Note: Descriptions are shown in the official language in which they were submitted.
CA 022~6716 1998-11-26
wo 98/09934 PCTIUS97/14859
-I -
~SATRDX ~nFrALLOPROTEnNASE nNHIBr~RS AND THEDR THERAPEUllC USES
FIELD OF THE ~NVENTION
The present invention relates to a method of inhibiting matrix
metalloproteinases using compounds that are dibenzofuran sulfonamide
derivatives. More particularly, the present invention relates to a method of treating
diseases in which matrix metalloproteinases are involved such as multiple
sclerosis, atherosclerotic plaque rupture? restenosis? aortic aneurism? heart failure,
periodontal disease, corneal ulceration? burns? decubital ulcers? chronic ulcers or
wounds, cancer metastasis, tumor angiogenesis, arthritis, or other autoimmune orinfl~mm~tory diseases dependent upon tissue invasion by leukocytes.
BACKGROUND OF THE INVENTION
The compounds of the present invention are inhibitors of matrix
metalloproteinases, e.g., stromelysin-l and gelatinase A (72 kDa gelatinase).
Stromelysin-1 and gelatinase A are members of the matrix
metalloprotçin~e~ (MMP). Other members include fibroblast collagenase,
neutrophil collagenase, gelatinase B (92 kDa gelatinase)? stromelysin-2,
stromelysin-3, matrilysin, collagenase 3, and the newly discovered membrane-
associated matrix metalloproteinases (Sato H., Takino T.? Okada Y.? Cao J.,
Shinagawa A., Yamamoto E., and Seiki M., Nature, 1994;370:61-65).
Stromelysin-1 is also known as MMP03 and gelatinase A is known as
MMP02. In addition, several other matrix metalloproteinases are known:
MMP01 - Fibroblast collagenase;
MMP07 - Matrilysin;
MMP09 Gelatinase B; and
MMP 13 - Collagenase -3.
The catalytic zinc in matrix metalloproteinases is typically the focal point
for inhibitor design. The modification of substrates by introducing chelating
groups has generated potent inhibitors such as peptidehydrox~m~t~s and thiol-
cont~ining peptides. Peptide hydrox~m~tes and the natural endogenous inhibitors
CA 022~6716 1998-11-26
WO 98/09934 PCT/US97/14859
of MMPs (TIMPs) have been used successfully to treat animal models of cancer
and inflamm~tion.
The ability of the matrix metalloproteinases to degrade various
components of connective tissue makes them potential targets for controlling
pathological processes. For example, the rupture of atherosclerotic plaques is the
most common event initiating coronary thrombosis. Destabilization and
degradation of the extracellular matrix surrounding these plaques by MMPs has
been proposed as a cause of plaque fissuring. The shoulders and regions of foam
cell accumulation in human atherosclerotic plaques show locally increased
expression of gelatinase B, stromelysin- 1, and interstitial collagenase. In situ
zymography of this tissue revealed increased gelatinolytic and caseinolytic activity
(Galla Z.S., Sukhova G.K., Lark M.W., and Libby P., "Increased expression of
matrix metalloproteinases and matrix degrading activity in vulnerable regions ofhuman atherosclerotic plaques", J. Clin. Invest., 1994;94:2494-2503~. In addition,
high levels of stromelysin RNA message have been found to be localized to
individual cells in atherosclerotic plaques removed from heart transplant patients
at the time of surgery (Henney A.M., Wakeley P.R., Davies M.J., Foster K.,
Hembry R., Murphy G., and Humphries S., "Localization of stromelysin gene
expression in atherosclerotic plaques by in situ hybridization," Proc. Nat'l. Acad.
Sci., 1991 ;88:8154-8158).
Inhibitors of matrix metalloproteinases will have utility in treating
degenerative aortic disease associated with thinning of the medial aortic wall.
Increased levels of the proteolytic activities of MMPs have been identified in
patients with aortic aneurisms and aortic stenosis (Vine N. and Powell J.T.,
"Metalloproteinases in degenerative aortic diseases," Clin. Sci., 1991;81:233-239).
Heart failure arises from a variety of diverse etiologies, but a common
characteristic is cardiac dilation which has been identified as an independent risk
factor for mortality (Lee T.H., Hamilton M.A., Stevenson L.W., Moriguchi J.D.,
Fonarow G.C., Child J.S., Laks H., and Walden J.A., "Impact of left ventricular
size on the survival in advanced heart failure," Am. J. Cardiol., 1993;72:672-676).
This remodeling of the failing heart appears to involve the breakdown of
extracellular matrix. Matrix metalloproteinases are increased in patients with both
CA 022~6716 1998-11-26
WO 98/09934 PCT/US97/14859
idiopathie and ischemie heart failure (Reddy H.K., Tyagi S.C., Tjaha I.E.,
Voelker D.J., Campbell S.E., and Weber K.T., "Activated myocardial collagenase
in idiopathic dilated cardiomyopathy," Clin. Res., 1993;41:660A; Tyagi S.C.,
Reddy H.K., Voelker D., Tjara I.E., and Weber K.T., "Myocardial collagenase in
failing human heart," Clin. Res., 1993;41:6XlA). Animal models of heart failure
have shown that the induction of gelatinase is important in cardiac dilation
(Armstrong P.W., Moe G.W., Howard R.J., Grima E.A., and Cruz T.F.,
"Structural remodeling in heart failure: gelatinase induction," Can. J. Cardiol.,
1994;10:214-220), and cardiac dilation precedes profound deficits in cardiac
function (Sabbah H.N., Kono T., Stein P.D., Mancini G.B., and Goldstein S.,
"Left ventricular shape changes during the course of evolving heart failure,"
Am. J. PhYsiol., 1992;263:H266-H270). Neointimal proliferation, leading to
restenosis, frequently develops after coronary angioplasty. The migration of
vascular smooth muscle cells (VSMCs) from the tunica media to the neointima is
a key event in the development and progression of many vascular diseases and a
highly predictable consequence of mechanical injury to the blood vessel
(Bendeck M.P., Zempo N., Clowes A.W., Galardy R.E., and Reidy M., "Smooth
muscle cell migration and matrix metalloproteinase expression after arterial injury
in the rat," Circulation Research, 1994;75:539-545). Northern blotting and
zymographic analyses indicated that gelatinase A was the principal MMP
expressed and excreted by these eells. Further, antisera capable of seleetively
neutralizing gelatinase A activity also inhibited VSMC migration across basementmembrane barrier. After injury to the vessel, gelatinase A activity increased more
than 20-fold as VSCMs under~vent the transition from a quiescent state to a
proliferating, motile phenotype (Pauly R.R., Passaniti A., Bilato C., Monticone R.,
Cheng L., Papadopoulos N., Gluzband Y.A., Smith L., Weinstein C., Lakatta E.,
and Crow M.T., "Migration of cultured vascular smooth muscle cells through a
basement membrane barrier requires type IV collagenase activity and is inhibitedby cellular differentiation," Cireulation Research, 1994;75:41-54).
Collagenase and stromelysin activities have been demonstrated in
fibroblasts isolated from inflamed gingiva (Uitto V.J., Applegren R., and
Robinson P.J., "Collagenase and neutral metalloproteinase activity in extracts
. . .. ~ . . ~ ........... . .
CA 022~6716 1998-11-26
W 098/09934 PCT~US97/14859
from inflamed human gingiva," J. Periodontal Res., 1981;16:417-424), and
enzyme levels have been correlated to the severity of gum disease (Overall C.M.,Wiebkin O.W., and Thonard J.C., "Demonstrations of tissue collagenase activity
in vivo and its relationship to infl~mm~tion severity in human gingiva,"
J. Periodontal Res., 1987;22:81-88). Proteolytic degradation of extracellular
matrix has been observed in corneal ulceration following alkali burns (Brown S.I.,
Weller C.A., and Wasserman H.E., "Collagenolytic activity of alkali burned
corneas," Arch. Opthalmol., 1969;81 :370-373). Thiol-cont~ining peptides inhibitthe collagenase isolated from alkali-burned rabbit corneas (Burns F.R.,
Stack M.S., Gray R.D., and Paterson C.A., Invest. Opththamol.,
1989;30: 1569-1575).
Stromelysin is produced by basal keratinocytes in a variety of chronic
ulcers (Saarialho-Kere U.K., Ulpu K., Pentland A.P., Birkedal-Hansen H.,
Parks W.C., Welgus H.G., "Distinct populations of basal keratinocytes express
stromelysin-l and stromelysin-2 in chronic wounds," J. Clin. Invest.,
1994;94:79-88).
Stromelysin- 1 mRNA and protein were detected in basal keratinocytes
adjacent to but distal from the wound edge in what probably represents the sites of
proliferating epidermis. Stromelysin-l may thus prevent the epidermis from
healing. Davies, et al., (Cancer Res., 1993;53:2087-2091) reported that a peptide
hydroxamate, BB-94, decreased the tumor burden and prolonged the survival of
mice bearing human ovarian carcinoma xenografts. A peptide of the conserved
MMP propeptide sequence was a weak inhibitor of gel~tin~e A and inhibited
human tumor cell invasion through a layer of reconstituted basement membrane
(Melchiori A., Albili A., Ray J.M., and Stetler-Stevenson W.G., Cancer Res.,
1992;52:2353-2356), and the natural tissue inhibitor of metalloproteinase-2
(TIMP-2) also showed blockage of tumor cell invasion in in vitro models
(DeClerck Y.A., Perez N., Shimada H., Boone T.C., Langley K.E., and
Taylor S.M., Cancer Res., 1992;52:701-708). Studies of human cancers have
shown that gelatinase A is activated on the invasive tumor cell surface
(Strongin A.Y., Marmer B.L., Grant G.A., and Goldberg G.I., J. Biol Chem.,
1993;268:14033-14039) and is retained there through interaction with a
CA 022~6716 1998-11-26
WO 98/09934 PCT/USg7/14859
receptor-like molecule (Monsky W.L., Kelly T., Lin C.-Y., Yeh Y.,
Stetler-Stevenson W.G., Mueller S.C., and Chen W.-T., Cancer Res.,
1993 ;53 :3159-3164). Inhibitors of MMPs have shown activity in models of tumor
angiogenesis (Taraboletti G., Garofalo A., Belotti D., Drudis T., Borsotti P.,
Sc~n7i~ni E., Brown P.D., and Giavazzi R., Journal of the National Cancer
Institute, 1995;87:293; and Benelli R., Adatia R., Ensoli B.,
Stetler-Stevenson W.G., Santi L., and Albini A., Oncolo~ Research,
1994;6:251 -257).
Several investigators have demonstrated consistent elevation of
stromelysin and collagenase in synovial fluids from rheumatoid and osteoarthritis
patients as compared to controls (Walakovits L.A., Moore V.L., Bhardwaj N.,
Gallick G.S., and Lark M.W., "Detection of stromelysin and collagenase in
synovial fluid from patients with rheumatoid arthritis and post-traumatic knee
injury," Arthritis Rheum., 1992;35:35-42; Zafarullah M., Pelletier J.P.,
Cloutier J.M., and Marcel-Pelletier J., "Elevated metalloproteinases and tissue
inhibitor of metalloproteinase mRNA in human osteoarthritic synovia,"
J. Rheumatol., 1993;20:693-697). TIMP-1 and TIMP-2 prevented the formation of
collagen fragments, but not proteoglycan fragments, from the degradation of boththe bovine nasal and pig articular cartilage models for arthritis, while a synthetic
peptide hydroxamate could prevent the forrnation of both fragments
(Andrews H.J., Plumpton T.A., Harper G.P., and Cawston T.E., A~ents Actions,
1992;37:147-154; Ellis A.J., Curry V.A., Powell E.K., and Cawston T.E.,
Biochem. BiophYs. Res. Commun., 1994;201:94-101).
Gijbels, et al., (J. Clin. Invest., 1994;94:2177-2182) recently described a
peptide hydroxamate, GM6001, that suppressed the development or reversed the
clinical expression of experimental allergic encephalomyelitis (EAE) in a dose
dependent manner, suggesting the use of MMP inhibitors in the treatment of
autoimmune inflAmm~tory disorders such as multiple sclerosis. A recent study by
Madri has elucidated the role of gelatinase A in the extravasation of T-cells from
the blood stream during infl~mm~tion (Ramanic A.M. and Madri J.A., "The
Induction of 72-lcD Gelatinase in T Cells upon Adhesion to Endothelial Cells is
VCAM-1 Dependent," J. Cell Biolo~, 1994;125:1165-1178). This tr~n~migration
.. ., ~ ,.. .. . ....
CA 022~6716 1998-11-26
W O 98/09934 PCT~US97/148S9
-6-
past the endothelial cell layer is coordinated with the induction of gelatinase A and
is mediated by binding to the vascular cell adhesion molecule-1 (VCAM-1). Once
the barrier is compromised, edema and infl~mm~tion are produced in the CNS.
~eukocytic migration across the blood-brain barrier is known to be associated with
the infl~mm~tory response in EAE. Inhibition of the metalloproteinase
gelatinase A would block the degradation of extracellular matrix by activated
T-cells that is necessary for CNS penetration.
These studies provided the basis for the belief that an inhibitor of
stromelysin-1 and/or gelatinase A will treat diseases involving disruption of
extracellular matrix resulting in infl~mm~tion due to Iymphocytic infiltration,
inappropriate migration of metastatic or activated cells, or loss of structural
integrity necessary for organ function.
We have identified a series of tricyclic aromatic sulfonamide compounds
that are inhibitors of matrix metalloproteinases, particularly stromelysin-1 andgelatinase A, and thus useful as agents for the treatment of multiple sclerosis,atherosclerotic plaque rupture, restenosis, aortic aneurism, heart failure,
periodontal disease, corneal ulceration, burns, decubital ulcers, chronic ulcers or
wounds, cancer metastasis, tumor angiogenesis, arthritis, or other autoimmune orinfl~mm~tory diseases dependent upon tissue invasion by leukocytes.
SUMMARY OF THE INVENTION
The present invention provides a method of inhibiting a matrix
metalloproteinase in a patient in need of matrix metalloproteinase inhibition
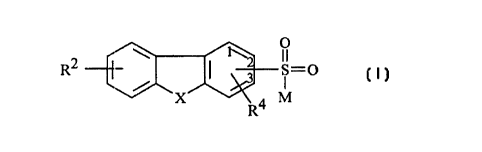
comprising ~tlmini~tering to the patient a therapeutically effective amount of acompound of Formula I
RZ ~ J~~
CA 022~6716 1998-11-26
WO 98/09934 PCT/US97/14859
wherein M is a natural (L) alpha amino acid derivative having the structure
COR
--N--H
H R
X is O, S, S(O)n~ CH2, CO, or NRQ;
RQ is hydrogen, C1-C6 alkyl, or -C1-C6 alkyl-phenyl;
R is a side chain of a natural alpha amino acid;
R1 is C1-Cs alkoxy, hydroxy, or-NHOR5;
R2 and R4 are independently hydrogen, -C 1 -C5 alkyl, phenyl -NO2, halogen,
-OR5, -CN, -Co2R5, -So3R5, -CHO, -CoR5, -CoNR5R6,
-(CH2)nNR5R6, -CF3, or-NHCOR5;
each R5 and R6 are independently hydrogen or C1-Cs alkyl; and
n is 0 to 2, and the pharmaceutically acceptable salts, esters, amides and prodrugs
thereof.
In one embodiment of the invention of Formula I, X is O.
In another embodiment of the invention of Formula I, X is S.
In another embodiment of the invention of Formula I, X is CH2.
In another embodiment of the invention of Formula I, X is NRQ.
In a plef~lied embodiment of the invention of Formula I, X is O and R2
and R4 are hydrogen.
In another embodiment of the invention of Formula I, X is CO.
In another embodiment of the invention of Formula I, X is S(O)n.
In another preferred embodiment of the invention of Formula I, R1 is
hydroxy, C1-Cs alkoxy, -NHOH, or-NHObenzyl.
In still another preferred embodiment, R is the side chain of the natural
alpha amino acid glycine, alanine, valine, leucine, isoleucine, cysteine, aspartic
acid, or phenyl~l~nine.
CA 022~6716 1998-11-26
W O 98109934 PCT~US97114859
In another embodiment, the present invention provides a method of
inhibiting a matrix metalloproteinase in a patient in need of matrix
metalloproteinase inhibition, the method comprising ~f~mini~tering to the patient a
therapeutically effective amount of a compound of Formula II
R' ~ S = O
wherein Z is a natural (L) amino acid derivative having the structure
cORa
- HN H
Rb
R2 and R4 are independently hydrogen, -Cl-Cs alkyl, phenyl -NO2, halogen,
-OR5, -CN, -CO2R5, -SO3R5, -CHO, -COR5, -CoNR5R6,
-(CH2)nNR5R6, -CF3, or-NHCOR5;
each R5 and R6 are independently hydrogen or C1-Cs alkyl;
Ra is C1-Cs alkoxy, hydroxy, or -NHORC;
Rb is a side chain of a natural alpha amino acid; and
Rc is hydrogen, C1-Cs alkyl, or -CH2 phenyl; and
n is 0 to 2, and the pharrnaceutically acceptable salts, esters, amides, and prodrugs
thereof.
In a plc;f~,,ed embodiment of the method comprising Formula II, the group
o
-S=O is located at the 2-position of the phenyl ring.
CA 022~6716 1998-11-26
WO 98/09934 PCT/US~7/14859
In another preferred embodiment of the method comprising Formula II, the
o
group -S=O is located at the 3-position of the phenyl ring.
Also provided is a method of inhibiting a matrix metalloproteinase in a
patient in need of matrix metalloproteinase inhibition, the method comprising
~lmini~tering to the patient a therapeutically effective amount of a compound of
Formula III
R2 --~ SJ~s=o III
wherein Z is a natural (L) amino acid derivative having the structure
cORa
H
Rb
R2 and R4 are independently hydrogen, -C 1 -C5 alkyl, phenyl -NO2, halogen,
-oR5,-CN,-Co2R5,-So3R5,-CHo~-coR5~-coNR5R
-(CH2)nNR5R6,-CF3, or-NHCORS;
each RS and R6 are independently hydrogen or Cl-Cs alkyl;
Ra is C 1 -C5 alkoxy, hydroxy, or -NHORC;
Rb is a side chain of a natural alpha amino acid; and
Rc is hydrogen, Cl-Cs alkyl, or -CH2 phenyl; and
n is 0 to 2, and the ph~ eutically acceptable salts, esters, amides, and prodrugs
thereof.
. . .
CA 022~6716 1998-11-26
WO 98/09934 PCT/US97/14859
-10-
Also provided is a method of inhibiting a matrix metalloproteinase in a
patient in need of matrix metalloproteinase inhibition, the method comprising
~(lmini~tering to the patient a therapeutically effective amount of a compound of
Formula IV.
R2 ~'CH~ I =~ IV
wherein Z is a natural (L) amino acid derivative having the structure
cORa
--N H
H Rb
R2 and R4 are independently hydrogen, -C I -Cs alkyl, phenyl -NO2, halogen,
-oR5, -CN, -Co2R5, -So3R5, -CHO, -CoR5, -CoNR5R6,
-(CH2)nNR5R6, -CF3, or-NHCoR5;
each R5 and R6 are independently hydrogen or Cl-Cs alkyl;
Ra is C1-Cs alkoxy, hydroxy, or -NHORC;
Rb is a side chain of a natural alpha amino acid; and
Rc is hydrogen, Cl-Cs alkyl, or -CH2 phenyl; and
n is 0 to 2, and the pharmaceutically acceptable salts, esters, amides, and prodrugs
thereof.
Also provided is a method of inhibiting a matrix metalloproteinase in a
patient in need of matrix metalloproteinase inhibition, the method comprising
a~lmini~tering to the patient a therapeutically effective amount of a compound of
Forrnula V
.. .. .... ... . , . . . . ..
CA 022~6716 1998-11-26
W O 98/09934 PCTrUS97/148~9
~C~IIl=O V
wherein Z is a natural (L) amino acid derivative having the structure
cORa
H
Rb
R2 and R4 are independently hydrogen, -C 1 -C5 alkyl, phenyl -NO2, halogen,
-oR5, -CN, -Co2R5, -So3R5, -CHO, -CoR5, -CoNR5R6,
-(CH2)nNR5R6, -CF3, or-NHCOR5;
each R5 and R6 are independently hydrogen or C1-Cs alkyl;
Ra is Cl-Cs alkoxy, hydroxy, or -NHORC;
Rb is a side chain of a natural alpha amino acid; and
Rc is hydrogen, C I -Cs alkyl, or -CH2 phenyl; and
n is 0 to 2, and the ph~ ceutically acceptable salts, esters, amides, and prodrugs
thereof.
Also provided is a method of inhibiting a matrix metalloproteinase in a
patient in need of matrix metalloproteinase inhibition, the method comprising
~lmini.ctering to the patient a therapeutically effective amount of a compound of
Formula VI
R2 ~ =0 VI
CA 022~6716 1998-11-26
WO 98/09934 PCT/US97/14859
-12-
wherein Z is a natural (L) amino acid derivative having the structure
cORa
H
Rb
R2 and R4 are independently hydrogen, -C 1 -C5 alkyl, phenyl -NO2, halogen,
-OR5, -CN, -CO2RS, -So3R5, -CHO, -CoR5, -CoNR5R6,
-(CH2)nNR5R6, -CF3, or-NHCOR5;
each R5 and R6 are independently hydrogen or Cl-Cs alkyl;
Ra is Cl-Cs alkoxy, hydroxy, or -NHORC;
nisOto2;
Rb is a side chain of a natural alpha amino acid; and
Rc is hydrogen, Cl-Cs alkyl, or -CH2 phenyl; and
n is 0 to 2, and the pharmaceutically acceptable salts, esters, amides, and prodrugs
thereof.
Also provided is a method of inhibiting a matrix metalloproteinase in a
patient in need of matrix metalloproteinase inhibition, the method comprising
~lmini~tering to the patient a therapeutically effective arnount of a compound of
Formula VII
R2 ~NR~J~ 1=~ VII
wherein Z is a natural (L) arnino acid derivative having the structure
,~ . , , -- I
CA 022~6716 1998-11-26
WO 98/09934 PCT/US97114859
-13-
cORa
--N H
H Rb
R2 and R4 are independently hydrogen, -Cl-Cs alkyl, phenyl -NO2, halogen,
-oR5, -CN, -Co2R5, -So3R5, -CHO, -CoR5, -CoNR5R6,
-(CH2)nNR5R6, -CF3, or -NHCoR5;
S each R5 and R6 are independently hydrogen or C I -Cs alkyl;
Ra is C1-Cs alkoxy, hydroxy, or -NHORC;
RQ is hydrogen, C1-C6 alkyl, or C1-C6 alkyl-phenyl;
Rb is a side chain of a natural alpha arnino acid; and
Rc is hydrogen, C1-Cs alkyl, or -CH2 phenyl; and
n is 0 to 2, and the ph~ ceutically acceptable salts, esters, amides, and prodrugs
thereof.
In a most preferred embodiment, the compound of Formula I-VIII is:
(L)-2-(dibenzofuran-2-sulfonylamino)-4-methyl-pentanoic acid;
(L)-2-(dibenzofuran-2-sulfonylamino)-3-methyl-pentanoic acid;
(L)-2-(dibenzofuran-2-sulfonylamino)-3-phenyl-propionic acid;
(L)-2-(dibenzofuran-2-sulfonylamino)-propionic acid;
(L)-2-(dibenzofuran-2-sulfonylamino)-3-methyl-butyric acid;
(L)-2-(dibenzofuran-2-sulfonylamino)-acetic acid;
(L)-2-(dibenzofuran-2-sulfonylamino)-succinic acid;
(L)-2-(dibenzofuran-2-sulfonylamino)-3-tritylsulfanyl-propionic acid;
(L)-2-(dibenzofuran-2-sulfonylamino)-3-mercapto-propionic acid;
(L)-2-(dibenzofuran-2-sulfonylamino)-3-methyl-pentanoic acid
hydroxyamide;
(L)-2-(dibenzofuran-2-sulfonylamino)-acetic acid tert-butyl ester;
(L)-2-(diben~ofuran-2-sulfonylamino)-propionic acid tert-butyl ester;
(L)-2-(dibenzofuran-2-sulfonylamino)-propionic acid tert-butyl ester;
, , . ~ ... . .. .. .......
CA 022~6716 1998-11-26
W O 98109934 PCT~US97/14859
-14-
(L)-2-(dibenzofuran-2-sulfonylamino)-4-methyl-pentanoic acid tert-butyl
ester;
(L)-2-(dibenzofuran-2-sulfonylamino)-3-methyl-pentanoic acid tert-butyl
ester;
S (L)-2-(dibenzofuran-2-sulfonylamino)-3-methyl-pentanoic acid benzyloxy-
amide;
(L)-2-(dibenzofuran-2-sulfonylamino)-3-phenyl-propionic acid tert-butyl
ester;
(L)-2-(dibenzofuran-3-sulfonylamino)-3-methyl-butyric acid;
3-Methyl-2-(9-methyl-9H-carbazole-3-sulfonylamino)-butyric acid;
2-(9-Benzyl-9H-carbazole-3-sulfonylamino)-3-methyl-butyric acid;
(L)-2-(9H-Fluorene-2-sulfonylamino)-3-methyl-butyric acid;
(L)-2-(5 ,5-Dioxo-SH-5~6-dibenzothiophene-3-sulfonylamino)-3 -methyl-
butyric acid;
(L)-2-(Dibenzothiophene-2-sulfonylamino)-3-methyl-butyric acid;
(L)-2-(7-Bromo-dibenzofuran-2-sulfonylamino)-3-methyl-butyric acid;
(L)-3-Methyl-2-(7-phenyl dibenzofuran-2-sulfonylamino)-butyric acid; and
2-(9H-Carbazole-3-sulfonylamino)-3-methyl-butyric acid.
Also provided by the present invention is a method of treating multiple
sclerosis, the method comprising ~lmini~tçring to a patient having multiple
sclerosis a therapeutically effective amount of a compound of Formula I-VIII.
Also provided by the present invention is a method of treating
atherosclerotic plaque rupture, the method comprising ~1mini~tering to a patienthaving an atherosclerotic plaque at risk for rupture a therapeutically effectiveamount of a compound of Formula I-VIII.
Also provided by the present invention is a method of treating or
preventing restenosis, the method comprising ~(lmini~tering to a patient having
restenosis or at risk of having restenosis a therapeutically effective amount of a
compound of Formula I-VIII.
CA 022~6716 1998-11-26
WO 98/09934 PCT/US97/14859
-15-
Also provided by the present invention is a method of treating aortic
aneurism, the method comprising a(lmini.stering to a patient having aortic
aneurism a therapeutically effective amount of a compound of Formula I-VIII.
Also provided by the present invention is a method of treating heart failure,
the method comprising ~rlmini.~tering to a patient having heart failure a
therapeutically effective amount of a compound of Formula I-VIII.
Also provided by the present invention is a method of treating periodontal
disease, the method comprising ~(1mini~tering to a patient having periodontal
disease a therapeutically effective amount of a compound of Formula I-VIII.
Also provided by the present invention is a method of treating corneal
ulceration, the method comprising :~(lmini~tering to a patient having corneal
ulceration a therapeutically effective amount of a compound of Formula I-VIII.
Also provided by the present invention is a method of treating burns, the
method comprising ~(lmini.~tering to a patient having burns a therapeutically
l S effective amount of a compound of Formula I-VIII.
Also provided by the present invention is a method of treating decubital
ulcers, the method comprising a~mini~tering to a patient having decubital ulcers a
therapeutically effective amount of a compound of Formula I-VIII.
Also provided by the present invention is a method of treating chronic
ulcers or wounds, the method comprising a.~mini~tering to a patient having
chronic ulcers or wounds a therapeutically effective amount of a compound of
Formula I-VIII.
Also provided by the present invention is a method of treating cancer
met~ct~i.c, the method comprising ~iministçring to a patient having cancer
metastasis a therapeutically effective amount of a compound of Forrnula I-VIII.
Also provided by the present invention is a method of treating tumor
angiogenesis, the method comprising allmini~tering to a patient having tumor
angiogenesis a therapeutically effective amount of a compound of Formula I-VIII.Also provided by the present invention is a method of treating arthritis, the
method comprising ~lmini.~tering to a patient having arthritis a therapeuticallyeffective amount of a compound of Formula I-VIII.
CA 022~6716 1998-ll-26
W O 98/09934 PCTrUS97/14859
-16-
Also provided by the present invention is a method of treating autoimmune
or infl~mm~tory diseases dependent upon tissue invasion by leukocytes, the
method comprising ~lministering to a patient having autoimmune or infl~mm~tory
diseases dependent upon tissue invasion by leukocytes a therapeutically effective
S arnount of a compound of Forrnula I-VIII.
The present invention also provides compounds of Formula I
R2 ~3''X '~ I =~
wherein M is a natural (L) alpha amino acid derivative having the structure
cORa
--H H
Rb
X is S, S(O)n~ CH2, CO, or NRQ;
Rb is a side chain of a natural alpha arnino acid;
RQ is hydrogen, C1-C6 alkyl, or -Cl-C6 alkyl-phenyl;
Ra is Cl-Cs alkoxy, hydroxy, or -NHoR5;
R2 and R4 are independently hydrogen, -C 1 -C5 alkyl, phenyl -NO2, halogen,
-oR5 , -CN, -CO2RS , -SO3RS , -CHO, -CORS , -CoNR5R6,
-(CH2)nNR5R6, -CF3, or-NHCORS;
each R5 and R6 are independently hydrogen or Cl-Cs alkyl; and
n is 0 to 2, and the ph~nTI~re~ltically acceptable salts, esters, amides, and prodrugs
thereof.
The present invention also provides compounds of Forrnula VIII
CA 02256716 1998-11-26
WO 98/09934 PCT/US97/14859
R2~o~O Vlll
wherein M is a natural (L) alpha amino acid derivative having the structure
cORa
--NH H
Rb
R2 and R4 are indepen~le~tly hydrogen, -C I -Cs alkyl, phenyl -N02, halogen,
5-oR5, -CN, -CO2RS, -So3R5, -CHO, -CoR5, -CoNR5R6, -(CH2)nNR5R6,
-CF3, or -NHCoR5;
Rb is a side chain of a natural alpha amino acid;
Ra is Cl-Cs alkoxy, hydroxy, or -NHoR5;
R2 and R4 are independently hydrogen, -C1-Cs alkyl, phenyl -NO2, halogen,
10-oR5 , -CN, -Co2R5 , -So3R5 , -CHO, -CoR5, -CoNR5R6,
-(CH2)nNR5R6, -CF3, or-NHCOR5;
each R5 and R6 are independently hydrogen or Cl-Cs alkyl; and
n is 0 to 2, and the ph~ ceutically acceptable salts, esters, amides, and prodrugs
thereof.
15DETAILED DESCRIPTION OF THE INVENTION
The present invention provides a method of inhibiting a matrix
metalloproteinase in a patient in need of matrix metalloproteinase inhibition
comprising ~lmini~tçring to the patient a therapeutically effective amount of a
compound of Forrnula I
CA 022~6716 1998-11-26
W O 98/09934 PCTrUS97/14859
-18-
R2 ~1~ ~ --~u
wherein M is a natural (L) alpha amino acid derivative having the structure
coRl
--N H
H R
X is O, S, S(O)n~ CH2, CO, or NRQ;
R is a side chain of a natural alpha amino acid;
R1 is Cl-Cs alkoxy, hydroxy, or -NHoR5;
R2 and R4 are independently hydrogen, -C I -Cs alkyl, phenyl -NO2, halogen,
-OR5, -CN, -Co2R5, -So3R5, -CHO, -CoR5, -CoNR5R6,
-(CH2)nNR5R6, -CF3, or-NHCOR5;
each R5 and R6 are independently hydrogen or C 1 -C5 alkyl; and
n is 0 to 2, and the pharmaceutically acceptable salts, esters, amides, and prodrugs
thereof.
The term "alkyl" means a straight or branched chain hydrocarbon.
Represçnt~tive examples of alkyl groups are methyl, ethyl, propyl, isopropyl,
isobutyl, butyl, tert-butyl, sec-butyl, pentyl, and hexyl.
The term "alkoxy" means an alkyl group attached to an oxygen atom.
Representative examples of alkoxy groups include methoxy, ethoxy, tert-butoxy,
propoxy, and isobutoxy.
The term "halogen" includes chlorine, fluorine, bromine, and iodine.
The term "phenyl" also includes substituted phenyl wherein one or more
hydrogen on the phenyl ring is replaced with an organic radical. Examples of
CA 022~6716 1998-11-26
W O 98/09934 PCTrUS97tl4859
-19-
suitable substituents include, but are not limited to, halogen, C 1 -C6 alkoxy, -CF3,
-NO2, -NH2, -NH(CI-C6 alkyl), or -N(Cl-C6 alkyl)2.
The symbol "-" means a bond.
The term "side chain of a natural alpha amino acid" means the group Q in
a natural amino acid of formula H2N-CH(Q)-COOH. Examples of side chains of
natural alpha amino acids include those of alanine, arginine, asparagine, aspartic
acid, cysteine, glutarnic acid, glycine, histidine, isoleucine, leucine, Iysine,methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine and
vallne.
A natural alpha arnino acid is an amino acid found in a living org;~ni~m
Examples of such amino acids include glycine, alanine, valine, leucine, isoleucine,
phenylalanine, proline, serine, threonine, tyrosine, asparagine, glutamine, Iysine,
arginine, tryptophan, histidine, cysteine, methionine, aspartic acid, and glutamic
acid.
The functional groups in the amino acid side chains can be protected. For
example, carboxyl groups can be esterified, amino groups can be converted to
amides or carbamates, hydroxyl groups can be converted to ethers or esters, and
thiol groups can be converted to thioethers or thioesters.
The compounds of Formula I-VIII can be a-lmini~tered to a patient either
alone or as part of a pharmaceutically acceptable composition. The compositions
can be ~tlmini~tered to patients such as humans and ~nim~l~ either orally, rectally,
parenterally (intravenously, intramuscularly or subcutaneously), intracisternally,
intravaginally, intraperitoneally, intravesically, locally (powders, ointments or
drops), or as a buccal or nasal spray.
Compositions suitable for parenteral injection may comprise
physiologically acceptable sterile aqueous or nonaqueous solutions, dispersions,suspensions or emulsions, and sterile powders for reconstitution into sterile
injectable solutions or dispersions. Exarnples of suitable aqueous and nonaqueous
carriers, diluents, solvents, or vehicles include water, ethanol, polyols
(propyleneglycol, polyethyleneglycol, glycerol, and the like), suitable mixturesthereof, vegetable oils (such as olive oil), and injectable organic esters such as
CA 022~6716 1998-11-26
WO 98/09934 PCT/US97/14859
-20-
ethyl oleate. Proper fluidity can be m~int~ined, for example, by the use of a
coating such as lecithin, by the m~inten~nce of the required particle size in the
case of dispersions, and by the use of surfactants.
These compositions may also contain adjuvants such as preserving,
wetting, emulsifying, and dispensing agents. Prevention of the action of
microorg~ni~m.~ can be ensured by various antibacterial and antifungal agents, for
example, parabens, chlorobutanol, phenol, sorbic acid, and the like. It may also be
desirable to include isotonic agents, for example sugars, sodium chloride, and the
like. Prolonged absorption of the injectable pharmaceutical form can be brought
about by the use of agents delaying absorption, for example, aluminum
monostearate and gelatin.
Solid dosage forms for oral a~lmini~tration include capsules, tablets, pills,
powders, and granules. In such solid dosage forms, the active compound is
admixed with at least one inert customary excipient (or carrier) such as sodium
citrate or dicalcium phosphate or (a) fillers or extenders, as for example, starches,
lactose, sucrose, glucose, mannitol, and silicic acid, (b) binders, as for example,
carboxymethylcellulose, ~lign~tes, gelatin, polyvinylpyrrolidone, sucrose, and
acacia, (c) humectants, as for example, glycerol, (d) disintegrating agents, as for
example, agar-agar, calcium carbonate, potato or tapioca starch, alginic acid,
certain complex silicates, and sodium carbonate, (e) solution retarders, as for
example paraffin, (f) absorption accelerators, as for example, quaternary
ammonium compounds, (g) wetting agents, as for example, cetyl alcohol and
glycerol monostearate, (h) adsorbents, as for example, kaolin and bentonite, and(i) lubricants, as for example, talc, calcium stearate, magnesium stearate, solid
polyethylene glycols, sodium lauryl sulfate or mixtures thereof. In the case of
capsules, tablets, and pills, the dosage forms may also comprise buffering agents.
Solid compositions of a similar type may also be employed as fillers in soft
and hard-filled gelatin capsules using such excipients as lactose or milk sugar as
well as high molecular weight polyethyleneglycols, and the like.
Solid dosage forms such as tablets, dragees, capsules, pills, and granules
can be prepared with coatings and shells, such as enteric coatings and others well-
known in the art. They may contain opacifying agents, and can also be of such
' T
CA 022~6716 1998-11-26
WO 98/09934 PCT/US97/14859
composition that they release the active compound or compounds in a certain partof the intestinal tract in a delayed manner. Examples of embedding compositions
which can be used are polymeric substances and waxes. The active compounds
can also be in micro-encapsulated form, if al)plopl;ate, with one or more of theS above-mentioned excipients.
Liquid dosage forms for oral a-lmini~tration include pharmaceutically
acceptable emulsions, solutions, suspensions, syrups, and elixirs. In addition to the
active compounds, the liquid dosage forms may contain inert diluents commonly
used in the art, such as water or other solvents, solubilizing agents and emulsifiers,
as for example, ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate,
benzyl alcohol, benzyl benzoate, propyleneglycol, 1,3-butyleneglycol,
dimethylformamide, oils, in particular, cottonseed oil, groundnut oil, corn germoil, olive oil, castor oil and sesame oil, glycerol, tetrahydrofurfuryl alcohol,polyethyleneglycols, and fatty acid esters of sorbitan or mixtures of these
substances, and the like.
Besides such inert diluents, the composition can also include adjuvants,
such as wetting agents, emulsifying and suspending agents, sweetening, flavoring,
and perfuming agents.
Suspensions, in addition to the active compounds, may contain suspending
agents, as for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol
and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide,
bentonite, agar-agar and tr~g~ nth, or mixtures of these substances, and the like.
Compositions for rectal atlmini~trations are preferably suppositories which
can be prepared by mixing the compounds of the present invention with suitable
non-irritating excipients or carriers such as cocoa butter, polyethyleneglycol, or a
suppository wax, which are solid at ordinary tempeldlu,es but liquid at body
tell~pelal~lre and therefore, melt in the rectum or vaginal cavity and release the
active component.
Dosage forms for topical ~lmini~tration of a compound of this invention
include ointment~, powders, sprays, and inh~l~nt~. The active component is
admixed under sterile conditions with a physiologically acceptable carrier and any
preservatives, buffers, or propellants as may be required. Ophthalmic
CA 022~6716 1998-11-26
WO 98/09934 PCT/US97/148S9
formulations, eye ointments, powders, and solutions are also contemplated as
being within the scope of this invention.
The compounds of the present invention can be ~ mini~tered to a patient at
dosage levels in the range of about 0.1 to about 1,000 mg per day. For a normal
human adult having a body weight of about 70 kg, a dosage in the range of about
0.01 to about 100 mg/kg of body weight per day is preferable. The specific dosage
used, however, can vary. For example, the dosage can depend on a numbers of
factors including the requirements of the patient, the severity of the conditionbeing treated, and the pharmacological activity of the compound being used. The
determin~tion of optimum dosages for a particular patient is well-known to thoseskilled in the art. The term "patient" includes humans and animal.
The term "ph~rm~eutically acceptable salts, esters, amides, and prodrugs"
as used herein refers to those carboxylate salts, amino acid addition salts, esters,
amides, and prodrugs of the compounds of the present invention which are, withinthe scope of sound medical judgment, suitable for use in contact with the tissues
of patients without undue toxicity, irritation, allergic response, and the like,commensurate with a reasonable benefit/risk ratio, and effective for their intended
use, as well as the zwitterionic forms, where possible, of the compounds of the
invention. The term "salts" refers to the relatively nontoxic, inorganic and organic
acid addition salts of compounds of the present invention. These salts can be
prepared in situ during the final isolation and purification of the compounds or by
separately reacting the purified compound in its free base form with a suitable
organic or inorganic acid and isolating the salt thus formed. Representative salts
include the hydrobromide, hydrochloride, sulfate, bisulfate, nitrate, acetate,
oxalate, valerate, oleate, palmitate, stearate, laureate, borate, benzoate, lactate,
phosphate, tosylate, citrate, maleate, fumarate, succinate, tartrate, naphthylate
mesylate, glucoheptonate, lactobionate and laurylsulphonate salts, and the like.These may include cations based on the alkali and ~lk~line earth metals, such assodium, lithium, potassium, calcium, m~gn.o~ium, and the like, as well as nontoxic
ammonium, quaternary ammonium, and amine cations including, but not limited
to ammonium, tetramethylammonium, tetraethylammonium, methylamine,
dimethylamine, trimethylarnine, triethylamine, ethylamine, and the like. (See, for
..
CA 022~6716 1998-11-26
WO 98109934 PCT/US971148S9
-23-
example, S. M. Berge, et al., "Pharmaceutical Salts," J. Pharm. Sci.,
1977:66(1-19) which is incorporated herein by reference.)
Examples of ph~ eutically acceptable, nontoxic esters of the
compounds of this invention include C I to C6 alkyl esters wherein the alkyl group
S is a straight or branched chain. Acceptable esters also include Cs to C7 cycloalkyl
esters as well as arylalkyl esters such as, but not limited to benzyl. C I to C4 alkyl
esters are preferred. Esters of the compounds of the present invention may be
prepared according to conventional methods.
Examples of pharmaceutically acceptable, nontoxic amides of the
compounds of this invention include amides derived from ammonia, primary
C I to C6 alkyl amines, and secondary C l to C6 dialkyl amines wherein the alkyl
groups are straight or branched chain. In the case of secondary amines, the amine
may also be in the form of a 5- or 6-membered heterocycle cont~ining one
nitrogen atom. Amides derived from ammonia, Cl to C3 alkyl primary amines and
C 1 to C2 dialkyl secondary arnines are preferred. Amides of the compounds of the
invention may be prepared according to conventional methods.
The term "prodrug" refers to compounds that are rapidly transformed
in vivo to yield the parent compound of the above formula, for example, by
hydrolysis in blood. A thorough discussion is provided in T. Higuchi and
V. Stella, "Pro-drugs as Novel Delivery Systems," Vol 14 of the
A.C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, ed.
Edward B. Roche, American Pharmaceutical Association and Pergamon Press,
1987, both of which are incorporated herein by reference.
In addition, the compounds of the present invention can exist in unsolvated
as well as solvated forms with pharmaceutically acceptable solvents such as water,
ethanol, and the like. In general, the solvated forms are considered equivalent to
the unsolvated forms for the purposes of the present invention.
The compounds of the present invention are ~rimini.~tered to a patient in
need of matrix metalloproteinase inhibition. In general, patients in need of matrix
metalloproteinase inhibition are those patients having a disease or condition inwhich a matrix metalloproteinase plays a role. Examples of such diseases include,
CA 022~6716 1998-11-26
WO 98/09934 PCT/US97/14859
-24 -
but are not limited to, multiple sclerosis, atherosclerotic plaque rupture, restenosis,
aortic aneurism, heart failure, periodontal disease, corneal ulceration, burns,
decubital ulcers, chronic ulcers or wounds, cancer metastasis, tumor angiogenesis,
arthritis, or other autoimmune or infl~mm~tory diseases dependent upon tissue
invasion by leukocytes.
In a preferred embodiment, the matrix metalloproteinase is
stromelysin-l or gelatinase-A.
A "therapeutically effective amount" is an amount of a compound of
Formula I-VIII that when ~tlmini.~tered to a patient having a disease that can be
treated with a compound of Formula I-VIII ameliorates a symptom of the disease.
A therapeutically effective arnount of a compound of Formula I-VIII is readily
determined by one skilled in the art by ~llmini~tering a compound of
~ormula I-VIII to a patient and observing the results.
The following examples illustrate particular embodiments of the invention
and are not intended to limit the scope of the specification, including the Claims,
in any manner.
EXAMPLES
General Dibenzofuran Sulfonamide Synthesis
The compounds of the present invention can be synthesized using a
number of different synthetic routes. Referring to the General Synthetic Scheme,the common starting materials are the sulfonyl chlorides (1). These are easily
synthesized by one skilled in the art by sulfonation of the parent heterocycle. Some
representative procedures are as follows. For dibenzofuran (1, X=O) and
dibenzothiophene (1, X=S), the parent heterocycle is sulfonated at the 2-position
using one equivalent of chlorosulfonic acid in chloroform at 0~C according to the
method of Bassin, et al., (Phosphorus, Sulfur and Silicon, 1992;72:157-170). Thesulfonic acid is then converted to the corresponding sulfonyl chloride (1, X=O,S)
by tre~tment with phosphorus pentachloride at 170-1 80~C. For carbazole
(1, X=NH), the parent heterocycle is sulfonated at the 3-position using sulfuric
CA 022~6716 1998-ll-26
W 098/09934 PCT~US97/14859
acid at 100~C followed by neutralization with barium carbonate to yield the
barium salt of the corresponding sulfonic acid according to the method of Loza,
et al., (Sb. Mater. Nauch.-Tekh. Konf. Ukrain. Zaoch. Poitekh. Inst. Vith,
Kharkov, 1966:202-205). The sulfonic acid is then converted to the correspondingsulfonyl chloride (1, X=NH) by treatment with phosphorus pentachloride at
170-180~C or reaction with either phosphoryl chloride, thionyl chloride, or oxalyl
chloride. For fluorene (1, X=CH2), according to the method of Chrzaszczewska
et al., (~odz. Tow. Nauk., Wydz 3, Acta Chim., 1966;11:143-155) the parent
carbocycle is sulfonated at the 2-position using one equivalent of chlorosulfonic
acid in chloroform at 0~C followed by neutralization with potassium hydroxide togive the potassium salt of the corresponding sulfonic acid. This fluorene derivative
can then be oxidized using aqueous potassium pern ~ng;ln~te at 80~C to the
corresponding fluorenone derivative (1, X=CO). The sulfonic acid salts are then
converted to the corresponding sulfonyl chloride ( I, X=CH2,CO) by treatment
with phosphorus pentachloride and phosphoryl chloride in chloroforrn.
In Method A, the sulfonyl chloride (1) is condensed directly with a natural
amino acid using a base such as triethylarnine (TEA) in a mixture of
tetrahydrofuran (THF) and water (3:5) at 10~C to yield the desired compound (2).The corresponding hydroxarnic acid (5) can be conveniently prepared by coupling
the acid (2) with an O-protected (usually benzyl) hydroxylamine using
dicyclohexylcarbodiimide (DCC) as the coupling agent in dichloromethane at
temperatures ranging from -(10) to 0~C. The protecting group can be removed
from compound (4) by catalytic hydrogenolysis using hydrogen gas at 50 psi and
Pd/BaSO4 in aqueous methanol to yield the hydroxamic acid derivative (5).
In Method B, the sulfonyl chloride (1) is conden.~ed with a suitably
C-protected (usually tertiary butyl ester) amino acid using a base such as
N-methylmorpholine (NMM) in a solvent such as dichloromethane at 0~C to yield
compound (3). The protecting group can be removed from the carboxylic acid by
tre~tmçnt with trifluoroacetic acid in dichloromethane at 25-35~C using anisole as
a carbocation scavenger to yield (2).
CA 02256716 1998-11-26
W O 98t09934 PCTrUS97/14859
-26-
O 5
Z Z
o= ~= o ~ o= ~= o -~ ,~ ~= "'= ~ ~t
~X ~ ~ ~X ~ ~ ~X
X
._ . \
Z~ ~ _
= V~= O ~
~X -- L ~X
X / ~o
C
Z ~
CA 022~6716 1998-11-26
W O 98/09934 PCT~US97114859
-27-
Examples Prepared by Method A
EXAMPLE 1
(L)-2-fDibenzofuran-2-sulfonylamino)-4-methvl-pentanoic acid
Step (a) (L)-2-(Dibenzofuran-2-sulfonylamino)-4-methyl-pentanoic acid,
S tert.-butyl ester
To a dichloromethane solution (20 mL) of (L)-leucine, tert.-butyl ester
(2.1 g, 0.0099 mol) and N-methylmorpholine (2.2 mL, 0.0199 mol) at 0~C under
an inert nitrogen atmosphere was added a dichloromethane solution (10 mL) of
dibenzofuran-2-sulfonyl chloride (1.0 g, 0.00375 mol) with stirring. The resulting
solution was stirred at 0~C for 4 hours and then partitioned with water (30 mL).The organic layer was separated and washed with water (2 x 30 mL) and brine
(2 x 30 mL). This was then dried over anhydrous magnesium sulfate, filtered, andthe solvent removed under reduced pressure. The residue was then flash
chromatographed on silica gel and the title product (1.0 g, 64%) was eluted with20% ethyl acetate/hexane; melting point = 106-109~C.
Step fb) (L)-2-(Dibenzofuran-2-sulfonYlamino)-4-methyl-pentanoic acid
(Example 1)
To a dichloromethane solution (5 mL) of the material obtained in step (a)
(0.5 g, 0.00119 mol) and anisole (0.5 mL) at room temperature with stirring was
added trifluoroacetic acid (5 mL). The resulting solution was stirred at room
temperature for 24 hours and then concentrated in vacuo. The residue was
triturated with a mixture of ethyl acetate/hexane to yield the title compound
(0.14 g, 33%); melting point = 75-80~C.
IH NMR (CDC13: ~ 8.4 (s, lH), 8.0 (d, IH), 7.9 (d, IH), 7.4-7.6 (m, 4H), 5.0 (d,IH), 3.9 (m, IH), 1.8 (m, IH), 1.4 (m, 2H), 0.9 (d, 3H), 0.8 (d, 3H) ppm.
Following the general procedure of Example 1, the following compounds were
obtained:
CA 022~6716 1998-11-26
WO 98/09934 PCT/US97/14859
-28-
EXAMPLE 2
(L)-2-(Dibenzofuran-2-sulfonylamino)-3-methYI-pentanoic acid
1H NMR (DMSO-D6): ~ 8.6 (s, lH), 8.3 (d, lH), 8.1 (d, lH), 7.8-7.9 (m, 3H),
7.6 (tr, lH), 7.5 (tr, lH), 3.7 (m, lH), 3.4 (s, lH), 1.7 (m, lH), 1.1-1.4 (m, 2H),
0.75-0.85 (m, 6H) ppm.
EXAMPLE 3
(L)-2-(Dibenzofuran-2-sulfonylamino)-3-phenyl-propionic acid; melting
point = 196- 198~C.
Examples prepared by Method B
EXAMPLE 4
(L)-2-(Dibenzofuran-2-sulfonylamino)-propionic acid
To a THF/water (5:3, 8 mL) solution of (L)-alanine (0.3 g, 0.0034 mol)
and triethylamine (1 mL) at 10~C was added dibenzofuran-2-sulfonyl chloride
(1.0 g, 0.00375 mol) in one portion with stirring. The resulting solution was stirred
at room temperature for 24 hours. The solution was then concentrated in vacuo
and the residue redissolved in water (10 mL). This solution was cooled in an icebath and then acidified with lN HCl. A white solid was deposited which was then
filtered and washed with water. This solid was recrystallized from aqueous ethanol
to give the title product (0.6 g, 50%); melting point = 158-163~C.
Following the general procedure of Exarnple 4, the following compounds
were obtained:
EXAMPLE 5
(L)-2-(Dibenzofuran-2-sulfonylamino)-3-methvl-butyric acid; melting
point = 163-165~C
EXAMPLE 6
(Dibenzofuran-2-sulfonylamino)-acetic acid; melting point = 208-210~C
.... , .. ., .. , ~ I
CA 022~6716 1998-11-26
W O 98/09934 PCT~US97/14859
-29-
EXAMPLE 7
(L)-2-(Dibenzofuran-2-sulfonylamino)-succinic acid; melting point = 165- 168~C
EXAMPLE 8
(L)-2-(Dibenzofuran-2-sulfonylamino)-3-tritYlsulfanYI-propionic acid
S INMl~ (DMSO-D6): o 8.5 (s, IH), 8.2 (m, 2H), 7.1-7.9 (m, l9H), 3.6 (m, lH),
3.5 (m, IH), 2.3 (d, 2H) ppm.
EXAMPLE 9
(L)-2-(Dibenzofuran-2-sulfonylamino)-3-mercapto-propionic acid
To a dichloromethane solution (10 mL) of (L)-2-(Dibenzofuran-
2-sulfonylamino)-3-tritylsulfanyl-propionic acid (Example 8, 1.0 g, 0.00168 mol)at room temperature was added trifluoroacetic acid (10 mL). A deep red/orange
solution resulted. To this solution was added triethylsilane (0.33 mL,
0.00202 mol), the color was immediately discharged, and the resulting clear
solution was stirred at room temperature for 3 hours. The solution was then
concentrated in vacuo and the residue redissolved in ether (10 mL) which was then
removed in vacuo. This procedure was repeated three times. The residue was
recryst~lli7Pd from ethyl acetate/hexane (1 :1) to yield the title compound (0.23 g,
39%); melting point = 164-166~C.
EXAMPLE 10
(L)-2-(Dibenzofuran-2-sulfonylamino)-3-methyl-Pentanoic acid hydroxyamide
Step (a) (L)-2-(Dibenzofuran-2-sulfonylamino)-3-methYI-pentanoic acid
benzyloxy-amide
To a THF solution (50 mL) of (L)-2-(Dibenzofuran-2-sulfonylamino)-
3-methyl-pentanoic acid (Example 2, 0.55 g, 0.0015 mol) and carbonyldiimidazole
(0.26 g, 0.0016 mol) at room temperature under an inert nitrogen atmosphere was
added O-benzylhydroxylamine (0.23 g, 0.0018 mol) in one portion. This solution
was then heated to reflux for 72 hours and then allowed to stir at room
temperature for 24 hours. The mixture was then concentrated in vacuo and flash
CA 022~6716 1998-11-26
WO 98/09934 PCTtlJS97/14859
-30-
chromatographed on silica gel eluting with ethyl acetate/hexane (1 :4) to yield the
title compound (0.27 g, 38%); melting point = 207-209~C.
Step (b) (L)-2-(Dibenzofuran-2-sulfonylamino)-3-methyl-pentanoic acid
hydroxvarnide (Example 10)
S A THF (2 mL)/methanol (10 mL) solution of the material obtained above
in step (a) (0.037 g, 0.0000793 mol) was hydrogenolyzed using hydrogen gas at
50 psi with a Pd/BaS04 catalyst at room temperature for 1 hour. The catalyst wasremoved by filtration and the solution concentrated in vacuo. The residue was
triturated with ether to yield the title compound (0.022 g, 74%).
1H NMR (CDC13): ~ 8.6-7.2 (m, 8H), 5.1 (m, lH), 4.1 (m, lH), 1.9-1.2 (m, 3H),
O.9(m,3H),0.85(m,3H)ppm.
EXAMPLE 11
(L)-2-(dibenzofuran-3-sulfonylamino)-3-methyl-butyric acid
Step (a) (Dibenzofuran-3-sulfonYI chloride)
3-Arninodibenzofuran (10 g, 54.6 mol) was diazotized by dissolving in
180 mL glacial acetic acid, 50 mL water, and 14 mL concentrated hydrochloric
acid at 0~C and adding 15 mL of a 5.5 M aqueous solution of sodium nitrite. The
resulting mixture was stirred for 1 hour before pouring into a solution of
copper(II)chloride (2.0 g, 14.9 mmol) in 240 mL of a 1: 1 mixture of benzene andglacial acetic acid saturated with sulfur dioxide. This mixture was allowed to
warm to room temperature and stirred for 16 hours. The reaction was partitioned
between water and chloroform. The chloroforrn layer was washed with water,
dried over m~gn.qcium sulfate, filtered, and concentrated to give the title
compound as a yellowish solid; melting point = 142-144~C.
Step (b)
Using the procedure of Example 1, (L)-leucine, tert.-butyl ester is replaced
with (L)-valine, tert.-butyl ester and dibenzofuran-2-sulfonyl chloride is replaced
CA 022~6716 1998-11-26
WO 98/09934 PCTtUS97/14859
with dibenzofuran-3-sulfonyl chloride, the title compound is obtained; melting
point= 197-200~C.
EXAMPLE 12
(L)-2-(9H-Fluorene-2-sulfonylamino)-3-methyl-butYric acid
S When in the procedure of EXAMPLE 1, (L)-leucine, tert.-butyl ester isreplaced with (L)-valine, tert.-butyl ester and dibenzofuran-2-sulfonyl chloride is
replaced with 9H-fluorene-2-sulfonyl chloride, the title compound is obtained.
EXAMPLE 13
(L)-2-(5,5-Dioxo-SH-5~6-dibenzothiophene-3-sulfonylamino)-3 -methyl-butyric
l 0 acid
When in the procedure of EXAMPLE 1, (L)-leucine, tert.-butyl ester is
replaced with (L)-valine, tert.-butyl ester and dibenzofuran-2-sulfonyl chloride is
replaced with 5,5-dioxo-5H-5~6-dibenzothiophene-3-sulfonyl chloride, the title
compound is obtained; melting point = 85-90~C.
EXAMPLE 14
(L)-2-(Dibenzothiophene-2-sulfonylamino)-3-methyl-butyric acid
When in the procedure of EXAMPLE I, (L)-leucine, tert.-butyl ester is
replaced with (L)-valine, tert.-butyl ester and dibenzofuran-2-sulfonyl chloride is
replaced with dibenzothiophene-2-sulfonyl chloride, the title compound is
obtained; melting point = 150-155~C.
EXAMPLE 15
(L)-2-(5 ,5-Dioxo-SH-5~6-dibenzothiophene-2-sulfonylamino)-3 -methyl-butyric
acld
To a glacial acetic acid (30 mL) solution of the material obtained in
EXAMPLE 14 Step (a) (1.5 g, 0.0036 mol) was added 10 mL of 30% hydrogen
peroxide. The resulting solution was heated to reflux for 2.5 hours, cooled to room
temperature and stirred for 16 hours and then f1ltered to give the crude product as
CA 022~6716 1998-11-26
WO 98109934 PCT/US97114859
-32-
a white solid. The solid was washed with water and boiling ether to yield the title
compound; melting point = 216-218~C.
EXAMPLE 16
(L)-2-(7-Bromo-dibenzofuran-2-sulfonylamino)-3-methYI-butyric acid
Step(a) 3-Bromo-dibenzofuran
3-Amino-dibenzofuran (15 g, 81.9 mmoles) was added in portions to a
suspension of cupric bromide (21.9 g, 98.2 mmoles) and tert.-butyl nitrite
(12.66 g, 122.8 mrnoles) in 350 mL of acetonitrile. This mixture was heated to
reflux for 2 hours and then stirred for 16 hours at room temperature. The reaction
was partitioned between 1 M HC 1 and diethyl ether. The diethyl ether layer was
washed with brine, dried over magnesium sulfate, filtered, and concentrated to
give an oily solid. Chromatography gave the title compound as a yellowish solid.
Step (b) 7-Bromo-dibenzofuran-2-sulfonyl chloride
Chlorosulfonic acid (3.75 mL, 56 mmoles) was added dropwise to a
solution of 3-bromo-dibenzofuran (9.21 g, 37.3 rnmoles) in 150 mL of chloroform
at room temperature. The reaction was stirred for 5 hours, cooled to 0~C, filtered,
and washed the solid with cold dichloromethane. This solid (6.12 g, 18.7 mmoles)was mixed with phosphorous pentachloride (12.9 g, 61.7 mrnoles) and the mixture
was heated to 110~C for 4 hours. The mixture was cooled to room temperature and
quenched with ice water. Filtered the resulting suspension to give the title
compound as a white solid.
Step (c) (L)-2-(7-Bromo-dibenzofuran-2-sulfonYlamino)-3-methyl-butyric
acid
When in the procedure of EXAMPLE 1, (L)-leucine, tert.-butyl ester is
replaced with (L)-valine, tert.-butyl ester and dibenzo~uran-2-sulfonyl chloride is
replaced with 7-bromo-dibenzofuran-2-sulfonyl chloride, the title compound is
obtained; melting point = 191 -193~C.
CA 022~67l6 l998-ll-26
W 098/09934 PCTrUS97/14859
-33-
EXAMPLE 17
(L)-3-Methyl-2-(7-phenvl-dibenzofuran-2-sulfonYlamino)-butyric acid
Step (a) (L)-2-(7-Bromo-dibenzofuran-2-sulfonvlamino)-butyric acid. tert.-
butyl ester
S When in the procedure of EXAMPLE 1, Step (a), (L)-leucine, tert.-butyl
ester is replaced with (L)-valine, tert.-butyl ester and dibenzofuran-2-sulfonylchloride is replaced with 7-bromo-dibenzofuran-2-sulfonyl chloride, the title
compound is obtained.
Step (b) (L)-3-Methvl-2-(7-phenyl-dibenzofuran-2-sulfonylamino)-butyric
acid, tert.-butyl ester
(L)-2-(7-Bromo-dibenzofuran-2-sulfonylamino)-3-methyl-butyric acid,
tert.-butyl ester (1.0 g, 2.0 mmoles) and phenyl boronic acid (0.3 g, 2.5 mmoles)
were mixed with 10 mL toluene with 5 mL water the 0.5 g sodium carbonate.
Tetrakis(triphenylphosphine) palladium (0) (0.15 g, 0.1 mmoles) was added and
the resulting mixture was heated to reflux for 6 hours. Another 0.15 g of the
palladium catalyst was added and reflux was continued for 16 hours. The reactionwas cooled to room temperature and partitioned between 1 M HC 1 and ethyl
acetate. The organic layer was dried over magnesium sulfate and concentrated to
give the title compound as a white solid.
Step(c) (L)-3-Methyl-2-(7-phenyl-dibenzoduran-2-sulfonylamino)-butyric
acld
(L)-3-Methyl-2-(7-phenyl-dibenzofuran-2-sulfonylamino)-butyric acid,
tert.-butyl ester (0.94 g, mmoles) was dissolved in concentrated trifluoroaceticacid and stirred for 2 hours. Concentrated in vacuo and triturated the residue with
diethyl ether to give the title compound as an off-white solid, melting
point= 254-255~C.
CA 022~6716 1998-11-26
W O 98/09934 PCTrUS97114859
-34-
rNHIBITION STUDIES
Experiments were carried out which demonstrate the efficacy of
compounds of Formula I and II as potent inhibitors of stromelysin-1 and
gelatinase A. Experiments were carried out with the catalytic domains, i.e.,
Table 1 shows the activity of the Examples with respect to both stromelysin-1 and
gelatinase A, GCD (recombinant gelatinase A catalytic domain); SCD
(stromelysin-l catalytic domain). ICso values were determined using a
thiopeptolide substrate, Ac-Pro-Leu-Gly-thioester-Leu-Leu-Gly-OEt (~e Q.-Z.,
Johnson L.L., Hupe D.J., and Baragi V., "Purification and Characterization of the
Human Stromelysin Catalytic Domain Expressed in Escherichia coli,"
Biochemistry, 1992;31:11231-11235). MMP01, MMP07, MMP09, and MMP13
activity was assayed in a method similar to MMP02 and MMP03 (SCD and
GCD). MMP01 and MMP09 can be obtained from Washington University School
of Medicine, St. Louis, Missouri. MMP07 can be obtained in accordance with the
known procedure set forth by Ye Q-Z, Johnson L.L., and Baragi V., "Gene
Syntheses and Expression in E. coli for PUMP, a Human Matrix
Metalloproteinase" Biochem. and BiophYs. Res. Comm., 1992;186:143-149.
MMP13 can be obtained in accordance with the known procedure set forth by
Freije J.M.P., et al., "Molecular Cloning and Expression of Collegenase-3, a
Novel Human Matrix Metalloproteinase Produced by Breast Carcinomas" J. Bio.
Chem., 1994;269:16766-16773.
Thiopeptolide Assay
Hydrolysis of the thiopeptolide substrate Ac-Pro-Leu-Gly-thioester-Leu-
Leu-Gly-OEt (Bachem) is used as the primary screen to determine ICso values for
MMP inhibitors. A 100 ~L reaction contains 1 mM 5,5'-dithiobis(2-nitroben~oic
acid) (DTNB), 100 ,~LM substrate, 0.1% Brij, enzyme, and inhibitor in the
~plopl;ate reaction buffer. Activated full-length enzymes are assayed at 5 nM,
Stromelysin Catalytic Domain (SCD) at 10 nM, and Gelatinase A Catalytic
Domain (GaCD) at 1 nM. Inhibitors are screened from 100 ~M to 1 nM.
CA 022~6716 1998-11-26
WO 98/09934 PCT/US97/14859
-35-
Full-length enzymes are assayed in 50 mM HEPES, 10 mM CaC12, pH 7.0; SCD
in 50 mM MES, 10 mM CaC12, pH 6.0; and GaCD in 50 mM MOPS, 10 mM
CaC12, 10,uM ZnC12, pEI 7Ø The change in absorbance at 405 nm is monitored
on a TherrnoMax microplate reader at room temperature continuously for
20 minutes.
HEPES is 4-(2-hydroxylethyl)-piperazine-1-ethane sulfonic acid;
MES is 2-morpholinoethane sulfonic acid menohydrate;
Ac is acetyl;
Pro is proline;
Leu is leucine;
Gly is glycine;
Et is ethyl; and
MOPS is 3-morpholinopropane sulfonic acid.
Soluble Proteoglycan Assay (stromelysin natural substrate assay) SCD (PG)
Solubilized proteoglycan substrate is prepared from bovine cartilage
powder (Sigma) using the method described by Nagase and Woessner in Anal.
Biochem., 1980;107:385-392. A 100 ~lL reaction contains 10 ,ug/mL proteoglycan,
enzyme, and inhibitor in 50 mM MES, 10 mM CaC12, pH 6Ø Activated full-
length stromelysin or stromelysin catalytic domain (SCD) is assayed at 100 nM.
Inhibitors are screened from 100 ~M to 1 nM. The reaction is incubated at 37~C
for 3 hours then stopped with the addition of 1,10-phenanthroline at a final
concentration of 1 mM. Reaction products are separated from undigested substrateusing ultrafree-MC polysulfone microcons with a 300,000 molecular weight cut-
off membrane (Millipore) and quantified using a modified l,9-dimethylene blue
(DMB) assay described by Farndale, Sayers, and Barren in Connective Tissue
Research, 1982;9:247-248. Absorbance is measured at 518 nm using 32 llg/mL
DMB in a 1 mL reaction. The standard curve is constructed from 0 to 100 ,ug
shark cartilage chondroitin sulfate C (Sigma).
CA 022~6716 1998-11-26
Wo 98l09934 PCT/US97l14859
-36-
Gelatin Assay (~elatinase natural substrate assaY) (Gel)
Rat tail Type I collagen (Sigma) is denatured by heating at 95~C for
20 minutes to prepare the gelatin substrate. A 50 ~L reaction contains 1.12 mg/mL
substrate, enzyme, inhibitor, and 80 ~g/mL soy bean trypsin inhibitor as an inert
internal standard in 50 mM MOPS, 10 mM CaC12, 10 ,uM ZnC12, pH 7Ø
Activated full-length gelatinase A is assayed at 1 nM and gelatinase A catalyticdomain (GaCD) at 10 nM. Inhibitors are screened from 100 ~M to 1 nM. The
reactions are incubated at 37~C for 30 minutes then stopped with 50 ~lL at 2X
Tricine gel loading buffer (Novex). Reaction products are separated from
undigested substrate by electrophoresis on Tricine-SDS 10-20% polyacrylamide
gradient gels (Novex). Protein bands are stained with Coomassie Brilliant Blue Rand quantified using a Bio Image densitometer (Millipore). ICso values are
calculated from the disappearance of substrate using the sum of the top three
bands of each reaction after norm~li7~tion with the internal standard.
MMP Inhibitor Bioassay
Animals are dosed by gavage with either vehicle or compound at 2, 10, or
50 mg/kg. Blood samples are collected from 3 to 4 ~nim~l~ from each dosing
group at 1, 2, 4, 6, and 24 hour postdose, centrifuged, and the plasma immediately
frozen at -20~C. Plasma protein is precipitated with an equal volume of
acetonitrile and separated by centrifugation at room temperature. The supernate is
evaporated to dryness and reconstituted to the original plasma volume with
50 mM Tris, pH 7.6. Ten-fold serial dilutions of the reconstituted plasma samples
are prepared in 50 mM Tris, pH 7.6 for dose response assays using the ~ropl;ate
thiopeptolide assay. The concentration of plasma which yields 50% inhibition of
enzyme is determined and used to calculate the inhibitor plasma level from the
known ICso value. To demonstrate that the compound can be quantitatively
extracted from plasma as active inhibitor, controls for each inhibitor include
normal rat plasma, normal rat plasma spiked with compound, and buffer dilutions
T
CA 022~6716 1998-11-26
WO 98/09934 PCTIUS97/14859
-37-
of compound. All control samples are subjected to acetonitrile precipitation andanalyzed with the thiopeptolide assay.
TABLE 1
ExampleMMPO I (GCD) (SCD) MMP07 MMP09 MMP 13
Number MMP02 MMP03
66 0.32 1.18 -- 100 --
2 100 ~.3 1.5 -- 100 --
3 100 0.9 0.72 -- 100 --
4 -- 1.7 5.4 -- --
19 0.0840.23 -- 100 --
6 100 0.73 4.8 -- 100 --
7 -- 1.2 1.0 -- -- --
8 -- 9.4 14.4 --
9 -- 4.5 0.69 -- --
-- 35 100 -- -- --
11 1.8 0.00450.015 5.0 -- 0.047
12 32.3 0.0490.185 10.8 100 0.34
13 ---- ---- ---- ----
14 100 0.61 0.69 27 -- 2.6
100 100 100 100 -- 100
16 -- 0.47 0.75 --
17 100 0.36 0.062 6 -- 0.69
CA 02256716 1998-11-26
Wo 98/09934 PCT/US97/14859
-38-
TABLE 2
Example 5
SCD (IC50) 0.233 ~M
SCD (PG) (IC50) 8.9 ,uM
GCD (IC50) 0.084 ~M
Gel (Ic50) 0.58 ,uM
Bioassay (50 mg/kg)
Peak 82 ~LM
24 Hours 0.18 ,uM
,, .