Note: Descriptions are shown in the official language in which they were submitted.
CA 0224780~ 1998-09-03
W097/3~ 1 PCT/CA97/~167
ExPression and Secretion of Heteroloqous PolYpe~tides
from Caulobacter
Field of Invention
This invention relates to the expression and secretion
of heterologous peptides, from Caulobacter wherein the
heterologous polypeptide is fused with the surface layer
protein (S-layer protein) of the bacterium, or a portion of
the S-layer protein.
Background of the Invention
Bacterial surface proteins have been used as carriers
for foreign (heterologous) polypeptides (particularly in
Salmonella and E. coli) for various purposes, including the
development of live vaccines. In some instances, the
heterologous material is expressed as a fusion product with
a surface protein of the bacterium. Generally, the use of
such surface proteins as a vehicle for expression and/or
presentation of heterologous polypeptides has been limited
by the characteristics of a particular surface protein.
The lipopolysaccharide layer of a bacterium, which tends to
stimulate a strong immune response, covers the integral
outer membrane proteins of the organism and potentially
affects efficient presentation of a cloned epitope. Where
the surface protein is functional (for example, as part of
a filamentous portion of a bacterial cell surface) there
will be limited opportunities to express a fusion product
and still retain the surface protein's function.
Generally, the organisms that have been used for these
purposes have been chosen because of the advantages
presented in respect of the organism's relationship to its
host.
Many genera of bacteria assemble layers composed of
repetitive, regularly aligned, proteinaceous sub-units on
the outer surface of the cell. These layers are
essentially two-dimensional paracrystalline arrays, and
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CA97/00167 -- 2
being the outer molecular layer of the organism, directly
interface with the environment. Such layers are commonly
known as S-layers and are found on members of every
taxonomic group of walled bacteria including:
Archaebacteria; Chlamydia; CYanobacteria; Acinetobacter;
Bacillus; A~uaspirillum; Caulobacter; Clostridium;
Chromatium. Typically, an S-layer will be composed of an
intricate, geometric array of at least one major protein
having a repetitive regular structure. In many cases, such
as in Caulobacter, the S-layer protein is synthesized by
the cell in large quantities and the S-layer completely
envelopes the cell and thus appears to be a protective
layer.
Caulobacter are natural inhabitants of most soil and
freshwater environments and may persist in waste water
treatment systems and effluents. The bacteria alternate
between a stalked cell that is attached to a surface, and
an adhesive motile dispersal cell that searches to find a
new surface upon which to stick and convert to a stalked
cell. The bacteria attach tenaciously to nearly all
surfaces and do so without producing the extracelluar
enzymes or polysaccharide "slimes" that are characteristic
of most other surface attached bacteria. They have simple
requirements for growth. The organism is ubiquitous in the
environment and has been isolated from oligotrophic to
mesotrophic situations. Caulobacters are known for their
ability to tolerate low nutrient level stresses, for
example, low phosphate levels. This nutrient can be
limiting in many leachate waste streams, especially those
with high levels of iron or calcium.
All of the freshwater Caulobacter that produce an S-
layer are similar and have S-layers that are substantially
the same. Such S-layers appear similar by electron
microscopy with the layer being hexagonally arranged in all
cases with a similar centre-centre dimension (see: Walker,
CA 0224780~ 1998-09-03
W O 97134000 PCT/CA97/00167
-- 3
S.G., et al. (1992). "Isolation and Comparison of the
Paracrystalline Surface Layer Proteins of Freshwater
Caulobacters" J. Bacteriol. 174: 1783-1792). 16S rRNA
sequence analysis of several S-layer producing Caulobacter
strains suggest that they group closely (see: Stahl, D.A.
et al (1992) "The Phylogeny of Marine and Freshwater
Caulobacters Reflects Their Habitat" J. Bacteriol.
174:2193-2198). DNA probing of Southern blots using the S-
layer gene from C. crescentus CB15 identifies a single band
that is consistent with the presence of a cognate gene
(see: MacRae, J.D. and, J. Smit. (1991) "Characterization
of Caulobacters Isolated from Wastewater Treatment Systems"
Applied and Environmental Microbiology 57:751-758).
Furthermore, antisera raised against the S-layer protein of
C. crescentus strain CB15 reacts with S-layer proteins from
other Caulobacter (see: Walker, S.G. et al (1992) [supra]).
A11 S-layer proteins isolated from Caulobacter may be
substantially purified using the same extraction method (pH
extraction) which would not be expected to be a general
purpose method for other bacterial membrane or surface
associated proteins. All strains appear to have a
polysaccharide reactive with antisera reactive against CB15
lipopolysaccharide species which appears to be required for
S-layer attachment (see: Walker, S.G. et al (1992)
[supra]).
The S-layer elaborated by freshwater isolates of
Caulobacter are visibly indistinguishable from the S-layer
produced by Caulobacter crescentus strains CB2 and CB15.
The S-layer proteins from the latter strains have
approximately 100,000 m.w. although sizes of S-layer
proteins from other species and strains will vary. The
protein has been characterized both structurally and
chemically. It is composed of ring-like structures spaced
at 22nm intervals arranged in a hexagonal manner on the
outer membrane. The S-layer is bound to the bacterial
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CAg7100167
-- 4
surface and may be removed by low pH treatment or by
treatment with a calcium chelator such as EDTA.
The similarity of S-layer proteins in different
strains of Caulobacter permits the use of a cloned S-layer
protein gene of one Caulobacter strain for retrieval of the
corresponding gene in other Caulobacter strains (see:
Walker, S.G. et al (1992) [supra]; and, MacRae, J.D. et al
~1991) [supra].
Expression, secretion and optionally, presentation, of
a heterologous polypeptide as a fusion product with the S-
layer protein of Caulobacter provides advantages not
previously seen in systems using organisms such as E. coli
and Salmonella where fusion products of other kinds of
surface proteins have been expressed. All known
Caulobacter strains are believed to be harmless and are
nearly ubiquitous in aquatic environments. In contrast,
many Salmonella and E. coli strains are pathogens.
Consequently, expression and secretion of a heterologous
polypeptide using Caulobacter as a vehicle will have the
advantage that the expression system will be stable in a
variety of outdoor environments and may not present
problems associated with the use of a pathogenic organism.
Furthermore, Caulobacter are natural biofilm forming
species and may be adapted for use in fixed biofilm
bioreactors. The quantity of S-layer protein that is
synthesized and is secreted by Caulobacter is high,
reaching 12~ of the cell protein. The unique
characteristics of the repetitive, two-dimensional S-layer
would also make such bacteria ideal for use as an
expression system, or as a presentation surface for
heterologous polypeptides. This is desirable in a live
vaccine to maximize presentation of the antigen or
antigenic epitope. In addition, use of such a presentation
surface to achieve maximal exposure of a desired
polypeptide to the environment results in such bacteria
CA 0224780~ 1998-09-03
W 0 97/34000 PCT/CA97100167
-- 5
being particularly suited for use in bioreactors or as
carriers for the polypeptide in aqueous or terrestrial
outdoor environments.
SummarY of Invention
This invention pertains to the discovery that the
C-terminal region of the Caulobacter S-layer protein is
essential for secretion of the S-layer protein. The
inventors have determined that the 3' region of the gene
which encodes the C-terminal region of the S-layer protein
is conserved among different strains of Caulobacter.
This invention provides a method of expressing and
presenting to the environment of a Caulobacter, a
polypeptide that is heterologous to the S-layer protein of
the Caulobacter, which comprises inserting a coding
sequence for the heterologous polypeptide in-frame into a
S-layer protein gene of Caulobacter, or a portion of said
S-layer protein gene, whereby the polypeptide is expressed
and secreted by the Caulobacter as a chimeric protein
comprising the heterologous protein and all or part of the
S-layer protein.
This invention provides a DNA construct for the
aforemention chimeric protein, and a bacterium comprising
such a DNA construct, wherein the DNA construct encodes all
or part of a S-layer protein, and one or more in-frame
sequences encoding one or more heterologous proteins.
This invention provides a DNA construct comprising one
or more restriction sites for facilitating insertion of DNA
into the construct and, DNA encoding at least the 82
C-terminal amino acids of Caulobacter S-layer protein.
Preferably, the C-terminal amino acids are or correspond to
amino acids 944 or 945-1026 of the RsaA protein of C.
crescentus.
CA 0224780~ 1998-09-03
W097/34~0 PCT/CA97/~167
-- 6
This invention provides a DNA construct comprising DNA
encoding a heterologous polypeptide sequence not present in
a Caulobacter S-layer protein upstream from and in-frame
with DNA encoding at least the 82 C-terminal amino acids of
Caulobacter S-layer protein. Preferably, the C-terminal
amino acids are or correspond to amino acids 944 or 945-
1026 of the rsaA protein of C. crescentus.
This invention also provides a secreted protein
obtained from the cell surface or cell medium of a
Caulobacter cell expressing the aforementioned DNA
constructs wherein the secreted protein comprises the
heterologous polypeptide and at least the 82 C-terminal
amino acids of a Caulobacter S-layer protein. Preferably,
the C-terminal amino acids are or correspond to amino acids
944 or 945-1026 of the RsaA protein of C. crescentus.
Description of the Drawinqs
For better understanding of this invention, reference
may be made to the preferred embodiments and examples
described below, and the accompanying drawings in which:
Figure 1 is the sequence of a Carrier cassette which
may be cloned into the PstI/BamHI site ~f E~ to deliver
a gene sequence of interest to sites within a Caulobacter
crescentus S-layer protein (rsaA) gene (SEQ ID NO:1).
Figure 2 is a restriction map of a plasmid based
promoter-less version of the rsaA gene (pTZ18U:rsaA~P)
containing restriction sites and which may be used to
accept heterologous DNA of interest.
Figure 3 is the nucleotide sequence of linker BamHI-
7165K (SEQ ID NO:2; and SEQ ID NO:3) carried in plasmid
pUC9B (pUC7165K), which may be used for mutagenesis at
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CA97/00167
-- 7
sites created in rsaA by a specific or non-specific
endonuclease.
Figure 4 is the nucleotide sequence a linker BamHI-
6571K (SEQ ID NO:4; and SEQ ID NO:5) carried in plasmid
pTZ19 (pTZ6571K) which may be used for mutagenesis at sites
created in rsaA by a specific or non-specific endonuclease.
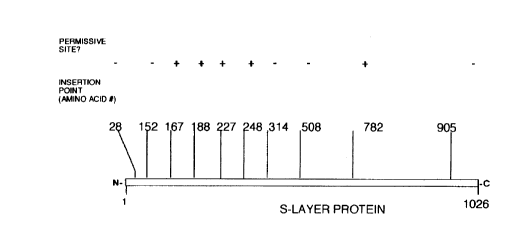
Figure 5 is a map of insertion events at TaqI sites in
the rsaA ~qene identified by amino acid number of the
insertion site in the S-layer protein and scored according
to whether the S-layer is produced in the modified
organism.
Figure 6 (comprising figures 6a, b, and c) shows the
complete nucleotide sequence of the C. crescentus S-layer
(rsaA) gene (SEQ ID NO:6) and the predicted translational
product in the single letter amino acid code. The -35 and
-10 sites of the promoter region as well as the start of
transcription and the Shine-Dalgarno sequence are
indicated. Partial amino acid sequences determined by
Edman degradation of rsaA protein and of sequenced peptides
obtained after cleavage with V8 protease are indicated by
contiguous underlining. The putative transcription
terminator palindrome is indicated with arrowed lines. The
region encoding the glycine-aspartate repeats is indicated
by underlined amino acid code letters. This region
includes five aspartic acids that may be involved in the
binding of calcium ions.
Figure 7 is a bar graph showing the approximate
location by amino acid block of 54 permissive sites in the
rsaA gene corresponding to ~3~I, HinPI, AclI, and MspI
sites described in Example 3.
Figure 8 is a portion of an amino acid sequence ~SEQ
ID NO: 8) from P. aeruqinosa PAK pilin in which the 12
CA 0224780~ 1998-09-03
W097/34~0 PCT/CA97/00167
- 8 -
amino acid pilus peptide epitope referred to in Example 5
is identified by superscript numerals 1-12.
Figure 9 is the nucleotide coding sequence and
corresponding amino acid sequence (SEQ ID NO:9) in respect
of the 184 amino acid sequence corresponding to amino acids
270-453 of the IHNV surface glycoprotein described in
Example 6.
Figure 10 is the amino acid sequence of the synthetic
cadmium binding peptide referred to in Example 4. The
cadmium binding site is shown in the figure.
Figure 11 shows locations of some of the sites in rsaA
in which single and multiple copies of the pilus peptide
described in Example 5 was expressed and secreted as part
of a chimeric rsaA protein.
Figure 12 shows a portion of pUC8 containing various
C-terminal fragments of rsaA as described in Example 7.
DescriPtion of the Preferred Embodiments
The preferred organism for use in this invention is
Caulobacter, particularly C. crescentus. While similarity
of the S-layer gene and S-layer secretion systems permits
the use of any S-layer protein producing Caulobacter in
this invention, C. crescentus strains CB2 and CB15 and
variants of those strains which contain homologs of the
gene encoding the 1026 amino acid paracrystalline S-layer
protein described in: Gilchrist, A. et al. 1992.
"Nucleotide Sequence Analysis Of The Gene Encoding the
Caulobacter crescentus Paracrystalline Surface Layer
Protein". Can. J. Microbiol. 38:193-208, are referred to
in the examples described below.
Caulobacter strains which either are incapable of
forming an S-layer, including those which shed the S-layer
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CAg7/00167
g
protein upon secretion, may be used in this invention.
Examples are the variants CB2A and CB15AKSac described in
Smit, J., and N. Agabian. 1984. "Cloning of the Major
Protein of the Caulobacter crescentus Periodic Surface
Layer: Detection and Other Characterization of the Cloned
Peptide by Protein Expression Assays". J. Bacteriol.
160:1137-1145.; and, Edwards, P., and J. Smit. 1991. "A
Transducing Bacteriophage for Caulobacter crescentus Uses
the Paracrystalline Surface Layer Protein as Receptor". J.
Bacteriol. 173, 5568-5572. Examples of shedding strains
are CB15Ca5 and CB15CalO described in Edwards and Smit
(1991) [supra], and the smooth lipopolysaccharide deficient
mutants described in Walker, S.G. et al. 1994.
"Characterization of Mutants of Caulobacter crescentus
Defective in Surface Attachment of the Paracrystalline
Surface Layer". J. Bacteriol. 176:6312-6323.
A heterologous polypeptide referred to herein may be
any peptide, polypeptide, protein or a part of a protein
which is desired to be expressed in Caulobacter and which
may be secreted by the bacterium. The heterologous
polypeptide includes enzymes and other functional sequences
of amino acids as well as ligands, antigens, antigenic
epitopes and haptens. The size of the heterologous
polypeptide will be selected depending upon whether an
intact S-layer is to be produced in the Caulobacter or
whether the chimeric protein to be recovered from the
bacterial medium as described below. Preferably, the
cysteine content of the heterologous polypeptide and the
capacity for formation of disulphide bonds within the
chimeric protein will be kept to a minimum to minimize
disruption of the secretion of the chimeric protein.
However, the presence of cysteine residues capable of
forming a disulphide bond which are relatively close
together, may not affect secretion.
CA 0224780~ 1998-09-03
W 097/34000 PCT/CA97/00167
- 10 -
Once a particular bacterium's S-layer protein gene is
characterized, this invention may be practised by
implementing one or more known methods to insert a selected
heterologous coding sequence into all or part of the S-
layer protein gene so that both the S-layer protein and the
heterologous sequence are transcribed ''in-framell.
Knowledge of an S-layer protein gene sequence permits one
to identify potential sites to install the heterologous
genetic material. The repetitive nature of the protein in
the S-layer permits multiple copies of a heterologous
polypeptide to be presented on the surface of the cell.
The following general procedure lays out courses of
action and specifies particular plasmid vectors or
constructions that may be used to accomplish fusion of an
S-Layer protein with a polypeptide of interest. The
following description uses the rsaA (S-layer) gene of C.
crescentus as an example (see Figure 6 and SEQ ID NO:6).
The latter gene sequence is characterized in Gilchrist, A.
et al (1992) [supra).
The general procedure includes detailed steps allowing
for the following possibilities:
1) use of a collection of potentially permissive
sites in the S-layer gene to install the genetic
information for a polypeptide of interest;
2) use of a Carrier cassette for delivering a gene
of interest to sites within the S-layer gene (the cassette
offers several advantages over direct modification of a
gene of interest, in preparation for insertion);
3) creation of a collection of random insertion
sites based on a restriction enzyme of choice, if the
available collection of potentially permissive sites is for
some reason unsuitable; and,
4) preparation of DNA coding for a polypeptide of
interest for direct insertion into permissive sites (ie,
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CAg7/00167
not using the Carrier cassette) by a method best suited for
the particular case (several options are suggested).
The general procedure involves the following steps and
alternative courses of action. As a first step the
practitioner will choose an appropriate region (or specific
amino acid position) of the S-layer for insertion of a
desired polypeptide. Second, the practitioner will create
a unique restriction site (preferably hexameric) in the
rsaA (S-layer) gene at a position within the gene encoding
that region (or corresponding to a specific amino acid)
using either standard linker mutagenesis (regional) or site
directed mutagenesis (specific amino acid). The unique
restriction site will act as a site for accepting DNA
encoding the polypeptide of interest. The plasmid-based
promoter-less version of the rsaA gene (pTZ18U:rsaA~P)
shown in Figure 2 may be used because it contains an
appropriate combination of 5' and 3' restriction sites
useful for subsequent steps (see: Gilchrist, A. et al
(1992) [supra]). The restriction site should not occur in
rsaA, its carrier plasmid or the DNA sequence coding for
the polypeptide of interest.
If it is unclear which region of the S-layer would be
suitable for insertion of a polypeptide of interest, a
random linker mutagenesis approach is used to randomly
insert a unique linker-encoded restriction site (preferably
hexameric) at various positions in the rsaA gene. Sites
for insertion of the linker are created using an
endonuclease, either of a sequence specific nature ~e.g.
tetrameric recognition site restriction enzyme) or sequence
non-specific nature (e.g. Deoxyribonuclease I [DNase I]).
A particularly suitable method is the generalized
selectable linker mutagenesis approach based on any desired
restriction site of: Bingle, W.H., and J. Smit. 1991
"Linker Mutagenesis Using a Selectable Marker: A Method
for Tagging Specific Purpose Linkers With an Antibiotic-
CA 0224780~ 1998-09-03
W 097/34000 PCT/CA97/00167
- 12 -
Resistance Gene". Biotechniques 10: 150-152. Because
endonuclease digestion is carried out under partial
digestion conditions, a library of linker insertions at
different positions in rsaA is created. Partial digestion
with Ms~I, HinPI and Aci:I can create 150 potential sites
for insertion of a Bam HI linker such as:
5'-CGACGGATCCGT
TGCCTAGGCAGC-5'
(SEQ ID. NO:10).
If restriction endonucleases are used to create sites
for subsequent insertion of a linker encoding a hexameric
restriction site, mutagenesis may also be done with a
mixture of 3 different linkers incorporating appropriate
spacer nucleotides in order to satisfy reading frame
considerations at a particular restriction site (only 1 of
the 3 linker insertions will be useful for subse~uent
acceptance of DNA encoding the polypeptide of interest).
With DNase I, only one linker is needed, but again only 1
of 3 linker insertions may be useful for accepting DNA
encoding the polypeptide of interest depending on the
position of the DNase I cleavage with respect to the 3
bases of each amino acid codon.
Next, a linker tagged with a marker is used to insert
DNA of interest at a restriction site. For example, if
BamHI sites are appropriate as sites for the introduction
of DNA encoding a polypeptide of interest, BamHI linkers
tagged with a kanamycin-resistance gene for selectable
linker mutagenesis may be used. One such 12-bp linker
carried in plasmid pUC1021K was described by Bingle and
Smit (1991) [Supra]. Two additional 15-bp linkers
(pUC7165K and pTZ6571K) constructed for creating the other
2 possible translation frames within the linker insert
itself are described in Figures 3 and 4 (SEQ ID NO: 2; SEQ
ID NO: 3; SEQ ID NO: 4; and, SEQ ID NO: 5). Any one of the
above three kanamycin-resistance tagged BamXI linkers is
CA 0224780~ 1998-09-03
W O 97/34000 rCT/CAg7/00167
- 13 -
suitable for mutagenesis at sites created in rsaA by
DNase I. As outlined above, a mixture of all three linkers
is preferably used for mutagenesis at sites created in rsaA
by restriction enzyme digestion.
Once a library composed of linker insertions encoding
desired hexameric restriction site at different positions
in rsaA has been created, the DNA encoding a polypeptide of
interest is inserted into the sites en masse (the library
of mutated rsaA genes may be manipulated as one unit). The
library is digested with the restriction enzyme specific
for the newly-introduced linker encoded restriction site
and ligated to a DNA fragment encoding the polypeptide of
interest and carrying the appropriate complementary
cohesive termini. The DNA specifying the polypeptide of
interest can be prepared by a number of standard methods,
which may include oligonucleotide synthesis of 2 anti-
complementary strands, polymerase chain reaction (PCR)
procedures, or addition of linkers whose termini are
compatible with the introduced sites in rsaA to a suitably
modified segment of DNA.
In order to facilitate the rapid recovery of useful
rsaA genes carrying newly inserted DNA at BamHI sites
encoding the polypeptide of interest, the Carrier
oligonucleotide shown in Figure 1 may be used. The Carrier
is designed to accept DNA (including multiple copies and
mixtures) prepared by PCR or annealed synthesized
oligonucleotides and controls direction of insertion of the
foreign segment into a rsaA gene through use of a
promoterless drug resistance marker. The DNA of interest
is first directionally cloned, if possible, using the XhoI,
StuI, or SalI sites or non-directionally cloned using any
one of the sites in the same orientation as a promoterless
chloramphenicol resistance (CmR) gene. To do this the DNA
of interest must be provided with the appropriate termini
for cloning and spacer nucleotides for maintaining correct
CA 0224780~ 1998-09-03
WO97/3~N~ PCT/CA97/~167
- 14 -
reading frame within the cassette and should not contain a
BglII site. For insertion into the BamHI linker library,
the DNA of interest is recovered as a BamHI fragment tagged
with a CmR gene. When ligated to the BamHI digested rsaA
linker library, only those colonies of the bacterium (eg.
E. coli) used for the gene modification steps that are
recovered will be those carrying insertions of the desired
DNA in the correct orientation, since the promoter on the
plasmid is 5' to rsaA~P and the CmR gene. This eliminates
screening for DNA introduction and increases the recovery
of useful clones by 100~ (1 of 3 versus 1 of 6). While
still manipulating the library as one unit, the CmR gene is
removed using BglII. The carrier oligonucleotide also
provides the opportunity to add DNA 5' or 3' to the DNA of
interest at SalI, XhoI or StuI sites providing the DNA of
interest does not contain any of these sites. This allows
some control over spacing between rsaA sequences and the
sequence of the DNA of interest.
Next, the rsaA genes carrying the DNA of interest in
the correct orientation is excised from the plasmid (eg.
from the pTZ18U:rsaA~P plasmid) and is transferred to a
suitable vector providing a promoter recognized by
Caulobacter. Such vectors include pWB9 or pWB10 (as
described in Bingle, W.H., and J. Smit. 1990). "High Level
Plasmid Expression Vectors for Caulobacter crescentus
Incorporating the Transcription and Transcription-
Translation Initiation Regions of the Paracrystalline
Surface Layer Protein Gene". Plasmid 24: 143-148) with
EcoRI/SstI sites. The ~NA of interest should not contain
the same restriction sites present in the vector. The
latter vectors allow expression of rsaA hybrids in S-layer
negative mutants of Caulobacter such as CB15KASac.
Those Caulobacter surviving transfer are ~ml ned for
chimeric protein secretion, S-layer assembly and
presentation of the new polypeptide activity, antigenicity,
CA 0224780~ 1998-09-03
W 097134000 PCT/CA97/00167
- 15 -
etc. by methods specific to the needs of the investigator
or the capabilities of the inserted sequence. Many of the
sites created are "benign" as they have no effect on the
functional regions of the protein involved with export,
self assembly, etc. However, not every site that results
in an absence of functional disruption of the S-layer is
best for insertion of new activities. Some sites may not
be well exposed on the surface of the organism and other
sites may not tolerate insertion of much more DNA than the
linker sequence.
By selecting the site of insertion of the heterologous
material, it is possible to express heterologous
polypeptides of up to about 60 (preferably less than 50)
amino acids in a S-layer chimeric protein which will
assemble as an S-layer on the cell surface. Single or
multiple insertions of smaller polypeptides (eg. 10-20
amino acids) at a wide range of the permissive sites in the
S-layer gene will permit S-layer formation. Some sites, as
reported herein, are sensitive to even small insertions
resulting in the chimeric protein being released into the
medium. Release may also be deliberately effected by use
of a shedding strain of Caulobacter to express the chimeric
protein or by physical removal of the S-layer from whole
cells.
Where S-layer formation is not required, this
invention permits the expression of quite large
polypeptides (eg. about 200 amino acids) as part of the S-
layer protein. Expressing a chimeric protein containing a
S-layer protein component having substantial deletions, as
described below, may increase the size of the heterologous
polypeptides that will be expressed and secreted by
Caulobacter.
The preceding methods describe insertion of linkers
in-frame into an rsaA gene (eg. a promoterless version of
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CA97/00167 - 16 -
the gene). The sites that are introduced allow subsequent
insertion of foreign DNA in-frame into the full length rsaA
gene. This invention also includes the construction of
chimeric S-layer protein genes and the resulting production
of chimeric S-layer proteins wherein the S-layer gene
component is highly modified by deleting large portions of
that gene which reduces the amount of Caulobacter protein
present in the secreted chimeric protein.
Generally, large deletions throughout the S-layer gene
will result in a chimeric protein that is not capable of
forming an S-layer. Attachment of the S-layer to the cell
is abolished if about the first 29 N-terminal amino acids
of the S-layer protein are deleted. Deletion of the first
776 amino acids from the N-terminal region will still
result in a chimeric protein that is secreted from the cell
but having a S-layer protein component of only the 250 C-
terminal amino acids. It has also been found that only the
extreme C-terminal region corresponding to approximately
amino acids 945-1026 of RsaA is required for secretion of
an S-layer chimeric protein from Caulobacter. Thus the
chimeric protein need only have the 82 amino acid C-
terminal region of the S-layer protein to be secreted from
the cell. Furthermore, use of the C-terminal region
corresponding to about amino acids 850-1026 (or more) of
RsaA not only permits the cell to transport the chimeric
protein outside of the cell, but also promotes spontaneous
aggregation of much of the secreted chimeric protein in the
cell medium and formation of a macroscopic precipitate that
may be collected with a course mesh or sheared to micron-
sized particles which may be ideal for vaccinepresentation. Yields of up to 250 mg. (dry weight) of
protein per liter of cells may be possible.
Sequence analysis of the 3' region of the S-layer
genes from different strains of Caulobacter shows that the
portion of the gene encoding the C-terminal region of the
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CA97/00167 - 17 -
S-layer protein is highly conserved along with the
immediate downstream non-translated and translated region.
Sequence analysis of the S-layer genes and downstream
regions in CB15 and CB2A (which are readily distinguishable
strains) shows identical DNA sequences coding for the last
118 C-terminal amino acids of the S-layer protein and the
downstream non-translated region. Sequencing of the next
downstream translated gene to amino acid 97 of the gene
product shows only a single base pair change between CB15
and CB2A, resulting in a conservative amino acid
substitution in the translation product. Conservation of
the C-terminal region of Caulobacter S-layer protein and
associated coding regions shows that this invention may be
carried out using any Caulobacter producing a S-layer
protein.
This invention may be practised as shown in the
Examples by expression of modified S-layer genes borne on
plasmids that are broad host range vectors capable of being
expressed in Caulobacter. Such plasmids are readily
constructed and introduced to Caulobacter by
electroportation. Typically, the plasmid is maintained in
the Caulobacter by antibiotic selection. Highly modified
rsaA genes with attached heterologous sequences may also be
introduced into Caulobacter on a plasmid that is not
replicated by Caulobacter. At a low but practicable
frequency, homologous recombination of the incoming
modified S-layer gene with the chromosome-resident copy of
the S-layer gene in the cell will result in a gene rescue
or transfer event. In some cases it may be desirable to
obtain a stable cell line in which the chimeric S-layer
gene is chromosomal. Various protocols for creating
chromosomal insertions are set out in the Examples.
Use of the S-layer protein as a vehicle for production
of a heterologous polypeptide has several advantages.
Firstly, the S-layer protein is synthesized in large
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CAg7/00167
- 18 -
quantities and has a generally repetitive sequence. This
permits the development of systems for synthesis of a
relatively large amount of heterologous material as a
fusion product with an S-layer protein (chimeric protein).
It may be desirable to retain the chimeric protein as part
of the bacterial cell envelope or, the fusion product may
be separated from the organism, such as by the method
described in: Walker, S.G., et al. 1992. "Isolation and
Comparison of the Paracrystalline Surface Layer Proteins of
Freshwater Caulobacters". J. Bacteriol. 174:1783-1792.
Alternatively, the Caulobacter strain that is used to
express the fusion product may be derived from a strain
such as CB15Ca5 that sheds its S-layer.
This invention is particularly suited for use in a
bioreactor systems. An example would be the use of a
modified Caulobacter expressing a polypeptide having
activity similar to that of a metallothionein in a
bioreactor, to bind toxic metals in sewage, waste water
etc. Caulobacters are ideal candidates for fixed-cell
bioreactors, the construction of which is well known. An
example of such a bioreactor is a rotating biological
contactor. Although other bacteria are found in the
environment that are capable of binding metals, they often
do so by producing copious polysaccharide slimes that
quickly plug filtration systems. In some cases, the
bacteria are not surface-adherent or the bacteria do not
show selectivity towards key toxic metals. By taking
advantage of the natural bio-film forming characteristics
of Caulobacter, bioreactors may be formed comprising a
substrate and a single layer of cells adhered thereon, with
the cells distributed at high density. A variety of
substrates may be used such as a column of chemically
derivatized glass beads or a porous ceramic material such
as ceramic foam.
CA 0224780~ 1998-09-03
W 097/34000 PCT/CA97/00167
-- 19 -
Another advantageous application for this invention is
in the production of batch cultures of modified Caulobacter
wherein the S-layer protein is a fusion product with an
enzyme. For example, such Caulobacter could be grown in
wood pulp suspensions at an appropriate juncture of the
pulping process in order to provide for enzymatic
decomposition of the wood-pulp structure ~e.g. with an
enzyme having an activity like xylanase or cellulase).
Such an application may permit more effective penetration
of bleaching agents in the wood-pulp bleaching process
thereby reducing the use of chlorine-based bleaching
agents.
Examples of enzymes that may be expressed as chimeric
S-layer proteins include alkaline phosphatase (eg. by
expression of the E~ A gene of E. coli; see: Hoffman,
C.S., and Wright, A. 1985. "Fusions of Secreted Protein to
Alkaline Phosphatase: An Approach for Studying Protein
Secretion". Proc. Natl. Acad. Sci. U.S.A. 82:5107-5111;
Bingle, W.H., et al. 1993." An "All Purpose" Cellulase
Reporter for Gene Fusion Studies and Application to the
Paracrystalline Surface (S)-Layer Protein of Caulobacter
crescentus". Can.J. Microbiol.39: 70-80; and Bingle, W.H.
and Smit, J. 1994. "Alkaline Phosphatase and a Cellulase
Reporter Protein Are Not Exported From the Cytoplasm When
Fused to Large N-terminal Portions of the Caulobacter
crescentus Surface (S)-Layer Protein". Can.J. Microbiol.
40:777-782.) and, cellulase (eg. by expression of the CenA
gene of Cellulomonas fimi; see: Bingle, W.H. et al. (1993)
[supra]; and Bingle, W.H. and Smit, J. (1994) [supra]).
Another advantageous application of this invention is
the production of organisms that secrete and optionally
present vaccine-candidate epitopes. For example, modified
Caulobacter may be readily cultured in outdoor freshwater
environments and would be particularly useful in fish
vaccines. The two-dimensional crystalline array of the S-
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CA97/00167
- 20 -
protein layer of Caulobacter, which has a geometrically
regular, repetitive structure, provides an ideal means for
dense packing and presentation of a foreign epitope to an
immune system in cases where the epitope is part of an
intact S-layer in the bacterial cell surface.
This invention also provides an efficient expression
system for polypeptides that may be harvested in large
quantities relatively free of cont~m'n~nts and protein of
Caulobacter origin. Expression of a heterologous
polypeptide fused with sufficient C-terminal amino acids of
the S-layer protein to promote secretion of the
heterologous polypeptide results in the accumulation of
large quantities of secreted protein in the cell medium.
In such cases, the chimeric protein does not have to be
released from the cell surface. Furthermore, adjustment of
the size of the S-layer protein portion can dictate whether
the secreted chimeric protein is soluble or will
precipitate in the cell medium. This embodiment may also
be useful in cases where the Caulobacter is to express a
foreign antigenic component and it is desired to minimize
the amount of Caulobacter protein that is associated with
the foreign antigen secreted by the Caulobacter.
Example 1: Production of Permissive Insertion Sites in
C.crescentus
Using the restriction enzyme TaaI, a partial digestion
of the rsaA gene in pTZ18U:rsaA~P produced a group of
linearized segments with random ~3~I sites cleaved. The
linearized segments were modified by use of the tagged
linker mutagenesis procedure of Bingle and Smit (1991)
[supra], using the 12-bp BamHI linker carried in plasmid
pUC102K discussed in the general procedure above. Those
products that produced a full-length protein in E. coli
were ultimately transferred to pWBI (a minor variation of
pWB9 that is replicated by Caulobacter), as described in
the general procedure. The resulting construction was
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CA97/00167 - 21 -
introduced into a C; crescentus strain. Distinguishable
events were retrieved and analyzed for the ability to
produce a full-length protein in C. crescentus and to
produce the crystalline S-layer on their surface and the
approximate location of the insertion. Cells were screened
for the presence of a S-layer protein of approximately
lOOkDa that is extracted from the surface of whole cells by
100 mM HEPES at ph2. The results of this screening
together with the approximate positions of five successful
events (and subsequently determined exact or specific
insertion positions) are illustrated in Figure 5.
The above-described five positive events represent
cases where the 4-amino acid insertion is tolerated with no
effect on the S-layer function. The S-layers of the
modified Caulobacter were indistinguishable from a wild-
type S-layer. Thus, they have a higher potential for
tolerating the addition of more foreign peptide material
than less characterized sites. By producing 3 versions of
the gene of interest, representing each possible reading
frame (using standard linker addition technology), one may
test each of these sites for suitability in expressing the
desired activity. Also, by using restriction enzymes other
than TaqI (such as AciI, HinPI or MspI) a larger library of
BamHI insertions may be created.
Example 2: Insertion of Cadmium binding PolYpePtides Into
Specific Sites
An insertion of the above described 12 bp linker was
made at the ~I site that corresponds to amino acid #188,
frame #3 (see Figure 6; SEQ ID NO:6; and, SEQ ID NO:7).
This created a unique BamHI site at that position. Because
the precise position of the ~3~I site could be assessed
from the DNA sequence information available for the rsaA
gene, the necessary translation frame was known and thus a
single construction of a metallothionein gene was made.
CA 0224780~ 1998-09-03
W097/3~X~ PCT/CA971~167
- 22 -
This was done by excision of the coding sequence of monkey
metallothionein II peptide (60 amino acids comprising l0
cysteine residues and having a molecular weight of about
5000) at known restriction sites and adapting the gene ends
with BamHI linkers with appropriate base pair spacers for
the needed translation frame.
After insertion into the BamHI site created at
position 188, frame 3, several clones were exAmlned by
determining whether they could bind elevated levels of
cadmium by the assay described below. The assay was
necessary because the segment had equal probability of
being inserted backwards. One clone that gave positive
results was ~Am;ned by electron microscopy and the
presence of a normal S-layer was confirmed. The plasmid in
the clone that gave positive results was also examined by
DNA sequencing analysis, sequencing across the junction
between the position 188 site and the 5' side of the
metallothionein gene. The sequence data confirmed correct
orientation.
The plasmid-containing clone and relevant control
strains were ~Am~ned for the ability to bind several
metals known to be bound by native metallothionein. This
was done by growing the strains of bacteria in the presence
of the metals at a concentration of 5ug/ml. After
extensive washing of the cells to remove unbound metal, the
cells were ashed by treatment at 5000C and the residue was
dissolved in dilute nitric acid and examined for metal
content by atomic absorption spectroscopy. The results
from one round of data collection is shown in Table l. In
the case of cadmium and copper, an elevated level of bound
metal is noted in the metallothionein-expressing strains.
CA 0224780~ l998-09-03
W 097/34000 PCT/CA97/00167
~ 23 ~
Table 1
n~t~l lon 1~-3eed (u~ et~llOD Ul-it of c~lL~
coPper C~ldmlum zin~
~rial 1 2
C~15 ~,79 1.0 0.71 ~.15
d~ a - l~y-r
C--15~BAC 2.11~ 1.33 1.07 4.0
1 0 (S-l~y r ~-g~tlv- ~tr~l~)
e~r/pl88.3 2.01 1.30 11.1 3.66
(cont~3~ 5-l~y r
wlth ll~-r ~-rt only)
r~l~-r~/pl88.3~T 2.79 3.09 19.1 3.00
(S-l~y-r w~
Example 3: Investiqation of Other Permissive Sites in rsaA
Gene
A library of 240 BamHI linker insertions was created
using the procedures of Example 1. Of the 240 insertions,
target sites in the rsaA gene were made with ~saI. 34
of the latter insertions were discarded because the clones
contained deletions of rsaA DNA as well as the linker
insertions. The remaining 11 resulted in 5 non-permissive
and the 6 permissive sites described in Example 1. The
remaining 195 insertions in the library were made using the
enzymes HinPI, AciI, and MspI to create target sites as
outlined in Example 1. Of the latter 195 insertions, 49
permissive sites were located for a total of 55. Of those
sites scored as non-permissive, some may have had deletions
of rsaA DNA at the linker insertion site. One BamHI linker
insertion at a TaqI site thought to be permissive was later
found by nucleotide sequencing to be located outside the
rsaA structural gene reducing the total number of
permissive sites to 54 from 55.
Figure 7 illustrates the approximate location by
restriction mapping of 54 permissive sites. The results
show that sites that will accept 2-4 amino acids while
CA 0224780~ 1998-09-03
W 0 97134000 PCT/CAg7/00167
- 24 -
still allowing the protein to be made and assembled into an
S-layer are scattered up and down the protein.
Furthermore, there is an unexpectedly high proportion of
sites at which such insertions do not prevent expression
and assembly of the S-layer. The results indicate that
approximately 25-50~ of in-frame linker insertions will be
tolerated by the S-layer protein and the Caulobacter and
that diverse regions of the protein will tolerate
insertions. Thus, Caulobacter is an ideal candidate for
expression of polypeptides fused with the S-layer and the
presence of multiple permissive sites extending along the
rsaA gene will permit the insertion of a plurality of the
same or different peptides into the same RsaA protein
molecule and expressed on the surface of a single
Caulobacter.
Exam~le 4: Further Studies with Cadmium bindinq
Pol~ePtides
The results described for Example 3 indicated that it
would be possible to insert metallothionein at multiple
places in the RsaA protein and thereby enhance the metal
binding capacity of such a transformed Caulobacter.
However, when the procedures of Example 2 were repeated to
insert the metallothionein coding sequence into others of
the 54 permissive sites identified in the preceding Example
in each case, the transformed Caulobacter did not secrete
a chimeric protein and did not synthesize an S-layer.
Furthermore, the transformed Caulobacter of Example 2 was
stable as long as the transformants were frozen immediately
after isolation. When continuously cultured for
approximately one week, the transformants deleted the
metallothionein portion of the S-layer and the S-layer
protein returns to its normal size.
Consideration of the predicted amino acid sequence of the
rsaA protein shows that the latter protein lacks cysteine
residues whereas metallothionein has a high cysteine
CA 0224780~ 1998-09-03
W 0 97/34000 PCT/CA97/00167
- 25 -
content. It thus appeared that for secretion and long term
expression of a RsaA chimeric protein, the heterologous
polypeptide portions of the chimeric protein should not
have high cysteine content and preferably, not be capable
of forming multiple disulphide bonds in the chimeric
protein in an aerobic environment.
Following the foregoing procedures, single and multiple
copies of DNA encoding the synthetic cadmium binding
peptide shown in Figure 10 ~SEQ ID NO:ll) was synthesized,
inserted at the amino acid 277 site of rsaA using the above
described Carrier cassette and was expressed in C.
crescentus. The peptide has a single cysteine residue.
Mild acid extracts of whole cells expressing the modified
rsaA gene were subjected to SDS-PAGE for identification of
S-layer proteins. The S-layer protein was expressed and
secreted when there was from 1 to 3 copies of the cadmium
binding peptide present at RsaA amino acid position 277.
Insertion of 4 or more copies resulted in a dramatic
reduction of S-layer protein released from the whole cells
by mild acid treatment to barely detectable levels.
Detection by autoradiography of RsaA protein in vivo
labelled with 35 S-cysteine and ln vitro with 125 I-
iodoacetamide confirmed that the cadmium binding peptide
was part of the chimeric RsaA protein. This demonstrates
that Caulobacter crescentus is capable of secretion of a
chimeric rsaA protein having a limited cysteine content and
a limited capacity for disulphide bond formation within the
chimeric protein.
Example 5: ExPression and Presentation of Antiqenic
Epitopes on Caulobacter Cell Surface
Using the library of the 49 permissive sites other
than those made with ~I described in Example 3, the
coding sequence for the 12-amino acid pilus peptide epitope
lacking cysteine residues from Pseudomonas aeruqinosa PAK
pilin was inserted at the sites using the procedures
CA 0224780~ 1998-09-03
W O 97/34000 PCTtCA97/00167 - 26 -
described above and employing the Carrier cassette shown in
Figure 1. Positioning of the added DNA between the first
Bam HI site and the Bql II site permitted use of the latter
site for making repeated insertions of DNA. The coding
sequence for the peptide shown in Figure 8, including both
cysteine residues was also inserted in separate
experiments.
DNA coding for the peptide shown in Figure 8 (SEQ ID NO:8)
was prepared by oligonucleotide synthesis of two anti-
complementary strands. The transformed bacteria werescreened for both production and presentation of the
epitopes by the transformed Caulobacter by using standard
Western immunoblot analysis ~see: Burnette, W. N. 1981.
"Western Blotting; Electrophoretic Transfer of Protein
from Sodium Dodecyl-Polyacrylamide Gels to Unmodified
Nitrocellulose and Radiographic Detection Antibody and
Radioiodinated Protein A". Analytical Biochemistry 112:195-
203) and by colony immunoblot tests in which the cells were
not disrupted (see: Engleberg, N.C., et al. 1984. "Cloning
an Expression of ~eqionella pneumo~hilia Antigens in
Escherichia coli". Infection and Immunity 44:222-227).
Anti-pilus monoclonal antibody obtained from Dr. Irvin,
Dept. of Microbiology, University of Alberta (Canada) was
used in the immunoblot analyses to detect the presence of
the pilus epitope insert. The antibody (called PK99H) was
prepared using purified Pseudomonas aeruqinosa PAK pilin as
the antigen and the monoclonal antibody against the 12
amino acid epitope was isolated by standard techniques
using BALB/C mice as a source of ascites fluid. Reaction
with the antibody in the whole cell colony immunoblot assay
shows that the epitope is not only expressed in the
transformed Caulobacter but is exposed on the S-layer
surface overlying the cell in such a way that the epitope
is available to the antibody. When the two cysteine
residues of the pilin epitope were incorporated in the
CA 0224780~ 1998-09-03
WOg7/34~ PCT/CA97/~167
- 27 -
chimeric protein, the protein was still expressed and
secreted at normal levels.
Of the organisms screened, insertions of the pilus epitope
at the following sites in the rsaA gene as determined by
nucleotide sequencing resulted in a positive reaction with
the antibody in the whole cell Colony immunoblot analysis:
69, 277, 353, 450, 485, 467, 551, 574, 622, 690, 723, and
944. The results show that the permissive sites that will
accept polypeptides of the size of the pilus epitope are
numerous and scattered across the rsaA gene.
Further studies with the pilus peptide resulted in
successful expression and secretion of RsaA chimeric
proteins have single copies of the peptide at the locations
shown in Figure 11. Also, four and seven copies of the 12
amino acid pilus peptide were expressed and secreted as a
RsaA chimeric protein when inserted at amino acids 277 and
551 respectively of the RsaA protein. However, insertions
of the pilus peptide at amino acids 69, 277, 450, 551 and
622 resulted in a chimeric protein that did not attach to
the cell surface and was released into the culture medium.
Example 6: Insertion of Larqe Polype~tides
Bacterial surface proteins from organisms other than
Caulobacter described in the prior art are generally not
known to accept polypeptides larger than about 60 amino
acids within the structure of the surface protein. The
procedures of the preceding Example were carried out in
order to insert the coding sequence of a 109 amino acid
epitope from IHNV virus coat glycoprotein at insertion
sites identified in the preceding Example. The IHNV
epitope was prepared by PCR and had the portion of the
sequence shown in Figure 9 (SEQ ID N0:9) which is
equivalent to amino acid residues 336-444 of the IHNV
sequence described in: Koener, J.F. et al. 1987.
"Nucleotide Sequence of a cDNA Clone Carrying the
CA 0224780~ l998-09-03
W O 97/34000 PCT/CA97/00167
- 28 -
Glycoprotein Gene of Infectious Hematopoietic Necrosis
Virus, a Fish Rhabdovirus". Journal of Virology 61:1342-
1349. Anti-IHNV polyclonal antibody against whole IHNV
obtained from Dr. Joann Leong, Dept. of Microbiology,
Oregon State University, U.S.A. (see: Xu, L. et al. 1991.
"Epitope Mapping and Characterization of the Infectious
Hematopoietic Necrosis Virus Glycoprotein, Using Fusion
Proteins Synthesized in Escherichia coli". Journal of
Virology 65:1611-1615) was used in the immunoblot assays
described in the preceding Example to screen for
Caulobacter that express and present the IHNV sequence on
the surface of the S-layer of the Caulobacter. Reaction in
the whole cell colony immunoblot assay was positive in
respect of insertions at sites 450 and 551, and negative at
a site which was at approximately amino acid 585.
The IHNV insert contains a single cysteine residue and is
an extremely large insert for successful expression as a
fusion product with a bacterial surface protein.
In further studies, the same 109 amino acid portion of the
IHNV glycoprotein was inserted at amino acid 450 of the
RsaA protein. The chimeric protein expressed and secreted
by Caulobacter crescentus and was recovered from the cell
culture medium. SDS-PAGE analysis of the recovered
proteins showed that some of the chimeric proteins were
smaller than the predicted rsaA chimeric protein but still
bound anti-IHNV antibody. Analysis of these proteolytic
products showed that cleavage of the chimeric protein
occurred at an Arg residue encoded by the gene transfer
cassette shown in Figure 1. Thus in some cases, adjustment
of the nucleotide sequence at the interface of the
polypeptide and rsaA coding sequences may be necessary to
prevent expression of an arginine residue.
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CA97/00167
- 29 -
Exam~le 7
Methods are described above for the insertion of
12-bp BamHI linker sites into a promoterless version of the
rsaA gene. Because linker insertions involve the insertion
of 12 bp (i.e. a multiple of three) an in-frame linker
insertion resulted in every case. These linker sites are
introduced to allow subsequent insertion of DNA encoding
foreign peptide/proteins. Expression of such chimeric
genes leads to the production of an entire full-length RsaA
protein carrying the inserted heterologous amino acid
sequence of interest. A number of BamHI site positions
were identified above precisely by nucleotide sequencing.
Four of the sites in the rsaA gene correspond to amino acid
positions 188, 782, 905, 944 in the RsaA protein. For this
example, an additional linker insertion was created at
amino acid position 95 of the native gene (i.e. this gene
carried its own promoter) using the same methodology. All
five in-frame BamHI linker insertion sites were inserted in
the rsaA so that the nucleotides of the linker DNA were
read in the reading frame GGA/TCC (Figure 12).
Because all BamHI linker nucleotides were read in
the same reading frame, the 5' region of one rsaA gene
carrying a BamHI linker insertion at one position could be
combined with the 3' region of an rsaA gene carrying
another of the BamHI linker insertions to create in-frame
deletions with a BamHI site at the joint between adjacent
regions of rsaA. Using such a method, in-frame deletions
of rsaA (aAA95-782) and rsaA(AAA188-782) were created.
DNA fragments encoding various C-terminal portions
of the 1026 amino acid RsaA protein were isolated using the
newly inserted BamHI linker sites as the 5'-terminus of the
fragment and a HindIII site as the 3' terminus of the
fragment. These BamHI fragments were transferred to the
BamHI/HindIII sites of pUC8 (J. Vieira, and J. Messing.
1982." The pUC Plasmids, an M13mp7-Derived System for
Insertion Mutagenesis and Sequencing With Synthetic
Universal Primers" Gene 19:259-268) creating "rsaA C-
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CA97/00167 - 30 -
terminal Segment Carrier plasmids" (Figure 12). The
insertion into pUC8 also resulted in the creation of an in-
frame fusion between the first 10 N-terminal amino acids of
LacZa and the various C-terminal fragments (AA782-1026,
AA905-1026 or AA944-1026) of RsaA. These LacZa:rsaA fusion
proteins can be produced in C. crescentus using the lacZa
transcription/translation initiation signals when
introduced on appropriate plasmid vectors or direct
insertion into the chromosome (see: W.H. Bingle, et al.
1993. "An All-Purpose Cellulase Reporter for Gene Fusion
Studies and Application to the Paracrystalline Surface (S)-
Layer Protein of Caulobacter crescentus." Can. J.
Microbiol. 39:70-80).
Both types of constructions (i.e., the deletion
versions and the C-terminal only segments) result in the
production of proteins that are secreted in Caulobacter
strains as highly modified RsaA proteins. The gene
segments can also facilitate the secretion of heterologous
polypeptides by insertion or fusion of appropriate DNA
sequences at the unique BamHl site that exists in each of
the constructions. The following describes specific
methods for doing so to create chimeric proteins capable of
secretion in C. crescentus.
A- Creatinq fusions of desired se~uences with C-terminal
portions of rsaA -Method 1
The general process is as follows:
1) Insertinq the desired se~uence into the Carrier
cassette. The following describes the specific manner in
which heterologous sequences may be introduced into the
Carrier cassette of Figure 1.
a) Insertion of a single copy of the desired gene
segment.
CA 0224780~ l998-09-03
WO 97/34000 PCT/CA97/00167 - 31 -
Depending upon the length of the gene segment, two
methods of construction may be used. For segments of up to
about 30 amino acids, two oligonucleotides of appropriate
sequence are chemically synthesized, annealed by mixing,
heating and slow cooling and then ligated into the Carrier
cassette. The oligonucleotides will also contain
additional base pairs that recreate "sticky ends" of
appropriate restriction endonuclease sites at each end of
the duplex DNA that results from the annealing process.
For longer segments, PCR is used to amplify a
region of a target DNA sequence. Oligonucleotides are
synthesized that have sequence complementary to the
boundaries of the desired sequence and which contain
additional base pairs that recreate a "sticky end" of an
appropriate restriction endonuclease site. In the present
example oligonucleotides are made to produce products with
the appropriate restriction endonuclease site for
directional cloning into the Carrier cassette. PCR
amplification of the desired sequence is then done by
standard method~.
For both methods, the sticky ends prepared must be
appropriate for an XhoI site at the 5' terminus of the
desired DNA sequence and StuI or SalI sites at the 3'
terminus; this places the desired gene segment in the
correct orientation within the Carrier cassette. Reading
frame continuity is maintained by appropriate design of the
oligonucleotides used for the PCR step.
b) Preparation of multiple copies of the desired
gene segment.
The Carrier cassette also allows production of
multiple insert copies. A BqlII site in the cassette is
restored after removal of the promoterless antibiotic
resistance gene; that site can be used to insert an
additional copy of the Carrier/desired sequence insertion,
using the terminal BamHI sites, because the "sticky ends"
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CA97/00167 - 32 -
produced by both BamHI and BqlII are the same. This
~piggy-back" insertion still maintains the correct reading
frame throughout the construction. Any number of
additional cycles of "piggy-backing" can be done because
the BamHI/BqlII ligation results in sequence which is no
longer a substrate for either enzy~me. The result is the
production of cassettes of multiple copies of the desired
sequence which can be transferred to appropriately modified
S-layer protein genes with the same ease as a single copy.
An additional feature of this method is that different
heterologous sequences can be paired together in this
multiple copy cassette with the same ease as multiple
copies of the same heterologous sequence.
Example 7a. Insertion of an 109 amino acid segment of the
IHNV surface glycoprotein to Carrier cassette.
Using the methods described, a PCR product was made
that contained the DNA coding for amino acids 336 to 444
(Figure 9) of the major surface glycoprotein of the
Infectious Hematopoietic Necrosis Virus (IHNV), which
infects Salmonid fish.
Exam~le 7b. Insertion of an 184 amino acid segment of the
IHNV surface glycoprotein to Carrier cassette.
Using the methods described a PCR product was made
that contained the DNA coding for amino acids 270 to 453 of
the IHNV glycoprotein segment shown in Figure 9..
ExamPle 7c. Insertion of single and multiple copies and an
epitope of the Pseudomonas aeruginosa PAK pilus gene to
Carrier cassette.
Oligonucleotides were constructed to code for the
pilus epitope described in Example 5, which corresponds to
a sequence at the extreme C-terminus of the pilus protein.
Using the methods outlined in part A(l)(b) of this Example,
3 tandem copies were prepared.
CA 0224780~ 1998-09-03
W O 97134000 PCT/CAg7/00167
- 33 -
2) Transfer of Carrier cassette to the rsaA C-terminal
Segment Carrier ~lasmids. The constructions described in
examples 7a and 7b above are then transferred to the rsaA
C-terminal Segment Carrier plasmids, described above,
resulting in an in-frame fusion of: a) a very short section
of the betagalactosidase protein (10 amino acids), b) the
desired sequence flanked by 2-3 amino acids derived from
Carrier cassette sequence and c) the appropriate rsaA
C-terminal segment. In some cases, the first codon of the
rsaA C-terminal segment is converted to a different codon
as a result of the fusion. For example, while the rsaA
C-terminal segment may have coded for amino acids 944-1026
of RsaA, the resulting chimeric protein may only have amino
acids 945-1026 native to RsaA.
Exam~le 7d. Fusion of Carrier/109 AA and 184 IHNV segments
to C-terminal rsaA segment AA782-1026.
This was done using the Carrier cassettes described
in Examples 7a and 7b above and the AA782-1026 rsaA C-
terminal Segment Carrier plasmid described above.
Exam~le 7e. Fusion of Carrier/109 AA and 184 AA IHNV
segments to C-terminal rsaA segment AA905-1026.
This was done using the Carrier cassettes described
in Examples 7a and 7b above and the AA905-1026 rsaA C-
terminal Segment Carrier plasmid described above.
Exam~le 7f. Fusion of Carrier/109 AA and 184 AA IHNV
segments to C-terminal rsaA segment AA944-1026.
This was done using the Carrier cassettes described
in Examples 7a and 7b above and the AA944-1026 rsaA C-
terminal Segment Carrier plasmid described above.
Exam~le 7q. Fusion of Carrier/3x Pilus Epitope segment to
C-terminal rsaA segment AA782-1026.
CA 0224780~ 1998-09-03
W 097~4000 PCTICA97/00167
- 34 -
This was done using the Carrier cassettes described
in Example 7c above and the AA782-1026 rsaA C-terminal
Segment Carrier plasmid described above.
3) Expression of the desired fusion in an ap~ropriate
Caulobacter host strain.
a) Plasmid-based expression.
To create plasmid vectors that can be introduced
and maintained in appropriate Caulobacter strains, the
entire rsaA C-terminal Segment Carrier plasmids were fused
to broad host range vectors pKT215 or pKT210 (see: M.
Bagdasarian, et al. 1981." Specific-Purpose Cloning
Vectors. II. Broad-Host-Range, High Copy Number RSF1010-
Derived Vectors, and a Host-Vector System for Gene Cloning
in Pseudomonas." Gene 16:237-247~ using the unique HindIII
restriction site present in each plasmid. The resulting
plasmid is introduced into Caulobacter by conjugation or
electroporation methods and is maintained by appropriate
antibiotic selection.
The fusions described in examples 7d-7g were expressed
in Caulobacter. In each case expression and secretion of
the chimeric S-layer protein was detected by Western
immunoblot analysis of electrophoretic gels of the cell
culture supermutant employing the monoclonal antibody for
each of the polypeptide epitopes. The transporter signal
for secretion from Caulobacter must be in the C-terminal
region of amino acids 945-1026 of the S-layer protein as
all chimeric proteins in the examples were secreted.
Precipitation of the chimeric protein occurred with the use
of rsaA segment AA782-1026 but not AA944-1026. Recovery of
precipitate using AA905-1026 was reduced as compared to
AA782-1026.
CA 0224780~ 1998-09-03
W O 97134000 PCT/CAg7~0167
- 35 -
b) Selection of appropriate Caulobacter host
strains
In nearly all cases the use of a S-layer negative
host strain is appropriate. C. crescentus strain CB2A and
strain CB15aKSac fulfil this requirement. If it is
important to ensure that all fusion protein is no longer
attached to the cell surface, the use C. crescentus strains
CB15Ca5KSac or CB15CalOKSac are appropriate. These strains
have additional mutations that result in the loss of the
production of a specific species of surface
lipopolysaccharide that has been demonstrated to be
involved with the surface attachment of native S-layer
protein as a 2-dimensional crystalline array (see: Walker
S.G. et al 1994. "Characterization of Mutants of _.
crescentus Defective in Surface Attachment of the
Paracrystalline Surface Layer". J. Bacteriol. 176:6312-
6323). Most often with the highly modified versions of the
S-layer gene, this precaution is not necessary since
virtually all regions of the gene that may have a role in
the attachment process have been removed.
There are two types of growth media well suited to both
propagation of Caulobacter for general purposes, including
cloning steps, and also to produce the secreted and
aggregated chimeric proteins. Example of the two types
are: 1) PYE medium, a peptone and yeast extract based
medium described in Walker et al, ~1994) [supraJ, and 2)
M6HiGG medium, a defined medium described in: Smit, J., et
al 1981. "Caulobacter crescentus Pilin: Purification,
Chemical Characterization and Amino-Terminal Amino Acid
Sequence of a Structural Protein Regulated During
Development". J. Biol. Chem. 256, 3092-3097. The latter
medium is especially appropriate for preparation of the
aggregated chimeric proteins since it permits growth to
higher densities (therefore maximizing protein yield) and
results in purer aggregated proteins since there are no
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CAg7/00167
- 36 -
medium derived proteins to contaminate the chimeric
proteins retrieved.
B- Creatinq Fusions of desired sequences with C-terminal
portions of rsaA -Method 2.
Methods other than the use of the Carrier cassette
plasmids are possible to create heterologous insertions
into deletion versions of a S-layer gene or to create
fusions with C-terminal portions of the S-layer protein.
PCR may be used although other known methods may also be
used. The general procedure is as follows:
1) Use PCR to prepare aPPropriate seqments:
a) Preparation of amplified segment with
appropriate ends is carried out in a manner similar to that
described part A(l)(a) above. ~ligonucleotides are
designed and synthesized such that they will anneal to
appropriate regions of the desired heterologous DNA and
also contain "sticky ends" of appropriate sequence and
frame so that the resulting PCR product can be directed
inserted into appropriate modified S-layer genes.
b) Transfer to appropriate C-terminal rsaA segments
is carried out by inserting the PCR products into the C-
terminal segments AA782-1026, AA905-1026, or AA944-1026, as
described in Examples 7d-7g above. In addition to the BamHI
site described, the EcoR1 restriction site could also be
used as the 5' terminus of the incoming PCR segment, since
this site is also available in the pUC8 vector and not in
the S-layer gene, so long as the correct reading frame was
maintained when designing the oligonucleotides used to
prepare the PCR product.
2) Expression of the desired fusion in an appropriate
Caulobacter host strain is carried out using the procedures
outlined in part A(3) above.
CA 0224780~ 1998-09-03
W O 97/34W0 PCT/CA97/00167
- 37 -
C- Creatinq insertions of desired seauences into versions
of a S-layer qene havinq larqe internal in-frame deletions.
The general process is as follows:
1) Creating appropriate in-frame deletions.
rsaA (~AA95-782~ and rsaA(~AA188-782) were prepared
as described above. Because most of the BamH1 linker
insertion sites are in the same reading frame with respect
to each other, it is possible to combine other pairs of 5'
and 3' segments using the same general method, with the
same result of maintenance of correct reading frame
throughout. These deletion versions must then be tested
individually to ensure that S-layer protein is still
secreted by the Caulobacter.
2) Insertion of a Gene Seqment Carrier cassette containinq
the desired sequences: as described at part A(1) above,
carried out using the procedure described in part A(2)
above.
Exam~le 7h. Insertion of the 109 AA IHNV segment into rsaA
(~AA95-782) and insertion of the 109 AA IHNV segment into
rsaA(~AA188-782) is carried out as in Examples 7d-7g above.
Expression of the desired genetic construction in
appropriate C. crescentus strains is done using the
procedures outlined in part A(3) above.
3) Alternate PCR ~rocedures: can be used to prepare a
heterologous segment for direct insertion into the BamHI
site with the deletion versions of the rsaA gene. The
procedure is essentially the same as described in part B(1)
above.
CA 0224780~ 1998-09-03
W O 97/34000 PCT/CAg7/00167
- 38 -
ExamPle 8. (Transfer to the native S-la~er qene
chromosomal site as a sinqle crossover event).
The fusion of the Carrier cassette with appropriate
heterologous DNA segments to a C-terminal S-layer protein
segment plasmid results in a pUC8-based plasmid that is not
maintained in Caulobacter. Selection for the antibiotic
marker on the plasmid results in detection of the rescue
events. Most commonly these are single crossover
homologous recombination events. The result is a direct
insertion of the entire plasmid into the chromosome. Thus
the resident copy of the S-layer gene remains unchanged as
well as the incoming highly modified S-layer gene. In such
cases it may be desirable to use Caulobacter strains in
which the resident S-layer gene is inactivated in known
ways. One example is the use of C. crescentus strain
CB15AKSac; this strain has an antibiotic resistance gene
cassette introduced at a position in the S-layer gene about
25~ of the way from the 5' terminus.
Example 9. (Transfer to the native S-layer qene
chromosomal site as a double crossover event).
In certain cases it may be desirable to completely
exchange the resident S-layer gene copy with the incoming
highly modified version. One method is the incorporation
of a sacB gene cassette (Hynes, M.F., et al. 1989. "Direct
Selection for Curing and Deletion of Rhizobium Plasmids
Using Transposons Carrying the Bacillus subtilis sacB
Gene." Gene 78: 111-119) into the pUC8 based plasmids
carrying the desired chimeric gene construction. This
cassette contains a levansucrase gene from Bacillus
subtilis that, in the presence of sucrose, is thought to
result in the production of a sugar polymer that is toxic
to most bacteria when expressed inside the cell. One first
selects for the single crossover event as described in
Example 8. Subsequent growth on sucrose-containing medium
results in the death of all cells except those that lose
the offending sacB gene by homologous recombination within
CA 0224780~ 1998-09-03
W O 97/~U~ PCT/CA97/00167
- 39 -
the 2 adjacent gene copies. Two events are possible;
restoration of the resident copy of the S-layer gene or
replacement of the resident copy with the incoming modified
gene (the latter is the desired event). A screen with
insertion DNA as probe or antibody specific to the
heterologous gene product identifies successful gene
replacement events. The method requires that the S-layer
gene sequence or native sequence immediately ad~acent to
the S-layer gene be on both sides of the heterologous
sequence (ie, Carrier cassette sequence plus heterologous
DNA) and in the present case is best suited for the
deletion versions of the S-layer gene.
Other methods are available for the delivery of
genes to the chromosome of a Caulobacter. Methods
involving the use of the transposons Tn5 and Tn7 as a means
of delivery of genes to random chromosome locations are
available (see: Barry, G.F. 1988 "A Broad-Host-Range
Shuttle System for Gene Insertion into the Chromosomes of
Gram-Negative Bacteria." Gene 71:75-84.). The use of the
xylose utilization operon as a target for chromosome
insertion have also been described. This method involves
the incorporation of a portion that operon into the pUC8
based plasmid constructions described above. This allows
homologous recombination within the xylose operon as a
means of plasmid rescue. Loss of the the ability to use
xylose as a nutrition source is used as the means of
confirming the rescue event.
This invention now being described, it will be
apparent to one of ordinary skill in the art that changes
and modifications can be made thereto without departing
from the spirit or scope of the appended claims.