Note: Descriptions are shown in the official language in which they were submitted.
CA 02250930 1998-09-25
WO 97/35858 PCT/US97104773
4-((THtEN-3-YL)METHYL] tMtDAZOLE DERTVATIVES HAVING a~-ADRENOCEPTOR AGONtS-
T1C ACTIVT1Y
The present invention relates to a2-adrenoceptor agonists having
analgesic activity. More particularly, the present invention relates to 4-
[{thien-3-yl)methyl]-imidazoles having improved analgesic activity.
BACKGROUND OF THE INVENTION
Clonidine is a centrally acting a2-adrenoceptor agonist with wide
clinical utility as an antihypertensive agent. Clonidine is believed to act by
inhibiting the release of norepinephrine from sympathetic nerve terminals
via a negative feedback mechanism involving a2-adrenoceptors located on
the presynaptic nerve terminal. This action is believed to occur in both the
central {CNS) and peripheral (PNS) nervous systems. More recently, the
role of a2-adrenoceptor agonists as analgesic agents in humans and
antinociceptive agents in animals has been demonstrated. Clonidine and
other a2-adrenoceptor agonists have been shown to produce analgesia
through a non-opiate mechanism and, thus, without opiate liability.
However, other behavioral and physiological effects were also produced,
including sedation and cardiovascular effects.
I H
I N~N
c, NJ
clonidine
Medetomidine and detomidine are a2-adrenoceptor agonists widely
used clinically in veterinary medicine as sedatives/hypnotics for pre-
anaesthesia. These compounds are hypotensive in animals and in humans,
but the magnitude of this cardiovascular effect is relatively insignificant.
Me Me Me H
M I \ I \~ M I \ I \J
~'N ~ ~N
a
H H
medetomidine detomidine
CA 02250930 1998-09-25
WO 97J35858 PCT/US97J04773
U.S. Pat. No. 3,574,844, Gardocki et al., teach 4-[4(or 5}-
imidazolyimethyl]-oxazoies as effective analgesics. The disclosed
compounds are of the general formula:
H R
N
N
N
R
Compounds of this type are insufficiently active and suffer from unwanted
side effects.
U.S. Pat. No. 4,913,207, Nagei et al., teach aryithiazolylimidazoles as
effective analgesics. The disclosed compounds are of the general formula:
S
Ar~ / CN1
I
--N
~H
Compounds of this type are insufficiently active and suffer from unwanted
side effects.
W092/14453, Campbell et al., teach 4-[(aryl or heteroaryl)methyl]-
imidaioles as effective analgesics. The disclosed compounds are of the
general formula:
R N R is H or alkyl
A~~ A is aryl or heteraaryl
N
H
The disclosed compounds are insufficiently active and suffer from unwanted
side effects.
Kokai No. 1-242571, Kihara et al., disclose a method to produce
imidazole derivatives for use, among other uses, as antihypertensive agents.
Z is H or phenyl
H-N~ N ~ JX R is H , alkyl or halogen
Z R~or2 X is S or O
-2-
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
A single mixture of compounds meeting the above formula was reportedly
produced by the inventive method. This was a mixture of 4-(2-thienyl)-
methylimidazole and 4-(3-thienyl)-methylimidazole represented by the
following formula:
_ .rS
H_N'%N ~ /
The disclosed compounds are insufficiently active and suffer from unwanted
side effects.
It is an object of the present invention to produce 4-[(thien-3-
yl)methyl]-imidazoles having improved analgesic activity.
It is another object of the present invention to produce 4-[(thien-3-
yl)methyl)-imidazole analgesics having reduced side effects.
SUMMARY OF THE INVENTION
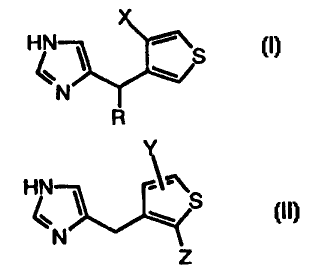
Briefly, there is provided by the present invention compounds having
improved analgesic activity of the formulae:
HN
\ S
N
R
wherein
R is hydrogen or methyl, and
X is C~-4alkyl, bromine or chlorine; or
Y
HN 1 ~~S
N
Z
wherein
Y is hydrogen, C1-4alkyl, bromine or chlorine, and
Z is C1-Qalkyl, bromine or chlorine.
- 3 -
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
DETAILED DESCRIPTION OF THE INVENTION
The compounds of the present invention may be made in basically a
two step process. In the first step, an appropriately substituted precursor
thiophene is obtained having hydrogen, C1_4alkyl, bromine or chlorine
substituents as desired and in the required positions. This precursor
thiophene will have an electrophilic carbon substituent at the 3-position. In
the second step, a precursor imidazole having an anion at the 4-position
capable of reacting with the electrophilic carbon of the precursor thiophene
to leave a carbon bridge residue, is reacted with the precursor thiophene to
produce the target skeleton followed by deoxygenation of the carbon bridge
residue. Of course, many variations are possible. It may be desirable to
substitute the thiophene initially, as described, or to modify the
substitution
on the thiophene following the formation of the base structure of the final
compound. Also, in compounds where it is desirable to have methyl
substitution on the carbon bridge residue, additional steps will be necessary.
Herein, a Grignard reaction is favored for use in the second step to
join the thienyl moiety and the imidazolyl moiety. Thus, it is preferred that
the precursor imidazole be substituted at the 4-position as a Grignard
reagent and that the precursor thiophene is substituted at the 3-position with
a carbonyl, such as, formyl or N,O-dimethylcarboxamido group.
The preferred precursor imidazole has the formula:
TAN
~'~MgX~
where X1 is iodo, bromo or chloro. This compound may be made by
methods well known to the art, i.e., reaction between alkyl Grignard or
magnesium and imidazolyl halide in dry, alcohol-free ether or THF or
dichloromethane.
The preferred precursor thiophenes have the formula:
Y
S
S
OHC
OHC pr Z pr MeO~ Me
AA BB CC
- 4 -
CA 02250930 1998-09-25
W0 97/35858 PCT/US97104773
where X, Y and Z are defined above. As starting materials to make the
preferred precursor thiophenes AA, BB and CC, the preparation of various
brominated and methyiated thiophenes is well known from the literature.
Precursor thiophenes of type AA may be produced from 3-bromo-4-
methylthiophene or 3-bromo-4-(bromo or chloro)thiophene by use of
halogen metal exchange. In a first step, the compound is treated with an
organo-alkali compound such as n-butyllithium, the product of which is
reacted, in a second step, in situ with DMF. The reaction is quenched with
aqueous ammonium chloride. The resultant compound is 4-methyl-
thiophene-3-carboxaldehyde or 4-(bromo or chloro)-thiophene-3-
carboxaldehyde. Precursor thiophenes of type BB, may be produced by
much the same method as those of type AA with the use of different starting
materials. The method just described to produce precursor thiophenes of
type AA may be employed to produce those of type BB where the starting
material is not 2-bromo or 5-bromo substituted. Thus, the halogen metal
exchange may be employed with 2-(methyl or chloro}-3-bromo-4-(methyl or
chloro or bromo) thiophene or 2-(methyl or chloro)-3-bromo-5-(methyl or
chloro) thiophene to produce type BB precursor thiophenes which are 2-
(methyl or chloro)-4-(methyl or chloro or bromo)-thiophene-3-
carboxaldehyde or 2-(methyl or chloro)-5-(methyl or chloro)-thiophene-3-
carboxaidehyde. Precursor thiophenes of type CC may be produced from 2-
(methyl or chloro or bromo)-4-(methyl or chloro or bromo)-thiophene-3-
carboxyiate or 2-(methyl or chloro or bromo)-5-(methyl or chloro or bromo)-
thiophene-3-carboxylate by two methods. In the first method, the
carboxylate starting material is converted to the acid chloride and reacted
with N,O-dimethylhydroxylamine to produce the Weinreb amide, thiophene
type CC. In the second method, the carboxylate is reacted with N,O-
dimethylhydroxylamine and an appropriate coupling agent, such as, DCC or
CDI, to produce the Weinreb amide.
The precursor imidazole may be reacted with any of the precursor
thiophenes of types AA or BB or CC by use of the Grignard Reaction. Where
the precursor thiophene is of type AA or BB, a solution of the thiophene
precursor is combined with a solution of the imidazole precursor at room
temperature and the reaction is quenched with aqueous ammonium chloride
solution to produce an imidazo thienyi methanol. The carbinol is
deoxygenated to final product, where R is hydrogen, by use of a reducing
agent, such as borane methyl sulfide in combination with TFA. Alternatively,
- 5 -
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
the methanol is catalytically deoxygenated to final product, where R is
hydrogen, by heating with Peariman's catalyst and an equivalent of acid. To
produce final product where R is methyl, the methanol is oxidized to the
corresponding ketone with an oxidizing agent, such as Mn02 or Jones
Reagent and the resulting ketone is reacted with methyl Grignard to produce
a carbinol which is deoxygenated as described immediately above. Where
the precursor thiophene is of type CC, a solution of the thiophene precursor
is combined with a solution of the imidazole precursor at room temperature
and the reaction is quenched with aqueous ammonium chloride solution to
produce an imidazo thienyl ketone. To produce final product, the ketone is
reduced to the carbinol by use of a reducing agent, such as, sodium
borohydride or lithium aluminum hydride and thereafter the carbinol is
deoxygenated as described immediately above.
The protecting group on the precursor imidazole is exemplified herein
as trityi, which is preferred. However, a person skilled in the art will
readily
recognize that other protecting groups are suitable. Suitable protecting
groups include dimethylsulfamoyl or methoxymethyl. The trityl group is
removed in the deoxygenation to final product or upon heating in a dilute
acid and alcoholic solvent.
The most preferred compounds of the present invention are shown in
Table I:
- fi -
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
M M
HN HN ~ HN
M ~ ~S ~ ~S
CAN , ~ S H N ' ' S ~N 1 ~ ~N ,
Me ~N Me Et
Cp-1 Cp-2 Cp-3 Cp-4
Me Et
HN HN .~~ H ~ HN M S
~~N 1 ~ S ~N ' ~ S ~~N , ~ S ~~N , ( /
Me Et Et Me
Cp-5 Cp-6 Cp-7 Cp-8
n-Pr i-Pr
HN , I S HN 1 I ~ HN 1 ~ S HN ' ~ S
v v
C / <
N N N N
Et Me Et n-Pr i-Pr
Cp-9 Cp-10
The activity of compounds of the invention as analgesics may be
demonstrated by the in vivo and in vitro assays as described below:
,adreneroi~eotor binding as av
Male, Wistar rats (150-250 g, VAF, Charles River, Kingston, NY) are
sacrificed by cervical dislocation and their brains removed and placed
immediately in ice cold HEPES buffered sucrose. The cortex is dissected
out and homogenized in 20 volumes of HEPES sucrose in a Teflon~-glass
homogenizer. The homogenate is centrifuged at 1000 g for 10 min, and the
resulting supernatant centrifuged at 42,000 g for 10 min. The resulting pellet
is resuspended in 30 volumes of 3 mM potassium phosphate buffer, pH 7.5,
preincubated at 25 °C for 30 min and recentrifuged. The resulting
pellet is
resuspended as described above and used for the receptor binding assay.
Incubation is pertormed in test tubes containing phosphate buffer, 2.5 mM
MgCl2, aliquots of the synaptic membrane fraction, the ligand 3H-para-
aminoclonidine and test drug at 25 °C for 20 min. The incubation is
terminated by filtration of the tube contents through glass fiber filter
sheets.
_7_
CA 02250930 1998-09-25
WO 97/35858 PCT/CTS97/04773
Following washing of the sheets with 10 mM HEPES buffer, the adhering
radioactivity is quantified by liquid scintillation spectrometry.
Binding of the test drug to the receptor is determined by comparing
the amount of radiolabeied ligand bound in control tubes without drug to the
amount of radiolabeled ligand bound in the presence of the drug. Dose-
response data are analyzed with LIGAND, a nonlinear curve fitting program
designed specifically for the analysis of ligand binding data. This assay is
described by Simmons, R. M. A., and Jones, D. J., Binding of [3H-]prazosin
and [3H-]p-aminoclonidine to a-Adrenoceptors in Rat Spinal Cord, Brain
Research 445:338-349, 1988.
Mouse Acetylcholine Bromide-Induced Abdominal Constriction Assay
The mouse acetylcholine bromide-induced abdominal constriction
assay, as described by Collier et al. in Brit. J. Pharmacol. Chem. Ther., 32:
295-310, 1968, with minor modifications was used to assess analgesic
potency of the compounds herein. The test drugs or appropriate vehicle
were administered orally (p.o.) and 30 minutes later the animal received an
intraperitoneal (i.p.) injection of 5.5 mg/kg acetylcholine bromide (Matheson,
Coleman and Bell, East Ruthertord, NJ). The mice were then placed in
groups of three into glass bell jars and observed for a ten minute
observation period for the occurrence of an abdominal constriction response
(defined as a wave of constriction and elongation passing caudally along
ttie abdominal wall, accompanied by a twisting of the trunk and followed by
extension of the hind limbs). The percent inhibition of this response to a
nociceptive stimulus (equated to % analgesia) was calculated as follows:
The % Inhibition of response, i.e., % analgesia is equal to the difference
between the number of control animals response and the number of drug-
treated animals response times 100 divided by the number of control
animals responding.
At least 15 animals were used for control and in each of the dnrg
treated groups. At least three doses were used to determine each dose
response curve and ED50 (that dose which would produce 50% analgesia).
The ED50 values and their 95% fiducial limits were determined by a
computer assisted probit analysis.
_g_
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
Table II
Mouse Abdominal Constri
tion
~moound Ki(nm) % Inhibition ED~n
CP-1 0.44 100% @30 mpk
CP-2 0.47 0.4 mpk
Cp-3 0.39 0.4 mpk
Cp-4 0.97 100% @30 mpk
Cp-5 0.69 1.3 mpk
CP-6 0.4 3.7 mpk
Cp-7 0.07 0.4 mpk
Cp-8 0.10 100% @30 mpk
Cp-9 0.29 100% @30 mpk
Cp-10 0.28 100% @30 mpk
Me
~s
N
Me Me 6.1 100% @30 mpk
~s
N 3.1 6.4 mpk
\ S
N
Me Me 2.3 1.6 mpk
1 ~s
N
Me 2.5 100% @30 mpk
M
\s
N '~'
Me Me 33.5 7.8 mpk
_g_
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
Et
HN
N
Me Et 11.4 67% ~a 30 mpk
Cv
N
Me Et 1.1 100% @30 mpk
Br
Bf
HN
N
0.98 27% [7a 30 mpk
Based on the above results, invention compounds of the present
invention may be used to treat mild to moderately severe pain in warm-
blooded animals, such as, humans by administration of an analgesically
effective dose. The dosage range would be from about 10 to 3000 mg, in
particular about 25 to 1000 mg or about 100 to 500 mg, of active ingredient 1
to 4 times per day for an average (70 kg) human although it is apparent that
activity of individual compounds of the invention will vary as will the pain
being treated. Pharmaceutical compositions of the invention comprise the
formula (I) compounds as defined above, particularly in admixture with a
pharmaceutically-acceptable carrier.
To prepare the pharmaceutical compositions of this invention, one or
more compounds of the invention or salt thereof as the active ingredient, is
intimately admixed with a pharmaceutical carrier according to conventional
pharmaceutical compounding techniques, which carrier may take a wide
variety of forms depending of the form of preparation desired for
administration, e.g., oral or parenteral such as intramuscular. In preparing
the compositions in oral dosage form, any of the usual pharmaceutical
media may be employed. Thus, for liquid oral preparations, such as for
example, suspensions, elixirs and solutions, suitable carriers and additives
include water, glycols, oils, alcohols, flavoring agents, preservatives,
coloring agents and the like; for solid oral preparations such as, for
example,
powders, capsules and tablets, suitable carriers and additives include
starches, sugars, diluents, granulating agents, lubricants, binders,
- 10 -
CA 02250930 1998-09-25
WO 97135858 PCT/US97/04773
disintegrating agents and the like. Because of their ease in administration,
tablets and capsules represent the most advantageous oral dosage unit
form, in which case solid pharmaceutical carriers are obviously employed. if
desired, tablets may be sugar coated or enteric coated by standard
techniques. For parenterals, the carrier will usually comprise sterile water,
through other ingredients, for example, for purposes such as aiding solubility
or for preservation, may be included. Injectable suspensions may also be
prepared, in which case appropriate liquid carriers, suspending agents and
the like may be employed. The pharmaceutical compositions herein will
contain, per dosage unit, e.g., tablet, capsule, powder, injection,
teaspoonful
and the like, an amount of the active ingredient necessary to deliver an
effective dose as described above.
The pharmaceutically acceptable salts referred to above generally
take a form in which the imidazolyl ring is protonated with an inorganic or
organic acid. Representative organic or inorganic acids include
hydrochloric, hydrobromic, hydroiodic, perchloric, sulfuric, nitric,
phosphoric,
acetic, propionic, glycolic, lactic, succinic, malefic, fumaric, malic,
tartaric,
atric, benzoic, mandeiic, methanesulfonic, fiydroxyethanesulfonic,
benezenesulfonic, oxalic, pamoic, 2-naphthalenesulfonic, R-toluenesulfonic,
cyclohexanesulfamic, salicylic or saccharic.
The following Examples illustrate the invention:
_ 11 _
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
EXAMPLE 1
HO
Me
To a solution of 3-bromo-2-methyl thiophene (4.2 g, 24 mmol) in 50
mL of dry Et20 cooled to -78°C was added n-BuLi (15.0 mL, 24 mmol)
dropwise. The bath temperature was allowed to rise to -20°C and DMF
(2.3
mL, 30 mmol) was added. The reaction mixture was allowed to warm to
room temperature overnight. The reaction was quenched with NH4CI (aq)
and extracted with Et20. The organic layer washed twice with water and
brine and then dried (MgS04). After evaporation of solvent, the crude
product was purified on flash silica gel (95:5 hexane/Et20) to afford 2-
methylthiophene-3-carboxaldehyde, ~1, as a light yellow oil (1.5 g, 50%).
tH NMR (CDCI3) supported the assigned structure.
ste~B
S
N
OH Me
To a solution of N-trityl-4-iodo-imidazole (11.8 g, 27 mmol) in dry
CH2CI2 (75 mL) was added EtMgBr (10.0 mL, 3.0 M in Et20) and the solution
was stirred for 3 hrs. Then a solution of 2-methylthiophene-3-
carboxaldehyde (3.3 g, 27 mmol) in CH2CI2 (20 mL) was added and the
reaction mixture was stirred at room temperature overnight. The reaction
was quenched with NH4CI (aq) and the mixture was transferred to a
separatory funnel. The aqueous layer was extracted with a second portion
of CH2CI2. The extracts were combined and washed with a small portion of
water, dried (Na2S04}, and filtered. The solvent was evaporated in vacuo to
give a thick syrup which was triturated with Et20 to give a solid which was
_ 12 _
CA 02250930 2004-10-15
recrystallized with charcoal treatment from EtOAc to give (2-methylthien-3-
yl)-1-tntyl-imidazol-4-yl-methanol, ~. ~H NMR (CDCI3) supported the
assigned structure.
v
Me
A solution of (2-methylthien-3-yl)-1-trityl-imidazol-4-yl-methanol (1.5
g, 3.5 mmol) was combined with HCI (3.4 mmol) and Pd(OH)2 (1.5 g) in
EtOH and hydrogenated (55 psi) at 55°C for 48 hrs. The catalyst
was
removed by filtration through Dicalite""and the solvent was evaporated in
vacuo. The residue was dissolved in water, washed twice with Et20, and
then basified with Na2C03 and extracted twice with EtOAc. The combined
extracts were dried (K2C03), filtered and solvent evaporated. The residue
was chromatographed on flash silica gel (99:0.75:0.25
EtOAc/MeOH/NH40H) to give a thick syrup which was dissolved in 2-PrOH
and combined with fumaric acid (116 mg). The solvent was evaporated and
the residue recrystallized from acetone to give the target compound, m.p.
140-141 °C. ~ H NMR (DMSO-ds) supported the assigned structure: S 2.3
(s,
3H), 3.75 (s, 2H), 6.6 (s, 2H), 6.75 (s, 1 H), 6.85 (d, J = 5.3 Hz, 1 H), 7.15
(d,
1 H), 7.65 (s, 1 H). Elemental Analysis: Calc. for C9H1oN2S~CaH404 C,
53.05; H, 4.79; N, 9.52. Found C, 53.22; H, 4.87; N, 9.50.
- 13 -
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
EXAMPLE 2
M HO
r~
s
To a solution of 3-bromo-4-methylthiophene (5.3 g, 30 mmol) in 100
mL of dry Et20 cooled to -78°C was added n-BuLi (20.0 mL, 32 mmol)
dropwise. The reaction mixture was allowed to slowly warm to -20°C and
was maintained at this temperature for 30 min. DMF (4.6 mL, 60 mmol) was
added and, the reaction mixture was allowed to come to ambient
temperature overnight. The reaction was quenched with aqueous
ammonium chloride, and the mixture was extracted twice with Et20. The
organic layers were combined and washed twice with water and then brine
and dried (MgS04). After filtration, the solvent was evaporated in vacuo.
The residue was chromatographed on flash silica gel (98/2 hexane : Et20) to
give 4-methylthiophene-3-carboxaldehyde, ~, (1.9 g, 50 %) as a fight
yellow oil. ~H NMR (CDCI3) supported the assigned structure.
M
v ~s
N
OH
To a solution of N-trityl-4-iodo-imidazole (11.8 g, 27 mmol) in dry
CH2Cl2 (75 mL) was added EtMgBr (10.0 mL, 3.0 M in Et20), and the
solution was stirred for 3 hrs. Then a solution of 4-methylthiophene-3-
carboxaldehyde (3.3 g, 27 mmol) in CH2CI2 (25 mL) was added. The
reaction mixture was stirred at room temperature overnight and then was
quenched with aqueous NH4CI. The mixture was transferred to a separatory
funnel, and the aqueous layer was extracted with a second portion of
- 14
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
CH2Cl2. The combined extracts were washed with a small portion of water,
dried (Na~S04), and filtered. The solvent was evaporated in. vacuo to give a
thick syrup which was triturated with Et20 to give a solid which was
recrystallized with charcoal treatment from EtOAc to give (4-methylthien-3-
yl)-1-trityl-imidazol-4-yl-methanol, ~. ~H NMR (CDCI3) supported the
assigned structure.
M
HN
~s
N
A solution of (4-methylthien-3-yl)-1-trityl-imidazol-4-yl-methanol (2.5
g, 5.7 mmol) was combined with 1~[ HCI (6 mL) and Pd(OH)2 (1.25 g) in
EtOH and hydrogenated (55 psi) at 50°C for 48 hrs. The catalyst
was
removed by filtration through Dicalite, and the solvent was evaporated in
vacuo. The residue was dissolved in water, washed twice with Et20, and
then basified with Na2C03 and extracted twice with EtOAc. The combined
extracts were dried (K2C03), filtered, and the solvent was evaporated. The
residue was dissolved in 2-PrOH and combined with fumaric acid (0.57 g, 1
eq.). After standing overnight a white solid was collected and recrystallized
from acetone to give the title compound m.p. 142-144°C. ~ H NMR (DMSO-
ds) supported the assigned structure: b 2.15 (s, 3H), 3.75 (s, 2H), 6.6 (s,
2H),
6.75 (s, 1 H), 7.05 (m, 1 H), 7.15 (m, 1 H), 7.65 (s, 1 H). Elemental
analysis:
Calc. for C9H~oN2S~C4H404 C, 53.05; H, 4.79; N, 9.52. Found C, 53.03; H,
4.73; N, 9.38.
- 15 -
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
EXAMPLE 3
4-f,1-(4-Methylthien-3-y)~~ -1 H-imida nla Fnmarata
M
C. v ~ s
N
O
To a solution of (4-methylthien-3-yl)-1-trityl-imidazol-4-yl-methanol,
~2, (6.5 g , 14.9 mmol) in 100 mL of CH2CI2 was added Mn02 (13 g). The
mixture was stirred at room temperature for 3 hr and then filtered through
Dicalite and the solvent was evaporated in vacuo to give (4-methylthien-3-
yl}-1-trityl-imidazol-4-yl-methanone, ~. 1H NMR (CDC13) supported the
assigned structure.
M
~ S
C ~ ~,
N
Me COH
To a solution of (4-methylthien-3-yl)-1-trityl-imidazol-4-yl-methanone,
~, (6.5 g, 14.9 mmol) in 75 mL of THF was added MeMgBr (3.0 M in Et20)
until TLC indicated complete reaction of starting material. The reaction was
quenched with aqueous NH4CI and extracted twice with EtOAc. The organic
extracts were combined, washed with water, then dried (Na2S04) and
filtered. The solvent was evaporated in vacuo and the residue was triturated
with Et20 to give 1-[{4-methylthien-3-yl)-1-trityl-imidazol-4-yl)-ethanol, ~.
~H NMR (CDCI3) supported the assigned structure.
- 16 -
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
ste~C
M
v ~s
N
Me
A solution of 1-((4-methylthien-3-yl)-i-trityl-imidazol-4-yl]-ethanol, ~,
(3.1 g, 6.9 mmol), 1 N HCI (7.1 mL) and Pd(OH)2 (1.75 g) in 50 mL of EtOH
was hydrogenated (60 psi) at 50°C, for 60 hrs. After cooling, the
catalyst
was removed by filtration and solvent evaporated in vacuo. The residue was
dissolved in water, and was washed twice with Et20, then basified with
Na2C03 and extracted twice with EtOAc. The organic extracts were
combined, dried (K2C03), and filtered. The solvent was evaporated in vacuo
and the residue was combined with fumaric acid (0.73 g, 1 eq) in 2-PrOH. A
white solid was collected and recrystallized from acetone to give the target
compound (1.8 g, 51 %) as a white crystalline solid, m.p. 132-134°C. ~
H
NMR (DMSO-ds) supported the assigned structure. 81.5 (d, J = 7.1 Hz, 3H),
2.1 (s, 3H), 4.05 (q, 1 H), 6.6 (s, 2H), 6.65 (s, 1 H), 7.1 (s, 2H), 7.65 (s,
1 H).
Elemental analysis: Calc. for C~oH~2N2S~C4H404 C, 54.53; H, 5.23; N, 9.08.
Found C, 54.44; H, 5.37; N, 9.00.
EXAMPLE 4
M
TrN
\ 's
N
EtxOH
A4
To a solution of (4-methylthien-3-yl)-1-trityl-imidazol-4-yl-methanone,
~, (2.1 g, 4.8 mmol) in 35 mL of THF was added 4.0 mL of EtMgBr (3.0 M in
Et20). The reaction was quenched with aqueous NH4C1 and extracted twice
with Et20. The organic extracts were combined, washed with water and
brine and then dried (Na2S04) and filtered. The solvent was evaporated in
_ 17 _
CA 02250930 1998-09-25
WO 97/35858 PCTILTS97/04773
vacuo , and the residue (1-[(4-methylthien-3-yl)-1-trityl-imidazol-4-yl]-
propanol), ~, was used directly in the next step.
M
N
Et
A solution of 1-[(4-methylthien-3-yl)-1-trityl-imidazol-4-yl]-propanol,
~, 1 N HCI (5.0 mL) and Pd(OH)2 (1.5 g) in 40 mL of EtOH was
hydrogenated (60 psi) at 50°C overnight. An additional 0.5 g of
catalyst was
added and hydrogenation was resumed overnight once again. After
cooling, the catalyst was removed by filtration and solvent evaporated in
vacuo. The residue was dissolved in water, and was washed twice with
Et20, then basified with Na2C03 and extracted twice with EtOAc. The
organic extracts were combined, dried (K2C03), and filtered. The solvent
was evaporated in vacuo, and the residue was chromatographed on flash
silica gel (99:0.75:0.25 EtOAc/MeOH/NH40H) to yield the title compound as
a free base (0.38 g, 38% for 2 steps). This was combined with fumaric acid
(0.21 g) in 2-PrOH and the solvent was evaporated in vacuo. The residue
was recrystallized from acetone to give the target compound (0.30 g) as a
white crystalline solid, m.p. 101-105°C. ~H NMR (DMSO-ds) supported the
assigned structure. b 0.85 (t, 3H), 1.95 (m, 2H), 2.15 (s, 3H), 3.85 (t, 1 H),
6.6
(s, 2H), 6.75 (s, 1 H), 7.05 (m, 1 H), 7.15 (m, 1 H), 7.65 (s, 1 H). Elemental
analysis: Calc. for C»H~4N2S~C4H4O4 C, 55.89; H, 5.63; N, 8.69. Found C,
55.87; H, 5.69; N, 8.56
_ 18 _
CA 02250930 1998-09-25
WO 97/35858 PCT/LTS97/04773
EXAMPLE 5
X3.5 dimethylthi .n- -yjlmet yll-1H-imid,~gle F~ma_r~,tp
Me
TrN
' S
N ~ 1
OH Me
g~
To a solution of N-trityl-4-iodo-imidazole (11.8 g, 27 mmol) in dry
CH2CI2 (200 mL) was added EtMgBr (11.0 mL, 3.0 M in Et20). After
complete halogen-metal exchange this solution was cannulated into a
solution of 2,5-dimethylthiophene-3-carboxaldehyde (3.5 g, 25 mmol) in 50
mL of CH2CI2. The reaction mixture was stirred at room temperature for 1 hr
and then quenched with aqueous NH4CI. The mixture was transferred to a
separatory funnel and the aqueous layer was extracted with a second
portion of CH2CI2. The combined extracts were dried (MgS04) and filtered.
The solvent was evaporated in vacuo to give a thick syrup which was
triturated with Et20 to give a solid which was recrystallized from acetone to
give (2,5-dimethytthien-3-yl)-1-trityl-imidazol-4-yl-methanol, ~. ~H NMR
(CDCI3) supported the assigned structure.
Me
HN
N
Me
A solution of [(2,5-dimethylthien-3-yl)-1-trityl-imidazol-4-ylJ-methanol,
~, (3.4 g, 6.9 mmol), concentrated HCI (0.31 g) and Pd(OH)2 (1.75 g) in 40
mL of 95% EtOH was hydrogenated (60 psi) at 50°C, for 60 hrs. After
cooling, the catalyst was removed by filtration and solvent evaporated in
vacuo. The residue was dissolved in water, and was washed twice with
Et20, then basified with Na2C03 and extracted twice with EtOAc. The
_ 19 _
CA 02250930 1998-09-25
WO 97/35858 PCT/US97104773
organic extracts were combined, dried (K2C03), and filtered. The solvent
was evaporated in vacuo and the residue was combined with fumaric acid in
2-PrOH. A white solid was collected and recrystallized from acetone to give
the target compound (0.63 g, 27%) as a white crystalline solid m.p. 148-
149°C. ~H NMR (DMSO-ds) supported the assigned structure. b 2.37 (s,
3H), 2.39 (s, 3H), 3.65 (s, 2H), 6.6 (s, 2H), 6.7 (s, 1 H), 7.65 (s, 1 H).
Elemental analysis: Calc. for C~oH~2N2S~C4H404 C, 54.53; H, 5.23; N, 9.08.
Found C, 54.74; H, 5.10; N, 9.00.
EXAMPLE 6
4-((2.5-diethylthien-3-,yf)met!b,yll-1 H-imi azole Fumarate
0
To a mixture of 2-ethylthiophene (56.1 g, 0.5 mol) and acetic
anhydride (59 mL) cooled in an ice bath was added 1 mL of HC104, The
reaction mixture became quite dark and there was a vigorous exothermic
reaction. After 1 hr, the mixture was diluted with CH2CI2 and poured onto
ice/NaHC03. This mixture was transferred to a separatory funnel, and the
organic Payer was washed with an additional portion of dilute NaHC03,
water, dried (MgS04) and filtered. The solvent was evaporated in vacuo to
give a brown oil which was distAled in vacuo (6-8 mm Hg). The product, ~,
was collected at 120-121 °C as a colorless liquid. ~ H NMR (CDCI3)
supported the assigned structure.
- ZO -
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
Et S Et
5-Ethyl-2-acetythiophene, ~, (18.5 g, 0.12 mol) was added to
hydrazine hydrate (30.0 mL) in 75 mL of ethylene glycol, and the mixture
was heated in an oil bath to 170°C. The excess hydrazine and water were
distilled out of the reaction mixture. After cooling to room temperature, KOH
(24.9 g, 0.44 mof) was added and again the mixture was heated in an oil
bath to 120°C, at which point a vigorous reaction and gas evolution
began.
Heating at 120-130°C was continued as the product was distilled
from the
reaction mixture. The distillate was extracted twice with Et20. The extracts
were then combined and washed with 3N HCI, water and finally brine and
then dried (MgS04) and filtered. The solvent was evaporated in vacuo and
the residue was distilled at ambient pressure (175-177°C) to give 2,5-
diethylthiophene, ~, (10.3 g, 61%) as a colorless liquid. ~H NMR (CDCI3)
supported the assigned structure.
Br
Et S Et
A solution of Br2 (6.4 g, 40.0 mmol) in CHC13 (25 mL) was added
dropwise to a solution of 2,5-diethylthiophene, ,~, (5.6 g, 40 mmol) in CHCI3
(75 mL). The reaction was stirred for 2 hrs at room temperature and then
poured onto ice/NaHS03. The organic layer was then washed with
saturated NaHC03, and then water and then dried (MgS04) and filtered.
The solvent was evaporated in vacuo and the residue was distilled at
reduced pressure (1 mm Hg) and 3-bromo-2,5-diethylthiophene, ~, was
collected as 5.0 g (57%) of a clear liquid b.p. 79-81°C @ 1 mm Hg. 1H
NMR
(CDC13) supported the assigned structure.
_ 21 _
CA 02250930 1998-09-25
WO 97/35858 PCT/ITS97/04773
HO
~ ~-Et
Et S
To a soi:~;ion of 3-bromo-2,5-diethyl thiophene, ~, (9.1 g, 41 mmol)
in 100 mL of ary Et20 cooled to -78°C was added n-BuLi (26.2 mL, 42
mmol)
dropwise. The bath temperature was allowed to rise to -20°C and DMF
(6.3
mL, 82 mmol) was added. The reaction mixture was allowed to warm to
room temperature overnight. The reaction was quenched with NH4CI (aq)
and extracted with Et20. The organic layer washed twice with water and
brine and then dried (MgS04). After evaporation of solvent, the crude
product was purified on flash silica gel (98:2 hexane/Ei20) to afford 2,5-
diethylthiophene-3-carboxaldehyde, Due, as a light yellow oil (5.0 g, 72%).
~H NMR (CDCI3) supported the assigned structure.
stp,~
Et
TrN
\ S
\Et
To a solution of N-trityl-4-iodo-imidazole (13.5 g, 31 mmol) in dry
CH2CI2 (75 mL) was added EtMgBr (10.0 mL, 3.0 M in Et20) and the solution
was stirred for 3 hrs. Then a solution of 2,5-diethylthiophene-3-
carboxaldehyde, ~, (5.0 g, 30 mmol) in CH2CI2 (20 mL) was added, and the
reaction mixture was stirred at room temperature overnight. The reaction
was quenched with NH4CI (aq) and the mixture was transferred to a
separatory funnel. The aqueous layer was extracted with a second portion
of CH2CI2. The extracts were combined and washed with a small portion of
water, dried (Na2S04), and filtered. The solvent was evaporated in vacuo to
give a thick syrup which was triturated with Et20 to give a solid which was
recrystallized from EtOAc to give (2,5-diethylthien-3-yl)-1-trityl-imidazol-4-
yl-
methanol, ~. iH NMR (CDC13) supported the assigned structure.
- 22 -
CA 02250930 1998-09-25
WO 97135858 PCT/CTS97/04773
Et
~ S
Et
To a solution of TFA (9.2 mL, 120 mmol) in dry CH2C12 (50 mL) cooled
in an ice bath, was added BH3~Me2S (90.0 mL, 1.0 M in CH2CI2) dropwise.
This was stirred at 0°C for an additional 90 min and then [(2,5-
diethylthien-3-
yi)-1-trityl-imidazol-4-yl]-methanol, ~, (1.4 g, 3 mmol) in CH2CI2 (25 mL)
was added in one portion and reaction mixture was allowed to come to room
temperature overnight. The reaction was quenched by the addition of 100
mL of 3:1 MeOH/3N HCI followed by refluxing for 2 hrs. Most of the solvent
was then evaporated in vacuo. The residue was dissolved in water and
washed twice with Et20, then basified with Na2C03 and extracted twice with
EtOAc. The organic extracts were combined, dried (K2C03) and filtered.
The solvent was evaporated in vacuo to give a syrup (0.69 g), which was
combined with fumaric acid (0.36 g) in MeOH. The solvent was evaporated
and the residue was recrystaUized from acetone to give the title compound
(0.70 g, 70%) as a white solid, m.p. 115-116.5°C. ~H NMR (DMSO-ds)
supported the assigned structure: 1.2 (m, 6H), 2.7 (m, 4H), 3.7 (s, 2H), 6.55
(s, 1 H), 6.6 (s, 2H), 6.75 (s, 1 H), 7.6 (s, 1 H). Elemental Analysis: Calc
for
2O C~2H~6N2S-C4H4O4 C, 57.13; H, 5.99; N, 8.33. Found C, 57.06; H, 6.06; N,
8.27.
- 23 -
CA 02250930 1998-09-25
WO 97/35858 PCT1US97/04773
EXAMPLE 7
Br
Et
To a mixture of 2-ethylthiophene (11.2 g, 0.100 mol) and sodium
acetate (16.4 g, 0.200 mol) in 75 mL of water was added bromine (32.0 g,
0.200 mol). The reaction mixture was stirred for 2 days. GC analysis
indicated that some monobrominated material was left so additional bromine
(7.75 g) and sodium acetate (5.00 g) were added. After a few hours of
stirring, GC analysis indicated that the monobromo material was gone so
zinc (19.6 g, 0.30 mol) was added in portions. The reaction mixture was
then refluxed for 25 h. The product was distilled out of the reaction mixture.
The distillate was extracted with ether twice. The ether extracts were
combined, washed with aqueous sodium bicarbonate, water, and brine, and
then dried (MgS04). The solution was concentrated in vacuo, and then
distilled under reduced pressure to provide 10.1 g (53%) of 3-bromo-2-
ethylthiophene, ~, b.p. 49-50°C[~4mmHg. The ~H NMR in CDCI3
supported the desired product structure.
step g
HO
Et
A solution of 3-bromo-2-ethylthiophene, A7, (8.9 g, 0.0465 mol) in 50
mL of diethyl ether was cooled to -78°C, and a solution of n-BuLi (29.0
mL,
1.6M) in hexanes was added dropwise. When the addition was complete,
the reaction was stirred at -78°C for 5 min. Then DMF (5.1 g, 0.070
mol) was
cannuiated into the reaction mixture which was allowed to warm to ambient
temperature and was stirred overnight. The reaction was quenched with
- 24 -
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
water and extracted twice with diethyl ether. The organic extracts were
combined, washed with twice with water and then brine and dried (MgS04).
The solution was filtered and concentrated to provide an oil which was
purified on flash silica gel with 97.5:2.5 hexanes:diethyl ether to give 1.66
g
(25%) of 2-ethylthiophene-3-carboxaldehyde, ~. The ~H NMR in CDCI3
supported the desired product structure.
TrN Et g
N
OH
To a solution of N-trityl-4-iodo-imidazole (4.1 g, 0.0095 mot) in dry
CH2CI2 (75 mL) was added a solution of MeMgBr (4.0 mL, 3.0 M) in diethyl
ether and the solution was stirred for 3 hrs. Then a solution of 2-
ethylthiophene-3-carboxaldehyde, ~, (1.66 g, 0.0087 mol) in CH2CI2 (20
mL) was added and the reaction mixture was stirred at room temperature
overnight. The reaction was quenched with aqueous NH4CI and the mixture
was transferred to a separatory funnel. The aqueous layer was extracted
with a second portion of CH2CI2. The extracts were combined and washed
with a small portion of water, dried (Na2S04), and filtered. The solvent was
evaporated in vacuo, and the residue was triturated with Et20 to give (2-
ethyithien-3-yl)-1-trityl-imidazol-4-yl-methanol, ~, as a beige solid which
was taken on to the next step directly.
stel~D,
N " 1
Et
A solution of (2-methylthien-3-yl)-1-trityl-imidazol-4-yl-methanol, ~,
(0.9 g, 0.00199 mol) was combined with 1 N HCI (2.0 mL) and Pd(OH)2 (1.5
g) in EtOH and hydrogenated (55 psi) at 50°C for 48 hrs. The catalyst
was
removed by filtration through Dicalite, and the solvent was evaporated in
- 25 -
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
vacuo. The residue was dissolved in water, washed twice with Et20, and
then basified with Na2C03 and extracted twice with EtOAc. The combined
extracts were c!ned (K2C03), filtered and solvent evaporated. The residue
was dissolved in 2-PrOH and combined with fumaric acid. The solvent was
evaporated anc! the residue recrystallized from acetone to provide 4-[(2-
ethylthien-3-yl)m~ethylJ-1 H-imidazole fumarate, ~, as a white solid, m.p.
132-134°C. ~ H NMR (DMSO-ds) supported the assigned structure: 61.20
(t,
J = 7.5 Hz, 3H), 2.8 (q, J = 7.5 Hz, 2H), 3.80 (s, 2H), 6.65 (s, 2H), 6.70 (s,
1 H),
6.80 (d, 1 H), 7.20 (d, 2H), 7.60 (s, 1 H). Elemental Analysis: Calc. for
1 O C~pH~2N2S~C4H4O4 C, 54.33; H, 5.23; N, 9.09. Found C, 54.42; H, 5.17; N,
9.02.
EXAMPLE 8
~(f2.4-Dimethylthien- ~rj)m t yll-1 H-imidazole Fumarate
Br Me
Me /S"Br
8$
A solution of 2,4,5-tribromo-3-methylthiophene (50.2 g, 0.15 mol;
Gronowitz, S.; Moses, P. Hakansson Arkiv. f. Kemi. 1960, 14, 267) in 400
mL of diethyl ether was cooled to -78°C, and a solution of nl3uLi (100
mL,
1.6M) was added dropwise. The starting material precipitated out of
solution, but when n-BuLi added, the reaction mixture became stirrable
again. When the addition was complete, the reaction mixture was stirred for
20 min, and then a solution of dimethyl sulfate (75.7 g, 0.600 mol) in 200 mL
of diethyl ether which was cooled to -50°C was added by cannulation.
When addition was complete, the reaction mixture was allowed to warm to
ambient temperature and was stirred overnight. The reaction was quenched
with 100 mL of 6N NaOH solution and stirred for 2h. The mixture was
transferred to a separatory funnel, and the aqueous layer was separated
and extracted with additional ether. The organic layers were combined,
washed with water and brine and dried (MgS04). The suspension was
filtered and concentrated to give an oil. Vacuum distillation provided 22.1 g
- 26 -
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
(55%) of 2,4-dibromo-3,5-dimethyithiophene, g$, b.p. 71-72°C a~ 0.4
mmHg.
The ~H NMR in CDCI3 supported the assigned structure.
Br Me
Me
A solution of 2,4-dibromo-3,5-dimethylthiophene, ~, (22.0 g, 0.081
moi) in 250 mL of THF was cooled to -78°C. Then a solution of n-BuLi
(53
mL, 1.6M) in hexanes was cooled to -78°C and added via cannutation. The
reaction was stirred for 3 h, and then was quenched with aqueous
ammonium chloride. The mxiture was extracted twice with diethyl ether.
The organic layers were combined, washed with water and brine and dried
(MgS04). The suspension was filtered and concentrated to give an oil.
Vacuum distillation provided 7.6 g (49%) of 3-dibromo-2,4-dimethyl
thiophene, ~$, b.p. 77-79°C @ 5 mmHg, as a nearly colorless liquid. The
1H
NMR in CDCI3 supported the assigned structure.
CHO Me
Me g
A solution of 3-bromo-2,4-dimethylthiophene, ~, (5.9 g, 0.031 mol) in
200 mL of diethyl ether was cooled to -78°C, and a solution of n-BuLi
(25.0
mL, 1.6M) in hexanes was added dropwise. When the addition was
complete, the reaction was stirred at -78°C for 4 h. TLC analysis
indicated
very tittle conversion so reaction mixture was warmed slowly to -25°C.
Then
a solution of DMF (4.5 g, 0.062 mol) in 25 mL of ether was cannuiated into
the reaction mixture which was allowed to warm to ambient temperature and
was stirred overnight. The reaction was quenched with water and extracted
twice with diethyl ether. The organic extracts were combined, washed with
twice with water and then brine and dried (MgS04). The solution was
- 27 -
CA 02250930 1998-09-25
WO 97/35858 PCT/L1S97104773
filtered and concentrated to provide an amber oil which was dissolved in
hexane. The solution was treated with charcoal, filtered through Dicalite,
and concentrated to give 2,4-dimethylthiophene-3-carboxaldehyde, ~$,
which was used directly in the next step.
step
Tr Me g
1 I
N 1' \
OH Me
J~
To a solution of 4-iodo-1-trityl imidazole (11.8 g, 0.027 mol) in 75 mL
of dry dichloromethane under nitrogen was added dropwise a solution of
methyl magnesium bromide in diethyl ether (9.0 mL, 3.OM). When addition
was complete, the reaction mixture was stirred for 1 h at 25~C. Then, 2,4-
dimethylthiophene-3-carboxaldehyde, ~$, (3.8 g, 0.027 mol) was added as
a solution in 20 mL of dichloromethane. After overnight stirring at ambient
temperature, the reaction was quenched with saturated ammonium chloride
solution. The layers were separated, and the aqueous layer was extracted
again with dichloromethane. The organic layers were combined, dried
(Na2S04), and concentrated in vacuo. The residue was triturated with ethyl
acetate to provide (2,4-dimethylthieno-3-yl}-1-trityl-imidazol-4yl-methanol,
p$, as an off-white solid which was taken on directly in the next step.
- 28 -
CA 02250930 1998-09-25
WO 97135858 PCT/US97/04773
N Me S
N
Me
A solution of BH3~Me2S (120 mL, 1.0 M) in dichloromethane was
added dropwise to a solution of TFA (18.2 g, 0.16 mol) in 50 mL of dry
dichloromethane at 0°C. When the addition was complete, the reaction
mixture was stirred for 2 h. Then the carbinol, j~, (1.8 g, 0.040 mol) was
added, and the reaction mixture was warmed to ambient temperature and
stirred overnight. The reaction was quenched with 100 mL of 1.5N HCI, and
then the mixture was refluxed on a steam bath for 2 h. The solution was
cooled and then concentrated in vacuo to provide a brown oil. The residue
was dissolved in water. This solution was washed twice with ether, basified
with Na2C03 and extracted with ethyl acetate. The ethyl acetate extracts
were combined, dried (Na2S04), and concentrated in vacuo. The residue
was purified on flash silica gel using 97.5:2.5 chloroform: 10% ammonium
hydroxide in methanol. The isolated material was dissolved in isopropanol,
and fumaric acid was added. The solvent was removed under reduced
pressure, and the residue was recrystallized from acetone to provide 0.274 g
of 4-[-(2,4-dimethylthien-3-yl)methyl)-1 H-imidazole fumarate, ~, as a
white solid, m.p. 160-162°C. The ~ H NMR in DMSO-ds supported the
assigned structure: 62.10 (s, 3H, Me), 2.40 (s, 3H, Me), 3.70 (s, 2H, CH2),
6.55 (s, 1 H), 6.65 (s, 2H), 6.85 (s, 1 H), 7.60 (s, 1 H). Elemental analysis:
Calculated for CiaH~2N2S~C4H404: C, 54.54; H, 5.23; N, 9.08. Found C,
54.45; H, 5.26; N, 9.06.
- 29 -
CA 02250930 1998-09-25
WO 97135858 PCT/US97/04773
EXAMPLE 9
4-j(4-E~hvfthien-3 yl meth~i!]-1 H-imidazole Fumarate
Br E1
Br S Br
To an ice-cooled solution of 3-ethylthiophene (25.75 g, 0.23 mol) in
75 mL of chloroform was added bromine (111.87 g, 0.7 mol). The reaction
mixture was allowed to warm to ambient temperature and was left to stir
overnight. Analysis by GC indicated that >90% of a single product was
present so the reaction mixture was poured onto ice. The mixture was
transferred to a separatory funnel and diluted with additional chloroform.
The layers were separated and the organic layer was washed with 200 mL
of 10% NaHS03 solution, water, and brine, and dried (MgS04). After
filtration, concentration in vacuo provided a dark oil which was distilled
under reduced pressure to provide 2,3,5-tribromo-4-ethylthiophene, $~. GC
analysis indicated that the product was reasonably pure and it was taken on
directly.
std. ,~B
Br Et
S
B~
A suspension of zinc (24.5 g, 0.375 mol} in 250 mL of 10% aqueous
acetic acid was placed in a round bottom flask fitted with a mechanical
stirrer. The suspension was heated at reflux, and 2,3,5-tribromo-4-
ethylthiophene, Ate, (26.1 g, 0.0750 mol) was added in portions. Reflux was
continued overnight, and then the product was removed by steam
distillation. The distillate was transferred to a separatory funnel and
extracted twice with ether. The ether layers were combined, washed with
saturated sodium bicarbonate solution, and dried (MgS04). After filtration,
- 30 -
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
the solution was concentrated to give 7 g of a clear oil. The original
reaction
pot was resubjected to steam distillation to provide a second batch of
product. Both batches contained a mixture of desired product and dibromo
compound. These were purified on flash silica gel with pentane as eluant to
provide 3.4 g of 3-bromo-4-ethylthiophene, ~, as a clear liquid. This
material was taken on directly in the next step.
CH Et
S
A solution of 3-bromo-4-ethylthiophene, B~, (3.4 g, 0.018 mol) in 40
mL of diethyl ether was cooled to -78°C, and a solution of n-BuLi (12.0
mL,
1.6M) in hexanes was added dropwise. The solution was allowed to warm
to -20°C, and DMF (1.46 g, 0.020 mol) was added. The reaction mixture
was
allowed to warm to ambient temperature and was stirred overnight. The
reaction was quenched with aqueous ammonium chloride solution and
extracted twice with diethyl ether. The organic extracts were combined,
washed with twice with water and then brine and dried (MgS04). The
solution was filtered and concentrated to provide 4-ethylthiophene-3-
carboxaldehyde, ~, which was used directly in the next step.
step p
TrN S
N
OH Et
To a solution of 4-iodo-1-trityl imidazoie (3.9 g, 0.0090 mol) in 40 mL
of dry dichioromethane under nitrogen was added dropwise a solution of
ethyl magnesium bromide in diethyl ether (3.0 mL, 3.OM). When addition
was complete, the reaction mixture was stirred for 1 h at 25~C. Then, 4-
ethyl-thiophene-3-carboxaldehyde, ~, (1.2 g, 0.0086 mol) was added as a
solution in 20 mL of dichloromethane. After overnight stirring at ambient
- 31 -
CA 02250930 1998-09-25
WO 97135858 PCT/US97/04773
temperature, the reaction was quenched with saturated ammonium chloride
solution. The layers were separated, and the aqueous layer was extracted
again with dichloromethane. The organic layers were combined, dried
(Na2S04), and concentrated to provide an orange-yellow solid. This
material was recrystallized from ethyl acetate to provide (4-ethylthiophen-3-
yt)1-trityl-imidazo-4yl-methanol, ~,Q, which was taken on directly in the next
step.
S
~\ I~
N " 1
Et
A solution of (4-ethylthien-3-yl)-1-trityl-imidazol-4-yl-methanol, ~, in
40 mL of ethanol containing 1 N hydrochloric acid (1.7 mL) and palladium
hydroxide (0.75 g) was shaken with hydrogen at 60 psi at 50~C on a Parr
hydrogenator for 3 days. The solution was cooled and filtered to remove the
catalyst. The filtrate was concentrated under reduced pressure. The residue
was dissolved in water and extracted twice with Et20 then basified with
sodium carbonate and extracted with ethyl acetate. The organic layers were
combined, dried (K2C03), and concentrated in vacuo. The residue was
dissolved in 2-propanol and fumaric acid was added. The solution was
concentrated in vacuo, and the residue was recrystallized from acetone to
provide 0.245 g of 4-[(4-ethylthien-3-yl)methyl]-1 H-imidazole fumarate, ~,
as a white solid, m.p. 142-144°C. The ~ H NMR in DMSO-d6 supported the
assigned structure: 61.20 (t, 3H, Me), 3.8 (s, 2H), 6.65 (s, 2H), 6.67 (s, 1
H),
7.05 (m, 1 H), 7.10 (m, 1 H), 7.60 (s, 1 H), 7.55 (s, 1 H). Elemental
analysis:
Calculated for C~pH~2N2S~C4H404: C, 54.53; H, 5.23; N, 9.08. Found C,
54.58; H, 5.33; N, 9.03.
- 32 -
CA 02250930 1998-09-25
WO 97135858 PCT/US97/04773
EXAMPLE 10
4-i(4-Ethvlthien-'~-yllet y~l-1 H-imida nlP Fur
S
N ~ \
O Et
To a solution of the carbinol, j,~, (1.17 g, 2.6 mmol) in 50 mL of
dichloromethane was added Mn02 (5.0 g). The reaction mixture was stirred
overnight and then was filtered. The filtrate was concentrated in vacuo to
provide 4-(4-ethylthien-3yl)-1-trity!-imidazol-4yl methanone, ~, which was
used directly in the next step.
S
N x
HO Me Et
A solution of methylmagnesium bromide (1.0 mL, 3.OM) in diethyl
ether was added to an ice-cooled solution of 4-(4-ethylthien-3-yl)-1-trityl-
imidazol-4yl methanone, ~,Q, (1.17 g, 0.0026 mol) in 25 mL of THF. After 30
min of stirring, TLC analysis indicated that some starting material was left
so
an additional 1.0 mL of methylmagnesium bromide was added, and the
reaction mixture was stirred over the weekend. The reaction was quenched
with aqueous ammonium chloride solution, and the resulting mixture was
extracted twice with ethyl acetate. The ethyl acetate extracts were
combined, washed with water and brine, dried (Na2S04), and filtered.
Concentration provided 1-(4-ethylthien-3-yl)-1-trityl-imidazol-4-yl ethanol,
~1Q, as an oil which crystallized on standing. This material was taken on
directly in the next step.
- 33 -
CA 02250930 1998-09-25
WO 97/35858 PCT/US97/04773
S
N
Me Et
A solution of 1-{(4-ethylthien-3-yl)-1-trityl-imidaz-4-ylJ-ethanol, ~Q, in
40 mL of ethanol containing 1 N hydrochloric acid (2.5 mL) and palladium
hydroxide (1.0 g) was shaken with hydrogen at 60 psi at 50~C on a Parr
hydrogenator for 3 days. The solution was cooled and filtered to remove the
catalyst. The filtrate was concentrated under reduced pressure. The residue
was dissolved in water and extracted twice with Et20, then basified with
sodium carbonate and extracted with ethyl acetate. The organic layers were
combined, dried (K2C03), and concentrated in vacuo. The residue was
purified on a silica gel column on a Foxy apparatus using 99:0.75:0.25 ethyl
acetate:methanol:ammonium hydroxide as eluant to provide a glass. This
material was dissolved in 2-propanol and fumaric acid was added. The
solution was concentrated in vacuo, and the residue was recrystallized from
acetone to provide 0.323 g of 4-[1-(4-ethylthien-3-yl)ethyl]-1 H-imidazole
fumarate, ~Q, as a white solid, m.p. 145-147°C. The ~ H NMR in DMSO-
ds supported the assigned structure: 61.50 (t, 3H, Me), 1.50 (d, 2H, Me), 4.05
(q, 1 H, CH), 6.60 (s, 3H), 7.05 (d, 1 H), 7.15 (d, 1 H), 7.55 (s, 1 H).
EIemeMal
analysis: Calculated for C> > H~ 4N2S~C4H4O4: C, 55.89; H, 5.63; N, 8.69.
Found C, 55.79; H, 5.47; N, 8.59.
- 34 -