Note: Descriptions are shown in the official language in which they were submitted.
CA 02244862 2005-04-04
71416-154
1
DESCRIPTION
SUBSTITUTED AMIDE DERIVATIVES
The present invention relates novel substituted
amide derivatives. More particularly, the substituted
amide derivatives of the present invention inhibit
protein-farnesyl transferase (PFT) in vivo thereby to
suppress function of oncogene protein Ras and thus
present antitumor activities, etc., and they are thus
useful in the pharmaceutical field.
The ras oncogene is activated by mutation, and its
translation product Ras protein plays an important role
in transformation of normal cells to cancer cells. Such
activation of ras oncogene is observed in many cancers
such as colorectal cancers or pancreatic cancers, and the
proportion thereof is reported to reach about 20$ of the
total human cancers. Accordingly, it is expected that
canceration can be suppressed and antitumor effects can
be obtained by suppressing such activation of ras
oncogene, by inhibiting the function of Ras protein as
its product.
Recently, it has been found that farnesyl-
modification of Ras protein itself is essential for
function of Ras protein, and it is. possible to suppress
localization of Ras protein at the plasma membrane by
CA 02244862 2005-04-04
71416-154
2
inhibiting this farnesyl-modification and thereby to
inhibit transformation to cancer cells. The protein-
farnesyl transferase (PFT) is an enzyme which catalyses
this farnesyl-modification of Ras protein, and by
inhibiting this enzyme, it is possible to suppress
function of carcinogenic Ras protein. Further, this
enzyme contributes to farnesyl-modification of only very
limited proteins in vivo. Accordingly, the inhibitor for
such an enzyme is expected to be a safe and highly
selective antitumor agent. From such a viewpoint, many
PFT inhibitors have been developed in recent years (Cell,
vol. 57, pp. 1167-1177 (1989); Proc. Natl. Acad. Sci.,
vol. 86, pp. 8323-8327 (1989); ditto, vol. 90, pp. 2281-
2285 (1993); Science, vol. 245, pp. 379-385 (1989);
ditto, vol. 260, pp. 1934-1937 (1993); ditto, vol. 260,
pp. 1937-1942 (1993); J. Biol. Chem., vol. 266, pp.
15575-15578 (1991); J. Antibiotics, vol. 46, pp. 222-227
(1993); Natur Medicine, vol. 1, pp. 792-797 (1995); JP-A-
5-201869; JP-A-5-213992).
Further, it has recently been found by a research by
the present inventors that these PFT inhibitors can block
the reactivation of static viruses by suppressing
development of matured Ras proteins and are useful as
anti-AIDS (HIV) agents (WO 96/17623).
However, up to now, all of the reported PFT
inhibitors have had some problems for development as
medicines, such that the activities are low in cells, and
CA 02244862 1998-07-29
r
t
r
..
the effects in vivo are inadequate_
It is an object of the present invention to provide
a novel antitumor agent or an anti-AIDS agent which
inhibits the protein-farnesyl transferase (PFT) thereby
to inhibit functional manifestation of oncogene protein
Ras and which thus provides antitumor or anti-AIDS
effects .
The present inventors have found that a compound
represented by general formula (I) or a pharmaceutically
acceptable salt or ester thereof:
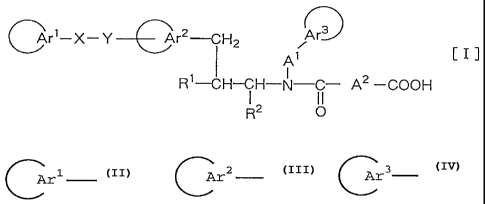
~r~-X-Y---~r2~H2 Ar
A, CIJ
R'-CH- i H-N- j- A2 -COOH
R2 O
[wherein each of
Crl Cr2 and ( Ar3
which may be the same or different, is an aryl group or
an aromatic heterocyclic group which may have
substituent(s) selected from the group consisting of a
halogen atom, a hydroxyl group, an amino group, a nitro
group, a cyano group, a carboxyl group, a lower alkyl
group, a lower alkenyl group, a lower alkoxy group, a
lowerhydroxyalkyl group, a lower fluoroalkyl group, a
lower alkoxycarbonyl group, a carbamoyl group and a lower
CA 02244862 1998-07-29
i
r
4
alkylcarbamoyl group; A1 is a C1_6 chain hydrocarbon group
which may have substituent(s) selected from the group
consisting of a halogen atom, a lower alkyl group, an oxo
group, a lower hydroxyalkyl group and a lower alkoxy
group or a-group represented by -A'-a-Wl-Alb- (wherein Ala is
a Cs_5 chain hydrocarbon group which may have
substituent(s) selected from the group consisting of a
halogen atom, a lower alkyl group, an oxo group, a lower
hydroxyalkyl group and a lower alkoxy group; Alb is a
single bond or a C1_4 chain hydrocarbon group which may
have substituent(s) selected from the group consisting of
a halogen atom, a lower alkyl group, an oxo group, a
lower hydroxyalkyl group and a lower alkoxy group; W1 is
an oxygen atom, a sulfur atom, an ethynylene group, a
cyclopropylene group or a group represented by -NRW-; and
RW is a hydrogen atom or a lower alkyl group) ; Aa is a C1_$
chain hydrocarbon group which may have substituent(s)
selected from the group consisting of a halogen atom, a
lower alkyl group, a hydroxyl group, a lower hydroxyalkyl
group, a lower alkoxy group, a carboxyl group, a lower
alkoxycarbonyl group, a lower alkenyloxycarbonyl group, a
lower carboxyalkyl group, an aryl group and an aralkyl
group; R1 is a lower alkyl group or a lower alkenyl group
which may have substituent(s) selected from the group
consisting of a hydroxyl group, a lower alkoxy group, a
lower alkoxycarbonyl group, a carbamoyl group, a lower
alkylcarbamoyl group and a lower alkylhydrazinocarbonyl
CA 02244862 1998-07-29
i
r
group, a lower alkoxy group, a carboxyl group, a lower
alkoxycarbonyl group, a carbamoyl group, alower
alkylcarbamoyl group, a group represented by RaC00-
(wherein Ra is a hydrogen atom, an amino group, a lower
5 alkyl group, a lower alkoxy group or a lower alkylamino
group) or by RbCONR'- (wherein Rb is a hydrogen atom, an
amino group, a lower alkyl group, a lower alkoxy group or
a lower alkylamino group; and Rc is a hydrogen atom or a
lower alkyl group) or an alicyclic group which may have
substituent(s) selected from the group consisting of a
halogen atom, a hydroxyl group, an amino group, a nitro
group, a cyano group, a carboxyl group, a lower alkyl
group, a lower alkenyl group, a-lower alkoxy group, a
lower hydroxyalkyl group, a lower fluoroalkyl group, a
lower alkoxycarbonyl group, a carbamoyl group and a lower
alkylcarbamoyl group and may have one or two oxygen atoms
and/or one or two nitrogen atoms; R~ is a lower alkyl
group; and each of X and Y which may be the same or
different, is an oxygen atom, a sulfur atom, a carbonyl
group, a group represented by -CHRd- (wherein Rd is a
hydrogen atom or a lower alkyl group) or by -NRe-
(wherein Re is a hydrogen atom or a lower alkyl group),
or X and Y together represent a vinylene group or an
ethynylene group, provided that when one of X and Y is an
oxygen atom, a sulfur atom or a group represented by
-NRe- (wherein RC is the same as defined above), the
other is a carbonyl group or a group represented by
CA 02244862 1998-07-29
6
-CHRd- (wherein Rd is the same defined above)], inhibits
the protein-farnesyl transferase (PFT) thereby to
suppress function of oncogene protein Ras, and thus is
useful as an antitumor agent or an anti-AIDS agent. The
present invention has been accomplished on the basis of
this discovery.
Thus, the present invention relates to a compound of
the formula (I), or its pharmaceutically acceptable salt
or ester, as well as its application.
Symbols and terms used in this specification will be
explained.
The aryl group means a phenyl group, a naphthyl
group or an anthryl group. A phenyl group or a naphthyl
group is preferred.
The aromatic heterocyclic group means a 5-membered
or 6-membered monocyclic aromatic heterocyclic group
containing one or two hetero atoms, which are the same or
different, selected from the group consisting of an
oxygen atom, a nitrogen atom and a sulfur atom, or a
fused aromatic heterocyclic group having such a
monocyclic aromatic heterocyclic group fused with the
above-mentioned aryl group or having the same or
different such monocyclic aromatic heterocyclic groups
fused with each other, which may, for example, be a
pyrrolyl group, an imidazolyl group, a pyrazolyl group, a
pyridyl group, a pyrazinyl gro-up, a pyrimidinyl group, a
pyridazinyl group, an oxazolyl group, an isoxazolyl
CA 02244862 1998-07-29
s
7
group, a furyl group, a thienyl group, a thiazolyl group,
an isothiazolyl group, an indolyl group, a benzofuranyl
group, a benzothienyl group, a benzimidazolyl group, a
benzoxazolyl group, a benzisoxazolyl group, a
benzothiazolyl group, a benzisothiazolyl group, an
indazolyl group, a purinyl group, a quinolyl group, an
isoquinolyl group, a phthalazinyl group, a naphthylidinyl
group, a quinoxalinyl group, a quinazolinyl group, a
cinnolinyl group or a pteridinyl group. Among them, a
furyl group, a thienyl group, a pyridyl group, a
pyrimidinyl group, an oxazolyl group, an isoxazolyl
group, a thiazolyl group, a benzofuranyl group, a
benzothienyl group, a benzimidazolyl group, a
benzoxazolyl group, a benzothiazolyl group or a quinolyl
group is preferred_
The halogen atom may be a fluorine atom, a chlorine
atom, a bromine atom or an iodine atom. For example, a
fluorine atom or a chlorine atom is preferred.
The lower alkyl group means a C1_6 linear or branched
alkyl group, which may, for example, be a methyl group,
an ethyl group, a propyl group, an isopropyl group, a
butyl group; -a--sec-butyl.- group;- a ter t-biltyi group-; -a -
pentyl group or a hexyl group. Among them, a methyl
group or an ethyl group is preferred.
The lower alkenyl group means a Ca_6 linear or
branched alkenyl group, such as a vinyl group, anallyl
group, an isopropenyl group, a 3-butenyl group, a 2-
CA 02244862 1998-07-29
_ ,
8
butenyl group, a 1-butenyl group, a 1-methyl-2-propenyl
group, a 1-methyl-1-propenyl group, a 1-ethyl-1-ethenyl
group,a 2-methyl-2-propenyl group, a 2-methyl-1-propenyl
group, or a 4-pentenyl group.
The lower alkoxy group means a C1_6 alkoxy or
alkylenedioxy group, which may, for example, be a methoxy
group, an ethoxy group, a propoxy group, an isopropoxy
group, a butoxy group, a tent-butoxy group, a
methylenedioxy group, an ethylenedioxy group or a
trimethylenedioxy group. Among them, a methoxy group, an
ethoxy group or a methylenedioxy group is preferred.
The lower hydroxyalkyl group means the above
mentioned lower alkyl group having a hydroxyl group, i.e.
a C1_6 hydroxyalkyl group, such as a hydroxymethyl group,
a hydroxyethyl group, a hydroxypropyl group or a
hydroxybutyl group. Among them, a hydroxymethyl group or
a hydroxyethyl group is preferred.
The lower fluoroalkyl group means the above-
mentioned lower alkyl group having fluorine atom(s), i.e.
a C1_6 fluoroalkyl group, such as a fluoromethyl group, a
difluoromethyl group, a trifluoromethyl group, a 1-
fluoroethyl group, a 2-fluoroethyl group, a 2,2,2-
trifluoroethyl group or a pentafluoroethyl group.
The lower alkoxycarbonyl group means a Cl_~
alkoxycarbonyl group, such as a methoxycarbonyl group, an
ethoxycarbonyl group, a propoxycarbonyl group, a
butoxycarbonyl group, a pentoxycarbonyl group, an
CA 02244862 1998-07-29
x
K
9
isopropoxycarbonyl group, an isobutoxycarbonyl group, a
tert-butoxycarbonyl group, an isopentoxycarbonyl group, a
neopentoxycarbonyl group, cyclopropoxycarbonyl group, a
cyclobutoxycarbonyl group, a cyclopentoxycarbonyl group,
a cyclohexyloxycarbonyl group, a 1-
methylcyclopropoxycarbonyl group, a 1-
methylcyclopentoxycarbonyl group, a 1-
methylcyclobutoxycarbonyl group, a 2-
methylcyclobutoxycarbonyl group, a 1,1-
dimethylpropoxycarbonyl group, a 1,2-
dimethylpropoxycarbonyl group, a 1-ethylpropoxycarbonyl
group, a 1,2-dimethylcyclopropoxycarbonyl group, a 2,2- _
dimethylcyclopropoxycarbonyl group, a 2,3-
dimethylcyclopropoxycarbonyl group, a 1,2-
dimethylcyclobutoxycarbonyl group, a 1,3-
dimethylcyclobutoxycarbonyl group, a 2,2-
dimethylcyclobutoxycarbonyl group, a 2,3-
dimethylcyclobutoxycarbonyl group, a 3,3-
dimethylcyclo-butoxycarbonyl group, a 2,4-
dimethylcyclobutoxycarbonyl group, a 2,3-
dimethylcyclopentoxycarbonyl group, a 2,4-
dimethylcyclopentoxycarbonyl group or a 2,5-
dimethylcyclopentoxycarbonyl group. Among them, a
methoxycarbonyl group, an ethoxycarbonyl group, a
propoxycarbonyl group, an isopropoxycarbonyl group, a
tent-butoxycarbonyl group, a cyclopropoxycarbonyl group,
a cyclobutoxycarbonyl group, a cyclopentoxycarbonyl
CA 02244862 1998-07-29
f
group, a 1-methylcyclopropoxycarbonyl group, a 1-
methylcyclopento-xycarbonyl group, a 1-
methylcyclobutoxycarbonyl group, a 1,1-
dimethylpropoxycarbonyl group or a 1,2-
5 dimethylpropoxycarbonyl group is preferred_
The lower alkylcarbamoyl group means a carbamoyl
group mono-substituted or di-substituted by the above-
mentioned lower alkyl group, such as a methylcarbamoyl
group, an ethylcarbamoyl group, a dimethylcarbamoyl group
10 or a diethylcarbamoyl group.
The chain hydrocarbon group means a linear saturated
aliphatic hydrocarbon group, or a linear unsaturated
aliphatic hydrocarbon group having one or more,
preferably one or two double bonds, at optional positions
on the carbon chain.
The saturated aliphatic hydrocarbon group may, for
example, be a methylene group, an ethylene group, a
trimethylene group, a tetramethylene group, a
pentamethylene group, a hexamethylene group,a
heptamethylene group or an octamethylene group.
The unsaturated aliphatic hydrocarbon group may, for
example, be a vinylene group, a propenylene group, a 1-
butenylene group, a 2-butenylene group, a 1,3-
butadienylene group, a 1-pentenylene group, a 2-
pentenylene group, a 1,3-pentadienylene group, a 1,4-
pentadienylene group, a 1-hexenylene group, a 2-
hexenylene group, a 3-hexenylene group, a 1,3-
CA 02244862 1998-07-29
y ' .
11
hexadienylene group, a 1,4-hexadienylene group, a 1,5-
hexadienylene group, a 1,3,5-hexatrienylene group, a 1-
heptenylene group, a 2-heptenylene group, a 3-heptenylene
group, a 1,3-heptadienylene group, a 1,4-heptadienylene
group, a 1,5-heptadienylene group, a 1,6-heptadienylene
group, a 1,3,5-heptatrienylene group, a 1-octenylene
group, a 2-octenylene group, a 3-octenylene-group, a 4-
octenylene group, a 1,3-octadienylene group, a 1,4-
octadienylene group, a 1,5-octadienylene group, a 1,6-
octadienylene group, a 1,7-octadienylene group, a 2,4-
octadienylene group, a 2,5-octadienylene group, a 2,6-
octadienylene group, a 3,5-octadienylene group, a 1,3,5-
octatrienylene group, a 2,4,6-octatrienylene group or a
1,3,5,7-octatetraenylene group.
The lower alkenyloxycarbonyl group means a C3_~
alkenyloxycarbonyl group, such as an allyloxycarbonyl
group, a 2-butenyloxycarbonyl group, a 1-methyl-2-
propenyloxycarbonyl group, a 3-methyl-2-
butenyloxycarbonyl group, a 1-cyclobutenyloxycarbonyl
group, a 2-cyclobutenyloxycarbonyl group, a 1-
cyclopentenyloxycarbonyl group or a 2-
cyclopentenyloxycarbonyl group. Among them, an
allyloxycarbonyl group or a 1-methyl-2-
propenyloxycarbonyl group is preferred.
The lower carboxyalkyl group means the above-
mentioned lower alkyl group having a carboxyl group, i.e.
a Ci_~ carboxyalkyl group, such as a carboxymethyl group,
CA 02244862 1998-07-29
' .
12
a carboxyethyl group, a carboxypropyl group or a
carboxybutyl group. Among them, a carboxymethyl group or
a carboxyethyl group is preferred.
The aralkyl group means the above-mentioned lower
alkyl group having the above-mentioned aryl group, such
as a benzyl group, a phenethyl group, a 3-phenylpropyl
group, a 1-naphthylmethyl group, a 2-naphthylmethyl group
or a 1-(2-naphthyl)ethyl group. Among them, a benzyl
group, a phenethyl group or a 2-naphthylmethyl group is
preferred.
The lower alkylamino group means an amino group
mono-substituted or di-substituted by the above-mentioned
lower alkyl group, such as a methylamino group, an
ethylamino group, a dimethylamino group or a diethylamino
group.
The lower alkylhydrazinocarbonyl group means a
hydrazinocarbonyl group mono-substituted or di-
substituted by the above-mentioned lower alkyl group,
such as a methylhydrazinocarbonyl group, an
ethylhydrazinocarbonyl group, a dimethylhydrazinocarbonyl
group or a diethylhydrazinocarbonyl group.
The alicyclic group which may contain one or two
oxygen atoms and/or one or two nitrogen atoms, means a 3-
membered to 7-membered saturated or unsaturated aliphatic
carbocyclic group, or a 3-membered to 7-membered
saturated or unsaturated aliphatic oxygen-containing
heterocyclic group which contains one or two oxygen atoms
CA 02244862 1998-07-29
' .
13
and/or one or two nitrogen atoms, such as a cyclopropyl
group, a cyclobutyl group, a cyclopentyl group, a
cyclohexyl group, a cycloheptyl group, _an oxiranyl group,
an oxetanyl group, an oxolanyl group, an oxanyl group, an
oxepanyl group, a 1,3-dioxetanyl group, a 1,3-dioxolanyl
group, a 1,3-dioxanyl group, a 1,4-dioxolanyl group, a
1,3-dioxepanyl group, a 1,4-dioxepanyl group, a
cyclobutenyl group, a cyclopentenyl group, a cyclohexenyl
group, a cycloheptenyl group, an oxirenyl group, an
oxetyl group, a 2,3-dihydrofuranyl group, a 2,5-
dihydrofuranyl group, 3,4-dihydropyranyl group, a 5,6-
dihydropyranyl group, a 2,3-dihydrooxepinyl group, a 4,5-
dihydrooxepinyl group, a 2,5-dihydrooxepinyl group, a
cyclopentadienyl group, a 1,3-cyclohexadienyl group, a
1,4-cyclohexadienyl group, a 2H-pyranyl group, a 4H-
pyranyl group, a 2,3,4,5-tetrahydrooxepinyl group, a
2,3,4,7-tetrahydrooxepinyl group, 2,3,6,7-
tetrahydrooxepinyl group, a 1,3-dioxorenyl group, a 1,3-
dioxinyl group, a 1,4-dioxinyl group, a dihydro-1,4-
dioxinyl group, a 6,7-dihydro-1,3-dioxepinyl group-, a
4,7-dihydro-1,3-dioxepinyl group, a 5,6-dihydro-1,4-
dioxepinyl group, a 2,3-dihydro-1,4-dioxepinyl group, a
1,3-dioxepinyl group, a 1,4-dioxepinyl group, an
aziridinyl group, an azetidinyl group, an azolidinyl
group, a perhydroazinyl group, a 1,3-diazolidinyl group,
a perhydro-1,3-diazinyl group, a perhydro-1,4-diazinyl
group, a 3,4-dihydro-2H-azolyl group, a 2,3-dihydro-1H-
CA 02244862 1998-07-29
14
azolyl group, a 3,4,5,6-tetrahydro-1,3-diazinyl group, a
1,2,5,6-tetrahydro-1,3-diazinyl group, a 1,2,3,4-
tetrahydro-1,4-diazinyl group, a 1,4-dihydro-1,4-diazinyl
group, a-1,3-oxazolidinyl group, a 4,5-dihydro-1,3-
oxazolyl group, a perhydro-1,3-oxazinyl group, a
perhydro-1,4-oxazinyl group, a 4,5-dihydro-6H-1,3-
oxazinyl group, a 5,6-dihydro-2H-1,3-oxazinyl group, a
3,4-dihydro-2H-1,3-oxazinyl group, a 2,3-dihydro-6H-1,4-
oxazinyl group or a 5,6-dihydro-4H-1,4-oxazinyl group-
The salt of the compound of the formula (I) may be a
pharmaceutically acceptable common salt, which may, for
example, be a base-addition salt of a carboxyl group, or
an acid-addition salt of an amino group when the compound
has such an amino group, or of a basic heteroaromatic
ring when the compound has such a basic aromatic
heterocyclic ring-
The base-addition salt may, for example, be an
alkali metal salt such as a sodium salt or a potassium
salt; an alkaline earth metal salt such as a calcium salt
or a magnesium salt; an ammonium salt; or an organic
amine salt such as a trimethylamine salt, a triethylamine
salt, a dicyclohexylamine salt, an ethanolamine salt, a
diethanolamine salt, a triethanolamine salt, a procaine
salt or an N,N'-dibenzylethylenediamine salt.
The acid-addition salt may, for example, be an
inorganic acid salt such-as a hydrochloride, a sulfate, a
nitrate, a phosphate or a perchlorate; an organic acid
CA 02244862 1998-07-29
salt such as a maleate, a fumarate, a tartrate, a
citrate, an ascorbate or a trifluoroacetate; or a
sulfonate such as a methanesulfonate, anisethionate, a
benzenesulfonate or a p-toluenesulfonate_
5 The ester of the compound of the formula ~I) means a
pharmaceutically acceptable common ester of a carboxyl
group adjacent toA2, of a carboxyl group if such a
carboxyl group or a lower carboxyalkyl group is present
on the chain hydrocarbon group as Aa, of a carboxyl group
10 as R1, or of a carboxyl group if such a carboxy group.is
present on the group represented by
Crl Cr2 or ( Ar3 . _
It may, for example, be an ester with a lower alkyl group
such as a methyl group, an ethyl group, a propyl group,
an isopropyl group, a butyl group, a sec-butyl group, a
15 tent-butyl group, a pentyl group, an isopentyl group, a
neopentyl group, a cyclopropyl group, a cyclobutyl group
or a cyclopentyl group, an ester with an aralkyl group
such as a benzyl group or a phenethyl group, an ester
with a lower alkenyl group such as an allyl group or a 2-
butenyl group, an ester with a lower alkoxyalkyl group
such as a methoxymethyl group, a 2-methoxyethyl group or
a 2-ethoxyethyl group, an ester with a lower
alkanoyloxyalkyl group such as an acetoxymethyl group, a
pivaloyloxymethyl group or a 1-pivaloyloxyethyl_group, an
ester with a lower alkoxycarbonylalkyl group such as a
CA 02244862 1998-07-29
16
methoxycarbonylmethyl group or an
isopropoxycarbonylmethyl group, an ester-with a lower
carboxyalkyl group such as a carboxymethyl group, an
ester with a lower alkoxycarbonyloxyalkyl group such as a
1-(ethoxycarbonyloxy)ethyl group or a 1-
(cyclohexyloxycarbonyloxy)ethyl group, an ester with a
lower carbamoyloxyalkyl group such as a
carbamoyloxymethyl group, an ester with a phthalidyl
group, or an ester with a (5-substituted-2-oxo-1,3-
dioxol-4-yl)methyl group such as a (5-methyl-2-oxo-1,3-
dioxol-4-yl)methyl group.
Further, when a hydroxyl group is present at the y-
or 8-position of the terminal carboxyl group or of a
carboxyl group when such a carboxyl group or a lower
carboxylalkyl group is present on the chain hydrocarbon
group represented by A2, such a hydroxyl group and a
carboxyl group may form an intramolecular ester i.e. a 5-
membered or 6-membered lactone ring_
Further, the compound of the present invention may
have stereoisomers such as optical isomers, diastereomers
or geometrical isomers, depending upon the form of its
substituents_ The compound of the present invention
includes all of such stereoisomers and their mixtures.
Among them, a compound represented by general formula (I'
-1).
CA 02244862 1998-07-29
17
Are-X-Y Ar2-CH2 H Ar3 ,
C ~ - i [I - I)
R~~-C N--C- A2 -COOH
~.._.:'-.,, O
R2 H
or general formula -(I'-2).
Are-X-Y Ar2-CH2 H Ar3 ,
~ I -2
~.:A~ [ )
R~~~ N-C- A2 -COOH
,,.s::.~ O
R2 H
[wherein
r1 C r2 r3
, ,
Al , A' , Rl , R? , x and y are as de f fined above ] i s
preferred.
Each of
r1 r2 and Ars
which may be the same or different, means an aryl group
or an aromatic heterocyclic group which may have
substituent(s) selected from the group consisting of a
halogen atom, a hydroxyl group, an amino group, a nitro
group, a cyano group, a carboxyl group, a lower alkyl
group, a lower alkenyl group, a lower alkoxy group, a
lower hydroxyalkyl group, a lower fluoroalkyl group, a
lower alkoxycarbonyl group, a carbamoyl group and a lower
CA 02244862 1998-07-29
18
alkylcarbamoyl group.
The aryl group or the aromatic heterocyclic group
which may have substituent(s) selected from the group
consisting of a halogen atom, a hydroxyl group, an amino
group, a vitro group, a cyano group, a carboxyl group, a
lower alkyl group, a lower alkenyl group, a lower alkoxy
group, a lower hydroxyalkyl group, a lower fluoroalkyl
group, a lower alkoxycarbonyl group, a carbamoyl group
and a lower alkylcarbamoyl group, means the above-
mentioned aryl group or the above-mentioned aromatic
heterocyclic group which is unsubstituted, or the above-
mentioned aryl group or the above-mentioned aromatic
heterocyclic group which has substituent(s) at optional
positions) for substitution, and said substituent(s) may
be one or more, preferably one or two, which may be the
same or different and which are selected from the group
consisting of a halogen atom, a hydroxyl group, an amino
group, a vitro group, a cyano group, a carboxyl group, a
lower alkyl group, a lower alkenyl group, a lower alkoxy
group, a lower hydroxyalkyl group, a lower fluoroalkyl
group, a lower alkoxycarbonyl group, a carbamoyl group
and a lower alkylcarbamoyl group_
Among the compounds represented by general formula
(I), compounds wherein
r1
is a phenyl group or a thienyl group, compounds wherein
CA 02244862 1998-07-29
19
Ar2
is a phenyl group, a thienyl group, a furyl group, a
thiazolyl group, an oxazolyl group, an isoxazolyl group,
a pyridyl group or a pyrimidinyl group and compounds
wherein
r3
is a phenyl group, a thienyl group, a naphthyl group, a
benzothienyl group or a benzofuranyl group, are
preferred.
A1 is a Cl_$ chain hydrocarbon group which may have
substituent(s) selected from the group consisting of a
halogen atom, a lower alkyl group, an oxo group, a lower
hydroxyalkyl group and a lower alkoxy group or a group
represented by -Ala-W -Alb- (wherein Ala is a C1_5 chain
hydrocarbon group which may have substituent(s) selected
from the group consisting of a halogen atom, a lower
alkyl group, an oxo group, a lower hydroxyalkyl group and
a lower alkoxy group; Alb is a single bond or a Cl_4 chain
hydrocarbon group which may have substituent(s) selected
from the group consisting of a halogen atom, a lower
alkyl group, an oxo group, a lower hydroxyalkyl group and
a lower alkoxy group; W'~ is an oxygen atom, a sulfur
atom, an ethynylene group, a cyclopropylene group or a
group represented by -NRw-; and RW is a hydrogen atom or
a lower alkyl group).
The Ci_~ chain hydrocarbon group which may have
CA 02244862 1998-07-29
substituent(s) selected from the group consisting of a
halogen atom, a lower alkyl group, an oxo group, a lower
hydroxyalkyl group and a lower alkoxy group, means the
above-mentioned chain hydrocarbon group having from 1 to
5 6 carbon atoms, which is unsubstituted, or the above-
mentioned chain hydrocarbon group having from 1 to 6
carbon atoms, which has substituent(s) at optional
positions) for substitution, and said substituent(s) may
be one or more, preferably from one to three, which are
10 the-same or different and which are selected from the
group consisting-of a halogen atom, a lower alkyl group,
an oxo group, a lower hydroxyalkyl group and a lower
alkoxy group.
Ala means a C1_5 chain hydrocarbon group which may have
15 substituent(s) selected from the group consisting of a
halogen atom, a lower alkyl group, an oxo group, a lower
hydroxyalkyl group anda lower alkoxy group_
The Cl_5 chain hydrocarbon group which may have
substituent(s) selected from the group consisting of a
20 halogen atom, a lower alkyl group, an oxo group, a lower
hydroxyalkyl group and a lower alkoxy group, means the
above-mentioned chain hydrocarbon group having from 1 to
5 carbon atoms, which is unsubstituted, or the above-
mentioned chain hydrocarbon group having from 1 to 5
carbon atoms, which has substituent(s) at optional
positions) for substitution, and the substituent(s) may
be one or more, preferably from one to three, which are
CA 02244862 1998-07-29
t " .
21
the same ordifferent and which are selected from the
group consisting of a halogen atom, a lower alkyl group,
an oxo group, a lower hydroxyalkyl group and a lower
alkoxy group_
A~ is a single bond or a Cl_4 chain hydrocarbon group
' which may have substituent(s) selected from the group
consisting of a halogen atom, a lower alkyl group, an oxo
group, a lower hydroxyalkyl group and a lower alkoxy
group.
The CI_4 chain hydrocarbon group which may have
substituent(s) selected from the group consisting of a
halogen atom, a lower alkyl group, an oxo group, a lower
hydroxyalkyl group and a lower alkoxy group, means the
above-mentioned chain hydrocarbon group having from 1 to
4 carbon atoms, which is unsubstituted, or the above-
mentioned chain hydrocarbon group having from 1 to 4
carbon-atoms, which has substituent(s) at optional
positions) for substitution, and the substituent(s) may
be one or more, preferably from 1 to 3, which are the
same or different and which are selected from the group
consisting of a halogen atom, a lower alkyl group, an oxo
group, a lower hydroxyalkyl group and a lower alkoxy
group.
W1 is an oxygen atom, a sulfur atom, an ethynylene
group, a cyclopropylene group or a group represented by
-NR"-, and preferably an oxygen atom, an ethynylene group
or a group represented by -NRw-.
CA 02244862 1998-07-29
22
Rw is a hydrogen atom or a lower alkyl group, and
preferably a hydrogen atom.
As A1, for example, a group represented by -CHa-,
-CHaCHaCHa- , -CHzCH=CH- , -CHZCH20- or -CHIC = C- is
preferred_ _
Aa means a C1_8 chain hydrocarbon group which may have
substituent(s) selected from the group consisting of a
halogen atom, a lower alkyl group, a hydroxyl group, a
lower hydroxyalkyl group, a lower alkoxy group, a
carboxyl group, a lower alkoxycarbonyl group, a lower
alkenyloxycarbonyl group, a lower carboxyalkyl group, an
aryl group and an aralkyl group_
The C1_$ chain hydrocarbon group which may have
substituent(s) selected from the group consisting of a
halogen atom, a lower alkyl group, a hydroxyl group, a
lower hydroxyalkyl group, a lower alkoxy group, a
carboxyl group, a lower alkoxycarbonyl group, a lower
alkenyloxycarbonyl group, a lower carboxyalkyl group, an
aryl group and an aralkyl group, means the above-
mentioned chain hydrocarbon group having from 1 to 8
carbon atoms, which is unsubstituted, or the above-
mentioned chain hydrocarbon group having from 1 to 8
carbon atoms, which has substituent(s) at optional
positions) for substitution, and the substituent(s) may
be one or more, preferably from 1 to 3, which are the
same or different and which are selected from the group
consisting of a halogen atom, a lower alkyl group, a
CA 02244862 1998-07-29
23
hydroxyl group, a lower hydroxyalkyl group, a lower
alkoxy group, a carboxyl group, a lower alkoxycarbonyl
group, a lower alkenyloxycarbonyl group, a lower
carboxyalkyl group, an aryl group-and an aralkyl group.
Compounds wherein AZ is a group represented by
formula (a):
R4
(CH2)m i H- C- CH -(CH2)~ [a]
R3 R5 Rs
(wherein R3 is a hydrogen atom, a hydroxyl group, a lower
hydroxyalkyl group, a lower alkoxy group or a carboxyl
group; R4 is a hydrogen atom, a halogenatom, a hydroxyl
group, a lower alkoxy group, a carboxyl group or a lower
carboxyalkyl group; RS is a hydrogen atom, a lower
hydroxyalkyl group, a carboxyl group, a lower
alkoxycarbonyl group or a lower alkenyloxycarbonyl group;
R6 is a hydrogen atom, a halogen atom, a hydroxyl group
or a carboxyl group; and each of m and n which may be the
same or different is an integer of from 0 to 2) and
compounds wherein A2 is a group represented by formula
(b)
OH
I
- (CH2) p C = j - (CH2) q- i H- (CH2) ~ -
Rs Rs
(wherein RS is a hydrogen atom, a lower hydroxyalkyl
group, a carboxyl group, a lower alkoxycarbonyl group or
a lower alkenyloxycarbonyl group; R6 is a hydrogen atom,
CA 02244862 1998-07-29
24
a hydroxyl group or a carboxyl group;p is 0 or 1; and
each of q and r which are the same or different is an
integer of from 0 to 2) are preferred.
When AZis represented by formula (a), R3 is
preferably a hydrogen atom, ahydroxyl group or a
carboxyl group, R4 is preferably a carboxyl group or a
lower carboxyalkyl group such as a carboxymethyl group,
RS and R6 are preferably hydrogen atoms or carboxyl
groups, and each of m and n which are the same or-
different is preferably 0 or 1_
When AZ is represented by formula (b), RS is a lower
hydroxyalkyl group such_as a hydroxymethyl group, a
carboxyl group, a lower alkoxycarbonyl group such as an
ethoxycarbonyl group or a lower alkenyloxycarbonyl group
such as an allyloxycarbonyl group, R6 is preferably a
hydrogen atom, and p, q and r are preferably 0_
Further, it is well known that in the case of a
compound having a partial structure represented by
formula (b), there exist enol form and keto form
tautomers, as shown below. The compound of the present
invention includes such enol form and keto form isomers
and their mixtures.
CA 02244862 1998-07-29
OH
I
(CHz) p C = C- (CH2) q-CH- (CHZ) ~ - [b]
R5 Rs
(Enol form)
5
O
I I
- (CH2) p C - CH- (CH2) q-CH- (CH2) r - [b'~
R5 Rs
(Keto form)
10 (wherein R5, R6, p, q and r are thesame as defined
above ) .
R1 is a lower alkyl group or a lower alkenyl group
which may have substituent(s) selected from the group
consisting of a hydroxyl group, a lower alkoxy group, a
15 lower alkoxycarbonyl group, a carbamoyl group, a lower
alkylcarbamoyl group and a lower alkylhydrazinocarbonyl
group, a lower alkoxy group, a carboxyl group, a lower
alkoxycarbonyl group, a carbamoyl group, a lower
alkylcarbamoyl group, a group represented by RaC00-
20 (wherein Ra is a hydrogen atom, an amino group, a lower
alkyl group, a lower alkoxy group or a lower alkylamino
group) or by RbCONRc- (wherein Rb is a hydrogen atom, an
amino group, a lower alkyl group, a lower alkoxy group or
a lower alkylamino group; and R' is a hydrogen atom or a
25 lower alkyl group) oran alicyclic group which may have
substituent(s) selected from the group consisting of a
halogen atom, a hydroxyl group, an amino group, a nitro
CA 02244862 1998-07-29
26
group, a cyano group, a carboxyl group, a lower alkyl
group, a lower alkenyl group, a lower alkoxy group, a
lower hydroxyalkyl group, a lower fluoroalkyl group, a
lower alkoxycarbonyl group, a carbamoyl group and a lower
alkylcarbamoyl group and may have one or two oxygen atoms
and/or one or two nitrogen atoms-
The lower alkyl group or the lower alkenyl group
which may have substituent(s) selected from the group
consisting of a hydroxyl group, a lower alkoxy group, a
lower alkoxycarbonyl group, a carbamoyl group and a lower
alkylcarbamoyl group means the above-mentioned lower
alkyl group or the above-mentioned lower alkenyl group,
which is unsubstituted, or the above-mentioned lower
alkyl group or the above-mentioned lower alkenyl group,
which has substituent(s) at optional positions) for
substitution, and said substituent(s) may be one or more,
preferably one or two, which are the same or different,
and which are.selected from the group consisting of a
hydroxyl group, a lower alkoxy group,a lower
alkoxycarbonyl group, a carbamoyl group and a lower
alkylcarbamoyl group-
The alicyclic group which may have substituent(s)
selected from the group consisting of a halogen atom, a
hydroxyl group, an amino group, a nitro group, a cyano
group, a carboxyl group, a lower alkyl group, a lower
alkenyl group, a -lower alkoxy group, a lower hydroxyalkyl
group, a lower fluoroalkyl group, alower alkoxycarbonyl
CA 02244862 1998-07-29
27
group, a carbamoyl group and a lower alkylcarbamoyl group
and may have one or two oxygen atoms and/or one or two
nitrogen atoms means the above-mentioned alicyclic group
which may have one or two oxygen atoms and/orone or two
nitrogen atoms and is unsubstituted, or the above-
mentioned alicyclic group which may-have one or two
oxygen atoms and/or one or two nitrogen atoms and has
substituent(s) at optional positions) for substation,
and said substituent(s) may be one or more, preferably
one or two which are the same or different and which are
selected from the group consisting of a halogen atom, a
hydroxyl group, an amino group, a nitro group, a cyano
group, a carboxyl group, a lower alkyl group, a lower
alkenyl group, a lower alkoxy group, a-lower hydroxyalkyl
group, a lower fluoroalkyl group, a lower alkoxycarbonyl
group, a carbamoyl group and a lower alkylcarbamoyl_
As R1, a lower alkoxycarbonyl group such as an
ethoxycarbonyl group is preferred.
Ra means a lower alkyl group, but a methyl group, an
ethyl group or a propyl group is preferred.
Particularly, a methyl group or an ethyl group is
preferred.
The group represented by
1
Cr -X -Y -
can be present as a substituent at any optional position
for substitution on the group represented by the formula
CA 02244862 1998-07-29
28
r2
Each of X and Y which are the same or different, is
an oxygen atom, a sulfur atom, a carbonyl group, a group
represented by -CHRd- (wherein Rd is a hydrogen atom or a
lower alkyl group) or by -NRe- (wherein Re a hydrogen
atom or a lower alkyl group), or X and Y together.
represent a vinylene group or an ethynylene group. When
one of X and Y is an oxygen atom, a sulfur atom or a
group represented by -NRe-.(wherein Re is the same as
defined above), the other is a carbonyl group or a group
represented by -CHRd- (wherein Rd is the same as defined
above ) _
Among the compounds represented by general formula
(I), compounds wherein X is a group represented by -NRe-
(wherein Re is the same as defined above), and Y is a
carbonyl group, compounds wherein X is an oxygen atom,
and Y is a group represented by -CHRd- (wherein Rd is the
same as defined above), compounds wherein X is a group
represented by -CHRd- (wherein Rd is the same as defined
above), and Y is an oxygen atom, compounds wherein both X
and Y are groups represented by -CHRa- (whereinRd is the
same as defined above) or compounds wherein X and Y
together represent a vinylene group are preferred.
Now, processes for producing the compound of the
present invention will be described.
The compound represented by general formula (I) of
CA 02244862 1998-07-29
29
the present invention can be prepared, for example, by
the following process 1, 2, 3, 4, 5, 6 or 7.
A compound represented by general formula (I) can be
prepared by reacting a compound represented by general
formula (II):
Are p-X°-Yp Ar2p-CH '4r3p
2 /
Atp
[II]
R' °-CH-CH-NH
R2
[wherein each of
Arl~' Ar2p and ~r3~
which are the same or different, is an aryl group or an
aromatic heterocyclic group which may have substituent(s)
selected from the group consisting of a halogen atom, a
nitro group, a cyano group, a lower alkyl group, a lower
alkenyl group, a lower alkoxy group, a lower fluoroalkyl
group, a lower alkoxycarbonyl group, a carbamoyl group
and a lower alkylcarbamoyl group, and a hydroxyl group,
an amino group, a carboxyl group and a lower hydroxyalkyl
group which may be protected; Alp is a C~_s chain
hydrocarbon group which may have substituent(s) selected
from the group consisting of a halogen atom, a lower
alkyl group, an oxo group, a lower hydroxyalkyl group
which may be protected and a lower alkoxy group or a
CA 02244862 1998-07-29
group represented by -AlaP-WlP-Albp- (wherein AlaP is a C1_s
chain hydrocarbon group which may have substituent(s)
selected from the group consisting of a halogen atom, a
lower alkyl group, an oxo group, a lower hydroxyalkyl
5 group which may be protected and a lower alkoxy group;
Albp is a single bond or a Cl_4 chain hydrocarbon group
which may have substituent(s) selected from the group
consisting of a halogen atom, a lower alkyl group, an oxo
group, a lower hydroxyalkyl group which may be protected
10 and a lower alkoxy group; Wlp isan oxygen atom, a sulfur
atom, an ethynylene group, a cyclopropylene group or a
group represented by -NRwp-; and Rwp is a hydrogen atom, a
lower alkyl group or a protecting group for an imino
group); Rlp is a lower alkyl group or a lower alkenyl
15 group which may have substituent(s) selected from the
group consisting of a hydroxyl group which may be
protected, a lower alkoxy group, a lower alkoxycarbonyl
group, a carbamoyl group, a lower alkylcarbamoyl group
and a lower alkylhydrazinocarbonyl group, a lower alkoxy
20 group, a carboxyl group which may be protected, a lower
alkoxycarbonyl group, a carbamoyl group, a lower
alkylcarbamoyl group, a group represented by R$COO-
(wherein Ra is a hydrogen atom, an amino group, a lower
alkyl group, a lower alkoxy group or a lower alkylamino
25 group) or by RbCONR°- (wherein Rb is a hydrogen atom, an
amino group, a lower alkyl group, a lower alkoxy group or
a lower alkylamino group; and R' is a hydrogen atom or a
CA 02244862 1998-07-29
31
lower alkyl group) or-an alicyclic group which may have
substituent(s) selected from the group consisting of a
halogeh atom, a nitro group, a cyano group, a lower alkyl
group, a lower alkenyl group, a lower alkoxy group, a
lower fluoroalkyl group, a lower alkoxycarbonyl group, a
carbamoyl group and a lower alkylcarbamoyl group and a
hydroxyl group, an amino group, a carboxyl group and a
lower hydroxyalkyl group which may be protected, and may
have one or two oxygen atoms and/or one or two nitrogen
atoms; Ra is a lower alkyl group; and eachof Xp and Yp
which are the same or different, is an oxygen atom, a
sulfur atom, a carbonyl group, a group represented by
-CHRd- (wherein Rd is a hydrogen atom or a lower alkyl
group) or by -NRep- (wherein Rep is a hydrogen atom, a
lower alkyl groupor a protecting group for an imino
group), or Xp and Yp together represent a vinylene group
or an ethynylene group, provided that when one of Xp and
Yp is an oxygen atom, a sulfur atom or a group
represented by -NRep- (wherein Rep is the same as defined
above), the other is a carbonyl group or a group
represented by -CHRd- (wherein Rd is the same as defined
above)] with a carboxylic acid represented by general
formula (III) or its reactive derivative:
HO- i-A2p-COORp [ I I I ]
O
[wherein AZp is a Cl_8 chain hydrocarbon group which may
CA 02244862 1998-07-29
32
have substituent(s) selected from the group consisting of
a halogen atom, a lower alkyl group, a lower alkoxy
group, a lower alkoxycarbonyl group, a lower
alkenyloxycarbonyl group, an aryl group and an aralkyl
group, and a hydroxyl group, a lower hydroxyalkyl group,
a carboxyl group and a lower carboxyalkyl group- which may
be protected; and RP is a hydrogen atomor a protecting
group for a carboxyl group], to obtain a compound
represented by general formula (IV):
r ~ P_XP _YP~ r2P~ H2
Ar3P
P [ I V~
~ P I 2P- P
R -CH- i H-N- i -A COOK
R2 O
[wherein
Crap Crzp ~r3~?
' ,
Alp, A2p, Rlp, R2, Rp, Xp and Yp are the same as defined
above], and, if necessary, removing any protecting
groups.
As the reactive derivative of the carboxylic acid
represented by general formula (III), an acid halide, a
mixed acid anhydride, an active ester or an active amide
may, for example, be used.
then the carboxylic acid represented by general
formula (III) is used, it is preferred to conduct the
reaction in the.presence of a condensing agent such as
CA 02244862 1998-07-29
33
N,N'-dicyclohexylcarbodiimide, 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimideor 2-chloro-1,3-
dimethylimidazolyl chloride.
The reaction of the compound represented by general
formula (II) with the carboxylic acid represented by
general formula (III) or its reactive derivative, is
conducted usually by using 1 mol or an excess molar
amount, preferably from 1 to 5 mols, of the carboxylic
acid represented by general formula (III) or its reactive
derivative, per mol of the compound represented by
general formula (II).
The reaction is conducted usually in an inert
solvent_ The inert solvent may, for example, be a
halogenated hydrocarbon such as methylene chloride,
chloroform, carbon tetrachloride, dichloroethane or
trichloroethylene; an ether such as ethyl ether,
tetrahydrofuran or dioxane; an aromatic hydrocarbon such
as benzene, toluene, chlorobenzene or xylene; an aprotic
polar solvent such as dimethylformamide, acetonitrile,
acetone, ethyl acetate or hexamethylphosphoric triamide,
or a mixture of such solvents_
The reaction temperature is usually from -70°C to the
boiling point of the solvent used for the reaction,
preferably from -20°C to 100°C_
The reaction time is usually from 5 minutes to 7
days, preferably from 10 minutes to 24 hours.
The above reaction can be conducted in the presence
CA 02244862 1998-07-29
34
of a base to facilitate the reaction.
As such a base, it is preferred to conduct the
reaction in the presence of an inorganic base such as
sodium hydroxide, potassium hydroxide, calcium hydroxide,
sodium carbonate, potassium carbonate or sodium
hydrogencarbonate, or an organic base such as
triethylamine, N-ethyldiisopropylamine, pyridine, 4-
dimethylaminopyridine or N,N-dimethylaniline_
Such a base is used usually in an amount of 1 mol or
an excess molar, amount, preferably from 1 to 5 mols, per
mol of the reactive derivative of the carboxylic acid
represented by general formula (III).
The acid halide of the compound represented by
general formula (III) can be obtained by reacting the
carboxylic acid represented by general formula (III) with
a halogenating agent in accordance with a conventional
method. As the halogenating agent, thionyl chloride,
phosphorus trichloride, phosphorus pentachloride,
phosphorus oxychloride, phosphorus tribromide, oxalyl
chloride or phosgene may, for example, be used.
The mixed acid anhydride of the compound represented
by general formula (III) can be obtained by reacting the
carboxylic acid represented by general formula (III) with
an alkyl chlorocarbonate such as ethyl chlorocarbonate or
with an-aliphatic carboxylic acid chloride such as acetyl
chloride, inaccordance with a conventional method.
Further, an intramolecular acid anhydride may be formed
CA 02244862 1998-07-29
between carboxyl groups at both terminals, or when a
carboxyl group is present on the chain hydrocarbon group
represented as AZp in general formula (III), an
intramolecular acid anhydride may be formed between such
5 a carboxyl group and a carboxyl group to be involved in
the reaction, to constitute a reactive derivative of the
carboxylic acid_
The active ester of the compound represented by
general formula (III) can be prepared by reacting the
10 carboxylic acid represented by general formula (III) with
an N-hydroxy compound such as N-hydroxysuccinimide, N-
hydroxyphthalimide or 1-hydroxybenzotriazole; or a phenol
compound such as a 4-nitrophenol, 2,4-dinitrophenol,
2,4,5-trichlorophenol or pentachlorophenol, in the
15 presence of a condensing agent such as N,N'-
dicyclohexylcarbodiimide or 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimide in accordance with a
conventional method_
The active amide of the compound represented by
20 general formula (III) can be prepared by reacting the
carboxylic acid represented by general formula (III) with
e_g_ 1,1'-carbonyldiimidazole or.l,1'-carbonylbis(2-
methylimidazole) in accordance with a conventional
method_
25 In the above reaction, when a hydroxyl group, an
amino group, an imino group or a carboxyl group which
will not be involved in the reaction, is present in a
CA 02244862 1998-07-29
36
reactant, it is preferred to conduct the reaction after
protecting such a hydroxyl group, an amino group, an
imino group or a carboxyl group appropriately by a
hydroxyl-protecting group, an amino- or imino-protecting
group or a carboxyl-protecting group and remove the
protecting group after the reaction.
The hydroxyl-protecting group may, for example, be a
lower alkylsilyl group such as a trimethylsilyl group or
a tert-butyldimethylsilyl group; a lower alkoxymethyl
group such as a methoxymethyl group or a 2-
methoxyethoxymethyl group; a tetrahydropyranyl group; an
aralkyl group such as a benzyl group, a p-methoxybenzyl_
group, a p-nitrobenzyl group or a trityl group; or an
acyl group such as a formyl group or an acetyl group.
Particularly preferred is a methoxymethyl group, a
tetrahydropyranyl group, a trityl group, a tert-
butyldimethylsilyl group or an acetyl group.
The amino- or imino-protecting group may, for
example, be an aralkylidene group such as a benzylidene
group, a p-chlorobenzylidene group or a p-
nitrobenzylidene group; an aralkyl group such as a benzyl
group, a p-methoxybenzyl group, a p-nitrobenzyl group, a
benzhydryl group or a trityl group; a lower alkanoyl
group such as a formyl group, an acetyl group, a
propionyl group, a butyryl group or a pivaloyl group; a
lower haloalkanoyl group such as a trifluoroacetyl group;
a lower alkoxycarbonyl group such as a methoxycarbonyl
CA 02244862 1998-07-29
37
group, an ethoxycarbonyl group, a propoxycarbonyl group
or a tert-butoxycarbonyl group; a lower
haloalkoxycarbonyl group such as a 2,2,2-
trichloroethoxycarbonyl group; an alkenyloxycarbonyl
group such as a 2-propenyloxycarbonyl group; an
aralkyloxycarbonyl group such as a benzyloxycarbonyl
group or a p-nitrobenzyloxycarbonyl group; or a lower
alkylsilyl group such as a_trimethylsilyl group or a
tert-butyldimethylsilyl group. Particularly preferred is
an acetyl group, a trifluoroacetylgroup, a tert-
butoxycarbonyl group or a benzyloxycarbonyl group.
The carboxyl-protecting group may, for example, be a
lower alkyl group suchas a methyl group, an ethyl group,
a propyl group, an isopropyl group or a tert-butyl group;
a lower haloalkyl group suchas a 2,2,2-trichloroethyl
group; a lower alkenyl group such as 2-propenyl group; or
an aralkyl group such as a benzyl group, a p-
methoxybenzyl group, a p-nitrobenzyl group, a benzhydryl
group or trityl group- Particularly preferred is a
methyl group, an ethyl group, a tert-butyl group, a 2-
propenyl group, a benzyl group, a p-methoxybenzyl group
or a benzhydryl group.
After completion of the reaction, conventional
treatment is conducted to obtain a crude product of the
compound represented by general formula (IV)- The
compound represented by general formula (IV) may or may
not be purified in accordance with a conventional method,
CA 02244862 1998-07-29
38
and if necessary, reactions for removing protecting
groups for a hydroxyl group, an amino group and a
carboxyl group may be carriedout in a proper Combination
to obtain a compound represented by general formula (I).
Removal o~protecting groups may vary depending upon
their types, but can be conducted in accordance with the
methods disclosed in a literature [Protective Groups in
Organic Synthesis, T.W. Greene, John Wiley & Sons (1981)]
or methods similar thereto, for example by solvolysis
employing an acid or a base, by chemical reduction
employing ametal hydride complex or by catalytic
reduction employing a palladium-carbon catalyst or Raney
nickel_
A compound represented by general formula (I-a):
Are-X-Y Ar2 -CH2 Ar3
i CI -aJ
A'
f- 1 ~ ~ 2
R C - Q -CH- i H-N- i -A -COOH
O R2 O
[wherein
r1 ~ r2 ~r3
' ,
Al, Aa, R~, X and Y are the same as defined above, Ql is
an oxygen atom or -NReq-; and Rf is a hydrogen atom, an
amino group, a lower alkyl group, a lower alkoxy group or
a lower alkylamino group] can be prepared by reacting a
compound represented by general formula (V):
CA 02244862 1998-07-29
39
r~ P-XP-YP~ r2P~H2 i
Ar3P
A,P Cv]
1 ~ I 2P P
H-Q -CH- i H-N-C-A -COOK
R2 O
[wherein
Arlp Ar2~' Cr3p
Alp, A2p, Q1, R2, Rp, Xp and Yp are the same as defined
above] with a compound represented by general formula
( VI )
Rf - C - Z
II (vI )
O
[wherein Rf is the same as defined above; and Z is a
leaving group] to obtain a compound represented by
general formula (IV-a):
Are P-Xp-YP Ar2P-CH Ar3P
[I V-a]
AP
1 ~ 2P P
R C - Q -CH-CH-N-C-A -COOK
R
[wherein
Crlp Cr2p ~r3p
.
.
Alp, AZp, Ql, R2, Rf, Rp, Xp and Yp are the same as defined
above], and if necessary, removing any protecting group.
Process 2 is a process for preparing a compound
represented by general formula (I) of the present
CA 02244862 1998-07-29
invention wherein R1 is a group represented by
Rf - ~~ - Q1 -
O
(wherein Q1 and Rf are the same as defined above), i_e. a
compound represented by general formula (I-a).
5 The reaction of the compound represented by general
formula (V) with the compound represented by general
formula (VI) is conducted usually by using 1 mol or an
excess molar amount, preferably from 1 to 3 mols, of the
compound represented by general formula (VI) per mol of
10 the compound represented by general formula (V).
The reaction is conducted usually in an inert
solvent_ The inert solvent may, for example, be a
halogenated hydrocarbon such as methylene chloride,
chloroform, carbon tetrachloride, dichloroethane or
15 trichloroethylene; an ether such as ethyl ether,
tetrahydrofuran or dioxane; an aromatic hydrocarbon such
as benzene, toluene, chlorobenzene or xylene; an aprotic
polar solvent such as dimethylformamide, acetonitrile,
acetone, ethyl acetate or hexamethylphosphoric triamide,
20 or a mixture of such solvents.
The reaction temperature is usually from -70°C to the
boiling point of the solvent used for the reaction,
preferably from -20~ to 100.
The reaction time is usually from 5 minutes to 7
25 days, preferably from 10 minutes to 24 hours.
CA 02244862 1998-07-29
a1
The above reaction is preferably conducted in the
presence of a base to facilitate the reaction_
Especially, unless Q1 in general formula (V) is a group
represented by -NReq-, it is essential to conduct the
reaction in the presence of an inorganic base such as
sodium hydride, n-butyllithium, sodium hydroxide,
potassium hydroxide, calcium hydroxide, sodium carbonate,
potassium carbonate or sodium hydrogencarbonate, or an
organic base such as triethylamine, N-
ethyldiisopropylamine, pyridine, 4-dimethylaminopyridine
or N,N-dimethylaniline.
Such a base is used usually in an amount of 1 mol or
an excess molar amount, preferably from 1 to 5 mols, per
mol of the compound represented by general formula (VI).
The leaving group represented by Z in general
formula (VI) may, for example, be a halogen atom such as
a chlorine atom, a bromine atom or an iodine atom, or an
organic sulfonyloxy group such as a methanesulfonyloxy
group, a p-toluenesulfonyloxy group or a
benzenesulfonyloxy group.
Inthe above reaction, when a hydroxyl group, an
aWirio - g r oup Or -a Car b'vx°yl gr o up W7.i e~'t wi i i iio t be
involved in the reaction is present in a reactant, it is
preferred to carry out the reaction after protecting such
a hydroxyl group, an amino group or a carboxyl group
appropriately by a hydroxyl-protecting group, an amino-
protecting group or a carboxyl-protecting group, and
CA 02244862 1998-07-29
42
remove any protecting group after the reaction_
The hydroxyl-protecting group, the amino-protecting
group and the carboxyl-protecting group may be the
protecting groups mentioned above with respect to process
1.
After completion of the reaction, conventional
treatment may be carried out to obtain a crude product of
the compound represented by general formula (IV-a). The
compound represented by general formula (IV-a) thus
obtained may or may not be purified by a conventional
method, and if necessary, reactions for removing
hydroxyl-, amino- and carboxyl-protecting groups may be
carried out in a proper combination to obtain a compound
represented by general formula (I-a).
The method for removing a protecting group varies
depending upon the type of the protecting group and the
stability of the desired compound (I-a). However,
removal of protecting groups can be suitably conducted in
accordance with the methods disclosed in the above-
mentioned literature or methods similar thereto.
Further, a compound represented by general formula
(i-) wherein-i-~l is a lover- aiicoacy group can lie prepared by
similarly reacting a compound represented by general
formula (V) wherein Q1 is an oxygen atom with a compound
represented by general formula Rfa-Z [wherein Rfa is a
lower alkyl group; and Z is the same as defined above]
instead of a compound represented by general formula
CA 02244862 1998-07-29
43
(VI) _
A compound represented by general formula (I-b):
Are-X Y Ar2 -CH Ar3
[I -b~
A'
s- 1- ~ I 2
R Q C -CH- i H-N- i -A -COOH
O R2 O
[wherein
Cri Cr2 Cr3
' ,
Al, A~, Ql, RZ, X and Y are the same as defined above; and
Rg is a hydrogen atom or a lower alkyl group] can be
prepared by reacting a compound represented by general
formula (VII):
Ar~p-X°-Yp Ar2p~H2 Ar3p
[V I I
App
~ I 2p-
Z- C - CH-CH-N-C-A COOK
o I Ii
R2 O
[wherein
rlp rZg ~r3~
Alp, A2p, R2, Rp, Xp, Yp and Z are the same as defined
above] with a compound represented by general formula
(VIII):
Rg-Ql-Fi (VIII)
[wherein Rg and Ql are the same as defined above] to
CA 02244862 1998-07-29
44
obtain a compound represented by general formula (IV-b):
Ar~p-Xp-Yp Ar2°~H
[I V-b]
A, p
R9-Q~- C -CH- i H-N- i -A2p-COORp
O 2
R O
[wherein
Arlp Ar2p Ar3p
Alp, A2p, Q1, R2, Rg, Rp, Xp and Yp are the same as defined
above], and, if necessary, removing any protecting group.
Process 3 is a process for producing a compound
represented by general formula (I) of the present
invention wherein R1 is a group represented by:
Rs - Q1 - C -
O
(wherein Q1 and RQ is the same as defined above), i.e. a
compound represented by general formula (I-b).
The process is usually carried out in an inert
solvent, preferably in the presence of a base, by using 1
mol or an excess molar amount, preferably from 1 to 3
mols, of the compound represented by general formula
(VIII), per mol of the compound represented by (VII).
With respect to the inert solvent, the base and the
reaction conditions, those described above for process 2
can be employed. Accordingly, the reaction and post-
treatment are preferably carried out in accordance with
CA 02244862 1998-07-29
process 2.
A compound represented by general formula (I-c):
Are-X Y Ar2 -CH Ars
5 ~ ~ 2 i CI-c7
Rh
~C=CH-CH-CH-N-C-A2 -COOH
R' / ~ 2 O
R
[wherein
r1 C r2 Cr3
' ,
Al, A2, R2, X and Y are the same as defined above; Rh is a
10 hydrogen atom or a lower alkyl group; and Rl is a
hydrogen atom, a lower alkyl group, a lower alkoxy group,
a lower alkoxycarbonyl group, a carbamoyl group, a lower
alkylcarbamoyl group or a lower alkylhycirazinocarbonyl
group] can be prepared by reacting a compound represented
15 by general formula (IX):
Ar3p
~r~P-XP-YP~r2P~H2 / [ x X]
A'
H- C - CH- i H-N- i -A2p-COORp
O R2 O
20 [wherein
rlp rep ~r3~
Alp, Azp, Ra, Rp, Xp and Yp are the same as defined above]
with a compound represented by general formula (X):
Rn
R~/CH-T [X]
CA 02244862 1998-07-29
a~
[wherein Rh and Rl are the same as defined above; and T
is a triphenylphosphonio group, a dimethoxyphosphoryl
group or a diethoxyphosphoryl group], to obtain a
compound represented by general formula (IV-c).
Ar3P
r~ P_XP-YP~ rzP-CH2 , [ I V - c
,, ASP
R ~ ~ 2P P
C=CH-CH-CH-N-C-A -COOK
R'~ I II
R2 O
[wherein
~ry Cr2p ~r3~
' ,
Alp, AZp, R2, Rh, R1, Rp, XP and Yp are the same as defined
above], and if necessary, removing any protecting group.
Process 4 is a process for producing a compound
represented by general formula (I) of the present
invention wherein R1 is a group represented by:
fi
R~
/C=CH-
R'
(wherein Rh and R1 are the same as defined above), i.e. a
compound represented by general formula (I-c).
The reaction of a compound represented by general
formula (IX) with a compound represented by general
formula (X) is carried out usually by employing-equimolar
amounts of the two reactants or using a slightly excess
amount of one of them.
The reaction is carried out usually in an inert
CA 02244862 1998-07-29
47
solvent. Such an inert solvent may, for example, be an
ether such as ethyl ether, tetrahydrofuran or dioxane; an
aromatic hydrocarbon such as benzene, toluene,
chlorobenzene or xylene; an aprotic polar solvent such as
dimethylformamide, ethyl acetate or hexamethylphosphoric
triamide; or a mixture of such solvents.
The reaction temperature is usually from -100°C to
the boiling point of the solvent used for the reaction,
preferably from -70°C to 50°C _
The reaction time is usually from 5 minutes to 7
days, preferably from 10 minutes to 24 hours_
Further, it is preferred to carry out the above
reaction in the presence of a base. The base may, for
example, be sodium hydride, n-butyllithium, sodium
methoxide, potassium tert-butoxide, sodium hydroxide or
potassium hydroxide_
Such a base is used in an amount of one mol or an
excess molar amount, preferably from 1 to 5 mols, per mol
of the compound represented by general formula (X).
After completion of the reaction, conventional
treatment may be carried out as it is when no protecting
group is present in the product, or after removing any
protecting group, when such a protecting group is present
in the product, to obtain a compound represented by
general formula (I-c).
Removal of protecting groups and post-treatment can
be carried out by the methods described in the above
CA 02244862 1998-07-29
48
process 1.
A compound represented-by general formula (I-d):
Are-X-Y Ar2 -CH Ar3
__~ 2 ~ [ I - d ~
Rh ~ At
R~ jCH-CH2-CH- i H-N- i -A2 -COOH
R2 O
[wherein
ri r2 r3
~ ' ,
Al, Aa, R2, R'', Rl, X and Y are the same as defined above]
can be prepared by reducing a compound represented by
general formula (IV-c):
Ar3P
r~ P_XP-YP___~ r2P~H2 ~ [ I V - c
Rh A ~ P
2P P
R~/C=CH-CH-- i H-N- i -A -COOK
2 O
R
[wherein
Crsp ~r2p ~r3~
' ,
Alp, AZp, R2, Rh, Rl, Rp, Xp and Yp are the same as defined
above] and, if necessary, removing any protecting group.
Process 5 is a process for producing a compound
represented by general formula (I) of the present
invention wherein R1 is a group represented by:
Rt,
jCH-CH2
R'
CA 02244862 1998-07-29
49
(wherein Rh and Rl are the same as defined above), i.e_ a
compound represented by general formula (I-d).
The reduction of a compound represented by general
formula (IV-c) is usually preferably carried out in an
inert solvent and by catalytic reduction using a
palladium-carbon.catalyst, a Raney nickel catalyst or a
platinum catalyst.
Such an inert solvent may, for example, be an
alcohol such as methanol, ethanol or propanol or acetic
acid.
The reaction temperature is usually from -20~ to
100°C, preferably from 0°C to room temperature. _
The reaction time is usually from 5 minutes to 7
days, preferably from 10 minutes to 24 hours.
The preferable hydrogen pressure for the catalytic
reduction is usually from atmospheric pressure to 5 atm.
The catalyst is usually used in an amount of from 0.01 to
1 mol, preferably from 0.05 to 0_2 mol, per mol of the
starting compound (IV-c).
After completion of the reaction, conventional
treatment may be carried out as it is when no protecting
group is present in the product, or after removing any
protecting group, when such a protecting group is present
in the product, to obtain a compound represented by
general formula (I-d).
Removal of protecting groups and post-treatment can
be carried out by the methods described in the above
CA 02244862 1998-07-29
c
process 1_
A compound represented by general formula (I-e):
Are-X-Y Ar2 -CH Ar3
5 ~ ~ 2 i [I _ e~
A~Q3 CH -CH- i H-N- i -A2 -COOH
R2 O
[wherein A3 is a C1_4 chain hydrocarbon group which may
have substituent(s) selected from the group consisting of
10 a halogen atom, a hydroxyl group, an amino group, a nitro
group, a cyano group, a carboxyl group, a lower alkyl
group, a lower alkenyl group, a lower alkoxy group, a
lower hydroxyalkyl group, a lower fluoroalkyl group, a
lower alkoxycarbonyl group, a carbamoyl group and a lower
15 alkylcarbamoyl group; each of QZ and Q3 which are the
same or different, is an oxygen atom or a group
represented by -NRj- (wherein Rj is a hydrogen atom or a
lower alkyl group); and
~r1 ~rz Cr3
' ,
Al , A~ , RZ , X and Y are the s ame as def fined above ] can be
20 prepared by reacting a compound represented by general
formula (IX):
Are p-Xp Y° Ar2°-CH ~ Ar3p
i [ I X~
A~°
H- C - CH- i H-N- i -A2p-COORp
O R2 O
CA 02244862 1998-07-29
a
51
[wherein
Arlp Ar2p Cr3P
' ~ '
Alp, A2p, R2, Rp, Xp and Yp are the same as defined above]
with a compound represented by general formula (XI):
HQZ-A3p-Q3H ( XI )
[wherein A3p is a C1_4 chain hydrocarbon group which may
have substituent(s) selected from the group consisting of
a halogen atom, a nitro group, a cyano group, a lower
alkyl group, a lower alkenyl group, a lower alkoxy group,
a lower fluoroalkyl group, a lower alkoxycarbonyl group,
a carbamoyl group and a lower alkylcarbamoyl group, and a
hydroxyl group, an amino group, a carboxyl group and a
lower hydroxyalkyl group which may be protected; and Qa
and Q3 are the same as defined above] to obtain a
compound represented by general formula (IV-e):
3P
~r~P_XP_YP~r2P~H2 Ar [ I V - a
A' P
2P P
A~ 3iCH-CH- j H-N- i-A -COOK
O
R
[wherein
Crlp ~rzp Crap
1p 2p 3p' 2 2 3 p p p
A , A , A , R , Q , Q , R , X and Y are the same as
defined above] and, if necessary, removing any protecting
group_
CA 02244862 1998-07-29
.
52
Process 6 is a process for producing a compound
represented by general formula (I) of the present
invention wherein R1 is a group represented by:
~Q2
A3 3 CH -
~-Q
(wherein A3, Qz and Q3 are the same as defined above), i.e.
a compound represented by general formula (I-e).
The reaction of the compound represented by general
formula (IX) with the compound represented by general
formula (XI) is conducted usually by using 1 mol or an
excess molar amount, preferably from 1 to 5 mols, of the
compound represented by general formula (XI), per mol of
the compound represented by general formula (IX).
The reaction is conducted usually in an inert
solvent_ The inert solvent may, for example, be a
halogenated hydrocarbon such as methylene chloride,
chloroform, carbon tetrachloride, dichloroethane or
trichloroethylene; an ether such as ethyl ether,
tetrahydrofuran or dioxane; an aromatic hydrocarbon such
as benzene, toluene, chlorobenzene or xylene, or a
mixture of such solvents.
The reaction temperature is usually from -70°C to the
boiling point of the solvent used for the reaction,
preferably from -20~ to 120°C_
The reaction time is usually from 5 minutes to 7
days, preferably from 10 minutes to 24 hours.
CA 02244862 1998-07-29
53
Further, the above reaction can be carried out in
the presence of an acid to facilitate the reaction_
As such an acid, it is preferred to conduct the
reaction in the presence of an organic acid such as p-
toluenesulfonic acid, methanesulfonic acid or pyridinium
p-toluenesulfonate; a Lewis acid such as aluminum
chloride, boron trifluoride-ether complex or zinc
chloride or an inorganic acid such as hydrochloric acid
or sulfuric acid
Such an aci-d is used usually in an amount of from
0_001 to 0_1 mol, preferably from 0_001 to 0_01 mol, per
mol of the compound represented by general formula (IX).
After completion of the reaction, the product is
subjected to usual treatment after removing any
protecting group if such a protecting group is present or
directly if no such protecting group is present, to
obtain a compound represented by general formula (I-e)_
Removal of the protecting group and the post
treatment may be conducted by the methods described with
respect to the above Process 1_
Process 7
A compound represented by general formula (I-f):
~r~-X-Y--~r2 -CH2 Ar3 ~I - f
t
A OH
R~-CH-CH-N-i -(CH2)p C= C- (CH2)q- i H- (CH2)r- COOH
R2 O R5 R6
[wherein
CA 02244862 1998-07-29
54
Arlp Ar2p Ar3p
' ,
Al, Rl, Ra, R5, R6, p, q, r, X and Y are the same as
defined above] can be prepared by oxidizing a compound
represented by general formula (IV-f):
Crt p Xp-Yp~r'2p-CH2 Ar3p
[IV - f)
p p OH
Rip-CH--i H-N- i-(CH2)p CH- CH- (CH2)q- i H- (CH2)r- COOR°
R2 O R5p R6p
[wherein RSp is a hydrogen atom, a lower alkoxycarbonyl
group or a lower alkenyloxycarbonyl group, or a lower
hydroxyalkyl group or a carboxyl group which may be
protected, R6p is a hydrogen atom, or a hydroxyl group or
a carboxyl group which may be protected; and
rlp ~ r2g ~r3~-
' ,
Alp, Rlp, R2, Rp, p, q, r, Xp and Yp are the same as defined
above] and, if necessary, removing any protecting group.
Process 7 is a process for producing a compound
represented by general formula (I) of the present
invention wherein A2 is a group represented by formula
(b)
OH
I
2 0 - ~CH2) p C = C- (CH2) q-CH- (CHZ) r - [b]
R5 Rs
(wherein R5, R6, p, q and r are the same as defined
above), i_e_ a compound represented by general formula
(I-f) .
CA 02244862 1998-07-29
The reaction of oxidizing the compound represented
by general formula (IV-f) is usually preferably carried
out in an inert solvent by using so-called Dess-Martin
oxidation employing 12-I-5 triacetoxyperiodinane; so-
5 called Swern oxidation employing oxalyl chloride and
dimethyl sulfoxide; a sulfur trioxide-pyridine complex;
pyridinium chlorochromate; active manganese dioxide; or
tetra-n-propylammonium perruthenate.
The inert solvent may, for example, be a halogenated
10 hydrocarbon such as methylene chloride, chloroform or
dichloroethane; anether such as ethyl ether,
tetrahydrofuran or dioxane; an aprotic polar solvent such
as acetonitrile, acetone, ethyl acetate or dimethyl
sulfoxide; or a mixture of such solvents.
15 The reaction temperature varies depending upon the
type of the oxidizing agent, etc. However, it is usually
from -100 to the boiling point of the solvent used for
the reaction, preferably from -70°C to 100°C.
The reaction time is usually from 5 minutes to 7
20 days, preferably from 10 minutes to 24 hours_
After completion of the reaction, the product is
subjected to usual treatment after removing a protecting
group when such a protecting group is present, or
directly when no such protecting group is present, to
25 obtain the compound represented by general formula (I-f).
The removal of the protecting group and the post-
treatment may be conducted in the same manner as
CA 02244862 1998-07-29
56
described above with respect to process 1_
Further, a compound corresponding to the compound
represented by general formula (IV-f) to be used as the
starting material in the above process 7, can be prepared,
for example, by hydrolyzing a compound represented by
general formula (IV-f-1).
[IV-f-1]
Xp-Yp~r2p-CHZ Ar3p
A
R~ p--CH--~ H-N-C-(CH2)p CH- CH- (CH2)Q- i H-(CH2)r
R2 O R5p R6p
[wherein
rip Crzp ~r3~
Alp, Rlp, R2, Rsp, Rgp, Rp, p, q, r, Xp and Yp are the same as
defined above] inthe presence of a base, to obtain a
compound represented by general formula (IV-f-2).
Xp-Yp-E- Ar2p-CH2 Ar3p
[IV-f-2]
OH
R~ p ~ H -N-C- CH I H- -CH- CH - COOM
-~ H II ( 2)p C ~ H- (CH2)q ~ ( 2)r
R2 O R5p R6p
[wherein M is a hydrogen atom or an alkali metal atom;
Crip Cr2p ~rs~
1p 1p 2 ' Sp 6p p p
A , R , R , R , R , p, q, r, X and Y are the same as
defined above] and then reacting thereto a diazo compound
represented by the general formula
CA 02244862 1998-07-29
57
Rgp - N = N
(wherein RPp is a lower alkyl group, a lower alkenyl
group, an aralkyl group or a lower alkoxycarbonylalkyl
group), or an alkylating agent represented by the general
formula RPp-Z (wherein RpP and Z are the same as defined
above ) .
Isolation and purification of the compound
represented by general formula (I), (I-a), (I-b), (I-c),
(I-d), (I-e) or (I-f) obtained by the above process can
be conducted by a single use or a proper combination of
conventional separating means such as column
chromatography employing silica gel, adsorbent resin,
etc_, liquid chromatography, solvent extraction and
recrystallization-reprecipitation_
The compound represented by general formula (I), (I-
a), (I-b), (I-c), (I-d), (I-e) or (I-f) can be converted
to a pharmaceutically acceptable salt or ester by a
conventional method. Reversely, the conversion from the
salt or ester to a-free carboxylic acid can also be
conducted by a conventional method.
The compounds represented by general formulas (II),
(III), (V), (VI), (VII), (VIII), (IX), (X) and (XI) may
be commercially available or can be prepared in
accordance with the methods disclosed in literature (J_
Med. Chem., 1_Q, 717 (1967); ibid., 725; Chem. Lett., 191
(1980); ibid., 93 (1982); ibid., 375 (1984); J_ Chem. Soc.
CA 02244862 1998-07-29
58
Chem. Commun., 579 (1984); Tetrahedron Letters,-~, 7459
(1995); Helvetica Chimica Acta, ~, 375 (1984)) or
methods similar thereto, or in accordance with the
following processes or the methods disclosed in Reference
Examples.
Rlp-CHZ C-~2 I
O
CArlp-Xp Yp~Ar2ECH2-Z
base
2
rlp Xp Yp~ r2p-CH2
Rip ~ H-C-R2 3
i1
O
I ) ~ r3E A i p-NH2 4
2) reduction
Arl~Xp Yp Ar2p CH Ar3p
2
Alp [II]
Rlp ~ H-CH-NH
R2
[wherein
Crlp ~r2p Ar3~-
' '
Alp, Rlp, RZ , Xp, Yp and Z are the same as def fined above ]
CA 02244862 1998-07-29
59
By this process, the desired compound (II) can be
prepared by reacting a ketone compound .1. represented by
general formula ~ with an alkylating agent represented by
general formula ~ to produce a compound represented by
general formula ~, then reacting the compound ~ with an
amine compound represented by general formula $., followed
by reduction_
The above reaction steps will be described in detail
referring to suitable reaction conditions, etc_
The first step of preparing the compound represented
by general formula ~ from the ketone compound 1., can be
conducted by reacting an equimolar amount or an excess
molar amount, preferably from 1 to 2 mols, of the
alkylating agent represented by general formula ~ to the
ketone compound 1 in the presence of a base in an inert
solvent which does not adversely affect the reaction or
without using any solvent_
The inert solvent may, for example, be an ether such
as ethyl ether, tetrahydrofuran or dioxane; an aromatic
hydrocarbon such as benzene, toluene or xylene; an
aprotic polar solvent such as dimethylformamide, dimethyl
sulfoxide or hexamethylphosphoric triamide, or a mixture
of such solvents_
The base to be used for this reaction, may, for
example, be an alkali metal hydride such as sodium
hydride, lithium hydride or potassium hydride; a lithium
amide such as lithium amide, lithium diisopropylamide or
CA 02244862 1998-07-29
lithium bis(trimethylsilyl)amide; an alkyllithium such as
methyllithium, butyllithium or tert-butyllithium; an
alkali metal alkoxide such as sodium methoxide, sodium
ethoxide or potassium tert-butoxide; or an alkali metal
5 hydroxide such as sodium hydroxide, potassium hydroxide
or lithium hydroxide.
The base is used usually in an amount of 1 mol or an
excess molar amount, preferably from 1 to 5 mols, per mol
of the starting material alkylating agent ~.
10 The reaction temperature is usually from -100°C to
the boiling point of the solvent used for the reaction,
preferably from -80°C to 100. The reaction time is
usually from 10 minutes to 48 hours, preferably from 30
minutes to 24 hours.
15 The step of preparing the desired compound(II) from
the compound represented by general formula ~ can be
conducted usually in an inert solvent such as methanol,
ethanol, benzene, ethyl ether or tetrahydrofuran by
reacting 1 mol or an excess molar amount, preferably from
20 1 to 2 mols, of the amine compound represented by general
formula ~ with 1 mol of the compound represented by
general formula ~ to preliminarily form an imine, which
is subsequently reduced_
The reaction temperature in the process for forming
25 the above imine is usually from 0°C to the boiling point
of the solvent used for the reaction, preferably from
room temperature to 100°C. The reaction time is usually
CA 02244862 1998-07-29
~r
61
from-5 minutes to 48 hours, preferably from 30 minutes to
24 hours_ After the formation of the imine, the reaction
solution may be used as it is to the subsequent step of
the reduction reaction, or the reaction solution may be
distilled or subjected to a conventional separation means
to isolate the imine compound, which is then subjected to
the subsequent reduction_
The reduction can be carried out by using a metal
hydride complex such as sodium borohydride, sodium
cyanoborohydride or lithium aluminum hydride, or by
catalytic reduction employing a palladium-carbon catalyst
or a Raney nickel catalyst_
When a metal hydride complex is used as a reducing
agent, the reducing agent is used usually in an amount of
1 mol or an excess molar amount, preferably from 1 to 5
mols, per mol of the above imine_
For the reduction, an inert solvent, for example, an
alcohol such as methanol or ethanol; an ether such as
dimethyl ether, ethyl ether, diisopropyl ether, dibutyl
ether, dimethoxyethane, dioxane, tetrahydrofuran or
diglyme; an aliphatic hydrocarbon such as pentane, hexane,
heptane or cyclohexane; or an aromatic hydrocarbon such
as benzene or toluene; or a mixture of such solvents, can
be used appropriately as a solvent depending upon the
type of the reducing agent.
The reaction temperature is usually from 0~ to room
temperature, and the reaction time is usually from 1 hour
CA 02244862 1998-07-29
62
to 6 hours.
The compounds represented by general formulas .~,
and ,~. may be commercially available or can be produced by
a proper combination, as the case requires, of the
methods disclosed in Reference Examples, or conventional
methods or methods similar thereto.
Process B
Cpi-lp Xp Yp~ rzp--CHz
R 1 p ~ H-C-Rz 3
I I
O
reduction
rlp Xp Yp~ rzp--CHz
5
Rlp ~ H-CH - OH
Rz
1 ) CH3SOZC1/TEA ~' (or PBr3)
~r3pAlp-I'THz 4
Arl~Xp Yp Arzp CH Ar3p
z
Aip
Rlp- ~ H--CH-NH CII]
Rz
x~ triethylamine
[wherein
CA 02244862 1998-07-29
63
Arlp ~ r r .
Al~, Rlp, R2, XP and Yp are the same as defined above
According to this process, the desired compound (II)
can be prepared by reacting a reducing agent such as a
metal hydride complex to a compound represented by
general formula ~. to obtain an alcohol compound ~ and
reacting an amine compound represented by general formula
to the alcohol compound .~..
The above reaction steps will be described in detail
referring to suitable reaction conditions, etc_
The step of reducing the compound represented by
general formula .~ to the alcohol compound ~ can be
conductedusually by using a metal hydride complex such
as sodium borohydride, diisobutylaluminum hydride,
lithium aluminum hydride or lithium tri-sec-
butylborohydride (L-selectride~), or by catalytic
reduction employing e.g. a palladium-carbon catalyst or a
Raney nickel catalyst, in an inert solvent which does not
adversely affect the reaction.
Tr~hen the metal hydride complex is used as the
reducing agent, such a reducing agent is used usually in
an amount of 1 mol or an excess molar amount, preferably
from 1 to 5 moll, per mol of the compound represented by
general formula
The inert solvent to be used in this reaction may be
suitably selected depending upon the type of the reducing
CA 02244862 1998-07-29
64
agent.
For example, when the reducing agent is sodium
borohydride, an inert solvent, such as an alcohol such as
methanol or ethanol; an ether such as dimethoxyethane,
dioxane, tetrahydrofuran or diglyme; an aprotic polar
solvent such as dimethylformamide or dimethylacetamide,
or water, or a solvent mixture thereof, may be used, and
particularly preferred is an alcohol such as methanol or
ethanol.
For example, when the reducing agent is
diisobutylaluminum hydride, an inert solvent, such as an
ether such as dimethyl ether, ethyl ether, diisopropyl
ether, dibutyl ether, dimethoxyethane, dioxane,
tetrahydrofuran or diglyme; an aliphatic hydrocarbon such
as pentane, hexane, heptane or cyclohexane; an aromatic
hydrocarbon such as benzene or toluene; or methylene
chloride, or a solvent mixture thereof, may be used, and
particularly preferred is toluene or methylene chloride.
For example, when the reducing agent is lithium
aluminum hydride or lithium tri-sec-butylborohydride, an
inert solvent, such as an ether such as dimethyl ether,
ethyl ether, diisopropyl ether, dibutyl ether,
dimethoxyethane, dioxane, tetrahydrofuran or diglyme; an
aliphatic hydrocarbon such as pentane, hexane, heptane or
cyclohexane; or an aromatic hydrocarbon such as benzene
or toluene, or a solvent mixture thereof, may be used,
and particularly preferred isethyl ether or
CA 02244862 1998-07-29
tetrahydrofuran_
For the catalytic reduction, the solvent is
preferably an alcohol such as methanol or ethanol.
The reaction temperature and the reaction time vary
5 depending upon the stability andthe susceptibility to
the reduction reaction of the starting material ketone
compound ~, the type of the reducing agent and the type
of the solvent. However, the reaction temperature is
usually from -80°C to 100°C, preferably from -70°C to
40°C,
10 and the reaction time is usually from 5 minutes to 2 days,
preferably from 30 minutes to 24 hours.
The step of preparing the desired compound (II) from
a compound represented by general formula .~. can be
carried out by reacting a sulfonating agent such as
15 methanesulfonyl chloride to the alcohol compound
represented by general formula .~ in the presence of a
base; or reacting a halogenating agent such as thionyl
chloride or phosphorus tribromide therewith, to convert
the hydroxyl group in the formula to a leaving group,
20 followed by reacting an amine compound represented by
general formula
The reaction for introducing the leaving group can
be conducted usually by reacting 1 mol or an excess molar
amount, preferably from 1 to 2 mols, of a sulfonating
25 agent and a base such as triethylamine to 1 mol of the
alcohol compound ~ in an inert solvent such as methylene
chloride, chloroform, benzene, tetrahydrofuran or ethyl
CA 02244862 1998-07-29
66
acetate, or using 1 mol or an excess molar amount,
preferably from 1 to 5 mols, of a halogenating agent.
The reaction temperature is usually from -70°C to the
boiling point of the solvent used for the reaction,
preferably from -20°C to 80~, and the reaction time is
usually from 5 minutes to 48 hours, preferably from 30
minutes to 24 hours-
Then, the step of reacting an amine compound ~ to
the. compound having the leaving group introduced,
obtained by the above reaction, can be conducted usually
by employing 1 mol or an excess molar amount, preferably
from 1 to 50 mols, of the amine compound ~ per mol of the
starting compound having the leaving group, in an inert
solvent such as methylene chloride, chloroform, benzene,
ethyl ether or tetrahydrofuran.
If necessary, this reaction can be conducted in the
presence of a base other than the amine compound
represented by general formula
As such a base, an inorganic base such as sodium
hydroxide, potassium hydroxide, calcium hydroxide, sodium
carbonate, potassium carbonate or sodium
hydrogencarbonate, or an organic base such as
triethylamine, N-ethyldiisopropylamine, pyridine or N,N-
dimethylaniline may, for example, be mentioned-
Such a base is used usually in an amount of 1 mol or
an excess molar amount, preferably from 1 to 5 mols, per
mol of the starting material compound-
CA 02244862 1998-07-29
67
The reaction temperature is usually from -50°C to
150°C, preferably from -20~ to 100°C, and the reaction
time is usually from 5 minutes to 7 days, preferably from
minutes to 24 hours.
CA 02244862 1998-07-29
68
rlp Xp Yp~ r2p--CHZ
Rlp ~ H-CH-OH
R2
1 ) DEAD ~' 1, Ph3P, Phthalimide (or HNg or DPPA ~' 3)
or i) CH3SOZCl, TEA * 2,
( ii)Phthalimide(or NaN3)
2) NH2NH2 (or reduction)
rlp Xp Yp~ r2p--CH2
6
Rlp ~ H--CH - NH2
R2
Crap Alp-Z 7
Arl~Xp Yp Ar2p CH Ar3p
2
A
Rlp- ~ H-CH-NH CII]
R2
x~ 1 diethyl azodicarboxylate
x~ 2 triethylamine
x~ 3 diphenylphosphoryl azide
CA 02244862 1998-07-29
69
[wherein
Arlp Ar2p Cr3p
' ~ '
Alp, RlP, RZ, XP, Yp and Z are the same as defined above]
According to this process, the desired compound (II)
can be prepared by reacting diethylazodicarboxylate,
triphenylphosphine and phthalimide (or hydrogen azide or
diphenylphosphoryl azide) or reacting a sulfonylating
agent such as methanesulfonyl chloride in the presence of
a base such as triethylamine, then reacting phthalimide
(or sodium azide) in the presence of a base, to the
alcohol compound represented by general formula ~, to
obtain a phthalimide-protected form (or an azide
compound) of the amine compound ~, then reacting
hydrazine (or a reducing agent) to remove the phthalimido
group (or reduce the azido group) to obtain an amine
compound represented by general formula ~, and finally
reacting a compound represented by general formula 7 to
the compound ~.
The above reaction steps will be described in detail
referring to suitable reaction conditions, etc_
For the step of producing the compound represented
by general formula ~ from the alcohol compound ~.' various
synthetic methods and reaction conditions well known in
organic synthetic chemistry for converting alcohol
compounds to amines, may be employed. For example, it is
preferred to employ the so-called Mitsunobu reaction
CA 02244862 1998-07-29
using diethyl azodicarboxylate, triphenylphosphine and
phthalimide (or hydrogen azide or diphenylphosphoryl
azide) or a method which comprises sulfonylation with a
sulfonylating agent such as methanesulfonyl chloride in
5 the presence of a base such as triethylamine, then
reacting phthalimide (or sodium azide) in the presence of
a base, and then treating (or reducing) the obtained
phthalimide (or azide) compoundwith hydrazine.
The above reactions are conducted usually in a
10 solvent inert to the reaction. The inert solvent may,
for example, preferably be tetrahydrofuran,
dimethoxyethane, benzene or toluene in the case ofthe _
above-mentioned Mitsunobu reaction; methylene chloride,
chloroform, tetrahydrofuran, benzene, ethyl acetate or
15 dimethylformamide in the case of the sulfonylation
followed by the reaction with phthalimide (or sodium
azide); an alcohol such as methanol or ethanol in the
next step of the phthalimide-removing reaction with
hydrazine; an ether such as ethyl ether or
20 tetrahydrofuran in the case where a metal hydride complex
is used as the reducing agent in the reduction reaction
of the azide compound; hydrous tetrahydrofuran in the
case where phosphine reduction is conducted with
triphenylphosphine or the like; and an alcohol such as
25 methanol or ethanol in the reductionby catalytic
reduction
With respect to the amounts of the reagents to be
CA 02244862 1998-07-29
71
used, in the above Mitsunobu reaction, each of diethyl
azodicarboxylate, triphenylphosphine and phthalimide (or
hydrogen azide or diphenylphosphoryl azide) is used in an
amount of 1 mol or an excess molar amount, preferably
from 1 to 5 mots, per mol of the starting material
alcohol compound ~. In the reaction with the phthalimide
(or sodium azide) after the sulfonylation, the
sulfonylating agent such as methanesulfonyl chloride is
used in an amount of 1 mol or an excess molar amount,
preferably from 1 to 2 mols, per mol of the alcohol
compound ~, and the base such as triethylamine used at
that time is usually in an amount of 1 mol or an excess_
molar amount, preferably from 1 to 2 mols, per mol of the
sulfonylating agent. In the next step of the reaction
with phthalimide (or sodium azide) in the presence of a
base, 1 mol or an excess molar amount, preferably from 1
to 5 moll of each of phthalimide and the base (or sodium
azide) is used per mol of the sulfonylating agent. Here,
the base to be used together with phthalimide is
preferably sodium carbonate or potassium carbonate.
Otherwise, without using such a base, a sodium salt or a
potassium salt of phthalimide may be used by itself_
Then, in the reaction for removing the phthalimido group
with hydrazine, hydrazine is used in an amount of 1 mol
or an excess molar amount, preferably from 1 to 10 mols,
per mol of the phthalimide compound as the starting
material compound. In the reduction of the azide
CA 02244862 1998-07-29
72
compound with a metal hydride complex or with
triphenylphosphine, the reducing agent is used usually in
an amount of 1 mol or an excess molar -amount, preferably
from 1 to 2 mots, per mol of the azide compound.
In the case of the above Mitsunobu reaction, the
reaction temperature is usually from -70~ to 100°C,
preferably from -20°C to 50~, and the reaction time is
usually from 5 minutes to 48 hours, preferably from 30
minutes to 24 hours. In the reaction for removing the
phthalimido group by hydrazine, the reaction temperature
is usually from 0~ to the boiling point of the solvent
used for the reaction, preferably from room temperature
to 100, and the reaction time is usually from 5 minutes
to 48 hours, preferably from 30 minutes to 24 hours. In
the reactio-n for converting the azide compound to the
amine compound by reduction, when a metal hydride complex
is used as the reducing agent, the reaction temperature
is usually from -70~ to 150°C, preferably from -20°C to
50°C, and the reaction time is usually from 5 minutes to
48 hours, preferably from 10 minutes to 10 hours. When
triphenylphosphine is used as the reducing agent, the
reaction temperature is usually from room temperature to
the boiling point of the solvent used for the reaction,
preferably from 30°C to 100°C, and the reaction time is
usually from 10 minutes to 48 hours, preferably from 30
minutes to 24 hours. Further, in the case of the
reduction by catalytic reduction, the reaction
CA 02244862 1998-07-29
73
temperature is usually from 0°C to 100°C, preferably from
room temperatureto 50°C, and the reaction time is
usually from 10 minutes to 48 hours, preferably from 10
minutes to 24 hours.
The step of producing the desired compound (II) from
the compound represented by generalformula ~ is carried
out usually in a solvent inert to the reaction and can be
carried out by reacting 1 mol or an excess molar amount,
preferably from 1 to 2 mots, of the alkylating agent
represented by general formula 2 to 1 mol of the amine
compound ~ in the presence of a base.
Such an inert solvent may, for example, be an ether.
such as ethyl ether, tetrahydrofuran or dioxane; an
aromatic hydrocarbon such as benzene, toluene or xylene;
an aprotic polar solvent such as dimethylformamide,
dimethyl sulfoxide or hexamethylphosphoric triamide, or a
mixture of such solvents.
The base to be used in this reaction, may, for
example, be an alkali metal hydride such as sodium
hydride, lithium hydride or potassium hydride; a lithium
amide such as lithium amide, lithium diisopropylamide or
lithium bistrimethylsilylamide; an alkyllithium such as
methyllithium, butyllithium or tert-butyllithium; an
alkali metal alkoxide such as sodium methoxide, sodium
ethoxide or potassium tert-butoxide; an alkali metal
hydroxide such as sodium hydroxide, potassium hydroxide
or lithium hydroxide; or an alkali metal carbonate such
CA 02244862 1998-07-29
74
as sodium carbonate or potassium carbonate_
Such a base is used usually in an amount of 1 mol or
an excess molar amount, preferably from 1 to 5 mols, per
mol of the starting material alkylating agent 2_
The reaction temperature is usually from -100°C to
the boiling point of the solvent used for the reaction,
preferably from -80~ to 10-0°C, and the reaction time is
usually from 10 minutes to 48 hours, preferably from 30
minutes to 24 hours_
Further, the compound represented by general formula
7 may be commercially available or can be produced by a
proper combination, as the case requires, of the methods
disclosed in Reference Examples, or conventional methods
or methods similar thereto_
Process D
rlp_XT'_Yp-~ r2p--CHZ
Rip ~ H-CH - NH2 6
R2
1) ~Ar3~Alpa-CORX 8
2) reduction
r
Ar3p
CrlP..Xp Yp~rZp CH2 RX Alpa
Rlp ~ H-CH-~ CII - aJ
R2
CA 02244862 1998-07-29
[wherein Alp8 is a C1_5 chain hydrocarbon group which may
have substituent(s) selected from the group consisting of
a halogen atom, a lower alkyl group, an oxo group, a
lower hydroxyalkyl group which may be protected and a
5 lower alkoxy group or a group represented by -Alcp-Wlp-A~p-
(wherein Alcp is a Cl_4 chain hydrocarbon group which may
have substituent(s) selected from the group consisting of
a halogen atom, a lower alkyl group, an oxo group, a
lower hydroxyalkyl group which may be protected and a
10 lower alkoxy group; Albp is a single bond or a Cl_4 chain
hydrocarbon group which may have substituent(s) selected
from the group consisting of a halogen atom, a lower
alkyl group, an oxo group, a lower hydroxyalkyl group
which may be protected and a lower alkoxy group; Wp is
15 an oxygen atom, a sulfur atom, an ethynylene group, a
cyclopropylene group or a group represented by -NR''p-;
and R"p is a hydrogen atom, a lower alkyl group or a
protecting group for an imino group); R" is a hydrogen
atom or a lower alkyl group; and
Crlp ~r2~ ~r3~-
' ,
20 Alp, Rlp, R2, Xg and Yp are the same as defined above]
According to this process, the desired compound (II-
a) can usually be prepared by preliminarily forming an
imine by reacting 1 mol or an excess molar amount,
preferably from 1 to 2 mols of the compound represented
25 by general formula $ to 1 mol of the amine compound
CA 02244862 1998-07-29
76
represented by general formula $ in an inert solvent such
as methanol, ethanol, benzene, ethyl ether or
tetrahydrofuran, and then reducing it.
This reaction can be carried out in the same manner
as the step for producing the desired compound (II) from
the compound represented by general formula $ in the
above process A_ Accordingly, with respect to the
reaction conditions, etc., similar conditions may be
employed_
Further, the compound represented by general formula
$ may be commercially available or can be produced by a
proper combination, as the case requires, of the methods
disclosed in Reference Examples, or conventional methods
or methods similar thereto.
CA 02244862 1998-07-29
77
rlp Xp Yp~ rzp CHz
5-a
RpOOC ~ H-CH - OH
Rz
1) reduction
2) E - Cl, base
~~'ip Xp Yp~ rzp CHz
E-O- CHz ~ H-CH -OH
i Rz 9
1 ) DEAD * 1, PhgP, Phthalimide (or HNg or DPPA * 3)
or 1) CH3SOZCl, TEA * z,
ii)Phthalimide(or hIaNg)
2) NHzNHz (or reduction)
rlp Xp Yp-~Arzp--CHz
E-O- CHz ~ H-CH - NHz
Rz 10
~Ar3~Alp-Z 7
Arl~Xp Yp , ArZp CH Ar3p
2 p
11
E-O-CHz ~ H-CH-NH
R2
x~ 1 diethyl azodicarbohylate
x~ 2 triethylamine
~ 3 diphenylphosphoryl azide
CA 02244862 1998-07-29
78
ArlL.Xp Yp Ar2p CLI Ar3p
2
Alp 11
i
E-O- CH2-CH-- i H-NH
R2
HO - C-A2p COORp [III]
O
Arlp-Xp Yp Ar2p ' CH Ar3p
P
E=O-CH2 ~ H-CH-N-C-A2p COORp 12
I O
R2
1) Hf or F-
2) oxidation
Ari'LXp Yp Ar2p CH Ar3p
2
[IX]
H- C ~ H-CH-N- C-AZp COORp
O IZ O
R
[wherein E is a trityl group or a tent-butyldimethylsilyl
group; and
rlp ~ r2p ~r3~
Alp, A2p, R2, Rp, Xp, Yp and Z are the same as defined
above]
According to this process, the desired compound (IX)
can be prepared by reducing a compound represented by
general formula 5-as to a diol compound, selectively
CA 02244862 1998-07-29
79
protecting only the primary hydroxyl group of the diol
compound to obtain a compound represented by general
formula ~, and then reacting the compound ~ with diethyl
azodicarboxylate, triphenylphosphine and phthalimide (or
hydrogen azide or diphenylphosphoryl azide) or reacting
the compound ~ with a sulfonylating agent such as
methanesulfonyl chloride in the presence of a base such
as triethylamine, and then reacting phthalimide (or
sodium azide) in the presence of a base, to obtain a
phthalimide-protected form (or an azide compound) of the
amine compound ~,Q, then reacting hydrazine (or a reducing
agent) to remove the phthalimido group (or reduce the _
azido group) to obtain an amine compound represented by
general formula .1~Q., reacting a compound represented by
general formula 2 to the compound ~ to obtain a compound
represented by general formula ~, reacting a carboxylic
acid represented by general formula (III) or its reactive
derivative to the compound to obtain a compound
represented by general formula ~, then removing the
protecting group for the primary hydroxyl group of the
compound 1~ and then oxidizing the compound 12..
The above reaction steps will be described in detail
referring to suitable reaction conditions, etc_
The step of reducing the compound represented by
general formula 5-as to the diol compound ~ can be
conducted usually by using a metal hydride complex such
as sodium borohydride, lithium borohydride,
CA 02244862 1998-07-29
diisobutylaluminum hydride, lithium aluminum hydride or
lithium tri-sec-butylborohydride (L-selectride~) in an
inert solvent which does not adversely affect the
reaction_
5 The reducing agent is used usually in an amount of 1
mol or an excess molar amount, preferably from 1 to 5
mols, per mol of the compound represented by general
formula 5-aa_
The inert solvent to be used in this reaction may be
10 suitably selected depending upon the type of the reducing
agent.
For example, when the reducing agent is sodium
borohydride or lithium borohydride, an inert solvent,
such as an alcohol such as methanol or ethanol; an ether
15 such as dimethoxyethane, dioxane, tetrahydrofuran or
diglyme; an aprotic polar solvent such as
dimethylformamide or dimethylacetamide, or water, or a
solvent mixture thereof, may be used, and particularly
preferred is an alcohol such as methanol or ethanol.
20 For example, when the reducing agent is
diisobutylaluminum hydride, an inert solvent, such as an
ether such as dimethyl ether, ethyl ether, diisopropyl
ether, dibutyl ether, dimethoxyethane, dioxane,
tetrahydrofuran or diglyme; an aliphatic hydrocarbon such
25 as pentane, hexane, heptane or-cyclohexane; an aromatic
hydrocarbon such as benzene or toluene; methylene
chloride, or a solvent mixture thereof, may be used, and
CA 02244862 1998-07-29
81
particularly preferred is toluene or methylene chloride_
For example, when the reducing agent is lithium
aluminum hydride or lithium tri-sec-butylborohydride, an
inert solvent, such as an ether such as dimethyl ether,
ethyl ether, diisopropyl ether, dibutyl ether,
dimethoxyethane, dioxane, tetrahydrofuran or diglyme; an
aliphatic hydrocarbon such as pentane, hexane, heptane or
cyclohexane; organ aromatic hydrocarbon such as benzene
or toluene, or a solvent mixture thereof, may be used,
and particularly preferred is ethyl ether or
tetrahydrofuran_
The reaction temperature and the reaction time vary_
depending upon the stability and the susceptibility to
the reduction reaction of the starting material compound
5aa, the type of the reducing agent and the type of the
solvent_ However, the reaction temperature is usually
from -80°C to 100°C, preferably from -70°C to
40°C, and
the reaction time is usually from 5 minutes to 2 days,
preferably from 30 minutes to 24 hours.
The next step of producing the compound .2 by
selectively protecting the primary hydroxyl group of the
diol compound obtained by the reduction can be carried
out by using a trityl group or a tert-butyldimethylsilyl
group as a protecting group and by reacting 1 mol or an
excess molar amount, preferably from 1 to 1_5 mots of
trityl chloride or tert-butyldimethylchlorosilane, per
mol of the diol compound in an inert solvent which does
CA 02244862 1998-07-29
82
not adversely affect the reaction in the presence of a
base.
The inert solvent may, for example, be methylene
chloride, tetrahydrofuran or dimethylformamide, and the
base may, for example, be triethylamine, 4-
dimethylaminopyridine or imidazole
The reaction temperature is usually from -20~ to the
boiling point of the solvent used for the reaction,
preferably from 0°C to room temperature, and the reaction
time is usually from 10 minutes to 7 days, preferably
from 30 minutes to 24 hours_
Each of the steps of producing the compound
represented by general formula 11. from the compound
represented by general formula ~ can be carried out in
the same manner as in each of the steps of producing the
compound represented by general formula (II) from the
alcohol compound represented by general formula ~ in the
above process C. Accordingly, with respect to each of
the reaction conditions, etc_, similar conditions may be
employed respectively.
The step of producing a compound represented by
general formula ~ from the compound represented by
general formula 11, can be carried out in the same manner
as in the reaction of the compound represented by general
formula (II) with the carboxylic acid represented by
general formula (III) or its reactive derivative in the
above Process 1. Accordingly, with respect to the
CA 02244862 1998-07-29
83
reaction conditions, etc., similar conditions may be
employed.
The step of removing the protecting group for the
primary hydroxyl group represented as E from the compound
represented by general formula .1.2. can be usually carried
out in a solvent inert to the reaction depending on the
kind of the protecting group, for example, by treating
with an acid such as acetic acid, formic acid,
trifluoroacetic acid, hydrochloric acid, sulfuric acid or
p-toluenesulfonic acid when the protecting group is a
trityl group, or with a similar acid or a fluoride salt
such as tetrabutylammonium fluoride or potassium fluoride
when the protecting group is a tert-butyldimethylsilyl
group.
The reaction solvent varies depending on the kind of
the reaction and the stability of the compound. However,
as the solvent, for example, methylene chloride,
methanol, ethanol, tetrahydrofuran or a solvent mixture
of such a solvent and water is preferred in the case of
treatment with an acid, and especially, when acetic acid,
formic acid or trifluoroacetic acid is used as the acid,
it is preferred to conduct the reaction by using the acid
itself or a mixture of the acid and water as the solvent.
Further, when a floor-ide salt such as tributylammonium
fluoride or potassium fluoride is used, for example,
tetrahydrofuran, methylene chloride or dimethylformamide
is preferred_
CA 02244862 1998-07-29
84
The reaction temperature is usually from -20°C to the
boiling point ofthe solvent to be used for the reaction,
preferably from -20°C to 50~, and the reaction time is
usually from 10 minutes to 48 hours, preferably from 30
minutes to 24 hours, in both case of treatment with an
acid and use of a fluoride salt.
The step of oxidizi-ng the primary alcohol compound
obtained by the above reaction to the desired compound
(IX) is usually carried out in an solvent inert to the
reaction by using, for example, pyridinium
chlorochromate, pyridinium dichromate, a sulfur trioxide-
pyridine complex, oxalyl chloride and dimethyl sulfoxide
(under conditions for the so-called Swern oxidation) or
12-I-5 triacetoxyperiodinane (Dess-Martin reagent) as an
oxidizing agent.
As the inert solvent, for example, a halogenated
hydrocarbon such as methylene chloride or chloroform is
usually preferred. The amount of an oxidizing agent is
usually 1 mol or an excess molar amount, preferably from
1 to 2 mols, per mol of the starting material alcohol.
The reaction temperature is usually from -80~ to the
boiling point of the solvent used for the reaction,
preferably from -80~ to 0~ when oxalyl chloride and
dimethyl sulfoxide are used as the oxidizing agent, and
from -20°C to room temperature when other oxidizing agent
is used
The reaction time is usually from 10 minutes to 48
CA 02244862 1998-07-29
hours, preferably from 30 minutes to 24 hours
irrespective of the kind of the oxidizing agent_
The compound represented by general formula 5-as is a
compound represented by general formula ~ wherein Rlp is
5 RpOOC-, and therefore can be prepared by the previously
mentioned process_
CA 02244862 1998-07-29
86
Process F
HO-CH2-CH -NHZ 13
R2
1) ~Ar3~Alp-Z
_7
2) BoczO
Ar3p
Aip
HO-CHZ- j H -N- Boc I4
R2
oxidation
Ar3p
Ap
OHC--CH -N- Boc 15
R2
~Arl~Xp Yp-~r~ECH2-Z SmI2
2
Arip-Xp Yp Ar2p CH Ar3p
C ~ Z Aip
16
HO--CH-CH-N- Boc
R2
Arl~Xp Yp Ar2p CH p,r3p
Aip
17
E 1- O ~ H-CH-NH
R2
CA 02244862 1998-07-29
87
1~ P P 2p CH Ar3P
CAr X Y --~Ar
A1P 17
El- O ~ H-CH-NH
R2
1) HO-O AZP-COORP Cue] 2) Protectingrgroupl of
1~ 'p p r2P CH Ar3P
r X Y ~A
Aip CV - a]
i
HO -CH-- i H-N- C -A2p COORp
- R2 O
Req-NHZ 18
~
P P 3P
r1 X Y ~Ar2P CH2 Ar
Aip CV - b]
HN-CH-CH-N-C-A2p COORP
O
R
[wherein Boc is a tert-butoxycarbonyl group; E1 is a
protecting group for a hydroxyl group; and -
Crlp Cr2p ~r3~
' ,
Alp, A2p, RZ , Req, Rp, Xp, Yp and Z are the same as def fined
above]
According to this process, a desired compound (V-a)
can be prepared by reacting a compound represented by
general formula ~ with a compound represented by general
CA 02244862 1998-07-29
88
formula 7, protecting the amino group with a tert-
butoxycarbonyl group to obtain a compound represented by
general formula .1~4., oxidizing the compound .1~4. to an
aldehyde compound ~, condensing the aldehyde compound
witha compound represented by general formula 2. in the
presence of samarium iodide to obtain a compound
represented by general formula .1~Z, protecting the
hydroxyl group of the compound, removing the tert-
butoxycarbonyl group to obtain a compound represented by
general formula .~,7, reacting the compound with a
carboxylic acid represented by general formula (III) or
its reactive derivative, and selectively removing the
protecting group represented as E1_
Another desired compound (V-b) can be prepared by
converting the hydroxyl group of the compound (V-a)
obtained by the above-mentioned process into a leaving
group and reacting an amine represented by general
formula .1$_
The above reaction steps will be described in detail
referring to suitable reaction conditions, etc_
The step of reacting a compound represented by
general formula .1.~. with a compound represented by general
formula 2 can be carried out in the same manner as the
step of producing a compound represented by general
formula (II) from an amine compound represented by
general formula ~ in the above process C_ Accordingly,
with respect to the reaction conditions, etc., similar
CA 02244862 1998-07-29
89
conditions may be employed.
The step of protecting the amino group of the
compound obtained by the above reaction can be conducted
by methods well known in synthetic organic chemistry, for
example, by the known methods disclosed in the literature
mentioned for process 1.
The'step of oxidizing a compound represented by
general formula ~ to an aldehyde 1~ is usually
preferably carried out in an inert solvent by using the
so-called Dess-Martin oxidation employing 12-I-5
triacetoxyperiodinane; the so-called Swern oxidation
employing oxalyl chloride and dimethyl sulfoxide; a
sulfur trioxide-pyridine complex; pyridinium
chlorochromate; active manganese dioxide; or tetra-n-
propylammonium perruthenate_
The inert solvent may, for example, be a halogenated
hydrocarbon such as methylene chloride, chloroform or
dichloroethane; an ether such as ethyl ether,
tetrahydrofuran or dioxane; an aprotic polar solvent such
as acetonitrile, acetone, ethyl acetate or dimethyl
sulfoxide; or a mixture of such solvents.
The reaction temperature varies depending upon the
type of the oxidizing agent,etc_ However, it is usually
from -100°C to the boiling point of the solvent used for
the reaction, preferably from -70~ to 100°C.
The reaction time is usually from 5 minutes to 7
days, preferably from 10 minutes to 24 hours.
CA 02244862 1998-07-29
The step of producing a compound represented by
general formula .~. by condensing an aldehyde represented
by general formula ~ with a compound represented by
general formula ~. in the presence of samarium iodide is
5 carried out usually by employing equimolar amounts of the
two reactants or using a slightly excess amount of one of
them in the presence of an equimolar or slightly excess
amount of samarium iodide, in an inert solvent.
Such an inert solvent may, for example, be an ether
10 such as ethyl ether, tetrahydrofuran or dioxane; an
aromatic hydrocarbon such as benzene, toluene,
chlorobenzene or xylene; an aprotic polar solvent such as
dimethylformamide or acetonitrile or a mixture of such
solvents.
15 The reaction temperature is usually from -100 to
the boiling point of the solvent used for the reaction,
preferably from -70°C to 50~ _
The reaction time is usually from 5 minutes to 7
days, preferabl-y from 10 minutes to 24 hours.
20 The step of producing a compound represented by
general formula ~ by protecting the hydroxyl group of a
compound represented by general formula ~ and removing
the tent-butoxycarbonyl group can be carried out by
methods well known in synthetic organic chemistry, for
25 example, by known methods disclosed in the literature
mentioned for the above process 1_
The hydroxyl-protecting group may be the protecting
CA 02244862 1998-07-29
91
group mentioned above with respect to process 1_
In the step of producing the desired compound (V-a)
from the compound represented by general formula .~7, the
reaction of the compound represented by general formula
.~7 with the carboxylic acid represented by general
formula (III) or its reactive derivative, can be carried
out in the same manner as the reaction of the compound
represented by general formula (II) with the carboxylic
acid represented by general formula (III) or its reactive
derivative in the above process 1_ Accordingly, with
respect to the reaction conditions, etc., similar
conditions may be employed.
For the step of selectively removing the protecting
group represented as E1 from the compound obtained by the
above reaction, various methods may suitably be selected
depending upon the type and the characteristics of the
protecting group.- Namely, utilizing the difference in
the stability against an acid, a base or reduction
between E1 and other protecting groups, the protecting
group can selectively be removed by a conventional means
such as an acid, a base or reduction- With respect to
specific conditions for such a reaction, the methods
disclosed in the known literature mentioned above for
process 1.
The step of producing another desired compound (V-b)
by converting the hydroxyl group of the compound (V-a)
obtained by the above-mentioned process into a leaving
CA 02244862 1998-07-29
92
group and then reacting an amine represented by general
formula ~$. can be carried out in the same manner as the
step of producing a compound represented by general
formula (II) from an amine compound represented by
general formula ~ in the above process B_ Accordingly,
with respect to the reaction conditions, etc., similar
conditions may be employed.
Further, the compound represented by general formula
may be commercially available, or may be produced by a
proper combination, as the case requires, of the methods
disclosed in Reference Examples, or conventional methods
or methods similar thereto.
Arl~Xp Yp Arzp CH Ar3p
C --~ Z A _
[IV g]
HO- C ~ H-CH-N- C -AZp COORp
O ~2 O
R
Introduction of leaving group
Arl~Xp Yp ArZp CH Ar3p
2 p
[VII]
~ - C -CH-CH-N- C -A2p COORp
o ~ ii
R2 O
CA 02244862 1998-07-29
r ~ .
93
[wherein
Arlp Ar2p Ar3p
, ,
AlP, A2p, Rp, Xp, and Z are the same as defined
R2, Yp
above]
According to this process, the desired compound
(VII) can be prepared by introducing a leaving group to a
compound represented by general formula (IV-g).
The reaction can be carried out i-n the same manner
as in the method of introducing a leaving group to the
compound represented by general formula .~. in the above
process B by using, for example, a halogenating agent
such as thionyl chloride, phosphorus trichloride,
phosphorus pentachloride, phosphorus oxychloride,
phosphorus tribromide, oxalyl chloride or phosgene, or a
sulfonating agent such as methanesulfonyl chloride, p-
toluenesulfonyl chloride or benzenesulfonyl chloride.
Accordingly, with respect to the reaction conditions,
etc., similar conditions may be employed.
R~ ,COOR° Rk Rn
RkCH2-COORrn -i- HC base Rm 00C- CH- CIA CIA COOR°
1COORq COORq
19 20 21
Rk Rn
1 I
HOOC-CH-CH-CH- COOR°
deprotection COORq [III - a]
CA 02244862 1998-07-29
94
[wherein each of R'' and Rn which are the same or
different, is a hydrogen atom, a lower alkyl group, an
aryl group or an aralkyl group; each of R and Rq which
are the same or different, is a carboxyl-protecting
group; and Rm is a tert-butyl group, a benzyl group, a
benzhydryl group or a trityl group]
Process H is a process for preparing a carboxylic
acid derivative represented by general formula (III-a)
among the above-mentioned compounds represented by
general formula (III).
According to this process, the desired carboxylic
acid derivative (III-a) can be prepared by conducting a
so-called Michael addition reaction which comprises
reacting a malefic acid derivative or a fumaric acid
derivative represented bygeneral formula ?~Q to an ester
derivative having a readily removable carboxyl-protecting
group Rm, represented by general formula 1~, in the
presence of a base, and then removing the carboxyl-
protecting group Rm from the-obtained Michael addition
product 2~. under a mild condition.
As the carboxyl-protecting groups represented as R°
and Rg, a lower alkyl group such as a tert-butyl group,
or a benzhydryl group, is preferred.
The protecting group -Rs is preferably a protecting
group which can readily be removed under a-mild condition
of catalytic reduction or ~ae_akly acidic condition and
which is stable under the-Michael addition reaction
CA 02244862 1998-07-29
-.
condition, such as a tert-butyl group, a benzyl group, a
benzhydryl group or a trityl group.
The above Michael addition reaction can be conducted
by reacting the compound represented by general formula
5 ~Q roan amount of 1 mol or an excess molaramount,
preferably from 1 to 2 mots, to 1 mol of the compound
represented by general formula ~ in the presence of a
base such as sodium hydride, butyllithium, lithium
diisopropylamide or lithium bis(trimethylsilyl)amide
10 usually in an inert solvent such as benzene, ethyl ether
or tetrahydrofuran_
Such a base is used usually in an amount of 1 mol or
a slightly excess molar amount, preferably from 1 to 1_5
mols, per mol of the compound represented by general
15 formula ?~Q_
The reaction temperature is usually from -100°C to
100°C, preferably from -80~ to room temperature, and the
reaction time is usually from 5 minutes to 24 hours,
preferably from 10 minutes to 10 hours.
20 The reaction conditions for the reaction for
removing the protecting group from the compound
represented by general formula 2~.. to form the desired
carboxylic acid derivative (III.-a), vary depending upon
the type of the protecting group, etc. For example, when
25 the protecting group is a tert-butyl group, a benzhydryl
group or a trityl group, a method may be employed wherein
the compound is treated with an acid such as acetic acid,
CA 02244862 1998-07-29
f ' .
96
formic acid, trifluoroacetic acid or hydrochloric acid,
preferably within a temperature range of from -20°C to
50°C for from 10 minutes to 24 hours in the absence of a
solvent or usually in an inert solvent such as methylene
chloride, anisole, tetrahydrofuran, methanol or ethanol
or a solvent mixture thereof with water_
For example, when the protecting group is a benzyl
group, a benzhydryl group or a trityl group, a method may
be employed wherein the compound is catalytically reduced
with a catalyst such as a palladium-carbon catalyst or a
Raney nickel catalyst preferably under a hydrogen
pressure of from 1 to 20 kg/cm' preferably within a
temperature range of from 0~ to 40~ for from 10 minutes
to 24 hours usually in an inert solvent such as methanol,
ethanol, dioxane, water or acetic acid, or a solvent
mixture thereof.-
Among compounds represented by general formula (III-
a), an optically active compound represented by general
formula (III-1b).
HOOC-CH2 ~ -CH2-COOR8
H COOR7 C I I I - 1b]
or general formula (III-2b):
HOOC-CHZ-C\ H2-COOR8
H COOR7 C I I I - 2b ]
[wherein each of R' and R$ which are the same or
CA 02244862 1998-07-29
x
97
different, is a carboxyl-protecting group] can be
obtained by reacting a racemic mixture of the compound
represented by general formula (III-b):
HOOC-CH2~H-CH2-COOR8 [ I I I - b ~
COOR7
[wherein each of R' and R8 which are the same or
different, is a carboxyl-protecting group] with
cinchonidine or quinine to obtain a mixture of two
diastereomers, then separating and collecting either one
of the diastereomers by utilizing the difference in the
solubility between the two diastereomers, followed by
recovering the free carboxylic acid by treating with an
acid_
Separation of the diastereomer mixture may be
conducted in an organic solvent such as carbon
tetrachloride or isopropyl ether_ Usually, the mixture
of the diastereomers is dissolved in a solvent in a hot
state, and the solution is gradually cooled to utilize
the solubility difference for separation of the
diastereomers_
Further, either one of the diastereomers thus
obtained is treated with an acid such as hydrochloric
acid to obtain an optically active compound represented
by general formula (III-1b) or (III-2b)_
The compounds represented by general formula .1~ and
~Q may be commercially available or can be produced by a
CA 02244862 1998-07-29
x ' ,
proper combinatio-n, as the case requires, of the methods
disclosed in Reference Examples, or conventional methods
or methods similar thereto.
~rlP XP YP~r2p~Hz
5
Rlp ~ H-CH-OH
R2
RS~OCORt
22
r1P Xp Yp-~~ArZp~H2
(R)
Rlp ~ H-CH-0CORt
23 I z
R
hydrolysis
CArlp Xp Yp~ r2p CH2
(S>
Rlp- ~ H-CH-OH
2 0 24 I
R"
rZp Xp Yp-~Ar2p CHZ
(R)
Rlp- ~ H-CH-OH
I 2
R
25 [wherein Rs is a hydrogen atom or a methyl group; R' is a
lower alkyl group, an aryl group or an aralkyl group; and
CA 02244862 1998-07-29
,.
99
w
Ar 1p Ar 2p
RlP, R2, Xp and Yp are the same as defined above]
Process I is a process for preparing an optically
active substance ~4. or ,~ of a compound represented by
general formula .~_
According to this process, the desired optically
active alcohol compounds 2~. and ~ can be prepared by
reacting a vinyl ester derivative represented by general
formula 2~ to a racemic alcohol derivative represented by
general formula ~ in the presence of a lipase, separating
the obtained optically active ester derivative ~ and the
optically active alcohol derivative, and then hydrolyzing
the ester group with respect to the optically active
ester derivative .2~_
R' of the vinyl ester derivative represented by
general formula ~ is preferably a lower alkyl group such
as a methyl group or an ethyl group; an aryl group such
as a phenyl group or a naphthyl group; or an aralkyl
group such as a benzyl group or a 2-phenylethyl group.
Particularly preferred is a methyl group, i_e_ a case
wherein the compound represented by general formula ~ is
vinyl acetate or isopropenyl acetate_
The above optical resolution reaction by lipase can
be conducted usually in an inert solvent such as
methylene chloride, chloroform, ethyl ether,
tetrahydrofuran, benzene, toluene, hexane, heptane or
CA 02244862 1998-07-29
100
acetonitrile, or by using the starting material vinyl
ester derivative represented by general formula 2~ itself
as the solvent.
The vinyl ester derivative .~.2. is used usually in an
amount of 1 mol or in a large excess molar amount,
preferably from 1 to 100 mols, per mol of the starting
material compound ~, and the amount of the lipase as the
catalyst is from 0.01 to 100, preferably from 0.1 to
20~, by weight, relative to the compound
The type of the lipase is preferably a lipase
derived from Pseudomonas sp. such as Toyothium LIPS
(manufactured by Toyobo?.
Further, the above enzymatic reaction tends to be
accelerated, when the reaction is carried out in the
presence ofa base. As a base to be used for this
purpose, an organic base such as triethylamine or
diisopropylethylamine, is preferred.
The base is used usually in an amount of 0_01 mol or
a slightly excess molar amount, preferably from 0_1 to
1_5 mols, relative to the starting material compound
The reaction temperature is usually from 0~ to 50°C,
preferably from room temperature to 40~. The reaction
time is usually from 30 minutes to 7 days, preferably
from 1 hour to 48 hours.
The hydrolytic reaction of the ester represented by
general formula 2.~ can be conducted by a common method
well known in the synthetic organic chemistry under an
CA 02244862 1998-07-29
s ' .
101
acidic or basic condition_
To demonstrate the usefulness of the present
invention, 50~ inhibitory concentrations (ICso values) of
the compounds of the present invention against the
protein-farnesyl transferase (PFT) activities, were
obtained.
(1) Preparation of PFT
PFT was separated in such a manner that a soluble
fraction of rat's brain was fractionated by means of 30~-
50~ saturated ammonium sulfate,further dialyzed and then
subjected to column chromatography by Q-sepharose~
(manufactured by Pharmacia) (Reiss et al., Cell, vol_ 62,
pp. 81-88 (1990)).
(2) Method for measuring PFT activities
Measurement of PFT activities was conducted by
using, as a prenyl acceptor, H-ras protein or a peptide
corresponding to the C-terminal 7 amino acid residues of
K-rasB protein which had biotin added to the N-terminal
(biotin-added Lys-Thr-Ser-Cys-Val-Ile-Met) and, as a
prenyl donor, [3H]-labeled farnesyl pyrophosphate (FPP)
(Reiss et a1_, Methods: A Companion to Methods in
Enzymology, vol. 1, No_ 3, pp_ 241-245 (1990)).
The [3H]-labeled farnesyl pyrophosphate (22_5
Ci/mmol) was purchased from New England Nuclear Co_ Non-
labeled farnesyl pyrophosphate was chemically synthesized
CA 02244862 1998-07-29
102
from ditriethylammonium phosphate, trans-trans-farnesol
and trichloroacetonitrile and purified by a XAD-2-resin
column and diethylaminoethylcellulose (Cornforth et al.,
Methods inEnzymology, vol. 15, pp. 385-390 (1969)).
H-ras protein was expressed in Escherichia coli and
purified (Gibbs et a1_, Proc. Natl. Acad. Sci_, vol. 81,
pp. 5704-5708 (1984)).
The PFT reaction solution containing H-ras protein
as the prenyl acceptor was 25 ,u1, and its composition
was 50 mM Hepes pH7.5/50 ~cM ZnCl2/5 mM MgCla/20 mM
KCl/5mM DTT/0.6 ,uM all trans [3H]-farnesyl
pyrophosphate/25 ,uM H-ras protein/PFT derived from rat
brain (Q-sepharose fraction). The reaction temperature
was 37°C, the preincubation time was 10 minutes, and the
reaction time was 20 minutes_
The PFT reaction solution containing biotin-added
Lys-Thr-Ser-Cys-Val-Ile-Met as the prenyl acceptor, was
,u1, and its composition was 50 mM tris-Cl pH7_5/50 ,uM
ZnCl2/5 mM MgCla/20 mM KC1/1mM DTTlO .2~ n-octyl- (3 -D-
20 glucopyranoside/0.6 ~ M all trans [3H]-farnesyl
pyrophosphate/3_6 ,uM biotin-added Lys-Thr-Ser-Cys-Va1-
Ile-Met/PFT derived from rat brain (Q-sepharose
fraction). The reaction temperature was 37°C, the
preincubation time was 10 minutes, and the reaction time
25 was 20 minutes.
The enzymatic reaction product containing H-ras
protein as the prenyl acceptor, was analyzed by SDS-PAGE
CA 02244862 1998-07-29
~ .
103
(sodium dodecyl sulfate/polyacrylamide gel
electrophoresis). The [3H]-labeled enzymatic reaction
product was boiled for 3 minutes in a buffer solution
containing 2$ SDS/50 mM Tris-Cl pH6.8/10~ sucrose/5~ 2-
5 mercaptoethanol, then subjected to 12~ polyacrylamide
slab gel electrophoresis whereby the [3H]-labeled H-ras
protein was fluorography-enhanced by EN3HANCE~
(manufactured by New England Nuclear Co_) and then
visualized by autoradiography (James et a1_, Science,
vol. 260, No_ 25, pp. 1937-1942 (1993)).
The measurement of PFT activities using H-ras
protein as the prenyl receptor, was also analyzed by a
rapid separate method. The mixed solution for
measurement wherein no prenyl donor was present, was
preincubated, and a prenyl group transferring reaction
was initiated by an addition of [3H]-FPP and terminated
at an appropriate time by an addition of 0_5 ml of 4~
SDS. Further, 0_5 ml of 30~ trichloroacetic acid was
added thereto and thoroughly mixed. Then, the reaction
solution was left to stand at 4°C for 60 minutes to let
H-ras protein precipitate_ This reaction solution was
filtered under reduced pressure through a Whatman GF/B
filter_ The filter was washed 6 times with 2 ml of 6~
trichloroacetic acid, and mixed with 8 ml of
scintillation cocktail (Clearsol Ice, manufactured by
Nacalai Tesque Co_)_ Then, counting was carried out by a
Beckmann TRI-CARB2500TR scintillation counter_
CA 02244862 1998-07-29
104
Measurement of PFT activities was also carried out
by using biotin-added Lys-Thr-Ser-Cys-Val-Ile-Met as the
prenyl acceptor_ The mixed solution for measurement
containing biotin-added Lys-Thr-Ser-Cys-Va1-Ile-Met as
the prenyl acceptor and containing no prenyl donor, was
preincubated, and then a prenyl group transferring
reaction was initiated by an addition of [3H]-FPP and
terminated at an appropriate time by an addition of 0_2
ml of 2 mg/ml bovine serum albumin/2~ sodium dodecyl
sulfate/150 mM NaCl_ Further, 0_02 ml of avidin agarose
(Pierce) was added thereto, and the mixture was shaked
for 30 minutes to let the [3H]-farnesyl group-transferred
biotin-added Lys-Thr-Ser-Cys-Val-Ile-Met sufficiently
bond to the avidin agarose_ Then, the avidin agarose was
washed four times with 1 ml of 2 mg/ml bovin serum
albumin (BSA)/4~ sodium dodecyl sulfate/150 mM NaCl, and
mixed with 1 ml of scintillation cocktail (Clearsol Ice,
manufactured by Nacalai Tesque). Then, counting was
carried out by a Beckmann TRI-CARB2500TR scintillation
counter_
The biotin-added Lys-Thr-Ser-Cys-Val-Ile-Met
heptapeptide used as an artificial substrate, was
synthesized in a solid phase by an Applied biosystems
model 431A peptide synthesizer, and an a-amino terminal
of the solid phase Lys-Thr-Ser-Cys-Val-Ile-Met
heptapeptide which was bound to a resin, was biotin-
modifiedby N-hydroxysuccinimide biotin, then cut off
CA 02244862 1998-07-29
a
105
from the resin and purified by reversed phase high
performance liquid chromatography (HPLC).
The addition of the compound of the present
invention to the PFT reaction system was carried out by
preliminarily adding dimethyl sulfoxide in an amount of
1~ by volume (0.25 ,u1) of the reaction solution.
The 50~ inhibitory concentrations (ICso values) of
the compounds of the present invention against PFT
activities, are shown in Table 1_
Table 1 50~ inhibitory concentrations
against PFT activities -
Compound ICso (nM)
Example 2 3_1
Example 4 7.8
From the foregoing results, the compounds of the
present invention have excellent inhibitory activities
against protein-farnesyl transferase (PFT) and thus
useful as antitumor agents, for example, against colon
cancer, pancreatic cancer, myloid leukemia, lung cancer,
carcinoma cutaneum or thyroid gland cancer, particularly
against pancreatic cancer_
Further, the protein-farnesyl transferase (PFT)
inhibitor of the present invention is capable of
inhibiting transfection of ras and capable of inhibiting
CA 02244862 1998-07-29
106
reactivation of HIV gene transformed into the host cells,
and thus is useful also as an anti-HIV agent.
The compound represented by general formula (I) of
the present invention can be orally or parenterally
administered, and it may be formulated into a formulation
suitable for such administration, so that it can be used
as an antitumor agent or an anti-HIV agent. To use the
compound of the present invention for clinical purpose,
it may be formulated into various formulations by an
addition of pharmaceutically acceptable additives to meet
the type of administration and then administered_ As
such additives, various additives which are commonly used
in the field of drug formulations, may be used,
including, for example, gelatin, lactose, saccharose,
titanium oxide, starch, crystalline cellulose,
hydroxypropylmethylcellulose, carboxymethylcellulose,
corn starch, microcrystalline wax, white petrolatum,
magnesium metasilicate aluminate, anhydrous calcium
phosphate, citric acid, trisodium citrate,
hydroxypropylcellulose, sorbitol, sorbitan fatty acid
ester, polysorbate, sucrose fatty acid ester,
polyoxyethylene hardened castor oil,
polyvinylpyrrolidone, magnesium stearate, light silicic
anhydride, talc, vegetable oil, benzyl alcohol, gum
arabic, propylene glycol, polyalkylene glycol,
cyclodextrin and hydroxypropylcyclodextrin, etc.
A drug formulation to be prepared as a mixture with
CA 02244862 1998-07-29
107
such additives, may, for example, be a solid formulation
such as a tablet, a capsule, a granule, a powder or a
suppository; or a liquid formulation such as a syrup, an
elixir or an injection drug_ These formulations can be
prepared in accordance with conventional methods commonly
employed in the field of drug formulations_ Further, in
the case of a liquid formulation, it may be of the type
which is to be dissolved or suspended in water or in
other suitable medium at the time of its use.
Particularly, in the case of an injection drug, it may be
dissolved or suspended in a physiological saline or in a
glucose solution, and a buffering agent or a preserving
agent may further be added_
These formulations may contain the compound of the
present.invention in a proportion of from 1_0 to 100 wt~,
preferably from 1_0 to 60 wt~ of the total amount_ These
formulations may further contain other therapeutically
effective compounds_
When the compound of the present invention is used
as an antitumor agent or an anti-HIV agent, its.dose and
the frequency of administration vary depending upon the
sex, the age, the body weight and the diseased degree of
the patient and the type and the range of the intended
treating effects. However, in the case of an oral
administration, it is preferred to administer from 0.01
to 20 mg/kg per day for an adult all at once or in a few
times in a divided fashion. In the case of parenteral
CA 02244862 1998-07-29
108
administration, it is preferred to administer from 0.002
to 10 mg/kg per day for an adult all at once or in a few
times in a divided fashion.
The other therapeutically effective compounds may,
for example, be drugs which bring about a decrease of
farnesyl pyrophosphate in vivo.
The drugs which bring about a decrease of farnesyl
pyrophosphate in vivo, are not particularly limited so
long as they are drugs having such activities and which
are acceptable as pharmaceuticals. However,
biosynthesis-inhibitors against farnesyl pyrophosphate,
for example, preferred. Among them, drugs which inhibit
the biosynthesis of farnesyl pyrophosphate, such as
hydroxymethylglutaryl CoA synthase-inhibitors or
hydroxymethylglutaryl CoA reductate-inhibitors
represented by e.g. lovastatin, simvastatin, pravastatin
and fluvastatin disclosed, for example,. in Nature, vol.
343, pp. 425-430 (1990), are preferred. Particularly
preferred are hydroxymethylglutaryl CoA reductase-
inhibitors such as lovastatin, simvastatin, pravastatin
and fluvastatin.
The composition comprising the compound of the
present invention and the above drug, can be formulated
in the same manner as in the case where the compound of
the present invention is used as a single drug. Such a
formulation may contain the protein-farnesyl transferase
inhibitor and a drug which brings about a decrease of
CA 02244862 1998-07-29
109
farnesyl pyrophosphate in vivo, as active ingredients, in
an amount of from 1.0 to 100 wt~, preferably from 1_0 to
60 wt~, of the entire drug.
Further, the weight ratio of the protein-farnesyl
transferase inhibitor and the drug which brings about a
decrease of farnesyl pyrophosphate in vivo, may be from
0.001:1 to 1000:1_ However, the weight ratio is
particularly preferably from 0_01:1 to 100:1__
Now, the present invention will be described in
further detail with reference to Examples and Reference
Examples, but the present invention is by no means
restricted by such Examples.
EXAMPLE 1
PrexLration of 3-(ethoxvcarbonyl)-4-hydroxv-4-fN-
f(1RS,2RS)-2-(methoxvcarbonyl)-1-methyl-3-~5-
(phenylcarbamoyl ) -2-fury~propyl 1 -N- l2-
naphthylmethyl)carbamoyll-'~-b~ noic a-id
(1) Preparation of N-(2-naphthylmethyl)-{(1RS,2RS)-2-
(methoxycarbonyl)-1-methyl-3-{5-(phenylcarbamoyl)-2-
furyl}propylamine hydrochloride
180 mg of (2RS,3RS)-3-{N-(tert-butoxycarbonyl)-N-(2-
naphthylmethyl)amino}-2-{5-(phenylcarbamoyl)-2-
furylmethyl}butanoic acid prepared in Reference Example 1
in 5 ml of ethyl acetate was stirred together with a
slight excess of diazomethane in ethyl ether for 5
minutes. The reaction solution was evaporated to dryness
CA 02244862 1998-07-29
110
under reduced pressure, and the residue was dissolved in
ml of 4N hydrochloric acid-dioxane and stirred at room
temperature for 1 hour The reaction solution was
evaporated to dryness under reduced pressure again, and
5 the residue was purified by silica gel column
chromatography [hexane/ethyl acetate = 1/1] to give 140
mg (yield. 92~) of the title compound as a colorless oily
substance_
(2) Preparation of tert-butyl 3-(ethoxycarbonyl)-4-[N-
[(1RS,2RS)-2-(methoxycarbonyl)-1-methyl-3-{5-
(phenylcarbamoyl)-2-furyl}propyl]-N-(2-
naphthylmethyl)carbamoyl]-4-(methoxymethyloxy)-3-
butenoate
163 mg of 1-benzyl 3-tert-butyl 2-ethyl 1-
(methoxymethyloxy)-1-propene-1,2,3-tricarboxylate
prepared in Reference Example 5 and 1 ml of 1,4-
cyclohexadiene were dissolved in 10 ml of ethanol and
refluxed under heating with 100 mg of a 10~ palladium-
carbon catalyst for 20 minutes_ The catalyst was
separated by filtration through celite, and the filtrate
was evaporated to dryness under reduced pressure to give
129 mg of 3-tert-butyl 2-ethyl 1-(methoxymethyloxy)-1-
propene-1,2,3-tricarboxylate as a pale brown oily
substance_
129 mg of the carboxylate thus obtained, 140 mg of
N-(2-naphthylmethyl)-{(1RS,2RS)-2-(methoxycarbonyl)-1-
methyl-3-{5-(phenylcarbamoyl)-2-furyl}propylamine
CA 02244862 1998-07-29
111
hydrochloride [the compound prepared in Example 1(1)] and
255 u1 of triethylamine were dissolved in 3 ml of
chloroform, and 104 mg of 2-chloro-1,3-
dimethylimidazolinium chloride in 2 ml of chloroform was
added under cooling with ice_ The resulting solution was
stirred at the same temperature for 2 hours. The
reaction solution was poured into water and extracted
with ethyl acetate. The organic layer was dried over
anhydrous magnesium sulfate. The desiccant was filtered
off, and the solvent was distilled off under reduced
pressure. The residue was purified by silica gel thin
layer chromatography [Kieselgel~ 60Fa54, Art~5717;
hexane/ethyl acetate = 1/2] to give 196 mg (yield: 84~)
of the title compound as a white solid.
(3) Preparation of 3-(ethoxycarbonyl)-4-hydroxy-4-[N-
[(1RS,2RS)-2-(methoxycarbonyl)-1-methyl-3-{5-
(phenylcarbamoyl)-2-furyl}propyl]-N-(2-
naphthylmethyl)carbamoyl]-3-butenoic acid
30 mg of tert-butyl 3-(ethoxycarbonyl)-4-[N
[(1RS,2RS)-2-(methoxycarbonyl)-1-methyl-3-{5-
(phenylcarbamoyl)-2-furyl}propyl]-N-(2-
naphthylmethyl)carbamoyl]-4-(methoxymethyloxy)-3-
butenoate in 0.5 ml of chloroform was stirred with 52 ~1
of bromotrimethylsilane at -30°C for 30 minutes and then
at 0~ for 5 hours. After addition of 1N hydrochloric
acid, the reaction solution was extracted with ethyl
acetate. The organic layer was dried over anhydrous
CA 02244862 1998-07-29
112
magnesium sulfate, and the desiccant was filtered off.
The solvent was distilled off under reduced pressure, and
the residue was purified by silica gel thin layer
chromatography [Kieselgel~ 60F254, Art~5744;
chloroform/methanol = 9/1~ to give 25 mg (yield: 98~) of
the title compound as a white solid.
1H-NMR (CDC13) 8 : 1. 10- I. 40 (6I-I, m) , 2. 55- 3. 7 0 (8H, m) ,
4. 00-4. 90(6H, m), 5. 50-5. 60 and 5. 95-6. 05 (total 1H, each m),
6. 75-7. 70(13H, m), 7. 92-8. 40(1H, m).
FAB-MS:657(M-i-H)
EXAMPLE 2
f(1RS.2RS)-2-liso~ o~ycarbonyl)-1-methyl-3-~5-
lphenylcarbamoyl)-2-fL~ry~ ropvll-N-(2-
naylmethyl)carbamoyll-3-butenoic acid
(1) Preparation of N-(2-naphthylmethyl)-{(1RS,2RS)-2-
(isopropoxycarbonyl)-1-methyl-3-{5-(phenylcarbamoyl)-2-
furyl}propylamine hydrochloride
35 mg of (2RS,3RS)-3-{N-(tert-butoxycarbonyl)-N-(2-
naphthylmethyl)amino}-2-{5-(phenylcarbamoyl)-2-
furylmethyl}butanoic acid prepared in Reference Example
1, 5_8 mg of isopropyl alcohol and 7.9 mg of 4-
dimethylaminopyridine were dissolved in 1 ml of
chloroform and stirred together with 25 mg of 1-ethyl-3-
(3-dimethylaminopropyl)carbodiimide hydrochloride at room
temperature for 16 hours. The reaction solution was
diluted with ethyl acetate and washed with 1N
CA 02244862 1998-07-29
113
hydrochloric acid, saturated aqueous sodium hydrogen
carbonate and saturated aqueous sodium chloride
successively, and the organic layer was dried over
anhydrous magnesium sulfate_ The desiccant was filtered
off, and the solvent was distilled off under reduced
pressure. The residue was purified by silica gel thin
layer chromatography [Kieselgel~ 60Fa54, Art~5744;
hexane/ethyl acetate = 1/1], and 30 mg of the resulting
compound was dissolved in 1 ml-of 4N hydrochloric acid-
dioxane and stirred at room temperature for 1.5 hours.
The reaction solution was evaporated to dryness under
reduced pressure to give 25 mg (yield: 80~) of the title
compound as a colorless oily substance_
(2) Preparation of 3-(ethoxycarbonyl)-4-hydroxy-4-[N-
[(1RS,2RS)-2-(isopropoxycarbonyl)-1-methyl-3-{5-
(phenylcarbamoyl)-2-furyl}propyl]-N-(2-
naphthylmethyl)carbamoyl]-3-butenoic acid
The reactions in Example 1(2) and (3) were carried
out by using N-(2-naphthylmethyl)-{(1RS,2RS)-2-
(isopropoxycarbonyl)-1-methyl-3-{5-(phenylcarbamoyl)-2-
furyl}propylamine hydrochloride instead of N-(2-
naphthylmethyl)-{(1RS,2RS)-2-(methoxycarbonyl)-1-methyl-
3-{5-(phenylcarbamoyl)-2-furyl}propylamine hydrochloride
used as the starting material in Example 1(2), to give
the title compound.
1H-NMR(CDC13) 8 :1. 00-1. 30(12I~, m), 2. 55-3. 80(5H, m),
4. 00-4. 95(7H, m), 5. 55-5. 70 and 5. 95-6. 05 (total 1H, each m),
CA 02244862 1998-07-29
114
6. 80-7. 80(13H, m), 7. 90-8. 40(11-1, m).
FAB-MS: 685 (M-E-H)
EXAMPLE 3
~ethox~rca-rbonyl)-1-methyl-3-~5-(phenylcarbamQy~~-2-
furvl~~oro~vll-N-(2-na~hthvlmethyl)ca-rbamoyll-4-by ro r-3-
butenoic acid
The title compound was prepared in the same manner
as in Example 2 except that ethanol was used instead of
isopropyl alcohol.
1H-NMR(CDC13) 8 : 1. 00- 1. 30 (9H, m) , 2. 70-4. 40 and 4. 50-
5. 00 (total 13H, each m), 5. 70-6. 20(1H, m), -6. 80-8. 00(I3H, m).
FAB-MS:671 (M-I-H)
EXAMPLE 4
Pre»aration of 3-lethoxvcarh~ny~-4-fN-(1R~ .R~ '~ )-4-
(ethoxvcarbonyl)-1-methyl-2-~5-(phenyl~arhamnyl_)2-
f"rvl-metl_,yl-~-3-buteny~ 1 -N- (2-naphthylmefil',yl 1 c~ar]~ moy~ 1 -4-
(1) Preparation of tert-butyl 3-(ethoxycarbonyl)-4-[N-
[(1RS,2RS)-2-(hydroxymethyl)-1-methyl-3-{5-
(phenylcarbamoyl)-2-furyl}propyl]-N-(2-
naphthylmethyl)carbamoyl]-4-(methoxymethyloxy)-3-
butenoate
400 mg of tert-butyl 3-(ethoxycarbonyl)-4-[N-
[(1RS,2RS)-2-(tert-butyldimethylsilyloxymethyl)-1-methyl-
3-{5-(phenylcarbamoyl)-2-furyl}propyl]-N-(2-
naphthylmethyl)carbamoyl]-4-methoxymethyloxy-3-butenoate
CA 02244862 1998-07-29
115
' prepared by the same reaction as in Example 1(2) by using
N-(2-naphthylmethyl)-[(1RS,2RS)-2-(tert-
butyldimethylsilyloxymethyl)-1-methyl-3-{5-
(phenylcarbamoyl)-2-furyl}propyl]amine prepared in
Reference Example 4 was dissolved in 4 ml of
tetrahydrofuran and stirred together with 0_95 ml of 1_0M
tetrabutylammonium fluoride in tetrahydrofuran solution
at room temperature for 2 hours. The reaction solution
was poured into water and~extracted with ethyl acetate,
and the organic layer was washed with saturated aqueous
ammonium chloride and saturated aqueous sodium chloride
and dried over anhydrous magnesium sulfate. The
desiccant was filtered off, and the solvent was distilled
off under reduced pressure. The residue was purified by
column chromatography [hexane/ethyl acetate = 1/1] to
give 147 mg (yield. 43~) of the title compound as a
colorless oily substance.
(2) Preparation of tert-butyl 3-(ethoxycarbonyl)-4-[N-
[(1RS,2RS)-2-formyl-1-methyl-3-{5-(phenylcarbamoyl)-2-
furyl}propyl]-N-(2-naphthylmethyl)carbamoyl]-4-
(methoxymethyloxy)-3-butenoate
124 mg of tert-butyl 3-(ethoxycarbonyl)-4-[N-
[(1RS,2RS)-2-(hydroxymethyl)-1-methyl-3-{5-
(phenylcarbamoyl)-2-furyl}propyl]-N-(2-
naphthylmethyl)carbamoyl]-4-(methoxymethyloxy)-3-
butenoate was dissolved in 3 ml of chloroform and stirred
with 288 mg of the Dess-Martin reagent (periodinane) at
CA 02244862 1998-07-29
T 4
i
116
room temperature for 30 minutes. The reaction solution
was poured into a mixture of saturated aqueous sodium
hydrogencarbonate and saturated aqueous sodium
thiosulfate and extracted with ethyl acetate_ The
organic layer was washed with saturated aqueous sodium
chloride and dried over anhydrous magnesium sulfate. The
desiccant was filtered off, and the solvent was distilled
off under reduced pressure. The residue was purified by
silica gel thin layer chromatography [Kieselgel~ 60F254~
Art~5744; hexane/ethyl acetate = 1/2] to give 65 mg
(yield: 53~) of the title compound as a white solid.
(3) Preparation of tert-butyl 3-(ethoxycarbonyl)-4-[N-
[(1RS,2RS,3E)-4-(ethoxycarbonyl)-1-methyl-2-f5-
(phenylcarbamoyl)-2-furylmethyl}-3-butenyl]-N-(2-
naphthylmethyl)carbamoyl]-4-(methoxymethyloxy)-3-
butenoate
17 mg of tert-butyl 3-(ethoxycarbonyl)-4-[N-
[(1RS,2RS)-2-formyl-1-methyl-3-{5-(phenylcarbamoyl)-2-
furyl}propyl]-N-(2-naphthylmethyl)carbamoyl]-4-
(methoxymethyloxy)-3-butenoate and 12 mg of
(carboethoxymethylene)triphenylphosphorane were dissolved
in 1 ml of chloroform and stirred at room temperature for
3 days_ The solvent was distilled off under reduced
pressure, and the residue was purified by silica gel thin
layer chromatography [Kieselgel~ 60Fa54, Art~5744;
hexane/ethyl acetate = 1/1] to give 16 mg (yield: 85~) of
the title compound as a colorless oily substance.
CA 02244862 1998-07-29
x
117
(4) Preparation of 3-(ethoxycarbonyl)-4-[N-
[(1RS,2RS,3E)-4-(ethoxycarbonyl)-1-methyl-2-{5-
(phenylcarbamoyl)-2-furylmethyl}-3-butenyl]-N-(2-
naphthylmethyl)carbamoyl]-4-hydroxy-3-butenoic acid
The title compound was prepared by the same reaction
as in Example 1(3) by using tert-butyl 3-
(ethoxycarbonyl)-4-[N-[(1RS,2RS,3E)-4-ethoxycarbonyl-1-
methyl-2-{5-(phenylcarbamoyl)-2-furylmethyl}-3-butenyl]-
N--(2-naphthylmethyl)carbamoyl]-4-(methoxymethyloxy)-3-
butenoate as the starting material.
iH-NMR(CDC13) 8 : I. I0- I. 40 (9H, m) , 2. 40-3. 70 (5H, m) ,
3. 90-4. 90(8H, m), 5. 53-5. 78 and 5. 90-6. 03 (total 2H, each m),
6. 48-6. 85(IH, m), 6. 90-7. 80(I3H, m), 7. 90-8. 52(IH, m).
FAB-MS: 697 (M-I-H)
EXAMPLE 5
(te-rt-bmtoxvcarbonyl)-1-methyl-3-f5-lx~henylcarbamovl)-2--
furyl~~rol2yll-N-~ (E)-3-phenyl-2-pro~yl~ca-rbamo5rl 1-4-
hvdroxv-3-butenoic acid
(1) Preparation of tert-butyl (2RS,3RS)-2-[N-[(1RS,2RS)-
2-(tert-butoxycarbonyl)-1-methyl-3-{5-(phenylcarbamoyl)-
2-furyl}propyl]-N-f(E)-3-phenyl-2-propenyl}carbamoyl]-5-
oxotetrahydrofuran-3-carboxylate
86 mg of tert-butyl (2RS,3RS)-2-{5-
(phenylcarbamoyl)-2-furylmethyl}-3-{(E)-3-phenyl-2-
propenylamino}butanoate prepared in Reference Example 3,
46 mg of (2RS,3RS)-3-(tert-butoxycarbonyl)-5-
CA 02244862 1998-07-29
Y
! t
118
- oxotetrahydrofuran-2-carboxylic acid prepared in
Reference Example 6 and 140 ~l of triethylamine were
dissolved in 2 ml of chloroform and stirred with 41 mg of
2-chloro-1,3-dimethylimidazolinium chloride in 1 ml of
chloroform under cooling with ice for 10 minutes and then
at room temperature for 1 hour_ The reaction solution
was poured into water and extracted with ethyl acetate,
and the organic layer was dried over anhydrous magnesium
sulfate_ The desiccant was filtered off, and the solvent
was distilled off under reduced pressure. The residue
was purified by silica gel column chromatography
[hexane/ethyl acetate = 10/1 ~ 2/1] to give 124 mg
(stoichiometric yield) of the title compound as a
colorless oily substance.
(2) Preparation of methyl (3RS,4RS)-3-(tert-
butoxycarbonyl)-4-[N-[(1RS,2RS)-2-(tert-butoxycarbonyl)-
1-methyl-3-f5-(phenylcarbamoyl)-2-furyl}propyl]-N-{(E)-3-
phenyl-2-propenyl}carbamoyl]-4-hydroxybutanoate
104 mg of tert-butyl (2RS,3RS)-2-[N-[(1RS,2RS)-2-
(tert-butoxycarbonyl)-1-methyl-3-f5-(phenylcarbamoyl)-2-
furyl}propyl]-N-~(E)-3-phenyl-2-propenyl}carbamoyl]-5-
oxotetrahydrofuran-3-carboxylate was dissolved in a
mixture of 3 ml of tetrahydrofuran and 1 ml of water and
stirred together with 1.0 ml of 1N aqueous sodium
hydroxide at room temperature for 18 hours. The reaction
solution was acidified with 1N hydrochloric acid and
extracted with ethyl acetate. The organic layer was
CA 02244862 1998-07-29
~ .
119
dried over anhydrous magnesium sulfate. The desiccant
was filtered off, and the solvent was distilled off under
reduced pressure. The resulting carboxylic acid was
dissolved in 10 ml of ethyl acetate, and a slight excess
of diazomethane in ethyl ether was added at room
temperature. The solvent was distilled off under reduced
pressure, and the residue was purified by silica gel
column chromatography [hexane/ethyl acetate = 10/1 -->
2/1] to give 109 mg (stoichiometric yield) of the title
compound as a colorless oily substance_
(3) Preparation of methyl 3-(tert-butoxycarbonyl)-4-[N-
[(1RS,2RS)-2-(tert-butoxycarbonyl)-1-methyl-3-~5-
(phenylcarbamoyl)-2-furyl}~ropyl]-N-{(E)-3-phenyl-2-
propenyl}carbamoyl]-4-hydroxy-3-butenoate
92 mg of methyl (3RS,4RS)-3-(tert-butoxycarbonyl)-4-
[N-[(1RS,2RS)-2-(tert-butoxycarbonyl)-1-methyl-3-{5-
(phenylcarbamoyl)-2-furyl}propyl]-N-{(E)-3-phenyl-2-
propenyl}carbamoyl]-4-hydro~rbutanoate was dissolved in 3
ml of acetonitrile and stirred with 56 mg of
tetrapropylammonium perruthenate at room temperature for
2 hours. The solvent was distilled off under reduced
pressure, and the residue vas roughly purified by silica
gel short column chromatography [chloroform ~ methanol]
and purified again by silica gel column chromatography
[hexane/ethyl acetate = 2J1 ~ 3/2] to give 70 mg (yield.
76~) of the title compound as a colorless oily substance.
(4) Preparation of 3-(tern-_butoxycarbonyl)-4-[N-
CA 02244862 1998-07-29
120
[(1RS,2RS)-2-(tert-butoxycarbonyl)-1-methyl-3-{5-
(phenylcarbamoyl)-2-furyl}propyl]-N-{(E)-3-phenyl-2--
propenyl}carbamoyl]-4-hydroxy-3-butenoic acid
68 mg of methyl 3-(tert-butoxycarbonyl)-4-[N-
[(1RS,2RS)-2-(tert-butoxycarbonyl)-1-methyl-3-{5-
(phenylcarbamoyl)-2-furyl}propyl]-N-{(E)-3-phenyl-2-
propenyl}carbamoyl]-4-hydroxy-3-butenoate was dissolved
in a mixture of 3 ml of tetrahydrofuran and 1 ml of water
and stirred together with 1 ml of 1N aqueous sodium
hydroxide at room temperature for 40 hours. The reaction
solution was acidified with 1N hydrochloric acid and
extracted with ethyl acetate. The organic layer was
dried over anhydrous magnesium sulfate. The desiccant
was filtered off, and the solvent was distilled off under
reduced pressure_ The residue was purified by silica gel
column chromatography [chloroform/methanol = 20/1 -~
10/1.] to give 44 mg (yield: 66~) of the title compound as
a pale yellow solid.
1H-NMR(CDC13) 8 :I. I0-1. 50(21H, m), 2. 80-3. 50 and 3. 60-
4. 65 (total 9H, each m), 6. 05-6. 45 and 6. 50-6. 65 (total 3H, each
m), 7. 00-7. 40(9H, m), 7. 65-7. 80(2H, m), 7. 95-8. 60(lI-I, m).
FAB-MS:703(M+I-I)
REFERENCE EXAMPLE 1
P rex~aration of l2RS 3RS)-3-(N- pert-butoxvcarh~nyll-N-l2-
aph~hymeth yl) amino-2-I5- (phenyl ca-rt-~amnyl 1 -2-
r~
-f p_ry~methyl~butanoic acid
(1) Preparation of tert-butyl 2-{5-(ethoxycarbonyl)-2-
CA 02244862 1998-07-29
121
furylmethyl}-3-oxobutanoate
A solution of 1_78 g of potassium tert-butoxide in
60 ml of tent-butyl alcohol was stirred together with
3_00 g of tert-butyl acetoacetate at 60°C -for 30 minutes
under heating and then cooled to room temperature. 3_40
g of ethyl 5-(chloromethyl)-2-furancarboxylate was added
dropwise, and the solution was stirred at room
temperature for 16 hours= The reaction solution was
poured into water and extracted with ethyl acetate, and
the organic layer was washed with saturated aqueous
sodium chloride and dried over anhydrous magnesium
sulfate. The desiccant was filtered off, and the solvent
was distilled off under reduced pressure. The residue
was purified by silica gel column chromatography
[hexane/ethyl acetate = 9/1 ~ 5/1] to give 2.26 g of the
title compound as a colorless oily substance.
(2) Preparation of tert-butyl (2RS,3SR)-2-{5-
(ethoxycarbonyl)-2-furylmethyl}-3-hydroxybutanoate
To 2_26 g of tert-butyl 2-{5-(ethoxycarbonyl)-2-
furylmethyl}-3-oxobutanoate in 50 ml of tetrahydrofuran,
7_7 ml of 1M lithium tri-sec-butylborohydride in
tetrahydrofuran solution was added at -78°C under
stirring, and the resulting reaction solution was stirred
at the same temperature for 2 hours. After addition of
water, the reaction solution was stirred at room
temperature for 30 minutes and extracted with ethyl
acetate. The organic layer was washed with saturated
CA 02244862 1998-07-29
122
aqueous sodium chloride and dried over anhydrous
magnesium sulfate. The desiccant was filtered off, and
the solvent was distilled off under-reduced pressure.
The residue was purified by silica gel column
chromatography [hexane/ethyl acetate = 12/1 -~ 4/1] to
give 1_65 g of the title compound.
(3) Preparation of tert-butyl (2RS,3RS)-3-amino-2-{5-
(ethoxycarbonyl)-2-furylmethyl}butanoate
To 1_65 g of tert-butyl (2RS,3SR)-2-{5-
(ethoxycarbonyl)-2-furylmethyl}-3-hydroxybutanoate in 40
ml of tetrahydrofuran, 2.32 g of triphenylphosphine, 1_40
ml of diethyl azodicarboxylate and 2_43 g of
diphenylphosphoryl azide were added successively under
cooling with ice under stirring, and the resulting
reaction solution was stirred at room temperature for 18
hours. The reaction solution was evaporated to dryness
under reduced pressure, and the residue was purified by
silica gel column chromatography [hexane/ethyl acetate =
12/1 ~ 3/1]_ The resulting azide was refluxed together
with 2_33 g of triphenylphosphine in 33 ml of 10~ hydrous
tetrahydrofuran under heating for 3 hours. The reaction
solution was cooled to room temperature, and the solvent
was distilled off under reduced pressure. The residue
was purified by silica gel column chromatography
[hexane/ethyl acetate = 1/1 --> ethyl acetate/methanol =
10/1] to give 686 mg of the title compound as a pale
yellow oily substance.
CA 02244862 1998-07-29
123
(4) Preparation of tert-butyl (2RS,3RS)-2-{5-
(ethoxycarbonyl)-2-furylmethyl}-3-{(2-
naphthylmethyl)amino}butanoate
500 mg of tert-butyl (2RS,3RS)-3-amino-2-{5-
(ethoxycarbonyl)-2-furylmethyl}butanoate in 15 ml of
methanol was stirred together with 251 mg of 2-
naphthaldehyde at 50~ for 2 hours under heating. The
reaction solution was cooled to 0°C and stirred with 61
mg of sodium borohydride at the same temperature for 15
minutes. Ethyl acetate and water were added to the
reaction solution for extraction, and the organic layer
was washed with saturated aqueous sodium chloride and
then dried over anhydrous magnesium sulfate. The
desiccant was filtered off, and the solvent was distilled
offunder reduced pressure_ The residue was purified by
silica gel column chromatography [hexane/ethyl acetate =
6/1].to give 555 mg of the title compound as a colorless
oily substance_
(5) Preparation of tert-butyl (2RS,3RS)-3-{(2-
naphthylmethyl)amino}-2-{5-(phenylcarbamoyl)-2-
furylmethyl}butanoate
538 mg of tert-butyl (2RS,3RS)-2-{5-
(ethoxycarbonyl)-2-furylmethyl}-3-{(2-
naphthylmethyl)amino}butanoate was dissolved in a mixture
of 5 ml of tetrahydrofuran and 5 ml of methanol and
stirred together with 5 ml of 1N aqueous sodium hydroxide
at room temperature for 2 hours. The reaction solution
CA 02244862 1998-07-29
_" .
Y
124
was acidified by 1N hydrochloric acid and then extracted
with ethyl acetate. The extract was dried over anhydrous
magnesium sulfate. The desiccant was filtered off, and
the solvent was distilled off under reduced pressure.
S The residue was dissolved in 5 ml of dimethylformamide
and then stirred together with 219 mg of aniline and 339
mg of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
hydrochloride at room temperature for 17 hours. After
addition of water, the reaction solution was extracted
with ethyl acetate, and the organic layer was washed with
1N aqueous sodium hydroxide and saturated aqueous sodium
chloride and then dried over anhydrous magnesium sulfate.
The desiccant was filtered off, and the solvent was
distilled off under reduced pressure. The residue was
purified by silica gel column chromatography
(hexane/ethyl acetate = 3/1] to give 473 mg of the title
compound as a colorless foamy substance.
(6) Preparation of (2RS,3RS)-3-{N-(tert-butoxycarbonyl)-
N-(2-naphthylmethyl)amino}-2-f5-(phenylcarbamoyl)-2-
furylmethyl}butanoate
846 mg of tert-butyl (2RS,3RS)-3-f(2-
naphthylmethyl)amino}-2-{5-(phenylcarbamoyl)-2-
furylmethyl}butanoate in 20 ml of formic acid was stirred
at room temperature for 3 hours, and the formic acid was
distilled off under reduced pressure. The residue was
dissolved in 20 ml of 4N hydrochloric acid-dioxane and
stirred at room temperature for 15 minutes. The reaction
CA 02244862 1998-07-29
.. ~ ,
125
solution was evaporated to dryness under reduced pressure
to give 814 mg of an amine hydrochloride as a white
solid. 814 mg of the amine hydrochloride, 8.5 ml of 1N
aqueous sodium hydroxide and 8.5 ml of dioxane were mixed
and stirred vigorously together with 1.19 g of di-tert-
butyl Bicarbonate at room temperature for 18 hours.
After addition of 1N hydrochloric acid, the reaction
solution was extracted withethyl acetate, and the
organic layer was washed with 1N hydrochloric acid, water
and saturated aqueous sodium chloride successively and
then dried over anhydrous magnesium sulfate. The
desiccant was filtered off, and the solvent was distilled
off under reduced pressure. The residue was purified by
silica gel column chromatography [chloroform/methanol =
50/1] to give 755 mg of the title compound as a white
solid.
REFERENCE EXAMPLE 2
Preparation of 2-(chloromethy~)-5-(x~henylcarbamoyl)fm ran
(1) Preparation of 5-(phenylcarbamoyl)-2-furfural
diethyl acetal
To 20.0 g of 2-furfural diethyl acetal in 200 ml of
ethyl ether, 75 ml of 1_68M n-butyllithium in hexane
solution was added at -78°C under a nitrogen atmosphere,
and the resulting solution was stirred at the same
temperature for 30 minutes and then at room temperature
for 30 minutes. The reaction solution was cooled to
-78°C again, and 14.0 ml of phenyl isocyanate was added_
CA 02244862 1998-07-29
126
The reaction solution was stirred at room temperature for
17 hours. After addition of saturated aqueous ammonium
chloride, the reaction solution was extracted with ethyl
acetate, and the organic layer was washed with 1N
hydrochloric acid, water, saturated aqueous sodium
hydrogencarbonate and saturated aqueous sodium chloride
successively and then dried over anhydrous magnesium
sulfate. The desiccant was filtered off, and the solvent
was distilled off under reduced pressure_ The residue
was purified by silica gel column chromatography
[hexane/ethyl acetate = 10/1 ~ 1/1] to give 25.9 g of
the title compound as a colorless oily substance.
(2) Preparation of 5-(phenylcarbamoyl)-2-furfural
50 ml of trifluoroacetic acid was added to 25.2 g of
5-(phenylcarbamoyl)-2-furfural diethyl acetal in 200 ml
of chloroform under cooling with ice, and the resulting
solution was stirred at the same temperature for 3 hours.
The reaction solution was diluted with chloroform, then
washed with water, saturated aqueous sodium
hydrogencarbonate and saturated aqueous sodium chloride
successively, and dried over anhydrous sodium sulfate_
The desiccant was filtered off, and the solvent was
distilled off under reduced pressure. The residue was
purified by silica gel column chromatography
[hexane/ethyl acetate = 10/1 --> 1/1] to give 12.5 g of
the title compound as a colorless oily substance.
(3) Preparation of 2-(chloromethyl)-5-
CA 02244862 1998-07-29
127
(phenylcarbamoyl)furan
3.5 g of sodium borohydride was added to 12.4 g of
5-(phenylcarbamoyl)-2-furfural in 200 ml of methanol
under cooling with ice, and the resulting solution was
stirred at the same temperature for 10 minutes and then
at room temperature for 1 hour. The reaction solution
was mixed with water under cooling withice and stirred
at room temperature for 30 minutes. The solvent was
distilled off under reduced pressure, and ethyl ether was
added to the residue. The resulting solution was washed
with 1N hydrochloric acid, water, saturated aqueous
sodium hydrogencarbonate and saturated aqueous sodium
chloride successively and then dried over anhydrous
sodium sulfate. The desiccant was filtered off, and the
solvent was distilled off under reduced pressure. The
residue was purified by silica gel column chromatography
[hexane/ethyl acetate = 1/1 -~ 1/2] to give 10_3 g of an
alcohol.
5_40 g of the alcohol was dissolved in 100 ml of
chloroform and 2_0 ml of thionyl chloride was added under
cooling at -60~_ The reaction solution was stirred at
0°C for 1 hour, then mixed with water and extracted with
ethyl acetate. The organic layer was washed with water,
saturated aqueous sodium hydrogencarbonate and saturated
aqueous sodium chloride successively and then dried over
anhydrous sodium sulfate. The desiccant was filtered
off, and the solvent was distilled off under reduced
CA 02244862 1998-07-29
r '
128
pressure. The residue was purified by silica gel column
chromatography [hexane/ethyl acetate = 3/1] to give 4_55
g of the title compound as a colorless oily substance.
REFERENCE EXAMPLE 3
Prex_~aration of tert-butyl l2RS,3RS)-2-~5-
(phenylcarbamoyl)-2-furylmethyl~-3-~(E)-3-phenyl-2-
pro~n_ylamino ~ butanoate
(1) Preparation of tert-butyl 3-oxo-2-{5-
(phenylcarbamoyl)-2-furylmethyl}butanoate
To 9.6 g of tert-butyl acetoacetate was dissolved in
80 ml of tetrahydrofuran, and 2_33 g of 60~ oily sodium
hydride was added under cooling with ice under stirring.
The resulting solution was stirred at the same
temperature for 10 minutes. After addition of 13_6 g of
2-(chloromethyl)-5-(phenylcarbamoyl)furan obtained in
Reference Example 2 in 80 ml of tetrahydrofuran, the
solution was stirred at room temperature for 10 minutes
and then at 60°C for 13 hours under heating. Water and
ethyl acetate were added to the reaction solution for
extraction, and the organic layer was washed with water,
saturated aqueous sodium hydrogencarbonate and saturated
aqueous sodium chloride successively and then dried over
anhydrous sodium sulfate. The desiccant was filtered off,
and the solvent was distilled off under reduced pressure.
The residue was purified by silica gel column
chromatography [hexane/ethyl acetate = 10/1 -~ 3/1] to
give 14.0 g of the title compound as a colorless oily
CA 02244862 1998-07-29
129
substance.
(2) Preparation of tent-butyl (2RS,3SR)-3-hydroxy-2-{5-
(phenylcarbamoyl)-2-furylmethyl}butanoate
43 ml of 1M lithium tri-sec-butylborohydride in
tetrahydrofuran solution was added to 14.0 g of tert-
butyl 3-oxo-2-{5-(phenylcarbamoyl)-2-
furylmethyl}butanoate in 100 ml of tetrahydrofuran at
-78~C under stirring, and the resulting reaction solution-
was stirred at the same temperature for 1 hour. While
the reaction solution was cooled with ice under stirring,
50 ml of 4N aqueous sodium hydroxide was added, and then
35 ml of 35~ aqueous hydrogen peroxide was gradually
added dropwise. The reaction solution was stirred at
room temperature for 1 hour. Ethyl ether and water were
added to the reaction solution for extraction, and the
organic layer was washed with saturated aqueous sodium
thiosulfate and saturated aqueous sodium chloride and
then dried over anhydrous sodium sulfate_ The desiccant
was filtered off, and the solvent was distilled off under
reduced pressure. The residue was purified by silica gel
column chromatography [hexane/ethyl acetate = 5/1 ~ 1/1]
to give 8.44 g of the title compound as a colorless oily
substance.
(3) Preparation of tert-butyl (2RS,3RS)-2-{5-
(phenylcarbamoyl)-2-furylmethyl}-3-{(E)-3-phenyl-2-
propenylamino}butanoate
3.0 g of tert-butyl (2RS,3SR)-3-hydroxy-2-{5-
CA 02244862 1998-07-29
_ r f
130
(phenylcarbamoyl)-2-furylmethyl}butanoate in 50 ml of
tetrahydrofuran was mixed with 3.25 g of
triphenylphosphine, 2.0 ml of diethyl azodicarboxylate
and 2.7 ml of diphenylphosphoryl azide under cooling with
ice under stirring and then stirred at room temperature
for 18 hours. The reaction solution was evaporated to
dryness under reduced pressure, and the residue was
purified by silica gel column chromatography
[hexane/ethyl acetate = 10/1 ~ 2/1]_ The resulting azide
was refluxed together with 3.25 g of triphenylphosphine
in 55 ml of 10~ hydrous tetrahydrofuran for 2 hours under
heating_ The reaction solution was cooled to room
temperature, and the solvent was distilled off under
reduced pressure. The residue was purified by silica gel
column chromatography [hexane/ethyl acetate = 1/1 --j
ethyl acetate -> chloroform/methanol = 10/1 -> 5/1]. The
resulting amine and 1.6 ml of trans-cinnamaldehyde were
dissolved in 50 ml of methanol and stirred at 70°C for
1.5 hours under heating. The reaction solution was
cooled to 0°C and then stirred with 800 mg of sodium
borohydride at the same temperature for 15 minutes and
then at room temperature for 24 hours. water was added
under cooling with ice, and the reaction solution was
stirred at room temperature for 30 minutes_ The solvent
was distilled off under reduced pressure. The residue
was mixed with ethyl ether and washed with 1N
hydrochloric acid, water, saturated aqueous sodium
CA 02244862 1998-07-29
~ T
131
hydrogencarbonate and saturated aqueous sodium chloride
successively and then dried over anhydrous sodium sulfate_
The desiccant was filtered off, and the solvent was
distilled off under reduced pressure. The residue was
purified by silica gel column chromatography
[hexane/ethyl acetate = 2/1 ~ 1/1] to give 1_05 g of the
title compound as a yellow oily substance.
REFERENCE EXAMPLE 4
Prex~aration of N-(2-naphthylmethyl)-f(1RS.2RS)-2-ltert-
~yldimethylsilylox-~rmethyl)-1-methyl-3-~5-
(x~henylca-rbamoy~)-2-furvl~8rogvllamine
(1) Preparation of (2RS,3SR)-3-(hydroxymethyl)-4-{5-
(phenylcarbamoyl)-2-furyl}butan-2-of
3.0 g of tert-butyl (2RS,3SR)-3-hydroxy-2-{5-
(phenylcarbamoyl)-2-furylmethyl}butanoate [the compound
prepared in Reference Example 3(2)] in 30 ml of
tetrahydrofuran was stirred together with 1.09 g of
lithium borohydride at room temperature for 2 hours_ The
reaction solution was cooled to 0~, mixed with 2N
hydrochloric acid and extracted with ethyl acetate, and
the organic layer was dried over anhydrous magnesium
sulfate. The desiccant was filtered off. and tl,P ~~~~TP"t
was distilled off under reduced pressure to give 1.56 g
of the title compound as a colorless oily substance.
(2) Preparation of (2RS,3SR)-3-(tert-
butyldimethylsilyloxymethyl)-4-{5-(phenylcarbamoyl)-2-
furyl}butan-2-of
CA 02244862 1998-07-29
- ,
132
645 mg of (2RS,3SR)-3-(hydroxymethyl)-4-{5-
(phenylcarbamoyl)-2-furyl}butan-2-of in12 ml of
dimethylformamide was mixed with 305 mg of imidazole and
372 mg of tert-butyldimethylsilyl chloride under cooling
with ice and stirred at the same temperature for 1 hour_
Saturated aqueous ammonium chloride and ethyl acetate
were added to the reaction solution for extraction, and
the organic layer was washed with water and saturated
aqueous sodium chloride and then dried over anhydrous
magnesium sulfate. The desiccant was filtered off, and
the solvent was distilled off under reduced pressure_
The residue was purified by silica gel column
chromatography [hexane/ethyl acetate = 3/1] to give 805
mg of the title compound as a colorless oily substance.
(3) Preparation of N-(2-naphthylmethyl)-[(1RS,2RS)-2-
(tert-butyldimethylsilyloxymethyl)-1-methyl-3-{5-
(phenylcarbamoyl)-2-furyl}propyl]amine
The title compound was prepared by the same
reactions as in Reference Example 1(3) and (4) by using
(2RS,3SR)-3-(tert-butyldimethylsilyloxymethyl)-2-hydroxy--
4-{5-(phenylcarbamoyl)-2-furyl}butane instead of tert-
butyl (2RS,3SR)-2-{5-(ethoxycarbonyl)-2-furylmethyl}-3-
hydroxybutanoate used as the starting material in
Reference Example 1(3)_
REFERENCE EXAMPLE 5
CA 02244862 1998-07-29
~, i ~ T
133
(1) Preparation of 3-tert-butyl 1,2-diethyl (1S,2R)-1-
hydroxy-1,2,3-propanetricarboxylate
31 ml of 1_69M n-butyllithium in hexane was
dissolved in 30 ml of tetrahydrofuran, stirred with 7.1
ml of diisopropylamine under cooling with ice for 30
minutes and then cooled to -78°C. 4_94 g of diethyl (S)-
malate in 20 ml of tetrahydrofuran was added dropwise at
-50°C or below, and the reaction solution was stirred at
-20~ for 1_5 hours. The reaction solution was cooled to
-78~, and 5_58 g of tert-butyl bromoacetate and 4_66 g
of hexamethylphosphoryl triamide in 20 ml of
tetrahydrofuran were added dropwise at -50~ or below.
The reaction solution was stirred at room temperature for
1 hour. The reaction solution was poured into 150 ml of
0_5N hydrochloric acid and extracted with diethyl ether,
and the organic layer was washed with saturated aqueous
sodium hydrogencarbonate and saturated aqueous sodium
chloride and then dried over anhydrous magnesium sulfate.
The desiccant was filtered off, and the solvent was
distilled off under reduced pressure. The residue was
purified by silica gel column chromatography
[hexane/ethyl acetate = 5/1 ~ 4/17 to give 3_88 g of the
title compound as a yellow oily substance.
(2) Preparation of 1-benzyl 3-tert-butyl 2-ethyl
(1S,2R)-1-hydroxy-1,2,3-propanetricarboxylate
913 mg of 3-tert-butyl 1,2-diethyl (1S,2R)-1-
hydroxy-1,2,3-propanetricarboxylate was dissolved in a
CA 02244862 1998-07-29
~a ~ b
134
mixture of 21 ml of tetrahydrofuran and 9 ml of water and
stirred with 3 ml of 1N aqueous sodium hydroxide at room
temperature for 3 days. The reaction solution was
acidified with 1N hydrochloric acid and then extracted
with ethyl acetate, and the organic layer was dried over
anhydrous magnesium sulfate. The desiccant was filtered
off, and the solvent was distilled off under reduced
pressure. The residue was purified by silica gel column
chromatography [chloroform/methanol = 50/1 ~ 10/1] to
give 820 mg of 3-tert-butyl 2-ethyl (1S,2R)-1-hydroxy-
1,2,3-propanetricarboxylate.
820 mg of the carboxylate was dissolved in 20 ml of
toluene and stirred with 1.05 g of N,N'-diisopropyl-O
benzylisourea at 100°C for 2 hours under heating. After
addition of 120 mg acetic acid, the reaction solution was
cooled to 0°C, and the insolubles were filtered off and
washed with iced toluene. The filtrate and the washing
liquid were combined and evaporated under reduced
pressure The residue was purified by silica gel column
chromatography [hexane/ethyl acetate = 9/1 --> 3/1] to
give 837 mg of the title compound as a colorless oily
substance.
(3) Preparation of 1-benzyl 3-tert-butyl 2-ethyl 1-oxo-
1,2,3-propanetricarboxylate
1.50 g of 1-benzyl 3-tert-butyl 2-ethyl (1S,2R)-1-
hydroxy-1,2,3-propanetricarboxylate in 30 ml of
chloroform was stirred with 2_35 g of the Dess-Martin
CA 02244862 1998-07-29
~° a
135
reagent (periodinane) at room temperature for 1_5 hours.
The reaction solution was poured into a mixture of
saturated aqueous sodium hydrogencarbonate and saturated
aqueous sodium thiosulfate and extracted with ethyl
acetate_ The organic layer was washed with saturated
aqueous sodium chloride and then dried over anhydrous
magnesium sulfate. The desiccant was filtered off, and
the solvent was distilled off under reduced pressure_
The residue was purified by silica gel column
chromatography [hexane/ethyl acetate = 9/1] to give 1.37
g of the title compound as a colorless oily substance.
(4) Preparation of 1-benzyl 3-tert-butyl 2-ethyl 1-
(methoxymethyloxy)-1-propene-1,2,3-tricarboxylate
992 mg of 1-benzyl 3-tert-butyl 2-ethyl 1-oxo-1,2,3-
propanetricarboxylate in 10 ml of dimethylformamide was
stirred together with 131 mg of 60~ oily sodium hydride
at room temperature for 1 hour. After addition of 273 mg
of chloromethyl methyl ether, the reaction solution was
stirred for another 45 minutes, then poured into water
and extracted with ethyl ether. The organic layer was
dried over anhydrous magnesium sulfate_ The desiccant
was filtered off, and the solvent was distilled off under
reduced pressure_ The residue was purified by silica gel
column chromatography [hexane/ethyl acetate = 5/1] to
give 962 mg of the title compound as a colorless oily
substance.
REFERENCE EXAMPLE 6
CA 02244862 1998-07-29
., v
136
preparation of (2RS 3RS)-3-(tert-buto~cycarbony~)-5-
~xnt-Pt-rar~~3.rnfuran-2-carboxyl ~ c acid
(1) Preparation of (2RS,3SR)-2-(benzyloxycarbonyl)-5-
oxotetrahydrofuran-3-carboxylic acid
5.2 g of (2RS,3SR)-5-oxotetrahydrofuran-2,3-
dicarboxylic acid in 88 ml of acetone was stirred with
6_5 g of 1,1'-dicyclohexylcarbodiimide at room
temperature for 2 hours. After addition of 3.26 ml of
benzyl alcohol, the reaction solution was stirred at the
same temperature for 12 hours. The insolubles were
filtered off, and the filtrated was concentrated under
reduced pressure_ The residue was purified by silica gel
column chromatography [hexane/ethyl acetate = 4/1 -~
chloroform/methanol = 5011] to give 7.93 g of the title
compound as a yellow solid_
(2) Preparation of 2-benzyl 3-tert-butyl (2RS,3RS)-5-
oxotetrahydrofuran-2,3-dicarboxylate
7.93 g of (2RS,3SR)-2-(benzyloxycarbonyl)-5-
oxotetrahydrofuran-3-carboxylic acid in 75 ml of
chloroform was mixed with 5_5 g of 4-
dimethylaminopyridine, 8.6 g of 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimide hydrochloride and 5.7 ml
of tert-butyl alcohol successively and stirred at room
temperature for 60 hours. The reaction solution was
poured into 1N hydrochloric acid under cooling with ice
and extracted with ethyl acetate. The organic layer was
dried over anhydrous magnesium sulfate_ The desiccant
CA 02244862 1998-07-29
.a . x
,.
137
was filtered off, and tha solvent was distilled off under
reduced pressure. The residue was purified by silica gel
column chromatography [hexane/ethyl acetate = 5/1] to
give 6_48 g of the title compound as a white solid.
(3) Preparation of (2RS,3RS)-(3-tert-butoxycarbonyl)-5-
oxotetrahydrofuran-2-carboxylic acid
4_92 g of 2-benzyl 3-tert-butyl (2RS,3RS)-5-
oxotetrahydrofuran-2,3-dicarboxylate in 50 ml of ethyl
acetate was catalytically reduced with 500 mg of a 10~
palladium-carbon catalyst at room temperature for 3 hours
under atmospheric pressure of hydrogen. The catalyst was
filtered off, and the filtrate was evaporated to dryness
to give 3_44 g of the title compound as a white solid_
REFERENCE EXAMPLE 7
prPr,arat-; on of (2S, 3S) -3- (tert-butoxtrcarbony'1 ) -5-
~x~t-Prra1-,Lrdr~f"ran-2-carboxylic acid
(1) Preparation of (2S,3R)-5-oxotetrahydrofuran-2,3-
dicarboxylic acid
16_49 g of 3-tert-butyl 1,2-diethyl (1S,2R)-1-
hydroxy-1,2,3-propanetricarboxylate prepared in Reference
Example 5(1), 100 ml of acetic acid and 50 ml of
concentrated hydrochloric acid were mixed and stirred at
70°C for 5 hours. The acetic acid and the hydrochloric
acid were distilled off under reduced pressure, and the
residue was dissolved in 100- ml of acetic acid and 50 ml
of concentrated hydrochloric acid again and stirred at
70°C for 12 hours. The acetic acid and the hydrochloric
CA 02244862 1998-07-29
4 ~ 1
a c
138
acid were distilled off under reduced pressure, and the
residue was mixed with 100 ml of trifluoroacetic acid and
stirred at 60°C for 5 hours. The trifluoroacetic acid
was distilled off under-reduced pressure, and the residue
was crystallized from hexane-ethyl acetate to give 9.38 g
of the title compound as a white powder_
(2) Preparation of (2S,3S)-3-(tert-butoxycarbonyl)-5-
oxotetrahydrofuran-2-carboxylic acid
The title compound was prepared in the same manner
as in Reference Example 6 by using (2S,3R)-5-
oxotetrahydrofuran-2,3-dicarboxylic acid as the starting
material_
T'f~TT~Z7~TRTAT~ APPLICABILITY
The compound of the present invention has excellent
protein-farnesyl transferase (PFT) inhibitory activities
and is useful as an antitumor agent or an anti-HIV agent_