Note: Descriptions are shown in the official language in which they were submitted.
CA 02247900 2005-07-28
METHODS AND APPARATUS FOR PARTICLE PRECIPITATION AND
COATING USING NEAR-CRITICAL AND SUPERCRITICAL ANTISOLVENTS
FIELD OF THE INVENTION
The present invention relates to a method and an apparatus for extremely small
particle precipitation, wherein a fluid dispersion containing a substance to
be precipitated
is contacted with a supercritical fluid (SCF) antisolvent such as carbon
dioxide under
near-or supercritical temperature and pressure conditions for maximizing small
particle
formation. The invention provides spray techniques wherein the interphase mass
transfer
rate is maximized between small droplets of the dispersion and antisolvent so
as to
generate precipitated particles having an average diameter of from about 0.1-
10 ~,m. The
invention also includes supercritical fluid coating techniques wherein
fluidized core
particles are coated with precipitated particles in a SCF antisolvent
precipitation chamber.
DESCRIPTION OF THE PRIOR ART
A number of industries have experienced a long-felt need for particle
micronization and nanonization. The need for an apparatus or method capable of
producing sub-micron and nano-sized particles is particularly pronounced in
the field of
pharmaceutics. Conventional techniques for particle-size reduction currently
practiced
suffer from many disadvantages. These conventional methods involve either
mechanical
comminution (crushing, grinding, and milling) or recrystallization of the
solute particles
from liquid solutions. The limitations of mechanical comminution for particle-
size
reduction are the shock sensitivity associated with the solid, thermal
degradation due to
heat generation during mechanical comminution, lack of brittleness
1
CA 02247900 1998-08-31
WO 97/31691 PCT/iIS97/03207
of some solids (e.g., most polymers}, and chemical degradation due to exposure
to the
atmosphere.
Conventional recrystallization of solutes from liquid solutions exploits the
dependence of a compound's solubility on temperature and/or mixture
composition. By
changing the temperature, or adding antisolvents to selectively remove the
solvent in
which the solid is solubilized, the desired material may be precipitated or
crystallized
from solution to form particles. Crystallization by either solvent evaporation
or solvent
extraction of a solute usually requires the use of toxic organic antisolvents,
surfactants
and oils, and yields wet particles that require further drying to remove
traces of
i 0 adsorbed solvent residues. Freeze drying tends to produce particles with
broad size
distribution that require further drying. Spray drying usually requires
evaporation of
solvent in a hot fluidized air bed. The high temperatures can degrade
sensitive drugs
and polymers. Monodisperse particle-size distribution with consistent crystal
structure
and crystalline properties is also difficult to attain using the above-noted
techniques.
Within the last decade, processes for the production of micron and sub-micron
sized particles have emerged that use either a supercritical fluid (i.e., a
fluid whose
temperature and pressure are greater than its critical temperature (T~) and
critical
pressure (P~)), or compressed fluids in a liquid state. A characteristic of a
substance
above its critical temperature is that it cannot be condensed regardless of
the exerted
pressure. It is well known that at near-critical temperatures, large
variations in fluid
density and transport properties from gas-like to liquid-like can result from
relatively
moderate pressure changes around the critical pressure (0.9-1.5 P~). While
liquids are
nearly incompressible and have low diffusivity, gases have higher diffusivity
and low
solvent power. Supercritical fluids can be made to possess an optimum
combination
of these properties. The high compressibility of supercritical fluids
(implying that large
changes in fluid density can be brought about by relatively small changes in
pressure,
making solvent power highly controllable} coupled with their liquid-like
solvent power
and better-than-liquid transport properties (higher diffusivity, lower
viscosity and lower
surface tension compared with liquids), provide a means for controlling mass
transfer
(mixing) between the solvent containing the solutes (such as a drug or
polymer, or both)
and the supercritical fluid.
Two processes that use supercritical fluids for particle formation are: ( 1 )
Rapid
Expansion of Supercritical Solutions CRESS) (Tom, J.W. Debenedetti, P.G.,
1991, The
Formation of Bioerodible Polymeric Microspheres and Microparticles by Rapid
Expansion ofSupercritical Solutions. BioTechnol. Prog. 7:403-411), and (2} Gas
Anti-
Solvent (GAS) Recrystallization (Gallagher, P.M., Coffey, M.P., Krukonis,
V.J., and
Klasutis, N., 1989, Gas Antisolvent Recrystallization: New Process to
Recrystallize
Compounds in Soluble and Supercritical Fluids. Am. Chem. Sypm. Ser., No. 406;
U.S.
2
CA 02247900 1998-08-31
WO 97/31691 PCTlUS97/03207
Patent No. 5,360,478 to Krukonis et al.; U.S. Patent No. 5,389,263 to
Gallagher et al.).
w See also, PCT Publication WO 95/01221 and U.S. patent No. 5,043,280 which
describe
additional SCF particle-forming techniques.
In the RESS process, a solute (from which the particles are formed) is first
solubilized in supercritical COZ to form a solution. The solution is then
sprayed through
a nozzle into a lower pressure gaseous medium. Expansion of the solution
across this
nozzle at supersonic velocities causes rapid depressurization of the solution.
This rapid
expansion and reduction in COz density and solvent power leads to
supersaturation of
the solution and subsequent recrystallization of virtually contaminant-free
particles.
The RESS process, however, is not suited for particle formation from polar
compounds
because such compounds, which include drugs, exhibit little solubility in
supercritical
CO2. Cosolvents (e.g., methanol) may be added to COZ to enhance solubility of
polar
compounds; this, however, affects product purity and the otherwise
environmentally
benign nature of the RESS process. The 12ESS process also suffers from
operational
I S and scale-up problems associated with nozzle plugging due to particle
accumulation in
the nozzle and to freezing of COZ caused by the Joule-Thompson effect
accompanying
the large pressure drop.
The relatively low solubiiities of pharmaceutical compounds in unmodified
carbon dioxide are exploited in the second process wherein the solute of
interest
{typically a drug, polymer or both) is dissolved in a conventional solvent to
form a
solution. The preferred ternary phase behavior is such that the solute is
virtually
insoluble in dense carbon dioxide while the solvent is completely miscible
with dense
carbon dioxide at the recrystallization temperature and pressure. The solute
is
recrystallized from solution in one of two ways. In the first method, a batch
of the
solution is expanded several-fold by mixing with dense carbon dioxide in a
vessel.
Because the carbon dioxide-expanded solvent has a lower solvent strength than
the
pure solvent, the mixture becomes supersaturated forcing the solute to
precipitate or
crystallize as microparticles. This process was termed Gas Antisolvent (GAS)
recrystallization (Gallagher et al., 1989).
The second method involves spraying the solution through a nozzle into
compressed carbon dioxide as fine droplets. In this process, a solute of
interest
(typically a drug, polymer or both) that is in solution or is dissolved in a
conventional
solvent to form a solution is sprayed, typically through conventional spray
nozzles,
such as an orifice or capillary tube(s), into supercritical COZ which diffuses
into the
spray droplets causing expansion of the solvent. Because the CO1-expanded
solvent
has a lower solubilizing capacity than pure solvent, the mixture can become
highly
supersaturated and the solute is forced to precipitate or crystallize. This
process has
been termed in general as Precipitation with Compressed Antisolvents
(PCA)(Dixon,
3
CA 02247900 1998-08-31
WO 97/31691 PCT/US97/03207
D.J.; Johnston, K.P.; Bodmeier, R.A. AIChE J. 1993, 39, 127-139.) and employs
either liquid or supercritical carbon dioxide as the antisolvent. When using a
supercritical antisolvent, the spray process has been termed Supercritical
Antisolvent
(SAS) Process (Yeo, S.-D.; Debenedetti, P. G.; Radosz, M.; Schmidt, H.-W.
Macromolecules 1993, 26, 6207-6210.) or Aerosol Spray Extraction System CASES)
Miiller, B. W.; Fischer, W.; Verfahren zur Herstellung einer mindestens einen
Wirkstoff and einen Trager umfassenden Zubereitung, German Patent Appl. No. DE
3744329 A1 1989).
PCT Publication WO 95/01221 teaches the use of a coaxial nozzle for the
IO co-introduction into a vessel of a supercritical fluid and solutions in
concurrent
directions of flow. Such nozzles achieve solution breakup through the
impaction of the
solution by a relatively higher velocity fluid. The high velocity fluid
creates high
frictional surface_forces causing solution disintegration into droplets. Any
potential
high energy waves generated with nozzles described in the prior art are random
and
I 5 originate from impaction and frictional effects of the high velocity fluid
on the solution
or secondary impaction of multiple vehicle droplets. For purposes of clarity,
such high
energy waves are defined as Type I waves.
High frequency sound waves can be generated via various types of transducers
such as piezoelectric, magnetostrictive, electromagnetic, pneumatic devices
(so-called
20 whistles similar to the common whistle based on the organ-pipe effect) and
other
mechanical transducers. The use of sound waves produced by one or more of
these
devices to generate droplets from Liquid surfaces or to atomize liquid spray
jets has been
known for more than half a-century (see Ensminger, "Ultrasonics: Fundamentals,
Technology, Applications ", 2d Ed., Marcel Dekker, 1988 for numerous
examples).
25 One of the earliest 'pneumatic devices' used to generate sound waves
employed
a jet of air impinging on a cavity to generate sound waves -- the so-called
Hartmann
whistle (J. Hartmann, "Construction, Performance and Design of the Acoustic
Air-Jet
Generator ", Journal of Scientific Instruments, 16, 140-149, 1939). In the
Hartmann
whistle, a jet of air, with velocities reaching Mach I, is directed into a
hollow cavity.
30 The impact at the bottom of the cavity causes a rise in pressure, which in
turn causes
a counter flow of the energizing gas. The momentum of this counterflow of
fluid
causes a pressure rarefaction in the cavity. When the force of the jet
overcomes the
momentum, the flow direction again reverses toward the bottom of the cavity to
complete the pressure cycle and propagating a sound wave. Focusing the
generated
35 sound waves on spray jets has been employed to atomize liquid spray jets.
It must be
appreciated that because the smallest practical focal region of a wave is a
sphere one
wave-length in diameter, the focal region of a focused sound wave is
relatively large as
compared to a focused light wave.
4
CA 02247900 1998-08-31
WO 97!31691 PCT/US97/03207
Whistle-type devices have been used to generate high-intensity sound waves in
w both air and liquids. A practical upper frequency for applications using air
is
approximately 30 kHz. Using helium or hydrogen, such whistles are capable of
generating ultrasonic energy in air up to 500 kHz. It is generally recognized
in the field
that the effectiveness of the device in an application correlates with the
frequency (or
inversely with the wavelength). The efficiency (ratio of radiated power to the
power
delivered to the transducer) for such has been reported between < 5% to 14%.
It is also
recognized in the field that the effectiveness of the device does not
necessarily correlate
with its efficiency. In other words, a low-efficiency nozzle can be highly
effective in
producing desired droplet sizes.
Whistles for generating sound waves in liquid have also been developed for
industrial use. Since the velocity of sound is considerably higher in liquids
than in
gases, jet velocities equal to the velocity of sound are impractical in
liquids. The
whistles of W. Janovsky and R. Pohlmann (Zeitschrift fiir Angewandte Physik,
1, 222,
1948) operate on the jet-edge principle, wherein a high-pressure jet of the
liquid or
liquids is impinged on the edge of a thin plate which is mounted at the
displacement
nodes. The plate vibrates in flexure at resonance producing low-frequency
waves,
typically on the order of magnitude of 5000 Hz. Such "liquid whistles" have
been used
to produce emulsions or dispersions of one dense medium in another dense
medium
(oil/water, mercury/water, etc.)
In many instances, especially in the pharmaceutical industry, it is desired to
coat
core particles or medicaments. Generally, such coating has been carried out
using
techniques such as electrolysis, vapor deposition, and fluidized bed or air
suspension
techniques. However, these methods all suffer from various drawbacks, e.g.,
the
difficulty in maintaining aseptic conditions, the inability to generate
extremely fine
particles for coating purposes and solvent emission control.
SUMMARY OF THE INVENTION
The present invention provides improved near- or supercritical fluid processes
for the precipitation of extremely small particles having average diameters
(inferred
from SEM photographs) on the order of from about 0.1-10 p.m and most
preferably up
to about 0.6 lzm. The methods of the invention find particular utility as
methods for
particle rnicronization and nanonization, particularly in the field of
pharmaceutics.
However, the methods of the invention can also be used in other fields such as
those
related to foods, chemicals, polymers, pesticides, explosives, coatings and
catalysts
wherein benefits are obtained from a decrease in particle sizes and
concomitant
increases in particle surface areas.
5
-_
CA 02247900 1998-08-31
WO 97/31691 PCT/iTS97/03207
Broadly speaking, the methods of the invention involve precipitation of
extremely small particles which can be recovered as particles, or deposited on
core
particles to form composite products. In all cases however, the methods of the
invention involve contacting a fluid dispersion (e.g., a gas or liquid
solution or
suspension) including a continuous phase dispersant with at least one
substance (e.g.,
a medicament such as a drug) dispersed in the dispersant with an antisolvent
at near-or
supercritical conditions for the antisolvent, so as to cause the antisolvent
to deplete the
dispersant and precipitate the substance as extremely small particles.
Conditions are
established during the contacting step so as to enhance the mass transfer rate
between
the antisolvent and the dispersant so that particle nucleation and
precipitation occur
rapidly.
In most cases, the fluid dispersions of the invention would be in the form of
liquid solutions, i.e., the dispersant is a solvent and the substance to be
precipitated is
a solute dissolved in the solvent. Moreover. the disnersants Shnnhi nnmr~rieP
a+ lPac+
1 S about 50% by weight (and more preferably at least about 90% by weight) of
the overall
dispersions. The conditions established during the dispersion/antisolvent
contacting
step are typically in the range of from about 0.7-1.4 T~ and from about 0.2-7
P~ of the
antisolvent; more preferably, these ranges are from about 1-1.2 T~ and from
about 0.9-2
P~ of the antisolvent. Preferably, the conditions during contact are
maintained so that
the dispersant and antisolvent are essentially completely miscible in aII
proportions
thereof.
The antisolvents used in the invention are normally selected from the group
consisting of carbon dioxide, propane, butane, isobutane, nitrous oxide,
sulfur
hexafluoride and trifluoromethane, with carbon dioxide being the single most
preferred
antisolvent for reasons of cost and ease of processing. In all cases, the
antisolvent
should be substantially miscible with the dispersant while the substance or
medicament
to be precipitated should be substantially insoluble in the antisolvent, i.e.,
the substance
or medicament, at the selected dispersionlantisoivent contacting conditions,
should be
no more than about 5% by weight soluble in the antisolvent, and preferably is
essentially completely insoluble.
In one preferred aspect of the invention, improved spray processes are
provided
for precipitation of extremely small particles. For example, use may be made
of
specialized nozzles for creating extremely fine droplet sprays of the fluid
dispersions
into a precipitation zone containing antisolvent. Using such equipment, the
methods
of the invention involve passing the fluid dispersion through a first
passageway and first
passageway outlet into the precipitation zone containing the antisolvent and
maintained
at the above-defined near- or supercritical conditions of temperature and
pressure for
the antisolvent. Simultaneously, an energizing gas stream is passed along the
second
6
CA 02247900 1998-08-31
WO 97J3169i PCTlUS97/03207
passageway and through a second passageway outlet proximal to the first fluid
-- dispersion outlet. The passage of such an energizing gas stream through the
second
outlet generates high frequency waves of the energizing gas adjacent the first
passageway outlet in order to break up the fluid dispersion into extremely
small
droplets. This causes the antisolvent in the precipitation zone to deplete the
dispersant
and rapidly precipitate small particles of the substance.
The preferred process of the invention involves deliberate generation of high
energy sonic waves (Type II waves) in addition to and substantially
independent of any
impaction and frictional forces typical of prior art nozzles (Type I waves).
Type II
sonic waves may be generated in the energizing gas stream or in the dispersion
itself.
In the former situation, specialized nozzles as described below are used, and
in the
latter, a starting dispersion may be sprayed onto a sonicating surface coupled
to a
transducer (e.g., piezoelectric, magnetostrictive, or electromagnetic), and
the resultant
particles are contacted with turbulent SCF fluid.
1 S In preferred forms, the specialized nozzle is of the type commercialized
by
Sonimist of Farmingdale, NY as Model 600-1. This nozzle includes an elongated
body
presenting a central tube which serves as the primary spray nozzle for the
dispersions
of the invention. The nozzle structure also includes a secondary passageway in
surrounding relationship to the central tube for passage of the energizing gas
along the
length of the central tube and out the nozzle outlet. The secondary passageway
for the
energizing gas is configured to present a converging section defining a
restricted throat,
with a diverging section downstream from the throat and leading to the nozzle
outlet.
In addition, the diverging portion of the secondary passageway is equipped
with a
radially expanded, annular resonator cavity for reflecting sound waves. The
outlet end
of the central tube is located downstream of the constricted throat.
Use of nozzles of this type serves to generate and focus the preferred high
frequency sonic waves of energizing gas which has been shown to maximize the
production of extremely small dispersion droplets in the precipitation zone,
thereby
leading to the precipitation of the very small particles of the invention. The
frequency
of the generated waves of energizing gas could range anywhere from 0.5 kHz to
300
kHz, and more preferably from about 10-100 kHz. It is believed that the
inherent
kinetic energy of the energizing gas stream is converted to acoustic energy by
virtue of
passage of the energizing gas stream through the restricted throat, resonator
cavity and
outlet of the nozzle. Generally, at least about 1 % (more preferably from
about 2-14%)
of the kinetic energy of the energizing gas stream is converted to acoustic
energy.
In preferred forms, the energizing gas is the same as the selected
antisolvent, and
in most cases carbon dioxide is used both as the antisolvent and energizing
gas. More
broadly however, the energizing gas may be selected from the group consisting
of air,
7
CA 02247900 1998-08-31
WO 97/31691 PCTlLTS97/03207
oxygen, nitrogen, helium, carbon dioxide, propane, butane, isobutane,
trifluoromethane,
w nitrous oxide, sulfur hexafluoride and mixtures thereof.
Where it is desired to coat core particles with a desired substance, a fluid
dispersion of the type described is sprayed into an enclosed precipitation
zone
containing a quantity of antisolvent at near-or supercritical conditions for
the
antisolvent. Simultaneously, a turbulent fluidized flow of the core particles
is created
within the precipitation zone by passing a fluidizing gas stream comprising
the selected
antisolvent into the precipitation zone. Conditions are maintained in the zone
so that
the antisolvent rapidly depletes the dispersant and precipitates the substance
as small
particles onto the fluidized core particles. Although the specialized nozzle
arrangement
described above can be used in the coating methods of the invention, such is
not
generally required. That is, in coating applications, the coating particles
can typically
be relatively Larger without detracting from the usefulness of the final
product;
accordingly, conventional capillary nozzles and the Like can be used to good
effect in
such coating processes.
In one form, the fluidized flow of core particles is established by passing
the
fluidizing gas stream in a direction which is substantially countercurrent to
the direction
of fluid dispersion spray into the precipitation zone. However, where modified
Wurster
coating devices are employed, the fluidizing gas stream is directed co-current
relative
to the direction of fluid dispersion spray. It is important that at least a
part of the
fluidizing gas stream be made up of the antisolvent, particularly where
countercurrent
fluidizing stream flow is employed. In any case, however, the fluidizing gas
stream
should normally have a concentration of antisolvent therein of at least about
70% by
weight and more preferably the fluidizing gas stream consists essentially of
antisolvent.
A wide variety of core particles can be used in the invention but generally
these
should have a maximum dimension of up to about 15 mm, and more preferably up
to
about T mm. Core particles such as glass or sugar beads can be used and in
pharmaceutical applications, it is contemplated that medicament tablets or
other discrete
solid dosage forms can be coated. The final coated products can range from
micron-
sized to several millimeters. In the case of medicaments, depending upon the
application, the final coatings would typically have a thickness of from about
0.1 ~cm
to 2 mm {more preferably from about I-500 pm), and the coating would be from
about
1-30% (more preferably from about 5-15%) by weight of the final coated
product.
In another aspect of the invention, a process is provided for the preparation
and
administration to a patient of a particulate medicament without the necessity
of _
transfernng the medicament between containers, i.e., the
dispersion/antisolvent
precipitation is carried out in a final use container which is subsequently
sealed and
permits withdrawal of medicament dose{s) from the use container. Generally,
this
8
CA 02247900 1998-08-31
WO 97/31691 PCT/US97/03Z07
method involves lyophobic precipitation of medicaments which may be performed
in
- a batch or semi-batch mode.
In preferred forms, the following general steps are performed: (a) the
medicament is dissolved in an organic solvent to form a solution or
suspension; (b) the
solution or suspension is sterile filtered; {c) the solution or suspension is
either metered
into the final use container prior to contact with the supercritical fluid
(batch mode) or
continuously as a spray with supercritical fluid contact (semi-batch mode);
(d) the
medicament suspension or solution in the container is contacted with the
supercritical
fluid until, at a predetermined concentration of supercritical fluid the
mixed, expanded
liquid is no longer a solvent fox the medicament and particle precipitation is
effected;
(e) the use container is purged with supercritical fluid until the organic
solvent is
completely depleted from the system; and (f) the finished solid particulate
medicament
is aseptically sealed in the use container. Thereafter, when it is desired to
use the
medicament, a liquid Garner may be placed in the use container to form a
mixture,
which can then be administered by injection or the like.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 is a schematic of the apparatus for the conventional SAS
recrystallization from organic solutions.
Fig. 2 is a schematic representation of an apparatus useful in the practice of
the
invention.
Fig. 3 is a schematic cross-sectional view of the nozzle employed in the
practice
of the invention.
Fig. 4 is an SEM micrograph ( 1 O,OOOX magnification) of hydrocortisone
micronized by recrystallization from a 5 mg/ml DMSO solution using the
conventional
SAS process with a 100 pm capillary nozzle.
Fig. 5 is an SEM micrograph of hydrocortisone micronized by recrystallization
from a 30 mg/ml DMSO solution using the conventional SAS process with a 100
~.m
capillary nozzle.
Fig. 6 is a GC-FID analysis of hydrocortisone recrystallized from a 30 mg/ml
DMSO solution using the conventional SAS process with a 100 um capillary
nozzle.
Figs. 7a and 7b are a pair of SEM micrographs (S,OOOX and 9,900X
magnification, respectively) of hydrocortisone nanonized by recrystailization
from a 30
mg/ml DMSO solution using the nozzle of the present invention (compressed COZ
is
used as energizing gas and as antisolvent).
Fig. 8 is an SEM micrograph (3,OOOX magnification) of hydrocortisone
micronized by recrystallization from a 30 mg/ml DMSO solution using the nozzle
of
9
CA 02247900 1998-08-31
WO 97/31691 PCT/US97l03207
the present invention (He is used as energizing gas and compressed COZ is used
as
w antisolvent).
Figs. 9a and 9b are a pair of SEM micrographs (500X and 1,OOOX
magnification, respectively) of polylactic-glycolic acid polymer (RG503H)
micronized
by recrystallization from a i 0 mg/ml ethyl acetate solution using the
conventional SAS
process with a 100 ~cm capillary nozzle.
Fig. 10 is an SEM micrograph (1,000X magnification) of RG503H micronized
by recrystallization from a 10 mg/ml ethyl acetate solution using the nozzle
of the
present invention (compressed COz is used as energizing gas and as
antisolvent).
Figs. lla and Ilb are a pair of SEM micrographs (1,000X and 10,000X
magnification, respectively) of ibuprofen nanonized by recrystallization from
a 30
mg/ml DMSO solution using the nozzle of the present invention (compressed CO~
is
used as energizing gas and as antisolvent).
Fig. 12 is an SEM micrograph {1,000X magnification) of micronized
camptothecin by recrystallization from a 5 mglml DMSO solution using the
nozzle of
the present invention {compressed CO~ is used as energizing gas and as
antisolvent).
Fig. I3a and 13b are a pair of SEM micrographs (2,OOOX and 15,OOOX
magnification, respectively) of camptothecin nanonized by recrystallization
from a 5
mg/ml DMSO solution using the nozzle of the present invention {compressed COZ
is
used as energizing gas and as antisolvent);
Fig. I4 is a schematic view of a modified precipitation vessel specifically
adapted for the coating of core particles in the overall apparatus of Fig. l;
Fig. 15 is an SEM photograph of an uncoated nonpareil sugar bead used in
Example 5;
Fig. I6 is a SEM photograph of a final RG503H-coated nonpareil sugar bead;
Fig. 17 is a SEM photograph of a RG503H-coated glass bead;
Fig. 18 is a SEM photograph of a RG503H-coated nonpareil sugar bead
produced in accordance with Example 7;
Fig. 19 is a SEM photograph of a RG503H-coated nonpareil sugar bead
produced in accordance with Example 7;
Fig. 20 is a SEM photograph of a hydrocortisone-coated glass bead produced
in accordance with Example 8;
Fig. 21 is a schematic representation of apparatus useful in the lyophobic
precipitation aspects of the present invention;
Fig. 22a is a differential scanning calorirnetry thermogram of unprocessed
phenytoin; and
Fig. 22b is a differential scanning calorimetry thermogram of phenytoin
processed by lyophobic precipitation in accordance with Example 10.
CA 02247900 1998-08-31
WO 97/31691 PCT/US97/03207
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
The following Examples set forth techniques, compositions, and system
parameters, as well as test results, demonstrating various aspects of the
present
invention. Examples 1-4 relate primarily to the particle micronization and
nanonization
aspects of the invention, whereas Examples 5-8 pertain to particle coating;
the
remaining examples illustrate production of finished products by lyophobic
precipitation. It is to be understood, however, that these examples are
presented by way
of illustration only and that nothing therein should be taken as a limitation
upon the
overall scope of the invention.
PARTICLE MICRONIZATION AND NANONIZATION
Equipment and Experimental Procedures, for Examples 1-4
Fig. I shows a schematic of the apparatus 10 used for particle
recrystallization
from organic solvents using the conventional SAS process. The experimental
unit 10
allowed SAS experiments to be conducted in either batch or semi-continuous
mode at
pressures up to 5,000 psi and temperatures up to 70°C. The mixing of
solvent and
antisolvent occurred at two different locations I2, I4 within the unit. Unit I
O provided
versatility in setting the operating parameters.
The unit 10 was built around a 65 ml high pressure 3erguson gauge (Burlington,
MA) view cell 16. The cell 16 was equipped with a sapphire window that allowed
viewing of the expansion and crystallization process. The cell 16 was housed
in a
heated, isothermal, transparent acrylic water bath 18. This water bath 18 was
used for
maintaining the cell 16 at a desired temperature (20-70°C). When the
bath temperature
was stable at a desired value, COa was pumped through the top side port 20 of
the cell
I6 with an ISCO (Lincoln, NE) 260D syringe pump 22 at a constant rate
(typically 5
ml/min. of liquid COZ) until the pressure in the cell 16 reached a desired
level {1,500
psi). When temperature and pressure in the cell 16 were stabilized, the
organic solution
(DMSO or ethyl acetate-solution of drug and/or polymer) was metered from the
top
central port 24 of the cell 16 through a stainless steel, 1/16" O.D., 100 l.un
LD. capillary
nozzle tubing 26 using a Milton Roy (Riviera Beach, FL) 396-89 minipump 28. It
was
found that a minimum solution flow rate of 2.5 ml/min. was needed to
consistently
obtain a jet spray. Both fluids were preheated to operating temperature by
passing
through heat exchangers 30, 32 housed together with the cell 16 in the water
bath 18.
Fresh COz and the organic solution streams mixed at location 14, which is just
downstream of the nozzle tip 33 at the top of the cell 16. A cloudy zone about
1 cm
long was seen to form in this area indicating intimate mixing of the fluids
and particle
- formation. Solvent depletion from the spray droplets causes the drug and/or
polymer
Il
CA 02247900 1998-08-31
WO 97/3i69i PCTIUS97/03207
dissolved in the organic solvent to nucleate. The resulting particles
descended down
the cell. Alternatively, the streams can be premixed prior to reaching the
nozzle tip 33
using the two-way valve 34.
Particles descending down the cell 16 either adhered to the cell walls or were
collected on a 6" long glass rod 36. Particles Larger than 0.5 ~.m leaving the
view cell
chamber were retained on a 0.5 p.m stainless steel frit housed in the T-shaped
fitting at
the central bottom port 38. A thermocouple inserted through this fitting was
used to
monitor the cell temperature. The drug andlor polymer depleted mixture of C02
and
organic solution flowed through a step-motor controlled, heated micrometering
valve
assembly 40. Upon expansion to a subcritical pressure (typically close to
atmospheric
pressure), the mixture separated into an organic liquid phase and a COZ gas
phase.
Phase separation took place in the flash drum 42; the organic solution flowed
through
a micrometering valve 44 and was collected in a vessel 46. The solution was
then
analyzed for drug and polymer content. COZ was vented through a second
micrometering valve 48, a rotameter 51, and an electronic mass flowmeter 50.
Typically, the solution was pumped for IS minutes in order to produce a
statistically representative sample of drug and/or polymer microparticles.
Following
this, the flow of organic solution was stopped while the CO~ flow was
continued for
another I .5 hours in order to flush out any organic phase left in the cell,
and to dry the
- collected particles. It was found that flowing COa at 1,500 psig for 1.5
hours was
adequate for flushing out the organic solvent present in the cell and for
drying the
particles. Following the drying period, the pressure was decreased to
atmospheric level
at a rate of -50 psi/min. Particle samples were collected from the cell
window, the
porous frit, and the glass tube, and were analyzed by scanning electron
microscopy
{SEM) to estimate particle size and morphology.
Accurate pressure control was essential in the highly compressible near-
critical
region. Pressure fluctuations in this region have a strong effect on the Level
of
expansion of the organic solution and thus on the level of supersaturation
and,
consequently, on crystal growth and crystal size distribution. Pressure
control in the
= cell I6, along with monitoring of pressure, temperature, and flow rate, were
accomplished using the Camile~ (Midland, MI) 2500 Data Acquisition and Control
system. A 100 steps/revolution stepping motor, operating at 200 half
steps/revolution,
was used to actuate the heated micrometering valve 54. Pressure control was
achieved
using an HC-ll microprocessor that interprets the output from the Camile PID
controller and acts as a step-motor controller. The software program allowed
the
microprocessor to seek a window wherein the valve will operate to provide
pressure
control within transducer precision (t10 psi).
12
CA 02247900 1998-08-31
WO 97131691 PCTlUS97/03207
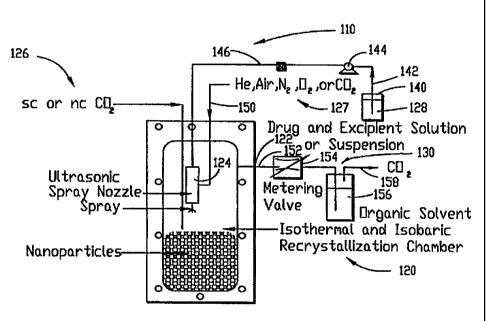
Fig. 2 shows schematically an apparatus I 10 according to the present
invention.
Apparatus 110 is identical to apparatus 10 with the exception that the view
cell serving
as crystallization chamber is replaced with a larger (450 ml), stainless steel
vessel that
can house the nozzle. Here again, the crystallization chamber was housed in an
isothermal water bath, and pressure is controlled as described previously with
regard
' to the conventional SAS process (Fig. 1). In apparatus 110, an organic
solvent such as
dimethyl sulfoxide (DMSO), in which solutes such as drug, polymer, and/or
excipient
w materials are solubilized, is also sprayed as a fine mist into a chamber
containing a near-
critical or supercritical antisolvent.
I 0 In more detail, apparatus 110 of the present invention includes an
isothermal and
isobaric recrystallization chamber 120, a spray nozzle 124, a source of
supercritical (sc)
or near-critical (nc) COZ 126, a source of compressed gas 127 which serves to
energize
the nozzle 124, a drug and excipient solution 128, an organic solvent
collection vessel
156, and a COZ outlet header 130.
The drug and excipient solution is drawn from vessel 140 through line 142 by
pump 144 and is discharged through line 146 into chamber 120 through line 146
as
shown in Fig. 2. The nozzle 124 is attached to the end of line 146 within
chamber 120.
Energizing gas for the nozzle consisting of He, N2, O~, air, C02, other
supercritical
fluids, or a mixture thereof, from source 127 is admitted through line 1 SO
into chamber
I20, as shown in Fig. 2. The near-critical or supercritical fluid
(antisolvent) is admitted
from source I26. Alternatively, if the energizing gas is supercritical (or
near-critical),
source 127 also can be used for admitting the supercritical fluid into chamber
120;
source 126 then may be either not employed, or used for admitting a
supercritical fluid
in the same composition as in source 124, or a supercritical fluid of
different
composition. This latter alternative can be used for either increasing or
decreasing the
concentration gradients between the antisolvent phase and the buffer zone. The
solute
depleted organic solvent and solvent-loaded COZ are removed from chamber 120
via
outlet I22 through line 152 and metering valve 154 into flash drum 156, in
which COZ
is allowed to separate from the Liquid organic solvent. The COz is allowed to
vent from
vessel 156 through vent line 158.
Fig. 3 is a schematic of a nozzle (Sonimist, Farxningdale, N.Y., Model 600-1 )
employed in apparatus 110. This nozzle N is of the convergent-divergent type
and
includes a central capillary-type tube T presenting an outlet O. The nozzle N
fiuther
includes a surrounding passageway P presenting an inlet I for energizing gas.
The
passageway P includes a converging section C presenting a restricted throat TH
and a
downstream diverging section D. The section D includes a radially expanded,
annular
resonator cavity CV. It is to be noted that the outlet O of the tube T is
positioned
downstream of the throat TH. The nozzle N is energized by compressed gas
13
CA 02247900 1998-08-31
WO 97/31691 PCT/US97/03207
(conventionally a light gas such as air, He, Oz, or NZ and in the present
invention
preferably through use of antisolvent gas). A sonic field (Type II waves) is
created at
the throat TH of the nozzle N as the energizing gas accelerates and reaches
the velocity
of sound or greater. These high frequency waves impinge upon the entrance of a
resonator cavity CV, and the latter serves to produce high frequency waves of
the
energizing gas, producing a chopping effect that breaks up the liquid jet
comprising the
solute-loaded solvent into extremely small droplets.
In the Fig. 3 device, the generated sonic waves are focused on the dispersion
spray in order to facilitate enhanced atomization of the spray. For
precipitation to
occur, the dispersant from the droplets must be transferred to the antisolvent
phase
surrounding the droplets. In addition to enhancing atomization, the
concomitant
increase in the mass transfer surface area produced by the sonic waves
enhances the
mass transfer rate between the droplets formed and the surrounding fluid
medium,
thereby increasing the rate of solid precipitation.
When spraying into ambient air, with 20-100 prig back pressure of energizing
gas, the Sonimist nozzle produces a fine, evenly dispersed spray of droplets
having
diameters in the range of 0. I-50 p.m depending on operating conditions. Mean
droplet
diameters of 1-10 pm are obtained when spraying water into ambient atmosphere.
If
interphase mass transfer does not significantly interfere with the atomization
process,
droplet sizes are expected to be even smaller when spraying into a higher
pressure
gaseous environment or when using organic solvents with lower surface tension
and
viscosity than water.
When using this nozzle, the flow rate of the energizing gas should be such
that
sonic velocities are attained by the energizing gas at the throat of the
nozzle. While in
conventional use of this nozzle, a pressure differential of 2 atm between the
energizing
gas and ambient atmosphere is sufficient to achieve this sonic velocity, this
pressure
differential is not sufficient when operating at near-critical or
supercritical conditions.
The following equation is used to estimate whether a discharge velocity is
subsonic
(Perry and Chilton, 1973, Chemical Engineer's Handbook, 5th Ed., McGraw Hill,
Chap. S):
PZ/Po > [2/(k+1)]'~-'
where Po is the energizing gas pressure, Pz is the nozzle outlet pressure, and
k is the
ratio of heat capacities at constant pressure and constant volume of the gas
(i.e., Cp/C").
For instance, for a pressure of 1,500 psig at the nozzle exit and an
energizing gas (C02)
pressure of 6,000 psig, P.,/Pa = 1,500/6,000 = 0.25; Cp/C" at 1,500 psig =
4.81; and
[2/(k+1)I~'~'-1 = 0.26. Hence P~/Po < [2/(k+1)]~k-i and the velocity can be
sonic.
14
CA 02247900 1998-08-31
WO 97/31691 PCT/US97I03207
While the above example illustrates conditions under which sonic velocities
may be estimated, such high velocities may not be required for all
applications. For
instance, it has been found that using a chamber pressure of 1,250 psig and an
energizing gas pressure of 1,850 psig provides enough energy to reduce
particle size
substantially. A one order of magnitude reduction in particle size (when
compared to
results obtained by conventional SAS recrystaliization) was also observed when
using
only 100 psig pressure differential between the chamber held at 1,500 psig and
the
energizing gas (COZ). Thus, the nozzle illustrated in Fig. 3 can be used in a
wide range
of operating conditions in order to substantially reduce particle size and to
increase
.10 surface area. Broadly speaking, the energizing gas should be delivered to
the nozzle N
at a pressure of from about 1100-6000 psig, more preferably from about 1500-
2500
psig, and at a temperature such that upon expansion, the energizing gas
attains the
desired temperature of the recrystallization chamber. The frequency of the
waves of
antisolvent created at the nozzle outlet should be at least about 0.5 kHz and
more
preferably from about 10-100 kHz.
Furthermore, the invention may be practiced without the use of the nozzle
illustrated in Fig. 3. The invention may be practiced with any nozzle that
provides a
means for using a gaseous (or near-critical or supercritical fluid) stream as
energizing
medium to atomize the sprayed solution into smaller droplets and/or to create
turbulence around the spray droplets which increases the mass transfer rates
between
the droplet and antisolvent phases. Both converging-diverging nozzles as well
as
converging nozzles may be employed in the present invention.
Examples 1-4
Comparison of Particles Produced by the Conventional
SAS Process and the Process of the Present Invention
In these examples, the recrystallization of hydrocortisone, poly (D,L-lactide-
glycolide) copolymer (RG503H}, ibuprofen, and camptothecin was studied. The
recrystallization of hydrocortisone and RG503H was performed using both the
conventional SAS process as well as the present invention.
Hydrocortisone is a common anti-inflammatory agent and ibuprofen is a
common pain reliever. They were acquired from Sigma Chemical Co., St. Louis,
MO,
and were used without further purification. Camptothecin is an anti-cancer
drug with
a very low aqueous solubility; reduction in its particle size or an increase
in its particle
surface area can substantially increase its dissolution rate and render it
therapeutically
more useful. RG503H was acquired from Henley, Montvale. N.J. It contains a l:l
molar ratio of iactide and glycolide and has an inherent viscosity in
chloroform of 0.3.
CA 02247900 1998-08-31
WO 97/31691 PCT/US97103207
RG503H is FDA approved for administration to humans, is non-toxic, non-tissue
reactive, biodegrades to non-toxic products, and is particularly suited for
surgical
sutures. PLGA copolymers have been the subject of intense micronization and
microencapsulation studies.
Certified grade DMSO and ethyl acetate (99.9% purity, Fisher Scientific,
Fairlawn, N.J.), bone dry COZ (99.8% purity, Genex, Kansas City) were used
without
furkher purification. Particles were collected on a double-sided carbon tape
applied to
an aluminum SEM tab that was placed in the crystallization chamber prior to
each
experiment. Particles that were deposited on the cells walls were also
collected for
I0 analysis. Particle morphology was determined by SEM (Hitachi, Model S-570).
Particle size was also estimated by SEM. The SEM samples were sputter-coated
with
Au/Pd alloy.
Hydrocortisone particles were redissolved in ethyl acetate, and analyzed by GC
FID for trace DMSO contamination. Effluent solutions recovered in the flash
drum
i S were also analyzed for hydrocortisone content.
The results of repeat conventional SAS recrystallization experiments are
compared in Table 1. Particle size for whisker particles refers to their
thickness or
width. The data in Table 1 demonstrate that the average particle size for all
solutes
studied, including HYAFF-7 (the ethyl ester of hyaluronic acid) is
reproducible,
20 indicating that the SAS spray technique is a controllable and reproducible
recrystallization technique.
16
CA 02247900 1998-08-31
WO 97/31691 PCT/US97/03207
Table 1. Reproducibility of Morphology and Size of Particles Formed by the
Conventional SAS Recrystailization Method, as Estimated from SEM Micrographs.
P = 1,500 psig; COZ Flow Rate = 5 ml/min.; Solution Flow Rate = 2.5 rnl/min;
Capillary Nozzle LD. = 100 ~,m.
Average
Run Solvent Solute ConcentrationTemp. Particle Particle
(mg/mi) (C) MorphologySize
(pm)
4-I2 DMSO Hydrocortisone30 35 Whisker 1
4-14 DMSO Hydrocortisone30 35 Whisker 1
4-I6 DMSO Hydrocortisone30 35 Whisker I
12-1 Ethyl AcetateRG503H IO 35 Microsphere5-50
5-8 Ethyl AcetateRG503H 10 35 Microsphere10-20
12-I6 DMSO RG503H 2 35 Tubes/Fiakes25-100
12-20 DMSO RG503H 2 35 Tubes/Flakes25-100
6-6 DMSO HYAFF-7 0.5 40 Resin >100
6-8 DMSO HYAFF-7 0.5 40 Resin >100
Example 1
Com orison of~es_u_-Z-t~s nf_R_Prrv.c ~71»nt~
~~ J J~~»~~~wvl~w
ofHydrocortisone from DMSO Solutions
Hydrocortisone Particles Produced Using the Conventional SAS Process
Fig. 4 shows the SEM micrograph of hydrocortisone particles recrystailized
from a 5 mg/ml DMSO solution using the 100 ~.m capillary nozzle (P = 1,500
psi; T =
35°C; CO., flow rate = 5 ml/min.; solution flow rate = 2.5 ml/min.).
Particles are
agglomerated, nearly spherical, and range in size from 0.5-1 ~tm.
Recrystallization of
hydrocortisone from a 30 mg/m1 DMSO solution yielded long (up to 1 mm), 1 ~,m
thick, whisker-shaped particles shown in Fig. S (P = 1,500 psi; T =
35°C; COZ flow rate
= 5 mI/min.; solution flow rate = 2.5 ml/min.; capillary LD. = 100 p.m). Note
that the
magnification level in the upper part of micrograph (b) is five-fold greater
when
compared to the lower micrograph. Greater nucleation rates should result at
this higher
concentration, which should lead to the formation of smaller particles
(Gallagher et al.,
19$9); however, it appears that the increase in viscosity at higher solute
concentrations
and the premature onset of nucleation, and crystallization prior to secondary
atomization hinder the atomization process, resulting in the formation of
elongated,
whisker-like particles. Indeed, the increase in particle size with an increase
in solute
17
CA 02247900 1998-08-31
WO 97/31691 ~ PCT/US97/03207
concentration was observed for all solutes recrystallized using the
conventional SAS
process.
Particles size is fairly reproducible. For three runs under these same
conditions
(30 mg/ml), particle thickness is narrowly distributed and is in the order of
1 pm. The
amount of DMSO in the hydrocortisone particles was below the detection limit
of the
GC-FID (= 10 ppm) (Fig. 6). It thus appears that the particles are virtually
solvent-free.
hydrocortisone Particles Produced using the Present Invention in which
Compressed
C01 Ylras Used as Energizing Gas and as Antisolvent
For a nozzle exit pressure of 1,500 psig and a temperature of 35°C,
calculations
indicate that an energizing pressure of roughly 6,000 psig at 55°C is
needed to obtain
sonic velocities at the nozzle exit. COZ must be pumped at a rate such that a
4,500 psig
back pressure is established. An experiment using 100 prig back pressure (i.e.
1,600
psig C02 supply pressure and 1,500 psig at the nozzle exit, corresponding to a
COz flow
rate of25 mI/min.) yielded hydrocortisone particles consisting of nearly
spherical, 0.5-1
p.m in size, and whisker-shaped particles, roughly 1 um wide and 10 p.m long.
These
results suggest that production of smaller particles can be achieved by using
COz at
even sub-sonic velocities to energize the nozzle. Hence, while near-sonic,
sonic, and
supersonic compressed gas velocities are preferred for production of
nanoparticles, even
lower compressed gas flow rates can significantly reduce particle size when
compared
to the conventional SAS process where the antisolvent phase is nearly-
stagnant.
Figs. 7a and 7b show a pair of SEM micrographs of hydrocortisone particles
recrystallized from a 30 mg/ml DMSO solution using the nozzle in Fig. 3, and
COZ as
energizing gas, In the recrystallization chamber, P = I,250 psig; T =
35°C; and the
solution flow rate = 2.5 mI/min. During the period when the solution was
pumped
{roughly 1 minute), the pressure of CO., at line 50 (Fig. 2) was equal to
1,850 psig,
thereby providing 600 psi of back pressure. COz temperature in source 24 (Fig.
2) was
brought up to 50°C, so that upon expansion from 1,850 psig to 1,250
psig, the
temperature decreased to nearly 35°C, the temperature in the
crystallization chamber.
This back pressure translated to a COZ flow rate of 90 ml/min. during the
atomization
phase. It is observed that the particles are discrete, nearly spherical, and
appear to be
narrowly distributed around 500 nanometers (nm). Nearly all particles are
smaller than
600 nm. These results are in contrast to the I um wide and nearly I mm long
fibers
observed previously (Fig. 5) when using the 100 ~m capillary nozzle. Hence, a
18
CA 02247900 1998-08-31
WO 97/31691 ~ PCT/IJS97/03207
significant decrease in the average particle size is observed with the use of
the present
invention.
Hydrocortisone Particles Produced Using the Present Invention in which He Was
Used
as Energizing Gas and Compressed COZ Was Used as Antisolvent
The 30 mg/ml DMSO solution of hydrocortisone was also recrystallized using
He at 1,600 psig as energizing gas and COZ at 1,500 psig, 35°C as
antisolvent. Fig. 8
demonstrates that it is possible to use a light gas to energize the nozzle.
Although these
conditions are not optimum, the process still produces particles that are
relatively small.
Some particles appear to be even smaller than 1 ~Cm. The merits of using He as
opposed
to C02 as energizing gas are not evident from Fig. 8; however, it is
anticipated that as
the solute concentration and viscosity of the solution is increased, it may be
necessary
to introduce a gaseous buffer such as He to avoid premature nucleation. When
using
a light gas to energize the nozzle, the flow rate of the supercritical fluid
relative to that
of the light gas should be high enough to provide sufficient antisolvent power
for the
supercritical fluid/light gas mixture. Use of COZ as both antisolvent and
energizing gas,
when possible, is advantageous over the use of a light gas as energizing gas
because (a)
chances for contamination are reduced, (b) the antisolvent power of COz is not
diminished, (c) required COZ flow rates are lower, and (d) solvent recovery is
efficient.
Example 2
Comparison ofResults ofRecrystallization ofRG503H
Particles Produced Using the Conventional SAS Process
RG503H was recrystallized from solutions of DMSO and ethyl acetate at a
pressure of 1,500 psig and a temperature of 35°C using a 100 ~.m
capillary nozzle. Neat
RG503H particles, as supplied by the vendor, are relatively Large,
agglomerated
precipitates (> 50 um). Table 2 depicts the effect of RG503H concentration on
size and
morphology of RG503H recrystallized from solution. RG503H in DMSO appears to
recrystallize as tubules at low concentrations, as a mixture of flakes and
tubules at
medium concentrations, and as precipitates of large amorphous material at
higher
concentrations.
Pre-mixing of CO~ with the DMSO solution prior to expansion, aimed at
- improving mass transfer efficiency, had little effect on particle size and
morphology,
but caused the formation of bubbles on the surface of the flakes. The
formation of
relatively large, agglomerated particles at increased polymer concentrations
parallel
19
CA 02247900 1998-08-31
WO 97131691 PCT/US97/03207
those of Dixon, D.J., Johnston, K.P. and Bodmeier, R.A., 1993, Polymeric
materials
formed by precipitation with a compressed antisolvent. Amer. In t. Chem. -
n~.~T.
39:127-139; Randolph et al.(1993); and Bodmeier, R., H. Wang, D.J. Dixon, S.
Mawson, and K.P. Johnston, 1995, Polymeric microspheres prepared by spraying
into
S compressed carbon dioxide. Pharm. Res. 12:1211-1217. As in the previous
example,
these results also demonstrate the increasing difficulty of atomization and
particle
micronization with increasing polymer concentration due to both an increase in
solution
viscosity and to premature mass transfer between the solution and COz. This
observation is fiutl2er corroborated in Figs. 9 a and 9b, which show that a
reduction in
the viscosity and/or surface tension of the solution through a change of
solvent, i.e.
from DMSO (1.9 cp and 41 dyn/cm) to ethyl acetate {0.46 cp and 24 dyn/cm) led
to the
formation of discrete microspheres (in Figs. 9 a and 9b, the sprayed solution
is 10
mg/ml RG503H in ethyl acetate; P=1,500 psi; T = 35°C; CO., flow rate =
5 ml/min.;
solution flow rate = 2.5 ml/min.; capillary LD.=100 pm). The inability to
attain sub-
I S micron particles of average size smaller than 0.6 pm using the
conventional SAS
process is attributed to mass transfer limitations. These are overcome in the
present
invention as explained earlier and as demonstrated in the following example.
Table 2. Micronization of RG503H by Conventional SAS Recrystallization. P =
1,500
psig; T = 35°C; C02 flow rate = 5 mI/min.; Solution Flow Rate = 2.5
ml/min.;
Capillary LD.= 100 pm; Solvent is DMSO except for Run 6.
Run Shape Particle [RGS03-1]
# Size (mg/ml)
(gym)
I whiskers 1 S O,S
2.S 2 whiskers/flakes I S/SO 2.0
3 whiskers/flakes 2S/> l OD 2.0
4 flakes 100 10.0
S amorphous >S00 100.0
6 hollow micro- <SO 10.0*
spheres
7 flakes with >S00 10.D~
bubbles
*: Solvent is ethyl acetate. ~: Premixing of solvent and CO,.
CA 02247900 1998-08-31
WO 97/31691 - PCT/LTS97/03207
RG503HParticles Produced Using the Present Invention in which Compressed COZ
is used as Energizing Gas and as Antisolvent
Fig. 10 shows an SEM micrograph of RG503H particles recrystaliized from a
mg/ml ethyl acetate solution. These particles are compared with particles
shown in
5 Figs. 9a and 9b, which are obtained using the conventional SAS process. Both
experiments were conducted at identical conditions of pressure, temperature,
and
solution flow rate (I,500 psig, 35°C, and 2.5 mI/min, respectively)
within the
crystallization chamber, except that the particles shown in Fig. 10 were
obtained using
the present invention in which compressed COZ was used as energizing gas. The
COZ
10 supply pressure was 1,600 psig. Similar to the particles seen in Figs. 9a
and 9b, the
RG503H particles in Fig. 10 are also nearly spherical; however, the particles
obtained
using the present invention appear more discrete and are an order of magnitude
smaller
than particles in Figs. 9a and 9b. As with the results obtained in the
previous example,
particle diameter is again narrowly distributed around 1 Vim. Thus, the
present
invention produces smaller particles than the conventional process with less
agglomeration, a property that is desirable, especially in the pharmaceutical
industry.
Example 3
Recrystallization o, f Ibuprofen from a DMSO Solution Using the Present
Invention
in which Compressed COa Was Used as Energizing Gas and as Antisolvent
Figs. 1 I a and 1 I b show a pair of SEM micrographs of Ibuprofen particles
recrystallized from a 30 mg/ml DMSO solution under the same operating
conditions as
in Example 2. Once again, particles appear to be discrete, particle sizes are
small and,
except for a fraction of micron-sized particles, most particles are smaller
and in the
range of 0.6 ~rn or less.
Example 4
Recrystallization of Camptothecin from a DMSO Solution
Using the Present Invention is which Compressed COz
Was used as Energizing Gas and as Antisolvent
Camptothecin, as supplied by the vendor, appears as amorphous particles with
diameters ranging from I-10 pm. Fig. 12 is an SEM micrograph of camptothecin
particles recrystallized from a 5 mg/mI DMSO solution under the same operating
conditions as in Example 2, (i.e., P = 1,500 psig, 35°C with a CO.,
back pressure of
- 35 roughly 100 psig). Particles are nearly spherical and discrete. Although
relatively large
21
CA 02247900 1998-08-31
WO 97/31691 PCT/US97/03207
in size (5-20 ~.m), these particles appear to be porous. The relatively high
surface area
of these particles should increase their dissolution rate and bioavailability.
Figs. 13a and 13b show a pair of SEM micrographs of camptothecin particles
recrystallized from a 5 mg/mI DMSO solution under the same operating
conditions as
in Example 1, Figs. 7 and 8 (i.e., P = 1,250 psig, T = 35°C, with a COZ
back pressure .
of 600 psig). Because of the higher expansion and velocities of the compressed
gas
(from 1,850 psig to 1,250 psig compared to 1,600 psig to 1,500 psig in the
previous ,.
experiment), smaller particles are formed. As seen in Fig. 13b, particles are
non-
agglomerated with the average diameter in the range of 0.5 Vim. Here again, as
in
Example 1 where favorable operating conditions were used, nanoparticles were
produced.
Alternative Embodiments
Note that in an alternative process, the chamber contains liquid COz or other
liquid antisolvent as opposed to supercritical COZ or another antisolvent in
its
supercritical form. In this case, the volume above the liquid phase (i.e., the
vapor
phase) contains mostly the light gas or the antisolvent which powers the
nozzle of the
present invention, and recrystallization takes place in either the Liquid
phase (when a
light gas is used to power the spray nozzle) or in both phases (when an
antisolvent is
used to power the spray nozzle). In the case where the antisolvent itself is
used to
power the nozzle, operating conditions are such that the energizing gas at its
near-
critical or supercritical state will nearly attain the conditions in the
recrystallization
chamber upon expansion through the nozzle. This alternative process is
attractive for
applications where containment of the recrystallized particles in the
crystallization
chamber is difficult because of entrainment in the supercritical phase. The
lower
buoyancy of liquids compared to supercritical fluids can minimize losses of
small
micro-sized or nano-sized particles.
Other Applications For The Inventive Method And Apparatus Disclosed Herein
This invention finds application in areas where reduction in particle size to
below 1 prn is desired for the purpose of increasing the surface area, the
rate of
dissolution, reactivity, or bioavailability.
The disclosed invention also finds application in areas where
recrystallization
of microparticles or nanoparticles from organic solutions is desirable. These
applications can find use in the production of foods, electronic equipment,
explosives,
22
CA 02247900 1998-08-31
WO 97!31691 ~ PCT/US97/03207
pharmaceutical products or intermediates (micronization, nanonization,
coating,
microencapsulation, lyophilization, and co-precipitation), catalysts
(micronization and
nanonization to increase the surface area of active sites or support),
explosives
(improved reactivity), coating (finer coatings), polymers (micronization and
nanonization), pesticides (micronization, nanonization, and
microencapsulation), and
other chemicals (micronization, nanonization, and microencapsulation).
Antisolvents useful in the application of this invention include, but are not
limited to, CO2, propane, butane, isobutane, CHF3, SF6 , and I~ O. Organic
solvents may
be either of the class of aromatic hydrocarbons, alcohols, esters, ethers,
ketones, amines,
or nitrated or chlorinated hydrocarbons. Preferred solvents include acetone,
ethanol,
methanol, dichloromethane, ethyl acetate and DMSO.
Conclusion
The method and apparatus of the present invention overcome the disadvantages
associated with conventional SAS processes in several ways. The high-velocity
wave-
front and/or turbulence established at the exit of the nozzle by the
energizing gas breaks
up the solution exiting the nozzle into a fine spray of droplets. The mass
transfer rate
between the spray droplets and the surrounding antisolvent phases is
essentially
proportional to the surface area of the spray droplets, and the antisolvent
and solute
concentration gradients. Use of the nozzle of the present invention provides a
means for
enhancing mass transfer rates through an increase in both the surface area of
the spray
and the interphase concentration gradients.
One effect of the creation of the small size droplets is to increase the
specific
surface area of the droplets, that in turn increases the rate of mass
transfer. Also, in
contrast to the electrically energized nozzle which produces a relatively low
velocity
spray, the compressed energizing gas passes the atomized droplets as it enters
the
supercritical antisolvent at high velocity and thereby creates a turbulence
which
prevents a build-up of depleted solvent in the proximity of the atomized
spray. An
increase in the concentration gradients between the droplet phase and the
antisolvent
nhacP nrnvirir c an inere~acPr~ r~rivina fnrra fnr intPrnhacP rr,ace tranefPr
.. r--._..- ra... ~ -...,.. »~~ ~-~.,...»...... ~... . ~.~b ~....... ~...
~~..,.:. y.~..~~. ..~..."~ ~~.a~~
Other advantages of the compressed gas-powered nozzle of the present
invention over other nozzles in their use for recrystallization of solutes
from organic
solutions or suspensions are:
23
CA 02247900 1998-08-31
WO 97/31591 ~ PCT/LTS97/03207
I . The relatively large size of the line through which the solution flows
through the nozzle compared to either capillary or micro-orifice nozzles
allows for
higher solution throughput and reduces the probability of nozzle plugging.
2. The same fluid can be used for both energizing the spray nozzle as well
as an anti-solvent.
3. The high velocity of the energizing gas stream imparts a high velocity
to the spray droplets, and therefore reduces the tendency for droplet
coalescence which
can lead to the formation of larger particles.
4. The high velocity of the gas or supercritical fluid energizing stream
provides a buffer zone at the tip of the nozzle that is either a gas or a low-
density
supercritical fluid. If the gas has little or no antisolvent power, the buffer
zone at the
tip of the nozzle serves to delay recrystallization until after secondary
atomization of
the spray has been achieved. This case is most attractive when using highly
viscous or
concentrated (nearly saturated or supersaturated) solutions.
I S If the energizing gas is itself a supercritical fluid antisolvent, the
buffer zone is
a highly turbulent zone of nearly pure antisolvent, thereby maximizing mass
transfer
rates between the droplets and the antisolvent while minimizing the droplet
coalescence
rate. 'This case is most attractive when recrystallizing drugs or polymers
from solutions
with Iow solute concentrations. Use of a compressed gas with intermediate
antisolvent
power (i.e. a mixture of light gas and antisolvent) provides a means for
controlling
interphase mass transfer rates, and therefore means for controlling particle
size.
The teachings of all references cited herein and those cited in the
Provisional
Application Serial No. 60/012,593 (identified above), and all references cited
therein,
are incorporated herein by reference.
Particle Coating
In Examples 5-8, coating of model core materials ( i .5 mm nonpareil sugar
beads
and 2 mm glass beads) with either a drug {hydrocortisone) or a polymer
(poly{D,L-
lactide-glycolide, RG503H) was investigated. Hydrocortisone was acquired from
Sigma Chemical Co., St. Louis, MO and was used with no further purification.
The
polymer was acquired from the Henley Co., Montvale, NJ and contained a 1:1
molar
ratio of lactide to glycolide and had an inherent viscosity in chloroform of
0.3 cps.
RG503H is FDA approved for administration to humans, is non-toxic, non-tissue
reactive, biogrades to non-toxic products, and is suited for surgical sutures.
24
CA 02247900 1998-08-31
WO 97!31691 PCTIUS97/03207
Certified grade ethylacetate and DMSO (99.9% purity, Fisher Scientifc,
Fairlawn, NJ), bone dry COZ (99.8% purity, Genex, Kansas City) were used with
no
further purification. Recrystallized microparticles were collected on glass
beads or
nonpareil sugar beads. Particles that deposit on the cell walls were also
collected for
analysis. Particle morphology and coating uniformity were evaluated by SEM
(Hitachi,
Model S-570). Particle size was also estimated by SEM. The SEM samples were
sputter coated with Au/Pd alloy.
Fig. 14 is a schematic view of a modified view cell used in the Fig. 1
apparatus
in the coating experiments. Specifically, the Fig. i apparatus was employed
except that
the modified view cell 16a was used in lieu of the cell 16. The cell 16a in
the
experiments was equipped with an internal, 16 cm-long, 8 mI glass tube 36a in
place
of the rod 36 of Fig. l, a COz extension Iine 20a leading from port 20 to the
bottom of
tube 36a, and the capillary nozzle tubing 26a was extended downwardly to a
point
adjacent the open end of tube 36a.
In use, the 16-cm long, 8 ml glass tube 36a is frst charged with nonpareil
sugar
beads or glass beads, and then fitted at the bottom of the view cell as shown
in Fig. 14.
When the bath temperature is stable at a desired value, COZ is pumped through
the line
20a at a constant rate (typically 5 mL/min. of liquid COZ) until pressure in
the cell
reaches a desired level ( 1500 psi). When temperature and pressure in the cell
are
stabilized, the organic solution (DMSO or ethyl acetate solution of drug
and/or
polymer) is metered through capillary nozzle tubing 26a. Both the organic
mixture and
COZ are preheated to operating temperature by passing through heat exchangers
housed
together with the cell in the adjacent water bath (see Fig. I ). in order to
establish
countercurrent flow and fluidize the beads, as described the CO, was
introduced at the
bottom of the tube through port line 20a while the organic solution of the
coating
material was sprayed from about 2 inches above. It is found that a minimum
solution
flow rate of 2.5 mL/min. is needed to consistently obtain a jet spray.
Fresh COz and the organic solution streams thus mixed within the glass tube.
Solution expansion caused the drug and/or polymer dissolved in the organic
solvent to
nucleate and the particles to crystallize and descend down the tube.
Recrystallized particles adhered either to the glass tube walls or deposited
on the
beads. Any particles escaping retention within the view cell chamber were
retained on
the steel frit housed in the T-shaped fitting at the central bottom port 38
{Fig. 1). A
thermocouple inserted through this fitting was used to monitor the cell
temperature. the
drug/polymer depleted mixture of CO= and organic solution flowed through the
step-
CA 02247900 1998-08-31
WO 97/31691 ~ PCTlLTS97/03207
motor controlled, heated micrometering valve assembly 40. Upon expansion to a
subcritical pressure (typically close to atmosphere pressure), the mixture
separates into
an organic liquid phase and a COZ gas phase. Phase separation took place in
flash drum
42; the organic solution flowed through the micrometering valve 44 and was
collected
in vessel 46. The solution was then analyzed for drug and polymer content. COZ
was
vented through a second micrometering valve 48, rotameter 59 and an electronic
mass
flowmeter 50 (all as shown in Fig. 1).
After the flow of organic solution was stopped, C02 flow was continued for
another 1-1/2 hours in order to flush out any organic solvent left in the
cell, and to dry
the collected particles. It was found that flowing COa at 1500 psig for 1-1/2
hours
(roughly seven times the view cell volume} was adequate for flushing out the
organic
solvent present in the cell and far drying the particles. It was observed that
no
recrystallized particles could be recovered when the drying periods were
shorter than
one hour; in this case, particles adhering to the tube walls redissolved in
the organic
solvent during pressure reduction as the organic solvent condensed out of the
CO~
phase. Clearly an increase in COZ flow rate will reduce the required drying
time; the
COZ flow rate can also be set high enough so that the coating process can
operate
continuously while keeping the steady state concentration of solvent in the
coating
chamber at a low enough level that the mixture is always supercritical and no
solvent
condensation in the coating chamber takes place.
Following the drying period, the pressure was decreased to atmospheric Ievel
at a rate of -50 psi/min. The coated beads were discharged from the glass tube
and
analyzed by scanning electron microscopy (SEM).
Equipment and Experimental Procedures for Examples 5-8
Example 5
Coating ofNonpareil Sugar Beads and Glass Beads with RG503H
Nonpareil sugar beads and glass beads I .5 mm and 2 mm diameter respectively
were first charged into a 16-cm Iong, 8 mL glass tube. The tube was then
fitted at the
bottom of the view cell (Fig. I4) and the cell was brought up to operating
pressure with
COZ. A 10 mg/ml ethyl acetate solution of RG503H was them pumped into the
glass
tube for 5 minutes. In order to establish countercurrent flow and fluidize the
beads, CO.,
was introduced at the bottom of the tube while the suspension was introduced
from
about 2 inches above. Capillary nozzle LD., temperature, pressure, solution
flow rate
26
CA 02247900 1998-08-31
WO 97!31691 ~ PCT/U597J03207
and COZ rate were 100 ~.m, 35°C, 1500 psig, 2.5 cc/min. and 5 mL/min.
ofchilied
liquid COZ respectively.
Fig. 15 is a micrograph of an uncoated nonpareil bead. Figs. 16 and 17 show
micrographs of a resulting coated nonpareil bead and glass bead respectively.
The
nonpareil bead is nearly uniformly coated with a layer of mostly microspheres
of
RG503H. Coating on the glass bead is less uniform possibly due to its larger
size which
reduces its mobility within the glass tube. The recrystaliized microspheres
(Fig. 16) are
of similar size (roughly 10 ~.m) to those obtained in runs at identical
conditions with th_e_
same solution in the absence of the beads (see Table 1 ).
In this experiment, constraining of the expansion to within the glass tube and
reduction of the efficiency of the atomization process by virtue of pumping
the solution
into the relatively small volume glass tube caused the solution to expand as a
pseudo-
liquid phase rather than as microdroplets. The recrystallized polymer
microparticles
were thus not entrained in the SCF, and were able to coat the beads. As
evidence of this
observation is the fact that upon removal of the glass tube from the view
cell, only the
bottom half of the tube visibly contained polymer particles. The upper half,
which was
not reached by the solution upon expansion appeared polymer-free.
Operation under conditions of higher COZ flow rates (25 cc/min. as liquid) to
improve the efficacy of the atomization step did eliminate the formation of
the
expanded liquid phase, but little coating was deposited on the beads due to
entrainment
of the recrystallized polymer microparticles by the high velocity SCF into the
view cell,
outside the glass tube, thereby reducing their probability of contacting the
beads.
Microparticle entrainment away from the region where the core material is
confined can
be avoided by eliminating the use of the glass tube and loading the core
particles into
the entire view cell. Alternatively, use of a modified cell approximating a
Wurster
coater would provide adequate conditions for antisolvent, solution or
suspension, and
substrate distribution within the coating chamber.
Example 6
3 0 Concentration Effects on Coating o, f Nonpareil Sugar Beads with RG503H
In this study, a solution of 25 mg/ml of RG503H in ethyl acetate was
recrystallized under the same conditions as in Example 6. Fig. 18 is an SEM
micrograph of a coated nonpareil bead. Coating is less uniform than on beads
coated
as described in Example 5 using a 10 mg/ml ethyl acetate solution of RG503H.
The
increase in concentration appears to increase the size of the recrystallized
particles and
27
CA 02247900 1998-08-31
WO 97/31691 PCT/US97/03207
reduce the uniformity of the coating. The increase in recrystallized
microparticle size
was also observed in the absence of the beads.
Example 7
Temperature Effects on Coating of Nonpareil Sugar Beads with RG503H ,
Fig. 19 shows an SEM micrograph of nonpareil sugar beads coated with
RG503H recrystallized under the same conditions as in Example 6 except than
the view ,
cell temperature was held at 40°C. Under these conditions, the polymer
is seen to
deposit on the sugar beads as a continuous film. Thus, a small increase in
temperature
(from 35°C to 40°C) can be sufficient to change the texture of
the coating layer.
Agglomeration of the beads can be avoided by improving the conditions under
which
the fluidization is carried out. Alternatively, use of a modified cell
approximating a
VVurster coater would provide adequate conditions for antisolvent, solution or
suspension, and substrate distribution within the coating chamber.
Example 8
Coating of glass Beads with Hydrocortisone
Fig. 20 shows a micrograph of a glass bead collected from a run where a
suspension of hydrocortisone in ethyl acetate was sprayed into COz using the
capillary
nozzle. This suspension was prepared by filtering a 10 mg/mL suspension of
hydrocortisone in ethyl acetate filtered through a 2 um porosity paper. The 16-
cm Long,
8 mI glass tube was first charged with 1 gram of roughly 2 mm diameter glass
beads,
and then fitted at the bottom of the view cell. Operating temperature,
pressure, solution
flow rate and COz flow rate were 35°C, 1500 psig, 2.5 mL/min. of liquid
CO ,
_ respectively. The suspension was pumped for five minutes. As illustrated in
Fig. 20,
the beads can be nearly uniformly coated with a thin film of hydrocortisone.
Formation
of a film, as opposed to microparticles was expected as our studies and those
of
previously referenced investigators indicate that an increase in organic
solution
saturation level or concentration leads to the formation of amorphous
particles with
agglomerate to form films and porous structures.
Alternative Embodiments
Because of its environmentally benign nature compared to alternative organic '
solvent-based coating process, and its greater potential to form thin film
coatings, the
present coating process provides an attractive alternative to the powder
coating
28
CA 02247900 1998-08-31
WO 97/31691 ~ PCT/US97/03207
electrostatic spraying technique. The instant process also provides an
alternative to the
Wurster coater technique. Alternatively, the core materials may be tumbled
down a
conveyor belt disposed in a high pressure COZ chamber while a solution or
suspension
is continuously sprayed on the core materials. Another alternative is to use
this process
. 5 for coating larger objects than drug tablets or pesticide granules.
Because
recrystallization can occur almost as soon as the spray exits the nozzle in
the SAS
process, a technique can be employed whereby a nozzle scans the surface of the
object,
and microparticles are rapidly deposited on the surface upon which the nozzle
is
spraying the solution. This process could be particularly useful for
efficiently painting
large surfaces. Another alternative is to coat a large object by merely
expanding the
solution spray onto a chamber containing the object without necessarily
scanning the
surface of the object.
Alternatively, adhesives or plasticizers can be added to the organic solution
to
facilitate adherence of the recrystallized particles to the surface of the
substrate or to
improve on the physical properties of the coating. Excipients such as
colorants may
also be added to the organic solution to enhance the aesthetic or functional
properties
of the coating.
Other Applications jor the Coating Method and Apparatus
This invention finds application in all areas where particle coating by
recrystallization of the shell material from an organic phase is desirable.
These
applications can find use, but are not limited to, in coating of-.
pharmaceutical tablets,
granules, pellets or capsules; pesticides; fertilizers; catalysts; seeds;
salts; circuit boards;
wires, containers and lids.
Antisolvents useful in the application of this invention include, but are not
limited to, CO2, propane, butane, isobutane, CHF3, SF6 and N20. Organic
solvents may
be either of the class of aromatic hydrocarbons, alcohols, esters, ethers,
ketones, amines,
or nitrated or chlorinated hydrocarbons. Preferred solvents include acetone,
methanol,
ethanol, propanol, isopropanol, dichioromethane, ethyl acetate and DMSO.
Blends of
these solvents may also be used.
Coating materials useful for this application include sugars, polymers such as
poly-lactide glycolide copolymers (PLGA), PLA, PGA, polyvinylpyrrolidone,
polyethylene glycols and methacrylic acid ester. The largest group of film
forming
resins are the cellulose ethers, especially the hydroxypropylmethyl cellulose.
Other
29
CA 02247900 1998-08-31
WO 97/31691 PCT/US97/03207
cellulose ethers include hydroxypropyl cellulose, methyl hydroxypropyi
cellulose,
methyl cellulose and ethyl cellulose.
Plasticizers may also be added to the coating solution or suspension if
properties
of the polymeric coating are not adequate. These plasticizers are used to
modify the
properties of the coating material through a reduction in its glass transition
temperature.
This can result in a less brittle, softer and more mechanical-stress resistant
coating.
Plasticizers can also decrease the permeability of the film to moisture and
enhance the
stability of the product. Common plasticizers include, but are not limited to,
phthalate
esters, castor oils, acetylated monoglycerides, triacetin, glycerin,
propyleneglycol, and
I0 -- polyethylene glycols.
Colorants such as dyes and pigments including iron oxides and titanium
dioxide,
may also be added to enhance the aesthetic appeal or the physical properties
of the
coating.
Lyophobic Precipitation
In Examples 9-12, the Iyophobic precipitation of drugs (hydrocortisone,
phenytoin, ibuprofen) in containers was investigated. Hydrocortisone,
phenytoin and
ibuprofen were acquired from Sigma Chemical Co., St. Louis, MO and were used
without further purification. Certified grade acetone, DMSO (99.9% purity,
Fisher
Scientific, Fairlawn, NJ) and bone dry COZ (99.8% purity, Air Products,
Lenexa) were
used.
The Fig. 1 apparatus was employed in Example 9 except the modified view cell
16 was fitted with a 15 cm long glass tube sealed on one end with a 4 ~m fi-
it. The tube
mimicked a specialized use container for the purposes of these experiments.
For
examples I0, 1 l and 12 the Fig. I apparatus was employed except that view
cell I6 was
replaced by 95 mL view cell 16b of Fig.21. The cell 16b was equipped with two
carbon
dioxide input lines Cr and C2, each having a corresponding valve V, and V.,
interposed
therein; the valves were in turn coupled to a common source of COZ as shown in
Fig.
21. The view cell 16b was also equipped in these examples with 12 x 75 mm
borosilicate tubes T" which mimicked final use containers. As illustrated in
Fig. 21, the
C02 line C1 extended downwardly into the interior of the tube T" beneath the
level of
liquid therein.
CA 02247900 1998-08-31
WO 97!31691 PCT/US97/03207
Example 9
Batch precipitation of hydrocortisone from a 200 mg/mI DMSO solution was
undertaken. A 1 cc aliquot of solution was pumped into the fritted glass tube
positioned
inside view cell 16. Pressure and temperature were maintained at 1,575 psig
and 31 °C.
Twelve standard liters of CO3 were introduced from the bottom end of the tube
and through the frit to expand the solvent and recrystallize the drug.
Following this
expansion period, 300 standard liters of COZ were introduced from the top end
of the
glass tube to "push" the expanded solution out of the tube through the glass
frit and to
dry the particles for one hour. This method is attractive because it provides
a means for
rapidly expanding the solution and recrystallizing the drug, while preventing
the
solution to expand over the upper rim of the glass tube (or dispensing
container).
Example 10
1 mL of a 24.1 mg/mL solution of phenytoin in acetone was transferred into the
borosiiicate tube. The tube was placed in view cell 16b of Fig 21. The Line C~
extended
through the phenytoin solution to the bottom of the borosilicate tube T". The
cell was
quickly pressurized to 800 psig with CO, at 40°C through line CZ. It is
noted that the
COZ introduction rate via line C, must be Buff ciently slow to prevent the
forceful
ejection of solution. Therefore, initial pressurization can be conducted more
quickly
using line C2.
Following initial pressurization, the valve VZ is closed and CO~ was
introduced
through line C, at 20 g/min for 9 minutes. The solution expanded and the drug
was
observed to precipitate. When the expanded solution reached the top of the
borosilicate
tube the COa flow rate in line C, was decreased to 4.5 g/min to minimize drug
loss as
the expanding solvent overflowed the top of the test tube. After 8 minutes,
the pressure
within cell 16b reached 1,300 psig. Total C02 introduction via bubbling
through line
C1 was 200 g. The cell was then depressurized and the borosilicate tube
containing
product was retrieved.
The precipitated phenytoin (Fig. 22b) was compared to the starting material
(Fig. 22a) by Differential Scanning Calorimetry (DSC) and found to exhibit
enthalpic
transitions consistent with the starting material.
Example 11
1 mL of a 30 mg/mL solution of ibuprofen in DMSO was transferred into the
- ~ 35 borosilicate tube. The tube was placed in the view cell 16b of Fig 21.
Line Ci extended
31
CA 02247900 1998-08-31
WO 97/31691 PCT/LTS97/03207
through the ibuprofen solution to the bottom of the borosilicate tube. The
cell was
quickly pressurized to 620 psig with CO, at 40°C through Iine C2. The
valve VZ on line
CZ was closed and CO ,was then introduced through Iine C ~t flow rates ranging
between 9 and 36 g/min.
The solution expanded and the drug was observed to precipitate. When the
expanded solution reached the top of the borosilicate tube the COZ flow rate
in Iine C,
was decreased to 0.9 g/min for minute 12 minutes. No solvent was observed to
remain
in the test tube. Precipitate was observed on the tube walls. This was a total
COz
introduction via bubbling through line C, of 125 g. Flow was then increased in
line C,
to 18 g/min for I.S minutes. The cell was depressurized and the tube
containing
product was retrieved.
Example 12
I mL of a 12.6 mg/mL solution of phenytoin in acetone was transferred into the
borosilicate tube T" . The tube was placed in view cell of Fig 21. The view
cell I6b
was placed in a solid state ultrasonic bath (Fisher Scientific). The cell was
quickly
pressurized to 900 psig with COZ at 40°C through line C2. The
ultrasonic bath was
energized to produce ultrasonic energy at 43 kHz. After one hour, the solution
had
expanded to three times its initial volume and the drug was observed to
precipitate.
Upon depressurization, the precipitated drug redissolved in the acetone.
Continued
processing as described or direct sonication of the drug solution container
would
ultimately result in the isolation of solid drug.
32