph ~N,r~
O O NHtBu
102 -Ph
Ph ~ N~ O NHtBu
o
103 Ph rS
¢~C.s ~ O~NHtBu
0- ''O
T~RT.R 4
O
- A ,,Jl~,z
Cmpd No. A Z
04 .--Ph rS
N N ~
~ O NHtBu
~M~NDED St~EET
CA 02243l2l l998-07-l4
- 100 -
05HN--I' ~Ph rS
~N~ ,N~
~ O NHt9u
106 .- Ph rS
~N_ ~N~
o O NHt8u
07 Plh rS
H N--~ -"' ,N ~
~N ~ O~N HtB u
0 8 ~ ~Ph ,Nr~)
O O NHtBu
109 .-Ph ,NrS
Ph ~ O~NHtBu
o
0 Ph rS
~S O~NHtBu
O'' ''O
111 ~ ,N,r~
O NHtBu
~N_
2 ~ rS
,N~
,,,~ O NHtBu
o
A~,~
CA 02243121 1998-07-14
- 101 -
3 ~ ,N,r~)
~\o O NHt8u
4 [~ ~Nr~
~ O NHtBu
~~¢N~
[~ ~
O NHtBu
~N~
2 5 7 0~ ~Nr~
- Q NHtBu
MeO~
s ~sa
AMENDED SHE~
CA 0 2 2 4 3 1 2 1 1 9 9 8 - 0 7 - 1 4
- 102 -
T~RR~! 5
OH
A~ Z
Cl~lL~d No. A Rl z
116 .--Ph H
N ~l,N ~ Bn~N ~n~~~Q
117 ~-~ ~Ph BnO ~>
118 ~ N~Ph Bn,N~O~_~
119 ~o~--N~'~h Bn
12 0 ~ - ~Ph Bn
121 ~N~ Bn
122 ~ Bn H
9 S~
CA 02243l2l l998-07-l4
W O 97/27180 PCT~US97/01610
- 103 -
The preferred compounds of this invention are
- compound numbers (as in Tables 1-5): 1, 2, 3, 4, 7, 8,
9, 13, 14, 16, 17, 18, 20, 23, 24, 25, 26, 32, 35, 38,
44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 62, 63, 72,
75, 76, 78, 80, 82, 83, 91, 92, 94, 95, 96, 101, 102,
109, 121, 122, 123, 124, 126, 127, 128, 129, 131, 132,
133, 134, 135, 137, 138, 140, 141, 145, 146, 147, 149,
150, 155, 156, 160, 161, 162, 164, 165, 170, 171, 175,
176, 177, 179, 180, 185, 186, 190, 191, 192, 194, 195,
200, 201, 208, 219, 220, 228 and 264. More preferred
are compound numbers: 2, 7, 8, g, 14, 18, 2~), 25, 26,
32, 38, 45, 47, 48, 49, 50, 51, 53, 54, 62, 63, 72, 82,
83, 91, 92, 94, 95, 96, 123, 126, 140, 141, 219, 220,
228 and 264. Even more preferred are compound numbers:
7, 8, 9, 20, 45, 50, 51, 53, 54, 82, 83, 92, 94, 96,
219, 220, 228 and 264.
In an alternate embodiment, this invention
also relates to novel methods for preparing compounds
and intermediates of the following structures.
One embodiment relates to a process
The compounds of this invention may be
synthesized using conventional techniques.
Advantageously, these compounds are conveniently
synthesized from readily available starting materials.
Although the syntheses of ~he compounds of
this invention are known to those of skill in the art,
the following general schemes are set forth to
illustrate these methods. These schemes should not be
viewed as limiting the scope of this invention in any
way.
- Using standard techniques, compounds of the
present invention having the general formula I may be
obtained as described in the following schemes:
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 104 -
~iCHEME 1
R~
R2 2 'N
~ ~ ~Ph3 P 2~
P 'N J Nxll~o ,R 1 'N J~CO2M o
El EV
1- d~lprot~ct ¦ M9 I M-OH
2. b~se / h~lat
R --OH
R~?~ H ~NH 2~ O-a,ctiva,ti"on
Ell EVII EVI
1 1. protect 1. ~oc20, DMAP
2. base, ~E~
-N--I R1 3, d~protect
R227~N H R 1
E1 E--~NH
o
E3
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 105 -
tiCHEME 1 (cont'd)
R1 . R1
N~ HN~
1. MoONHMe l EDCI
~1 2 reduce
h 1~ ~ 3 TMSCH2MgBr
n-BuLi 4 8P3 otherate
2. reduction 5 AcSH I hv
6. Cl2 / AcOH / HCI
R1 R1
H S ,OM e H o,,S~'~
Elll~ Elllb
1. doprotect
2 cyclke
1. d-protect
R 1 2. cycleo
~,\N H
0"5~'0
EIV
1. protect
2. base, ~E~
3. dcprotect
R1
E~,NH
O"S~O
E2
CA 02243121 1998-07-14
W O 97127180 PCT~US97/01610
- 106 -
SCHEME 2
1. R2NH2 1. R2NH2
2. ', ~ Rl 2 ~' R1
H 03. r dudionR2NH/~NH 3. r dudion N'~f
EVIII
f~ SOC~ ox llyl chlonde
or SOlm2 \,~
R1 "
2N~\NH O
E6
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 107 -
.. .
SI~Hr ME 3
O O
~, R1R ~ Rl o R2X~
H bnse H R2 X~ o chv~tion R
Route A Routc B
EIX
1. doprotoct
2. baso / heat
R ~NH
R O
E7
1. deprotect
2. baso I hnat
o ,Rl
R2~ N ~OT8DM S
Brp2 H
EX
1. TBDMSCI O
2. deprotect Br 11
Route C 3. base, R2>~~M c
R1
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 108 -
SCHEME IV
t. N~H; ~R72
z 2. doprot~c~X OH
(Z ~ E1 - E7)
R12~,
~O"S,O~OMe ~ O
EXI~ EXlb
N~ ~,N.
~ C~2 N HtBu
EXld
CA 02243l2l l998-07-l4
W O 97/27180 PCTrUS97/01610
- -- 109 --
SCHEME 5
1. allyl bromide
(Z ~ E1 - E6) ~ Z ~~ ~ 2. epo~adee z
HN(R2 / \ 1, (R1)NH2
)2/ ~2. R4X-Act
OH R2 ~ OH R
Z ~N ~R2 Z ~N 'XR~
E10 E9
(X~ C(O) ~rS02
SCHEME 6
Rl 1- O- prote~
ç~ 0~ 2 base /;E- u
EXIV E1 1
Methods for producing the compounds of this
invention are well known in the art of organic
synthesis. Several intermediates are commercially
available, e.g. from Aldrich Chemical Company, Inc.,
Milwaukee, WI. The synthesis of heterocycles El-E6
(Schemes 1 and 2) begins with any protected amino
aldehyde, the preparations for which are well known in
the art from suitably protected amino acids, esters or
alcohols. In the case of the this intermediate,
transient protection of the amino group may be
accomplished by means known in the art (see, e.g. T.W.
Greene and P.G.M. Wuts "Protective Groups in Organic
Synthesis", Second Edition, pp. 309-405 ~1991 John
Wiley and Sons, Inc. New York, NY and E. Gross and J.
CA 02243l2l l998-07-l4
W O 97/27180 PCTAUS97/01610
-- 110 --
Meinhofer "The Peptides, Vol. 3: Protection of
Functional Groups in Peptide Synthesis" pp. 3-88;
~1981 Academic Press, Inc. New York, NY). Carbamates
such as Boc, Fmoc, Alloc and Cbz are particularly
convenient protecting groups, the introduction and
removal of are described in the above references.
The synthesis of E1 is illustrated in Scheme
1. The protected amino aldehyde is treated with an
alpha substituted or alpha, alpha disubstituted amino
ester under typical reductive amination conditions well
known in the art, such ~s sodium cyanoborohydride in a
~solvent mixture of DMF/Acetic acid. The resulting
compound EI is then deprotected and free based with
either a tertiary amine base or potasium carbonate in
methanol to effect cyclization to form EII The
resulting secondary amine may the be protected with
groups (detailed in the references above) such as
benzyl or t-butyloxycarbonyl ~Boc) utilizing conditions
well known in the art to form analogs of E1.
Preparation of E2 is achieved by reaction of
a startlng aldehyde with ethyl
diethylphosphoranylmethanesulfonate and subsequent
reduction of the double bond (see: Gennari et al.,
Angew. Chem. Int. Ed. Engl., 33, pp. 2067-69 (1994)) to
yield compound EIIIa. Cyclization may then be achieved
by deesterification and activation of the sulfonate
moiety as described in Gennari, followed by
deprotection of the nitrogen protection group to yield
the cyclized product EIV. Alternatively, an amino acid
3C may be converted to compound EIIIb using standard
synthetic methods illustrated in Scheme 1. Compound
EIIIb can be cyclized to afford compound EIV. Compound
EIV may then be N-protec~ed, for example, in the
presence of Boc anhydride and DMAP (see: Flynn et al.,
-
CA 02243121 1998-07-14
W O 97127180 PCT~US97/01610
. Or~. Chem. 48, pp. 2~24-26 (1983)), and treated wlth
a non-nucleophilic base such as LDA or
hexamethyldisilazane to generate the anion at the
center alpha to the SO2 moiety. This anion may then be
quenched with a variety of electrophiles and
subsequently deprotected to form the desired analogs of
E2. Alternatively, this anion may be quenched with an
aldehyde to form (after subse~uent dehydration, i.e.,
an aldol-type condensation) an exo-methylene compound
which may then be reduced (i.e., hydrogenation) to form
the desired analogs of E2. Analogously, preparation of
E3 results from a Wittig reaction using
methyl(triphenylposphoranylidene) acetate followed by
simultaneous reduction of the double bond and
cyclization using magnesium metal in methanol (Wei et
al., Tetrahedron Lett., 34(28), pp. 4439-42 (1993)). A
similar N-protection, deprotonation, quench and N-
deprotection scheme, or condensation-reduction scheme,
as described in the preparation of E2, results in
desired analogs of E3. Alternatively, E3 may be
prepared from commercially available EVI. The hydroxyl
group may be activated using commonly available
reagents such as methanesulfonyl chloride or para-
toluenesulfonyl chloride in the presence of a tertiary
amine base. The addition of a nucleophile to displace
the mesylate or tosylate yields EVII (Ackermann et al.,
Helv. Chim. Acta, 73, pp. 122-32 (1990)) which may be
treated as described above to obtain E3.
Methods for the preparation of compounds E4-
E6 are also well known in the art and stem from readily
available protected amino aldehydes. Treatment of
these aldehydes with a variety of amines under
reductive amination conditions well known in the art,
such as sodium cyanoborohydride using DMF/Acetic acid
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 112 -
as a solvent mixture, followed by deprotection of the
primary amine yields diamine EVIII. Intramolecular
cyclization with a variety of activated carbonyl,
dicarbonyl or sulfuryl equivalents in the presence of a
tertiary amine base yields compounds E4-E6. Examples
of activating reagents include but are not limited to
carbonyldiimidazole, phosgene, sulfuryldichloride,
sulfuryldiimidazole, sulfonyl diimide, and oxalyl
chloride.
Methods leading to the production of analogs
of compound E7 are also known in the art (McManus et
~al., J. Med. Chem., 8, pp. 766-76 (1965)3. Scheme 3
exemplifies several potential routes to the synthesis
of compound E7. Any protected amino alcohol may be
deprotonated to form the alkoxide which may be reacted
with a substituted alpha bromo ester to form ether EIX
(route A). Alternatively (route B), EIX may be formed
from activation of a protected amino alcohol with, for
example, methanesulfonyl chloride or para-
toluenesulfonyl chloride in the presence on a tertiaryamine base and subsequent addition of a nucleophile
such as an alkoxide from an alpha hydroxy acid to
displace mesylate or tosylate to yield EIX. Compound
EIX can then be deprotected, free based with a tertiary
2S- amine base or potassium carbonate in methanol, and
heated to effect cyclization to form E7. Alternatively
(route C), E7 may be prepared from a protected amino
alcohol by protection of the hydroxyl group with, for
example, t-butyldimethyl silyl chloride/imidazole to
afford the silyl ether. Subse~uent nitrogen
deprotection and acylation with a alpha bromo acid in
the presence of any number of available coupling agents
(for example dicylcohexylcarbodiimide, other related
carbodiimide reagents or isobutyl chloroformate) or
-
CA 02243121 1998-07-14
W O 97/27180 PCTMS97/01610
- 113 -
acylation with an alpha bromo acid chloride provides
compound EX. Desilylation using, for example,
tetrabutylammonium formate in THF followed by formalion
of the alkoxide with base affords cyclization to E7.
Alternatively, E7 may be prepared from the
corresponding a-methylene compound (i.e., both R2 are H
in E7, the nitrogen may be protected if necessary) by a
multiple deprotonation-alkylation sequence to give an
E7 wherein each R2 is inserted in an independent
alkylation step and each R may be attached to form a
spirocyclic product (i.e., alkylation with a
dihaloalkane).
Schemes 4-6 describe methods for converting
the cyclic compounds E1-E7 into compounds of this
invention. For example, compounds of the type Z,
exemplified by compounds E1-E7, may be deprotonated
and reacted with a functionalized epoxide to generate
the desired compounds as described in Scheme 4.
Several of the described epoxides are readily
synthesized via methods well known in the art (Maligres
et al,, Tetrahedron Lett., 36, pp. 2195-98 (1995)).
optionally, further modification of the compounds may
be performed subsequent to epoxide opening using
reactions and materials well known in the art. For
example, subsequent to epoxide opening utilizing
example EXIb deprotection of the carbamate allows
further modification of the unmasked amine.
Alternatively, as shown in Scheme 5,
compounds EZ may be converted to the desired products
in a more stepwise fashion. Compounds EZ may be
deprotonated using, for example, sodium hydride in DMF
and treated with a three carbon based epoxide to
generate epoxide EXII. Examples of such reagents
include, but are not limited to, epibromohydrin,
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 114 -
epichlorohydrin and glycidyl tosylate. Several other
potential methods for preparing compounds o~ the type
EXII are well known in the art, for example, the anion
of z may be reacted with allyl bromide or allyl iodide
to form an allyl intermediate, which may subse~uently
be oxidized to form the desired epoxide. Several
epoxidation conditions for the generation of either
racemic or chiral epoxides are well known in the art.
Epoxide EXII may then treated with an amine and
suse~uently carbonylated or sulfonated using activated
species well known in the art to generate final
~compounds of the type E9. Alternatively EXII may be
reacted with a functionalized secondary amine followed
by optional manipulation of R2 to produce compounds of
the type E10. One example of such manipulation is
reaction of EXII with the known Boc piperazine EXIII
(Dorsey et al., J. Med. Chem.,37, pp. 3443-51 (1999)).
Subsequent to epoxide opening, the Boc group may be
removed and the unmasked secondary amine may be further
manipulated by reaction with various electrophiles to
form the desired product.
Scheme 6 describes a method for introduction
of electrophiles into comounds of the type EXIV. Said
compounds may be protected with a variety of protecting
groups, for example t-butyldimethylsilyl triflate, to
mask the secondary hydroxyl group ~ollowed by treatment
with a non-nucleophilic base such as lithium
diisopropylamide or hexamethyldisilyzane to generate
the anion alpha to the carbonyl. Various electrophiles
may then be added to substitute the position alpha to
the carbonyl, or alternatively an aldol-type
condensation-reduction scheme may be employed.
Deprotection of the secondary hydroxyl then yields the
desired product.
CA 02243l2l l998-07-l4
W O 97/2718~ PCTAUS97/01610
- 115 -
As can be appreciated by the skilled artisan,
the above synthetic schemes are not intended to
comprise a comprehensive list of all means by which the
compounds described and claimed in this application may
be synthesized. Further methods will be evident to
those of ordinary skill in the art.
Moreover, the determination of the optimum
overall scheme, as well as the choice of reagents and
reactions used to carry out the various steps in a
given scheme will be based upon factors that are
readily apparent to those of skill in the art. These
factors include the identity o~ the compound to be
produced, the efficiency of the individual steps and
schemes in producing that compound in terms of overall
15 yield, time, and cost and availability of reagents. It
will therefore be apparent that some routine
experimentation may be required in determining the
optimum scheme to produce certain compounds of this
invention.
It should be understood that the compounds of
this invention may be modified by appending appropriate
functionalities to enhance selective biological
properties. Such modifications are known in the art
and include those which increase biological penetration
into a given biological compartment (e.g., blood,
lymphatic system, central nervous system), increase
oral availability, increase solubility to allow
administration by injection, alter metabolism and alter
rate of excretion.
The compounds of this invention are
characterized by a superior ability to inhibit protease
activity and viral replication, particularly aspartyl
protease activity. These compounds are especially well
suited for inhibiting HIV aspartyl protease. We
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 116 -
believe that this activity is due to specific steric
and electronic interactions between the protease and
compounds of this invention. This belief stems from
our analysis of the structural basis for the activity
of compounds of this invention, in view of the known
crystal structures of HIV protease and bound
inhibitors, such as the structure reported in Miller et
al. "Structure of Complex of Synthetic HIV-1 Protease
with a Substrate-Based Inhibitor at 2.3 ~ Resolution",
Science, vol. 246, pp. 1149-1152 (1989), which is
incorporated herein by reference, as well as structures
determined in our laboratories.
The novel compounds of the present invention
are excellent ligands for aspartyl proteases,
particularly HIV-1 and HIV-2 proteases. Accordingly,
these compounds are capable of targeting and inhibiting
late stage events in HIV replication, i.e., the
processing of the viral polyproteins by HIV encoded
proteases. Such compounds inhibit the proteolytic
processing of viral polyprotein precursors by
inhibiting aspartyl protease. Because aspartyl
protease is essential for the production of mature
virions, inhibition of that processing effectively
blocks the spread of virus by inhibiting the production
2~ of infectious virions, particularly from chronically
infected cells. Compounds according to this invention
advantageously inhibit the ability of the HIV-l virus
to infect immortalized human T cells over a period of
days, as determined by an assay of extracellular p24
antigen -- a specific marker of viral replication.
Other anti-viral assays have confirmed the potency of
these compounds.
~ The compounds of this invention may be
employed in a conventional manner for the treatment of
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 117 -
viruses, such as HIV and HTLV, which depend on aspartyl
proteases for obligatory events in their life cycle.
Such methods of treatment, their dosage levels and
requirements may be selected by those of ordinary skill
in the art from available methods and techniques. For
example, a compound of this invention may be combined
with a pharmaceutically acceptable adjuvant for
administration to a virally-infected patient in a
pharmaceutically acceptable manner and in an amount
effective to lessen the severity of the viral infection
or to alleviate pathological effects associated with
HIV infection or i~mllnosuppression such as
opportunistic infections or various cancers, tumors,
CMV retinitis, candida infections, maternal fetal
transmission, and AIDS related dementia,.
Alternatively, the compounds of this
invention may be used in prophylactics and methods for
protecting individuals against viral infection during a
speci~ic event, such as childbirth, or over an extended
period of time. The compounds may be employed in such
prophylactics either alone or together with other
antiretroviral agents to enhance the efficacy of each
agent. As such, the novel protease inhibitors of this
invention can be administered as agents for treating or
preventing HIV in~ection in a mammal.
The compounds of formula I, especially those
having a molecular weight of less than about 700
g/mole, may be readily absorbed into the bloodstream of
mammals upon oral administration. Compounds of formula
I having a molecular weight of less than about 600
- g/mole and aqueous solubility of greater than or equal
to 0.1 mg/mL are most likely to demonstrate high and
consistent oral availability. This surprisingly
impressive oral availability makes such compounds
CA 02243121 1998-07-14
W O 97/27180 PCTAU~97/01610
- 118 -
excellent agents for orally-administered treatmen~ and
prevention regimens against HIV infection.
The compounds o~ this invention may be
administered to a healthy or HIV-infected patient
either as a single agent or in combination with other
anti-viral agents which interfere with the replication
cycle of HIV. By administering the compounds of this
invention with other anti-viral agents which target
different events in the viral life cycle and which
target different viral substrains with varying
susceptability to specific agents, the therapeutic
effect of these compounds is potentiated. For
instance, the co-administered anti-viral agent can be
one which targets early events in the life cycle of the
virus, such as cell entry, reverse transcription and
viral DNA integration into cellular DNA. Anti-HIV
agents targeting such early life cycle events include,
didanosine (ddI), dideoxycytidine (ddC), d4T,
zidovudine ~AZT), 3TC, 935U83, 1592U89, 524W91,
polysulfated polysaccharides, sT4 (soluble CD4),
ganiclovir, trisodium phosphonoformate, eflornithine,
ribavirin, acyclovir, alpha interferon and tri-
methotrexate. Additionally, non-nucleoside inhibitors
of reverse transcriptase, delavirdine (U90) or
2~ nevirapine, may be used to potentiate the effect of the
compounds of this invention, as may viral uncoating
inhibitors, inhibitors of trans-activating proteins
such as tat or rev, or inhibitors of the viral
integrase.
3Q Combination therapies according to this
invention exert an additive or synergistic effect in
inhibiting HIV replication because each component agent
of the combination acts on a different site of HIV
replication or on different strains of virus present in
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 119 -
an infectious population. The use of such combination
therapies may also advantageously reduce the dosage of
a given conventional anti-retroviral agent which would
be required for a desired therapeutic or prophylactic
effect, as compared to when that agent is administered
as a monotherapy. Such combinations may reduce or
eliminate the side effects of conventional single anti-
retroviral agent therapies, while not interfering with
the anti-retroviral activity of those agents. These
combinations reduce potential of resistance to single
agent therapies, while minimizing any associated
~toxicity.
Advantages of combining HIV protease
inhibitors may include viral population effects,
whereby certain members of a virus population which
shpw reduced sensitivity to one protease inhibitor may
be fully sensitive to another inhibitor or may in fact
have enhanced sensitivity to the second inhibitor.
Alternatively or in addition, administration of two or
more different inhibitors may be used to reduce
specific toxicities associated with a single agent.
This advantage of combination therapy also applies to
co-administration of the protease inhibitor of this
invention with other antiviral agents. Alternatively
or in addition, co-administration of more than one
protease inhibitor may lower the rate of metabolic
inactivation of the compounds of this invention, for
- instance, by inhibiting enzymatic systems such as
cytochrome P450, or esterases or the like. In
particular, co-administration of compounds of this
~ invention with protease inhibitors such as ritonavir or
other agents such as ketoconazole, grapefruit juice and
antiulcer medications such as ~2-blockers, which
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 120 -
inhibits cytochrome P450 3A4, may advantageously enhance
their biological half-life.
These combinations may also increase the
efficacy of the conventional agent without increasing
the associated toxicity. Compounds of this invention in
combination with other anti-HIV agents may act in an
additive or synergistical manner in preventing the
replication of HIV in human T cells. Preferred
combination therapies include the administration of a
compound of this invention with AZT, ddI, ddC, d4T,
3TC, 935U83, 1592U89, 524W91 or a combination thereof.
Alternatively, the compounds of this
invention may also be co-administered with other HIV
protease inhibitors such as VX-g78 (Vertex, also known
as 141W94 (Glaxo-Wellcome) and KVX-478 (Kissei)),
- sa~uinavir (Ro 31-8959, Roche), indinavir (L-735,524,
Merck)), ritonavir (~3T 538, Abbott), nelfinavir (AG
1343, Agouron), palinavir (Bila 2011 BS), U-103017
(Upjohn), XM 412 (DuPont Merck), XM 450 (DuPont Merck),
BMS 186318 (Bristol-Meyers S~uibb), CPG 53,437 (Ciba
Geigy), CPG 61,755 (Ciba Geigy), CPG 70,726 (Ciba
Geigy), ABT 378 (Abbott), GS 3333 (Gilead Sciences), GS
3403 (Gilead Sciences), GS 4023 (Gilead Sciences), GS
4035 (Gilead Sciences), GS 4145 (Gilead Sciences), GS
4234 (Gilead Sciences), and GS 4263 (Gilead Sciences)
or prodrugs of these or related compounds to increase
the effect of therapy or prophylaxis against various
viral mutants or members of HIV quasi species.
We prefer administering the compounds of this
invention as single agents or in combination with
retroviral reverse transcriptase inhibitors, such as
nucleoside derivatives, or other HIV aspartyl protease
inhibitors, including multiple combinations comprising
from 3-5 agents. We believe that the co-administration
~.
CA 02243l2l l998-07-l4
W O 97/27180 PCT~US97/01610
- 121 -
of the compounds of this invention with retroviral
reverse transcriptase inhibitors or HIV aspartyl
protease inhibitors may exert a substantial addi'ive or
synergistic effect, thereby preventing, substantially
reducing, or completely eliminating viral replication
or infection or both, and symptoms associated
therewith. Particularly preferred is administration of
a combination of a compound of formula I, 3TC and
zidovudine ~AZT). Also preferred are administrations
of combinations of a compound of formula I and 1592U89,
or of compounds of formula I with VX-478, optionally
with one or more reverse transcriptase inhibitors,
paarticularly, AZT, 3TC and 1592U89.
The compounds of this invention can also be
administered in combination with immunomodulators and
immunostimulators (e.g., bropirimine, anti-human alpha
interferon antibody, IL-2, GM-CS~, interferon alpha,
diethyldithiocarbamate, tumor necrosis factor,
naltrexone, tuscarasol, and rEPO); and antibiotics
(e.g., pentamidine isethiorate) to prevent or combat
infection and disease associated with HIV infections,
such as AIDS, ARC and HIV-associated cancers.
When the compounds of this invention are
administered in combination therapies with other
agents, they may be administered sequentially or
concurrently to the patient. The additional agents may
be administered separately, as part of a multiple dose
regimen, from the compounds of this invention.
Alternatively, those agents may be part of a single
dosage form, mixed together with the compounds of this
~ invention in a single composition. The pharmaceutical
compositions according to this invention may comprise a
combination of an aspartyl protease inhibitor of this
CA 02243121 1998-07-14
W O 97127180 PCT~US97/01610
- 122 -
invention and one or more therapeutic or prophylactlc
agents.
Although this invention focuses on the use of
the compounds disclosed herein for preventing and
treating HIV infection, the compounds of this invention
can also be used as i~hibitory agents for other viruses
which depend on similar aspartyl proteases for
obligatory events in their life cycle. These viruses
include other AIDS-like diseases caused by
retroviruses, such as simian immunodeficiency viruses,
HTLV-I and HTLV-II. In addition, the compounds of this
invention may also be used to inhibit other aspartyl
proteases, such as renin, pepsin, cymosin, RSV
protease, AMV protease, SIV protease and FIV protease,
and in particular, other human aspartyl proteases,
including renin, and aspartyl proteases that process
endothelin precursors.
Pharmaceutical compositions of this invention
comprise any of the compounds of the present invention,
and pharmaceutically acceptable salts thereof, with any
pharmaceutically acceptable carrier, adjuvant or
vehicle. Pharmaceutically acceptable carriers,
adjuvants and vehicles that may be used in the
pharmaceutical compositions of this invention include,
but are not limited to, ion exchangers, alumina,
aluminum stearate, lecithin, self-emulsifying drug
delivery systems ~SEDDS) such as d-~-tocopherol
polyethyleneglycol lO00 succinate, surfactants used in
pharmaceutical dosage forms such as Tweens or other s
3C similar polymeric delivery matrlces, serum proteins,
such as human serum albumin, polyethyleneglycol
polymers such as PEG-400, buffer substances such as
phosphates, glycine, sorbic acid, potassium sorbate,
partial glyceride mixtures of saturated vegetable fatty
. .
-
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 123 -
- acids, water, salts or electrolytes, such as protamine
sulfate, disodium hydrogen phosphate, potassium
- hydrogen phosphate, sodium chloride, zinc salts,
colloi~al silica, magnesium trisilicate, polyvinyl
pyrrolidone, cellulose-based substances, polyethylene
glycol, sodium carboxymethylcellulose, polyacrylates,
waxes, polyethylene-polyoxypropylene-bloc~ polymers,
polyethylene glycol and wool fat. Cyclodextrins such
as ~-, ~-, and y-cyclodextrin, or chemically modlfied
derivatives such as hydroxyalkylcyclodextrins,
including 2- and 3-hydroxypropyl-~-cyclodextrins, or
~ other solublized derivatives may also be advantageously
used to enhance delivery of compounds of formula I.
The pharmaceutical compositions of this
invention may be administered orally, parenterally, by
- inhalation spray, topically, rectally, nasally,
buccally, vaginally or via an implanted reservoir. We
prefer oral administration or administration by
injection. The pharmaceutical compositions of this
invention may contain any conventional non-toxic
pharmaceutically-acceptable carriers, adjuvants or
vehicles. In some cases, the pH of the formulation may
be adjusted with pharmaceutically acceptable acids,
bases or buffers to enhance the stability of the
formulated compound or its delivery form. The term
parenteral as used herein includes subcutaneous,
intracutaneous, intravenous, intramuscular, intra-
articular, intrasynovial, intrasternal, intrathecal,
4 intralesional, and intracranial injection or infusion
techniques.
The pharmaceutical compositions may be in the
form of a sterile injectable preparation, for example,
às a sterile injectable aqueous or oleaginous
suspension. This suspension may be formulated
CA 02243121 1998-07-14
W O 97/27180 PCTr~S97/01610
- 124 -
according to techniques known in the art using suitable
dispersing or wetting agents (such as, for example,
Tween 80) and suspending agents. The sterile
injectable preparation may also be a sterile injectable
solution or suspension in a non-toxic parenterally-
- acceptable diluent or solvent, for example, as a
solutlon in 1,3-butanediol. Among the acceptable
vehicles and solvents that may be employed are
mannitol, water, Ringer~s solution and isotonic sodium
chloride solution. In addition, sterile, fixed oils
are conventionally employed as a solvent or suspending
medium. For this purpose, any bland fixed oil may be
~ employed including synthetic mono- or diglycerides.
Fatty acids, such as oleic acid and its glyceride
derivatives are useful in the preparation of
injectables, as are natural pharmaceutically-acceptable
oils, such as olive oil or castor oil, especially in
their polyoxyethylated versions. These oil solutions
or suspensions may also contain a long-chain alcohol
diluent or dispersant such as carboxymethyl cellulose
or similar dispersing agents which are commonly used in
the formulation of pharmaceutically acceptable dosage
forms such as emulsions and or suspensions. Other
commonly used surfactants such as Tweens and Spans
and/or other similar emulsifying agents or
bioavailability enhancers which are commonly used in
the manufacture of pharmaceutically acceptable solid,
liquid, or other dosage forms may also be used for the
purposes of formulation.
The pharmaceutical compositions of this
invention may be orally administered in any orally
acceptable dosage form including, but not limited to,
hard or soft gelatin capsules, tablets, emulsions and
aqueous suspensions, dispersions and solutions. In the
CA 02243l2l l998-07-l4
W O 97127180 PCT~US97/01610
- 125 -
case of tablets for oral use, carriers which are
commonly used include lactose and corn starch.
_ Lubricating agents, such as magnesium stearate, are
also typically added. For oral administration in a
capsule form, useful diluents include lactose and dried
corn starch. When aqueous suspensions and/or emulsions
are administered orally, the active ingredient may be
suspended or dissolved in an oily phase combined with
emulsifying and/or suspending agents. If desired,
certain sweetening and/or flavoring and/or coloring
agents may be added.
The pharmaceutical compositions of this
invention may also be administered in the form of
suppositories for rectal administration. These
compositions can be prepared by mixing a compound of
this invention with a suitable non-irritating excipient
which is solid at room temperature but liquid at the
rectal temperature and therefore will melt in the
rectum to release the active components. Such
materials include, but are not limited to, cocoa
butter, beeswax and polyethylene glycols.
Topical administration of the pharmaceutical
compositions of this invention is especially useful
when the desired treatment involves areas or organs
readily accessible by topical application. For
application topically to the skin, the pharmaceutical
composition should be formulated with a suitable
ointment containing the active components suspended or
dissolved in a carrier with suitable emulsifying
agents. Carriers for topical administration of the
c compounds of this invention include, but are not
limited to, mineral oil, liquid petroleum, white
petroleum, propylene glycol, polyoxyethylene
polyoxypropylene compound, emulsifying wax and water.
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 126 -
Alternatively, the pharmaceutical composition can be
formulated with a suitable lotion or cream containing
the active compound suspended or dissolved in a
carrier. Suitable carriers include, but are not
limited to, mineral oil, sorbitan monostearate,
polysorbate 60, cetyl esters wax, cetearyl alcohol,
2-octyldodecanol, benzyl alcohol and water. The
pharmaceutical compositions of this invention may also
be topically applied to the lower intestinal tract by
rectal suppository formulation or in a suitable enema
formulation. Topically-transdermal patches are also
included in this invention.
The pharmaceutical compositions of this
invention may be administered by nasal aerosol or
inhalation. Such compositions are prepared according
to techniques well-known in the art of pharmaceutical
formulation and may be prepared as solutions in saline,
employing benzyl alcohol or other suitable
preservatives, absorption promoters to enhance
2D bioavailability, fluorocarbons, and/or other
solubilizing or dispersing agents known in the art.
Dosage levels of between about 0.01 and about
100 mg/kg body weight per day, preferably between about
0.5 and about 75 mg/kg body weight per day of the
active ingredient compound are useful in the prevention
and treatment of viral infection, including HIV
infection. Typically, the pharmaceutical compositions
of this invention will be administered from about 1 to
about 5 times per day or alternatively, as a continuous
3~ infusion. Such administration can be used as a chronic
or acute therapy. The amount of active ingredient that
may be combined with the carrier materials to produce a
single dosage form will vary depending upon the host
treated and the particular mode of administration. A
CA 02243121 1998-07-14
W O 97/27180 PCT~US97101610
- 127 -
typical preparation will contain from about 5~ to about
95~ active compound (w/w). Preferably, such
preparations contain from about 2C% to about 80~- active
compound.
Upon improvement of a patient's condition, a
maintenance dose of a compound, composition or
combination of this invention may be administered, if
necessary. Subsequently, the dosage or frequency of
administration, or both, may be reduced, as a function
of the symptoms, to a level at which the improved
condition is retained when the symptoms have been
-alleviated to the desired level, treatment should
cease. Patients may, however, require intermittent
treatment on a long-term basis upon any recurrence of
disease symptoms.
As the skilled artisan will appreciate, lower
or higher doses than those recited above may be
required. Specific dosage and treatment regimens for
any particular patient will depend upon a variety of
factors, including the activity of the specific
compou~d employed, the age, body weight, general health
status, sex, diet, time of administration, rate of
excretion, drug combination, the severity and course of
the infection, the patient's disposition to the
infection and the judgment of the treating physician.
The compounds of this invention are also
useful as commercial reagents which effectively bind to
aspartyl proteases, particularly HIV aspartyl protease.
As commercial reagents, the compounds of this
invention, and their derivatives, may be used to block
- proteolysis of a target peptide or may be derivatized
to bind to a stable resin as a tethered substrate for
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 128 -
affinity chromatography applications. For example, a
compound of formula I may be tethered to an affin ty
column to purify recombinantly produced HIV protease.
Derivatization of the compounds of this invention to
produce affinity chromatography resins and the methods
used to purify proteases using such resins are well
known and within the skill of the art. These and other
uses which characterize commercial aspartyl protease
inhibitors will be evident to those of ordinary skill
in the art. (See: Rittenhouse, J. et al. Biochem.
Biophys. Res. Co~lln. 171, p. 60 (1990) and Heimbach,
- J.C. et al. Ibid 164, p. 955 (1989)).
In order that this invention be more fully
understood, the following examples are set forth.
These examples are for the purpose of illustration only
and are not to be construed as limiting the scope of
the invention in any way.
General M~terials ~nd Metho~s
All temperatures are recorded in degrees
Celsius. Thin layer chromatography (TLC) was carried
out using 0.25 mm thick E. Merck silica gel 60 F254
plates and elution with the indicated solvent system.
Detection of the compounds was carried out by treating
the plate with an appropriate visualizing agent, such
as 10% solution of phosphomolybdic acid in ethanol or a
0.1% solution of ninhydrin in ethanol, followed by
heating, and/or by exposure to W light or iodine
vapors when appropriate. Thick layer silica gel
chromatography was also carried out using E. Merck 60
F254 plates ("prep plates") of 0.5, 1.0, or 2.0 mm
thickness. Following development of the plate, the
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 129 -
band of silica containing the desired compound was
isolated and eluted with-an appropriate solvent.
Analytical HPLC was carried out using a Water's Delta
Pak, 5 ~M silica, C18 reversed-phase column, 3.9 mm ID
x 15 cm L with a flow rate of 1.5 mL/min using the
following table:
Mobile phase: A = 0.1% CF3C02H in H20
B = O. 1% CF3C02H in CH3CN
Gradient: T = 0 min., A (95~), B (5%)
T = 20 min., A (0%), B (100~)
T = 22.5 min., A (0~), B (100%)
Preparative HPLC was also carried out using C18
reversed-phase media. HPLC retention times were
recorded in minutes. NMR spectral data was recorded
using a Bruker AMX500, equipped with either a reverse
or QNP probe, at 500 MHz, and was taken in the
indicated solvent.
We have measured the inhibition constants of
each compound against HIV-l protease using the method
described essentially by M.W. Pennington et al.,
Pe~ti~es 1990, Giralt, E. and D. Andreu, Eds., Escom,
Leiden, Netherlands (1991); and the method described
essentially by Partaledis et al., J. Virol., 69, pp.
5228-35 (1995).
Compounds of invention were tested for their
antiviral potency in several virological assays. In
the first assay, the compounds were added as a solution
in dimethylsulfoxide (DMSO) to a test cell culture of
~ CCRM-CEM cells, a strain of CD4 human T-cell lymphoma
cells, previously acutely infected with HIVIIIb using
CA 02243121 1998-07-14
W O 97127180 PCTrUS97/01610
- 130 -
standard protocols (see Meek, T. D. et al., "Inhibition
of HIV-1 protease in infected T-lymphocytes by
synthetlc peptide analogues", Nature, 343, p. 90
(1990).
The effect of the compounds on inhibiting the
replication of the virus was measured by determining
the HIV extracellular p24 antigen concentration using a
commercial enzyme immunoassay (obtained from Coulter
Corporation, Hialeah, FL).
Antiviral activity may also be measured in a
separate assay in MT4 cells. Antiviral HIV activity
and compound-induced cytotoxicity were measured in
parallel by means of a propidium iodide based procedure
in the human T-cell lymphotropicvirus transformed cell
line MT4. Aliquots of the test compounds were serially
diluted in medium (RPMI 1640, 10% fetal calf serum
(FCS), and gentamycin) in 96-well plates (Costar 3598)
using a Cetus Pro/Pette. Exponentially growing MT4
cells were harvested and centrifuged at 1000 rpm for 10
minutes in a Jouan centrifuge (model CR 4 12). Cell
pellets were resuspended in fresh medium ~RPMI 1640,
20~ FCS, 20% IL-2, and gentamycin) to a density of 5 x
105 cells/ml. Cell aliquots were infected by the
addition of HIV-1 (strain IIIB) diluted to give a viral
multiplicity of infection of 100 x TCID~0. A similar
cell aliquot was diluted with medium to provide a
mockinfected control. Cell infection was allowed to
proceed for 1 hour at 37 ~C in a tissue culture
incubator with humidified 5~ CO2 atmosphere. After the
1 hour incubation the virus/cell suspensions were
diluted 6-fold with fresh medium, and 125 ~1 of the
cell suspension was added to each well of the plate
.
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 131 -
containing prediluted compound. Plates were then
placed in a tissue culture incubator with humidi ied 5
C~2 for 5 days. At the end of the incubation period,
27 ul of 5% Nonidet-40 was added to each well of the
incubation plate. After thorough mixing with a Costar
multitip pipetter, 60 ul of the mixture was transferred
to filter-bottomed 96-wellplates. The plates were
analyzed in an automated assay instrument (Pandex
Screen Machine, ~axter Biotechnology Systems). The
assay makes use of a propidium iodide dye to estimate
the DNA content of each well. The antiviral effect of
a test compound is réported as an IC50, i.e. the
inhibitory concentration that would produce a 50~
decrease in the HIV induced cytopathic effect. This
effect is measured by the amount of test compound
required to restore 50% of the cell growth of HIV-
infected MT-4 cells compared to uninfected MT-4 cell
controls.
References:
1. Averett, D.R. 1989. Anti-HIV compound assessment
by two novel high capacity assays. J. Virol. Methods
23: 263-276.
2. Schwartz, O., et al. 1988. A rapid and simple
colorimetric test for the study of anti-HIV agents.
~IDS Res. and Hl~m~n Retroviruses, 4 (6): 441-447.
3. Daluge, S.M., et al. 1994. 5-chloro-2',3'-
- dedeoxy-3'fluorouridine (935U83), a selective anti-
human immunodeficiency virus agent with an improved
.
CA 02243121 1998-07-14
W O97/27180 PCTrUS97/01610
- 132 -
. .
metabolic and toxicological profile. Antimicro.
Aaents ~n~ Chemother., 38(7):1590-1603.
4. Dornsife, R.E., et al. 1991. Anti-human
immunodeficiency virus synergism by zidovudine (3'-
azidothymidine) and didanosine (dideoxyinosine)
contrasts with their additive inhibition of normal
human marrow progenitor cells. Pnti mi cro. Agents and
Chemother., 35~2): 322-328.
Depending on the cell type and the desired
lQ readout, syncytia formation, reverse-transcriptase (RT)
activity, or cytopathic effect as assayed by a dye
uptake method may also be used as readouts of antiviral
activity. See H. Mitsuya and S. Broder, "Inhibition of
the Ln vitro infectivity and cytopathic effect of human
T-lymphotropic virus type III/lymphoadenopathy-
associated virus (HTLV-III/LAV) by 2',3'-
dideoxynucleosides", Proc. Natl. Acad. Sci. USA,
vol. 83, pp. 1911-1915 (1986).
Insofar as the compounds of this invention
are able to inhibit the replication of the HIV virus in
CD4 cells of human lineage, they are of evident
clinical utility for the treatment of XIV infection.
These tests are predictive of the compounds ability to
inhibit HIV protease n vivo.
CA 02243l2l l998-07-l4
W O 97/27180 PCTrUS97/01610
- 133 -
Synthetic Ex~m~rles
.
= Fx~mrle 1
A.
oxalyl chloride [~
-- DMSO
~NHB~c ~ ~~NHBoc
~:)H -78~C h
N-(t-butoxycarbonyl)-L-phenylalaninol;
251.3 g/Mol lO.Og 39. 8 mmol
DMSO 78 g/Mol 3.80mL 49.0 mmol
oxalyl chloride 126.9 g/Mol 3.82mL 43.8mmol
triethylamine lOl g/Mol 23.OmL 160mmol
methylene chloride 200 mL
The oxalyl chloride was added dropwise to a
solution of DMSO in methylene chloride at -78 ~C.
After stirring for 10 minutes, the alcohol was added as
a solution in methylene chloride. The reaction was
then stirred at -78 ~C for 45 minutes. At this time
the triethylamine was added and a white precipitate
formed. The reaction was then stirred 45 minutes at
-78 ~C and 45 minutes at O ~C. The reaction was then
quenched by the addition of a solution of 90g of citric
acid in 300 mL of water. The organic portion of the~ 20 reaction was then washed by (2 x 80 mL) of both
saturated sodium bicarbonate and brine. The combined
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 134 -
organic layers were then dried over sodium sulfate,
filtered and concentrated in vacuo to leave a white
solid. The aldehyde was then used without further
purification in the reductive amination.
B.
~NH2 ~,
~ NaCNBH; ,~--N----NHBoc
H 1 HOAc/DMF 2
allyl amine 57 g/Mol 6.0 mL 160 mmol
aldehyde est. 39. 8 mmol
sodium cyanoborohydride 62.8g/Mol 4.0g 6.4 mmol
DMF - 180 mL
acetic acid (glacial) 1.8 mL
The aldehyde of Example lA was dissolved in 180 mL
of DMF at 25 ~C. This was followed by addition of the
aldehyde and 1.8 mL of acetic acid respectively. After
2 hours sodium cyanoborohydride was added, as a solid.
The reaction was then stirred at 25 ~C for 12 hours.
The reaction was then quenched by the addition of 50 mL
of saturated sodium bicarbonate, and after 10 min.
diluted by 100 mL of diethyl ether. The organic
portion was then washed by (2 x 50 mL) of both
saturated sodium bicarbonate and brine. The combined
organic layers were then dried over magnesium sulfate,
filtered and concentrated in vacuo. The crude oil was
CA 02243l2l l998-07-l4
W O 97/27180 PCTrUS97/01610
- 135 -
- purified by silica gel chromatography eluting with 30 ~
ethyl acetate: hexane to provide 8.8 g of product (29.8
- mmol, 75~).
.~ ".-~
~N~NHBoc 1) HCI ~Nr~
2) TEA, C~l o 2
Boc amine 291 g/Mol 6.8g 23.4 mmol
HCl/dioxane 4 N HCl 15 mL
deprotected diamine-2HCl 3.83g 14.7 mmol
carbonyl diimidazole 162.15g/Mol 2.77g 17.1 mmol
triethylamine 12.7mL 179 mmol
methylene chloride 550mL 0.03 M
The Boc amine of Example lB was stirred in 15 mL
of 4N HCl at 25 ~C for 1.5 hours. The reaction mixture
was then concentrated in vacuo to provide a white
foaming solid. 3.83 mg of the deprotected diamine was
dissolved in 500 mL of methylene chloride. To this,
triethyl amine was added. After stirring for 20
minutes, CDI was added ~solid). The reaction was then
stirred for 24 hours. This was followed by
concentration in vacuo. The crude material was
purified by silica gel chromatography, eluting with
ethyl acetate, to provide 2.15 g (67 %) of the desired
allyl urea.
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 136 -
~ xam~le 2
A.
O Ph O tohens:CH2C12 0 Ph
H~H 2: 3 Ph~OJ~¢oMe
~ 1 2
1 aldehyde 1.0 equiv.,
2 methyl 1.05 eq.
(triphenylphosphoranyli
dene)acetate
3 toluene 80mL
4 methylene chloride 120mL
Combine 7.9g of (S)-N-Boc-amino-3-phenyl-1-propanal,
40mL of anhydrous toluene and 60mL of anhydrous
methylene chloride. Add 9.8g of the ylide followed by
20mL o~ toluene and 60mL of methylene chloride. Stir
overnight at room temperature. After approximatly 18
hours the solvent was removed in vacuo and the residue
was purified by flash chromatography (EtOAc/Hexane) to
give 7.lg(77%) o~ the desired ester.
B.
Ph~ O ~ N ~ OM Mg(tumln9s)HCI /~~
CA 02243l2l l998-07-l4
W O 97/27180 PCTrUS97/01610
- 137 -
1 ester 4.5g, 1.0 equiv.
2 magnesium turnings (Aldrich) 3. 2g 10.0 ea
' 3 2N HCl @ 10 eq.
s
To a solution of ester 1 in anhydrous methanol at 0 ~C
was added Mg turnings with stirring under N2. Bubbling
became evident within 1 hour. The reaction was then
stirred at 0 ~C for -2. 5 hours then allowed to warm to
RT over~ight (TLC (95: 5, CH2C12:MeOH) showed reaction
complete. st. mat. Rf = .84, prod. Rf = .25). The
reaction was cooled to 0 ~C, neutralized with 2 1~ HCl,
diluted with water, and the volume reduced in vacuo.
The remaining aqueous layer was extracted with 3
portions of methylene chloride and the combined organic
layers were washed with brine, dried (MgSO4), filtered,
and concentrated in vacuo.. The residue was then
purified by silica get flash chromatography (CH2C12 --
>3% MeOH/CH2C12) to yield desired lactam product
(1.74g, 75~ yield).
Literature reference: Tetrahedron. Lett., 1993, 34
20(28), pp. 4439-4442.
Boc anhydride
-- TEA, DMAP .--
~NH c~t2cl2lcH3cN ~\NBoc
O O
1 2
CA 02243121 1998-07-14
W O97/27180 PCTrUS97/01610
- 138 -
1 lactam from 2B 1.0 equiv., 1.7g
2 BOC anhydride 2.5 equiv., 5.2g
3 triethylamine 2.0 equiv, 2.7mL
4 DMAP 1.2 equiv, 1.4g
., ,
Lactam 1 was dissolved in methylene chloride ~20mL) and
to this solution was added a solution of Boc anhydride
2 in CH2Cl2 (10 ml)followed by triethylamine (2 eq) and
DMAP (1.2 eq). After stirring for 4 hours at room
temperature the reaction was refluxed for 4 hours and
lD after this time, an additional 1.0g of Boc anhydride in
acetonitrile (20mL) and 700uL of triethylamine were
added. The reaction was stirred for 15 hours at room
temperature. (TLC ~95:5, CH2Cl2 : MeOH) Rf (st mat.) =
.31. Rf(prod) = .66.) The solvent was then removed
in vacuo and the residue was partitioned between
methylene chloride and water. The organic layer was
washed with water and brine, dried (MgSO4) and
filtered. The dried organic layer was then
concentrated in vacuo and the residue was puri~ied by
silica gel chromatagraphy (CH2C12) to yield desired boc
lactam 2 (2.3g, 86%).
-- LDA
Allylbromide r-~
THF ~ NBoc
O O
1 2
CA 02243l2l l998-07-l4
W O 97127180 PCTrUS97/01610
- 139 -
1 BOC-lactam from 2C 1.0 ëquiv.,85 mg
2 Allyl Bromide, (Aldrich) 1.8 equiv. , 51uL
3 LDA, 1. 29M (Aldrich) 2.0 equiv , 420 uL
Boc-lactam 1 was dissolved in dry THF and cooled to
-78 ~C and to this solution was added LDA via syringe.
After stirring for 40 min. at -78 ~C, allyl bromide
was added via syringe and the reaction was stirred for
3 hours after which time an additional amount of allyl
bromide (17 ul) was added. The reaction was then
stirred at -78 ~C for 4 hours (TLC (5:95, MeOH:CH2Cl2)
Rf (st mat.) = .34. Rf(2 diast.) = .55 and.61). The
reaction was then quenched with lmL saturated NaCl
solution, and partitioned between saturated sodium
bicarbonate and ethyl acetate. The organic layer was
then washed with water and brine, dried (MgSO4),
filtered and concentrated in vacuo. The residue was
purified by silica get chromatography to yield
allylated product 2 (47mg, 48% yield).
E.
D 1' LDA, benzyl bromide 0~
A mixture of diisopropylamine (4.6 mL, 3 eq) and THF
- (10 mL) was cooled to -78 ~C, and to this solution was
added n-butyl lithium (1.4 eq) via syringe. This
CA 02243121 1998-07-14
WO 97/27180 PCTrUS97/01610
- 140 -
mixture was warmed to -10 ~C and stirred for 40 min,
after which time the mixture was cooled back to -78 ~C
A solution o~ Boc lactam 1 (3.0 g, 1 eq) in THF (15 mL
total) was added. The reaction mixture was then
stirred at -78 ~C for 40 ~in ~ollowed by the addition
of benzyl bromide (1.45 mL, 1.1 eq) via syringe . After
stirring for 2.5 hours at -78 ~C, the reaction was
warmed to -45 ~C and stirred an additional 1 hour. The
reaction was then quenched at -78 ~C, with 0.5 mL
saturated NaCl solution. The reaction was warmed to
room temperature, diluted with ethyl acetate and the
organic layer was washed with water and satuated NaCl,
dried (MgSO4) and concentrated in vacuo. The residue
was then dissolved in methylene chloride (50 mL) and to
this solution was added triflouroacetic acid (8 mL,
excess). After 4 hours the reaction was concentrated
ln vacuo, and partitioned between a saturated solution
of sodium bicarbonate and ethyl acetate. The organic
layer was washed with water and brine and then dried
20c (MgSO4) and concentrated in vacuo. The resulting
residue was purified by flash silica get chromatography
to give 726 mg (30%) of the desired benzyl lactam
product 2 as a mixture of diastereomers.
Ex~le 3
A.
2-bromo-propionyl bromide ~ NaH ,~
HO' HO ~ ,~Br ~l,Nil
2 0
CA 02243l2l l998-07-l4
W O 97t27180 PCT~US97/01610
- 141 -
S~nthesis of 2-oxo-3-~ethyl-6-phenylmethylmorDholine.
~ Dissolve S-(-)-2-Amino-3-phenyl-l-propanol (1.51 g, lQ
mmol) in THF (10 ml). To 0 ~C solution add (rac)-2--
bromopropionyl bromide (1.04 ml, 10 mmol), followed by
a dropwise addition of diisopropylethylamine (1.73 ml,
10 mmol). ~arm up to rt and continue stirring for 90
min. Remove solvents in vacuo and remove salts by
ethyl acetate/water extraction (3X). Following
magnesium sulfate drying, the ethyl acetate layer is
evaporated and residue redissolved in anhydrous THF.
To 0 ~C solution of intermediate 2 add 13 mM of NaH
(from 60% mineral oil dispersion, removed by washing,
with hexane). Solution was warmed up to rt and
reaction terminated (MeOH) after 1 hr. Residue left
after solvents removal was again partitioned between
ethyl acetate/water (2X), organic phases combined,
dried with magnesium sulfate, filtered and evaporated,
resulting in 1.20 g crude product. Silica gel
chromatography (ethyl acetate) yielded 0.70 g of
pure product, 34% yield. H NMR (CDC13): 7.25 (m, 5H),
6.75 (broad s, lH), 4.19 (q, lH, J=7.0 Hz), 3.76 (2H,
d, J=7.5 Hz), 3.57 (lH, m), 2.90 (2H, m), 1.49+1.46
(both s, total integration 3H). CHN: 70.0 (calc:
70.2), 7.3 (7.4), 6.8 (6.8). Mass Spec. (API-)=204 (M-
1). Silica gel plates: R~=0.19 (1/1 ethyl
acetate/hexane). HPLC at 220 nm (YMC 0.46 cm x 25 cm
C18 reverse phase) t=11.47 min (single peak), gradient:
0-100%B/30 min, 1.5 ml/min, A=O.I% TFA in water, B=0.1%
TFA in acetonitrile.
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 142 -
2-brom~i~b~lbro~de ~ NaH 0 ~ r
1-10 - ' HO N~ ~
H 0
1 2 3
Synthesis of 2-oxo-3 3-~;~ethyl6-
phenylmethylmorpholine.
Dissolve 3.02g (20 mM) of S-(-)-2-Amino-3-phenyl-1-
propanol in lO ml THF. To 0 ~C solution add 2-
Bromoisobutyryl bromide (2.47 ml, 20 mmol), followed by
dropwise addition of diisopropylethylamine (3.47 ml,
20 mmol). Warm up to rt and continue stirring for
90 min. Remove solvents in vacuo and remove salts by
ethyl acetate/water extraction (3X). Following
magnesium sulfate drying, the ethyl acetate layer is
evaporated and residue redissolved in anhydrous THF.
Following silica gel chromatography (1/l ethyl
acetate/hexane), 1.20 g of intermediate 2 is isolated
from mixture containing overacylation product.
To 0 ~C solution of 2 in 4 ml of anhydrous DMF add 4 mM
of NaH (from 60~ mineral oil disperslon, removed by
washing with hexane).
After 14 hrs at rt, the solvent was removed and solid
residue partitioned between ethyl acetate/water (2X),
organic phases combined, filtered, evaporated and
(silica gel) chromatographed with ethyl acetate,
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 143 -
.
resulting in 0.20 g of product homogenous ~y TLC, but
heterogeneous by HPLC.
C.
Ph Ph Ph Ph Ph
X--CI, I
1 2 34 5 6
Synthesis of 2-oxo-3 3-spirocvclohexvl6-
phenylmethyl~r~holine via multiple deprotonation-
alkylation route.
A solution of 1 ~5.73 g) was dissolved in 5 ml of
anhydrous DMF, cooled down to 0~ C and 0.72 g of NaH
was added portionwise. After stirring for 15 min at
room temperature, the solution was cooled to 0~ C and
4.70 g of p-methoxy-benzyl chloride was added. The
reaction was then stirred at room temperature for two
hours, followed by silica gel purification, yielding
4.72 g (51%) of 2.
M (AP+) =312.1 (M+1). lH NMR (CDC13)=7.26-6.87 (9H,m),
5.42 (lH,d), 3.85 (lH,d), 4.34 (lH,d), 4.20 (d,lH),
3.79 ~s,3H), 3.68 (lH,d), 3.42 (lH,d), 3.26 (lH,m),
2.95 (2H, m).
~ 20 4.70 g of 2 was dissolved in 10 ml of anhydrous THF,
cooled to -78 ~C and 9.8 ml of 2M LDA in
heptane/THF/ethylbenzene was added. After 15 min, 4.56g
~ of 1-chloro-5-iodopentane was added dropwise and the
CA 02243121 1998-07-14
W O97/2718~ PCTrUS97/01610
- 144 -
reaction carried out at -78 ~C for 1 hr and then
quenched. The solvents were removed and the material
was purified by silica gel (2.6g, 41 4~). The resulting
compound (3) was ca l:l mixture of two diastereomers.
MS (API~)=416.2 (M+l). lH NMR (CDCl3)= 7.4-6.9 (9H, m),
5.40 (lH), 4.23 (lH), 3.83 (lH), 3.80 (s,3H), 3.75
(lH), 3.55 (3H), 3.36 (lH), 3.12 (lH), 2.96 (lH), 1.88
(m,4H), 1.58 (m,4H).
2.6 g of 3 was dissolved in 5 ml of acetone. 1.87 g of
sodium iodide was added and refluxed overnight. Acetone
was then removed in vacuo and the crude material
purified by ethyl acetate/aqueous extraction, resulting
in 2.8g of 4 (88.3%).
MS (API+)=508.1 (M+1), 530.1 (M+Na).
2.8g of 4 was dissolved in 40 ml of anhydrous THF,
cooled down to -78 ~C, and 3.6 ml of 2M LDA was added.
The reaction was allowed to progress for 2 hrs, with
gradual temperature increase to room temperature. The
residue was quenched with water, THF was evaporated and
2G the crude material desalted between ethyl
acetate/water, resulting in 1.90 g of 5.
lH NMR (CDCl3)=7.35-6.83 (m,9H), 5.35 (d,lH), 3.79
(s,3H), 3.76 (d,lH), 3.55 (m,2H), 3.23 (m,lH), 3.0
(m,2H), 2.0-1.05 (m,10H).
2~ 1.90g of 5 was deprotected by 9.61 g of CAN in 3/1
(v/v) acetonitrile/water overnight at room temperature.
The product 6 (0.50g) was purified on silica using
EtOAc/hexane/methanol gradient.
.
CA 02243121 1998-07-14
W O97/27180 PCTAUS97/01610
- 145 -
~~ M (AP+) =259 (M+1). lH NMR (CDC13)=7.22 (m,5H), 6.96
-(s,lH), 3.82 (m,lH), 3.67 ~m,lH), 3.60 (m,lH), 2.83
(m,2H), 2.0-1.20 (m,10H).
~amDle 4
A.
1) T~ tll": 1~1 r~ sium f ~
bromide THF ~_
-2) boru,.t.ill.loride ~therate
~NHCbz ~NHCbz
7.0g of the aldehyde 1 was dissolved in 40 mL of THF
and added dropwise to a cooled (-78~) solution of 128
mL ~128mMol) of lM trimethylsilyl methylmagnesium
bromide in ether. The resulting mixture was allowed to
warm to rt and poured into water. After diluting with
ethyl acetate and lN HCl, the layers were separated and
the organic layer was washed with 10% aqueous sodium
bicarbonate. Drying over magnesium sulfate and removal
of the solvent in vacuo gave a viscous oil, which was
re-dissolved in 150 mL of dichloromethane and treated
dropwise with 15.6 mL of borontrifluoride etherate.
The resulting mixture was stirred for 5 days at rt and
then quenched with 10% NaOH. The organic layer was
dried and evaporated and the residue was
chromatographed on silica gel (20% ethyl
acetate/hexanes) to give 5.2g of a yellow solid.
Recrystallization from hexane yielded 4.6g of the
desired alkene as a white solid in three crops.
-
CA 02243121 1998-07-14
WO 97/27180 PCTtUS97/01610
-- 146 --
B.
Thicacebc acid Q
- AIBN ~
~NHCbz ~S----NHCbZ
2,0g (7.1mMol) of the alkene from the previous step
were mixed with 10 mL of carbon tetrachloride and 1.4
mL (20mMol) of thioacetic acid. A spatula tip of AIBN
was added and the mixture was irradiated in a ~uartz
vessel at 254nm for 2h. The resulting mixture was
diluted with dichloromethane and extracted with satd.
aqueous sodium bicarbonate. Drying and removal of the
solvent, followed by chromatography on silica gel ~15
ethyl acetate/hexane) gave the desired thioaceta~e
(2~.0g) as a pale yellow liquid which solidified on
standing.
O ~ acetic acid ~ HBrtHOAc C~N
NHCbz ClO2S'--NHCbz O O
A solution of 0.85g of the thioacetate from the
previous step in 30 mL of acetic acid and 15 mL of lN
HCl was cooled on ice and exposed to a stream of
chlorine gas for 2h. Ethyl acetate was added and the
organic layer was separated, dried and co-evaporated
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 147 -
~~ with toluene to give the desired sulfonyl chloride as a
- white solid (1.05g).
0.7g of the sulfonyl chloride 2 obtained in the
previous step were dissolved in 30 mL of 30% HBr in
acetic acid. After 2h, the volatiles were removed in
vacuo, the gummy residue was redissolved in 100mL of
chloroform and the solution was treated with lmL of
triethylamine. The mixture was stirred for lh and then
extracted with lN HCl and 10% aqueous sodium
bicarbonate. Drying over magnesium sulfate and removal
of the solvent gave a brown oil which was
chromatographed on silica gel (2% MeOH/dichloromethane)
to give the desired sulfonamide as an off-white solid
(0.305g). lH-NMR (CDC13): 2.20 (lH,m), 2.48 (lH,m),
2.89 (2H,m), 3.10 (lH,m), 3.23 lH,m), 3.84 (lH, m),
4.18 (lH, bs), 7.30 (5H,m). 13C-NMR (CDC13): 28 8,
42.0, 47.8, 56.2, 127.8, 129.1, 129.3, 136.6.
Fxam~le 5
Synthesis of Sulfamate
A.
1) ",t:tl"~l~.., .e, ~DCI, DMF
- 2) H2/ 5%Pd-C/",eti,al,ol
--NHCbz q--NHz
OH N2HCH3
A solution of 30g of Cbz-(L)-phenylalanine, 6 8g of
methylamine hydrochloride, 14 8g of
hydroxybenzotriazole and 22 mL of N-methylmorpholine in
CA 02243121 1998-07-14
W O 97127180 PCT~US97/01610
- 148 -
300 mL of dimethylformamide was cooled on an ice-bath
and treated with 19.2g of ~DCI. The mixture was
allowed to reach rt overnight and then poured into 2000
mL of water. The product was collected by filtration,
dried and redissolved in 500mL of methanol and 300 mL
of THF. lg of 5~ palladium on carbon was added and the
mixture was stirred under hydrogen for 36h Filtration
and removal of the solvent, followed by short plug
filtration through silica gel (5% MeOH(2M NH3)/
dichloromethane) gave the desire amine as a pale yellow
solid (17g).
~_ 1 ) LiBH4, TMS-CI, THF -
2) slJIrv,.~l; ' ":-e pyridine O~S~O
NI~ICH3 2
A solution of 1.22g (56 mMol) of lithiumborohydride in
28 mL of THF was treated with 14.2 mL (112mMol) of
chlorotrimethyl silane. The resulting mixture was
treated scoopwise with 5g (28mMol) of the amide from
the previous step. After stirring at rt for 24h, 40 mL
of methanol were added carefully, followed by 10 mL of
2Q acetic acid. Repeated evaporation from methanol gave a
colorless glass, which was dissolved in 100mL of 20~
NaOH. Extraction with 4x50mL of chloroform, followed
by drying and removal of the solvent gave a yellow oil
which was chromatographed on silica gel (20~
methanol(2M ammonia)/dichloromethane to give 1.5g of
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 149 -
,~ the desired diamine as a colorless oil, and 2.0g of
- recovered starting material.
0.15g of the diamine from the previous step were
dissolved in 0.5 mL of pyridine and added dropwise to a
refluxing solution of O.lg of sulfonyldiimide in 1.5mL
of pyridine. Reflux was continued for 24h and the
volatiles were removed in vacuo. The resulting brown
oil was chromatographed on silica gel ~20~ methanol(2M
ammonia)/dichloromethane) to give the desired
sulfonylurea as a yellow oil (0.04g). H-NMR (CD30D):
2.60 (3H,s), 2.86 (lH,dd), 2.96 (lH,dd), 3.15 (lH,dd),
3.47 (lH,dd), 4.18 (lH, m), 7.22 (5H,m),
7.38 (lH,d). 13C-NMR (CD30D): 31.8, 39.9, 50.0, 57.8,
126.5, 128.2, 129.0, 136.6
~xample 6
Ph LDA Ph
~oc EtO~ C~,C12 Eti~
Boc lactam 1 (1.27 g, leq) was dissolved in TH~ (27 mL)
and cooled to -78 ~C. To this solution was added LDA
(Aldrich, 1.5 M in hexane, 3.7 mL, 1.2 eq) via syringe
over 3 minutes. After stirring for 85 minutes at -
78 ~C, a solution of ethyl iodoacetate (600 uL, 1.1
eq) in THF (13 mL) was added via syringe over 6
minutes. The reaction was then stirred at -78 ~C for
- 4. 5 hours, then at 1.5 hours at -40 ~C. The reaction
was then cooled back to -78 ~C and quenched with 2.5 mL
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97101610
- 150 -
saturated NaCl solution, and partitioned between -.
saturated sodium bicarbonate and ethyl acetate. The
organic layer was then washed with brine, dried
(MgSO4), filtered and concentrated in vacuo. The
residue was purified by flash silica get chromatography
eluting with 5~ EtOAc/ C~2C12 to give 1.67g of
substituted lactam product 2 contaminated with a minor
amount of lactam starting material 1. HPLC showed 52%
product and 28~ starting material. This mixture was
then dissolved in methylene chloride (45 mL) and cooled
to 0 ~C. To this solution was added trifluoroacetic
acid (2 mL) and the reaction was stirred at room
temperature for 1.5 hr. TLC showed no BOC material
and the reaction was concentrate in vacuo and
partitioned between saturated bicarbonate solution and
ethyl acetate. The organic was washed with water,
brine and dried (MgSO4). The organic layer was
evaporated in vacuo, and the residue was purified ~y
flash chromatography eluting with 3:1 EtOAc/ hexane to
give 770mg of pure lactam product 2.
Fx~m~le 7
-- TMEDA ~
~\NH -150 C, 15 min ll--\NH
2) 12, -15 ~ C to 0 ~ C, 30min
A solution of 5-benzyl-pyrrolidinone 1 (1.5 gr, 8.86
mmol) was dissolved at ambient temperature under
CA 02243l2l l998-07-l4
W O 97/27180 PCT~US97/01610
- 151 -
- nitrogen in anhydrous dichloromethane (40 mL). TMEDA
(6.5 mL, 42.8 mmol) was added via pipette and the
solution was cooled and maintained at -20 ~C. TMSI
(2.33 mL, 17.12 mmol) was added via pipette and the
mixture was stirred for 15 min. Solid iodine (4.345 g,
17.12 mmol) was added and the mixture was stirred
vigorously for 15 minutes and then quenched by rapid
addition of the reaction mixture into aqueous 10%
sodium sulfite solution ~100 mL). The mixture was
10 transferred to a separatory funnel and the layers were
separated. The organic layer was washed with lN
NaHS04, water, and then dried over MgS04. The solution
was then diluted in half with methanol and stirred
overnight under a nitrogen atmosphere. The solvent was
removed ln vacuo and the residue was purified by flash
chromatography, eluting with ethyl acetate : hexane
(7:3). Pure iodo lactam product 2 was recovered as a
solid (2.11 g).
~x~le 8
A.
Bn Bn
Bn,N OH 1) Ms-CI, TEA, C~CI2 Bn'~ OMs
[3~ 2
To a solution of dibenzylphenylalinol l (100 mmol) in
methylene chloride (lOOmL), was added triethylamine
~15Q mmol). The mixture was cooled to O ~C and
: methanesulfonyl chloride (110 mmol) was slowly added.
The mixture was stirred at O ~C for one hour and then
-
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 152 -
- poured into a beaker containing diethyl ether (400mL). ~~
The mixture was filtered and washed with more diethyl
ether and the filtrate was washed with water, saturated ,'
NaHCO3 and saturated brine. The organic layer was then
dried (MgS04), filtered and concentrated to yield 41 g
of crude mesylate product 2 as a light yellow-brown
thick oil, which was used as is in subsequent steps.
Bn Bn
Bn'N~OMs K CO3 ~ Bn~l~Et
80 ~ C ~ Et
Diethyl malonate (300 mmol) was dissolved in
acetonitrile (250 mL) and to this solution was added
potassium carbonate (300 mmol); the suspension was
stirred overnight at room temperature. Mesylate l (100
mmol) in acetonitrile (60mL) was then added to the
reaction mixture which was then heated to 80 ~C and
stirred overnight. The reaction mixture was then
filtered and concentrated in vacuo. Addition of hexane
to the residue formed a precipitate, which was filtered
as pure malonate product 2 (19.5 g). Material was used
as is.
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 153 -
Bn 1 ) NaH, TtiF, O-C- RT
~ 1 C:'3
Malonate 1 ~10.6 mmol) was dissolved in dry THF (40 mL)
and cooled to O ~C. To this solution, sodium hydride
(17 mmol) was added in portions and the suspension was
stirred for 1.5 hr at O ~C. The triflate 2 (12 mmol)
.in dry THF (lOmL) was then slowly added to the reaction
mixture and after complete addition the reaction was
allowed to warm to room temperature and was stirred
overnight. The reaction was then diluted with water
(lOOmL) and extracted with diethyl ether (3x50 mL).
The combined organic layers were then washed with
saturated brine, dried over MgS04, filtered and
concentrated in vacuo. The crude product was purified
by mplc (eluted with a gradient of 9:1 hexane:ethyl
acetate up to 4:1 hexane:ethyl acetate to yield product
3 (4.2 g, 73 %).
N ~CO2Et 1) HCI/H2 Pd/C 0
Bn~ W 2) CH2CI2 sat. NaHCO3 /~~ .~
90% yield U~NH
The subsituted malonate 1 (1.62 mmol) was suspended in
ethanol and to this was added conc. HCl (0.24 mL, 2.4
CA 02243121 1998-07-14
W O97/27180 PCTnUS97/01610
- 154 -
mmol) and 10~ palladium on Carbon (0.162 mmol). This
mixture was then stirred under a balloon of hydrogen
gas at room temperature overnight. The reaction was
then filtered through Celite and to the filtrate was
added triethylamine (10 mL, excess) followed by solid
sodium bicarbonate (excess). The mixture was stirred
for 0.5 hr, filtered and concentrated to yield a yellow
- solid. This residue was then dissolved in ethyl
acetate and washed with water, 0.5N HCl, saturated
sodium bicarbonate, and brine. The organic layer was
dried (MgSO4), filtered, and dried to yield crude
~lactam product 2, which was used as is.
1)KOH,e~anDI
~ 2)TfiOH.DM50,80-C ~ ~
Lactam 1 (1.18 mmol) was dissolved in ethanol (5mL) and
to this solution was added KOH (10 mmol). The mixture
was stirred for 3 hr at room temperature and then
concentrated to dryness. The residue was dissolved in
water and washed with diethyl ether. The aqueous layer
was then acidified with HCl and extracted with ethyl
acetate. The organic layer was dried (MgSO4), filtered
and concentrated ln vacuo to yield 341 mg of a light
yellow solid. The residue was dissolved in DMSO (3mL)
and to this solution was added p-toluenesulfonic acid
mono- hydrate, and the mixture was heated to 80 ~C
CA 02243121 1998-07-14
W O 97/27180 PCT~US97101610
- 155 -
rc ove-rnight. The mixture was diluted with water (15 mL)
and extracted with ethyl acetate. The organic layer
was washed with saturated sodium carbonate and brine
followed by drying with MgSO4. The organic layer was
then filtered and concentrated in vacuo to yield the
~HF substituted lactam product t245 mg, 77% from ester)
which was used as is in the next step without further
purification.
~x~m~le 9
~A.
---Ph NaH, Ph OC~
JX~IJ~ chloride ~/
o O ~ C--~RT
0
Sodium hydride (60% dispersion in mineral oil, 4.0 g,
1.17 eq) was washed with 4 x 25 mL portions of hexanes
to remove the mineral oil, then suspended in 25 mL of
DMF and cool-ed to 0 ~C. A solution of lactam 1 (15g, 1
eq) in dry DMF (25 mL) was then added dropwise via
canula into the cold NaH suspension over 40 min. An
additional 65 mL of DMF was then added to aid stirring.
After stirring the anion for 1 hour, p-methoxybenzyl
chloride (14.5 mL, 1.26 eq) was added over 5 min at
0 ~C. The reaction was then allowed to warm to room
temp. An additional amount of p-methoxybenzyl chloride
~ was added to drive the reaction to completion. TLC
(Et~AC) Rf lactam 1 = 0.21. Rf product 2 = 0.43.
CA 02243l2l l998-07-l4
W O 97/27180 PCTnUS97/01610
- 156 -
After 3. 5 hours, the reaction was poured into cold ~-
water and extracted twlce with ethyl acetate. The
- combined organic layers were washed with water (5X),
brine, dried (MgSO4) and filtered. Concentration in
vacuo, afforded a crude solid which was purified by
crystallization( 7:1 hexane:EtOAc) to yield the
protected lactam product 2 ~19g, 75%).
B.
TMSI ¢~
OM 7MEDA OMe
To protected lactam 1 (328 mg, 1.11 mmol) and
N,N,N1,Nl-tetramethylethylenediamine (Aldrich, 5.0
equiv., 5.55 mmol, 645 mg, 838 ml) in 15 ml
dichloromethane at -15 ~C, was added
iodotrimethylsilane rAldrich, 1.0 equiv., l.11 mmol,
222 mg, 158 ml). After 15 min, iodine ~Aldrich, 1.2
equiv., 1.33 mmol, 338 mg) was added in one portion and
- the reaction warmed to 0 ~C. After 3t) min the reaction
was quenched with 5 ml each of 10% aqueous sodium
sulfite and saturated aqueous sodium chloride. The
orgnic layer was separated, dried over magnesium
2Q sulfate, filtered and concentrated in vacuo.
Purification by flash column chromatography (silica
gel, 2. 5 x 10 cm, 2. 596 diethylether in dichloromethane)
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97101610
- lS7 -
yielded 322 mg of diastereomeric iodolactam 2 as a
white solid.
-
C .
~1}NnH~ ~
t10 C, l~h ~OMe
To iodolactam 1 (1.18g, 2.91 mmol) and methyl vinyl
sulfone (Aldrich, 6.0 equiv., 17 mmol, 1.82 g, 1.5 ml)
in 25 ml refluxing toluene was added tributyltin
hydride (Aldrich, 1.3 equiv., 3. 79 mmol, 1.10 g, 1.0
ml) and AIBN (P~altz & Bauer, 0.12 equiv., 0.35 mmol,
57 mg) as a solution in 5 ml toluene over 1.2 h. After
16 h the solvent was removed in vacuo, and the residue
taken up in 200 ml diethyl ether and stirred with 20 ml
10~ a~ueous potassium fluoride (wt/v) at ambient
temperature. After 3 h the orgnic layer was separated,
dried over magnesium sulfate, filtered and concentrated
in vacuo . Purification by flash column chromatography
(silica gel, 5 x 20 cm, 2:1 ethyl acetate/hexanes)
yielded 0.31g of diastereomeric sulfone 2 as a white
solid.
CA 02243121 1998-07-14
. W O 97/27180 PCT~US97/01610
- 158 -
Fx~m~le 10
A.
O NHCbz
\ / NaCNEi~
HCI ~ O~ 19~ acatic acid/DMF HN/ \NI ICbz
O ~rCO2C~
1 2
~To a solution of solution of Cbz-L-phenylalinal (13 g,
45.9 mmol) in 1%AcOH/DMF (200 mL) mL was added
aminoisobutyic acid methyl ester hydrochloride 1 (8.5
g, 55.1 mmol) with stirring at room temperature. Once
homogeneous, solid sodium cyanoborohydride ( 8. 6 g,
137.6 mmol) was added in one portion. Some bubbling
was evident and the reaction was stirred over~ight at
room temperature. The reaction was quenched with water
(20 mL) and concentrated in vacuo to about 100 mL. The
concentrate was diluted with ethyl acetate and washed
with water and brine followed by drying (MgSO4). The
organic layer was evaporated in vacuo to yield a yellow
residue which was purified by MPLC (elutant 1:2 ethyl
acetate : hexane) to afford amine product 2 (11.6 g,
66%).
-
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 159 -
B.
~ 1 ) 30% HBr/HOAc HN~ "
H~ NHC~ 2) DlE~ dl-ol >~NH
~CO2CH3 11
1 2
To a solution of amine l (1.41 gr, 3.7 mmol) in
methylene chloride (25 mL) was added 30% HBr in acetic
'acid (6 mL) via pipet. Vigorous gas evolution occurred
and the reaction was allowed to stir overnight at room
temperature. The mixture was then evaporated in vacuo
and dried under high vacuum. The residue was then
dissolved in methanol (25 mL) and to this solution was
added diisopropylethylamine (5eq) and the reaction was
stirred at room temperature overnight. The solvent was
removed in vacuo and the residue was taken up in ethyl
acetate and washed with water, saturated NaHC03 and
brine. The organic layer was dried (MgS04) filtered
and concentrated in vacuo to yield crude product .
Flash silica gel chromatography (8% methanol /
methylene chloride) afforded pure piperazinone product
~ (556 mg, 70 %).
CA 02243121 1998-07-14
W O97/27180 PCTAUS97/01610
- 160 -
Benzyl bromide
HN I ' K2C03
~NH Ac~ul l- ile
O O
To a solution of piperazinone 1 ~556 mg, 2.55 mmol) and
potassium carbonate (1.06 g, 7.6 mmol) in acetonitrile
was added benzyl bromide (364 uL, 3 mmol) and the
reaction was stirred at room temperature overnight.
The reaction was then filtered and concentrated in
vacuo. The residue was dissolved in ethyl acetate,
washed with water and brine and dried IMgSO4). The
organic layer was then removed in vacuo and the residue
was flash chromatographed (3% methanol in methylene
chloride) to yield pure benzyl protected piperazinone
product ~ (589 mg, 75%).
~x~le 11
A.
1 ) Benzylamine
O Ph EDCI, HOBT, Ph
I I NMM, DMF
HO~ ~ "",1 , ~N ""'
2) 30% HBr/HOAc ll H
~-Cbz 3) Borane-THF ~ ~
A solution of C~z-(l)-Phenylalanine (15 gr, 50 mmol),
HOBT (7.4g, 50mmol), N-methyl morpholine 15.5 mL, 50
mmol) and benzylamine (6 mL, 55 mmol) in 250 mL of DMF
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 161 -
was cooled to 0 ~C and treated with EDCI (9. 6 g, 50
mmol). The resulting mixture was stirred at 25 ~C for
- 12h and the volatiles were removed in vacuo.
Partitioning between ethyl acetate and lN hydrochloric
acid, followed by extraction with 10~ sodium
bicarbonate, drying over magnesium sulfate and
evaporation of the solvent afforded the desired amide
as a white solid (19.5g).
l9g of the above material were dissolved in 280mL of
30% hydrogen bromide in acetic acid and stirred at
25 ~C for 3h. The volatiles were removed and the
~residue was partitioned between water and ether. The
aqueous layer was treated with excess 6N sodium
hydroxide and extracted twice with ethyl acetate.
Drying over magnesium sulfate and evaporation of the
solvent afforded the desired amine as a pale yellow oil
~14.0g), which was redissloved in 200 mL of
tetrahydrofuran and treated with 200 mL of lM borane-
THF in tetrahydrofuran. The mixture was stirred at
25 ~C for 72h and then heated to reflux for 4h. The
solution was cooled and treated with lO0 mL of methanol
under vigorous gas evolution. The volatiles were
removed and the resulting residue was dissolved in lS0
mL of concentrated hydrochloric acid. After refluxing
for lh, the volatiles were removed and the residue was
dissolved in 300 mL of 3N sodium hydroxide. Extraction
with 3 times 250 mL of dichloromethane, drying over
magnesium sulfate and chromatography on 2 inches of
silica gel (2~ methanol-dichloromethane) gave the
desired diamine as a pale yellow honey (9.2g).
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 162 -
Ph sulfonyldiimide [~3
[3~--H--1 pyridine 0 A
o"'b
A solution of sulfonyldiimide (3. 6 g, 36 mmol) in 100
mL of pyridine was heated to reflux and treated
dropwise with a solution of the diamine 1 (7. 2 g, 30
mmol) from the previous step in 20 mL of pyridine.
After 2h of reflux, 15 mL of triethylamine and 0.4g of
4-dimethylaminopyridine were added and heating was
continued for 12h. The volatiles were evaporated and
the residue was partltioned between lN hydrochloric
acid and ethyl acetate. Extraction of the organic
layer with saturated sodium bicarbonate, drying over
magnesium sulfate and chromatography on silica gel (1:1
ethylacetate - hexanes~ afforded the desired cyclic
sulfamate 2 as a white solid (6.0g).
lH-HMR (CDC13): 2. 80(lH, dd), 2. 96(lH,dd), 2.98(lH,dd),
3.32(lH,dd), 3.95(1H,m), 4.04(lH,d), 4.24(lH,d),
4.40(lH,d), 7.18 (2H, d), 7.2-7.4(8H)
13C-NMR(CDC13): 41.5, 50.0, 52.7, 53.8, 127.5, 128.0,
128.2, 128.3, 28.4, 128.5, 135.5, 136.0
CA 02243l2l l998-07-l4
W O 97/27180 PCTrUS97/01610
- 163 -
Fxample 12
A.
O~;;~NH Cbz ~ ~3~NH NH Cbz
The Cbz-phenylalaninol mesylate 1 (280 mg, 0.77 mmol)
was stirred in acetonitrile (5 mL) containing benzyl
amine (413 mg, 3.85 mmol) and sodium iodide (115 mg,
0.77 mmol). The reaction was then refluxed for 29
hours. The reaction was then cooled to 25 ~C and
concentrated in vacuo. The crude oil was then purified
by silica gel chromatography, eluting with CH2Cl2 with
a gradient up to 1:1 CH2Cl2:EtOAc to provide 120 mg of
the desired diamine 2.
Ph Ph
[3~NH I~Ct~ HO~ 1~NH NH2
The Cbz protected diamine 1 ( 120 mg, 0. 32 mmol)
was stirred in 2.0 mL of 30 ~o HBr in acetic acid for
one hour. This was followed by concentration in vacuo.
~ The crude oil was then dissolved into toluene and
concentrated in vacuo two times followed by evacuation
at approx. 1 mm Hg. The crude diamine was then
r purified by silica gel chromatography, eluting with
. .
CA 02243121 1998-07-14
W O97/27180 PCT~US97/01610
- 164 -
95:5:1, CH2Cl2:MeOH:NH4OH to provide 71 mg ( 90 ~) of
the desired diamlne 2.
Ph '
The diamine 1 (56 mg, 0.23 mmol) was dissolved in
~3.0 mL of CH2Cl2. This was followed by the addition of
TEA (66 uL, 0.25 mmol) and then CDI (32 mg, 0.25 mmol).
A new spot was observed by tlc after 2-3 hours (Rf =
0.29 in EtOAc on SiO2~. The reaction mixture was then
concentrated and the residue was purified by silica gel
chromatography, eluting with EtQAc, to provide 32 mg
(52~) of the desired benzyl urea 2.
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 165 -
.x~m~le 13
-
Synthesis of Compoun~ 1
+ I~N~ ~OMo N H ~ r
o ~ ~C--~RT N~2
2 3 OAII~
allyl urea216 g/Mol lOOmg 0.46 mmol
NaH, (60% in oil) 24 g/Mol 140.0 mg 9.7 mmol
epoxide325.4 g/Mol 150.0 mg 0.96 mmol
DMF 2.0 mL
The urea of Example lC was dissolved in 1.0 mL of
anhydrous DMF and cooled to 0 ~C. This was followed by
the addition of 140 mg NaH. The reaction turned darker
over the next hour at 0 ~C. This was followed by the
dropwise addition of the epoxide as a solution in DMF
~0.6 mL), washing with 300 uL of DMF. The reaction was
then stirred one hour at 0 ~C, followed by warming to
25 ~C. Tlc indicated nearly complete conversion to two
new products (Rf = 0.4 and 0.45 on SiO2 with 2:1
hexane: ethyl acetate, between that of the epoxide and
the urea). The reaction was then cooled to 25 ~C and
quenched by the addition of 3 mL of saturated sodium
bicarbonate. The reaction mixture was then diluted by
lS mL of methylene chloride and washed by both
saturated sodium bicarbonate and brine, (2 x 15 mL
each). The organic portions were then dried over
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 166 -
sodium sulfate, filtered and concentrated in vacuo.
The crude product was then purified by silica gel
chromatography, eluting with 80% ethyl acetate: hexane
to provide 35.0 mg of the desired alcohol. r
~x~le 14
A.
o~ 0~C-->R~
OMe
1 lactam 1.0 equiv., 295mgg
2 sulfonamide epoxide 1.1 equiv., 520mg
3 NaH, 60% in oil (Aldrich) 1.5 equiv, 102mg
10 4 DMF 8 mL
Lactam 1 was dissolved in 3mL of DMF and cooled to
0~ C. To this solution was then added sodium hydride
as a solid and the reaction was stirred for 40 min. at
0 ~C. The anion solution was canulated into a solution
of epoxide 2 in 3 mL of DMF. The reaction was stirred
at 0 ~C for 5 minutes, then warm to room temperature
and stirred overnight (TLC (95:5, CH2Cl2 : MeOH) Rf (st
mat.) = .26. Rf(prod) = .46). After 22 hours, the
reaction was cooled to 0 ~C, and quenched with
2Q H20/EtOAc. The organic layer was washed with water(5X)
and brine, dried (MgSO4), filtered, and concentrated in
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 167 -
- vacuo. The residue was then purified by silica gel
chromatography (40% ether/ CH2Cl2) to yield product 3
- (310mg,37%).
~,
F.x;~rn.,~le 1 S
A.
OH /~ TMS-tnflate <~ O ~0
~\ ~ DMF ~\ [~~2
OMe OMe
1 lactam 1.15g, 1.0 equiv.
t-butyldimethylsilyl 1.5 equiv. + .5 eq.,
trifluoromethanesulfonate (1.06mL)
imidazole 2.5 equiv + .5 eq, (470mg)
Lactam 1 was dissolved in 5mL of DMF and cooled to
0 ~C. To this solution was then added imidazole
followed by TBDMS-triflate. The reaction was then
allowed to warm to room temperature. After
approximatly 2 hours, an additional .5 eq.(80mg) of
TBDMS-triflate and .5 eq.(265uL) of imidazole was added
and the reaction was stirred overnight. The reaction
was quenched with saturated Na~CO3 solution and
partitioned between H20/EtQAc. The organic layer was
washed with water(5X) and brine, dried (MgSO4),
,.
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 168 -
filtered, and concentrated in vacuo to yield product 2
(1.5 gr, 37%) which was used as is.
Fx~le 16
Synthesis of Co~ound 7
--5i-- ~0 Ailyl bromid~ ~
~ 2) TBAF ~ ~ c~ ~'>
OMB OMe
1 silyl-lactam 1.0 equiv.,23mg
Allyl Bromide, (Aldrich) 2.1 equiv. , 7 uL
LDA, 1.29M (Aldrich) 1.25 equiv , 36U~.
TBAF, 1.0~, (Aldrich) 2.5 equiv., 95uL:
Silyl protected lactam 1 was dissolved in THF and
cooled to -78 ~C. To this solution, was added LDA
(1.25 eq) via syringe. After stirring for 30 minutes
at -7 8 ~C, allyl bromide was added via syringe. After
2 hours an additional 2ul of allyl bromide was added
and the reaction was stirred at -78 ~C for 2.5 hours,
then warmed to room temp for 17 hours (TLC (2:8,
ether:CH2Cl2) Rf (st mat.) = .56. Rf(silyl-prod) =
.72). After this time, TBAF (lM in THF) was added
and the reaction was stirred at room temperature for 7
hours (TLC (1:9, ether:CH2Cl2 ) Rf(prod) = .20). The
reaction mixture was then partitioned between H20/EtOAc
CA 02243l2l l998-07-l4
W O 97/27180 PCT~US97/01610
- 169 -
~ and the organic layer was washed with water and brine,
dried (MgS04) and filtered concentrated in vacuo. The
; residue was then purified by silica gel chromatography
(10% etherJmethylene chloride) to yield product 2
(6mg,30% yield).
~x~le 17
Synthesls of Co~polln~ ~0
--Si-- ~0 Benzyl bromide
A ~ 2) TBAF ~ ~~ OH
~N N'SO2 ~ ~N N~So2
O ~ O
OMe O~b
silyl-lactam 1.0 equiv., 122mg
benzyl bromide, (Aldrich) 1.5 equiv. 42uL
LDA, 1.29M (Aldrich) 1.4 equiv , 275ul
TBAF, l.OM, (Aldrich) 2.5 equiv., 625uL
Silyl lactam l was dissolved in dry THF (6mL) and
cooled to -78 ~C. To this solution was then added LDA
and the reaction was stirred for 30 minutes at -78 ~C
after which time benzyl bromide was added via syringe.
The reaction was stirred at -78~C until reaction was
complete (1.5 hours, TLC (1:9, ether CH2Cl2 ) Rf (st
mat.) = .29. Rf(silyl-prod) = .62. Rf(BzBr) = .79).
The reaction was then quenched at -78 ~C with 6uL water
and then TBAF (lM in THF was added and the reaction was
warmed to room temperature and stirred for 3 hours (TLC
~.
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
-- 170 --
(1:9, ether:C~2Cl2 ) Rf(prod) = .28). The reaction was
partition betweem H20/EtOAc and the organic layer was
washed with with water and brine, driçd (MgS04) and
filtered and concentrated in vacuo. The residue was
purified by silica gel chromatography (10~ ether/
CH2Cl2) to yield benzyl product 2 (71mg, 48% ).
Fx~mrle 18
Synthesis of Com~ound 16
-si- ~ Mdhyliodide
~"~,N 2)T}AF ' r>
OMe OMe
1 silyl-lactam 1.0 equiv.,66mg
Methyl iodide, (Aldrich) 1.6 equiv., 16uL
LDA, 1.29M ~Aldrich) 1.3 equiv , llOuL
TBAF, l.OM, (Aldrich~ 3.0 equiv., 325uL:
The reaction for the above methylated compound was
carried out as per the procedure described for compound
20 (Example 17) substituting methyl iodide for benzyl
bromide on the scale described in the above table. The
final compound was purified by silica gel
chromatography using 10% ether/ CH2Cl2 to yield
methylated product 2 (33mg, 60% yield).
CA 02243l2l l998-07-l4
W O 97/27180 PCTrUS97/~1610
- 171 -
- ~x~m~le l9
~_ A.
7r'~ IlGtl~ j",
~NHO ~C ~RT ~\N~
O O
1 2
1 lactam 1.0 equiv., 400mg
epibromohydrin 1.5 equiv., 280uL
sodium hydride, 80% oil disp. 2.0 equiv, 126mg
DMF 15mL
Lactam l was dissolved in dry DMF (15 mL) and
cooled to 0 ~C under a nitrogen atmosphere. To this
solution was added sodium hydride (2 eq) in one portion
and the reaction was stirred at 0 ~C for 1 hour after
which, epibromohydrin was added via syringe. After
stirring for 5 min. at 0 ~C the reaction was warmed to
room temperature (TLC (EtOAc) Rf (st mat.) = .16.
Rf(prod) = .23). After 1.5 hours at room temperature
the reaction was quenched with saturated NH4Cl and
extracted with CH2Cl2. The organic layer was then
washed with water(4X) and brine, dried (MgSO4) and
filtered, and concentrate in vacuo. The residue was
~ then purified by silica gel chromatagraphy (3:1
EtOAC:hexane) to yield 315mg(60%) of epoxide product 2
which was used as is in the next step.
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 172 -
B.
~, CEytoHp~~ ylamine
o 80~C ~ N ~ i
O O
1 2
1 lactam 1.0 equiv., 315mg
cyclopentylmethylamine 5.75 equiv., 775mg
anhy. EtOH 3mL
Epoxide l was dissolved in 3 mL of EtOH and to
this solution was added cylcopentylmethylamine. The
reaction was heated to 80 ~C for 2.5 hours lTLC
(9:1,CH2Cl2:MeOH) Rf (st mat.) = .56. Rf(prod) = .13).
The solvent was removed in vacuo and the residue was
purified by silica gel chromatagraphy (3~MeOH/ CH2Cl2
to 10%MeOH/ CH2Cl2) to yield 224mg(50%) of amine product
1~ 2.
C. Synthesis of Com~ound 15
1)TMS-CI,TEA
~~ 2) ~,"_ ~o,.~L~ lfonyl chloride -~
~N ~NH , ~N~-'N--S02
0 2 [~3
OMB
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 173 -
~ 1 lactam l.O equiv., 315mg
chlorotrimethylsilane 2.2 equiv., ''2uL
triethylamine 5.0 equiv., 280uL
4-methoxybenzenesulfonylchloride 1.5 equiv., 124 mg
TBAF, l.OM 4.4 equiv., 1. 7 8mL
Amine l from Example l9B was dissolved in
methylene chloride and cooled to O ~C. To this
solution was added triethylamine (2.5 eq) followed by
chlorotrimethylsilane. The reaction was then warmed to
room temperature and stirred under nitrogen for 2.0
hours. An additional amount of triethylamine was added
(2.5 eq) and 4-methoxybenzenesulfonyl chloride was
added. The reaction was stirred at room temperature
for 3 hours. After this time, TBAF (lM in THF) was
added and the reaction stirred at room temperature for
1 hour. The solvent was removed in vacuo. and the
residue partitioned ~etween ethyl acetate and aqueous
saturated bicarbonate solution. The organic layer was
washed with water, brine, dried MgS04 , filtered and
the solvent removed in vacuo. (TLC (8:2, CH2Cl2:
ether), Rf(upper diast.) = .21 Rf(lower diast.) = .12).
The residue was purified by silica gel chromatagraphy
(25% ether/CH2Cl2) to yield 52 mg(26%) of (upper
diastereomer). The lower diastereomer was further
purified by preperative TLC (1:1, ether:CH2Cl2) to give
23mg~12~) of the lower diastereomer.
CA 02243121 1998-07-14
WO 97/27180 PCT~US97/01610
- 174 -
Ex~m~le 20
Synthesis of compollnd 47
~ o ~ 0~C-~RT
- ONb
Morpholinone 1 was dissolved in 1 ml of anhydrous DMF,
cooled to 0 C and to this solution was added 4.4 mg o~
NaH. The solution was brought to room temperature for
30 min and then cooled down to OC before adding 0.20 g
of epoxide 2. After heating for 5 hrs at 45 ~C, the
solvent was removed in vacuo and puri~ied on silica gel
yielding 111 mg o~ final product 2 (compound 47). M
10 (ES+) =585 (M+1), 607.1 (M+Na). lH NMR (CDC13)= 7.52
(d, 2H), 7.30 ~m, 5H), 6.95 (d, 2H), 4.05 (m, lH), 3.87
(3H, s), 3.60 Im, ZH), 3.16 (m, 4H), 3.0 ~m, 4H), 2.18
(lH, m), 1.97 ~m, 2H), 1.60 (m, 14H), 1.23 (m, 4H).
CA 02243121 1998-07-14
WO 97/27180 PCT~US97/01610
- 175 -
xam~Dle 21
Synthesis of Co~ound 109
~ 1) NaH DMF, 80-C
- 1.,.,,;,.,~, : ,' acrylic acid ~ - o
~NH 2) Ozone ~OH
b 3) Dimethyl sulfide 2 O O
11 ,iu~. ', t butyl amide, ~ O ~S
EDC, HOBT, NMM, DMF ~p~N~
To a cooled solution (-78 ~C) of benzyl lactam 1
(0.150g, 0.57 mmol) and bromomethyl acrylic acid
(0.094g, 0.57mmol) in anhydrous THF (4.OmL) was added
NaH (60%, 0.046g, 1.14 mmol) with stirring. The
solution was allowed to gradually warm to room
temperature and stir for 1.5h. The reaction mixture was
then diluted with ethyl acetate (60mL) and washed with
l.ON HCl (2 x lOmL) and brine (2 xlOmL). The organic
layer was dried (magnesium sulfate), filtered, and
evaporated to give an off white solid. This solid was
dissolved in methylene chloride/methanol (80/20, lOmL)
and through the cooled solution(-78 ~C) was bubbled
ozone for lOmin. The solution was flushed with oxygen,
warmed to O ~C, and methyl sulfide (2.OmL) was added at
O ~C. The mixture was allowed to warm to room
temperature and stand for l.Oh. Evaporation of the
solvent afforded crude product 2 as a yellow oil. To a
solution of the acid 2 in anhydrous DMF (3.OmL) was
added thioproline-t-butylamide (O.llg, 0.57mmol),
hydroxybenzotriazole (0.77g, 0.57 mmol), N- methyl-
CA 02243121 1998-07-14
w o 97n7180 PCT~US97/01610
- 176 -
morpholine (0.62mL, 0.57mmol) and EDCI (O.llg, 0.57
mmol) respectively with stirring at room temperature.
After 24h. at room temperature, the reaction mixture
was evaporated and the residue was dissolved in ethyl
acetate (lOOmL). The solution was washed with 1 ON HCl
(2 x 20mL), 10~ sodium carbonate (2 x 20mL), water (1 x
lOmL), brine( 1 x lOmL), filtered and evaporated to
give 0.210g of a yellow oil. The oil was purified by
column chromatography; hexane~ethyl acetate (60J40) to
give compound 3 (0.050g, 18%) MS: M+l= 522; H NMR
(chloroform-d) 1.35(d, g~); 1.85(m,2H); 2.6(m, 3H);
2.85(m,lH); 3.15(m,2H); 3.40(m,lH); 3.8(m,lH); 4.1(m,
2H); 4.4(m,lH); 4.70(m,lH); 4.95(m, lH); 6.1(d, lH);
7.1(m,4H); 7.25(m,6H).
~x~le 22
Synthesis of Compound 80
1 ) NaH, DMF, 80'C <~ ~ ~
ccnc. HCI /~ OH 1' H OH
~NH ~ , ~,N J~N.",~
~N.~
2 ~>
0.80g of allyl lactam 1 was dissolved in 1 ml of DMF,
cooled to O ~C and 89.5 mg of sodium hydride was then
added. The solution was then brought up to ambient
temperature for 30 min, again cooled down to O ~C and
1.4 g of epoxide 2 was added. The reaction was warmed
to 50 ~C under N2 blanket for 3 hrs. The resulting
crude mixture was then chromatographed on silica gel
!~
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 177 -
~ yielding 1.4g of 3 (63.7~). This amount was treated
with 12 ml of 4N HCl in dioxane and 2 ml water for 30
~ min. The product was then chromatographed on C18rphplc,
yielding 0.36g of two diastereomers, su~jected to
5 chiral separation, which resulted in 138 mg of pure
diastereomer 3. MS (ES- 551.3 IM-1)), ES+, 553.3 (M+1)
and 575.3 (M+Na). lH NMR (CDCl3)= 7.20 ~m, 14H), 6.26
(m, lH), 5.62 (m, lH), 5.24 (m, lH), 4.97 (m, 2H), 4.23
(m, lH), 3.83 (m. 2H), 3.61 ~m, lH), 2.95 (m, lOH),
2.40 (m, lH), 2.24 (m, lH), 2.04 (m, lH), 1.95 (m, 2H),
1.70 (m, 2H).
F.x~Tr~l e 23
Synthesis of Compound 91
1) NaH, DMF, 80-C ~ ~0
NH c~nc. HCI ~ H O
A solution of cyclic sulfamate 1 (O.lg, 0.33mmol) in 2
mL of dimethyl formamide was cooled to 0 ~C and treated
with of 60% sodium hydride (0.005g, 0.13 mmol) in oil.
The mixture was stirred at 25 ~C for 1.5h and treated
with of epoxide 2 (0.125g, 0.33mmol) The resulting
mixture was stirred at 60 ~C for 3h, more sodium
hydride (0.005g) was added and heating was continued
over night. The volatiles were removed in vacuo and
the residue was dissolved in 2 mL of 4M hydrogen
CA 02243121 1998-07-14
W O 97/27180 PCTnUS97/01610
- 178 -
.
chloride in 1,4-dioxane. Water (0.5 mL) was added and
the mixture was stirred for 6h at 25 ~C. The reac~ion
mixture was diluted with ethyl acetate and extracted
with 10~ soduim bicarbonate. Drying over magnesium
sulfate and removal of the solvents gave a yellow gum,
which was sub~ected to C-18 preparative HPLC
~acetonitrile-water gradient). The desired material 3
was isolated as a minor fraction (9 mg) as a white
solid
lH-NMR(CDCl3): 2.10(2H), 2.70(2H), 2.8-3.2(8H),
3.4(1H), 3.58(lH), 4.02(1~), 4.15(1H), 4.22(2H),
5.30(lH), 5.86(lH), 7.06(2H), 7.1-7.4(16H).
F.Xi~ e 24
Synthes;s o~ Co~ollnd 83
1) N~H, DMF, 80-C
conc. HCI ~ OH ~ H OH
H ~ ~\N~5N"~
~N.,Q
2 ~
To a cooled solution (O ~C) of compound 1 (0.19Og,
C.72mmol) in anhydrous DMF (lOmL) was added NaH(60%,
0.028g, 0.72mmol) with stirring. The solution was
allowed to warm to room temperature and stir for l.Oh.
Compound 2 (O.Z75g, 0.73mmol) was added at room
temperature and the mixture was heated at 60 ~C for
5.Oh. The solution was evaporated and the reside was
partioned between ethyl acetate (150mL) and water
-
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 179 -
(30mL). The organic layer was washed with water (2 x
10mL), brine (25mL), dried (MgSO4), filtered, and
~ evaporated to give a grey oil. The oil was purified by
column chromatography: hexane/ethyl acetate (60/40) to
give 0.23g (50~) of the acetonide protected product.
The acetonide (0.185g, 0.29mmol) was dissolved in
isopropanol (lOmL) and treated with conc. HCl (3.0mL)
at room temperature. After 1.5h., the solution was
adjusted to pH 11 with 3.ON NaOH and then concentrated.
The aqueous solution was extracted with ethyl acetate
(3 x75mL). The ethyl acetate was dried (MgSO4) and
evaporatated to give a clear film. The crude product
was purified ~y column chromatography: hexane/ethyl
acetate (45/55) to give the product as a white solid
(0.090g, 50%). Preparative HPLC on chiral phase
(isopropanol-hexane gradient) yielded the desired
diastereomer 3 (10mg) along with a 1:1 mixture of the
desired diastereomer and an additional epimer (50mg).
MS: M+1= 603 H NMR (chloroform-d) 1.80(m, 6H);
2.50(m,lH); 2.60(m, 2H); 3.0(m,8H); 3.60(m,lH),
3.70(m,lH); 3.95(m,lH); 4.25(m,lH); 5.30(m,lH);
6.00(m,lH); 7.05(m,4H); 7.25(m,15H).
CA 02243121 1998-07-14
W O97/27180 PCT~US97/01610
- 180 -
Fxample 25
Synthesis of ComDol~nd 8
A.
Ph Ph
NaH,
(s) &~ h'
~,1~ aMF ~ J~, o
1 2
Allyl lactam 1 (443 mg, 2.06 mmol) was dissolved in DMF
(2 mL) and to this solution was added sodium hydride
(2.2 mmol). The reaction mixture was stirred at room
temperature for 1 hr after which (s)-epichlorohydrin
(172 ul, 2.2 mmol) was added neat. The reaction was
stirred at room temperature for 4 hr, diluted with
10- water (20 mL) and extracted with ethyl acetate. The
organic layer was then washed with water, brine and
dried (MgSO4) and filtered. Concentration in vacuo
afforded crude epoxide product 2 which was used without
further purification.
1~ B.
~J~,N NNX ~J~N~ ,~ )
Lactam epoxide 1 (180 mg, 0.66 mmol) and
decahydroisoquinoline 2 (160 mg, 0.66 mmol) were heated
CA 02243l2l l998-07-l4
W O 97/27180 PCTrUS97/01610
- 181 -
~ to 80 ~C in isopropanol. After three hours the
reaction was cooled to 25 ~C and stirred for 48 hours
~ at room temperature. The reaction was then
concentrated in vacuo. Purified by silica gel
chromatography, eluting with 25 % EtOAc : Hexanes,
providing 90 mg (90% pure by HPLC) of desired product
3.
~ x~mple 26
Synthesis of Compol~nd 9
A.
~ ~Ph
~N,Boc ~~ ~
HN 'rJ O 2 ~l,NJ
O~ INHi-PrOH, 75~C3 o~NH
t Bu t Bu
The Boc protected piperazine 1 ~21.4 mg, 0.081 mmol),
was dissloved in 1.5 mL of i-PrOH. This was followed
by the addition of the lactam epoxide 2 ( 18. 3 mg, 0.068
mmol). The reaction vessel was then fitted with a
reflux condenser and heated to 75 ~C for 16 hours. TlC
indicated complete consumption of both starting
materials and formation of a new material. The
reaction was then cooled to 25 ~C and concentrated in
vacuo. The complete consumption of epoxide was
confirmed by both tlc and lH NMR. The crude addition
product was then used without further purification.
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 18Z -
B.
~N' 1) HC!'~ N ~,
~J 2) Picolyl G~l~.iJ. HCI
~ o~NH TEA, DMF24hr ~ o~NH
t Bu 2t Bu
The Boc protected piperazine addition product l ~rom
the previous step was stirred for 2 hours in 1.0 mL of
4N HCl/dioxane. This was followed by concentration in
vacuo. The crude solid was then dissolved in 10 mL of
'CH2Cl2 and washed by 2 x 10 mL of each saturated a~ueous
sodium bicarbonate and saturated aqueous brine. The
combined organic portions were then dried over MgSO4,
filtred and concentrated in vacuo to provide the
freebase of the desired intermediate. The crude amine
was then dissolved in 1.0 mL of DMF at 25 ~C. This was
followed by the addition of the hydrochloride salt of
3-picolyl chloride (0.081 mmol). After stirring 5
minutes triethylamine (300 uL, mmol) was added. The
reaction was then stirred ~or 36 hours the reaction was
quenched by the addition o~ 1.0 mL o~ saturated a~ueous
sodium bicarbonate. The reaction mixture was then
diluted by the addition of 10 mL of diethyl ether and
washed by 2 x 10 mL of each saturated aqueous sodium
bicarbonate and saturated aqueous brine. The combined
organic portions were then dried over MgS04, filtered
and concentrated in vacuo to provide the crude product.
Puri~ication of the crude solid was carried out by
silica gel chromatography ~1000 uM SiO2 prep. plate)
eluting with 20 ~ MeOH/CH2Cl2. This provided 3.1 mg of
the desired product 2, with 96 % purity by HPLC. The
CA 02243121 1998-07-14
W O 97/27180 PCTrUS9710161
- 183 -
overall yield for addition, deprotection of N-Bo_ and
coupling with 3-picolyl chloride was 9 ~.
-
.x~le 27
Svnthesis of Compound 3
A.
Ph 1) NsH, DMF .. ~ Ph
~ TsO~O ~ 2
Allyl urea 1 (195.2 mg, 0.09 mmol) was dissolved in 6.0
mL of DMF and cooled to 0 ~C. This was followed by the
addition of NaH ~54 mg, 1.0 mmol). The glycidyl
tosylate (410 mg, mmol) was then added as a solià. The
reaction was stirred for 4 hours at 25 ~C and then
quenched by the addition of 4 mL of saturated aqueous
sodium bicarbonate. The reaction was then extrac~ed by
10 mL of Et20. The organic layer was then washed by 10
mL o~ saturated aqueous sodium bicarbonate and 2 x 10
mL of saturated brine. The combined organic portions
were then dried over MgSO4, filtered and concentrated
in vacuo to provide the desired epoxide 2 (180 mg, 73
yield). The epoxide was then used without furthe-
purification.
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/O161O
~ 184 ~
".. ~_Ph ~
~ N~B~ ~~~ ~ ~ ~ ~ ~BOC
H ~ ' ~_~
~ ;-PrOH,7S-C 18 hr ll ~
o~NH o~NH
t~U 3 tBU
Piperazine 1 (25.7 mg mmol) and epoxide 2 (22.6 mg,
mmol) were heated to 75 ~C in 1.5 mL of i-PrOH for 18
hours. After cooling to 25 ~C the crude reaction
mixture was concentrated in vacuo. Complete
consumption of the epoxide was apparent by both tlc and
1H NMR.
e~ ~O
OH f~N' 1) HCI/dioxane I I OH ~N~ N
2) Picolyl Chloride-HCI ~
o~NH TEA, DMF 24 hr ~o~NH
~ tBU 2 tBU
The Boc protected piperazine 1 from the previous step
was stirred for 1.5 hours in 1.0 mL of 4 N HCl in
dioxane. This was followed by concentration in vacuo.
The crude hydrochloride salt was then dissolved in 10
mL of CH2C12 and washed by 10 mL of both saturated
sodium bicarbonate and saturated brine. The organic
portion was then dried over MgSO4, filtered and
concentrated in vacuo. The free amine was then taken
up in 1 mL of DMF. This was followed by the addition
OL 3-picolyl chloride HCl salt (50 mg, mmol) and
;
.;~
CA 02243121 1998-07-14
W O 97/27180 PCTAJS97/01610
- 185 -
~ triethyl amine (300 uL), respectively. The reaction
was then stirred at 25 ~C for 30 hours. The reaction
was then quenched by the addition of 2 mL of saturated
sodium bicarbonate and diluted by 10 mL of Et2O. The
organic portion was then washed by 10 mL of saturated
sodium bicarbonate and 2 X 10 mL of saturated brine.
The combined organic portions were then dried over
MgSO4, filtered and and concentrated in vacuo. The
crude material was purified by silica gel
chromatography (1000 uM prep. plate) eluting with 3:1,
CH2Cl2:MeOH to provide 8. 8 mg of the desired product 2.
The overall yield for addition, deprotection of the N-
Boc and reaction with 3-picolyl chloride was 19.3~.
Fx~m~le 28
Synthesis of Compound 62
<~_ 1) NaH, e"i~ lu,~ OH ~H
O i~uuluuallul, heat ~ o~N~
The THF lactam 1 (O.4 mmol) was dissolved in dry DMF at
0 ~C and to this solution was added sodium hydride
(0.47 mmol). After 30 min of stirring, (s)-
epichlorohydrin (0.47 mmol) was added and the reaction
was allowed to warm to room temperature and stir
overnight. The reaction was then diluted with water
and extracted with ethyl acetate. The organic layer
was washed sequentially with 0. 5N HCl, saturated NaHCO3
- and brine, followed by drying (MgSO4), filtration and
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 186 -
concentration in vacuo to yield product (118 mg, crude)
which was used as is. The lactam-epoxide (0.4 mmo;,
crude) was dissolved in isopropanol (2 mL), and to this
solution was added decahydroisoquinoline t-butylamide
(0.7 mmol). The mixture was then heated to 80 ~C and
stirred overnight. The reaction mixture was cooled and
concentrated to dryness in vacuo, the residue of which
was applied to a preperative TLC plate and eluted with
100% ethyl acetate to yield pure product (88 mg, 42~)
as a mixture of diastereomers.
~x~le 29
Synthesis of Compound 32
A.
1 LDA/THF/-78jC
~B~ 2 ~ ~ O ~H
-78~C---R~
A stirred, cooled (-78 ~C ) solution of 1.4 g (5.0
mmol) of pyrrolidinone in 35 mL of anhydrous
tetrahydrofuran was treated in a dropwise fashion with
3.6 mL (7.2 mmoL) of lithium diisopropylamide. The
resultant solution was stirred for 70 min, and
subsequently treated with 0.57 mL (6.0 mmoL) o~ 3-
pyridine carboxaldehyde. The homogenous solution wasailowed to ambiently warm to room temperature (RT), and
stirring was continued overnight. The reaction mixture
was diluted with 400 mL of dichloromethane, washed once
.
CA 02243l2l l998-07-l4
W O 97/27180 PCT~US97/01610
- 187 -
with 150 mL of water, dried (magnesium sulfate),
filtered, concentrated, and purified on silica gel
using 3:1 ethyl acetate/hexanes as the eluent,
affording 0.6 g (46%) of the desired compound as a
golden oil which solidified upon standing.
1H NMR (d6-DMSO, 400MHz) 8.65 ~s, lH); 8.47 (m, 2H);
7.83 (d, J = 8.0 Hz, lH); 7.41 (m, lH); 7. 23 (m, 5H);
7. 03 (t, J = 2.7 Hz, lH); 3.g6 (m, lH); 3.07 (m, lH);
2.89 - 2.65 ( series of m, 3H ). M+H (265.2).
B.
H2 / 10% Pd-C
H CH~OH / RT / lh H
o
A vigorously stirred suspension of 330 mg ( 1. 25 mmoL
of eneamide and 80 mg of 10~ palladium on carbon
(Degussa) in 12mL of anhydrous methanol was
hydrogenated (Hydrogen balloon) for 1 h. The mixture
was diluted with 100 mL of methanol, carefully
filtered, concentrated, and purified on silica gel
using ethyl acetate as the eluent, affording 2 95 mg
(899~) of an isomeric mixture of the desired compounds
as a golden oil which solidified upon standing.
H NMR (d6-DMSO, 400MHz) 8. 36 (s, 2H); 7.88 (s, lH);
7.56 (d, J = 7.9 Hz, lH)i 7.27 - 7.12 (m, 7H); 3.66
(m, lH)i 2.96 - 2.37 (series of m, 7H) . M~H (267.2);
M+Na (289.2)
-
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 188 -
1 ) NaH, DMF, 80'C trans-isomer 1
~H conc. HCI
0~ ,
~8
cis-isome~2
The lactam obtained above was coupled to the
corresponding epoxide according to the protocol used
for Example 24. The final purification was performed
on silica gel ~2% 2M ammonia-methanol in
dichloromethane) to give the cis- and the trans-lactam
diastereomers each as white solids.
trans-isomer 1: Rf: 0.20 H NMR (CDC13,. 400MHz) :
1.62~2H,m), 1.86(4H,m), 2.19tlH,m), 2.63(2H,m), 2.78-
3.10(8H,m), 3.65(lH,m), 3.75(lH,bt), 3.95(lH,t),
4.27(lH,t), 5.24(lH,m), 6.32(lH,d), 7-7.4(14H,m),
8.22(lH,s), 8.34(lH,s). M~H (604)
cis-isomer 2: Rf: 0.18. H NMR (CDC13,. 400MHz) :
1.35(1H,m), 1.60(2H,m), 1.95(2H,m), 2.19(1H,dd),
CA 02243121 1998-07-14
W O97/27180 PCTrUS97/01610
- 189 -
., .
~ 2.48(lH,dd), 2.60(lH,m), 2.8-3.05(5H,m), 3.10(lH,dd),
3.26(lH,dd), 3.60(lH,m), 3.78(lH,m), 3.99(lH,m),
4.15(1H,bs), 4.24(lH,t), 5.24(lH,m), 6.18(lH,d),
7.02(2H,d), 7-7.3(lOH,m), 7.41(lH,d), 8.25(lH,s),
58.40(lH,d). M~H (604)
Ex~m~le 30
'
NH + ~ NH2 DIEA 70~C H
1 2 3
The iodolactam 1 (0.43 mmol) was dissolved in dry
acetonitrile in a high pressure tube and to this
solution was added diisopropylethylamine (Pierce, 0.65
mmol) followed by aniline 2 (Aldrich, 0.47 mmol). The
tube was sealed and the reaction heated to 70 ~C with
stirring overnight. The reaction was cooled to ambient
temperature, solvent removed in vacuo, and the residue
taken up in ethyl acetate/water. The organic layer was
washed sequentially with saturated aqueous NaHCQ3 and
brine, followed by drying (MgSO4), filtration and
concentration in vacuo. The crude residue was purified
by flash silica gel chromatography eluting with 1:1
ethyl acetate/hexanes to give 61 mg of product 3; TLC
Rf = 0.29 (l:l ethyl acetate/hexanes); HPLC Rt = 12.6
min (96%); MALDI-TOF MS m/z 267 (M ).
-
CA 02243121 1998-07-14
W O97/27180 PC~US97/01610
-- 190 --
F~mrle 31
~ THF
PMB lactam 1 (1.5 g, 5.07 mmol) was dissolved in THF
~12 mL), cooled to -78 ~C, and to this solution was
added LDA (6.6 mmol , 1.3 eq.), over 7 minutes to give
a greenish-brown anion. The reaction mixture was
stirred at -78 ~C for 55 minutes after which a solution
of bromoacetonitrile (400 ul, 0.75 mmol, 1.1 eq.) was
added over 2 minutes while keeping the internal
reaction temperature at <-65 ~C. The reaction was
stirred at -78 ~C ~or 2 hours, then warmed to room
temperature and stirred for an additional 16 hours.
The reaction was cooled to -50 ~C and quenched with
saturated ammonium chloride solution. The reaction was
partitioned between ethyl acetate and a saturated
bicarbonate solution. The aqueous layer was extracted
with ethyl acetate. The combined organic layers were
then washed with water, brine and dried (MgS04) and
filtered. Concentration in vacuo afforded 1.6g of
crude material, which was purified by silica gel
chromatography to give 640 mg (38~) of the desired
material 2.
lH NMR (CDC13) d 7.31 (m, 3H), 7.18 ld, 2 H), 7.09 (d,
2H), 6.90 (d, 2H), 5.08 (d, lH), 3.92 (d, lH), 3.81 (s,
CA 02243121 1998-07-14
WO 97/27180 PCT~US97/01610
-- 191 --
~ 3H), 3.70 (m, lH), 2.92 ~dd, lH), 2.72 ~m,2H), 2.55
(dd,lH), 2.42 (m,lH), 2.19 (dd,lH), 1.81 (m, lH).
B.
Ph Ph
~OC~ (N~l4)2ce(N~)6
NC ~ C~CN:water
1 2
PMB lactam 1 (640mg, 1.9 mmol) was dissolved in CH3CN
(9 mL). 1 mL of water was added followed by 3.1 g of
cerium ammonium nitrate. The reaction went from dark
amber to light orange within 5 minutes and was stirred
at room temperature for 18 hours. The reaction was
concentrated in vacuo and the residue was partitioned
between ethyl acetate and a saturated bicarbonate
solution. The aqueous layer was extracted with ethyl
acetate. The combined organic layers were then washed
with saturated bicarbonate solution, water, brine,
dried (MgSO4) and filtered. Concentration in vacuo
afforded 590 mg of crude material, which was purified
by silica gel chromatography (9:1 CH2Cl2 : EtOAc) to
give 285 mg (70%) of the desired material 2. HPLC
suggests 2 diastereomers, retention time 9.95
min.(major) and 10.17 min. (minor).
H NMR (CDCl3) d 7.37 (m, 2H), 7.28 (m, 1 H), 7.20 (m,
2H), 5.74 (br s, lH), 3.95 (m, lH), 2.85 (dd, lH),
2.79-2.65 (m, 3H), 2.55 (dd, lH), 2.27 (m,2H).
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 192 -
F.~;?7r~rl e 32
- OMe 1) LDA, THF / = ~ OMe
~\N~
2 3
The PMB lactam 1 (O.46 mmol) was dissolved in dry THF
at -78 ~C and to this solution was added lithium
diisopropylamide (Aldrich, 1.5 M in cyclohexane, 0.65
mmol). The solution was stirred for 15 minutes at
-78 ~C and 4-(Chloromethyl)-3,5-dimethylisoxazole 2
(Acros Organics, 0.56 mmol) was added. The cooling
bath was removed and the solution warmed to room
temperature and stirred overnight. The reaction was
diluted with water and extracted with ethyl acetate.
The organic layer was washed sequentially with
saturated aqueous NaHCO3 and brine, followed by drying
(MgSO4), filtration and concentration in vacuo. The
crude residue was purified by flash silica gel
chromatography eluting with 10% diethyl
ether/dichloromethane to give 53 mg of product 3 as a
mixture of diastereomers.
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 193 -
B.
6~ ~e
. CAN ~ ~ H
7:3CH3CN/~20 O
1 2
Lactam 1 (0.13 mmol) was dissolved in 7:3
acetonitrile/water. Ceric ammonium nitrate (Aldrich,
0.26 mmol) was added and the mixture was stirred at
ambient temperature until the starting material was no
longer evident by TLC. Acetonitrile was removed in
vacuo, and the residue taken up in ethyl acetate/water.
The~organic layer was washed sequentially with
saturated aqueous NaHC03 and brine, followed ~y drying
(MgS04), filtration and concentration in vacuo. The
crude residue was purified by flash silica gel
chromatography eluting with 8~ MeOH in dichloromethane
to give 21 mg of product 2; TLC Rf = 0.47 (8%
MeOH/CH2C12 ) .
-
CA 02243121 1998-07-14
W O 97/27180 PCT~S97/01610
- 194 -
~x~m~le 33
A.
NPMB 2)~ru,l,.aldeh~de ~ NPMB
b 3)~BS,PPh3 O
1 2
Lactam 1 (1.43 mg, 4.86 mmol) was dissolved in
anhydrous THF (25 mL) and cooled to -78 ~C. This was
followed by the addition of 3.9 mL of L~A (5.83 mmol,
1.2 eq.). The anion solution was stirred at -78 ~C for
45 minutes and then cannulated into a -78 ~C solution
of p-formaldehyde (437 mg) in 25 mL of THF, washing
with 1 mL of THF. The reaction was warmed to room
temperature over 4 hr and stirred overnight. The
reaction was quenched by the addition of 10 mL of a
saturated sodium bicarbonate, and concentrated in vacuo
to remove the THF. The crude reaction mixture was
partitioned between ethyl acetate and saturated sodium
bicarbonate. The aqueous layer was extracted with
ethyl acetate. The combined organic layers was then
washed with water, brine and purified by silica gel
chromatography (gradient of 50 to 75 % ethyl acetate:
hexanes), to provide 584 mg (45%) of the desired
alcohol, as well as 265 mg of recovered starting
material.
CA 02243121 1998-07-14
W O 97127180 PCT~US97/01610
- 195 -
The alcohol (316mg, 0.979 mmol) was then dissolved in 3
mls of CH2Cl2 and added to a 0 ~C solution of triphenyl
phosphine (734 mg, 2.8 EQ.) and NBS (534 mg, 3 EQ.) in
3 mls of CH2Cl2. After l hour the reaction was
quenched by the addition of 10 mL of Et20. The organic
layer was then filtered and the filtrate washed with
saturated sodium bicarbonate, brine, dried (MgSO4) and
filtered. Concentration in vacuo afforded the crude
product which was purified by silica gel chromatography
(CH2Cl2) to provide 151 mg (40% of the bromide.
The bromide (87.2 mg, 0.28 mmol) was dissolved in 2 mL
of benzene and treated with imidazole (46mg, 3 EQ.).
After heating to 125 ~C for 20 hours the reaction was
cooled to 25 ~C and concentrated in ~acuo. The crude
product which was purified by silica gel chromatography
(5 % MeOH/CH2Cl2), to provide the addition product
(50%) and the elimination product (2) in a 50 % yield.
'
.--~ 1 ) CAN -~
~N PMB 2) imidazole N~;~N ~NH
The lactam 1 (621 mg, 2.02 mmol) was dissolved in 7 mL
acetonitrile, followed by the addition of H2O (3 mL).
This was followed by the addition of CAN, 3.32 g (6.06
mmol, 3 EQ.). The reaction was stirred at 25 ~C for 1
CA 02243121 1998-07-14
W O97/2718~ PCTr~S97/01610
-- lg6 --
hour. After concentrating the reaction in vacuo, the
crude material was resuspended in ethyl acetate and
washed with saturated sodium bicarbonate, brine, dried
(MgSO4) and filtered. Concentration in vacuo afforded
the crude product which was purified by silica gel
chromatography (3% methanol:CH2C12) to procide the
desired unprotected lactam (122 mg, 32 ~)
The ~,~-unsaturated lactam (55 mg, 0.29 mmol) was then
heated to 130 ~C in 2 mL of benzene containing
imidazole (30 mg, 0.44 mmol) for 24 hours. After
cooling to 25 ~C, the reaction mixture was concentrate
in vacuo. The crude material was purified by silica
gel chromatography, eluting with 5~ methanol:CH2C12 to
provide 46.7 mg of the desired addition product ~63 ~)
as well as 15.7 mg of recovered starting olefin (29 %).
~xam~le 34
S~H + ~ H CH3CN , ~N~NH
o DIEA, 70~C
2 3
The iodolactam 1 (0.45 mmol) was dissolved in dry
acetonitrile in a high pressure tube and to this
solution was added diisopropylethylamine (Pierce, 1.35
mm-ol) followed by indoline 2 (Aldrich, 0.54 mmol). The
-
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 197 -
tube was sealed and the reaction heated to 70 ~C with
stirring overnight. The reaction was cooled to ambient
temperature, solvent removed in vacuo, and the residue
taken up in ethyl acetate/water. The organic layer was
washed sequentially with saturated aqueous NaHCO3 and
brine, followed by drying (MgSO4), filtration and
concentration in vacuo. The crude residue was purified
by flash silica gel chromatography eluting with ethyl
acetate to give 113 mg of product 3; TLC Rf = 0.39
(ethyl acetate); HPLC Rt = 13.1 min (92%); MALDI-TOF
MS m/z 293 ~M ).
,~
Fx~le 35
A.
Ph~2~0Me EtNOz ~ OMe
In an oven-dried 100 mL round-bottomed flask, the vinyl
sulfone PMB lactam 1 (1.2126 g, 2.55 mmol) was
dissolved in 50 mL of C6H6. Phenyl isocyanate (2.0 mL,
18.4 mmol) was added via syringe followed by the
dropwise addition of nitroethane (0.4 mL, 5.56 mmol).
Triethylamine (2.0 mL, 14.3 mmol) was added dropwise.
The solution was refluxed for 15 minutes and cooled. A
white solid precipitated during the heating period.
The mixture was cooled, poured into water and extracted
,.
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 198 -
. with CH2Cl2 The organic extract was dried (MgS04) and
~ evaporated in vacuo to afford a brown oil that was
chromatographed to afford the isoxazole PM.3 lactam 2
(901 mg, 90%) as a light yellow oil.
B.
OMe
~ N ~ 70% CH3CN-H20
- 1 2
In a 25 mL round-bottomed flask, isoxazole PMB lactam 1
(900 mg, 2.30 mmol) was dissolved in 14 mL of 70%
CH3CN-H2O. Ceric ammonium nitrate (3.607 g, 6.58 mmol)
was added forming a dark orange solution. The mixture
was stirred until the starting material was no longer
evident by TLC (10% EtOAc/CH2C12). The light yellow
solution was diluted with CH2Cl2 and washed with water.
The organic layer was separated, dried (MgS04), and
evaporated in vacuo to afford a brownish-red oil that
was chromatographed (10% EtOAc/CH2Cl2) to produce the
lactam 2 (300.3 mg, 48%) as a colorless oil.
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 199 -
~x~mnle 36
NH ~ CH3CN ~ ~ NH
1 2 3
The iodolactam 1 (0.78 mmol) was dissolved in dry
acetonitrile in a high pressure tube and to this
solution was added diisopropylethylamine (Pierce, 2.35
mmol) followed by N-methylaniline 2 ~Aldrich, 0.94
mmol). The tube was sealed and the reaction heated to
70 ~C with stirring overnight. The reaction was cooled
to ambient temperature, solvent removed in vacuo, and
the residue taken up in ethyl acetate/water. The
organic layer was washed sequentially with saturated
aqueous ~aHCO3 and brine, followed by drying (MgSO4),
filtration and concentration in vacuo. The crude
residue was purified by flash silica gel chromatography
eluting with 2:1 ethyl acetate/hexanes to give 134 mg
of product 3; TLC Rf = 0.24 (2:1 ethyl
acetate/hexanes); HPLC Rt = 12.7 min (80%); MALDI-
TOF MS m/z 282 (M ).
CA 02243121 1998-07-14
W O 97/27}80 PCTnUS97/01610
- 200 -
F.x~ ~ le 37
[3~ 0~
'''~ ~ ~ ~ dioxane10~C ~ ~
NaH (0.96 g, 40 mmol) was suspended into 20 mL of
dioxane. This was followed by the addition of diethyl
malonate (4.6 mL, 40 mmol), then phenyl iodide (2.2 mL,
20 mmol) and finally copper (I) iodide (7.6g, 40 mmol).
The reaction was then heated to 100 ~C for 14 hours.
The reaction was then quenched with water and diluted
with ethyl acetate, the organic layer was washed with
water and saturated NaCl, dried (MgSO4) and
concentrated in vacuo. The crude product was further
purified by MPLC (SiO2~ eluting with 4:1, toluene:
ethyl acetate to provide 1.21 g of product (29 %
isolated yield).
B.
~0~~~ ~J EtOO~
CsCO3 MeCN
CA 02243121 1998-07-14
W O 97/27180 rCT~US97/01610
- 201 -
- The alkylated malonic ester (1, 227 mg, 1.09 mmol) was
stirred for 14 hours in acetonitrile (2.5 mL)
containing; cesium carbonate (710 mg, 2.18 mmol) and
the bromide (516 mg, 1.31 mmol). The reaction was then
concentrated to dryness in vacuo. After re-suspension
of the reaction mixture in ethyl acetate, the reaction
mixture was washed with water, saturated NaHC03 and
saturated NaCl, dried (MgS04) and concentrated in
vacuo. The crude product was further purified by MPLC
(SiO2) to provide 200 mg of the desired product 35.2
yield).
EtO~ [~ H2 ( approx. 1 ATM) ~NH
PdlC, HCI, EtOH EtO O
0 1 2
To the malonate (1, 200 mg) in ethanol (3 mL~ was added
concentrated HCl (lOOuL) and an excess of 5~ Pd / C
(approx. 50 mg). The reaction was then fitted with a
balloon of H2 and hydrogenated for 14 hours. After
purging the reaction mixture of H2 triethylamine (1
' mL, 7 mmol, excess) and an excess of solid NaHC03 was
_ added. After stirring for 30 minutes the reaction was
filtered and concentrated in vacuo. The yellow oil was
.,
CA 02243121 1998-07-14
W O97/27180 PCT~US97/01610
- 202 -
then re-dissolved in ethyl acetate and the reaction
mixture was washed with water, saturated NaHCO3 and
saturated NaCl, dried ~MgSO4) and concentrated in vacuo
= to provide the desired product. The 1H NMR was
consistent with the desired material.
~x~le 38
A.
\ OMe Ph
-~' ~ 1)LDA,THF "
2) propargyl bromide
O O
1 2
In an oven-dried 25 mL round-bottomed flask, the PMB-
lactam 1 (563.7 mg, 2. 75 mmol) was dissolved in 10 mL
of THF. The solution was cooled to -7 8 ~C and l. 5M LDA
(2.0 mL, 3.00 mmol) was added dropwise via syringe
producing the yellow color o~ the enolate. The
solution was stirred for 15 minutes at -78 ~C and
propargyl bromide (310 uL, 3.48 ~ Lol) was added
dissipating the yellow color. The cooling bath was
removed and the solution was warmed to room temperature
and stirred overnight. The solution was poured into lN
HCl and extracted with CH2Cl2. The organic extracts
were combined and washed with saturated aqueous
NaHCO3. The organic layer was separated, dried (MgSO4)
and evaporated in vacuo to afford a brown oil that was
chromatographed (90~ CH2Cl2/hexane) to produce the
propargyl lactam 2 (577 mg, 8696) as a colorless oil.
CA 02243121 1998-07-14
W O 97127180 PCTrUS97/01610
- 203 -
Ph OMe
70% C~3CN20 ~ ~;b" IN~H
In a 25 mL round-bottomed flask, propargyl PMB lactam 1
(358.2 ~g, 1.08 mmol) was dissolved in 6 mL of 70~
CH3CN-H20. Ceric ammonium nitrate (1.321 g, 2.41 mmol)
was added for~ing a dark orange solution. The mixture
was stirred until the starting material was no longer
evident by TLC (10% EtOAc/CH2C12). The light yellow
solution.was diluted with EtOAc and washed with water.
The organic layer was separated, dried (MgSO4), and
evaporated in vacuo to afford a yellow oil that was
chromatographed (10% EtOAc/CH2C12) to produce the
propargyl lactam 2 (145 mg, 63%) as a colorless oil.
F.x~le 39.
NH DIEA 70~C- ~ N ~ .'~H
1 2 3
The iodolactam 1 (1.38 mmol) was dissolved in dry
acetonitrile in a high pressure tube and to this
CA 02243121 1998-07-14
W O 97/27180 rCTrUS97/01610
- 204 -
solution was added diisopropylethylamine (Pierce, 4.15 -
mmol) followed by tetrahydroquinoline 2 ~Aldrich, 1.66
mmol). The tube was sealed and the reaction heated to .~
70 ~C with stirring overnight. The reaction was cooled
to ambient temperature, solvent removed in vacuo, and
the residue taken up in ethyl acetate/water. The
organic layer was washed sequentially with saturated
aqueous NaHCO3 and brine, followed by drying (MgSO4),
filtration and concentration in vacuo. The crude
residue was purified by flash silica gel chromatography
eluting with 1:1 ethyl acetate/hexanes to give 233 mg
of product 3; TLC Rf = 0.21 (1:1 ethyl
acetate/hexanes); HPLC Rt = 14.0 min (85~); MA1DI-
TOF MS m/z 307 (M ).
Fx~m~le 40
- A.
Ph OMe Ph OMe
1 ) PhSCI
3) DBU O
In an oven-dried 250 mL round-bottomed flask, N-
chlorosuccinimide (2.5177 g, 18.9 mmol) was dissolved
in 75 mL of CH2C12. The solution was cooled to 0 ~C and
20 thiophenol (1.90 mL, 18.5 mmol) was added dropwise via
syringe causing an immediate formation of a yellow
color and an exotherm. The orange solution of PhSCl
was stirred for 30 minutes at room temperature and a
-
CA 02243121 1998-07-14
W O 97t27180 PCT~US97/01610
- 205 -
~ solution of the allyl lactam 1 (6.156 g, 18.4 mmol! was
added dropwise dissipating the orange color. The light
~ yellow solution was stirred for two hours and the
solvent was removed in vacuo. CCl4 was added to the
yellow oll that remained and the undissolved
succinimide was removed by filtration. The filtrate
was evaporated in vacuo to afford the diastereomeric
chlorosulfides as a yellow oil that was chromatographed
(CH2C12) rapidly to remove low Rf impurities. The two
highest Rf spots were the chlorosulfide diasteromers.
The purified mixture of chlorosul~ides was dissolved in
CH2C12 and m-chloroperbenzoic acid (2.0 g, 11.6 mmol)
was added with cooling from an ice-bath. The mixture
was stirred for 10 minutes and filtered. The filtrate
was evaporated in vacuo to afford a yellow oil (8.125
gj 86%) that produced two low Rf spots (CH2Cl2) using
thin-layer chromatography for the two chlorosulfone
diastereomers. The oil was redissolved in CH2C12 and
DBU (2.7 mL, 18.1 mmol) was added dropwise at room
temperature. The solution was heated for 15 minutes
causing the solution to turn dark yellow. The solution
was cooled and the solvent was evaporated in vacuo.
The residue was chromatographed (CH2Cl2) to afford the
pure vinyl sulfone 2 (4.805 g, 55%) as a colorless oil.
B.
Ph OMe TMSCHN2 ~MS .~ OMe
PhOa Sw~ BuLi
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 206 -
- In an oven-dried 25 mL round-bottomed flask,
trimethylsilyl diazomethane (140 uL, 0.280 mmol) was
dissolved in 5 mL of THF. The bright yellow solution
was cooled to -78 ~C and n-BuLi (320 uL, 480 mmol) was
added. In a separate oven-dried 25 mL round-bottomed
flask, the vinyl sulfone PMB lactam 1 (108 mg, 0.227
mmol) was dissolved in 5 mL of THF and added dropwise
via syringe at -78 ~C to the lithiate solution. The
resulting solution was stirred for 1 hour at -78 ~C and
then two hours at 0 ~C. The mixture was acidified with
lN HCl and extracted with CH2Cl2. The organic extract
was dried (MgS04) and evaporated in vacuo to afford a
cloudy, colorless oil that was chromatographed (20~
. EtOAc/CH2Cl2) to produce the TMS pyrazole PMB lactam 2
(88.4mg, 87%) as a clear, colorless oil.
C.
~MS~ OMe TBAF Ph OMe
CH3CN ~N~3
1 2
In an oven-dried 25 mL round-bottomed flask, the TMS
pyrazole PMB lactam 1 (1. 1345 g, 2. 53 mmol) was
dissolved in 110 mL of 91~ CH3CN/H20.
Tetrabutylammonium fluoride ~2.7 mL of a l.OM solution
in THF, 2. 70 mmol) was added dropwise via syringe. The
reaction was refluxed for 4 8 hours and cooled. The
solvent was evaporated in vacuo and the residue was
-
CA 02243l2l l998-07-l4
W O 97/27180 PCTrUS97/01610
- 207 -
dissolved in CH2Cl2. The organlc solution was washed
with lN HCl solution, dried (MgSO4), and evaporated in
- vacuo to afford a yellow oil that was chro~atographed
(20~ EtOAc/CH2C12) to afford the pyrazole (688 mg, 72~) -
as a light yellow oil.
D.
OMa 1)NaH ~ ~ONb
In an oven-dried 100 mL round-bottomed flask, the
pyrazole PMB lactam 1 (588 mg, 1.57 mmol) was dissolved
in 25 mL of THF. NaH (50 mg of a 60% dispersion in
mineral oil, 2. 08 mmol) was added. Gas evolution was
observed. Methyl chloroformate (140 uL, 1.81 mmol) was
added and the reaction was stirred at room temperature
overnight. The mixture was acidified with lN HCl and
extracted with CH2Cl2. The organic extract was dried
(MgSO4), and evaporated in vacuo to afford the pyrazole
carbamate PMB lactam 2 (588 mg, 87%) as a light yellow
oil.
E.
Ph~ OMe Ph~
M ~N3J~ 70~/Oace~rlibi~~/WJt~Me--O~ ~N~H
.
CA 02243121 1998-07-14
W O 97127180 PCTnUS97/01610
- 208 -
In an oven-dried 100 mL round-bottomed flask, the
pyra~ole carbamate PMB lactam 1 (577 mg, 1.33 mmol) was
dissolved in 30 mL of 70~ CH3CN-H2O. Ceric ammonium
nitrate (2.5123 g, 4.58 mmol) was added. The orange
solution was-stirred at room temperature until the
starting material was no longer evident by TLC (1 hr).
The light yellow solution was poured into water and
extracted with EtOAc. The organic extract was dried
(MgSO4) and evaporated in vacuo to afford the pyrazole
carbamate lactam 2 (228 mg, 55%) as a clear, colorless
oil.
FX~ ~ le 41
-
B 35nN3 ~ OMe
~ 205~C
1 2
In a heavy-walled screw-top test tube, the propargyl
lactam 1 (1.111 g, 3.33 mmol) was dissolved in 7 mL of
xylene. Tributyltin azide (1.965 g, 5.92 mmol) was
added, the tube was sealed and heated to 205 ~C
overnight. The dark brown solution was cooled and
directly chromatographed using a gradient from CH2C12
to 50~ EtOAc/CH2C12 to afford the triazole PMB lactam 2
(827 mg, 66~) as a light yellow oil.
. .
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 209 -
H Ph OMe ~Ph Ph OMe
~ ~ 2) PhCI'2Br /~,
In an oven-dried 100 mL round-bottomed flask, the
triazole PMB lactam 1 ~827 mg, 2.20 mmol) was dissolved
in 40 mL of THF. NaH (124 mg of a 60% dlspersion in
mineral oil, 5.17 mmol) was added. Gas evolution was
observed. Benzyl bromide (400 uL, 3.36 mmol) was
added. The reaction was stirred at reflux until the
starting material was not longer evident by thin-layer
chromatography (50% EtOAc/CH2Cl2). The mixture was
acidi~ied with lN HCl and extracted with CH2Cl2. The
organic extract was dried (MgSO~), evaporated ln vacuo
to af~ord a dark yellow residue that was
chromatographed (20% EtOAc/CH2Cl2) to produce the
benzyl triazole PMB lactam 2 (740 mg, 72%) as a light
yellow oil.
~ ~ ~ a 70%C~CN-~O ~
3 In an oven-dried 50 mL round-bottomed flask, the benzyl
triazole PMB lactam 1 (740 mg, 1.59 mmol) was dissolved
.
CA 02243121 1998-07-14
W O 97/27180 PCTAJSg7/01610
- 210 -
in 22 mL of 70~ CH3CN-H2O. Ceric ammonium nitrate (2.1
g, 3.83 mmol) was added. The orange solution was
stirred at room temperature until the starting material -~
was no longer evident by TLC (1 hr). The mixture was
poured into water and extracted with EtOAc. The
organic extract was dried (MgSO4) and evaporated in
vacuo to afford the benzyl triazole lactam 2 (336 mg,
61%) as a clear, colorless oil.
Fx~m~le 42
1) LDAtTHF
2) acetone .-'
BOC 3~ Ma in's rgt ~N_H
4) H2/10% Pd-C ~
MeOH 2
5) TFA/Ct~CI2
BOC-lactam 1 (1.8 g, 6.6 mmol) was dissolved in THF (50
mL) and cooled to -78 ~C. To this solution was added
LDA (Aldrich, 1.5 M in cyclohexane, 5.3 mL, 7.9 mmol)
via syringe over lQ minutes. After stirring for 60 min
at -78 ~C, acetone (4.9 mL, 66 mmol) was added via
syringe over 1 minute. The reaction was stirred for an
additional 40 minutes before being quenched with lN HCl
(15 mL~. Ethyl acetate (100 mL) was added and the
layers were partitioned. The organic layer was washed
with brine, dried over magnesium sulfate, ~iltered, and
concentrated in vacuo to a yellow oil that slowly
.
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 211 -
- crystallized. The crude alcohol was dissolved in
dichloromethane (50 mL) and Martin's sulfurane
~- (Aldrich, 7.5 g, 11 mmol) was added in one portion.
The reaction was stirred for 36 h at room temperature
before being concentrated in vacuo. Flash
chromatography over silica gel (3:1 hexane:ethyl
acetate) provided the alkene as a mixture of isomers.
The alkene, 10% Pd-C (1.0 g), and methanol (40 mL) were
combined in a Parr bottle and pressurized to 50 psi of
hydrogen gas. After 4 h of agitation, the reaction
vessel was evacuated and filtered through a plug of
Celite. The cake was washed with ethyl acetate (20 mL)
and the combined filtrate was concentrated in vacuo to
give the isopropyl BOC-lactam as a pale yellow oil.
The lactam was dissolved in dichloromethane (20 mL) and
trifluoroacetic acid (10 mL) was added slowly. The
reaction was stirred at room temperature for 24 h
before being diluted with ethyl acetate (100 mL) and
carefully neutralized with 10~ sodium carbonate to pH
7. The layers were partitioned and the organic layer
was dried over magnesium sulfate, filtered, and
concentrated in vacuo. Flash chromatography over
silica gel (3:1 ethyl acetate:hexane) gave the
isopropyl lactam as a white powder. MS (ES+) = 240
(M+Na)
CA 02243121 1998-07-14
W O97/27180 PCTrUS97/01610
- 212 -
Fx~le 43
.'
Boc 1- LDA/THF/-78~,C ~N~H
-78~C--- RT
A stirred, cooled ~-78 ~C) solution of 1.4 g (5.0 mmol)
of pyrrolidinone in 35 mL of anhydrous tetrahydrofuran
was treated in a dropwise fashion with 3.6 mL ~7.2
mmoL) of lithium diisopropylamide. The resultant
solution was stirred for 70 min, and su~sequently
treated with 0.57 mL (6.0 mmoL) of 3-pyridine
carboxaldehyde. The homogenous solution was allowed to
ambiently warm to RT, and stirring was continued
overnight. The reaction mixture was diluted with 400
mL of dichloromethane, washed lX with 150 mL of water,
dried (magnesium sulfate), filtered, concentrated, and
purified on silica gel using 3:1 ethyl acetate/hexanes
as the eluent affording 0.6 g (46~) of the desired
compound as a golden oil which solidified upon
standing. H NMR (d6-DMSO, 400MHz) 8.65 (s, lH); 8.47
(m, 2H)i 7.83 (d, J = 8.0 Hz, lH); 7.41 (m, lH); 7.23
(m, 5H); 7.03 ~t, J = 2.7 Hz, lH); 3.96 (m, lH); 3.07
(m, lH); 2.89 - 2.65 (series of m, 3H). M+H (265.2).
.
CA 02243l2l l998-07-l4
W O 97/27180 PCT~US97/01610
- 213 -
xample 44
~~ ~ I) LDA
- pyn~n~-3~bo~1dehvde G~N
Ç~NBOC ~2/Pd-C ~NH
O O
1 2
The first step of the sequence was performed as for
F.x~mrl e 43. The olefin was carried forward as follows:
step 2
A vigorously stirred suspension of 330 mg (1.25 mmoL)
of eneamide and 80 mg of 10% palladium on carbon
(Degussa) in 12mL of anhydrous methanol was
hydrogenated (Hydrogen ~alloon) for 1 h. The mixture
was diluted with 100 mL of methanol, carefully
filtered, concentrated, and purified on silica gel
using ethyl acetate as the eluent affording 295 mg
(89~) of an isomeric mixture of the desired compounds
as a golden oil which solidified upon standing. lH NMR
(d6-DMSO, 400MHz) d 8.36 (s, 2H); 7.88 (s, lH); 7.56
(d, J = 7.9 Hz, lH); 7.27 - 7.12 (m, 7H); 3.66 (m,
lH); 2.96 - 2.37 (series of m, 7H). M+H (267.2); M+Na
(289.2)
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 214 -
F.x~le 45
1) LDA
.~pyri~ine-2-carb~-Y~l~Phyde ~
~\NBoc-78~ C , ~N
2) ~d-C
~ 1 2
The synthesis of the 2-pyridyl methylpyrrolidone was
carried out as shown in Example 44.
~x~le 46
I) LD
, ~rli~,e 1 carboYaldehyde N~
2) H2/Pd-C i~/~ ~NH
O O
The 4-pyridylmethylpyrrolidone was prepared following
procedures outline for Example 44.
CA 02243l2l l998-07-l4
W O 97127180 PCT~US97/01610
- 215 -
~ Fx~le 47
c 1 ~eo~pocH2c ~ buIN~
- 2 H2/PdC ~ NH
Boc-Phenyl~l~nin~l ' X~
4 TF~nPl-tr~li7r X=O N-Bn
X = N-Bn
A.
A solution of 5.06g (20 mmol, 1 equiv) of tert-Butyl-
P, P-dimethylphosphonoacetate in 15 mL THF cooled to
0 ~C was treated with 0. 528g of NaH at 0 ~C and then
warmed up to room temperature for 30 min. Next,
solution of 5. Og (20 mM, 1 equiv) of Boc-Phenylalaninal
in 5 mL THF was added dropwise at 0~C and the reaction
continued for 2 h. The crude product was diluted with
ethyl acetate and partitioned with aqueous citric acid
(2x), sodium bicarbonate (2x), organics collected and
dried over magnesium sulfate. The product was then
dissolved in 100 mL methanol, added 0. 6g 10% Pd/C and
hydrogenated at 25 psi overnight, and the desired
compound purified on a silica column using 1/4 ethyl
acetate/hexane. Yield 3.8g (51.4%).
H NMR (CDCL3, 300 MHz) ~ (broad signals and
conformational averaging) 7.20 (m, 5H), 4.46 (m. 0.5H),
3.79 (m, 0.5H), 3.72 (s, 0.5H), 2.80 (m, 0.5H), 2.46
(m. 0.5H), 2.27 (m, lH), 1.78 (m, lH), 1.50 (m. lH),
{1.44, 1.42, 1.41, 1.38} (all s, total 18H). Low
resolution MS m/e 372.2 (M+Na ).
i
CA 02243l2l l998-07-l4
WO 97/27180 PCT~US97/01610
- 216 -
A solution o~ 4.63 g (13.25 mmol, 1 equlv) of the above
ester in 200 mL of THF was treated with 40 mL (39.75
mmol, 3 equiv) of lM lithium bis(trimethylsilyl) amide
in THF at -78 ~C. After 90 min at -78 ~C, the solution
was added 5.5g (13.25 mmol, 1 equiv) of N-benzyl-N-
bis(iodoethane) in 10 mL of THF and the reaction
continued for 6 hours during which it reached the room
temperature. The reaction was quenched wlth 10% aqueous
solution of citric acid and extracted to ethyl acetate,
and the product treated with 1:1 (v/v) DCM/TFA (40 mL)
~or 40 min, after which solvents were removed and the
crude purified to homogeneity by RP HPLC with total
. yield of 14 2%. The resulting TFA salt was then
neutralized with triethylamine, extracted between ethyl
acetate/water, organics collected and dried, thus
yielding a free base form of the spiropyrrolidone
product which is used in subsequent coupling to the
epoxide. 1H NMR (T~A salt, CDCL3, 300 MHz~ ~ 7.30 (m,
lOH), 5.85 (m, lH), 4 .16 (m, 2H), 3. 86 (m, lH), 3. 68
(m, lH), 3.36 (m, 3H), 2.88 (dd, lH), 2. 62 (dd, lH),
1.7-2.2 ~m, 6H). Low resolution MS m/e 335. 2 ~M~H
Fx~rr~l e 48
Spirocycle X=O was synthesized according to
bisalkylation protocol o~ Example 47 above except that
bis-0-(iodoethyl) ether was used in reaction step B
(1.26g, 3, 87 mmol, l equiv). H NMR (d6-DMSO, 300 MHz)
~ 7.79 (s, lH), 7.22 ~m, 5H), 3.73 (m, 3H), 3.24 (m,
2H), 2.88 (dd, lH, J=4.8, 13.4), 2.57 ~dd, lH, J=8.4,
30 13.4), 2.03 (m., lH), 1.76 tm, lH), 1.55 (m, 2H), 1.22
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 217 -
- (m, lH), 1.01 (m, lH). Low resolution MS m/e 246 2
(M+H ).
Fx~le 49
1 ~eO)2POCH2COOtbu/N~
2.H2/Pd-C ~ ~
3. LE~S /7
Boc-Phenyl~ ninz~
X=CE~
Spirocycle X=CH2
A.
A solution of 1.36g (3.88 mmol, 1 equiv) of the ester
from Example 47 step A in 5 mL of THF was cooled to -
78 ~C and treated with 9.32 mL ~9.32 mmol, 2.4 e~uiv)
of lM lithium bis(trimethylsilyl) amide in THF. After 1
h at -78 ~C, 0.992g (4.27 mmol, l.l equiv) of 1,5-
iodochloropentane was added and the reaction allowed to
progress at -15 ~C for 1 h, quenched with 10% aqueous
citric acid, and extracted to ethyl acetate, resulting
in 1.60g of product. Low resolution MS m/e 476.2
(M+Na )
A solution of 1.6g (3.53 mmol, l equiv) of the above
chloride in 30 mL acetone was treated with 5.29 g (35.3
mmol, 10 equiv) of NaI and refluxed overnight. Solvents
were then removed and the residue partitioned between
ethyl acetate/water. Organics were dried with magnesium
CA 02243121 1998-07-14
W O 97~7180 PCT/US97/01610
- 218 -
sulfate and purified on silica gel using 1/3 ethyl
acetate/hexane resulting in 1.2 g of the desired iodide
(62.4~ yield a~ter chromatography). _Y
lH NMR (CDCL3, 300 MHz) ~ 7.20 (m, 5H), 4.38 (m, lH),
3.79 (m, lH), 3.13 (t, 2H, J=6.9), 2.73 (m, 2H), 2.25
~m, lH), 1.76 (m, 2H), 1.43 (s, 9H), 1.38 rS, 9H), 1.2-
1.7 (m, 7H). Low resolution MS m/e 568 (M+Na ), m/e
362.2 (M+H )
A solution 1.15g ~2.1 mM, 1 equiv) of the above product
in 20 mL of anhydrous THF was cooled to -78 ~C and
treated with 3.2 mL (3 mmol, 1.5 equiv) of lM lithium
bis(trimethylsilyl) amide in THF. The reaction was then
allowed to warm up to room temperature, solvents
removed and the crude product purified on preparative
HPLC. H NMR (CDCL3, 300 MHz) ~ 7.32 (m. 4H), 7.19 (d,
12H), 3.88 (m, lH), 2.82 (m, 2H), 2.24 (dd, lH), 1.2-
1.8 (m, llH). Low resolution MS m/e 384.2 (M+Na ), m/e
362.2 (M+H ).
Fxample 50
1) LDA/TltF ,--~'
~) CHO ~N_H
3. H2 . Pd-C
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 219 -
A.
The Boc-pyrrolidone (4.4 g, 16 mmol) was dissolved in
THF (40 mL) and cooled to -78 ~C. To this solution was
added LDA (Aldrich, 1.5 M in cyclohexane, 12.8 mL, 19
~ .
mmol) via syringe over 10 minutes. After stirring for
60 min at -78 ~C, 3-formyl-5,6-dihydro-2~-pyran (US
Patent 4,532,337) (1.8 g, 16 mmol) in T~F (5 mL) was
added via syringe over l minute. The reaction was then
allowed to reach room temperature and stir for 20 h
before being quenched with saturated ammonium chloride
~15 mL). Ethyl acetate (50 mL) was added and the
layers were partitioned. The organic layer was washed
with brine, dried over magnesium sulfate, filtered, and
concentrated in vacuo. The residue was purified by
flash chromatography over silica gel (95:5
chloroform:methanol) to give dihydropyran lactam as a
beige powder. MS (ES+) = 270 (M+1), 292 (M+Na)
B.
The dihydropyran obtained above (1.2 g, 4.4 mmol), 10%
Pd-C (0.2 g), and methanol (35 mL) were combined in a
Parr bottle and pressurized to 50 psi of hydrogen gas.
After 3 h of agitation, the reaction vessel was
evacuated and filtered through a plug of Celite. The
cake was washed with methanol (20 mL) and the combined
filtrate was concentrated in vacuo. Flash
chromatography over silica gel (95:5
chloroform:methanol) gave the tetrahydropyran lactam 2
~ as a white powder. MS (ES+) = 274 (M+1), 296 (M+Na)
CA 02243121 1998-07-14
W O97/27180 PCTrUS97/01610
- 22~ -
Fx~m~le 51
. ~ . ~ , '
Boc ~V~NH
A.
A solution of 2.6g (8.24 mmol, 1 equiv) of the allyl-
pyrrolidone in 80 mL tetrahydrofuran and 25 mL water
was cooled to 0 ~C and treated with 5.29 g (24.7 mmol,
3 equiv) NaIO4, followed by the addition of 838 mg of
2. 596 solution of osmium tetroxide in 2-methyl-2-
- propanol. The reaction was continued for 2 h at room
temperature, solvents removed and the residue
partitioned between ethyl acetate and water. Ethyl
acetate was then dried over MgSO4, resulting in 3.0 g
of the crude aldehyde.
H NMR (CDCL3, 300 MHz) ~ 9.75 (s, lH), 7.22 (m, 5H),
4.32 (m, lH), 3.05 (m, 2H), 2.82 (m, 3H), 2.53 ~m, lH),
2.22 (m, lH), 1.58 (s, 9H). Low resolution MS m/e
356.1 (M+Na ); m/e 689.3 (2M+Na ~.
A solution of 2.88g of the above aldehyde in 10 mL
methanol was cooled to 0 ~C and sodium borohydride was
added over 2 h, until all the starting material
(Rf=0.55, Merck Kiselgel 60, 0.25 mm, 1:1 ethyl
acetate/hexane) was consumed. The title compound had
Rf=0.30 (same conditions). Solvents were then removed,
and the residue was extracted between ethyl acetate and
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97101610
- 221 -
-~ 10~ aqueous citrlc acid. Organic fractions were washed
~ with water and dried over magnesium sulfate.
~, Purification on a silica column (1:1 ethyl
acetate/hexane) afforded 1.5 g ~57% yield) of the
alcohol. Low resolution MS m/e 342.2 (M+Na ); m/e
661.4 (2M+Na ~.
A solution of 0.46g (1.44 mmol, 1 equiv) of the above
alcohol in 4 mL tetrahydrofuran was treated with 0.215
g (1.875 mmol, 1.3 equiv) of mesyl chloride and 0.242g
(1.875 mmol, 1.3 equiv) of diisopropylethylamine. The
reaction was allowed to proceed for 30 min at room
temperature, solvents removed and the residue
partitioned between ethyl acetate and water. Organics
were dried with magnesium sulfate and purified on a
silica column (1/1 ethyl acetate/hexane), yielding 0.50
g (87.3%) of the desired mesylate. Rf=0.57 (Merck
Kiselgel 60, 0.25 mm, 1:1 ethyl acetate/hexane). H
NMR (CDCL3, 300 MHz) ~ 7.22 (m, 5H), 4.39 (m, 3H), 3.09
(dd, lH, J=6.4, 13.2), 2.98 (s, 3H), 2.76 (dd, lH,
J=8.g, 13.2), 1.64 (m, 2H), 1.57 (s, 9H).
A solution of 0.33g (0.831 mmol, 1 equiv) of the above
mesylate in 3 mL DMF was cooled to 0 ~C and treated
with 26 mg (1.080 mmol, 1.3 equiv) of sodium hydride.
After 3h at room temperature the reaction was quenched
with aqueous citric acid and purified on silica gel
using 1:3 ethyl acetate/hexane (v:v). The resulting
product (0.18g, 72.0% yield) was then treated with 1:1
dichloromethane/ trifluoroacetic acid (5 ml) for 1/2h,
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 222 -
r~,u'ting in 0.12g (71.8%, based on mesylate) of the '-
d~ :ed product. H NMR (CDCL3, 300 MHz) ~ 7.23 (m,
5H), 7.04 (broad s, lH), 3.99 (m, lH), 2.85 (m, 2H),
2.26 (dd, lH, J=8.1, 12.9), 1.92 (dd, lH, J-5.0, 12.9),
1.10 (m, 2H), 0.72 (m, 2H). Low resolution MS m/e
342.2 ~M+Na );
~m~le 52
1 aLDA/THF/-78~C
b ~etone
~c 3 Et2~CN
4 TFA
A solution of 1.5g (5.4 mMol) of the pyrrolidinone in
25 mL of tetrahydrofuran was cooled to -78 ~C and
treated with 4.3 mL (6.5 mMol) of lithiumdiisopropyl
amide (2M in THF). After stirring for 0.25h, acetone
(2.8g (50 mMol) was added, the reaction mixture was
kept at -78 ~C for 2 h and then quenched with lN
hydrochloric acid. Extraction with ethyl acetate,
drying over magnesium sulfate and removal of the
solvent in vacuo a~forded the crude product which was
redissolved in 25 mL of dichloromethane and treated
wi h 8g of Martin's sulfurane. After stirring for 12h
at _5 ~C, the mixture was participated between ethyl
acetate and lN hydrochloric acid. Drying over
magnesium sulfate and removal of the solvent gave the
desired alkene. 0.755 g of the crude alkene were
dissolved in 15 mL of toluene and treated with 3 mL (3
mMol) of diethyl aluminumcyanide (lm in toluene) and
the resulting mixture was stirred at 25 ~C for 5 h.
CA 02243121 1998-07-14
W O 97/27180 PCTrU~97/01610
- 223 -
-~ The solvent was removed and the residue was
chromatographed on silica gel (209~ ethylacetate-
~,, hexanes) to give the desired nitrile (0.4g) as a
colorless oil. Deprotection with trifluoroacetic acid-
dichloromethane (1:1) for 3h at 25 ~C followed by
chromatography on silica gel gave the desired lactam
(0.22g) as a white solid. M+H: 243
Fx~m~le 53
1)sodiumazide
'' 3)4~ o~- ~ ~
O tritylchloride ~ H O
A.
A solution of 3-iodo-5-benzyl-pyrrolidinone (2.67 g,
8.87 mmol) and sodium azide (0.69 g, 10.61 mmol) in
dimethylformamide (20 mL) was stirred at ambient
temperature under a nitrogen atmosphere for 18h. The
solvent was evaporated using a stream of nitrogen, and
the residue was dissolved in ethyl acetate, washed with
water and ~rine, and concentrated in vacuo to give a
yellow solid. Chromatography on silica gel, eluting
with hexane:ethyl acetate (4:1), gave 1.82 g of the
product as a 1:1 mixture of diastereomers which was
used without separation in the next reaction. MS:
ES+, 239 (M+Na). The chromatography also gave 0.12 g
or the trans isomer as a colorless oil and 0.43 g of
the cis isomer as a colorless oil which crystallized
-
CA 02243121 1998-07-14
W 097/27180 PCTAJS97/01610
- 224 -
-
upon standing. TLC (hexane : ethyl acetate (1:1)) Rf ~-
trans isomer =0.6 and Rf cis isomer = 0.5.
,~ .
B.
A mixture of the above azide (0.575 g, 2.66 mmol) and
5% palladium on carbon (0.030 g) in methanol (20 mL)
was stirred under 40 psi of hydrogen for 18h at ambient
temperature. The mixture was filtered through a pad of
Celite to remove the catalyst, followed by filtration
through 5 g of silica gel, washing with
chloroform:methanol (9:1). The filtrate was
concentrated in vacuo to give 0.46 g (90%) of the
product as a mixture of diastereomers. MS: ES+, 191
(M+1) and 213 (M~Na).
A solution of the above amine (0.44 g, 2.3 mmol), 4-
anisylchlorodiphenylmethane (0.71 g, 2.3 mmol) and
triethylamine (0.5 mL, 3.5 mmol) in dic~loromethane (20
mL) was stirred under a nitrogen atmosphere at ambient
temperature for l~h. The solutlon was washed with
water (2x50 mL) and brine, dried (MgS04), and
concentrated in vacuo. The residue was purified by
chromatography on silica gel, eluting with hexane:ethyl
acetate (7:3) then with hexane:ethyl acetate (1:1), to
give 0.41 g of the cis isomer as a yellow solid and
0.19 g of the trans isomer as a white solid. TLC
(hexane : ethyl acetate (7:3)) Rf cis isomer =0.5 and
Rf trans isomer = 0.4.
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 225 -
Fx~le 54
~ .
.' NH2 DMF/70~C I--\
¢~F Na~CO3 ~N~f
Iodolactam 1 (prepared as described previously in
Example 7) (0.55 g, 1.8 mmol) was dissolved in DMF (5
mL) and treated with 2-fluoroaniline (Aldrich, 0.20 g,
1.8 mmol) and solid sodium carbonate (0.39 g, 3.7
mmol). The reaction was then heated to 70 ~C for 24 h
before the solvent was removed in vacuo. Ethyl acetate
(50 mL) and water (20 mL) were added and the layers
were partitioned. The organic layer was dried over
sodium sulfate, ~iltered, and concentrated in vacuo.
Flash chromatography over silica gel (1:1 hexane:ethyl
acetate) gave the anilinolactam 2 as a pale yellow
foam. MS (AP+) = 285 (M+1), 307 (M+Na)
Fxample 55
~ NH2 DMF/70~C /~
,NH + ~ Na2CO3 ~H~
J 1 2
. .
- CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 226 -
Using the procedure described in Example 54, the
anilinolactam was prepared, purified, and isolated as a
beige foam. MS ~AP+) = 285 (M+l), 307 (M+Na)
F.x~nu?Ie 56
NH2 DMF~0~C ~ ~ H
CN NC
1 2
Using the procedure described in Example 54, the
anilinolactam was prepared, purified, and isolated as a
beige ~oam. MS (AP+) = 292 (M+1), 314 (M+Na)
Fx~le 57
~' ~,N~2 EtOH/~
~ ,~,NH + ~ J ~o3 ~H~
Iodolactam 1 (prepared as described previously in
Example 7) (0.77 g, 2. 6 mmol) was dissolved in absolute
ethanol (10 mL) and treated with 3-aminopyridine (0.26
g, 2.8 mmol) and solid sodium carbonate (0.40 g, 3.8
mmol). The reaction was then heated at reflux for 24 h
before the solvent was removed in vacuo. Chloroform (50
-
CA 02243121 1998-07-14
W O 97127180 PCT~US97/01610
- 227 -
" .
mL) and water (20 mL) were added and the layers were
partitioned. The organic layer was dried over sodium
sulfate, filtered, and concentrated in vacuo.
Preparatory silica gel TLC (95:5 chloroform:methanol)
gave the pyridylaminolactam 2 as a red oil. MS (AP+) =
268 (M+1), 290 (M+Na)
Fx~le 58
A.KCN,DMF
B. ~12, PdlC, HCI
~NH--~ ~ Ph~l~N S~\NH--
A.
To a solution of iodolactam 1 (13.43 g, 44.6 mmol, 1
eq) in dimethylformamide (60 mL) under nitrogen was
added potassium cyanide (3.49 g, 1.2 eq). After
stirring at ambient temperature for 24 h, the reaction
mixture was evaporated in vacuo and the residue was
partitioned between ethyl acetate, saturated aqueous
brine and water. The layers were separated and the
aqueous layer was back-extracted twice with ethyl
acetate. The com~ined organic layers were washed with
saturated aqueous brine, dried over anhydrous magnesium
sulfate, filtered and evaporated in vacuo. The residue
was purified by flash silica gel chromatography eluting
with hexane : acetone (3:1). Fractions containing the
product were combined, evaporated in vacuo to provide
CA 02243l2l l998-07-l4
W O 97/27180 ~CTrUS97/01610
- 228 -
5.89 g (66%) of cyanolactam as a mixture of
diastereomers. MS (APCI): M+Na -- 223.
A solution of cyanolactam (5.78 g, 28.9 mmol) from step
A in absolute ethanol (233 mL) under Nitrogen was
com~ined with 10 wt.% Palladium on charcoal (2.33 g)
and concentrated hydrochloric acid (9.31 mL, 4 eq.).
The mixture was reduced under hydrogen gas at 50 psi
for 16 h. The reaction was purged with nitrogen,
filtered and evaporated in vacuo. The residue was
combined with toluene (~ 100 mL) and concentrated in
vacuo to a residue to remove residual water. The
azeotropic removal with toluene was repeated four times
leaving a residue which was dried under high vacuum to
provide the crude amine a-s a gum (7.18 g, 1039~). MS
(ESI): M+l = 205.
The crude amine (7.16 g, 29.8 mmol, l eq) from step B
was combined under argon in dichloromethane (100 mL)
with diisopropylethylamine (13 mL, 74.4 mmol, 2.5 eq)
and triphenylmethylchoride (9.13 g, 32. 7 mmol, 1.1 eq).
After stirring at ambient temperature for 16 h, the
reaction mixture was treated with 5% w/v aqueous
potassium carbonate and transferred to a separatory
funnel. After separating the layers, the aqueous layer
was back-extracted with dichloromethane and the
combined organic layers were dried over anhydrous
sodium sulfate and evaporated in vacuo to proved a
crude mixture of diastereomers. The mixture was
_L
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 229 -
purified by flash silica gel chromatography eluting
with ethyl acetate : hexane (3:7). Fractions
containing the less polar diastereomer were combined
and evaporated in vacuo to provide 3.52 g (26 ~) o
trityl protected amine as a crystalline solid. MS
(APCI): M+Na = 469.
F.x;lTr~l e 59
An alternate procedure for the synthesis of the
benzyllactam:
A.
~Ph ~Ph
~f ~Ph +OHC~NHBOC ~ lî NHBOC
~,I~,Ph
A mixture of methyl 2-(triphenylphosphoranylidene)-
hydrocinn~m~te (13.20 g, 31.1 mmol, 1.15 eq) and N-
tertbutoxycarobonyl-L-phenylalanal (6.76 g, 27.1 mmol,
1 eq) were combined in 200 mL chloroform and allowed to
stir at ambient temperature over 64 h. The reaction
was concentrated in vacuo and the residue was purifled
by flash silica gel chromatography eluting with 85:15
hexane : ethyl acetate. Fractions containing the
product were combined and evaporated in vacuo to
provide the olefin as a crystalline solid (9.38 g,
77~). MS (ESI): M + Na = 418.
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 230 -
Ph 1. Pd/C.~2 Ph
- 3 DIEA, K2C0 3
A solution of the olefin (9.30 g, 23.5 mmol, leq) from
step A in absolute ethanol (250 mL) was combined under
nitrogen with Palladium on carbon (10 wt%, 1.90 g) and
reduced under a ~alloon of Hydrogen gas over 16 h. The
reaction mixture was purged with nitrogen, diluted with
dichloromethane, filtered, and evaporated in vacuo to
low volume. The solution was diluted with
dichloromethane and filtered through a pad of
diatomaceous earth washing with dichloromethane. The
filtrate was evaporated in vacuo and dried under vacuum
to provide a 5:1 mixture of diastereomers of the BOC-
amino ester as an oil ~9.68 g, 104 %). MS ~ESI): M +
Na = 420.
The oil was dissolved in dichloromethane (25 mL) and
treated with trifluoroacetic acid (25 mL) under Argon.
After stirring for Q.5 h at ambient temperature, the
reaction mixture was evaporated in vacuo. The residue
was dissolved in methanol (50 mL) and treated with
diisopropylethyl amine ~17 mL) followed by anhydrous
potassium carbonate (13.49 g, 98 mmol, 4 eq) and
stirred for 16 h at ambient temperature under an Argon
atmosphere. The mixture was evaporated in vacuo and
the residue was partitioned between dichloromethane and
water. The layers were separated and the aqueous layer
-
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 231 -
was back-extracted three times with dichloromethane.
The combined organic layers were washed with aqueous
hydrochloric acid (lN) and the layers were separated.
. The aqueous layer was back-extracted with
dichloromethane and the combined organic layers were
dried over anhydrous magnesium sulfate and evaporated
in vacuo to a residue. The crude product was purified
by flash silica gel chromatography eluting with a
gradient of 45-60 % ethyl acetate in hexane. Fractions
containing the less po~ar diastereomer were combined
and concentrated in vacuo to a solid and dried under
high vacuum to provide the enantiopure lactam as a
white crystalline solid (4.48 g, 72%). MS (ESI): M
Na = 288. H NMR (CDCl3): 1.90 (m, lEI); 2.01 (m, lH);
2.67 (m, 4H); 3. 16 (m, lH); 3. 65 (m, lH); 5.70 (s, lH);
7.18 (m, lOH).
Fx~m~le 60
Synthesis of Compound 123
o~J< 0
//\~O CH3CN N
H H
1 2 3
To a suspension of (2S)-(+)-glycidyl 3-
nitrobenzenesulfonate 1 (Aldrich, 19.47 mmol) and
~=
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/~1610
- 232 -
potassium carbonate (Baker, 38.93 mmol) in dry
acetonitrile was added ~S)-t-butyl decahydro-3-
isoquinoline carboxamide 2 (NSC Technologies, 21.41
mmol) and the reaction stirred at ambient temperature
overnight. The solvent was removed in vacuo, and the
residue taken up in ethyl acetate/water, the organic
layer was washed sequentially with saturated aqueous
NaHC03 and brine, followed by drying ~MgS04),
- ~iltration and concentration in vacuo. The crude
residue was purified by flash silica gel chromatography
eluting with 10% diethyl ether/dichloromethane to give
3.62 g of product 3; HPLC Rt = 9.2 min (100~), TLC
Rf = 0.26 (10~ diethyl ether/dichloromethane); 1H NMR
(CDC13) d 6.59 (br s, 1 H), 3.00 (d, 1 H), Z.97 ~m, 1
H), 2.89 (dd, lH), 2.73 ~m, lH), 2.65 (m, 1 H), 2.57
(m, 1 H), 2.22 ~dd, lH), 2.08 ~dd, lH), 1.81-1.70 ~m, 4
H), 1.65-1.19 ~m, 8 H), 1.38 (s, 9 H).
B.
o~N~ ~ N ~
Pl,u~,uhc,~ne Bsse P4-t-Bu 3 H
THF, -78~C to r.t.
2-pyridylmethyl lactam 1 ~35 mg, 0.13 mmol) was
dissolved in anhydrous THF (1 mL) and cooled to -
78 ~C. Phosphazene Base P4-t-Bu (Fluka, l.OM in
hexane, 130 uL, 0.13 mmol.~ was added to give an
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 233 -
~ orangish brown anion. The anion solution was sti~red
at -78 ~C for 35 minutes and was then cannulated unàer
nitrogen over 30 seconds into a -78 ~C solution of 2
. (39 mg, 0.13 mmol) in lmL of THF and was washed in with
0.5 mL of THF. The reaction was gradually warmed to
room temperature over 4 hr, then stirred at room
temperature for 3 days. The reaction was cooled to -
78 ~C, quenched with 0.5 mL of a saturated ammonium
chloride solution, and concentrated in vacuo to remove
the THF. The residue was partitioned between ethyl
acetate and saturated bicarbonate solution and the
aqueous layer was extracted with ethyl acetate. The
com~ined organic layers were then washed with water,
brine and dried ~MgSO4) and filtered. Concentration in
vacuo afforded 75 mg crude material which was purilied
via silica gel to give 18 mg(25%) of 3. Maldi MS: M +
H = 561.5 (M~ = 560.79). TLC (EtOAc) Rf = 0.19 (major
diast.) & 0.29 (minor diast.). TLC (5% MeOH/EtOAc) Rf
= 0.28 (major diast.) & 0.36 (minor diast.). HPLC
retention times were 11.24 min. (major) & 11.32 min.
(minor).
H NMR (CDCl3) d 8.52 (m, lH), 7.61 (m, 1 H), 7.34-7.10
(~" 7H), 6.10-5.95 (m, lH), 4.11 (m, lH), 3.96-3.73 (m,
3H), 3.46-2.74 (m, 6H), 2.65-2.47 (m, 2H), 2.23 (m,
2H), 2.10-1.15(m, 15H), 1.37 (s,9H).
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97101610
- 234 -
Fx~nle 61
-
Synthesis of Compo~-nd 72
A.
Ph Ph
""1 (Ns)a8,~: ~,lo,~ rdrin ,~ "~
DMF i ~N
3-pyridylmethyl lactam 1 (85 mg, 0.32 mmol) was
dissolved in ~MF ~1.5 mL), cooled to O ~C, and to this
solution was added sodium hydride (0.48mmol) to give a
yellow anion. The reaction mixture was stirred at O ~C
for 70 minutes after which (s)-epichlorohydrin (35 ul,
0.45 mmol) was added neat. The reaction was stirred at
0 ~C ~or 5 minutes, then warmed to room temperature and
stirred for 24 hours. The reaction was cooled to O ~C
and quenched with O.5 mL of a saturated ammonium
chloride solution. The reaction was partitioned
between ethyl acetate and a saturated bicarbonate
solution. The aqueous layer was extracted with ethyl
acetate. The combined organic layers were then washed
with water, brine and dried (MgS04) and filtered.
Concentration in vacuo afforded 49 mg of crude epoxide
which was used without further purification.
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 235 -
.,
B.
~,~N '~ H~ ~1 ~N~
NH O
2 ~ 3 J<
Crude lactam epoxide 1 (49 mg) and
decahydroisoquinoline 2 (91 mg, 0.38 mmol) were heated
to 65-70 ~C in isopropanol. After 90 hours the
reaction was cooled to 25 ~C and stirred for 1 hour at
room temperature. The reaction was then concentrated
in vacuo, and purified by silica gel chromatography,
eluting with 5 96 MeOH : EtOAc, providing 30 mg ~87S~
pure by HPLC) of desired product 3 as a mixture of 4
diastereomers. HPLC shows 2 split peaks 11.30 min. &
11.04 min.. TLC (5% MeOH/CH2C12) Rf = 0.27. TLC (10
MeOH/CH2C12) Rf = 0.45.
H NMR (CDC13) d 8.45--8.35 (m, 2H), 7.48 (m, 1 H),
7.35-7.09 (m, 6H), 6.63-5.94 (m, lH), 3.98-3.63 (m,
3H), 3.42-2.73 (m, 5H), 2.70-2.11 (m, 5H), 2.07-1.20 (m,
16H), 1.36 (s,9H).
CA 02243121 1998-07-14
WO 97/27180 PCT~US97/01610
- 236 -
F~x~ e 6
Synthesis of Com~ollnd 54
H ~--~NJ<
Pl ~5ph~c-~~ Base P4-t-Bu 3 H
THF, -7~C to r.t.
4-pyridylmethyl lactam 1 ( 33 mg, Q.12 mmol) was
- dissolved in anhydrous THF (1 mL) and cooled to
-78 ~C. Phosphazene Base P4-t-Bu (Fluka, l.OM in
hexane,125 uL, 0.125 mmol) was added to give a brown
anion. The anion solution was stirred at -78 ~C for 35
minutes and was then cannulated under nitrogen over 30
seconds into a -78 ~C solution of 2 (39 mg, 0.13 mmol)
in lmL of THF and was washed in with 0.5 mL of THF.
The reaction was gradually warmed to room temperature
over 4 hr, then stirred at room temperature for 3 days.
The reaction was cooled to -78 ~C, quenched with 0.5 mL
of a saturated ammonium chloride solution, and
concentrated in vacuo to remove the THF. The residue
was then partitioned between ethyl acetate and
saturated bicarbonate solution. The aqueous layer was
extracted with ethyl acetate and the combined organic
layers were then washed with water, brine and dried
(MgSO4) and filtered. Concentration in vacuo afforded
83 mg crude material which was purified via silica gel
-
CA 02243l2l l998-07-l4
W O 97/27180 PCT~US97/01610
- 237 -
to-give 11mg~16%) of 3. Maldi MS: M + H = 560.4. ~MW
= 560.79).TLC (EtOAC) Rf = 0.08 (major diast.) & 0.16
~minor diast.). TLC (5% MeOH/EtOAc) Rf = 0.18 ~major
diast.) & 0.26 (minor diast.). HPLC retention time was
11.05 min.
H NMR (CDCl3) d 8.50 (m, 2H), 7.35-7.02 (m, 7H), 5.89
(m, lH), 4.05-3.78 ~m, 3H), 3.37-2.69 (m, 5H), 2.62-
2.45 (m, 4H), 2.26 (m, 2H), 2.08-1.16(In, 15H), 1.38
(s,9H).
Fxample 63
Synthesis of Com~ol1nd 130
1) ~ ~ Ph H
O
~NH
In an oven-dried 25 mL round-bottomed flask, alkyne
lactam 1 (54. 6 mg, 0.682 ~r~nol) was dissolved in 5 mL of
DMF. Sodium hydride ( 34. 4 mg of a 60% dispersion in
15 mineral oil, 0. 860 mmol) was added with cooling using
an ice-bath. Gas evolution was observed. (S)-
Epichlorohydrin ( 60 uL, 0.765 mmol) was added.- The
mixture was stirred overnight at room temperature, then
the decahydroisoquinoline amide (182 mg, 0.770 mmol)
20 was added. The mixture was heated to 8Q ~C overnight.
The mixture was cooled, poored into water, and
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 238 -
extracted with CH2Cl2. The organic extract was washed
several times with water, dried (MgS04), and evapora~ed
in vacuo to afford a yellow residue that was purifieà
by preparative HPLC to afford the a diastereomeric
mixture of alkyne DHIQ lactam 2 (120 mg, 34~) as a
light yellow oil. H.PLC: retention times of 13.57,
13.67, 13.87 minutes in a 5:1:1 ratio respectively. H
~NMR : d 1.3-1.4 three singlets in a 2:1:1 ratio; 1.4-
2.7 ~several overlapping multiplets, 2.8-2.95
lQ (multiplet), 3.0-3.7 (multiplet), 3.8-4.1 (multiplet),
5.95-6.05 (multiplet), -6.1, 6.18, 6.32, 6.4 (broad
singlets in a l:l:l:i ratio), 6.2-6.3 (doublet of
- doublets), 7.1~-7.35 (multiplet). MALDI-MS: peak at
506.3 (M + H ).
~ m~le 64
Synthesis of Compollnd 124
H ~ H ~ N
1 Pl,o~haz~neBaseP4-t-Bu 3 H
THF-78~Ctor.t.
The lactam 1 (0.13 mmol) was dissolved in dry THF at
-78 ~C and to this solution was added Phosphazene Base
P4-t-Bu (Fluka, l.OM in hexane, 0.14 mmol). After
stirring 15 minutes the anion solution was added via
cannula to a solution of epoxide 2 (0.13 mmol)
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 239 -
dissolved in dry THF at -78 ~C and the reaction was
allowed to warm to room temperature and stir overnight.
The reaction was then diluted with water and extracted
with ethyl acetate. The organic layer was washed
sequentially with saturated aqueous NaHCO3 and brine,
followed by drying (MgSO4), filtration and
concentration in vacuo. The crude residue was taken up
in dichloromethane and filtered through a plug of
silica gel eluting with 8% MeOH in dichloromethane.
Product containing fractions were concentrated in vacuo
and the resultant residue further purified by
preparative HPLC (column: Delta-Pak C18 15mm lCoA
l9x300 mm. Gradient: 20% to 100% acetonitrile in
water with 0.1% TFA. Flow rate: 20 ml/min.
lS Detection: 214 nm) to yield 3 mg of product 3 as a
mixture of diastereomers; TLC Rf = 0.44 (8%
MeOH/CH2Cl2); HPLC Rt = 14.8, 14.9 min (95%); MALDI-
TOF MS m/z 561 (M ); 1H ~MR (CDCl3) d 7.35-7.10 (m, 7
H), 6.73 (m, 1 H), 6.58 (d, 2 H), 5.82 (br s, 1 H),
4.12-3.85 (m, 4 H), 3.51 (m, lH), 3.30 (m, lH), 2.92
(m, 1 H), 3.63-2.20 (m, 4 H), 2.Q5-1.12 (m, 18 H), 1.38
(s, 9 H).
CA 02243121 1998-07-14
W O 97127180 PCTrUS97/01610
- 240 -
F.X~ ~ le 6S
Synthesis of Compol-nd 127
NC ,S~H e ~ J~< NC~p~
Phosl~h.. ~. i,e Base P4-t-Bu 3 H
THF, -78~C to r.t.
Cyanomethyl lactam 1 (82 mg, 0.38 mmol) was dissolved
in anhydrous THF (2 mL) and cooled to -78 ~C.
Phosphazene Base P4-t-Bu ~Fluka, l.OM in hexane, 380
uL, 0.38 mmol) was added to give a yellow anion. The
anion solution was stirred at -78 ~C for 35 minutes and
was then cannulated under nitrogen over 30 seconds into
a -78 ~C solution of 2 (112 mg, 0.38 mmol) in 2mL of
1~- THF and was washed in with 0.5 mL of THF. The reaction
- was gradually warmed to room temperature over 4 hr,
then stirred at room temperature for 3 days. The
reaction was cooled to -78 ~C, quenched with 0.5 mL of
a saturated ammonium chloride solution, and
concentrated in vacuo to remove the THF. The residue
was then partitioned between ethyl acetate and
saturated bicarbonate solution and the aqueous layer
was extracted with ethyl acetate. The combined organic
layers were then washed with water, brine and dried
(MgS04) and filtered Concentration in vacuo afforded
375 mg crude material which was purified via silica
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 241 -
gel (8 : 2, ethyl acetate: CH2C12, to give 118 mg(61~)
of 3 that was < 80% pure. 58 mg was purified via prep.
HPLC to give 10 mg of pure material as a 2: 1 mixture
of diastereomers. HPLC retention times were 12.73 min.
t67%) & 12.86 min. ~33%). Maldi MS: M + H = 510.47 (MW
= 508.71). TLC ( EtOAc) Rf = 0.37 & 0.31.
~H NMR ~CDC13) d 7.38-7.13 ~m, 5H), 6.09-5.82 (~r s,
lH), 4.29-3.96 (m, 3H), 3.84 (m, lH), 3.49-2.91 (m,
5H), 2.77-2.18 (m, 9H), 2.10-1.20(m, llH), 1.39 (s,9H).
Fx~mnle 66
Synthesis of Compollnd 131
H ~ N~
Ph~sphc~ e Base P4-t-3u 3 H
THF, -7~C to r.t.
The lactam 1 (0.061 mmol) was dissolved in dry THF at
-78 ~C and to this solution was added Phosphazene Base
P4-t-Bu (Fluka, l.OM in hexane, 0.067 mmol). After
stirring 15 minutes the anion solution was added via
cannula to a solution of epoxide 2 (0.061 mmol)
dissolved in dry THF at -78 DC and the reaction was
allowed to warm to room temperature and stir overnight.
The reaction was then diluted with water and extracted
with ethyl acetate. The organic layer was washed
sequentially with saturated aqueous NaHC03 and ~rine,
CA 02243l2l l998-07-l4
W O 97/27180 PCTAUS97/01610
- 242 -
followed by drying (MgSO4), filtration and
concentration in vacuo. The crude residue was purlfied
by flash silica gel chromatography eluting wlth 3~ MeOH
in dichloromethane to give 2.1 mg of product 3 as a 1:1
mixture of diastereomers; TLC Rf 1 0.14 ~2:1 ethyl
acetate/hexanes); HPLC Rt = 13.6, 13.8 min (68%);
MALDI-TOF MS m/z 580 (M ); lH NMR (CDC13) d 7. 32-7.08
~(m, 5 H), 5.86 ~br s, 1 H), 4.08-3.73 (m, 4 H), 3.65-
3.14 (m, 4H), 3.00-2.~9 (m, 8H), 2.41--0.92 (m, 13 H),
10 2.27 (s, 1.5 H), 2.22 (s, 1.5 H), 2.16 (s, 1,5 H), 2.11
(s, 1.5 H), 1.46 (s, 9 H).
F.xi~r~le 67
Svnthesis of Co~olln~ 126
Pi,os~,l.~,.. ~ Base P4-t-Bu 3 H
THF, -78 ~C to r.t.
The lactam 1 (O. 20 mmol) was dissolved in dry THF at
15 -78 ~C and to this solution was added Phosphazene Base
P4-t-Bu (Fluka, l.OM in hexane, O. 21 mmol). After
stirring 15 minutes the anion solution was added via
cannula to a solution of epoxide 2 (0.20 mmol)
dlssolved in dry THF at -78 ~C and the reaction was
20 allowed to warm to room temperature and stir overnight.
The reaction was then diluted with water and extracted
-
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 243 -
with ethyl acetate. The organic layer was washed
sequentially with saturated aqueous NaHCO3 and brine,
followed by drying ~MgSO4), filtration and
concentration in vacuo. The crude residue was taken up-
in dichloromethane and filtered through a plug ofsilica gel eluting with 3% MeOH in dichloromethane.
Product containing fractions were concentrated in vacuo
and the resultant residue further purified by
preparative HPLC (column: Delta-Pak C18 15mm 100A
l9x300 mm. Gradieht: 20% to 100% acetonitrile in
water with 0.1% TFA. Flow rate: 20 ml/min.
Detection: 214 nm) to yield 2.5 mg of product 3 as a
mixture of diastereomers; TLC Rf = 0.21 (3%
MeOH/CH2Cl2); HPLC Rt = 14.8 min (98%); MALDI-TOF MS
m/z 588 (M ).
Fx~m~le 68
Synthesis of Compound 132
~ NH H ~
In an oven-dried 25 mL round-bottomed flask, isoxazole
lactam 1 (54.6 mg, 0.201 mmol) was dissolved in 3 mL of
THF. (S)-Epichlorohydrin (20 uL, 0.255 mmol) was
added. P-4-tBu phosphazene base (210 uL, 0.210 mmol)
CA 02243l2l l998-07-l4
W O97/~7180 PCTrUS97101610
- 244 -
was added dropwise via syringe initially producing a
dark orange-brown color that faded The mixture was
stirred for 30 minutes at room temperature and the
mixture was poured into water and extracted with
CH2Cl2. The organic extract was dried (Na2SO4) and
evaporated in vacuo. The residue was dissolved in
anhydrous CH3CN and the decahydroisoquinoline amide
(54.4 mg, 0.230 mmol) was added. The mixture was
refluxed overnight. The solvent was evaporated and
10- the residue was puri~ied by preparative HPLC to afford
the isoxazole DHIQ lactam 2 (38 mg, 3496) as a light
yellow oil. HP1C: retention times of 12.28, 12.86,
13. 68 minutes at 93% purity. H NMR : d 1. 3-1 4 three
singlets in a 4:4:1 ratio; 1. 4-2.7 ( several overlapping
multiplets, 1.4-2.3 (several overlapping multiplets),
2.45-3.35 (several overlapping multiplets), 3.35-4.1
(several multiplets), 4.3-4 4 (doublet), 5.8
(multiplet), 5.9, 6.0, and 6.3 (three broad singlets in
a ratio of 4: 4:1 ratio), 7.1-7. 2 (multiplet), 7.2-7. 4
2Q (multiplet). M~IDI-MS: calc'd: 564.9; found 565.5 (M +
H ).
. .
CA 02243l2l l998-07-l4
W O 97127180 PCTrUS97/01610
- 245 -
~ Fx~m~le 69
~~ Svnthesis of Compo1~nd 125
N ~ ~ ~ ~ ~ N ~ ~,~,N
1 rl,u~hdzeneBaseP4-t-Bu 3 H
THF~-78~Ctor.t
The lactam 1 (0.12 mmol) was dissolved in dry THF at
-78 ~C and to this solution was added Phosphazene Base
P4-t-Bu ~Fluka, l.OM in hexane, 0.13 mmol). After
stirring 15 minutes the anion solution was added via
cannula to a solution of epoxide 2 (0.12 mmol)
dissolved in dry THF at -78 ~C and the reaction was
allowed to warm to room temperature and stir overnight.
The reaction was then diluted with water and extracted
with ethyl acetate. The organic layer was washed
sequentially with saturated aqueous NaHCO3 and brine,
followed by drying ~MgSO4), filtration and
concentration in vacuo. The crude residue was taken up
in dichloromethane and filtered through a plug of
silica gel eluting with 3% MeOH in dichloromethane.
Product containing rractions were concenLraied ir, -vacuo
and the resultant residue further purified by
preparative HPLC ~column: Delta-Pak C18 15mm 100A
l9x300 mm. Gradient: 20% to 100% acetonitrile in
water with 0.1~ TFA. Flow rate: 20 ml/min.
Detection: 214 nm) to yield 1.5 mg of product 3 as a
CA 02243l2l l998-07-l4
W O 97/27180 pcTrus97lol6lo
- 246 -
single diastereomer; TLC Rf = 0.27 (3~ MeOH/CH2Cl2);
HPLC Rt = 14.7 min ~100~)i ~ALDI-TOF MS m/z 576
(M ); H NMR (CDCl3) d 7.40-7.15 (m, 7 H), 6.70 (m, 1
H), 6.55 (d, 2 H), 5.80 ~br s, 1 H), 4.28 (m, 1 H),
4.05-3.90 (m, 2 H), 3.70-3 38 (m, ZH), 3.20 (m, lH),
3.00-2.75 (m, 2 H), 2.70 (s, 3 H), 2.55 (m, 2H), 2.30
(m, 2H), 2.20-0.80 (m, 14 H), 1.35 (s, 9 H).
~x~m~le 70
Sllnthesis of Com~ound 128
A.
~l
1~~~
O ~ NH
Pl,ospha2~neBase ~ OH
1 2 ~
The lactam 1 (90 mg, 0.28 mmol) was dissolved in THF (3
mL) and cooled to -78 ~C. This was followed by the
addition of the phosphazene base (Fluka; lM in hexane,
0.28 mL, 0.28 mmol). After stirring at -78 ~C ~or one
hour the epoxide was added as a solution in 1 mL THF.
The reaction was then warmed to 25 ~C and stirred for
an additional 3 hours. The reaction was then quenched
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 247 -
by the addition of water and extracted by ethyl
acetate. The organic portion was then dried over
MgSO4, filtered and concentrated in vacuo. The crude
oil was purified by silica gel chromatography, eluting
with 1:1, ethyl acetate:hexanes, this provided the two
major products (HPLC indicated two components for each
isolate).
B.
'
OH ~ LiOH ~ OH ~Ç
ON~JNH ~ O~NH
~ 2 ~
To the elaborated lactam 1 ( 40 mg) in 2:1, THF:H2O (5
mL) was added LiOH (2 eq.). The reaction was then
stirred at 40 ~C for 16 hours. TLC indicated the
formation of a new component. The reaction was diluted
by ethyl acetate, after which the organic portion was
separated, dried over MgSO4, filtered and concentrated
in vacuo. to yield product 2 as a mixture of
diastereomers.
1H NMR (CDC13) : d 7.10-7.50 (m, 10 H), 5.90-6.15 (m,
lH), 3.90-4.40 (m, ZH), 3.20-3.70 (m, 3H), 2.80-3.10
(m, 2H), 2.60-2.70 (m, 2H), 2.20-2.60 (m, 3H), 1.60-
2.10 (m, 9H), 1.40 (q, 15H), 1.20-1.40 (m, 8H).
~.
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 248 -
Fx~m~le 71
Synthesis of Co~olln~ ~59 ~-
,~NH OlN~ o~l~N;~
~ 2 N
ospha~eneBaseP4-t-Bu 3 H
THF,-78~Ctor.t
The lactam 1 ~0.11 mmol) was dissolved in dry THF at
-78 ~C and to this solution was added Phosphazene Base
P4-t-Bu (Fluka, 1.0M in hexane, 0.12 mmol). After
stirring 15 minutes the anion solution was added via
cannula to a solution of epoxide 2 (0.11 mmol)
dissolved in dry THF at -78 ~C and the reaction was
allowed to warm to room temperature and stir overnight.
The reaction was then diluted with water and extracted
with ethyl acetate. The organic layer was washed
sequentially with saturated aqueous NaHCO3 and brine,
followed by drying ~MgSO4), filtration and
concentration in vacuo. The crude residue was taken up
in dichloromethane and filtered through a plug of
silica gel eluting with 5% MeOH in dichloromethane.
Product containing fractions were concentrated in vacuo
and the resultant residue further purified by
preparative HPLC (column: Delta-Pak C18 15mm lo0A
l9x300 mm. Gradient: 20% to 100% acetonitrile in
water with 0.1~ TFA. Flow rate: 20 ml/min.
-
CA 02243121 1998-07-14
W 097/27180 PCTrUS97/01610
- 249 -
Detection: 214 nm) to yield 12 mg of product 3; TLC
Rf = 0.50(8% MeOH/CH2C12); HPLC Rt -- 12 8 min (100~);
M~LDI-TOF MS m/z 541 (M ); 1H NMR (CDC13) d 7.35-7.16
(m, 5 H), 5. 86 (br s, 1 H), 4 .08-3.76 (m, 4 H), 3 .49-
3.22 (m, 4 H), 2.89 (br s, 1 H), 2.50 (m, 2 H), 2.25
(br s, lH), 2.14-1.11 (m, 22 H), 1.38 (s, 9 H) .
Fx;~Tr~le 72
Synthesis of Compound 260
~ ~H
0 ~ NiH
In an oven-dried 25 mL round-bottomed ~lask, triazole
lactam 1 (124 mg, 0.358 mmol) was dissolved in 5 mL of
THF. (S) -Epichlorohydrin (50 UL, O. 639 mmol) was
added. P-4-tBu phosphazene base ( 370 UL of l.OM
solution in hexane, 0. 370 mmol) was added dropwise via
syringe initially producing a dark orange-brown color
that faded. The mixture was stirred for 30 minutes at
room temperature and the decahydroisoquinoline amide
(124 mg, 0. 525 mmol) was added. The mixture was
refluxed overnight. The solvent was evaporated and
the residue was purified by preparative HPLC to afford
the triazole DHIQ lactam (189.1 mg, 84%). HPLC:
retention times of 12. 94, 14.Z2 minutes at 99g6 purity.
CA 02243l2l l998-07-l4
W O 97/27180 PCT~US97/01610
- 250 -
H NMR : dl. 3-1. 4 two singlets in a 1:1 ratio; 1. 4-3.1
(several overlapping multiplets, 1. 4-2.3 (several
overlapping multiplets), 2.45-3.35 (several overlapping ~~
multiplets~, 3.2-4 .2 (several multiplets), 5. 4-5.6
(multiplet), 6.1 and 6.45 (two broad singlets in a 1:1
ratio), 5.9, 6.0, and 6.3 (three broad singlets in a
ratio of 4:4:1 ratio), 7.1 (doublet), 7.2-7.5
(multiplet). MALDI-MS: calc'd (-DHIQ): 401.2; found
403.6 (M - DHIQ + 2H ).
~x~n~le 73
Synthesis of Co~rol7nd 1~9
Ph ~ ~ H Ph~
, ~ H tributyl tin azide ~O~NH
In a heavy-walled screw-top test tube, the alkyne
lactam 1 (83 mg, 0.164 mmol) was dissolved in 5 mL of
xylene. Tributyltin azide (200 mg, 0. 602 mmol) was
added, the tube was sealed and heated to 205~ C
overnight. The dark brown solution was cooled and
directly chromatographed using a gradient from CH2C12
to 50% EtOAc/MeOH to afford the triazole product 2 (14
mg, 2.59~) as a light yellow oil. HPLC: retention times
of 12.01, 12.44, 13.01, 13.22 minutes in an 8:4:1:1
ratio at 99% purity. MALDI-MS: calc'd (-DHIQ): 55Q.4;
found 552.9 (M + 2H ).
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 251 -
x~m~le 74
.
3 Svnthesis of Com~ound 227
7PC ~ ~ "
3.) iautJl u~ )c. HCI rt
To a cooled solution (-78 ~C) of lactam 1 (O.lOg,
0.46mmol) in anhydrous THF (l.OmL) was added
phosphazene base P4 t-butyl solution (l.OM in hexanes,
- 0.46mL,0.46mmol) with stirring. After a 15 min.
stirring period, epoxide 2 (0.173g,0.46mmol) was added
in one portion and the reaction was allowed to slowly
warm to rt. After 0.5h at rt, l.OM HCl (lO.OmL) was
added and the solution was diluted with ethyl acetate
(60mL). The ethyl acetate was washed with sat. NaHC03
(l x lOmL), brine (l x lOmL) dried (MgS04), filtered,
and evaporated to give a brown foam. The crude
acetonide (0.270g, 0.46mmol) was dissolved in
isopropanol (lOmL) and treated with conc. HCl (3.0mL)
at rt. After 2.Oh., the solution was adjusted to pH 11
with 3.ON NaOH and extracted with ethyl acetate (3
x75mL) . The ethyl acetate was dried(MgS04) and
evaporated to give the crude product which was purified
by column chromatography: methylene chloride/methanol
(98/2) to give the product as an off white solid
(0.09Og, 36%). MS: crude acetonide: M+Na = 617;
CA 02243121 1998-07-14
WO 97/27180 PCT~US97/01610
- 252 -
product: M+Na = 577 lH NMR (CDCl3) O.90(m, 6H); 1.15(m,
lH); 1.40(m, lH); 1.5Q-1.80(m, 2H); l.90(m,lH); 2.18(m,
2.25H); 2.30-2.50(m, lH); 2.60(m, 0.75H); 2.80-3.10(m, ~
4H); 3.30(m, 2H); 3.60(m, 1.25H); 3.80(m, 1.75H);
3.95(m, lH); 4.25(m, lH); 4.40(m, 0.75H); 5.00(m,
0.25H); 5.25(m, lH); 5.95(d, 0.25H); 6.10(d, 0.75H);
7.00-7.40(m, 14H)
Fx~le 75
Svnthesis of Compolln~ 232
I) N H,Dl~,80~C ~< ~il
O r~ conc. HCI O OH ~ OH
O~LO
Prepared using the procedure outlined in Example 24.
~he acetonide was purified by column chromatography:
60/40 hexane/ethyl acetate. MS: M+NA = 647. The product
was purified by column chromatography: 98/2
CH2Cl2/MeOH. MS: M+H = 585 lH NMR (CDC13) 1.70(m, 2H);
1.80(m, lH); l.90(m, lH); 2.10(m, lH); 2.40-3.10(m,
lOH); 3.60(s, 3H); 3.75(m, lH); 3.90(m, lH); 4.0(m,
lH); 4.30(m, 3H); 5.30(m, lH); 6.10(d, lH); 7.00- -
7.40(m, lgH). r
CA 02243121 1998-07-14
W O 97/2718Q PCT~US97/01610
- 253 -
~~ F.x~le 76
~ Synthesis of Compound 231
<~ I) NaE~, Dl~, 80~C ~ ~
~,'' conc. HCI~ " OH~ OH
~,NH ~ ~3
Prepared using the procedure outlined in Example 24.
The acetonide was purified by column chromatography:
S 98/2 CH2Cl2/MeOH. MS: M+H = 642. The product was
puri~ied by column chromatography: 96/4 CH2Cl2/MeOH.
MS: M+H = 602 H NMR (CDCl3) 1.50-2.50(m, 6H~; 2.50-
3. 40(m, 6H); 3. 50-4.40(m, 7H); 5. 25 (m, lH); 5.95(m,
lH); 7.00-7.60(m, 18H).
~x~m~le 77
Svnthesis of Compound 216
<~ 1 ) NaH, DMF, 80~C
.~ 2 8
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 254 -
Prepared using the procedure outlined in FY~mrle 24.
The acetonide was purified by column chromatography:
50/50 hexane~ethyl acetate. MS: M~NA = 645. The product
was purified by column chromatography: 96/4
CH2C12/MeOH. MS: M+NA = 605 H NMR (CDCl3) 1.10-1.40~m,
2H); 1.70(m, 2H); 1.80-2.10(m, 4H); 2.35(m, 0.5H);
2.50(m, lH); 2.65(m,0.5H)i 2.80-3.10~m, 4H); 3.20(m,
2H); 3.30-3.55(m, 3H); 3.70(m, lH); 3.80-4.00(m, 4H);
4.25(m, lH); 4.37(m, lH); 5.27(m, lH); 6.15(d, lH);
7.10-7.40(m, 14H).
Fx~le 78
Synthesis of Cnm~ollnd ~21
I) NA~ DMF, 80~C ~ ,~
conc. HCI ~ OH ~ H OH
NH ~ ?N
Prepared using the procedure outlined in ~Y~mrle 24.
The acetonide was not purified by column
chromatography. MS: (crude) M+H = 644. The product was
purified by column chromatography: 96/4 CH2Cl2/MeOH.
MS: M+H = 604 lH NMR (CDCl3) 1.40-2.20(m, 6H); 2.30(m,
lH); 2.50-3.40(m, 9H); 3.75(m, 2H); 4.00(m,lH); 4.25(m,
lH); 5.30(m, lH); 6.35(d, 0.5H); 6.50~d, 0.5H); 7.00-
7.40(m, 14H); 7.50(m, 2H); 8.50(m, 2H).
CA 02243l2l l998-07-l4
W O 97/27180 PCTrUS97/01610
- 255 -
Fx~le 79
.
Y Synthesis of ComDound ~23
" ~
I) N~H.DM~.~0~C
~\NH collc HCI N-o ' OH ~ OH
~ ~L ~ ~N ,~
O~N.~ ~ 3
2 ~
Prepared using the procedure outlined in F~ m~ e 24.
The acetonide was not purified by column
chromatography. MS: (crude) M+NA = 670. The product was
purified by column chromatography: 97/3 CH2C12/MeOH.
MS:-M+NA = 630 lH NMR (CDCl3) 1.40tm, lH); 1.30-1.80(m,
2H); 1.95(m, lH); 2.10(m, lH); 2.25(m, 2H); 2.30-
3.40(m, 7H); 3.60-3.80(m, 2H); 3.85(m,lH); 4.00(m, lH);
4.25(m, lH); 4.45(m,lH); 5.30(m, lH); 5.80(m,lH);
6.15(d, lH); 7.10-7.40(m, 14H).
CA 02243121 1998-07-14
W O97/27180 PCTrUS97/01610
- 256 -
~x~le 80
Synthesis of Com~o~lnd 730
<~ ~ I) N~ D~, 80~C
~J~NH
Prepared using the procedure outlined in FX~r7e 24.
The acetonide was not purified by column
chromatography. MS: (crude) M~NA = 7~ 6. The product was
purified by column chromatography: 97/3 CH2C12/MeOH.
MS: M+NA - 706.
~x~m~le 81
Synthesis of Compound 224
~ I) N-l~, D~, 8PC ~ f~
O~;, conc HCI
~3 .
. .
CA 02243121 1998-07-14
W O 97127180 PCT~US97/~1610
- 257 -
Prepared using the procedure outlined in Example 24.
The acetonide was not purified by column
chromatography. MS: (crude) M+NA = 673. The produc. was
purified by column chromatography: 96/4 CH2C12/MeOH.
MS: M+NA = 633 H NMR (CDC13) 0.090-1.30(m, 4H); 1.40-
1.80(m, 4H); 1.90-2.35(m, 3H); 2.45 (m, lH); 2.65~m,
lH); 2.70-3.10(m, 6H); 3.25(m, 3H); 3.60-4.00~m, 6H);
4.25(m, lH); 4.35(m, 0.5H); 4.75(m, 0.5H); 5.25(m, lH);
6.20(m, lH); 7.10-7.40(m, 14H).
~xam~le 82
Svnthesis o~ Co~round 225
1)pl,~ F-78~C ~ (CZ-
3.) ;~u~nu~al~ùl~ n~c. HCI rt
Prepared using the procedure outlined in Example 74.
The acetonide was not purified by column
chromatography. MS: (crude) 2M+NA = 1179. The product
was purified by column chromatography: 80/20 ethyl
acetate/hexane. MS: M+H = 539 H NMR (CDC13) 0. 55 (m,
lH); 0.659m, lH); 0.95(m, lH); 1.05(m, lH); 1.75(m,
2H); 1.95(m, lH); 2.20 ~dd, lH); 2.65(dd, lH); 2.70-
3.10(m, 6H); 3.20(d, lH); 3.65(dd, lH); 3.95m, 2H);
4.25(t, lH); 5.25(m, lH); 5.95(d, lH); 7.10-7.40(m,
14H).
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 258 -
Fx~m~le 83
Svnthesis of Com~olln~ 226
I) N~ Dl~, 80~C
CODC- HCi ~3~ .~ ;,~
Prepared using the procedure outlined in Example 24.
The acetonide was purified by column
chromatography:60/40 hexane/ethyl acetate. MS: M+NA =
666-. The product was purified by column chromatography:
40/60 hexane/ethyl acetate. MS: M+H = 604 H NMR l. 55
(m, 0.5H); 1.70m, 0.5H); 1.95(m, lH); 2.50 (m, lH);
2.70-3 10(m, 7.5H); 3.15(dd, lH); 3.30(m, lH); 3.40(m,
lH); 3.75(m, lH); 3.80-4.10(m, 2H); 4.25(m, 2.5H);
4.45(m, 0.5H); 5.25(m, lH); 6.15(m, lH); 6.45(d, lH);
6.55(q, lH); 6.70(q, lH); 7.10-7.40(m, 16H).
, . .
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 259 -
~, Fx~le 84
-
Svnthesis of Compound 229
.u.~ , THF, -78~C NC~
3 ) ~u~ ~Uwn~,, HCI rt NC~
isom~r 2
- Prepared using the procedure outlined in FY~mrle 74 The
acetonide was purified by column chromatography and the
diasteriomers were isolated separately. MS: (isomer 1)
M+NA - 642; (isomer 2) M+NA = 642. The individual
diastereomers were deprotected and purified by column
chromatography: 98/2 CH2Cl2/MeOH to give isomer 1 and
isomer 2. MS: (isomer 1) M+NA = 602; (isomer 2) M+NA =
602. H NMR (CDCl3) isomer 1: 1.05 (d, lH); 1.35(s,
3H); 1.45 (s, 3H); 1.75(m, lH); 1.90-2.20(m, 3H);
2.65(m, lH); 2.70-3.10(m, 8H); 3.70(m, lH); 3.95(m,
2H); 4.20(m, lH); 4.35(m, lH); 5.25(m, lH); 6.05(d,
lH); 7.10-7.40(m, 14H). H NMR (CDCl3) isomer 2: 1.10
(d, lH); 1.40(s, 3H); 1.50 (s, 3H); 1.75(m, lH);
1.95(m, lH); 2.15(m, lH); 2.50(m, 2H~; 2.80-3.10(m,
6H); 3.35(m, 2H); 3.65(m, lH); 3.80(m, lH); 4.00(m,
2H); 4.25(m, lH); 5.25(m, lH); 5.95(d, lH); 7.10-
7.40(m, 14H).
CA 02243l2l l998-07-l4
WO 97/27180 rCT~US97/01610
- 260 -
Fx~le 85
Svnthesis of Compolln~ ~61
~N~ OH
b~"~ ce, THF, -78~C H3~ isomer 1
~ ~ 2 ~? OH
3.) i~.,,.~,. '' ,1_. HCI rt H
isom~r 2
Prepared using the procedure outlined in Ex~mple 74.
The acetonide was purified by column chromatography:
60/40 hexane/ethyl acetate and the diasteriomers were
isolated separately. MS: (isomer 1) M+H = 658; (isomer
2) M+H = 658. The individual diastereomers were
deprotected and purified by column chromatography:
40/60 hexane/ethyl acetate to give isomer 1 and isomer
2. MS: (isomer 1) M+H = 618; (isomer 2) M+NA = 640. H
NMR (CDCl3) isomer 1: 1.75(m, lH)i 1.90-2.20(m, 3H);
2.70(s, 3H); 2.75-3.15(m, 6H); 3.75(m, lH); 4.00(m,
3H); 4.25(m, lH); 4.65~m, lH); 5.25(m, lH); 6.05(d,
lH); 6.55(dd, 2H); 6.70(m, lH); 7.00-7.40(m, 16H). lH
NMR (CDCl3) isomer 2: 1.70~m, 2H); 1.~5~m, lH); 2.25~m,
lH); 2.55~m, lH); 2.70~s, 3H); 2.80-3.10~m, 8H);
3.35~dd, lH); 3.40(dd, lH); 3.75(m, lH); 3.80(m, lH);
4.Q5(m, lH); 4.25(m, lH); 4.55(t, lH); 5.30(m, lH);
6.05(d, lH); 6.70(m, 2H); 7.10-7.40(m, 17H).
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 261 -
xam~le 86
Synthesis of Co~nound 228
3 . .
~ ~ ZO~pd/C ~
~N~
separahd t~ .o N J~3
N' --N ~ ~N
isomer 2
The benzyl triazole from Fx~mrle 80 was puri~ied (and
diastereomers isolated) by column chromatography: 97/3
CH2Cl2/MeOH. MS: M+NA = 706. The individual benzyl
protected diastereomers were dissolved in MeOH and
combined with Z0% Pd/C (cat.). Each solution was
hydrogenated under pressure(50 psi) at rt for 5 days
and the resulting crude product was purified by column
chromatography 96/4 CH2Cl2/MeOH to give isomers 1 and
2. MS: (isomer 1) M+H = 594; (isomer 2) M+NA = 616. lH
NMR (CDCl3) isomer 1: 1.60(m, lH); 1.80(m, 2H); 2.40(s,
lH); 2.60-3.15(m, 10H); 3.65(m, lH); 3.80(m, lH);
4.00(m, lH); 4.20(m, lH); 5.25(m, lH); 6.90(m, lH);
7.00-7.40(m, 14H). H NMR (CDCl3) isomer 2: 1.30(m,
~ lH); 1.75(m, lH); 1.95(m, 2H); 2.35(m, lH); 2.50(m,
lH); 2.80-3.10(m, 8H); 3.25(d, lH); 3.65(m, lH);
3.80(m, lH); 4.05(m, lH); 4.30(m, lH); 4.50(m, lH);
5.25(m, lH); 6.75(m, lH); 7.10-7.40(m, 14H).
CA 02243121 1998-07-14
W O 97~7~80 PCTrUS97/01610
- 262 -
Fx~le 87
Svnthesis of Com~ound 219
~ ~H OH
separated~i~,t~;~o",~,~ isomers1,2,3
Isomer 1: Prepared using the procedure outlined in
Example 24. The acetonide was purified by column
chromatography 30/70 hexane/ethyl acetate MS: M+NA =
645. The product was purified by column chromatography:
30/70 hexane/ethyl acetate MS: M+NA = 605 H NMR
(CDC13) 1.45~m, lH); 1.70(m, lH); 1.80-2.05(m, 4H);
2.25 (q, lH); 2.35(q, lH); 2.65(m, lH); 2.75-3.10(m,
8H); 3.60(m, 3H); 3.75(m,lH); 3.85(m, lH); 3.95~m, 2H);
4.25Im, lH); 5.25(m, lH); 6.05(m, lH); 7.10-7.40(m,
14H).
Isomers 2,3:(Chiral center within THF ring has opposite
configuration to that of isomer 1 above). Prepared
using the procedure outlined in FY~mrle 74. The
acetQnide was purified (diastereomers isolated) by
column chromatography 30/70 hexane/ethyl acetate MS:
M+NA = 645. The individual diastereomers were purified
by column chromatography: 30J70 hexane/ethyl acetate
MS: (isomer 2) M+NA = 605 (isomer 3) M+NA = 605 lH NMR
(CDC13) (isomer 2) 1.45~m, 2H); l.90(m, 2H); 2.10(m,
lH); 2.30 (m, lH); 2.35(m, lH); 2.45(m, lH); 2.75-
3.10(m, 6H)i 3.25(m, 2H); 3.65(m, 3H); 3.75(m,3H);
3.95(m, 2H); 4.25(m, 2H); 5.25(m, lH); 6.10(m, lH);
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 263 -
A~ 7.10-7.40(m, 14H). lH NMR (CDCl3) (isomer 3) 1.15(m,
lH); 1.80(m, lH); 1.95(m, 2H); 2.10(m, lH); 2.25 (m,
lH); 2.40(m, lH); 2.60(m, lH); 2.75-3.10(m, 8H);
3.40(m, lH); 3.60-4.00(m, 6H); 4.25(m, lH); 5.25(m,
5 lH); 6.05(m, lH); 7.10-7.40(m, 14H);
Fx~le 88
Synthesis of Co~ound 233
OH
,1., base,THF,-7e~c ~i ~ O N
~NH 2.) [~ iwmer 1
3~ v~ ,N
isomer 2
Prepared using the procedure outlined in Example 74.
The acetonide was purified (diastereomers isolated) by
column chromatography 45/55 hexane/ethyl acetate MS:
M+NA = 692. The individual diastereomers were purified
by column chromatography: 35/65 hexane/ethyl acetate
MS: (isomer 1) M+H = 630 (isomer 2) M+H = 630 lH NMR
(CDCl3) (isomer 1) 1.7S(m, lH); 1.95(m, lH); 2.10(m,
2H); 2.75-3.10(m, 8H); 3.15(d, 2H); 3.30(m, 2H);
3.80(m,2H); 4.00(m, 2H); 4.25(m, lH); 5.25(m, lH);
6.00(m, lH); 6.20(d, lH); 6.60(t, lH); 6.95-7.40(m,
18H). H NMR (CDCl3) (isomer 2) 1.75(m, lH); 1.95(m,
2H); 2.15(m, lH); 2.55 (m, lH); 2.75-3.10(m, 8H); 3.20-
CA 02243121 1998-07-14
W O97/27180 PCTrUS97/01610
- 264 -
3.50(m, 4H); 3.75 (m, lX); 3.85~m,lH)4.00(m, lH);
- 4,25(m, lH); 4.35(m, lH); 5~25(m, lH); 6.10(m, lH);
6.30(m, lH); 6.56(t, lH);7.10-7.40(m, 18
H)-
5_ ~x~m~le 89
Svnthesis of Co~ol~nd ~34
base, THF, -7~C ~ OH H OH
O O O
3 ) i~ hl,. HCI rt
Prepared using the procedure outlined in Example 74.
The acetonide was purified by column chromatography:
97/3 CH2Cl2~MeOH. MS: M+H = 633. The product was not
purified. MS: M+H = 593.
-
CA 02243121 1998-07-14
W O 97/27180 PCT~US97J01610
- 265 -
i.
Fxam~le 90
Svnthesis of Compound 235
.~
A.
I-bCO ~< ~ N~}~DMF~0~C
~ o~ C C~ ~
A mixture of the trans isomer of the lactam above
(0.125 g, 0.27 mmol) and 60% sodium hydride (0.010 g,
0.25 mmol) in dimethylformamide (4 mL) was stirred
under a nitrogen atmosphere for 30 min. The epoxide
(0.113 g, 0.30 mmol) was added and the mixture was
heated at 60 ~C for 4h. The mixture was re-charged
with 60~ sodium hydride (0.015 g, 0.37 r[ur~ol) heated at
60 ~C for an additional 1.5h, and stirred at ambient
temperature for 18h. The mixture was diluted with
dichloromethane, washed with brine, dried ~MgSO4), and
concentrated in vacuo. The residue was purified }:y
chromatography on silica gel, eluting with hexane:ethyl
acetate (7:3) then with hexane:ethyl acetate (1:1) to
give O.llg (48%) of product as a brown oil. MS: AP+,
862 (M+Na) and AP-, 874 (M+Cl).
B.
A solution of the acetonide (O.llg, 0.13 mmol) in 2-
propanol (7 mL) and concentrated hydrochloric acid (3
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 266 -
mL) was stirred at ambient temperature for 3h, ~-
neutralized with 2N sodium hydroxide, and extracted
with diethyl ether. ~he extracts were dried (MgSO4),
filtered, and concentrated in vacuo to give 0.040 g
(58%) yield of the crude product, which was used
without further purification. MS: ES+, 550 (M+Na) and
ES-, 562 (M+Cl).
C.
~ A solution of the amine (0.14 g, 0.27 mmol),
methylchloroformate (0.023 mL, 0.30 mmol) and Et3N
(0.05 mL, 0.36 mmol) in dichloromethane (2 mL) was
stirrred at ambient temperature under a nitrogen
atmosphere for 18h. The volatiles were removed in
vacuo, and the residue was purified by reverse phase
preparative HPLC to give a tan oil. Lyophilization
gave 0.012 g (8%) of the product as a white solid. MS:
ES+,608 (M+Na). H NMR (CDCl3) 1.71 (m, lH); 1.96 (m,
lH); 2.11 (m, lH3; 2.26 (m,lH); 2.71-3.05 (m, 8H); 3.50
(s, 3H); 3.65 (m, lH); 3.82 ~m, lH); 4.00-4.39 (m, 5H);
5.28 (m, lH); 5.43 (s, lH); 6.40 (d,lH); 7.08-7.33 (m,
14H).
..
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 267 -
- F.xam~le 91
~ Synthesis of Compound 239
1.) ~ u~ e, TffF, -78 ~C ~ OH f[~3 OH
F H~ 2-) ~ F H~ ~N ,~
3.) iw,~ ,ul~u,,.,. HCI r~
~ Prepared as described in Ex~mple 74 with the exception
. that water, rather than 1.0 N HCl was used to quench
the reaction. MS (AP-) of the acetonide = 660 (M-1).
MS-(AP+) of the product = 644 (M+Na). 1HNMR of the
product (CDC13): d 1.68 (m, 3H), 2.07 (m, 3H), 2.54
(m, 2H), 2.92 (m, 6H), 3.43 (m, lH), 3.78 (m, lH), 4.00
(m, 2H), 4.50 (m, lH), 5.34 (m, lH), 6.10 (m, lH), 6.70
10(m, lH), 7.24 (m, 18H)
F.xample 92
Svnthesis of Compound 238
F~ ~ 1 ) pl ~ , THF -7 FC ,~
3.) iSO~Iu~a~ IU. HCI rt
CA 02243l2l l998-07-l4
W O 97/27180 PCT~US97/01610
- 268 -
Prepared as described in FY~mrle 74 with the exception ~-
that water, rather than 1.0 N HCl was used to quench
the reaction. MS (AP+) of the acetonide = 684 tM+Na).
MS (AP+) of the product = 644 (M+Na). lHNMR of the
product (CDC13): d 1. 62 (m, 3H), 2.00 (m, 3H), 2.50
(m, 2H), 2.80 (m, 6H~, 3.30 (m, 2H), 4.00 (m, 2H), 4.34
(m, lH), 5.33 (m, lH), 6.14 (m, lH), 6.30 (m, lH), 7.24
(m, 18H)
F:x~Tr~le 93
Synthesis of Co~pollnd ~40
NC~ 1.~ ~'1 ~ b~se, THF, -78~C ,[~ ~ OH
N ~UH 2 ) ~N ~ N 8
3,) :. ,~....~ ,..1!~"" HCI rt
Prepared as described in F~mr1e 74 with the exception
that water, rather than 1.0 N HCl was used to quench
the reaction. MS (AP+) of the acetonide = 691 (M+Na).
MS (AP+) of the product = 651 (M+Na). HNMR of the
product (CDC13): d 1. 66 (m, 3H), 2.08 (m, 3H), 2.59
(m, 2H), 2.95 (m, 6H), 3.40 (m, lH), 3.85 (m, lH), 4.14
(m, 2H), 4.27 (m, lH), 5.32 (m, lH), 6.22 (m, lH), 6.73
(m, lH), 7.25 (m, 18H).
CA 02243l2l l998-07-l4
WO 97/27180 PCT/US97/01610
- 269 -
-~ Fx~Tr~le 94
-
Synthesis of Compound 241
bas~, THF, -78~C ~ OH ~ OH
N~\NH 2 )
3.) i~ a~ ,. HC~ r~
Prepared as described in Example 74 with the exception
that water, rather than 1.0 N HCl was used to quench
the reac.tion. MS (AP+) of the acetonide = 645 (M+l).
MS (AP+) of the product = 627 (M+Na). lHNMR of the
product (CDCl3): d 1 . 70 (m, 3H), 2.00 (m, 3H), 2 . 58
(m, 2H), 2 . 97 (m, 6H), 3 . 40 (m, lH), 3 . 85 (m, lH), 4 . 10
~m, 2H), 4 . 32 (m, lH), 5 . 33 (m, lH), 6. 30 (m, lH), 6.80
(m, lH), 7 . 25 (m, 17H), 8.01 (m, lH) .
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 270 -
F.x~m~le 95
Synthesis of Com~oun~ 208
Ph ,~
A. NaH, DMF, F3CO2S~ ~O~=~ Ph
~I B LiOH, THF, DME I Ph
P~CHN Ç; C TBDMSCI, Im. Ph3CHN~
OH
HN
_~ ~,~ CH30(CO)CI, DIEA
A.-
The lactam (1.20 g, 2.69 mmol, 1 eq) was dissolved in
anhydrous dimethylformamide (8 mL) under Argon and
cooled with an isopropanol dry ice bath to -40 ~C. A
solution of sodium bis~trimethylsilyl)amide (l.OM in
THF, 2.69 mL, 2.69 mmol, 1 eq) was added dropwise via
syringe and the reaction was stirred for 15 min.
1~ maintaining the bath temp between -40 - -50 ~C.
Dihydro-5(S)-[[[(tri~luoromethyl)sulfonyl]oxy]methyl]-
3(R)-(phenylmethyl)-3(2H)-furanone (J. Med. Chem.,
1994, Vol. 37, No. 21, 3443-51; 1.00 g, 2.96 mmol, 1.1
eq) was added as a solid and the reaction was stirred
vigorously for 10 min. and then quenched with several
drops of glacial acetic acid. The reaction mixture was
evaporated in vacuo to a residue and partitioned
between ethyl acetate, saturated aqueous brine, and
water. After separating the layers, the aqueous layer
=
;
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- - 271 -
~ was back-extracted with ethyl acetate. The combined
organic layers were washed with saturated aqueous
Y brine, dried over anhydrous magnesium sulfate,
evaporated in vacuo and purified by flash silica gel
chromatography eluting with ethyl acetate : hexane
(3:7). Fractions con~aining the alkylated lactam were
combined, evaporated in vacuo to provide 0.883 g (52 ~)
of product as a foam. MS (ESI): M+Na = 657.
The butyrolactone (1.202 g, 1.89 mmol, 1 eq) ~rom step
A was dissolved at ambient temperature in
dimethoxyethane (20 mL) and cooled with an ice water
bath. Aqueous lithium hydroxide (1.0 N, 4.75 mL, 4.75
mmol, 2. 5 eq) was added via pipette and the mixture
~ was stirred for 0. 5 h. The reaction was warmed to
ambient temperature and stirred for an additional 1 h.
Aqueous citric acid (10~ w/v) was added to reach an
acidic pH and the mixture was evaporated in vacuo. The
residue was partitioned between ethyl acetate : diethyl
ether (4:1) and aqueous citric acid (10~ w/v). After
separating the layers, the aqueous layer was back-
extracted with ethyl acetate. The combined organic
layers were washed with water, saturated aqueous brine,
dried over anhydrous magnesium sulfate, evaporated in
vacuo and dried under high vacuum to provide the acid
(1.32 g, 10696) as a foam. MS (APCI): M - 1 = 651.
C.
The acid (1.28 g, 1.97 mmol, 1 eq) from step B in 5 mL
anhydrous dimethylformamide under Argon was combined
CA 02243121 1998-07-14
WO 97/27180 PCT~US97/01610
- 272 -
with imidizole (1.472 g, 21.6 mmol, 11 eq) followed by ~-
tertbutyldimethylsilyl chloride (2.96 g, 19.7 mmol, 10
eq) and stirred at a~r~ient temperature for 16 h. The
reaction was quench by addition of methanol ~15 mL) and
stirred ~or an additional 45 min. Aqueous lithium
hydroxide (1.0 N, 2.0 mL, 1 eq) was added and the
mixture was evaporated in vacuo. The residue was
partitioned between ethyl acetate and aqueous sodium
hydrogen sulfate (1.0 N). After separating the layers,
the aqueous layer was back-extracted with ethyl
acetate. The combined organic layers were washed with
saturated aqueous brine, dried over anhydrous magnesium
sulfate, evaporated in vacuo and dried under high
vacuum to provide the silyl protected acld (1.46 g,
1~ 97%) as a foam. MS (APCI): M - 1 = 766.
D.
The silyl protected acid (1.32 g, 1.72 mmol, 1 eq) from
step C in anhydrous dimethylformamide (7 mL) under
Argon was treated consecutively with diisopropylethyl
amine (0.316 mL, 1.81 mmol, l.D5 eq), 1-
hydroxybenzotriazole (0.244 g, 1.81 mmol, 1.05 eq),
(lS,2R)-(-)-1-amino-2--indanol(0.283 g, 1.90 mmol, 1.1
eq~, and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide
hydrochloride (0.347 g, 1.81 mmol, 1.05 eq). After
stirring at ambient temperature for 3 h, the reaction
mixture was evaporated in vacuo, and partitioned
between ethyl acetate, saturated aqueous brine, and
water. After separating the layers, the aqueous layer
was back-extraçted with ethyl acetate. The combined
organic layers were washed with saturated aqueous
I
CA 02243121 1998-07-14
W O 97/27180 PCTAUSg7/01610
- 273 -
-~ brine, dried over anhydrous magnesium sulfate,
evaporated in vacuo and purified by flash silica gel
chromatography eluting with ethyl acetate : hexane
(3:7). Fractions containing the product were combined,
evaporated in vacuo and dried under high vacuum to
provide the protected amide (1.06 g, 69%) as a foam. MS
(ESI): M+Na = 920.
The protected amide (1.035 g, 1 15 mmol, 1 eq) from
step D was dissolved in trifluoroacetic acid (15 mL)
and stirred under Argon for 15 min. The reaction was
evaporated in vacuo and trituratated with diethyl
ether/hexane. After decanting the mother liquor, the
residual solid was dried under high vacuum to provide a
partially deprotected product. The crude material was
dissolved again in trifluoroacetic acid (15 mL) and
stirred for 20 min. under Argon. The reaction mixture
was evaporated in vacuo to a residue and triturated
with hexane/diethyl ether. The slurry was filtered,
washed with hexane and dried under high vacuum to
provide the deprotected amine (0.607 g, 83%) as a
trifluoroacetic acid salt. MS (ESI): M+1 = 542.
The amine (0.025 g, 0.038 mmol, 1 eq) from step E was
combined with diisopropylethylamine (0.0146 mL, 0.038
mmol, 2.2 eq) in dichloromethane (1.5 mL) under Argon.
The solution was treated with methylchloroformate
(0.0028 mL, 0.0362 mmol, 0.95 eq). After stirring for
approximately lO min., the reaction mixture was applied
CA 02243l2l l998-07-l4
W O 97/27180 PCT~US97/01610
- 274 -
directly to a 20x20 cm (500 uM, silica gel GF) --
preparative thin layer chromatography plate and eluted
with 95: 5 dichloromethane : methanol. The product band
was removed from the plate and the product was washed
from the silica gel with 85:15 dichloromethane
methanol (10 mL). The solution was evaporated in
vacuo, triturated with hexane, evaporated in vacuo, and
dried under high vacuum to provide the car~amate as a
white solid (0. 0164 g, 72 %). The product was
lQ lyophilized from acetonitrile : water (1:1). MS (APCI):
M + Na = 622. H NMR (CDCl3 + NaOD): 1. 66 (m, lH);
1.90 (m, 3H); 2.29 (m, lH); 2.64 (m, lH); 2.92 (m, 7H);
3.18 (m, lH); 3.40 (m, lH); 3.56 ~s, 3H); 3.66 (m, lH);
3.85 (m, lH); 3.58 (m, lH); 4.26 (m, lH); 5.27 (m, lH),
6.07 (d, lH, J=7.8); 7.14 ~m, 6H); 7.28 (m, 8H) .
~.x~le 96
Synthesis of Compound 236
Ph Ph Ph Ph
~FA~H2N~ /~ OH ( H OH CF3SO2NH_~ ~ f N
~ Cf:3S02~20, DIEA
The aminomethyl pyrolidinone (0. 025 g, 0.038 mmol,
eq) was combined with diisopropylethylamine ~0.0146 mL,
0.038 mmol, 2.2 eq) in dichloromethane (1.5 mL) and
cooled to -78 ~C with a dry ice acetone bath. The
solution was treated with trifluoromethane sulfonic
anhydride (0.0064 mL, 0. 038 mmol, 1 eq) in
dichloromethane (0. 5 mL). The reaction mixture was
CA 02243l2l l998-07-l4
W O 97/27180 PCTrUS97101610
- 275 -
then allowed to warm to room temperature and applied
directly to a 20x20 cm (500 uM, silica gel GF)
preparative thin layer chromatography plate and eluted
with 95: 5 dichloromethane : methanol. The product band
was removed from the plate and the product was washed
from the silica gel with 85 :15 dichloromethane
methanol ~10 mL). The solution was evaporated in vacuo
.to a residue and lyophilized from acetonitrile : water
(1:1) to provide the desired product as a white
lyophile (0.008 g, 31 96). MS ~APCI): M + Na = 696. H
NMR (CDCl3 + NaOD): 1. 64 (m, lH); 1.98 ~m, 3H)i 2.Z2
(m, lH); 2.72 (m, lH); 2.91 (m, 8H); 3.47 (m, lH); 3.78
(m, lH); 3.97 (m, lH); 4.09 (m, lH); 4.31 (m, lH); 5.23
(m, lH); 6.17 (d, lH, J=8.7); 7.21 (m, 14H).
.
Fx~m~le 97
Synthesis of Compound 211
lFA~ ~H o ~ ~ <~0~ i H o
The aminomethyl pyrolidinone (0.030 g, 0.046 mmol, 1
e~) was combined with diisopropylethylamine (0. 0175 mL,
O.10 mmol, 2.2 eq) and 3-(R) -hydroxy-tetrahydro~uran-N-
hydroxysuccinimide carbonate (W093-US8458, 0.016 g,
0.046 mmol, 1 eq) in dichloromethane (1.5 mL) and
allowed to stir for 16 h at ambient temperature. The
dichloromethane was removed in vacuo and replaced with
acetonitrile (2 mL). The mixture was heated at reflux
CA 02243121 1998-07-14
W O97/27180 PCTrUS97/01610
- 276 -
~or 20 min. and then cooled and evaporated in vacuo.
The residue was dissolved in dichloromethane (~0.5 mL),
applied directly to a 20x20 cm (500 uM, silica gel GF)
preparative thin layer chromatography plate and eluted
with 9:1 dichloromethane : methanol. The product ~and
was removed from the plate and the product was washed
from the silica gel with 85 :15 dichloromethane
methanol (10 mL). The solution was evaporated in vacuo
to a residue and lyophilized from acetonitrile : water
(1:1) to provide the desired product as a white
lyophile (0.022 g, 73 %). MS (ESI): M + Na = 678. H
NMR (CDC13 + NaOD): 1.65 (m, lH); 1.93 (m, 5H); 2.32
(m, lH); 2.65 (m, lH); 2.90 (m, 7H); 3. 22 (m, lH); 3. 37
(m, lH); 3. 54 (m, 2H~; 3. 79 (m, 4H); 3.97 (m, lH); 4. 22
(m, lH); 5.11 (m, lH); 5.27 (m, lH); 6. 34 (d, lH,
J=8.9); 7. 22 (m, 14H).
~x~ le 98
Synthesis o~ Compound 215
I.NaH/THF
6~ o\¦-- MeO ~ OH
2.HCI
~he starting cyclic urea was obtained ~ollowing
procedures outlined in Examples 11 and 12. Coupling
with the epoxide followed the protocol detailed in
Example 24.
CA 02243l2l l998-07-l4
W 097/27180 PCT~US97/~1610
- 277 -
~~ . F.x~le 99
Synthesis of Co~oun~ 242
~ . ~ I.~A ~ ~
O.3g of the protected intermediate obtained in FX~nrle
98 was treated with lO mL of TFA over 5 h at room
temperature. The reaction was quenched by removing the
TFA, and the resulting crude treated with excess of
sodium carbonate in methanol/water for 10 minutes. The
solvents were removed, product extracted between ethyl
acetate/water, organics combined, dried with magnesium
sulfate, removed in vacuo, and purified by preparative
HPLC, resulting in 0.15g (76.7%) of product 2. lH NMR
(CDCl3, 300 MHz) ~ 8.10 (lH, d, J=8.4), 7.24 (lOH, m),
7.05 (5H, m), 5.28 (m, lH), 4.10 (lH, t, J=4.2), 3.97
(lH, t, J=4.9), 3.53 (lH, m), 3.39 (2H, m), 2.95 (5H,
m), 2.69 (m, 2H), 2.54 (lH, dd), 2.17 (lH, m), 1.92
(lH, m), 1.78 (m, lH). Low resolution MS m/e 514.1
(M+H ), m/e 536. 2 (M+Na ).
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 278 -
FxillT~le lO0
- Svnthesis o~ Compollnc~ 243
3 OH KOtBu
~ ~N R-halide ~ ~ ~N
A solution of 20 mg (0.039 mmol) of the urea obtained
Example 99 in 1 mIJ DMF was treated with potassium t-
butoxide ~26.3 mg, 0.234 mmol, 6 equiv) and
equilibrated at room temperature for 10 min. Next, 6.3
mg of 3-picolyl chloride in 1 mL DMF was added and the
reaction quenched after 20 min. Solvent were then
removed and the residue purified on preparative RP
HPLC resulting in 14.2 mg (60.20%) of the product. H
NMR (d6-acetone, 400 MHz) ~ 8.57 (d, lH, J=5.3), 8.42
(s, lH), 8.01 (d, lH, J=8.0), 7.80 (t, lH, J=5.9), 7.20
(rn, 14H), 6.92 ~d, lH, J=8.8), 5.23 (m, lH), 4.29 (d,
lH, J=16.2), 4.29 (m, lH), 4.11 (d, lH, J=16.2), 3.98
15~ (m, 2H), 3.48 (dd, lH), 3.18 (m, 2H), 3.00 (m, 4H),
2.75 (m, 3H), 1.93 (m, lH), 1.88 (m, lH), 1.66 (m, lH).
Low resolution MS m/e 605.4 (M+H ), m/e 627.4 (M+Na ).
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 279 -
~x~m~le 101
Svnthesis of Co~pound ~44
OH ~ OH KOtBu
~ 3 R-halide ~ ~N. 8
This compound was synthesized using the protocol
outlined for Example lO0 starting from 51 mg (0.1 mM)
of cyclic urea and 3-methylbenzyl bromide (18.5 mg, 0.1
mmol, 1 equiv), resulting in 6.2 mg of the product
after preparative HPLC purification. H NMR (d6-DMSO,
300 MHz) ~_7.68 (lH, d, J=8.5), 7.24 (19H, m), 5.18
(lH, m), 4.26 (lH, m), 4.16 (lH, d, J=15.7), 4.01 (lH,
d, J=15.7), 3.79 (2H, m), 3.33 (lH, m), 3.05 (6H, m),
2.78 ~m, 2H), 2.62 (2H, m), 2.24 (s, 3H), 1.80 (lH, m),
1.38 (lH, m). Low resolution MS m/e 618.2 (M+H ).
Fxample 102
Synthesis of Com~ound 245
~OH ~ OH KOtBu ~ ~ ~ OH
-halide ~ N.. 8
-
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/01610
- 280 -
This compound was synthesized using the protocol ~-
outlined for ~.x~m~l e 100 starting from 51 mg (C.1 mM)
of cyclic urea and 3-fluorobenzyl bromide (18.9 mg, 0.1
mmol, 1 e~uiv), resulting in 7.6 mg of the product
after preparative HPLC purification. H NMR (d6-DMSO,
- 300 MHz) ~ 7.71 (lH, d, J=8.5), 7.24 (19H, m), 5.18
(lH, m), 4.28 (lH, m), 4.19 (lH, d, J=15.7), 4.03 (lH,
d, J=15.7), 3.82 (2H, m), 3.33 (lH, dd), 3.05 (6H, m),
2.80 (m, 2H), 2.59 (2H, m), 1.79 (lH, m), 1.38 (lH, m).
Low resolution MS m/e 622.1 (M+H ).
F,X~ e 103
Synthesis of Com~ol~nd 262
OH KOtBu
N ~ R-nalide ~ ~ ~N 8
Obtained following the protocol outlined for F~mrle
100 using 2-picolyl chloride.
LC/MS-MH 605.
.
CA 02243l2l l998-07-l4
W O 97127180 PCTrUS97/01610
- 281 -
F,x~mnle 1 04
~~ Synthesis of Compolln~ 213
KOtl3u o f3 a~
N~ 8 R-halide ~ N 8
Obtained following the protocol outlined for Example
100 using 3,4,5-trimethoxybenzyl chloride.
lH NMR (DMSO)d6 1.35 (t,lH),1.78 ~t,lH), 2.45 ~m,2H),
2.62 (m,2H), (s,6H), 3.8 (m,2H), 4.1 (q,2H), 4.28
(t,2H), 5.18 (m,2H), 6.45 (s,2H), 6.93-7.38 (m,16H)
7.68 (d,2H), LC/MS-MH 694.
Fx~nle 105
Synthesis of Co~otl~d 246
R-halide 09{~
Obtained following the protocol outlined for Example
100 using 4-amidobenzyl chloride.
H NMR (DMSOd6) 1.35 (t,lH),1.78 (t,lH),2.45
(m,2H),2.62 (m, (s,6H), 3.8 (m,2H), 4.1(q,2H), 4.28
CA 02243121 1998-07-14
W O 97/27180 PC~rUS~7/01610
- 282 -
(t,2H), 5.18 (m,2H), 6.45 (s,2H~, 6.93-7.38 (m,16H)
7.68 (d,2H), LC/MS-MH 694.
F.x~r~le 10 6
Synthesis of Compound 257
u~ lL~la~,~yli~, acid/ ~3
McOOC~ ] 2 eq. Na~, THF MeOOC~NrS
3- EDCI~ HOBT- H~ ~ ~H~
H~-bu
S Following the procedure outlined in Example 21 the
desired ketoamide was obtained as a white fluffy solid
after purification on reversed phase HPLC. M+H: 504
1H NMR: 1.38 and 1.48 (9H,s), 1.8-3.0(ca 7H,m), 3.72
and 3.73 (3H,s), 3.5(1H,m), 3.8(1H,m), 4.0(2H,m), 4.2-
4.8(3H,m) 7.2-7.4(5H,m). Note: Complex NMR signals due
to rotational isomers, diastereomers and ketone-hydrate
equillibria.
Fx~m~le 107
Svnthesis of Compound 2~8
Ph~ Ph
~ HOBTIEDCI/NMMIDMAP ' ,~ c~
Ph~ ,~OH , Pn~$
O N
-
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 283 -
The procedure was followed as described in Example 21
except that instead of thioproline-t-~utylamide bçing
coupled to the ketoacid 1, thioproline-dimethyl
propargylamide 2 was used. This compound was made by
treating a 0 ~C solution of N-BOC-4-thio-L-proline
(Sigma, 2.0 g, 8.6 mmol) in THF (40 mL) with
diisopropylethylamine (4.5 mL, 26 mmol) followed ~y
dropwise addition of isobutyl chloroformate (1.1 mL,
8.6 mmol) via syringe. The reaction was stirred for 30
minutes at 0 ~C before the dropwise addition of 90~
1,1-dlmethylpropargylamine (Aldrich,-1.0 mL, 8.6 mmol).
After stirring for 17 h at room temperature, the
reaction was concentrated in vacuo. Ethyl acetate (70
mL) and water (35 mL) were added to the residue and the
layers were partitioned. The organic layer was dried
over sodium sulfate, filtered, and concentrated in
vacuo. The crude residue was then dissolved in
dichloromethane (20 mL) and treated slowly with
tri~luoroacetic acid (20 mL). The reaction was stirred
~or 24 h before being diluted with ethyl acetate (70
mL) and carefully neutralized with 10~ sodium carbonate
to pH 7. The layers were partitioned and the organic
layer was dried over sodium sulfate, filtered, and
concentrated in vacuo. Flash chromatography over
silica gel (1:1 hexane:ethyl acetate) gave amide 2 as a
white foam. MS (ES+) = 199 (M+l). Coupling of
ketoacid 1 (300 mg, 0.854 mmol) with amide 2 (170 mg,
0.854 mmol) gave ketoamide 3 (84 mg, 0.211 mmol, 25~)
after preparatory silica gel TLC (3:1 ethyl
acetate:hexane). MS (AP+) = 532 (M+l), 554 (M+Na);
HNMR (CDC13): d 1.66 (s, 3H), 1.69 (s, 3H), 1.94 (m,
2H), 2.37 (d, lH), 2.51 (m, 3H), 2.92 (m, lH), 3.21 (m,
CA 02243121 1998-07 14
W O 97t27180 PCT~US97/01610
- 284 -
2H), 3.52 (m, lH), 3.83 ~m, lH), 4.22 (m, 2H), 4.44 (m,
lH), 4.81 (m, lH), 5.00 (m, lH), 6.57 (d, lH), 7.1 ~m,
4H), 7.22 (m, 6H).
F.x;?Tr~rle 108
Svnthesis of C~ound 263
f=\
- 1.) sodium hydride DMF .. ~J
2)~ ' ~CY{
3.) i~ HCI rt
Prepared using the procedure outlined in Example 24.
The acetonide was purified by column chromatography:
65/35 hexane/ethyl acetate. MS: M+Na - 643. The product
was puri~ied ~y column chromatography: 40/60
hexane/ethyl acetate. MS: M+Na = 603 lH NMR (CDC13)
1.05(m, lH); 1.10-1.40(m, 6H); 1.50-1.75(m, 4H); 1.80-
2.00(m, 2H); 2.45(m, lH); 2.80-3.10(m, 4H); 3.20(m,
2H); 3.30(m, lH); 3.45(s, lH); 3.65(m, lH); 3.80(m,
lH); 3.90(m, lH); 4.25(m, lH); 4.60(m, lH); 5.27(m,
lH); 6.00(d, lH); 7.10-7.40(m, 14H).
.
CA 02243121 1998-07-14
W O97127180 PCTnUS97/01610
- 285 -
x~m~le 109
Svnthesis of Compound 206
~-1~ o~ ,SiR3 ,~0 o ~
~ ~N~
OCH3
A solution of 22.3g (0.147 mol, 1 equiv) of S(-)-2-
Amino-3-phenyl-1-propanol in 30 mL THF, cooled to 0 ~C,
was treated with 25.5 mL (0.147 mol, 1 equiv) of DIEA,
followed by addition of 11.7 mL (0.147 mmol, 1 equiv)
of chloroacetyl chloride. After 1 hr at room
temperature, 18.0g (0.16 mol) of potassium -tert-
10 butoxide was added at 0 ~C, the reaction warmed up to
room temperature and allowed to proceed for 15 min.
Solvents were then removed and the crude residue
partitioned between ethyl acetate/water, organics dried
over MgSO4 resulting in 23.8g (85%) of the desired
15 product. H NMR (CDCL3, 300 MHz) ~ 7.20 (m, 5H), 6.67
(s, lH), 4.15 (m, 2H), 3.75 (m, lH), 3.86 (dd, lH,
J=11.6, 3.7), 3.55 (dd, lH, J=11.6, 6.3), 2.82 (m, 2H).
Low resolution MS m/e 192.1 (M+H )
20 A solution of 0.477g (2.5 mmol, 1 equiv) of the
morpholinone above in 1 mL of anhydrous DMF was treated
with 12 mg (0.5 mmol, 0.2 equiv) of sodium hydride
-
CA 02243121 1998-07-14
W O 97/27180 PCTAUS97/0161
- 286 -
(95%) at O C. The reaction was continued at room
temperature for 10 min, and then cooled down tG O ' C,
followed by addition of 0.813g (2.5 mmol, 1 equiv) of
epoxide in 1 mL DMF. The reaction was then carried out
at 50 C for 5 h. Following ethyl acetate/water
extraction, the organics were combined and dried
resulting in 1.18 g of crude product, used further
without purification. Low resolution MS m/e 539.0
(M+Na ) .
C.
A solution of 1.18g ~2.287 mol, 1 equiv) of the above
crude in 4 mL anhydrous THF was treated with 0.44g
(3.43 mol, 1.5 equiv~ of DIEA, followed by 0.907g (3.43
mmol, 1.5 equiv) of TBDMS triflate. After 1 h at room
temperature, the product was purified on silica gel
(Rf=0.26, 1:3 ethyl acetate/hexane), yielding 0.85g of
the TBDMS ether (59.0%). H NMR (CDCL3, 300 MHz) ~ 7.50
(d, 2H, J=8.9), 7.24 (m, 5H), 6.97 (d, 2H, J=8.9), 4.40
(~, lH), 4.21 (d, lH, J=6.3), 4.17 (d, lH, J=6.3), 3.82
(s, 3H), 3.65 (m, 2H), 3.54 (m, lH), 3.35 (m, lH), 3.17
(m, lH), 3.00 (m, 4H), 2.77 (m, lH), 2.21 (m, lH), 1 79
(m, lH), 1.57 (m, 5H), 1.24 (m, lH), 1.03 (m, lH), 0.86
(s, 9H), 0.05 (s, 3H), 0.02 (s, 3H). Low resolution MS
m/e 653.1 (M+Na ), m/e 631.1 (M+H ).
D.
A solution of 0.12g (0.19 mmol, 1 equiv) of the
precursor above in 1.5 mL THF was cooled to -78 C and
added 0.25 mL (0.25 mmol, 1.3 equiv) lithium
bis(trimethylsilyl)amide (lM solution in THF). After 20
min, 0.029 mL (0.248 mmol, 1.3 equiv) of benzyl bromlde
~.
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 287 -
was added and reaction allowed to proceed at room
temperature for additional 1 h. Purification on silica
gel (mixture of diastereomers, Rf=0.46, 0. 51 in 1:3
ethyl acetate/hexane) provided 44 mg (32.2~) of the
TBDMS-protected product. Low resolution MS m/e 1464.6
(2M+Na ), m/e 721.1 (M+H ).
A solution of 40 mg of the silylated product above in
0.3 mL THF was treated with 0.3 mL of lM TBAF in THF
for 25 min at room temperature and purified on a silica
column, resulting in 30 mg of the final product.
Rf=0.38 and 0.34 (2/5/0.3 ethyl acetate: hexane:
methanol). H NMR (CDCL3, 300 MHz) shows both
diastereomers and integrates as expected. Low
resolution MS m/e 629. 3 (M+Na ).
Fx~le 110
Synthesis of Compound 205
O ~ OH
- N~2
OCH3
i
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/~1610
- 288 -
A solution of 0.12g (0.19 mmol, 1 equiv) of ~he
compound prepared in Example lO9C was dissolved ir 1.5
mL THF and was treated with 0.30 mL (0.30 mmol, 1.5
equiv) of lithium bis~trimethylsilyl)amide (lM solution
in THF) at -78 ~C. After 20 min, 0.023 mL (0.266 mmol,
1.4 equiv) of allyl bromide was added, reaction allowed
to warm up to the room temperature and carried out for
additional 1 h. The reaction was then quenched with
aqueous ammonium chloride and both diastereomers
separated on a silica gel. The ~lower) R~=0.50
diastereomer (1:3 ethyl acetate/hexane) was then
treated with 10-fold excess of TBAF (lM in THF) for 25
min at room temperature, followed by another silica
purification, which provided 14 mg of the desired
allylated product. 1H NMR (CDCL3, 300 MHz) ~ 7.73 (d,
2H, J=9.0), 7.24 (m, 5H), 6.99, (d, lH, ~=8.9), 5.84
(m, lH), 5.12 (m, 2H), 4.27 (m, lH), 4.10 (m, lH), 3.86
(s, 3H), 3.84 (m, 3H), 3.58 (m, lH), 2.8-3.3 (m, 7H),
2.62 (m, 2H), 2.09 (m, lH), 1.60 (m, 6H), 1.24 (m, 2H).
Low resolution MS m/e 579.3 (M+Na ), m/e 1135.4
(2M+Na ).
;
.
CA 02243121 1998-07-14
W O 97/27180 PCT~US97/01610
- 289 -
m~le 111
Synthesis of Com~ound 207
~ ~ ~ o~C-~RT
NO2
A solution of 0.092g of the morpholinone described in
Example 20 (0.35 mmol, lequiv) in 1.5 mL anhydrous DMF
was cooled to 0 ~C and added 9.6 mg (0.4 mmol, 1 equiv)
o_ NaH. After 1/2h 0.13g (0.32 mmol) of the epoxide 2
was added and reaction carried out at room temperature
for lO h, quenched with lN HClaq~ and purified on
preparative RP ~PLC. Yield 70 mg (36.5%). Low
resolution MS m/e 622.1 (M+Na ), m/e 1221.1 (2M+Na )
ExAmnle 112
Using the methods described by Pennington et al.
and Partaledis et al. (supra), we obtained inhibition
15 constants for the following compounds of this
invention:
Com~ound Ki (nM)
- 1 160
2* 180
3* 1,800
5* >10,000
CA 02243121 1998-07-14
W 097/27180 PCT~US97/01610
.
- 290 -
6* >10,000
7 9
8~ 5
9~ 90
>10,000
11 >10,000
12 >10,000
13 225
14 16
550
16 56
- 17 115
18 15
19 3,000
1,5
21 >20,000
22 600
23 70
24 350
83
26 58
27 3,000
28 1,400
>15,000
31 390
32 160
33 1,100
34 950
130
36 >20,000
37 >20,000
38 17
CA 02243l2l l998-07-l4
W O 97/27180 PCTrUS97/01610
- 291 -
39 600
>20,000
41 >20,000
42 330
43 >lO,000
44 120
46 >10,000
47 20
50* 100
51* 90
52* 1,100
54* 12
55* 30
56* 280
57* 400
58* 5,800
59* >8,000
60* 170
61* >1,000
62* 120
63* 200
64* >5,000
65* 2,gO0
66* 1,300
67* 3,900
68* >10,000
69* >10,000
70* 790
71* 2,500
- 72* 85
73* 190
CA 02243l2l l998-07-l4
W O 97127180 PCTrUS97/01610
- 292 -
74* 1, 200
76* 250
77~ 560
78* 10
79* >3,000
80* 3
82* 15
83* 0.50
8 5* 2,600
87* 15
88* - 270
90* 220
91* 12
92* ( isomer 1) 3.0
92* (isomer 2) 300
93* 420
95* - 10
96* 4
98* >10,000
102* 1,200
105* >10,000
109* 250
111* >10, 000
112* 8,600
113* >10,000
: 114* ~1,000
115* >10,000
123* (isomer 1) 300
123* (isomer 2) 13
129* (isomer 1) 800
124* (isomer 2) 1900
125* (isomer 1) 400
..
CA 02243121 1998-07-14
W O 97/27180 PCTrUS97/01610
- 293 -
125* (isomer 2) 1000
126* 86
127* . 92
128* 96
129* 400
130* 100
131* (isomer1) 42
131* ( isomer 2) 52
132* 60
133* (isomer1) 24
133* (isomer2) 120
208* 100
209* 2,200
210* 100
211* 5,600
- 212* 5,900
213* 3,100
214* 240
215* 10,000
216* 1,000
217* >10,000
218* 700
219* ~isomer1) 20
219* (isomer2) 54
219* (isomer3) 330
220* 7
221* 50
223* 18
- 224* 90
225* 370
- 226* 29
227* 100
CA 02243121 1998-07-14
W O 97127180 PCTnUS97/01610
- 294 -
228* 16
- 229* . 28
232* 500
233* (isomer 1) 23
233* (isomer 2) 1200
235* 270
236* 3.6
* Inhibition constant measured at pH 6Ø
~D F~x~m~l e 113
Using the MT4 cell assay method (supra),
we measured the antiviral activity for the following
compounds of this invention:
Co ~ ol~nd 15~50 (uM)
26 16
54 1.0
83 0.32
92 (isomer l) 0.21
96 0.40
123 (isomer 1) 0.90
123 (isomer 2) 0.74
127 0 85
130 1.0
131 (isomer 1) 2.4
-
131 (isomer 2~ 2.9
132 0.75
.
CA 02243121 1998-07-14
WO 97/27180 PCTrUS97/01610
- 2g5 -
214 2.1
219 (isomer 1) 0.4
~ 219 (isomer 2) 1.7
219 (isomer 3) 6.0
220 0.10
223 0.68
224 2.0
225 2.0
226 3.5
227 2.75
228 0.48
229 0.79
232 2.47
233 (isomer 1) 3.7
233 (isomer 2) 1.6
~ 236 5 0
The above data show that each of the
tested compounds inhibits HIV aspartyl protease.
~hile we have described a number of
embodiments of this invention, it is apparent that our
basic constructions may be altered to provide other
embodiments which utilize the products and processes of
this invention. Therefore, it will be appreclated that
the scope of this invention is to be defined by the
appended claims, rather than by the specific
embodiments which have been presented by way of
-example.