Note: Descriptions are shown in the official language in which they were submitted.
CA 02252229 2002-04-15
69562-22
SPECIFICATION
Novel Pyridoneearboxylic Acid Derivatives or Their Salts,
and Antibacterial Agents Containing the Same
as -Their Effective Components
Technical Field
The present invention relates to novel pyridone-
carboxylic acid derivatives or their salts having excellent
io antibacterial activities and oral absorption, and'
antibacterial agents containing the,_same.
Background Technology
Many compounds having a basic skeleton of pyridone-
carboxylic acid are known to be useful synthetic
~ntibacterials for their excellent antibacterial activities
and wide antibacterial spectrum. Among such compounds,
norfloxacin (Japanese Patent Application.Laid-Open No.
53-141286), enoxacin (Japanese Patent Application Laid-Open ~'
2o No. 55-31042), ofloxacin (Japanese Patent Application
Laid-Open No. 57-46986), ciprofloxacin (Japanese Patent
Application Laid-Open No. 58-74667) , tosufloxacin (Japanese
Patent Application Laid-Open No. 60-228479), and the like are
' widely used in clinical practice for treating infections.
These compounds, however, need further improvements in
antibacterial activities, intestinal absorption, metabolic
stability, and side effects, and in particular, in
phototoxicity and cytotoxicity.
3o Summary of the Invention
The present invention has been completed in view of such
situation, and an object of the present invention is to
provide novel pyridonecarboxylic acid derivatives or the
salts thereof having satisfactory antibacterial activities,
intestinal absorption and metabolic stability, and reduced
CA 02252229 1998-10-16
- 2 -
side effects, in particular, phototoxicity and cytotoxicity;
and antibacterial agents containing the same.
In order to achieve the above object, the inventors of
the present invention have made an intensive study to find
compounds which would be excellent synthetic antibacterial
agents in clinical practice, and found that novel compounds
represented by the following general formula (1) have good
antibacterial activities to gram negative and positive
bacteria as well as an extremely low toxicity, and therefore,
1o would be very useful synthetic antibacterial agents. The
present invention has been accomplished on the bases of such
a finding.
According to the present invention, there is provided
pyridonecarboxylic acid derivatives or their salts
represented by the following formula (1):
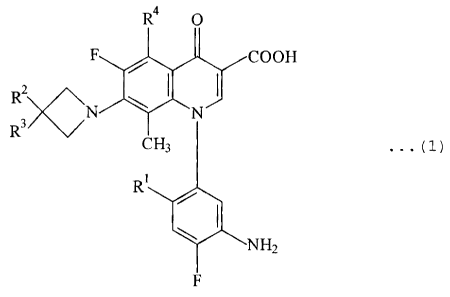
R4 O
F / COOH
R3 N \ ~ N
CH3 ...
(1)
R'
\ NHZ
F
[In the formula, R1 represents hydrogen atom, a halogen atom
or a lower alkyl group; RZ represents hydrogen atom or a
lower alkyl group; R' represents substituted or unsubstituted
amino group or hydroxyl group; and R' represents hydrogen
atom, a lower alkyl group, amino group or nitro group.]
According to the present invention, there is also provided
3o antibacterial agents containing the pyridonecarboxylic acid
derivatives or their salts as their effective components.
Best Mode for Carrying Out the Invention
The novel pyridonecarboxylic acid derivatives of the
present invention are represented by the general formula (1)
CA 02252229 2005-07-20
69562-22
- 3 -
as shown above, and the term "lower" used for the
substituents of the pyridonecarboxylic acid derivatives
represented by the general formula (1) designates that the
substituent comprises 1 to 7 carbon atoms, and preferably
1 to 5 carbon atoms in the case of a linear substituent, and
that the substituent comprises 3 to 7 carbon atoms in the
case of a cyclic substituent.
The halogen atoms represented by R1 of the general
formula (1) include fluorine, chlorine, bromine and iodine
atoms, among which fluorine and chlorine atoms are
preferred, and fluorine atom is most preferred.
The lower alkyl groups represented by R1, RZ and R9
include methyl group, ethyl group, n-propyl group, i-propyl
group, n-butyl group, i-butyl group, t-butyl group, pentyl
group, hexyl group, and heptyl group, among which methyl
group is preferred.
The substituents of the substituted amino group
represented by R3 include lower alkyl groups containing
1 to 7 carbon atoms such as methyl group, ethyl group,
n-propyl group, i-propyl group, n-butyl group, i-butyl
group, t-butyl group, pentyl group, hexyl group, and heptyl
group; lower alkenyl groups containing 2 to 7 carbon atoms
such as vinyl group, allyl group, 1-propenyl group, butenyl
group, pentenyl group, hexenyl group, and heptenyl group;
aralkyl groups containing 7 to 11 carbon atoms such as
benzyl group and 1-phenylethyl group; aryl groups containing
6 to 14 carbon atoms such as phenyl group and naphthyl
group; lower alkanoyl groups containing 1 to 4 carbon atoms
such as formyl group, acetyl group, propionyl group, butyryl
group and isobutyryl group; lower alkoxycarbonyl groups
CA 02252229 2005-07-20
69562-22
- 3a -
containing 2 to 3 carbon atoms such as methoxycarbonyl group
and ethoxycarbonyl group, aroyl groups containing
7 to 15 carbon atoms such as benzoyl group and naphthoyl
group; amino acid residues such as natural amino acid
residues or oligopeptide residues containing 2 to 3 natural
amino acid residues such as glycyl, leucyl, valyl, alanyl,
phenylalanyl, alanyl-alanyl, glycyl-valyl, and glycyl-
glycyl-valyl, and the amino acid residues or the
oligopeptide residues wherein the functional
CA 02252229 2005-07-20
695'62-22
- 4 -
group thereof is protected with an acyl, a lower aralkyl, or
other protective groups which is commonly used in peptide
chemistry; and cyclic amino group. One or two substituents
which may be the same or different may be selected from the
substituents as described above. The compound protected with
the amino acid residue or the oligopeptide residue is
expected to have an improved water solubility.
Preferable groups represented by R' include amino group,
lower alkylamino groups, di-lower alkylamino groups, lower
1o alkanoylamino groups, amino acid-substituted amino groups,
oligopeptide-substituted amino groups, and hydroxyl group.
More preferable groups of R' include amino group, methylamino
group, ethylamino group, dimethyamino group, formylamino
group, glycyl-amino group, leucyl-amino group, valyl-amino
group, alanyl-amino group, alanyl-alanyl-amino group, and
hydroxyl group among which the amino group being the most
preferred.
Preferable combinations of the R2 and R' are methyl group
or hydrogen atom for the RZ and amino group, methylamino
2o group or hydroxyl group for the R'.
Preferable combinations of the R1, RZ and R' in the
general formula (1) is fluorine atom, chlorine atom or methyl
group for the R1, hydrogen atom or methyl group for the R~,
and amino group, methylamino group or hydroxyl group for the
R', and the most preferable combination is methyl group for
the R1, hydrogen atom for the Rz and amino group for the R'.
The pyridonecarboxylic acid derivatives of the formula
(1) or the salts thereof as described above may form either
an acid adduct salt or a base adduct salt. The salts are preferably
pharmaceutically acceptable salts. The term "salt" used herein also
includes a chelate salt with a boron compound. Exemplary acid adduct
salts include (A) salts with a mineral acid such as hydrochloric acid
or sulfuric acid; (B) salts with an organic carboxylic acid such as
formic acid, citric acid, trichloroacetic acid, trifluoroacetic acid,
fumaric acid, or malefic acid; and (C) salts with a
CA 02252229 2005-07-20
695'62-22
- s -
sulfonic acid such as methanesulfonic acid, benzenesulfonic
acid, p-toluenesulfonic acid, mesitylenesulfonic acid, or
naphthalenesulfonic acid; and exemplary base adduct salts
include (A') salts with an alkaline metal such as sodium or
potassium; (B') salts with an alkaline earth metal such as
calcium or magnesium; (C') ammonium salts; (D') salts with a
nitrogen-containing organic base such as trimethylamine,
triethylamine, tributylamine, pyridine, N,N-dimethylaniline,
N-methylpiperidine, N-methylmorpholine, diethylamine,
1o cyclohexylamine, procaine, dibenzylamine, N-benzyl- p -
phenethylamine, 1-ephenamine, or N,N'-dibenzylethylene-
diamine. Exemplary boron compounds include boron halides
such as boron fluoride, and lower acyloxyborons such as
acetoxyboron_.
The pyridonecarboxylic acid derivatives of the formula
(1) or the salts thereof may also be in the form of hydrates
or solvates, preferably pharmaceutically acceptable solvates, in
addition to the non-solvated forms. Accordingly, the compounds of
the present invention include all of the crystalline forms, the
hydrate forms, and the solvate forms.
The pyridonecarboxylic acid derivatives of the formula
(1) or the salts thereof as described above may also be
present in the form of optically active substances, and such
optically active substances are also within the scope of the
compounds of the present invention. Still further, the
compounds of the formula (1) may be present in the form of
(cis or trans) stereoisomers, and such stereoisomers are also
within the scope of the compounds of the present invention.
The pyridonecarboxylic acid derivatives of the formula
(1) or the salts thereof of the present invention as described
above may be produced by any procedure appropriately selected
in accordance with such factors as the type of the substituents,
and an exemplary procedure is as described below.
The compounds represented by the general formula (1) may
be produced, for example, by the procedure 1 or procedure 2
CA 02252229 1998-10-16
- 6 -
represented by the reaction scheme as described below:
(Procedure 1)
R" 0
F COOLS 1 ) 1~5-CH FOR' ( s )
/I
LZ \ L, R.
CH3 2 ) F ~ ~ NHz ( c )
(A)
to R~ 0 R4 0
F / COOLS F / ( COOLS
LZ \ L~ ~H LZ \ NJ
CH3 NH cyclization CH3
I R~ / I
\ \
F F
(D) (E)
R' 0 R' 0
2 o F / I I COOLS F / I I COOLS
LZ \ NJ LZ \ NJ
nitration CH3 reduction CHa
R~ / I R~ /
\ NOz \ NHZ
25 F
F
(F) (~)
R~ 0 R4 0
F / I I COOH F / COOH
3o Lz \ R3 N \ I N
av
CH3 R3 ~NH ( M ) CH3
hydrolysis R~ R' /
I
\ I NH \ NHz
Z
ss F F
(H) (1)
CA 02252229 1998-10-16
[wherein R5 represents a lower alkoxy group or -NRBR' group
(wherein RB and R9 independently represent a lower alkyl
group); R6 and R' independently represent a lower alkyl group;
and L1 and LZ independently represent the same or different
halogen atom; L' represents a lower alkyl group; and R1, Rz, R'
and R' are as defined above.]
More illustratively, the compound (1) of the present
invention is produced by reacting compound (A) with an
orthoformate (B) such as ethyl orthoformate or methyl
l0 orthoformate and then, with compound (C) to produce compound
(D); cyclizing the compound (D) to produce compound (E);
nitrating the compound (E) to produce compound (F); reducing
the compound (F) to produce compound (G); hydrolyzing the
compound (G) to produce compound (H); and aminating the
compound (H) by using an azetidine derivative (M).
The reaction between the compound (A) and the
orthoformate (B) is generally carried out at 0 to 160°C, and
preferably 50 to 150°C usually for a reaction period of 10
minutes to 48 hours, and preferably for 1 to 10 hours. In
this case, especially not limited, carboxylic anhydride such
as acetic anhydride is desirably added in the above reaction.
The orthoformate (B) is used in equimolar amount or more to
the compound (A), and preferably, in about 1 to 10 times the
molar amount to the compound (A).
The reaction with the compound (C) may be effected
without any solvent or in a solvent. The solvent used in
this reaction may be any solvent as long as the reaction is
not affected by the solvent, and the exemplary solvents
include aromatic hydrocarbons such as benzene, toluene, and
xylene; ethers such as diethylether, tetrahydrofuran,
dioxane, monoglyme, and diglyme; aliphatic hydrocarbons such
as pentane, hexane, heptane, and ligroin; halogenated
hydrocarbons such as methylene chloride, chloroform, and
carbon tetrachloride; nonprotonic polar solvents such as
dimethylformamide and dimethylsulfoxide; and alcohols such as
CA 02252229 1998-10-16
g _
methanol, ethanol and propanol. This reaction is generally
conducted at 0 to 150°C, and preferably at 0 to 100°C usually
for a reaction period of 10 minutes to 48 hours. The
compound (C) is used in equimolar amount or more to the
compound (A), and preferably, in 1 to 2 times the molar
amount to the compound (A).
Alternatively, compound (A) may be reacted with an
acetal such as N,N-dimethylformamide dimethylacetal or
N-dimethylformamide diethylacetal, and then, with compound
(C) to produce the compound (D). The solvent used in the
reaction with the acetal may be any solvent as long as the
reaction is not affected by the solvent, and the exemplary
solvents are those described in the foregoing. This reaction
is generally conducted at 0 to 150°C, and preferably at room
temperature to 100°C generally for a reaction period of 10
minutes to 48 hours, and preferably for 1 to 10 hours.
Next, the cyclization of the compound (D) into the
compound (E) is conducted in an adequate solvent either in
the presence or absence of a basic compound. The solvent
2o used in this reaction may be any solvent as long as the
reaction is not affected by the solvent, and the exemplary
solvents include aromatic hydrocarbons such as benzene,
toluene, and xylene; ethers such as diethylether,
tetrahydrofuran, dioxane, and monoglyme; halogenated
hydrocarbons such as methylene chloride, chloroform, and
carbon tetrachloride; alcohols such as methanol, ethanol,
propanol, and butanol; and nonprotonic polar solvents such as
dimethylformamide and dimethylsulfoxide. Exemplary basic
compounds used are alkaline metals such as metal sodium and
metal potassium; metal hydrides such as sodium hydride and
calcium hydride; inorganic salts such as sodium hydroxide,
potassium hydroxide, sodium carbonate, and potassium
carbonate; alkoxides such as sodium methoxide, sodium
ethoxide, and potassium t-butoxide; metal fluorides such as
sodium fluoride and potassium fluoride; organic salts such as
CA 02252229 1998-10-16
- 9 -
triethylamine and 1,8-diazabicyclo[5.4.0]undecene (DBU).
This reaction is conducted at a reaction temperature of 0 to
200°C, and preferably, at room temperature to 180°C, and the
reaction is generally completed in 5 minutes to 24 hours.
The basic compound is used in equimolar amount or more to the
compound (B), and preferably, in 1 to 2 times the molar
amount to the compound (B).
The nitration of the compound (E) into the compound (F)
may be conducted by the procedure generally used in the
to nitration of aromatic compounds, and mixed acid of nitric
acid or nitrate (such as potassium nitrate) in combination
with sulfuric acid or acetyl nitrate is used for the
nitrating agent. The amount of the mixed acid used in the
reaction is such that 1 equivalent or more of sulfuric acid
and 1 equivalent or more of nitric acid are used with 1
equivalent of the compound (E), and the reaction is conducted
by adding the compound (E) to the mixed acid. The reaction
is preferably conducted at a reaction temperature of -10°C to
80°C for a period of 5 minutes to 5 hours.
The reduction of the compound (F) into the compound (G)
may be conducted by any common procedure such as molten metal
reduction wherein zinc, iron, tin, tin chloride (II) or the
like is used in an acidic solution; the reduction wherein a
sulfate such as sodium sulfate, sodium hydrosulfide, or
sodium dithionate is used; and catalytic reduction wherein
platinum, Raney nickel, platinum black (Pt-C) or palladium
carbon (Pd-C) is used. When formic acid-iron is used, the
compound (G) obtained is in the form of a formylamino
compound.
The hydrolysis of the compound (G) into the compound (H)
may be conducted under any conditions commonly used in the
hydrolysis, for example, in the presence of a basic compound
such as sodium hydroxide, potassium hydroxide, sodium
carbonate, and potassium carbonate, a mineral acid such as
hydrochloric acid, sulfuric acid, and hydrobromic acid, or an
CA 02252229 1998-10-16
- 10 -
organic acid such as p-toluenesulfonic acid, and in a solvent
such as water, an alcohol such as methanol, ethanol or
propanol, or an ether such as tetrahydrofuran or dioxane, a
ketone such as acetone or methylethylketone, acetic acid, or
a mixture of such solvents. The reaction is generally
conducted at room temperature to 180°C, and preferably, at
room temperature to 140°C usually for a reaction period of 1
to 24 hours.
The reaction of the compound (H) with an azetidine
1o derivative (M) for amination of the compound (H) into the
compound of formula (1) of the present invention may be
conducted in a solvent which does not affect the reaction.
Exemplary such solvents include aromatic hydrocarbons such as
benzene, toluene, and xylene; alcohols such as methanol and
ethanol; ethers such as tetrahydrofuran, dioxane, and
monoglyme; halogenated hydrocarbons such as methylene
chloride, chloroform, and carbon tetrachloride; and
nonprotonic polar solvents such as dimethylformamide,
dimethylsulfoxide, N-methylpyrroridone; acetnitrile,
2o pyridine, and the like. The reaction may be conducted in
optional presence of neutralizer such as sodium carbonate,
calcium carbonate, sodium hydrogencarbonate, triethylamine,
and 1,8-diazabicyclo[5.4.0]undecene (DBU), and at a reaction
temperature of from room temperature to 160°C, and the
reaction is generally completed in several minutes to 48
hours, preferably in 10 minutes to 24 hours. The compound
(M) is used in equimolar amount or more to the compound (H),
and preferably, in 1 to 5 times the molar amount to the
compound (H).
The compound (H) as described above may also be prepared
by the following procedure, namely, by the procedure 2.
CA 02252229 1998-10-16
- 11 -
(Procedure 2)
R' 0 NHS
F / COOL' R'
I
L'~ \ I L' ~ + \ I NHR"'
CHI ~R~ F
(I) (J)
R" 0
F / ( I COOLS
t o L2 \ N J
condensation
CH3 deprotection
cyclization R,
\ NHR'
is F
(K)
R4 0
F / I I COOH
LZ \ NJ
2 o CH3
R' /
NHZ
F
25 (H)
[wherein Rl° is a protective group for the amino group; and
R1, R9 , R6 , L1, Lz and L' are as de f fined above . ]
More illustratively, compound (A) is reacted with
orthoformate (B) to produce acrylate (I), and the acrylate
30 (I) is reacted with phenylene diamine (J) instead of the
compound (C) in the above-described procedure 1 for
condensation and cyclization to produce compound (K). The
lower alkyl group L' and the protective group R1° for the
amino group are then cleaved to obtain compound (H).
CA 02252229 1998-10-16
- 12 -
The series of steps in the procedure 2 from the compound
(I) to the compound (H) may be conducted under the conditions
similar to the reactions of the procedure 1 as described
above wherein the compound (G) is produced from the compound
(A). In the procedure 2, the cleavage of the lower alkyl
group L' and the protective group R1° for the amino group
(which are typically an acyl group and carbamoyl group,
respectively) may be conducted by hydrolysis with an acid or
an alkali under the conditions similar to the conditions used
to in the hydrolysis of the compound (G) into the compound (H)
in the procedure 1 as described above.
When amino group, imino group, hydroxyl group, mercapto
group, carboxyl group or the like which is not involved in
the reaction is present in the starting materials of the
procedure 1 or 2 as described above, such group may be
protected during the reaction, and the protective group may
be eliminated after the completion of the reaction by a
conventional method. The protective group used in such a
case may be any group as long as the compound of the present
2o invention produced by the reaction can be deprotected with no
decomposition in its structure, and any group commonly used
in the field of peptide, amino sugar, and nucleic acid
chemistry can be used.
2,4,5-trifluoro-3-methylbenzoyl acetate (A') which is a
typical starting compounds (A) for the procedures 1 and 2 as
described above may be produced by series of steps in the
procedure 3 as described below.
35
CA 02252229 1998-10-16
- 13 -
(Procedure 3)
COOR" HzC~C00R'z ( ~ ) COOR"
F / I COOR' ' ~ COOR'' F / I COOR"
F \ F base F \ F
F R'z00C COOR''
(N) (P)
hydrolysis and COOH
decarboxylation F / ~ COOH heating
to F \ F base
HOOCCHZ
(Q)
0
F / I COOH a . g . Procedure 4 F / COOL'
is
F \ F F \ ( F
CH3 CH3
(S) (A')
[wherein R11 and RlZ represent lower alkyl groups, and L' is as
2o deffined above.]
Compound (N) is reacted with a malonate (0) in the
presence of a basic compound to produce compound (P), and the
compound (P) is hydrolyzed and decarboxylated to produce
compound (Q). The compound (Q) is heated in the presence
25 of a basic compound to produce compound (S). The thus
produced compound (S) is converted into the starting compound
(A) by known reactions such as procedure 4 as described
below.
The reaction of the compound (N) with the malonate (0)
30 is conducted in the presence of a basic compound and in an
appropriate solvent which does not affect the reaction.
Exemplary such solvents are aromatic hydrocarbons such as
benzene, toluene, and xylene; ethers such as diethylether,
tetrahydrofuran, dioxane, monoglyme, and diglyme; aliphatic
35 hydrocarbons such as pentane, hexane, heptane, and ligloin;
CA 02252229 1998-10-16
- 14 -
halogenated hydrocarbons such as methylene chloride,
chloroform, and carbon tetrachloride; and nonprotonic polar
solvents such as dimethylformamide and dimethylsulfoxide; and
alcohols such as methanol, ethanol, and propanol. Exemplary
basic compounds used are alkaline metals such as metal sodium
and metal potassium; metal hydride such as sodium hydride and
calcium hydride; inorganic salts such as sodium hydroxide,
potassium hydroxide, sodium carbonate, and potassium
carbonate; alkoxides such as sodium methoxide, sodium
1o ethoxide, and potassium t-butoxide; metal fluorides such as
sodium fluoride and potassium fluoride; and organic salts
such as triethylamine and 1,8-diazabicyclo[5.4.0]undecene
(DBU), among which use of metal oxides such as sodium hydride
and calcium hydride are preferred.
The reaction conditions are not particularly limited.
The reaction, however, is generally conducted under heating
for a reaction period of about 30 minutes to 48 hours. The
compound (0) is used in an equimolar amount or more to the
compound (N), and preferably, in about 1 to 5 times the molar
2o amount to the compound (N).
The hydrolysis and decarboxylation reactions of the
compound (P) into the compound (Q) are conducted in the
presence of an acid such as sulfuric acid, hydrochloric acid,
hydrobromic acid, and acetic acid, typically, by adding such
acid to the compound (P) and refluxing for 1 to 4 days.
The reaction from the compound (Q) to the compound (S)
is conducted in the presence of a basic compound, and
generally, by heating the reaction system to a temperature in
the range of from room temperature to 180°C for 1 to 48
hours, and preferably, to a temperature of from 100 to 140°C
for 1 to 24 hours. The basic compounds used in this reaction
may be the same as those described for the reaction of the
compound (N) and the malonate (O), and use of triethylamine
is most preferred. Exemplary solvents used are
N-methylpyrrolidone, dimethylformamide and dimethylsulfoxide,
CA 02252229 1998-10-16
- is -
and N-methylpyrrolidone is most preferred.
The compound (S) is converted into the starting compound
(A') by a known process such as procedure 4 as described
below. In the procedure 4, the compound (S) is reacted with
a chlorine compound such as oxalyl dichloride, phosphorus
pentachloride, phosphorus trichloride, phosphoryl chloride,
thionyl chloride, or sulfuryl chloride to produce acid
chloride (T), and the acid chloride (T) is reacted with a
malonate (U) in the presence of magnesium and ethanol to
produce compound (v), and the compound (v) is converted into
the starting compound (A) by refluxing with addition of
paratoluene sulfonic acid and water, as known in the art.
(Procedure 4)
is F / COOH chlorine compound F / COC1
F \ I F F \ F
CH3 CH3
(s) (T)
~ COOLS O
zo HZC~COOL3 ( U ) F
/ C-CH COOLS
~COOL3
Mg F \ I F
CzH50H CH3
(v)
is HZO O
p-toluenesulfonic acid F / COOLS
refluxing F \ I F
CH3
(A')
30 [wherein L' is as defined above.]
The process of the procedure 3 is most preferred for
producing the starting compound (A') since the intermediate
(S) can be synthesized by relatively small number of steps at
relatively high yield. In addition, the reagents used in the
35 procedure 3 are relatively easy to handle, and the reaction
CA 02252229 1998-10-16
- 16 -
apparatus and treatments are relatively simple. The starting
compound (A) may be produced by a process other than the
procedure 3, for example, by the process described in the
documents as described below or its modification.
1) J. Heterocyclic Chem. 22, 1033 (1985)
2) Liebigs Ann. Chem. 29 (1987)
3) J. Med. Chem. 31, 991 (1988)
4) J. Org. Chem. 35, 930 (1970)
5) Japanese Patent Application Laid-Open No. 62-246541
l0 6) Japanese Patent Application Laid-Open No. 62-26272
7) Japanese Patent Application Laid-Open No. 63-145268
8) J. Med. Chem. 29, 2363 (1986)
9) J. Fluorin Chem. 28, 361 (:1985)
10) Japanese Patent Application Laid-Open No. 63-198664
11) Japanese Patent Application Laid-Open No. 63-264461
12) Japanese Patent Application Laid-Open No. 63-104974
13) European Patent Application No. 230948
14) Japanese Patent Application Laid-Open No. 2-282384
15) Published Japanese Translation of PCT International
Publication for Patent Application No. 3-502452
16) J. Het. Chem. 27, 1609 (1990)
17) Japanese Patent Application Laid-Open No. 7-215913
For example, the production process described in the
document 17) is the process wherein the starting compound (A)
is obtained by the procedure 5 as described below.
(Procedure 5)
F / N~R'4 1 ) LiN(Si(CH3)3)Z F / NJ'R"
.0 v0
F ~ ~ F 2 ) methylating agent F ~ F
(W) CH3 (x)
0
hydrolysis F / I COOH a , g ~ procedure 4 F / COOLS
F F F ~ I F
3 5 CH3 I
( s ) CH3 ( A , )
CA 02252229 1998-10-16
- is -
[wherein Rl' and Rl' represent lower alkyl groups, and L' is as
defined above.]
In this procedure, compound (W) is first deprotonated
with lithium bis(trimethylsilyl)amide and reacted with a
methylating agent such as methyl iodide or methyl bromide to
produce compound (X), and the compound (X) is hydrolyzed by
heating under reflux with an acid such as hydrochloric acid,
sulfuric acid or hydrobromic acid to produce compound (S).
The compound (S) is then converted into the starting compound
to (A') as in the above-described process, for example, by
procedure 4 as described above.
In this case, lithium bis(trimethylsilyl)amide used as
the deprotonating agent in the reaction from the compound (W)
to the compound (X) is generally prepared from hexamethyl-
disilazane and n-butyllithium with no purification. The
deprotonation and the methylation reactions are conducted by
using an appropriate solvent at a relatively low temperature
of from -10°C to room temperature.
The process of procedure 5 is relatively preferable
among the known processes since the synthesis can be
accomplished by relatively small number of steps at a
relatively high yield. The process of procedure 5, however,
is not fully satisfactory in view of industrial scale
production since n-butyllithium used in the preparation of
lithium bis(trimethylsilyl)amide, the deprotonating agent, is
inconvenient to handle since it undergoes spontaneous
ignition by contact with oxygen or moisture, and the
deprotonation and the methylation reactions have to be
conducted at low temperature. Other known processes also
suffer from drawbacks such as too many number of steps,
complicated reactions, insufficient yield, or the like. The
process of procedure 3 is preferable in view of industrial
scale production.
The thus obtained compounds of the present invention are
isolated and purified in accordance with standard methods.
CA 02252229 1998-10-16
- lg -
The compounds obtained may be in the form of salts, free
carboxylic acids, or free amines depending on the conditions
of the isolation and the separation. The forms of the
compounds are mutually convertible, and the compounds of the
present invention in desired form may be produced.
The compounds (1) or the salts thereof of the present
invention have excellent antibacterial activities, intestinal
absorption, and metabolic stability, and suffer from reduced
phototoxicity, cytotoxicity and other side effects, and
1o therefore, they can be advantageously used as an effective
component in antibacterial agents.
The compounds represented by the general formula (1)
above or the salts thereof may be formulated into an
antibacterial composition with a pharmaceutically acceptable
carrier adapted for parenteral administration such as
injection, transrectal administration, or eye drop, or oral
administration in solid or liquid form.
When the antibacterial composition of the present
invention is in the form of an injection, it may be in the
2o form of a solution, a suspension or an emulsion in a
pharmaceutically acceptable sterilized water or a non-aqueous
medium. Examples of appropriate non-aqueous carriers,
diluents, media, and vehicles include propylene glycol,
polyethylene glycol, vegetable oils such as olive oil, and
organic esters adequate for injection such as ethyl oleate.
Such composition may also contain additives such as a
preservative, a wetting agent, an emulsifier and a
dispersant. The composition may be sterilized, for example,
by filtration through a bacteria-removing filter, or by
incorporating a sterilizer in the form of a sterilizer or a
sterile solid composition soluble in a sterilizable medium
for injection just before its use.
A preparation for eye drop administration may preferably
contain a solubilizer, a preservative, an isotonizing agent,
a thickening agent, and the like in addition to the compounds
CA 02252229 1998-10-16
1<~
of the present invention.
Solid preparations for oral administration include
capsules, tablets, pills, powders, and granules. In
preparing such solid preparations, the compounds of the
present invention are typically mixed with at least one inert
diluent such as sucrose, lactose or starch. The preparation
may also contain substances other than the inert diluents
such as lubricant (for example, magnesium stearate etc.). In
the case of capsules, tablets or pills, the preparation may
to also include a buffer. The tablets and the pills may have an
enteric coating.
Liquid preparations for oral administration include
pharmaceutically acceptable emulsions, solutions,
suspensions, syrups and elixirs containing an inert diluent
normally used in the art such as water. In addition to such
inert diluent, the composition may also contain additives
such as a wetting agent, an emulsifying agent, a suspending
agent as well as a sweetener, a seasoning, and a flavor.
Preparations for enteral administrations may preferably
contain an excipient such as cacao butter or suppository wax
in addition to the compound of the present invention.
The dose of the compounds (1) of the present invention
varies depending on the nature of the compound administered,
route of administration, the desired treatment period, and
other factors. The compounds of the present invention,
however, are typically administered at about 0.1 to 1000
mg/kg per day, and in particular, at about 0.5 to 100 mg/kg
per day. If desired, such dose may be administered in 2 to 4
portions.
3o The compounds (1) and the salts thereof of the present
invention exhibit extremely strong antibacterial activities
simultaneously with reduced phototoxicity and cytotoxicity,
and therefore, would be widely applicable as pharmaceuticals
for human and other animals as well as pharmaceuticals for
fishes, pesticides, food preservatives, and the like. The
CA 02252229 1998-10-16
- 20 -
compounds of the present invention are also expected to
exhibit antivirus properties, and especially, anti-HIV (human
immunodeficiency virus) actions, and to be effective in
preventing and treating AIDS.
Next, the present invention is described in further
detail by referring to Examples, Comparative Examples, and
Reference Examples, which by no means limit the scope of the
present invention.
[Reference Example 1]
(1) Preparation of diethyl 2-(2,5,6-trifluoro-3,4-bis-
(ethoxycarbonyl) henyl)malonate
To 60 ml of tetrahydrofuran was suspended 8 g of sodium
hydride from which oil content had been removed by petroleum
ether, and 30 ml of tetrahydrofuran solution of 32 g of
diethyl malonate was added dropwise to this suspension in an
ice bath, and the mixture was stirred at room temerature for
minutes. 50 ml of tetrahydrofuran solution of 26.6 g of
20 diethyl 3,4,5,6-tetrafluorophthalate was added dropwise to
the rea,ption solution in an ice bath, and the mixture was
stirred at 60°C for 1 hour. The reaction solution was
allowed to cool, and 8 ml of acetic acid was added to the
solution and the solution was stirred at room temperature for
20 minutes. To the solution was added chloroform and water
to extract the organic layer. The extract was dried over
magnesium sulfate and the solvent was distilled off to obtain
50 g of the title compound in crude form.
(2) Preparation of 2-(2,5,6-trifluoro-3,4-dicarboxyphenyl)-
3o acetic acid
To 50 g of diethyl 2-(2,5,6-trifluoro-3,4-bis(ethoxy-
carbonyl)phenyl)malonate were added 60 ml of conc.
hydrochloric acid and 60 ml of acetic acid, and the mixture
was refluxed overnight. The ethyl acetate in the reaction
solution was distilled off under reduced pressure, and the
CA 02252229 1998-10-16
- 21 -
solution was refluxed for 2 days. The acid in the reaction
solution was distilled off, and hexane was added to the
residue. The solid content was collected by filtration and
dried to obtain 27 g of the title compound in crude form.
Characteristic features: colorless powder
1HNMR ( de-DMSO ) b ;
3.78 (s, 2H)
(3) Preparation of 3-methyl-2,4,5-trifluorobenzoic acid
To 27 g of 2-(2,5,6-trifluoro-3,4-dicarboxyphenyl)acetic
to acid was added 28 ml of N-methylpyrrolidone and 10 ml of
triethylamine and the mixture was stirred at 140°C for 2
days. The reaction solution was poured into 6N hydrochloric
acid with cooling, and separated by adding diethylether and
aqueous solution of sodium hydroxide. The aqueous layer was
acidified and extracted with diethylether.
The organic layer was dried over magnesium sulfate and
the solvent was distilled off. Hexane was added to the
residue., and the solid content was collected by filtration to
obtain 12 g of the title compound in powder form.
2o Characteristic features: colorless needle crystals
Melting point: 97-100°C
1HNMR (CDC1,) b ;
2.29 (d, J=3Hz, 3H), 7.70 (dd, J=llHz, l6Hz, 1H)
[Reference Example 2]
Preparation of ethyl 2,4,5-trifluoro-3-methylbenzoylacetate
2.4 g of magnesium, 10 ml of ethanol, and 0.4 m1 of
carbon tetrachloride were stirred in a three necked flask at
room temperature for activation, and 15 ml of diethyl
3o malonate and 40 ml of tetrahydrofuran were gradually added
dropwise. The mixture was stirred at 80°C for 4 hours. The
reaction solution was allowed to cool, and then cooled to
-40°C. To 50 ml solution in methylene chloride of 15.5 g of
2,4,5-trifluoro-3-methylbenzoic acid obtained in Reference
Example 1(3) were added 8 ml of oxalyl dichloride and 5 drops
CA 02252229 1998-10-16
- 22 -
of N,N-dimethylformamide, and the mixture was stirred at room
temperature for 3 hours. The solvent was distilled off, and
tetrahydrofuran was added for azeotropic distillation. The
residue was dissolved in 40 ml of tetrahydrofuran, and this
solution was gradually added dropwise to the above-described
reaction solution at -40°C. After the dropwise addition, the
reaction solution was stirred at room temperature for 3 days,
and the solvent was distilled off. 50 ml of 12N hydrochloric
acid was added to the solution to about pH 2, and the
1o solution was extracted with ch:Loroform, and solvent was
distilled off. To the residue were added 30 ml of water and
0.6 g of p-toluenesulfonic acid, and the mixture was stirred
under reflux for 6 hours. The reaction solution was allowed
to cool, extracted with chloroform, and washed with water and
5~ aqueous solution of sodium hydrogencarbonate successively.
The organic layer was dried over magnesium sulfate and
concentrated under reduced pressure to obtain the title
compound as 5.5 g of powder and 10.2 g of brown oil.
[Reference Example 3]
Preparation of ethyl 1-(3-tert-butoxycarbonylamino-4,6-
difluorophenyl)-6,7-difluoro-8-methyl-1,4-dihydro-4-
oxoquinoline-3-carboxylate
To 8.2 g of ethyl 2,4,5-trifluoro-3-methylbenzoylacetate
obtained in the Reference Example 2 were added 18.9 g acetic
anhydride and 7.0 g of triethy:L formate, and the mixture was
heated under reflux for 3 hours. The solvent was distilled
off, and the residue was azeotropically distilled by adding
toluene. To half of the residue was added 10 ml of
3o chloroform, and 3.7 g of N-(tert-butoxycarbonyl)-4,6-
difluoro-m-phenylenediamine dissolved in 10 ml of chloroform
was added dropwise at 0°C. The mixture was stirred at room
temperature for 30 minutes. The solvent was distilled off,
and the solid content was collected from the residue by
filtration and washed with ethanol to obtain 3.8 g of ethyl
CA 02252229 1998-10-16
- 23 -
2-(2,4,5-trifluoro-3-methylbenzoyl)-3-(3-tert-
butoxycarbonylamino-4,6-difluorophenylamino)acrylate.
To solution of the resulting ethyl 2-(2,4,5-
trifluoro-3-methylbenzoyl)-3-(3-tert-butoxycarbonylamino-
4,6-difluorophenylamino)acrylate in 10 ml of N,N-dimethyl-
formamide was added 1.2 g of potassium carbonate, and the
mixture was stirred at 100°C for 1 hour. The reaction
solution was extracted by adding water and ethyl acetate,
and organic layer was dried over magnesium sulfate, and the
solvent was distilled off. The solid content was collected
from the residue by using ethanol and washed with
diethylether to obtain 2.9 g of the title compound.
Characteristic features: pale yellow powder
Melting point: 183-185"C
1HNMR (CDC1,) S ;
1.40 (t, J=7Hz, 3H), 1.51 (s, 9H), 1.82 (d, J=3Hz, 3H),
4.39 (q, J=7Hz, 2H), 6.81 (brs, 1H),
7.10 (t, J=lOHz, 1H), 8.25 (t, J=lOHz, 1H),
8.29-8.40 (m, 2H)
[Reference Example 4]
Preparation of 1-(3-amino-4,6-difluorophenyl)-6,7-difluoro-
8-methyl-1,4-dihydro-4-oxoguinoline-3-carboxylic acid
To 2.8 g of ethyl 1-(3-tert-butoxycarbonylamino-4,6-
difluorophenyl)-6,7-difluoro-8-methyl-1,4-dihydro-4-
oxoquinoline-3-carboxylate obtained in the Reference Example
3 was added 10 ml of 12N hydrochloric acid, and the mixture
was heated under reflux for 4 hours. The reaction solution
was allowed to cool, and the solid precipitate was collected
3o by filtration and washed with ethanol and diethylether
successively to obtain 1.9 g of the title compound.
Characteristic features: pale yellow powder
Melting point: 266-267°C
CA 02252229 1998-10-16
- 24 -
'HNMR ( de-DMSO ) b ;
1.86 (d, J=3Hz, 3H), 7.13 (t, J=8Hz, 1H),
7.46 (t, J=llHz, 1H), 8.25 (t, J=9Hz, 1H), 8.68 (s, 1H)
[Example 1]
7-(3-aminoazetidin-1-yl)-1-(3-amino-4,6-difluorophenyl)-6-
fluoro-8-methyl-1,4-dihydro-4-oxoguinoline-3-carboxylic acid
Solution of 70 mg of 3-aminoazetidine~dichloride, 200 mg
of 1,8-diazabicyclo[5.4.0]undecene, and 300 mg of pyridine
was stirred at 100°C, and to this solution was added
1-(3-amino-4,6-difluorophenyl)-6,7-difluoro-8-methyl-
1,4-dihydro-4-oxoquinoline-3-carboxylic acid obtained in the
Reference Example 4. The mixture was heated with stirring at
110°C for 60 minutes. Addition of diethylether to the
reaction solution followed by decantation was repeated three
times, and 5 ml of ethanol was added with heating to 70°C.
The solution was allowed to stand, and the solid precipitate
was collected by filtration to obtain 50 mg of the title
compound.
Characteristic features: pale yellow powder
Melting point: 213-218°C
1HNMR ( ds-DMSO ) b ;
1.61 (s, 3H), 3.66-3.81 (m, 2H), 3.82-3.95 (m, 1H),
4.36-4.52 (m, 2H), 5.47 (brs, 2H), 7.04 (t, J=9Hz, 1H),
7.40 (t, J=lOHz, 1H), 7.75 (d, J=l4Hz, 1H), 8.48 (s, 1H)
[Example 2]
1-(3-amino-4,6-difluorophenyl)-6-fluoro-8-methyl-7-(3-methyl-
aminoazetidin-1-yl)-1,4-dihydro-4-oxoguinoline-3-carboxylic
acid
The procedure of Example 1 was repeated except that
3-aminoazetidine~dichloride was replaced with 110 mg of
3-N-methylaminoazetidine~dichloride to obtain the title
compound.
Characteristic features: pale yellow powder
CA 02252229 1998-10-16
- 25 -
Melting point: 189-194°C
1HNMR ( de-DMSO ) b ;
1.64 (s, 3H), 2.20 (s, 3H), 3.78-3.90 (m, 1H),
3.90-4.01 (m, 1H), 4.33-4.50 (m, 2H), 5.50 (brs, 2H),
7.02 (t, J=9Hz, 1H), 7.42 (t, J=llHz, 1H),
7.76 (d, J=l4Hz, 1H), 8.45 (s, 1H)
[Example 3]
7-(3-amino-3-methylazetidin-1-yl)-1-(3-amino-4,6-difluoro-
phenyl)-6-fluoro-8-methyl-1,4-dihydro-4-oxoguinoline-3-
carboxylic acid
The procedure of Example 2 was repeated except that
3-aminoazetidine~dichloride was replaced with 110 mg of
3-amino-3-methylazetidine~dichloride to obtain the title
compound.
Characteristic features: pale yellow powder
Melting point: 209-222°C
1HNMR ( ds-DMSO ) b ;
1.37 (s, 3H), 1.63 (s, 3H), 3.83-3.94 (m, 1H),
3.95-4.13 (m, 3H), 5.46 (brs, 2H), 7.04 (t, J=9Hz, 1H),
7.40 (t, J=llHz, 1H), 7.76 (d, J=l4Hz, 1H), 8.48 (s, 1H)
[Reference Example 5]
Preparation of ethyl 1-(4-fluoro-2-methylphenyl)-6,7-
difluoro-8-methyl-1,4-dihydro-4-oxoguinoline-3-carboxylate
The procedure of Reference Example 3 was repeated except
that N-tert-butoxycarbonyl-4,6-difluoro-m-phenylenediamine
was replaced with 1.4 g of 4-fluoro-2-methylaniline to obtain
the title compound.
3o Characteristic features: pale yellow powder
Melting point: 187-189°C
iHNMR ( CDC1, ) b ;
1.40 (t, J=7Hz, 3H), 1.64 (d, J=3Hz, 3H), 2.08 (s, 3H),
4.39 (q, J=7Hz, 2H), 7.06-7.17 (m, 2H),
7.34 (t, J=5Hz, 1H), 8.22-8.35 (m, 2H)
CA 02252229 1998-10-16
- 26 -
[Reference Example 6]
Preparation of ethyl 1-(4-fluoro-2-methyl-5-nitrophenyl)-
6,7-difluoro-8-methyl-1,4-dihydro-4-oxoguinoline-3-carboxylate
To solution of 1.7 g of ethyl 1-(4-fluoro-2-methyl-
phenyl)-6,7-difluoro-8-methyl-:1,4-dihydro-4-oxoquinoline-3-
carboxylate obtained in the Reference Example 5 in 5 ml of
sulfuric acid was incrementally added 480 mg of potassium
nitrate on ice. After the addition, the mixture was stirred
at room temperature overnight and the reaction solution was
poured into ice water and stirred. Chloroform and water were
added for separation, and the organic layer was dried over
magnesium sulfate and the solvent was distilled off. The
solid precipitate was collected by filtration and washed with
ethanol to obtain 1.6 g of the title compound.
Characteristic features: pale yellow powder
Melting point: 211-214°C
1HNMR ( CDCl, ) S ;
1.40 (t, J=7Hz, 3H), 1.69 (d, J=3Hz, 3H), 2.20 (s, 3H),
4.40 (q, J=7Hz, 2H), 7.39 (d, J=llHz, 1H),
8.19 (d, J=7Hz, 1H), 8.23-8.35 (m, 2H)
(Reference Example 7]
Preparation of ethyl 1-(4-fluoro-3-formylamino-6-methyl-
phenyl)-6,7-difluoro-8-methyl-1,4-dihydro-4-oxoguinoline-
3-carboxylate
16 m of formic acid was added to 1.6 g of ethyl
1-(4-fluoro-2-methyl-5-nitrophenyl)-6,7-difluoro-8-methyl-
1,4-dihydro-4-oxoquinoline-3-carboxylate obtained in the
Reference Example 6 and dissolved. 2.1 g of iron powder was
3o added, and the mixture was stirred at 70°C for 90 minutes.
The reaction solution was allowed to cool, and the catalyst
was filtered off by celite, and the solvent was distilled off
the filtrate. The solid precipitate was collected by
filtration and washed with ethanol to obtain 1.6 g of the
title compound.
CA 02252229 1998-10-16
- 27 -
Characteristic features: pale brown powder
Melting point: 246-249°C
[Reference Example 8]
Preparation of 1-(3-amino-4-fluoro-6-methylphenyl)-6,7-
difluoro-8-methyl-1,4-dihydro-4-oxoquinoline-3-carboxylic
acid
The procedure of Reference Example 4 was repeated except
that ethyl 1-(3-tert-butoxycarbonylamino-4,6-difluorophenyl)
6,7-difluoro-8-methyl-1,4-dihydro-4-oxoquinoline-3
carboxylate was replaced with 1.6 g of ethyl 1-(4-fluoro-
3-formylamino-6-methylphenyl)-6,7-difluoro-8-methyl-1,4-
dihydro-4-oxoquinoline-3-carboxylate obtained in the
Reference Example 7 to obtain the title compound.
Characteristic features: colorless powder
Melting point: 248-250°C
1HNMR ( ds-DMSO ) 6 ;
1.76 (d, J=3Hz, 3H), 1.83 (s, 3H), 6.98 (d, J=8Hz, 1H),
7.16 (d, J=l2Hz, 1H), 8.27 (t, J=9Hz, 1H), 8.50 (s, 1H)
[Example 4]
7-(3-aminoazetidin-1-yl)-1-(3-amino-4-fluoro-6-methyl-
phenyl)-6-fluoro-8-methyl-1,4-dihydro-4-oxoguinoline-3-
carboxylic acid
The procedure of Example 1 was repeated except that
1-(3-amino-4,6-difluorophenyl)-6,7-difluoro-8-methyl-
1,4-dihydro-4-oxoquinoline-3-carboxylic acid was replaced
with 110 mg of 1-(3-amino-4-fluoro-6-methylphenyl)-6,7-
difluoro-8-methyl-1,4-dihydro-4-oxoquinoline-3-carboxylic
3o acid obtained in the Reference Example 8 to obtain the title
compound.
Characteristic features: pale yellow powder
Melting point: 162-169°C
1HNMR ( ds-DMSO ) b ;
1.55 (s, 3H), 1.79 (s, 3H), 3.67-3.78 (m, 2H),
CA 02252229 1998-10-16
- 28 -
3.83-3.94 (m, 1H), 4.35-4,50 (m, 2H), 5.41 (brs, 2H),
6.92 (d, J=8Hz, 1H), 7.09 (d, J=l2Hz, 1H),
7.79 (d, J=l4Hz, 1H), 8.34 (s, 1H)
[Example 5]
7-(3-amino-3-methylazetidin-1-yl)-1-(3-amino-4-fluoro-6-
methylphenyl)-6-fluoro-8-methyl-1,4-dihydro-4-oxoguinoline-
3-carboxylic acid
The procedure of Example 1 was repeated except that
l0 1-(3-amino-4,6-difluorophenyl)-6,7-difluoro-8-methyl-
1,4-dihydro-4-oxoquinoline-3-carboxylic acid was replaced
with 180 mg of 1-(3-amino-4-fluoro-6-methylphenyl)-6,7-
difluoro-8-methyl-1,4-dihydro-4-oxoquinoline-3-carboxylic
acid obtained in the Reference Example 8, and
3-aminoazetidine~dichloride was replaced with 110 mg of
3-amino-3-methylazetidine~dichloride to obtain the title
compound.
Characteristic features: pale yellow powder
Melting point: 242-247°C
1HNMR ( de-DMSO ) S ;
1.36 (s, 3H), 1.54 (s, 3H), 1.78 (s, 3H),
3.77-3.85 (m, 1H), 3.95-4.09 (m, 3H), 5.44 (brs, 2H),
6.93 (d, J=9Hz, 1H), 7.09 (d, J=l2Hz, 1H),
7.88 (d, J=l4Hz, 1H), 8.34 (s, 1H)
[Example 6]
1-(3-amino-4-fluoro-6-methylphenyl)-6-fluoro-8-methyl-7-
(3-methylaminoazetidin-1-yl)-1,4-dihydro-4-oxoguinoline-3-
carboxylic acid
3o The procedure of Example x was repeated except that
1-(3-amino-4,6-difluorophenyl)-6,7-difluoro-8-methyl-
1,4-dihydro-4-oxoquinoline-3-carboxylic acid was replaced
with 180 mg of 1-(3-amino-4-fluoro-6-methylphenyl)-6,7-
difluoro-8-methyl-1,4-dihydro-4-oxoquinoline-3-carboxylic
acid obtained in the Reference Example 8, and
CA 02252229 1998-10-16
- 29 -
3-aminoazetidine~dichloride was replaced with 110 mg of
3-N-methylaminoazetidine~dichloride to obtain the title
compound.
Characteristic features: pale yellow powder
Melting point: 235-237°C
1HNMR ( ds-DMSO ) b ;
1.56 (s, 3H), 1.80 (s, 3H), 2.20 (s, 3H),
3.77-3.90 (m, 1H), 3.90-4.02 (m, 1H), 4.31-4.59 (m, 2H),
5.44 (brs, 2H), 6.91 (d, J=8Hz, 1H), 7.10 (d, J=llHz, 1H),
7.89 (d, J=l4Hz, 1H), 8.34 (s, 1H)
[Reference Example 9]
Preparation of ethyl 1-(2-chloro-4-fluorophenyl)-6,7-
difluoro-8-methyl-1,4-dihydro-4-oxoquinoline-3-carboxylate
The procedure of Reference Example 3 was repeated except
that N-tert-butoxycarbonyl-4,6-difluoro-m-phenylenediamine
was replaced with 1.6 g of 2-chloro-4-fluoroaniline to obtain
the title compound.
Characteristic features: pale yellow powder
Melting point: 188-191°C
1HNMR ( CDCl, ) s ;
1.40 (t, J=7Hz, 3H), 1.77 (d, J=3Hz, 3H),
4.39 (q, J=7Hz, 2H), 7.34-7.47 (m, 3H),
8.23 (t, J=lOHz, 1H), 8.31 (m, 1H)
[Reference Example 10]
Preparation of ethyl 1-(2-chloro-4-fluoro-5-nitrophenyl)-
6,7-difluoro-8-methyl-1,4-dihydro-4-oxoguinoline-3-
carboxylate
The procedure of Reference Example 6 was repeated except
that ethyl 1-(4-fluoro-2-methylphenyl)-6,7-difluoro-8-methyl-
1,4-dihydro-4-oxoquinoline-3-carboxylate was replaced with
1.8 g of ethyl 1-(2-chloro-4-fluorophenyl)-6,7-difluoro-
8-methyl-1,4-dihydro-4-oxoquinoline-3-carboxylate obtained in
the Reference Example 9 to obtain the title compound.
CA 02252229 1998-10-16
- 30 -
Characteristic features: pale yellow powder
Melting point: 253-256°C
1HNMR (CDCls ) b ;
1.40 (t, J=7Hz, 3H), 1.83 (d, J=2Hz, 3H),
4.39 (q, J=7Hz, 2H), 7.62 (d, J=9Hz, 1H),
8.13 (d, J=7Hz, 1H), 8.23 (t, J=9Hz, 1H), 8.29 (s, 1H)
[Reference Example 11]
Preparation of ethyl 1-(6-chloro-4-fluoro-3-formylamino-
to phenyl)-6,7-difluoro-8-methyl-1,4-dihydro-4-oxoquinoline-3-
carboxylate
The procedure of Reference Example 7 was repeated except
that ethyl 1-(4-fluoro-2-methyl-5-nitrophenyl)-6,7-difluoro-
8-methyl-1,4-dihydro-4-oxoquinoline-3-carboxylate was
replaced with 1.6 g of ethyl 1-(2-chloro-4-fluoro-5-nitro-
phenyl)-6,7-difluoro-8-methyl-1,4-dihydro-4-oxoquinoline-3-
carboxylate obtained in the Reference Example 10 to obtain
the title compound.
Characteristic features: pale brown powder
2o Melting point: 261-263°C
1HNMR ( ds-DMSO ) b ;
1.26 (t, J=7Hz, 3H), 1.78 (s, 3H), 4.23 (q, J=7Hz, 2H),
7.94 (d, J=lOHz, 1H), 8.08 (d, J=9Hz, 1H),
8.34-8.47 (m, 2H), 8.50 (s, 1H)
[Reference Example 12]
Preparation of 1-(3-amino-6-chloro-4-fluorophenyl)-6,7-
difluoro-8-methyl-1,4-dihydro-4-oxoquinoline-3-carboxylic
acid
3o The procedure of Reference Example 4 was repeated except
that ethyl 1-(3-tert-butoxycarbonylamino-4,6-difluorophenyl)-
6,7-difluoro-8-methyl-1,4-dihydro-4-oxoquinoline-3-
carboxylate was replaced with 1.7 g of ethyl 1-(6-chloro-4-
fluoro-3-formylaminophenyl)-6,7-difluoro-8-methyl-1,4-
dihydro-4-oxoquinoline-3-carboxylate obtained in the
CA 02252229 1998-10-16
- 31 -
Reference Example 11 to obtain the title compound.
Characteristic features: pale yellow powder
Melting point: > 258°C (decomposed)
1HNMR ( ds-DMSO ) S ;
1.18 (d, J=3Hz, 3H), 5.60-5.91 (br, 2H),
7.10 (d, J=7Hz, 1H), 7.59 (d, J=lOHz, 1H),
8.26 (t, J=9Hz, 1H), 8.71 (s, 1H)
[Example 7]
l0 7-(3-aminoazetidin-1-yl)-1-(3-amino-6-chloro-4-fluoro-
phenyl)-6-fluoro-8-methyl-1,4-dihydro-4-oxoguinoline-3-
carboxylic acid
The procedure of Example 1 was repeated except that
1-(3-amino-4,6-difluorophenyl)-6,7-difluoro-8-methyl-
1,4-dihydro-4-oxoquinoline-3-carboxylic acid was replaced
with 110 mg of 1-(3-amino-6-chloro-4-fluorophenyl)-6,7-
difluoro-8-methyl-1,4-dihydro-4-oxoquinoline-3-carboxylic
acid obtained in the Reference Example 12 to obtain the title
compound.
Characteristic features: pale yellow powder
Melting point: > 175°C (decomposed)
1HNMR ( ds-DMSO ) b ;
1.65 (s, 3H), 3.69-3.81 (m, 2H), 3.84-3.96 (m, 1H),
4.39-4.50 (m, 2H), 5.65 (brs, 2H), 7.05 (d, J=7Hz, 1H),
7.54 (d, J=lOHz, 1H), 7.77 (d, J=l4Hz, 1H), 8.50 (s, 1H)
[Example 8]
1-(3-amino-4,6-difluorophenyl)-6-fluoro-7-(3-hydroxy-
azetidin-1-yl)-8-methyl-4-oxo-1,4-dihydroquinoline-3-
3o carboxylic acid
70 mg of 3-hydroxyazetidine~monochloride, 200 mg of
1,8-diazabicyclo[5.4.0]-7-undecene, and 200 mg of pyridine
were stirred at 80°C, and to this mixture was added 150 mg of
1-(3-amino-4,6-difluorophenyl)-6,7-difluoro-8-methyl-4
oxo-1,4-dihydroquinoline-3-carboxylic acid. The mixture was
CA 02252229 1998-10-16
- 32 -
stirred at 90°C for 10 minutes. Diethylether was added to
the reaction solution, and the solution was decanted. 1 ml
of ethanol was added to the residue and the solid precipitate
was collected by filtration and dried to obtain 70 mg of the
title compound.
Characteristic features: pale yellow powder
Melting point: 206-208°C
1HNMR ( db-DMSO ) b ;
1.63 (s, 3H), 3.88-3.95 (m, 1H), 3.95-4.02 (m, 1H),
4.44-4.56 (m, 3H), 5.48 (brs, 2H), 5.70 (brs, 1H),
7.02 (t, J=9Hz, 1H), 7.42 (t, J=llHz, 1H),
7.78 (d, J=l3Hz, 1H), 8.49 (s, 1H)
[Example 9]
1-(3-amino-4-fluoro-6-methylphenyl)-6-fluoro-7-(3-hydroxy-
azetidin-1-yl)-8-methyl-4-oxo-1,4-dihydroquinoline-3-
carboxylic acid
70 mg of 3-hydroxyazetidine~monochloride, 200 mg of
1,8-diazabicyclo[5.4.0]-7-undecene, and 200 mg of pyridine
were stirred at 80°C, and to this mixture was added 150 mg of
1-(3-amino-4-fluoro-6-methylphenyl)-6,7-difluoro-8-methyl-
4-oxo-1,4-dihydroquinoline-3-carboxylic acid. The mixture
was stirred at 90°C for 20 minutes. Diethylether was added
to the reaction solution, and the solution was decanted. 1
ml of ethanol was added to the residue and the solid
precipitate was collected by filtration and dried to obtain
70 mg of the title compound.
Characteristic features: pale yellow powder
Melting point: 147-151°C
1HNMR ( d6-DMSO ) b ;
1.55 (s, 3H), 1.82 (s, 3H), 3.85-3.95 (m, 1H),
3.95-4.03 (m, 1H), 4.40-4.53 (m, 3H), 5.43 (brs, 2H),
5.68 (brs, 1H), 6.91 (d, J=8Hz, 1H),
7.1-0 (d, J=lOHz, 1H), 7.81 (d, J=l4Hz, 1H), 8.35 (s, 1H)
CA 02252229 1998-10-16
- 33 -
[Reference Example 13]
Preparation of 2,4,5-trifluoro-3,6-dimethylbenzoic acid
70 ml of 1.69M n-hexane solution of n-butyllithium was
added dropwise to solution of 18 ml of diisopropylamine in 75
ml of tetrahydrofuran at -65°C under nitrogen stream, and the
mixture was stirred at the same temperature for 15 minutes.
To the solution was added dropwise solution of 9.5 g of
2,4,5-trifluoro-3-methylbenzoic acid in 75 ml of
tetrahydrofuran at -60°C, and the mixture was stirred at the
same temperature for 15 minutes. To the solution was added
dropwise 9.5 ml of methyl iodide at -70°C, and the mixture
was stirred at the same temperature for 30 minutes and
overnight at room temperature. Diethylether and water were
added for separation, and the aqueous layer was collected.
The aqueous layer was acidified by adding conc. hydrochloric
acid, and extracted with diethylether. The organic layer was
dried over ahnydrous magnesium sulfate, and the solvent was
distilled off under reduced pressure to obtain 6.6 g of the
title compound.
2o Characteristic features: pale yellow powder
Melting point: 118-119°C
1HNMR (CDC1, ) b ;
2.23 (s, 3H), 2.40 (s, 3H)
[Reference Example 14]
Preparation of ethyl 2,4,5-trifluoro-3,6-dimethylbenzoyl
acetate
To 400 mg of magnesium were added 1.5 ml of ethanol and
0.05 ml of carbon tetrachloride, and the mixture was stirred
3o at room temperature. To the thus activated solution was
added solution of 2.7 ml of ethyl malonate in 5 ml of
tetrahydrofuran, and the mixture was stirred at 80°C for 4
hours. After allowing to cool, the solution was cooled to
-40°C. To solution of 3.3 g of the 2,4,5-trifluoro-3,6-
dimethylbenzoic acid obtained in the Reference Example 13 in
CA 02252229 1998-10-16
- 34 -
ml of methylene chloride were added 1.5 ml of oxalyl
chloride and 3 drops of N,N-dimethylformamide and the mixture
was stirred at room temperature for 3 hours. The solvent and
the reagent were distilled off under reduced pressure.
5 Toluene was added for azeotropic distillation, and solution
of the residue in 5 ml of tetrahydrofuran was added dropwise
to the above-described reaction solution at -40°C. After the
dropwise addition, the reaction solution was stirred
overnight at room temperature, and the solvent was distilled
off. To the residue was added 3 ml of 12N hydrochloric acid
to adjust pH to about 2, and the solution was extracted with
chloroform. The solvent was distilled off and 10 ml of water
and 100 mg of p-toluenesulfonic acid were added to the
solution. The solution was stirred under heating and reflux
condition for 5 hours. After allowing to cool, the solution
was extracted with chloroform, and the organic layer was
washed with water, dried over anhydrous magnesium sulfate,
and concentrated under reduced pressure. The residue was
subjected to silica gel column chromatography and 3.2 g of
the title compound as a pale brown oil was obtained from the
fractions eluted with ethyl acetate: hexane of 1:10.
[Reference Example 15]
Preparation of ethyl 1-(3-tert-butoxycarbonylamino-4,6-
difluorophenyl)-6,7-difluoro-5,8-dimethyl-4-oxo-1,4-
dihydroguinoline-3-carboxylate
To 3.2 g of the ethyl 2,4,5-trifluoro-3,6-dimethyl-
benzoyl acetate obtained in the Reference Example 14 were
added 7.8 g of anhydrous acetic acid and 2.8 g of triethyl
orthoformate, and the mixture was heated under reflux for 4
hours. The reaction solution was allowed to cool, and the
reagents and the like were distilled off at reduced pressure.
Toluene was added for azeotropic distillation. 10 ml of
chloroform was added to the residue, and solution of 2.7 g of
N-t-butoxycarbonyl-4,6-difluorophenylenediamine in 5 ml of
CA 02252229 1998-10-16
- 35 -
chloroform was added dropwise. The mixture was stirred
overnight at room temperature, and the solvent was distilled
off. The residue was subjected to silica gel column
chromatography and 4.0 g of aminoacrylate compound as a pale
yellow solid product was obtained from the fractions eluted
with ethyl acetate: hexane of 1:20.
To the solution of all of the thus obtained
aminoacrylate compound in 20 m1 of N,N-dimethylformamide was
added 1.0 g of potassium carbonate, and the mixture was
stirred at 70°C for 3.5 hours. Ethyl acetate and water were
added, and the organic layer separated was collected and
dried over anhydrous magnesium sulfate. The solvent was
distilled off and the residue was subjected to silica gel
column chromatography to obtain 3.0 g of the title compound
from the fractions eluted with ethyl acetate: hexane of 1:2.
Characteristic features: pale yellow powder
Melting point: 141-142°C
1HNMR ( CDC13 ) b ;
1.38 (t, J=7Hz, 3H), 1.51 (s, 9H), 1.76 (d, J=2Hz, 3H),
2.86 (d, J=3Hz, 3H), 4.38 (q, J=7Hz, 2H),
6.80 (brs, 1H), 7.08 (t, J=lOHz, 1H), 8.17-8.30 (m, 2H)
[Reference Example 16]
Preparation of 1-(3-amino-4,6-difluorophenyl)-6,7-difluoro-
5,8-dimethyl-4-oxo-1,4-dihydroguinoline-3-carboxylic acid
To 1.8 g of the ethyl 1-(3-tert-butoxycarbonylamino-
4,6-difluorophenyl)-6,7-difluoro-5,8-dimethyl-4-oxo-1,4-
dihydroquinoline-3-carboxylate obtained in the Reference
Example 15 was added 10 ml of 12N hydrochloric acid, and the
mixture was stirred under heating and reflux condition for 4
hours. After allowing to cool, the solid precipitate was
collected by filtration, washed with water and ethanol
successively, and dried to obtain 1.2 g of the title
compound.
Characteristic features: colorless powder
CA 02252229 1998-10-16
- 36 -
Melting point: > 281°C (decomposed)
1HNMR ( ds-DMSO ) 6 ;
1.80 (s, 3H), 2.82 (s, 3H), 7.06 (t, J=8Hz, 1H),
7.46 (t, J=llHz, 1H), 8.62 (s, 1H)
[Example 10]
7-(3-aminoazetidin-1-yl)-1-(3-amino-4,6-difluorophenyl)-
6-fluoro-5,8-dimethyl-4-oxo-1,4-dihydroquinoline-3-carboxylic
acid
1o To 100 mg of the 1-(3-amino-4,6-difluorophenyl)-
6,7-difluoro-5,8-dimethyl-4-oxo-1,4-dihydroquinoline-3-
carboxylic acid obtained in the Reference Example 16 were
added 300 mg of pyridine, 70 mg of 3-aminoazetidine~
dichloride, and 150 mg of 1,8-diazabicyclo[5.4.0]-7-undecene,
and the mixture was heated and stirred at 40°C for 24 hours.
After allowing to cool, the solvent and the like were
distilled off under reduced pressure. To the residue was
added 1 ml of ethanol, and the solution was allowed to stand
for 5 days. The solid content was collected by filtration,
washed with ethanol and dried to obtain 15 mg of the title
compound.
Characteristic features: pale yellow powder
Melting point: > 235°C (decomposed)
1HNMR ( ds-DMSO ) b ;
1.60 (s, 3H), 2.73 (s, 3H), 3.99-4.08 (m, 1H),
4.08-4.18 (m, 1H), 4.23-4.32 (m, 1H), 4.42-4.56 (m, 2H),
5.49 (brs, 2H), 7.02 (t, J=8Hz, 1H),
7.39 (t, J=llHz, 1H), 8.33 (s, 1H), 8.46 (s, 1H)
[Reference Example 17]
Preparation of 2,4,5-trifluoro-3-methyl-6-nitrobenzoic acid
To 100 ml of conc. sulfuric acid was dissolved 31 g of
2,4,5-trifluoro-3-methylbenzoic acid, and to this solution
was incrementally added 19.5 g of potassium nitrate on ice.
The solution was stirred at room temperature for 3 days, and
CA 02252229 1998-10-16
- 37 -
another 1.4 g of potassium nitrate was added on ice. After
stirring for 6 hours, the solution was poured into ice water,
and the solid precipitate was collected by filtration. The
precipitate was dissolved in diethylether and washed with
water. The organic layer was dried over anhydrous magnesium
sulfate, and the solvent was distilled off. The solid
precipitate was collected by filtration to obtain 21 g of the
title compound.
Characteristic features: pale yellow powder
1HNMR (CDC1,) S ;
2.38 (s, 3H)
[Reference Example 18]
Preparation of ethyl 2,4,5-trifluoro-3-methyl-6-nitrobenzoyl
acetate
To 2.2 g of magnesium were added 8 ml of ethanol and 0.4
ml of carbon tetrachloride, and the mixture was stirred at
room temperature. To the thus activated solution was added
solution of 14 ml of ethyl malonate in 40 ml of
2o tetrahydrofuran, and the mixture was stirred at 80°C for 4
hours. After allowing to cool, the solution was cooled to
-40°C. To solution of 20 g of the 2,4,5-trifluoro-3-methyl-
6-nitrobenzoic acid obtained in the Reference Example 17 in
40 ml of methylene chloride were added 8.4 ml of oxalyl
chloride and 3 drops of N,N-dimethylformamide and the mixture
was stirred at room temperature for 3 and a half hours. The
solvent and the reagent were distilled off under reduced
pressure. Toluene was added for azeotropic distillation, and
solution of the residue in 15 ml of tetrahydrofuran was added
dropwise to the above-described reaction solution at -40°C.
After the dropwise addition, the reaction solution was
stirred overnight at room temperature, and the solvent was
distilled off. To the residue was added 20 ml of 12N
hydrochloric acid to adjust pH to about 2, and the solution
was extracted with chloroform. The solvent was distilled off
CA 02252229 1998-10-16
- 38 -
and 50 ml of water and 400 mg of p-toluenesulfonic acid were
added to the solution. The solution was stirred under
heating and reflux condition for 5 hours. After allowing to
cool, the solution was extracted with chloroform, and the
organic layer was washed with water, dried over anhydrous
magnesium sulfate, and concentrated under reduced pressure.
The residue was subjected to silica gel column chromatography
and 8.0 g of the title compound as a red oil was obtained
from the fractions eluted with ethyl acetate: hexane of 1:10.
to
[Reference Example 19]
Preparation of ethyl 1-(3-tert-butoxycarbonylamino-4,6-
difluorophenyl)-6,7-difluoro-8-methyl-5-nitro-4-oxo-1,4-
dihydroguinoline-3-carboxylate
To 4 g of the ethyl 2,4,5-trifluoro-3-methyl-6-
nitrobenzoyl acetate obtained in the Reference Example 18
were added 8.5 g of anhydrous acetic acid and 3.2 g of
triethyl orthoformate, and the mixture was heated under
reflux for 2 hours. The reaction solution was allowed to
cool, and the reagents and the like were distilled off at
reduced pressure. Toluene was added for azeotropic
distillation. 10 ml of chloroform was added to the residue,
and solution of 3.7 g of N-t-butoxycarbonyl-4,6-difluoro-
phenylenediamine in 20 ml chloroform was added dropwise. The
mixture was stirred at room temperature for 3 days, and the
solvent was distilled off to obtain 4.8 g of aminoacrylate
compound as a pale yellow oil.
To the solution of all of the thus obtained
aminoacrylate compound in 15 ml of N,N-dimethylformamide was
3o added 1.4 g of potassium carbonate, and the mixture was
stirred at 70°C for 30 minutes. The solution was allowed to
cool, and ethyl acetate and water were added to the solution.
The organic layer separated was collected and dried over
anhydrous magnesium sulfate. The solvent was distilled off
and the residue was subjected to silica gel column
CA 02252229 2005-07-20
695'62-22
- 39 -
chromatography to obtain 4.0 g of the title compound from the
fractions eluted with ethyl acetate: hexane of 1:2.
Characteristic features: pale yellow powder
Melting point: 188-189°C
1HNMR ( CDC1, ) 6
1.36 (t, J=7Hz, 3H), 1.52 (s, 9H), 1.88 (d, J=3Hz, 3H),
4.38 (q, J=7Hz, 2H), 6.94 (brs, 1H),
7.15 (t, J=lOHz, 1H), 8.29-8.44 (m, 2H)
[Reference Example 20]
Preparation of ethyl 5-amino-1-(3-amino-4,6-difluorophenyl)-
6,7-difluoro-8-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylate
To solution of 1.0 g of the ethyl 1-(3-tert-butoxy-
carbonylamino-4,6-difluorophenyl)-6,7-difluoro-8-methyl-5-
nitro-4-oxo-1,4-dihydroquinoline-3-carboxylate obtained in
the Reference Example 19 in 5 ml of acetic acid was added 900
mg of iron powder, and the mixture was heated and stirred at
90°C for 4 hours and 40 minutes. The catalyst in the
reaction solution was filtered off by using Celite*, and the
2o solvent in the residue was distilled off. The residue was
subjected to silica gel column chromatography, and ethanol
was added to the oil obtained from the fractions eluted with
ethyl acetate:hexane of 1:1. The powder precipitate was
collected by filtration to obtain 200 mg of the title
compound.
Characteristic features: pale yellow powder
Melting point: 134-135°C
1HNMR ( d6-DMSO ) b ;
1.24 (t, J=7Hz, 3H), 1.57 (d, J=2Hz, 3H),
4.19 (q, J=7Hz, 2H), 5.45 (brs, 2H),
6.95 (t, J=8Hz, 1H), 7.39 (t, J=llHz, 1H), 8.20 (s, 1H)
*Trade-mark
CA 02252229 1998-10-16
- 40 -
[Reference Example 21]
Preparation of 5-amino-1-(3-amino-4,6-difluorophenyl-
2-yl)-6,7-difluoro-8-methyl-4-oxo-1,4-dihydroquinoline-3-
carboxvlic acid
To 200 mg of ethyl 5-amino-1-(3-amino-4,6-difluoro-
phenyl)-6,7-difluoro-8-methyl-4-oxo-1,4-dihydroquinoline-3-
carboxylate obtained in the Reference Example 20 was added 5
ml of 12N hydrochloric acid, and the mixture was heated under
reflux for 10 hours. The reaction solution was allowed to
l0 cool, and the solid precipitate was collected by filtration.
The precipitate was washed with ethanol, and then with
diethylether to obtain 140 mg of the title compound.
Characteristic features: yellow powder
Melting point: > 290°C
1HNMR ( ds-DMSO ) b ;
1.61 (d, J=2Hz, 3H), 7.02 (t, J=8Hz, 1H),
7.42 (t, J=llHz, 1H), 8.47 (s, 1H)
[Test 1]
Antibacterial activity
Minimum growth inhibitory concentration (MIC, a g/ml)
was evaluated in accordance with the standard method of Japan
Chemotherapy Society (Chemotherapy 29(1), 76, 1981). The
results are shown in Table 1. It should be noted that
tosufloxacin was also evaluated for its minimum growth
inhibitory concentration for the purpose of comparison. The
results are also shown in Table 1.
35
CA 02252229 1998-10-16
- 41 -
Table 1
S.aureus 209P P.aeruginosa IF03445
Compound of Ex. 1 0.006 0.05
Compound of Ex. 2 0.006 0.1
Compound of Ex. 3 0.006 0.1
Compound of Ex. 4 0.003 0.1
Compound of Ex. 5 0.013 0.2
Compound of Ex. 6 0.013 0.2
Compound of Ex. 7 0.013 0.1
Compound of Ex. 8 0.006 0.2
Compound of Ex. 9 0.013 0.2
Compound of Ex. 10 0.003 0.1
Tosufloxacin 0.05 0.39
[ Test 2 ]
Phototoxicity test
Female ICR mice (5 to 6 week old) were intravenously
administered with the test compound (40 mg/kg/10 ml), and
irradiated with UVA (320 to 400 nm, 1.8 mW/cmz/sec) for 4
hours. Abnormality in the ears was monitored at 0 hour
immediately after the irradiation and after 24 hours. The
ear abnormality was evaluated by the following criteria: no
abnormality (0 point), very slight erythema (1 point), well
defined erythema (2 points), moderate to severe erythema or
edema formation (3 points). The results are shown in Table
2. Tosufloxacin which is a conventional known antibacterial
agent was also tested in a similar way for the purpose of
comparison. The results are also shown in Table 2.
CA 02252229 1998-10-16
- 42 -
m~L1 ., "I
0 hour 24 hours
(point, (point,
occurrence) occurrence)
Compound of Ex. 1 0, 0/3 0, 0/3
Compound of Ex. 2 0,
0/3 0, 0/3
Compound of Ex. 3 0, 0/3 0, 0/3
Compound of Ex. 4 0, 0/3 0, 0/3
Compound of Ex. 5 0, 0/3 0, 0/3
Compound of Ex. 6 0, 0/3 0, 0/3
Compound of Ex. 7 0, 0/3 0, 0/3
Compound of Ex. 8 0, 0/3 0, 0/3
Compound of Ex. 9 0, 0/3 0, 0/3
Compound of Ex. 10 0, 0/3 0, 0/3
Tosufloxacin 1.8, 4/5 0.8, 4/5
[ Test 3
Absorption and excretion
A compound of the present invention was measured for its
urine and bile recovery rates :in rats after its oral
administration to evaluate absorption and excretion of the
compounds.
(1) Urine recovery rate
Male SD rats of 6 week old which had been fasted
overnight were administered orally with 0.5~ methylcellulose
suspension of the test compounds (20 mg/10 ml/kg). The urine
3o collection was continued until 24 hours after the
administration. The concentration of the test compound in
the urine was measured by paper disc method using Bacillus
subtilis ATCC6633 for the test bacterium to determine the
urine recovery rate.
CA 02252229 1998-10-16
- 43 -
(2) Bile recovery rate
Into choledoch duct of male SD rats of 6 week old which
had been fasted overnight was inserted a polyethylene tube
under etherization. After arousing, the rats were forcedly
administered orally with the test compounds as in the case of
the above (1), and bile collection was continued until 24
hours after the administration. The bile with no further
treatment and the bile alkaline hydrolyzed (with O.1N NaOH,
37°C, 1 hour) were measured for their concentration of the
test compound by the same procedure as in the case of the
above (1) to determine the bile recovery rate.
The results are shown in Table 3.
Table 3
Recovery
rate (24
hours,
$)
Urine Bile Bile* Total
recovery rate
Compound of Ex. 3.9 10.4 26.7 30.6
3
* Recovery rate for the bile after alkaline hydrolysis
30