Note: Descriptions are shown in the official language in which they were submitted.
CA 02251060 1998-10-07
WO 97/37988 PCT/EP97/OI475
FILE, P~-~! TI1B~ A~FE
T-L~-r 'i'~: ANSLA'1'I~N
IS
ACYLATED N-HYDROXYMETHYL THALIDOMIDE PRODRUGS
HAVING AN IMMUNOMODULATORY EFFECT
This invention relates to thalidomide prodrugs, to a method of producing them,
and to
the use of the same as a pharmaceutical active ingredient.
The excessive formation of the cytokinin TNF-a. (tumour necrosis factor a)
plays a
central part in the pathogenesis of graft-versus-host syndrome, of multiple
sclerosis, of
transplant rejection, aphthous stomatitis, erythema nodosum leprosum, morbus
Boeck,
rheumatoid arthritis and a series of other diseases which are associated with
inflammatory symptoms. One basis for the therapy of these diseases consists of
the
targeted suppression of the release of TNF-a,, by administering
immunomodulating
active ingredients, such as dexamethasone, pentoxifylline or thalidomide for
example.
In the treatment of aphthous stomatitis, thalidomide has proved to be superior
to
classical immunosuppressants. Other examples of diseases in which thalidomide
has
exhibited good efficacy without resulting in a general immunosuppression
include
CA 02251060 1998-10-07
WO 97/37988 PCT/EP97/01475
2
cutaneous lupus erythematosus, pyoderma gangrenosum and orogenital ulcers with
morbus Beh~et, as well as ulcerations in HIV-infected patients, which do not
differ
histologically from aphthous ulcers and in which - in contrast to the majority
of HIV-
associated mucocutaneous lesions - no microbial instigators can be detected.
As
distinct from stomatitis aphthosa, these lesions, which can be characterised
as major
aphthae, occur in the entire digestive tract, and when located in the
pharyngeal space or the
oesophagus make the absorption of food difficult, and also make the taking of
oral
medication difficult, due to the pain which they cause. The pathogenetic
factors are
endogenous mediators which have effects on the endothelium and on circulating
leukocytes. Under the influence of locally-formed TNF-a and other cytokinins,
there is
a marked increase in the adhesiveness of the endothelium in relation to
leukocytes,
which makes a definitive contribution to the development of venous vasculitis.
Substances which, like thalidomide, suppress this alteration of the
endothelium without
at the same time blocking the specific cellular immune defence, can constitute
an
I S important advance in therapy.
In severe cases of pharyngeal or oesophageal ulcers, in which the taking of
oral
medication is made difficult, or in which this may even be impossible, and in
cases of
HIV-associated pathology in which severe symptoms of diarrhoea make the use of
oral
medication unpredictable, the parental administration of active ingredients is
appropriate. However, the low solubility of thalidomide in water (0.012 mg/ml;
Arch.
Pharm. 321, 371 (1988)) constitutes an obstacle to the parenteral application
of this
active ingredient.
Thalidomide derivatives are known from DE 42 11 812 which comprise a
benzoyloxymethyl group with an amine-bearing substituent on the nitrogen atom
of the
glutarimide residue. These thalidomide derivatives have a considerably higher
solubility
in water than that of thalidomide. The pH values of aqueous solutions of these
compounds are considerably lower than the physiological pH range, however, so
that it
is necessary to increase the plI before their application. In the course of
this procedure,
the corresponding bases are precipitated, with the consequence that the
advantage of
higher solubility in water is eliminated to a considerable extent or even
completely.
The underlying object of the present invention was to develop thalidomide
prodrugs
CA 02251060 1998-10-07
WO 97/37988 PCT/EP97/01475
3
which are water-soluble within the physiological pH range. The object was also
that
the compounds to be developed should not give rise to any toxicological
effects due to
the separation of non-physiological prodrug residues.
It has been found that the requirements imposed on the compounds to be
developed
are fulfilled by selected thalidomide prodiugs.
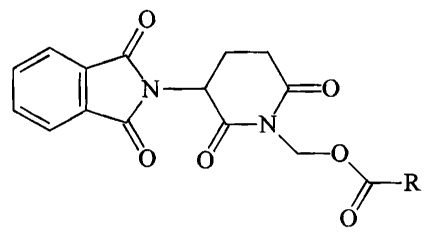
Accordingly, the present invention relates to thalidomide prodrugs of formula
I
0
i \
N ~O
N
0 0 ~-o~
h---R
~~O
wherein R denotes -CHR'-NHRZ or -(CHZ)"COOH, R' denotes H or C1_.~ alkyl, Rz
denotes H, C,_~ alkyl, C(O)-CHZ-NHR~ or an amino-protective group, R~ denotes
H or
I S an amino-protective group, and n is an integer between 2 and 4, in the
form of their
bases or salts of physiological acids.
The definition of the R' wherein R' is a C,_4 alkyl radical includes both
straight chain
and branched hydrocarbon radicals, which may be substituted with OH, COOH,
-C(O)NH2, NH2, -NHC(O)NH2, -NHC(NH)NHZ or S-C,_3-alkyl, or with a substituted
or unsubstituted phenyl group.
The definition of Rz wherein RZ is a C,_~ alkyl radical includes straight
chain and
branched hydrocarbon radicals.
Compounds which are suitable as thalidomide prodrugs of formula I, wherein R
denotes -CHR'-NHR2, are those in which the R' radical denotes H, CHI, -
CH(CH~)z,
-CHZCH(CH~)2 or -CH(CH~)CHZCH~, particularly those compounds in which the R'
CA 02251060 1998-10-07
WO 97/37988 PC'1'/EP97/01475
4
radicals denote H, CHI or -CH(CH~)2 and RZ denotes H, CH3,
C(O)-OC(CH3)3 or C(O)-O-CHZ-C~HS .
Of the thalidomide prodrugs of formula I in which R denotes -(CHz)"COOH, the
compound in which n is 2 is particularly suitable.
The present invention further relates to a method of producing thalidomide
prodrugs of
formula I, wherein R denotes -CHR'-NHR2, R' denotes H or C~_a alkyl, RZ
denotes I-I,
C~_~ alkyl, C(O)-CHZ-NHR~ or an amino-protective group, R~ denotes H or an
amino-
protective group, and R~ denotes H or an amino-protective group, which is
characterised in that N-hydroxymethylthalidomide is reacted with an amino
acid, the
amino function of which is protected, in the presence of a carbodiimide or
carbonyldiimidazole, and the protective group for the amino function is
subsequently
split ofd' by acidolysis if desired.
20
The t-butyloxycarbonyl radical and the benzyloxycarbonyl radical are
particularly
suitable as a protective group for the amino function. The reaction of N-
hydroxymethylthalidomide with a protected amino acid is conducted in the
presence of
equimolar to double equimolar amounts of a carbodiimide, for example
dicyclohexylcarbodiimide, or of a carbonyldiimidazole, in an organic solvent,
for
example dichloromethane, chloroform, acetone, dimethylformamide and/or
pyridine.
The reactions can be conducted in the presence of a catalyst, for example 4-
pyrrolidinopyridine or 4-dimethylaminopyridine.
Acidolytic cleavage of the amino-protective group is preferably effected with
trifluoroacetic acid, optionally in the presence of an organic solvent, for
example
dichloromethane.
The present invention also relates to a method of producing thalidomide
prodnigs of
formula I wherein R denotes -(CHz)"COOH, which is characterised in that N-
hydroxymethylthalidomide is reacted with an acid anhydride in the presence of
an
amore.
Succinic anhydride is preferably used as the acid anhydride. Triethylamine
and/or
CA 02251060 1998-10-07
WO 97/37988 PCT/EP97/01475
pyridine are suitable as the amines which are usually used in equimolar to
double
equimolar amounts with respect to N-hydroxymethylthalidomide. The reactions
are
usually conducted in an organic solvent, for example dichloromethane,
chloroform,
pyridine and/or dimethylformamide, in the presence of a catalyst, for example
4
5 pyrrolidinopyridine or 4-dimethylamino-pyridine.
Salts of compounds according to the invention with physiologically compatible
acids,
for example hydrochloric acid, hydrobromic acid, sulphuric acid,
methanesulphonic
acid, formic acid, acetic acid, oxalic acid, succinic acid, tartaric acid,
mandelic acid,
fumaric acid, lactic acid, citric acid, glutamic acid and aspartic acid, are
obtainable
either from the corresponding bases or front trifluoroacetates. For the
production of
hydrochlorides, the corresponding trifluoroacetates are preferably converted
into
hydrochlorides with the aid of a weakly basic anion exchanger. Short chain
aliphatic
alcohols, for example methanol, are preferred as solvents.
The compounds according to the invention can be applied as substances which
are
soluble in water in the physiological pH range between 7.0 and 7.5, and are
toxicologically harmless. Accordingly, the present invention also relates to
the use of a
thalidomide prodrug of formula I as an active ingredient in drugs, which are
preferably
administered parenterally.
In addition to at least one thalidomide prodrug of formula I, drugs according
to the
invention contain support materials, fillers, solvents, diluents, colorants
and/or binders.
The selection of these adjuvant substances and of the amounts to be used
depends on
whether the drug is to be administered intravenously, intraperitoneally,
intradermally,
intramuscularly, intranasally or locally.
The amount of active ingredient to be administered to the patient, which
depends on
the weight of the patient, on the type of parenteral application, on the
indication and on
the degree of severity of the illness, is usually between 0.1 and 10 mg/kg of
a
thalidomide prodrug of formula I.
Due to their pronounced immunomodulatory action, which does not result in a
general
immunosuppression, thalidomide prodrugs of formula I are suitable for the
treatment
CA 02251060 1998-10-07
WO 97/37988 PC'T/EP97/01475
G
of all diseases which are characterised by a high level of TNF-a, formation or
by focal
vasculides.
CA 02251060 1998-10-07
WO 97/37988 PCT/EP97/01475
7
Examples
Preparation of compounds according to the invention
t-butyl oxycarbonyl-protected amino acids were prepared by the method
described in
Hoppe-Seyler's Z. physiol. Chem. 357, 1651 (1976).
Column chromatography separations were performed using silica gel of particle
size
0.05 - 0.2 mm supplied by the Merck company.
The solubility in water was determined by UV spectroscopy at 300 nm and
25°C.
Example 1
N-t-butyl oxycarbonyl-2-aminoacetic acid-[3-(1,3-dihydro-1,3-
dioxo-2H-isoindol-2-yl)-2,G-dioxopiperidin-1-yl] methyl ester (1)
2.88 g ( 10 mmoles) N-hydroxymethylthalidomide, I .75 g ( 10 mmoles) N-t-butyl
oxycarbonyl-glycine, 2.06 g ( 10 mmoles) dicyclohexylcarbodiimide and 0.15 g (
1
mmole) 4-pyrrolidinopyridine were stirred in 50 ml of dry dichloromethane for
24
hours at room temperature. The precipitated dicyclohexylurea was subsequently
filtered off and the filtrate was shaken first with acetic acid and then with
water. After
drying the organic phases over magnesium sulphate and removal of the solvent
by
distillation, the residue was recrystallised from ethanol. 2.59 g (58 %
theoretical) of
compound I were obtained, with a melting point of 108 to I 1 1 °C.
Example 2
2-aminoacetic acid-[3-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)-2,G
-dioxo-piperidin-1-yl] methyl ester hydrochloride (2)
1.78 g (4 mmoles) of compound 1 prepared as in Example 1 were stirred for 1
hour at
room temperature in 20 ml of a mixture of 25 volume % trifluoroacetic acid and
75
volume % dichloromethane. The solvent was subsequently removed under vacuum
and
CA 02251060 2005-06-20
24272-73
8
the residue obtained was co-evaporated with dichloromethane. The
trifluoroacetate
obtained was in dissolved in 160 ml methanol and was converted into the
TM
hydrochloride by means of ion exchange chromatography (Amberlite IR 45, which
had
previously been converted into the C1 form with 1 N hydrochloric acid, was
used as the
ion exchanger). After removing the solvent by distillation, the residue
obtained was
suspended in boiling ethanol and was treated drop-wise with water in order to
form a
clear solution. 0.81 g (53 % theoretical) of compound 2 was obtained; this had
a
melting point of 227 to 232°C and a solubility in water of 49.7 mg/ml,
corresponding to
33.6 mg thalidomide/ml.
Example 3
N-t-butyloxycarbonyl-2-methylaminopropionic acid-(3-(1,3-dihydro-
1,3-dioxo-2H-isoindol-2-yl)-2,6-dioxo-piperidin-1-yl) methyl ester (3)
4.32 g ( 15 mmoles) N-hydroxymethylthalidomide, 3.05 g ( 15 mmoles) N-t-butyl-
oxycarbonyl-L-N-methylalanine, 3.09 g ( 15 mmoles) dicyclohexylcarbodiimide
and
0.22 g ( 1.5 mmoles) 4-pyrrolidinopyridine were stirred in 75 ml dry
dichloromethane
for 24 hours at room temperature. The precipitated dicyclohexylurea was
filtered off
and the filtrate was shaken first with acetic acid and then with water. After
drying the
organic phases with magnesium sulphate, removing the solvent by distillation
and
purifying the residue by column chromatography using dichloromethane/acetone
(in a
volume ratio of 9 : 1 ), 4.44 g (63 % theoretical) of compound 3 with a
melting point of
123 to 125°C were obtained.
CA 02251060 1998-10-07
WO 97/37988 PCT/EP97/01475
9
Example 4
2-methylaminopropionic acid-[3-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)-
2,G-dioxopiperidin-1-yl] methyl ester hydrochloride (4)
Compound 3, which was obtained as in Example 3, was converted into compound 4
under the conditions given in Example 2. Crystallisation from methanol/diethyl
ether
gave 1.05 g (64 % theoretical) of compound 4 with a melting point of 147 to
151 °C
and a solubility in water of >300 mg/ml, corresponding to > 190 mg
thalidomide/ml.
Example 5
N-t-butyloxycarbonyl-2-amino-3-methylbutyric acid-[3-(1,3-dihydro-
1,3-dioxo-2H-isoindol-2-yl)-2,G-dioxo-piperidin-1-yl] methyl ester (5)
3.85 g (53 % theoretical) of compound 5 with a melting point of 82 to
85°C were
obtained, under the conditions given in Example 3, from 4.32 g (IS mmoles) N-
hydroxymethylthalidomide, 3 .26 g ( 15 mmoles) N-t-butyloxycarbonyl-L-valine,
3.09
g ( 15 mmoles) dicyclohexylcarbodiimide and 0.22 g ( 1.5 mmoles) 4-
pyrrolidinopyridine.
Example G
2-amino-3-methylbutyric acid-[3-(1,3-dihydro-1, 3-dioxo-2H-isoindol-2-yl)-2,G-
dioxopiperidin-1-yl] methyl ester hydrochloride (6)
1.95 g (4 mmoles) of compound 5 obtained as in Example 5 were converted into
compound 6 under the conditions given in Example 2. Crystallisation from
ethanol/diethyl ether gave 1.01 g (60 % theoretical) of compound 6 with a
melting
point of 145 to I50°C and a solubility in water of >300 mg/ml,
corresponding to >190
mg thalidomide/ml.
Example 7
CA 02251060 1998-10-07
WO 97/37988 PCT/EP97/01475
2-(N-t-butyloxycarbonyl-aminomethylcarbonylamino)-acetic acid-[3-
(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)-2,6-dioxopiperidin-1-yl] methyl
ester (7)
5 3.67 g (73 % theoretical) of compound 7 with a melting point of 178 to 181
°C were
obtained, under the conditions given in Example I, from 2.88 g (10 mmoles) N-
hydroxymethylthalidomide, 2.32 g (10 mmoles) N-t-butyl oxycarbonyl-glycyl-
glycine, 2.06 g ( I 0 mmoles) dicyclohexylcarbodiimide and 0.15 g ( 1 mmole) 4-
pyrrolidinopyridine.
Example 8
2-(aminomethylcarbonylamino) acetic acid-[3-(1,3-dihydro-1,3-
dioxo-2H-isoindol-2-yl)-2,6-dioxo-piperidin-1-yl] methyl ester hydrochloride
(8)
1.35 g (77 % theoretical) of compound 8 with a solubility in water of >300
mg/ml,
corresponding to > 190 mg thalidomide/ml, were obtained, under the conditions
given
in Example 2, from 2.01g (4 mmoles) of compound 7 prepared as in Example 7.
Example 9
[3-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)-2,6-dioxo-piperidin-1-yl]-
methoxycarbonylpropionic acid (9)
2.88 g (10 mmoles) N-hydroxymethylthalidomide, 2 g (20 mmoles) succinic
anhydride,
2.8 ml (20 mmoles) triethylamine and 0.15 g ( 1 mmole) 4-pyrrolidinopyridine
were
stirred in 50 ml dry dichloromethane for 24 hours at room temperature. The
reaction
mixture was subsequently shaken firstly with 5 % hydrochloric acid and then
with
water. After drying the organic phases over magnesium sulphate and removing
the
solvent by distillation, the residue obtained was treated with a small amount
of ethyl
acetate and the precipitated crystals were recrystallised from ethanol. 2.66 g
(68
theoretical) of compound 9 were obtained, with a melting point of 143 to
146°C and a
solubility in water of 16.7 mg/ml, corresponding to I 1.1 mg thalidomide/ml.
CA 02251060 1998-10-07
WO 97/37988 PCT/EP97/01475
Pharmacological investigations
The release of TNF-oc was investigated in vitro on human mononuclear cells of
the
peripheral blood (T cells, B cells and monocytes), after stimulation with
lipopolysaccharide (LPS). LPS is a constituent of the bacterial cell wall and
stimulates
monocytes and macrophages.
Mononuclear cells were obtained from the heparin-treated blood of at least
three
volunteer donors. For this purpose, 20 ml blood in each case were separated by
known
methods via a Ficoll-Paque gradient, and the cells were harvested and washed
three
times with a cell culture medium. The cell culture medium which was used
consisted of
RPMI 1640 medium, supplemented with 2 mM glutamine (Life Technologies,
Eggenstein), 10 % foetal calf serum (Life Technologies), 50 pg/l streptomycin
(Sigma,
Deisenhofen), 50 IU/ml penicillin (Sigma, Deisenhofen) and 100 ~rM /3-
mercaptoethanol (Merck, Darmstadt). The mononuclear cells were subsequently
taken
up in 15 ml cell culture medium and were divided into 1 ml batches in sterile
24-hole
incubation plates (Sigma). 1 trl dimethylsulphoxide (DMSO, Merck) was added to
the
1 ml batches which were used as the control batch. 1 girl of a solution of a
compound
according to the invention (in DMSO; final concentration in the test: 0.5; 5;
12.5 and
50 ~rg/ml) was added to the test batches. The batches were incubated for one
hour in a
COZ incubation cabinet (5 % CO2, 90 % atmospheric humidity). 2.5 trg LPS (from
1i:.
coli 0127: B8, Sigma, Deisenhofen) was subsequently added as a stimulant to
each
batch with the exception of the control batches. Incubation of the cultures
was
continued for 20 hours. Following the incubation, the concentration of TNF-a
in the
cell culture supernatant liquors was determined by ELISA tests (Boehringer
Mannheim). The magnitude of the inhibition of the release of TNF-a was
calculated
from the measured values of the control batches and of the batches incubated
with the
compounds according to the invention. The concentrations which resulted in 50
inhibition of the release of TNF-a (the ICS" values) were calculated with the
aid of a
linear regression line.
The following Table shows the inhibiting effect of the compounds according to
the
invention on the LPS-induced release of TNF-a:
CA 02251060 1998-10-07
WO 97/37988 PCT/EP97/01475
12
Compound according to ~ Inhibition of the release of ~ ICso [pg/ml]
the invention I TNF-a at a final concentration
of 50 pg/nll
1 78 % 2.7
2 54
6 97 % 2.0
9 35
Efficacy of compounds according to the invention in an animal model
For the in oioo characterisation of the immunopharmacological effects of the
compounds according to the invention, a test model was selected in which T-
lymphocytes were stimulated. The relevance of this immunopharmacological
animal
model results from the requisite cell-cell cooperation for the initiation of
an immune
response. The prerequisite for activation and clonal expansion of the T cells
is first
created by the interaction between antigen-presenting cells, e.g. monocytes,
and T
cells. This is a characteristic both of defence against infection and of
autoimmune
aggression or transplant rejection or graft vs. host reaction (graft vs. host
disease).
Lymphocyte infiltrates constitute the initial stage of chronic graft vs. host
reactions, for
example, and also constitute the initial stage of acute aphthous lesions in
the mucous
membrane of the mouth, such as those which occur in morbus Beh~et or in what
are
termed idiomatic aphthae.
The stimulation of T cells was effected by the intravenous application of the
staphylococci enterotoxin B (SEB; 200 yg) to balb/c mice which had been
pretreated
with galactosamine. SEB is a superantigen, which firstly binds MHC molecules
to
antigen-presenting cells, and secondly binds invariable structures to certain
T cell-
receptor families. This results in the activation both of T cells and of
monocytes. The
concentration of the cytokin interleukin-2 (IL-2) in the serum was determined,
as a
parameter for T cell activation, by means of a commercial ELISA test which
specifically detects murine IL-2. The injection of SEB resulted in a time-
dependent
increase in the serum IL-2 level, with a clear maximum two hours after SEB
CA 02251060 1998-10-07
WO 97/37988 PCT/EP97/01475
13
application. The origin of the IL-2 from T cells which was measured in the
serum
could be verified by the application of SEB to T cell-deficient SCID mice,
which
thereupon formed no IL-2.
The compounds according to the invention were dissolved in 1 % aqueous
carboxymethyl cellulose (CMC) and were administered intraperitoneally to the
animals,
in doses of 100 to 400 mg/kg and in a volume of 1 ml, 30 minutes before the
SEB
application. The animals of the control group received 1 ml/kg of a 1 %
aqueous CMC
solution, applied intraperitoneally. The serum IL-2 concentrations were
determined 2
hours after the application of SEB. The maximum inhibiting effects (in %) on
the
serum IL-2 level in the groups treated with a compound according to the
invention
compared with the control group are given in the following Table. The
percentages
quoted represent the average values of 6 to 8 separate tests in each case. The
doses
which resulted in a 40 % reduction in serum IL-2 concentrations (ED4~, values)
were
calculated via a regression line.
Compound according to ~ Max. inhibition of the ~ EDa" [mg/kg]
the invention ~ increase in serum IL-2 at a
dose of 400 mg/kg
1 34
2 68 % 230
4 26
6 52
Comparative investigations showed that, in contrast to glucocorticoids, the
compounds
according to the invention even inhibited the increase in serum IL-2 when they
were
administered 30 minutes before stimulation by SEB. To achieve an inhibiting
effect by
glucocorticoids, however, it was necessary for these substances to be applied
18 hours
before stimulation by SEB.