Note: Descriptions are shown in the official language in which they were submitted.
o CA 02257825 1998-09-04
T
- 1 -
New Processes For Pre arina
Pesticidal Intermediates
This invention relates to a process far preparing
certain cyanomethylpropane derivatives and the use of
these compounds in the synthesis of pesticides and
pesticide intermediates.
W094/21606 describes the use of dicyanoproprionate
reacting with para SF5 aniline derivatives to
provide, with further reaction, cyanopyrazoles.
Ethyl 2,3-dicjranopropionate was first prepared and
characterised by Higson and Thorpe (J.Chem.Soc. 89,
1460 (1906)) who obtained the material in good yield
(70-81~) by reaction of formaldehyde cyanohydrin with
the sodium salt of ethyl cyanoacetate. Dickinson [J.-
Am. Chem. Soc. 82, 6132 (1960)} repeated this work.
This method of preparing the dicyanopropionate
suffers from a significant drawback in that it is
first necessary to isolate the intermediate
formaldehyde cyanohydrin. This highly water soluble
cyanohydrin is obtained by lengthy continuous
extraction and has a limited stability, often
decomposing violer_t1y upon attempted distillation.
Furthermore, this reaction requires care given the
risk of formation of dimeric side-products. The
preparation of dicyanopropionates has also been
described by Whiteley and Marianelli (Synthesis
(1978), 392) with the process leading to 2,3-
disubstituted succinodinitriles from the
cyanoacetate, an aldehyde (a 1 to 3 carbon
alkylaldehyde or benzaldehyde) and potassium cyanide
via 3-substituted-2,3- dicyanopropionates (which were
not isolated). However, the yield decreases
dramatically from isobutyraldehyde to acetaldehyde.
In the same manner Smith and Horwitz (J. Am. Chem.
Soc. 1949, 71, 3418) described the same reaction with
a ketone with a yield of 70~. This prior art
therefore teaches that yields improve with increasing
size of group adjacent to the carbonyl group.
In one aspect the present invention seeks to
provide a process for preparing cyanomethyl propane
derivatives satisfying one or more of the following
criteria.
- avoiding the use of formaldehyde cyanohydrin;
- avoiding the dimerisation side reaction;
CA 02257825 1998-09-04
WO 97/32843 PCT/EP97/01036
2
- obtaining the required product directly in high yield and
with high purity.
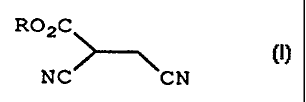
Therefore the present invention provides a process for preparing
a compound of formula (I):
R02C
I
NC CN
wherein
R represents straight- or branched- chain alkyl having up to 18
carbon atoms; or a salt thereof;
which comprises the reaction of a cyanoacetate of formula (II}:-
to R02C-CH2CN II
wherein R is as defned above, with a cyanide salt and formaldehyde
or a source thereof.
Preferably R represents straight- or branched-chain alkyl having
from i to 6 carbon atoms and most preferably R represents ethyl.
15 Suitable salts of cyanide include metal salts and organic salts
(e.g. tetra-alkylammonium cyanides such as tetrabutylammonium
cyanide). Preferably the cyanoacetate of formula (II) reacts with an
alkali or alkaline earth metal cyanide salt, with alkali metal cyanide
salts being especially suitable for use in the present invention,
2o particularly potassium cyanide or sodium cyanide. The product may
conveniently be isolated as the alkaline earth metal or alkali metal
salt. Alternatively, the reaction mixture is acidified, for example with
a mineral acid such as sulphuric acid or hydrochloric acid, to give the
compound of formula (I). Where a compound of formula (I) above is
25 desired (rather than a salt thereof] high yieids are generally obtained
when the reaction mixture is acidified without the addition of water.
Whilst formaldehyde itself can be used in the reaction it is more
convenient to use the polymerised form known as paraformaldehyde
[(HCOH)n3, available for example from Aldrich Chemical Company.
30 The reaction is generally performed using about I molar
equivalent of a compound of formula (11]; about 0.95 to 1.0 molar
CA 02257825 1998-09-04
WO 97/32843 PCT/EP97/Og036
3
equivalents of cyanide salt; and about l molar equivalent of
formaldehyde compound (based on the formaldehyde content).
The reaction may be carried out in the presence of a solvent.
Preferably, the reaction is performed in a solvent medium which is
usually an alcoholic medium or dimethyl formamide (DMF), N-
methyl pyrrolidone (NMP), dioxan, tetrahydrofuran (THF) or
dimethoxyethane. Especially preferred solvents are CI-Cg alcohols
such as methanol or, most preferably, anhydrous ethanol. Although
the temperature of the reaction is not critical the reaction will
to normally be performed from about 0 to about 120oC or at the reflux
temperature of the solvent. Generally best results are obtained by
introducing the formaldehyde source after the other reactants have
been combined.
The reaction generally takes place under substantially
anhydrous conditions (it being understood that the reaction proceeds
with the formation of one equivalent of water), as in the event of
prolonged exposure to aqueous conditions, there is a risk that the
ester group of the compound of formula (I) will undergo hydrolysis
(due to the basic conditions arising during the reaction) to the
2o corresponding acid of formula (I) (in which R is replaced by
hydrogen) and subsequently undergo decarboxylation to give
1,2-dicyanoethane.
The compound of formula (I) is useful in the preparation of
pesticidally active compounds, for example as described in European
Patent Publication Nos. 02951 I7 and 02341 I9, and W~93/06089.
In particular, the process of the invention may form part of an in
w situ preparation of another pesticidal intermediate and in a ftu~ther
aspect the invention provides a process for the preparation of a
compound of formula (III):
CA 02257825 1998-09-04
WO 97/32843 PCT/EP97/0i036
4
R=
HEN ~ ~N
N
R2 ~ ~ W
R3
(IIn
wherein Rl is cyano; W is nitrogen or -CR4; R2 and R4
independently represent halogen; and R3 represents halogen,
haloalkyl (preferably trifluoromethyl), haloalkoxy (preferably
trifluoromethoxy) or -SFS; which process comprises:
(a) reacting a cyanoacetate of formula (II) as defined above,
1.
with a cyanide salt and formaldehyde or a source thereof, to give a
compound of formula (I) as defined above; and
1o (b) reacting the compound of formula (I) thus obtained with
the diazonium salt of a compound of formula (IV):
NHZ
R2 ~ ~ W
R3
(
wherein W, R2 and R3 are as defined above, to give a
i5 compound of formula (V):
CA 02257825 1998-09-04
WO 97/32843 PCT/Eg'97/i~1036
S
~R1
NC
'~C02R
,N
N
I
R2 ~tnl
I
R3
wherein W, R, R1, R2 and R3 are as defined above, followed by
the cyclisation of said compound of formula (V}.
Compounds of formula (~ above possess a chiral centre giving
rise to different enantiomers, and also may exist as different
geometric isomers or mixtures thereof. All such forms are embraced
by the present invention. In this process, the product of reaction step
(a) is generally acidified with an alcoholic solution of a mineral acid,
l0 preferably an ethanolic solution of hydrogen chloride. This also
ensures that any acid by-product of the reaction step {a) (leading to
the corresponding compound of formula (I} in which R is replaced by
hydrogen) is re-esterif ed. For these reasons it is also preferred that in
this process, reaction step (a} takes place under substantially
15 anhydrous conditions.
Reaction step (b) is generally performed in the presence of an
inert solvent, for example water, acetonitrile, dichloromethane or
DMF, or more preferably an alcoholic solvent (e.g. methanol or
ethanol) and is optionally buffered (e.g. with sodium acetate). The
20 diazonium salt of a compound of formula (N} may be prepared using
diazotising agents known in the literature and is conveniently
prepared with a molar equivalent of sodium nitrite and a mineral acid
(e.g. hydrochloric or sulphuric acid}, at a temperature of from about
-IO°C to about SO°C, more preferably from about 0°C to
about 5°C.
2s The diazonium salt of the compound of formula {IV} is generally
prepared in situ as solvents such as alcohols tend to reduce diazonium
salts quickly. In the present reaction, the reaction of the diazonium
CA 02257825 1998-09-04
WO 97/32843 PCT/EP97/01036
6
salt of the compound of formula (IV) to give a compound of formula
(V) above generally occurs faster than the reduction of the diazonium
salt.
Subsequent hydrolysis, preferably using mild conditions with a
base such as aqueous sodium hydroxide, sodium carbonate or
ammonia, may be necessary to effect the cyclisation of the compound
of formula (V) to a compound of formula (III).
The molar ratio of the compounds of formula (II):(IV) is
generally from about 1.5:1 to about 1:4, preferably from about 1.3:1
to to about 1:1, more preferably about 1.1:1.
Compounds of formula (III) and (IV) above are described in the
literature, for example see EP-AI-02951 I7. Compounds of formula
(V) above are novel and thus constitute a further feature of the present
invention.
z5 The following non-limiting examples illustrate the invention.
Exam~ie 1
Preparation of ethyl 2,3-dicyanopropionate.
Potassium cyanide (13.0g, 0.2M) was stirred in absolute ethanol
2o and ethyl cyanoacetate (22.6g, 0.2M} and paraformaldehyde (6.0g,
0.2m) were added at ambient temperature. After S minutes the white
suspension was heated under reflux conditions for 12 minutes, and
the orange solution was evaporated to dryness in vacuo at below
25°C to give a buff solid. The solid (the potassium salt) was
25 dissolved in water (400m1), acidified to pH 5 with 2M hydrochloric
acid solution, giving a red oil. This mixture was extracted with
dichloromethane and the extracts dried and evaporated to dryness in
vacuo to give the title compound as a red oil (23.5g), 1H NMR
(CDCl3) d 4.3 (2H, q), 3.95 (1H, t), 3.0 (2H, d), 1.35 (3H, t);
3o identical with an authentic sample. Distilled material had b.p. 132-
136oC at O.SmmHg.
Yield : 77%.
Comparative example according to J. Chem. Soc. 89. 1460 (19061
CA 02257825 1998-09-04
WO 97/32843 PCT/EP97/01036
7
A solution of sodium ethoxide [prepared from sodium ('_'5.2g,
1.15M) and absolute ethanol f 6~Oml)] was stirred under an inert
atmosphere and treated with ethyl cyanoacetate (/?7.7m1, 1.2M) over
20 minutes. The solution was cooled to below lOoC and then added
slowly to a solution of formaldehyde cyanohydrin (freshly prepared,
70g, 0.2M) in absolute ethanol (200m1) at 5oC over 55 minutes.
After standing overnight the mixture was poured into ice-water ( 1 L)
and acidifed to pH 1-2 with concentrated hydrochloric acid. This
was extracted into dichloromethane, dried over anhydrous magnesium
l0 sulphate and evaporated to give a dark orange oil (150.6g). This was
distilled in vacuo collecting the title compound (73.6g) as a colourless
oil, b.p. 144-148°C/lmbar. Yield : 40%
The superiority of the process of the invention is thus clearly
demonstrated over this prior art.
Exampae 2
Process for the preparation of 5-amano-3-cyano-
1-(2,6-dichloro-4-trifluoromethyI-phenyl)pyrazote.
Sodium cyanide (20g, 0.408M) and ethyl cyanoacetate (46g,
0.408M) were dissolved in absolute ethanol (300m/) under an inert
atmosphere. Paraformaldehyde (12.2g, 0.408M) was added,
producing an exotherm, and the temperature was maintained below
50°C. The reaction mixture was then stirred at ambient temperature
for between 5 and 7 hours, cooled to between 0 and 5°C, and an
ethanolic solution containing hydrogen chloride (0.45M) was added,
maintaining the temperature below 5°C. The reaction mixture was
left overnight and 11 lml of a solution of hydrochloric acid (0.73M) in
ethanol was added to the suspension thus obtained at about 5°C.
2,6-Dichloro-4-trifluoromethylaniline (84.44g, 0.367M) was added at
this temperature followed by sodium nitrite (35.84g, 0.514M)
resulting in the formation of ethyl 2,3-dicyano-2-[2,6-dichloro-4-
trifluoromethylphenyl)azo~propionate, which may be isolated by
column chromatography, eluting with a pentane%ther solution and/or
reverse phase chromatography with an acetonitrile-water solution; or
CA 02257825 1998-09-04
WO 97/32843 PCT/EP97/OI036
8
by removing the ethanol by distillation. dissolving the reaction
mixture in toluene, washing the toluene solution with water and
evaporating the toluene to dryness. 'H NMR (CDCI3) I.37(t,3H),
3.55(s,2H), 4.43(q,2H), 7.65(s,2H).
Ammonia gas (9.68, 0.56M) was then bubbled into the reaction
mixture at 0°C. The ethanol was evaporated from the reaction
mixture under reduced pressure and the concentrated liquors were
taken up into a mixture of toluene and ethyl acetate. This solution
was washed with water and after concentration of the toluene phase at
i0 80°C, the solution was cooled to give the title compound as a
crystalline solid, and the liquors were then concentrated and cooled to
give a second crop of recrystallised product, m.p. 141 - 142°C
(combined weight of title compound 87.548; yield based on aniline
starting material = 78%).
I5