Note: Descriptions are shown in the official language in which they were submitted.
CA 022~1292 1998-10-09
W097/38705 PCT~S97/05744
N-FORMYL HYDROXYLAMINE CONTAINING COMPOUNDS
USEFU~ AS ACE INHIBITORS AND/OR NF.P INHIBITORS
Summarv of the Invention
This invention is directed to novel
compounds possessing angiotensin converting enzyme
(ACE) inhibitory activity and/or neutral
endopeptidase (NEP) inhibitory activity and methods
of preparing such compounds. This invention is
also directed to pharmaceutical compositions
cont~in;ng such ACE and/or NEP inhibiting compounds
or pharmaceutically acceptable salts thereof and
the method of using such compositions.
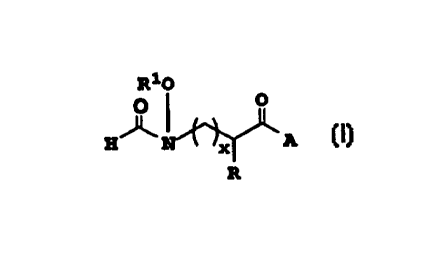
The compounds of this invention are those
of the formula (I)
R~O
J~ I ~A
including a pharmaceutically acceptable salt
- thereof where:
x is 0 or l;
R is H, alkyl, alkenyl, aryl-(CH2)p-,
heteroaryl-(CH2)p-, cycloheteroalkyl-(CH2)p-~ or
R can be joined together with the carbon to
which it is attached to form a 3 to 7 membered ring
which may optionally be fused to a benzene ring;
Rl is H or -COR2 where R2 is alkyl, aryl-
(CH2)p-, cycloheteroalkyl-(CH2)p-~ heteroaryl-
(CH2)p-, alkoxy, or cycloalkyl- (CH2)p-;
p is O or an integer from l to 8; and
~ - A is a dipeptide derived from one or two
non-proteinogenic amino acid or is a
conformationally restricted dipeptide mimic as
described below.
A is a dipeptide derivative of the
structure
CA 022~1292 1998-10-09
W O 97/38705 PCT~US97/05744
A(l) ~ 7 ~ R2~
Jr~ ~ N ~ R2b
H
O CoR4
where Rla, Rlb, R2a and R2b are independently
selected from H, alkyl, aryl-(CH2)p-~ cycloalkyl,
S cycloheteroalkyl-(CH2)p-, heteroaryl-(CH2)p-,
biphenylmethyl, or
Rla and Rlb or R2a and R2b may be joined
together to the carbon to which they are attached
to form a 3 to 7 membered ring, optionally fused to
R3 R5
1, ';."" R2b
a benzene ring; and CoR4 refers to an
optional 5 or 6 membered ring contAining a single
hetero atom and which may optionally include an R5
substituent (as shown) which is H, alkyl, aryl-
(CH2)p or cycloalkyl-(CH2)p~ cycloheteroalkyl-
(CH2)p, or cycloheteroaryl-(CH2)p-;
R3 is H, alkyl or aryl -(CH2)p-;
R4 is OH, Oalkyl, O-(CH2)paryl- or NRl(R2)
where Rl and R2 are independently H, alkyl, or
aryl(CH2)p or heteroaryl-(CH2)p-;
with the proviso that in A(l) at least one
of
R,3 R5
Rla Rlb I ~ ~
~ N ~ ~ and ~ ~
O CoR4
is other than a natural a-amino acid, and thus
must be other than valine, leucine, phenylalanine,
t~ro-sine, serine, cysteine, threonine, methionine,
aspartic acid, glutamic acid, arginine, lysine or
proline.
In addition, A can be a conformationally
restricted dipeptide mimic which has the structure
CA 02251292 1998-10-09
W O 97/38705 PCT~US97/05744
A(2 )
N~
H ¦¦ l
O CoR4
and is a non-proteinogenic dipeptide.
Thus, the compound of formula I include
R10
:rA ~ ~l Rl~ Rlb IR R5
H N--( CH2 ) x~ ~, N'~;~Rab
H
S R O CoR4
and
IA(2) 1 0 ~
I _(C}~2)x~ N~S
O R O CoR4
The term "conformationally restricted
dipeptide mimic" refers to a structural skeleton
which has the attributes of a conventional
dipeptide
R O R O
l 11 1 11
--NH--CEI-C--NEI--CH--C--
but having enhanced biological properties due to
additional bonds which limit the rotational
freedom.
Examples of the A(2) dipeptide mimics
include any of the conformationally restricted
dipeptide mimics set out below.
CA 0225l292 l998-lO-09
W 0 97/38705 PCTrUS97/05744
".5~ f '~ R~
H o CoR4 n O r 1~ CoR4 ~ CoR4
where Y - O S CH2 where X = CH2 and or S(0)o~1~2
'' andX=O,Swhenn=1
,Xl A~8) X
A(6) ~ A(7) ~ ~
H~\ ~ ~CoR4
where y1 = o, S, NH
where X1 = H, Ph, or S(O)n,
NHSo2R5 where X1 = H, Ph,
(R5 H) NHSo2R5
(Rs H
A(9) ~ A(10)
O~COR~ ~~CO~ COJ~
where Y = O, S, CH2 where Z = O or H, H
or S(0)o,l,2
A(12) ~Z A(13) ~ A(14) H~
N~, R7 IS~ ~ N~ R7 ~ N~J
H o CoR4 ~ CoR4 ~ CoR4
where Z = O or H, H where Y = O, S, CH2
or S(O)0,1,2
A(~ A(1~ 5 A(17)~
H O CoR4 H ~ CoR4 H O CoR4
whereY=O,S,
or S(~)0,1,2
A(1Y) ~3 A(19~ ~R 17 ~J\( ~0,
--~b' N~ R7 5S~ ~, N~ R7 ~ ~,N~ R7
H ~ CoR4 ~ CoR4 H O CoR4
where Y = O, S, CH2
CA 022~1292 1998-10-09
W097/38705 PCT~S97/05744
A(21) ~ A(22) ~
~N~ R7 ~' N~ ~ R7
H ~ CoR4 H o CoR4
A(23) ~_
whereY=O,S,CH20rs(O)o~1~2
H o CoR4 X =O~S(O)o~1~2~cH2
With respect to A(5), R11 and R12 are
independently selected from hydrogen, alkyl,
substituted alkyl, alkenyl, substituted alkenyl,
cycloalkyl -(CH2)m-, aryl -(CH2)m-, substituted
aryl -(CH2)m-, and heteroaryl -(CH2)m-, or R11 and
R12 taken together with the carbon to which they
are attached complete a saturated cycloalkyl ring
of 3 to 7 carbons, or R11 and R12 taken together
with the carbon to which they are attached complete
a keto substituent, i.e.,
/c - o
- with respect to A(13) R8, R9 and R7 are
independently selected from hydrogen, alkyl,
substituted alkyl, alkenyl, substituted alkenyl,
cycloalkyl ~(CH2)m~, aryl-(CH2)m~, substituted
aryl-(CH2)m-, and heteroaryl-(CH2)m~;
R10 and R6 are independently selected from
hydrogen, alkyl, substituted alkyl, alkenyl,
substituted alkenyl, cycloalkyl -(CH2)m-, aryl-
(CH2)m, substituted aryl -(CH2)m-, and heteroaryl-
(CH2)m-, or R6 and R10 taken together with the
carbon to which they are attached complete a
saturated cycloalkyl ring of 3 to 7 carbons, R6 and
R8 taken together with the carbon to which they are
attached complete a saturated cycloalkyl ring of 3
to 7 carbons, or R9 and R10 taken together with the
carbon to which they are attached complete a
saturated cycloalkyl ring of 3 to 7 carbons;
CA 022~1292 1998-10-09
W O 97/38705 PCTAUS9710~744
m is zero or an integer from 1 to 6;
R4 is OH, Oalkyl, O-(CH2)m-heteroaryl,
Il oJ~o
--CH-O-C-R15
Rl4 , O-(CH2)m-aryl, or -o-c~2 Rl6
NR1(R2);
S where R1 and R2 are independently H, alkyl,
aryl(CH2)p, aryl or heteroaryli
R14 is hydrogen, lower alkyl, cycloalkyl,
or phenyli
R15 is hydrogen, lower alkyl, lower alkoxy
or phenyli
R16 is alkyl or aryl-(CH2)m~; and
R17 is hydrogen, alkyl, substituted alkyl,
alkenyl, substituted alkenyl, cycloalkyl-(CH2)m-,
aryl-(CH2)m~, substituted aryl-(CH2)m-, or
IS heteroaryl-(CH2)m~-
R18 is H, alkyl or alkenyl, and R18 and R17
may be taken together with the carbon and nitrogen
to which they are attached to complete a saturated
N-cont~; n; ng ring of 5 or 6 ring members.
R19 is H or an alkyl, and in A(4), R19 and X
(which is CH2) together with the carbons to which
they are attached may form an aromatic ring of
carbons (as in A(15).
The starting compounds H-A(l) and H-A(2)
are described in the literature or are obtained by
modifications of known procedures. For example,
the starting compounds of formula H-A(l) or H-A(2)
wherein A(1) or A(2) is as defined in formulas
A(5-), A(13), A(16), A(21), where Y (where present)
is CH2 are disclosed by Thorsett et al., J. Med.
Chem., 29, p. 251 - 260 (1988), Harris et al. in
U.S. Patents 4,587,050, 4,587,238, 4,629,787 and
Yanagisawa et al. in U.S. Patent 4,734,410.
CA 022~l292 Isss-lo-os
W 097/38705 PCT~US97/05744
The starting compounds of formula H-A(l) or
H-A(2) wherein A(l) or A(2) is as defined in
formulas A(3) and A(13) where Y is S(O)n are
disclosed by Yanagisawa et al., J., Med. Chem., 30,
p. 1984 - 1991 (1987) and 31, p. 422 - 428 (1988),
Karanewsky in U.S. Patent 4,460,579, Cheung et al.
in U.S. Patent 4,594,341, and Yanagisawa et al. in
U.S. Patent 4,699,905.
The starting compounds of formula H-A(l) or
H-A(2) wherein A(l) or A(2) is as defined in
formula A(5) are disclosed by Karanewsky in U.S.
Patents 4,460,579 and 4,711,884.
The starting compounds of formula H-A(l) or
H-A(2) wherein A(l) or A(2) is as defined in
formulas A(3) (Y is -CH2-, and A(21) are disclosed
by Watthey et al., J. Med. Chem., 28, p. 1511
1516 (1985) and Watthey in U.S. Patents 4,410,520,
4,470,988, 4,473,575, 4,537,885 and 4, 575,503 and
also by Parsons et al., Biochemical & Biophysical
Research Comm., 117, p. 108 - 113 (1983) and in
U.S. Patent 4,873,235.
The starting compounds of formula H-A(l) or
H-A(2) wherein A(l) or A(2) is as defined in
formula A(3) and Y is S or O are disclosed by Slade
et al., J. Med. Chem., 28, p. 1517 - 1521 (1985)
and in U.S. Patent 4,477,464 and Itoh et al., Chem.
Pharm. Bull., 34, p. 1128 - 1147 (lg86) and 34, p.
2078 - 2089 (1986) as well as Sugihara et al. in
U.S. Patent 4,548,932 (Y is O) and Katakami et al.
in U.S. Patent 4,539,150 (Y is S).
The starting compounds of formula H-A(l) or
H-A~2) wherein A(l) or A(2) is as defined in
formula A(16) can be prepared by reduction of the
corresponding starting compounds wherein A(l) or
A(2) is as defined in formula A(3).
The starting compounds of formula H-A(l) or
H-A(2) wherein A(l) or A(2) is as defined in
CA 022~1292 1998-10-09
W O 97/38705 PCTrUS97/0~744
formula A(22) are disclosed by Flynn et al in U.S.
Patent 4,973,585.
The starting compounds of formula H-A~l) or
H-A~2) wherein A(l) or A(2) is as defined in
formula A~10) and Y is S, -SO, or -SO2 are
disclosed by Harris et al. and Patchett et al. in
U.S. Patents 4,415,496 and 4,617,301.
The starting compounds of formula H-A(l) or
H-A(2) wherein A(l) or A(2) is as defined in
formula A(10) and Y is CH2, and is as defined in
formula A(23) where x2 is CH2 is disclosed by
Thorsett, Actual. Chim. Ther., 13, p. 257-268
(1986).
The starting compounds of formula H-A(l) or
H-A(2) wherein A(l) or A(2) is as defined in
formulas A(ll) and A(19) and A(20) are disclosed by
Attwood et al., Federation of European Biochemical
Studies, 165, p. 201-206 (1984) and in U.S. Patent
4,512,994 and Natoff et al., Drugs Of The Future,
12, p. 475-483 (1987).
The starting compounds of formula H-A(l) or
H-A(2) wherein A(l) or A(2) is as defined in
formula A(12) are disclosed by Huang et al. in U.S.
Patent 4,465,679.
The starting compounds of formula H-A(l) or
H-A(2) wherein A(l) or A(2) is as defined in
formula A(18) are disclosed by Bolos et al. in
Tetrahedron, 48, p. 9567-9576 ~1992).
The starting compounds of formula H-A(l) or
H-A(2) wherein A(l) or A(2) is as defined in
forml]lA.s A~4) and A~15) are disclosed in European
Patent Application 0629627A2.
The starting compounds of formula H-A~l) or
H-A(2) wherein A(l) or A(2) is as defined in
formula A(9) are disclosed in U.S. application
Serial No. 100,408 (file HA611a).
CA 022~1292 1998-10-09
W O 97/38705 rCT~US97/05744
The starting compounds of formula H-A(l) or
H-A(2) wherein A(l) or A(2) is as defined in
formulas A(7) and A(8) are disclosed in European
Patent Application 481,~22 (Flynn et al) and
European Patent Application 0534363A2 (Warshawsky
et al).
The starting compounds of formula H-A(l) or
H-A(2) wherein A(l) or A(2) is as defined in
formula A(14) are disclosed in U.S. application
Serial No. 153,854 (file HA615).
The starting compounds of formula H-A(l) or
H-A(2) wherein A(l) or A(2) is as defined in
formula A(17) are disclosed in European Patent
Application 0599444Al (Barrish et al).
In addition, in accordance with the present
invention, a pharmaceutical composition is provided
which includes a therapeutically effective amount
of compound I and a pharmaceutically acceptable
carrier therefor.
The pharmaceutical composition as defined
above will be useful in the treatment of
cardiovascular diseases such as hypertension and/or
congestive heart failure.
Furthermore, in accordance with the present
2S invention, a method is provided for treating a
cardiovascular disease such as hypertension and/or
congestive heart failure, as well as other diseases
as set out hereinafter, which includes the step of
~m;nistering to a m~mm~lian species, including
humans, dogs and cats, a therapeutically effective
amount of a composition as defined above.
Detailed Descri~tion Of The Invention
The term "alkyl" or "lower alkyl" refers to
straight or br~nche~ chain radicals having up to
and including ten carbon atoms, preferably up to
and including six carbon atoms, which may
g
CA 022~1292 1998-10-09
W O 97138705 PCTnUS97/05744
optionally include one, two, or three substituents
including a hydroxy, amino, alkyl, cycloalkyl,
aryl, halo, trifluoromethyl, cyano, -NH(lower
alkyl), -N(lower alkyl)2, lower alkoxy, lower
S alkylthio, carboxy or heteroaryl.
The term "alkenyl" refers to straight or
branched chain radicals of 3 to 10 carbon atoms
having one or two double bonds, preferably straight
chain radicals of 3 to 5 carbons having one double
bond, which may optionally be substituted with one,
two or three substituents including alkyl, aryl,
cycloalkyl, hydroxy, amino, halo, trifluoromethyl,
cyano, -NH(lower alkyl), -N(lower alkyl)2, lower
alkoxy, lower alkylthio, carboxy or heteroaryl.
The terms "alkoxy" or "lower alkoxy" and
"alkylthio~ or "lower alkylthio" refer to such
alkyl groups as defined above attached to an oxygen
or sulfur.
The term "cycloalkyl" refers to saturated
rings of 3 to 7 carbon atoms.
- The term "halo" refers to chloro, bromo,
fluoro, and iodo.
The term "aryl" refers to aromatic groups
contAin;ng 6 to 10 carbons, preferably phenyl, 1-
naphthyl, and 2-naphthyl, which may optionally
contain one, two or three substituents selected
from alkyl, alkoxy, alkylthio, halo, hydroxy,
trifluoromethyl, -SO2NH2, amino, -NH(lower alkyl),
or -N(lower alkyl)2, di- and tri-substituted
phenyl, l-naphthyl, or 2-naphthyl, wherein said
substituents are preferably selected from methyl,
methoxy, methylthio, halo, hydroxy, and amino.
The term "heteroaryl" refers to unsaturated
rings of 5 or 6 atoms cont~;n;ng one or two 0 and S
atoms and/or one to four N atoms provided that the
total number of hetero atoms in the ring is 4 or
less, which may optionally be substituted with one,
- 10 -
CA 022~1292 1998-10-09
W097/38705 PCT~S97/05744
two or three substituents which include alkyl,
aryl, cycloalkyl, alkoxy or halo. The heteroaryl
ring is attached by way of an available carbon or
nitrogen atom. Preferred heteroaryl groups include
2-, 3-, or 4-pyridyl, 4-imidazolyl, 4-thiazolyl, 2-
and 3-thienyl, and 2- and 3-furyl. The term
heteroaryl also includes bicyclic rings wherein the
five or six membered ring cont~; n; ng o, S, and N
atoms as defined above is fused to a benzene or
pyridyl ring. Preferred bicyclic rings are 2- and
3-indolyl and 4- and 5-quinolinyl. The mono or
bicyclic heteroaryl ring can also be additionally
substituted at an available carbon atom by a lower
alkyl, halo, hydroxy, benzyl, or cyclohexylmethyl.
Also, if the mono or bicyclic ring has an available
N-atom such N atom can also be substituted by an N-
protecting group such as
- CH2 - O- CH2 ~ ' -S02 ~ CH3
2,4-dinitrophenyl, lower alkyl, benzyl, or
benzhydryl.
The compounds of formula I of the invention
may be prepared as outlined in Reaction Scheme I
set out below (where x is 0 or 1).
CA 02251292 1998-10-09
W O 97/38705 PCT~US97/05744
Reaction Scheme I
Qq~C02H PG1-O-NH? PG o~ N~C02 reS~Iution HN~
3 (optically pure or
(Q is CH2, where x-1 and enriched)
Q is H, Br where x=0)
H-A(1) or H-A(2)
O ~ Acylation
o N~oR4 PG1 ~~ . H-C0-0-COR (4a)
standard peptide coupling R H ~ CoR4
~ ~C I removal of one orboth ~ J'C
pG1 Q ~ u~ 9 groups (PG1 HO'~ N~
~0 R 5 ~ coR4 and/or R ) ~ R H o CoR4
IA
As shown in Scheme I, acid 2 may be reacted
with a suitably O-protected (e.g. PGl is benzyl, p-
methoxybenzyl, tetrahydropyranyl, trityl,
benzhydryl, etc.) hydroxylamine to give the adduct
3. Compound 3 may be coupled directly with amine
H-A(l) or H-A(2) to give a mixture of
diastereomers which may be separated or preferably
compound 3 may be optically enriched or purified,
employing conventional techniques, to give 3*.
Subsequent coupling with H-A(l) or H-A(2) gives 4
in diastereomerically enriched or pure form.
Reaction of the hydroxylamine nitrogen of 4 with a
formylating agent affords 5. At this point one or
both protecting groups may be removed, either
se~uentially or simultaneously, to produce compound
of the invention IA. For example, when PGl is
benzyl and R4 is Obenzyl, both may be removed by
hydrogenolysis. When pGl is benzyl and R4 is
~Omethyl or ~Oethyl, the PGl group may be removed
by hydrogenolysis and the ester group may be
converted to the acid by base hydrolysis. pGl
CA 022~1292 1998-10-09
W O 97/3870~ PCTrUS97/05744
groups such as THP or trityl may be removed by
treatment with strong acid such as hydrogen
chloride or trifluoro acetic acid in a protic
solvent.
- 5 Alternately, compounds of the invention IA
may be obtained by the route depicted in Scheme II
- (where x is 0 or l).
Reaction Scheme II
Acylation
PG1 O~,C02H H.co-o-COR PG -Q~CO2H optionalchiral
3 O 7
H-A(1 ) or H-A(2)
~C I
PG~,CO2H ~N~C~oR4 PG1-~ ~ CoR4
standard peptide coupling 5
As seen in Reaction Scheme II, compound 3
may be formylated with an formylating agent 4a to
give acid compound 7. This acid may be coupled
with A~l) or A(2) directly or optically resolved
to give 7* and then coupled to give compound 5.
Compound 5 is then converted to compound of the
invention IA as described above.
The compounds of formula I of the invention
contain one or more asymmetric centers. Thus,
these compounds can exist in diastereoisomeric
forms or in mixtures thereof and all of such forms
àre within the scope o~ this invention. The above
described processes can utilize racemates,
enantiomers, or diastereomers as starting
materials. When diastereomeric compounds are
prepared, they can be separated by conventional
- 13 -
CA 022~1292 1998-10-09
W O 97~870~ PCTrUS97/05744
chromatographic or fractional crystallization
methods.
The compounds of formula I of the invention
can be isolated in the form of a pharmaceutically
acceptable salt. Suitable salts for this purpose
are alkali metal salts such as sodium and
potassium, alkaline earth metal salts such as
calcium and magnesium, and salts derived from amino
acids such as arginine, lysine, etc. These salts
are obtained by reacting the acid form of the
compound with an equivalent of base supplying the
desired ion in a medium in which the salt
precipitates or in aqueous medium and then
lyophilizing.
The compounds of formula I of the invention
are inhibitors of angiotensin converting enzyme
and/or neutral endopeptidase. Thus, the compounds
of formula I including their pharmaceutically
acceptable salts are useful in the treatment of
physiological conditions in which either
- angiotensin converting enzyme inhibitors or neutral
endopeptidase inhibitors have been shown to be
useful. Such conditions include cardiovascular
diseases, particularly, hypertension, congestive
heart failure, renal failure, and hepatic
cirrhosis, as well as analgesic activity. The
compounds of formula I are also inhibitors of other
metalloproteases such as the matrix
metalloproteases, for example, gelatinase,
collagenase and stromylysin and thus are useful in
the treatment of osteroarthritis, rheumatoid
arthritis, metastatic tumors, and angiogenesis.
Diuresis, natriuresis, and blood pressure
reduction are produced in a m~mm~l ian host such as
man by the ~m; nl stration of from about 1 mg. to
about 100 mg. per kg. of body weight per day,
preferably from about 1 mg. to about 50 mg. per kg.
- 14 -
CA 022~1292 1998-10-09
W097~8705 PCT~S97/05744
of body weight per day, of one or more of the
compounds of formula I or a pharmaceutically
acceptable salt thereof. The compounds of formula
I are preferably administered orally, but
parenteral routes such as subcutaneous,
intramuscular, and intravenous can also be
employed. The daily dose can be administered
singly or can be divided into two to four doses
administered throughout the day.
The ACE and/or NEP inhibitors of formula I
can be ~m; n; stered in combination with human ANF
99 - 126. Such combination would contain the
inhibitor of formula I at from about 1 to about 100
mg. per kg. of body weight and the human ANF 99 -
126 at from about 0.001 to about 0.1 mg. per kg. of
body weight.
The ACE and/or NEP inhibitors of formula I
can be administered in combination with other
classes of pharmaceutically active compounds. For
example, a calcium channel blocker, a potassium
channel activator, a cholesterol reducing agent,
etc.
The ACE and/or NEP inhibitors of formula I
or a ph~rm~ceutically acceptable salt thereof and
other pharmaceutically acceptable ingredients can
be formulated for the above described pharmacetical
uses. Suitable compositions for oral
administration include tablets, capsules, and
elixirs, and suitable compositions for parenteral
30 ~m; ni stration include sterile solutions and
suspensions. About ~0 to 500 mg. of active
ingredient is compounded with physiologically
acceptable vehicle, carrier, excipient, binder,
preservative, stabilizer, flavoring, etc., in a
unit dose form as called for by accepted
phArm~ceutical practice.
CA 022~1292 1998-10-09
W O 97/38705 PCTrUS97/05744
Preferred compounds of the invention are
those of formula I wherein
R1 is H,
x is 1,
S R is alkyl or arylalkyl, and
A is A(l), preferably
Rl~ Rlb
H ~
where ~ is preferably a non-proteinogenic
amino acid portion wherein,
R1a and R1b are each independently alkyl
such as methyl or ethyl, or arylalkyl such as
benzyl, or
R1a and R1b together with the carbon to
which they are attached form a 3-7 membered ring,
preferably a 5-membered ring, or
R1a and/or R1b is biphenylmethylene and the
other may be H.
Also preferred are compounds where A is
R13 ,R
N jj~ R2b
A(l~, preferably where COR" and is a non-
proteino-genic amino acid where R3 is H, alkyl,
such as methyl or ethyl, aryl such as phenyl, or
arylalkyl, such as benzyl,
R2a and R2b are independently selected from
H, alkyl, aryl, arylalkyl (with at least one of R2a
and R2b being other than H) or R2a and R2b together
with the carbon to which they are attached form a
3-7 membered ring, preferably 5- or 6-membered
~ing.
Also preferred are compounds where A is
A(2) wherein R4 is OH.
The following Examples represent preferred
embodiments of the present invention.
- 16 -
CA 022~1292 1998-10-09
W097/3870~ PCT~S97/0~744
Exam~le 1
0~
o y N ~ N ~ CO2H
H
A.
0~
Boc~
N
Atl).
o
H, ~ C- OC(CH3)3
--~ COOH
NO2
A solution of BOC-L-serine (24.3 g, 0.118
mole) in dry dimethylformamide (25 ml) was added
dropwise over a period of 1.0 hour to a cooled (0~,
ice-salt bath) suspension of 60% NaH (10.1 g, 0.25
mole) in dry dimethylformamide (200 ml) and
stirring was continued at 0~ until the frothing
subsided (ca. 2.0 hours). The reaction mixture was
treated dropwise with 1-fluoro-2-nitrobenzene (14.3
ml, 0.13 mole) over a period of 20 minutes, stirred
at 0~ under argon for 4.0 hours then poured into
ice-water (750 ml) and extracted with Et2O (2 x
100-ml). The a~ueous phase was brought to pH 1.0
with 6 N HCl (70 ml), extracted with EtOAc (3 x 500
ml) and the combined organic extracts were washed
with brine (100 ml), dried (anhydrous Na2SO4),
filtered, evaporated to dryness and dried in vacuo .
The crude product mixture was chromatographed on a
silica gel column (Merc~), eluting the column with
CA 022~1292 1998-10-09
WOg7/3870~ PCT~S97/05744
CH2Cl2:CH3OH:HOAc (100:5:0.2) to give title
compound as a thick yellow syrup (27.222 g, 70.7%)
with consistent lH-NMR and 13C-NMR spectral data.
TLC: Rf 0.27 (Silica gel; CH2C12:CH30H:HOAc-
100:5:0.5; W, PMA).
A(2).
o
H~ ~,N+C- OC(CH3)3
--COOH
NH2
A solution of Part A~1) compound (27.1 g,
83 mmoles) in dry methanol (500 ml) was treated
with 10% Pd/C (900 mg) and hydrogenated at 40 psi
for 2.0 hours. The reaction mixture was filtered
through a Celite~ pad in a millipore unit, washing
the pad well with CH3OH (5 x 100 ml). The dark
filtrate was evaporated to dryness and dried ln
vacuo to give a dark solid. The crude product was
triturated with CH2Cl2:Hexane (1:4) to give title
compound as a light tan solid (17.69 g, 71. %) with
consistent 1H-NMR and 13C-NMR spectral data. TLC:
Rf 0.15 (Silica gel; CH2C12:CH30H:HOAc- 20:1:1;
W) .
A(3)-
~0~
N~
H o
A solution of Part A(2) compound (16.69 g,
56.3 mmoles) in dry dimethyformamide (121 ml) was
treated with 1-ethyl-3-(3-dimethylaminopropyl)-
30 carbodiimide (10.64 g, 55.5 mmoles) and stirred at
room temperature for 3.0 hours. The reaction
- mixture was partitioned between EtOAc (2 x 492 ml)
- 18 -
CA 022~1292 1998-10-09
W O 97/38705 PCTAUS97/05744
and 1.0 N NaHCO3 (492 ml), and the combined organic
extracts were washed with H2O (3 x 492 ml), brine
(492 ml), dried (anhydrous MgSO4), filtered,
evaporated to dryness and dried in vacuo. The
crude product was chromatographed on a silica gel
column (Merck), eluting the column with
EtOAc:Hexane mixtures (1:4; 1:2; 1:1) to give title
compound as off-white crystals (10.5 g, 72.4%) with
consistent 1H-NMR and 13C-NMR spectral data. TLC:
Rf 0.40 (Silica gel; EtOAc:Hexane- 1:4i W).
~~~_D
B~N ~ ~
H o Co2~n
A solution of Part A compound (640 mg, 2.30
mmol) in dry THF (12 mL) at 0~C was treated with
LiN(TMS)2 (1.0 M in THF, 2.60 mL, 2.60 mmol)
followed approximately 30 seconds later with benzyl
bromoacetate (475 ,uL, 687 mg, 3.0 mmol). After 25
minutes, the mixture was quenched with saturated
NH4Cl, diluted with H2O, and extracted with EtOAc.
The EtOAc extract was washed with H2O and brine,
then dried (Na2SO4), filtered and stripped to give
a yellow oil. Flash chromatography (Merck SiO2,
3/7-EtOAc/hexanes as eluant) provided title
compound (967 mg, 98%) as a colorless oil/foam.
~0~
HCI-H2N~ ~
~ CO2Bn
-- 19 --
CA 022~1292 1998-10-09
WO 97/3870~ PCT/US97/05744
A solution of Part B compound (960 mg, 2.25
mmol) in 1,4-dioxane ~4 mL) was treated with a
solution of 4.0 M HCl in 1,4-dioxane (6 mL) at room
temperature. After 3 hours, the mixture was
5 concentrated in vacuo, triturated with Et2O to give
a solid and stripped to afford title compound (858
mg, 105% of theory).
m.p. 152-155~C.
I0 D.
O ~~
~~ ~ H ~ CO2Bn
0¢~
D(l).
~3
~ coo~
A solution of benzylmalonic acid (23.06 g,
0.12 mole) in H2O (200 mL) was treated with 37%
CH20 solution (278.4 mL) and 40% aqueous (CH3)2NH
(35 mL, 0 31 mole) then stirred overnight at room
20 temperature under argon. The clear solution was
heated to an internal temperature of 90~C for 2.0
hours (at which time gas evolution had ceased),
cooled and acidified to pH 1.0 with 12 N HCl (20
mL). The white precipitates were filtered off,
25 washed with H2O (3 x 25 mL) and dried in vacuo to
give title compound as a white solid (12.85 g,
66.6%) with consistent lH-N~ and 13C-N~. spectral
data. TLC: Rf 0.63 (Silica geli CH2C12:MeOH- 9:1;
W). m.p. 66-68~C.
- 20 -
CA 022~1292 1998-10-09
W097/38705 PCT~S9710~744
D(2). H
~ , COOH
(J. Med. Chem. 28, 1985, 1167)
s
A solution of Part D(l) compound (8.9 g,
54.9 mmoles) and O-benzylhydroxylamine (26.7 g,
0.23 mole) in absolute EtOH (9.0 ml) was refluxed
for 7 days, cooled to room temperature and
evaporated to dryness. The residual syrup was
dissolved in 1.0 _ NaOH ~55 ml), stirred for 15
minutes then extracted with EtOAc (4x 18 ml). The
organic phase was washed with H2O (3 x 10 ml) and
the a~ueous extracts were combined and acidified to
pH 2.0 with 1.0 N HCl (62 ml). The acidic aqueous
phase was then extracted with EtOAc (5 x 75 ml) and
the combined organic extracts washed with H2O (2 x
- 30 ml), dried (anhydrous Na2SO4), filtered,
evaporated to dryness and dried in vacuo. The
crude product (3.93 g, 25.1%) was triturated with
Et2O:Hexane (1:4; 2 x 25 ml) and all solids
obtained were dissolved in CH2Cl2 and filtered,
washing the insoluble precipitates with CH2C12.
The clear filtrate was evaporated and dried in
vacuo to give title compound as an opaque
colorless solid with consistent lH-NMR and 13C-NMR
spectral data.
TLC: Rf 0.33 (Silica gel; CH2C12:MeOH- 9:1; W,
~MA) .
M.p. 69-71~C.
- 21 -
.
CA 022~1292 1998-10-09
W O 97/38705 PCT~US97/05744
D(3)-
OHC
~ ~, COOH
A cooled (0~C, ice-salt bath~ mixture of
HCOOH (17.5ml) and acetic anhydride (AC2O) (1.75
ml) was stirred for 20 minutes, treated with Part
D~2) compound ~1.0 g, 3.5 mmoles) and stirring was
continued at 0~C for another 3.0 hours. The
reaction mixture was stripped to dryness,
evaporated from Et2O (2 x 25 ml), toluene (20 ml)
and hexane (2 x 50 ml) then dried in vacuo to give
title compound as a thick syrup (1.096 g, 100%
crude yield) with consistent
1H-NMR and 13C-NMR spectral data. TLC: Rf 0.23
(Silica geli CH2C12:MeOH- 9:1; W, PMA).
D(4)-
~~~_D
~ --~ NH~ N~
0~
A solution of Part D~3) compound (366 mg,
1.19 mmol) in CH2Cl2 (9 mL) at 0~C was treated with
HOBT hydrate (210 mg) followed by EDAC (230 mg,
1.20 mmol). After 20 minutes, the mixture was
treated with Part C amine hydrochloride 3 (390 mg,
1.07 mmol) followed by 4-methylmorpholine (200 ,uL,
184 mg, 1.8 mmol). The mixture was stirred at 0~C
for 1 hour and at room temperature for 2 hours.
The reaction was partitioned between EtOAc and 5%
- KHS04. The EtOAc extract was washed successively
.
CA 022~1292 1998-10-09
W097/38705 PCTNS97/05744
with H2O, 50% saturated NaHCO3 and brine, then
dried (Na2SO4), filtered and stripped. Flash
chromatography (Merck SiO2, 50% to 60% EtOAc in
hexanes as eluant) provided title compound (550 mg,
84%) as a white foam which was shown by NMR and
HPLC to be a 1:1 mixture of diastereomers.
O (~
~/ _ H O CO2H
H ~
W Isomer A
A solution of Part D compound (535 mg, 0.87
mmol) in MeOH (10 mL) was hydrogenated (balloon)
over 10% Pd/C (123 mg) at room temperature for 2.75
hours. The solvent was filtered through Celite and
the filtrate was stripped to give a diastereomeric
mixture of title Isomer A and Isomer B
o ~~
0~ N I H~
H ~
Isomer B
~ . Trituration of a solution
of the residue in MeOH with Et2O provided 350 mg of
the diastereomeric mixture. Approximately 255 mg
of this mixture was separated by preparative HPLC
('YM~ S5 ODS 30 x 250 mm column; flow rate 25 mL/min
detecting at 220 nm; 40 to 100% B over a 30 minute
linear gradient (solvent A: 90%H20-10% MeOH-0.1%
TFAi solvent B: 10% H2O-90% MeOH-0.1% TFA); title
Isomer A tR = 14.4 min; separation performed in
three runs). The desired fractions were stripped,
CA 02251292 1998-10-09
W O 97/38705 PCTnUS97/05744
azetroped with EtOAc, re-dissolved in EtOAc and
triturated with Et2O to give title Isomer A (105.5
mg) as an off-white solid.
S MS: (M+NH4)+ 459; (M-H)- 440
HPLC YMC S3 ODS column (6.0 x 150 mm); eluted with
B:A solvent mixture, 40 to 100% B over a 20 minute
linear gradient (solvent A: 90% H2O-10% MeOH-0.2%
H3PO4; solvent B:0% H2O-90% MeOH-0.2% H3PO4); flow
rate 1.5 mL/min detecting at 220 nm; tR=9.67 min
(96.0%).
Anal. Calc'd for C22H23N3O7-1.6H2O-0.lEtOAc-0.1Et2O
C, 56.29; H, 5.80; N, 8.64
Found: C, 56.21; H, 5.15; N, 8.29.
- 24 -
CA 022~1292 1998-10-09
W O 97/38705 PCT~US97/OS744
Exam~le 2
O ~~
~ ¦ H o CO2H
H ~
S A solution of Example 1 Part E Isomers A
and B (1:1 mixture of diastereomers, 535 mg, 0.87
mmol) in MeOH (10 mL) was hydrogenated (balloon)
over 10% Pd/C (123 mg) at room temperature for 2.75
hours. The solvent was filtered through Celite and
the filtrate was stripped to give a diastereomeric
mixture of Isomers A and B. Trituration of a
solution of the residue in MeOH with Et2O provided
350 mg of the diastereomeric mixture.
Approximately 255 mg of this mixture was separated
by preparative HPLC (YMC S5 ODS 30 x 250 mm column;
flow rate 25 mL/min detecting at 220 nm; 40 to 100%
B over a 30 minute linear gradient (solvent A:
90%H20-10% MeOH-0.1% TFA ; solvent B: 10% H2O-90%
MeOH-0.1% TFA); Isomer B tR = 18.6 min; separation
performed in three runs~. The desired fractions
were stripped, azetroped with EtOAc, re-dissolved
in EtOAc and triturated with Et2O to give Isomer B
(88.0 mg) as an off-white solid.
MS: (M+NH4)+ 459; (M-H)- 440
HPLC YMC S3 ODS column (6.0 x 150 mm); eluted with
B:A solvent mixture, 40 to 100% B over a 20 minute
linear gradient (solvent A: 90%H20-10% MeOH-0.2%
H3PO4; solvent B:0% H2O-90% MeOH-0.2% H3PO4); flow
rate 1.5 mL/min detecting at 220 nm; tR = 13.8 min
(94.0%).
CA 022~1292 1998-10-09
W O 97138705 PCTnUS97/05744
Anal. Calc'd for C22H23N3O7-1.5H2O-0 2Et2O
C, 56.66; H, 5.84i N, 8.69
Found: C, 56.84; H, 5.22; N, 8.42.
Exam~le 3
~ ~Me
y _ O CO2H
H ~
~ CO2H
-- t1R. 2S)~ eph~d~i"e
¢~/ salt
A solution of Example 1 Part D(l) compound
' ~, CO2H ~
(2.563 gm, 8.98 mmol) in CH3CN (20
mL) was treated with (lR,2S)~ ephedrine (1.522
gm, 9.2 mmol) and stirred until homogeneous. Most
of the solvent was removed by rotary evaporation
and the residue was dissolved in Et2O (25 mL) and
treated with hexane (16 mL) in portions until the
mixture was slightly turbid. The solution was
seeded and let stand overnight at room temperature.
The precipitate was collected by filtration and
r,insed with 1:1 Et2O:hexanes and dried to afford
2.101 gm of white crystals ([a]D = -16.4~ (c 0.6,
CH2C12)). The solid (2.087 gm) was dissolved in
CH2C12, concentrated and diluted with Et2O (18 mL)
and hexane (8 mL) and seeded. The precipitate was
collected by filtration and washed with 1:1-
Et2O:hexanes followed by hexanes to give title
- 26 -
CA 022~1292 1998-10-09
W 0 97~8705 PCT~US97/05744
compound (1.995 gm) which was diastereomerically
enriched in one isomer but not diastereomerically
pure ([a]D = -17.0~ (c 0.6, CH2C12)).
mp 110-114~C
Material suitable for x-ray crystallographic
analysis was obtained by repeated recrystallization
of the solid from CH3CN. mp 117-119~Ci
([a]D = -19.7~ (c 0.4, CH2C12)).
O
CO2Et
B(1) .
r OH
(Pht is phthaloyl)
Pht=N CO2H
To a stirred solution of L-(+)-hydroxynor-
leucine (75 g, 509. 6 mmole) and sodium carbonate
(54 g, 509. 6 mmole) in water (900 ml) at room
temperature under argon was treated with N-ethoxy-
carbonyl-phthalimide (111. 7 g, 509.6 mmole). After
being stirred for 2.0 hours, the resulting solution
was filtered through a pad of celite. The filtrate
25 was cooled in an ice bath and carefully acidified
to pH=3 with 6N HCl solution. The white solid
which had precipitated was filtered and dried over
P2Os ln vacuo to afford Compound 1 (124. 5 g) in
88.1% yield.
M.P. 162~C
Hl-NMR (DMSO): d = 1.32 (m, 6H), 2.13 (m, 2H),
4.38 (s, OH), 5.75 (m, lH), 7.92 (m, 4H) ppm
- 27 -
CA 022~l292 l998-lO-09
W 097t38705 PCT~US97/05744
B(2).
rOH
Pht=N CO2Bn
S To a stirred slurry of Part B(l) compound
(124.5 g, 0.449 mole) and cesium carbonate (73.2 g,
0.225 mole) in DMF (1.25 L) at room temperature
under argon was added benzyl bromide (98.4 g, 0.575
mole). After 2.5 hours, the resulting solution was
poured into EtOAc (3.0 L), washed with water (3X),
5% LiCl solution and brine, dried over anhydrous
Mg2SO4 and evaporated i vacuo to afford title
compound (142 g) as an oil in 86.1% yield.
H1-NMR (CDC13): d = 1.50 (m, 4H), 2.32 (m, 2H),
3.62 (m, 2H), 4.91 (dd, lH), 5.22 (d, 2H), 7.31 (m,
5H), 7.77 (m, 2H), 7.86 (m, 2H) ppm
C13-N~. (CDC13): 22.62, 28.46, 31.91, 52.32,
62.32, 67.46, 123.55, 128.06, 128.31, 128.53,
131.77, 134.23, 135.28, 167.76, 169.25 ppm
B(3)-
Pht=N CO2Bn
- To a stirred and chilled (-78~C, Dry ice-
IPA bath) oxalyl chloride solution (2.0 M solution
in CH2C12, 16.3 ml, 32.6 mmole) under argon was
added dropwise a solution of dimethyl sulfoxide
(4.64 ml, 65.32 mmole) in dry CH2Cl2 (10 ml).
After the addition was complete, the solution was
CA 022~1292 1998-10-09
W O 97/38705 rCTnUS97/05744
stirred at -78~ for 15 minutes, then treated with a
solution of Part B(2) compound (lOg, 27.22 mmole)
in dry CH2Cl2 (70 ml), stirred at -78~ for another
15 minutes and slowly treated with triethylamine
(16 ml). The resulting solution was stirred at
-78~ for 15 minutes, gradually warmed up to 0~,
poured into 1:1 EtOAc-Et2O t500 ml), washed with
1.0 N HC1 solution, water and brine, dried over
anhydrous Mg2SO4 and evaporated in vacuo to afford
title compound (10 g) as a light yellow oil in 100%
yield.
H1-NMR (CDCl3): d = 1.66 (m, 2H), 2.40 (m, 4H),
4.90 (dd, lH), 5.18 (d, 2H), 7.35 (m, 5H), 7.74 (m,
IS 2H), 7.86 (m, 2H), 9.72 (s, lH) ppm
C13-NMR (CDCl3): 18.66, 27.99, 42.87, 51.83,
67.47, 123.50, 128.00,128.26, 128.44, 131.58,
134.21, 135.04, 167.55, 168.80, 201.31 ppm
B(4).
~ OH
>
Pht=N CO2Bn
A stirred and chilled (0~C, ice bath)
solution of Part B(3) compound (10.1 g, 27.64
mmole) in dry CH2C12 (100 ml) under argon was
treated with a solution of trimethylaluminum (2.0 M
solution in hexane, 23.4 ml, 46.8 mmole). The
resulting solution was stirred for 45 minutes,
quenched with 100 ml of a saturated NH~Cl solution
(foaming) and partitioned between 1:1 Et2O-water
(400 ml). The organic layer was separated and the
aqueous layer was.re-extracted with EtOAc (2x150
ml). The organic extracts were combined, washed
- 29 -
CA 022~1292 1998-10-09
W097/38705 PCT~S97tO5744
with brine, dried over anhydrous Mg2SO4 and
evaporated in vacuo to afford title compound (10.3
g) as a gum in 98.7% yield.
TLC: Silica gel, 6:4 EtOAc-hexane, Rf = 0.42, W
and PMA.
H1-NMR (CDCl3): d = 1.12 (d, 3H), 1.43 (m, 4H),
3.73 (m, 2H), 4.90 (dd, lH), 5.19 (d, 2H), 7.30
IO (m, 5H), 7.76 (m, 2H), 7.86 (m, 2H) ppm
C13-NMR (CDC13): 22.5, 23.40, 28.47, 28.59, 38.20,
38.34, 52.20.67.35, 67.S1, 123.43, 127.94, 128.19,
128.41, 131.65, 134.11, 135.16, 167.62, 167.67,
15 169.13 ppm
B(5)-
,.~
- Pht=N CO2Bn
To a stirred and chilled (-78~C, Dry ice-
IPA bath) oxalyl chloride solution (2.0 M solution
in CH2C12, 257.3 ml, 514.6 mmole) under argon was
added CH2C12 (300ml). To this solution, a solution
of dimethyl sulfoxide (80.4 g, 1.03 mole) in dry
25 CH2C12 (30 ml) was added dropwise. After the
addition was complete, the reaction mixture was
stirred at -78~ for 20 minutes, treated with a
solution of Part B(4) compound (151 g, 395.88
~mmo~le) in dry CH2Cl2 (700 ml), stirred at -78~C for
another 20 minutes and slowly treated with
triethylamine (300 ml). The resulting solution was
stirred at -78~ for 15 minutes, gradually warmed up
to 0~, poured into 1:1 EtOAc-Et2O (3 L), washed
with l.O N HCl solution, water and brine, dried
over anhydrous Mg2SO4 and evaporated ln vacuo to
- 30 -
CA 022~1292 1998-10-09
W O 97/38705 rCTrUS97/05744
afford title compound (149.4 g) as a yellow oil in
99.5% yield.
TLC: Silica gel, 6:4 EtOAc-hexane, Rf=0.5, W and
PMA.
Hl-NMR ~CDCl3): d = 1.60 (m, 2H), 2.10 (s, 3H),
2.26 (m, 2H), 2.47 (m, 2H),, 4.90 (dd, lH), 5.19
(d, 2H), 7.30 (m, 5H), 7.74 (m, 2H), 7.84 (m, 2H)
ppm
C13-NMR (CDCl3): 20.15, 27.93, 29.84, 42.47,
51.89, 67.40, 123.46, 127.97, 128.23, 128.43,
131,61, 134.17, 135.10, 167.57, 168.93, 207.80 ppm
B(6).
~ OH
Pht=N C02~n
A chilled (-78~C, Dry ice-IPA Bath) and
stirred solution of titanium(IV) chloride (112.05
g, 590.65 mmole) in CH2C12 (1.5 L) under argon was
treated with methylmagnesium chloride (3 M solution
in THF, 196.9 ml, 590.65 mmole). The black
solution was allowed to warm up to -35~C and a
solution of Part B(5) compound (149.4g, 393.77
mmole) was added dropwise. After the addition was
complete, the resulting solution was allowed to
warm up to 0~C, stirred at 0~C for 2 hours and
quenched with saturated NH4Cl solution. The CH2Cl2
layer was separated. The aqueous layer was
extracted with CH2C12 (2x700 ml). The CH2C12
extracts were combined, washed with brine, dried
over anhydrous Mg2SO4 and evaporated ln vacuo. The
black residue was passed through a pad of silica
CA 022~1292 1998-10-09
W097l38705 PCT~S97/05744
gel (E. Merck, 230-400 mesh, 900 g) eluting with
EtOAc-hexane (1:1) to afford a tlc-homogeneous
title compound ~144.8 g) as a yellow oil in 93~ in
yield.
s
TLC: Silica gel, 1:1 EtOAc-hexane, Rf=0.4, W and
PMA.
Hl-NMR (CDCl3): d=1.14 (s, 6H), 1.45 (m, 4H), 2.30
(m, 2H), 4.90 (dd, lH), 5.19 (d, 2H), 7.30 (m, 5H),
7.74 (m, 2H), 7.86 (m, 2H) ppm
C13-NMR (CDCl3): 20.88, 29.00, 29.17, 42.78,
52.13, 67.35, 70.47, 123.44,127.95, 128.19, 128.41,
131.66, 134.11, 167.66, 169.14 ppm
B(7)-
"~
Pht=N CO2Bn
A stirred solution of Part B(6) compound
(44.3 g, 364.89 mmole) and azidotrimethylsilane
(63.06 g, 547.34 mmole) in dry CH2Cl2 (2.2 L) at
room temperature under argon was treated with boron
trifluoride diethyl etherate (67.32 g, 474.36
mmole). After being stirred for 5 days, the
resulting solution was quenched with water (1.5 L).
The organic layer was separated, washed with
saturated NaHCO3 solution, water and brine, dried
aver anhydrous Mg2SO4 and evaporated in vacuo. The
residue was chromatographed on a column of silica
gel (E. Merck, 230-400 mesh, 700 g) eluting with
EtOAc-hexane (1:3) to afford a tlc-homogeneous
title compound (124.9 g) as a light yellow oil in
81.3% yield.
- 32 -
CA 022~1292 1998-10-09
W O 97/38705 PCT~US97/05744
TLC: Silica gel, 3:7 EtOAc-hexane, Rf=0.5, UV and
PMA.
S H1-MMR (CDC13): d=1.20 (s, 6H), 1.45 (m, 4H), 2.30
(m, 2H), 4.90 (dd, lH), 5.19 (d, 2H), 7.30 (m, 5H),
7.74 (m, 2H), 7.86 (m, 2H) ppm
C13-NMR (CDC13): 20.97, 25.67, 25.92, 28.80,
40.53, 52.02, 61.16, 67.40, 123.47, 127.97, 128.23,
128.43, 131.66, 134.14, 135.12, 167.60, 169.01 ppm
B(8).
Pht=N ~ I~
0
A solution of Part B(7) compound (124.8 g,
296.81 mmole) and 10% Pd/C (32g) in dry DMF (2.0 L)
was hydrogenated for 24 hours. After completion,
argon was bubbled through the reaction mixture to
remove excess hydrogen and methyl sulfide (2.6 ml)
was added to poison the palladium. To this
solution 1-hydroxybenzotriazole hydrate (46.74 g)
was added and followed by ethyl-3(3-dimethylamino)-
propylcarbodiimide hydrochloride salt (68.74 g).
The resulting solution was stirred at room
temperature under argon for 3.5 hours, diluted with
EtOAc (2 L) and filtered through a pad of celite.
The filtrate was washed with 0.5 N HCl solution,
saturated NaHCO3 solution, and brine, dried over
anhydrous Mg2SO4 and evaporated in vacuo to give a
gum. This was triturated with Et2O-hexane (2:1) to
afford a tlc-homogeneous title compound (74.5 g) as
a white solid in 87.7% yield.
TLC: Silica gel, 3:7 EtOAc-CH2C12, Rf=0.35, W and
PMA.
- 33 -
CA 022~1292 1998-10-09
W O 97/38705 PCT~US97/05744
H1-NMR (CDC13): d=1.30 (s, 3H), 1.45 (s, 3H), 1.74
(m, 2H), 1.96 (m, 3H), 2.74 (m, lH), 4.98 (d, lH),
6.00 (s, lH), 7.20 (m, 2H), 7.85 (m, 2H) ppm
s
C13-NMR (CDC13): 23.89, 26.65, 29.58, 33.32,
40.68, 52.69, 54.51, 123.34, 123.15, 133.87,
168.06, 171.03 ppm
B(9).
Ph3C~ N~(~
A stirred solution of Part B(8) compound
(74.5 g, 260.19 mmole) in a mixture of CH30H (900
ml) and CH2C12 (250 ml) at room temperature under
argon was treated with hydrazine monohydrate (18.24
g, 364.26 mmole). After 48 hours, the solid was
filtered off and the filtrate was evaporated ln
vacuo to give a solid (41 g).
To a stirred solution of the above solid
(41 g) in CH2C12 (2 L) at room temperature under
argon was added triethylamine (50 ml) and
triphenylmethyl chloride (83.41 g). After 1.5
hours, the resulting slurry was diluted with EtOAc,
washed with water and brine, dried over anhydrous
Mg2SO4 and evaporated in vacuo to give a gum. This
was triturated with Et2O-pentane to give title
compound (100.1 g) as a white solid in 96.5% yield.
~LC: Silica gel, 6:4 EtOAc-hexane, Rf=0.53, W and
PMA.
H1-NMR (CDCl3): d=1.00 (s, 3H), 1.10 (s, 3H), 1.46
(m, 6H), 3.36 (m, lH), 4.03 (m, lH), 5.20 (d, lH),
6.00 (s, lH), 7.20 (m, 2H), 7.85 (m, 2H) ppm
- 34 -
~ , . . .
CA 022~1292 1998-10-09
W O 97~870S PCTrUS97/05744
C13-NMR (CDCl3): 22.86, 25.81, 33.50, 34.23,
40.16, 51.97, 55.60, 71.89, 126.22, 127.61, 128.96,
146.48, 176.71 ppm
B(10).
Me
Me
H2N~ N~l
~ CO2Et
To a stirred solution of Part B(9) compound
l0 (50 g, 125 mmole) in dry THF (1020 ml) at room
temperature under argon was added simultaneously
(at same rate) a solution of lithium
bis(trimethylsily)amide (1.0 M solution in THF,
627.3 ml, 627.3 mmole) and a solution of ethyl
bromoacetate (104.8 g, 627.3 mmole) in THF (523 ml)
over the period of 1.0 hour. After the addition
was complete, the solution was stirred for 30
hours, quenched with saturated NH4Cl solution (1.0
- liter) and extracted with EtOAc (3x700 ml). The
EtOAc extracts were combined, washed with saturated
NaHCO3 solution and brine, dried over anhydrous
Mg2SO4 and evaporated in vacuo to afford a black
oil. The experiment was repeated on the same scale
to give a similar result. The combined black oils
was chromatographed on a column of silica gel (E.
Merck, 230-400 mesh, 1.6 kg) eluting with EtOAc-
hexane (1:4) to give a light yellow oil. This was
dissolved in dry CH2Cl2 (2 L) and treated with
trifluoroacetic acid (78 ml). The solution was
stirred at room temperature under argon for 1.0
hour and then evaporated in vacuo at 30~. The
residue was diluted with 1.0 N HCl solution (400
ml) and washed with Et2O (2x400 ml). The a~ueous
was carefully neutralized to pH=7-8 with solid
NaHCO3 (foaming) and extracted with CH2Cl2 (3x1.2
CA 022~1292 1998-10-09
W O 97138705 PCTrUS97/05744
L). The CH2C12 extracts were combined, dried over
anhydrous Na2SO4 and evaporated in vacuo to afford
a tlc homogeneous title compound (51.5 g) as a
light brown oil in 84.7% yield.
s
TLC: Silica gel, 8:1:1 CH2C12-CH3OH-AcOH, Rf=0.3,
PMA and Ninhydrin.
Hl-NM~ (CDC13): d=1.28 (t, 3H), 1.36 (s, 3H), 1.38
(s, 3H) 1.60 (m, lH), 1.90 (m, 5H), 3.75 (m, lH),
4.00 (d, lH), 4.22 (q, 2H), 4.28 (d, 2H) ppm
C13-NMR (CDCl3): 14.00, 20.06, 28.19, 30.07,
32.29, 39.98, 46.87, 53.20, 58.38, 60.73, 170.35,
177,06 ppm
~ Me
HN ~ N ~ ~
H o C02Et
Part A compound (641 mg, 1.42 mmol) was
partitioned between EtOAc and 5% KH2PO4 (adjusted
to pH 2.5 with H3PO4). The layers were separated
and the aqueous layer was back-extracted with
EtOAc. The pooled EtOAc extracts were washed with
brine, dried (Na2SO4), filtered and stripped to
give an oil (assume 1.42 mg). The oil was
dissolved in CH2Cl2 (10 mL) and the resulting
solution was treated with Part B amine (364 mg,
1.50 mmol) in CH2Cl2 (2 mL) and cooled to 0~C. The
mixture was subsequently treated with HOBT hydrate
(195 mg) followed by EDAC (285 mg, 1.48 mmol).
After stirring at 0~C for 45 minutes and at room
temperature for 45 minutes, the mixture was
- 36 -
, . ..... ...
CA 022~1292 1998-10-09
W O 97~8705 PCTrUS97/05744
partitioned between EtOAc and 5% KH2PO4 (adjusted
to pH 2.5 with H3PO4). The EtOAc extract was
washed successively with H2O, 50% saturated NaHCO3
and brine, then dried (Na2SO4), filtered and
stripped. The residue was flash chromatographed
(Merck SiO2, 7/3-EtOAc/hexanes as eluant) to obtain
title compound (427 mg, 59%, TLC Rf 0.37 (8/2-
EtOAc/hexanes)) as a diastereomerically pure
compound. In addition, the minor diastereomer was
isolated from the column (66 mg, 9%, TLC Rf O . 27
(8/2-EtOAc/hexanes)). NMR of this material was
consistant with an isomer of the title compound.
~M e
~ ~ M e
y - H O CO2Et
~
Acetic anhydride (500 ~L) was added to
formic acid (5.0 mL) at 0~C and the mixture was
stirred for 30 minutes. Approximately 2.6 mL of
this solution was added to a solution of Part C
compound (208 mg, 0.413 mmol) in THF (1.1 mL) at
0~C. After 30 minutes, most of the solvent was
removed by rotary evaporation and the residue was
partitioned between EtOAc and saturated NaHCO3.
The EtOAc extract was washed with brine, dried
(Na2SO4), filtered and stripped to give title
compound (216 mg, 97%) as an oily foam which was
used directly in the next reaction without futher
purification.
TLC Rf 0.37 (EtOAc)
HPLC YMC S3 ODS column (6.0 x 150 mm); eluted with
B:A solvent mixture, 40 to 100% B over a 20 minute
- 37 -
CA 02251292 1998-10-09
W O 97/38705 PCTrUS97/05744
linear gradient (solvent A: 90%H20-10% MeOH-0.2%
H3PO4; solvent B:0% H2O-90% MeOH-0.2% H3PO4); flow
rate 1.5 mL/min detecting at 220 nm; tR = 17.2 min
100%).
E.
~M e
~ ~'b' Me
y _ O CO2Et
H ~
A solution of Part D compound (216 mg,
0.402 mmol) in absolute EtOH (5 mL) was
hydrogenated (balloon) over 10% Pd/C (33 mg) at
room temperature for 2 hours. The mixture was
filtered through Celite, stripped, and azeotroped
twice with EtOAc/Et2O/hexanes to give title
compound (174 mg, 97%) as an off-white foam.
TLC Rf 0.33 (5/95-HOAc/EtOAc)
HPLC YMC S3 ODS column (6.0 x 150 mm); eluted with
B:A solvent mixture, 40 to 100% B over a 20 minute
linear gradient (solvent A: 90%H20-10% MeOH-0.2%
H3PO4; solvent B:0% H2O-90% MeOH-0.2% H3PO4); flow
rate 1.5 mL/min detecting at 220 nm; tR = 12.8 min
( 100%).
F.
~' ~ OyN~~cMMOeeH
H ~
-- 38 --
CA 022~1292 1998-10-09
W097/38705 PCT~S97/05744
A stirred solution of Part E compound (168
mg, 0.376 mmol) in MeOH (3 mL) at room temperature
was treated with aqueous 1 N NaOH (3 mL). An
additional portion of aqueous 1 N NaOH (3 mL) was
added after 3.5 hours. After a total of 6 hours,
the mixture was made acidic with 5% KHSO4 and
extracted twice with EtOAc. The EtOAc extract was
washed with brine, dried (Na2SO4), filtered and
stripped. The residue was dissolved in a small
amount of MeOH and EtOAc and triturated with
Et2O/hexanes to give title compound (134 mg, 86%~
as an off-white solid/foam
([a]D = +18.0~ (c 0.5, CH2Cl2)).
I5 TLC Rf 0.10 (5/95-HOAc/EtOAc)
HPLC YMC S3 ODS column (6.0 x 150 mm)i eluted with
B:A solvent mixture, 40 to 100% B over a 20 minute
linear gradient (solvent A: 90%H20-10% MeOH-0.2%
H3PO4; solvent B:0% H2O-90% MeOH-0.2% H3PO4); flow
rate 1.5 mL/min detecting at 220 nm; tR = 9~~~
min (>97.4%).
Anal. Calc'd for C21H29N3O6-0.75H2O-0-3Et2O
C, 58.57; H, 7.42; N, 9.23
Found C, 58.31i H, 7.20i N, 8.99.
Exam~le 4
~S-(R*,R*)]-3-[[3-(Formylhydroxyamino)-l-oxo-2-
(phenylmethyl)propyl]amino]-2,3,4,5-tetrahydro-2-
oxo-lH-benzaze~ine-l-acetic acid
N~~ N~ N~
HO - H ~ COOH
13'
- 39 -
CA 022~1292 1998-10-09
W O 97/3870~ PCTrUS97/05744
~ N~, COOC2H5
H2N ll
A~l).
Z~o
H
Solid sodium azide (26.0 g., 0.2 mole) was
introduced into a 3-neck round-bottom flask with an
overhead stirrer, made into a paste with warm water
(26 ml), layered with chloroform (160 ml) and
cooled down to 0~ (ice-salt bath). The mixture was
treated dropwise with concentrated sulfuric acid
(11.2 ml, 0.5 eq.) over a period of 10 minutes,
stirred for an additional 10 minutes then decanted
into a flask cont~lnlng anhydrous sodium sulfate.
The dried solution was filtered through a glass
wool plug in a funnel into a 500-ml round-bottom
flask. Titration of an aliquot (1.0 ml) with 1.0 N
NaOH using phenolphthalein as an indicator gave a
normalitity of 1.7 N for the hydrazoic acid.
Tetralone (15.94 g, 0.108 mole) was added
to the hydrazoic acid solution (0.136 mole or 1.25
eq.), heated to 40-45~ (oil bath) then treated
dropwise with 36.0 N H2SO4 (28.7 ml, 5 eq.) over a
period of 1.0 hour. (Intense bubbling took place
with each drop added for the first 30 minutes).
The reaction mixture was cooled down to room
temperature, poured into H2O (720 ml) and stirred
for 5 minutes. The solution was then extracted
with EtOAc (3 x 250 ml) and the combined organic
extracts were washed with brine (100 ml), dried
(anhydrous MgSO4), filtered, evaporated to dryness
and dried in vacuo . The crude product (17.819 g)
- 40 -
CA 022~1292 1998-10-09
W097/38705 PCT~S97/05744
was recrystallized from CH2C12 (70 ml) and Hexane
(400 ml) to give title compound as off-white
precipitates (10.017 g, m. pt. 138-140~C) with
consistent lH-NMR and 13C-NMR spectral data.
The mother liquor was chromatographed on a
silica gel column (Merck, 240 g), eluting the
column with EtOAc:Hexane (1:4) to give an
additional amount of 5.058 g (total yield= 15.075
g, 85.6 %).
TLC: Rf 0.37 (Silica geli EtOAc:Hexane-l:li W).
A(2).
~ ~ Br
A solution of Part A(l) compound (1.0 g,
6.20 mmoles) in dry CHC13 (15 ml) was cooled down
to 0~C (ice-salt bath), treated with PCls (1.5 g,
7.20 mmoles) followed by I2 (15 mg) then stirred at
- 0~C under argon for 30 minutes. The yellow
solution was treated with Br2 (0.39 ml or 1.2 g,
7.51 mmoles), warmed up to room temperature and
refluxed under argon for 4.0 hours. The mixture
was then poured into ice-water (20 g), stirred and
the phases were separated, washing the a~ueous
phase with CHC13 (25 ml). The combined organic
extracts were washed with H2O (5.0 ml), dried
(anhydrous MgSO4), filtered, evaporated to dryness
and dried in vacuo. The crude product mixture was
chromatographed on a silica gel column (Merck, 70
g),-eluting the column with EtOAc:Hexane (1:9) to
give title compound as off-white precipitates
(1.137 g., m.pt. 170-172~, 70.1 %) with consistent
H-NMR and 13C-NMR spectral data. TLC: Rf 0.13
(Silica geli EtOAc:Hexane -1:4; W).
- 41 -
CA 022~1292 1998-10-09
W O 97/38705 PCT~US97/05744
A(3)-
A solution of Part A(2) compound (936 mg,
3.9 mmoles) and NaN3 (300 mg, 4.6 mmoles) in dry
dimethylsulfoxide (20 ml) was stirred at 60~ (oil
bath) under argon for 6.0 hours. The reaction
mixture was cooled down to room temperature, poured
into cold water (125 ml), stirred for 15 minutes
and filtered, washing the solids formed with water.
The crude product was dried in vacuo at 60~ over
drierite for 24 hours to give title compound (725
mg, m.pt. 150-152~, 91.9 %) as an off-white solid
with consistent lH-NMR and 13C-NMR spectral data.
TLC: Rf 0.58 (Silica gel; EtOAc:Hexane- 1:4 then
1:1; W).
A(4).
~ ~ ~ N3
cooC2H5
A solution of Part A(3) compound (10.858
g, 53.7 mmoles) in dry tetrahydrofuran (100 ml) was
treated with Bu4NBr (1.791 g, 5.56 mmoles) and
powdered KOH (3.937 g, 70.2 mmoles) followed by
ethyl bromoacetate (6.8 ml, 61.3 mmoles). The
reaction mixture was stirred at room temperature
under argon for 1.5 hours then partitioned between
H2O (196 ml) and CH2C12 (2 x 375 ml). The combined
organic extracts were washed with H2O (2 x 196 ml)
and brine (100 ml), dried (anhydrous Na2SO4),
filtered, evaporated to dryness and dried in vacuo .
The crude product was combined with the crude
~ product mixture from a previous run (2.936 g, 12.86
- 42 -
CA 022~1292 1998-10-09
W O 97138705 PCT~US97/05744
mmole scale) and chromatographed on a silica gel
column (Merck), eluting the column with
Toluene:EtOAc (98.2) and EtOAc:Hexane (1:9) to give
title compound as a solid (15.48 g, 93.5%)1 with
consistent lH-NMR and 13C-NMR spectral data.
TLC: Rf 0.63 (Silica geli EtOAc:Hexane- 1:2; W).
A(5)-
NH2
COOC2H5
A solution of Part A(4) compound (8.95 g,
31.0 mmoles) in absolute ethanol (50 ml) was
treated with 10% Pd/C (443 mg) and hydrogenated at
45 psi for 3.5 hours, venting the Parr bottle every
30 minutes for the first 1.5 hours. The mixture
was filtered through a Celite~ pad in a millipore
unit, w~.~h;ng the pad well with absolute ethanol (3
x 50 ml). The clear filtrate was evaporated to
dryness and dried in vacuo to give title compound
as a thick yellow syrup (7.929 g, 97.5%) with
consistent 1H-NMR and 13C-NMR spectral data. TLC:
Rf 0.45 (Silica gel; CH2Cl2:CH30H- 9:1; W).
A(6)-
H2N~ N~COOC2H5
0
A solution of Part A(5) compound (14.8 g,
56.4 mmoles) and L-tartaric acid (8.50 g) in hot
absolute ethanol (118 ml) was kept overnight at 0~,
at room temperature for 3 days and then at 0~ for
another 2 days. The solid that formed was
recrystallized from absolute ethanol (118 ml~ two
- 43 -
CA 022~1292 1998-10-09
W097/38705 PCT~S97/OS744
more times until a consistent specific rotation was
obt~'ne~. The precipitates (6.319 g) from the
second recrystallization was then suspended in
EtOAc (100 ml), treated with 10% NH40H (12 ml) and
stirred for 5 minutes. The organic phase was
separated, washed with 10% NH40H (10 ml) and brine
(15 ml), dried (anhydrous Na2S04), filtered,
evaporated to dryness and dried in vacuo to give
title compound as a white solid (3.927 g, m.pt.
105-107~, 26.5%) with consistent lH-NMR and 13C-NMR
spectral data.
[a]D = -277~ (c 0.99, EtOH). TLC : Rf 0.45 (Silica
gel; CH2C12:CH30H- 9:1; W).
I5 B.
,N ~ N
~~ -- H ~
Example 3 Part A ephedrine salt (414 mg,
0.93 mmole), was partitioned between 5 % KH2P04
~adjusted to pH 2.5; 4.0 ml) and EtOAc ( 2 x 20 ml)
and the combined organic extracts were washed with
brine (4.0 ml), dried (anhydrous Na2S04), filtered,
evaporated to dryness and dried in vacuo to give
the free acid of the Example 4 Part A compound as
a clear syrup (286.6 mg, 100 % crude yield).
A solution of the above free acid (286.6
~g,~0.93 mmole) in dry CH2C12 (6.0 ml) was cooled
to 0~C (ice-salt bath) and treated sequentially
with a solution of the above free amine (271 mg) in
dry CH2Cl2, HOBT-H20 (126.1 mg, 0.93 mmole) and
EDAC (185.4 mg, 0.97 mmole). The reaction mixture
was stirred at 0~C for 1.0 hour, at room
CA 022~1292 1998-10-09
W097/38705 PCT~S97/057
temperature for 2.0 hours, then partitioned between
EtOAc (2 x 20 ml) and H2O (4.0 ml). The organic
extracts were washed with 5% KH2PO4 (adjusted to pH
2.5; 4.0 ml), H2O (4.0 ml), saturated NaHCO3 (4.0
ml) and brine (4.0 ml), dried (anhydrous Na2SO4),
filtered, evaporated to dryness and dried in vacuo .
The crude product was chromatographed on a silica
gel column (Merck, 70 g.), eluting the column with
EtOAc:Hexane mixtures (1:3i 1:1) to give pure title
compound (202 mg) and impure product. A second
chromatography gave title compound as a syrup
(total of 292.1 mg, 59.3~) with consistent lH-NMR
and
13C-NMR spectral data. TLC: Rf 0.32 (Silica gel;
EtOAc:Hexane -1:1; W).
OHC N~J~ , N~ COOC2H5
A cooled solution of HCOOH (5.0 ml) was
treated with acetic anhydride (Ac2O) (0.5 ml) and
stirred at 0~C for 30 minutes. A solution of Part
B compound (288 mg, 0.54 mmole) in dry THF (1.5 ml)
was cooled to 0~C (ice-salt bath), treated with the
above Ac2O/HCOOH mixture (3.4 ml) and stirred at
0~C for 1.0 hour. The reaction mixture was
èvaporated to dryness and the residual syrup was
dissolved in EtOAc (40 ml), washed with saturated
NaHCO3 (5.0 ml) and brine (5.0 ml), dried
(anhydrous Na2SO4), filtered, evaporated to
dryness, evaporated from toluene and dried in vacuo
to give title compound as a syrup (311.3 mg, 100 %
- 45 -
CA 022~1292 1998-10-09
W O 97/38705 PCTrUS97/05744
crude) with consistent lH-NMR and 13C-NMR spectral
data. TLC: Rf 0.18 (Silica gel; EtOAc:Hexane (1:1;
W) .
S D.
N--J~ N$~CooC2H5
HO - H ~
A solution of Part C compound (311 mg) in
CH30H (10 ml) was treated with 10% Pd/C (53 mg) and
hydrogenated (balloon) at room temperature for 2.0
hours. The reaction mixture was diluted with CH30H
(10 ml) and filtered through a Celite~ pad in a
millipore unit, washing the pad well with CH30H (3
x 10 ml). The clear riltrate was evaporated to
dryness and dried in vacuo to give title compound
as a syrup (256.7 mg, 100% crude) with consistent
lH-NMR and 13C-NMR data. TLC: Rf 0.25 (Silica gel;
CH2Cl2:MeOH- 9:1; W).
E. [S-(R*,R*)]-3-[[3-(Formylhydroxyamino)-
l-oxo-2-~phenylmethyl)propyl]amino]-
2,3,4,5-tetrahydro-2-oxo-lH-benzazepine-l-
acetic acid
A solution of Part D compound (256.7 mg) in
CH30H (3.5 ml) was treated with 1.0 _ NaOH (2.17
ml, 4 e~) and stirred at room temperature for 1.0
hour under argon. The reaction mixture was brought
to pH 1.0 with 5% KHS04 (9.45 ml), extracted with
EtOAc (40 ml) and the organic extract washed with
brine (5.0 ml), dried (anhydrous Na2S04), filtered,
evaporated to dryness and dried in vacuo. The
crude product was triturated with CH2Cl2 Hexane
- 46 -
CA 02251292 1998-10-09
W O 97/38705 PCT~US97/05744
~1:4-25 ml) and hexane (20 ml) then dried in vacuo
to give title compound as an amorphous off-white
solid (215.6 mg, 90.4%) with consistent MS, IR, lH-
NMR and analytical data. TLC: Rf O.30 (Silica gel
EtOAc:HOAc- 95:5i W).
[a] D = -332.8~ (c 0.558, CH30H)
HPLC: tR= 5.21 min (95.8% R isomer); tR =9.58 min
(3.59% S isomer); YMC S3 ODS-A 150 x 6 mm; 220 nm,
flow rate = 1.5 ml/min; 56% (10% H20- 90% CH30H-
0.2% H3P04)/44% (90% H20- 10% CH30H-0.2% H3P04),
isocratic.
Anal. Calc'd for C23H25N306:
C, 62.86i H, 5.73; N, 9.56
Found: C, 62.88i H, 5.98; N, 9.20.
ExamDle 5
O ~U
OHC ~ N ~ll' N~ ~J
H ~ COOH
A.
OH
H f
Pht=N ~ ~~
O COOCH3
A solution of L-hydroxynorleucine (2.0 g,
13.6 mmoles) in dry methanol (70 ml) was saturated
with HCl gas until a clear yellow solution was
obt~'ne~. The reaction mixture was cooled to room
temperature, stirred for 2.0 hours, evaporated to
- 47 -
CA 022~1292 1998-10-09
W097l38705 PCT~S97/05744
dryness, evaporating the syrup once from toluene
(100 ml) then evaporated in vacuo to give the ester
as a yellow oil. The crude ester was dissolved in
dry CH2C12 (50 ml) and dry DMF (15 ml), treated
with NMM (2.5 ml, 22.7 mmoles) and cooled to 0~C
(ice-salt bath). The mixture was treated with N-
phthaloyl-L-phenylalanine (4.0 g, 13.6 mmoles),
HOBt-H2O (1.89 g, 13.99 mmoles) and EDAC (2.87 g,
14.98 mmoles), stirred at 0~C for 25 minutes and at
room temperature for 2.0 hours.
The reaction mixture was partitioned
between EtOAc (2 x 200 ml) and H2O (60 ml) and the
combined organic extracts were washed sequentially
with 0.5 N HCl (60 ml), H2O (60 ml), 1/2 saturated
NaHCO3 (60 ml) and brine (60 ml), dried (anhydrous
Na2SO4), filtered, evaporated to dryness and dried
in vacuo . The crude product mixture was
chromatographed on a silica gel column (Merck, 200
g), eluting the column with EtOAc to give the
desired product as a syrup (4.0 g). An additional
321 mg was obtained on re-chromatography of the
impure fractions to give title compound (4.32 g,
73%) with consistent 1H-NMR and 13C-NMR spectral
data.
TLC: Rf 0.43 (Silica gel; EtOAci W).
O
H //
Pht=N ~ ~~
O COOCH3
A solution of oxalyl chloride (1.02 ml,11.7
mmoles) in dry CH2C12 (56 ml), was cooled to -78~C
(dry-ice-acetone bath), treated with a solution of
dry DMSO (1.67 ml, 21.6 mmoles) in CH2Cl2 (2.0 ml)
- 48 -
.. . , . ~
CA 022~1292 1998-10-09
W O 97/38705 PCTrUS97/05744
and stirred at -78~C for 20 minutes. The mixture
was treated with a solution of Part A compound
(4.29 g, 9.78 mmoles) in dry CH2Cl2 (22 ml),
stirred at -78~C for another 15 minutes, then
treated with triethyl-amine (8.4 ml). The reaction
mixture was stirred at -78~C for 5.0 minutes,
allowed to come to room temperature over a period
of 45 minutes, then partitioned between EtOAc (200
ml) and 0.5 N HCl (2 x 20 ml). The organic phase
was washed with brine (40 ml), dried (anhydrous
Na2SO4), filtered, evaporated to dryness and dried
in vacuo to give title compound as a thick syrup
(4.428 g, 100% crude yield), with consistent lH-NMR
and 13C-NMR spectral data.
TLC: Rf 0.73 (Silica gel; EtOAc; W).
¢~
Pht= ~ ~
o COOCH3
A mixture of Part B compound (4.428 g, 9.78
mmoles) and TFA (0.20 ml, 2.6 mmoles) in dry CH2C12
(62 ml) was refluxed under argon for 2.0 hours.
The reaction mixture was cooled to room
temperature, washed with 1/2 saturated NaHCO3 (20
ml) and brine (20 ml), dried (anhydrous Na2SO4),
filtered, evaporated to dryness and dried in vacuo.
The crude product mixture was chromatographed on a
sil~ica gel column (Merck, 200 g), eluting the
column with CH2C12:EtOAc (9:1) to give the desired
product as a syrup. The syrup was triturated with
Et2O:Hexane ~2:1-60 ml) to give title compound as
a white precipitate (2.92 g, 72%; m.p. 141-143~C)
with consistent lH-NMR and 13C-NMR spectral data.
- 49 -
CA 022~1292 1998-10-09
W097/38705 PCT~S97/OS744
TLC: Rf 0.67 (Silica gel; CH2C12:EtOAc-9:li W).
Pht= N~ ll
A solution of Part C compound (2.923 g,
6.99 mmoles) in dry CH2C12 (14 ml~ was treated with
triflic acid (4.15 ml, 6.7 eq) and the resulting
yellow solution was stirred at room temperature for
20 hours. The reaction mixture was then poured
into ice-water (100 ml), extracted with EtOAc (3 x
100 ml) and the combined organic extracts washed
with H2O (2 x 25 ml) and brine (25 ml), dried
(anhydrous Na2SO4), filtered, evaporated to dryness
and dried in vacuo . The crude product mixture was
chromatographed on a silica gel column (Merck),
eluting the column with EtOAc:Hexane mixtures (1:1;
2:1) and EtOAc:HOAc (100:1). The desired fractions
were combined, evaporated to dryness and dried in
20 vacuo to give impure title compound as a solid
foam (1.238 g, 42%) with consistent lH-NMR and 13C-
NMR spectral data. TLC : Rf 0.73 (Silica gel;
EtOAc:HOAc-95:5; W).
E.
Pht=N~
o COOBn
A solution of Part D compound (1.238 g,
3.06 mmoles) in dry DMF (3.5 ml) was treated
- 50 -
CA 022~1292 1998-10-09
W O 97/38705 PCTAUS97/05744
sequentially with benzyl bromide (0.35 ml, 2.94
mmoles) and Cs2CO3 (450 mg, 1.38 mmoles) then
stirred at room temperature for 3.0 hours. The
mixture was diluted with EtOAc (50 ml), washed with
H2O (5.0 ml), 0.5 _ HCl (5.0 ml) and brine (5.0
ml), dried (anhydrous Na2SO4), filtered, evaporated
to dryness and dried in vacuo. The crude product
(1.63 g) was chromatographed on a silica gel column
(Merck), eluting the column with EtOAc:Hexane (1:3)
to give title compound as a syrup (586.4 mg, 39%)
with consistent lH-NMR and 13C-NMR spectral data.
TLC: Rf 0.45 (Silica geli EtOAc:Hexane-l:l; W).
H2N O cooen
A solution of Part E compound (586 mg,
1.18 mmoles) in dry methanol (15 ml) was treated
with NH2NH2-H2O (66 ,ul, 1.2 eq) and stirred at room
temperature for 48 hours. The reaction mixture was
diluted with Et2O (50 ml) and filtered through a
millipore unit, washing the solids well with Et2O
(40 ml). The clear solution was evaporated to
dryness and the solids obtained were suspended in
CH2C12 (90 ml) and the solution filtered through a
millipore unit, washing the solids well with CH2C12
(40 ml). The combined organic extracts were washed
with brine (15 ml), dried (anhydrous Na2SO4),
filtered, evaporated to dryness and dried in vacuo
to give title compound as a thick syrup (351 mg, 82
%) with a consistent lH-NMR spectrum.
TLC: Rf 0.42 (CH2Cl2 MeOH-9:1; W, Ninhydrin)
- 51 -
CA 022~1292 1998-10-09
W O 97/38705 PCTnUS97105744
~'
BnO _ H O COOBn
Example 3 Part A ephedrine salt (538 mg,
5 1.2 mmoles), was partitioned between 5% KH2PO4
(adjusted to pH 2.5; 5.4 ml) and EtOAc (2 x 22 ml)
and the combined organic extracts were washed with
brine (5.4 ml), dried (anhydrous Na2SO4), filtered,
evaporated to dryness and dried in vacuo to give
10 the free acid of the ephedrine salt as a clear
syrup (323 mg, 100% crude yield).
A solution of the free acid in dry
CH2Cl2 (8.0 ml) was cooled to 0~C (ice-salt bath)
and treated sequentially with a solution of Part F
compound (351 mg, 0.96 mmole) in dry CH2C12 (2.0
ml), HOBT-H2O (163 mg, 1.2 mmoles) and EDAC t 240
mg, 1.25 mmoles). The reaction mixture was stirred
at 0~C for 1.0 hour, at room temperature for 1.5
hours, then partitioned between EtOAc (40 ml) and
H2O (5.0 ml). The organic extracts were washed
with 5 % KH2PO4 (adjusted to pH 2.5i 5.0 ml), H2O
(-5.0 ml), saturated NaHCO3 (5.0 ml) and brine (5.0
ml), dried (anhydrous Na2SO4), filtered, evaporated
to dryness and dried in vacuo. The crude product
(810 mg) was chromato-graphed on a silica gel
column (Merck), eluting the column with
EtOAc:Hexane (1:3) to give pure title compound
(494 mg, 65%) as a solid foam with consistent lH-
N~ and 13C-NMR spectral data.
TLC: Rf 0.45 (Silica gel; EtOAc:Hexane -1:1; W).
- 52 -
CA 022~1292 1998-10-09
W O 97/38705 PCTrUS97105744
O ~
BnO H O COOBn
A cooled solution (0~C, ice-salt bath) of
HCOOH (5.0 ml) was treated with Ac20 (0.5 ml) and
stirred at 0~C for 30 minutes. A solution of Part
G compound (493 mg, 0.78 mmole) in dry THF (2.2 ml)
was cooled to 0~C (ice-salt bath), treated with the
above Ac20/HCOOH mixture (4.9 ml) and stirred at
0~C for 1.5 hours. The reaction mixture was
evaporated to dryness, evaporated from Et20 (50 ml)
and the residual syrup was dissolved in EtOAc (60
ml), washed with saturated NaHC03 (7.0 ml) and
brine (7.0 ml), dried (anhydrous Na2S04), filtered,
evaporated to dryness, evaporated from toluene and
dried in vacuo to give title compound as a syrup
(558.3 mg, 100 % crude) with consistent lH-NMR and
13C-NMR spectral data.
TLC: Rf 0.2 (Silica gel; EtOAc:Hexane-l:l; W).
I.
OHC~N,J~ N~
~ H ~ COOH
A solution of Part H compound (535 mg, 0.78
mmole) in CH30H (15 ml) was treated with 10 % Pd/C
(83 mg) and hydrogenated (balloon) at room
- 53 -
CA 022~1292 1998-10-09
W O 97/38705 PCTnUS97tO5744
temperature for 4.0 hours. The reaction mixture
was diluted with CH30H (15 ml) and filtered through
a celite pad in a millipore unit, washing the pad
well with CH30H (3 x 15 ml). The clear filtrate
was evaporated to dryness and dried in vacuo to
give a syrup (354.8 mg) which was triturated with
CH2C12:Hexane (1:5-30 ml) and hexane (25 ml) then
dried in vacuo. Title compound was obtained as an
off-white solid foam (348.5 mg, 90%).
TLC: Rf 0.38 (Silica geli CH2Cl2:MeOH- 9:1; W).
MS (M+H)+ = 480
[a]D = +44.6~ (c 0.52, CH30H)
15 HPLC : tR= 11.72 min (95.9% ); YMC S3 ODS-A 150 x 6
mm; 220 nm, flow rate = 1.5 ml/mini 55% (10% H20-
90% CH30H- 0.2% H3P04)/ 45% (90% H20- 10% CH30H-
0.2% H3P04), isocratic.
20 Anal. Calc~d for C26H2gN306-0.4 H20-0.14 Hexane
- (Eff. Mol. Wt. = 497.08):
C, 64.63; H, 6.83; N, 8.46
Found: C, 64.24i H, 6.43; N, 8.12
The following are examples of additional
compounds of the invention which may be prepared
employing procedures set out hereinbefore and in
the working Examples.
- 54 -
CA 02251292 1998-10-09
PCT~US97/05744
W O 97~870S
R10
~R ~ A
Example
No. R-- _ ~ A
6 H 1CH2Ph r )~
~ CO2H
7 H 1 CH2Ph 5--N$~
~ CO2H
~ H
8 H 1CH2CH(CH3)2~--N$~)
~ CO2H
9 H 1 CH2Ph ~ $
~ CO2H
~--N
H 1CH2CH(CH3)25--N~N~
~ CO2H
11 H 1 CH2Ph ~--N~N~Me
~ CO2H
12 H 1 CH2Ph
5--N~ ~--J
~ CO2H
CA ~2251z92 1998 1o og
WO 97/3870S
PcTlus97/o5744
13 H I ~
~Nle
"f~ Ph
14 H 1 CH(CH3)2 5--~ ,, C~2H
H 1 CH(CH3)2 ~--~N~--c02H
H O 2=
16 H 1 CH(cH3)2 HN$~ N~ Co2H
,.~
~OH
17 H 1 CH2Ph
HN~ CO2H
\~ Ph
-- 56 --