Note: Descriptions are shown in the official language in which they were submitted.
CA 02255615 2003-12-24
COMPOSITIONS COMPRISING CONJUGATES OF CIS-DOCOSAHEXAENOIC ACID AND
DOCETAXEL
Background of the Invention
Taxol~ (paclitaxel) was first isolated in 1971 from the bark of Taxus
brevifolia and was
approved in 1992 by the US Food and Drug Administration for treatment of
metastasic ovarian cancer
and later for breast cancer. Its mechanism of action is believed to involve
promoting formation and
h~rstabilization of microtubules, thereby preventing the disassembly of
microtubules necessary for
completion of cell division. It also has been reported that Taxol induces
expression of cytokines,
affects the activity of kinases and blocks processes essential for metastasis,
in as yet uncharacterized
mechanisms of action.
Taxol has attracted unusually strong scientific attention, not only because of
its unique
antiproliferative mechanism of action, but also because it is active against
nearly all cancers against
which it has been tested and because it has been discovered to be an analog of
numerous closely
related compounds occurring naturally. These compounds, taxanes, are now
recognized as a new
class of anticancer compounds.
Taxol's strength against cancers of diverse tissue origin also represents a
significant drawback.
An ideal anticancer agent has tissue specificity, thereby reducing side-
effects on normal (dividing)
cells. Taxo1 analogs with tissue specificity therefore are desired. Another
drawback of Taxol is its
extreme insolubility. Taxol can be administered effectively in a solvent
including cremophorTM,
which combination can provoke severe hypersensitive immune responses. As a
result of these
drawbacks, and also as a result of the potential for modifying Taxol at
numerous sites as
demonstrated by other naturally-occurring taxanes with anticancer activity, a
search for more
selective taxanes was launched.
To date, more than 200 taxanes have been synthesized (or isolated) and tested
in vitro or in vivo
for anticancer activity. The results, however, have been so disappointing that
the National Cancer
Institute (NCn generally no longer is interested in testing Taxol analogs. In
general with Taxol
analogs, the solubility problems remain, and/or potency is sharply reduced,
and/or selectivity is not
improved, and/or the ratio of the median toxic dose to the median effective
dose ("therapeutic index")
is unacceptably reduced.
CA 02255615 1998-11-18
WO 97/44026 PCT/US97/08866
-2-
Taxol has the following formula:
O Ac0 O OH
P' N H O -~ B C
Ph~O'~~ ~ _ H - pOH
OH HO
PhC02 OAc
Taxanes have the basic three ring structure (A, B and C), substituted or
unsubstituted.
Taxol's carbons are numbered conventionally as follows:
18 10 9 19
11
12 i 1 $ 8
A, 16 9 C 5
13 17
4
1a 1 2 H
Based upon the taxanes tested to date, as many questions have been raised as
leave been
answered, and general rules have not been fashioned easily in predicting
selectivity, activity and
solubility. Firstly, no rules have emerged regarding selectivity. Those
taxanes that are strongly
i 5 active appear to have activity as broad as Taxol's activity, and no
headway appears to have been
made in terms of developing a more selective Taxol analog.
Some information about activity has emerged. Numerous substitutions have been
made at
C7, C9, C 10, C 19, R, and combinations thereof while retaining significant,
but usually reduced,
activity. Substitutions at C2, C4 and 2'OI-I, however, are generally not
tolerated. These conclusions
2o are only generalities, for example, because some substitutions at C9-C 10
(cyclic derivatives) are not
tolerated and some substitutions at C2 (meta substitutions on the phenyl) are
tolerated. Likewise,
the C 13 side chain and, in particular, the 2'OH are required, although the
minimum structural
requirements of the side chain have not been calculated for therapeutic
efficacy.
Attempts to improve Taxol's solubility have not resulted in successful
clinical products. One
approach has been to manufacture prodmgs of Taxol, which prodrugs undergo in
vivo transformation
into Taxol and some other product. Attempts were made to esterify the C7
hydroxy and 2' hydroxy
groups, with the hope that the bond would be stable in solution (to permit
preferred administration
modes -i.v. over at least 24 hours) but would cleave readily in vivo. The
groups tested were all
hydrophilic and included amines, short carboxylic acids (using e.g. succinic
anhydride and glutaric
anhydride), sulfonic acids, amino acids and phosphates. Generally, activity
was reduced although
some success was obtained with certain derivatives. Again, no particular
pattern emerged permitting
one to predict reliably which groups could be substituted on Taxol to yield a
therapeutically useful
CA 02255615 2003-12-24
-3-
product, although it was suggested that the 2' OH derivatives may cleave more
easily than the C7 OH
derivatives.
Several other factors add to the problem of predicting which Taxol analogs
will be effective.
Multiple mechanisms of action have been proposed in the literature, and a
change in one position may
have no effect on activity on one such mechanism but may eliminate activity on
another mechanism.
In addition, changes that favorably influence activity may unfavorably
influence bioavailability. For
example, Taxol affects microtubule formation inside a cell, but a change in
structure that increases
intracellular activity may adversely affect the ability of Taxol to gain entty
into a cell. Taxol also is
known to bind to proteins, and the effect on activity that results from a
change in Taxol's binding to
protein (in terms of conformation, cellular absorption and solubility) is
unknown.
It has been reported that Taxol does not get into the brain, apparently
excluded by the blood
brain burner. It is not known why this is so, as Taxol is lipophilic, gets
into cells and might be
expected to cross the blood brain barrier.
The most promising of the two hundred analogs tested is TaxotereTM
(docetaxel), because of
its slightly increased activity and solubility. Oddly, however, TaxotereTM
differs from Taxol at sites
which typically do not have a strong influence on activity, and one would not
predict the
improvements in TaxotereTM from these differences, even in hindsight.
TaxotereTM has the following formula:
O " '
~O'~NH O
~ ~ ",. ,
Ph~O'~ :1-I
OH H~ O OAc
O~Ph
DHA (docosahexaenoic acid) is a 22 carbon naturally-occurring, unbranched
fatty acid that
previously has been attached to drugs to help deliver them across the blood
brain barrier. DHA is
attached via the acid group to hydrophilic drugs and renders these drugs more
hydrophobic
(lipophilic). DHA is an important constituent of the brain and recently has
been approved as an
additive to infant formula. It is present in the milk of lactating women. The
mechanism of action by
which DHA helps drugs conjugated to it cross the blood brain barrier is
unknown.
CA 02255615 2003-12-24
-q,_
Summary of the Invention
The present invention involves the unexpected finding that conjugates of
TaxotereTM and a
highly lipophilic group, a C22 unbranched carbon chain, have a different
selectivity relative to
TaxotereTM. The conjugates, in general, are believed to render the activity of
the taxanes selective for
colon cancer, breast cancer, and central nervous system cancer ("targeted
cancers"). The conjugates,
also unexpectedly, restrict the activity of the taxanes even within these
three categories of cancer
relative to that of TaxotereTM. The conjugates further unexpectedly, reduce
sharply the activity of the
taxanes relative to that of TaxotereTM in most cell lines of tissue types
other than colon, breast, and
central nervous system, thereby reducing potential side effects of the
conjugates versus those of
TaxotereTM. The therapeutic index of the conjugates is improved, versus that
of TaxotereTM for
targeted cancers.
According to one aspect of the invention, a composition of matter is provided.
The
composition is a covalent conjugate of cis-docosahexaenoic acid and
TaxotereTM. Preferably, the
conjugate consists of cis-docosahexaenoic acid and TaxotereTM, wherein the cis-
docosahexaenoic
acid is conjugated directly to Taxotere, free of a linker, for example via the
carboxylic acid group of
the cis- docosahexaenoic acid and a hydroxyl group of TaxotereTM.
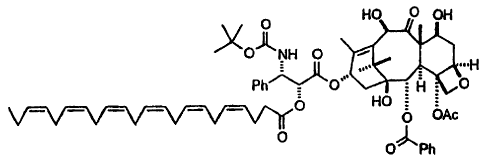
1n one embodiment the coniugate is:
O HO O OH
2~ ~O~NH O
,,, ..,H
Ph~O'~~ ~ : li : O
HO ~ OAc
O
O O~Ph
In another embodiment the conjugate is:
v ~ OH
O
~O~NH O
~ ~ ,...
,..
Ph' v'O :H,
OH HO __
Ph
CA 02255615 1998-11-18
WO 97144026 PCT/US97/08866
-5-
In another embodiment the conjugate is:
O
fO O O
o
~O~NH O -'
Ph~O'~~ : Fi = ~OH
OH HO
p OAc
O~ Ph
In another embodiment the conjugate is:
O
to
o Ho 0 0
~O~NH O
Ph~O'~' = Fi - OH
OH HO O
OAc
O~ Ph
The conjugates of the invention can be isolated conjugates. An isolated
conjugate is one
which is separated from other different taxane conjugates.
Pharmaceutical preparations containing one or more of the foregoing conjugates
also are
provided. The pharmaceutical preparations preferably include a sterile,
pharmaceutically acceptable
2o carrier. The pharmaceutical preparations also can contain other anti-cancer
agents.
The foregoing compositions of matter and pharmaceutical preparations are
useful for treating
cancer, preferably breast cancer, colon cancer and central nervous system
cancer.
Brief Description of the Drawings
Figure 1 is a graph plotting concentration of conjugate 1 versus percent
growth of leukemia
cells.
Figure 2 is a graph plotting concentration of conjugate 1 versus percent
growth of non-small
cell lung cancer cells.
Figure 3 is a graph plotting concentration of conjugate 1 versus percent
growth of colon
cancer cells.
higure 4 is a graph plotting concentration of conjugate I versus percent
growth of CNS
cancer cells.
I~igure 5 is a graph plotting concentration of conjugate 1 versus percent
growth of melanoma
CA 02255615 1998-11-18
WO 97/44026 PCT/US97/08866
-6-
cells.
Figure 6 is a graph plotting concentration of conjugate 1 versus percent
growth of ovarian
cancer cells.
Figure 7 is a graph plotting concentration of conjugate 1 versus percent
growth of renal
cancer cells.
Figure 8 is a graph plotting concentration of conjugate I versus percent
growth of prostate
cancer cells.
Figure 9 is a graph plotting concentration of conjugate I versus percent
growth of breast
cancer cells.
1o Figure 10 is a graph plotting concentration of conjugate 2 versus percent
growth of leukemia
cells.
Figure 1 I is a graph plotting concentration of conjugate 2 versus percent
growth of non-small
cell lung cancer cells.
Figure 12 is a graph plotting concentration of conjugate 2 versus percent
growth of colon
~ 5 cancer cells.
Figure I3 is a graph plotting concentration of conjugate 2 versus percent
growth of CNS
cancer cells.
Figure 14 is a graph plotting concentration of conjugate 2 versus percent
growth of
melanoma cells.
2o Figure 15 is a graph plotting concentration of conjugate 2 versus percent
growth of ovarian
cancer cells.
Figure lb is a graph plotting concentration of conjugate 2 versus percent
growth of renal
cancer cells.
Figure 17 is a graph plotting concentration of conjugate 2 versus percent
growth of prostate
25 cancer cells.
Figure 18 is a graph plotting concentration of conjugate 2 versus percent
growth of breast
cancer cells.
Figure I 9 is a graph plotting concentration of Taxol versus percent growth of
leukemia cells.
Figure 20 is a graph plotting concentration of Taxol versus percent growth of
non-small cell
30 lung cancer cells.
Figure 21 is a graph plotting concentration of Taxol versus percent growth of
colon cancer
cells.
CA 02255615 1998-11-18
WO 97/44026 PCT/US97/08866
_7_
Figure 22 is a graph plotting concentration of Taxol versus percent growth of
CNS cancer
cells.
Figure 23 is a graph plotting concentration of Taxol versus percent growth of
melanoma
cells.
Figure 24 is a graph plotting concentration of Taxol versus percent growth of
ovarian cancer
cells.
Figure 25 is a graph plotting concentration of Taxol versus percent growth of
renal cancer
cells.
Figure 26 is a graph plotting concentration of Taxol versus percent growth of
prostate cancer
i o cells.
Figure 27 is a graph plotting concentration of Taxol versus percent growth of
breast cancer
cells.
Detailed Descriyrtion of the Invention
cis-docosahexaenoic acid (DHA) is a naturally occurring fatty acid. Ii is an
unbranched chain
fatty acid with six double bonds, all cis. Its structure is as follows:
O
OH
DHA can be isolated, for example, from fish oil or can be chemically
synthesized. These
2o methods, however, can generate traps isomers, which are difficult and
expensive to separate and
which may present safety problems in humans. The preferred method of
production is biological
synthesis to produce the all cis isomer. The preferred source of DI1A is from
Martck I3iosciences
Corporation of Columbia, Maryland. Martek has a patented system for
manufacturing DHA using
microalgae which synthesize only a single isomer of DI-IA, the all cis isomer.
Martek's patents
include U.S. Pat. Nos. 5,374,657, 5,492,938, 5,407,957 and 5,397,591.
DHA also is present in the milk of lactating women, and Martek's licensee has
obtained
approval in Europe of DI-IA as a nutritional supplement for infant formula.
It is known that DI-IA cw be unstable in the presence of oxygen. To stabilize
DI-IA and its
conjugates it is important to add anti-oxidants to the material after it is
synthesized. One method of
3o stabilization is to make-up the newly synthesized material in the following
solution: 100 g neat
DI-IA-taxol plus 100 g of vehicle (100 ml propylene glycol, 70 mg alpha-
tocopherol, 5 mg
dilaurylthiodipropionic acid, 50 mg ascorbic acid) prepared and held under
argon in amber, scaled
CA 02255615 1998-11-18
WO 97/44026 PCT/LTS97/08866
_g_
vials and stored at four degrees centigrade. The following anti-oxidants may
also be employed:
ascorbic acid, ascorbyl palmitate, dilauryl ascorbate, hydroquinone, butylated
hydroxyanisole,
sodium meta bisulfate, t-~3 carotene and a-tocopherol. A heavy metal chelator
such as
ethylenediamine tetra-acetic acid (EDTA) may also be used.
Paclitaxel was first isolated from the bark of Taxus revifoli (Wani et al., J.
Am. Chem.
~c,, 93, 2325, 1971). Its isolation and synthesis have been reported
extensively in the literature.
Applicants obtained paclitaxel from a commercial source, Hauser Laboratories,
of Boulder,
Colorado.
Example 1
o TAXOL
Ac0 OH
O
Ph~NH O
,,,
Ph I v -O ~~~ : I-I =
HO O
O ~ OAc
O O Ph
conjugate 1
To synthesize DHA-Taxol, a solution of Taxol (41 pmol) in methylene chloride
(2.SmL)
under argon was mixed with 4-dimethylaminopyridine (41 ~mol),
dicyclohexylcarbodiimide
(82~mo1), and DHA (4i ~mol) and the reaction mixture was stirred at ambient
temperature for two
hours. Following dilution with ether, the reaction mixture was washed with 5%
hydrochloric acid,
water, saturated aqueous sodium chloride, dried, and concentrated. Radial
chromatography of the
residue produced 45ms (94%) of crystalline Taxol-DI-IA conjugate 1.
CA 02255615 2003-12-24
9 _ o Aco O off
Ph ~ NH O
TAXOL ~ ph ~O "~ ~~~~ H ~ ..,H
O
osiEt3 Ho o
OAc
O~ Ph
A
O
to O Ac0 O O
Ph~NH O
~ ~ ,,., ..,H
Ph~O'~ z H ; O
OSiEt3 HO O
OAc
O~.Ph
O
Ac0 O O
Ph ~ NH O r
Ph~Or s H = O
,. ~H
..
~ ~ ~ OAc
O ~ conjugate 2
The production of conjugate 2 involves several steps including a number of
protection-
acylation-deprotection steps. A solution of Taxol (59pmo1) in methylene
chloride (2.5mL) was
mixed at ambient temperature under argon with imidazole (147 pmol) and
triethylsilyl chloride
(147p,rrrol). The reaction mixture was stirred for thirty minutes, diluted
with additional methylene
chloride, washed with water, saturated aqueous sodium chloride, dried, and
concentrated
Chromatography of the residue produced 50mg (88%) of intermediate A plus 5mg
of the 2', 7-
di(triethylsilyl) ether derivative. A solution of intermediate A (52pmo1) in
methylene chloride (3mL)
was mixed at ambient temperature under argon with 4-dimethylaminopyridine
(521rmo1),
dicyclohexylcarbodiimide (104pmo1), and DHA (52p,mo1). The reaction mixture
was stirred for ten
hours, diluted with ether, passed through celiteT'", and concentrated.
Chromatography of the residue
produced 65.9mg of intermediate B. A solution of intermediate B (51 pmol) in
acetonitrile (2mL) at
0°C under argon was mixed with 49% aqueous HF (0.2mL) and the reaction
mixture was stirred for
one hour. After dilution with ether, the reaction mixture was washed with
water, saturated aqueous
sodium chloride, dried, and concentrated. Radial chromatography of the residue
produced 44.6mg
(75%) of TaxolT"'-D)aA conjugate 2.
CA 02255615 1998-11-18
WO 97/44026 PCT/US97/08866
-10-
Conjugates 1 and 2 were sent to the United States National Cancer Institute
(NCI) for
screening in the NCI's anticancer screening program. The conjugates were
provided in ethanol
(approximately 40mg analog/2m1 ethanol). The conjugates were sealed in vials
under argon to avoid
exposure of the conjugates to oxygen because the conjugates were believed to
be sensitive to
oxygen. Instructions were provided to store at 4°C and to open the
vials only when ready for
immediate experimental use. Instructions also were provided to use the ethanol
solutions containing
the conjugates directly or to dissolve the analogs further in DMSO
(dimeihylsulfoxide) at
appropriate concentrations, with vortexing if necessary for adequate
dispersal.
t0 The activities of conjugates 1 and 2 were tested against 57 cancer cell
lines. The results are
presented in Figs. 1-9 for conjugate 1, Figs. 10-18 for conjugate 2 and Figs
19-27 for Taxol. To
understand the data, reference is made to the guides provided by the NCI,
excerpted as follows:
t5 The measured effect of the compound on a cell line is currently calculated
according to one or the
other of the following two expressions:
If (Mean OD,~, -Mean OD,~) >_ 0, then
Zo PG =100 x (Mean OD,a, -Mean OD,u,°)/(Mean ODG,, -Mean Od,~)
If (Mean ODD" -Mean OD,~) < 0, then
PG =100 x (Mean OD,a, -Mean Od,~ro)/Mean Od,
25 Where:
Mean OD""o ~ The average of optical density measurements of SRB-derived color
just before
exposure of cells to the test compound.
Mean OD,~" - The average of optical density measurements of SRI3-derived color
3o after 48 hours exposure of cells to the test compound.
Mean ODD,, - The average of optical density measurements of SRB-derived color
after 48 hours with no exposure of cells to the test compound.
CA 02255615 1998-11-18
WO 97/44026 PCT/US97/08866
-11-
Experimental data was collected against each cell line. ... Each concentration
is expressed as the
logo (molar or p,g/ml). ... The response parameters GI50, TGI, and LC50 are
interpolated values
representing the concentrations at which the PG is +S0, 0, and -50,
respectively. Sometimes these
response parameters cannot be obtained by interpolation. If, for instance, all
of the PGs in a given
row exceed +50, then none of the three parameters can be obtained by
interpolation. In such a case,
the value given for each response parameter is the highest concentration
tested. ... This practice is
extended similarly to the other possible situations where a response parameter
cannot be obtained
by interpolation.
Dose-Response Curves:
The dose-response curve page of the data package is created by plotting the
PGs against the logo of
the corresponding concentration for every cell line. The cell line curves are
grouped by subpanel.
Horizontal lines are provided at the PG values of +50, 0, and -50. The
concentrations corresponding
to points where the curves cross these lines are the GI50, TGI and LC50,
respectively.
. Several important distinctions are apparent from the data. Most important,
the patterns of
anticancer actively for conjugates 1 and 2 differ from that of Taxol. In one
sense, conjugates l and
2 are effective anticancer agents against a more restricted set of cancer cell
lines. For example,
2o conjugates 1 and 2 were not very effective against any of the six leukemia
cancer cell lines tested,
whereas Taxol was somewhat effective against all four leukemia cell lines
against which Taxol was
tested. (See Figs. 1, 10 and 19.)
The relative activity against members within a class of cancers also was
altered. For
example, at TGI (horizontal line at zero in the graphs), Taxol was more
effective against non-small
cell lung cancer line H522 than against H460 (by about 3 logs), whereas
conjugates 1 and 2 were
slightly more effective against H460 than H522. As another example, Taxol was
least effective at
TGI against CNSU251, whereas conjugate 1 was most effective against CNSU251
and conjugate
2 was also very effective against CNSU251{relative to other CNS cell lines).
As a further example,
Taxol was equivalent in activity toward MDA-N and MDA-MB-435 breast cancer
cell lines at all
3o concentrations tested, whereas conjugates 1 and 2 were more effective
against MDA-N than
MDA-MB-435 at all concentrations tested.
To further illustrate the differences in the activity of conjugates 1 and 2
versus that of Taxol,
SUBSTITUTE SHEET (RULE 26)
CA 02255615 1998-11-18
WO 97/44026 PCT/US97/08866
- 12-
the NCI subjected the data to a statistical analysis designed by the NCI to
reflect differences in the
pattern of activity of anticancer agents. Conjugate I and conjugate 2 were
determined to be
statistically different in their pattern of activity versus Taxol in this
unique measurement by the
NCI.
It also is to be noted that, in general, conjugates 1 and 2 were one thousand
to ten thousand
times less potent than Taxol for many cell lines tested. This reduction in
activity is important,
especially since conjugates 1 and 2 maintained strong activity against some
cell lines. Conjugates
1 and 2 will be sufficiently active against certain cell lines, but will have,
on average, a substantially
and disproportionately lower activity against other cell lines, reducing
potential side effects. For
to example, the TGI for Taxol against CNS SF-539 is -6.95, and the TGI for
conjugate 1 against this
cell line is -5.13 and for conjugate 2 is -5.53. (In other words, the activity
of the analogs was reduced
versus that of Taxol by less than 2 logs). The GI50 for Taxol against CNS SF
539 is -7.52, whereas
the GISOs for conjugates 1 and 2 are -6.22 and -5.56, respectively (again less
than 2 logs difference).
In contrast, Taxol has a GI50 for cell line CNSSF 268 of less than -I0.0,
whereas conjugates 1 and
2 have GISOs for CNSSF 268 of 5.36 and 5.28, respectively. This represents a
reduction of activity
in the conjugates vs. that of Taxol by at least about 5 logs activity! On
average, the GI50 for Taxol
across all cell lines tested is at least -9.19. (It is probably much higher
since concentrations less than
-10 were not tested, and if Taxol was active at -10.0, -I O (instead of the
actual lower value) was
used ~in calculating the average of -9.19. There were 27 instances when this
occurred.) The average
2o GISOs for conjugates 1 and 2 , on the other hand, were 5.49 and 5.22,
respectively. Therefore, the
average difference in activity for Taxol vs. the conjugates is at least
between 3 and 4 logs. Thus,
the sharp reduction in the activity of the conjugates against many cell lines
vs. a lesser reduction for
other cell lines is expected to reduce the potential side effects of the
conjugates versus those of Taxol
at effective doses:
Cancers other than CNS, breast and colon cancer can be treated. Por example,
there was
activity against non-small cell lung cancer cells, melanoma cells and ovarian
cancer cells. However,
the activity was relatively reduced and was extremely specific, limiting the
utility of the conjugates
for treating generally such cancers. In any event, cancer patients could be
evaluated to determine
if a conjugate is strongly active against the patient's cancer prior to
selecting the conjugate as the
3o anti-cancer agent of choice for that patient.
The foregoing experiments establish that DI-IA conjugates have altered
specificity versus that
of Taxol for cancer cell lines. Because of this altered specificity, it also
is clear that the conjugates
CA 02255615 2003-12-24
-13-
themselves are gaining access into the target cells (as opposed to simply
releasing Taxol into the
environment outside of the cell). Thus, the DHA moiety appears to selectively
target certain cell
types as opposed to others. The ability of the conjugates to gain entry into
the targeted cells was
unknown prior to the invention, and the ability of the DHA moiety to
selectively target certain cell
types was unexpected.
The same is true of DHA-TaxotereTM covalent conjugates, examples of which are
presented
below. TaxotereTM's synthesis has been reported extensively in the literature.
One example is
Kanazawa, A, et al., J. Organic Chem. 1994, Vol. 59, pp. 1238-1240.
Example 4
TAXOTERET~"
HO O OH
O
UHA / dicyclohexylca~bodiimide ~O'~ NH O
....H
4-dimethylamit~opyridine . ~ ~ .,.~ ~ O
Pt~~O~~. : ti
p HO ~ OAc
O
O~ Ph
conjugate 3
A solution of TaxotereTM in methylene chloride under argon is mixed with
4-dimethylaminopyridine, dicyclohexylcarbodiimide, and DHA. The reaction
mixture is stirred
at ambient temperature. Radial chromatography of the residue is performed to
produce
TaxotereTM-DHA conjugate 3.
CA 02255615 2003-12-24
-14-
HO O OSi(CH2CH~3
O
th uiethylsilyl chloride ~O~NH O
TAXOTERE 'rcriidazole ~= ~ .... ; ....H
Ph' v"O M H c O
(~aCHtI~O HO p
OAt
O
C
O
O . O O OSi(CH~CH~~
DHA I dicycbhoxykkarbodiimide ''
~O~NH O
4~dimethylarrwt~opyridir~e ~ ~_ ~ ,,.. _ ....H
Ph~O'~ : H : O
(CH~Chiz)~SiO HO ~ OAc
O~ Ph
HF
conjugate 4
A solution of TaxotereTM in dimethylformamide is mixed at ambient temperature
under argon
with imidazole and triethylsilyl chloride. The reaction mixture is stirred at
ambient temperature,
diluted with methylene chloride, washed with water, saturated aqueous sodium
chloride, dried and
concentrated. Radial chromatography of the residue is performed to produce
intermediate C. A
solution of intermediate C in methylene chloride is mixed at ambient
temperature under argon with 4
dimethylaminopyridine, dicyclohexylcarbodiimide, and DHA. The reaction mixture
is stirred at
ambient temperature, diluted with ether, passed through celiteTM, and
concentrated. Radial
chromatography of the residue is performed to produce intermediate D. A
solution of intermediate
CA 02255615 2003-12-24
- I S-
D in acctonitrile at 0°C under argon is mixed with 49% aqueous HF and
the reaction mixture is
stirred at the same temperature. After dilution with ether, the reaction
mixture is washed with water,
saturated aqueous sodium chloride, dried, and concentrated Radial
chromatography of the residue is
performed to produce TaxotereTM-DHA conjugate 4.
p ~~ ~ c~11
Ie~t~utytdicnethylsilyl ct~lo~ide ~p~NH O
imida:ole ph~p~' ~~~~ ii : o
TAXOTERETM ten.C,F'IaI~H~ySiO HO p
OAc
to E
1 eqwvaleN DHA
dicydohoxyka~bodiimide
4~dirtl°lhylaminopytidif~°
~V~o~s HF
--
wuJu~uw t
A solution of TaxotereTM in dirnethylforcnamide is mixed at ambient
temperature under argon
with imidazole and tent-butylydimethylsilyl chloride. The reaction mixture is
stirred at ambient
temperature, diluted with methylene chloride, washed with water, saturated
aqueous sodium chloride,
dried, and concentrated. Radial chromatography of the residue is performed to
produce intermediate E.
A solution of intermediate E in methylene chloride is mixed at ambient
temperature under argon with 4-
dimethylaminopyridine, dicyclohexylcarbodiimide, and 1 equivalent of DHA. The
reaction mixture is
stirred at ambient temperature, diluted with ether, passed through celiteTM,
and concentrated. Radial
chromatography of the residue is performed to produce intermediate F.
(Intermediate H is also obtained
and used in Example 8 below.) A solution of interntediate F in acetonitrile at
0°C under argon is mixed
with aqueous HF and the reaction mixture is stirred at the same temperature.
After dilution with ether,
the reaction mixture is washed with water, saturated aqueous sodium chloride,
dried, and concentrated
Radial chromatography of the residue is performed to produce TaxotereTM-DHA
conjugate 4.
INf~C~1'1~1(CrH~t~
CA 02255615 2003-12-24
-16-
Ezam~rle 7
O
tM.bulyldimethylsilyt chloride ~p~NH 0
_ N
TAXOTERETM imidezole ~ ~
Ph'
icrl~C,Hy(CH~r~O
p''~ Ph
O O
O O O
io O
DHI1I dicyclohexyylca~odi~e ~O~ NH O ~ " H
4-dimethylaminopyddiee Ph~O~° ~~~~ i lO
ic~tL,H;.(CH~)?Si0 HO n OAc
O~ Ph
aqueous HF O
~O~NH O
OH
O'~Ph
A solution of TaxotereT"' in dimethylformamide is~mixed at ambient temperature
under
argon with imidazole and tert-butylydimethylsilyl chloride. The reaction
mixture is stirred at
ambient temperature, diluted with methylene chloride, washed with water,
saturated aqueous
sodium chloride, dried, and concentrated. Radial chromatography of the residue
is performed to
produce intermediate E. A solution of intermediate it in methylene chloride is
mixed at
ambient temperature under argon with 4-dimethylaminopyridine,
dicyclohexylcarbodiimide,
and DHA. The reaction mixture is stirred at ambient temperature, diluted with
ether, passed
through celiteTM, and concentrated. Radial chromatography of the residue is
performed to
produce intermediate G. A solution of intermediate G in acetonitrile at
0°C under argon is
CA 02255615 2003-12-24
-17-
mixed with aqueous HF and the reaction mixture is stirred at the same
temperature. After
dilution with ether, the reaction mixture is washed with water, saturated
aqueous sodium
chloride, dried, and concentrated. Radial chromatography of the residue is
performed to
produce TaxotereTM-DHA conjugate 5.
Ezamule 8
HO O OH
O
~O~NH O
teef~bcAyidimelh~lyt ctioride ~= ~ ,... ~~~H
TAXOTERETM ~ Ph~O'M = H = O
im~dazole tert~C~Hy(CHal2S~ HO O OJ~c
O~Ph
E
~ ~~~t OHA ~O~NH
dicydohexy~atbodiimide Ph''~
4'YPf~tfert~.hiefCH~l~Sit
o~'Ph
aqueous HF
coajapte f
A solution of TaxotereTM in dimethylformamide is mixed at ambient temperature
under
argon with imidazoIe and tert-butylydimethylsilyl chloride. The reaction
mixture is stirred at
ambient temperature, diluted with methylene chloride, washed with water,
saturated aqueous
sodium chloride, dried, and concentrated. Radial chromatography of the residue
is performed to
produce intermediate E. A solution of intermediate E in methylene chloride is
mixed at ambient
temperature under argon with 4-dimethylaminopyridine,
dicyclohexylcarbodiimide, and 1
equivalent of DHA. The reaction mixture is stirred at ambient temperature,
diluted with ether,
passed through celiteTM, and concentrated. Radial chromatography of the
residue is performed to
produce intermediate H and
CA 02255615 2003-12-24
-18-
intermediate F which was used above in Example 6. A solution of intermediate H
in acetonitrite
at 0°C under argon is mixed with aqueous HF and the reaction mixture is
stirred at the same
temperature. After dilution with ether, the reaction mixture is washed with
water, saturated
aqueous sodium chloride, dried, and concentrated. Radial chromatography of the
residue is
performed to produce TaxotererM-DHA conjugate 6.
The compounds useful in the invention may be delivered in the form of anti-
cancer cocktails.
An anti-cancer cocktail is a mixture of any one of the compounds useful with
this invention with
another anti-cancer agent such as an anti-cancer drug, a cytokine, andlor
supplementary potentiating
agent(s). The use of cocktails in the treatment of cancer is routine. In this
embodiment, a common
l0 administration vehicle (e.g., pill, tablet, implant, injectable solution,
etc.) would contain both the
conjugate useful in this invention and the anti-cancer drug and/or
supplementary potentiating agent.
Anti-cancer agents include anti-cancer drugs. Anti-cancer drugs are well known
and include:
Acivicin; Aclarubicin; Acodazole Hydrochloride; Acronine; Adozelesin;
Aldesleukin; Altretamine;
Ambomycin; Ametantrone Acetate; Aminoglutethimide; Amsacrine; Anastrozole;
Anthramycin;
Asparaginase; Asperlin; Azacitidine; Azetepa; Azotomycin; Batimastat;
Benzodepa;
Bicalutamide; Bisantrene Hydrochloride; Bisnafide Dimesylate; Bizelesin;
Bleomycin;
Bleomycin Sulfate; Brequinar Sodium; Bropirimine; Busulfan; L-Buthiamine
Sulfoxide;
Cactinomycin; Calusterone; Camptothecin; Caracemide; Carbetimer; Carboplatin;
Carmustine;
Carubicin Hydrochloride; Carzelesin; Cedefingol; Chlorambucil; Cirolemycin;
Cisplatin;
Cladribine; Crisnatol Mesylate; Cyclophosphamide; Cytarabine; Dacarbazine;
Dactinomycin;
Daunorubicin Hydrochloride; Decitabine; Dexormaplatin; Dezaguanine;
Dezaguanine
Mesylate; Diaziquone; Docetaxel; Doxorubicin; Doxorubicin Hydrochloride;
Droloxifene;
Droloxifene Citrate; Dromostanolone Propionate; Duazomycin; Edatrexate;
Eflornithine
Hydrochloride; Elsamitrucin; Enloplatin; Enpromate; Epipropidine; Epirubicin
Hydrochloride;
Erbulozole; Esorubicin Hydrochloride; Estramustine; Estramustine Phosphate
Sodium;
Etanidazole; Etoposide; Etoposide Phosphate; Etoprine; Fadrozole
Hydrochloride; Fazarabine;
Fenretinide; Floxuridine; Fludarabine Phosphate; Fluorouracil; Flurocitabine;
Flutamide;
Fosquidone; Fostriecin Sodium; Gallium Nitrate; Gemcitabine; Gemcitabine
Hydrochloride;
Hexamethylmelamine; Hydroxyurea; Idarubicin Hydrochloride; Ifosfamide;
Ilmofosine;
Interferon Alfa-2a; Interferon Alfa-2b; Interferon Alfa-nl; Interferon Alfa-
n3; Interferon Beta-1
a; Interferon Gamma-I b; Interleukin 2; Iproplatin; Irinotecan Hydrochloride;
Lanreotide
Acetate; Letrozole; Leuprolide Acetate; Liarozole Hydrochloride; Lometrexol
Sodium;
Lomustine; Losoxantrone Hydrochloride; Masoprocol; Maytansinc; Mechlorethamine
Hydrochloride; Megesterol; Megestrol Acetate; Melengestrol Acetate; Melphalan;
Menogaril;
Mercaptopurine;
CA 02255615 2003-12-24
-19-
MethotrexateTM; MethotrexateT"' Sodium; Methyl Glyoxal Bis-Guanylhydrazone,
Metoprine;
Meturedepa; Mitindomide; Mitocarcin; Mitocromin; Mitogillin; Mitoguazone;
Mitomalcin;
Mitomycin; Mitosper; Mitotane; Mitoxantrone Hydrochloride; Mycophenolic Acid;
Nocodazole; Nogalamycin; Octreotide; Ormaplatin; Oxisuran; Paclitaxel;
Pegaspargase;
Peliomycin; Pentamustine; Pentostantin; Peplomycin Sulfate; Perfosfamide;
Pipobroman;
Piposulfan; Piroxantrone Hydrochloride; Plicamycin; Plomestane; Porfimer
Sodium;
Porfiromycin; Prednimustine; Prednisone; Procarbazine Hydrochloride;
Puromycin; Puromycin
Hydrochloride; Pyrazofurin; Riboprinc; Rogletimide; Safingol; Safingol
Hydrochloride;
l0 Semustine; Simtrazene; Sparfosate Sodium; Sparsomycin; Spirogermanium
Hydrochloride;
Spiromustine; Spiroplatin; Streptonigrin; Streptozocin; Sulofenur;
Talisomycin; Tamoxifen
Citrate; Tamoxifen Methiodide, Taxanes; Taxoids; Tecogalan Sodium; Tegafur;
Teloxantrone
Hydrochloride; Temoporfin; Teniposide; Teroxirone; Testolactone; Thiamiprine;
Thioguanine;
Thiotepa; Tiasofuran; Tiazofurin; Tirapazamine; Topotecan Hydrochloride;
Toremifene Citrate;
Trestolone Acetate; Triciribine Phosphate; Trimetrexate; Trimetrexate
Glucuronate;
Triptorelin; Tubulozole Hydrochloride; Uracil Mustard; Uredepa; Vapreotide;
Verteporfm;
Vinblastine Sulfate; Vincristine Sulfate; Vindesine; Vindesine Sulfate;
Vinepidine Sulfate;
Vinglycinate Sulfate; Vinleurosine Sulfate; Vinorelbine Tartrate; Vinrosidine
Sulfate;
Vinzolidine Sulfate; Vorozole; Zeniplatin; Zinostatin; Zorubicin
Hydrochloride.
Other anti-cancer drugs include: 20-epi-1,25 dihydroxyvitamin D3; 5-
ethynyluracil;
abiraterone; aclarubicin; acylfulvene; adecypenol; adozelesin; aldesleukin;
ALL-TK
antagonists; altreiamine; ambamustine; amidox; amifostine; aminolevulinic
acid; amrubicin;
amsacrine; anagrelidc; anastrozole; andrographolide; angiogenesis inhibitors;
antagonist D;
antagonist G; antarelix; anti-dorsalizing morphogenetic protein-1;
antiandrogen, prostatic
carcinoma; antiestrogen; antincoplaston; antisense oligonucleotides;
aphidicolin glycinate;
apoptosis gene modulators; apoptosis regulators; apurinic acid; ara-CDP-DL-
PTBA; arginine
deaminase; asulacrine; atamestane; atrimustine; axinastatin 1; axinastatin 2;
axinastatin 3;
azasetron; azatoxin; azatyrosine; baccatin III derivatives; balanol;
batimastat; BCR/ABL
antagonists; benzochlorins; benzoylstaurosporine; beta lactam derivatives;
beta-alethine;
betaclamycin B; betulinic acid; bFGF inhibitor; bicalutamide; bisantrene;
bisaziridinylspermine; bisnafide; bistratene A; bizelesin; breflatc;
bropirimine; budotitane;
buthionine sulfoximine; calcipotriol; calphostin C; camptothecin derivatives;
canarypox IL-2;
capecitabine; carboxamidc-amino-triazole; carboxyamidotriazole; CaRcst M3;
CARN 700;
CA 02255615 2003-12-24
- 19a -
cartilage derived inhibitor; carzelesin; casein kinase inhibitors (ICOS);
castanospermine;
cecropin B; cetrorelix; chlorins; chloroquinoxalinc sulfonamide; cicaprost;
cis-porphyrin;
cladribine; clomifenc analogues; clotrimazole; collismycin A; collismycin B;
combretastatin
A4; combretastatin
CA 02255615 1998-11-18
WO 97/44026 PCT/US97/08866
-20-
analogue; conagenin; crambescidin 816; crisnatol; cryptophycin 8; cryptophycin
A derivatives;
curacin A; cyclopentanthraquinones; cycloplatam; cypemycin; cytarabine
ocfosfate; cytolytic factor;
cytostatin; dacliximab; decitabine; dehydrodidemnin B; deslorelin;
dexifosfamide; dexrazoxane;
dexverapamil; diaziquone; didemnin B; didox; diethylnorspermine; dihydro-5-
azacytidine;
dihydrotaxol, 9-; dioxamycin; diphenyl spiromustine; docosanol; dolasetron;
doxifluridine;
droloxitene; dronabinol; duocarmycin SA; ebselen; ecomustine; edelfosine;
edrecolomab;
eflornithine; elemene; emitefur; epirubicin; epristeride; estramustine
analogue; estrogen agonists;
estrogen antagonists; etanidazole; etoposide phosphate; exemestane; fadrozole;
fazarabine;
fenretinide; filgrastim; finasteride; flavopiridol; flezelastine; fluasterone;
fludarabine;
o fluorodaunorunicin hydrochloride; forfenimex; formestane; fostriecin;
fotemustine; gadolinium
texaphyrin; gallium nitrate; galocitabine; ganirelix; gelatinase inhibitors;
gemcitabine; glutathione
inhibitors; hepsulfam; heregulin; hexamethylene bisacetamide; hypericin;
ibandronic acid;
idarubicin; idoxifene; idramantone; ilmofosine; ilomastat; imidazoacridones;
imiquimod;
immunostimulant peptides; insulin-like growth factor-I receptor inhibitor;
interferon agonists;
~ 5 interferons; interleukins; iobenguane; iododoxorubicin; ipomeanol, 4-;
irinotecan; iroplact;
irsogladine; isobengazole; isohomohaiicondrin B; itasetron; jasplakinolide;
kahalalide F; lamellarin-
N triacetate; lanreotide; leinamycin; lenograstim; lentinan sulfate;
leptolstatin; letrozole; leukemia
inhibiting factor; leukocyte alpha interferon; leuprolide + estrogen +
progesterone; leuprorelin;
levamisole; liarozole; linear polyamine analogue; lipophilic disaccharide
peptide; lipophilic platinum
2o compounds; lissoclinamidc 7; lobaplatin; lombricine; lometrexol;
lonidaminc; losoxantrone;
lovastatin; loxoribine; lurtotecan; lutetium texaphyrin; Iysofylline; lytic
peptides; maitansine;
mannostatin A; marimastat; masoprocol; maspin; matrilysin inhibitors; matrix
metalloproteinase
inhibitors; menogaril; merbarone; meterelin; methioninasc; metoclopramide; MIF
inhibitor;
mifcpristone; miltefosinc; mirimostim; mismatched double stranded RNA;
mitoguazone; mitolactol;
25 mitomycin analogues; mitonafide; mitotoxin fibroblast growth factor-
saporin; mitoxantrgne;
mofarotenc; molgramostim; monoclonal antibody, human chorionic gonadotrophin;
monophosphoryl lipid A + myobacterium cell wall sk; mopidamol; multiple drug
resistance gene
inhibitor; multiple tumor suppressor 1-based therapy; mustard anticancer
agent; mycaperoxide B;
mycobacterial cell wall extract; myriaporone; N-acetyldinalinc; N-substituted
bcnzamides; nafarclin;
3o nagrcstip; naloxone + pcntazocine; napavin; naphtcrpin; nartograstim;
nedaplatin; nemorubicin;
neridronic acid; neutral endopcptidasc; nilutamidc; nisamycin; nitric oxide
modulators; nitroxide
antioxidant; nitrullyn; 06-benzylguanine; octreotide; okicenone;
oligonucleotides; onapristone;
CA 02255615 1998-11-18
WO 97/44026 PCTIUS97/08866
-21 -
ondansetron; ondansetron; oracin; oral cytokine inducer; ormaplatin;
osaterone; oxaliplatin;
oxaunomycin; paclitaxel analogues; paclitaxel derivatives; palauamine;
palmitoylrhizoxin;
pamidronic acid; panaxytriol; panomifene; parabactin; pazelliptine;
pegaspargase; peldesine;
pentosan polysulfate sodium; pentostatin; pentrozole; perflubron;
perfosfamide; perillyl alcohol;
phenazinomycin; phenylacetate; phosphatase inhibitors; picibanil; pilocarpine
hydrochloride;
pirarubicin; piritrexim; placetin A; placetin B; plasminogen activator
inhibitor; platinum complex;
platinum compounds; platinum-triamine complex; porfimer sodium; porfiromycin;
propyl bis-
acridone; prostaglandin J2; proteasome inhibitors; protein A-based immune
modulator; protein
kinase C inhibitor; protein kinase C inhibitors, microalgaI; protein tyrosine
phosphatase inhibitors;
purine nucleoside phosphorylase inhibitors; purpurins; pyrazoloacridine;
pyridoxylated hemoglobin
polyoxyethyIene conjugate; raf antagonists; raltitrexed; ramosetron; ras
farnesyl protein transferase
inhibitors; ras inhibitors; ras-GAP iWibitor; retelliptine demethylated;
rhenium Re 18b etidronate;
rhizoxin; ribozymes; RII retinamide; rogletimide; rohitukine; romurtide;
roquinimex; rubiginone B 1;
ruboxyl; safingol; saintopin; SarCNU; sarcophytol A; sargramostim; Sdi 1
mimetics; semustine;
senescence derived inhibitor 1; sense oligonucleotides; signal transduction
inhibitors; signal
transduction modulators; single chain antigen binding protein; sizofiran;
sobuzoxane; sodium
borocaptate; sodium phenylacetate; solverol; somatomedin binding protein;
sonermin; sparfosic acid;
spicamycin D; spiromustine; splenopentin; spongistatin 1; squalamine; stem
cell inhibitor; stem-cell
division inhibitors; stipiamide; stromelysin inhibitors; sulfinosine;
superactive vasoactive intestinal
2o peptide antagonist; suradista; suramin;~ swainsonine; synthetic
glycosaminoglycans; tallimustine;
tamoxifcn mcthiodidc; tauromustinc; tazarotenc; tccogalan sodium; cegafur;
tellurapyrylium;
telomerase inhibitors; temoporfin; temozolomide; teniposide;
tetrachlorodecaoxide; tetrazomine;
thaliblastine; thalidomide; thiocoraline; thrombopoietin; thrombopoietin
mimetic; thymalfasin;
thymopoietin receptor agonist; thymotrinan; thyroid stimulating hormone; tin
ethyl etiopurpurin;
tirapazamine; titanocene dichloride; topotecan; topsentin; toremifene;
totipotent stem cell facjor;
translation inhibitors; tretinoin; triacetyluridine; triciribine;
trimetrexate; triptorelin; tropisetron;
turosteride; tyrosine kinase inhibitors; tyrphostins; UBC inhibitors;
ubenimex; urogenital sinus-
derived growth inhibitory factor; urokinase receptor antagonists; vapreotide;
variolin B; vector
system, erythrocyte gene therapy; velaresol; veramine; verdins; vcrteporfin;
vinoreibine; vinxaltine;
3o vitaxin; vorozole; zanoterone; zeniplatin; zilascorb; zinostatin
stimalamer.
Supplementary potentiating agents likewise are well characterized and include:
Tricyclic
anti-depressant drugs (e.g., imipramine, desipramine, amitryptyline,
clomipramine, trimipramine,
CA 02255615 1998-11-18
WO 97/44026 PCT/US97/08866
- 22 -
doxepin, nortriptyline, protriptyline, acnoxapine and maprotiline); non-
tricyclic anti-depressant drugs
(e.g., sertraline, trazodone and citalopram); Ca++ antagonists (e.g.,
verapamil, nifedipine, nitrendipine
and caroverine); Calmodulin inhibitors (e.g., prenylamine, trifluoroperazine
and clomipramine);
Amphotericin B; Triparanol analogues (e.g., tamoxifen); antiarrhythmic drugs
(e.g., quinidine);
antihypertensive drugs (e.g., reserpine); Thiol depleters (e.g., buthionine
and sulfoximine) and
Multiple Drug Resistance reducing agents such as Cremaphor EL.
The compounds of the invention also can be administered with cytokines such as
granulocyte
colony stimulating factor.
The compounds of the invention, when used in cocktails, are administered in
therapeutically
o effective amounts. A therapeutically effective amount will be determined by
the parameters
discussed below; but, in any event, is that amount which establishes a level
of the drugs) in the area
of the tumor which is effective in inhibiting the tumor growth.
When administered, the formulations of the invention are applied in
pharmaceutically
acceptable amounts and in pharmaceutically acceptable compositions. Such
preparations may
routinely contain salts, buffering agents, preservatives, compatible carriers,
and optionally other
therapeutic ingredients. When used in medicine the salts should be
pharmaceutically acceptable, but
non-pharmaceutically acceptable salts may conveniently be used to prepare
pharmaceutically
acceptable salts thereof and are not excluded from the scope of the invention.
Such
pharmacologically and pharmaceutically acceptable salts include, but are not
limited to, those
prepared from the following acids: hydrochloric, hydrobromic, sulphuric,
nitric, phosphoric, malefic,
acetic, salicylic, p-toluene sulfonic, tartaric, citric, methane sulfonic,
formic, malonic, succinic,
naphthalene-2-sulfonic, aid benzene sulfonic. Also, pharmaceutically
acceptable salts can be
prepared as alkaline metal or alkaline earth salts, such as sodium, potassium
or calcium salts.
Suitable buffering agents include: acetic acid and a salt (1-2% WN); citric
acid and a salt
(1-3% WN); boric acid and a salt (0.5-2.5% WN); and phosphoric acid and a salt
(0.8-2% W/,V).
Suiiablc preservatives include bcnzalkonium chloride (0.003-0.03% W/V);
chlorobutanol
(0.3-0.9% WN); parabens (0.01-0.25% WN) and thimerosal (0.004-0.02% WN).
'I he active compounds of the present invention may be a pharmaceutical
composition having
a therapeutically effective amount of a conjugate of the invention optionally
included in a
3o phannaceuticaliy-acceptable carrier. The term "pharmaceutically-acceptable
carrier" as used herein
means one or more compatible solid or liquid filler, dilutants or
encapsulating substances which are
suitable for administration to a human or other animal. The term "carrier"
denotes an organic or
CA 02255615 2003-12-24
-23-
inorganic ingredient, natural or synthetic, with which the active ingredient
is combined to facilitate
the application. The components of the pharmaceutical compositions are capable
of being
commingled with the molecules of the present invention, and with each other,
in a manner such that
there is no interaction which would substantially impair the desired
pharmaceutical efficacy.
Compositions suitable for parenteral administration conveniently comprise a
sterile
preparation of the conjugates of the invention. This preparation may be
formulated according to
known methods. Formulations for taxanes can be found in Chapter 9 of Taxol:
Science and
Ap-plications, CRC Press, Inc., 2000 Corporate Boulevard, N.W., Boca Raton, FL
33431. In general,
Taxol has been formulated as a 6 mg/ml cremophorTM EL (polyoxyethylated castor
oil)/ethanol
mixture, which is diluted to final volume with normal saline or 5% o dextrose.
A l5mg/ml solution
of TaxotereTM has been formulated in polysorbate 80 (polyoxyethylene
sorbitanmonooleate)lethanol
mixture, diluted with 5% dextrose.
The sterile preparation thus may be a sterile solution or suspension in a non-
toxic
parenterally-acceptable diluent or solvent. In addition, sterile, fixed oils
are conventionally employed
as a solvent or suspending medium. For this purpose any bland fixed oil may be
employed including
synthetic mono or di-glycerides. In addition, fatty acids such as oleic acid
find use in the preparation
of injectables. Carrier formulations suitable for oral, subcutaneous,
intravenous, intramuscular, etc.
can be found in Remin~ton's Pharmaceutical Sciences, Mack Publishing Company,
Euston, PA.
The invention is used in connection with treating subjects having, suspected
of having,
developing or suspected of developing cancer. A subject as used herein means
humans, primates,
horses, cows, pigs, sheep, goats, dogs, cats and rodents.
The conjugates of the invention are administered in effective amounts. An
effective amount
means that amount necessary to delay the onset of, inhibit the progression of
or halt altogether the
onset or progression of the particular condition being treated. In general, an
effective amount will be
that amount necessary to inhibit mammalian cancer cell proliferation in-situ.
When administered to a
subject, effective amounts will depend, of course, on the particular condition
being treated; the
severity of the condition; individual patient parameters including age,
physical condition, size and
weight; concurrent treatment; frequency of treatment; and the mode of
administration. These factors
are well known to those of ordinary skill in the art and can be addressed with
no more than routine
experimentation. It is preferred generally that a maximum dose be used, that
is, the highest safe dose
according to sound medical judgment.
CA 02255615 1998-11-18
WO 97/44026 PCT/US97/08866
-24-
Dosage may be adjusted appropriately to achieve desired drug levels, locally
or systemically.
Generally, daily oral doses of active compounds will be from about 0.01 mg/kg
per day to 1000
mg/kg per day. It is expected that IV doses in the range of about 1 to 1000
mg/m2 per day will be
effective. In the event that the response in a subject is insufficient at such
doses, even higher doses
(or effective higher doses by a different, more localized delivery route) may
be employed to the
extent that patient tolerance permits. Continuous IV dosing over, for example
24 hours or multiple
doses per day are contemplated to achieve appropriate systemic levels of
compounds.
A variety of administration routes are available. The particular mode selected
will depend
of course, upon the particular drug selected, the severity of the disease
state being treated and the
t o dosage required for therapeutic efficacy. The methods of this invention,
generally speaking, may
be practiced using any mode of administration that is medically acceptable,
meaning any mode that
produces effective levels of the active compounds without causing clinically
unacceptable adverse
effects. Such modes of administration include oral, rectal, sublingual,
topical, nasal, transdermal
or parenteral routes. The term "parenteral" includes subcutaneous,
intravenous, intramuscular, or
t 5 infusion. Intravenous routes are preferred.
The compositions may conveniently be presented in unit dosage form and may be
prepared
by any of the methods well known in the art of pharmacy. All methods include
the step of bringing
the conjugates of the invention into association with a carrier which
constitutes one or more
accessory ingredients. In general, the compositions are prepared by uniformly
and intimately
2o bringing the compounds into association with a liquid carrier, a finely
divided solid carrier, or both,
and then, if necessary, shaping the product.
Compositions suitable for oral administration may be presented as discrete
units such as
capsules, cachets, tablets, or lozenges, each containing a predetermined
amount of the active
compound. Other compositions include suspensions in aqueous liquors or non-
aqueous liquids such
25 as a syrup, an elixir, or an emulsion.
Other delivery systems can include time-release, delayed release or sustained
release delivery
systems. Such systems can avoid repeated administrations of the active
compounds of the invention,
increasing convenience to the subject and the physician. Many types of release
delivery systems are
available and known to those of ordinary skill in the art. 'they include
polymer based systems such
3o as polylactic and polyglycolic acid, polyanhydrides and polycaprolactone;
nonpolymer systems that
are lipids including sterols such as cholesterol, cholesterol esters and fatty
acids or neutral fats such
as mono-, di and triglycerides; hydrogel release systems; silastic systems;
peptide based systems;
CA 02255615 1998-11-18
WO 97144026 PCT/US97108866
- 25 -
wax coatings, compressed tablets using conventional binders and excipients,
partially fused implants
and the like. In addition, a pump-based hardware delivery system can be used,
some of which are
adapted for implantation.
A long-term sustained release implant also may be used. "Long-term" release,
as used
herein, means that the implant is constructed and arranged to deliver
therapeutic levels of the active
ingredient for at least 30 days, and preferably 60 days. Long-term sustained
release implants are
well known to those of ordinary skill in the art and include some of the
release systems described
above. Such implants can be particularly useful in treating solid tumors by
placing the implant near
or directly within the tumor, thereby affecting localized, high-doses of the
compounds of the
invention.
The analogs of the invention also are useful, in general, for treating
mammalian cell
proliferative disorders other than cancer, including psoriasis, actinic
keratosis, etc.
Those skilled in the art will be able to recognize with no more than routine
experimentation
numerous equivalents to the specific products and processes described above.
Such equivalents are
intended to be included within the scope of the appended claims.
We claim as follows: