Note: Descriptions are shown in the official language in which they were submitted.
CA 022467~6 1998-08-18
WO 97/31910 PCT/US97/02712
~ TITLE OF THE rNV~NTION
FIBRINOGEN RE(~EPTOR ANTAGONISTS
CROSS-REF~RENCE TO RELATE;D APPLICAT~ONS
S This application claims priority to U.S. provisional
application No. 60/012,380, filed February 28, 1996.
BACKGROUND OF THE rNVENTION
The invention relates generally to modulating cell adhesion
and to inhibiting the binding of fibrinogen and other proteins to blood
platelets, and inhibiting the aggregation of blood platelets specifically to
the gp IIb/IIIa fibrinogen receptor site. Fibrinogen is a glycoprotein
present in blood plasma that participates in platelet aggregation and in
fibrin forrnation. Platelets are cell-like anucleated fragments, found in
the blood of all m~mm~l~, that also participate in blood coagulation.
Interaction of fibrinogen with the IIb/IIIa receptor site is known to be
essential for normal platelet function.
When a blood vessel is damaged by an injury or other
causative factor, platelets adhere to the disrupted subendothethial
surface. The adherent platelets subsequently release biologically active
constituents and aggregate. Aggregation is initiated by the binding of
agonists, such as thrombin, epinephrine, or ADP to specific platelet
membrane receptors. Stimulation by agonists results in exposure of
latent fibrinogen receptors on the platelet surface, and binding of
fibrinogen to the glycoprotein IIb/IIIa receptor complex.
Attempts have been made to use natural products and
synthetic peptides to deterrnine the mech~ni~m of adhesion and platelet
aggregation. Por example, Rouslahti and Pierschbacher in Scienc~, 23~,
491-497 (1987), describe adhesive proteins such as fibronectin,
vitronectin, osteopontin, collagens, thrombospondin, fibrinogen, and
von Willebrand factor that are present in extracellular matrices and in
blood. The proteins contain the tripeptide arginine-glycine-aspartic acid
(RGD) as their glycoprotein IIb/II~a recognition site. These arginine-
glycine-aspartic acid cont~ining tripeptide~s are recognized by at least
CA 022467~6 1998-08-18
WO 9'7/31910 PCT/US97/02712
one member of a family of structurally related receptors, integrins,
which are heterodimeric proteins with two membrane-spanning
subunits. The authors state that the conformation of the tripeptide
sequence in the individual proteins may be critical to recognition
5 specificity.
Cheresh in P~oc. Nat'l Acad. Sci. U.S.A., 84, 6471-6475,
(1987), describes an Arg-Gly-Asp directed adhesion receptor expressed
by human endothethial cells that is structurally similar to the IIb/IIIa
complex on platelets but is antigenically and functionally distinct. This
10 receptor is directly involved in endothelial cell attachment to fibrinogen,
von Willebrand factor, and vitronectin.
Pierschbacher and Rouslahti, in J. of Biol. Chem., 262,
(36), 1729~-17298 (1987) hypothesized thatthe Arg-~ly-Asp sec~uence
alone would be a sufficient signal for receptor recognition and binding
15 and that, therefore, the conformation of the tri-peptide sequence would
be determinative. Various synthetic peptides were produced and the
authors concluded that the sterochemical conformation of Arg-Gly-Asp
as influenced by enantiomeric substitutions or additions to this se~uence
significantly in~luenced receptor-ligand interaction. The authors further
20 showed that cyclization of a decapeptide by forming a disul~ide bridge
between non-terminal residues Pen and Cys, rendered the peptide much
less effective at inhibiting attachment to fibronectin.
In Proc. Nat'l Acad. Sci. U.S.A., 81, 5985-5988 (1984), the
same authors describe tetrapeptide variants of the cell recognition site of
25 fibronectin that retain attachrnent-promoting activity. Peptides having a
tetrapeptide recognition site are described in U.S. Pat. Nos. 4,589,881
and 4,614,517. A number of large polypeptide fragments in the cell-
binding domain of fibronectin have cell-attachment activity. For
example, see U.S. Pat. Nos. 4,517,686, 4,661,I l l and U.S. Pat. No.
30 4,578,079.
Ruggeri et al., P~ c. Na~'l Acad. Sci. U.S.A., 83, 5708-
5712 ~1986) explore a series of synthetic peptides designed in ~engths to
16 residLIes, that contain RGD and a valine attached to the aspartic acid
CA 022467~6 1998-08-18
WO 97/31910 PCTIUS97/02712
residue of RC~D that inhibit fibrinogen binding to platelets. See also
Koczewiak et al., Biochem. 23, 1767-1774 (1984); Ginsberg et al.,
J. Biol. Chem. 260(7), 3931-3936 (1985); and Haverstick et al., Blo~d
66(4), 946-952 (1985). Other inhibitors are disclosed in Eur. Pat. App.
Nos. 275,748 and 298,820.
A number of low molecular weight polypeptide factors
have been isolated from snake venom. These factors apparently have
high affinity for the gp IIb/IlIa complex. For example, ~uang et al., J.
Biol Chem., 262, 16157-16163 (1987); Huang et al., Biochemistry, 28,
661-666 (1989) describe the primary structure of the venom trigramin
which is a 72 amino acid polypeptide that contains the RGD subunit.
Echistatin is another compound which has high affinity for the gp
IIb/IIIa complex. This polypeptide contains 49 amino acids and has the
~GD subunit and various disulfide bridges. Gan et al., J. Biol. Chem.,
263, 19827-19832 (1988). See also, Dennis et al., Proc. Nat'l Acad. S~.
USA, 87, 2471-2475 (1989). However, these snake venom factors also
have high affinity for other members of the adhesive protein receptor
family including the vitronectin and fibronectin receptors so are not
selective for the gp IIb/IIIa complex.
While it is known that the tripeptide sequence Arg-Gly-Asp
is present in certain polypeptides that can duplicate or inhibit the cell
attachment-promoting effects of fibronectin and vitronectin, the
tripeptide Arg-Gly-Asp has low activity. At present, there is little
understanding of how other amino acids coupled to this sequence
influence binding specificity. U.S. Patent No 5,023,233 discloses small
cyclic hexapeptides which contain the sequence Arg-Gly-Asp and are
useful platelet aggregation inhibitors. U.S. Patent No. 5,037,808
discloses the use of indolyl platelet-aggregation inhibitors which are
believed to act by antagonizing interactions between fibrinogen and/or
extracellular matrix proteins and the platelet gp IIb/IIIa receptor. U.S.
Patent No. 5,037,808 discloses guanidino peptide mimetic compounds
that retain an Asp residue which inhibit platelet aggregation.
WO9014103 describes the use of antibody-poly-peptide conjugates
wherein said polypeptides contain the Arg-Gly-Asp (RGD) sequence.
CA 022467~6 1998-08-18
WO 97/31910 PCT/US97/02712
WO9111458 discloses the use of large cyclic peptide.s
cont~ining RGD flanked by proline residues which are platelet
aggregation inhibitors. WO91()1331 discloses small cyclic platelet
aggregation inhibitors which are synthetic cyclic pentapeptides
5 conf~ining the tripeptide sequence Arg-Gly-Asp and a thioether linkage
in the cycle. U.S. Patent No. 5,051,405 also discloses the use of peptides
and pseudopeptides such as N-amidino-piperidine-3-carboxylglycyl-L-
aspartyl-L-valine that inhibit platelet aggregation and thrombus
formation in m~mm~lian blood. EP 445 796 discloses linear compounds
10 which can include internal piperazinyl or piperidinyl derivatives.
EP437 367 discloses linear polypeptide fibrinogen receptor antagonists.
U.S. Patent No. 5,256,812 discloses compounds of the R l-A-(W)a-X-
(~H2)b-(Y)c-B-Z-~OOR wherein Rl is a guandidino or arnidino moiety
and A and B are chosen from specific monosubstituted aryl or
15 heterocyclic rnoieties.
VVhile a multitude of compounds or peptide analogs
believed to inhi~it platelet aggregation by inhibiting binding to a blood
platelet by fibrinogen are known, the present invention provides novel
fibrinogen receptor antagonists that have significant binding activity and
20 are, therefore, useful for the reasons stated herein. A number of very
serious diseases and disorders involve hyperthrombotic complications
which lead to intravascular thrombi and emboli. Myocardial infarction,
stroke, phlebitis alld a number of other serious conditions create the
need for novel and effective fibrinogen receptor antagonists.
SUMMARY OF THE rNVENTION
One object of this invention is to provide novel compounds
which are active as fibrinogen receptor antagonists. Fibrinogen
receptor antagonists of this invention have the general formula:
X-A-Y-Z-B I~
and include, for example, the compounds of formula
CA 022467~6 1998-08-18
WO 97/31910 PCT/US97/02712
R3 O
/ \ ~ ~ ,CH--C_R6
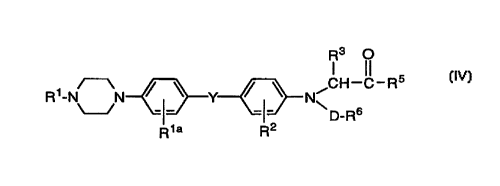
R1a R2 IV,
wherein the variable groups are defined in detail below.
Compounds of the invention are useful for inhibiting the
binding of fibrinogen to blood platelets and for inhibiting the
aggregation of blood platelets. Therefore, it is another object of this
invention to provide methods of inhibiting the binding of fibrinogen to
blood platelets, inhibiting the aggregation of blood platelets, treating
thrombus formation or embolus formation, or preventing thrombus or
embolus formation in a mammal, preferably a human, using the instant
compounds. Combination therapies are also described which employ the
instant compounds with other active agents such as a thrombolytic agent,
an anticoagulant agent, and/or an antiplatelet agent. ~ further object of
this invention is to provide pharmaceutical compositions which are
useful in the above-described methods. Further objects of this invention
will be apparent from the disclosure herein.
DETAILED Dl~SCRIPTION OF THE INVlENTION
The present invention provides compounds having the
formula I
X-A-Y-Z-B
and the pharmaceutically acceptable salts, esters, solvates and
stereoisomers thereof wherein:
X is heterocycle;
25 heterocycle is selected from:
(1 ) a five or six membered saturated, partially unsaturated or
aromatic ring which consists of carbon atoms and one, two
or three heteroatoms selected from the group -O-, -N-,
-N(R 1 ) and -S-, wherein one of the carbon atoms may be
substituted with a member selected from R 1 a and -NHR 1,
CA 02246756 1998-08-18
WO 97/31910 3PCT/US97/02712
. .
(2) an eight to ten membered bicyclic ring system which is
saturated, or completely or partially unsaturated, and which
consists of carbon atoms and one, two or three heteroatoms
selected from the group -O-, -N-, -N(~ arld -S-,
wherein one of the carbon atoms may be substituted with a
member selected from R 1 a and -NHR 1,
(3) a thirteen to fourteen membered tricyclic ring system
which is saturated, or completely or partially unsaturated,
and which consists of carbon atoms and one, two or three
heteroatoms selected from the group -O-, -N-, -N(RI)- and
-S-, wherein one o~ the carbon atoms may be substituted
with a member selected from R 1 a and -NHR l;
A is a bond between X and Y or is selected from:
( 1 ) phenyl substituted with R 1 a,
(2) -N(R1)-, and
/ /
(3) --N
R1a
Y is selected from:
( 1 ) -C 1-8 aL~cyl-,
(2) -C4 1 0 cycloalkyl-,
(3) -Co g aL~cyl-NRl-CO-Co 8 aLkyl-,
(4) -Co g aLkyl-CONR1-Co 8 alkyl-,
(S) -Co g aLkyl-O-Co 8 all;:yl-,
(6) -Co g alkyl-SOp-co-g alkyl-,
(7) -(CH2)0 8-aryl-(CH2)0-8-,
(8) (-CH2)0-6-aryl-sop-
(9) -(CH2)0 8-aryl-CO-(CH2)0-8-,
( 10) -(cH2~o-6-aryl-sop-(cH2)o-6
(11) -(CH2)0 6-NR1-(CH2)0 6-,
( 12) -(cH2)o-6-aryl-cH(oH)-(cH2)o-6
( 13 ) -(cH2)o-~-aryl-coNH-(cH2)o-8-~
CA 022467~6 1998-08-18
WO 97131910 PCT/US97/02712
(14) -Co g alkyl-SOp-NR l-Co g alkyl-,
(15) -Co g alkyl-CO-Co 8 alkyl-, and
~ (16) -Co g alkyl-CH(OH)-Co g-alkyl-;
5 p is an integer selected from 0, 1 and 2;
Z is selected from aryl and heterocycle;
aryl is a 5- or 6-membered aromatic ring system which is unsubstituted
10 or mono-, di- or tri-substituted with R2;
B is
R3 l l
CH--C ~ R5
-(CH2~m--N~ 4
R
wherein m is an integer selected from 0 and 1;
R ~ and R3 are independently selected at each oc~urrence from:
( 1 ) hydrogen,
(2) C 1-10 alkyl-,
(3) C3-8 cycloalkyl-,
(4) aryl-Co 8 alkyl-,
(5) amino-C0 g alkyl-,
(6) C1 6 alkylamino-Co g alkyl-,
(7) C1 6 dialkylamino-Co 8 alkyl-,
(8) Cl 3 acylamino-Co g alkyl-,
(9) C1 4 alkoxy-Co 6 alkyl-,
(10) -C0-6 alkyl-CO2H,
( 1 1 ) -C0-6 alkyl-CO2Cl -3 alkyl,
(12) -O-C0-6 alkyl-CO2H and
( 13) hydroxy-Co-6 alkyl-;
Rla is independently selected at each occurrence from:
( l ) hydrogen,
CA 02246756 1998-08-18
WO 97/31910 rcT/uss7/o27l2
(2) halogen,
(3) C1-10 alkyl-,
(4) C3-8 cycloaLkyl-,
(S) aryl-Co 8 alkyl-,
(6) amino-C0 8 alkyl-,
(7) C1 6 aLkylamino-Co 8 alkyl-,
(8) C1 6 dialkylamino-Co 8 alkyl-,
(9) Cl 3 acylamino-Co 8 alkyl-,
( 10) C 1 4 alkoxy-Co-6 alkyl-,
(1 1 ) -C0-6 alkyl-C02H,
(12) -C0-6 alkyl-C02C1 3 alkyl,
( 13 ) -0-C0-6 alkyl-C02H,
(14) hydroxy C0-6 alkyl and
( 1 5) oxo (=0);
1~
R2 is independently selected at each occurrence from:
( 1 ) hydrogen,
(2) halogen,
(3) Cl 1o alkyl-,
(4) C3-8 cycloalkyl-,
(5) aryl-Co 8 alkyl-,
(6) amino-C0 8 alkyl-,
(7) C1 6 alkylamino-Co 8 aLkyl-,
(8) C 1 6 dialkylamino-Co-8 alkyl-,
(9) Cl 3 acylamino-Co 8 alkyl-,
(10) Cl 4 alkoxy-Co 6 alkyl-,
(1 1 ) -C0-6 alkyl-C02H,
(12) -C0-6 alkyl-C02Cl 3 aLIcyl,
(13) -0-C0-6 alkyl-C02H, and
( ~ 4) hydroxy C0-6 alkyl;
R4 is ~elected from
( 1 ) -(CH2)p-D-R6 wherein p i.s defined above,
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
(2) ~ , and
O D-
(3) G~'
D- or
(4) when Z is unsubstituted or substituted phenyl and m is zero,
R4 together with the nitrogen to which it is attached can
S form a bicyclic structure with Z (phenyl) as follows:
~0
CH-C-R5
~\J 2 R3 o
R
D is selected from -SO2- and -C(O)-;
R5 is selected from:
(1) -OH,
(2) C1 g alkyloxy-,
(3) aryl C0-6 aLkyloxy-,
(4) C 1 8 alkylcarbonyloxy C1 4 alkyloxy-,
(5~ aryi C1 8 aikyicarbonyloxy Cl 4 aLkyloxy-, and
(6) L- or D-amino acid joined by an amide linkage and
wherein the carboxylic acid moiety of said amino acid is as
the free acid or is esterified by C1 6 alkyl; and
R6 is selected from:
(1) -Cl 6alkyl, unsusbstituted, mono- or di-substituted with
Rla,
CA 02246756 1998-08-18
WO 97131910 PCT/US97102712
- ~0 -
(2) -(Co 6aL~cyl)aryl, wherein the aLkyl group is unsusbstituted,
mono- or di-substituted with Rla,
(3) -(Co 6alkyl)heterocycle, wherein the aLkyl group is
unsusbstituted, mono- or di-substituted with Rla,
(4) -NRl(Cl 6aLkYl), wherein the aLkyl group is unsusbstituted,
mono- or di-substituted with R1a,
(S) -NRl(Co 6alkylaryl), wherein the aLkyl group is
unsusbstituted, mono- or di-substituted with Rla,
(6) -NRl(Co 6aLlcylheterocycle), wherein the aLkyl group is
unsusbstituted, mono- or di-substituted with R1a,
(7) -C3-6 cycloalkyl, and
(8) -CF3.
In a one embodiment of this invention are compounds of
15 formula I which have the particular formula II:
Rl 3 l l
CH--C- R5
X-A-Y-Z--N~
D-R6 II
and the pharmaceutically acceptable salts, solvates and stereoisomers
20 thereof wherein:
X-A- together represent a group selected from:
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 11 -
R 1--N3 R 1--N~N-- R 1_ N3~
R1a
R1_N N~\ N~N
\ I , ~I ,
R1a R1a
/~ R1--NH N , ~(R1~_,
(C~ ~ R ~ (C~N
n is an integer selected from 2, 3, 4, and 5;
Q is selected from -N(R1)-, -S- and -0-;
S Y is selected from:
(1) -Co g aLkyl-NRI-Co-co-8 alkyl-,
(2) -Co g a~kyl-CONRl-Co g alkyl-,
(3) -C0-8 aLkyl-O-Co 8 alkyl-,
(4) -Co g aLkyl-SOp-co-g alkyl-,
(5) -(C H 2)0-6-N Rl-(C H 2)0 6-,
(6) -C0-8 alkyl-S Op-~nRl-Co-8 alkyl-,
(7) -Co g aLkyl-CO-CO 8 alkyl-, and
(8) -Co g alkyl-CH(OH)-Co g-alkyl-;
15 p is an integer selected from 0, 1 and 2;
Z is selected from
(1) aryl and
(2) a five or six membered saturated, partially unsaturated or
aromatic heterocyclic ring which is unsubstituted, or
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/OZ712
.
monosubstituted or disubstituted with R 1 a, which consists of
carbon atoms and one, two or three heteroatoms selected from the
group -O-, -N-, -N(Rl~- and -S-, and which may be fused to a
benzene ring to form a bicyclic structure, for example,
--N~ or ~ ~
aryl is a 5- or 6-membered aromatic carbon ring which is unsubstituted
or mono-, di- or tri-substituted with R2;
Rl is independently selected at each occurrence from -H, Cl lo alkyl,
C3-8 cycloalkyl-, aryl-Co 8 aLkyl- and hydroxy-Co 6 alkyl-;
Rla is independently selected at each occurrence from -H, halogen,
-Cl-10 aLkyl, C3-8 cycloalkyl-, aryl-Co 8 aLkyl-, and
amino-C0 8 alkyl-;
R2 is independently selected at each occur~ence ~rom -H7 halogen,
-Cl lo aLkyl, C3-8 cycloalkyl-, aryl-Co 8 alkyl- and
Cl 4 alkoxy-Co 8 aLkyl-;
R3 is independently selected at each occurrence from -H, -~1-10 alkyl,
C3-8 cycloaLkyl- and aryl-Co 8 aLkyl-;
R5 is selected from:
( 1 ) -OH,
(2) C1 8 alkyloxy-,
(3) aryl-Co 6 aLkyloxy-,
(4) C1 8 aLkylcarbonyloxy-Cl 4 alkyloxy-, and
(S) aryl-C 1-8 alkylcarbonyloxy-C 1-4 alkyloxy-;
D is selected from -SO2- and -C(O)-; and
R6 is selected from:
(1 ) -Cl 6aLkyl, unsusbstituted, mono- or di-substituted with
Rla,
(2) -(Co 6aL~cyl)aryl, wherein the aLkyl group is unsusbstituted,
mono- or di-substituted with Rla,
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 13 -
(3) -(Co 6alkyl)heterocycle, wherein the aLkyl group is
unsusbstituted, mono- or di-substituted with Rla,
(4) -NRl(Cl 6aL~cyl), wherein the alkyl group is unsusbstituted,
mono- or di-substituted with R1a,
(5) -NRl(Co 6alkylaryl~, wherein the aLkyl group is
unsusbstituted, mono- or di-substituted with R1a,
(6) -NRl(~0 6aLkylheterocycle), wherein the aLkyl group is
unsusbstituted, mono- or di-substituted with R1a,
(7) -C3-6 cycloaLkyl,
(8) -CF3,
(9) ~~ ~ (10)~?
O CH2 , and Cl H2 o OF
~1 l ) when Z is unsubstitu~d or substituted phenyl, D-R6
lS together with the nitrogen to which it is attached can form a bicyclic structure with Z (phenyl) as follows:
~0
'CH-C-R5
~\iJ~ R3 1 1
R2 ; and
heterocycle is selected from a five or six membered saturated, partiall~
unsaturated or aromatic ring which is unsubstituted, or
monosubstituted or disubstituted with Rla, and which consists of
carbon atoms and one or two heteroatoms selected from the group
-O-, -N-, -N(R1)- and -S-;
and wherein the rem~ining variables are as defined above in formula I.
CA 022467=,6 1998-08-18
WO 97/31910 PCT/US97/02712
- 14 -
In a second, further embodiment of this invention are
compounds of formula II and the pharmaceutically acceptable salts,
solvates and stereoisomers thereof wherein X-A- together represent a
group selected from:
Rl-N N~ R1-
R1a R1a
N3N/~} , and
S R
and the rem~inin~ variable are as defined above in formula ~.
In a third, fur~er embodiment of this invention are
10 compounds of formula II and the ph~ eeutically acceptable salts,
solvates and stereoisomers thereof wherein X-A- together represent a
group selected from:
R1-N~ N~ R1_N3
R1a R1a
3 /~}, and /~
R1a
15 and the pha~naceutically acceptable salts, hydrates and stereoisomers
thereof wherein:
Q is selected form -NH-, -O- and -S-;
Y is selected from:
( 1 ) -NR I -CO-,
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
(2) -CONR I -,
(3) -O-,
(4) -SOp
(5) -NRl
(6) -SOp-NR 1 ,
(7) -CO-, and
(8) -CH(OH)-;
p is an integer selected from 0, 1 and 2;~0 Z is selected from:
( 1 ) phenyl,
(2) phenyl which is mono-, di- or tri-substituted with R2,
(3) thienyl,
--N~
(4) and (5) o
R1 is independently selected at each occurrence from -H and
-C1-10 alkyl;
R 1 a is independently selected at each occurrence from -H, halogen and
-Cl 1o alkyl,
R2 is independently selected at each occurrence from -H, halogen and
-C l l o aIkyl;
R3 is independently selected at each occurrence from -H and
-Cl lo alkyl;
25 R5 is selected from:
( 1 ) -OH,
(2) C1 8 alkyloxy-,
(3) aryl-Co 6 aLkyloxy-,
(4) C 1-8 alkylcarbonyloxy-C l 4 alkyloxy-, and
(5) aryl-Cl 8 aLkylcarbonyloxy-Cl 4 aLkyloxy-;
D is selected from -SO2- and -C(O)-; and
CA 02246756 1998-08-18
WO 97/31910 PCT/US97102712
- 16 -
R6 is selected from:
(1) -Cl 6aL~cyl, unsusbstituted, mono- or di-substituted with
~la,
(2) -(Co 6aL~cyl)aryl, wherein the alkyl group is unsusbstituted,
mono- or di-substituted with Rla,
(3) -(Co 6alkyl)heterocycle, wherein the aLkyl group is
unsusbstituted, mono- or di-substituted with Rla,
(4) -NRl(Cl 6aLkyl), wherein the aLkyl group is unsusbstituted,
mono- or di-substituted with Rl a,
(5) -NRl(Co 6alkylaryl), wherein the aLkyl group is
unsusbstituted, mono- or di-substituted with Rla,
(6) -NRl(Co 6aLkylheterocycle), wherein the aLlcyl group is
unsusbstituted, mono- or di-substituted with Rla,
(7) -( ~3-6 cycloaLkyl,
(8) -CF3,
(g) ~ (10)~
O CH2 , and Cl H2 ~ or
(1 ~ ) when Z is unsubstituted or substituted phenyl, D-R6
together with the nitrogen to which it is attached can form a
bicyclic structure with Z (phenyl) as follows:
~0
CH- ICI -R~
R2 ; and
heterocycle is selected from a five or six membered saturated, partially
,
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
-
unsaturated or aromatic ring which is unsubstituted, or
monosubstituted or disubstituted with Rla~ and which consists of
carbon atoms and one or two heteroatoms selected from the group
-O-, -N-, -N(Rl)- and -S-;
and any rem~ining variables are as defined in follnula II.
In a fourth, further embodiment of this invention are
compounds of formula II having the particular formula III
R1-N~N~ \D-R6
R1a III
and the ph~ ceutically acceptable salts, solvates and stereoisomers
thereof wherein:
15 Y is selected from:
(1) -NR1 CO,
(2) -CONR 1 ,
(3) -O-,
(4) -SOp
(5) -NR ~ -,
(6) -SOp-NR 1 ,
(7) -CO-, and
(8) -CH(OH)-;
25 p is an integer selected from 0, 1 and 2;
Z is selected from:
( 1 ) phenyl,
(2) phenyl which is mono-, di- or tri-substituted with R2,
(3) thienyl,
CA 02246756 1998-08-18
WO 517/31910 PCT/US97/02712
- 18 -
--N~ , N
(43 and (S) o
Rl is independently selected at each occurrence from -H and
10 alkyI;
5 R I a is independently selected at each occurrence from -H, halogen and
-Cl lo alkyl;
R2 is independently selected at each occurrence from -H, halogen and
-Cl 1o alkyl;
R3 is independently selected at each occurrence from -H and
-C~l-10 alkyl;
RS is selected from:
( 1 ) -OH,
(2) C 1-8 alkyloxy-,
(3) aryl-Co 6 aLkyloxy-,
(4) C1 8 aLkylcarbonyloxy-Cl 4 aLkyloxy-, and
(S) aryl-C 1-8 aLkylcarbonyloxy-C 1-4 aLkyloxy-;
D is selected from -SO2- and -C(O)-; and
R6 is selected from:
(1) -Cl 6aLkyl, unsusbstituted, mono- or di-substituted with
Rla,
(2) -(Co 6aLkyl)aryl, wherein the aLkyl group is unsusbstituted,
mono- or di-substituted with ~la,
(3) -(Co 6alkyl)heterocycle, wherein the alkyl group is
unsusbstituted, rnono- or di-substituted with Rl a,
(~) -NRl(Cl 6alkyl), wherein the aLkyl group is unsusbstituted,
mono- or di-substituted with R 1 a,
(5) -NRl((:~0 6alkylaryl), wherein the alkyl group is
unsusbstituted, mono- or di-substituted with R1a,
(6) -NRl(Co 6aLkylheterocycle), wherein the aLkyl group is
unsusbstituted, mono- or di-substituted with R1a,
(7) -C3-6 cycloaLkyl~
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 19 -
(8) -CF3,
(g) ~ (10) ~
O CH2 , and I , or
S (11) when Z is unsubstituted or substituted phenyl, D-R6
together with the nitrogen to which it is attached can form a
bicyclic structure with Z (phenyl) as follows:
~0
'CH-C-R5
~\iJ~ R3 o
R2 ; and
heterocycle is selected from a five or six membered saturated, partially
unsaturated or aromatic ring which is unsubstituted, or
monosubstituted or disubstituted with R1a, and which consists of
carbon atoms and one or two heteroatoms selected from the group
-O-, -N-, -N(R1)- and -S-;
and the rem~ining variables are as defined in formula II.
In a fifth, further embodiment of this invention are
compounds of formula III having the particular formula IV
R3 0
Rl-N N~Y~ CH~ R5
D R6
Rla R2 IV
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- - 20 -
and the ph~ ceutically acceptable salts, solvates and stereoisomers
thereo~ wherein:
Y is selected from -C(O)-N(R 1 )- and -N(R 1 )-C(O)-;
R1 is independently selected at each occurrence from -H and
S -C~ lo aLkyl;
Rla is independently selected at each occurrence from -H, halogen and
-Cl 10 alkyl;
R2 is independently selected at each occurrence from -H, halogen and
-Cl lo alkyl;
R3 is independently selected at each occurrence from -H and
-~1-10 alkyl;
RS is selected from:
(1) -OH,
(2) C1 8 alkyloxy-,
(3) aryl-Co-6 alkyloxy-,
(4) C1 8 alkylcarbonyloxy-Cl 4 aLkyloxy-, and
(5) aryl-Cl g aLkylcarbonyloxy-Cl 4 aLkyloxy-;
D is selected from -SO2- and -C~O)-; and
R6 is selected from:
(1 ) -C~1 -6alkYl~ unsusbstituted, mono- or di-substituted with
Rla,
(2) -(Co 6aLkyl)aryl, wherein the aLkyl group is unsusbstituted,
mono- or di-substituted with Rl a,
(3) -(Co 6alkyl)heterocycle, wherein the aLkyl group is
unsusbstituted, mono- or di-substituted with Rla,
(4) -NRl(CI 6alkyl), wherein the aLkyl group is unsusbstituted,
mono- or di-substituted with R 1 a,
(S) -NRl(Co 6alkylaryl), wherein the aLkyl group is
unsusbstituted, mono- or di-substituted with Rl a,
(6) -NRl(Co 6aLkylheterocycle), wherein the alkyl group is
unsusbstituted, mono- or di-substituted with Rla,
(7) -C3-6 cycloalkyl,
(8) -CF3,
CA 022467~6 1998-08-18
WO 97/31910 PCT/US97/02712
~ <
(g) ~ (10)~
OCH2 , and I , or
(1 1 ) when Z is unsubstituted or substituted phenyl, D-R6
together with the nitrogen to which it is attached can form a
bicyclic structure with Z (phenyl) as follows:
~0
J~ R3 li
R2 ; and
heterocycle is selected from a five or six membered saturated, partially
unsaturated or aromatic ring which is unsubstituted, or
monosubstituted or disubstituted with Rla, and which consists of
carbon atoms and one or two heteroatoms selected from the group
-O-, -N-, -N(R1)- and -S-;
and any rem~ining variables are as defined in formula III.
In a sixth, further embodiment of this invention are
compounds of formula I~ wherein Y is selected from -C(O)-NH- and
-NH-C~(O)-; and R6 is selected from (1) unsubstituted, mono and di-
substituted phenyl, (2) methyl, (3) benzyl wherein the aryl portion may
be unsubstituted, mono or di-substituted, and (4) thienyl; and any
remaining variables are as defined in formula IV.
Tn one class of the instant six embodiments are compounds
of formulas I, II, III, and IV as defined in the general definition
wherein Y is -C(O)-NH-. Compounds in this class are exemplified, but
not limited to, those of formula V as defined in Table I. The carbons
around the phenyl rings in formula V have been arbitrarily numbered in
order to clearly identify the substituent positions, where necessary.
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
TABLE I
HN~N~ C-NH--~ D-R6
R1a R2
V,
R1a R_ R_ D R_
a) H H OCH2CH3 -C(O)- phenyl
b) H H OH -C(O)- -CH3;
c) H H OH -C(O)- phenyl;
d) lH H OCH3 -C(O)- -CH3;
e) H H OH -C(O)- 2-F-phenyl;
f) H H OH -C(O)- cyclopropyl;
g) H H OH -C(O)- 3-pyridinyl;
h) H H OH -C(O)- 4-pyridinyl;
i) 6-CH3 H OH -C(O)- phenyl;
j) H 6-CH3 OH -C(O)- phenyl;
k) H S-CH3 OH -C(O)- phenyl;
1) H H OH -SO2- -CF3;
m~ H H O~ -SO2- 2-F-phenyl;
n) H H OC2HS -S02- 2-thienyl;
o) H H OH -so2- 2-thienyl;
p) H H OH -so2- phenyl;
q) H 6-CH3 O~ -SO2- phenyl;
r) H 2-Br OH -SO2- 2-F-phenyl;
s) H H OH -S~2-
o CH2
CA 02246756 l998-08-l8
WO 97/31910 PCT/US97/02712
- 23 -
t) H H OH -S02- G~?
CH2 o
; and
u) H H OH -C(O)- -CH2-CH2- bondedto
D and the carbon
denoted with * to form
~0
~ CHz-~
In a second class of the instant embodiments are compounds
of formulas I, II, m, and IV wherein Y is -NH-C(O)-. Compounds in
this class are exemplified, but not limited to, those of formula VI as
S defined in Table II.
TABLE II
~ 1~ CH2 B- R5
HN N~NH-C--Z--N
\=/ SO2-R6
Vl
Z R_ R6
a) 1,4-phenyl OH CH3;
b) 1,4-phenyl OCH3 CH3;
c) 1,4-phenyl OCH(CH3)2 CH3;
d) 1,4-phenyl OH phenyl
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 24 -
e) 1,4-phenyl OH 4-Br-phenyl;
f) 1,4-phenyl OH benzyl; and
g) 2,5-thienyl OH phenyl.
Additional examples of compounds within the scope of this
~nvention are shown below but a~e not limited to, those of formula VII
as defined in Table III.
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
T~BLE III
Cl 12-C-OH
X~A~Y~Z~(CH2)n--N~ /=\
Vll SO2~
X-A Y Z n
N~3 3 -C(O)NH- 1,4-phenyl 0
HN~ -C(O)NH- 1,4-phenyl 0
c) ~ ~ -C(O)NH- 1,4-phenyl 0
HN~
HN
HN~ CH2)2-
HN~ } -(CH2)3-0- 1,4-phenyl
s
Wlhen any substituent (e.g., Rl, R2, etc.) occurs more than
one time in any constituent, its definition on each occurrence is
independent of its definition at any other occurrence. Also,
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 26 -
combinations of substitutents and/or variables are pe~nissible only if
such combinations result in stable compounds.
As used herein "aLkyl" is intended to include bo~
branched- and straight-chain saturated aliphatic hydrocarbon groups
S having the specified number of carbon atoms, e.g., methyl (Me),
ethyl (Et), propyl (Pr), butyl (Bu), pentyl, hexyl, heptyl, octyl, and
the isomers thereof such as isopropyl (i-Pr), isobutyl (i-Bu), secbutyl
(s-Bu), tertbutyl (t-Bu), isopentyl, isohexyl and the like. The term
"cycloaLkyl" is intended to include cyclized alkyl chains having the
10 specified number of carbon atoms, e.g., cyclopropyl, cyclobutyl,
cyclopentyl, cyclohexyl and cycloheptyl. "Alkoxy" or "alkyloxy"
represents an alkyl group having the indicated number of carbon
atoms attached through an oxygen bridge, e.g., methoxy, ethoxy,
propyloxy, iso-propoxy, n-butoxy, iso-butoxy, sec-butoxy, t-butoxy
15 and the like.
The specified number of carbon atoms in the
appropriate groups described herein may include a zero in the range,
e.g., C0-6 or C0 8. When a zero is in the specified range, it means
that a bond is present in place of that carbon group.
The term halo or halogen is meant to include fluoro,
chloro, bromo and iodo. The term "oxy" means an oxygen (O)
atom.
~e term "aryl" is defined above in the definition of
Formula I; unsubstituted, mono-, di- and tri-substituted phenyl (Ph)
2~ is preferred.
The tellll heteroaryl is defined above in the definition of
Formula I. The term heteroaryl encompasses a five or six-
membered heteroaryl ring as defined in formula I ~used to a
benzene, pyridine or pyrimidine ring. Examples of heteroaryl
30 groups include pyrrolyl, triazolyl, pyrazolyl, imidazolyl, pyridyl,
pyrimidinyl, pyrazinyl, furanyl, pyranyl, thienyl, oxazolyl,
isooxazolyl, thiazolyl, indolyl, benzimidazolyl, benzofuranyl,
benzopyranyl, benzothienyl, quinolyl, isoquinolyl and the like. The
heteroaryl ring may be attached within structural Formula I at any
CA 022467~6 l998-08-l8
WO 97/31910 PCT/US97/02712
- 27 -
heteroatom or a carbon atom in the ring which results in the creation
of a stable structure. Preferred heteroaryl groups include pyridyl,
~ thiazolyl, oxazolyl, thienyl, indolyl, benzofuranyl, and benzothienyl.
The term "C0-6 alkylaryl" as used herein includes an alkyl
5 group as defined above bonded to an aryl group as defined above. The
C0-6 designation refers to the alkyl component of the alkylaryl unit.
Examples of C0-6 aL~cylaryl include phenyl-, benzyl-, fluorobenzyl-,
chlorobenzyl-, phenylethyl-, phenylpropyl-, fluorophenylethyl-, and
chlorophenylethyl-.
The term "C0-6 alkylheterocycle" as used herein includes
an alkyl group as defined above bonded to a heterocycle group as
defined above. The C0-6 designation refers to the aL~cyl component of
the alkylheterocycle unit. Examples of C0-6 aLkylheterocycle include
thienyl-, thienylmethyl-, thienylethyl-, and thienylpropyl-.
Arnino acids suitable for compounds of the present
invention include naturally occurring L- or D-amino acids, for exarnple,
those naturally occurring L-amino acids present in hllm~n~, e.g., protein
amino acids, including L-alanine, L-arginine, L-asparagine, L-aspartic
acid, L-cysteine, L-glllt~mine, L-glutamic acid, L-glycine, L-histidine,
L-isoleucine, L-leucine, L-lysine, L-methionine, L-phenyl~l~nine, L-
proline, L-serine, L-threonine, L-tryptophan, L-tyrosine, and L-valine,
and those naturally occurring D-amino acids which are non-protein
arnino acids, such as those found, for example, in antibiotic substances
produced by bacteria and fungi, including D-valine, D-asparagine, D-
glllt~m~te, D-ornithine, D-phenyl~l~nine, D-leucine, D-cysteine, and D-
aspartate. (see Zubay "BIOCHEMI~TRY" Addison-Wesley Publi~hing
Company, Inc. (Reading, MA) 1983 pp. 867-870 and Stryer
"BIOCHEMISTRY" W.H. Freeman and Company (New York, NY) 3rd
Edition 1988 pp. 16-21).
The term "pharmaceutically acceptable salts" shall mean
non-toxic salts of the compounds of this invention which are generally
prepared by reacting the free base with a suitable organic or inorganic
acid. Representative salts include the following salts:
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 28 -
acetate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate,
borate, bromide, calcium edetate, camsylate, carbonate, chloride,
clavulanate, citrate, dihydrochloride, edetate, edisylate, estolate, esylate,
fumarate, gluceptate, gluconate, glllt~m~le, glycollylarsanilate,
5 hexylresorcinate, hydrabamine, hydrobromide, hydrochloride,
hydroxynapthoate, iodide, isothionate, lactate, lactobionate, laurate,
~ te, maleate, mandelate, mesylate, methylbromide, methylnitrate,
methylsulfate, mucate, napsylate, nitrate, oleate, oxalate, pamaote,
palmitate, panthothenate, phosphate/diphosphate, polygalacturonate,
10 salicylate, stearate, subacetate, succinate, t~nn~te, tartrate, teoclate,
tosylate, triethiodide, valerate.
Prodrugs, such as ester derivatives of described
compounds, are compound derivatives which, when absorbed into the
bloodstream of a warm-blooded ~nim~l, anabolize or cleave in such a
15 manner as to release the drug fo~n and permit the drug to afford
improved therapeutic efficacy.
The compounds of the present invention are chiral and
the present compounds may occur as racemates, racemic mixtures
and as individual diasteriomers or enantiomers with all such isomeric
20 forms being included within the scope of this invention.
Furthermore, some of the crystalline forms for compounds of the
present invention may exist as polymorphs and as such are intended
to be included in the present invention. In addition, some of the
compounds of the instant invention may form solvates with water or
25 common organic solvents. Such solvates and hydrates, as well as
anhydrous compositions, are encompassed within the scope of this
invention.
The term "therapeutically effective amount" shall mean that
amount of a drug or pharmaceutical agent that will elicit the biological
3(~ or medical response of a tissue, system, ~nim~l or human that is being
sought ~y a researcher, veterinarian, medical doctor or o~er clinician,
which includes alleviation of the symptoms oi~ the disease being treated.
- The term "m~mm~l" includes hllm~ns.
CA 022467~6 1998-08-18
WO 97/31910 PCT/US97102712
- 29 -
The term "anti-coagulant" shall include heparin, and
warfarin. The term "thrombolytic agent" shall include agents such as
- streptokinase and tissue pl~minogen activator. The term "platelet anti-
aggregation agent" shall include agents such as aspirin and
S dipyridamole.
The compounds of the present invention can be
~1mini.~tered in such oral fo~ns as tablets, capsules (each of which
includes sustained release or timed release formulations), pills, powders,
granules, elixirs, tinctures, suspensions, syrups, and emulsions.
10 Likewise, they may be ~-lministered in intravenous (bolus or infusion),
intraperitoneal, subcutaneous, or intramusculsar form, all using forms
well known to those of ordinary skill in the pharmaceutical arts. An
effective but non-toxic amount of the compound desired can be
employed as an anti-aggregation agent.
(~ompounds of the invention may be ~clmini~tered to
patients where prevention of thrombosis by inhibiting binding of
fibrinogen to the platelet membrane glycoprotein complex IIb/I~Ia
receptor is desired. They are useful in surgery on peripheral arteries
(arterial grafts, carotid endarterectomy) and in cardiovascular surgery
20 where manipulation of arteries and organs, and/or the interaction of
platelets with artificial surfaces, leads to platelet aggregation and
consumption. The aggregated platelets may form thrombi and
thromboemboli. Compounds of this invention may be ~1mini~tered to
these surgical patients to prevent the formation of thrombi and
2~ thromboemboli.
Extracorporeal circulation is routinely used for
cardiovascular surgery in order to oxygenate blood. Platelets adhere to
surfaces of the extracorporeal circuit. Adhesion is dependent on the
interaction between gp IIb/~IIa on the platelet membranes and
30 fibrinogen adsorbed to the surface of the circuit. (Gluszko et al., Amer.
J. Physi~ol., 252(H), 615-621 (1987)). Platelets released from artificial
surfaces show impaired hemostatic function. Compounds of the
invention may be a~lrnini~tered to prevent adhesion.
CA 02246756 1998-08-18
WO 97/31910 PCTJUS97/02712
- 30 -
Other applications of these compounds include prevention
of platelet thrombosis, thromboembolism and reocclusion during and
after thrombolytic therapy and prevention of platelet thrombosis,
thromboembolism and reocclusion after angioplasty or coronary artery
5 bypass procedures. They may also be used to prevent myocardial
infarction.
The dosage regimen l~tiTi~ing the compounds of the present
invention is selected in accordance with a variety of ~actors including
type, species, age, weight, sex and medical condition of the patient; the
10 severity of the condition to be treated; the route of a~lmini~tration; the
renal and hepatic function of the patient; and the particular compound
or salt thereof employed. An ordinarily skilled physician or
veterinarian can readily determine and prescribe the effective amount of
the drug required to prevent, counter, or arrest the progress of the
1 5 condition.
Oral dosages of the present invention, when used for the
indicated effects, will range between about 0.01 mg per kg of body
weight per day (mg/kg/day) to about 100 mg/lcg/day and preferably
0.01-100 mg/kg/day and most preferably 0.01-20 mg/kg/day.
20 Typically, oral dosages for an adult patient are, for example, 1 mg, lû
mg or 100 mg. Intravenously, the most preferred doses will range
from about 1 to about 10 mg/kg/minllte during a constant rate infusion.
Advantageously, compounds of the present invention may be
~clmini~tered in divided doses of two, three, or four times daily.
25 Furthermore, preferred compounds for the present invention can be
~lmini~tered in intranasal form via topical use of suitable intranasal
vehicles, or via transdermal routes, using those ~orms of transdermal
skin patches well known to those of ordinary skill in that art. To be
atlmini~tered in the form of a transdermal delivery system, the dosage
30 a-1mini~tration will, or course, be continuous rather that inteImittent
throughout the dosage regirne.
In the methods of the present invention, the compounds
herein described in detail can form the active ingredient, and are
typically aflmini~tered in admixture with suitable pharmaceutical
CA 022467~6 1998-08-18
WO 97/31910 PCT/US97/02712
- 31 -
diluents, excipients or carriers (collectively referred to herein as
"carrier" materials) suitably selected with respect to the intended form
Of ~lministration, that is, oral tablets, capsules, elixirs, syrups and the
like, and consistent with convention pharmaceutical practices. As such,
5 a therapuetically effective amount of a compound of formula I can be
used for the preparation of a medicament useful for inhibiting the
binding of fibrinogen to blood platelets, inhibiting the aggregation of
blood platelets, treating thrombus formation or embolus formation, or
preventing thrombus or embolus formation in a m:~mm~l For example,
10 the medicament may be comprised of from 1 mg to 100 mgs of a
compound of formula I, or more particularly, it may contain 1 mg, 10
mgs, 50 mgs, or 100 mgs of said compound.
Therapeutically effective amounts of a compound of
formula I together with another active agent such as an anticoagulation
15 agent or a thrombolytic agent can be used for the preparation of a
medicament useful for inhibiting the binding of fibrinogeh to blood
platelets, inhibiting the aggregation of blood platelets, treating thrombus
formation or embolus formation, or preventing thrombus or embolus
formation in a m~mm~l. Examples of other active agents which may be
20 used include pl~minogen activators or streptokinase, heparin, aspirin,
warfarin, ticlopidine and/or clopidogrel.
For instance, for oral ~(lministration in the form of a tablet
or capsule, the active drug component can be combined with an oral,
non-toxic, pharmaceutically acceptable, inert carrier such as lactose,
25 starch, sucrose, glucose, methyl cellulose, magnesium stearate,
dicalcium phosphate, calcium sulfate, mannitol, sorbitol and the like; for
oral a~lminietration in li~uid form, the oral drug components can be
combined with any oral, non-toxic, ph~ ceutically acceptable inert
carrier such as ethanol, glycerol, water and the like. Moreover, when
30 desired or necessary, suitable binders, lubricants, distintegrating agents
and coloring agents can also be incorporated into the mixture. Suitable
binders include starch, gelatin, natural sugars such as glucose or beta-
lactose, corn-sweeteners, natural and synthetic gums such as acacia,
tragacanth or sodium alginate, carboxymethylcellulose, polyethylene
CA 02246756 1998-08-18
WO 97131910 PCT/US97/02712
glycol, waxes and the like. Lubricants used in these dosage fo~ns
include sodium oleate, sodium stearate, m~nesium stearate, sodium
benzoate, sodium acetate, sodium chloride and the like. Disintegrators
include, without limit~tion, starch methyl cellulose, agar, bentonite,
5 xanthan gum and the like.
The compounds of the present invention can also be
~1m;ni~tered in the form of liposome delivery systems, such as small
unilamellar vesicles, large llnil:~mellar vesicles and multilamellar
vesicles. Liposomes can be formed from a variety of phospholipids,
10 such as cholesterol, stearylamine or phosphatidylcholines.
Compounds of the present invention may also be delivered
by the use o~ monoclonal antibodies as individual carriers to which the
compound molecules are coupled. The compounds of the present
invention may also be coupled with soluble polymers as targetable drug
15 carriers. Such polymers can include polyvinylpyrrolidone, pyran
copolymer, polyhydroxy-propyl-methacrylamide-phenol, polyhydroxy-
ethyl-aspartamide-phenol, or polyethyleneoxide-polylysine substituted
with palmitoyl residues. Furthermore, the compounds of the present
invention may be coupled to a class of biodegradable polymers useful in
20 achieving controlled release of a drug, for example, polylactic acid,
polyglycolic acid, copolymers of polylactic and polyglycolic acid,
polyepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters,
polyacetals, polydihydropyrans, polycyanoacrylates and cross linked or
amphipathic block copolymers of hydrogels.
2~ The compounds of the present inven~ion can also be co-
~lmin;~tered with suitable anticoagulation agents or thrombolytic agents
such as pl~minogen ac~ivators or streptokinase in the treatment of
various vascular pathologies. They may also be combined with hepann,
aspirin, warfarin, ticlopidine and/or clopidogrel. Co~(lmin;~tration
30 includes ~lmini~tration together at essentially the same time in a single
dosage form or in separate dosage forms, or each agent ~lministered at
separately staggered times in order to achieve beneficial thrombosis
prevention or thrombolysis.
CA 022467~6 1998-08-18
WO 97/31910 PCT/US97/OZ712
The compounds of the present invention can be prepared
readily according to the following Schemes and Examples or
modifications thereof using readily available starting materials,
reagents and conventional synthesis procedures. Those skilled in the
5 art will readily understand that known variations of the conditions
and processes of the following preparative procedures can be used to
prepare these compounds. The examples are not intended to be
limitations on the scope of the instant invention in any way, and they
should not be so construed. Furthermore, the compounds described
10 in the following examples are not to be construed as forming the
only genus that is considered as the invention, and any combination
of the compounds or their moieties may itself form a genus. Specific
definitions of variables in the Schemes are illustrative only, and are
not intended to limit the procedures described, unless otherwise
15 noted. All temperatures are degrees Celsius unless otherwise noted.
Some abbreviations used herein are as follows: TBAF is
tetrabutylammonium fluoride; DEAD is diethyl azodicarboxylate;
PPh3 is triphenyl phosphine. Many of the compounds described in
the examples were analyzed by FAB mass spectroscopy (FABMS),
20 and MS values are denoted.
In the schemes and examples below, various reagent
symbols have the following meanings:
Ac: acyl (CH3-C(O)-)
BOC (or Boc): t-butyloxycarbonyl
BOC2O: di-t-butyl dicarbonate
BOP: Benzotriazol-1-yloxytris(dimethylamino)phosphonium,
hexafluorophosphate
Bn: benzyl
n-BuLi: n-butyllithium
t-BuLi: tert-butyllithium
CBZ: Carbobenzyloxy
CH2C12: Methylene chloride
- CHC13: chloroform
Pd-C: Palladium on activated carbon catalyst
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 34 -
DMF: Dimethylformarnide
DMSO: Dimethylsulfoxide
~DC: 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide
hydrochloride
S EtOAc: ethyl acetate
EtOH: ethanol
HOAc: acetic acid
LDA: Lithium diisopropylamide
MeOH: methanol
NMM: N-methyl morpholine
NMP: N-methyl pyrrolidine
Oxone: potassium peroxymonosulfate
PYCLU: chloro N,N,N',N'-bis(pentamethylene)formamidinium
hexafluorophosphate
RT: room temperature
TBDMS or TMS: t-butyldimethyl silyl
TFA: trifluoroacetic acid
THF: tetrahydrofuran;
TLC: thin layer chromatography
CA 02246756 1998-08-18
WO 97/31910 PCTMS97/02712
- 35 -
SCHEME 1
BOC--N = I
+ DMF _ ~'O~CHO
HO~CHO ~ 3
1 2 HCI-H2N--CO2CH3
1 -4
NaCNBH3, NaOAc
4A sieves, CH30H
BOC--N=o~3 CH2NH/~CO2CH3
PhSO2CI
Pyridine
CH2CI2, 0~C ~ RT
BOC--N ~ ~0~ S02Ph
1 N NaOH
EtOH
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 36 -
~CHEME 1 CONTINUED
BOC--N >~--o~3CH2N--co2H
TFA/CH2CI2
HN~--O~ CH2N--C02H
EXAMPLE 1
Step 1: 4-13-(N-Boc-Piperidin-4-yl~propyloxylbenzaldehyde (1-3)
A solution of 3-(N-Boc-piperidin-4-yl)propyl iodide 1 1
(5.0 g, 14.2 mmol) (preparation described in EP 478,328), 4-
10 hydroxybenzaldehyde 1 2 (1.73 g, 14.2 mmol), Cs2CO3 (9.2 g, 28.4
mmol) and DMF (50 ml) was stirred at ambient temperature for 2.0
hours. The reaction mixture was diluted with ethyl acetate and then
washed with H2O, sat. NaHCO3, 10% KHSO4, brine, dried (MgSO4)
and concentrated. Flash chromatography (silica, 10% EtOAc/hexane)
15 provided 1 3 as a white solid.
TLC Rf 0.80 ~silica, 40% EtOAc/hexane)
lH NMR (300 MHz, CDCl3) o 9.88 (s, lH), 7.83 (d, J=9Hz, 2H), 6.99
(d, J=9Hz, 2H), 4.11 (m, 2H), 4.04 (t, J=6Hz, 2EI), 2.69 (bt, 2H), l.84,
(m, 2H), 1.69 (bd, 2H), 1.46 (m, 12H), 1.14 (m, 2H).
Step 2: N~ (N-Boc-Piperidin-4-yl)propyloxy)phen-4-yl-
methyllglycine methvl ester (1-5)
To a st~rred suspension of 4A molecular sieves (4 g) and
CH30H (30 ml) was added NaOAc (1.18 g, 14.5 mmol), compound 1 3
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
~ (1.0 g, 2.89 mmol), amine 1-4 (363 mg, 2.89 mmol)-and NaCNBH3
(546 mg, 8.67 mmol). After 18 h, the reaction mixt~lre was filtered
- through a celite pad. The pH of the resulting solution was adjusted to ~2
by the dropwise addition of conc. HCI to decompose excess hydride.
The pH was adjusted to ~12 by the addition of K2C03. The mixture
was extracted with EtOAc. The organic portion was washed with brine,
dried (MgS04) and concentrated. Flash chromatography (silica, 80%
EtOAc/provided) amine 1 5 as a colorless oil.
TLC Rf 0.32 (silica, EtOAc)
lH NMR (300 MHz, CD30D) ~ 7.22 (d, J=9Hz, 2H), 6.85 (d, J=9Hz,
2H), 4.10 (bd, 2H), 3.94 (t, J=6Hz, 2H), 3.70 (m, SH), 3.36 (s, 2H), 2.77
(m, 2H), 1.77 (m, 4H), 1.43 (m, 12H), 1.07 (m, 2H).
Step 3: N-~(3-(N-Boc-Piperidin-4-yl)propyloxy)phen-4-yl)-
methyll-N'-phenylsulfonyl ~Iycine methyl ester (1-6)
To a stirred solution of amine 1-5 (900 mg, 2.15 mrnol),
pyridine (347 ,ul, 4.30 mmol) and CH2C12 (20 ml) at 0~C was added
phenylsulfonylchloride (300 ,ul, 2.37 mmol) followed by the removal of
the cooling bath. After 18 h, the reaction was diluted with EtOAc and
then washed with H20, 10% KHS04, brine, dried (MgS04) and
concentrated. Flash chromatography (silica, 20% EtOAc/hexane) gave
ester 1-6 as a~ colorless oil.
TLF Rf 0.82 (silica, 50% EtOAc/hexane)
1H NMR (300 MHz, CDC13) ~ 7.88 (d, J=9H, 2H), 7.56 (m,3H), 7.14
(d, J=9Hz, 2H), 6.81 (d, J=9Hz, 2H), 4.43 (s, 2H), 4.12 (m, 2H), 3.91
(m, 4H), 3.53 (s, 3H), 2.68 (bt, 2H), 1.79 (m, 2H), 1.68 (bd, 2H), 1.46
(m, 12H), 1.10 (m, 2H).
Step 4: N-[(3-(N-Boc-Piperidin-4-yl)propyloxy)phen-4-yl-methyl]-
N'-phenylsulfonyl glycine (1-7)
A solution of ester 1 6 (900 mg, 1.61 mmol), lN NaOH (2
ml) and EtOH (5 ml) was stirred at ambient temperature for 30 minlltes.
The reaction mixture was then acidified with 10% KHS04, followed by
extraction with EtO~c. The organic portion was washed with brine,
-
CA 02246756 1998-08-18
WO g7/31910 PCT/US97/02712
- 38 -
dried (MgS04) and concentrated to give the carboxylic acid 1 7 as a
white solid.
TLC Rf 0.48 (silica, 9:0.5:0.5 CH2C12/MeOH/AcOH)
lH NMR (300 MHz, CD30D) ~ 7.87 (d, J=8Hz, 2H), 7.59 (m, 3H), 7.09
S (d, J=8Hz, 2H), 6.82 (d, J=9H, 2H), 4.41 (s, 2H), 4.04 (bd, 2H), 3.93 (t,
J=6Hz, 2H), 3.84 (s, 2H), 2.76 (m, 2H), 1.79 (m, 4H), 1.44 (m, 12H),
1.04 (m, 2H).
Step 5: N-[(3-(Piperidin-4-yl)propyloxy)phen-4-yl-methyl]-N'-
phenylsulfonyl glycine (1-8)
A solution of acid 1 7 (400 mg, 0.7334 mrnol), TFA (3 ml)
and CH2C12 (3 ml) was stirred at ambient temperature for 1.0 hour.
The solution was concentrated and then azeotroped with toluene. Flash
chromatography (silica, 20:1 :1 EtOH/NH40H/H20) furnished amine
~ as a white solid.
TLC Rf 0.41 (silica, 20:1:1 l~tOH/NH40H/H20)
lH NMR (300 MHz, NaOD/D20) ~ 7.79 (d, J=8Hz, 2H), 7.66 (d,
J=7Hz, lH), 7.56 (t, J=8Hz, 2H), 7.06 (d, J=8Hz, 2H), 8.45 (d, J=9Hz,
2H), 4.42 (s, 2H), 4.01 ~t, J=8Hz, 2H), 3.76 (s, 2H), 2.91 (m, 2H), 2.47
(t, J=12Hz, 2H), 1.74 (m, 4H), 1.32 (m, 3H), 1.06 (m, 2H).
CA 02246756 1998-08-18
WO 97/31910 PCT/~JS97/02712
- 39 -
SCHEME 2
Br~ NaN3 . ~Ç \
~ benzene u
2-1 ~ O 1,
NaN(TMS)2, DMF
-1 5~C
Boc--N~ I
-1 5~C ~ RT
Boc--N~
tBuLi DMF, THF
-78~C
Boc--N ~ O C H0
HCI~H2N--CO2CH3
NaCNBH3, NaOAc
4A sieves, CH30H
CA 02246756 l998-08-l8
WO 97/31910 PCT/US97/02712
- 40 -
SCHEME 2 CONTINUED
~ --NH C02CH3
Boc--N~/ N~
2-6
PhSO2CI
Pyridine, CH2CI2
0~C ~ RT
~f I ~Co2cH3
E~oc--N~--N~ SO2Ph
O \ EtOAC
2~ HCl(g)\
'~ ~N CO2CH3
HCI~HN~--N~ SO2Ph
1 N NaOH ~
EtOH 2-1 0
~N~CO2H
Boc-N~ O SO2Ph
2-8
HCI (g)
EtOAc
HCI-HN~ O SO2Ph
2-~
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 41 -
EXAMPLE 2
- Step 1: 6-Bromo-3~4-dihydroisoquinolin-1-one (2-2)
To a stirred solution of bromide 2-1 (20.0 ~, 94.8 mmol),
H2SO4 (25.4 ml) and benzene (130 ml) at ambient temperature was
added NaN3 (8.88 g, 136.6 mmol) portionwise over a 30 minute period.
After 1.0 hour, the reaction was diluted with EtOAc and then washed
with H20, brine, dried (MgSO4) and concentrated. Flash
chromatography (silica, 40% EtOAc/hexanes ~ EtOAc) fuInished
bromide 2-2 as an orange solid.
TLC Rf 0.31
1H NMR (300 MHz, CDCl3) o 7.93 (d, J=8Hz, lH), 7.49 (d, J=8Hz,
lH), 7.40 (s, lH), 6.32 (bs, lH), 3.57 (m, 2H), 2.99 (t, J=7Hz, 2H).
Step 2: 6-Bromo-N-[(N-Boc-piperidin-4-yl)ethyl]-3,4,-
dihydroisoquinolin-l-one (2-4)
To a stirred solution of bromide 2-2 (1.33 g, 5.88 mmol)
and DMF (30 ml) at -15~C was added NaN (TMS)2 (1.0 M/THF; 6.4
ml) dropwise over a 10 minute period. After 10 minlltes, a solution of
iodide ~ (2.0 g, 5.88 mmol) and DMF (4 ml) was added to the
reaction mixture followed by the removal of the cooling bath. After 1.0
hour, the solution was diluted with LtOAc and then washed with 10%
KHSO4, brine, dried (MgSO4) and concentrated. Flash
chromatography (silica, 50% EtOAc/hexanes) afforded bromide 2-4 as a
colorless oil.
TLC Rf 0.23 (silica, 50% EtOAc/hexanes)
H NMR (300 MHz, CDC13) ~i 7.93 (d, J=8Hz, lH), 7.46 (d, J=8Hz,
lH), 7.35 (s, lH), 4.12 (m, 2H), 3.56 (m, 4H), 2.97 (t, J=7Hz, 2H), 1.69
(m, 3H), 1.54 (m, 2H), 1.45 (s, 9H), 1.13 (m, 2H).
~tep 3: 6-Formyl-N-[(N-Boc-piperidin-4-yl)ethyl]-3,4-
dihydroisoquinolin-1-one (2-5)
To a stirred solution of bromide 2-4 (1.90 g, 4.34 mmol),
DMF (342 ~1, 4.34 mmol) and THP (15 ml) at -78~C~ was added t-BuLi
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 42 -
(1.7 M/pentane; 7.66 ml, 13.02 mmol) dropwise over a 5 minute period.
After 1.0 hour, the reaction was quenched with AcOH. The solution
was diluted with EtOAc and then washed with 10% KHSO4, sat.
NaH~03, brine, dried (MgSO4) and concentrated. Flash
S chromotography (silica, 30% ~ 50% EtOAc/hexanes) gave aldehyde
~i as a colorless oil.
TLC Rf 0.13 (silica, 50% EtOAc/hexanes)
1H NMR (300 MHz, CD~13) ~ 10.06 (s, lH), 8.24 (d, 8Hz, IH), 7.84 (d,
J=8Hz, lH), 7.72 (s, lH), 4.12 (m, 2H), 3.61 (m, 4H), 3.08 (t, J=7Hz,
10 2H), 2.69 (t, 13Hz, 2H), 1.75 (d, J=13Hz, 2H), 1.59 (m, 2H), 1.45 (s,
10H), 1.19 (m, 2H).
Step 4: N-(N-[(N-Boc-Piperidin-4-yl)ethyl]-3,4-dihydro-
isoquinolin-l-one-6-yl-methyl)~lycine methyl ester (2-6~
To a stirred suspension of 4A molecular sieves (3.60 g) and
CH30H (30 ml) was added NaOAc (1.06 g, 12.95 mmol), benzaldehyde
2-5 (1.0 g, 2.59 mmol), amine 1 4 (326 mg, 2.59 mmol) and NaCNBH3
(490 mg, 7.77 mmol). After 1.0 h, the reaction mixture was filtered
through a celite pad. The pH of the resulting solution was adjusted to ~2
20 by the dropwise addition of lN HCl. After 10 minutes, the pH was
adjusted to ~12 by the addition of K2~03. The mixture was extracted
with EtOAc. The organic portion was washed with brine, dried
(MgSO4) and concentrated. Flash chromatography (silica, EtOAc)
afforded amine 2-6 as a white solid.
25 TT.C Rf 0.09 (silica, EtOAc)
1H NMR (300 MHz, CD3OD) 7.89 (d, 3=8Hz, lH~, 7.33 (d, J=8~z, lH),
7.26 (s, lH), 4.07 ~m, 2H), 3.81 (s, 2H), 3.72 (s, 3H), 3.60 (m, 4H),
3.40 (s, 2H), 3.00 (t, J=7Hz, 2H), 2.73 (bt, 2H), 1.78 (bd, J=12Hz, 2H),
1.61 (m, 2H), 1.44 (s, 10H), 1.10 (m, 2H).
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 43 -
- Step 5: N-(N-[(N-Boc-Piperidin-4-yl)ethyl]3,4-dihydro-
isoquinolin-l-one-6-yl-methyl)-N'-phenylsulfonyl glycine
- methyl ester (2-7)
To a stirred solution of amine 2-6 (800 mg, 1.75 mmol),
S NMM (488 ,ul, 3.50 mmol) and CH2Cl2 (10 ml~ at 0~C was added
benzenesulfonyl chloride (243 ~11, 1.93 mmol) in a single portion
followed by the removal of the cooling bath. After 48 h, the
heterogeneous mixture was diluted with EtOAc and then washed with
H2O, sat. NaHCO3, 10% KHSO4, brine, dried (MgSO4) and
concentrated. Flash chromatography (silica, 50% ~ 60%
EtOAc/hexanes) furnished ester 2-7 as a white solid.
TLC Rf 0.16 (silica, 50% EtOAc/hexanes)
1H NMR (300 MHz, CD30D) ~ 7.86 (m, 3H), 7.65 (d, J=7Hz, lH), 7.59
(d, J=9Hz, 2H), 7.20 (d, J=8Hz, lH), 7.14 (s, lH), 4.52 (s, 2H), 4.07 (m,
2H), 3.98 (s, 2H), 3.59 (m, 4H), 3.52 (s, 3H), 2.94 (t, J=8Hz, 2H), 2.76
(m, 2H), 1.78 (bd, J=13Hz, 2H), 1.57 (m, 2H), 1.44 (s, 10H), 1.15 (m,
2H).
Step 6: N-(N-[(N-Boc-Piperidin-4-yl)ethyll-3,4-dihydro-
isoquinolin-1-one-6-yl-methyl)-N'-phenylsulfonyl glycine
(2-8)
A solution of ester 2-7 (600 mg, 1.00 mmol), IN NaOH
(2.0 ml) and EtOH (5 ml) was stirred at ambient temperature for 1.5
hours. The reaction mixture was then acidified with 10% KHSO4
followed by extraction with E~tOAc. The organic portion was washed
with brine, dried (MgSO4~ and concentrated to give acid 2-8 as a white
solid.
1H NMR (300 MHz, CD30D) ~ 7.86 (m, 3H), 7.64 (d, J=7Hz, lH), 7.57
(d, J=9Hz, 2H), 7.21 (d, J=8Hz, lH), 7.13 (s, lH), 4.54 (s, 2H), 4.04
~ 30 (bd, J=l lHz, 2H), 3.93 (s, 2H), 3.59 (m, 4H), 2.94 (t, J=6Hz, 2H), 2.73
(m, 2H), 1.76 (bd, J=13Hz, 2H), 1.57 (m, 2H), 1.44 (s, 10H), 1.13 (m,
2~).
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 44 -
HCI~HN~N~X~ I CO2H
o
Step 7: N-(N-[(Piperidin-4-yl)ethyl]-3,4-dihydroisoquinolin-1-one-
6-yl-methyl)-N'-phenylsulfonyl glycine ~2-9)
S EtOAc (8 ml) at 0~C was treated with HC~l (g) until
saturated. A suspension of acid 2-8 (300 mg, 0.5124 mmol) and ~tOAc
(2 ml) was added in a single portion. After 30 minlltes, argon was
bubbled ~rough the solution. The solvent was removed in vacuo to
give amine 2-9 as a white solid.
lH NMR (300 MHz, D20) ~ 7.79 (d, J=8Hz, 2EI), 7.74 (m, 2H), 7.57
(m, 2H), 7.18 (d, J=8Hz, lH), 7.01 (s, lH), 4.46 (s, 2H), 4.08 (s, 2H),
3.56 (m, 4H), 3.42 (bd, J=14Hz, 2H), 2.96 (t, J=13Hz, 2H), 2.86 (t,
J=7Hz, 2H), 2.03 (d, J=14Hz, 2H), 1.64 (m, 3H), 1.45 (m, 2H).
EXAMPLE 2A
N-(N-[(Piperidin-4-yl)ethyl~ -3,4-dihydroisoqllinolin- 1 -one-6-yl-
mçthyl)-N'-phenylsulfonyl glycine methyl ester hydrochloride (2- 10)
EtOAc (8 ml) at 0~C was treated with HCI (g) until
saturated. A suspension of ester 2-7 (300 mg, 0.5004 mmol) and EtOAc
(2 ml) was added in a single po~ion. After 30 minutes, argon was
bubbled through the solution. The solvent was removed in vacuo to
give amine 2-I0 as a white solid.
lH NMR (D20) ~; 7.82 (d, J=8Hz, 2H), 7.74 (m, 2H), 7.60 (t, J=8Hz,
2~ 21H), 7.21 (d, J=8Hz, lH), 7.07 (s, lH), 4.47 (s, 2H), 4.14 (s, 2H), 3.56
(m, 7H), 3.42 (d, J=13Hz, 2H), 2.90 (m, 4H), 2.03 (d, J=14Hz, 2H),
1.65 (m, 3H), 1.45 (m, 2H).
CA 02246756 l998-08-l8
WO 97/31910 PCT/US97/02712
- 45 -
SCHEME 3
~--N~ 1. Boc20, Et3N, DMF ~ J3~NH2
HNJ 2. H2, Pd/C, EtOH BocNJ
3-1 3-2
Cl~ N~2
o ;~
~, NO2
H
I~--N
BocN J
3-4
1- H2, Pd/C, EtOH/
2. RSO2CI, pyridine
R' S, ~2
,~ N ~ ~ N~ H
BocNJ ~, R-Ph 2 aq. NaOH, TFA, CH2C12
~-5c, R=Me
~, R=4-Br-Ph
CA 02246756 1998-OX-18
WO g7/31910 PCT/US97/02712
- 46 -
SCHEME 3 CON~I~lED
SO2 ~
'J~OH
N
~N~ ~
HN J 3-6a, R=Ph
-6b, R=Bn
f'-6c, R=Me
~, R=4-Br-Ph
EXAMPLE 3A
Step 1: 4-(4-tert-Butyloxycarbonyl-piperazin-l-yl)aniline (3-2)
To a solution of 1-(4-nitrophenyl)piperazine C~) (9.8 g,
47.29 mmol) and triethylamine (7.3 mL, 52.37 mmol) in dichloro-
10 methane (175 mL~ at RT, Boc2O (11.38 g, 52.14 mmol) was added
portionwise. The resultant mixture was stirred at RT for 2 h. The
product mixture was concentrated under vacuum, and the residue
triturated with hexane (150 mL). The resultant yellow powder was
obtained by filtration.
Without further puri~ication, the yellow solid was dissolved
ethanol f~200 mL) and shaken under an atmosphe~e of hydrogen gas at
50 psi in the presence of 0.8 g of 5% Pd/C for 18 h at RT. The product
solution was filtered through a pad of Celite, and the filtrate was
concentrated under vacuum to give 2 as pink solid.
Step 2: 4-[4-(4-tert-Butyloxycarbonyl-piperazin-l-yl)phenylamino-
carbonyll-nitrobenze f3-4)
To a suspension of 4-nitrobenzoic acid (4.06 g, 24.29
mmol) in dichloromethane (40 mL) and DMF (200 ~lL) at RT, oxalyl
25 chloride (2.8 mL, 32 mmol) was added dropwise with a syringe pump
CA 022467~6 l998-08-l8
WO 97/31910 PCT/US97/OZ712
- 47 -
over a period of 1 h. The resultant solution was stirred at RT for O.S h,
and concentration under vacuum. The residue was dissolved in benzene
and concentrated under vacuum to remove residua} oxalyl chloride.
The resultant 4-nitroben~oyl chloride (~) was dissolved in dichloro-
methane (25 mL) and was added dropwisely to a cold (0~C) solution of
~niline 3-2 (6.12 g, 22.1 mmol) and DMAP (135 mg) in a mixture of
dichloromethane (30 mL) and pyridine (4.5 mL) over a period of 30
min. The resultant slurry was diluted with dichloromethane (60 mL)
and stirred at 0~C for 1 h. The product mixture was further diluted
with dichloromethane (600 mL), washed successively with sat. a~.
sodium bicarbonate, sat aq. potassium hydrogen sulfate, water, and then
brine until the aqueous extract was neutral. The dichloromethane
solution was loaded directly onto a column of silica gel and the colurnn
eluted with ethyl acetate. Collection and concentration of appropriate
lS fractions provided the amide 3-4.
Step 3: N-Phenylsulfonyl-4-[4-(4-ter~ utyloxycarbonyl-piperazin-
1-yl)-phenylaminocarbonyllaniline (3-Sa)
A suspension of the nitrobenzene 3-4 (6.9 g, 16.18 mmol)
and 5% Pd/C (0.93 g) in ethanol (110 mT .) was shaken under an
atmosphere of hydrogen gas at 50 psi overnight at RT. The resultant
mixture was filtered through a plug of Celite, and washed repetitively
with methanol (500 mL). The filtrate was concentrated under vacuum.
The residue was treated with toluene and concentrated under vacuum to
remove residual alcohols. The resultant aniline (4.49 g, 11.32 rnmol~
was treated with phenylsulfonyl chloride (1.75 mL, 13.7 mmol) in
pyridine ~lS mL) at 100~C for 2 h. The resultant mixture was
concentrated under vacuum. The residue was dissolved in methanol and
concentrated onto silica gel. The resultant solid was loaded onto a
column of silica gel and eluted with 80% ethyl acetate in hexane.
Collection and concentration of appropriate fractions provided the
sulforlamide 3-Sa.
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- ~8 -
Step 4: N- { 4-[4-(Piperazin- 1 -yl)phenylaminocarbonyl3phenyl } -N-
phenyl-sulfonylglycine (3-6a)
To cold (0~C) solution of the sulfonamide 3-5a (2.02 g,
3.76 mmol) in DMF (15 mL), sodium hydride (96 mg, 3.99 mmol) was
added. The mixture was stirred at RT ~or lS min. The resultant
solution was cooled back to 0~C, and methyl bromoacetate (380 ,uL,
4.01 mmol) was added, and stirred at RT overnight. The product
nnixture was concentrated under vacuum, and the residue dissolved in
ethyl acetate. The organic fraction was washed successively with aq.
sodium bicarbonate and brine, dried over magnesium sulfate, filtered
and concentrated under vacuum. The residue was subjected to column
chromatography on silica gel eluting with 60% ethyl acetate in hexane.
Collection and concentration of ~loL~riate fractions provided N-4-
[4(4-tert-butyloxycarbonyl-piperazin- 1 -yl)phenylaminocarbonyl]phenyl-
l 5 N-phenylsulfonylglycine methyl ester.
To a solution of the glycine methyl ester (430 mg, 0.7
mmol) in a mixture of methanol (10 mL) and ethanol (3 mL~ at RT, aq.
sodium hydroxide (2.8 mL, 1 M, 2.8 mmol) was added. The resultant
mixture was stirred at RT for 5 h. The product mixtlIre was
concentrated, acidified with 1 M hydrochloric acid, and extracted with
ethyl acetate (3 x 70 mL). The organic extracts were combined, dried
over sodium sulfate, filtered and concentrated under vacuum. The
residue was dissolved in dichloromethane (7 mL) and treated with
trifluoroacetic acid (3 mL) at RT for 2.5 h. The product solution was
concentrated under vacuum. The residue was dissolved in water, frozen
and Iyophilized overnight to provide the sulfonylglycine ~ as a white
fluffy solid.
lH NMR (CD30D): ~ 7.84 (2H, d, J=8.6Hz), 7.71-7.49 (7H, m), 7.36
(2H, d, J=8.6Hz), 7.03 (2H, J=9.0 Hz), 4.51 (2H, s), 3.38 (8H, br s).
Analysis calculated for C25H26N4OsS- 1.30 TFA-0.20 H2O
C, 51.28; H, 4.32; N, 8.67
Found: C, 51.29; H, 4.31; N, 8.90
CA 022467~6 1998-08-18
WO 97/31910 PCT/US97/02712
- 49 -
~XAMPLE 3B
Step 1: N-Benzylsulfonyl-4-[4-(4-tert-butyloxycarbonyl-piperazin-
1-yl)-phenylaminocarbonyll-aniline (3-5b)
S Following the procedure described for 3-5a, but
substituting benzylsulfonyl chloride for phenylsulfonyl chloride, 3-5b
was prepared.
Step 2: N- { 4-[4-(Piperazin- 1 -yl)phenylaminocarbonyl]phenyl } -N-
benzyl-sulfonylglycine (3-6b)
Following the procedure described for 3-6a, but starting
with N-benzylsulfonyl-4-[4-(4-tert-butyloxycarbonyl-piperazin-1-yl)-
phenylaminocarbonyl~nilin~ (;~), 3-6b was prepared.
Analysis calculated for C26H2gN4OsS- 1.40 TFA-0.44 H2O
C, 51.16; H, 4.51; N, 8.29
Found: C, 51.16; H, 4.51; N, 8.55
EXAMPLE 3C
~0 Step 1: N-Methylsulfonyl-4-[4-(4-tert-butyloxycarbonyl-piperazin-
1-yl)-phenylaminocarbonyllaniline (3-5c)
Following the procedure described for 3-5a, but
subsLiLu~ g methanesulfonyl chloride for phenylsulfonyl chloride, 3-5c
was prepared.
Step 2: N- { 4-[4-(Piperazin- 1 -yl)phenylaminocarbonyl3phenyl } -N-
methylsulfonyl~lycine (3-6c)
Following the procedure described for 3-6a, but starting
with N-methylsulfonyl-4-[4-(t-tert-butyloxycarbonyl-piperazin-1-
30 yl)phenylaminocarbonyl]~nilin~ (3-5c), 3-6c was prepared.
Analysis calculated for C2oH24N4oss-l.48 TFA-0.14 H2O
C, 45.67; N, 4.30; N, 9.28
Found: C, 45.67; H, 4.30; N, 9.49
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
. ,
- 50 -
EXAMPLE 3D
Step 1: N-4-bromo-phenylsulfonyl-4-r4-(4-tert-butyloxycarbonyl-
piperazin-l-yl)-phenylamino-carbonyll-aniline (3-5d)
Following the procedure described for 3-Sa, but
substituting 4-bromophenylsulfonyl chloride ~or phenylsulfonyl
chloride, 3-Sd was prepared.
Step 2: N- { 4-[4-(piperazin- 1 -yl)phenylaminocarbonyl]phenyl } -N-
4-bromo-phenyl-sulfonylglycine (3-6d)
Following the procedure described for 3-6a, but starting
with N-4-bromo-phenylsulfonyl-4-~4-(4-tert-butyloxycarbonyl-
piperazin-1-yl)-phenylamino-carbonyl3-aniline (3-5d), 3-6d was
prepared.
Analysis calculated for C25H25BrN405S ~ 1.58 TFA ~ 0.34 H20
C, 44.52; H, 3.62; N, 7.37
Found: C, 44.51; H, 3.62; N, 7.50
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
SCHEME 4
1. RSO2CI,R-S02'N~oEt
[~ 2. NaH,
NO2 BDrMCFH2CO2Et, ~ Rl
NO2 4-8
1. H2, Pd/C, EtOH O
l N Cl
BocN J
r ~
SO2 ~
O ~N~OEt
~N~ ~
BocN J 4-10
1. aq. NaOH
2. HCI, EtOAc
SO~
HJ~
HN J 4-11
CA 02246756 l99X-08-18
WO ~7/31910 PCT/US97/OZ712
SCHEME 4 COWED
wherein the precursors (4-8, 4-10) and the corresponding final products
(4-11) have lR and Rl defined as follows:
Compound R R 1
a) Ph H
b) 2-thienyl H
c) 2-F-Ph H
d) 3-F-Ph H
e) 4-F-Ph H
f) Ph CH3
g) 2-F-Ph B r
h) CF3 H
i) (R)- H
camphor
j) (S)-camphor H
EXAMPLE 4A
~SO2 ~
~ ~OEt
02N~
4-8a
Step 1: N-4-nitrophenyl-N-phenylsulfonylglycine ethyl ester ~4-8a)
A solution of 4-nitroaniline (~Z) (13.8 g, 100 mrnol) and
phenylsulfonyl chloride (14 rnL, 100 mmol) in pyridine (50 mL) was
heated at 1 00~C for 2 h. The resultant solution was concentrated, and
the residue dissolved in ethyl acetate. The organic extract was washed
CA 022467~6 1998-08-18
WO 97/31910 PCT/US97/027~2
- 53 -
successively with 2 M hydrochloric acid, sat. aq. sodium bicarbonate
brine, dried over magnesium sulfate, filtered and concentrated under
- vacuum. The residue was dissolved in a minimum amount of ethyl
acetate with warning, and hexane added until the solution turned cloudy.
S The mixture was allowed to cool slowly to RT and then chilled at 0~C.
The yellow solid precipitated was obtained by filtration. Further drying
under vacuum overnight provide N-phenylsulfonyl-4-nitroz~ni1ine.
A cold (0~C) solution of N-phenylsulphonyl-4-nitro~niline
(7.39 g, 30 mmol) in DMF (65 mL) was treated with sodium hydride
(0.76 g, 32 mmol) portionwise over a period of 1.5 h. A solution of
ethyl bromoacetate (4 mL) in DMF (10 mL) was added, and the
resultant mixture stirred at RT overnight. The product mixture was
concentrated under vacuum, and the residue dissolved in ethyl acetate.
The organic extract was washed with brine, dried over magnesium
15 sulfate, filtered and concentrated under vacuum. The residue was
subjected to column chromatography on silica gel eluting with 0.5%
methanol in chloroform. Collection and concentration of appropriate
fractions provided N-4-nitrophenyl-N-phenylsulfonylglycine ethyl ester
(4-8a) as a clear gum.
~SO2 ~
o ~--N~J~o~t
,¢~ H
BocN J 4-1 oa
Step 2: N- { 4-[4-(4-tert-Butyloxycarbonylpiperazin- l -
yl)phenylcarbonyl-amino]phenyl } -N-phenylsulfonylglycine
ethvl ester (4- l Oa)
A solution of 4-8a (8.9 g, 24.4 mmol) in a mixture of
ethanol (100 rnL) and ethyl acetate (25 mL) was hydrogenated under an
atmosphere of hydrogen gas at 45 psi in the presence of 5% Pd/C (0.89
CA 02246756 1998-08-18
WO 97/31910 PCTIUS97102712
- 54 -
g) at RT for 1.5 h. The resultant mixture was filtered through a plug of
Celite, and the filtrate concentrated under vacuum. The residue was
redissolved in toluene and concentrated under vacuum to provide the
corresponding aniline.
A solution of 4-[4-(tert-butyloxycarbonyl)piperazin- 1 -yl]-
benzoic acid (4-9 wherein R2 = H) (1.0 g, 3.3 mmol) in
dichloromethane (25 mL) and DMF (3 drops) at RT was treated with
oxalyl chloride (0.43 mL, 4.9 mmol) over a period of 10 min. The
resultant solution was stirred at RT for I h, and concentrated under
10 vacuum. The residue was dissolved in toluene and concentrated to
remove residual oxalyl chloride. The resultant acid chloride 4-9 was
redissolved in dichloromethane (5 mL~, and added to a cold (0~C)
solution the above ~niline (1.1 g, 3.3 mmol) and DMAP (0.48 g, 3.9
mmol) in dichloromethane (25 mL). The resultant mixture was stirred
1~ at RT overnight, diluted with dichloromethane and washed successively
with 10% aq citric acid, sat. sodium bicarbonate, and brine. The
organic extract was dried over magnesium sulfate, filtered and
concentrated under vacuum. The residue was subjected to column
chromatography on silica gel eluting with 50% ethyl acetate in hexane.
20 Collection and concentration of appropriate fractions provided 4-lOa as
a gum.
~SO2 ~
O ,~N~JI--OH
,~NH~J
~N
HN~J 4-11a
Step 3: N- { 4-[4-~Piperazin- l -yl)phenylcarbonylarnino]phenyl } -N-
2~ phenyl-sulfonyl~lvcine (4-lla~
CA 022467F76 1998-08-18
WO 97/31910 PCT/US97/02712
- 55 -
- To a solution of the glycine ethyl ester 4-lOa (260 mg, 0.41mmol) in methanol (2.5 mL), aq. sodium hydroxide (0.85 mL, 1 M, 1.7
mmol) was added. The resultant mixture was stirred at RT for 2 h.
The product mixture was concentrated, acidified, and extracted into
5 dichloromethane. The combined organic extract were dried over
sodium sulfate, filtered and concentrated under vacuum. The residue
was dissolved in ethyl acetate (30 rnL), cooled to 0~C, and treated with a
steady stream of anhydrous hydrogen chloride gas for 10 min. The
resultant solution was stirred at 0~C for 1 h, and concentrated under
10 vacuum. The residue was dissolved in water, frozen and lyophilized
overnight to provide glycine 4-1 la as a white fluffy solid.
1H NMR (CD30D): ~ 7.89 (2H, d, J=8.8Hz), 7.69-7.50 (7H, m), 7.17
(2H, d, J=8.8Hz), 7.11 (2H, d, J=9.OHz), 4.42 (2H, s), 3.6 (4H, m), 3.4
(4H, m).
Analysis calculated for C2sH26N40sS-0.15 EtOAc
C, 51.34; H, 5.00; N, 9.35
Found: C, 51.34; H, 4.96; N, 9.30
EXAMPLE 4B
Following the procedures described in Example 4A, Steps
1-3, but substituting the applol~liate reagents as described below, the
following compounds were made:
25 (1) N- { 4- [4-(Piperazin- 1 -yl)phenylcarbonylamino]phenyl } -N-
thienylsulfonylglycine (4-1 lb)
This product was prepared using 2-thiophene sulfonyl
chloride in place of phenylsulfonyl chloride in Step 1.
Analysis calculated for C2sH26N405S- 1.40 TFA-0.15 H20
C, 46.74; H, 3.91; N, 8.45
Found: C, 46.73; H, 3.92; N, 8.72
(2) N- { 4-[4-(Piperazin- 1 -yl)phenylcarbonyl~mino]phenyl } -N-
2-fluoro-phenyl-sulfonylglycine (4- l l c)
This product was prepared using 2-fluorophenyl-sulfonyl
chloride in place of phenylsulfonyl chloride in Step 1.
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 56 -
Analysis calculated for C25H2sFN405S ~ 1.60 TF~
C, 48.74, H, 3.86; N, 8.06
Found: C, 48.37; H, 3.92, N, 8.46
(3) N- { 4-~4-(Piperazin- 1 -yl)phenylcarbonylamino]phenyl } -N-
3-fluoro-phenylsulfonylglycine (4-1 1 d)
This product was prepared using 3-fluorophenylsulfonyl
chloride in place of phenylsulfonyl chloride in Step 1.
Analysis calculate for C25H25FN405S ~ 1.30 TFA ~ 0.35 H20
C, 49.69; H, 4.08; N, 8.40
Found: C, 49.68; H, 4.04; N, 8.44
(4) N- { 4-[4-(Piperazin- 1 -yl)phenylcarbonylamino]phenyl } -N-
4-fluorophenyl-sulfonylglycine (4- l 1 e)
This product was prepared using 4-fluorophenylsulfonyl
chloride in place of phenylsulfonyl chloride in Step 1..
Analysis calculated for C25H25FN405S ~ 1.35 TFA ~ 0.35 H20
C, 49.45; H, 4.05, N, 8.33
Found: C, 49.45; H, 4.00; N, 8.40
(5) N- ~ 4-~4-(Piperazin-1 -yl)phenylcarbonylamino] -3 -
methylphenyl ~-N-phenylsulfonylglycine (4-1 1 f)
This product was prepared using 3-methyl-4-nitro~niline in
place of 4-nitroaniline in Step 1.
Analysis calculated for C26H2gN405S ~ 1.40 TFA ~ 0.42 H20
C, 51.19; H, 4.51; N, 8.29
Found: C, 51.17; H, 4.51; N, 8.53
(6) N- { 4-[4-(Piperazin- 1 -yl)phenylcarbonylamino]-3 -bromo-
phenyl ~ -N-2-fluoro-phenylsulfonyl~lycine (4- l l g)
This product was made using 3-bromo-4-nitroaniline and 2-
fluorophenylsulfonyl chloride in place of 4-nitroaniline and
phenylsulfonyl chloride, respectively, in ~tep 1.
Analysis calculated for C25H24BrFN4oss ~ 1.50 TFA ~ 0.35 ~I20
CA 02246756 l998-08-l8
WO 97/31910 PCT/[~S97/02712
- 57 -
- C, 43.75; H, 3.44; N, 7.29
Found: C, 43.76; H, 3.41; N, 7.53
(7) N- { 4-t4-(Piperazin- 1 -yl)phenylcarbonylamino]phenyl } -N-
trifluoro-methylsulfonylglycine (4- 11 h)
This product was prepared using trifluoromethanesulfonic
anhydride and tert-butyl bromoacetate in place of phenylsulfonyl
chloride and ethylbromo acetate, respectively, in Step 1.
Analysis calculated for C20H2lF3N4oss ~ 1.25 TFA ~ 0.35 H20
C, 42.54; H, 3.64; N, 8.82
Found: C, 42.55; H, 3.60; N, 9.05
0~
SO2 ~
,3~N~'oH
f~--N 4-11 i
HNJ
(8) N- { 4-[4-(Piperazin- 1 -yl)phenylcarbonylamino~phenyl ~ -N-
(1 R)- 10-camphorsulfonylsulfonyl~;lycine (4- 11 i)
This product was prepared using (lR)-(-)-10-
camphorsulfonyl chloride in place of phenylsulfonyl chloride in Step 1.
~ Analysis calculated for C29H36N406S ~ 1.50 TFA ~ 0.15 H20
C, 51.77; H, 5.13; N, 7.55
Found: C, 51.74; H, 5.13; N, 7.63
CA 02246756 1998-08-18
WO 97131910 PCT/US97102712
- 58 -
~0
SO~2~'
~N ~i
HN ~J
(9) N- { 4-[4-(Piperazin- 1 -yl)phenylcarbonylamino lphenyl } -N-
(lS)-10-camphorsulfonyl-sulfonyl-~lycine (4-1 lj)
This product was prepared using (lS)-(-)-10-
camphorsulfonyl chloride in place of phenylsulfonyl chloride in Step 1..
Analysis calculated for C29H36N406S ~ 1.50 TFA ~ 0.10 H20
C, 51.83; H, 5.12; N, 7.56
Found: C, 51.84; H, 5.14; N, 7.72
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
_ 59 _
SCHEME S
NH2 1. R3COCI, ~ O
R~ 'OEt
NO2 BrcH2co2Et~ O N R1 5-13
4-7
1. H2, Pd/C, o
EtoH ll
~NJ~ ~c
BocN J
(4-9)
~ ~
,~ ~ H ~;
BocN J R2 5-14
1. aq. NaOH
2. TFA. CH2C12
R3~o
O N~JI'OH
~ ~J'' H
HN J 5-15
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 60 -
SCH~ME 5 CON~I~UED
wherein the precursors (5-13, 5-14 and 4-9) and the corresponding final
products (5-15) have R1, R2 and R3 defined as follows: ~
s
Cmpd RI R2 R3
a) H H Ph
b) H H 2-F-Ph
c) H H 3-F-Ph
d) H H 4-F-Ph
e) H H 2-pyridyl
f) H H 3-pyridyl
g) H H 4-pyridyl
h) H H ~H3
i) H H ~
J) H H CH2ocH2ph
k) H H CH2OH
1) 3-CH3 H Ph
m) 2-CH3 H Ph
n) H 3-CH3 Ph
o) H 2-CH3 Ph
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/OZ712
- 61 -
~ EXAMPL~ SA
~00
~ ~OEt
02N J'~ 5-1 3a
5 ~tep 1: N-4-Nitrophenyl-N-benzoyl~lycine ethyl ester (5-13a)
A mixture of 4-nitroaniline (4-7) (10.0 g, 72 mmol) and
anhydrous potassium carbonate (20 g, 1.44 mmol) in anhydrous THF
(240 mL) and benzoyl chloride (9.1 mL, 78 mmol) was stirred at RT
overnight. The resultant solution was poured into 10% aq. HCl (200
10 mL) and cooled to 0~C. The yellow solid precipitated was filtered,
washed successively with water (200 mL) and hexane (200 mL).
Further drying under vacuum overnight provided the required 4-nitro-
N-benzoylaniline.
A cold (0~C) solution of the above nitro aniline (4.8 g, 19.8
15 mrnol) in DMF (40 mL) was treated portionwise with sodium hydride
(0.48 g, 20 mmol). After the mixture was stirred at 0~C for 0.5 h, a
solution of ethyl bromoacetate (2.6 mL, 23.4 mmol) in DMF (20 mL)
was added. The resultant mixture was stirred at 50~C overnight and
concentrated under vacuum. The residue was partitioned between ethyl
20 acetate and water. The organic extract was washed with brine, dried
over magnesium sulfate, filtered, and concentrated under vacuum to
provide the nitrophenyl glycine ester 5-13a.
_
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 62 -
~00
,~ ~ N
BocN J 5-1 4a
Step 2: N- { 4-L4-(4-tert-Butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonyl-amino]phenyl } -N-benzoylglycine ethyl
ester (5-14a)
Following the procedure described for 4-lOa. but
sub~lilulillg N-4-nitrophenyl-N-benzoyl-glycine ethyl ester (5-13a) for
N-4-nitrophenyl-N-phenylsulfonyl-glycine ethyl ester (4-8a), 5-14a was
prepared.
~0 0
~ ~N~J~'OH
~N~
~ NJ~J
HN I ~-1 5a
Step 3: N- { 4-t4-(Piperazin- 1 -yl)phenylcarbonylamino]phenyl } -N-
benzoyl-glycine (5-15a)
lS Following the procedure described for 4-1 la, but
substituting N- ~4-[4-(4-tert-butyloxycarbonylpiperazin- l-yl)phenyl-
carbonylamino~phenyl } -N-benzoylglycine ethyl ester (5-1 4a~ ~or N- { 4-
CA 02246756 1998-08-18
WO 97131910 PCT/US97/02712
- 63 -
~ [4-(4-tert-butyloxycarbonylpiperazin- 1 -yl)phenylcarbonyl-
arnino]phenyl}-N-phenylsulfonylglycine ethyl ester (4-lOa~, 5-15a was
- prepared.
Analysis calculated for C26H26N404 ~ 1.35 TFA ~ 0.30 H20
C~, 55.79; H, 4.56; N, 9.07
Found: C, 55.76, H, 4.57; N, 9.29
EXAMPLE SB
I~F
~ O
~ ~OEt
02N~ S-13b
Step 1: N-4-Nitrophenyl-N-2-fluorobenzoyl-~1~,7cine ethyl ester ~5-13b~
Following the procedure described for 5-13a, but
substituting 2-fluorobenzoyl chloride for benzoyl chloride, 5-13b was
1 S prepared.
~00
o ~ N~OEt
~NJi ~
BocN ~ 5-1 4b
.
CA 02246756 1998-08-18
WO g7/31910 PCT/US97/02712
- 64 -
Step 2: N- { 4-~4-(4-tert-Butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonyl-amino]phenyl }-N-2-fluorobenzoylglycine
ethyl ester (5-14b)
Following the procedure described for 4-lOa, but
S substituting N-4-nitrophenyl-N-2-fluorobenzoyl-glycine ethyl ester (~
13b) for N-4-nitrophenyl-N-phenylsulfonylglycine ethyl ester (~), 5-
14b was prepared.
~F
~0 0
O ~[3~N~'oH
~N
HN ~ 5-1 5b
Step 3: N- { 4-~4-(Piperazin- 1 -yl)phenylcarbonylamino~phenyl } -N-
2-fluoro-benzoyl~lycine (5-1 Sb)
Following the procedure described for 4-1 la, but
substituting N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
15 phenylcarbonylamino~phenyl}-N-2-fluorobenzoyl-glycine ethyl ester
(5-1 4b) for N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonylamino]phenyl}-N-phenylsulfonylglycine ethyl ester
(4- l Oa), 5- l Sb was prepared.
Analysis calculated for C26H2sFN404 ~ 1.35 TFA ~ 0.15 H20
C, 54.45; H, 4.24; N, 8.85
Found: C, 54.45; H, 4.26; N, 8.78
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/û2712
- 65 -
~ EXAMPLE SC
F
~~O
~ ~OEt
02N~ 5-13c
Step 1: N-4-Nitrophenyl-N-3-fluorobenzoyl~lycine ethyl ester (5-13c)
Following the procedure described for 5-13a, but
5 substituting 3-fluorobenzoyl chloride for benzoyl chloride, 5-13c was
prepared.
O ~N~OEt
~N~
BocN J 5-1 4c
Step 2: N- { 4-[4-~4-tert-Butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonyl-amino]phenyl }-N-3-fluorobenzoylglycine
ethyl ester (5-14c)
Following the procedure described for 4-lOa, but
substituting N-4-nitrophenyl-N-3-fluorobenzoyl-glycine ethyl ester
(5-13cl for N-4-nitrophenyl-N-phenylsulfonylglycine ethyl ester (4-8a),
15 5-14c was prepared.
CA 02246756 l998-08-l8
WO 97/31910 PCT/US97/02712
- 66 -
F
~~O
~ ~ H
HN~J 5-15c
Step 3: N- { 4-[4-(piperazin- 1 -yl)phenylcarbonylamino]phenyl } -N-
3-iluoro-benzoylglycine (5-lSc)
Following the procedure described for 4-1 la. but
5 substituting N- { 4-L4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonylamino]phenyl } -N-3-fluorobenzoyl-glycine ethyl ester
(5-1 4c) for N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonylarnino]phenyl}-N-phenylsulfonylglycine ethyl ester
(4- l Oa), S- 1 5c was prepared.
Analysis calculated for C26H2sFN4o4 ~ 1.70 T~A ~ 0.35 H20
C, 52.19; H, 4.08; N, 8.28
Found: C~ 52.19; H, 4.08; N, 8.42
EXAMPLE SD
F~
~0 0
,~ ~OEt
02N 5-1~d
- Step 1: N-4-Nitrophenyl-N-4-fluorobenzoylglycine ethyl ester ~S-l 3d)
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 67 -
- Following the procedure described for 5-13a. but
substituting 4-fluorobenzoyl chloride for benzoyl chloride, 5-13d was
prepared.
F~
~00
o ~.,~ N~J~oEt
r~N~
~N~
5-14d
BocN
Step 2: N- { 4-r4-(4-tert-Butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonyl-amino]phenyl } -N-4-fluorobenzoylglycine
ethyl ester (5-14d~
Following the procedure described for 4-lOa, but
10 substituting N-4-nitrophenyl-N-4-fluorobenzoylglycine ethyl ester
(5-13d) for N-4-nitrophenyl-N-phenylsulfonylglycine ethyl ester (4-8a),
5-14d was prepared.
F~
~0 0
O ~N~J~OH
f N'¢ ~
HN ~,J 5-1 5d
15 Step 3: N-{4-[4-(Piperazin-1-yl)phenylcarbonylamino]phenyl}-
N-4-fluoro-benzoylglycine (5-lSd)
CA 02246756 1998-08-18
WO 97/31910 PCTIUS97/02712
.
- 68 -
Following the procedure described for 4-lla, but
substituting N- { 4-~4-(4-fert-butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonylamino]phenyl } -N-4-fluorobenzoyl -glycine ethyl ester
(5-1 4d) for N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
S phenylcarbonylamino]phenyl}-N-phenylsulfonylglycine ethyl ester
(4- l Oa), S- l Sd was prepared.
Analysis calculated for C26H25FN404 ~ 1.40 TFA ~ 0.30 H20
C, 53.92; H, 4.24; N, 8.73
Found: C, 53.90; H, 4.23; N, 8.83
EXAMPLE SE
1~ IN
~0 0
,~ ~OEt
02N 5-1 3e
Step 1: N-4-Nitrophenyl-N-picolinoyl-~lycine ethyl ester (5-13d)
lS Following the procedure described for 5-13a, but
substituting picolinoyl chloride hydrochloride for benzoyl chloride, S-
I3d was prepared.
I~IN
~0 0
~N~oEt
1/' N
BocN ~,J 5-1 4e
CA 02246756 1998-08-18
WO 97/31910 PCT/US97102712
- 69 -
Step 2 N- {4-[4-(4-tert-Butyloxycarbonyl-piperazin- l -yl)-
phenylcarbonyl-amino]phenyl ~-N-picolinoylglycine ethyl
ester (5-14e)
Following the procedure described for 4-lOa. but
5 substituting N-4-nitrophenyl-N-picolinoyl-glycine ethyl ester (5-13d~
for N-4-nitrophenyl-N-phenylsulfonylglycine ethyl ester (~), 5-14e
was prepared.
I~N
~0 0
O ~N'J~OH
~ N
HNJ 5-15e
Step 3 N- { 4-[4-(Piperazin- 1 -yl)phenylcarbonylamino]phenyl } -
N-picolinoyl-glycine (5- 15e)
Following the procedure described for 4-1 la. but
substituting N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
lS phenylcarbonylamino]phenyl}-N-picolinoylglycine ethyl ester (5-14e)
for N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-phenylcarbonyl-
amino]phenyl}-N-phenylsulfonylglycine ethyl ester
(4-lOa), 5-15e was prepared.
Analysis calculated for C25H25N504 ~ 2.10 TFA ~ 0.20 H20
C, 49.92; H, 3.95; N, 9.97
~ Found: C, 49.88; H, 3.95; N, 10.04
CA 02246756 1998-08-18
WO 97/31910 PCTtUS97/02712
- 70 -
EXAMPLE SF
~00
,¢~ ~OEt
02N 5-13f
Step 1: N-4-Nitrophenyl-N-nicotinoyl-glycine ethyl ester (5-13f)
S Following the procedure described for 5-13a. but
substituting nicotinoyl chloride hydrochloride for benzoyl chloride, S-
13f was prepared.
~00
O ~ ~OEt
,¢~ H
I N 5-14f
BocN ~,J
~0 Step 2: N- { 4-~4-(4-tert- Butyloxycarbonyl-piperazin- I -yl)-
phenylcarbonyl amino]phenyl } -N-nicotinoylglycine ethyl
ester (5-14f)
Following the procedure described for 4-lOa. but
substituting N-4-nitrophenyl-N-picolinoyl-glycine ethyl ester (S-L3f) for
lS N-4-nitrophenyl-N-phenylsulfonylglycine ethyl ester (4-8a), 5-14f was
prepared.
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
~ ~I
~0 o
,~ HN J~
~f
HN
Step 3 N- { 4-~4-(Piperazin- 1 -yl)phenylcarbonylamino]phenyl ~ -N-
nicotinoyl-,elycine (5-lSf)
Following the procedure described for 4-1 la. but
substituting N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonylamino]phenyl } -N-nicotinoylglycine ethyl ester (~
for N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-phenylcarbonyl-
amino]phenyl}-N-phenylsulfonylglycine ethyl ester (4-lOa), 5-lSf was
1 0 prepared.
Analysis calculated for C25H25N504 ~ 2.20 TFA ~ 0.35 H20
C, 49.27; H, 3.92; N, 9.77
Found: C, 49.25; H, 3.92; N, 10.12
l S EXAMPLE 5G
N~
~0 0
~ ~OEt
02N~ 5-139
Step 1: N-4-Nitrophenyl-N-isonicotinoyl-glycine ethyl ester (5-13~)
CA 02246756 l998-08-l8
WO g7/31910 PCT/US97/02712
- 72 -
Following the procedure described for 5-13a, but
substituting isonicotinoyl chloride hydrochloride for benzoyl chloride,
5-13g was prepared.
N~
~0 0
,¢~ N
BocN J 5-14~
Step 2: N- { 4-[4-(4-tert-Butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonyl-arnino~phenyl }-N-isonicotinoylglycine
ethyl ester (5-14g)
Following the procedure described for 4-lOa, but
sub~ u~ g N-4-nitrophenyl-N-isonicotinoyl-glycine ethyl ester (5-13g)
for N-4-nitrophenyl-N-phenylsulfonylglycine ethyl ester (4-8a), 5-14
was prepared.
N~
~00
--' N
5-1 59
lS HN
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 73 -
- Step 3: N-{4-~4-(Piperazin-l-yl)phenylcarbonylamino]phenyl~-N-
isonicotin-oyl-glycine (5-lSg)
Following the procedure described for 4-1 la, but
substituting N- ~ 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -
5 yl)phenylcarbonylamino]phenyl }-N-isonicotinoylglycine ethyl ester
(5- 14g~ for N- { 4-[4-(4-fert-butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonylamino}phenyl}-N-phenylsulfonylglycine ethyl ester
(4-lOa), 5- 15~ was prepared.
Analysis calculated for C25H25N504 ~ 2.35 TFA ~ 0.65 H20
C, 4~.26; H, 3.91; N, 9.47
Found: C, 48.25; H, 3.90; N, 9.57
EXAMPLE 5H
~o o
,~ ~OEt
02N 5-1 3h
Step 1: N-4-Nitrophenyl-N-acetyl-~lycine ethyl ester (5-13h)
Following the procedure described for 5-13a, but
substituting acetyl chloride for benzoyl chloride, S-13h was prepared.
~o o
O ~ N~OEt
~N~
~N~
5-14h
BocN
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 74 -
Step 2: N- { 4-1 4-(4-tert-Butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonyl-amino~phenyl } -N-acetylglycine ethyl ester
(5-14h)
Following ~e procedure described for 4-lOa~ but
S substituting N-4-nitrophenyl-N-acetyl-glycine ethyl ester (5-13h) for N-
4-nitrophenyl-N-phenylsulfonylglycine ethyl ester (4-8a), 5-14h was
prepared.
~ O
o ~ N~I~oH
~NJ~
HN
Step 3: N- { 4-[4-(Piperazin- 1 -yl)phenylcarbonylamino]phenyl } -N-
acetyl-~lycine (5-lSh)
Following the procedure described for 4-1 la, but
substituting N- ~ 4-L4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
15 phenylcarbonylamino3phenyl}-N-acetylglycine ethyl ester (5-14h) for
N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-phenylcarbonyl-
amino]phenyl}-N-phenylsulfonylglycine ethyl ester (4-lOa), S-lSh was
prepared.
Analysis calculated for C21H24N404 ~ 1.30 TFA ~ 0.55 H20
C, Sl.l l; H, 4.80; N, 10.10
Found: C, 51.09; H, 4.74; N, 10.25
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- FXAMPLE 5I
~0
o
~ ~OEt
02N~ 5-13i
Step 1: N-4-Nitrophenyl-N-cyclopropanecarboxylglycine ethyl ester (S-1 3i)
Following the procedure described for 5-13a~ but
substituting cyclopropanecarbonyl chloride for benzoyl chloride, 5-13i
was prepared.
~0 0
,3J HN~
BocN ~,J 5-1 4i
Step 2: N- { 4-[4-(4-terf-Butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonyl-amino]phenyl } -N-cyclopropane-
ca~xylg~ycine e.hyl es.er (5-14i)
Following the procedure described for 4-lOa. but
15 substituting N-4-nitrophenyl-N-cyclopropanecarboxylglycine ethyl ester
(5-13i) for N-4-nitrophenyl-N-phenylsulfonylglycine ethyl ester (~),
5-14i was prepared.
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 76 -
~0 0
N
HNJ 5-15i
Step 3: N- { 4-[4-(Piperazin- 1 -yl)phenylcarbonylamino]phenyl } -3!1-
cyclopropanecarboxylglycine (5- 15i3
Following the procedure described for 4-3 la, but
substituting N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonylaminolphenyl}-N-cyclopropanecarboxylglycine ethyl
ester (~) for N-~4-[4-(4-tert-butyloxycarbonyl-piperazin-1-yl)-
phenylcarbonylamino~phenyl}-N-phenylsulfonylglycine ethyl ester
10 (4- 1 Oa), 5- 1 Si was prepared.
Analysis calculated for C23H26N404 ~ 1.38 TFA ~ 0.62 H20
C, 52.35; H, 4.88; N, 9.48
Found: C, 52.35; H, 4.86; N, 9.65
~XAMPLE 5J
~0 ~~ O
02N ~i
Step 1: N-4-Nitrophenyl-N-benzyloxyacetyl-glycine ethyl ester (5-13j)
Following the procedure described for 5-13a, but
substitllting ben~;yloxyacetyl chloride for benzoyl chloride, 5-13; was
20 prepared.
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
.
~~~~ ~
O ~ N~OEt
J3JI' H
--N
BocN J ~
Step 2: N-{4-[4-(4-tert-Butyloxycarbonyl-piperazin-1-yl)-
phenylcarbonyl-amino]phenyl ~ -N-benzyloxyacetylglycine
ethyl ester (5-14j)
Following the procedure described for 4-lOa, but
substituting N-4-nitrophenyl-N-benzyloxyacetylglycine ethyl ester
(5-13j) for N-4-nitrophenyl-N-phenylsulfonylglycine ethyl ester
(4-8a), 5-14j was prepared.
~~~~ ~
o ~N~I'oH
~NJ~
HN ~J ~-1 5j
Step 3: N- { 4-[4-(Piperazin- 1 -yl)phenylcarbonylamino]phenyl } -N-
benzyloxy-acetylglycine (5- l Sj)
Following the procedure described for 4-1 la, but
substituting N- ~ 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonylamino~phenyl } -N-benzyloxy-acetylglycine ethyl ester
(5-1 4j) for N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonylamino]phenyl } -N-phenylsulfonylglycine ethyl ester
20 (4- lOa), ~ was prepared.
CA 02246756 1998-08-18
WO g7/31910 PCT/US97/02712
- 78 -
Analysis calculated for C2gH30N4O5 ~ 1.56 TFA ~ 0.30 H2O
C, 54.50; H, 4.73; N, 8.17
found: C, 54.49; H, 4.73; N, 8.51
EXAMPL33 5K
HO'--f5 O
O ~N'~OH
~N~
H
~N
HN~
N- { 4- [4-(Piperazin- 1 -yl)phenylcarbonylamino]phenyl } -N-hydroxy-
acetylglycine (5-lSk)
A mixture of N-{4-[4-(piperazin-1-yl)phenylcarbonyl-
aminolphenyl}-N-benzyloxyacetylglycine (S-lSj, 100 mg), 5% Pd (10
mg) on charcoal, TFA (1 mL) and ethanol (10 mL) was stirred under a
balloon of hydrogen gas for 18 h at RT. The resultant mixture was
concentrated, and the residue subjected to column chromatography on a
15 reverse phase C-18 column to provide ~.
Analysis calculated for C21H24N4O5 ~ 1.45 TFA ~ 0.40 H2O
C, 49.07; H, 4.52; N, 9.58
Found: C, 49.08; H, 4.47; N, 9.68
CA 02246756 l998-08-l8
WO 97/31910 PCT/US97/02712
- 79 -
EXAMPLE 5L
~0 0
~ ~OEt
02N~
c~3
Step 1: N-3-Methyl-4-nitrophenyl-N-benzoyl-glycine ethyl ester (5-131)
Following the procedure described for 5-13a. but
sub~liluting 3-methyl-4-nitroaniline for 4-nitroaniline, 5-131 was
prepared.
~00
O ~ N~OEt
~N'¢~ CH3
BocN J 5-141
Step 2: N- { 4-[4-(4-tert-Butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonyl-amino]-3-methyl-phenyl }-N-
benzoyl~lycine ethyl ester (5-141)
Following the procedure described for 4-lOa, but
15 substituting N-3-methyl-4-nitrophenyl-N-benzoyl-glycine ethyl ester
CA 02246756 1998-08-18
WO g7/3~910 PCT/US97/02712
- 80 -
(5-131) for N-4-nitrophenyl-N-phenylsulfonylglycine ethyl es~er (4-8a),5-141 was prepared.
~0 0
~ N ~:~J''' C H3
HN ~,J
5-15i
Step 3: N- { 4-[4-(Piperazin- 1 -yl)phenylcarbonylaminol -3 -methyl-
phenyl~-N-2-benzoyl~lycine (5-151)
Following the procedure described for 4-1 la, but
substituting N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
10 phenylcarbonylamino]-3-methyl-phenyl}-N-benzoylglycine ethyl ester
(5-141) for N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonylamino]phenyl}-N-phenylsulfonylglycine ethyl ester
(4-IOa), 5-151 was prepared.
Analysis calculated for C27H2gN404 ~ 1.52 TFA ~ 0.54 H20
C, 55.04; H, 4.70; N, 8.55
found: C, 55.03; H, 4.71; N, 8.84
CA 02246756 l998-08-l8
WO 97/31910 PCT/IJS97/027~2
- 81 -
EXAMPLE 5M
~0 0
,¢~ ~OEt
02N CH3
5-1 3m
Step 1: N-2-Methyl-4-nitrophenyl-N-benzoyl-glycine ethyl
ester (5-13m)
Following the procedure described for 5-13a~ but
substituting 2-methyl-4-nitroaniline for 4-nitroaniline, 5-13m was
prepared.
~00
o ~N~OEt
,¢~ tNi ~ C H3
f N
BocN 'J
~-1 4m
Step 2: N - { 4- [4-(4-tert-Butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonyl-amino]-2-methyl-phenyl }-N-
benzoylglycine ethyl ester (5-14m)
Following the procedure described for 4-lOa. but
substituting N-2-methyl-4-nitrophenyl-N-benzoylglycine ethyl ester
CA 02246756 1998-08-18
WO 97/31910 PCTIUS97/02712
- 82 -
(5-13m) for N-4-nitrophenyl-N-phenylsulfonylglycine ethyl ester
(~), 5-14m was prepared.
~00
~ ,¢~ ~OH
HNJ
5-t~m
Step 3: N- { 4-[4-(Piperazin- 1 -yl)phenylcarbonylamino} -2-methyl-
phenyl ~ -N-2-benzoyl~lycine (S- l Sm)
Following the procedure described for 4-1 la. but
substituting N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
lO phenylcarbonylamino3-2-methyl-phenyl}-N-benzoylglycine ethyl ester
(S- l 4m) for N- { 4-r4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonylamino]phenyl}-N-phenylsulfonylglycine ethyl ester
(4- l Oa), 5-1 5m was prepared.
Analysis calculated for C27H2gN40~ ~ 1.56 TFA ~ 0.58 H20
lS C~ 54.74; H, 4.69; N, 8.48
~ound: C, 54.75, H, 4.69; N, 8.74
CA 02246756 l998-08-l8
WO 97/31910 P~'r/US97/OZ712 .
- 83 -
~ EXAMPLE SN
~~ O
O ~ N~OEt
H J~
I--N
BocN J CH3
~-14n
Step 1: N-{4-~4-(4-tert-Butyloxycarbonyl-piperazin-1 -yl)-3-
methylphenyl-carbonylamino3-phenyl } -N-benzoylglycine
ethyl ester ~5-14n)
Following the procedure described for 4-lOa, but
substituting N-4-nitrophenyl-N-benzoylglycine ethyl ester (5-13a) for
N-4-nitrophenyl-N-phenylsulfonylglycine ethyl ester (4-8a), and
sub~liLu~ g 4-[4-(tert-butyloxycarbonyl)piperazin-1-yl]-3-
methylbenzoic acid for 4-[4-(tert-butyloxycarbonyl~piperazin-1-yl]-
benzoic acid, 5-14n was prepared.
~,00
N
H
~N' ~
HN ''J CH3 5-1 5n
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 84 -
Step 2: N- ~ 4-[4-(Piperazin- 1 -yl)-3-methyl-phenylcarbonylamino]-
phenyl ~ -N-benzoylglycine (5- 15n~
Following the procedure described for 4-l la, but
5 substituting N- { 4-~4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-3 -
methylphenylcarbonylamino] -phenyl } -N -benzoylglycine ethyl ester
(5-14n) for N-{4-[4-(4-tert-butyloxycarbonyl-piperazin-1-yl)-
phenylcarbonylamino]phenyl}-N-phenylsulfonylglycine ethyl ester
(4- 1 Oa), 5- 15n was prepared.
10 AnaIysis calculated for C27H28N404 ~ 1.42 TFA ~ 0.44 H20
C, 55.79; H, 4.75; N, 8.72
Found: C, 55.80; H, 4.76; N, 8.78
EXAMPLE 50
~00
o ,~N~OEt
~H
~N Clt3
BocN ~J
5-14c
Step 1: N- {4-[4-(4-tert-Butyloxycarbonyl-piperazin- 1 -yl)-2-
methylphenyl-carbonylamino]-phenyl } -N-benzoylglycine
ethyl ester (5-140)
Following the procedure described for 4-lOa, but
substituting N-4-nitrophenyl-N-benzoylglycine ethyl ester (5-13a) for
N-4-nitrophenyl-N-phenylsulfonylglycine ethyl ester ~4-8a), and
substituting 4-[4-(tert-butyloxycarbonyl)piperazin- 1 -yl 3 -2-methyl-
benzoic acid for 4-[4-(tert-butyloxycarbonyl)piperazin-1-yl3-benzoic
acid, 5-140 was prepared.
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
.
- 85 -
~00
,¢~ N J3'
~N CH3
HNJ
5-15c
Step 2: N- { 4-[4-(Piperazin- 1 -yl)-2-methylphenylcarbonylamino]-
S phenyl ~-N-benzoyl,~lYcine ~5-150)
Following the procedure described for 4-1 la, but
substituting N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-2-
methylphenylcarbonylamino] -phenyl } -N -benzoylglycine ethyl ester
(5-140) for N- { 4-[4-(4-tert-butyloxycarbonyl-piperazin- 1 -yl)-
10 phenylcarbonylamino]phenyl}-N-phenylsulfonylglycine ethyl ester
~4-lOa), S-lSo was prepared.
Analysis calculated for C27H2gN404 ~ 1.56 TFA ~ 0.46 H20
C, 54.92; H, 4.66; N, 8.51
Found: C, 54.93; H, 4.67; N, 8.58
CA 02246756 1998-08-18
WO 97/31910 PCT/lJS97102712
- 86 -
S(~HEl~E 6
HN~ BOC20 tBuO N~Br
1. CH3MgBr
2. tBuLi
3. C~
tBuO~N~, ~OH SO2Ph
0 6-3 H2N~N~,CO2Et/PYCLU
--~ SO2Ph 1. HCI/EtOAc
tBuO~N~ H H~N~CO2Et
~--~ SO2Ph
HN~N H~N~CO2H
6-5
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 87 -
E~AMPLE 6
tBuO~N~O N~CO2Et ~g
1 . HCI/EtOAc
2. LiOH
HN~O SO2Ph
S ~tep 1: 9-H-2-(1,1-Dimethylethoxycarbonyl)-7-bromo ,13-carboline
(6-2)
A suspension of 6-1, prepared by the method of Rinehart et
al., (JACS, 1987,109, p 3378-3387) (0.366 g, 1.46 mmol) in CH2C12
(8 mL) was treated with triethylamine (0.61 mL, 4.4 mmol) followed
by di-tert-butyldicarbonate (0.38 g, 1.7 mmol) for 1 hour at room
temperature. The solution was concentrated and the residue
chromatographed (20% EtOAc/Hexanes) to give 6-2 as a white solid.
Rf(20% EtOAc/Hexanes) 0.28
lH NMR (400 MHz, CDC13) ~ 8.0-7.6 (m, lH), 7.46 (s, lH), 7.33 (d,
lS lH), 7.2 (d, lH), 4.6 (bs, 2H), 3.78 (bs, 2H), 2.76 (bs, 2H), 1.5 (s, 9H).
Step 2: 9-H-2-(1,1 -Dimethylethoxycarbonyl)-,13-carbolin-7-yl
carboxylic acid (6-3)
A solution of 6-2 (0.26 g, 0.734 mmol) in THF (10 mL)
20 was cooled to 0~C and treated with methylmagnesium chloride (3.0 M in
THF, 0.29 mL, 0.87 mmol) to give a pale yellow solution. After 15
minutes the solution was cooled to -78~C and treated with t-BuLi (1.7M
in pentane, 4.35 mL, 7.39 mmol) to give a bright yellow solution.
After 10 minutes C02 gas was bubbled vigorously through the solution
CA 02246756 1998-08-18
WO 97/31910 PCTIUS97/02712
- 88 -
for I0 minutes. Saturated NH4CI, water and enough 6N NaOH to reach
pH12 were added and the solution extracted with EtOAc. The EtOAc
layer was back extracted with 0.5 NaOH and the aqueous layers
combined, acidfied to pH 7 and extracted with EtOAc, the EtOAc layer
S was dried (Na2S04) filtered and concentrated to give 6-3 as an off-
white solid.
Rf(75:25:1 CHCl3/MeOH~lIOAc)0.48.
lH NMR (400 MHz, DMSO-d6) ~ 12.0 (bs, lH), 11.2 (s, lH), 7.93 (s,
lH), 7.6 (d, lH), 7.45 (d, lH), 4.6 (s, 2H), 3.68 (m, 2H), 2.7 (m, 2H),
1.4 (s, 9H).
Step 3: N- f 4-{9-Boc-~B-carboline-7-yl)carbonyl~mino]phenyl } -N-
phenvl-sulfonyl glycine (6-4)
A solution of 6-3 (0.075 g, 0.24 mmol) and N-4-
aminophenyl-N-phenylsul~onylglycine ethyl ester (0.086 g, 0.28 mmol)
in CH2C12 (3 mL) was treated with diisopropyl~mine and PYCLU to
give ~ as a white solid a~ter chromatography in a gradient of 40 to
50% EtOAc/Hexanes.
Rf(40% EtOAc/Hexanes)0.13
lH NMR (400 MHz, CDC13) ~; 8.5-8.2 ~m, lH), 8.0 (d, lH), 7.2 (2s,
2H), 7.6 (2s, 2H), 7.58 (m, lH), 7.53 (s, 2H), 7.47 (m, 2H), 7.2 (d, 2H),
4.68 (bs, 2H), 4.4 (s, 2H), 4.13 (q, 2H), 3.8 (bs, 2H), 2.7 (bs, 2H), 1.5
(s, 9H), 1.2 (t, 3H).
Step: 4 N- { 4-[(9-H-~-Carboline-7-yl)carbonylamino]phenyl ~ -N-
phenyl-sulfonyl ~lycine (6-5)
A solution of 6-4 (0.088 g, 0.139 mmol) in EtOAc (15 mL)
was treated first with HCl gas, then with LiOH~H2O to give 6-5 as a
white solid after chromatography in 18:1:I EtOH/H2O/NH4OH.
Rf(l 8:1:1 EtOH/H2O/NH4OH) 0.43
lH NMR (400 MHz, D20) ~ 7.79 (s, IH), 7.6 (m, 1H), 7.52 (m, 2H),
7.46 (m, 4H), 7.32 (d, 2H), 7.08 (d, 2H), 4.08 (s, 2H), 3.84 (s, 2H), 2.95
(m, 2H), 2.63 (m, 2H).
CA 02246756 1998-08-18
WO 97/31910 PCT/lJS97/02712
- 89 -
SC~IEME 7
N~CH3 Z~
KMnO4/H2o
N~ CO2H 7-2
1. H2/PtO2/HOAc/MeOH
60 psi
2. BOC2O
BOCN~CO2H 7-3
so2C6H5 NJ~N PF6
H2N~N CO2Et [~ ~
i-Pr2NEt, CH2CI2
BOCN~ }~~ N ~,CO2Et
LiOH
CA 02246756 1998-08-18
WO 97131910 PCT/US97/02712
- 90 -
SCE~EME 7 CON~ED
BOCN~ SO2C6H~
H ~ N~,CO2H
HCUEtOAc
HN/~N~SO2CGH
7-6
EXAMPLE 7
Step 1: 4-(4-Pyridyl)phenylcarboxylate (7-2)
A slurry of 7-1 (5g, 29.6 mmol, prepared as described in
Chambron, J.C.; Sauvage, J.P., Tetrahedron, 1987, 895 and Comins,
D.L., Abdullah, A.H., J. Org. Chem., 1982, 47, 4315 method B) in 200
mL H20 was treated with 10% HCl until the solids dissolved. The
solution was treated with solid KMn04 in portions (11.2 g, 888 mmol),
stirred until the KMn04 had dissolved and heated to 90~C for 18 hr.
An additional 2 g of KMnO4 was added and the reaction was again
15 heated to 90~C for 2 hr. The reaction was cooled to ~60~C, filtered and
the solids were washed with warm water. The filtrate was evaporated
and the residue chromatographed (Silica gel, 10~ EtOH/H20/NH40H)
to give 7-2 as an off-white solid.
lH NMR (400 MHz, DMSO) ~ 8.61 (m, 2H), 8.0 (m, 2H), 7.72 (m, 4H).
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 91 -
~ Step 2: 4-(4-N-BOC-piperidinyl)phenylcarboxylate (7-3)
A solution of 7-2 (0.5 g7 2.5 mmol) in 20 mL 20%
HOAc/MeOH was treated with 250 mg PtO2 and hydrogenated at 50 psi
for 4 hr. The solution was filtered through SoLka Floc, evaporated and
S azeotroped with heptane to remove excess HOAc. The intermediate
amino acid acetic acid salt was obtained as a white solid.
Rf(10:1:1 EtOH/H2O/NH4OH) 0.3.
lH NMR (400 MHz, CD3OD) ~ 8.96 ~m, 2H), 7.35 (m, 2H), 3.5 (bd,
2H), 3.4 (m, 2H), 3.2 (m, 2H), 3.0 (m, 2H).
A slurly of the amino acid (0.5 g, 1.9 mmol) in 30%
H2O/dioxane (12 mL) was treated with 1 N NaOH (4.8 mL) and di-ter~-
butyldicarbonate (0.564 g, 2.58 mmol) at room temperature for 6 hr.
The reaction was acidified to pH S with 10% KHSO4 and extracted
several times with EtOAc.
The EtOAc layers were combined and evaporated to give 7-
3 as a white solid.
Rf(97:3:1 CHCl3/MeOH/HOAc) 0.39.
lH NMR (400 MHz, CD30D) ~i 7.95 (d, 2H), 7.33 (d, 2H), 4.2 (bd,
2H), 2.85 (b, 3H), 1.8 (bd, 2H), 1.6 (m, 2H), 1.48 (s, 9H).
Step 3: Ethyl 2-(1 -phenylsulfonamido-4-(4-(N-(1,1 -
dimethylethoxycarbonyl)-piperidin-4-yl)phenyl-
carboxamide~-phenyl)acetate (7-4)
A solution of ~ (0.457 g, 1.5 mmol) N-4-aminophenyl-N-
phenylsulfonylglycine ethyl ester (0.50 g, 1.5 mmol) in CH2CI2 (10
mL) was treated with diisopropylamine (0.287 mL, 1.65 mmol) and
PYCLU (0.594 g, 1.65 rnmol) and stirred at room temperature for 24
hours. The solution was diluted with EtOAc and washed with H2O,
10% citric acid, saturated NaHCO3 and brine, dried over MgSO4,
filtered and evaporated to give 7-4 as a tan oil. Rf(50%
EtOAc/Hexanes)0.16
1H NMR (400 MHz, CDC13) ~ 7.8 (d, lH), 7.7 (d, lH), 7.58 (m, 3H),
7.45 (m, 2H), 7.32 (d, lH), 7.2 (d, lH), 4.4 (s, 2H), 4.25 (m, 2H), 4.15
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 92 -
(q, 2H), 2.82 (m, 2H), 2.78 (m, lH), 1.82 (bd, 2H), 1.6 (m, 3H), 1.45
(s, 9H), 1.2 (t, 3H).
Step 4: N- { 4-[4-N-Boc-piperidin-4-yl)phenylcarboxylamino]-
phenyl ~-N-phenylsulfonyl~lycine (7-5~
A solution of 7-4 (0.7 g, 1.12 mmol) in 1:1:1
MeOH/H20/rrHF was treated with LiOH-H20 (0.097 g, 1.12 mmol) for
24 hours. The solution was diluted with EtOAc and ln% KHS04 and
the layers separated. The organic layer was dried over MgS04, filtered
and evaporated to give 7-5 as a tan solid.
Rf(9: I: 1 CH2Cl2/MeOH/HOAc)0.55
1H NMR (400 MHz, CD30D) ~ 7.89 (m, 2H), 7.65 (m, 5H3, 7.05 (m,
2H), 7.8 (d, 2H), 7.18 (d, 2H), 4.4 (s, 2H), 4.2 (bd, 2H), 2.85 (m, 3H),
2.83 (bd, 2H), 1.6 (m, lH), 1.48 (s, 9H).
Step 5: N-{4-[4-Piperidin-4-yl)phenylcarbonylamino]phenyl } -N-
phenyl-sulfonyl~lycine (7-6)
~ solution of 7-5 (0.4 g, 0.67 mmol) in EtOAc (5 mL) was
cooled to -78~C, saturated with HCl gas, warmed to 0~C and stirred for
1 hour, then concentrated at ambient temperature to give 7-6 as a white
solidafterchromatographyin 10:1:1 EtOH/H20/NH40H.
Rf(l o~ EtOH/H20/NH40H)0.34
~H NMR (400 MHz, ~2~ + NaOD) ~ 7.65-7.4 (m, 7H), 7.35 (d, 2H),
7.26 (m, 2H), 7.05 ~m, 2H), 4.08 (bs, 2H), 2.95 (m, 2H), 2.55 (m, 3H),
1.65 (m, 2H), 1.45 (m, 2H).
CA 02246756 1998-08-18
PCT/US97/02712
WO 97/31910
- 93 -
SCHEME 8
~ + ~OEt 1. N-Methylmorpholine, NMP
N HN 2. LiOH, H20
o t. (COCI)2
~OH SO2 O
f~N 2,~,N 'J~OEt
N 8-t H2N
SO2 ~ NaOH
O r~ ~OEt
~NH~
8-2
N
~'
SO2 ~
O ~N~J~OH
~N~
~,N
Il l 8-3
N ~
CA 02246756 1998-08-18
WO 97/31910 PCT/US97/02712
- 94 -
EXAMPLE 8
Step 1: 4-(Pyridyl)piperidin-4-yl-carboxylic acid (8-1)
Ethyl isonipecotate (6.0 g, 38.66 mmol), 4-chloropyridine
hydrochloride (5.9 g, 38.66 mmol) and N-methylrnorpholine ~9.3 mL,
85.0 mmol) were dissolved in N-methylpyrrolidinone (50 mL~ and the
resulting solution heated at 100~ for 48 h. The solution was
concentrated in vacuo and the residue dissolved in ethyl acetate (200
mL) and washed with water and brine (2 x 100 rnL), then dried
IO (Na2S04) and evaporated. The resulting residue was purified by flash
chromatography (5%MeOH/CH2Cl2) to afford ethyl 4-
(pyridyl)piperidin-4-yl-carboxylate as a crystalline solid.
A solution of the above ester (10 g, 42.7 mmol) in THF (50
mL) was treated with lN LiOH (47 mL, 47.0 mmol) and H20 (50 mL).
The resulting solution was concentrated and the aqueous residue cooled
to 0~C, then adjusted to p~ ~ 6 with IN HCI and the resulting solid 8-1,
collected by filtration.
Step 2: N- { 4-[N-(4-Pyridyl)-piperidinyl-4-carbonylamino]phenyl } -
N-phenvl-sulfonyl-~lycine methyl ester ~8-2)
Following the procedure described for 4-lOa. but
substituting N-(4-pyridyl)-piperidinyl-4-carboxylic acid (~) for 4-~4-
(tert-butyloxycarbonyl)-piperazin-l-yl]benzoic acid, 8-2 was prepared.
Step 3: N-{4-[N-(4-Pyridyl)-piperidinyl-4-carbonylamino]phenyl}-
N-phenylsulfonyl-glycine (8-3)
Following the procedure described for 4-1 la, N-{4-rN-~4-
pyridyl)-piperidinyl-4-carbonylamino]phenyl ~ -N-phenylsulfonylglycine
methyl ester 8-2 was hydrolyzed. HPLC purification provided 8-3.
Analysis calculated for C2sH2sN40sS ~ 1.40 TFA ~ 0.80 H20
C, 50.02; H, 4.23; N, 8.39
~ound: C, 50.00; H, 4.24; N, 8.65
CA 022467=,6 1998-08-18
WO 97/31910 PCT/US97/02712
- 95 -
SCHEME 9
~OH
~, NO2 ~N
~ BocN J
HN~
~N~ 1. H2, Pt/C, THF
--N 2. PhSO2CI, pyridine
BocN J 9-2
H ,~
,¢~N~N~o2 1. NaH, BrCH2CO2-t-Bu
2. TFA, CH2C12
f~N ~
BocNJ g
~SI02 ~
~N
HN
-
CA 02246756 1998-08-18
WO 97131910 PCTllJS97/02712
- 96 -
EXAMPLE 9
Step 1: N-4-(4-tert-Butyloxycarbonylpiperaziny- 1 -y)-benzoyl] -5-
S nitro-indoline (9-2)
Following the procedure described for 4-lOa, but
substituting 5-nitroindoline C~L) for N-4-aminophenyl-N-
phenylsulfonylglycine e~yl ester, 2~ was prepared.
Step 2: N-[4-(4-tert-Butyloxycarbonylpiperaziny-1-y)-benzoyl]-5-
phenyl-sulfonylamino-indoline (9-3)
Following the procedure described for ~, but subtituting
N-[4-(4-tert-butyloxycarbonylpiperaziny- 1 -y)-benzoyl~-5-nitro-indoline
(2~) for nitrobenzene 3-4, ~ was prepared. The catalytic
15 hydrogenation was carried out in the presence of Pt/C in THF.
~tep 3: N- { N-[4-(4-tert-Butyloxycarbonylpiperaziny- 1 -yl)-
benzoyll-5-indolinyl~-N-phenylsufonyl-glycine (9-4)
Following the procedure described ~or ~, but
20 substit-ltin~ N-[4-(4-tert-butyloxycarbonylpiperaziny- 1 -y)-benzoyl~ -5 -
phenylsulfonylamino-indoline ~) for sulfonamide 3-5a7 9-4 was
prepared.
Analysis calculated for C27H2gN405S ~ 1.50 TFA
~, 52.10, H, 4.30; N, 8.10
Folmd: C, 52.13; H, 4.26; N, 8.23
CA 02246756 l998-08-l8
WO 97/31910PCTIUS97/02712
- 97 -
SCHEME 10
H ~N~2 H2O2, NaC102 HO~No2
10-1 1 0-2
O O
, NH2 (COCI)2
--N
BocN J y
H ~NO2
1 . H2, Pt2S, MeOH ~ N ~S
2. PhSO2CI, pyridine ~N
BocN J 10-3
@'
SO2 O 1. NaH,
H ~N ~JI~OMe BrCH2CO2Me
,[3~N~S 2. aq NaOH; TFA,
~N
BocN J 10-4 ~
,02 ~
--OH
~N~S
~N~ ~
HNJ 10-5
CA 02246756 1998-08-18
WO 97/31910 PCT/IJS97tO2712
- 98 -
EXAMPLE 10
Step 1: 5-Nitro-thiophene-2-carboxylic acid (10-2)
S To a cold (0~C) mixture of S-nitro-thiophene-2-
carboxaldehyde Cl~.) (7.86 g, 50 mmol), NaH2P04 ~1.86 g, dissolved
in 20 mL water), 30% H2~2 (6 mL) m acetonitrile (S0 mL), a solution
of NaClO2 (8 g) in water (70 mL) was added over a period of I h.
After stirr~ng at RT for S h, the reaction mixture was treated with
10 sodium sulfite (S00 mg) and 1 M aq. HCl. The resultant mixture was
extracted with ethyl acetate (3X). The organic extracts were combined,
washed with brine, dried over anhdrous magnesium sulfate, filtered, and
concentrated under vacuum to provide acid 10-2.
~S Step 2: N-(S-Nitro-thiophene-2-carboxyl)-4-(4-tert-
butyloxycarbonyl-piperaziny-1-yl)-~niline (10-3)
Following the procedure described for 3-4, but substituting
S-nitro-thiophene-2-carboxylic acid ~10-2) for 4-nitroben~oic acid, 10-3
was prepared.
Step 3: N- { 2-r4-(4-tert-Butyloxycarbonylpiperazin-yl)-
phenylamino-carbonyllthien-5-vl ~-N-phenylsulfonyl-
glycine methyl ester (104)
Following the procedure described for 3-Sa, but
2~ substitu~ing N-(5-nitro-thiophene-2-carboxyl)-4-~4-tert-butyloxy-
carbonylpiperaziny-1-yl)-aniline ClQ~) for 3-4. The reduction was
carried out in methanol in the presence of Pt2S i~or three hour under a
balloon of hydrogen gas. Phenylsulfonylation, was described for 3-Sa
provided 10-4.
31)
CA 02246756 1998-08-18
WO 97/31910 PCT/~JS97/02712
_ 99 _
Step 4: N-{ 2-[4-(1 -Piperazin-yl)-phenylaminocarbonyl3thien-5-yl } -
N-phenvl-sulfonyl-glycine (10-5)
Following the procedure described for,3-6a, but
sub~liluLillg 10-4 for~, iO-5 was prepared.
S Analysis calculat~d for C23H24N405S2 ~ 1.72 TFA ~ 0.28 H20
C, 45.25; H, 3.77; N, 7.98
found: C, 45.25; H, 3.77; N, 8.13
CA 02246756 1998-08-18
WO 97/31910 PCTlaTS97/02712
- 100-
SCHEME 1 1
[~H HNO3 ~&~H
NaHMDS, BrCH2CO2Me
H2, Pd/C
~ 02N 11-3
f N
BocN ~J
O ~ ~OMe aq. NaOH;
H TFA, CH2CI2
~N
BocN J 11-4
~ ,~OH
HN J 11-5
CA 022467~6 l998-08-l8
WO 97/31910 PCT/US97/02712
- 101 -
EX~MPLE 1 1
Step 1: 6-Nitro-3.4-dihydroquinolin-2-(lH)-one (1 1-2)
S To a solution of 3,4-dihydroquinolin-2-(lH)-one (1.50 g) in
78% sulfuric acid (300 mL) at RT, a mixture of 69% nitric acid (0.84
mL) in 78% sulfuric acid (60 mL) was added. The reacting mixture
was stirred at RT for lS min, poured into ice-water, and extracted with
methylene chloride (3 x 200 mL). The organic extracts were combined,
washed with brine (4 x), dried over anhydrous m~gnesium sulfate,
filtered and concentrated onto silica gel. The residue was loaded onto a
column of silica gel, and eluted with 3% methanol in chloroform.
Collection and concentration of appropriate fractions provided the
nitroquinolinone 1 1-2.
Step 2: 6-Nitro- 1 -carbomethoxymethyl-3.4-dihydroquinolin-2-( 1 H)-one
(1 1-3)
Following the procedure described for 5-13a, but
substituting 6-nitro-3,4-dihydroquinolin-2-(lH)-one (~) for 4-nitro-
N-benzoyl~nil;ne, 1 1-3 was prepared.
Step 3: 6-~4-(4-tert-Butyloxycarbonyl-piperazin- 1 -yl)-
phenylcarbonylamino I-1 -carbomethoxymethyl-3,4-
dihydroquinolin-2-( 1 H3-one ( 1 1 -4)
Following the procedure described for 4-lOa. but
substituting 6-nitro- 1 -carbomethoxymethyl-3 ,4-dihydroquinolin-2-( 1 H)-
one (11-3) for N-4-nitrophenyl-N-phenylsulfonyl-glycine ethyl ester (~L
8a), 11-4 was prepared.
Step 4: 6-[4-(4-Piperazin-1-yl)-phenylcarbonylamino]-1-
carbohydroxymethyl-3.4-dihydroquinolin-2-( 1 H3-one ( 1 1 -5)
Following the procedure described for 4-l l a. but
substituting 6-[4-(4-tert-butyloxycarbonyl-piperazin-1-yl)-
CA 02246756 1998-08-18
WO g7/31910 PCT/US97/OZ712
- 102-
phenylcarbonylamino~ -1 -carbomethoxy-methyl -3 ,4-dihydroqu~nolin-2-
(lH)-one (11 -4) for 4- l Oa~ l 1 -5 was prepared.
Analysis calculated for C~22H24N404 ~ 1.22 TFA ~ 0.84 H20
C, 52.17; H, 4.82, N, 9.96
Found: C, 52.15; H, 4.82; N, 10.13
EXAMPLE 12
Tablet Preparation
Tablets containing 25.0, 50.0, and 100.0 mg., respectively,
of the active compound p) from Table I are prepared as illus~rated
below:
TABLE FOR DOSES CONTA~ING
FROM 25-lOOMG OF THE ACTIVE COMPOUND
Amount-m~
Active Compound 25:0 50.0 100.0
Microcrystalline cellulose 37.25 100.0 200.()
Modified food corn starch 37.25 4.25 8.5
Magnesium stearate 0.50 0.75 1.5
All of the active compound, cellulose, and a portion of the
com starch are mixed and gr~n~ ted to 10% corn starch paste. The
20 resulting granulation is sieved, dried and blended with ~e remainder of
the corn starch and the m~gn~sium stearate. The resulting gr~n~ tion is
then compressed into tablets containing 25.0, 50.0, and 100.0 mg,
respectively, of active ingredient per tablet.
CA 02246756 1998-08-18
WO 97/31910 PCTIUS97/02712
- 103 -
EXAMPLE 13
Intravenous fo~nulations
An intravenous dosage form of the above-indicated active
compound is prepared as follows:
Active Compound 0.5-10.Omg
Sodium Citrate 5-50mg
Citric Acid 1-15mg
Sodium Chloride 1 -8mg
Water for Injection (USP) q.s. to 1 L
Utilizing the above quantities, the active compound is
dissolved at room temperature in a previously prepared solution of
sodium chloride, citric acid, and sodium citrate in Water for Injection
(USP, see page 1636 of United States Phzlrm~copeia/National Formulary
for 1995, published by United States Pharmacopeial Convention, Inc.,
Rockville, Maryland, copyright 1~94.
Therapeutic Treatment
Compounds of the invention may be ~lministered to
patients where inhibition of hllm~n or m~mm~lian platelet aggregation
or adhesion is desired.
Compounds of the invention are useful in inhibiting platelet
aggregation and thus, they may find utility in surgery on peripheral
arteries (arterial grafts, carotid endaterectomy) and in cardiovascular
surgery where manipulation of arteries and organs, and/or the interation
of platelets with artificial surfaces, leads to platelet aggregation and
consumption. The aggregated platelets may form thrombi and
thromboemboli. Compounds of the invention may be ~flmini~tered to
these surgical patients to prevent the formation of thrombi and
thromboemboli.