Note: Descriptions are shown in the official language in which they were submitted.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
TITLE OF THE INVENTION
SUBSTITUTED IMIDAZOLES HAVING CYTOKINE INHIBITORY
ACTIVITY
BACKGROUND OF THE INVENTION
The present invention relates to substituted imidazole
compounds which have cytokine inhibitory activity. Cytokine mediated
diseases and cytokine inhibition, suppression and antagonism are used in
the context of diseases or conditions in which excessive or unregulated
production or activity of one or more cytokines occurs. Examples of
cytokines which are effected typically include Interleukin-1 (IL-1),
Interleukin-6 (IL-6), Interleukin-8 (IL-8) and Tumor Necrosis Factor
(TNF).
Interleukin-1 (IL-1) and Tumor Necrosis Factor (TNF} are
produced by a variety of cells which are involved in immunoregulation
and other physiological conditions.
There are many disease states in which IL-1 is implicated.
Examples are rheumatoid arthritis, osteoarthritis, endotoxemia, toxic
shock syndrome, acute and chronic inflammatory diseases, such as the
inflammatory reaction induced by endotoxin or inflammatory bowel
disease; tuberculosis, atherosclerosis, muscle degeneration, cachexia,
psoriatic arthritis, Reiter's syndrome, rheumatoid arthritis, gout, traumatic
arthritis, rubella arthritis and acute synovitis. Recent evidence also links
IL,-1 activity to diabetes.
Interleukin-1 has been demonstrated to mediate a variety of
biological activities thought to be important in immunoregulation and
other physiological conditions. [See, e.g., Dinarello et al., Rev. Infect.
Disease, 6, 51 (1984}]. The known biological activities of IL-1 include
the activation of T helper cells, induction of fever, stimulation of
prostaglandin or collagenase production, neutrophil chemotaxis,
induction of acute phase proteins and the suppression of plasma iron
levels.
Excessive or unregulated tumor necrosis factor (TNF)
production or activity has been implicated in mediating or exacerbating
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-2-
rheumatoid arthritis, rheumatoid spondylitis, osteoarthritis, gouty
arthritis, and other arthritic conditions, sepsis, septic shock, endotoxic
shock, gram negative sepsis, toxic shock syndrome, adult respiratory
distress syndrome, cerebral malaria, chronic pulmonary inflammatory
disease, silicosis, pulmonary narcosis, bone resorption diseases,
reperfusion injury, graft v. host rejection, allograft rejections, fever and
myalgia due to infection, cachexia secondary to infection or malignancy,
cachexia secondary to acquired immune deficiency syndrome (AIDS),
AIDS related complex (ARC), keloid formation, scar tissue formation,
Crohn's disease, ulcerative colitis and pyresis.
Monokines, such as TNF, have also been shown to activate
HIV replication in monocytes and/or macrophages [See Poli, et al., Proc.
Natl. Acad. Sci., 87:782-784 (1990)], therefore, inhibition of monokine
production or activity aids in limiting HIV progression. TNF has been
implicated in various roles with other viral infections, such as the
cytomegalovirus (CMV), influenza virus and the herpes virus.
Interleukin-6 (IL-6) is a cytokine effecting the immune system and
hematopoiesis. It is produced by several mammalian cell types in
response to agents such as IL-I, and is correlated with disease states such
as angiofollicular lymphoid hyperplasia.
Interleukin-8 (IL-R) is a chemotactic factor first identified
and characterized in 1987. Many different names have been applied to
IL-8, such as neutrophil attractant/activation protein-1 (NAP-1),
monocyte derived neutrophil chemotactic factor (MDNCF), neutrophil
activating factor (NAF), and T-cell lymphocyte chemotactic factor. Like
IL-1, IL-8 is produced by several cell types, including mononuclear cells,
fibroblasts, endothelial cells and ketainocytes. Its production is induced
by IL-1, TNF and by lipopolysaccharide (LPS). IL-8 stimulates a number
of cellular functions in vitro. It is a chemoattractant for neutrophils, T-
lymphocytes and basophils. It induces histamine release from basophils.
It causes lysozomal enzyme release and respiratory burst from
neutrophils, and it has been shown to increase the surface expression of
Mac-1 (CDl lb/CD 18) on neutrophils without ale novo protein synthesis.
CA 02257200 1998-12-02
WO 97/47618 PCT/ITS97/09888
There remains a need for compounds which are useful in
treating cytokine mediated diseases, and as such, inhibit, suppress or
antagonize the production or activity of cytokines such as IL-1, IL-6, IL-8
and TNF.
SUMMARY OF THE INVENTION
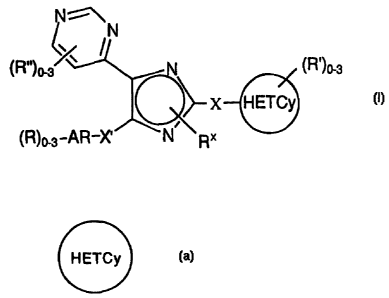
The present invention addresses a compound represented by
formula I:
N %\
~N
(R")o_3/ N (R')o-s
X HETCy
(R)o-3 a ~-X~ N
or a pharmaceutically acceptable salt thereof, wherein:
X and X' each independently represent -(CH2)m-Y-(CH2)n -,
wherein m and n represent integers within the range of from 0 - 4, such
that the sum of m and n is from 0 - 6; Y represents a member selected
from the group consisting of: a direct bond; O; S(O)y, with y equal to 0,
1 or 2; NRq~, with Rq~ as defined below; C(O); OC(O); C(O)O;
SOXNR9~ with x equal to 1 or 2 and R9~ as defined below; NRq~SOx ;
C(O)NR9~ and NRq~C(O) ;
HETCy
represents a 4 to 10 membered non-aromatic
heterocycle containing at least one N atom, and optionally containing 1-2
additional N atoms and 0-1 O or S atom;
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-4-
Rx represents H, Cl_6 alkyl(R9)3, C3_g cycloalkyl, OCl-6
alkyl(R9)3 or C(O)C 1 _6 alkyl(Rq)3;
each R independently represents a member selected from the
group consisting of: halo; hydroxy; C 1 _6 alkyl(R9)3; OC 1-6
alkyl(R9)3; C3_g cycloalkyl(R9)3; CN; CONH2; CONHCI-6
alkyl(R9)3; CON(Cl_~ alkyl(R~l)3)2; NH2; NHCI-b alkyl(R9)3;
NHC3_g cycloalkyl; N(C1_~ alkyl(R9)3)2; CON(C3_g cycloalkyl)(C1-6
alkyl(R9)3)); COZH; C02C1_6 alkyl(R9)3; C(O)Cl-6 alkyl(R9)3;
aryl(Rq)3; heterocyclyl(R9)3; heteroaryl(R9)3; CF3; SH; N02;
NHS02C1_6alkyl(R9)3, NHS02aryl(R9)3, NHS02heteroaryl(R9)3;
N(R9')C(O)Cl_6 alkyl(R9)3; NR~I'C(O)NH(Cl_6 alkyl(R9)3); C2_4
alkenyl(R9)Z_3 and CZ_4 alkynyl(R9)1-3;
each R" independently represents a member selected from
the group consisting of: halo; hydroxy; C 1 _6 alkyl(R9)3; OC 1-6
alkyl(R9)3; C3_g cycloalkyl(R9)3; CN; CONH2; CONHCI-6
alkyl(R9)3; CON(Cl_6 alkyl(R9)3)2; NH2; NHCI_6 alkyl(R9)3;
NHC3-g cycloalkyl; N(Cl_6 alkyl(R9)3)2; CON(C3_g cycloalkyl)(C1-6
alkyl(R9)3)); C02H; C02C 1 _~ alkyl(R~1)3; C(O)C 1 _6 alkyl(R9)3;
aryl(R9)3; heterocyclyl(R9)3; heteroaryl(R9)3; CF3; SH; N02;
SOyCI_6 alkyl(R9)3, with y as defined above; SO~1H2; S0~1HC1-6
alkyl(R9)3; SO~I(Cl-( alkyl(R9)3)2 ; NHS02C1_6alkyl(R9)3,
NHSOZaryI(R9)3, NHS02heteroaryl(R9)3; N(R9')C(O)C 1 _6
alkyl(R9)3; NR9'C(O)NH(Cl-6 alkyl(R9)3); CZ-q. alkenyl(Rq)2_3
and C2-q. alkynyl(R9)1-3;
each R' independently represents a member selected from the
group consisting of: CONH2; CONHC 1 _6 alkyl(R~1)3; CONIC 1-6
alkyl(R9)3)2; CONHC3_g cycloalkyl(R9)3; CON(C3_g
cycloalkyl(R9)3)2; CON(C3_g cycloalkyl)(C 1 _6 alkyl(R9)3)); C02H;
C02C1 _6 alkyl(R9)3; C(O)C 1 _6 alkyl(R9)3; C02C3_g cycloalkyl(R9)3;
C(O)C3_g cycloalkyl(R9)3; -[C(O)(CH2)j-CRSR6-(CH2)k-NR~Ip-RS;
-C(O)C3-g cycloalkyl{R9)3; -C(O)heterocyclyl(R9)3; -CON[Cl-
6alkyl(R9)3)[C3-8 cycloalkyl(R9)3]; -C(O)aryl(R9)3 ;
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
_5-
-C(O)heteroaryl(Rq)3 ; hydroxy; C 1 _6 alkyl(Rq)3; C3_g
cycloalkyl(Rq)3; OC 1 _6 alkyl(Rq)3; OC3_g cycioalkyl(Rq)3;
heterocyclyl(Rq)3; CN; NH(Rq"); NHC1_6 alkyl(Rq)3; N(Cl-6
alkyl(Rq)3)2; NHC3-g cycloalkyl(Rq)3; N(C3_g cycloalkyl(Rq)3)2;
CF3; SH; N02; C2_4 alkenyl(Rq)2_3 , aryl(Rq)3 ; heteroaryl(Rq)3 ;
C2_4 alkynyl(Rq) 1 _3 ; -OC(O) C3_g cycloalkyl(Rq)3; S02NH2;
S02NHC 1 _G alkyl{Rq)3; S02N(C 1 _G alkyl(Rq)3)2 ; NHS02C 1 _
(alkyl(Rq)3 ; NHS02ary1(Rq)3; NHS02heteroary(Rq)3;
-OC(O)heterocyclyl(Rq)3; N(Rq~)C(O)C 1 _6 alkyl(Rq)3;
NR9~C(O)NH(C 1 _6 alkyl(Rq)3); -OC(O)C 1 _6 alkyl(Rq)3;
-OC(O)aryl(Rq)3; -OC(O)heteroaryl(Rq)3; -C(=NRq~)NH2 ;
-C(=Nq~)NHC1_6 alkyl(Rq)3; -C(=Nq~)N(C1_6 alkyl(Rq)3)2;
-O~ (O~- UH2)1-CR5R6- (CH2)k' NR~J
and
LNR~OH2)k-CR5R6-~CH2)l.C(QJp OR9
wherein j and k independently represent integers of from 0 -
3;
RS and R6 are independently H, aryl, C 1 _6 alkyl(Rq)3, or
CRSR6 in combination represents a 3, 4, 5 or 6 membered cycloalkyl or
heterocyclyl group, an aryl group or a heteroaryl group;
p represents 1, 2 or 3, with the proviso that when p
represents 1, CRSR6 represents a 3, 4, 5 or 6 membered cycloalkyl group
or a heterocyclyl group, an aryl group or a heteroaryl group, and at least
oneofjandkis l,2or3;
R~ and RA are independently H, C 1 _6 alkyl or aryl;
R9 represents H, a negative charge balanced by a positively
charged group or a protecting group;
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-6-
R9 represents a member selected from the group consisting
of: R9~; halo; CN; C02H; C02C 1 _4 alkyl; C(O)C 1 _q. alkyl ; NH(Rq~~) ;
aryl{Ra)3; heteroaryl(Ra)3; NHCI-4 alkyl ; N(Cl_4 alkyl)2 ; CONH2 ;
SH ; S(O)yCI_6 alkyl(Ra)3; C(O)NHCI_6 alkyl(Ra)3; C(O)N(Cl-6
alkyl(Ra)3)2~ C3-8 cycloalkyl; NHC(NH)NH2 ; -heteroalkyl(Ra)3;
-NHC(O)NH2;
~Ra)3
N
and N
N N
wherein and independently represent mono or
bicyclic ring systems, non-aromatic or partially aromatic, containing from
5-10 ring atoms, 1-4 of which are N and 0-1 of which are O or S(O)y,
with y equal to 0, 1 or 2, optionally containing 1-2 carbonyl groups;
each Ra independently represents a member selected from
the group consisting of: H, C 1 _6 alkyl, OC 1 _( alkyl, aralkyl, substituted
aralkyl, heteroaralkyl, substituted heteroaralkyl, aralkoxy, substituted
aralkoxy, halo, hydroxy, CN, CONH2, CONHC 1-6 alkyl, CONIC 1-6
alkyl)2, C02H, C02C1_6 alkyl, C(O)C1_6 alkyl, phenyl, CF3, SH,
N02, SOyCI_6 alkyl, with y as defined above; S02NH2, S02NHCl_(
alkyl, NHSOZ(substituted aryl), NHS02(substituted heteroaryl),
NHS02C1_6alkyl, NHS02ary1, NHS02heteroaryl, NH2, NHC1_( alkyl,
N(Cl _6 alkyl)2, NHC(O)C 1-6 alkyl, NHC(O)NH(C 1-6 alkyl), CZ-4
alkenyl and C2_4 alkynyl;
Rq~ represents H, OH, C 1 _q. alkyl, -OC 1 _4 alkyl, aryl or
C(O)Cl_4 alkyl, and
Rq~~ represents H, OH or OC 1 _4 alkyl.
CA 02257200 1998-12-02
WO 97/47618 PCT/ITS97/09888
_7 _
A pharmaceutical composition is also included in the
invention described herein, which is comprised of a compound of formula
I as defined above in combination with a pharmaceutically acceptable
carrier.
Also included in the invention is a method of treating a
cytokine mediated disease in a mammal, comprising administering to a
mammalian patient in need of such treatment an amount of a compound
of formula I which is effective for treating said cytokine mediated
disease.
DETAILED DESCRIPTION OF THE INVENTION
The invention is described herein in detail using the terms
defined below unless otherwise specified.
The term "alkyl" refers to a monovalent alkane
(hydrocarbon) derived radical containing from 1 to 15 carbon atoms
unless otherwise defined. It may be straight or branched, and when of
sufficient size, e.g., C3_ 15 may be cyclic. Preferred straight or
branched alkyl groups include methyl, ethyl, propyl, isopropyl, butyl
and t-butyl. Preferred cycloalkyl groups include cyclopropyl,
cyclopentyl and cyclohexyl.
Alkyl also includes an alkyl group substituted with a
cycloalkyl group, such as cyclopropylmethyl.
Alkyl also includes a straight or branched alkyl group
which contains or is interrupted by a cycloalkylene portion. Examples
include the following:
and -(CH2)w ~ CH
- (CH2)x~ '~~1~ (CHZ)y~- ~ ( 2)z
wherein: x' and y' = from 0-10; and w and z = from 0-9.
The alkylene and monovalent alkyl portions) of the alkyl
group can be attached at any available point of attachment to the
cycloalkylene portion.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
_g_
When substituted alkyl is present, this refers to a straight,
branched or cyclic alkyl group as defined above, substituted with 1-3
groups as defined with respect to each variable.
Heteroalkyl means an alkyl group containing from 2-15
carbon atoms and being interrupted by from I -4 heteroatoms selected
from O, S and N.
The term "alkenyl" refers to a hydrocarbon radical
straight, branched or cyclic containing from 2 to 15 carbon atoms and
at /east one carbon to carbon double bond. Preferably one carbon to
carbon double bond is present, and up to four non-aromatic (non-
resonating) carbon-carbon double bonds may be present. Preferred
alkenyl groups include ethenyl, propenyl, butenyl and cyclohexenyl.
As described above with respect to alkyl, the straight; branched or
cyclic portion of the alkenyl group may contain double bonds and may
be substituted when a substituted alkenyl group is provided.
The term "alkynyl" refers to a hydrocarbon radical
straight, branched or cyclic, containing from 2 to 15 carbon atoms and
at least one carbon to carbon triple bond. Up to three carbon-carbon
triple bonds may be present. Preferred alkynyl groups include
ethynyl, propynyl and butynyl. As described above with respect to
alkyl, the straight, branched or cyclic portion of the alkynyl group may
contain triple bonds and may be substituted when a substituted alkynyl
group is provided.
Aryl refers to aromatic rings e.g., phenyl, substituted
phenyl and like groups as well as rings which are fused, e.g., naphthyl
and the like. Aryl thus contains at least one ring having at least 6
atoms, with up to two such rings being present, containing up to 10
atoms therein, with alternating (resonating) double bonds between
adjacent carbon atoms. The preferred aryl groups are phenyl and
naphthyl. Aryl groups may likewise be substituted as defined below.
Preferred substituted aryls include phenyl and naphthyl substituted
with one or two groups.
The term "heteroaryl" refers to a monocyclic aromatic
hydrocarbon group having S or 6 ring atoms, or a bicyclic aromatic
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-9-
group having 8 to 10 atoms, containing at least one heteroatom, O, S
or N, in which a carbon or nitrogen atom is the point of attachment,
and in which one additional carbon atom is optionally replaced by a
heteroatom selected from O or S, and in which from 1 to 3 additional
carbon atoms are optionally replaced by nitrogen heteroatoms. The
heteroaryl group is optionally substituted with up to three groups.
Heteroaryl includes aromatic and partially aromatic groups
which contain one or more heteroatoms. Examples of this type are
thiophene, purine, imidazopyridine, pyridine, oxazole, thiazole, oxazine,
pyrazole, tetrazole, imidazole, pyridine, pyrimidine, pyrazine and
triazine. Examples of partially aromatic groups are
tetrahydroimidazo[4,5-c]pyridine, phthalidyl and saccharinyl, as defined
below.
Each Ra independently represents a member selected from
the group consisting of: H, C 1 _6 alkyl, OC 1 _6 alkyl, aralkyl, substituted
aralkyl, heteroaralkyl, substituted heteroaralkyl, aralkoxy, substituted
aralkoxy, halo, hydroxy, CN, CONH2, CONHCI_6 alkyl, CON(Cl-6
alkyl)2, C02H, C02C1 _6 alkyl, C(O)C 1 _6 alkyl, phenyl, CF3, SH,
N02, SOyCI_6 alkyl, with y as defined above; S02NH2, S02NHCl-6
alkyl, NHS02(substituted aryl), NHS02(substituted heteroaryl),
NHS02C 1 _6alkyl, NHS02ary1, NHS02heteroaryl, NH2, NHC 1 _6 alkyl,
N(Cl _6 alkyl)2, NHC(O)C 1 _6 alkyl, NHC(O)NH(C 1 _~ alkyl), C2-4
alkenyl and CZ_4 alkynyl. In substituted aralkyl, substituted
heteroaralkyl and substituted aralkoxy, the aryl, heteroaryl or alkyl
portions thereof can be substituted as appropriate.
Substituted alkyl, aryl and heteroaryl, and the substituted
portions of aralkyl, aralkoxy, heteroaralkyl, heteroaralkoxy and like
groups are substituted with from 1-3 groups selected from the group
consisting of: halo, hydroxy, cyano, acyl, acylamino, aralkoxy,
alkylsulfonyl, arylsulfonyl, alkylsulfonylamino, arylsulfonylamino,
alkylaminocarbonyl, alkyl, alkoxy, aryl, aryloxy, aralkoxy, amino,
alkylamino, dialkylamino, and sulfonylamino.
The terms "heterocycloalkyl" and "heterocyclyl" refer to a
cycloalkyl group (nonaromatic) in which one of the carbon atoms in the
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-10-
ring is replaced by a heteroatom selected from O, S(O)y or N, and in
which up to three additional carbon atoms may be replaced by said
heteroatoms. When three heteroatoms are present in the heterocycle, they
are not all linked together.
Examples of heterocyclyls are piperidinyl, morpholinyl,
azetidinyl, pyrrolidinyl, tetrahydrofuranyl, imidazolinyl, piperazinyl,
pyrolidin-2-one, piperidin-2-one and the like.
Acyl as used herein refers to -C(O)Cl-( alkyl and -C(O)-
aryl.
Acylamino refers to the group -NHC(O)C1-6 alkyl and
-NHC(O)aryl.
Aralkoxy refers to the group -OC I -6 alkylaryl.
Alkylsulfonyl refers to the group -S02C1_6 alkyl.
Alkylsulfonylamino refers to the group -NHS02C1-(alkyl.
Arylsulfonylamino refers to the group -NHSOZaryI.
Alkylaminocarbonyl refers to the group -C(O)NHC 1 _6 alkyl.
Aryloxy refers to the group -O-aryl.
Sulfonylamino refers to the group -NHS03H.
Halo means Cl, F, Br and I selected on an independent basis.
~Ra)3
N/ i '
N
and are optional substituents linked
to the HETCy group.
N N
and independently represent mono or bicyclic
ring systems, non-aromatic or partially aromatic, containing from 5-10
ring atoms, 1-4 of which are N and 0-1 of which are O or S(O)y, with y
equal to 0, 1 or 2, and when partially aromatic, the non-aromatic portion
thereof optionally containing 1-2 carbonyl groups. Hence, these ring
N
systems can be heteroaryl or heterocyclic as defined above.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-11-
is linked to HETCy through a nitrogen atom contained in the ring system,
either directly or through a linking group which is part of R'. Examples
N
include phthalidyl and saccharinyl, as further defined below. is
likewise linked to HETCy, but through a carbon atom contained in the
ring system.
The term phthulidyl refers to the heteroaryl
O
-N
i
group: O
The term saccharinyl refers to the heteroaryl
N
group: S02
The group
-O~(O~-(CH2)1-CR5R6-(CH2)k'NR~J Rs
P
means -O-C(O~(CH2)WCR5R~-(CH2)k-NR~-R8
-O-C((~-(CH2)1-CR5R6-(CH2)k-NR~C(O)(CH2)j CR5R6(CH2)kNR~R8
and
-OC(O~CH2)jCR5R6(CH2)kNR7C(O)(CH2)jCR5R6(CH2)kNR~C(O)(CH2)jCR5R6(CH2)kNR~Rg
LNR~(CH2)k"CR5R6-(CH2)j.C(OJ OR9
Likewise, the group P
CA 02257200 1998-12-02
WO 97/47618 PCT/~JS97/09888
-12-
means -NRZ (CH2)k-CR5R6- (CH2)~-C(p)-OR9
-NRZ (CH2)k-CR~R6- (CH2)1-C(p?NR~(CH2)kCR5R6(CH2)1C02R9
or
-NR7(CH2)hCR5R6(CH2)~C(O)NR~(CH2)kCR5R6(CH2)~~C(O)NR~(CHZ)kCR5R6(CH2)~C02R9
The variables are determined independently within each
group. Thus, e.g., when more than one j is present, they may be the same
or different. The values of RS and R~ can be H, C l _6 alkyl(R9)3 or aryl,
or CRSR6 taken in combination represents a 3-6 membered cycloalkyl or
heterocyclyl group, an aryl group or a heteroaryl group. Examples of
suitable CRSR6 groups include:
The patterns of attachment noted above can also include heteroatoms as
appropriate. When an aryl or heteroaryl group is represented by CRSR6,
attachment cannot be through the same carbon atom.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-13-
The term "TNF mediated disease or disease state" refers to
disease states in which TNF plays a role, either by production or
increased activity levels of TNF itself, or by causing another monokine to
be released, such as but not limited to IL,-1 or IL-6. A disease state in
which IL-1, for instance is a major component, and whose production or
action, is exacerbated or secreted in response to TNF, would therefore be
considered a disease state mediated by TNF.
The term "cytokine" as used herein means any secreted
polypeptide that affects the functions of cells and is a molecule which
modulates interactions between cells in the immune, inflammatory or
hematopoietic response. A cytokine includes, but is not limited to,
monokines and lymphokines regardless of which cells produce them.
Examples of cytokines include, but are not limited to, Interleukin-1 (IL-
1 ), Interleukin-6 (IL-6), Interleukin-8 (IL-8), Tumor Necrosis Factor-
alpha (TNF-a) and Tumor Necrosis Factor-beta (TNF-b).
By the term "cytokine interfering or cytokine suppresive
amount" is mean an effective amount of a compound of formula I which
will cause a decrease in the in vivo activity or level of the cytokine to
normal or sub-normal levels, when given to the patient for the
prophylaxis or therapeutic treatment of a disease state which is
exacerbated by, or caused by, excessive or unregulated cytokine
production or activity.
The compounds of the present invention may contain one or
more asymmetric carbon atoms and may exist in racemic and optically
active forms. All are within the scope of the present invention.
One subset of compounds of particular interest is
described with respect to formula I wherein one or two R" groups are
present, and each independently represents NH2, NHC1-( alkyl(R9)3,
N(CI_6 alkyl)2, NHC3_R cycloalkyl, N(R9')C(O)C1.6 alkyl(R9)3,
C 1 _6 alkyl(R9)3, OC 1 _6 alkyl(R9)3, C02H, CONH2,
NRq~C(O)NHCI-6 alkyl(R~l)3 or heterocyclyl(R9)3. Within this
subset of compounds, all other variables are as previously defined.
Another subset of interest includes compounds of formula
I wherein HETCy represents a 5-6 membered non-aromatic
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-14-
heterocycle with 1-2 nitrogen atoms contained therein. HETCy is
preferably a pyrrolidinyl or piperidinyl group, and most preferably a
4-piperidinyl group. Within this subset of compounds, all other
variables are as previously defined.
Another subset of compounds is represented by formula I
wherein R' is selected from C 1 _6 alkyl(R9)3, OC 1-6 alkyl(R9)3,
-C(O)C 1 _~ alkyl(R9)3; CN, N02 and C02C 1 _~ alkyl(R9)3. Within
this subset of compounds, all other variables are as previously defined.
Another subset of interest includes compounds of formula
I wherein from 1-3 R groups are present and each independently
represents a member selected from the group consisting of: halo,
hydroxy, CI-( alkyl(R9)3, OCI_6 alkyl(R9)3 , NH2, NHCI-6
alkyl(Rq)3 , N(C1-( alkyl(R~l)3)2 and CF3. Within this subset of
compounds, all other variables are as previously defined.
More particularly, a subset of interest relates to
compounds of formula I wherein one or two R groups are present,
selected from halo and CF3. Within this subset of compounds, all
other variables are as previously defined.
Another subset of compounds is represented by formula I
wherein Rx is H, C3_g cycloalkyl or C 1-6 alkyl(R9)3. More
particularly, Rx represents H, CH3 , CH2CH3, CH2CH2CH3,
CHZCH2CH2CH3 ~ °~ ~H2~ . Within this subset of
compounds, all other variables are as previously defined.
Another subset of compounds is represented by formula I
wherein X' represents a direct bond. Within this subset of compounds,
all other variables are as previously defined.
Another subset of compounds is represented by formula I
wherein X represents -(CH2)m-Y-(CH2)n-, Y represents a direct
bond, O, S or C(O); m represents 0 or 1 and n represents 0 or 1.
Within this subset, X preferably represents a direct bond. Within this
subset of compounds, all other variables are as previously defined.
A preferred subset of compounds is represented by
formula I wherein:
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-15-
one or two R" groups are present, each independently
representing NH2, NHC I _6 alkyl(R9)3, N(C 1 _g alkyl~2,
NHC3_g cycloalkyl, N(R9')C(O)C 1 _6 alkyl{R9)3, C1 _6 alkyl(R9)3,
OC1_6 alkyl(R9)3, C02H, CONH2, NR9~C{O)NHCI_6 alkyl{R9)3
or heterocyclyl(R9)3;
HETCy represents a 5-6 membered non-aromatic
heterocyclyl with 1-2 nitrogen atoms contained therein;
R' is selected from C 1 _6 alkyl(R9)3, OC 1 _6 alkyl(R9)3,
-C(O)C 1 _6 alkyl{R9)3; CN; N02; and C02C 1 _6 alkyl(R9)3
; from 1-3 R groups are present and each independently
represents a member selected from the group consisting of: halo,
hydroxy, C 1 _6 alkyl(R9)3, OC 1 _~ alkyl(R9)3 , NH2, NHC 1-6
alkyl(R9)3 , N(C1_6 alkyl(R9)3)2 C02H and CF3 ;
Rx is H, C3_g cycloalkyl or C 1 _6 alkyl(R~l)3
X' represents a direct bond;
X represents -(CH2)m-Y-(CH2)n-, Y represents a direct
bond; m represents 0 or 1 and n represents 0 or 1.
A more preferred subset of compounds of the invention is
represented by formula I wherein:
one or two R" groups are present, selected from the
group consisting of: NH2, NHC 1 _~ alkyl(R~1}3, N(C 1 _6 alkyl)2, C 1-b
alkyl(R9)3, OCl_6 alkyl(R9)3 and heterocyclyl(R9)3;
HETCy represents a piperidinyl group;
zero or one R' is present, which is selected from C I -6
alkyl(Rq)3 , -C02C 1 _6 alkyl(R9)3 and -C(O)C 1 _6 alkyl(R9)3;
one or two R groups are present, selected from halo and
CF3;
Rx is H, C3_g cycloalkyl or C 1 _~ alkyl(R9)3; and
X and X' represent a direct bond.
A more preferred subset of compounds of the invention is
represented by formula I wherein:
one R" group is present and is selected from:
i
CA 02257200 1998-12-02
WO 97/47618 PCT/L1S97/09888
- I 6-
-NH2 ~ -NHCH3 -NH--Q -NHCH2CF3
,
-N(CH3)2 -NHCH2 ~ ~ OCH3 -N,
,
,
-NHCH2 ~ ~ ~ -NHCH ~ ~ ~ -NHCH2
CH3
F3C
C F3
-NHCH2 ~ ~ -NHCH2 ~ ~ CF3 and -OC~_6 alkyl .
,
HETCy represents a 4-piperidinyl group;
R' is absent;
one or two R groups are present selected from halo and
CF3;
CH2-
RX represents H, CH3 ~ or ;
and X and X' represent direct bonds.
Representative species falling within the present invention
include the following:
CA 02257200 1998-12-02
WO 97!47618 PCT/US97/09888
-17-
CF3 CF3
,-
\ /
/
N i H ~~ I / N ~ H ~N O
HN ~ HN ~ O
OCH3 ~ OCH3
CF3
\ /
/
N
N ~N H N O
NHCH3 O
CF3
\ /
N
N
N ~N H N O
NH O
H3C
H
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
- I 8-
CF3
N
IV ~ ~ O
a
NH O
H
CH3
CF3
N
N
N ~N H ~ ~
NH O
b
CF3
N
N
N ~N H ~ O
NH2 O
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-19-
CF3
N
I\ N 1 /I
NY N CH3 N~ O \
NH2 ~O
CF3
N
N ~N H ~ O \
OEt O
CF3 CF3
I N I N
I \ H ~ I \ H
N~N NCH N~N N\/CH3
NH NI ~H
O
w
/ OCH3 1 / OCH3
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-20-
CF3 CF3
\ ~ \
/ N / N
I \ H ~ I \ H
N~N N~N~CH3 N~ N NH
NH p CH3 NH
pCH3 ~ pCH3
CF3 CF3
\ / \ /
/ v / v
I \ N ~ I \ N
N\/ N H NH N\/ N H NH
TNHCH3 ~N' H2
CF3
CF3
\ N
\ /
/ \ N ~ N H NH
l
I \
N ~ N H NH N
N(CH3)2
CA 02257200 1998-12-02
WO 97/47618 PCT/I1S97/09888
-21-
CF3 CF3
\ / \ /
I ~' N I \ H
N~N H NH N~N NH
NH - NH
HC = ~ ~ H
3 ,~
H CHa
CF3
CF3
\ /
N \ / CH3
\ N I N
I
N\/N NH ~ \ N
NH N~ N NH
NH2
CF3
CF3 \ / H
/ N
\ / ~ \
N~ N NH
i \ N ~ NH
N\/ N CH NH \
3
NH2 CF3
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-22-
CF3 CF3
H ~ H
N / N
/N \ N
I 1 I
N~N NH NYN NH
NH ~ I NH ~ CF3
\ CFa \
CF3
C F3
H
I ~ / H
I \ N / N
N\/ N NH I \
N ~l
NHCH2CF3 NON NH
CF3 CF3
H \ / H
N / N
\ N I \ N
NYN NH N~N NH
C02H C(O)NH2
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-23-
CF3
N
I N
N~N CH3 NH
HN
H
CH3
As used herein, THF means tetrahydrofuran, DMF means
dimethylformamide and HOBT means hydroxybenzotriazole.
The compounds of the present invention are prepared by
procedures illustrated in the accompanying schemes. The general
method of preparing the imidazole nucleus is outlined in Scheme 1. 2-
Mercaptopyrimidine 1 (commercially available from Aldrich Chemicals
Inc) is reacted with a dialkyl dimethylformamide acetal such as dimethyl
formamide dimethyl acetal in solvent (e.g. toluene) in the presence of a
base e.g. diisopropylethylamine. Deprotonation of the product 2 with a
strong base such as lithium diisopropylamide and quenching of the anion
with an appropriately substituted N,O-dimethylhydroxamide 3 provides a
ketone 4. Formation of the oximoketone 5 may be carried out under
acidic conditions with either an inorganic nitrite (e.g. sodium nitrite) or
an organic nitrite (e.g. t-butyl nitrite). Reaction of the oximinoketone
with a suitably functionalized and protected amino aldehyde and an
ammonium salt (e.g. ammonium acetate) in acetic acid leads to a
hydroxyimidazole 6. Reduction to the imidazole 7 may be accomplished
by a number of methods described in the literature (hydrogenation,
reaction with PC13 or titanium trichloride). The imidazole may then be
alkylated either by reaction with an alkyl halide and base in solvent (e.g.
methyl iodide, cesium carbonate, DMF) or by heating with a
dimethylformamide dialkyl acetal neat or in a solvent (e.g.
CA 02257200 1998-12-02
WO 97/47618 PCT/ITS97/09888
-24-
dimethylformamide dimethyl acetal in toluene) to provide a mixture of
isomers 8 and 9, separable by chromatography.
As shown in Scheme 2, the methylthiopyrimidine can be
oxidized to the methylsulfonylpyrimidine, for example with oxone in
methanol and then the methylsulfonyl group displaced with an amine to
provide an aminopyrimidine derivative. Deprotection of the
benzyloxycarbonyl protecting group could then be accomplished by
treatment with hydrogen bromide in acetic acid or aqueous hydrochloric
acid.
Scheme 3 illustrates compounds that may be obtained after
the amine displacement reaction described in Scheme 2, where the amine
utilized was 4-methoxybenzylamine. Reduction of the intermediate 10
with lithium aluminum hydride affords the N-methypiperidine derivative
13, Treatment with hydrogen bromide in acetic acid provides the
piperidine 12 and treatment with hydrochloric acid at reflux leads to 11.
The piperidine of compound 12 can be further reacted for example by
reductive alkylation to provide 14 or by acylation to provide compounds
15.
Alternatively the methanesulfonyl group can be displaced
with cyanide as shown in Scheme 4 to afford 17. Compound 17 may then
be converted to a number of different products. Utilizing refluxing
hydrochloric acid leads to the parent pyrimidine 19, whereas hydrogen
bromide in acetic acid gives the carboxamide 20, which can be
hydrolyzed to the acid 21 under basic conditions, e.g. aqueous sodium
hydroxide solution.
Reactions on the piperidine nitrogen may also be carried out
prior to the displacemnt of the sulfonylpyrimidine as shown in Scheme 5.
The piperidine nitrogen of 22 may be acylated, for example employing
EDC, HOBt and triethylamine and an appropriate carboxylic acid in
DMF to give 23 and then the sulfonyl group displaced with an amine, e.g.
ammonia to provide 24.
CA 02257200 1998-12-02
WO 97147618 PCT/US97/09888
-25-
The aminopyrimidine can also be acylated, for example by
treatment with an appropriate acid chloride (Scheme 6) to give 26 from
which the protecting group can be removed with hydrogen bromide in
acetic acid, yielding compounds 27.
SCHEME 1
/CH3 dimethylformamide \ CH3
dimethyl acetal
N~ N - N ~ N
diisopropylethylamine
SH toluene heat SCH3
2
~R)o-s~ I ~R)o-3'
Strong Base \ O I
e.g. LDA \ O
Acid
O
\ RN02 \ NOH
~R)o-s~- \ NMeOMe N ~ N N ~ N
4
SCH3 SCH3 5
3
(R)o-a~
N O
N H40Ac
~N-~C
AcOH I \ N O
RCHO N ~ N OH
6
SCH3
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-26-
SCHEME 1 cont'd
(R)o-3~
/
reduction I N
-- ~ N
e.g. TiCl3/MeOH I \ H O
N~N
SCH3 7
alkyl halide/base
or
dimethylformamide
dialkyl acetal
heat
(R
(R)o-3~
N O /
~ ,N-~
\ N~ O
N ,N CHs +
8
SCH3
) 0-3\
NCH3 O
N-~
\ N O
NYN
SI CH3
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-27-
(R)o-s
N O
I ~ ~N--~
I \ N O
N~ N Rx
SCH3 I
Oxone ~ MeOH
(R)o-s~
N O
I ~ ~N ---~
I \ 1 O
N~N Rx
SO2CH3
NH2-R
(R)
N O /
,N --
N O
N~N Rx
HIN
R
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-28-
(R)
Scheme 2 cont'd
o-s~~-
N o
i ~ ~N--~
I ~ N\ O
N\//N RX
i
HN
'R
HBr/AcOH
or
HCI/H20 reflux
(R)
0-3\
N
I ~ NH
I ~ N~
N~N Rx
HN
~R
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-29-
(R)o-3
\ / N O
~ ~N~
H O
N\/ N
H~N ~ OCH3 10
LiAIH4 THF
HCI / H20/ hea
~R)o_s\
~R)o-s~
\ N
NH I ~ 'N-CH3
I ~ H \--~ I ~ ' H
N~ N
N~ N
NH2 11 HN \ / OCH3 i3
H B r/Ac0 H
~R)o-s~
N _
I ~ NH
~' H
N\ / N
HN~ ~ OCH3 12
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-30-
Scheme 3 cont'd
(R)o_3~
\ N ~I
.N
I ~'H
N~ N
HN ~ OCH3 (R)o-s~
N O
~ ,N-~
I ~ H R"
N\/ N
HN -~' OCH3
R" = CH3,
CH2N(CH3)2
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-31-
(R)o-3~
N O
~N--~
I \ ~ O
N~N Rx
SO2CH3
NaCN,DMF
(R)o 3' / HBrlAcOH
N O
I v N
\ N O
N ~ N Rx
17
CN
(R)o
NH
N YN Rx
S02CH3 18
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-32-
(R)o-sW
N O
I v~N~O
I ~'N x
N ~ N ~R
17
CN
AcOH
3N HCI
(R)
(R)o-sw''
N
N ~ ~?~N H
~~~N H N
I ~'N~ ~ N ~N Rx 20
x
NON R ONH2
19
NaOH/H20
(R)o-sW /
N _
NH
I
N~N Rx
COOH
21
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-33-
~R)o-s~
N _
I ~ NH
I ~ 'N
N\/N Rx
S102C H3 22
EDC, HOBT,Triethylamine
R2-COOH
~R)o-s~~
N ~ ,
N
( ~ N R2
N ~ N Rx
23
S02C H3
NH3
~R)o-s~~
N O
I ~ ~N --~
i N R2.
N i N Rx
R2~= alkyl, aryl
NH2 24
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-34-
(R
) 0-3\
N O
~ ,N -~
I ~ N O
N~N Rx R2~-COCI
N H2 25
(R)o-3y /
N O
~ ,N -~
I ~ N O
N i N Rx
R2~ = alkyl, aryl HBr/AcOH
HN
R2.
O
26
( R ) 0-3~~
N _
I ~ NH
I ~~j
N~ N Rx
HN Rz= alkyl, aryl
~'l- R2
O
27
The compounds of the present invention are useful in
various pharmaceutically acceptable salt forms. The term
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-35-
"pharmaceutically acceptable salt" refers to those salt forms which would
be apparent to the pharmaceutical chemist. i.e., those which are
substantially non-toxic and which provide the desired pharmacokinetic
properties, palatability, absorption, distribution, metabolism or excretion.
Other factors, more practical in nature, which are also important in the
selection, are cost of the raw materials, ease of crystallization, yield,
stability, hygroscopicity and flowability of the resulting bulk drug.
Conveniently, pharmaceutical compositions may be prepared from the
active ingredients in combination with pharmaceutically acceptable
carriers.
The pharmaceutically acceptable salts of the compounds
of formula I include conventional non-toxic salts or quarternary
ammonium salts of the compounds of formula I formed e.g. from non-
toxic inorganic or organic acids. For example, non-toxic salts include
those derived from inorganic acids such as hydrochloric, hydrobromic,
sulfuric, sulfamic, phosphoric, nitric and the like; and the salts
prepared from organic acids such as acetic, propionic, succinic,
glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic,
sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methane-
sulfonic, ethane disulfonic, oxalic, isethionic, trifluoroacetic and the
like.
The pharmaceutically acceptable salts of the present
invention can be synthesized by conventional chemical methods.
Generally, the salts are prepared by reacting the free base or acid with
stoiehiometric amounts or with an excess of the desired salt-forming
inorganic or organic acid or base, in a suitable solvent or solvent
combination.
The compounds of the present invention may have
asymmetric centers and occur as racemates, racemic mixtures, and as
individual diastereomers. All such isomers, including optical isomers,
being included in the present invention.
The compounds of formula 1 can be used in the prophylactic
or therapeutic treatment of disease states in mammals which are
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-36-
exacerbated or caused by excessive or unregulated cytokines, e.g., IL-l,
IL-6, IL-8 or TIVF.
Because the compounds of formula I inhibit cytokines, the
compounds are useful for treating diseases in which cytokine presence or
activity is implicated, such as rheumatoid arthritis, rheumatoid
spondylitis, osteoarthritis, gouty arthritis and other arthritic conditions.
The compounds of formula I are useful to treat disease states
mediated by excessive or unregulated TNF production or activity. Such
diseases include, but are not limited to sepsis, septic shock, endotoxic
shock, gram negative sepsis, toxic shock syndrome, adult respiratory
distress syndrome, cerebral malaria, chronic pulmonary inflammatory
disease, silicosis, pulmonary sarcoidosis, bone resorption diseases, such
as osteoporosis, reperfusion injury, graft v. host rejection, allograft
rejection, fever, myalgia due to infection, cachexia secondary to infection
or malignancy, cachexia secondary to acquired immune deficiency
syndrome (AIDS), AIDS, ARC (AIDS related complex), keloid
formation, scar tissue formation, Crohn's disease, ulcerative colitis,
pyresis, AIDS and other viral infections, such as cytomegalovirus
(CMV), influenza virus, and the herpes family of viruses such as Herpes
Zoster or Simplex I and II.
The compounds of formula I are also useful topically in the
treatment of inflammation such as in the treatment of rheumatoid
arthritis, rheumatoid spondylitis, osteoarthritis, gouty arthritis and other
arthritic conditions; inflamed joints, eczema, psoriasis or other
inflammatory skin conditions such as sunburn; inflammatory eye
conditions including conjunctivitis; pyresis, pain and other conditions
associated with inflammation.
The compounds of formula I are also useful in treating
diseases characterized by excessive IL-R activity. These disease ,states
include psoriasis, inflammatory bowel disease, asthma, cardiac and renal
reperfusion injury, adult respiratory distress syndrome, thrombosis and
glomerulonephritis.
The invention thus includes a method of treating psoriasis,
inflammatory bowel disease, asthma, cardiac and renal reperfusion injury,
CA 02257200 1998-12-02
WO 97/47618 PCTlUS97/09888
7_
adult respiratory distress syndrome, thrombosis and glomerulonephritis,
in a mammal in need of such treatment, which comprises administering to
said mammal a compound of formula I in an amount which is effective
for treating said disease or condition.
When administered to a patient for the treatment of a disease
in which a cytokine or cytokines are implicated, the dosage used can be
varied within wide limits, depending upon the type of disease, the age and
general condition of the patient, the particular compound administered,
the presence or level of toxicity or adverse effects experienced with the
drug and other factors. A representative example of a suitable dosage
range is from as low as about 0.01 mg/kg to as high as about 100 mg/kg.
However, the dosage administered is generally left to the discretion of the
physician.
The methods of treatment can be carried out by delivering
the compound of formula I parenterally. The term 'parenteral' as used
herein includes intravenous, intramuscular, or intraperitoneal
administration. The subcutaneous and intramuscular forms of parenteral
administration are generally preferred. The instant invention can also be
carried out by delivering the compound of formula I subcutaneously,
intranasally, intrarectally, transdermally or intravaginally.
The compounds of formula I may also be administered by
inhalation. By 'inhalation' is meant intranasal and oral inhalation
administration. Appropriate dosage forms for such administration, such
as an aerosol formulation or a metered dose inhaler, may be prepared by
convention techniques.
The invention also relates to a pharmaceutical composition
comprising a compound of formula I and a pharmaceutically acceptable
carrier. The compounds of formula I may also be included in
pharmaceutical compositions in combination with a second
therapeutically active compound.
The pharmaceutical carrier employed may be, for example,
either a solid, liquid or gas. Exemples of solid carriers include lactose,
terra alba, sucrose, talc, gelatin, agar, pectin, acacia, magnesium stearate,
stearic acid and the like. Exemples of liquid carriers are syrup, peanut
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-38-
oil, olive oil, water and the like. Examples of gaseous carriers include
carbon dioxide and nitrogen.
Similarly, the carrier or diluent may include time delay
material well known in the art, such as glyceryl monostearate or glyceryl
distearate, alone or with a wax.
A wide variety of pharmaceutical dosage forms can be
employed. If a solid dosage is used for oral administration, the
preparation can be in the form of a tablet, hard gelatin capsule, troche or
lozenge. The amount of solid carrier will vary widely, but generally will
be from about 0.025 mg to about 1 g. When a liquid dosage form is
desired for oral administration, the preparation is typically in the form of
a syrup, emulsion, soft gelatin capsule, suspension or solution. When a
parenteral dosage form is to be employed, the drug may be in solid or
liquid form, and may be formulated for administration directly or may be
suitable for reconstitution.
Topical dosage forms are also included. Examples of topical
dosage forms are solids, liquids and semi-solids. Solids would include
dusting powders, poultices and the like. Liquids include solutions,
suspensions and emulsions. Semi-solids include creams, ointments, gels
and the like.
The amount of a compound of formula I used topically will,
of course, vary with the compound chosen, the nature and severity of the
condition, and can be varied in accordance with the discretion of the
physician. A representative, topical, dose of a compound of formula I is
from as low as about 0.01 mg to as high as about 2.0 g, administered one
to four, preferably one to two times daily.
The active ingredient may comprise, for topical
administration, from about 0.001 % to about 10% w/w.
Drops according to the present invention may comprise
sterile or non-sterile aqueous or oil solutions or suspensions, and may be
prepared by dissolving the active ingredient in a suitable aqueous
solution, optionally including a bactericidal and/or fungicidal agent
and/or any other suitable preservative, and optionally including a surface
active agent. The resulting solution may then be clarified by filtration,
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-39-
transferred to a suitable container which is then sealed and sterilized by
autoclaving or maintaining at 9$-100°C for half an hour. Alternatively,
the solution may be sterilized by filtration and transferred to the container
aseptically. Examples of bactericidal and fungicidal agents suitable for
inclusion in the drops are phenylmercuric nitrate or acetate (0.002%),
benzalkonium chloride (0.01 %) and chlorhexidine acetate (0.01 %).
Suitable solvents for the preparation of an oily solution include glycerol,
diluted alcohol and propylene glycol.
Lotions according to the present invention include those
suitable for application to the skin or eye. An eye lotion may comprise a
sterile aqueous solution optionally containing a bactericide and may be
prepared by methods ,similar to those for the preparation of drops.
Lotions or liniments for application to the skin may also include an agent
to hasten drying and to cool the skin, such as an alcohol or acetone,
and/or a moisturizer such as glycerol or an oil such as castor oil or arachis
oil.
Creams, ointments or pastes according to the present
invention are semi-solid formulations of the active ingredient for external
application. They may be made by mixing the active ingredient in finely-
divided or powdered form, alone or in solution or suspension in an
aqueous or non-aqueous liquid, with a greasy or non-greasy base. The
base may comprise hydrocarbons such as hard, soft or liquid paraffin,
glycerol, beeswax, a metallic soap; a mucilage; an oil of natural origin
such as almond, corn, arachis, castor or olive oil; wool fat or its
derivatives, or a fatty acid such as stearic or oleic acid together with an
alcohol such as propylene glycol or macrogels. The formulation may
incorporate any suitable surface active agent such as an anionic, cationic
or non-ionic surfactant such as sorbitan esters or polyoxyethylene
derivatives thereof. Suspending agents such as natural gums, cellulose
derivatives or inorganic materials such as silicas, and other ingredients
such as lanolin may also be included.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-40-
EXAMPLE 1
4-METHOXYBENZYL-f4-(2-PIPERIDIN-4-YL-5-(3
TRIFLUOROMETHYLPHENYL)-3H-IMIDAZOL-4-YLl-PYRIMIDIN
2-YLIAMINE
C F3
NH
\ I N
N
I \,' H
NvN
HN' I ~ OCH3
Step 1-A
2-Methylthio-4-methy~wrimidine
To 2-mercapto-4-methylpyrimidine~HCl (SO.Og, 0.307mo1e)
in toluene (750 mL), under argon, was added diisopropylethylamine (80.0
mL, 0.461 mole} followed by N,N-dimethylformamide dimethyl acetal
( 100 mL) and the mixture heated to reflux for 4 hours. Upon cooling, the
reaction was concentrated in vacuo to an oil, dissolved in ether (400 mL),
and washed with water (2xS0 mL). The organic extract was dried over
anhydrous sodium sulfate, filtered and concentrated to an oil which was
vacuum distilled to give 2-methylthio-4-methylpyrimidine (36.4 g) as an
oil.
1 H NMR(CDC13) d 8.37 (d, 1 H, J = 7.SHz), 6.82 (d, 1 H, J = 7.SHz}, 2.55
(s 3H), 2.45 (s, 3H).
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-41-
Step 1-B
2-(2-Methylthiop~rimidin-4-yl~-1-(3-trifluoromethvlphenyl)ethanone
To a solution of diisopropylamine (7.9 mL, 0.056 mole) in
THF (100 mL) at -78°C under argon was added 2.5M N-butyllithium
(22.5 mL, 0.056 mole), followed after 5 minutes by a solution of 2-
methylthio-4-methylpyrimidine (5.27 g, 0.0376 mole) in THF (20 mL).
Upon stirring for 15 min. at -7$°C, a solution of N-methoxy-N-
methyl-3-
trifluoromethylbenzarnide (9.63 g, 0.041 mole) in THF (90 mL) was
added. The reaction was allowed to warm to 0°C and then quenched by
pouring into water (400 mL) and ethyl acetate (400 mL}. The layers were
separated and the aqueous layer washed with ethyl acetate (200 mL). The
ethyl acetate extracts were combined, dried over anhydrous sodium
sulfate, filtered, and concentrated to a solid ( 11.9 g). Trituration with
10% ether/hexane ( 100 mL) gave 9.5 g of the title compound.
1H NMR (CDC13) (mixture of keto-and enol tautomers) d 6.0-8.5 (m,
6H, rotamers), 2.4-2.7 (m, 3H), .
Step 1-C
1-(2-Meth, l~pyrimidin-4-yl)-2-(3-trifluoromethylphenyl)-ethane-1,2-
dione 1-oxime
To a mixture of 2-(2-methylthiopyrimidin-4-yl)-1-(3-
trifluoromethylphenyl)ethanone (4.5 g, O.OI44 mole) in acetic acid (67
mL), was added THF (54 mL) and water (9 mL). The mixture was
cooled to +5°C and a solution of sodium nitrite ( 1.34 g, 0.0194 mole)
added dropwise while maintaining the temperature below +10°C. Upon
completion of addition, the reaction was allowed to warm to room
temperature for 1 hour, diluted with water (200 mL) and ethyl acetate
(200 mL) and the pH adjusted to 7.5 with 3N NaOH. The layers were
separated and the aqueous layer washed with ethyl acetate ( 100 mL). The
organic layers were combined, dried over anhydrous sodium sulfate,
filtered and concentrated to an oil. Yield 4.9 g.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-42-
I H NMR (CDCl3) d 8.58 (d, I H, J = 5.4 Hz), 8. I 8 (s, 1 H), 8.06 (d, 1 H, J
= 8.6 Hz), 7.89 (d, I H, J = 8.6 Hz), 7.66 (t, 1 H, J = 8.6 Hz), 7.52 {d, I H,
J
= 5.4 Hz), 2.18 {s, 3H), 1.60 (brs, 1 H).
Step 1-D
4-f 1-H dy roxv-5-(2-meth lthiopyrimidin-4-vl)-4-(3-
trifluorometh~phenyi)-IH-imidazol-2-yli.piperidine-1-carboxylic acid
bent, 1 ester
To a mixture of 1-(2-methylthiopyrimidin-4-yl)-2-(3-
trifluoromethylphenyl)ethane-1,2-dione-1-oxime (5.0 g, 0.0146 mole)
and N-carbobenzyloxypiperidine-4-carboxaldehyde [Amici et al Eur. J.
Med. Chem. 26, 625-631 ( 1991 )] (4.70 g, 0.019 mole), in acetic acid ( 140
mL) was added ammonium acetate (23 g, 0.295 mole). The mixture was
heated to reflux for 1-1/2 hours, cooled and concentrated to remove most
of the acetic acid. The residue was dissolved in water (200 mL) and ethyl
acetate (400 mL) and the pH adjusted to 7.5 with 3N sodium hydroxide.
The ethyl acetate layer was removed, dried over Na2S04, filtered and
concentrated to give an oil (9.5 g crude product). The oil was used in the
next step without further purification.
Step 1-E
4-f 5-(2-Methylthiopyrimidin-4-yl)-4-('i-trifluoromethYl~hen~1)-1 H-
imidazol-2- ~~llpiperidine-I-carboxylic acid benzyl ester
To a stirring solution of 4-[1-hydroxy-5-(2-
methylthiopyrimidin-4-yI)-4-(3-trifluoromethylphenyl)-IH-imidazol-2-
yl]piperidine-1-carboxylic acid benzyl ester (9.5 g. crude, 0.0146 mole)
in methanol (130 mL) at 20°C was added titanium (III) chloride (25 mL,
0.029 mole, 15% wt in 20-30% HCl) dropwise over 10 minutes. The
reaction was allowed to stir for 3 hours then quenched by pouring slowly
into a mixture of 10% aqueous sodium bicarbonate ( 1.5 L) and ethyl
acetate (600 mL). After stirring for 30 minutes the organic Layer was
removed and aqueous extract washed with ethyl acetate (2x200 mL). The
organic extracts were combined, dried over anhydrous sodium sulfate,
and concentrated to an oil. The oil was chromatographed on silica using
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-43-
60% ethyl acetate/hexane to give 6.35 g of a yellow foam upon
concentration of product containing fractions.
1 H NMR (CDCl3) d 10.18 (brs, 1 H), 8.28 (d, 1 H, J = 5.5 Hz), 7.2-7.8 (m,
9H), 6.95 (d, 1 H, J = 5.5 Hz), 5.12 (s, 2H), 4.30 (brs, 2H), 3.0 (brm, 3H),
2.58 (s, 3H), 1.7-2.2 (m, 4H).
Step 1-F
4-f5-(2-Methvlsulfonylpvrimidin-4~ ly )i4-(.3-trifluoromethvlT, 1~)-1H-
imidazol-2-vllpioerT e-1-carboxylic acid benzvl ester
To a stirring solution of 4-[5-(2-methylthiopyrimidin-4-yl)-
4-(3-trifluoromethylphenyl}-1 H-imidazol-2-yl]piperidine-1-carboxylic
acid benzyl ester (2.5 g, 0.0045 mole) in methanol (75 mL) at 20°C was
slowly added an aqueous solution (75 mL) of OxoneO (8.32 g, 0.0135
mole). The reaction was allowed to stir for 4 hours, concentrated in
vacuo to remove methanol, diluted with 10% aqueous sodium
bicarbonate ( 100 mL), and extracted with ethyl acetate (2x 150 mL). The
organic extracts were combined, dried over anhydrous sodium sulfate,
filtered and concentrated to give the title compound (2.75 g).
1H NMR (CDC13) d 11.06 (s,lH), 7.5-8.6 (m, 6H), 7.35 (m, SH), 5.12 (s,
2H), 4.30 (brs, 2H), 3.35 (s, 3H), 2.7-3.1 (m, 3H), 1.8-2.2 (m, 4H).
Step 1-G
4-LS ~~4-Methoxvbenzylamino)-p~rrimidin-4-yl)-4-(3-
trifluoromethvlohenvl)-1H-imidazol-2-,~pi~eridine-1-carboxylic acid
benz, l
A mixture of 4-[5-(2-methylsulfonylpyrimidin-4-yl)-4-(3
trifluoromethylphenyl)-1H-imidazol-2-yl]-piperidine-1-carboxylic acid
benzyl ester (1.5 g, 0.00256 mole) and 4-methoxybenzylamine (3.51 g,
0.026 mole) was heated in a pressure tube at 140°C for 10 minutes. The
mixture was allowed to cool and the residue column chromatographed on
silica using 5% methanol/rnethylene chloride to give 1.52 g of a yellow
powder upon concentration in vacuo of product containing fractions.
CA 02257200 1998-12-02
WO 97147618 PCT/IJS97/09888
-44-
Anal. Calc'd for C35H33N6~3F3: C 65.41, H 5.18, N 13.08.
Found: C 65:12, H 5.29, N 12.98.
Step 1-H
4-Methoxvbenzyl-f4-f2-piperidin-4-yl-5-(3-trifluorometh lnhenyl)-3H-
imidazol-4-yll-wrimidin-2-yllamine
To a stirred solution of 4-~:5-[2-(4-methoxybenzylamino)-
pyrimidin-4-yl]-4-(3-trifluoromethylphenyl)-I H-imidazol-2-yl]-
piperidine-1-carboxylic acid benzyl ester (I.0 g, 0.00156 mole) in
methylene chloride ( 16 mL) under argon was slowly added 30%
hydrogen bromide in acetic acid ( I 6 mL). The mixture was stirred at
20°C for 30 minutes and then diluted with diethyl ether (160 mL). The
resulting mixture was stirred for 1 hour, filtered and solid washed with
ether (10 mL). The resulting solid was dissolved in 10% aqueous sodium
bicarbonate (40 mL) and methylene chloride (50 mL). The methylene
chloride was separated and the aqueous layer washed with methylene
chloride (25 mL). The organic extracts were dried over anhydrous
sodium sulfate, filtered and concentrated to a foam (0.80 g). The solid
was chromatographed on silica using methylene
chloride/methanol/aqueous ammonium hydroxide (90:10:2) to give upon
concentration of product containing fractions 180 mg of the title
compound.
1 H NMR (CDC13) d 9.8 (brs, I H), 8.12 (d, 1 H, J = 5.8 Hz), 7.90 (s, 1 H),
7.80 (d, 1 H, J = 8.1 Hz), 7.62 (d, 1 H, J = 8.1 Hz), 7.42 (t, 1 H, J = 8.1
Hz),
7.32 (d, 1 H, J = 8.1 Hz), 6.90 (d, 2H, J = 8. I Hz), 6.78 (brs, 1 H), 5.44
(brs, 1 H), 4.6 {brs, 2H), 3.80 (s, 3H), 3.22 (m, 2H), 2.95 (m, 1 H), 2.77
(m, 2H), 1.6-2.2 (m, 4H).
The following examples (2-17) were prepared as described
in Example 1 and were isolated either by silica gel chromatography to
give the free base, preparative HPLC to give the trifluoroacetic acid salt
upon lyophilization, or crystallization of the hydrobromide salts.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-45-
EXAMPLE 2
4-(5-(2-METHYLAMINOPYRIMIDIN-4-YL)-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl-
PIPERIDINE-I-CARBOXYLIC ACID BENZYL ESTER
C F3
O
N~ OCHp
H
N'/N
HN~ CH3
To a stirred solution of 4-[5-(2-methylsulfonylpyrimidin-4-
yl)-4-(3-trifluoromethylphenyl)-I H-imidazol-2-yl]-piperidine-1-
carboxylic acid benzyl ester (Example 1 F) (0.5 g, 0.8 mmole) in ethanol
(10 mL) in a pressure vessel (50 mL) was added 40% aqueous
methylamine (20 mL). The vessel was sealed and then heated at 100°C
for 3 hours. The reaction was cooled, concentrated in vacuo and
chromatvgraphed on silica using methylene chloride/methanol/aqueous
ammonium hydroxide (95:5:1 ) to give 0.42 g of product. Crystallization
from ether ( 15 mL) gave 0.30 g of the title compound as a white solid.
Anal. Calc'd for C28H27N602F3: C 62.68, H 5.07, N 15.66.
Found: C 62.89, H 5.05, N 15.92.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-46-
EXAMPLE 3
4-f 5-(2-METHYLAMINOPYRIMIDIN-4-YL)-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl-PIPERIDINE
CF3
N
'N H
I \ 'H
N ,N
Y
HN~CH3
The title compound was prepared from 4-[5-[2-
(methylamino)-pyrimidin-4-yl]-4-(3-trifluoromethylphenyl)-1 H-
imidazol-2-yl]-piperidine-1-carboxylic acid benzyl ester (Example 1 F as
described in Example 1 step H.
Anal Calc'd for C2pH21 N6F3: C 59.69, H 5.26, N 20.88
Found: C 59.62, H 5.31, N 20.9_5
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-47 -
EXAMPLE 4
4=(5-(2-DIMETHYLAMINOPYRIMIDIN-4-YL)-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl-PIPERff~INE
C F3
W
I
N ,N
H3C~N_CH3
N~ /~
N H
a
The title compound was prepared in a similar manner from
4-[5-(2-methylsulfonylpyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-
imidazol-2-yl]-piperidine-1-carboxylic acid benzyl ester (Example 1F) as
described in Example 1 steps G (replacing 4-methoxybenzylamine with
40% aqueous dimethylamine) and H.
1 H NMR (CDC13) d 9.9 (brs, 1 H), 8.15 (d, 1 H, J = 5.0 Hz), 7.9 (s, 1 H),
7.81 (d, 1 H, J = 7.4 Hz), 7.62 (d, 1 H, .l = 7.4 Hz), 7.54 (t, 1 H, J = 7.4
Hz),
6.54 (brs, 1 H), 3.2 (m, 6H), 3.0 {m, 1 H), 2.78 (m, 2H), 1.7-2.2 {m, 6H).
Mass Spectral Analysis - M+1 = 417.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-48-
EXAMPLE 5
4-IS-f 2-( 1-PIPERDINYL)-PYRIMIDIN-4-YLl-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl-PIPERIDINE
CF3
i
N
'N H
I ' 'H
N ,N
N
The title compound was prepared from 4-[5-(2-
methylsulfonylpyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-imidazol-
2-yl]-piperidine-1-carboxylic acid benzyl ester (Example 1F) as
described in Example 1 steps G {replacing 4-methoxybenzylamine with
piperidine) and H.
1 H NMR (CDC13) d 9.8 (brs, I H), 8.15 (d, I H, J = 5.5 Hz), 7.90 (s, I H),
1 S 7.80 (d, 1 H, J = 7.8 Hz), 7.62 (d, 1 H, J = 7.8 Hz), 7.52 (t, 1 H, J =
7.8 Hz),
3.80 (brs, 4H), 3.22 (m, 2H), 3.0 (m, 1 H}, 2.77 (m, 2H), I .4-2.2 (m, 1 OH).
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-49-
EXAMPLE 6
~R)-4-[5-(2-( I -PHENYLETHYLAMINO)-PYRIMIDIN-4-YL)-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl
PIPERIDINE-1-CARBOXYLIC ACID BENZYL ESTER
CF3
O
N~N~OCH2 ~
I \
N1YN
HN
H3C H
The title compound was prepared from 4-[5-(2-methylsulfonylpyrimidin-
4-yl)-4-(3-trifluoromethylphenyl)-1 H-imidazol-2-yl]-piperidine-1-
carboxylic acid benzyl ester (Example 1F) as described in Example 1
steps (replacing 4-methoxybenzylamine with R(+)-a-
methylbenzylamine).
Anal. Calc'd for C35H33N602F3~0.3H20: C 66.50, I~ 5.36, N 13.30
Found: C 66.52, H 5.27, N 13.32
MP 85-87°C
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-50-
EXAMPLE 7
f R)-4-f 5-(2-~~l -PHENYLETHYLAMINO)-PYRIMIDIN-4-YL)-4 ;~3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl PIPERIDINE
HYDROCHLORIDE
CF3
W
N
~~NH
~ H
N'/N
~HCI
HN
HaC H
The title compound was prepared from (R)-4-[5-(2-( 1-
phenylethylamino)-pyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1H-
imidazol-2-yl]-piperidine-I-carboxylic acid benzyl ester (Example 6) as
described in Example I step H.
MP = 155-160°C
[a]D = +165.1°(MeOH)
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-51-
EXAMPLE R
(S)-4-f 5-f 2-~l-PHENYLETHYLAMINO)-PYRIMIDIN-4-YLl-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl
PIPERIDINE-1-CARBOXYLIC ACID BENZYL ESTER
CF3
I, o
N
N OCH2-
I w
NY N
HIN
CH
The title compound was prepared from 4-[5-{2-
methylsulfonylpyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-imidazol-
2-yl]-piperidine-1-carboxylic acid benzyl ester (Example 1 F) as described
in Example 1 step G (replacing 4-methoxybenzylamine with S(-)-a-
methylbenzylamine).
Anal. Calc'd for C35H33N602F3~0.2H20: C 66.69, H 5.34, N 13.33
Found: C 66.74, H 5.3R, N 12.98
MP RS-R7°C.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-52-
EXAMPLE 9
(S)-4-f 5-(2-( 1-PHENYLETHYLAMINO)-PYRIMIDIN-4-YL)-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl-PIPERIDINE
CF3
i
N _
NH
\ H
NYN ~HCI
HN
H CH
The title compound was prepared from (S)-4-[5-[2-(1-
phenylethylamino)-pyrimidin-4-yl]-4-{3-trifluoromethylphenyl)-1 H-
imidazol-2-yl]-piperidine-1-carboxylic acid benzyl ester (Example 8) as
described in Example 1 step H.
MP 157-161°C.
[a] D = -165.1 °(MeOH)
CA 02257200 1998-12-02
WO 97/47618 PCT/iJS97/09888
-53-
EXAMPLE 10
4-f S-f 2-(CYCLOPROPYLAMINO~PYRIMIDIN-4-YL)-4-t3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl
PIPERIDINE-1-CARBOXYLIC ACID BENZYL ESTER
CF3
O
~N~OCH2 ~
I
N\ / N
NH
The title compound was prepared from 4-[5-(2-
methylsulfonylpyrimidin-4-yI)-4-(3-trifluoromethylphenyl)-1 H-imidazol-
2-yl)-piperidine-1-carboxylic acid benzyl ester (Example 1 F) as described
in Example 1 step G (replacing 4-methoxybenzylamine with
cyclopropylamine).
Mass Spectral Analysis = M+1 = _563.
CA 02257200 1998-12-02
WO 97/47618 PCT/ITS97/09888
-54-
EXAMPLE 11
4-f 5-(2-(CYCLOPROPYLAMINO)PYRIMIDIN-4-YL)-4~3
TRIFLUOROMETHYLPHENYL)-i H-IMIDAZOL-2-YLl-PIPERIDINE
CF3
N
~~N H
I
N\/N
NH
The title compound was prepared 4-[5-(2-
(Cyclopropylamino)pyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-
imidazol-2-yl]-piperidine-1-carboxylic acid benzyl ester (Example 10) as
described in Example 1 step H.
1 H NMR(CDC13) d 10.1 (brs, 1 H), 8.16 (d, 1 H, J = 5.3 Hz), 7.92 (s, 1 H},
7.80 (d, 1 H, J = 8.0 Hz), 7.62 (d, 1 H, J = 8.0 Hz), 7.54 (t, 1 H, J = 8.0
Hz),
6.62 (brs, 1 H}, 3.20 (m, 2H), 3.0 (m, 1 H), 2.75 (m, 2H), 1.7-2.2 (m, 5H),
0.5-1.0 (m, 4H).
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-55-
EXAMPLE 12
4-f 5-(2-AMINOPYRIMIDIN-4-YL)-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YL~'
PIPERIDINE-1-CARBOXYLIC ACID BENZYL ESTER
CF3
O
N
NH O-CH2
I ~ 'H
N\/ N
~2CF3COOH
NH2
To a 50 mL pressure vessel cooled to -50°C and containing 4-[5-(2-
methylsulfonylpyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-imidazol-
2-yl]-piperidine-1-carboxylic acid benzyl ester (Example 1 F) ( 100 mg,
0.176 mmole} was introduced liquid ammonia ( 10 mL). The vessel was
sealed, allowed to warm to 25°C and stirred for 18 hours. The ammonia
was allowed to evaporate and the residue purified by preparative HPLC
using a C-18 column and 0.1 % trifluoroacetic acid/water and acetonitrile
as eluants. The title compound was isolated from the product containing
fractions by lyophilization to yield 100 mg.
Mass Spectral Analysis - M+1 = 523
Anal. Calc'd for C27H25N602F3~2.OCF3COOH: C 49.60, H 3.63, N
11.20
Found: C 49.53, H 3.40, N 10.90
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-56-
EXAMPLE I3
4-f 5-(2-(2,2,2-TRIFLUOROETHYLAMINO)-PYRIMIDIN-4-YL)-4-(3-
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl-PIPERIDINE
CF3
N
'N H
\ 'H
N ,N
~3 HBr
NH
~ CF3
The title compound was prepared from 4-[S-(2-methylsulfonylpyrimidin-
4-yl)-4-(3-trifluoromethylphenyl)- I H-imidazol-2-yl]-piperidine- I -
carboxylic acid benzyl ester (Example 1 F) as described in Example 1
steps G (replacing 4-methoxybenzylamine with 2,2,2-
trifluoromethylamine and carrying out the reaction for 7 days at 140oC)
and H.
Mass Spectral Analysis - M+1 = 471
MP - 205-210°C
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-57-
EXAMPLE 14
4-TRIFLUOROMETHYLBENZYL-14-f 2-PIPERIDIN-4-YL-5-(3
TRIFLUOROMETHYLPHENYL)-3H-IMIDAZOL-4-YLl-PYRIMIDIN
2-YL,] AMINE
CF3
N _
NH
I
N\/ N
HN
CF3
The title compound was prepared from 4-[5-(2
methylsulfonylpyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-imidazol
2-yl]-piperidine-1-carboxyl is acid benzyl ester {Example I F} as described
in Example 1 steps G (replacing 4-methoxybenzylamine with 4-
trifluoromethylbenzylamine) and H.
Purification by preparative HPLC resulted in isolation of the
trifluoroacetic acid salt after lyophilization of product containing
fractions.
Anal Calc'd for C27H24N6F~~2.~CF3COOH: C 45.22, H 3.12, N 9.71
Found: C 45.48, H 3.22, N 9.13
MP 75-79°C
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-58-
EXAMPLE 15
3-TRIFLUOROMETHYLBENZYL-f4-f2-PIPERIDIN-4-YL-5-(3
TRIFLUOROMETHYLPHENYL)-3H-IMIDAZOL-4-YLl-PYRIMII7IN-
2-YLl AMINE
CF3
N
'N H
\ 'H
N\/N
NH
C F3
The title compound was prepared from 4-[5-(2
methylsulfonylpyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-imidazol
2-yl]-piperidine-1-carboxylic acid benzyl ester (Example 1 F) as described
in Example 1 steps G (replacing 4-rnethoxybenzylamine with 3-
trifluoromethylbenzylamine) and H.
Purification by preparative HPLC resulted in isolation of the
trifluoroacetic acid salt after lyophilization of product containing
fractions.
Anal. Calc'd for C27H24N6F~~2.~CF3COOH: C 45.22, H 3.12, N 9.71
Found: C 45.31, H 3.24, N 9.71
MP 68-74°C
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-59-
EXAMPLE 16
2-TRIFLUOROMETHYLBENZYL-f4-j2-PIPERIDIN-4-YL-5-(3
TRIFLUOROMETHYLPHENYL)-3H-LMIDAZOL-4-YLl-PYRIMIDIN
2-YLl AMINE
N
'N H
I ' _H
N ,N
HN
F3C
CF3
The title compound was prepared from 4-[5-(2
methylsulfonylpyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-imidazol
2-ylJ-piperidine-1-carboxylic acid benzyl ester (Example 1 F) as described
in Example 1 steps G (replacing 4-methoxybenzylamine with 2-
trifluoromethylbenzylamine) and H.
Purification by preparative HPLC resulted in isolation of the
trifluoroacetic acid salt after lyophilization of product containing
fractions.
Anal. Calc'd for C2'7H24N6F6~3.OCF3COOH: C 44.15, H 3.14, N 9.36
Found: C 44.16, H 3.15, N 9.21
MP 65-68°C
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-60-
EXAMPLE 17
4-15-(2-(2-INDANYLAMINO)-PYRIMIDIN-4-YL~-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl-PIPERIDINE
CF3
N
~N H
f \ 'H
N ,N
~3HBr
HN
The title compound was prepared from 4-[5-(2-
methylsulfonylpyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-imidazol-
2-yl]-piperidine-1-carboxylic acid benzyl ester (Example 1F) as described
in Example 1 steps G (replacing 4-methoxybenzylamine with 2-
aminoindane) and H.
The product was isolated as the trihydrobromide salt.
Anal. Calc'd for C28H27N(F3~3HBr, 1.5 H20: C 43.41, H 4.29, N
10.85
Found: C 43.78, H 4.59, N 10.49
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-61-
EXAMPLE 18
f 4-f 2-( 1-METHYL-PIPERIDIN-4-YL)-5-!3
TRIFLUOROMETHYLPHENYL)-3H-IMIDAZOL-4-YLl-PYRIMIDIN
2-YLl-4-METHOXYBENZYLAMINE
CF3
i
N
\ -N-CH3
\ 1H
N\/ N
HN
OCH3
To a stirred solution of 4-(5-(2-{4-methoxybenzylamino)-
pyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-imidazol-2-yl]-
piperidine-1-carboxylic acid benzyl ester (Example 1G) (0.227 g,
0.000353 mole) in THF (10 mL) was slowly added 1.OM lithium
aluminum hydride in THF (0.53 mL, 0.0053 mole). The reaction was
warmed to reflux for 30 min., cooled and water (200 mL) added
dropwise. The reaction mixture was diluted with THF (20 mL) and
sodium sulfate (1.0 g) added. The mixture was stirred well for 10
minutes, filtered, concentrated in vacuo, and chromatographed on silica
using methylene chloride/methanol/aqueous ammonium hydroxide
(90:10:2) to give 100 mg of the title compound upon concentration of
product containing fractions. The residue was triturated from 10%
ether/hexane to give 70 mg of the title compound as a white solid.
1 H NMR (CDC13) d 9.85 (brs, 1 H), 8.10 (brd, 1 H), 7.91 (s, 1 H), 7.80 (d,
1H, J = 8.1), 7.5-7.7 (m, 2H), 7.31 (d, 2H, J = 8.1 Hz), 6.90 (d, 2H, J =
8.1 ), 6.59 (brd, 1 H), 5.45 (brs, 1 H), 4.58 (d, 2H, J = 5.6Hz), 3.80 (s,
3H),
2.8-3.0 (m, 3H), 2.32. (s, 3H), 1.8-2.2 (m, 6H).
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-62-
Anal Calc'd for C2$H29N60F3~0.3 H20: C 63.69, H 5.65, N 15.92
Found: C 63.70, H 5.70, N 15.94
EXAMPLE 19
1-f 4-f 5-(2-(4-METHOXYB ENZYLAMINO)-PYRIMIDIN-4-YL)-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl
PIPERIDINE-YLl-ETHANONE
C F.~
N
N ~ N N C Hs
HN / \ OCH3 O
IO
To a stirred solution of 4-methoxybenzyl-[4-[2-piperidin-4-
yl-5-(3-trifluoromethylphenyl)-3H-imidazol-4-yl]-pyrimidin-2-ylJamine
(Example 1H) (0.150 g, 0.295 mmole) in DMF (4 mL) was added acetic
I S acid ( 1$ mL, 0.31 mmole), triethylamine (62 mL, 0.44 mmole), 1-
hydroxybenztriazole hydrate (68 mg, 0.44 mmole) and EDC (85 mg, 0.44
mmole). The mixture was stirred at 20°C for 18 hours then diluted with
10% aqueous sodium bicarbonate (20 mL) and ethyl acetate (30 mL).
The organic extract was removed, washed with water ( 10 mL), dried over
20 anhydrous sodium sulfate, filtered, and column chromatographed on
silica using methylene chloride/methanol/aqueous ammonium hydroxide
(95:5:1 ) to give 146 mg. Crystallization from ether gave 120 mg of the
title compound.
Anal Calc'd for C29H29N602F3: C 63.20, H 5.31, N 15.26
25 Found: C 62.$0, H 5.29, N 15.16
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/U9888
-63-
EXAMPLE 20
1-f 4f 5-(2-(4-METHOXYBENZYLAMINO)PYRIMIDIN-4-YL)-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl
PIPERIDINYLI-2-DIMETHYLAMINOETHANONE
CFs
I
i
N
i ~ H
N~ N N~ CHs
j( ~N~
HN / \ pCH ~ CHs
3
The following compound was prepared from 4-
methoxybenzyl-[4-[2-piperidin-4-yl-5-{3-trifluoromethylphenyl)-3H-
imidazol-4-yl]-pyrimidin-2-yl]amine (Example 1 H) as described in
example 19 using N'-N'-dimethylglycine in place of acetic acid to give
the title compound.
1 H NMR (CDC13) d 9.78 (brs, 1 H), 8.12 (d, 1 H, J = 5.8 Hz), 7.90 (s, 1 H),
7.80 (d, 1 H, J = 8.1 Hz), 7.64 (d, 1 H, J = 8.1 Hz), 7.54 (t, 1 H, J = 8.1
Hz),
7.30 (d, 2H, J = 8.5 Hz), 6.90 (d, 2H, J = 8.5 Hz), 6.58 {d, 1 H, J = 5.8
Hz), 3.8 (s, 3H), 2.3 (s, 6H).
Anal Calc'd for C31 H34N702F3: C 62.72, H 5.77, N 16.52
Found: C 62.57, H 5.95, N 16.76
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-64-
EXAMPLE 21
f 4-f 2-( 1-B ENZYLPIPERIDIN-4-YL)-5-,(3
TRIFLUOROMETHYLPHENYL)-3H-IMIDAZOL-4-YLl-PYRIMIDIN
2-YLl-4-METHOXYBENZYLAMINE
CF3
I w ~N
N
N
H
N~N
HN / \ pCH3
To a well stirred solution of 4-methoxybenzyl-[4-[2-piperidin-4-yl-5-(3-
trifluoromethylphenyl)-3H-imidazol-4-yl]-pyrimidin-2-yl]amine
(Example 1H) (100 mg, 0.197 mmole) and benzaldehyde (22 mg,0.21
mmole) in dichloromethane under argon was added sodium
triacetoxyborohydride (44 mg, 0.21 mmole). The mixture was stirred for
4 hours and then quenched by addition of 1 N HCl (5 mL). Aqueous
sodium bicarbonate (20 mL) and ethyl acetate (20 mL) were added and
the layers mixed well. The ethyl acetate layer was removed, concentrated
in vacuo and residue chromatographed on silica using methylene
chloride/methanol/aqueous ammonium hydroxide (97:3:1 ) to give upon
concentration of product containing fractions 94 mg of an oil.
Crystallization from ether/hexane (4 mL) gave RO mg solid.
1H NMR (CDC13} d 9.75 (brs, 1H), 8.10 (d, 1H, J = 5.5 Hz), 7.90 (s, 1H),
7.80 (d, 1 H, J = 8.7 Hz), 7.62 (d, 1 H, J = 8.7 Hz), 7.54 (t, I H, J = 8.7
Hz),
7.25-7.4 (m, 7H},6.90 (d, 2H, J = 8.7 Hz), 6.58 {d, i H, J = 5.5 Hz), 4.58
(d, 2H, 6.2 Hz), 3.78 (s, 3H), 3.53 (s, 2H), 3.0 (m, 2H), 2.85 (m, 1H), 1.8-
2.2 (m, 6H).
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-65-
Anal Calc'd for C34H33N60F3: C 68.21, H 5.56, N 14.04
Found: C 67.99, H 5.59, N 13.88
EXAMPLE 22
j4-f2-PIPERIDIN-4-YL-5-(3-TRIFLUOROMETHYLPHENYL~-3H-
IMIDAZOL-4-YL1-PYRIMIDIN-2-YLl AMINE
TRII-iYDROCHLOR1DE
CF3
N
NH
~' N
H
NYN .3 HCI
INH2
4-Methoxybenzyl-[4-[2-piperidin-4-yl-5-(3-
trifluoromethylphenyl)-3H-imidazol-4-yl]pyrimidin-2-yl]amine (Example
1H) (0.125 rng, 0.26 mmole) and 3N HCl (3S mL) were heated to 100°C
for 12 hours. The reaction was cooled, washed with diethyl ether ( I O
mL), and concentrated in vacuo to a solid. Trituration from $0%
ether/ethanol (10 mL) gave the title compound, 0.102 g, as a yellow solid.
MP - 225-230°C
1 H NMR {CD30D) d 8.19 (d, 1 H, J = 6.6Hz), R.0 (s, 1 H), 7.96 (d, 1 H, J
= 8.4 Hz), 7.89 (d, 1 H, J = 8.4 Hz), 7.77 (t, 1 H, J = 8.4 Hz), 7.02 (d, 1 H,
J
= 6.6 Hz), 3.2-3.6 (m, SH), 2.1 = 2.4 (m, 4H).
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-66-
EXAMPLE 23
4-f5-(2-AMINOPYRIMIDIN-4-YL)-1-METHYL-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl-
PIPERIDINE-1-CARBOXYLIC ACID BENZYL ESTER
CF3
i
N N O ~ I
N'
N N CHs O
NH2
Step 23A
4-f 5-(2-Methylthiopyrimidin-4-yl)-1-methyl-4-(3-trifluorometh,~nhenyl)-
1H-imidazol-2-vll-piperidine-1-carboxylic acid Benz, 1 ester
To a stirring solution of 4-[5-(2-methylthiopyrimidin-4-yl)-
4-(3-trifluoromethylphenyl)-1 H-imidazol-2-yl]-piperidine-1-carboxylic
acid benzyl ester (Example 1 E) (4.0 g, 7.22 mmole) in toluene ($0 mL)
was added dimethylformamide dimethyl acetal (4.0 mL) and the mixture
heated to reflux for 18 hours. The reaction was cooled, concentrated in
vacuo to a foam and chromatographed on silica using 5%
acetone/methylene chloride to give upon concentration of the product
containing fractions the title compound 2.78 g.
1 H NMR (CDC13) d 8.33 {d, 1 H, J = 6.0 Hz), 7.78 (s, 1 H), 7.5-7.6 (m,
2H), 7.3-7.45 (m, 6H):, 6.76 (d, 1 H, J = 6.0 Hz}, 5. I 3 (s, 2H), 4.35 (brs,
2H), 3.80 (S, 3H), 2.98 (m, 3H), 2.60 (s, 3H), 2.0 (m, 4H).
Additional elution of the above column chromatography
with 5% methanol/methylene chloride gave 4-[4-(2-methylthiopyrimidin-
4-yl)-1-methyl-5-(3-trifluoromethylphenyl)-1 H-imidazol-2-yl]-
piperidine-1-carboxylic acid benzyl ester.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-67-
1 H NMR (CDC13) d 8.39 (d, 1 H, J = 6.0 Hz), 7.5-7.74 (m, SH), 7.3 - 7.44
(m, SH), 5.18 (S, 2H), 4.35 (brs, 2H), 3.38 (.s, 3H), 2.7-3.1 (m, 3H), 1.9-
2.1 (m, 4H), 1.7 (S, 3H).
Step 23B
4-IS-!2-Methylsulfonylpyrimidin-4-yl)-1-meth,~(3-
trifluorometh~phenyl)-1 H-imidazol-2-~piperidine-1-carboxylic acid
benzyl ester
The title compound was prepared by oxidation of 4-[5-(2-
methylthiopyrimidin-4-yl)-1-methyl-4-(3-trifluoromethylphenyl)-1 H-
imidazol-2-yl]-piperidine-1-carboxylic acid benzyl ester as described in
Example 1 step F.
1 H NMR (CDC13) d 8.58 (d, 1 H, J = 5.7 Hz), 7.76 (S, 1 H), 7.60 (m, 2H, J
= 10.0 Hz), 7.48 (t, 1H, J = 10.0 Hz), 7.37 (m, SH), 7.24 (d, 1H, J = 5.7
Hz), 5.14 (S, 2H), 4.34 (brs, 2H), 3.98 (S, 3H), 3.40 (S, 3H), 3.01 (m,
3H), 1.90 - 2.0 (m, 4H).
Step 23 C
4-f 5-(2-Aminopyrimidin-4-Yl)-1-methyl-4-(3-trifluoromethyl~henyl)
-1 H-imidazol-2-yll-piperidine-1-carboxylic acid benzyl ester
The title compound was prepared from 4-[5-(2-
Methylsulfonylpyrimidin-4-yl)-1-methyl-4-(3-trifluoromethylphenyl)-
1H-imidazol-2-yl]-piperidine-1-carboxylic acid benzyl ester as described
in Example 12.
MP - 80-83°C
Mass Spectral Analysis - M+1 = 537
1 H NMR (CDC13) d 8.18 (d, 1 H, J = 5.9 Hz), 7.82 (S, 1 H), 7.61 {d, 1 H, J
= 8.6 Hz), 7.49 (d, 1 H, J = 8.6 Hz), 7.3-7.4 (m, 6H), 6.48 (d, 1 H, J = 5.9
Hz), 5.1-5.3 (m, 4H), 4.35 (brs, 2H), 3.74 (S, 3H), 2.95 (m, 3H), 1.8-2.1
(m, 4H).
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-68-
EXAMPLE 24
4-[5-(2-AMINOPYRIMIDIN-4-YL)-1-METHYL-4 ~3
TRIFLUOROMETHYLPHENYL)
-1 H-IMIDAZOL-2-YLl-PIPERIDINE
CFA
N _
NH
N
N . N CH3
~3 CF3COOH
NH2
The title compound was prepared from 4-[4-(2-
Aminopyrimidin-4-yl)-1-methyl-4-(3-trifluoromethylphenyl)-1 H-
imidazol-2-yl]-piperidine-1-carboxylic acid benzyl ester as described in
Example 1 step H, and isolated as the trifluoroacetic acid salt after
purification by preparative HPLC and lyophilization of the product
containing fractions.
MP = 57-60°C
I H NMR (CD30D) 8.06 (d, l H, J=6.5 Hz), 7.84 (s, 1 H), 7.72 (m, 2H),
7.62 (t, l H, J=8.1 Hz), 6.53 (d, 1 H, J=6.5Hz), 4.00 (s, 3H), 3.2-3.4 (m,
SH), 2.0-2.2 (m, 4H)
Mass Spectral Analysis - M+1 = 403
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-69-
EXAMPLE 25
4-f4-t2-AMINOPYRIMIDIN-4-YL)-1-METHYL-5-t3
TRIFLUOROMETHYLPHENYL)
-3H-IMIDAZOL-2-YL1PIPERIDINE-I-CARBOXYLIC ACID BENZYL
ESTER
CF3
CH3 /
N
I ~ ~N O
~N
N ,N O
NH2
The title compound was prepared from 4-[4-(2-
methylthiopyrimidin-4-yl)-I-methyl-5-(3-trifluoromethylphenyl)-3H-
imidazol-2-yl]piperidine-I-carboxylic acid benzyl ester as described in
Example 12.
MP 215-217°C
Anal Calc'd for C2gH27N602F3: C 62.68, H 5.07, N 15.66
Found: C 62.34, H 4.77, N 15.50
Mass Spectral Analysis - M+I = 537
CA 02257200 1998-12-02
WO 97/47618 PCT/LTS97/09888
-70-
EXAMPLE 26
4-f 4-(2-AMINOPYRIMIDIN-4-YL)-1-METHYL-5-(3
TRIFLUOROMETHYLPHENYL)
-3H-IMIDAZOL-2-YLl-PIPERIDINE
C F3
~ Hs
N
NH
'N
I \
N~ N 3.0 CF3COOH
NH2
The title compound was prepared from 4-[4-(2-
aminopyrimidin-4-yl)-1-methyl-5-(3-trifluoromethylphenyl)-3H-
imidazol-2-yl]-piperidine-1-carboxylic acid benzyl ester as described in
Example I step H.
Mass Spectral Analysis - M+1 = 403.
__~._ ..._._..u __.
CA 02257200 1998-12-02
WO 97!47618 PCTIUS97/09888
-71-
EXAMPLE 27
4-( f2-PIPERIDIN-4-YL]-5-(3-TRIFLUOROMETHYLPHENYL)-3H
IMIDAZOL-4-YL 1-PYRIMIDiNE-2-CARBOXAMIDE
TRIHYDROBROMIDE
CF3
N _
NH
~N
i ~ H
N .N
~3 HBr
O NH2
Step 27A
4-(5-f2-C.yano~yrimidin-4-yll-4-(3-trifluoromethvl)-1 H-imidazol-2-yll-
piperidine-1-carboxylic acid benzyl ester.
To a stirred solution of 4-[5-(2-methylsulfonylpyrimidin-4-
yl)-4-(3-trifluoromethylphenyl)-1 H-imidazo 1-2-yl)-piperidine-1-
carboxylic acid benzyl ester {Example 1 F) (0.50 g, 0.85 mmole) in
dimethyl sulfoxide (3 mL) was added sodium cyanide (0.087 g, 1.70
mmole) and the resulting mixture warmed to GO°C for 2 hours. The
reaction was cooled, diluted with water (30 mL) and extracted with ethyl
acetate (2x30mL). The ethyl acetate extracts were dried over anhydrous
sodium sulfate, filtered and concentrated to an oil. The residue was
chromatographed on silica using ethyl acetate/hexane to give upon
concentration of the product containing fractions a foam, (0.455 g).
Mass Spectral Analysis - M+1 = 533.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-72-
Sten 27B
4-(2-Piperidin-4-yl]-5-(3-trifluorometh~r~henyl)-3H-imidazol-4-yl)-
pyrimidine-2-carboxamide trihydrobromide
The title compound was prepared from 4-[5-[2-
cyanopyrimidin-4-yl]-4-(3-trifluoromethyl)-1 H-imidazol-2-yl]-
piperidine-1-carboxylic acid benzyl ester as described in Example 1 step
H.
Mass Spectral Analysis - M+1 = 417
MP - 235-240°C
EXAMPLE 28
4-((2-PIPERIDIN-4-YL)-S-(3-TRIFLUOROMETHYLPHENYL)-3H
IMIDAZOL-4-YL)1-PYRIMIDINE
C F3
N
'N H
'N
H
N ~ N ~3 CF3COOH
4-[5-[2-Cyanopyrimidin-4-yl]-4-{3-trifluoromethyl)-1 H-
imidazol-2-yl]-piperidine-1-carboxylic acid benzyl ester (0.05 g, 0.09
mmole) and 3N HCl {3 mL) were heated to 100°C for 72 hours. The
reaction was cooled and concentrated to a solid. The solid was purified
by preparative HPLC to give the title compound (40 mg) upon
lyophilization of the product containing fractions.
Mass Spectral Analysis - M+1 = 374.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-'
EXAMPLE 29
4-( f2-(PIPERIDIN-4-YL)-5-(3-TRIFLUOROMETHYLPHENYL)-3H
IMIDAZOL-4-YLl l-PYRIMIDIN-2-CARBOXYLIC ACID
CF3
N
NH
~N
i ~ H
N . N ~2 CF3COOH
O OH
To a stirred solution of 4- { [2-piperidin-4-yl]-5-(3-
trifluoromethylphenyl)-3H-imidazol-4-yl) } pyrimidine-2-carboxamide
trihydrobromide (0.120 g, 0.18 mmole) in methanol ( 1 mL) was added
SN sodium hydroxide ( 1 mL) and the reaction stirred for 18 hours. The
reaction was diluted with SN HCI ( 1 mL) and purified by preparative
HPLC to give the title compound ( 100 mg) upon lyophilization of the
product containing fractions.
Mass Spectral Analysis - M+1 = 418
MP 60-67°C
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-74-
EXAMPLE 30
1-f4-f 5-(2-AMINOPYRIMIDIN-4-YL)-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YLl-PIPERIDIN
4-YLl ETH ANONE
CF3
O
i ~
/ 'CH
N
~N
H
N~N
NH2
S~ 30A
4-f 5-(2-Methylsulfonylpyrimidin-4-yl )-4-(3-trifluoromethylphenyl)- I H-
imidazol-2-yllpiperidine
To a stirred solution of 4-[5-[2-methylsulfonylpyrimidin-4-
yl]-4-(3-trifluoromethylphenyl)-1 H-imidazol-2-yl]piperidine-1-
carboxylic acid benzyl ester (Example 1 F) (400 mg, 0.683 mmole) in
methylene chloride (6 mL), under argon was added dropwise 30%
hydrogen bromide /acetic acid {6.0 mL). The reaction was allowed to stir
for 1 hour, diluted with diethyl ether (60 mL) and the resulting solid
filtered to give 0.474 g of the dihydrobromide salt.
MP 125-I30°C
Mass Spectral Analysis - M+1 = 4.52.
Step 30B
1-[4-f 5-(2-Methylsulfonvlpyrimidin-4-yl)-4-(3-trifluorometh~l~henyl)-
1 H-imidazol-2-yll-piperidin-4-yllethanone
The title compound was prepared from 4-[5-(2-
methylsulfonylpyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-imidazol-
2-yl]piperidine as described in Example 19.
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
_75_
1H NMR (CD30D) Rotamer d 8.6-8.84 (brm, 1H), 7.6-8.2 (m, 5H), 4.60
(brd, 1 H, J = 18 Hz), 4.10 (d, 1 H, J = I 8 Hz), 2.7-3.4 (m, 6H), 2.26 (s,
3H), 1.8-2.2 (m, 4H).
Step 30C
1-f4-f5-(2-Aminopvrimidin-4- ly )-4-(3-trifluoromethvlphen 1~)-1H-
imidazol-2-~piperidin-4-yllethanone
The title compound was prepared from 1-[4-[5-(2-
methylsulfonylpyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-imidazol-
2-yl]piperidin-4-yl]ethanone described in Example 12.
1H NMR (CD30D) d 8.14 (d, 1H, J = 6.6 Hz), 7.94 (s, 1H), 7.91 (d, 1H,
J = 8.5 Hz}, 7.83 (d, 1 H, J = 8.5 Hz}, 7.73 (t, 1 H, J = 8.5 Hz), 6.94 (d, 1
H,
J = 6.6 Hz), 4.65 (d, 1 H, J = 18 Hz), 4.10 (d, 1 H, J = 18 Hz), 2.80 (m,
1H), 1.8-2.2 (m, 7H).
Anal Calc'd for C21 H21 N60F3~2.0 CF3COOH~ 1.0 H20: C 44.37, H
3.72, N 12.42
Found: C 44.16, H 3.51, N I 2.53
EXAMPLE 31
j4~_5-(2-AMINOPYRIMIDIN-4-YL)-4-(3
TRIFLUOROMETHYLPHENYL)-1 H-IMIDAZOL-2-YL1-PIPERIDIN-
I -YL1PHENYLMETHANONE
O
CF3 N
y w
N
N
H
N~ N
NH2
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-76-
The title compound was prepared from 4-[5-(2-
methylsulfonylpyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-imidazol-
2-yl]piperidine (Example 30A) as described in Examples 19 (replacing
acetic acid with benzoic acid) and 12
I H NMR (CD30D) d 8.14 (d, 1 H, J = 6.5 Hz), 7.95 (s, 1 H), 7.92 (d, 1 H,
J = 8.7 Hz), 7.84 (d, 1 H, J = R.7 Hz), 7.73 (t, 1 H, J = 8.7 Hz), 7.4-7.6 (m,
SH), 6.96 (d, 1 H, J = 6.5 Hz), 3.R5 (brm, 1 H), 3.05 (m, 1 H), 1.8-2.2 (rn,
6H).
Anal Calc'd for C26H23N64F3~2CF3COOH, O.SH20: C 49.2$, H 3.54,
N i 1.84
Found: C 49.38, H 3.59, N 11.52
EXAMPLE 32
N-( f5-(2-PIPERIDIN-4-YL)-4-(3-TRIFLUOROMETHYLPHENYL~
I H-IMIDAZOL-4-YL1PYRIMIDIN-2-YLl l ACETAMIDE
CF3
N _
NH
~N
i ~ H
N .N
~3 HBr
HN
,~T- CHs
O
Step 32A
4-LS-(2-Acetamidopyrimidin-4-yl)-4-(3-trifluoromethyphen, l
imidazol-2-,~[piperidine-1-carboxylic acid benz. Iy ester
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
To a stirred solution of 4-[5-(2-aminopyrimidin-4-yl)-4-(3-
trifluoromethylphenyl)-1H-imidazol-2-yl]-piperidine-1-carboxylic acid
benzyl ester (Example 12) (0.220 g, 0.421 mmole) in THF (4 mL) under
argon at 0°C was added diisopropylethylamine (0.22 mL, 1.26 mmole)
followed by acetyl chloride (0.036 mL, O.SO mmole). The reaction was
stirred at 20°C for 1 hour, diluted with water ( 10 mL) and product
extracted with ethyl acetate (2xS0 mL). The ethyl acetate extracts were
dried over anhydrous sodium sulfate, filtered, concentrated and
chromatographed on silica using 5% methanol/methylene chloride to give
the product (0.170 g) as an oil.
1 H NMR (CDC13) Rotamers d 10.2 (brs, 1 H), 8.56 (d, 1 H, J = 6.6 Hz),
7.2-8.0 (m, lOH), 5.12 (brs, 2H), 4.30 (brs, 2H), 2.9-3.1 (m, 3H), 2.32 (s,
3H), 1.8-2.2 (m, SH).
Example 32B
N-j5-(2-Piperidin-4-yl)-4-(3-trifluoromethylphen~l)-1 H-imidazol-4-
~lp~rimidin-2-yllacetamide
The title compound was prepared from 4-[5-(2-
acetamidopyrimidin-4-yl)-4-(3-trifluoromethyphenyl)-1 H-imidazol-2-
yl]piperidine-1-carboxylic acid benzyl ester as described in Example 30,
Step A.
Mass Spectral Analysis M+1 = 43 t
MP >200°C (Dec)
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
_'
EXAMPLE 33
N- ( 4-(2-PIPERIDIN-4-YL)-5-(3-TRIFLUOROMETHYLPHENYL)-1 H
IMIDAZOL-4-YLl-PYRIMIDIN-2-YL 1 BENZAM117E
CF3
N _
NH
'N
H
N .N
~3 HBr
HN
O '_'
The title compound was prepared from 4-[5-(2-
aminopyrimidin-4-yl)-4-(3-trifluoromethylphenyl)-1 H-imidazol-2-yl]-
piperidine-1-carboxylic acid benzyl ester as described in Example 32
(replacing acetyl chloride with) benzoyl chloride.
MP >260°C (Dec)
Mass Spectral Analysis - M+1 = 493
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-79-
EXAMPLE 34
(S)- ( 4-f 3-METHYL-2-PIPERIDIN-4-YL-5-(3
TRIFLUOROMETHYLPHENYL)-3H-IMIDAZOL-4-YLl-PYRIMIDIN
2-YL 1-( 1-PHENYLETHYL~AMINE
CF3
Iw
N
NH
~N
i
N . N CH3
~3.0 HCI
HN
/ \
H
CH3
STEP 34A
4-f 5-(2-MethYlsulfonylpyrimidin-4-vl)-1-methyl-4-(3-
trifluoromethvhhenvl)-1H-imidazol-2w11-piperidine-1-carboxylic acid
benz, l
The title compound was prepared using the procedure set
forth in Example 1, Steps 1 A to I E, and Example 23, Steps 23A and
23B.
STEP 34B
SS)-4-(5-(2-( 1-Phen, l~vlamino)pyrimidin-4-vl)-1-meth, 1-~, 4-(3=
trifluorometh~~phenyl)-1H-imidazol-2-vll-piperidine-1-carbox, ly 1C acid
benz, 1 ester
A mixture of 4-[5-(2-methylsulfonylpyrimidin-4-yl)-1-
methyl-4-(3-trifluoromethylphenyl)-1 H-imidazol-2-yl] -piperidine-1-
carboxylic acid benzyl ester ( 12.6 gm, 0.021 mole) and S(-)-(a)-
methylbenzylamine (25.0 gm, 0.21 mole) was heated to 100°C for 1 hour,
under argon. The mixture was cooled and chromatographed on silica {1
kg) using 40% ethyl acetate/ hexane to give (S)-4-[5-(2-( 1-
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-80-
phenylethylamino)-pyrimidin-4-yl]-1-methyl-4-(3-
trifluoromethylphenyl)-1H-imidazol-2-yl]-piperidine-1-carboxylic acid
benzyl ester {13.4 gms.)
STEP 34C
fS)-4-f 5-(2-( 1-Phenylethylamino)-pyrimidin-4-yl)-1-methyl-4-(~-
trifluoromethylphenvl)-1 H-imidazol-2-vllpioeridine
To a solution of (S)-4-[5-[2-(1-phenylethylamino)-
pyrimidin-4-yl)-1-methyl-4-(3-trifluoromethylphenyl)-1H-imidazol-2-ylJ-
piperidine-1-carboxylic acid benzyl ester(12.5 gm, 0.0195 mole) in
dichloromethane ( 170mL) at 0 °C, under argon, was slowly added 30 %
hydrogen bromide in acetic acid ( 170 mL). The solution was allowed to
stir for 1.5 h at OoC and then diluted with diethyl ether (2.0 L). The
resulting solid was filtered under argon, washed with diethyl ether (500
ml) and sucked dry under argon. The solid was then dissolved in
dichloromethane (500 mL) and 10 % aqueous sodium bicarbonate (500
mL) and mixed well. The dicholormethane solution was removed and the
aqueous layer washed with dichloromethane (200 mL). The
dichloromethane extracts were dried over anhydrous sodium sulfate,
concentrated and the resulting foam (9.6 gm) chromatographed on silica
using (dichloromethane/methanol/acetic acid/water- 90/10/1/1). The
resulting product containing fractions were washed with 10 % aqueous
sodium bicarbonate, dried over anhydrous sodium sulfate and
concentrated to give (S)-4-[5-[2-(1-phenylethylamino)-pyrimidin-4-yl]-1-
methyl-4-(3-trifluoromethylphenyl)-1H-imidazol-2-yl]-piperidine (9.4
gm). The solid was dissolved in ethyl acetate (400 ml) filtered and then
treated with a solution of hydrochloric acid/ethyl acetate ( 84 mL, 0.033
gm HCl/mL, 4 eq.). The resulting solid was filtered, dissolved in water
(100 ml) and lyophilized to give (S)-4-[5-[2-(1-phenylethylamino)-
pyrimidin-4-yl]-1-methyl-4-(3-trifluoromethylphenyl)-1H-imidazol-2-yl]-
piperidine trihydrochloride ( 12.0 gm).
Anal. Calc. for C2gH29N6F3 .3.0 HCI 2.7 H20. C, 50.57 ; H, 5.60 ; N,
12.57
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-81-
Found C, 50.39; H, 5.47; N, 12.33
1 H NMR (CD30D) d 8.27 (brs, I H,), 8.0 (s, 1 H), 7.91 (d, 1 H, J = 8.7
Hz), 7.82 (d, IH, J = 8.7 Hz), 7.73 (t, 1H, 3 = 8.7 Hz), 7.2-7.6 (m, 5H),
6.64 (d, 1H, J = 6.5 Hz), 5.2 (brs, 1 H), 3.2-4.0 (m, 8H), 2.2-2.4 (m, 4H),
1.65 (d, 3H, J = 7.2 Hz).
EXAMPLE 35
(S)-( 4-f 3-METHYL-2-( I -METHYLPIPERIDIN-4-YL)-5-(3
TRIFLUOROMETHYLPHENYL)-3H-IMIDAZOL-4-YLl
PYRIMIDINE-2-YL ~ -( 1-PHENYLETHYL)AMINE
CF3
3.0 CF3COOH
N
N
N~N CH3 NCH
3
HN
H CH ~
3
The title compound was prepared from (S)-4-[5-(2-(1-
phenylethylamino)pyrimidin-4-yl)-1-methyl-4-(3-trifluoromethylphenyl)-
IH-imidazol-2-yl]-piperidine-1-carboxylic acid benzyl ester as described
in example 18.
Anal. Calc'd for C29H31N(F3 3CF3COOH: C 48.73, H 3.97, N 9.74
Found: C 48.92, H 4.20, N 9.85
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-82-
EXAMPLE 36
CYCLOPROPYL-(4-f 3-METHYL-2-(PIPERIDIN-4-YL)-5-(3
TRIFLUOROMETHYLPHENYL -L3H-IMIDAZOL-4-YLl-PYRIMII7IN
2-YL ) AMINE
C F3
3 CF3COOH
N
N
N~N CH3 NH
HN\
The title compound was prepared from 4-[5-(2-
methylsulfonylpyrimidin-4-yl)-1-methyl-4-(3-trifluoromethylphenyl)-1 H-
imidazol-2-yl]-piperidine-1-carboxylic acid benzyl ester (example 23,
step B) as described in example 34, steps B (replacing S(-)-(a)-
methlybenzylamine with cyclopropylamine) and C.
Anal Calc'd C23H25N6F3 3.0 CF3COOH 1.0 H20: C 43.40, H 3.77, N
10.47
Found: C 43.41, H 3.74, N 10.53
__ __ _ _. ____._ _._ __ , _
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
_g3_
EXAMPLE 37
(S)- ( 4-f 3-METHYL-2-( 1-CYCLOPROPYLPIPERIDIN-4-YL)-5-(3
TRIFLUOROMETHYL
PHENYL)-3H-IMIDAZOL-4-YLl-PYRIMIDINE-2-YL 1-yl
PHENYLETHYL)AMINE
C F3
/ 3.0 CF3COOH
N
N~N CH3 N
HN
H ~ /
C H3
To a solution of (S)-{4-[3-methyl-2-piperidin-4-yl-5-(3-
trifluoromethylphenyl)-3H-imidazol-4-yl]-pyrimidin-2-yl }-( 1-
phenylethyl)amine (300 mg, O.S92 mmole) in methanol (5 ml) under
argon was added acetic acid (0.34 ml, 6.0 mmole) and [(1-
ethoxycyclopropyl)oxy]-trimethylsilane (0.6 ml, 3.0 mmole). After
stirring for 5 minutes sodium cyanoborohydride (150 mg, 2.30 mmole)
was added. The mixture was warmed to reflux for 4 hours cooled and
concentrated to an oil. The oil was dissolved in methylene chloride (30
ml) and water (20 ml) added. The pH was adjusted to 10.0 with 1 N
sodium hydroxide and the methylene chloride layer removed, dried over
sodium sulfate and concentrated to a foam. The foam was purified by
reverse phase preparative HPLC chromatography using 0.1 %
trifluoroacetic acid/water and acetonitrile to give upon lyophilization of
the product containing fractions 4R0 mg of the title compound.
Anal Calc'd for C31 H33N6F3 3.0 CF3COOH: C 50.00, H 4.08, N 9.46
Found: C 50.39, H 4.24, N 9.76
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-84-
EXAMPLE 38
(S)- ( 4-f2-( 1-CYCLOPROPYLMETHYL-PIPERIDIN-4-YL)-3
METHYL-5-(3-TRIFLUOROMETHYLPHENYL)-3H-IMIDAZOL-4
YLl-PYRIMIDIN-2-YL I -( 1-PHENYLETHYL)-AMINE
TRIHYDROCHLORIDE
C F3
3.0 HCI
1
I N
N ~ N CH
3
HN
H IC H \
3
The title compound was prepared from (S)-{4-[3-methyl-2-
piperidin-4-yl-5-(3-trifluoromethylphenyl)-3H-imidazol-4-yl]-pyrimidin-
2-yl)-(1-phenylethyl)amine (example 34) as described in example 21
(replacing benzaldehyde with cyclopropanecarboxaldehyde).
Anal Calc'd for C32H35N6F3 3.OHC1 2.55 H20: C 53.68, H 6.07, N
11.74
Found: C 53.79, H 6.24, N 11.34
M.P. 145-155°C
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-85-
EXAMPLE 39
~S~( 1-PHENYLETHYL)-( 4-f 2-PIPERIDIN-4-YL-3-PROPYL-5-(3
TRIFLUOROMETHYLPHENYL)-3H-IMIDAZOL-4-YLl-PYRIMIDIN
2-YL 1 AMINE TRIHYDROCHLORI17E
C F3
3.0 HCI
N
N
N ~ N NH
HN CH$
H
CH3
The title compound was prepared using the procedures set
forth in Example 1, Steps 1 A to 1 E, Example 23, Steps 23A (replacing
dimethylformamide dimethyl acetal with dimethylformamide dipropyl
acetal) and 23B and Example 34, Steps 34B and 34C.
Anal Calc'd for C3pH33N6F3 .3.OHC1 0.7 H20: C 54.87, H 5.74, N
12.80
Found: C 54.92, H 5.98, N 12.55
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-86-
EXAMPLE 40
f 4-f5-(3.4-DICHLOROPHENYL)-3-METHYL-2-PIPERIDIN-4-YL-3H-
IMIDAZOL-4-YLl-PYRIMIDIN-2-YL l -( 1-PHENYLETHYL)-AMINE
CI
CI
N
~N H
~N
N C H3
N
H3C~~~ 1NH
H
The title compound was prepared using the procedures set
forth in Example 1, Steps 1 A to 1 E (replacing N-methoxy-N-methyl-3-
trifluoromethylbenzamide with N-methoxy-N-methyl-3,4-
dichlorobenzamide), Example 23, Steps 23A and 23B and Example 34,
Steps 34B and 34C.
Anal Calc'd for C27H2gN~C12 0.6 CH3C12: C 59.36, H 5.27, N 15.05
Found: C 15.52, H 5.07, N 15.03
I5
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
_g7_
EXAMPLE 41
( 4-15-~3,4-DICHLOROPHENYL)-3-METHYL-2-PIPERIDIN-4-YL-3H
IMIDAZOL-4-YL,]-PYRIMIDIN-2-YL 1-(4-METHOXYBENZYL)
AMINE
c
NH
/ '~'(
vl N C H3
N
NH
OCH3
The title compound was prepared using the procedures set
forth in Example l, Steps 1 A to 1 E (replacing N-methoxy-N-methyl-3-
trifluoromethylbenzamide with N-methoxy-N-methyl-3,4-
dichlorobenzamide), Example 23, Steps 23A and 23B and Example 34,
Steps 34B (replacing S-(a)-methylbenzylamine with 4-
methoxybenzylamine) and 34C.
Anal Calc'd for C27H2gN6C120 ~ 0.60 CH2Cl2: C 57.71, H 5.12, N
14.63
Found: C 57.90, H 5.24, N 14.36
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
_gg_
EXAMPLE 42
~ 4-f 5-13.4-DICHLOROPHENYL)-3-PROPYL-2-PIPERIDIN-4-YL-3H-
IMIDAZOLE-4-YLl PYRIMIDIN-2-YL l -( I -PHENYLETHYL)-AMINE
CI
CI
N
'N H
~N
/N
NH
,,
The title compound was prepared using the procedures set
forth in Example 1, Steps 1 A to 1 E (replacing N-methoxy-N-methyl-3-
trifluoromethylbenzamide with N-methoxy-N-methyl-3,4-
dichlorobenzamide), Example 23, Steps 23A (replacing
dimethylformamide dimethyl acetal with dimethylformamide dipropyl
acetal) and 23B and Example 34, Steps 34B and 34C.
1H NMR (CDC13): d (8.14, J=5.13Hz, d, 1H); (7.64, s, 1H); (7.19 - 7.40,
m, 8H); (6.37, J=4.88Hz, d, 1 H); (5.78, bs, 1 H); (5.2I , J=6.84Hz, 13.92,
q, 1H); 3.91 - 4.OR, bm, 1H); (3.60 - 3.78, m, 3H); (3.47, J=6.6Hz, q, 1H);
(3.30 - 3.36, m, 2H); (2.78 - 2.86, m, 3H); (1.86 - 1.98, m, 2H); (1.58,
J=6.84,Hz d, 3H); ( 1.00 - 1.53, m, 3H); (0.60, bs,2H).
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-89-
EXAMPLE 43
CYCLOPROPYL- ( 4-f 5-(3.4-DICHLOROPHENYL)-2-PIPERI17IN-4-
YL-3-PROPYL-3H-IMIDAZOL-4-YLl-PYRIMIDIN-2-YL 1-AMINE
CI
CI
' NH
N~"
1N H
d
The title compound was prepared using the procedures set
forth in Example 1, Steps 1 A to 1 E (replacing N-methoxy-N-methyl-3-
trifluoromethylbenzamide with N-methoxy-N-methyl-3,4-
dichlorobenzamide), Example 23, Steps 23A (replacing
dimethylformamide dimethyl acetal with dimethylformamide dipropyl
acetal) and 23B and Example 34, Steps 34B (replacing S-(a)-
methylbenzylamine with cyclopropylamine) and 34C.
1H NMR (CDC13): d (8.19, J=4.64Hz, bd, 1 H); (7.68, s, 1 H}; (7.24 -
7.33, b, 2H); (6.44, J=5.12Hz, bd, 1 H); (5.47, s, 1 H); (4.26, bs, 2H);
(3.30, J=12.69Hz, bd, 2H); (2.76 - 2.Rg, m, 4H); (2.56, bs, 2H); ( 1.$8 -
2.17, m, 4H); ( 1.57 - 1.64, m, 2H); ( 1.25, s, 1 H); (0.83 - 0.98, m, 2H);
(0.60 - 0.66, bs, 2H).
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-90-
EXAMPLE 44
CYCLOHEXYL-14-f 5-~3 4-DICHLOROPHENYL)-2-PIPERIDIN-4-
YL-3-PROPYL-3H-IMIDAZOL-4-YLl-PYRIMIDIN-2-YL 1-AMINE
CI
CI
v N
~N H
~N
w
1
N~N
-=~N H
The title compound was prepared using the procedures set
forth in Example 1, Steps 1 A to 1 E (replacing N-methoxy-N-methyl-3-
trifluoromethylbenzamide with N-methoxy-N-methyl-3,4-
dichlorobenzamide), Example 23, Steps 23A (replacing
dimethylformamide dimethyl acetal with dimethylformamide dipropyl
acetal) and 23B and Example 34, Steps 34B (replacing S-(a)-
methylbenzylamine with cyclohexylamine) and 34C.
1H NMR (CDC13): d (8.14, J=4.RRHz, bd, 1H); (7.63, s, 1H); (7.23 -
7.32, m, 2H); (6.35, J=6. I Hz, d, 1 H); (5.13, J=7.81 Hz, bd, 1 H); (4.14 -
4.19, m, 2H); (3.$1 - 3.91, m, 1 H); (3.32, J=12.7Hz, bd, 2H); (2.71 - 2.97,
m, 4H); (2.17, s, 2H); ( I .01 - 2.17, m, 15H); (0.90 - 0.99, m, 3H).
The ability of compounds of the present invention to inhibit
the synthesis or the activity of cytokines can be demonstrated using the
following in vitro assays.
Lipopolysaccharide mediated production of cytokines
Human peripheral blood mononuclear cells (PBMC) are
isolated from fresh human blood according to the procedure of Chin and
___~_. _ .._ _T
CA 02257200 2005-10-27
-9 I -
Kostura, J. Immunol. 151, 5574-5585 (1993). Whole blood is collected
by sterile venipuncture into 60 mL syringes coated with 1.0 mL of
sodium- heparin (Upjohn, 1000 U/mL) and diluted 1:1 in Hanks Balanced
Salt Solution (Gibco). The erythrocytes are separated from the PBMC's
by centrifugation on a Ficoll-Hypaque lymphocyte separation media. The
PBMC's are washed three times in Hanks Balanced Salt Solution and
then resuspended to a final concentration of 2 x 106 cell/mL in RPMI
containing 10% fresh autologous human serum, penicillin streptomycin
(10 U/mL) and 0.05% DMSO. Lipopolysaccharide (Salmonella type
IO Re545; Sigma Chemicals) is added to the cells to a final concentration of
100 ng/mL. An aliquot (0.1 mL) of the cells is quickly dispensed into
each well of a 96 well plate containing O.I mL of the test compound, at
the appropriate dilution, and are incubated for 24 hours. at 37°C in 5%
C02 . At the end of the culture period, cell culture supernatants are
assayed for IL-lb , TNF-a, IL-6 and PGE2 production using specific
ELISA.
IL-1 mediated cytokine production
Human peripheral blood mononuclear cells are isolated from
fresh human blood according to the procedure of Chin and Kostura, J.
Immunol. 151, 5574-5585 ( 1993). Whole blood is collected by sterile
venipuncture into 60 mL syringes coated with 1.0 mL of sodium- heparin
(Upjohn, 1000 U/mL) and diluted 1:1 in Hanks Balanced Salt Solution
(Gibco). The erythrocytes are separated from the PBMC's by
centrifugation on a Ficoll-Hypaque lymphocyte separation media. The
PBMC's are washed three times in Hanks Balanced Salt Solution and
then resuspended to a final concentration of 2 x 106 cell/mL in RPMI
containing 10% fresh autologous human serum, penicillin streptomycin
(10 U/mL) and 0.05% DMSO. Endotoxin free recombinant human TL-lb
is then added to a final concentration of 50 pMolar. An aliquot (0.1 mL)
of the cells is quickly dispensed into each well of a 96 well plate
containing 0.1 mL of the compound at the appropriate dilution. and are
incubated for 24 hours. at 37°C in 5% C02 . At the end of the culture
Trademark*
CA 02257200 2005-10-27
-92-
period, cell culture supernatants are assayed for TNF-a, IL-6 and PGE2
synthesis using specific ELISA..
Determination of IL-1 b . TNF-a, IL-6 and prostanoid production from
LPS or IL-I stimulated PBMC's
IL-1 b ELISA
Human IL-lb can be detected in cell-culture supernatants or
whole blood with the following specific trapping ELISA. Ninety-six well
plastic plates (Immulori 4; Dynatech) are coated for 12 hours at 4°C
with
1 mg/mL protein-A affinity chromatography purified mouse anti-human
IL-Ib monoclonal antibody (purchased as an ascites preparation from
LAO Enterprise, Gaithersburg Maryland.) diluted in Dulbecco's~'
phosphate-buffered saline (-MgCl2, -CaCl2). The plates are washed with
PBS-Tweeri (Kirkegaard and Perry) then blocked with 1 % BSA diluent
and blocking solution (Kirkegaard and Perry) for 60 minutes at room
temperature followed by washing with PBS Tween. IL-lb standards are
prepared from purified recombinant IL-1 b produced from E. coli.. The
highest concentration begins at 10 ng/mL followed by 11 two-fold serial
dilutions. For detection of IL-1 b from cell culture supernatants or blood
plasma, 10 - 25 mL of supernatant is added to each test well with 75 - 90
mL of PBS Tween. Samples are incubated at room temperature for 2
hours then washed 6 times with PBS Tween on an automated plate
washer (Dennly). Rabbit anti-human IL-Ib polyclonal antisera diluted
1:500 in PBS-Tween is added to the plate and incubated for 1 hour at
room temperature followed by six washes with PBS-Tween. Detection
of bound rabbit anti-IL-1 b IgG is accomplished with Fab' fragments of
Goat anti-rabbit IgG-horseradish peroxidase conjugate (Accurate
Scientific) diluted 1:10,000 in PBS-Tween. Peroxidase activity was
determined using TMB peroxidase substrate kit (Kirkegaard and Perry)
with quantitation of color intensity on a 96-well plate Molecular Devices
spectrophotometer set to determine absorbance at 450 nM. Samples are
evaluated using a standard curve of absorbance versus concentration.
Trademark*
CA 02257200 1998-12-02
WO 97/47618 PCT1US97/09888
-93-
Four-parameter logistics analysis generally is used to fit data and obtain
concentrations of unknown compounds.
TNF-a ELISA
Immulon 4 (Dynatech) 96-well plastic plates are coated with
a 0.5 mg/mL solution of mouse anti-human TNF-a monoclonal antibody.
The secondary antibody is a 1:2500 dilution of a rabbit anti-human TNF-
a polyclonal serum purchased from Genzyme. All other operations are
identical to those described above for IL-lb. The standards are prepared
in PBS-Tween + 10% FBS or HS. Eleven 2 fold dilutions are made
beginning at 20 ng/mL TNF-a.
IL-6 ELISA
Levels of secreted human IL-6 are also determined by
specific trapping ELISA as described previously in Chin and Kostura, J.
Immunol. 151, 5574-5585 ( 1993). (Dynatech) ELISA plates are coated
with mouse anti-human IL-6 monoclonal antibody diluted to 0.5 mg/mL
in PBS. The secondary antibody, a rabbit anti-human IL-6 polyclonal
antiserum, is diluted 1:5000 with PBS-Tween. All other operations are
identical to those described above for IL-lb. The standards are prepared
in PBS-Tween + 10% FBS or HS. Eleven 2 fold dilutions are made
beginning at 50 ng/mL IL-6.
PGE~. production
Prostaglandin E2 is detected in cell culture supernatants
from LPS or IL-1 stimulated PBMC's using a commercially available
enzyme immunoassay . The assay purchased from the Cayman Chemical
(Catalogue number 514010) and is run exactly according to the
manufacturers instructions.
Interleukin8 (IL-8)
The present compounds can also be assayed for IL-8
inhibitory activity as discussed below. Primary human umbilical cord
endothelial cells {HUVEC) (Cell Systems, Kirland, Wa) are maintained
CA 02257200 1998-12-02
WO 97/47618 PCT/US97/09888
-94-
in culture medium supplemented with IS% fetal bovine serum and 1 %
CS-HBGF consisting of aFGF and heparin. The cells are then diluted 20-
fold before being plated (250 pl) into gelatin coated 96-well plates. Prior
to use, culture medium is replaced with fresh medium {200p1). Buffer or
test compound (25111, at appropriate concentrations) is then added to each
well in quadruplicate wells and the plates incubated for 6h in a
humidified incubator at 37°C in an atmosphere of 5% C02. At the end of
the incubation period, supernatant is removed and assayed for IL-8
concentration using an IL-R ELISA kit obtained from R&D Systems
(Minneapolis, MN). All data is presented as mean value (ng/mL) of
multiple samples based on the standard curve. IC50 values where
appropriate are generated by non-linear regression analysis.