Note: Descriptions are shown in the official language in which they were submitted.
CA 02248964 1998-09-14
W O 97/33588 PCTAJS97/03348
IRON CO~P~EXE8 OF NITROGEN-CON~TNTN~
M~r~YC~C LIGANDS EFFEC~I~E A8 CATALY8T8 FOR
DISMUTAT~G S~PEROXIDB
CRO88 REFERENC~ TO R~LATED APPL~CATION
This application is a continuation-in-part of
p~n~;~g application Serial No. 08/397,469, filed March
1, 1995, which is a continuation of pending application
Serial No. 08/231,599, ~iled April 22, 1994.
B~CRGRO~ND OF T~E lNV ~ ON
15 The present invention relates to compounds
effective as catalysts for dismutating superoxide and,
more particularly, relates to iron(II) or iron(III)
complexes of nitrogen-cont~; n; n~ fifteen-m~mh~ed
macrocyclic ligands which catalytically dismutate
superoxide. Application Serial No. 08/397,469 is hereby
incorporated by reference herein in its entirety.
The enzyme superoxide dismutase catalyzes the
conversion of superoxide into oxygen and hydrogen
peroxide according to e~uation (l) (hereinafter referred
to as dismutation). Reactive oxygen metabolites derived
from superoxide are postulated to contribute to the
tissue pathology in a number of
~2 ~ + ~2 ~ + 2H+ ~ O2 + H2O2 (1)
inflammatory diseases and disorders, such as reperfusion
injury to the ischemic myocardium, inflammatory bowel
disease, rheumatoid arthritis, osteoarthritis,
atherosclerosis, hypertension, metastasis, psoriasis,
organ transplant rejections, radiation-in~ injury,
asthma, influenza, stroke, burns and trauma. See, ~or
example, Bulkley, G.B., Reactive oxygen metabolites and
reperfusion injury: aberrant triggering of
SU~~ TESHEET(RULE26~
CA 02248964 l99X-09-l4
~CT~US97/03348
W097/3358~ _
reticuloendothelial function, T~e Lancet, Vol. 344, pp.
934-36, october 1, 1994; Grisham, M.B., Oxidants and
free radicals in inflammatory bowel disease, The Lancet,
Vol. 344, pp. 859-861, Septemhe~ 24, 1994; Cross, C.E.
Qt al., Reactive oxygen species and the lun~, The
Lancet, Vol. 344, pp. 930-33, October 1, 1994; Jenner,
P., Oxidative damage in neurodegenerative disease, The
Lancet, Vol. 344, pp. 796-798, September 17, 1994;
Cerutti, P.A., Oxy-radicals and cancer, The Lancet , Vol.
344, pp. 862--863,September 24, 1994 Simic, M. G., et
al, oxygen Radicals in Biology and Medicine, Basic Life
Sciences, Vol. 49, Plenum Press, New York and London,
1988; Weiss J. Cell. Biochem., 1991 SUppl. 15C, 216
Abstract CllO (1991); Petkau, A., Cancer Treat. Rev. 13,
17 (1986); McCord, J. Free Radicals Biol. Med., 2, 307
(1986); and Bannister, J.V. et al, Crit. Rev. Biochem.,
22, 111 (1987). The above-identi~ied re~erences from
T~e Lancet teach the nexus between free radicals derived
from superoxide and a variety of diseases. In
particular, the Bulkley and Gr;~h~ references
specifically teach that there is a nexus between the
dismutation of superoxide and the final disease
treatment.
It is also known that superoxide is involved in
the breakdown o~ endothelium-derived vascular relaxing
factor (EDRF), which has been identified as nitric oxide
(NO), and that EDRF is protected from breakdown by
superoxide dismutase. This suggests a central role for
activated oxygen species derived from superoxide in the
pathogenesis of vasospasm, thrombosis and
atherosclerosis. See, for example, Gryglewski, R.J. et
al , "Superoxide Anion is Involved in the Breakdown of
Endothelium-derived Vascular Relaxing Factor", Nature,
~ Vol. 320, pp. 454-56 ~1986) and Palmer, R.M.J. et al.,
"Nitric Oxide Release Accounts ~or the Biological
Activity of Endothel-ium Derived Relaxing Factor",
S~ TESH~ET(RULE26)
,
CA 02248964 l998-09-l4
07-21(12463)A
--3--
Nature, Vol. 3Z7, pp. 523-26 (1987).
Clinical trials and animal studies with natural,
recombinant and modi~ied superoxide dismutase enzymes
have been completed or are ongoing to demonstrate the
therapeutic efficacy o~ reducing superoxide levels in
the disease states noted above. However, numerous
problems have arisen with the use o~ the enzymes as
potential therapeutic agents, including lack o~ oral
activity, short hal~-lives in vlvo, immuno~enicity with
nonhuman derived enzymes, and poor tissue distribution.
The iron complexes o~ nitrogen-containing
~ifteen-membered macrocyclic ligands that are low
molecular weight mimics o~ superoxide dismutase (SOD)
are use~ul as therapeutic agents and avoid many o~ the
lS problems associated with SOD enzymes.
8UMM~RY OF T~E INVENTION
It is an object of the invention to provide iron
complexes o~ nitrogen-containing ~i~teen-membered
macrocyclic ligands that are low molecular weight mimics
o~ superoxide dismutase (SOD) which are useful as
therapeutic agents ~or in~lammatory disease states or
disorders which are medicated, at least in part, by
superoxide.
It is
yet a ~urther object of the invention to provide iron
complexes o~ nitroqen-containing ~i~teen-membered
macrocyclic ligands that have unexpectedly improved
stability compared to corresponding manganese complexes.
According to the invention, pharmaceutical
compositions in unit dosage ~orm use~ul ~or dismutating
3~ superoxide are provided comprising (a) a therapeutically
or prophylactically e~ective amount o~ an iron complex
~ME~D SffEF~
CA 02248964 1998-09-14
PCT/US97/03348
W097/33588
of the invention and (b) a nontoxic, pharmaceutically
acceptable carrier, adjuvant or vehicle.
Further according to the invention, a method of
preventing or treating a disease or disorder which is
5 medicated, at least in part, by superoxide is provided
co~prising a~m; n; stering to a subject in need of such
prevention or treatment, a therapeutically or
prophylactically effective amount of an iron complex of
the invention.
DE~AI~ED DE8CRIP~ION OF T~E lNv~NllON
The present invention is directed to iron
complexes of nitrogen-cont~i~ing fifteen-membered
macrocyclic ligands which catalyze the conversion of
superoxide into oxygen and hydrogen peroxide. These
complexes are represented by the formula:
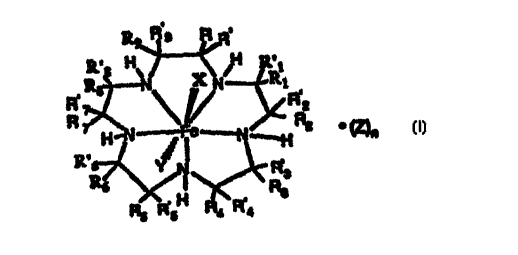
Rg~l9 ~
~H--N _ I c ~ ~Z)n
2 5 R' 7~ ~ R 3
wherein R, R', R" R'l, R2, R'2, R3, R'3, R4, R'~,
R5, R'5, R6, R'6, R~, R'" RR~ R'8, R9, and R'4
independently are selected from the group consisting of
hydrogen, alkyl, alkenyl, alkynyl, cycloalkyl,
cycloalkenyl, cycloalkylalkyl, cycloalkylcycloalkyl,
3~ cycloalkenylalkyl, alkylcycloalkyl, alkenylcycloalkyl,
alkylcycloalkenyl, alkenylcycloalkenyl, heterocyclic,
t~ ? i J~ hA- S~ TESHEET(RU~E26)
CA 02248964 1998-09-14
PCTrUS97/03348
W O 97133588
aryl and aralkyl radicals and radicals attached to the
~-carbon of ~-amino acids; or R~ or ~1 and R2 or R'2, R3
or R'3 and R4 or R'4, R5 or R'5 and R6 or R'6, R7 or R'7 and
R8 or R' 8~ and R9 or R'g and R or R' together with the
carbon atoms to which they are attached independently
- form a saturated, partially saturated or unsaturated
cyclic having 3 to 20 car~on atoms; or R or R' and Rl or
R'l, ~2 or R' 2 and R3 or R' 3, R4 or R'~ and R5 or R~s~ R6 or
R' 6 and R~ or ~' 7, and R8 or R' 8 and R9 or R' 9 together
with the carbon atoms to which they are attached
independently form a nitrogen containing heterocycle
having 2 to 20 carbon atoms provided that when the
nitrogen containing heterocycle is an aromatic
heterocycle which does not contain a hydrogen attached
to the nitrogen, the hydrogen attached to the nitrogen
as shown in the above formula, which nitrogen is also in
the macrocyclic ligand or complex, and the R groups
attached to the same carbon atoms of the macrocycle are
absent.
X, Y and Z represent suitable ligands or charge-
neutralizing anions which are derived from any
monodentate or polydentate coordinating ligand or ligand
system or the corresponding anion thereof (for example
benzoic acid or benzoate anion, phenol or phenoxide
anion, alcohol or alkoxide anion). X, Y and Z are
independently selected from the group consisting of
halide, oxo, aquo, hydroxo, alcohol, phenol, dioxygen,
peroxo, hydroperoxo, alkylperoxo, arylperoxo, ammonia,
- alkylamino, arylamino, heterocycloalkyl amino,
heterocycloaryl amino, amine oxides, hydrazine, alkyl
hydrazine, aryl hydrazine, nitric oxide, cyanide,
cyanate, thiocyanate, isocyanate, isothiocyanate, alkyl
nitrile, aryl nitrile, alkyl isonitrile, aryl
isonitrile, nitrate, nitrite, azido, alkyl sulfonic
acid, aryl sulfonic acid, alkyl sulfoxide, aryl
SU~S~ TESHEET~RULE26)
CA 02248964 1998-09-14
PC~nUS97/0334B
W O 97/33~88
sulfoxide, alkyl aryl sulfoxide, alkyl sulfenic acid,
aryl sulfenic acid, alkyl sulfinic acid, aryl sulfinic
acid, alkyl thiol carboxylic acid, aryl thiol carb-
oxylic acid, alkyl thiol thiocarboxylic acid, aryl thiol
thiocarbo~ylic acid, alkyl carboxylic acid (such as
acetic acid, trifluoroacetic acid, oxalic acid), aryl
carboxylic acid (such as benzoic acid, phthalic acid),
urea, alkyl urea, aryl urea, alkyl aryl urea, thiourea,
alkyl thiourea, aryl thiourea,alkyl aryl thiourea,
sulfate, sulfite, ~isulfate, bisulfite, thiosulfate,
thiosulfite, hydrosulfite, alkyl phosphine, aryl
phosphine, alkyl phosphine oxide, aryl phosphine oxide,
alkyl aryl phosphine oxide, alkyl phosphine sulfide,
aryl phosphine sulfide, alkyl aryl phosphine sulfide,
alkyl phosphonic acid, aryl phosphonic acid, alkyl
phosphinic acid, aryl phosphinic acid, alkyl phosphinous
acid, aryl phosphinous acid, phosphate, thiophosphate,
phosphite, pyrophosphite, triphosphate, hydrogen
phosphate, dihydrogen phosphate, alkyl guanidino, aryl
guanidino, alkyl aryl guanidino, alkyl carbamate, aryl
carbamate, alkyl aryl car~amate, alkyl thiocarbamate
aryl thiocar~amate, alkyl aryl thiocarbamate, alkyl
dithiocar~amate, aryl dithiocarbamate, alkyl aryl
dithiocarbamate, bicar~onate, carbonate, perchlorate,
chlorate, chlorite, hypochlorite, perbromate, bromate,
bromite, hypobromite, tetrahalomanganate,
tetrafluoroborate, hexafluorophosphate,
hexa~luoroantimonate, hypophosphite, iodate, periodate,
metaborate, tetraaryl borate, tetra alkyl borate,
tartrate, salicylate, succinate, citrate, ascorbate,
saccharinate, amino acid, hydroxamic acid,
thiotosylate, and anions of ion r~h~nge resins, or
systems where one or more o~ X,Y and Z are independently
attached to one or more of the "R" groups, wherein n is
an integer from O or 1. The preferred ligands from
which X, Y and Z are selected include halide, organic
S~ TESH~FT(RULE26)
CA 02248964 1998-09-14
PCTAJS97/03348
W O 97/33S88
acid, nitrate and bicarbonate anions.
As utilized herein, the term "alkyl", alone or in
combination, means a straight-chain or branched-chain
alkyl radical containing from 1 to about 22 carbon
atoms, preferably from about 1 to about 18 carbon atoms,
- and most preferably from about 1 to about 12 carbon
atoms. Examples of such radicals include, but are not
limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl,
isobutyl, sec-butyl, tert-butyl, pentyl, iso-amyl,
hexyl, octyl, nonyl, decyl, dodecyl, tetradecyl,
hexadecyl, octadecyl and eicosyl. The term "alkenyl",
alone or in combination, means an alkyl radical having
one or more double bonds. Examples of such alkenyl
radicals include, ~ut are not limited to, ethenyl,
propenyl, 1-butenyl, cis-2-butenyl, trans-2-butenyl,
iso-butylenyl, cis-2-pentenyl, trans-2-pentenyl,
3-methyl-~-butenyl, 2,3-dimethyl-2-butenyl, 1-pentenyl,
1-hexenyl, 1-octenyl, decenyl, do~c~nyl, tetradecenyl,
hexadecenyl, cis- and trans- 9-octadecenyl,
1,3-pentadienyl, 2,4-pentadienyl, 2,3-pentadienyl,
1,3-hexadienyl, 2,4-hexadienyl, 5,8,11,14-
= eicosatetraenyl, and 9,12,15-octadecatrienyl. The term
"alkynyl", alone or in combination, means an alkyl
radical having one or more triple bonds. Examples of
such alkynyl groups include, but are not limited to,
ethynyl, propynyl (propargyl~, 1-butynyl, 1-octynyl,
9-octadecynyl, 1,3-pentadiynyl, 2,4-pentadiynyl,
1,3-hexadiynyl, and 2,4-hexadiynyl. The term
- "cycloalkyl", alone or in combination means a cycloal~yl
radical containing from 3 to about lO, preferably from
3 to about 8, and most preferably from 3 to about 6,
carbon atoms. Examples of such cycloalkyl radicals
include, but are not limited to, cyclopropyl,
cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl,
cyclooctyl, and perhydronaphthyl. ~he term
"cycloalkylalkyl" means an alkyl radical as defined
SlJ~ 111 ~JTE SHEET (RULF 26)
CA 02248964 1998-09-14 t
PCTnUS97103348
W O 97/33~88
above which is substituted by a cyc7oalXyl radical as
defined above~ Examples of cycloalkylalkyl radicals
include, but are not limited to, cyclohexylmethyl,
cyclopentylme~hyl, (4-isopropylcyclohexyl~methyl,
~4-t-butyl-cyclohexyl)methyl, 3-cyclohexylpropyl,
2-cyclo-hexylmethylpentyl, 3-cyclopentylmethylhexyl,
1-(4-neopentylcyclohexyl)methylhexyl, and 1-(4-
isopropylcyclohexyl)methylheptyl. The term
"cycloalkylcycloalkyl" means a cycloalkyl radical as
defined abo~e which is substituted by another cycloalkyl
radical as defined above. Examples of
cycloalkylcycloalkyl radicals include, but are not
limited to, cyclohexylcyclopentyl and
cyclohexylcyclohexyl. The term "cycloalkenyl", alone or
in combination, means a cycloalkyl radical having one or
more double bonds. Examples of cycloalkenyl radicals
include, but are not l~mited to, cyclopentenyl,
cyclohexenyl, cyclooctenyl, cyclopentadienyl,
cyclohexadienyl and cyclooctadienyl. The term
"cycloalkenylalkyl" means an alkyl radical as defined
above which is substituted by a cycloalkenyl radical as
defined above. Examples of cycloalkenylalkyl radicals
include, but are not limited to, 2-cyclohexen-1-
ylmethyl, 1-cyclopenten-1-ylmethyl, 2-(1-cyclohe~n-1-
yl~ethyl, 3-(1-cyclopenten-1-yl)propyl,
1-~1-cyclohexen-1-ylmethyl)pentyl, 1-(1-cyclopenten-1-
yl)hexyl, 6-(1-cyclohexen-1-yl)hexyl, 1-(1-cyclopenten-
}-yl)nonyl and 1-~1-cyclohexen-1-yl~nonyl. The terms
"alkylcycloalkyl" and "alkenylcycloalkyl" mean a
cycloalkyl radical as defined above which is su~stituted
by an alkyl or alkenyl radical as defined above.
Examples of alkylcycloalkyl and alkenylcycloalkyl
radicals include, but are not limited to,
- 2-ethylcyclobutyl, 1-methylcyclopentyl,
l-hexylcyclopentyl, 1-methylcyclohexyl,
1-19-octadecenyl)cyclopentyl and 1-(9-
S~ TE SHEET ~R~LE26)
CA 02248964 1998-09-14
~CT~US97/03348
W O 97/33S88
octadecenyl3cyclohexyl. The terms "alkylcycloalkenyl"
and "alkenylcycloalkenyl" means a cycloalkenyl radical
as de~ined above which is substituted by an alkyl or
alkenyl radical as defined above. Examples of
alkylcycloalkenyl and alkenylcycloalkenyl radicals
~ include, but are not limited to, 1-methyl-2-cyclopentyl,
1-hexyl-2-cyclopentenyl, 1-ethyl-2-cyclohexenyl,
~ l-butyl-2-cyclohexenyl~ 1-(9-octadecenyl)-2-cyclohexenyl
and 1-(2-pentenyl)-2-cyclohexenyl. The term "aryl",
alone or in combination, means a phenyl or naphthyl
radical which optiona~ly carries one or more
substituents selected from alkyl, cycloalkyl,
cycloalkenyl, phenyl, naphthyl, heterocycle, alkoxyaryl,
alkaryl, alkoxy, halogen, hydroxy, amine, cyano, nitro,
alkylthio, phenoxy, ether, trifluoromethyl and the like,
such as phenyl, p-tolyl, 4-methoxyphenyl, 4-(tert-
butoxy)phenyl, 4-fluorophenyl, 4-chlorophenyl,
4-hydroxyphenyl, 1-naphthyl, 2-naphthyl, and the like.
The term 'laralkyl'l, alone or in combination, means an
alkyl or cycloalkyl radical as defined above in which
one hydrogen atom is replaced by an aryl radical as
defined above, such as benzyl, 2-phenylethyl, and the
like. The term "heterocyclic" means ring structures
containing at least one other kind of atom, in addition
to carbon, in the ring. The most common of the other
kinds of atoms include nitrogen, oxygen and sulfur.
- Examples of heterocyclics include, but are not limited
to, pyrrolidinyl, piperidyl, imidazolidinyl,
- tetrahydrofuryl, tetrahydrothienyl, furyl, thienyl,
pyridyl, ~uinolyl, isoquinolyl, pyridazinyl, pyrazinyl,
indolyl, imidazolyl, oxazolyl, thiazolyl, pyrazolyl,
pyridinyl, benzoxadiazolyl, benzothiadiazolyl, triazolyl
and tetrazolyl groups. The term "saturated, partially
saturated or unsaturated cyclic" means fused ring
structures in which 2 carbons of the ring are also part
of the fifteen ~~h~red macrocyclic ligand. The ring
S~ TESHEET (RULE 26)
,
CA 02248964 1998-09-14
PCTnUS97/~3348
W O 97/3358~
10--
.
structure can contain 3 to 20 carbon atoms, preferably 5
to 8 carbon atoms, and can also contain one or more
other kinds of atoms in addition to carbon. The most
common of the other kinds of atoms include nitrogen,
oxygen and sulfur. The ring structure can also contain
more than one ring. The term "nitrogen containing
heterocycle" means ring structures in which 2 carbons
and a nitrogen of the ring are also part of the fifteen-
-~h~ed macrocyclic ligand. The ring structure can
lo contain 2 to 20, preferable 4 to 10 carbon atoms, can be
partially or fully unsaturated or saturated and can also
contain nitrogen, oxygen and/or sul~ur in the portion of
the ring which is not also part of the fifteen-membered
macrocyclic ligand. The term "organic acid anion"
re~ers to car~oxylic acid anions ha~ing from about 1 to
about 18 carbon atoms. The term "halide" means chloride
or bromide.
The overall charge-type of the complex can be
varied ~rom negative to positive ~y carbon substitution
2~ of the appropriate charged groups on the macrocyclic
framework. By considering the dispositive nature of the
iron metal center, the overall charge on the complex can
be adjusted as needed to ~nh~nce desired pharmaceutical
properties such as osmolality, tissue distribution and
non-target clearance. For example, if the complex
carries only charge neutral functionality, such as
C-alkyl substitution, then the overall charge on the
complex will be dete~ ;ne~ by the iron center and will
be positive. Multi-positive complexes are available via
the incorporation of pendant cations such as protonated
aminoalkyl groups. These types of comp7exes can bind to
endogenous anions, anionic proteins, cell mèmbranes, and
the like. If pendant~anionic groups are attached, such
~ as carboxylates, phenolate, phosphonates, sulfonates and
the like, the overall charge on the complex can be
envisioned as zero or positive, i.e. an anionic complex
S~ UTESHEET(RULF26~
CA 02248964 1998-09-14
PCT/US97/03348
W097/33588
will result. The pendant groups may be designed to
axially chelate and formally displace the axial anions
or they may be designed specifically to not chelate but
retain a charge type.
Currently, preferred compounds are those wherein
- at least one, preferably at least two, of the "R" groups
represent alkyl, or alkyl substituted with
~ -ORlo or -NR~oRl1 wherein Rlo and Rl1 are independently
hydrogen or alkyl, and the r~r~ining R groups represent
hydrogen, a saturated, partially saturated or
unsaturated cyclic, or a nitrogen cont~;ning
heterocycle, more preferably hydrogen or a saturated,
partially saturated or unsaturated cyclic; those wherein
at least one, preferably at least two, of R~ or R'l and
R2 or R'2, R3 or R'3 and R4 or R'4, R5 or R'5 and R6 or R'6,
R~ or R', and R8 or R' 8t and R9 or R'9 and R or R' together
with the carbon atoms to which they are attached
represent a saturated, partially saturated or
unsaturated cyclic having 3 to 20 carbon atoms and all
the remaining "R" groups are hydrogen, nitrogen
containing heterocycle, alkyl or alkyl substituted with
-ORlo or -NRloRIl groups, more preferably hydrogen, alkyl
or alkyl substituted with -OR1o or -NRloRll groups; and
those wherein at least one, preferably at least two, of
25 R or R' and Rl or R'l, R2 or R' 2 and R3 or R' 3, R4 or R'4
and Rs or R'st R6 or R' 6 and R~ or R' 7, and R8 or R' 8 and R9
or R'9 together with the carbon atoms to which they are
attached are bound to form a nitrogen containing
heterocycle having 2 to 20 carbon atoms and all the
L ~ ~; n ing "R" groups are independently selected from
hydrogen, saturated, partially saturated or unsaturated
cyclic, alkyl or alkyl substituted with -~Rlo or -NRloR
groups. As used herein, "R" groups means all of the R
groups attached to the carbon atoms of the macrocycle,
i.e., R, R', Rl, R'l, R2, R'2, R3, R'3, R4, R'4, R5, R'5, R6,
SUt~ 111 UTE SHEET (P~ULE 26)
CA 02248964 1998-09-14
W O 97/33S88 PCTrUS97/03348
R'6, R~, R'7, R8, R' 8~ R9 and R' 9. Examples of complexes
of the invention include, but are not limited to,
compounds having the formulas:
~ ~CI ~ N ~ ~C
10~ N ~ ~ N ~
15O_~N/~N~ H?~ H
H' N rc N H H~
The macrocyclic ligand wherein all R's are H can
be prepared according to the general synthetic scheme A
set forth below utilizing methods known in the art for
preparation of certain intexmediates and certain
ligands. See, for example, Richman et al., ~. Am. Chem.
Soc., 96, 2268 (1974); Atkins et al. Org. synth ., 58, 86
(1978); and EP 287 465. Thus a triazaalkane is
tosylated in a suitable solvent system to produce the
corresponding tris(N-tosyl) derivative. Such derivative
is then treated with a suitable base to produce the
corresponding disulfonamide anion. The disulfonamide
anion is then reacted with a di-O-tosylated di-N-
tosylated diazaalkane diol to produce the corresponding
pentatosylpentaazacycloalkane. The tosyl yrOU~S are
then xe~oved and the resulting compound is reacted with
S~ TESHEET(RULE26)
CA 02248964 1998-09-14
W O 97/33588 PCT~US97/03348
-13-
an iron compound under essentially anhydrous and
anaerobic conditions to form the corresponding iron
pentaazacycloalkane complex.
The macrocyclic ligands useful in the complexes
of the present invention, wherein Rl, R'l~ R3, R'3, Rs~
- R~s~ R~, R'7, R9 and R'9 can be H or any functionality as
previously described, can be prepared according to the
- general peptide method shown in Scheme B set forth
below. The procedure for preparing the cyclic peptide
precursors from the corresponding linear peptides are
the same or significant modifications of methods known
in the art. See, for example, Veber, ~.F. et al., J.
Org. Chem., 44, 3101 (1979). The general method
outlined in Scheme B below is an example utilizing the
sequential solution-phase preparation of the
functionalized linear pentapeptide from N-terminus to
C-terminus. Alternatively, the reaction sequence to
prepare the linear pentapeptide can be carried out by
solid-phase preparation utilizing methods known in the
art. The reaction sequence could be conducted from
C-terminus to N-terminus and by convergent approaches
such as the coupling of di- and tri-peptides as needed.
Thus a Boc-protected amino acid is coupled with an amino
acid ester using standard peptide coupling reagents.
The new Boc-dipeptide ester is then saponified to the
free acid which is coupled again to another amino acid
ester. The resulting Boc-tri-peptide ester is again
saponified and this method is continued until the Boc-
- protected pentapeptide free acid has been prepared. The
Boc protecting group is removed under standard
conditions and the resulting pentapeptide or salt
thereof is converted to the cyclic pentapeptide. The
cyclic pentapeptide is then reduced to the
~ pentaazacyclopentadecane with lithium aluminum hydride
or borane. The final ligand is then reacted with an
iron compound under essentially anaerobic conditions to
SU~~ TESHEET (RULE 26)
CA 02248964 1998-09-14
W O 97/33588 PCTrUS97/03348
form the corresponding iron pentaazacyclopentadecane
complex.
The R groups in the macrocycles produced by the
cyclic peptide route, i.e., Rl~ R'1~ R3, R'3, Rs, R'5, R7,
R'~, R9 and R' 9, could be derived from the D or L ~orms
of the amino acids Alanine, Aspartic acid, Arginine,
Asparagine, ~ysteine, Glycine, Glutamic acid, Glut;~m;nf~,
Histidine, Isoleucine, Leucine, Lysine, Methionine,
Proline, Phenylalanine, Serine, Tryptophan, Threonine,
Tyrosine, Valine and/or the R groups of unnatural
~-amino acids such as alkyl, ethyl, butyl, tert-butyl,
cyc7oalkyl, phenyl, alkenyl, allyl, alkynyl, aryl,
heteroaryl, polycycloalkyl, polycycloaryl,
polycycloheteroaryl, imines, aminoalkyl, hydroxyalkyl,
hydroxyl, phenol, amine oxides, thioalkyl,
carboalkoxyalkyl, carboxylic acids and their
derivatives, keto, ether, aldehyde, amine, nitrile,
halo, thiol, sulfoxide, sulfone, sulfonic acid, sul~ide,
disulfide, phosphonic acid, phosphinic acid, phosphine
oxides, sulfonamides, amides, amino acids, peptides,
proteins, carbohydrates, nucleic acids, fatty acids,
lipids, nitro, hydroxylamines, hydroxamic acids,
thiocarbonyls, borates, boranes, boraza, silyl, siloxy,
silaza, and combinations thereof.
The macrocyclic ligands useful in the complexes
of the present invention can also be prepared by the
diacid dichloride route shown in Scheme C set forth
below. Thus, a triazaalkane is tosylated in a suitable
solvent system to produce the corresponding tris(N-
tosyl) derivative. Such a derivative is treated with a
suitable base to produce the corresponding disulfonamide
anion. The disulfonamide anion is dialkylated with a
suitable electrophile to produce a derivative of a
~ dicarboxylic acid. This derivative of a dicarboxylic
acid is treated to produce the dicarboxylic acid, which
is then treated with a suitable reagent to form the
S~ 111 UTE SHEEl- (RULE 26)
CA 02248964 1998-09-14
W O 97/33588 PCTrUS97103348
diacid dichloride. The desired vicinal diamine is
ob~ained in any of several ways. One way which is useful
is the preparation from an aldehyde by reaction with
cyanide in the presence of ammonium chloride followed by
treatment with acid to produce the alpha ammonium
- nitrile. The latter compound is reduced in the presence
of acid and then treated with a suita~le base to produce
- the vicinal diamine. Condensation o~ the diacid
dichloride with the vicinal diamine in the presence of a
suitable base forms the tris(tosyl)diamide macrocycle.
The tosyl groups are removed and the amides are reduced
and the resulting compound is reacted with an iron
compound under essentially anhydrous and anaerobic
conditions to form the corresponding substituted
pentaazacycloalkane iron complex.
The vicinal diamines have been prepared by the
route shown (known as the Strecker synthesis) and
vicinal diamines were purchased when commercially
available. Any method of vicinal diamine preparation
could be used.
The macrocyclic ligands useful in the complexes
of the present invention can also be prepared by the
bis(haloacetamide) route shown in Scheme D set forth
below. Thus a triazaalkane is tosylated in a suitable
solvent system to produce the corresponding tris(N-
tosyl) derivative. Such a derivative is treated with a
suitable base to produce the corresponding disulfonamide
anion. A bis(haloacetamide), e.g., a
- b,is(chloroacetamide), of a vicinal ~iA~ne is prepared
by reaction of the diamine with an excess of haloacetyl
halide, e.g., chloroacetyl chloride, in the presence of
a base. The disulfonamide anion of the tris(N-tosyl)
_ triazaalkane is then reacted with the
- bis(chloroacetamide) of the diamine to produce the
substituted tris(N-tosyl)diamide macrocycle. The tosyl
groups are removed and the amides are reduced and the
SU~ TrS~EET(RULF2~)
=
CA 02248964 l99X-09-14
W O 97/33588 PCT~US97tO3348
-16-
resulting compound is reacted with an ircn compound
under essentially anhydrous and anaerobic conditions to
form the corresponding substituted pentaazacycloalkane
iron complex.
The macrocyclic ligands useful in the complexes
of the present invention, wherein R" R 1~ R2, R 2 are
part of a C15- or trans- cycloalkyl ring system and R5,
.
R 5, R~, R ~ and R9, R g can be H or any functiona~ity
previously described, can be prepared according to the
pseudo-peptide method shown in Scheme E set forth below.
A cis-l, 2-Diaminocycloalkane or a trans-(R,R)-1,2-
diaminocycloalkane or trans-(s~s)-l~2-diaminocycloalkane
can be used in this method in combination with any amino
acids. This allows the relative stereo~he ;fitry of the
cycloalkane fused ring and substituent, Rs~ R 5, R~, R ~,
R9, R 9, functionality and stereo~-h~ try to be defined
in any manner. As an example trans-(R,R)-1,2-
diaminocyclhexane was monotosylated and reacted with Boc
anhydride to afford the differentiated N-Boc, N-tosyl
derivative. The sulfonamide was alkylated with methyl
bromoacetate using sodium hydride as the base and
saponified to the free acid. The cyclohexanediamine
containing N-tosylglycine serves as a dipeptide
surrogate in standard solution-phase peptide synthesis.
Thus, coupling with a functionalized amino acid ester
affords the corresponding pseudo-tripeptide. Two
se~uential TFA c~eavage-couplings affords the pseudo-
pentapeptide which can be N- and C-terminus deprotected
in one step using HCl/AcOH. DPPA mediated cyclization
followed by T;Al~or Borane reduction affords the
corresponding macrocylic ligand. This ligand system is
reacted with an iron compound, such as iron (III)
chloride under essentially anaerobic conditions to form
the corresponding functionalized iron (III)
pentaazacycloalkane complex.
SU~IllUTFSHEET(RULE26)
CA 02248964 l998-09-l4
W O 97133588 PCTrUS97/03348
-17-
The macrocyclic ligands useful in the complexes
of the present invention, wherein Rl, R " R~, R 2 and R~,
R 5, R6, R 6, are part of a cis- or trans- cycloalkyl
ring system and R9, R 9 can be H or any functionality
previously described, can be prepared according to the
- iterative pseudo-peptide method shown in Scheme F set
forth below. A cis-l, 2-Diaminocycloalkane or a trans-
~ (R,R)-1,2-diaminocycloalkane or trans-(S,S)-1,2-
diaminocycloalkane can be used in any combination with
each other using this method and in combination with any
amino acids. This allows the relative stereochemistry
of both cycloalkane fused rings and substituent, Rg, R 9,
functionality and stereochemistry to be defined in any
~-nn~r, Thus, the (S,S)-1,2-~;Am;nocyclohexyl-N-
tosylglycine dipeptide surrogate, prepared from (S,S)-
1,2-diaminocycloh~ne exactly as in Scheme E in the
case of (R,R)-1,2-~iA inocyclohexane, can be coupled
with a functionalized amino acid ester to afford the
corresponding pseudo-tripeptide. TFA cleavage affords
the pseudo-tripeptide TFA salt which is coupled with
(R,R)-diaminocyclohexyl-~N-tosylglycine. Saponification
and TFA cleavage a~fords the bis-cyclohexano cont~; n; ng
pseudo-pentapeptide. DPPA mediated cyclization followed
by T.;Al~4 or Borane reduction affords the corresponding
bis-cyclohexano-fused macrocylic ligand. This ligand
system is reacted with an iron compound, such as iron
- (III) chloride under essentially anaerobic conditions to
form the corresponding functionalized iron (III)
- pentaazacycloalkane complex.
The macrocyclic ligands use~ul in the complexes
of the present invention can also be prepared according
to the general procedure shown in Scheme G set forth
below. Thus, an amino acid amide, which is the
corresponding amide derivative of a naturally or non-
3~ naturally occurring ~-amino acid, is reduced to form the
corresponding substituted ethylen~ m; ne, Such amino
S~ TESHEET(RULE26)
CA 02248964 1998-09-14
PCT~US97/03348
W O 97/33S88
18
-
acid amide can be the amide deri~ative of any one of
many well known amino acids. Preferred amino acid
amides are those represented by the formula:
~ ~ ~
~ a
wherein R is as previously defined. Most preferred are
those wherein R represents hydrogen, alkyl,
cycloalkylalkyl, and aralkyl radicals. The diamine is
then tosylated to produce the di-N-tosyl derivati~e
which is reacted with a di-O-tosylated tris-N-tosylated
triazA~lk~ne diol to produce the corresponding
substituted
N-pentatosylpentaaZacycloalkane. The tosyl ~roups are
then removed and the resulting compound is reacted with
an iron compound under essentially anhydrous and
anaerobic conditions to form the correspondin~
substituted iron pentaazacycloalkane complex.
The complexes of the present invention, wherein
R9, and R2 are alkyl, and R3, R'3, Rq, R'4, R~, R'5, R6,
R'6, R~, R~7, R8 and R'8 can be alkyl, arylalkyl or
cycloal~ylalkyl and R or R' and Rl or R', together with
the carbon atoms they are attached to are bound to ~orm
a nitrogen con~;n; ng heterocycle, can also be prepared
according to the general procedure shown in Scheme H set
~orth below utilizing methods known in the art for
preparing the iron
pentaazabicyclo~12.3.1~octadecapentaene complex
precursor. See, for example, Alexander et al., Inorg.
S~ TESHEET(R~LE26)
-
.
CA 02248964 1998-09-14
W O 97/33588 PCT~US97/03348
_~9_
Nucl. Chem. Lett., 6, 445 (1970). Thus a 2,6-
diketopyridine is condensed with triethylene tetraamine
in the presence of an iron compound to produce the iron
pentaazabicyclo[12.3.1]octadecapentaene complex. The
iron pentaazabicyclo[12.3.1]octadecapentaene complex is
hydrogenated with 5% rhodium on carbon at a pressure of
1000 psi to give the corresponding iron
- pentaazabicyclo[12.3.1]octadecatriene co~plex.
The macrocyclic ligands useful in the complexes
of the present invention can also be prepared by the
pyridine diamide route shown in Scheme I as set forth
below. Thus, a polyamine, such as a tetraaza compound,
cont~ining two primary amines is condensed with dimethyl
2,6-pyridine dicarboxylate by heating in an appropriate
solvent, e.g., methanol, to produce a macrocycle
incorporating the pyridine ring as the 2,6-
dicarboxamide. The pyridine ring in the macrocycle is
reduced to the corresponding piperidine ring in the
macrocycle, and then the diamides are reduced and the
resulting compound is reacted with an compound under
essentially anhydrous and anaerobic conditions to form
the corresponding substituted pentaazacycloalkane iron
complex.
When the ligands or charge-neutralizing anions,
i.e. X, Y and Z, are anions or ligands that cannot be
introduced directly from the iron compound, the complex
with those anions or ligands can be formed by conducting
an ~h~nge reaction with a complex that has been
prepared by reacting the macrocycle with an iron
compound.
S~ UTESHEFT~RULE26)
~ CA 02248964 l998-09-l4
07-2~(12463)A
-20-
8C$EME
ty~ln~
tsHN ,~ r C~
0~ ~S~
N ~
~,/
OUF, ~ C
?
~ 50~ ~OO-C
e
AMENDED StlEE~
CA 02248964 1998-09-14
W O 97/33S88 PCT~US97/03348
-21-
~M~ B
R, R', UO ~ F,TE~RT R, R~, H~ o
- IbcNH>~ ~ ~OEt O ~ H~ ~OEI
¦¦ R~ Elhyl cl~te
O t R, C~IF,TE~, 0 ~C O ~. R,
R,R~, H o EDC-HCI. HOBT.
N~OH, H2~ ~N~H ,, H~ ~Oh DMF. TE.~ RT
CH~OH o ~1 ~ R~ ~ ~ F,TE~O~C
R, ~, H 1~l ~ R~ ~ NbOH, ~2~ ~ H O
o R ~ I ~ CH~OH BxNtH
R, R ~ ~ R, R', H O EDC-HCI HOBT, o
DMF, TEA, F~T
ElocNH ~ Y N ~ ~OEt ' H2N~
El~l .,1~ JFS OE~
o P~ R, .H ~ Rs ~ D~F. TEA. 0 ~C 1~ Rs
N-OH. 1~2~
C~
r
.
ff O P~ H O O EDC-HCI, HOBT,
D~ H2~ ~ W
o R, R, H O R', Rs R, R, DMF, TEA, 0 'C
-
SU~ 111 ~JTE SHEET (RULE 26)
~7-21(12463)A CA 02248964 1998'09-14
-22-
8C~ ~ E B ( Cont ' d)
N~OH.H~O
C~ ,OH
R~ R~i 0 3
o R~ o Rs Rs H O
~ C~Ct2 o~
O R~ H o Rr R ~ R~o
R~ R~ H It R ~ R~ H Cl DPi A i7uf. T~, R~--~HH Hi~l~o
oJ~O~, ~
THF
BH,,T~F
R',~ C ~, ~eC~ I P''~ ,~'~( H~ ~" .R,
Rs Rs ~ Rs Rs ~
~fE~ D S~f
; CA 02248964 1998-09-14
07-21(12463)A
-Z3 -
8r~F~M~ C
R~ NH2 H~N R~ V~ Ts~N N_~;
R~HR~ R" C~HsOH ~R$~
1. KCN. NH,~ B~CFlqC~
NH~OH, U~t ,
~~a ~~_QCl-l~ C~O_~O
~N R ~ H T~ s
H~N ~ a ~$~~
¦ H2. PK ~'2- Ha ~ QH
_2 H~
R~ R.R ~11 ~_QH HO_~
a H N NH~ C~
I CKXX~
Rb ~ R ~ t o
H~ NH2 ~ bas~ TRs _ N ~ ~5
P7 ~ R ~' ~ R~
T ~ N ~ S~ dmeor~t ~N
R~ ~4~R, --R~H~ ffR2
A~END~D St~EEr
, CA 02248964 l998-09-l4
07-21(12463)A
-24 -
8C~M~ D
Ts -N - ~L o - ~ ff~ ~ ~
R~TS~ ~ a~ RRt~ a
~/~, .
R ~ N ~ p~
s R~
'' \ LL~
FeC13
AMENGED SltEET
CA 02248964 1998-09-14
W O 97/33S88 PCTnUS97103348
25-
8C~EME E
NH2 TSC~ ~ ~NHTS (Boc)~O ~NHTS 8r ~ OCH
'NH2 ~ ~NH2 'NHBoc NaH,DMF
OCH~ ~ ~ ~ OH ~Rt2~N ~ OCH
'NHBoc 'NHBoc RS R's
o~N~JI~N~;~CH3 TFA ~N D~;;OCH3 BocR~N~OH
'NHBoc 'NH3~ CF3CC~'
ECC
R~ ~ ~CF~CO2 ~CCH~
EDC
aN~D~N~ocH~ HCUHOAC O~ ~H
~9 T97i ~ NR~3B~C R9 R'9~NH2Rt3-Ha
. . .
SU~ TE SHEET(RULE 26)
07-21(12463)A CA 02248964 1998-09-14
-Z6-
8C~E~e ~ (Co~'t.)
'
LIAIH" or BH~
c~_N~ ~ FeC13 ~N~N~
",, ,~- -N--~ MeOH ~ N >~
R~" R7 R~" R~
Rg R~g R9 R 9
Ah~END~D SHEET
CA 02248964 1998-09-14
W O 97/33S88 PCT~US97/03348
~C~EME F
T~S ~ ~! OE~ ~~N5 T~S ~ ~ ~
N OH IHCI-R~-HN ~ ~ NH3OC
NHBoc
TFA
Tls u~ R~o N D~ o~N~JI'
0~_ Cr Nff~oc NH2-TFA
N NltBoc
>'
KOH
O~ ~NR~ T'~F~o
NH ~' I Ts
1 Ts O~ N NH2-TFA
-o----N~NHBoc X
<_) \_/
~u~ 111 ~lTE SHEET tRULE 26)
~ CA 02248964 l998-09-l4
Q7-21(124~3)A
-28-
fiC~}~E F (Con't.
T~
T~ O R~ p ~ "N R", ~0
C~-- R~ DPPA, ~lH
~
o~ N NH2~rFA
--O
li~H" or BH,
C~ CH,OH C~
AMEhlE)ED S~E~
7 CA 02248964 1998-09-14
Q1--21 ( 12463 ) A
--29--
8CH ~ E G
O ~ R T ~ N~'~~'~NTs
~N ~H~ N-
LW~ O
R 0~ o . DM~:
T~ T~
Tsa ~o
Tsa / Et~N
R ~ c~a~
T~l~ NHT~ ~, Ts ~)
~/Dh~F.lOO~C
T~ T j
T~ ' T2
,
H~r c~ E~250~ ~
0}~
~ Y ~1 ~eO~ ~
AMENDED SHEF~
~ CA 02248964 l998-09-l4
O7-21(1~463)A
--30--
8 CHF~M ~
~ t2 H2N~
R ~ o~R2 ~ R7~N N~
P~ R, Rs
FeC13
~ec)H
R7~2
F~ Rs ~
5% Rh~C
. 1000 psi
. ~eC)H, 100~C
A~A~NG~D S~tEE~
~ CA 02248964 l998-09-l4
37-21(12463) A
--3 1--
8rFr~ I
~R, C~ 3
R R9 C~3C~
r~
R y
~2 P~2
CY.3Ci~ ~C)
R ~ ~ R7
R~ R
R LiAI~
eC
AMENDED SitE~
CA 02248964 1998-09-14
W 097133588 PCTAUS97/03348
The pentaazamacrocycles of the present invention
can possess one or more asymmetric car~on atoms and are
thus capable of existing in the form of optical isomers
as well as in the form of racemic or nonracemic mixtures
thereof. The optical isomers can be obtained by
resolution of the racemic mixtures according to
conventional processes, for example by formation of
diastereoisomeric salts by treatment with an optically
active acid. Examples of appropriate acids are
tartaric, diacetyltartaric, di~enzoyltartaric,
ditoluoyltartaric and camphorsulfonic acid and then
separation of the mixture of diastereoisomers by
crystallization followed by liberation of the optically
active bases from these salts. A different process for
separation of optical isomers involves the use of a
chiral chromatography column optimally chosen to
~;m; ze the separation of the enantiomers. Still
another available method involves synthesis of covalent
diastereoisomeric molecules by reacting one or more
secondary amine group(s) of the compounds of the
invention with an optically pure acid in an activated
form or an optically pure isocyanate. The synthesized
diastereoisomers can be separated by conventional means
such as chromatography, distillation, crystallization or
su~limation, and then hydrolyzed to deliver the
enantiomerically pure ligand. The optically active
compounds of the invention can likewise be obtained by
utilizing optically active starting materials, such as
~atural amino acids.
The compounds or complexes of the present
invention can be utilized to treat numerous inflammatory
disease states and disorders that are mediated, at least
in part, by superoxide. For example, reperfusion injury
to an ischemic organ, e.g., reperfusion injury to the
ischemic myocardium, surgically-induced ischemia,
inflammatory ~owel disease, rheumatoid arthritis,
rd~ SU~ ITF SHEET (RUI E 26)
CA 02248964 1998-09-14
PCT~US97/03348
W O 97/33588
osteoarthritis, psoriasis, organ transplant rejections,
radiation-induced injury, oxidant-induced tissue
in~uries and damage, athero5clerosis, thrombosis,
platelet aggregation, metastasis, stroke, acute
pancreatitis, insulin-dependent diabetes mellitus,
disseminated intravascular coagulation, fatty embolism,
adult and infantile respiratory distress, and
carcinogenesis.
Activity of the compounds or complexes of the
present invention for catalyzing the dismutation of
superoxide can be demonstrated using the stopped-flow
kinetic analysis technique as described in Riley, D.P.,
Rivers, W.J. and Weiss, R.H., I'Stopped-Flow Kinetic
Analysis for Monitoring Superoxide Decay in Aqueous
Systems," Anal. Biochem., 1~6, 344-349 (1991~, which is
incorporated by reference herein. Stopped-flow kinetic
analysis is an accurate and direct method for
quantitatively monitoring the decay rates of superoxide
in water. The stopped-~low kinetic analysis is suitable
for screening compounds for S0~ activity and activity of
the compounds or complexes of the present invention, as
shown by stopped-flow analysis, correlate to treating
the a~ove disease states and disorders.
Total dai}y dose administered to a host in single
or divided doses may be in amounts, for example, from
about 1 to about 100 mg/kg body weight daily and more
- usually about 3 to 30 mg/kg. Dosage unit compositions
may contain such amounts of submultiples thereof to ma~e
- up the daily dose.
The amount of active ingredient that may be
combined with the carrier materials to produce a single
dosage form will vary depending upon the host treated
and the particular mode of administration.
The dosage regimen for treating a disease
condition with the compounds and/or compositions of this
invention is selected in accordance with a variety of
SU~~ T~SHEET(RULE26)
CA 02248964 1998-09-14
W O 97/33588 PCT~US~7/03348
factors, including the type, age, weight, sex, diet and
medical sondition of the patient, the severity of the
disease, the route of administration, pharmacological
considerations such as the activity, efficacy,
pharmacokinetic and toxicology profiles of the
particular compound employed, whether a drug delivery
system is utilized and whether the compound is
~in;~tered as part of a drug combination. Thus, the
dosage regimen actually employed may vary widely and
therefore may deviate from the preferred dosage regimen
set ~orth above.
The compounds of the present invention may be
administered orally, parenterally, by in~lAtion spray,
rectally, or topically in dosage unit formulations
containing conventional nontoxic pharmaceutically
acceptable carriers, adjuvants, and vehicles as desired.
Topical ~m~istration may also involve the use of
transdermal a~; ni ctration such as trAn~ermal patches
or iontophoresis devices. The term parenteral as used
herein includes subcutaneous injections, intravenous,
intramuscular, intrasternal injection, or infusion
techniques.
Injectable preparations, for example, sterile
in~ectable aqueous or oleaginous suspensions may be
formulated according to the known art using suitable
dispersing or wetting agents and suspending agents. The
sterile injectable preparation may also be a sterile
injectable solution or suspension in a nontoxic
parenterally acceptable diluent or solvent, for example,
as a solution in 1,3-butanediol. Among the acceptable
vehicles and solvents that may ~e employed are water,
Ringer's solution, and isotonic sodium chloride
solution. In addition, sterile, fixed oils are
conventionally employed as a solvent or susp~n~
medium. For this purpose any ~land fixed oil may be
employed including synthetic mono- or diglycerides. In
SlJff~ ITE SHEET (RULE 26)
CA 02248964 1998-09-14
W O 97/33588 PCTnUS97/03348
,
.
addition, fatty acids such as oleic acid rind use in the
preparation of injectables.
suppositories for rectal administration of the
drug can be prepared by mixing the drug with a suitable
nonirritating excipient such as cocoa butter and
polyethylene glycols which are solid at ordinary
temperatures but liquid at the rectal temperature and
~ will therefore melt in the rectum and release the drug.
Solid dosage forms for oral ~inistration may
include capsules, tablets, pills, powders, granules and
gels. In such solid dosage forms, the active compound
may be admixed with at least one inert diluent such as
sucrose lactose or starch. Such dosage forms may also
comprise, as in normal practice, additional substances
other than inert diluents, e.g., lubricating agents such
as magnesium stearate. In the case of capsules,
tablets, and pills, the dosage forms may also comprise
buffering agents. Tablets and pills can additionally be
prepared with enteric coatings.
Liquid dosage forms for oral ~' ; ni ctration may
include pharmaceutically acceptable emulsions,
solutions, suspensions, syrups, and elixirs containing
inert diluents commonly used in the art, such as water.
Such compositions may also comprise ad~uvants, such as
wetting agents, emulsifying and suspending agents, and
sweetening, flavoring, and perfuming agents.
- While the compounds of the invention can be
a~;n;stered as the sole active pharmaceutical agent,
~ they can also be used in combination with one or more
compounds which are known to be effective against the
specific disease state that one is targeting for
treatment.
Contemplated equivalents of the general formulas
set forth above for the compounds and derivatives as
well as the intermediates are compounds otherwise
corresponding thereto and having the same general
S~ lUTFS~EET(RUEE26)
CA 02248964 1998-09-14
W O 97133588 PCTrUS97/03348
-36-
properties such as tautomers of the compo~nds and such
as wherein one or more of the various R groups are
simple variations of the substituents as defined
therein, e.g., wherein R is a higher alkyl group than
that indicated, or where the tosyl groups are other
nitrogen or oxygen protecting groups or wherein the
o-tosyl is a halide. Anions having a charge other than
1, e.g., carbonate, phosphate, and hydrogen phosphate,
can be used instead of anions having a charge of 1, so
long as they do not adversely affect the overall
activity of the complex. ~owever, using anions having a
charge other than 1 will result in a slight modification
of the general formula for the complex set forth above.
In addition, where a substituent is designated as, or
can be, a hydrogen, the exact chemical nature of a
substituent which is other than hydrogen at that
position, e.g., a hydrocarbyl radical or a halogen,
hydroxy, amino and the like functional group, is not
critical so long as it does not adversely affect the
overall activity and/or synthesis procedure. Further,
it is contemplated that iron (III) complexes will be
equivalent to the subject iron (III) comp~exes.
The chemical reactions described above are
generally disclosed in terms of their broadest
application to the preparation of the compounds of this
invention. Occasionally, the reactions may not be
applicable as described to each compound included within
the disclosed scope. The compounds for which this
occurs will be readily recognized by those skilled in
the art. In all such cases, either the reactions can be
successfully performed by conventional modifications
known to those skilled in the art, e.g., by appropriate
protection of interfering groups, by changing to
alternative conventional reagents, by routine
modification of reaction conditions, and the like, or
other reactions disclosed herein or otherwise
SU~IllUTESHEET(RULE26)
CA 02248964 1998-09-14
W O 97/33588 PCT~US97/03348
37-
conventional, will be applicable to the p~eparation of
the corresponding compounds of this invention. In all
preparative methods, all starting materials are known or
readily preparable from known starting materials.
Without further elaboration, it is believed that
one skilled in the art can, using the preceding
description, utilize the present invention to its
fullest extent. The following preferred specific
emh~i~cnts are, therefore, to be construed as merely
illustrative, and not limitative of the remainder of the
disclosure in any way whatsoever.
EXAMPLE8
All reagents were used as received without
purification unless otherwise indicated. All NMR
spectra were obtained on a Varian VXR-300 or VXR-400
nuclear magnetic resonance spe~LL~ ?ter. Qualitative
and quantitative mass spectroscopy was run on a Finnigan
MAT90, a Finnigan 4500 and a VG40-250T using m-
nitrobenzyl alcohol(NBA) or m-nitrobenzyl alcohol/LiCl
(NBA+Li). Melting points (mp) are uncorrected.
The following abbreviations relating to amino
acids and their protective g~oups are in accordance with
the recommendation by IUPAC-IUB Commission on
Biochemical Nomenclature (Biochemistry, 11, 1726 (}972))
and common usage.
SUBSTITUTE SHEET (RULE 26)
CA 02248964 1998-09-14
PCTrUS97/03348
W O 97/33~88
38-
Ala L-Alanine
DAla D-Alanine
Gly Glycine
ppg Propargylglycine
5 Tyr L-Tyrosine
Bzl Benzyl
Boc tert-Butoxycarbonyl
Et Ethyl
TFA Trifluoroacetate
10 DMF Dimethylformamide
HOBT-H20 1-Hydroxy-(lH)-benzotriazole
monohydrate
EDC-HCl 1-(3-Dimethylaminopropyl)-3-
ethylcarbodiimide
hydrochloride
TEA Triethylamine
DMSO Dimethylsulfoxide
THF Tetrahydrofuran
DPPA Diphenylphosphoryl azide
20 DMPU Dimethylpropyleneurea
c concentration, g/cc
DME 1,2-Dimethoxyethane
The abbreviation Cyc represents 1,2-
cyclohexan~ in~ (stereochemistry, i.e. R~R or SIS, is
indicated as such). This allows three letter code
peptide nomenclature to be used in pseudopeptides
containing the 1,2-cyclohexan~;~;ne "residue".
ExamplQ 1
A. Synthesis of 1 4.7-Tris(~-toluenesulfonvl)-1 4 7-
triazahe~tane
This compound was synthesized following the
procedure of Atkins, T. J.; Richman, J.E.; and
Oettle, w.F.; Org. Synth., 58, 86 - 98 (1978). To a
S~ TESHEET~RULE26~
CA 02248964 1998-09-14
W O 97133588 PCTrUS97/03348
.
39-
stirred solution of p-toluenesulfonyl chloride (618 g,
3.24 mole) in pyridine (1500 ml) at 0~C was added a
solution of 1,4,7-triazaheptane (95.5 g, 0.926 mole) in
pyridine (150 ml) under a dry argon atmosphere,
maintaining the temperature < 50~C. The addition
- required 30 minutes. After the mixture was allowed to
cool to room temperature slowly while stirring for 3 h,
- H2O(2 l) was slowly added to the cooled (ice bath)
mixture. The heavy white precipitate which formed was
filtered and washed thoroughly with H2O. The pale yellow
solid was dissolved in DMF (3 l) and 0.1 N HCl (4 l) was
slowly added at 5~C. The slurry was filtered and the
pale yellow solid was washed thoroughly with H2O and
dried in vacuo to give 486 g ~93% yield) of the product:
mp 180-1~C; lH NMR(DMSO-d6) ~ 2.39 (s,3 H), 2.40 (s, 6
H), 2.84 (m, 4 H), 3.04 (t, J-6.9 Hz, 4 H) 7.40 (d,
J=8.1 Hz, 4 H), 7.59 (d, J=8.3 Hz, 2 H), 7.67 (m, 6 H).
B. Svnthesis of 1 4.7-~risf~-toluenesulfonyl)-1 4 7-
triazaheptane-1 7-disodium Salt
This compound was synthesized following the
procedure of Atkins, T.J.; Richman, J.E., and Oettle,
W.F.; Org. Synth., 58 86-98 (1978). To a mechanically
stirred slurry of 1,4,7-tris(p-toluenesulfonyl)-1,4,7-
triazaheptane prepared as in Example lA (486 g, 0.859mole) in ethanol (1150 ml) heated to reflux under a dry
- argon atmosphere was added a solution of sodium ethoxide
(prepared by dissolving sodium metal (39.5 g, 1.72 mole)
- in absolute ethanol (1.0 1)) as rapidly as possible.
The clear brown solution which formed rapidly was
allowed to cool to room temperature and ethyl ether (1.0
l) was added. The crystals were filtered under a dry
argon blanket, washed with 3:1 ethanol:ethyl ether and
ethyl ether. The crystals were then dried in vacuo to
give 509 g (97% yield) of the product as a white powder:
H NMR (DMSO-d6) ~ 2.30 (s 6 H~, 2.36 (s, 3 H), 2.63 (t,
SU~:i 111 ~ITF SHEET (RULE 26)
CA 02248964 1998-09-14
W O 97/33588 PCT~US97/03348
40-
J=8.7 Hz, 4 H), 2.89 (t, J=7.2 Hz, 4 H) 7.11 (d, J=8.1
Hz, 4 H), 7.28 (d, ~=8.0 Hz, 2 H), 7.46 (m, 6 H).
C. SYnthesis of 3 6-Bis~-toluenesulfonYl)-3,6-
diazaoctane-1 8-di-P-toluenesulfonate
To a stirred solution of p-toluenesulfonyl
chloride (566 g, 2.97 mole) and triethylamine (300 g,
2.97 mole) in CH2Cl2 (2.0 l) at 0~C under a dry argon
atmosphere was added 3,6-diazaoctane-1,8-diol (100 g,
0.675 mole) in portions, maintaining the temperature
<10~C. The addition required 30 minutes. The mixture
was allowed to warm to room temperature while stirring
an additional 18 h and was then poured onto ice (1000
g). The CH2Cl2 layer was separated, washed with 10% ~Cl,
H20 and saturated NaCl solution, and dried (MgS04). The
solution was concentrated in vacuo to a volume of 1.5 l.
Crystallization by the addition of h~Y~ne (4 l) gave 477
g (92% yield) of the product as colorless needles: mp
151-3~C; lH NMR (CDCl3) 8 2.43 (s, 12 H), 3.29 (s, 4 H),
3.36 (t, J=5.2 Hz, 4 H) 4.14 (t, J=5.2 Hz, 4 H), 7.33
(d, J=7.8 Hz, 8 H), 7.71 (d, J=8.2 Hz, 4 H), 7 79 (d,
J=8.3 Hz, 4 H).
D. Synthesis of 1.~.7.10 13-Penta(p-toluenesulfonyl)-
l.4.7.10 13-~entaazacYclopentadecane
This compound was synthesized following the
procedure of Richman, J.E., and Atkins, T.J., ~. Am.
Chem. Soc., 96, 2268-70 (1974). To a stirred solution
of 1,4,7-tris(p-toluenesUlfonyl)-1,4,7-triazaheptane-
1,7-disodium salt prepared as in Example lB (146 g,
0.240 mole) in anhydrous DMF (2250 ml) was added
dropwise over 3 h to a solution of 3,6-~is(p-toluene-
sulfonyl)-3,6-diazaoctane-1,8-di-p-toluenesulfonate
prepared as in Example lC (184 g, 0.240 mole) in
anhydrous DMF (1020 ml) under a dry argon atmosphere,
maintaining the temperature at 100~C. After stirring an
SlJ~ IT~ SHEET (RULE 26)
CA 02248964 l998-09-l4
W O 97/33588 PCTrUS97/03348
41-
additional 1 h at 100~C, the solution was concentrated
in vacuo to a volume o~ 1.5 l. H2O (500 ml) was slowly
added at 80OC to crystallize the product. The resulting
slurry was slowly cooled to 0~C and additional H2O (1250
ml) added. The solid was filtered, washed thoroughly
- with ~2~ and then 90% ethanol and dried in vacuo. The
off-white solid was dissolved in CH2Cl2, insoluble
impurities were removed by filtration and the filtrate
was washed with H2O and then dried (MgSO4). The solvent
was removed in vaCuo to give a yellow solid which was
purified by recrystallization from CH2Cl2-hexane to give
164 g (69% yield) of the product as a white crystalline
solid: mp 290-3~C; lH NMR (CDCl3) ~ 2.44 (s, 15 H~ 3.27
(s, 20 H), 7.32 (d, ~=8.3 ~z, 10 H), 7.66 (d, J=8.3 Hz,
10 H).
E. SYnthesis of 1,4,7 10.13-Pentaazac~clo~entadecane
A mixture of 1,4,7,10,13-penta(p-
toluenesulfonyl3-1,4,7,10,13-pentaazacyclopentadecane
prepared as in Example lD (168 g, 0.170 mole) and
concentrated H2SO4 (500 ml) was heated at 100~C with
stirring under a dry argon atmosphere for 70 h. To the
resulting dark brown solution ethanol (500 ml) was added
dropwise with stirring at 0~C followed by ethyl ether
(3 l). The white solid was filtered and washed with
ethyl ether. The solid was then dissolved in H2O (500
~ ml) and the resulting solution washed with ethyl ether.
Upon reducing the volume of the solution in vacuo to 200
ml, the p~ was adjusted to 10-11 with 10 N NaOH and the
solvent was removed in vacuo. Ethanol (500 ml) was then
added and removed in vacuo to dryness. The resulting
tan oily solid was extracted with hot THF (2x500 ml) and
filtered at room temperature. The filtrates were
combined and the solvent removed in vacuo to give the
crude product as a yellow crystalline solid which was
SUBSTITUTESHEET(RULE26)
CA 02248964 1998-09-14
W O 97/33S88 PCT~US97/03348
42-
then redissolved in CH3CN and filtered to remove
insoluble impurities. Recrystallization from cold (-
20~C) CH3CN gave 11.3 g (31% yield) of the produc~ as
colorless needles: mp 108-9~C; lH NMR (CDC13) ~ 1.74 (br
s, 5 H), 2.73 (s, 20 H); Exact mass (M+~i)+: calcd,
222.2270; Found, 222.2269 tC~oH2sN5Li)~
~. SYnthesis of rIron(III)dichloro(1,4,7,10,13-
PentaazacycloPentadecane~1hexafluorophos~hate
Upon an inert atmosphere in a drybox, 108 mg
(0.50 mmol) of the ligand, 1,4,7,10,13-
tetraazacyclopentadecane, was dissolved in 15 ml of
anhydrous methanol. To this solution was added with
vigorous stirring 2 ml of a pyridine solution containing
0.50 mmol (80 mg) of anhydrous FeCl3. The resultant dark
solution was heated to reflux for two hours with
stirring and then allowed to cool to room temperature
and then filtered. To the filtrate was added 20 ml of a
clear methanolic solution of NH4PF6 (163) mg). A yellow
precipitate formed instantly and was collected by
filtration, washed with diethyl ether and dried in vacuo
overnight. The yield after drying was 170 mg (0.338
mmol) corresponding to a 68% theoretical yield. Anal.
Calc. for C1OH20NsCl2FeF6P CH30H: C, 25.07; H, 5.41: N,
13.92. Found: C, 25.18; H, 5.60; N, 13.89. Mass
spectrum (~AB, NBA matrix): m~z 3Q6 (~Fe(L)Cl+e]~ and m/z
341 ( r Fe(L)Cl2J+ were o~served.
Ex~mpl~ 2
A; Synthesis of N~ toluenesulfonyl)-(R,R)-1,2-
diaminocyclohexane
To a stirred solution of (R,R)-1,2-
~;A~;nocycl~h~Ane (300 g, 2.63 mole) in CH2Cl2 ~5.00 1)
at -10 C was added a solu~ion of
p-toluenesulfonylchloride (209 g, 1.10 mole) in CH2Clz
(5.00 1) dropwise over a 7 h period, maintaining the
S~ TESHEET~RULE26)
CA 02248964 l99X-09-14
PCTrUS97/03348
W O 97/3358~ _
-43-
.
temp at -5 to -10 C. The mixture was allowed to warm to
room temp while stirring overnight. The mixture was
c~ncentrated in vacuo to a volume of 3 1 and the white
solid was removed by filtration. The solution was then
washed with H20 (10 x 1 1) and was dried over MgSO4.
Removal o~ the solvent in vacuo gave 286 g (97.5% yield)
of the product as a yellow crystalline solid: lH NMR
(CDCl3) ~ 0.98 - 1.27 (m, 4 H), 1.54 - 1.66 (m, 2 H),
1.81 - 1.93 (m, 2 H), 2.34 (dt, J = 4.0, 10.7 Hz, 1 H),
2.42 ( s, 3 H), 2.62 (dt, J = 4.2, 9.9 Hz, 1 H), 7.29
(d, J = 8.1 Hz, 2 H), 7.77 (d, J = 8.3 Hz, 2 H); MS
(LRFAB - DTT - DTE) m/z 269 [M + H~+.
B. Svnthesis of N-(~-toluenesulfonYl)-N -(Boc)-(R R)-
1 2-diaminocyclohexane
To a stirred solution of N-(p-toluenesulfonyl)-
(R,R)--1,2--~1; A -~nocycl~he~rAnF~ prepared as in Example 2A
(2S6 g, 0.955 mole) in THF (1.15 1) was added a 1 N
solution of aqueous NaOH (1.15 1, 1.15 mole). Di-t-
butyldicarbonate (229 g, 1.05 mole) was then added andthe resulting mixture was stirred overnight. The layers
were separated and the a~ueous layer was adjusted to pH
2 with 1 N HCl and saturated with NaCl. The aqueous
solution was then extracted with CH2Cl2 (2 x 500 ml) and
the extracts and THF layer were combined and dried over
MgSO4. The solvent was removed in vacuo to give a yellow
solid. The crude product was purified by
crystallization ~rom a THF-ether-hexanes mixture to give
310 g (88.1% yield) of the product as a white
crystalline solid: mp:
137 - 139 C; lH NMR (CDCl3) ~ 1.04 - 1.28 (m, 4 H), 1.44
(s, 9 H), 1.61 - 1.69 (m, 2 H), 1.94 - 2.01 (m, 2 H),
2.43 (s, 3 H), 2.86 (brs, 1 H), 3.30 (br d, J = 9.6 Hz,
~ 1 H), 4.37 (br d, J = 6.7 Hz, 1 H), 5.48 (br d, J = 4.6
Hz, 1 H), 7.27 (d, J = 9.7 Hz, 2 H), 7.73 (d, J = 8.1
SUBSTITUTE SHErT (RULE 26)
CA 02248964 1998-09-14
W O 97/33588 PCTrUS97/03348
-44-
Hz, 2 H); MS (LRFAB, NBA - Li) m/z 375 [M + Li3+.
C. Svnthesis of Boc-(R.R)-CvctTs)-qly-OMe
To a stirred solution of N-(p-toluenesulfonyl)-
N -(soc)-(R~R)-l~2-~ nocyclohexane prepared as in
Example 2B (310 g, O.841 mole) in anhydrous DMF (3.11 l)
at O C was added NaH (37.4 g - 60 % in oil, 0.934 mole)
in portions and the resulting mixture was stirred for 30
min. Methyl bromoacetate (142 g, 0.925 mole) was then
added dropwise over 45 min and the mixture was allowed
to warm to room temp while stirring overnight. After
stirring for 17 h, the solvent was removed in vac~o and
the residue was dissolved in ethyl acetate(3 l) and H20
(1 l). The ethyl acetate solution was washed with
saturated NaHCO3 (1 l), saturated NaCl (500 ml) and was
dried over MgSO4. The solvent was re~v~d in v~cuo and
the resulting oil was dissolved in ether.
Crystallization by the addition of h~nes gave 364 g
(98 % yield~ of the product (TLC (98:2 C~Cl3-MeOH~silica
gel/W detn) showed that the product contained about 5
starting material) as colorless needles: mp o~ pure
sample 151 - 2 C , 1K N ~ (COCl3~ S 1.11 - 1.22 (m, 4 H),
1.45 (s, 9 ~), 1.64 - 1.70 (m, 3 H), 2.16 - 2.19 (m, 1
H), 2.43 (s, 3 H), 3.34 - 3.40 (m, 2 H), 3.~8 (s, 3 H),
25 4.06 (A8q, J - 18.5 Hz, ~ = 155 Hz, 2H), 4.77 (br s 1
H), 7.30 (d, J = 8.3 Hz, 2 H), 7.82 (d, J - 8.3 Hz, 2
H); MS (~RFAB, DTT - DTE~ m/z 441 ~M + H]+.
D. Synthesis of Boc-(R.R)-Cvc(Tsl-Gl~-OH
To a stirred solution of impure Boc-(R,R)-
Cyc(Ts)-Gly-OMe prepared as in Example 2C (217 g, 0.4g2
mole) in MeO~ (1.05 l) was slowly added a 2.5N solution
of aqueous NaOH (295 ml, 0.737 mole) and the resulting
solution was stirred for 2 h The solvent was removed
in vacuo and the residue was dissolved in H20 (1.5 l).
S~ TESHEET(RULE26)
CA 02248964 1998-09-14
W O 97/33588 PCT~US97/03348
-45-
The solution was filtered to remove a small amount of
solid and was washed with ether t7 x 1 1~ to remove the
impurity (compound lB) which upon drying of the combined
washes over MgS04 and removal of the solvent in vacuo
resulted in recovery of 8.37 g. The pH of the aqueous
~ solution was then adjusted to 2 with 1 N HCl and the
product was extracted with ethyl acetate (3 x 1 1).
- The extracts were combined, washed with saturated NaCl
(500 ml) and dried over MgSO4. The 501vent was removed
in vacuo and the residual ethyl acetate removed by
coevaporation with ether (500 ml) and then CH2Cl2 (500
ml) to give 205 g (97.6% yield) of the product as a
white foam: 'H NMR (CDC13) ~ 1.15 -- 1.22 (m, 4 H), 1.48
(s, 9 H), 1.55 - 1.68 (m, 3 H), 2.12 - 2.15 (m, 1 H),
2.43 (s, 3 H), 3.41 -- 3.49 (m, Z H), 3.97 (ABq, J = 17.9
Hz, ~ = 69.6 Hz, 2 H), 4.79 (br s, 1 H), 7.31 (d, J =
8.1 Hz, 2 H), 7.77 (d, J = 8.3 Hz, 2 H), 8.81 (br s,
1 H); MS (LRFAB, NBA -- Li) m/z 433 [M ~ Li]+.
20 E. S~nthesis of N-(p-toluenesulfonyl)--(S S)--1 2--
diaminoc~,rclohexane
To a stirred solution o~ (S,S)--1,2-
~inocyclohexane (300 g, 2.63 mole) in CH2C12 (5.00 1)
at -10 C was added a solution of
p-toluenesulfonylchloride (209 g, 1.10 mole) in CH2C12
(5.00 1) dropwise over a 8 h period, maintaining the
- temp at --5to -10 C. The mixture was allowed to warm to
R~r while stirring overnight. The mixture was
- concentrated in vacuo to a volume of 3 1 and the white
solid was removed by filtration. The solution was then
washed with H20 (10 x 1 1) and was dried over MgS04.
Removal of the solvent in vacuo gave 289 g (98.3~ yield)
of the product as a yellow crystalline solid: lH N~
(C~Cl3) ~ 0.98 - 1.27 (m, 4 H), 1.55 - 1.66 (m, 2 H),
1.81 -- 1.94 (m, 2 H), 2.32 (dt, ;1 = 4.0, 10.9 Hz, 1 H),
S~ lT~: SHEET(RUIE 26)
CA 02248964 1998-09-14
PCTAUS97/03348
W O 97t33588
-46-
2.42 (s, 3 H), 2.61 ~dt, J - 4.0, 9.9 ~z, 1 H), 7.30 (d,
J = 7.9 Hz, 2 H), 7.77 (d, J = 8.3 Hz, 2 H); MS
(LRFAB,GT - HCl) m/z 269 ~M + H]'.
F. SYnthesis of N-(~-toluenesulfonYl)-N -(Boc)-(S S)-
1 2-diaminoc~clohexane
To a stirred solution of N-(p-toluenesulfonyl)-
(S,S)-1,2-diaminocyclohexane prepared as in Example 2E
(289 g, 1.08 mole) in THF (1.29 l) was added a 1 N
solution o~ aqueous NaOH (1.29 l, 1.29 mole~. Di-t-
butyldicarbonate (258 g, 1.18 mole) was then added and
the resulting mixture was stirred overnight. The solid
was removed ~y filtration and washed with THF . The THF
layer was separated and the aqueous layer was ad~usted
to pH 2 with l N HCl and saturated with NaCl. The
a~ueous solution was then extracted with CH2Cl2 (2 x 500
ml) and the extracts and THF layer were com~ined, washed
with saturated NaCl (500 ml) and dried over MgSO4. The
solvent was removed in vacuo to give a yellow slurry.
Crystallization with the addition of ether gave 364 g
(91.9% yield) of the product as colorless needles: mp
137 - 139 C; lH NMR (CDCl3) ~ 1.06 - 1.31 (m, 4 H), 1.44
(s, 9 H), 1.60 - 1.69 (m, 2 H), 1.95 - 1.99 (m, 2 H),
2.42 (s, 3 H), 2.86 (br s, 1 H), 3.30 (br d, J = 2.6 Hz,
25 l H), 4.41 (br d, J = 7.3 Hz, l H), 5.54 (br d, J = 5.4
Hz, 1 H), 7.28 (d, ~ = 8.1 Hz, 2 ~), 7.73 (d, J = 8.3
Hz, 2 H); MS (LRF~B, NBA - HCl) m/z 369 rM + H]'.
G. Synt~esis of Boc-(S S)-Cvc~Ts)-~ly-OMe
To a stirred solution of N-(p-toluenesulfonyl)-
N -(Boc)-(srs)-l~2-~ nocycloh~yAne prepared as in
Example 2F (364 g, 0.989 mole) in anhydrous DMF (3.66 l)
at O C was added NaH (47.4 g - 60~ in oil, 1.19 mole) in
- portions and the resulting mixture was stirred for 1.5
h. The mixture was warmed to room temp and stirred an
additional 30 min and then cooled back to O C. Methyl
S~ U~E5HEET(RULE26)
CA 02248964 1998-09-14
W O 97/33S88 PCTnUS97/03348
47-
bromoacetate (189 g, 1.24 mole) was added dropwise over
30 min and the mixture was allowed to warm to RT while
stirring overnight. A~ter stirring for 17 h, the
solvent was removed in vacuo and the residue was
dissolved in a mixture of ethyl acetate(3 1) and H2O
(1 1). The layers were separated and the ethyl acetate
solution was washed with saturated NaHCO3 (1 1), H2O (1
- 1), saturated NaCl (2 x 500 ml) and was dried over MgSO4.
The solvent was removed in vacuo and the resu~ting oil
was dissolved in ether. Crystallization by the addition
of hexanes gave 290 g of the crude product as yellow
needles. Another 180 g was recovered from the filtrate
as an oil. TLC (98:2 CHCl3-MeOH/silica gel/ W detn)
showed that both the solid and the oil contained
starting material. ~H NMR (CDCl3) ~ 1.06 - 1.29 (m, 4
H), 1.44 (s, 9 H), l.S8 - 1.66 (m, 3 H), 2.17 - 2.19 (m,
1 H), 2.43 (s, 3 H), 3.28 - 3.43 (m, 2 H), 3.68 (s, 3
H), 4.25 (ABq, J = 18.5 Hz, ~ - 115 Hz, 2H), 4.76 (br
s 1 H), 7.31 (d, J = 8.3 Hz, 2 H), 7.83 (d, J = 8.3 Hz,
2 H); MS (LRFAB, NBA - Li) m/z 447 [M + H]+.
H. Svnthesis of Boc-fS S)-Cyc(Ts)-Gly-OH
To a stirred solution of impure Boc-(S,S)-
Cyc(Ts)-Gly-OMe prepared as in Example 2G (197 g, 0.447
25 mole) in MeOH ~925 ml) was 510wly added a 2.5N solution
of aqueous NaOH (268 ml, 0.670 mole) and the resulting
- solution was stirred for 2 h. The solvent was removed
in vacuo and the residue was dissolved in H2O (1 1). The
solution was washed with ether (4 x 1 1) to remove the
impurity (compound 2F) which upon drying of the combined
washes over MgSO4 and removal of the solvent in vacuo
resulted in recovery of 14.8 g. The pH of the a~ueous
solution was then adjusted to 2 with 1 N HCl and the
product was extracted with ethyl acetate (3 x 1 1).
The extracts were combined, washed with saturated NaCl
S~ TESHEET(RUEE26)
CA 02248964 1998-09-14
WO 97133588 PCTrUSs7/03348
-48-
and dried over MgSO4. The 501vent was removed in vacuo
to give 171 g (89.7~ yield) of the product as an oil
which crystallized on standing: IH NMR (CDCl3) ~ 1.10
-- 1.22 (m, 4 H), 1.45 (s, 9 H), 1.55 -- 1.68 (m, 3 H),
2.13 - 2.16 (mr 1 H), 2.43 (s, 3 H), 3.39 - 3.41 (m, 2
H), 4.00 (ABq, J = 18.1 Hz, ~ = 80.4 Hz, 2 H), 4.82
(br s, 1 H), 7.31 (d, J = 8.3 Hz, 2 H), 7.75 (d, J =
8.3 Hz, 2 H), 9.28 (br s, 1 H); MS (LRFAB, NBA - Li) m/z
433 ~M + Li~+.
I. Synthesis of ~oc-(S~S)-Cyc(Ts~-Gly-Gly-OEt
To a stirred solution of Boc-~S,S)-Cyc(Ts)-Gly-
OH prepared as in Example 2H (26.7 g, 62.5 mmole) in
degassed anhydrous DMF (690 ml) was added HOBT (10.1 g,
75.0 mmole) and EDC-HCl (14.4 g, 75.0 mmole). After the
resulting solution was stirred for 30 min, glycine ethyl
ester hydrochloride (9.60 g, 68.8 mmole) was added and
the pH adjusted to 8 with TEA. After stirring for 2.75
days the solvent was ~e~aved in vacuo. The residue was
dissolved in a mixture of ethyl acetate ~1 1) and H2O (1
1) and the layers were separated. ~he aqueous layer was
extracted with ethyl acetate (1 1) and the extracts were
combine. The ethyl acetate solution was washed with 0.1
N HCl (1 1), saturated NaHCO3 (1 1), saturated NaCl (500
ml) and was dried over MgSO4. The solvent was removed in
vacuo to give 30.2 g (94.4% yield) of the product as a
white foam: lH NMR (CDCl3) ~ 1.19 - 1.23 (m, 3 H), 1.28
~t, J =7.05 Hz, 3 H), 1.42 (s, 11 H), 1.63 - 1.71 (m, 2
H), 2.16 - 2.18 (m, 1 H), 2.43 (s, 3 H), 3.50 - 3.57 (m,
2 H), 3.83 (ABq, J = 17.7 Hz, delta v = 35.7 Hz, 2 H),
4.01 (dAB~, J = 6.05, 17.92 Hz, ~ = 28.9 Hz, 2 H),
4.20 (q, ~ = 7.3 Hz, 2 H), 4.88 (br s, 1 H), 7.31 (d, J
~ 8.3 Hz, 2 H), 7.36 (br s, 1 H), 7.73 (d, ~ = 8.3 Hz, 2
H); MS (LRFAB, NBA - HCl) m/z 512 [M + H~+.
S~ lUTFSHLET ~RULE ~)
CA 02248964 1998-09-14
W O 97/33588 PCT~US97/03348
-49-
J. Svnthesis of (S S)-Cyc(Ts)-Gl~-Gly-OEt TFA salt
To a stirred solution of Boc-(S,S)-Cyc(Ts)-Gly-
Gly-OEt prepared as in Example 2I (30.1 g, 58.8 mmole)
in CH2Cl2 (265 ml) was added TFA (63 ml) and the
resulting solution was stirred for 30 minutes. The
- solvent was removed in vacuo and residual TFA was
coevaporated with CH2Cl2 (2 x 1 l) and ether (l 1). The
- oil was then triturated with ether (2 x 1 1) and the
ether decanted. The resulting foam was dried in vacuo
to give 33.7 g (assumed quantitative yield) of the
product as a tan powder: lH NMR (CDCl3) ~ 0.96 - 1.23 (m,
4 H), 1.25 (t, J = 7.3 Hz, 3 H), 1.51 - 1.66 (m, 3 H),
2.12 - 2.26 (m, 1 H), 2.41 (s, 3 H), 2.98 - 3.10 (brs, 1
H), 3.67 - 3.71 (m, 1 H), 4.04 (ABq, J = 17.7 Hz, ~ u =
154 Hz, 2 H), 4.04 (d, J = 4.4 HZ, 2 H), 4.17 (q, J =
7.3 Hz, 2 H), 7.29 (d, J = 8.3 Hz, 2 H), 7.70 (d, J =
8.3 Hz, 2 H), 8.04 (br s, 1 H), 8.14 (br s, 3 H) MS
= (LRFAB, NBA - HCl) m/z 412 [M + H~+.
K. Synthesis of Boc-fR R3-Cvc(Ts)-Gl~-(S S)-Cyc(Ts)-
&l~-Glv-OEt
To a stirred solution of Boc-(R,R)-Cyc(Ts)-Gly-OH
prepared as in Example 2D (25.1 g, 58.8 mmole) in
degassed anhydrous DMF (650 ml) was added HOBT (9.54 g,
70.6 mmole) and EDC-HCl (13.5 g, 70.6 mmole). After the
resulting solution was stirred for 30 min (S,S)-Cyc(Ts)-
- Gly-Gly-OEt TFA salt prepared as in Example lJ (33.6 g,
58.8 mmole) was added and the pH was adjusted to 8 with
TEA. After stirring ~or 2.75 days, the solvent was
removed in vacuo. The residue was dissolved in a
mixture of ethyl acetate (1 l) and H2O (1 l) and the
layers were separated. The ethyl acetate solution was
washed with 0.1 N HCl (2 x 1 l), saturated NaHCO3 (2 x 1
l), saturated NaCl (500 ml) and was dried over MgSO~.
The solvent was removed in vacuo to give 47.5 g (98.4%
SUtsS 111 ~)TE SHEET (RULE 26)
CA 02248964 1998-09-14
W O 97133S88 PCTnUS97103348
-50-
-
yield~ o~ the product as a tan foam: IH NMR (CDCl3) ~
1.12 - 1.83 (m, 26 H), 2.21 - 2.24 ~m, 2 H1, 2.42 (s, 3
H), 2.43 (g, 3 H), 3.36 - 3.51 (br s, 2 H), 3.68 -3.96
tm, 6 H), 4.00 (d, J = 5.4 Hz, 2 H), 4.19 (q, J = 7.1
Hz, 2 H), 4.72 (br s, 1 H), 6.78 (br s, 1 H), 7.31 (d, J
- 8.1 Hz, 4 H), 7.46 (br s, 1 H), 7.79 (m, 4 H); MS
(LRFAB, NBA - ~Cl) m/z 820 [~ + H~+.
~. Svnthesis of Boc-~R R~-Cvcf TS) - G1Y-(S,S) - CYC(TS) -
10 G1Y--G1Y--OH
To a stirred solution of Boc-(R,R)-Cyc(~s)-Gly-
~S,S)-Cyc(Ts)-Gly-Gly-OEt prepared as in Example 2K
(47.4 g, 57.8 mmole) in MeOH (240 ml) was added a 2.5 N
solution of aqueous NaOH (34.7 ml, 86.7 mmole) and the
resulting solution was stirred for 2 h. The solvent was
Le~d in Yacuo and the residue was dissolved in H20
(1 1). The a~ueous solution was washed with ether
(2 x 1 l) and the pH was adiusted to 2 with 1 N HCl.
The solution was then saturated with NaCl and extracted
with ethyl acetate (3.x 1 l). The combined extracts
were dried over MgSO4 and the solvent was removed in
~acuo. The residual ethyl acetate was removed by
coevaporation with C~2Cl2 and the resulting foam was
dried in vacuo to give 4S.7 g (g9.7% yield) o~ the
product as a tan powder: IH NMR (CDCl3) 8 1.16 - 1.75 (m,
23 H), 2.~3 - 2.17 (m, 2 H), 2.41 (s, 3 H), 2.42 (s, 3
H), 3.49 - 4.16 (m, lO H), 4.53 (br s, 1 H), 7.01 (br s,
H), 7.30 ~d, J = 8.1 Hz, 4 H), 7.40 (br s, 1 ~), 7.79
~d, ~ - 8.1 Hz, 2 H), 7.86 (d, J = 7.7 Hz, 2 H), 10.40
(br s, 1 H); MS (LRFAB, NBA - HCl) m/z 792 ~M + H]'.
M . Svnthesis of (R.R~-cYc (TS ) -G1Y - ( S . S ) -CYC tTs)-Gly-
G1Y - OH TFA salt
To a stirred solution of Boc-(R,R)-Cyc(Ts)-Gly-
(S,S)-Cyc(Ts)-Gly-Gly-OH prepared as in Example 2L (45.5
-
SU~~ T~ SffEET(RUL~ 26)
CA 02248964 1998-09-14
W O 97/33588 PCT~US97/03348
51-
g, 57. 5 mmole) in CH2C12 (260 ml) was adde~ TFA ~60 ml).
The resulting solution was stirred for 30 min and the
solvent was removed in vacuo. Residual TFA was removed
by coevaporation with CH2Cl2 (3 x 1 1) and trituration of
the resulting foam with ether (1 1, 2 X 750 ml),
decanting the ether each time. ~fter desiccation in
vacr~o, 47. 4 g (100% yield) of the product was obtained
as an off white powder: lH NMR (CDCl3) S 1.05 -- 1.31 (m,
9 H), 1.48 - 1.63 (m, 5H), 2.11 - 2.21 (m, 2 H), 2.40
(s, 3 H), 2.42 (s, 3 H), 3.25 (br s, 1 H), 3.60 - 3.80
(m, 3 H), 3.83 - 4.19 (m, 6 H), 6.94 (br s, 1 H), 7.31
(m, 4 H), 7.69 (m, 4 H), 7.83 (br s, 3 H), 13.17 (br s,
2 H3; MS (LRFAB, DTT - DTE) m/z 692 [M + H]+.
N. S~rnthesis of CYClo-rtR R)-Cyc(Ts)--GlY-~S S)-Cyc(Ts)--
Gl'.t--Gly--~
To a stirred solution of (R,R) - Cyc(Ts) - Gly - (S,S) -
Cyc (Ts) - Gly-Gly-OH TFA salt prepared as in ~xample 2M
(32.2 g, 40.0 mmole) in degassed anhydrous DMF (10.0 1)
at -78 C was added DPPA (13 4 g, 48.8 mmole). The pH of
the solution was then adjusted to 8 with TEA and t~e
solution was allowed to stand for 6 h at -78 C. The pH
was readjusted to 8 with TEA and the solution was warmed
to -45 C for 24 h. After readjusting the pH as before,
the solution was allowed to warm to -40 C for 24 h. The
pH was adjusted as before and the solution was allowed
- to stand at -20 C for 24 h. The pH was readjusted as
before and the solution was allowed to warm to 2 C over
2-4 h. The pH had dropped only slightly. The pH was
readjusted as before and the solution was allowed to
stand at 2 C for another 24 h after which time the pH
had not changed. The solution was divided equally among
6 - 4 1 beakers and H20 ( 1. 1 1 ) was added to each. Then
added a total of 5.00 kg mixed-bed ion ~Y~-h~nge resin to
the solution (divided equally among the 6 beakers) and
stirred the mixtures for 6 h. The resin was then
S~ TESHEET(RULF26)
CA 02248964 1998-09-14
PCT/US97/0334
W097I33~8
-52-
filtered and wa5hed with DMF. The solvent was then
removed in vacuo and the solid residue was dissolved in
MeOH (100 ml) and filtered to remove finely divided
solids. The solution was then concentrated in vacuo to a
volume of 25 ml and ether was added periodically as the
crystallization proceeded to give 22.2 g (82.5 % yield)
of the product as colorless needles; mp 190 - 200 C; IH
NM~ (CDCl3) ~ 0.87 - 2.13 (m, 16 H), 2.41 (s, 3 H), 2.45
(s, 3 H), 3.56 - 3.97 (m, 10 H), 6.66 (br s, 1 H), 7.18
(br s, 1 H~, 7.34 (d, J 5 8.1 Hz, 4 H), 7.65 (br s, 1
H), 7.71 (d, J - 7.3 Hz, 2 H), 7.89 (d, J - 7.3 Hz, 2
H); MS tLRFAB, NBA - Li) m/z 680 tM ~ Li~'.
O. S~nt~esis of 2,3-(R,R)-8,9-(S,S)-Bis-cYclohexano-
1,4,7,10,13-~entaazacYClo~entadecane
To a stirred solution of Cyclo-[(R,R)-Cyc(Ts)-
Gly-(S,S)-Cyc(Ts)-Gly-Gly~ prepared as in Example 2N
(19.4 g, 28.8 mmole) in anhydrous THF (475 ml) was added
a solution of 1.0 M riAl~4 in THF (345 ml, 345 mmole)
dropwise over 30 min. The yellow homogeneous solution
was refluxed for 20 h (by which time it had become
heterogeneous) and was then cooled to 0 C. The mixture
was then quenched by the dropwise addition of a 1~% NaSO4
solution (~0 ml) while cooling in an ice bath. The
2~ solids were removed by filtration under an Ar blanket
and the THF was removed in vacuo to give an oil which
rapidly crystallized. The solids were then refluxed
with anhydrous THF (1 l) for 1 h and the mixture was
filtered and the solvent removed in vacuo as before.
The solids were then refluxed with a mixture of THF
(} l) and MeOH (500 ml) for 1 h and worked up as before.
The residues from the extractions were then dissolved in
- anhydrous THF, combined and solids were removed by
filtration. The solvent was removed in vacuo and the
yellow foam dried by azeotroping H2O with toluene
SUBSTITUTES~EET(RULE26)
CA 02248964 1998-09-14
W O 97/33588 PCTrUS97/03348
53-
.
(1.75 l) in vacuo at 90 C. Then refluxed the solids
with hexanes (1 l) for 30 min and transferred the hot
solution to a tared flask and removed the solvent in
vacuo to give 6.1 g of an oil which crystallized on
S standing. The remaining solids were refluxed with
hexanes as before and obtained 1.4 g of an oil which
crystallized on standing. The solids were then
dissolved in MeOH and toluene (1 l) was added. The
solvent was removed in vacuo and any remaining R2O was
removed by azeotroping with toluene (1 l) and then
hexanes (3 x 1 l). The resulting fine powder was
refluxed with hexanes (1 l) for 2 h under argon and
filtered into a tared flas~. The solvent was removed in
vacuo to give 1.7 g oil which crystallized on s~An~; n~,
The crystalline residues from the 3 extracts were
dissolved in hexanes and combined. A small ~ -u.lL of
haziness was removed by filtration and the solution was
concentrated to give 5.3 g (57% yield) of product as a
pale yellow crystalline solid. Recrystallization from
acetonitrile gave 4.47 g (48.0~ yield) of a colorless
crystalline solid: mp 107 - 8 C; lH NMR ~CDCl3) ~ 0.95
- l.ol (m, 4 H), 1.19 - 1.24 (m, 4 H), 1.70 - 1.73 (m, 4
H), 1.97 (br s, 5 H), 2.08 - 2.14 (m, 8 H), 2.49 - 2.68
(m, 6 H), 2.74 - 2.80 (m, 2 H), 2.85 - 2.90 (m, 2 H),
2.94 - 2.99 (m, 2 H); MS (LRFA~, NBA) m/z 324 [M + H~;
Anal. calcd. for C~8H37N5: C, 66.83; H, 11.53; N, 21.65.
- Found: C, 66.80; H, 11.44; N, 21.71.
P. SYnthesis of ~Iron fIII~ dichloro (2.3-(R.R)-8.9-
(S.S)-Bis-c~clohexano-1.4.7.10.13-
pentaazacyclo~entadecanel ch}oride
Under an inert atmosphere in a drybox, 199 mg
~0.615 mmol) of the ligand, 2,3-(R,R)-8,9-~S,S)-bis-
cyclohexano-1,4,7,10,13-tetraazacyclopent~ec~n~, was
dissolved in 10 ml o~ an anhydrous methanol solution
cont~;n;ng 0.6t5 mmol (100 mg) of anhydrous FeCl3. The
SU~i 1 1 1 ilTE SHEEl- (RULE 26)
CA 02248964 1998-09-14
PCT~US97/03348
WO g713358~
-54-
resultant dark yellow-orange solution was heated to
reflux for one-half hour with stirring and then allowed
to cool to room temperature and then ~iltered. The
filtrate was reduced to dryness and redissolved in 25 cc
of hot abs. Ethanol and then filtered through Celite~.
The ethanol solution was reduced to 10 ml volume. To
this warm ethanol solution was added diethyl ether to
the cloud point. The so}ution was allowed to sit
undisturbed for 16 hours upon which a yellow
microcrystalline precipitate had formed. The ye~low
solid was isolated by filtration, washed with diethyl
ether, and dried in vac~o overnight. The yield after
drying was 235 mg (0.486 mmol) corresponding to a 79~
theoretical yield. Anal. Calc. ~or ClOH20N5FeCl3CH3CH20H:
C, 45.25; H, 8.16: N, 13.19; Cl, 20.03. Found: C,
44.97; ~, 8.07; N, 13.01; Cl, 19.88. Mass spectrum
(FAB, NBA matrix~: m/z 449 ([Fe(L)Cl~e~+ and m/z 431
([Fe(L)Cl2~' were observed.
A. Svnthesis of N,N -Bis(chloroacetvl~lR,2R-
diaminocvclohexane
lR, 2R-(-3-Diaminocyclohexane (6.98 g, 61.13
mmol) was dissolved in 75 ml of alcohol free CHCl3 in a
4 neck 2000 ml round bottom flas}c along with 37 ml El2O
under argon. ~wo Normag dropping funnels were connected
to the reaction flask, and charged separately with,
ch}oroacetyl chloride (15 ml, 188.3 mmol) in alcohol
free CHCl3 ~88 ml~, and K2CO3 (24.1 g, 174.4 mmol) in ~18
m-l H20. An internal thermometer was inserted into th~
reaction flask. After cooling the two phase mixture in
the reaction flask to 0 ~C in an ice bath, the additions
from the dropping funnels were started in such a way as
to keep the proportion of each solution added
approximately equal over a 1 h 20 min period. During
the addition, an ice salt bath was used to moderate the
SU~ItlUT~SHEET(RUEE26~
CA 02248964 1998-09-14
W O 97/33588 PCT~US97/03348
5~
temperature, keeping it between 3 and -3 ~C. A shell of
ice formed on the inside of the reaction flask which
didn't seem to impede the stirring. The reaction flask
was removed ~rom the ice bath at the end of the addition
and was stirred for 2 h 20 min. The lower chloroform
- layer appeared to have a considerable quantity of a
light solid in it at ice bath temperature, but it
dissolved as the reaction warmed. The reaction mixture
was placed in a separatory funnel, some additional
chloroform added, and the layers were separated. The
aqueous layer was extracted with another portion of
CHCl3, and the combined chloroform layers were washed
with water, then saturated NaCl, dried (Na2S04) and
stripped down to a brownish white solid. This solid was
stirred overnight with about 450 ml of ether, then
filtered, much of the color staying in the ether, giving
a beige solid, 13.68 g, 51.60 mmol, 84.4% yield. lH NMR
(CDCl3, 400 MHz) d 1.34 (m, 4H), 1.80 (m, 2H), 2.08 (m,
2H), 3.74 (m, 2~), 3.99 (ABq, J = 15.1 Hz, dn =82H~,4H),
726(brs,2H);13C NMR(CDCl3,100 MH~)d 24.59, 32.07, 42.45,
53.94, 166.65; MS (FAB, NBA-LiCl matrix~: m/z (relative
intensity) 273 (100) [M+ Li]+, 275 (71) [M+ Li]+.
R. SYnthesis of N-Tosylqlvcvl-lR.2R-diaminocYclohexane
lR, 2R-Diaminocyclohexane (10.0 g, 87.57 mmol)
was dissolved in dry DMF (150 ml) under argon and cooled
to -10~C. Separately, N-tosylglycine (10.04 g, 43.62
mmol), 1-hydroxybenZotriazole (6.75 g, 44.08 mmol), and
l-ethyl-3-(3-dimethylaminopropyl)carbodiimide
hydrochloride (8.45 g, 44.05 mmol) were dissolved in dry
DMF (150 ml), and cooled to -10~C under argon. The
latter solution was added to the diaminocyclohexane
solution at -10~C via cannula. After 2 hours at this
temperature, water (8 ml) was added and the reaction was
allowed to warm to 0~C over one hour, then to room
S~ TE SHEET(RULE 26~
CA 02248964 1998-09-14
PCTrUS97103348
WO g7/33588
56-
temperature over the next half hour. The solvent was
removed on the rotary evaporator under reduced pressure.
T~e residue was heated to 40 to 42~C with water (150 ml)
added in small portions with stirring. After 25 minutes
this solution was filtered. The white precipitate was
largely the bis adduct (5.55 g). Exactly 68 ml of the
filtrate was worked up by repeated extraction with
dichloromethane (9 x 50 ml). The combined organic phase
was dried (sodium sulfate), filtered and the solvent was
removed. The resulting white solid which contained some
residual DMF was redissolved in dichloromethane (30 ml)
and added dropwise to a stirred solution of 9: 1 ether:
hexane (250 - 300 ml) giving an i ?~iate precipitate
which was stirred overnight and then filtered. This
procedure was repeated, stirring for three hours instead
of overnight. After drying the white product on the
vacuum line, 2.36 g, 7.25 mmol were obtained, equivalent
to a 36.7% yield for the entire reaction. IH NMR (CDCl3,
400 MHz) d 1.10 - 1.34 (m, 4H), 1.70 (d, J = 9.7 Hz,
2H), 1.81 ~ 7 (2 m, 2H), 2.41 (s, 3H), 2.51 (td, J -
10.2, 3.8 Hz, lH), 3.53 (m + ABq, J = 16.9 Hz, dn =
51.6 Hz, 3H), 3.69 (br s, 3H), 6.84 (d, J = 9.1 ~z, lH),
7.30 (d, J = 8 3 Hz, 2H), 7.73 (d, J = 8.3 Hz, 2H); 13C
NMR (C~Cl3, 100 MKz) d 21.48, 24.87, 24.97, 32.08, 35.16,
46.09, 54.85, 55.78, 127.15, 129.85, 136.02, 143.84,
168.69; MS (GTHCl): m/z 326 (100) rM+ H}+.
C. SYnthesis of 2R 3R 8R 9R-Bis(cYclohexano~-13-~-
t~luenesul~on~l-1 4 7 10.13-pentaazacYclo~entadecan-
6 11 15-trione
N-p-Toluenesulfonylglycyl-lR,2R-
~;~;~ocyclohexane (1.11 g, 3.42 mmol) and N,
N -bis(chloroacetyl)-lR,2R-diaminocyclohexane (0.913 g,
3.42 mmol) were combined in a one liter flask and dry
N,N-dimethylacetamide (650 ml) was added. The flask was
inerted. After 10 minutes, the sodium hydride was added
SUBSTITUTESHrET(RULE26)
-
CA 02248964 1998-09-14
W O 97/33S88 PCTnUS97/03348
directly to the homogeneous mixture. The reaction flask
was placed in a 70~C oil bath. After the internal
temperature reached 45-50~C, gas evolution became
constant. The oil bath temperature was stabilized at
about 65~C with some excursions from about 60 to 75~C.
Overnight, the reaction mixture became homogeneous.
After heating for 17 hours the reaction flask was
removed from the bath and allowed to cool. The solvent
was removed under reduced pressure, and t~e yellowish
oil was placed on the vacuum line. The residue was
treated with dichloromethane (300 ml) and washed with
water (40 ml) and twice with saturated sodium chloride
(40 ml each). After combining, the aqueous layers were
backwashed with dichloromethane (100 ml). The combined
organic layers were dried over sodium sulfate, ~iltered,
and stripped down to a viscous yellow oil which was
placed on the vacuum line, 2.14 g. This residue was
chromatographed using 0.5% NH40H/ 9% CH30H/ 90.5% CH2Cl2.
On tlc on silica using the same system, R~ = 0.25.
Fractions containing the correct spot were combined and
evaporated down to a slightly off white solid, 0.89 g,
1.71 mmol, 50.1% yield. lH NMR (CDCl3, 300 MHz) d 0.92 -
2.1 (several m, 15H), 2.27 (m, lH), 2.41 (s, 3H), 3.10
(ABq, J = 16 Hz, dn = 34.2 Hz, 2H), 3.39 (m, lH), 3.58
(m, 3H), 3.83 (m, lH), 4.08 (d, J = 17.6 Hz, lH), 4.39
(d, J = 17.4 Hz, lH), 7.30 (m, 3H), 7.44 (d, J = 5.9 Hz,
lH), 7.76 (d, J = 7.8 Hz, 2H), 8.05 (d, J = 8.4 Hz, lH);
13C NMR (CDC13, 100 MHz) d 21.39, 24.20, 24.69, 24.87
- (double intensity), 31.49, 31.54, 31.58, 32.43, 47.01,
52.19, 52.25, 52.49, 52.97, 55.63, 58.36, 127.65,
129.67, 135.28, 143.97, 167.52, 170.04, 172.84; MS
(FAB, NBA-LiCl matrix): m/z (relative intensity) 526
(100) {M+ Li~+, 370 (29) [M+ Li -Ts]+.
S~ lTI: SHEET(RULE 26)
CA 02248964 1998-09-14
PCTfUS97/03348
W O 97/33588
-58-
.
D. Synthesis of 2R,3R,8R, 9R-Bis(cyclohexano)-
1 4,7 10 13-pentaazacYcloPentadecane
2R,3~,8R,9R-Bis(cyclohexano)-13-p-
toluenesulfonyl-1,4,7,10,13-pentaazacyclopentadecan-
6,11,15-trione (4.072 g, 7.84 mmol) was placed in a 1
liter ~lask under an argon atmosphere, and dry 1,2-
dimethoxyethane (dme, 220 ml) was added. The powder
fused, and did not appreciably dissolve. It was
partially ~roken up with a spatula, and stirred in a
cold water bath while lithium aluminum hydride (0.5 M in
dme, 140 ml, 70 mmo~) was added in portions over a 10
minute period. Initially, the solution h~cA ~ cloudy,
and undissolved chllnk~ o~ compound were present. After
about 70 ml had been added, the solution was ~airly
homogeneous, wi~h only a few ~ olved pieces
r~ ~;n;ng, which appeared to dissolve with gas
evolution. Heating was started after a few minutes, and
the solution rapidly ~r~ - heterogeneous and yellow.
The reaction mixture was refluxed overnight. Reflux was
ended after 16.5 hours. The reaction mixture was cooled
in a cold water bath, then in a -18~C bath. Water (2.2
ml) was added ca~tiousl y in small quantities over a 5 to
10 minute period, followed more rapidly by lS% NaOH (2.2
ml), then by water (6.6 ml~. Stirring was continued for
2 hours in the ice bath. Tetrahydrofuran (thf, 210 ml)
was added and stirring was continued for about an hour.
The thick white suspension was allowed to settle, and
was filtered with a filter transfer device (~1 Whatman
paper). The filtrate was stripped. The white residue
was stirred with thf (150 ml) and filtered onto the
stripped first filtrate. The solvent was removed under
reduced pressure, and the residue was placed on the
vacuum line. The resulting yellow-white solid was
~ extracted with hot dry hexane (initially 70 ml,
65~C; then an additional 15 ml) and filtered through a
filter transfer device (#50 Whatman paper), and the
SUBSTITUTESHEET(RULF26)
CA 02248964 1998-09-14
W O 97/33588 PCTrUS97/~3348
-59 -
solvent was removed under reduced pressure. This crude
product, weight about 1.5 g, was dissolved in hot
(>70~C) dry acetonitrile (a~out 60 ml), filtered (filter
transfer device, #50 Whatman paper), concentrated by
more than half, reheated to dissolve all of the white
solid, then allowed to cool slowly to room temperature.
White crystals were obtained, 0.923 g, 2.85 mmol, 36.4
- yield. ~H NM~ (C6D6, 300 MHz) d 0.75 - 1.21 (several m,
8H), 1.23 - 2.19 (several m, 17H), 2.36 - 2.61 (several
~, 6H), 2.61 - 2.73 (m, 2H~, 2.74 - 2.85 (m, 2H), 2.90
(d, J -- 7.5 Hz, 2H); 13C NMR (C6D6, 75 MHz) d 25.48,
25.56, 32.41, 32.48, 46.50, 47.82, 49.56, 61.86, 62.88;
Anal. calcd. for Cl8H3~N5 C, 66.83; H, 11.54; N, 21.65.
Found: C, 66.66; H, 11.46; N, 21.78.
Example 3
~. Synthesis of r IronfIII)dichloro(2.3-(R.R)-8.9-fR.Rl-
bis-cyclohexano-1.4.7.10.13-~entaazacyclopentadecane)~
hexafluoro~hos~hate
Upon an inert atmosphere in a drybox, 97 mg (0.30
mmol) of the ligand, 2R, 3~, 8R, 9~-Bis(cyclohexano)-
1,4,7,10,13-tetraazacyclopentadecane, was dissolved in
15 ml of anhydrous methanol. To this solution was added
with ~igorous stirring 2 ml of a pyridine solution
cont~;n;ng 0.30 mmol (48 mg) of anhydrous FeCl3. The
resultant dark brown solution was heated to reflux for
three hours with stirring and then allowed to cool to
room temperature and then filtered. To the filtrate was
added 20 ml of a clear methanolic solution of NH4PF6
(120) mg). This solution was evaporated to dryness and
2 ml of anhydrous acetonitrile was added to the
resultant solid. This mixture was stirred vigorously
~ for two hours and then filtered and the resultant yellow
filtrate was evaporated to dryness. The resultant
yellow solid was dissolved in hot ethanol and fi~tered.
S~ TESHEET(RULE26)
CA 02248964 1998-09-14
PCTrUS97/03348
W O 97133S88
-60-
-
The solution was evaporated to dryness and the resultant
yellow solid collected by filtration from a diethyl
ether wash. The yellow precipitate was dried in vacuo
overnight. The yield after drying was 75 mg
corresponding to a 42% theoretical yield. Anal. Calc.
for C1~H3~NsCl2F6FeP: C, 36.35; H, 6.28: N, 11.78. Found:
C, 36.37; H, 6.34; N, 11.58.
Exampl~ 4
A. Synthesis of Boc-DAla-Ala-OEt
To a solution of Boc-DAla (25.0 g, 132.1 mmo~) in
DMF (1450 ml) was added HOBT-H2O (19.8 g, 129.3 mmol) and
EDC-HCl (28.0 g, 146.3 mmol) and the resulting solution
was allowed to stir at RT for 30 min. To this solution
was added Alanine ethyl ester hydrochloride (20.3 g,
132.1 mmol) and TEA (20.4 ml, 146.3 mmol) and the
reaction was allowed to stir for 3 days (for
convenience). The DMF was evaporated and the residue
was partitioned between water (500 ml) and ethyl acetate
(500 ml). The ethyl acetate solution was washed with lN
NaHSO4 (250 ml), water (250 ml), saturated NaHCO3 (250
ml), brine (250 ml) and dried over Na2SO4. Filtration
and concentration afforded 31.7 g (83% yield) of the
desired dipeptide as a white foam: IH NMR (DMSO-d6)
1.14 (d, J 5 7.4 Hz, 3 H), 1.16 (t, J - 7.4 Hz, 3 H),
1.24 (d, J 2 7.0 Hz, 3 H), 1.36 (s, 9 H), 3.96 - 4.09
(m, 3 H), 4.17 - 4.22 (apparent quintet, J = 7.4 Hz, 1
H), 6.77 ( d, J = 7.7 H2, 1 H), 8.09 (d, J 5 7.0 Hz, 1
H); MS (LRCI, CH4) m/z (relative intensity) = 317 (5) ~M
+ C2Hs]+~ 289 (60) [M + H3+.
B. SYnthesis of Boc-Ala-Ala-OH
~ To a suspension of the dipeptide (15.0 g, 93.6
mmol) in THF (192 ml) was added 0.5 N NaOH solution (192
SlJts::i 111 UTE S~IEET (RULE 26)
= CA 02248964 l998-09-l4
W O 97/33588 PCTAUS97/03348
-61-
-
ml). To the resulting solution was added
di-t-butyldicar~onate (26.6 g, 121.7 mmol) at once. The
pH o~ the reaction was maintained at -10 for 5 h and the
mixture was then allowed to stir overnight. The pH of
the reaction was again adjusted to -10 and the solution
was extracted with ethyl acetate (2 x 100 ml). The pH
of the aqueous layer was adjusted to -3.5 with aqueous
- potassium bisulfate and this mixture was extracted with
ethyl acetate (3 x 100 ml). The combined extracts were
dried (MgSO4), filtered and concentrated to afford 20.7 g
(85% yield) of the desired product as a white powder: 1H
NMR (DMSO-d6) ~ 1.16 (d, J = 6.8 Hz, 3 H), 1.28 (d, J =
7.3 Hz, 3 H), 1.38 ts, 9 H), 3.9~ - 4.09 (m, 1 H), 4.20
(quintet, J = 7.3 Hz, 1 H3, 6.87 (d, J = 8.0 Hz, 1 H),
8.00 (d, 7.3 Hz, 1 H); MS (HRFAB, NBA - Li) m/z =
267.1~7 [M + Li~+; 267.1532 calcd for CllH20N2O5Li.
C. SYnthesis of DAla-Ala-OEt-TFA
The protected dipeptide (31.4 g, 109 mmol) was
dissolved in methylene chloride (200 ml) and TFA (66 ml)
was added. The resulting solution was allowed to stir
for 30 min at ~T and concentrated. The residue was
coevaporated with methylene chloride (2 x 200 ml),
dissolved in ether and oiled out with the addition of
excess hexanes. The solvents were decanted and the
residue was pumped at high vacuum for 12 h to afford
39.6 g ~100% yield, contains residual TFA) of the
desired TFA salt as an orange oil: IH NMR (DMSO-d6)
- 1.16 (t, J = 7.0 Hz, 3 H), 1.28 (d, J = 7.0 Hz, 3 H),
1.34 (d, J = 7.0 Hz, 3 H), 3.86 (bs, lH), 4.07 (q, J =
7.0 Hz, 2 H), 4.26 (quintet, J = 7.0 Hz, 1 H), 8.21 (~s,
3 H), 8.86 (d, J = 7.4 Hz, 1 H); MS (LRC~, CH4) m~z
(relative intensity) 217 (~) [M + C2H5]+, 189 (40) tM+H]t.
-
S~3lll~TESHEET(R~EZ6)
CA 02248964 1998-09-14
W O 97/33588 PCTrUS97/03348
-62-
D. SYnthesis of Boc-Ala-Ala-DAla-Ala-oEt
To a solution of Boc--Ala--Ala--OH (20.1 g, 77.2
mmol) in DMF (850 ml~ was added HOBT-H2O (13.1 g, 85.4
mmol) and EDC-HCl (16.4 g, 85.4 mmol). To this solution
was added DAla-Ala-OEt-TFA (23.3 g, 77.2 mmol~ followed
by TEA (11.9 ml, 85.4 mmol) and the resulting mixture
was stirred for 12 h thereafter. The DMF was evaporated
and the residue was dissolved in ethyl acetate (300 ml)
and washed with 1 N potassium bisulfate (150 ml), water
{150 ml), saturated sodium bicarbonate (150 ml) and
brine (150 ml). The ethyl acetate layer was dried
(MgSO4), filtered and concentrated to half volume and
crystallization was allowed to proceed. Isolation by
filtration afforded 20.5 g (62~ yield) of the desired
tetrapeptide as a white solid: lH NMR (DMSO-d6) ~ 1.13
(d, J 5 7.0 Hz, 3 H), 1.17 (two coincidental d, J = 7.0
Hz, 6 ~), 1.25 (d, J = 7.4 Hz, 3 H), 3.91 - 4.30 (m, 6
H), 6.87 (d , 7.0 Hz, 1 H), 7.92 (d, J = 6.3 Hz, 1 H),
8.07 (d, J - 7.3 Hz, 1 H), 8.09 (d, J = 6.6 Hz, 1 H);
MS (HRFAB, NBA - Li) m/z = 437.2600 [M ~ Li]+; 437.2588
calcd for Cl9H34N4O~Li.
. Synthesis of Boc-Ala-A~a-DAla-Ala-OH
A solution of Boc-Ala-Ala-DAla-Ala-OEt (10.9 g,
25.3 mmo7) in methanol (100 ml) was treated with 2.5 M
sodium hydroxide (20.0 ml, 50.0 mmol) and the resulting
solution was allowed to stir for 2 h at RT. At this
time the pH of the solution was lowered to ~3 with the
addition of a~ueous potassium bis-lfate and the
resulting mixture was extracted w~h ethyl acetate (3 x
100 ml). ~he com~ined extracts were dried (MgSO4),
filtered and concentrated to afford 6.8 g (67% yield of
the desired acid as a white solid: IH NMR (DMSO-d6)
1.17 (d, J = 7.2 Hz, 3 H), 1.20 (two coincidental d, J =
7.1 Hz, 6 H), 1.28 (d, J = 1.3 Hz, 3 H), 1.38 (s, 9 H),
S~ TESHEET(~ULE26)
CA 02248964 l998-09-l4
WO 97/33588 PCTIUS97/03348
--63--
3 . 90 - 4 . 00 (m, 1 H), 4 . 17 - 4 . 30 (m, 3 H), 6. 93 (d, J =
6.7 Hz, 1 H3, 7.96 (d, J = 6.7 Hz, 1 H), 8.04 (d, J =
7 . 4 Hz , 1 H), 8 . 07 (d, J = 7 . 8 Hz , 1 H); MS (~IRFAB, NBA
- Li) m/z = 409.2331 [M + Li~i; 409.2353 calcd for
5 CI~H30N4o~Li.
F. Svnthesis of Boc-Ala-Ala-~Ala-Ala-DAla-OBzl
- To a solution of Boc-Ala-Ala-DAla-Ala-OH (6.5 g,
16. 3 mmol) in DMF (180 ml) was added HOBT-H20 (2. 86 g,
18 . 7 mmol) and EDC-HCl (3 . 58 g, 18.7 mmol). The
resulting solution was allowed to stir for 15 min at RT
and treated with DAla-OBzl p-toluenesulfonate salt (6. 57
g, 18.7 mmol) and TEA (2.6 ml, 18.7 mmol). This mixture
was allowed to stir for 12 h thereafter. The DMF was
evaporated and the residue was partitioned ~etween ethyl
acetate (300 ml) and water (300 ml). The ethyl acetate
layer was washed with 1 N potassium bisulfate (150 ml),
water (150 ml), saturated sodium bicarbonate (150 ml)
and brine (150 ml). The ethyl acetate layer was then
dried (MgSO4), filtered and concentrated to afford 9.0 g
(100% yield) of the desired compound as a white powder:
IH NMR (DMSO-d6) ~ 1.17 (d, J = 7.3 Hz, 3 H~, 1.21 (two
coincidental d, J = 7.0 Hz, 6 H), 1.22 (d, J = 7.0 Hz,
3 H), 1.32 (d, J = 7.3 Hz, 3 H), 1.37 (s, 9 H), 3.90 -
4.09 (m, 1 H), 4.18 - 4.34 (m, 4 H), 5.13 (ABq, J =
12.7, ~ = 10.5 Hz, 2 H), 6.94 (d, J = 7.3 Hz, 1 H),
7.30 - 7.41 ~m, 5 H), 7.97 (d, J = 7.0 Hz, 1 H), 8.10 -
8.18 (m, 2 H), 8.25 (d, J = 6.9 Hz, 1 H); MS (HRFAB,
- NBA - Li) m/z = 570.3140 ~M + ~i]+; 570.3115 calcd ~or
C2~H4~N5O8Li.
G. Svnthesis of Ala-Ala-DAla-Ala-DAla-HCl
Boc-Ala-Ala-DAla-Ala-DAla-OEt (10.4 g, 18.7 mmol)
was dissolved in acetic acid (225 ml) and treated with
concentrated hydrochloric acid (75 ml). The resulting
SUBSTITUTESHEET~RULE26)
CA 02248964 1998-09-14
PCT~US97103348
W O 97/33588
-64-
-
solution was allowed to stir at RT for 14 h thereafter.
~t this time the reaction was concentrated, coevaporated
with water (50 ml) and azeotropically dried by toluene
coevaportation (2 x 100 ml) to afford 7.8 g (96% yield)
of the deprotected pentapeptide hydrochloride as a white
powder: lH NMR (D20) 8 1.29 - 1.39 (m, 12H), 1.47 (d, J
= 7.0 Hz, 3 H), 4.06 (q, J = 7.0 Hz, 1 H), 4.18 - 1.38
(m, 4 H3; MS (LRFAB, NBA - HCl) 374 [M + H]'.
H. Synt~esis of Cyclo- (Ala-Ala-DAla-Ala-DAla-~
To a solution of Ala-Ala-DAla-Ala-DAla-HCl (7.8
g, 19.0 mmol) in DMF (2400 ml) at -40 C was added DPPA
(6.29 g, 22.8 mmol) and enough TEA to adjust the "pH" to
-8 (measured by spotting the reaction mixture on
moistened hydrion paper). This solution was allowed to
stand at -23 C for 48 hours and at 8 C for 48 hours.
During this time the "pH" was again maintained at -8
with the periodic addition of T~A. At the end of this
period the reaction mixture was poured into water (2400
ml~ and stirred with mixed-bed ion ~ch~nge resin (1200
g) for 6 h. The resin was removed by filtration and the
~iltrate was concentrated to a volume of - 100 ml.
Ether (500 ml) was added and the precipitated white
solid was isolated by filtration and washed with more
ether (250 ml). The solid was then trit~rated by
stirring with THF (100 ml) for 12 h (to remove traces of
DMF), filtered and thoroughly dried to afford 3.15 g
(47% yield) of the desired cyclic peptide as a fine
white powder: IH NMR (DMS0-d~) 8 1.08 - 1.25 (m, 12
H), 1.24 (d, J = 1.3 Hz, 3 ~), 4.00 - 4.~0 (m, 1 H~,
4.26 - 4.30 (m, 2 H), 4.34 (q, J = 7.2 Hz, 1 H), 4.41
(~, J - 7.6 Hz, 1 H), 7.58 (d, J = 7.0 Hz, 1 H), 7.83
(d, J = 8.4 Hz, 1 H), 8.22 (d, ~ = 6.2 Hz, 1 H), 8.33
~ ~ (d, J = 7.81, 1 H), 8.49 (d, J = 6.8 Hz, 1 H); MS
(HRFAB, NBA - HCl) m/z 356.1989 (M + H)'; 356.1934 calcd
for C1sH25Nso5 (M ~ H) -
Sl)~lll~JTF SHEET(RULE26)
CA 02248964 1998-09-14
W O 97133588 PCT~US97/03348
I. Svnthesis of (2S. 5R. 8S llR 14S)-Pentamethvl-
1 4 7 10.13-pentaazacYcloPentadecane
To a stirred suspension of cyclo- (Ala-Ala-DAla-
Ala-~Ala-) (3.10 g, 8.70 mmol) in THF (70 ml) at RT was
added lithium aluminum hydride (108 ml of a 1.0 M
solution in THF, 108 mmol). The resulting mixture was
stirred at ~T for 2 h and heated to reflux for 16 h
- thereafter. The mixture was then cooled to ~-20 C and
quenched with the dropwise addition of saturated sodium
sulfate (-30 ml). The resulting mixture was concentrated
to a dry white powder and this powder was triturated with
ether (2 x 150 ml). The combined triturates were
concentrated and recrystallized form acetonitrile to
afford l.10 g (44 % yield) of the desired ligand as a
white solid: IH NMR (CDCl3) ~ 0.96 (d, J = 5.2 Hz, 3 H),
1.00 ttwo coincidental d, ~ = 5.0 Hz, 6 H), 1.02 (two
coincidental d, J = 5.0 Hz, 6 H), 1.30 - 1.55 (bm, 2 H),
1.85 - 2.15 (bs, 3 H), 2.05 - 2.19 (m, 5 H), 2.42 -
3.00 (complex m, 12 H); MS (HRFAB, NBA - HCl) m/z =
286.3013 (M + H)~; 286.2971 calcd for C15H36N5.
J. SYnthesis of rIron (III)dichloro-(2S. 5R. 8S llR
14S3-Pentamethyl-1 4 7 10 13-
pentaazacvclopentadecanelhexafluorophosPhate
This complex was prepared in a fashion entirely
analogous to that described previously in Example 3.
After recrystallization of the crude yellow solid from
ethanol, yellow crystals were obt~;ne~ in a 40% yield.
Analysis calc. for Cl5H35N5Cl2FeF6P: C, 32.37; H, 6.34: N,
12.59. Found: C, 32.44; H, 6.30; N, 12.40.
Example 5
StopPed-Flow Kinetic AnalYsis
Stopped-flow kinetic analysis has been utilized to
determine whether a compound can catalyze the dismutation
SUBSTITUTE SltEET ~RULE 26~
CA 02248964 1998-09-14
W 097/33~88 PCTAUS97/03348
of superoxide (Riley, D.P., Rivers, W.J. and Weiss, R.H.,
"Stopped-Flow Kinetic Analysis for Monitoring Superoxide
Decay in Aqueous Systems," Anal. Biochem, 96, 344-349
tl991~). For the attainment of consistent and accurate
measurements all reagents were biologically clean and
metal-free. To achieve this, all buffers (Calbiochem)
were biological grade, metal-free buffers and were
handled with utensils which had been washed first with
0.1 N HCl, followed by purified water, followed by a
rinse in a 104 M EDTA bath at pH 8, followed by a rinse
with purified water and dried at 65~C for several hours.
Dry DMSO solutions of potassium superoxide (Aldrich) were
prepared under a dry, inert atmosphere of argon in a
Vacuum Atmospheres dry glovebox using dried glassware.
The DMSO solutions were prepared immediately before every
stopped-flow experiment. A mortar and pestle were used
to grind the yellow solid potassium superoxide (~100 mg).
The powder was then ground with a few drops of DMSO and
the slurry transferred to a flask containing an
additional 25 ml of DMSO. The resultant slurry was
stirred for 1/2 h and then filtered. This procedure gave
reproducibly ~2 mM concentrations of superoxide in DMSO.
These solutions were transferred to a glovebag under
nitrogen in sealed vials prior to loading the syringe
under nitrogen. It should be noted that the
D~SO/superoxide solutions are extremely sensitive to
water, heat, air, and extraneous metals. A fresh, pure
solution has a very slight yellowish tint.
Water for buffer solu~: -ns was delivered from an
in-house deionized water sys~ to a Barnstead Nanopure
Ultrapure Series 550 water sy~ em and then double
distilled, first from alkaline potassium permanganate and
then from a dilute EDTA solution. For example, a
~ solution containing 1.0 g of potassium permanganate, 2
liters of water and additional sodium hydroxide necessary
to bring the pH to 9.0 were added to a 2-liter flask
SU~IllUTESHEET(RULE26)
CA 02248964 1998-09-14
W O 97/33588 PCTrUS97/03348
-67-
fitted with a solvent distillation head. This
distillation will oxidize any trace of organic compounds
in the water. The final distillation was carried out
under nitrogen in a 2.5-liter flask containing 1500 ml of
water from the first still and 1.0 x 106M E~TA. This
step will remove remaining trace metals from the
ultrapure water. To prevent EDTA mist from volatilizing
over the reflux arm to the still head, the 40-cm vertical
arm was packed with glass beads and wrapped with
insulation. This system produces deoxygenated water that
can be measured to have a conductivity of less than 2.0
nanomhostcm2.
The stopped-flow spe~L~o cter system was designed
and manufactured by Kinetic Instruments Inc. (Ann Arbor,
MI) and was interfaced to a MAC IICX personal computer.
The software for the stopped-flow analysis was provided
by Kinetics Instrument Inc. and was written in QuickBasic
with MacAdios drivers. Typical injector volumes (0.10 ml
of ~uffer and 0.006 ml of DMS0) were calibrated so that a
large ~ of water over the DMS0 solution were mixed
together. The actual ratio was approximately 19/1 so
that the initial concentration of superoxide in the
aqueous solution was in the range 60-120 ~M. Since the
published extinction coefficient of superoxide in H20 at
2S 245 nm is ~2250 M~l cm-1 (1), an initial absorbance value
of approximately 0.3-0.5 would be expected for a 2-cm
path length cell, and this was observed experimentally.
Aqueous solutions to be mixed with the DMS0 solution of
superoxide were prepared using 80 mM concentrations of
the Hepes buffer, pH 8.1 (free acid + Na form). one of
the reservoir syringes was filled with 5 ml of the DMS0
solution while the other was filled with 5 ml of the
aqueous buffer solution. The entire injection block,
~ mixer, and spe~Ll-~eter cell were immersed in a
thermostatted circulating water bath with a temperature
of 21.0 + 0.5~C.
SU~IIlUTESHEET(RULE26)
CA 02248964 1998-09-14
W O 97/33588 PCTrUS97/03348
-68-
Prior to initiating data collection for a
superoxide decay, a baseline average was obtained by
injecting several 5hots of the buffer and DMSO solutions
into the mixing chamber. These shots were averaged and
storQd as the baseline. The first shots to ~e collected
during a series of runs were with aqueous solutions that
did not contain catalyst. This assures that each series
of tria}s were free of cont~inAtion capable of
generating f irst-order superoxide decay profiles. If the
decays observed ~or several shots o~ the buffer solution
were second-order, solutions of iron (III) complexes
could be utilized. In general, the potential SOD
catalyst was screened over a wide range of
concentrations. Since the initial concentration of
superoxide upon mixing the DMSO with the a~ueous buffer
was -1.2 x 10-4 N, we wanted to use a iron (III) complex
concentration that was at least 20 times less than the
substra~e superoxide. Co~equently, we generally
screened compounds for SOD activity using concentrations
ranging ~rom 5 x 10 '7 to 8 x 10-~ M. Data acquired from
the experiment was imported into a suitable math program
(e.g., Cricket Graph) so that stAn~d kinetic data
analyses could be performed. The cataly~ic rate constant
for ~i~ Lation of superoxide by the iron (III) complexes
of Examples 1-4 were determined from the linear plot of
observed rate constants (ko~) versus the concentration of
the iron (III) complexes. kob, values were o~tained from
the liner plots of ln absor~ance at 245 nm versus time
for the ~i ! ~ation of superoxide by the iron (III)
complex. The k~.t (M-lsecl) of the iro,n (III) complexes of
Examples 1-4 are shown in Table I.
The iron (III) complexes of the nitrogen-
cont~;n;ng macrocyclic ligands in Examples 1-4 are
~ effective catalysts for the dismutation of superoxide, as
can ~e seen from the kc~ data in Table I.
SUP'STITUTE SHEET (RULE 26)
CA 02248964 l998-09-l4
W O 97/33588 PCTAUS97/03348
-69-
.
TABLE I
Compound kC,I@ pH=7 . 6, 21~C
5ExamPle No. (M-l sec -1)
1 1. 06 x 107
- 2 0 . 96 x 107
3 1.60 X 107
4 2 . 94 x 107
SUBSr ~ ~3T~ SHEET (RULE 26)