Note: Descriptions are shown in the official language in which they were submitted.
CA 02252970 1998-11-30
- 1 -
Optimization of cells for endogenous gene activation
Description
The invention concerns a process for optimizing gene
expression in cells. A first aspect concerns a process
for changing the expression of a target gene that is
present endogenously in a eukaryotic cell by introducing
a heterologous expression control sequence or/and an
amplification gene into the genome of the cell by means
of homologous recombination and also concerns the
excision of the inserted foreign DNA mediated by a site-
specific recombinase and its replacement by other
heterologous expression control sequences or/and
amplification genes. The invention additionally concerns
the introduction of one or several nucleic acid
sequences to which an activator protein or an activator
protein complex e.g. a hypoxia-inducible factor (HIF)
binds, into the genome of a eukaryotic cell by
homologous recombination in order to change the
expression of a target gene. Furthermore the invention
concerns a method for testing the influence of non-
coding nucleic acid fragments on the 5' side or 3' side
on the expression of a target gene by determining the
expression of a reporter gene. In addition the invention
concerns a process for preparing a DHFR-negative
eukaryotic cell containing a recombinase target sequence
as well as the expression of a nucleic acid sequence
inserted into the recombinase target sequence.
Gene expression in a cell can take place constitutively
for example in so-called housekeeping genes or be
regulated. Regulated expression is particularly
necessary for genes which only have to be expressed in a
CA 02252970 1998-11-30
2 -
particular development stage of the cell or when there
is a change in the environmental conditions.
Expression is regulated at the transcription level by
the promoter that is operatively linked with the coding
nucleic acid sequence the activity of which can be
controlled by repressors and activators. Binding of
repressors or activators to non-coding nucleic acid
sequences of the gene can reduce or increase the
activity of the promoter (L. Stryer, Biochemie, Chapter
12, "Spektrum der Wissenschaft, Verlagsgesellschaft",
Heidelberg, 1990). The amount of repressors or
activators that are contained in a cell is in turn
regulated by factors such as for example environmental
conditions. Hypoxia-inducible factors (HIF) are an
example of activators which are induced by reduced 02
supply and lead to an increased expression of the
erythropoietin gene (Blanchard K.L. et al., Hypoxic
induction of the human erythropoietin gene: Cooperation
between the promoter and enhancer, each of which
contains steroid receptor response elements, (1992),
Mol. Cell. Biol. 12, 5373-5385; Wang G.L. and Semenza
G.L., Characterization of hypoxia-inducible factor 1 and
regulation of DNA binding activity by hypoxia, (1993),
J. Biol. Chem., 268, 21513-21518; Wang G.L. et al.,
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-
PA heterodimer regulated by cellular 02 tension, (1995),
Proc. Natl. Acad. Sci. USA, 92, 5510-5514).
Furthermore the amount of an expressed protein depends
on the stability of the mRNA. Recognition sequences for
mRNA degrading enzymes are located in the 3' region of
an mRNA which influence the stability of the mRNA and
thus the expression level (Shaw G. and Kamen, R., A
Conserved AU Sequence from the 3'Untranslated Region of
CA 02252970 1998-11-30
- 3 -
GM-CSF mRNA Mediates Selective mRNA Degradation, Cell
(1986), 659-667). In this connection the half-life of
the mRNA correlates with the amount of expressed
protein. A third level of expression regulation is
translation.
Hence the expression of a gene is subject to complex
regulation mechanisms which can differ greatly in
individual cases.
Proteins can be obtained with the aid of recombinant DNA
technology which utilizes knowledge on expression
regulation (Sambrook et al., 1989, Molecular Cloning, A
Laboratory Manual, Cold Spring Harbor). Vectors are used
for this which contain a nucleic acid sequence coding
for the corresponding protein under the control of a
suitable promoter as well as additional sequences that
are necessary to express the protein and replicate the
vector. The vector is then introduced into a host cell
by means of known methods, the cell is cultured and the
recombinant protein can be isolated from the cell or the
culture medium.
Prokaryotic or eukaryotic cells can be used as the host
cell. Prokaryotic cells, in particular E. coli cells,
are unproblematic to handle but have a number of
disadvantages when eukaryotic proteins are expressed
recombinantly.
Prokaryotes and eukaryotes differ in the expression
processing path, in the cell medium conditions, as well
as in the chaperones involved in protein processing.
Hence a eukaryotic protein produced in a prokaryote may
differ decisively from the corresponding native protein.
CA 02252970 1998-11-30
- 4 -
For example the protein folding pattern and the activity
of the protein may be modified. Also proteins in a
prokaryotic host cell are usually not glycosylated.
However, a correct glycosylation pattern is a crucial
characteristic in many cases for the effectiveness and
tolerance for example in the production of proteins for
a pharmaceutical formulation.
Glycosylated proteins are therefore produced by means of
eukaryotic host cells or cell lines, for example CHO
(Chinese Hamster Ovary) cells. Despite the use of
eukaryotic cells, changes in the recombinantly produced
protein can occur due to species differences for example
when expressing a human protein in non-human cells which
is why this method is unsuitable for many applications.
For the recombinant production of proteins, host cells
are transiently or stably transfected with expression
vectors, stably transfected cells being used in
particular for large scale production processes.
Unspecific, random integration of the expression vector
sequences into the genome of the host cell can lead to
cells with a low production capacity or to unstable
properties of the cells. For example the production
output can decrease during the course of a production
process or the ability of the cells to express the
recombinant protein can be completely lost.
A method for increasing gene expression is gene
amplification in which a nucleic acid sequence coding
for a protein is coupled to an amplification gene. A
multiplication of both sequences is achieved by a
selection step which leads to an increased expression
CA 02252970 1998-11-30
- 5 -
(Schimke, R.T. (Ed.) (1982), Gene amplification, Cold
Spring Harbor Lab., Cold Spring Harbor, NY).
A nucleic acid coding for a dihydrofolate reductase
(DHFR) can for example be used.as an amplification gene
(Kaufmann R.J., Sharp P.A. (1982), Amplification and
expression of sequences cotransfected with a modular
dihydrofolate reductase complementary DNA gene, J. Mol.
Biol. 159:601 ff).
A selection step carried out with methotrexate enables
cells to be obtained which are resistant to methotrexate
and contain in their genome the nucleic acid sequence
coding for a DHFR and the nucleic acid sequence coupled
thereto in a 20- to 50-fold amplification (R. Knippers,
1982, "Molekulare Genetik", Thieme, Stuttgart).
Such a gene amplification method is most effectively
carried out with a DHRF-negative cell. JP-62265992
describes for example a human DHFR-negative cell.
However, it does not mention a site-specific integration
of an expression vector by means of homologous
recombination and amplification of these sequences in
this cell.
Even when carrying out a gene amplification process the
disadvantages described above such as instability of the
cells can occur due to random integration of the
expression vector into the genome of the cell.
It is only possible to avoid the described disadvantages
when foreign DNA is site-specifically integrated at a
selected gene locus by homologous recombination which
leads to an endogenous gene activation. Corresponding
CA 02252970 1998-11-30
- 6 -
methods are known and are called gene targeting (WO
90/11354; WO 91/09955). In this process the cell is
transfected with a vector which contains a positive
selection marker gene flanked by nucleic acid sequences
which are homologous to a gene locus at which it is
intended to integrate the vector into the genome of the
cell. Between the homologous nucleic acid sequences
there is additionally a heterologous expression control
sequence in order to increase the expression of the
target gene in the cell and optionally an amplification
gene to increase the copy number of the target gene.
A disadvantage of previously known gene targeting
methods is that it is often extremely laborious to
produce cells with properties that enable the production
of a desired protein in an adequate amount and quality
for commercial purposes. In particular the selection of
optimal expression control sequences or/and
amplification genes for the expression of a desired
target protein often requires a very large series of
homologous recombination experiments which are extremely
time-consuming due to the complicated procedure for
isolating clones in which the desired recombination
event has taken place.
Homologous recombination can also be used to switch off
the expression of certain genes in a cell in order to
carry out protein function studies. For this knockout
mice are produced in which the gene coding for a protein
to be examined is switched off by homologous
recombination in embryonic stem cells. After carrying
out additional process steps, mice are obtained that
cannot express a functional protein from the start of
their development due to the inactivation of both
alleles of this gene (Thomas, K.R., Capecchi M.R.,
CA 02252970 1998-11-30
- 7 -
(1987), Site-directed mutagenesis by gene targeting in
mouse embryo-derived stem cells, Cell 51: 503-512).
The Cre-Lox system can be used to tissue-specifically
and time-specifically switch off a certain gene and to
examine it. For this purpose a nucleic acid fragment
flanked by two loxP sequences is introduced into the
genome of a cell by homologous recombination and can
subsequently be cleaved again from the genome by a Cre
recombinase that is expressed in the cell (Sauer B,
Henderson N (1989): Site-specific DNA recombination at
loxP sites placed into the genome of mammalian cells.
Nuc Acid Res 17:147-161; Sauer B., Henderson N. (1990),
Targeted insertion of exogenous DNA into the eukaryotic
genome by the Cre recombinase, New Biol. 5:441-449). The
prior art makes no mention of using the Cre-lox system
or another site-specific recombinase system for the
site-specific integration of expression control
sequences or amplification genes into the genome of
eukaryotic cells in -order to change endogenous gene
expression.
The object of the present invention was to provide a new
process for optimizing endogenous gene activation by
homologous recombination which at least partially
eliminates the disadvantages of the prior art.
This object is achieved by providing a new process and
vector constructs which considerably simplify an
optimization of the expression output of genes in
eukaryotic cells. A first aspect of the invention
concerns a process for changing the expression of a
nucleic acid sequence which is present endogenously in a
eukaryotic cell which is characterized in that
CA 02252970 1998-11-30
- 8 -
(a) the cell is transfected with a first vector comprising
(i) at least one sequence selected from a first
heterologous expression control sequence and a
first amplification gene,
(ii) a positive selection marker gene,
(iii) at least two target sequences for a site-specific
recombinase flanking the sequences (i) and (ii),
(iv) DNA sequences which flank the sequences (i), (i)
and (iii) and are homologous to a nucleic acid
section in the genome of the cell in order to
allow a homologous recombination
(b) the transfected cell is cultured under conditions under
which a homologous recombination of the vector takes
place and
(c) the cell obtained according to step (b) is isolated.
A cell is provided by the process according to the
invention which has an endogenous gene in operative
linkage with a heterologous expression control sequence
or/and an amplification gene and these sequences are
flanked by target sequences for a site-specific
recombinase e.g. the Cre recombinase. This cell is very
well-suited for investigations on the optimization of
the expression of the target gene since the presence of
the target sequences for the site-specific recombinase
enables a simple replacement of the first heterologous
expression control sequence or/and the first
amplification gene by a second heterologous expression
control sequence or/and a second amplification gene.
The term "site-specific recombinase" according to the
present invention encompasses proteins and protein
complexes which mediate DNA rearrangements on a specific
DNA target sequence including site-specific recombinases
of the integrase or resolvase invertase classes (Stark
CA 02252970 1998-11-30
- 9 -
et al., Trends Genet. 8 (1992), 432-439; Abremski and
Hoess, Protein Engineering 5 (1992), 87-91; Khan et al.,
Nucleic Acids Res. 19 (1991), 851-860) and site-specific
recombination mediated by intron-coded endonucleases
(Perrin et al., EMBO J. 12 (1993), 2939-2947). Preferred
recombinase proteins are selected from the group
comprising the FLP recombinase of the 2 p episome of
Saccharomyces cerevisiae (e.g. Falco et al., Cell 29
(1982), 573-584; Cox, Proc. Natl. Acad. Sci. USA 80
(1983) 4223-4227; Konsolaki et al., New Biologist 4
(1992), 551-557), the Cre recombinase of the E. coli
phage P1 (e.g. Sauer and Henderson (1989) supra), the
R-recombinase from the Zygosaccharomyces rouxii plasmid
pSR1 (Matsuzaki et al., J. Bacteriol. 172 (1990), 610-
618), the A recombinase from the Kluyveromyces
drosophilarium plasmid pKD1 (Chen et al., Nucleic Acids
Res. 14 (1986), 4471-4481), the A recombinase from the
Kluveromyces waltii plasmid pKW1 (Chen et al., J. Gen.
Microbiol. 138 (1992), 337-345), a component of the
2-int recombination system (Landy, Annu Rev. Biochem. 5
(1989), 913-949) and a component of the gin
recombination system of the phage p (Klippel et al.,
EMBO J. 12 (1993), 1047-1057). In addition the fusion
proteins described in the European Patent EP-B-0 707 599
composed of a site-specific recombinase and a nuclear
receptor or the ligand-binding domain thereof are also
suitable. Target sequences of the Cre recombinase i.e.
loxP sequences are particularly preferably used for the
process according to the invention.
In contrast to the recombinant production of proteins by
site-unspecific integration of heterologous genes and
their associated disadvantages, the process according to
the invention utilizes the advantages of site-specific
endogenous gene activation by homologous recombination.
CA 02252970 1998-11-30
- 10 -
A simplified selection of suitable combinations of
heterologous expression control sequences and
amplification genes enables optimized production clones
with stable properties to be obtained with high
probability which enable the production of a protein
which substantially corresponds to the native protein
with regard to its structure and activity.
The selection of suitable homologous sequences which
flank the heterologous expression control sequence, the
amplification gene, the. positive selection marker gene
and the recombinase target sequences is preferably
carried out according to the methods described in
W090/11354 and W091/09955.
In addition the homologous sequences may also contain
modifications which lead to mutations in the expressed
protein such as for example point mutations, insertions
or/and deletions of individual amino acids or whole
amino acid sections.
Thus the process according to the invention not only
enables the expression level of an endogenous nucleic
acid sequence to be changed in a single process step but
also simultaneously enables the introduction of a
mutation into the coding region of the endogenous
nucleic acid sequence. Hence the process according to
the invention is particularly advantageous for the
production of proteins for pharmaceutical applications.
Such proteins should have no further modifications
compared to native proteins apart from mutations to
increase the efficacy of the protein.
According to the invention it is possible to use any
CA 02252970 1998-11-30
- 11 -
eukaryotic cell, preferably a mammalian cell,
particularly preferably a human cell. The process
according to the invention can be carried out with non-
immortalized cells e.g. fibroblasts and also with
immortalized cells e.g. tumour cell lines. Immortalized
cells are preferred.
The solutions and media used to carry out the process
according to the invention are preferably selected such
that optimal conditions are present in the respective
process step. The cells are cultured using media which
contain all substances necessary for an adequate cell
growth and are optionally buffered. It is preferable to
use cells which can be cultured in serum-free medium.
The cell used is particularly preferably a Namalwa,
HT1080 or HeLa S3 cell.
The process according to the invention enables
optimization of the expression of a nucleic acid
sequence present endogenously in the cell i.e. of a
target gene by selection of an optimal expression
control sequence, an optimal amplification gene or/and
by selection of an optimal combination of expression
control sequence and amplification gene.
Any nucleic acid sequence can be used as the
heterologous expression control sequence which
influences the expression of the target gene after its
integration into the genome of the cell. This includes
nucleic acid sequences which can directly interact with
transcription components such as transcription
initiation factors or RNA polymerases and nucleic acid
sequences whose influence on transcription is mediated
by interactions with activators or repressors. The
CA 02252970 1998-11-30
- 12 -
heterologous expression control sequence preferably
contains a promoter/enhancer, particularly preferably
viral promoters and most preferably a CMV promoter.
The heterologous expression control sequence can also
include a 3' non-coding sequence. 3' non-coding
sequences can have a stabilizing or destabilizing effect
on an mRNA and thus increase or decrease its half-life.
The introduction of a sequence that stabilizes an mRNA
can increase the half-life of an mRNA and thus the yield
of its encoded protein.
In a preferred embodiment an endogenous expression
control sequence of the target gene is removed by the
homologous recombination. This is particularly
advantageous when the endogenous sequence contains a
repressor-binding sequence. The expression can also be
reduced by a 3' non-coding sequence which has a
destabilizing effect on the mRNA which results in a
decrease in the amount of translated protein.
In addition the process according to the invention
allows the selection of an optimal amplification gene.
The amplification gene is preferably used in an
expressible form i.e. in operative linkage with a
suitable promoter and is arranged in the vector such
that after homologous integration of the vector into the
genome of the eukaryotic cell it is located near to the
target gene. Carrying out an amplification step leads to
an increase in the number of copies of the target gene
in the cell. This can result in a further increase in
the expression of the endogenous nucleic acid sequence.
Examples of suitable amplification genes are
dihydrofolate reductase (DHFR), adenosine deaminase,
CA 02252970 1998-11-30
- 13 -
ornithine decarboxylase or muteins of these genes. The
amplification gene is preferably a DHFR gene or a
mutated form thereof (Simonsen et al., Nucleic Acids
Res. 1988, 16 (5): 2235-2246), especially in cells which
contain an endogenous DHFR gene.
Any suitable resistance gene for a eukaryotic cell which
leads to a selectable phenotype such as e.g. an
antibiotic resistance can be used as a positive
selection marker. The positive selection marker gene is
preferably a neomycin, kanamycin, geneticin or
hygromycin resistance gene. The positive selection
marker gene is preferably used in an expressible form
i.e. in operative linkage with a suitable promoter.
If a negative selection marker gene is used then a
second negative selection step is usually carried out in
addition to the positive selection step. The advantage
of this is that, after carrying out the selection steps,
the identified clones contain a lower proportion of
false-positive clones i.e. vectors that are randomly
integrated into the genome. The negative selection
marker gene is preferably a thymidine kinase gene (TK)
or/and a hypoxanthine-guanine-phosphoribosyl transferase
gene (HGPRT).
As a result of the presence of the target sequences of
the site-specific recombinase it is possible to cut out
nucleic acid sequences located between these sequences
from the genome of the cell using the site-specific
recombinase. The nucleic acid sequence located between
the target sequences is preferably cleaved from the
genome by transient activation of the corresponding
recombinase in the cell. This transient activation of
CA 02252970 1998-11-30
- 14 -
the recombinase can for example be carried out by
(a) transfecting the cell with a second vector
containing a nucleic acid sequence coding for the
recombinase operative linked with an expression
control sequence that is active or can be activated
in this cell and
(b) culturing the cell transfected in this manner under
conditions under which the recombinase is expressed
and is active and
(c) optionally isolating the cell.
If recombinase/nuclear receptor fusion proteins are
used, the transient activation of the cell can also be
carried out by the controlled addition of the ligand for
the nuclear receptor.
After removing the DNA located between the target
sequences, the remaining target sequence e.g. the loxP
sequence can be used for additional process steps.
In a further preferred embodiment the process is
characterized in that
(a) the cell is transfected with a third vector comprising
(i) at least one sequence selected from a second
heterologous expression control sequence and a
second amplification gene
(ii) a positive selection marker gene which preferably
differs from the positive selection marker gene
of the first vector and
(iii) at least two recombinase target sequences
flanking the sequences (i) and (ii)
(b) the transfected cell is cultured under conditions under
which the sequence flanked by the target sequences is
integrated into the target sequence in the genome of
CA 02252970 1998-11-30
- 15 -
the cell
(c) the cell obtained according to step (b) is isolated and
(d) optionally steps (a) to (c) are repeated at least once
with expression control sequences or/and amplification
genes which vary in each case.
Hence the process according to the invention enables
many expression control sequences, amplification genes
or combinations of expression control sequences and
amplification genes to be tested simply and rapidly.
Hence it is not necessary to carry out a time-consuming
and expensive site-specific integration for each
individual heterologous expression control sequence and
each individual amplification gene to determine an
optimal expression/amplification system for each
individual target gene.
The positive selection marker gene in a third vector
preferably differs from that of a first vector in order
to simplify the selection process and to minimize the
number of false positive clones.
The recombinase target sequences in the vector used
according to the invention can correspond to naturally
occurring target sequences or optionally have mutations
which do not impair the effectiveness of the site-
specific recombinase.
A further subject matter of the invention is a vector for
homologous recombination in particular for the site-
specific introduction of recombinase target sequences into
the genome of a cell comprising
(i) at least one sequence selected from an expression
control sequence and an amplification gene,
CA 02252970 1998-11-30
- 16 -
(ii) a positive selection marker gene,
(iii) at least two target sequences for a site-specific
recombinase which flank the sequences (i) and (ii),
(iv) DNA sequences flanking the sequences (i), (ii) and
(iii) which are homologous to a nucleic acid section
in the genome of a cell in order to allow a
homologous recombination and
(v) optionally a negative selection marker gene.
In addition all vectors according to the invention
preferably contain the necessary sequence elements for
propagation and multiplication in suitable host cells
such as origin of replication, selection marker genes
etc..
Yet a further subject matter of the invention is a
vector, in particular for introducing DNA into the
genome of a cell by means of a site-specific recombinase
system comprising
(i) at least one sequence selected from an expression
control sequence and an amplification gene,
(ii) a positive selection marker gene and
(iii) at least two recombinase target sequences flanking
the sequences (i) and (ii).
Yet a further subject matter of the present invention is
a eukaryotic cell, preferably a human cell, which is
obtainable by a process as described above. This cell,
e.g. a human cell, is preferably characterized in that
it
(a) contains at least one chromosomally located sequence
selected from a heterologous expression control
sequence and an amplification gene in operative
linkage with a nucleic acid sequence that is present
CA 02252970 1998-11-30
- 17 -
endogenously and
(b) this sequence is flanked by recombinase target
sequences.
A further aspect of the present invention concerns a
process for changing the expression of a nucleic acid
sequence that is present endogenously in a eukaryotic cell
which is characterized in that
(a) the cell is transfected with a vector comprising
(i) at least one nucleic acid sequence that binds an
activator protein e.g. a hypoxia-inducible
factor (HIF),
(ii) a positive selection marker gene,
(iii) DNA sequences flanking the sequences (i) and (ii)
which are homologous to a nucleic acid section in
the genome of the cell in order to allow a
homologous recombination,
(b) the transfected cell is cultured under conditions under
which a homologous recombination of the vector takes
place and
(c) the cell obtained according to step (b) is isolated.
Surprisingly the genomic integration of a nucleic acid
sequence which binds one or several activator proteins
(proteins which increase gene expression by binding to
the nucleic acid sequence) in the region of the
expression control sequence of a target gene in
particular in its regulatory regions, does not reduce
the expression of the target gene but in contrast it is
possible by selection of suitable culture conditions to
increase the expression of the endogenous target gene or
to induce the expression of a non-expressed endogenous
target gene.
CA 02252970 1998-11-30
- 18 -
Examples of suitable activator proteins are the hypoxia-
inducible factors HIF-1a and HIF-10 as well as the
interferon regulated factor 1 (IRF-1) which can increase
transcription by binding to the interferon consensus
sequence (ICE) (Tanaka N., Kawakami T., Taniguchi T.,
Mol. Cell. Biol. (1993), Aug; 13(8): 4531-4538).
After operatively linking one or several nucleic acid
sequences, that bind a HIF or other activator proteins,
to a target gene that is present endogenously, the
expression of the target gene can be regulated by
selecting suitable culture conditions. An advantage of
this, especially for a commercial scale production, is
that the expression of a protein can be induced at an
optimal time for the production process. This is
beneficial because the average residence time of the
synthesis product in the culture medium supernatant is
reduced. This also reduces the amount of undesired
degradation products of the protein. This has a positive
effect on the subsequent purification steps, reduces the
production costs and leads to a qualitatively improved
final product.
In order to carry out the process according to the
invention it is sufficient to operatively link one or
several activator-binding nucleic acid sequences with
the target gene. Preferably two HIF-binding nucleic acid
sequences are used. The HIF-binding nucleic acid
sequence is particularly preferably selected from the 53
bp sequence according to sequence ID NO.1, the 43 bp
sequence according to sequence ID NO.2, a sequence
homologous to these sequences or a sequence hybridizing
with these sequences under stringent conditions.
CA 02252970 1998-11-30
- 19 -
The use of two HIF-binding nucleic acid sequences
surprisingly leads to a synergistic effect. This leads
to a greater increase in the expression of endogenous
nucleic acids than when using each of these sequences
alone.
If necessary the expression of the activator protein
which binds to the activator sequences introduced in the
region of the target gene can be induced or/and
increased in the cell. This can for example be achieved
by transfecting the cell with a vector comprising
(i) a nucleic acid sequence coding for an activator
protein which is operatively linked with an active
expression control sequence in this cell and
(ii) optionally a positive selection marker gene.
Any nucleic acid sequence coding for an activator
protein can be used whose expression product can bind to
the activator-binding nucleic acid sequence integrated
into the genome. The activator protein is preferably a
HIF-la or/and HIF-1(3 protein. If the nucleic acid
sequence that is present endogenously already contains
activator-binding or preferably HIF-binding nucleic acid
sequences it may be sufficient to merely introduce a
vector into the cell containing a nucleic acid sequence
coding for an activator protein or preferably for a HIF
protein which is operatively linked with an active
expression control sequence in the cell and optionally a
positive selection marker gene.
The expression control sequence which is linked
operatively with the nucleic acid sequence coding for
the activator protein can be inducible which provides an
additional method for activation by suitable culture
CA 02252970 1998-11-30
- 20 -
conditions such as for example by addition of hormones
or heavy metals. This enables the expression of an
endogenous target gene to be induced at an optimal time
for the production process.
An advantage of using a constitutively active expression
control sequence is that the activator protein is
expressed constitutively independent of the addition of
activators into the culture medium.
If the activator protein-binding nucleic acid sequence
is a HIF-binding nucleic acid sequence, the expression
of the target gene can for example be induced by
suitable culture conditions e.g. at an 02 concentration
of 0.1 - 2 %.
A further subject matter of the present invention is a
vector for homologous recombination comprising
(i) at least one nucleic acid sequence which binds an
activator protein,
(ii) a positive selection marker gene,
(iii) DNA sequences flanking the sequences (i) and (ii)
which are homologous to a nucleic acid section in
the genome of the cell in order to allow a
homologous recombination.
Yet a further subject matter of the present invention is
a eukaryotic cell, preferably a human cell, which is
obtainable by one of the processes described above. This
cell is preferably characterized in that it contains at
least one heterologous, chromosomally located, activator
protein/complex-binding nucleic acid fragment
operatively linked with a gene that is present
endogenously in the cell. Activator protein-binding
CA 02252970 1998-11-30
- 21 -
nucleic acid fragments can be substituted in the genome
with the aid of a site-specific recombination system as
elucidated above which enables a simple identification
of an optimal activator sequence for a certain target
gene.
A further aspect of the present invention concerns a
process for testing the influence on its expression of non-
coding nucleic acid sequences from the region of a target
gene present endogenously in a eukaryotic cell which is
characterized in that
(a) the cell is transfected with a vector comprising
(i) a heterologous expression control sequence
that is active or can be activated in the cell
which is operatively linked with a reporter
gene and
(ii) non-coding nucleic acid fragments on the 5' side
or/and 3' side from the region of the target
gene,
(b) the cell is cultured under conditions under which the
expression control sequence is active and
(c) the expression of the reporter gene is measured.
It can be simply determined with the process according
to the invention how a heterologous expression control
sequence has to be placed in the region of the target
gene in the genome in order to achieve an optimal
expression rate of the target gene and what influence
the presence or the absence of 5' or/and 3' non-coding
sequences from the region of the target gene has on the
expression. The test vectors are preferably transiently
transfected into cells and the expression of the
reporter gene is determined. In this manner it is
possible to rapidly and cheaply test many arrangements
of a heterologous expression control sequence and a
CA 02252970 1998-11-30
- 22 -
target gene or many different expression control
sequences. The heterologous expression control sequences
include nucleic acid sequences which can directly
interact with transcription components such as
transcription initiation factors or RNA polymerases and
nucleic acid sequences whose influence on transcription
is mediated by interactions with activators or
repressors. The heterologous expression control sequence
is preferably a promoter/enhancer, particularly
preferably a viral promoter and most preferably a CMV
promoter. The process according to the invention
contributes to a strong cost reduction especially in
processes which contain further complicated process
steps. This is for example the case in the production of
transgenic animals such as mice, sheep or cows in which
it is intended to increase the expression of a
particular endogenous nucleic acid sequence in a certain
cell type.
The non-coding nucleic acid fragment 5' or 3' from the
target gene region is preferably arranged in the vector
according to it's genomic arrangement on the 5' side or
3' side of the reporter gene.
Any reporter gene known to a person skilled in the art
whose expression can be detected in the cell can be used.
A reporter gene is preferably used which codes for
chloroamphenicol-acetyl-transferase (CAT), 3-galactosidase
(13-Gal) or lacZ. On the other hand it is also possible to
use a reporter gene coding for a protein of interest e.g.
EPO, whose expression can be detected by immunological
methods e.g. ELISA.
In a preferred embodiment at least two vectors which
CA 02252970 1998-11-30
- 23 -
contain different 5' or/and 3' non-coding nucleic acid
fragments of the target gene are each transfected into
different cells and the expression of the reporter gene
in the different cells is determined with methods known
to a person skilled in the art. It can be easily
established with the process according to the invention
which arrangement of the heterologous expression control
sequence results in an optimal expression for a certain
host cell.
A further aspect of the invention concerns a process for
providing a DHFR-negative eukaryotic cell preferably a
mammalian cell and particularly preferably a human cell
which is characterized in that
(a) the cell is transfected with a first vector comprising
(i) at least one target sequence for a site-specific
recombinase,
(ii) DNA sequences flanking sequence (i) which are
homologous to a DHFR nucleic acid sequence that
is present endogenously in the cell in order to
allow a homologous recombination and
(iii) optionally a positive selection marker gene and
optionally a negative selection marker gene,
(b) the transfected cell is cultured under conditions under
which a homologous recombination of the vector takes
place and
(c) the cell obtained according to step (b) is isolated.
In the process according to the invention the
recombinase target sequences and the homologous
sequences are selected and used as explained above.
The positive selection marker gene - if present - is
arranged between the sequences that are homologous to a
CA 02252970 1998-11-30
- 24 -
DHFR gene. The negative selection marker gene - if
present - is arranged outside the homologous sequences.
After homologous recombination has taken place in the
DHFR locus, no functional DHFR protein can be
synthesized by the cell. In this case the sequences of
the vector can be arranged such that the promoter of the
DHFR gene is inactivated or/and such that a functional
DHFR protein can no longer be synthesized due to an
insertion or deletion in the coding sequence of the DHFR
gene.
In order to inactivate both alleles of a DHFR gene, the
cells are firstly transfected with a vector according to
the invention, then selected and isolated. One allele of
the DHFR gene is inactivated in these cells i.e. they
are heterozygous (+/-) for the DHFR gene. Then these
cells can be again transfected with a vector according
to the invention which preferably contains a positive
selection marker gene that is different from the first
vector. After a selection step cells are obtained in
which both DHFR alleles are inactivated. Alternatively
an increase of the selection pressure can lead to a gene
conversion and thus to an inactivation of both alleles
(cf. e.g. Mortensen et al., Mol. Cell. Biol. 12 (1992),
2391-2395).
The process according to the invention provides a DHFR
negative cell whose use in a gene amplification process
has the advantage that it does not synthesise an
endogenous DHFR protein. When a selection step is
carried out to amplify a heterologous nucleic acid
sequence which is coupled to a nucleic acid sequence
coding for a DHFR protein, the expression product of the
CA 02252970 1998-11-30
- 25 -
endogenous DHFR gene does not have an interfering
influence and thus there is an increase in the
efficiency of the gene amplification.
Any suitable selection marker gene that leads to a
selectable phenotype can be used as a positive selection
marker gene e.g. antibiotic resistance. The nucleic acid
sequence coding for the positive selection marker gene
is preferably a neomycin, kanamycin, geneticin or
hygromycin resistance gene.
Any negative selection marker gene known to a person
skilled in the art can be used, the nucleic acid
sequence coding for the negative selection marker gene
is preferably a thymidine kinase gene (TK) or/and
hypoxanthine-guanine-phosphoribosyl transferase gene
(HGPRT).
The sequence flanked by recombinase target sequences can
be cleaved out of the'genome of the cell by transient
activation of the corresponding recombinase e.g. by
(a) transfecting the cell with a vector containing a
nucleic acid sequence coding for a recombinase
operatively linked with an expression control
sequence that is active in this cell,
(b) culturing the cell transfected in this manner under
conditions under which the recombinase is expressed
and is active and
(c) optionally isolating the cell.
It is not only possible with the process according to
the invention to inactivate a DHFR gene but also to cut
out sequences of a DHFR gene which are located between
the recombinase target gene sequences as well as the
CA 02252970 1998-11-30
26 -
introduced selection marker gene from the genome of a
cell by a recombinase-mediated reaction.
If the sequence flanked by recombinase target sequences
contains a positive selection marker gene, the cell
containing this sequence is antibiotic resistant. Hence
it can be easily selected by methods known to a person
skilled in the art.
A further advantage of the DHFR-negative cell produced
by the process according to the invention is that its
properties can be characterized by methods known to a
person skilled in the art and the cells can be
subsequently used for other processes. Moreover the
recombinase target sequence introduced at the DHFR gene
locus enables the site-specific integration of nucleic
acid sequences into the genome.
A further preferred embodiment concerns a process for
introducing a heterologous DHFR gene into a eukaryotic
cell which is characterized in that a DHFR-negative cell
obtained by one of the processes described above
(a) is transfected with a third vector comprising
(i) optionally a positive selection marker gene
which preferably differs from the'positive
selection marker gene of the first vector,
(ii) a nucleic acid sequence coding for a DHFR,
(iii) a nucleic acid sequence to be amplified coding
for a protein in an expressible form in which
each of the nucleic acid sequences from the
partial sequences (i), (ii) and (iii) is
flanked on the 5' side and 3' side by at least
one recombinase target sequence
(b) the transfected cell is cultured under conditions
CA 02252970 1998-11-30
- 27 -
under which the nucleic acid sequence flanked by
recombinase target sequences is integrated into the
recombinase target sequence that is already present
in the genome of the cell and
(c) the cell obtained according to step (b) is isolated.
The positive selection marker gene, the DHFR gene and
the target gene coding for the desired protein are
preferably each operatively linked with an expression
control sequence that is active or can be activated in
the cell. A polycistronic construct with internal
ribosomal binding sites is also in principle possible.
The nucleic acid sequence to be amplified of the target
gene should, however, be driven by a separate promoter.
Particularly preferred expression control sequences are
viral promoters/enhancers. A CMV promoter is most
preferred for the expression of the protein.
It is advantageous to carry out the integration
according to the invention of heterologous sequences
into the genome of a cell in a site-specific manner and
thus exclude interferences of the heterologous sequences
with genomic sequences. Hence this avoids the resulting
disadvantages as described further above such as
unstable production clones.
In order to increase the expression rate of a
heterologous nucleic acid sequence coding for a protein,
it is possible to carry out an amplification step with
methotrexate by known process steps.
A further subject matter of the present invention is a
vector comprising
(i) optionally a positive selection marker gene,
CA 02252970 1998-11-30
- 28 -
(ii) a nucleic acid sequence coding for a DHFR and
(iii) a nucleic acid sequence in an expressible form
coding for a desired protein in which each nucleic
acid sequence from the partial sequences (i), (ii)
and (iii) is flanked on the 5' side and 3' side by
at least one recombinase target sequence.
Yet a further subject matter of the present invention is
a vector for homologous recombination comprising
(i) optionally a positive selection marker gene,
(ii) at least one recombinase target sequence in each
case which flanks the sequence (i) and
(iii) DNA sequences flanking the sequences (i) and (ii)
which are homologous to a DHFR nucleic acid
sequence that is present endogenously in a cell in
order to allow a homologous recombination and
(iv) optionally a negative selection marker gene
outside and preferably on the 3' side of the
homologous sequences (iii).
In addition the invention concerns a eukaryotic cell,
preferably a human cell, obtainable by one of the
processes described above. This cell is characterized in
that
(a) at least one endogenous nucleic acid sequence coding
for a DHFR is inactivated and preferably both
endogenous alleles and
(b) at least one recombinase target sequence is
integrated into the genome in the region of this
nucleic acid sequence coding for DHFR.
Finally, yet a further subject matter of the invention
is a eukaryotic cell, preferably a human cell, which is
characterized by a heterologous nucleic acid sequence in
CA 02252970 1998-11-30
- 29 -
the region of an endogenous DHFR gene locus comprising
(i) a nucleic acid sequence coding for DHFR,
(ii) a nucleic acid sequence coding for a desired
protein and
(iii) at least one recombinase target sequence.
The invention is illustrated by the following examples,
Figures and the sequence protocol.
Figure legends
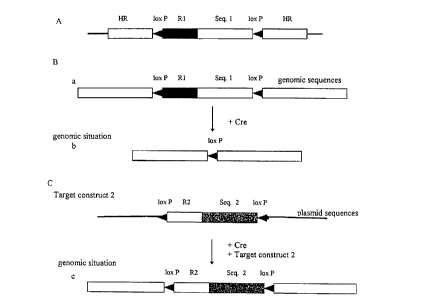
Figure 1
(A) shows a vector for the homologous recombination
which is used as the first vector. HR: homologous
sequence, Seq 1: first heterologous expression
control sequence, R1: positive selection marker
gene, loxP : loxP sequence with orientation,
(B) shows genomic sequences
(a) after completion of the homologous recombination,
(b) after excision of a sequence flanked by loxP
sequences catalysed by a Cre recombinase,
C) shows a vector for a Cre recombinase mediated
integration which contains a sequence arranged between
the loxP sequences
(c) shows genomic sequences after integration of a
second vector at the loxP sequence, R2: positive
selection marker gene which optionally differs from
R1, Seq 2: second heterologous expression control
sequence.
CA 02252970 1998-11-30
- 30 -
Figure 2
(A) shows a vector for the homologous recombination HR:
homologous sequence, R-box: positive and optionally
negative selection marker gene, loxP : loxP sequence
with orientation, HSV-tk: Herpes simplex thymidine
kinase;
(B) shows a vector for the homologous recombination with
one-sided homologous sequence.
Figure 3
shows the CMV promoter/HIF-controlled erythropoietin
(EPO) expression of HeLa S3 cells which were transfected
with the vectors pHYG, pHIF-la and pARNT (pHIF-13) and
whose EPO expression was measured in the cell
supernatants 3, 4 and 5 days after the transfection.
(erythropoietin concentration in gg/ml).
pHYG: control vector, pHIF-la: a HIF-la cDNA under the
control of an SRa promoter, pARNT: a HIF-13 cDNA under
the control of a CMV promoter.
Figure 4
shows 4 different vectors which each contain a CMV
promoter (C) and the reporter gene (3-galactosidase (B)
in which non-coding nucleic acid fragments of the target
gene (S) of different lengths have been inserted between
these sequences. The length of the non-coding nucleic
acid fragments is Okb in the vector A3-178, 2.5 kb in
the vector A3-177, 3.7 kb in the vector A3-175 and
5.7 kb in the vector A3-181. The control vector pNASS(3
CA 02252970 1998-11-30
- 31 -
contains the reporter gene P-galactosidase without a CMV
promoter.
Figure 5
shows a measurement of the expression of the reporter
gene j3-galactosidase after transfection of HeLa S3 cells
with the vectors of Figure 4 in a dilution series (1:2
to 1:128).
Figure 6
(A) shows the vector pNDI for homologous recombination
in a DHFR gene locus. A positive selection marker
gene (Neo) is flanked by two loxP sequences. The
sequences that are homologous to a DHFR gene (5',
3' DHFR region) are located on the 5' side of one
of the loxP sequences and on the 3' side of the
other loxP sequence.
(B) shows the vector pHDI for homologous recombination
in a DHFR gene locus. A positive selection marker
gene (Hyg) is flanked by two loxP sequences. The
sequences that are homologous to a DHFR gene (51,
3' DHFR region) are located on the 5' side of one
of the loxP sequences and on the 3' side of the
other loxP sequence.
Figure 7
(A) shows the genomic construction of a DHFR gene with
exon 1, exon 2 and exon 3 as well as the introns
that are located between them,
(B) shows a diagram of a target construct corresponding
CA 02252970 1998-11-30
- 32 -
to the vector of Figure 6,
(C) shows the genomic structure after completed
homologous recombination of the vector for the
homologous recombination in a DHFR gene. The
distance between the EcoRI cleavage sites is 2.9 kb
when using the vector pNDI and 3.7 kb when using
the vector pHDI. Neo: neomycin, Hyg: hygromycin,
kb: kilobases.
Figure 8
shows a vector which contains a nucleic acid sequence
coding for a protein X and a nucleic acid sequence
coding for a DHFR protein which each include regulatory
sequences and are flanked by two loxP sequences. This
vector can be used for Cre recombinase catalysed
integration into the genome in a loxP sequence.
SEQ ID NO.1 shows a first HIF-binding nucleotide sequence,
SEQ ID NO.2 shows a second HIF-binding nucleotide sequence
SEQ ID NO.3 shows a loxP sequence
EXAMPLES
Example 1 Expression of an erythopoietin gene under the
control of a CMV promoter and overexpression
of HIF
The vectors pHYG, pHIF-la and pARNT (cf Figure 3) are
transfected into genetically modified HeLa S3 cells. A
CMV (cytomegalovirus) promoter which controls the EPO
expression was introduced into the cells proximally to
the erythropoietin gene (EPO) translation start of an
CA 02252970 1998-11-30
- 33 -
EPO allele. The cells usually produce 1 pg
erythropoietin per 24 hours per 107 cells. 24 hours
before transfection they are passaged at a concentration
of 6 x 104 cells per 6 well plate. On the day of
transfection the cells are incubated with a DNA-DOTAP
mixture. The mixture contains 1.25 pg of the respective
vector, 10 Al DOTAP (Boehringer Mannheim 1202375) in a
final volume of 75 Al in 20 mM Hepes buffer per well.
The mixture is pre-incubated for 10-15 minutes at room
temperature. The cells are then incubated for 6 hours
with the DNA-DOTAP in 3 ml medium per well. Subsequently
the cells are washed twice with PBS buffer and cultured
in complete medium for 5 days. On day 3, 4 and 5 100 Al
supernatant was removed each time and analysed with an
erythropoietin ELISA. The assay is completed on day 5
and the cell count is determined. The amount of
erythropoietin per well is calculated relative to the
same cell count (cf. Figure 3).
The example shows that an induction of the
erythropoietin gene by HIF is still possible although a
heterologous expression control sequence (CMV promoter)
has been introduced into the promoter region of an
allele of the erythropoietin gene. The measured increase
in the erythropoitin concentration indicates a
synergistic effect of the hypoxia-induced factor or the
hypoxia-induced factors on both alleles.
It therefore becomes evident that the expression of an
endogenous nucleic acid sequence can be increased by
introducing a heterologous expression control sequence.
If an activator (HIF) is expressed in the cell for which
binding nucleic acid sequences are present in the
expression control sequence, then the expression of this
gene can be further increased. If corresponding
CA 02252970 1998-11-30
- 34 -
sequences are not present in this gene locus, they can
be specifically introduced into the genome by the
process according to the invention by means of
homologous recombination.
Example 2 Optimized arrangement of an expression control
sequence for increasing the expression of an
endogenous nucleic acid sequence
Sequences on the 5' side of an endogenous gene can
stimulate the expression as well as have repressing
properties. When a heterologous expression control
sequence is introduced into the genome on the 5' side of
a target gene, the expression level is influenced by the
endogenous 5' sequence. In order to achieve an optimal
expression of the target gene by means of a heterologous
expression control sequence, this must be arranged such
that the activity of the heterologous expression control
sequence is not reduced by non-coding sequences on the
5' side of the target gene. A specific arrangement would
be advantageous in order to achieve synergistic effects
of the individual sequence elements. In order to test
various arrangements of the heterologous expression
control sequence i.e. in order to for example determine
at which distance from the translation start of the
coding sequence of the target gene the heterologous
expression control sequence has to be integrated into
the genome of the cell, different vectors with different
5' non-coding nucleic acid fragments of the target gene
are tested (cf. Figure 4). The vectors described in
Figure 4 are transfected into HeLa S3 cells and the
expression of the reporter gene (3-galactosidase is
measured (cf Figure 5).
CA 02252970 1998-11-30
- 35 -
24 hours before the assay the cells are passaged at a
concentration of 1 x 106 cells per 10 centimetres petri
dish. On the day of transfection the cells are incubated
with a DNA-DOTAP mixture. The mixture contains 1 pmol of
the respective vector (A3-178, A3-177, A3-175, A3-181 or
pNASSf3, cf. Figure 4) in 60 gl DOTAP (Boehringer
Mannheim 1202375) made up to 300 Al with a 20 mM HEPES
buffer solution. The mixture is incubated for 10-15
minutes at room temperature. The cells are pre-incubated
for 6 hours with DNA-DOTAP in 6 ml serum-free medium per
petri dish. Afterwards the cells are washed twice with
PBS buffer and cultured in complete medium for 22 hours.
In order to measure the 0-galactosidase expression, the
cells are isolated in 200 Al PBS and lysed by freezing
at -20 C and thawing. 10 gl of the lysate is diluted
1:10 with substrate (3.29 mM chlorophenol red-(3-D
galactopyranoside (Boehringer Mannheim 884308), 100 mM
HEPES, 150 mM NaCl, 2 MM MgCl2, 1 % BSA, 0.1 % Triton-X
100, 1 % sodium azide, pH 7. The samples are diluted in
1:2 steps and incubated at 37 C in a 96 well plate until
a dark-red colour has formed. The samples are then
measured at 570/580 nm, or 550 nm.
As shown in Figure 5 the expression of the reporter gene
is highest in cells that have been transfected with the
vector A3-178. In this vector the heterologous
expression control sequence is proximal to the
translation start of the coding sequence.
Hence this method can be used to simply and rapidly
determine which arrangement of a heterologous expression
control sequence in the genome of a host cell has to be
selected in order to achieve an optimal expression of an
endogenous target gene.
CA 02252970 1998-11-30
- 36 -
Example 3 Production of DHFR-negative cells
In a first step vectors for recombination according to
the invention are prepared. These vectors are
transfected into human cell lines in a second step and
screened for homologous recombination events. In this
manner firstly one and then the second allele for the
DHFR gene can be inactivated.
DHFR vector for homologous recombination
The human DHFR gene is located on chromosome 5 and
comprises 30 kb which are arranged in 6 exons. A 1.8 kb
EcoRI fragment which contains parts of the promoter,
parts of exon 2 and the complete exon 1 is used to
prepare the vector for homologous recombination. Exon 1
is removed by an AapI digestion and the Neo (1.4 kb) or
Hyg (2.2 kb) resistance gene is inserted into the
resulting gap (0.45 kb) via linkers. These linkers
contain the minimal sequence TAT TG AAG CAT ATT ACA TAC
GAT ATG CTT CAA TA (loxP sequence) in addition to the
adaptor nucleotides. The linker sequences are arranged
in the same orientation and the resistance gene is
preferably antisense relative to the DHFR gene. After
the resistance gene has been inserted, the homology
region is enlarged. For this the vector is extended by
the EcoRI fragments from the 3' region (6.0 kb) (Fig.
6). In this manner one obtains the target constructs
pNDI (11.5 kb) and pHDI (12.3 kb) according to the
invention.
After completed homologous recombination the complete
exon 1 (amino acids 1-28) and parts from the promoter of
the DHFR gene have been removed. The cell can now no
CA 02252970 2008-06-17
- 37 -
longer express a functional DHFR protein.
Transfection of cells
The human cell lines used should not be polyploid for
chromosome 5 and should not have been kept under MTX
selection. In both cases more than 2 alleles would have
had to been inactivated.
HeLa S3 cells (ATCC CCL-2.2)
The cells are cultured in tissue culture flasks in RPMI
1640 medium, 10 % foetal calf serum, 2 mM L-glutamine
and 1 mM MEM (non-essential amino acids). The incubation
is carried out at 37 C and 5 % CO2. The electroporation
buffer contains 20 mM Hepes, 138 mM NaCl, 5 mM KC1,
0.7 mm Na2HPO4 6 mM D-glucose monohydrate, pH 7Ø 10 gg
linearized vector DNA (pNDI) is electroporated (Biorad
Gene Pulser) into 1 x 107 cells at 960 F and 250V.
After electroporation the cells are taken up in medium
containing 600 Ag/ml G418 (geneticin Boehringer
Mannheim) and cultured. After 10 days selection (medium
changed every 2 days) the positive clones are isolated
and expanded.
HT1080 cells (ATCC CCL-121)
The cells are cultured and selected as described for
HeLa S3 cells using DMEM medium containing 10 % foetal
calf serum, 2 mM L-glutamine and 1 mM sodium pyruvate.
* Trade-mark
CA 02252970 1998-11-30
- 38 -
Namalwa cells (ATCC CRL-1432)
This cell line is a suspension cell line and must be
cultured correspondingly. The medium corresponds to that
described for Hela S3 cells. After transfection the
cells are distributed among forty 96-well plates.
Positive clones are expanded in 48- 24- 12- and 6-well
plates. The selection is carried out with 1 mg/ml G418.
Detection of the DHFR-negative (+/-) cells
Insertion of the vector is detected by means of Southern
blot analysis or by PCR. If homologous recombination has
occurred correctly, a 2.9 kb band is detected after
EcoRl digestion which has been formed by the insertion
of the Neo gene in addition to a 1.8 kb band which
represents the intact DHFR gene (Figure 7c). Mixed
clones (unequal ratio of the band intensities in the
Southern blot) are separated by single cell deposition
in a FACS, subcloned and subsequently expanded. One
allele of the DHFR gene is inactivated in the clones
that have been identified as positive.
Production of DHFR-negative (-/-) cells
Cell clones in which a DHFR allele (+/-) is inactivated
can be subjected to a renewed homologous recombination.
For this they are transfected as described above with
gg linearized DNA of the vector pHDI. The selection is
carried out in medium containing 500 g/ml hygromycin B
(Boehringer Mannheim).
Increasing the G418 concentration in the medium
CA 02252970 1998-11-30
- 39 -
increases the selection pressure on DHFR+/- cells and
DHFR-/- cells are obtained. A genetic conversion leads
to an interchromosomal recombination which is why the
second DHFR allele is inactivated.
The DHFR-/- cells contain two inactivated DHFR alleles
and can no longer synthesize tetrahydrofolate. Therefore
thymidine, glycine and purine have to be added to the
medium (supplementation). Optionally the cells are
cultured in a' medium (Gibco BRL).
The DHFR-/- cells are detected as described above. In
homozygous DHFR-negative cells no wild-type band
(1.8 kb) is detectable. Cells that have been transfected
with pDHI exhibit a new 3.7 kb band in EcoRI Southern
blot after homologous recombination (Figure 7c).
Use of DHFR-negative cells (-/-)
The cells according to the invention can be used for the
large-scale production of proteins. For this a vector
according to the invention (according to Figure 8) and
an expression vector coding for a Cre recombinase are
transfected into the DHFR-/- cells. The Cre recombinase
removes the antibiotic resistance from the DHFR gene
locus and integrates the vector according to the
invention into the loxP sequence in the genome of the
DHFR-/- cell. The cells are again antibiotic sensitive
and independent of a thymidine, glycine and purine
supplementation.
The selection can be achieved by using a medium without
supplementation or by adding a suitable antibiotic to
the culture medium. In this case the antibiotic
CA 02252970 1998-11-30
- 40 -
corresponds to the resistance gene which has been
removed by the Cre recombinase from the genome of the
cell. If the vector integrated into the loxP sequence
contains a positive selection marker gene, the selection
can be carried out by adding this antibiotic to the
medium.
Increasing the production output by gene amplification
In order to increase the production output of the cells
for the recombinant protein, a methotrexate (MTX)
selection is carried out which amplifies the DHFR gene
introduced into the cell and the heterologous nucleic
acid sequence coding for a protein.
In order to achieve an amplification the cells are
cultured in the presence of increasing concentrations
(100-1000 mM) MTX. The degree of amplification is
monitored by densitometric evaluation of comparative
Southern blot (before, during and after MTX addition).
The cells according to the invention obtained after the
amplification step contain many copies of the introduced
DHFR gene and of the inserted heterologous nucleic acid
sequence at the loxP locus. They are characterized by a
high production output of the heterologous nucleic acid.
CA 02252970 1999-02-26
40a
SEQUENCE LISTING
(1) GENERAL INFORMATION:
(i) APPLICANT:
(A) NAME: Boehringer Mannheim GmbH
(B) STREET: Sandhofer Strasse 112-132
(C) CITY: Mannheim
(E) COUNTRY: DE
(F) POSTAL CODE (ZIP): D-68305
(ii) TITLE OF INVENTION: Optimization of cells for endogenous gene
activaton
(iii) NUMBER OF SEQUENCES: 3
(iv) CORRESPONDENCE ADDRESS:
(A) ADDRESSEE: SWABEY OGILVY RENAULT
(B) STREET: 1981 McGill College, Suite 1600
(C) CITY: Montreal
(D) STATE: Quebec
(E) COUNTRY: Canada
(F) ZIP: H3A 2Y3
(v) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
(C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: Patentln Release #1.0, Version #1.30 (EPO)
(vi) CURRENT APPLICATION DATA:
(A) APPLICATION NUMBER: 2,252,970
(B) FILING DATE: November 30, 1998
(C) CLASSIFICATION
(vi) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: 97 121 075.2
(B) FILING DATE: 1, DEC. 1997
(vii) ATTORNEY/AGENT INFORMATION:
(A) NAME: Kevin P. Murphy
(B) REGISTRATION NUMBER: 3302
(C) REFERENCE/DOCKET NUMBER: 4659-366 KPM/CC/LM
(viii) TELECOMMUNICATION INFORMATION:
(A) TELEPHONE: (514) 845-7126
(B) TELEFAX: (514) 288-8389
(2) INFORMATION FOR SEQ ID NO: 1:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 53 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: both
CA 02252970 1999-02-26
40b
(D) TOPOLOGY: linear
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 1:
CCTCTCCTCT AGGCCCGTGG GGCTGGCCCT GCACCGCCGA GCTTCCCGGG ATG 53
(2) INFORMATION FOR SEQ ID NO: 2:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 43 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: both
(D) TOPOLOGY: linear
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 2:
CTACGTGCTG TCTCACACAG CCTGTCTGAC CTCTCGACCC TAC 43
(2) INFORMATION FOR SEQ ID NO: 3:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 34 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: both
(D) TOPOLOGY: linear
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 3:
TATTGAAGCA TATTACATAC GATATGCTTC AATA 34