Note: Descriptions are shown in the official language in which they were submitted.
CA 02259183 2004-02-20
wo ~aioo3~s pcziQSS~~a.a.aas
sz~r~
P'LDORi~US RE71CTION ADIa SEpARATTON SYSTEMS
Related Reference
'rhe present application claims priority to U.S. Patent
No. 5,777,121, entitled Fluorous Reaction Systems and filed ,7une
28, 199fi.
Field of the Tnvention
the present invention relates to novel coxr~pasitions and
to methods of carrying out chemical reactions.
Dacls~round of the invention
Organic compounds are purified on a dai~.y basis in
uncounted numbers of research and commercial laboratories and
plants around the world. Purification costs account for a
significant fraction of the expenses for organic compounds
developed and sold by chemical, pharmaceutical, and other
industries. Chromatographic methods of purification are i.mmezlse~.y
~.~nportant, yet they are also expensive and time consuming.
Simpler but sometimes less effective methods are based on
technic,~ues of phase separation. Four phases are commonly used in
standard laboratory separat.ioxs methods: a gas phas0, a solid
phase, and two liquid phases - organic and aqueous. Among the
phase separation techniques, liquid-liquid extractions play a
time-honored role in the purificat~.on of
CA 02259183 1998-12-22
WO 98/00376 2 PCTlUS97/11215
organic compounds. These extractions are almost always
conducted with an organic solvent and water. Most frequently,
they are used to separate (that is, purify) organic compounds
from inorganic compounds. A less frequent but still important
application of organic-water extractions is an acid-base
extraction.
It is not widely recognized by synthetic organic
chemists that there is a "third liquid phase", the
fluorocarbon (or "fluorous") phase, whose members are not
miscible in either water or many organic solvents. See, for
example, Hudlicky, M. "Chemistry of Organic Fluorine
Compounds", Ellis Horwood: Chichester (1992). As used herein,
the term "fluorous liquid phase" refers to a liquid phase
comprising one or more solvents rich in carbon fluorine bonds.
A fluorous liquid phase is substantially immiscible with an
"organic phase" and forms a liquid-liquid biphasic mixture
with an organic phase.
As used herein, the term "fluorous", when used in
connection with an organic (carbon-containing) molecule,
refers generally to an organic molecule having a domain or a
portion thereof rich in carbon-fluorine bonds (for example,
fluorocarbons, fluorohydrocarbons, fluorinated ethers and
fluorinated amines). Such portion or domain may comprise part
of a fluorous compound or the entire fluorous compound. In
general, compounds comprising a relatively high weight
percentage of fluorine partition preferentially into the
fluorous liquid phase in a fluorous/organic liquid biphasic
mixture. See U.S. Patent No. 5,463,082. As used herein, the
terms "fluorocarbons" and "perfluorocarbons" include organic
compounds in which all hydrogen atoms bonded to carbon atoms
have been replaced by fluorine atoms. The terms
"fluorohydrocarbons" and "hydrofluorocarbons" include organic
compounds in which at least one hydrogen atom bonded to a
CA 02259183 1998-12-22
WO 98/00376 3 PCT/US97/11215
carbon atom has been replaced by a fluorine atom. Saturated
perfluorocarbon fluids have important applications in surface
chemistry, biology, and engineering. Most organic compounds
are completely or substantially insoluble in fluorocarbon
fluids, and many organic solvents are immiscible therein,
although this miscibility depends somewhat on the fluorous-
organic pairing. Solvents like carbon tetrachloride, ether,
and THF have the highest solubilities in fluorocarbon fluids,
and pairings of fluorocarbon fluids with these solvents are
either miscible or can be made miscible by slight warming.
There are a wide assortment of fluorocarbon fluids
commercially available under trade names like "FlutecT"" and
"Fluorinert''M". These fluids are made industrially by chemical
or electrochemical fluorination processes. Most of these are
mixtures of fluorocarbons with similar boiling points
(sometimes with small amounts of fluorinated ethers). These
mixtures are roughly analogous to the "petroleum ether"
solvents often used in organic chemistry. Fluorinated ethers
and fluorinated amines are also commercially available.
Although rarely referred to as such, these
fluorocarbon "fluids" are effectively solvents. The first
application of fluorocarbon solvents in the area of
traditional organic synthesis appeared in 1993 when D.W. Zhu
described a series of transesterification reactions in the
Fluorinert Fluid''" FC-77 (a fluorocarbon mixture containing
mostly isomers of CgFlg, by 97°C). Zhu, D.W., Synthesis, 953-
54 (1993). As illustrated in the following example, low
boiling alcohols were replaced by high boiling ones, and phase
separation was used at two stages.
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11215
4
FC-77
PhCO yC~H~ + PhCH yOH ~ PhCO pCHyPh + HOC aH~
~ alcohol separates in a "Dean-Stark" trap
~ product separates from FC-77 on cooling
First, an "inverse Dean-Stark" trap was used to separate the
low-boiling alcohol from the reaction mixture and thereby
drive the equilibrium. Second, the product ester separated
from the FC-77 on cooling. Another common fluorocarbon fluid
is FC-72''", a mixture of C6F14 isomers with a boiling point of
56°C. FC-72 and FC-77 are commercially available from 3M.
Shortly after the work of Zhu, Horvath and Rabai
described the synthesis of a "fluorous" phosphine ligand and
used this to generate a rhodium catalyst for a standard
hydroformylation reaction. Horvath, I. T. and Rabai, J.,
Science, 266, 72-75 (1994). See also U.S. Patent
No. 5,463,082; and Gladysz, J. A., Science, 266, 55 (1999).
The hydroformylation was conducted in a liquid biphasic
mixture of perfluoromethylcyclohexane (fluorous solubilizing
solvent) and toluene (organic solubilizing solvent) under a
CO/H2 atmosphere as illustrated below.
CGHScH,i~-CAF"cF;
CBH~~CH=CH2 + CO r CgH~~CH(CHO)CH s+ C~oHZ~CHO
(C6F,~CFI~CH2)3P
Rh(CO).,(acac)
~ two phase reaction
~ catalyst in fluorous phase
can be reused
The products were separated from the catalyst by separation of
the two liquid reaction phases, and the recovered catalyst
from the fluorinated phase was successfully reused in another
hydroformylation.
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11215
The distinctive physiochemical properties of a
fluorous liquid phase can be used advantageously to provide
unexpected solvent effects including altered and improved
product yields, reactivities and/or selectivities. Likewise,
5 physiochemical differences between fluorous molecules and
organic (that is, non-fluorous) molecules provide a valuable
tool to effect separation.
It is, therefore, very desirable to develop
additional fluorous reaction components, reaction systems,
reaction schemes and separation schemes.
Summary of the Invention
The term "reagent," as used herein in connection
with combinatorial syntheses, refers to a chemical entity that
is required for a reaction but contributes either an invariant
piece or no piece to the products of a combinatorial
synthesis. The term "reactant," as used herein in connection
with combinatorial synthesis refers generally to a type of
molecule that contributes a variable piece to the products of
a combinatorial synthesis. The distinction between the terms
"reactant" and "reagent" in "common" (non-combinatorial)
organic syntheses is vague, but those skilled in the art often
refer to a reaction component as a reagent if it contributes
no piece, a rather small piece, or a piece without carbon
atoms therein to the target product. As used herein, the term
"reagent" includes a catalyst if used in a substoichiometric
quantity.
As used herein, the term "substrate" refers
generally to a reaction component that is a major starting
material of a synthetic reaction, normally prepared in a prior
step. The term "target product" refers generally to the
CA 02259183 1998-12-22
WO 98/00376 6 PCT/US97/11215
target or desired molecules) of a transformation derived by
reaction of the substrate with the other reaction components)
in the medium. The terms "side product" or "byproduct" refer
generally to a product derived from any components) of the
reaction medium which is not the target product and is
preferably separated therefrom.
The term "organic/fluorous phase separation
technique" refers generally to any separation technique that
is based upon the presence of the fluorous groups) on a
fluorous component or molecule to establish a separation
between compounds bearing the fluorous groups) and compounds
not bearing the fluorous group(s). Such techniques include,
but are not limited to, simple extractions between a fluorous
liquid and an organic liquid. The fluorous phase can also
consist the of fluorous components) to be separated
themselves as, for example, a liquid or a solid phase. Also
included are techniques like countercurrent distribution and
"solid phase extraction." Solid phase extractions involve,
for example, the use of highly fluorinated polymers or
stationary phases in combination with organic or
organic/aqueous mobile phases. Chromatography materials
consisting of bonded stationary phases (for example, "S-
OSi(Me)2(CHZ)"Rf", where "S" is a standard support like silica
gel, "n" is 2 or 3, and Rf is a linear or branched
perfluoroalkyl group) are known. Some of these stationary
phases (that is, fluorous solid phases) are commercially
available (for example, Fluofix''M Columns) from companies like
Keystone Scientific (USA) and NEOS Co. (Japan). It is known
that highly fluorinated compounds are strongly retained by
these columns when they are eluted with organic or
organic/aqueous mobile phases, while organic compounds pass
through. See, for example, H. A. H. Billiet, et a1, J.
Chromat., 218, 443 (1981) and G. E. Berendsen, et al., Anal.
Chem., 52, 1990 (1980). However, elution with a fluorous
1
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11215
7
mobile phase will release all retained fluorous compounds. In
favorable cases, even standard "normal" (silica, alumina,
etc.) or "reverse phase" (C18) stationary phases can be used
because the presence of the fluorous group dramatically alters
the mobility of compounds bearing that group relative to
compounds not bearing that group. All preferred
organic/fluorous phase separation techniques enable relatively
simple separations of fluorous and non-fluorous components.
The terms "fluorous substrate," "fluorous reactant,"
"fluorous reagent" etc. (or, generally, "fluorous reaction
component") refer generally to a reaction component comprising
a portion rich in carbon-fluorine bonds. Fluorous reaction
components generally partition preferentially into (or onto)
the fluorous phase. The term "fluorous reaction component"
also includes, however, a reaction component that
(1) comprises a portion rich in carbon-fluorine bonds,
(2) does not partition preferentially into a fluorous phase,
but (3) forms fluorous products) and/or byproducts)
comprising such a portion rich in carbon-fluorine bonds during
reaction. The fluorous products and/or byproducts partition
preferentially into a fluorous phase. The terms "organic
substrate," "organic reactant," "organic reagent" etc. (or,
generally, "organic reaction component") refer generally to a
reaction component that does not comprise a portion rich in
carbon-fluorine bonds. Organic reaction components and
organic compounds generally partition preferentially into an
organic (that is, non-fluorous) phase (for example, into an
organic layer in an organic/fluorous liquid extraction).
- The present inventors have discovered that liquid
biphasic reaction systems comprising a fluorous phase and a
nonfluorous phase previously investigated by others are not
operable in a number of reaction systems. Indeed, in many
cases the partition coefficients for the reaction components
CA 02259183 1998-12-22
WO 98/00376 8 PCT/US97/11215
(that is, reagents, reactants and catalysts) may be such that
the phase separation between the liquid phases of biphasic
systems severely inhibits or prevents reaction. It has been
discovered that the processes of reaction and phase separation
(that is, for recovery of product) are preferably separated.
Therefore, in one embodiment of the present
invention, all of the reaction components of the reactions of
the present invention, including any reagents and reactants,
are preferably substantially soluble in an "organic/fluorous
solubilizing liquid phase" during the course of the reaction.
As used herein, the term "organic/fluorous solubilizing liquid
phase" refers to a liquid phase comprising a solvent system
adapted or selected to substantially solubilize both an
organic reaction components) and a fluorous reaction
component(s). It is not necessary that the reaction
components be completely soluble in the organic/fluorous
solubilizing liquid phase at any or all times during the
reaction. Each reaction component (organic or fluorous) has
at least approximately a 0.1 millimolar solubility therein
and, more preferably, at least approximately a 1 millimolar
solubility therein. The target product and/or any byproducts
need not be substantially soluble in the organic/fluorous
solubilizing liquid phase. Indeed, the target product and/or
any byproducts may, for example, form an immiscible liquid
phase or an insoluble solid phase.
For reaction in an organic/fluorous solubilizing
liquid phase, the organic/fluorous solubilizing liquid phase
may comprise: (i) an organic solvent or a mixture of organic
solvents (for example, carbon tetrachloride, THF and/or
ether); (ii) a homogeneous mixture of an organic solvent (or
solvents) with a fluorous solvent (or solvents) (for example,
FC-72 mixed with carbon tetrachloride, ether or THF); or a
hybrid organic/fluorous solvent (or solvents) used either
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11215
9
alone or in combination with either or both an organic solvent
(or solvents) and a fluorous solvent (or solvents). Solvent
systems as described above are known in the art.
The organic/fluorous solubilizing liquid phase is a
homogeneous liquid phase with respect to organic and fluorous
liquid phases. As used herein, the term "homogeneous liquid
phase" refers to a liquid phase in which no internal liquid-
liquid physical boundaries (for example, a meniscus) are
visible between an organic phase and a fluorous phase. See,
for example, CRC Handbook of Chemistry and Physics, 61St
Edition, C-691 (1980) (determining miscibility on the basis of
either observation or absence of an interfacial meniscus).
Thus, there are no internal fluorous-organic physical
boundaries observed in the organic/fluorous solubilizing
liquid phases of the present invention. Such organic/fluorous
solubilizing liquid phases may form a liquid-liquid physical
boundary with an aqueous phase in some reactions where water
is present, however.
As used herein, the term "hybrid organic/fluorous
solvent" refers to a solvent comprising both an organic (for
example, a hydrocarbon) portion or domain and a fluorous (for
example, a fluorocarbon or fluorohydrocarbon) portion or
domain. In general, hybrid organic/fluorous solvents will not
form a biphasic system or mixture when mixed with either
organic solvents or with fluorous solvents. Some hybrid
organic/fluorous solvents may form a biphasic mixture with an
organic solvent or a fluorous solvent (for example, FC-72 and
CF3CHzOH form a biphasic mixture), but such hybrid organic
fluorous solvents are still useful either alone or in
combination with other solvents for creating an
organic/fluorous solubilizing liquid phase. Examples of
hybrid organic/fluorous solvents include, but are not limited
to, benzotrifluoride (BTF; C6HSCF3) , trifluoroethanol, p-
CA 02259183 1998-12-22
WO 98100376 PCT/US97/11215
chlorobenzotrifluoride (C1C6H9CF3) , and
1,4-bis(trifluoromethyl)benzene (CF3C6H9CF3) . Examples of
homogeneous mixtures of hybrid organic/fluorous solvents with
organic solvents and/or fluorous solvents for use in the
5 present invention include BTF/CH2C12, H20/BTF/THF/acetone,
BTF/FC-72 and BTF/FC-72/ether. Hybrid organic/fluorous
solvents are somewhat analogous to hybrid organic/aqueous
solvents such as alcohols (for example, CH3CHzOH) which have an
organic portion and an aqueous (or water-like) portion and
10 generally do not form a biphasic mixture when mixed with
either organic solvents or water.
The present invention thus generally provides a
method for carrying out a chemical reaction comprising the
steps of forming an organic/fluorous solubilizing liquid phase
comprising a solvent system. The solvent system is selected
or adapted to substantially solubilize a fluorous reaction
component or components (that is, a fluorous reagent, a
fluorous catalyst and/or a fluorous reactant). The fluorous
reaction component is functionalized to comprise at least one
fluorous moiety having the formula -(R)d(Rf)e. (Rf)e is at
least one fluorous group and a is a whole number. (R) d is an
organic (for example, hydrocarbon) spacer group, which may be
present or absent, and d is an integer equal to at least zero.
The solvent system is also adapted to substantially solubilize
an organic reaction component or components convertible to an
organic product in a reaction scheme including one or more
reactions.
After synthesis of the organic product in the
organic/fluorous solubilizing liquid phase, an
organic/fluorous phase separation technique is used to effect
separation of an organic target product from any remaining
fluorous reaction components and/or any fluorous byproducts
formed in the reaction. In a preferred embodiment, a co-
T
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11215
11
solvent or co-solvents is preferably added to the
organic/fluorous solubilizing liquid phase to effect a phase
separation into at least a fluorous liquid phase and an
organic liquid phase. A solid phase, a gas phase and/or an
aqueous phase may also be present. In some cases, it may be
preferable to remove by evaporation part or all of the
organic/fluorous solubilizing liquid phase before addition of
the co-solvent or co-solvents.
The fluorous reaction components) thus preferably
comprises a sufficient number of fluorous moieties to render
any excess fluorous reaction components and fluorous
byproducts derived from the fluorous reaction components
readily separable in an organic/fluorous phase separation
technique. In an organic/fluorous liquid extraction, for
example, excess fluorous reaction components) and/or fluorous
byproducts) are preferentially partitionable into the
fluorous liquid phase after a single or a series of
extractions. Likewise, the organic product is preferentially
partitionable into the organic liquid phase after a single or
a series of extractions.
The organic spacer group (R)d may contain H and C, or
may contain groups containing O, N, S, P, As and Si in
addition to H and C in the backbone and/or as substituents.
In general, (R)d is rich in hydrogen atoms in comparison to
(Rf ) e. Preferably, d is an integer equal to at least zero or
any whole number. More preferably, d is a whole number less
than 4. Most preferably d is 0, l, 2 or 3. In many cases, an
organic spacer group is preferable or required because of the
strongly electron withdrawing nature of fluorous groups.
Addition of a hydrocarbon group (for example, a -CHZCHZ- group)
as a spacer group between the fluorous group and a reaction
component generally reduces the electron withdrawing effect of
the fluorous group on the reaction component. In some cases,
CA 02259183 1998-12-22
WO 98/00376 12 PCT/t1S97/11215
the electron withdrawing nature of the fluorous group may have
no effect or a beneficial effect upon the reaction component.
In such cases, the organic spacer group may be omitted (that
is, d = 0) .
The fluorous reaction components often may contain a
plurality of fluorous moieties (for example, Q-[(R)d(Rf)e]Z,
wherein Q represents a standard reaction component and Z > 1)
having a significant proportion of fluorine atoms as compared
to the molecular weight of the entire reaction component. The
fluorous moieties may be attached to the same atom on the
fluorous reaction components) or to different atoms thereon.
Sufficient fluorous moieties are preferably used such that any
fluoro.us reaction components and/or any fluorous byproducts
remaining after reaction are separable from the organic target
product via an organic/fluorous phase separation technique.
However, the chemical activity of underlying reaction
component Q is preferably changed little or not at all by
addition thereto of fluorous portion (Rf)e.
In cases in which the fluorous reaction components)
are not completely reacted, preferably, at least approximately
20 wt o to approximately 90 wt o, and, more preferably, about
50 wt o to 90 wt % of the total weight of a fluorous reaction
component comprises fluorine. In all such cases, sufficient
fluorine content and appropriate structure should be present
to render the fluorous reaction component separable in an
organic/fluorous phase separation technique (for example,
partitionable preferentially into the fluorous liquid phase
after phase separation) to enable separation thereof from the
organic target product.
In cases in which a fluorous reaction component is
used in such quantities that it is completely reacted, only
the resulting fluorous byproducts) must be separated from the
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11215
13
organic target product. In such cases, preferably, at least
approximately 20 wt o to approximately 90 wt ~, and, more
preferably, about 50 wt o to 90 wt o of the total weight of a
fluorous byproducts) comprise fluorine. As clear to one of
ordinary skill in the art, if the organic portion of the
fluorous reaction component was relatively large in comparison
to any organic portion of the corresponding fluorous
byproduct(s), the fluorine wt o of the fluorous reaction
component can be less than 20 wt o. As also clear to one of
ordinary skill in the art, the preferential partitioning of
the fluorous reaction component to the fluorous phase not
important in these cases. In general, any fluorous compound
to be separated from an organic compound in an
organic/fluorous phase separation technique preferably
comprises at least approximately 20 wt o fluorine, and, more
preferably, at least 50 wt o fluorine, to facilitate
separation.
Typically, known standard (non-fluorous) reactions
can be carried out under the present invention with one or
more fluorous functionalized reaction components within the
range of reaction conditions used in the corresponding
standard reactions. The present invention is equally
applicable, however, to newly developed organic reactions.
The fluorous reaction components can be prepared by
fluorination or fluoro-functionalization of a starting
reaction component, by modification of another fluorous
reaction component, or by total synthesis. For example,
fluorous tin reaction components can be made conveniently in
one or more steps. An illustrative method of synthesis of
fluorous tin reaction components is the combination of known
nucleophiles, for example Grignard reagents such as
RfCH2CH2MgBr, with known tin electrophiles, for example Cl3SnX.
This combination leads either directly or through the agency
CA 02259183 1998-12-22
WO 98/00376 14 PCT/US97/11215
of one or more additional transformations wherein one group X
is replaced by another to preparation of a large new class of
fluorous tin reaction components [RfCH2CH2]3SnX. The
interchange of groups X in organotin chemistry is well known
to those skilled in the art and can be accomplished by a large
class of reactions wherein a nucleophilic precursor of the
product X group replaces a leaving group X (for example, a
halogen or triflate) in the tin precursor (for example,
stannylation of an alcohol), by reactions wherein a tin
nucleophile (X - metal) adds to or displaces an electrophile
precursor of the product X group (for example, a substitution
reaction of a stannyl metal with an allyl halide), or by
reactions in which the tin SnX bond adds to a multiple bond
(for example, hydrostannylation of an alkene or a carbonyl
group). Similarly, the use of other standard classes of
nucleophiles and tin electrophiles allows entry into related
groups of reagents with other fluorous substituents on tin.
Analogous transformations can generally be applied to the
synthesis of related silicon and germanium reaction
components.
Transformations under the method of the present
invention generally parallel the transformations of known
"non-fluorous" reaction components with the advantages that
the fluorous reaction components and any fluorous byproducts
derived from the fluorous reaction components can be removed
from the organic products by one or more organic/fluorous
phase separation techniques (for example by organic/fluorous
liquid-liquid extraction). The recovered fluorous reaction
components can often either be reused directly or recycled by
standard reactions to reusable forms. These are significant
advantages compared to the standard reaction components.
The method of the present invention also offers
significant advantages over the current fluorous multiphase
CA 02259183 1998-12-22
WO 98/00376 15 PCT/ITS97/11215
reactions. See U.S. Patent No. 5,463,082. While there are
benefits to conducting some types of catalytic and other
organic reactions in multiphase systems, the vast majority of
organic reactions are preferentially conducted in liquid
phases in which the key reaction components have substantial
solubility. Separation into immiscible fluorous and organic
liquid phases is not expected to be beneficial for many
important reactions classes and may often be detrimental. In
the method of the present invention, fluorous reaction
components and organic reaction components react under
conditions in which both are substantially soluble in the same
organic/fluorous solubilizing liquid phase.
For example, organic reactions of tin, germanium,
and silicon reagents R3MX (where M = Si, Ge, Sn and X = an atom
or a group participating in a reaction with an organic
compound) are routinely used by those skilled in the art to
accomplish many different organic transformations. Most
reactions of these reaction components are preferentially
conducted in a homogeneous liquid phase. Reactions of the
fluorous analogs of these reaction components, [{Rf)e{R)d]3MX
are likewise preferentially conducted in a homogeneous liquid
phase. For example, the reagents [C6F13CHZCH~]3SiX where X - H
and C1 are known compounds that can be used by the methods
described herein to conduct reactions such as hydrosilylation
and reduction (X - H) or silylation (X - Cl) that are
analogous to the reactions of standard (non-fluorous) reagents
R3SiX where X = H or C1 and R = alkyl or cycloalkyl. Likewise,
fluorous allyl- and vinyltin and allyl- and vinylsilane
reaction components can be used for typical ionic allylations
and vinylations, and fluorous allyl- and vinyltin reaction
components can be used for typical radical allylations and
vinylations as well. These are but a few examples selected
from the rich, well known chemistry of tin, germanium and
silicon.
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/1I215
16
Fluorous compounds remaining after reactions of the
present invention may, for example, be separated from organic
compounds by a simple liquid-liquid extraction, thereby
providing a very substantive purification that for many
reactions would previously have required chromatography or
some other more demanding technique. The present invention
provides significant advantages in both "common" and
"combinatorial" organic synthesis.
In common organic synthesis, individual steps are
conducted sequentially until the final target molecule or
product is made. In combinatorial organic synthesis, the
target is not a single molecule but instead a "library" of
tens to millions of molecules. Multiple reactions are
conducted either together or in parallel to provide multiple
products as individual compounds or mixtures. The techniques
of combinatorial chemistry are becoming very popular in the
pharmaceutical industry as tools to discover and optimize new
drugs. In combinatorial synthesis, the premium of simple
methods of purification is even higher than in normal
synthesis; one cannot chromatograph hundreds or thousands of
samples. For this reason, combinatorial synthesis is now
conducted almost exclusively on the solid (polymeric) phase,
where purification can be effected simply by filtration.
Unfortunately, the purification attractions of the solid phase
turn into synthetic detractions. Conducting liquid phase
reactions can be difficult because the polymer never truly
dissolves in the reaction solvent.
Combinatorial syntheses are usually automated.
There are several features that favor the automation of
organic synthesis with substrates in the liquid phase rather
than on the solid phase. Four of these are briefly considered
below. First, there are more phases available. Counting
water as three phases (neutral, acidic, basic) provides seven
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11215
17
different phases. There is then much more flexibility to this
approach because there are more phases and more possible
separations. Second, in the liquid phase approach with a
fluorous reaction component, the substrate is not "affixed" in
any phase, so purification of products by "phase switching" is
now an option. Phase switching is simply modifying the
substrate so that it preferentially partitions out of one
phase and into another. Such phase switches can be envisioned
between several different phases and can be accomplished at
any point in a synthesis. Third, there is no need for
"attachment" and "detachment" of the substrate to the solid
phase. All concerns about stabilities of polymers and linkers
to reaction conditions are eliminated; the only concern is the
substrates. Fourth, many reactions are preferably conducted
in a homogenous liquid phase. This is in direct contrast to
solid phase syntheses, where true homogeneity is never
obtained.
The present invention, for example, also provides
substantially universal methods for synthesizing and
separating organic compounds. The methods are particularly
useful in combinatorial synthesis techniques, but find use in
substantially any reaction and/or separation requiring
separation of one organic compound from another organic
compound.
The present invention thus provides generally a
method of separating a first organic compound from a mixture
comprising at least a second organic compound. Under this
method, the first organic compound is first selectively
reacted with a fluorous reaction component to attach a
fluorous moiety or fluorous "phase tag" to the first organic
compound to result in a fluorous compound. The first organic
compound may be mixed with the second organic compound before
it is selectively reacted with the fluorous reaction
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11215
18
component. Alternatively, the second organic compound can be
formed or added after formation of the fluorous compound. The
fluorous moiety has a molecular weight sufficiently high to
render the fluorous compound separable from the second organic
compound via an organic/fluorous phase separation technique.
The fluorous compound is then separated from the second
organic compound via the organic/fluorous phase separation
technique. Preferably, the fluorous compound is partitionable
into a fluorous liquid phase to enable separation in an
organic/fluorous liquid-liquid extraction. The separated
fluorous compound may be reacted, if desired, to regenerate
the first organic compound.
The present invention also provides generally a
method of synthesizing an organic target product and
separating the organic target product from other organic
compounds including excess organic reaction components and/or
organic byproducts. Under this method, a first organic
compound is reacted with a first fluorous reaction component
to attach a fluorous moiety or fluorous phase tag to the first
organic compound to result in a second fluorous reaction
component. The fluorous moiety comprises sufficient fluorine
to render a fluorous target product produced in a reaction
scheme including a single reaction or series of reactions
separable via an organic/fluorous phase separation technique
from excess organic reaction components) and/or organic
byproducts}. Preferably, the fluorous target product
partitions preferentially into a fluorous liquid phase to
enable separation via organic/fluorous liquid-liquid
extraction.
The fluorous reaction component is reacted with at
least a second organic compound/reaction component to produce
the fluorous target product. One reaction with a single
'second" organic compound or numerous additional reactions
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11215
19
with other organic compounds can occur before synthesis of the
fluorous target compound is complete. The fluorous target
product is then separated from organic compounds including any
remaining second organic compound and/or any organic byproduct
via the organic/fluorous phase separation technique. The
separated fluorous target product is then reacted to cleave
the fluorous moiety and provide the organic target product.
The "fluorous phase tagging" reaction/separation methods are
equally useful in reactions occurring in an organic/fluorous
biphasic mixture and in reactions occurring in a
organic/fluorous solubilizing liquid phase.
The present invention also provides chemical
compounds of Si, Ge and Sn that are well suited for use as
fluorous reaction components and/or fluorous phase tags. In
that regard, the present invention provides generally a
chemical compound of the formula
XM[ (R) (Rf) J3,
wherein M is Si, Ge or Sn. X is H, F, Cl, Br, I, N3, OR1, OH,
OOH, OOR1 SRS, SeRl, CN, NC, NRlRz, a cyclic group (for example,
an aryl group), a substituted cyclic group (for example, a
substituted aryl group), a heterocyclic group (for example, a
heteroaryl group), or a substituted heterocyclic group (for
example, a substituted heteroaryl group). Such cyclic groups
are preferably of 5 to 25 carbon atoms.
X may also be a linear or branched alkyl group of 1
to 15 carbons. In the case that M is Sn or Ge, the linear or
branched alkyl group is preferably of 3 to 15 carbons.
Further, X may be a substituted linear or branched alkyl
group, an alkenyl group, a substituted alkenyl group, an
alkynyl group, a substituted alkynyl, an acyl group, or a
CA 02259183 1998-12-22
WO 98/00376 2 0 PCT/US9'7/11215
substituted acyl group. These groups preferably are of 1 to
20 carbon atoms.
X may also be M' ( (R' ) (Rf' ) ) 3, OM' ( (R' ) (Rf' ) ) 3 or
OOM' ( (R' ) Rf' ) ) 3, wherein M' is Si, Ge, or Sn. R1 and R2 are
each independently, the same or different, H, a linear or
branched alkyl group, a substituted linear or branched alkyl
group, a cyclic alkyl group, a substituted cyclic alkyl group,
an alkylsulfonyloxy group, a perfluoroalkylsulfonyloxy group,
an acyl group, a substituted acyl group, or a perfluoroacyloxy
group. R and R' are each independently, the same or
different, an alkylene group of 1 to 6 carbons or a
substituted alkylene group of 1 to 6 carbon atoms. Rf and Rf'
are each independently, the same or different, a linear
perfluoroalkyl group of 3 to 20 carbons, a branched
perfluoroalkyl group of 3 to 20 carbons, or a hydrofluoroalkyl
group of 3 to 20 carbons, wherein the hydrofluoroalkyl group
comprises up to one hydrogen atom for each two fluorine atoms
thereof.
The terms "alkyl", "aryl" and other groups refer
generally to both unsubstituted and substituted groups unless
specified to the contrary. Alkyl can be saturated or
unsaturated and branched or unbranched. Preferred
substituents of substituted groups include but are not limited
to groups containing C, H, C1, F, Br, I, N, S, P, As or Si.
The term "alkylene" refers to an acyclic carbon chain or a
saturated acyclic carbon chain represented by the formula
-C"HZn- (for example, -CHzCH2-) , wherein hydrogen may be
replaced by a monovalent substituent.
In a number of preferred embodiments wherein M is
Sn, X is preferably H, F, C1, Br, I, N3, Ofd, OSn (CHZCHZRf ) 3, an
allyl group, a phenyl group, a 4-methoxyphenyl group, a
2-pyridyl group or a 2-furyl group.
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11Z15
21
R is a preferably a linear alkylene group of 1 to 5
carbons. Rf is a preferably a linear perfluoroalkyl chain of
6 to 12 carbons. In general, the larger the molecule to be
made fluorous (that is, the higher the molecular weight) the
greater the fluorine content required.
In general, the present invention provides compounds
that are fluorous analogs of standard Sn, Ge and Si compounds.
Standard organometallic reaction components and reactions are
reviewed in Davis, A., ed., Comprehensive Or anometallic
Chemistry II, Pergamon Press, Oxford, Vol. 2, 217-304 (1995).
Brief Description of the Drawings
Figure 1 illustrates organic substrates reduced with
a novel fluorous reagent.
Figure 2A illustrates a current combinatorial
synthetic scheme.
Figure 2B illustrates an embodiment of a
combinatorial synthetic scheme of the present invention.
Figure 3 illustrates the results of a combinatorial
synthesis under the present invention.
Figure 4A illustrates a standard Stille coupling.
Figure 4B illustrates a Stille coupling under the
present invention.
Figure 4C illustrates several examples of Stille
couplings under the present invention with yields.
CA 02259183 1998-12-22
WO 98/00376 2 2 PCTIUS97/11215
Figure 5 illustrates a phase tagging scheme.
Figure 6 illustrates in situ method of nitrile oxide
generation and cycloaddition.
Figure 7 illustrates nitrile oxide cycloaddition
with fluorous-tagged allyl alcohols.
Figure 8 illustrates schematically an example of
fluorous phase switching.
Figure 9 illustrates purification of Grignard
products by fluorous phase switching.
Figure 10 illustrates fluorous tagging of residual
reaction components or byproducts.
Figure 11 illustrates the combination of a fluorous
reagent and a fluorous phase tag.
Figure 12 illustrates several examples of fluorous
Ugi reactions.
Figure 13 illustrates an example of a fluorous
analog of the Biginelli reaction.
Figure 14 illustrates a procedure and yields for the
reaction of Figure 13.
CA 02259183 1998-12-22
WO 98/00376 2 3 PCT/ITS97/11215
Detailed Description of the Invention
1. Synthesis In An Organic/Fluorous Solubilizing Liquid
Phase.
Synthesis in an organic/fluorous solubilizing liquid
phase under the present invention will be discussed in
connection with several examples of novel fluorous synthetic
schemes using reaction components (that is, reactants,
reagents, and catalysts) of the general formula:
XM( (R) (Rf) )3
In this general formula Rf is a fluorous group and,
preferably, a perfluorinated group having 3-20 carbons
(XM[(R)a(Rf)e])Z; wherein d = a = 1 and z = 3). (R) is a
hydrocarbon group and, preferably a -CHZCH~- group. M is
selected from the group consisting of silicon, germanium and
tin. X is an atom or a group that is involved in a reaction
with an organic substrate. These reaction components are used
in a number of different ways to synthesize and purify organic
molecules, as outlined below.
Reactions of organic substrates with fluorous reagents to
provide organic target products.
In this synthetic scheme, an organic substrate was
reacted with a fluorous reagent of the general formula
XM((R)(Rf))3, which can be used in excess if desired. After
reaction in an organic/fluorous solubilizing liquid phase,
organic-fluorous extraction/separation upon addition of an
appropriate co-solvent provides the target product in the
organic liquid phase, and the excess fluorous reagent and the
products derived therefrom in the fluorous liquid phase. The
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11215
24
method not only facilitates purification of the target product
relative to existing methods, but it also allows ready
recovery of a fluorous side product in a state suitable for
recycling to the original reagent for reuse. In some cases,
the original reagent is recovered directly. Thus, both
purification and disposal costs are reduced.
In one study, a fluorous reagent, tris (2-
(perfluorohexyl) ethyl) tin hydride 3 [ (C6F13CH2CH2) 3SnH] was
synthesized. The approved name of fluorous tin hydride
reagent 3 is tris (3,3,4,9,5,5,6,6,7,7,8,8,8-
tridecafluorooctyl)tin hydride. It has been discovered that
this reagent behaves very similarly to "standard" (that is,
nonfluorous) tin hydride reagents in radical reductions, yet
it has significant practical (and possibly also ecological)
advantages over the commonly used compounds, tributyltin
hydride, tris(trimethylsilyl)silicon hydride, and related
reagents. In the reactions studied, a hybrid organic/fluorous
solvent comprising benzotrifluoride (BTF, C6H5CF3,
trifluoromethyltoluene) or benzotrifluoride mixed with tert-
butanol, was used to provide a homogeneous reaction medium or
phase (the organic/fluorous solubilizing liquid phase).
Homogeneous liquid phase solvents comprising mixtures of
organic and fluorous solvents are known and can also be used
in the reactions of the present invention. Organic solvents
in which the fluorous reagent is substantially soluble (for
example, hexane, THF and/or ether) can also be used.
Benzotrifluoride (BTF) was selected in part because of its
favorable properties and low cost.
The equation below summarizes a preferred method for
preparing novel fluorous tin hydride reagent 3. Preparation
of the Grignard reagent from 2-perfluorohexyl-1-iodoethane and
quenching with phenyltrichlorotin provided the novel
intermediate product la. Brominolysis of the phenyl-tin bond
CA 02259183 1998-12-22
WO 98/00376 2 5 PCT/US97/11215
and reduction of the resulting novel tin bromide 2 with
lithium aluminum hydride in ether provided novel fluorous tin
hydride reagent 3. This product was isolated in 820 overall
yield as a clear liquid after purification by vacuum
distillation.
Rf(CH 2)2Mgl C~3SnP ~ (Rf(CH 2)2)3SnPh Br2 > (Rf(CH 2)2)aSnBr
Rf = C6F~3 1a 2
LiAlH4> (C6F~3CH2CH2)3SnH
3
Attempts to reduce a typical organic substrate, 1-
bromoadamantane, using fluorous tin hydride reagent 3 under
fluorous conditions like those used by Zhu resulted in
unacceptably slow reaction rates and unacceptably low yields.
Similarly, attempts under fluorous biphasic conditions like
those used by Horvath and Rabai or in normal organic solvents
like benzene also resulted in unacceptably slow reaction rates
and unacceptably low yields. It is believed that the
partition coefficients for the reaction components are such
that the phase separation prevents a radical chain from
propagating with bromoadamantane. Simple extractions provide
crude estimates of partition coefficients. Fluorous tin
hydride reagent 3 (1.0 g) was partitioned between PFMC (10 mL)
and an organic solvent (10 mL) by shaking for 5 min in a
separatory funnel. Evaporation of the organic layer provided
the following weights: benzene, 22 mg; MeOH, 30 mg; CH2C12,
47 mg; EtOAc, 104 mg; CHC13, 141 mg.
In contrast, treatment of perfluorodecyl iodide with
1.2 equiv of fluorous tin hydride reagent 3 and loo AIBN in
refluxing PFMC provided the corresponding reduced compound 4
in 72o yield as illustrated in the equation below. The
CA 02259183 1998-12-22
WO 98/00376 2 6 PCT/US97/11215
success of this fully fluorous reaction (that is, fluorous
solvent, fluorous reagent, fluorous substrate and fluorous
product) suggested that a homogeneous medium was important
thereto.
AIBN
C 10F21 I + (C 6F13CH 2CH 2)3SnH> C 1pF21 H
PFMC
reflux 4 (~2%)
AIBN
Ad-Br + (C 6F13CH 2CH 2)3SnH> Ad-H
reflux
3
equivale nts Solvent yields
1.2 BTF 90%
0.1 BTF/t BuOH (1/1) 92%
0.01 BTF/t BuOH (1/1) g5%
Adamantyl bromide was cleanly reduced over
approximately 3 hours with 1.2 equiv of fluorous tin hydride
reagent 3 in refluxing BTF (stoichiometric procedure). After
evaporation of the BTF and liquid-liquid extraction (PFMC-
CH2C12) to separate the tin products, adamantane was isolated
in 90o yield (as determined by GC integration). Under the
stoichiometric procedure, fluorous tin hydride reagent 3
reduces a number of other functional groups besides halides,
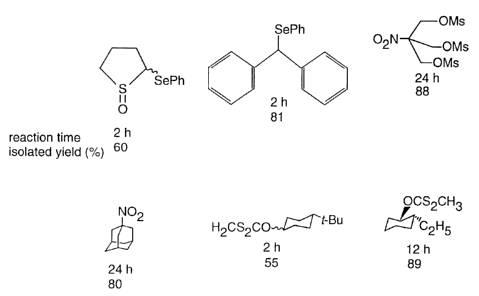
as shown in Figure 1. In these substrates, the nitro,
phenylseleno, or xanthate groups are replaced by hydrogen.
A catalytic procedure was also developed by using
loo fluorous tin hydride reagent 3 and 1.3 equiv of NaCNBH3 in
a 1/1 mixture of BTF and tert-butanol at reflux. This
procedure is analogous to the ~~standard" reaction developed by
Stork for nonfluorous tin hydrides. Stork, G. and Sher, P.
M., J. Am. Chem. Soc., 208, 303 (1986). After approximately
3 hours, the reduction of 1-bromoadamantane was complete.
T
CA 02259183 1998-12-22
WO 98/00376 2 ,~ PCT/US97/11215
After evaporation, the products were isolated by partitioning
between three liquid phases: water removes the inorganic
salts, methylene chloride extracts the adamantane (isolated in
92o yield), and perfluoromethylcyclohexane extracts the tin
products. Analyses by 1H NMR and 19F NMR (estimated detection
limit 1-2o) failed to detect any fluorinated products in the
residue from the methylene chloride phase, and likewise no
adamantane was detected in the fluorous extract. The residue
from the fluorous extract was reused five times to reduce
bromoadamantane by this catalytic procedure with no decrease
in yield. In separate experiments, successful reductions of
1-bromoadamantane were observed with as little as 10 of the
fluorous tin hydride reagent 3. A control experiment showed
that 1-bromoadamantane was not reduced by NaCNBH3 alone under
these conditions over approximately 2Q hours.
Synthetic chemists have long lauded the ienic~ and
radical reactivity profile of tributyltin hydride, but
bemoaned its separation and toxicity problems. The results of
the present studies indicate that fluorous tin hydride
reagent 3 retains the laudable reactivity profile of
tributyltin hydride. However, fluorous tin hydride reagent 3
can be separated from organic products by liquid-liquid
extraction. The ability to use fluorous tin hydride reagent 3
in catalytic amounts and to repeatedly reuse the fluorous
residue indicates that large scale applications of fluorous
tin hydride reagent 3 or a suitable relative are practical
because it is not necessary to synthesize or to dispose of
large quantities of tin. A family of related tin reagents can
provide similar practical benefits for other important
organotin reactions. A review of nonfluorous (standard) tin
reactions is provided in Pereyre, M.; Quintard, J. P. and
Rahm, A., Tin in Organic Synthesis, Butterworths: London;
(1986) .
CA 02259183 1998-12-22
WO 98/00376 2 8 PCT/US97/112I5
Fluorous reagents such as fluorous tin hydride
reagent 3 also have important applications in combinatorial
synthesis. Most current combinatorial synthetic strategies
place the substrate on a polymeric solid phase (P) (see
Figure 2A) so that it can be separated from other compounds in
the reaction mixture by the phase separation technique of
filtration. However, there are a number of synthetic
advantages to combinatorial strategies that place the
substrate in the organic liquid phase, especially for
syntheses of relatively small libraries (for example, tens to
hundreds of compounds). Fluorous reagents provide new options
for these types of syntheses because fluorous reagents and the
substrates (organic soluble) can be separated by the phase
separation technique of extraction. See Figure 2B.
To illustrate the possibilities, a
~~fluorou.s/organic" step was simulated in a homogeneous liquid
phase combinatorial synthesis by conducting a series of
radical additions in parallel. The results are illustrated in
Figure 3. Three halides were crossed with three alkenes, and
reductions were conducted simultaneously in nine individual
vials under the catalytic procedure. The nine products were
~~purified" by three-phase liquid-liquid extraction (conducted
in the original reaction vial) and evaporation. Yields were
then determined by recording NMR spectra in the presence of an
internal standard. The crude products were quite pure (no
significant starting materials or side products as assayed by
capillary GC), and could hypothetically be used directly in
the next step of a sequence. Automation of the extractions
will make more parallel reactions possible.
Combinatorial synthesis with substrates in the
organic liquid phase can already be conducted without
chromatography if all the other reagents are volatile, water
soluble, or on a solid phase. In the case of fluorous
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11215
29
reagents, the possibilities for liquid phase combinatorial
synthesis in a spatially separated mode are greatly expanded.
Like filtration, the phase separation techniques of extraction
and evaporation also allow ready separation of components, so
excesses of reagents can be used. The pairing of organic
substrates with fluorous reagents is expected to be especially
important since a full range of traditional (including
anhydrous) reactions can be conducted under homogenous liquid
phase conditions, yet the products and reagents can still be
separated by extraction. In short, the detractions to
synthesis posed by phase separation can be divorced from its
advantages in purification.
Reactions of organic substrates with fluorous reactants to
provide organic target products.
The features of this synthetic scheme are similar to
those described above, except that a fluorous reactant reacts
with an organic reaction component. The method of the present
invention is illustrated with a combinatorial Stille coupling
in Figure 9B. A standard Stille coupling is illustrated in
Figure 4A.
The standard Stille reaction as illustrated in
Figure 9A is an important member of a family of transition
metal catalyzed cross coupling reactions that is regularly
used in modern organic synthesis, and it has recently been
extended to solid phase combinatorial synthesis. Stille, J.
K., Angew. Chem. Int. Ed. Engl., 25, 508 (1986); Mitchell,
T.N., Synthesis, 803 (1992); Deshpande, M. S., Tetrahedron
Lett., 35, 5613 (1994). The characteristic feature of the
standard Stille reaction is that one of the coupling partners
is a trialkylorganotin compound (see Figure 4A). The alkyl
substituents are almost always methyl or butyl groups. The
Stille reaction is popular because the tin reagents are
CA 02259183 1998-12-22
WO 98/00376 3 ~ PCT/US97/11215
relatively air and moisture stable, can be easily synthesized
and purified, and tolerate a wide variety of both protected
and unprotected functional groups. After the Stille reaction,
the tin becomes a liability: trimethyltin byproducts are easy
to remove but toxic, while tributyltin compounds are less
toxic but difficult to remove.
The present inventors have discovered that compounds
of the general structure ArSn(CH2CH2C6F13)3 participate in
representative Stille couplings to make biaryls and
diarylmethanes, and that all the advantages of the fluorous
synthetic scheme of the present invention are exhibited. The
present studies teach new options for the emerging field of
liquid-phase combinatorial synthesis.
Fluorous phenyl tin reactant 1a served as one of the
reactants for a Stille coupling. Brominolysis of la as
described above provided the tin bromide 2, which served as
the precursor for preparing the p-methoxyphenyl- (lb), 2
furyl- (lc) and 2-pyridyl- (ld) fluorous tin reactants by
standard reactions with either aryllithium or aryl Grignard
reagents.
Stille reactions were conducted under the standard
set of conditions illustrated in Figure 4B. These conditions
were selected based on a number of trial experiments with
fluorous aryl tin reactants la-d. Because Stille reactions
are not generally conducted under biphasic conditions, a
solvent system that substantially solubilized both the organic
substrate and the fluorous tin reactant and that provided
clean transformations within a reasonable time frame was used.
DMF and THF were both useful, but reactions were rather slow
(approximately 2 days). Solvents comprising equal parts of
DMF/THF and solvents comprising equal parts of DMF/C6H5CF3
. ..... . _
CA 02259183 1998-12-22
WO 98/00376 31 PCT/US97/11215
both provided homogeneous liquid phase reactions (as
determined by observation) and reasonable reaction rates
(<22 hours) at 80°C. The DMF/THF mixture (1/1) was selected
for the standard experiments.
A mixture of 1.2 equiv fluorous tin reactant (la-d),
1 equiv halide or triflate (5a-e, 0.2 mmol), 2% PdCl2(PPh3)2.
and 3 equiv of LiCl in 1/1 DMF/THF (1 mL) was heated at 80°C.
Reactions were conducted in individual vessels in groups of
five (one tin reagent with all five partners). After
approximately 22 hours, each mixture was evaporated to remove
some of the solvent and then was partitioned in a three-phase
extraction between water (top), dichloromethane (middle) and
FC-72 (bottom). Evaporation of the FC-72 phase provided
fluorous tin chloride 8 (C6F13CHZCH~) 3SnC1 (80-900) . Most of
the residual 10-20o fluorous tin chloride 8 remained in the
organic phase. If desired, the residual amount can be removed
by washing with FC-72. Recovered fluorous tin chloride 8 was
routinely recycled. Evaporation of the organic phase provided
a crude organic product that was further purified by
preparative TLC to provide major cross-coupled biaryl or
diarylmethane 6 along with small amounts of symmetrical
biaryl 7 (5-100) derived from the tin reactant. The
symmetrical biaryl is a common byproduct in standard Stille
couplings.
Yields for the cross-coupled products are shown in
Figure 4C for fluorous tin reactants la-c. These reactants
gave very clean crude products, and isolated yields of target
product 6 were generally high (>80%), except for a few cases
with the furyl tin reagents where the products are somewhat
volatile. As in the case of the standard Stille coupling of
2-pyridyltributyltin reagent, the five crude products from the
pyridyl tin reagent ld were not very clean, so these reaction
mixtures were not fully purified. See Gronowitz, S. et al.,
CA 02259183 1998-12-22
WO 98/0037b 3 2 PCT/US97/11215
J. of Organometallic Chem., 460, 127 {1993). Significant
amounts of cross-coupled products (estimated 25 to 500) were
produced with p-nitrophenyl triflate and bromide and with
iodobenzene; however, yields of pure products were not
determined.
A preparative reaction was conducted with 0.40 g of
p-bromonitrobenzene (2 mmol) and 2.97 g of phenyltin
reactant 1a (2.4 mmol) in 10 mL of 1/1 DMF/THF at 80°C for
approximately 22 hours. Both reactants were consumed
according to TLC analysis. After azeotropic evaporation with
toluene at 75°C (to remove some of the solvent), a three-phase
extraction was conducted as described above. The methylene
chloride phase was then washed three more times with water and
FC-72 (together) to remove DMF and fluorous products. The
crude organic product was purified by flash chromatography to
provide 337 mg (85o) of 4-nitrobiphenyl and 17 mg (5s) of
biphenyl. The crude fluorous tin chloride (99%) from the FC-
72 phase was reacted with phenyl magnesium bromide to provide
2.85 g (960 overall) of the original tin reactant la after
purification by passing through a short column of neutral
alumina.
The success of the Stille reaction coupled with the
prior radical and ionic reactions of the analogous tin hydride
indicates that rendering other tin reactants fluorous can be a
general strategy to make the vast repertoire of organotin
chemistry more practical and more environmentally friendly.
2. Fluorous Phase Tagging In Synthesis And Separation.
The techniques discussed below rely on the ability
to render an organic molecule fluorous by addition of a
"fluorous phase tag" or "fluorous tag". The fluorous-tagged
___._~_...._ T
CA 02259183 1998-12-22
WO 98/00376 3 3 PCT/US97/11215
organic molecule is separable from organic (non-fluorous)
molecules via an organic/fluorous phase separation technique.
For example, the fluorous-tagged molecule may partition
preferentially into the fluorous liquid phase in a
fluorous/organic liquid-liquid extraction. The techniques
provide simple methods to separate organic molecules based on
the presence or absence of a fluorous tag.
As illustrated in Figure 5, an organic molecule 0
(or a library of organic molecules) bearing nucleophile Nu is
attached to a fluorous phase tag L[(R)d(Rf~e]g through the
intermediacy of a standard organic reaction with an organic
molecule and a fluorous tagging reagent. Figure 5 illustrates
this attachment via a nucleophilic substitution reaction.
This illustration is, however, only for the purposes of
example. Many other types of standard reactions can be used.
In some cases, the fluorous tag itself and the tagging process
can serve as a fluorous analog to existing "standard" (that
is, non-fluorous) protecting groups. Illustrative standard
protecting groups may be found, for example, in Greene, T. W.
and Wuts, P. G. M., Protective Groups in Organic Synthesis,
Wiley-Interscience: New York, (1991). The fluorous tagging
method of the present invention is not limited to this design,
however. For example, in another strategy, the fluorous tag
can be a surrogate for another atom or group in the final
product.
The fluorous phase tag, L[(R)d(Rf)e]g, comprises a
linker L that bridges a fluorous moiety (or moieties) (R)d(Rf)e
(wherein R, Rf, d and a are as described above; and g is an
integer greater than or equal to one) to organic molecule 0.
In some cases, it may be desirable to have more than one tag
per molecule, and these tags may be the same or different. R
can be present or absent as described above. Linker L can
likewise be present or absent, but its presence is often
CA 02259183 1998-12-22
WO 98/00376 3 4 PCT/US97/11215
preferable because it provides a ready means of attachment and
detachment of the fluorous tag. The linker L can be any
standard atom or functional group, either organic or fluorous,
that preferably can be attached and detached (if necessary)
under standard reaction conditions.
Reactions of fluorous substrates with organic reactants to
provide fluorous target products.
This synthetic scheme provides, for example, an
alternative to the now common use of polymers in large
molecule synthesis and combinatorial chemistry. The method of
the present invention has the advantage over such solid phase
techniques of allowing the routine use of standard liquid
phase reagents and reaction conditions.
To begin a synthesis, an organic substrate is
rendered fluorous by attachment to a fluorous group (for
example, a silyl or a stannyl group) that acts as a fluorous
phase tag. A reaction or sequence of reactions is then
conducted in which the products are purified by, for example,
phase separation techniques including liquid-liquid extraction
(for organic or water soluble reagents, reactants,
impurities), filtration (for polymeric or solid reagents,
reactants, impurities) or evaporation (for volatile reagents,
reactants, impurities). At the end of the synthesis, the
target organic product is released from the fluorous tag, and
then separated from all fluorous products by, for example,
organic/fluorous liquid-liquid extraction or filtration, if
the target product is a solid.
As a test reaction for fluorous liquid phase
synthesis, nitrile oxide cycloadditions to fluorous-tagged
derivatives of simple unsaturated alcohols were chosen. These
reactions occur in high yields with terminal alkenes and
_._... . T
CA 02259183 1998-12-22
WO 98/00376 3 5 PCT/LTS97/11215
alkynes and an interesting class of heterocycles is produced.
Nitrile oxide cycloadditions provide a good test reaction
because there are two common ways to produce nitrile oxides
that use different reagents and thus provide differing
purification challenges. These two methods - the Huisgen
method and the Mukaiyama method - are summarized in Figure 6.
See Carmella, P. and Grtinanger, P., 1,3-bipolar Cycloaddition
Chemistry, Wiley-Interscience, New York, Vol. 1, 291 (1984).
If the nitrile oxide precursor is used in excess, both methods
require that the product be separated from the nitrite oxide
dimer (a furoxan). Purification of reaction mixtures from the
more popular Mukaiyama method is especially challenging
because the two reagents (R3CHZN0z and PhNCO), the furoxan
dimer, and obligatory sym-Biphenyl urea (PhNHCONHPh) byproduct
are all organic compounds. Chromatographic procedures for
separation are usually used.
A protocol for the nitrite oxide cycloaddition
reactions is shown in Figure 7, using allyl alcohol as an
example. Five examples of the overall process (without
intermediate chromatography or characterization) as designed
for combinatorial synthesis are described herein. Additional
examples of the overall process with intermediate
chromatography and characterization are included in the
Experimental Examples set forth below. The phase tagging
reagent 9 was designed as a fluorous analog of the popular
trialkylsilyl class of protecting groups that is commonly used
in organic synthesis, and it is readily available on a multi-
gram scale. For phase tagging reagent 9, Br is a leaving
group for the purpose of attachment to an organic molecule.
Linker L is Si; R is CHZCH2; d is l; Rf is C5F13; a is 1; and g
is 3.
Silylation of allyl alcohol was performed by using
excess alcohol 10 (2-4 equiv) in THF under standard
CA 02259183 1998-12-22
WO 98/00376 3 6 PCT/US97/112I5
conditions. Workup and purification included evaporation and
a three-phase liquid extraction with water (top), CHzCl2
(middle), and FC-72 (bottom). The organic phase containing
the unreacted alcohol and the water phase containing the amine
hydrobromide (and probably also some alcohol in the case of
these substrates) were discarded, and the fluorous phase was
concentrated to provide the desired fluorous-tagged silyl
ether 11. At this stage, the strategy allows for ready
separation of unreacted organic substrates 10 from tagged
products 11 as demonstrated by the intentional use of excess
alcohol. In the case of some or all low molecular weight
alcohols, the excess alcohol was removed at the evaporation
stage.
Nitrite oxide cycloadditions were then conducted
under standard Mukaiyama (R3 - Pr and Me) or Huisgen (R3 - tBu
or Ph) conditions. To mimic the need to drive reactions to
completion and to deliberately generate impurities for
separation, all the reagents were used in four- to tenfold
excesses. The Huisgen reactions were conducted in CHZC12, a
solvent in which the fluorous substrates 11 were not
completely soluble (as determined by the naked eye) while the
Mukaiyama reactions were conducted in benzotrifluoride
(trifluoromethylbenzene, C6HSCF3), a solvent in which the
substrates 11 appear to fully dissolve. After the reactions,
three-phase extractions were conducted as described above
except this time the organic extraction solvent was benzene.
The water (middle) and organic (top) phases were again
discarded, and the evaporation of the fluorous phase {bottom)
then provided the cycloadducts 12 substantially free from
organic (and inorganic) impurities.
The fluorous tag was then removed by desilylation of
the products 12 with HF~pyridine in Et20 at room temperature.
After removal of the fluorous tag, the organic phase contained
. __...._
CA 02259183 1998-12-22
WO 98/00376 3 ~ PCT/US97/11215
the desired product in a final three-phase extraction between
aqueous ammonium chloride (top), CHZC12 (middle), and FC-72
(bottom). Evaporation of the CHZCIz phase then provided the
final products 13, which were analyzed for yield and purity
without any additional purification. The whole sequence
proceeded without crystallization or chromatography, and
overall isolated yields of 13a-a for the three step sequence
are shown under each of the final products in Figure 7. The
isolated yields are reasonable (in the case of 13d, material
loss due to evaporation contributes to a lower yield) and the
GC purities are quite good, especially considering that the
deliberate stoichiometry mistakes generated large amounts of
byproducts. Interestingly, the anti/syn ratio obtained in 13e
is virtually identical to the ratio obtained in nitrile oxide
cycloadditions with normal (non-fluorous) trialkylsilyl
ethers. See Houk, K. N., et al., J. Am. Chem. Soc., 106,
3880-82 (1989).
This one-step synthesis (not including attachment
and detachment of the phase tag) clearly illustrates the
potential for single-step and multi-step fluorous-phase
synthesis. The vast majority of the existing reagents and
reactants that are used in organic synthesis as well as the
byproducts that these reagents and reactants produce are
organic or inorganic molecules. Tagging a substrate as
fluorous differentiates (that is, renders relatively easily
separable) the tagged substrate and all its subsequent
products (until the tag is removed) from anything else that is
added to the reaction.
This tagging strategy provides a number of important
advantages over the technique of linking organic molecules to
polymers. First, it is difficult to envision a more robust
tag than a perfluorocarbon segment Rf. Perfluorocarbons are
among the most stable compounds of carbon that are known (See
CA 02259183 1998-12-22
WO 98/00376 3 8 PCT/US97/11215
Hudlicky, supra). Although the linker L and the group R (if
present) will not necessarily be as stable as fluorous
segment Rf, the linker requirements in liquid phase synthesis
are not as stringent as in solid phase synthesis. Indeed, the
fluorous tags of the present invention can often be viewed as
analogous to traditional organic protecting groups. Under
this view, the fluorous tags function as groups that both
protect functionality and serve to alter the phase preference
of the molecule. Thus, tags designed to double as protecting
groups are a valuable asset in a sequence of synthetic steps .
In contrast, the linkers and polymers used in solid phase
synthesis are assets in purification, but are frequently
liabilities in the actual synthetic steps.
As in solid phase synthesis, excess reagents and
reactants can be used to drive the reaction to completion
based on the substrate since the phase of the substrates
is
different from the phase of the reactants and reagents.
Indeed, it is crucial that the substrate be completely
consumed since the substrate has the same phase as the product
and therefore cannot be separated from it by a phase
separation technique. Herein lies the Achille s Heel of all
~~one-phase" techniques: the phases of the substrate,
the
desired product derived from the substrate, and any byproducts
derived from the substrate are the same. Thus, these
compounds cannot normally be separated by a simple phase
separation technique. ~~One phase" techniques do
not address
the larger separation problem in reactions that do not occur
in quantitative or near-quantitative yield based on substrate.
Thus combinatorial synthesis is limited because
general
classes of reactions that occur in quantitative
yield on a
diverse collection of substrates are still the exception
rather than the rule.
_ __
CA 02259183 1998-12-22
WO 98/00376 3 9 PCT/US97/11215
Phase Switching Separations Using Fluorous-Tagged Molecules
The present inventors have discovered that a
solution to this problem is to conduct a selective
"organic/fluorous phase switch". In this process, the phase
of one product (or a subset of products) is temporarily
changed so that it can be separated from other products of the
same phase. After the separation, the phase of the altered
product may be switched back, such that it is returned to its
original phase.
So-called acid/base extractions are an example of a
"phase switch". For example, organic amines can be separated
from other organic compounds by extraction into acid (phase
switches from organic to water) followed by neutralization,
which returns the amines to the organic phase. Unfortunately,
the switch that pKa provides is not sufficiently general. In
that regard, many classes of organic compounds cannot be
extracted into water at any pH where they are stable.
It has been discovered that organic/fluorous phase
switches are a substantially general and powerful method to
purify organic reactions or mixtures of organic compounds.
The trigger for the switch is the selective reaction of an
organic molecule with a fluorous tag. Tagged products are
then separated from non-tagged ones by an organic/fluorous
phase separation technique such as extraction. The fluorous
tag may then be removed to return the purified compound to its
original phase if desired. The method is illustrated in
Figure 8. The fluorous tags for the fluorous phase switch
have the same general features as described above. In this
example, selectivity is based on a target functional group
X " . The wealth of chemoselective transformations known in
organic synthesis forms the basis for designing fluorous phase
switches, which are much more general and useful than
CA 02259183 1998-12-22
WO 98/00376 4 O PCT/L1S97/11215
traditional acid/base switches for simple purification of
organic mixtures.
The technique is illustrated by the sequence shown
in Figure 9. A standard Grignard reaction of an aldehyde with
1.5 equiv of a Grignard reagent is followed by addition of
excess fluorous tag 9. The fluorous ether products 14 are
then separated by three-phase extraction. Treatment of these
crude products with cesium fluoride (CsF) followed by a second
three-phase extraction provides the alcohol products 15 in the
yields and purities indicated in Figure 9. These reactions
demonstrate that the products can be tagged and extracted into
the fluorous layer. The purification features of the
technique are also apparent. For example, when the aldehyde
is used in excess instead of the Grignard reagent, then the
unreacted aldehyde is left in the organic phase after the
first reaction. In the standard procedure where excess
Grignard reagent is used, the residual reagent presumably
reacts with an equivalent amount of the silylating agent to
form a silane RSi (CHZCHZC6F13) 3. This silane is extracted into
the fluorous phase with the desired silyl ether in the first
extraction. However, because it does not react with CsF, it
is left in the fluorous phase during the second extraction
when the alcohol switches back to the organic phase. In
effect, the silyl ether 14 is "temporarily fluorous" but the
silane is "permanently fluorous". There are thus two
different triggers that can be manipulated: one into and one
out of the fluorous phase.
Silylation is only one among many standard reactions
that can be used for fluorous tagging. Others include but are
not limited to acylations of organic alcohols, amines or other
groups with fluorous acyl groups, sulfonyl groups or related
groups (or the reverse), ketalization of organic aldehydes or
ketones with fluorous diols (or the reverse), alkylation of
_.
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11215
41
organic oxygen-, nitrogen-, or carbon-based nucleophiles with
fluorous halides, mesylates, or related alkylating agents (or
the reverse), and many more.
If the design is such that the desired products are
organic and the impurities are fluorous, there is no need to
pull the second trigger to complete the phase switch. This is
shown by the simple radical addition and nitrile oxide
cycloaddition reactions in Figure 10. In both cases, excess
alkene was used; this excess is essential for high yield in
the radical addition but not in this particular nitrite oxide
cycloaddition. After the reaction is complete, sufficient
fluorous tin hydride 3 is added to hydrostannylate all
unreacted alkene, and the reaction is partitioned into organic
and fluorous phases as usual. In both cases, the organic
phase contains the desired product free from unreacted alkene.
It is evident that this type of fluorous tagging strategy can
be extended to remove byproducts from the reaction components
(as opposed to the unreacted reaction components themselves).
The fluorous tags can be used in excess, since they are
fluorous and will be separated from the organic product either
in the first extraction or the second.
The fluorous tags in these types of experiments are
solely for the purposes of purification. However, for many
types of reactions it is possible to "retool" existing
reagents to directly integrate the tagging features. As an
illustration of this, tin azide 16 shown in Figure 11 was
prepared. Tin azide 16 is the fluorous analog of tributyltin
azide, which is a popular reagent for making tetrazoles from
nitrites. See, Akido, K. et al., J. Organometal. Chem., 33,
337 (1971). Reaction of an organic nitrite 17 used in twofold
excess (to simulate incomplete conversion) with fluorous
azide 16 in benzotrifluoride at 80°C for 12 h provided the
fluorous tetrazoles 18 after organic/fluorous extraction
CA 02259183 1998-12-22
WO 98/00376 4 2 PCT/IJS97/11215
(benzene/FC-72) to remove unreacted nitrile and any other
organic byproducts. Brief exposure of the fluorous
tetrazoles 18 to ethereal HC1 followed by fluorous/organic
extraction (acetonitrile/FC-72) and evaporation of the organic
{acetonitrile) phase provided the pure tetrazoles 19 in the
indicated yields. These yields follow the expected trends for
reactions with the reagent tributyltin azide. The above
examples show the power of the fluorous phase switch to
provide pure organic products even in reactions that do not
occur in quantitative yield, and they also show the ability to
remove fluorous reagents and byproducts from organic products.
The ultimate fluorous product from the reaction,
(C6F13CH2CH2) 3SnCl 8, can be readily retrieved from the final
fluorous phase and recycled in high yield.
A more common way to conduct these reactions would
be to use excess tin azide to drive the reaction to completion
based on the nitrite substrate. Indeed, the treatment of one
equivalent of p-tolunitrile with three equivalents on tin
azide 16 under the standard conditions provided pure tolyl
tetrazole 19 (R' - tot) in almost quantitative yield.
Finally, the phase switching method can also be used
to rectify problems in prior steps of a sequence of synthetic
steps. This was illustrated by a simple doping experiment.
Since nitrites are commonly prepared from halides, 1 equiv of
p-tolunitrile was doped with an additional equivalent of 4-
bromotoluene and then the reaction and extraction sequence in
Figure 11 was carried out. The 9-bromotoluene does not react
with the tin azide 16, and it partitions into the organic
layer in the first extraction. In the end, the desired
tetrazole 19 (R' - tot) was isolated in about the same yield
and purity as in the experiment without the bromide.
CA 02259183 1998-12-22
WO 98/00376 4 3 PCT/US97/11215
Fluorous Multicomponent Reactions
Multicomponent reactions combine a substrate and two
or more reactants to provide products in a single step. These
reactions are especially important in combinatorial synthesis
because structurally diverse libraries of products can be
quickly made simply by mixing and matching the various
reaction components. The fluorous techniques outlined herein
are readily adaptable to multicomponent reactions and provide
for simple purification of the products.
The "Ugi four component condensation" is an
important multicomponent reaction because it brings together
four diverse components to form amino acid amides. See, Ugi,
I., Angew. Chem., Int. Ed. Engl. 21, 810 (1982). Examples of
a fluorous variant of the Ugi four component condensation are
shown in Figure 12. Fluorous acid 20 is prepared by a
standard set of reactions as described in the section on
"Experimental Examples". Reaction of fluorous acid 20 with
large excesses (generally 17 equiv) of the other three
components - an amine R8NH2, an aldehyde R9CH0, and an
isonitrile R1°NC (the amine and the aldehyde are known to
condense to form an imine, so for the purposes of
illustration, preformed imines were also successfully used in
a few examples} - followed by two-phase extraction
(benzene/FC-72} and evaporation of the fluorous phase provided
a fluorous Ugi product (not shown). The unreacted or
partially reacted components and byproducts were left in the
organic phase. Without further purification, the fluorous Ugi
product was desilylated with tetrabutylammonium (TBAF).
Extractive purification separates the fluorous tag and any
other fluorous compounds from the organic Ugi product 21,
which is isolated by evaporation of the final organic phase.
Yields and purities of a series of products are listed Figure
12. These yields and purities are impressive, especially
CA 02259183 1998-12-22
WO 98/00376 4 4 PCT/US97/11215
considering that the products 21 derive from reaction of only
4 of the 52 total molar equivalents of reaction components in
the mixture. Thus, the unreacted components and byproducts
derived therefrom constitute by far the major part of the
original crude reaction product.
The Biginelli reaction is another important
multicomponent reaction that has recently. been transferred
from its conventional solution form to the solid phase. See,
Wipf, P.; Cunningham, A. Tetrahedron Lett., 36, 7819 (1995).
Figure 13 illustrates a strategy for conducting Biginelli
reactions by fluorous tagging. This method is compared with
the conventional Biginelli reaction illustrated in Figure 14.
Excess keto-ester and aldehyde (3 equiv) are used to drive the
conventional Biginelli reaction to completion, and the final
products 25 must be purified by chromatography to remove
excess reactants and byproducts.
As shown in Figure 13, the fluorous Biginelli
reaction requires no chromatography. Attachment of the urea
to the acid bromide derived from 22 provides the requisite
fluorous substrate 23, which is purified as usual by
fluorous/organic extraction. The Biginelli reactions were
again conducted with large excesses of keto ester and aldehyde
reactants (10 equiv) both to drive the reactions to completion
and to deliberately generate large amounts of organic reaction
components to separate. The components were heated for 3 d at
50°C in 2/1 THF/BTF containing HCl. The fluorous products 24
were then isolated by three-phase extraction. Desilylation
with tetrabutylammonium fluoride (TBAF) followed by extractive
purification provided the pure organic products 25, which were
identical to the products prepared by the conventional
procedure. Yields for the conventional procedure (yield 1)
and the fluorous procedure (yield 2) are provided in
CA 02259183 1998-12-22
WO 98/00376 4 5 PCT/US97I11215
Figure 14. Product purities were good over a diverse range of
absolute yields without any chromatographies.
These transformations illustrate the power of the
fluorous approach for conducting and purifying multicomponent
reactions. Beyond that, they also illustrate that medium-
sized "drug-like" organic molecules can be synthesized by
fluorous techniques. The final organic products of these
multicomponent reactions typically have molecular weights in
the range of 400-450. Yet the fluorous-tagged precursors of
these products were successively extracted into the fluorous
phase for purification. The acid 20 also provides an example
where the fluorous tag has a role not as a protecting group,
but instead as a surrogate for another atom or group; in this
case, the tag is a surrogate for hydrogen in the final
product.
The methods or strategies of fluorous phase tagging
synthesis and separation set forth above are not mutually
exclusive and can be readily integrated in multi-step
sequences. For example, an organic compound can be rendered
fluorous at an intermediate stage in a synthesis, and then one
or more additional transformations can be conducted on the
fluorous substrate prior to its return to the organic phase.
The fluorous methods of the present invention can also be
smoothly integrated with existing solid phase and liquid phase
methods.
In the future, combinatorial synthesis of small
molecule libraries by multi-step reaction sequences that do
not occur in quantitative yield will place an increased demand
on simple methods for purification of reaction mixtures.
Issues of purification previously considered technical must be
raised to the level of strategic planing on par with issues
like regio- and stereocontrol, protecting groups, and the
CA 02259183 1998-12-22
WO 98/00376 q 6 PCT/US97/11215
like. In this context, the fluorous methods presented herein
provide new strategic options for purification in
combinatorial and other synthesis.
EXPERIMENTAh EXAMPhES
1. Preparation of Iris (3,3,4,4,5,5,6,6,7,7,8,8,8-
Tridecafluorooctyl)phenyltin, (tris (2-Perfluoro-
hexylethyl)phenyltin) (la): To the Grignard reagent prepared
from 2-perfluorohexyl-1-iodoethane (100 g, 211 mmol) and
magnesium (6.53 g, 269 mmol) in dry ether (150 mL) was added
phenyltintrichloride (15.9 g, 52.7 mmol) dissolved in dry
benzene (100 mL). After refluxing for 4 h, the reaction was
stirred for 16 hours at 25°C. The reaction mixture was
hydrolyzed with NHqCl solution, and the organic phase was
washed with 5% Na2S203 solution and deionized water, and then
dried over anhydrous MgSOq. The solvent was evaporated to
dryness. After removal of the major byproduct bis (1,9-
perfluorohexyl)butane by vacuum distillation (87-92°C, 0.2 mm
Hg), the resulting residue was purified by column
chromatography on neutral alumina with hexane to give pure
compound la (56.1 g, 86%) as a colorless oil.
1H NMR (CDC13) d 7. 41 (s, 5 ~I) , 2. 31 (m, 6 H) , 1.31 (t, J =
8.3 Hz, 2J (119Sn-H) - 53.4 Hz, 6 H) ; 119Sn NMR (CDC13) -11.7
ppm; IR (thin film) 3100, 2950, 1238, 1190, 1149, 655 cm-l; MS
(mlz) 1161 (M+ - Ph), 891 (M+ - CH2CH2C6F13).
2. Preparation of Bromo Iris (3,3,4,4,5,5,6,6,7,7,8,8,8-
Tridecafluorooctyl) tin, (Bromo tris (2-
perfluorohexyl)ethyltin) (2): Bromine (5.83 g, 36.5 mmol) in
ether (10 mL) was added dropwise to an ice-cold solution of 1a
. T
CA 02259183 1998-12-22
WO 98/00376 4 7 PCT/US97/11215
(43.0 g, 34.8 mmol) in dry ether (80 mL). The mixture was
warmed to 25°C over 2 h with stirring. Removal of the ether,
bromobenzene, and excess of bromine by evaporation under
reduced pressure resulted in an orange oil. Purification by
vacuum distillation (150-152°C, 0.5 mm Hg) yielded compound 2
(42.4 g, 980) as a colorless oil.
1H NMR (CDC13) d 2.42 (m, 6 H), 1.56 (t, J - 8.3 Hz, 2J
(119Sn-H) - 53.4 Hz, 6 H); 119Sn NMR (hexane) 259.2 ppm (m);
IR (thin film) 3600, 1250, 1227, 1145, 534 cm-1; MS (m/z):
1161 (M+ - Br), 893 (M+ - CH2CH2C6F13)-
3. Preparation of tris (3,3,4,4,5,5,6,6,7,7,8,8,8-
Tridecafluorooctyl)tin Hydride, (tris (2-
Perfluorohexyl)ethyltin Hydride) (3): An ethereal solution of
LiAlH4 (0.8 mL, 0.8 mmol) (1M) was added dropwise to an ice-
cold solution of Iris (2-perfluorohexyl)ethyltin bromide (1.0
g, 0.8 mmol) in ether (20 mL) and the reaction mixture was
stirred for 3 h at 0°C. The reaction mixture was quenched by
slowly adding water (5 mL), followed by 20o sodium potassium
tartrate solution (20 mL). After separation of the ethereal
layer, the aqueous phase was extracted with ether (3 x 25 mL),
and the combined extracts were dried over anhydrous MgS04.
Removal of the ether by distillation yielded a slightly yellow
liquid which was fractionated under reduced pressure. The
fraction, boiling at 145-150°C, 3 mm Hg was collected yielding
910 mg (970) of the hydride 3 as a colorless oil.
1H NMR (CDC13) d 5.27 (s, 1 H), 2.35 (m, 6 H), 1.16 (t, J =
8. 1 Hz, 2J (119Sn-H) - 53.4 Hz, 6 H) ; 119Sn NMR (CDC13) - 84. 5
(1J (119Sn-H) - 1835 Hz); IR (thin film) 1842, 1197 cm-1. MS
(m/z) 1161 (M+ - H), 813 (M+ - CH2CH2C6F13).
CA 02259183 1998-12-22
WO 98/00376 4 8 PCT/US97/11215
4. Representative Stoichiometric Experimental Procedure for
Fluorous Tin Hydride Reductions: To a stirred solution of 1-
bromoadamantane (100 mg, 0.46 mmol) and tris (2-
perfluorohexyl)ethyltin hydride (640 mg, 0.55 mmol) in
benzotrifluoride (9.2 mL) was added a catalytic amount of
AIBN. The reaction mixture was heated at reflux temperature
for 3 h. The solvent was evaporated and the crude residue was
partitioned between dichloromethane (20 mL) and
perfluoromethylcyclohexane (10 mL). The two layers were
separated and the dichloromethane phase was concentrated
yielding adamantane as a pure compound (56 mg, 900).
5. Representative Catalytic Experimental Procedure for
Fluorous Tin Hydride Reductions: A suspension of 1-
bromoadamantane (347 mg, 1.60 mmol), bromo tris 2-
(perfluorohexyl)ethyltin (200 mg, 0.16 mmol), sodium
cyanoborohydride (138 mg, 2.1 mmol) and AIBN (in catalytic
amount) in benzotrifluoride (1.6 mL) and tent-butanol (1.6
mL) was heated in a sealed tube at reflux during 3 h. The
solvent was evaporated and the crude residue was partitioned
between water {10 mL), dichloromethane (15 mL) and
perfluoromethylcyclohexane (10 mL). The three layers were
separated and the dichloromethane phase was dried over MgS04
yielding, after evaporation, adamantane as a pure compound
(200 mg, 92 0 ) .
6. Representative Combinatorial Chemistry Experimental
Procedure for Fluorous Tin Hydride Reductive Additions: In a
typical experiment, a suspension of alkyl iodide (0.1 mmol),
olefin {0.5 mmol), bromo Iris 2-(perfluorohexyl)ethyltin
(12.4 mg, 0.01 mmol), sodium cyanoborohydride (9.6 mg, 0.13
mmol) and AIBN (in catalytic amount) in BTF (0.5 mL) and tert-
butanol (0.5 mL) was heated at reflux in a sealed vial for 12
h.
T
CA 02259183 1998-12-22
WO 98/00376 4 9 PCT/US97/11215
To the cooled reaction mixture, PFMC (2 mL) and
dichloromethane (1 mL) were added. After separation of the 2
phases, the dichloromethane phase was extracted another time
with PFMC (1 mL) and then with water (1 mL). The organic phase
was filtered through neutral alumina and evaporated to
dryness. The yields of this reactions were determined by 1H
NMR using CH2C12 and hexamethyldisiloxane as internal
standards (See Figure 3).
7. Representative Experimental Procedure for Fluorous Tin
Hydride Reductive Cyclizations: A suspension of hexenyl
bromide (0.32 mmol), bromo Iris (2-perfluorohexyl)ethyltin (40
mg, 0.032 mmol), sodium cyanoborohydride (28 mg, 0.42 mmol)
and AIBN (in catalytic amount) in BTF (3.2 mL) and tert-
butanol (3.2 mL} was heated at reflux in a sealed tube. The
progress of the reaction was monitored by TLC. The solvent was
evaporated and the crude residue was partitioned between water
{8 mL}, dichloromethane (15 mL) and FC-72 (12 mL). The three
layers were separated and the dichloromethane phase (middle
layer) was extracted twice with FC-72 (2 x 10 mL} , dried over
MgS04 yielding, after evaporation, the cyclopentane
derivative. Starting from 6-bromo-1,1-diphenylhexene and 7-
bromohept-2-enenitrile, diphenylmethylcyclopentane and
cyclopentaneacetonitrile were isolated in 75 arid 66~ yield
respectively.
8. Representative Procedure for Fluorous Tin Hydride Ionic
Reductions of Aldehydes: A solution of aldehyde {0.144 mmol),
zinc chloride (393 mg, 2.88 mmol), tris (2-
perfluorohexyl)ethyltin hydride (104 mg, 0.09 mmol) in ether
(2.9 mL) was heated at reflux in a sealed tube. The progress
of the reaction was monitored by TLC. The solvent was
evaporated and to the crude residue was added water (2 mL),
dichloromethane (5 mL) and PFMC (4 mL). The three resulting
layers were separated and the dichloromethane layer (middle
CA 02259183 1998-12-22
WO 98/00376 5 0 PCT/US97/11215
layer) was extracted twice with PFMC (2 x 5 mL), dried over
MgS04, filtered through silica and evaporated under reduced
pressure to yield the pure alcohol. In that way, benzyl
alcohol, p-nitrobenzyl alcohol and 3-phenyl-1-propanol were
obtained from benzaldehyde, p-nitrobenzaldehyde and 3-
phenylpropanaldehyde in 78, 64 and 68o yield respectively.
9. Preparation of tris (3,3,4,4,5,5,6,6,7,7,8,8,8-
Tridecafluorooctyl)(4'-methoxyphenyl)tin, (tris (2-
Perfluorohexylethyl)(4~-methoxyphenyl)tin) (lb): To the
Grignard reagent prepared from 4-bromoanisole (681 mg, 3.64
mmol) and magnesium (102 mg, 9.20 mmol) in dry ether (20 mL)
was added a solution of 2 (3.97 g, 2.80 mmol) in dry ether (10
mL). After refluxing for 1 h, the reaction was stirred for 16
h at 25°C. The reaction mixture was quenched with NH4C1
solution and diluted with ether, and the organic phase was
washed with deionized water then dried over anhydrous MgS04.
The solvent was evaporated to dryness. Purification by vacuum
distillation (166°C, 0.25 mm Hg) and then column
chromatography on neutral alumina with hexane yielded pure
compound lb (5.20 g, 740) as a colorless oil.
1H NMR (CDC13) d 7. 30 (d, J = 8. 3 Hz, 2 H) , 6. 98 (d, J = 8.3
Hz, 2 H), 3.82 (s, 3 H), 2.29 (m, 6 H), 1.27 (t, J = 8.3 Hz,
2J (119Sn-H) - 54.0 Hz, 6 H); 119Sn NMR (CDC13) 123.7 ppm; IR
(thin film) 1500, 1375, 1240, 1205, 1145, 1065, 745, 700 cm-1;
MS (m/z) 1267 (M+), 1161 {M+ - C6H40Me), 921 (M+ -
CH2CH2C6F13).
10. Preparation of tris (3,3,4,4,5,5,6,6,7,7,8,8,8-
Tridecafluorooctyl) (2 ' -furyl) tin, ( tris (2-
Perfluorohexylethyl) (2' -furyl) tin) (lc) : To a solution of
furan (667 mg, 9.80 mmol) in dry THF (25 mL) at 0°C was added
~. _._.~_.__
CA 02259183 1998-12-22
WO 9$/00376 51 PCT/US97/11215
a 1.5 M solution of LDA in cyclohexane (6.53 mL, 9.80 mmol}.
After stirring 1 h at 0°C, the resulting mixture was treated
with a solution of 2 (8.67 g, 7.00 mmol) in dry THF (15 mL).
The reaction mixture was warmed to 25°C over 1 h and then
stirred for 16 h at 25°C. The reaction mixture was quenched
with NH4C1 solution and diluted with ether. After separation,
the organic phase was washed with deionized water and then
dried over anhydrous MgS04. The solvent was evaporated to
dryness. Column chromatography on neutral alumina with hexane
yielded pure compound lc (2.44 g, 280) as a colorless oil.
1H NMR (CDC13) d 7.76 (s, 1 H), 6.63 (s, 1 H), 6.47 (s, 1 H),
2.35 (m, 6 H), 1.29 (t, J = 9.7 Hz, 2~7 (119Sn-H) - 56.8 Hz, 6
H}~ 119Sn NMR (CDC13) 100.7 ppm; IR (thin film} 1445, 1355,
1240, 1205, 1145, 1065, 745, 700 cm-l; MS (m/z} 1228 (M+),
1161 (M+ - furyl), 881 (M+ - CH2CH2C6F13).
11. Preparation of tris (3,3,4,4,5,5,6,6,7,7,8,8,8-
Tridecafluorooctyl)(2'-pyridyl)tin, (tris (2-
Perfluorohexylethyl) (2'-pyridyl)tin) (ld): To the Grignard
reagent prepared from 2-bromopyridine (822 mg, 5.20 mmol) and
magnesium (146 mg, 6.20 mmol) in dry ether (30 mL) was added a
solution of 2 (2.48 g, 2.00 mmol} in dry ether (5 mL). After
refluxing for 1 min., the reaction was stirred for 17 hours at
25°C. The reaction mixture was quenched with NH4C1 solution.
After separation, diluted with ether, and the organic phase
was washed with deionized water and then dried over anhydrous
MgS04. The solvent was evaporated to dryness, and the
resulting residue was partitioned between toluene and FC-72.
The two phases were separated. The FC-72 phase was washed with
toluene and concentrated to afford pure compound ld (2.18 g,
880) as a pale yellow oil.
CA 02259183 1998-12-22
WO 98/00376 5 2 PCT/US97/11215
1H NMR (CDC13) d 8.71 (d, J = 4 . 3 Hz, 1 H) , 7. (t, =
58 J 7.
7
Hz,1 H), 7.36 (d, J 7.3 Hz, 1 H), 8.20 (m, 1 H), 2.29 (m,
= 6
H) 1.34 (t, J = 8.2 Hz, 2J (119Sn-H) - 59.3 Hz, 6 H) 119Sn
, ;
NMR(CDC13) 88.6 ppm; IR (thin film) 1570, 1450, 1360, 1240,
1205, 1145, 1060, 735, 700 cm-1; MS (m/z) 1238 (M+), 1161 (M+
- pyridyl), 892 (M+ - CH2CH2C6F13).
12. General Procedure for the Stille Couplings: A sealed tube
under nitrogen was charged with tin reactant (0.24 mmol),
substrate (0.20 mmol), lithium chloride (25.4 mg, 0.60 mmol),
dichlorobis(triphenylphosphine)palladium(II) (2.8 mg, 0.004
mmol), dry DMF (0.5 mL), and dry THF (0.5 mL). The mixture was
heated at 80°C for 22 h. The solvent was evaporated and the
residue was partitioned between water (10 mL), dichloromethane
(15 mL), and FC-72 (10 mL). The three phases were separated
and the dichloromethane phase was dried over anhydrous MgS04.
Evaporation of the FC-72 phase provided chloro Iris
(3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)tin, (chloro
tris (2-perfluorohexyl)ethyltin) 8, which was routinely
recycled. Evaporation of the dichloromethane phase provided
crude organic product, which was further purified by silica
gel preparative TLC to provide the major cross-coupled product
6 (see yields in Figure 9C) and a small amount (5 - 100) of
the symmetrical biaryl 7 derived from the tin reactant.
13. Chloro tris (3,3,4,4,5,5,6,6,7,7,8,8,8-
Tridecafluorooctyl) Tin 8, (Chloro tris (2-
Perfluorohexyl)ethyltin 8): 1H NMR (CDC13) d 2.46 (m, 6 H),
1.53 (t, J = 7. 9 Hz, 2J (119Sn-H) - 47. 6 Hz, 6 H) ; 11'~Sn NMR
(CDC13) 273 ppm; IR (thin film) 1450, 1360, 1240, 1205, 1145,
1065, 735, 700 cm-1; MS (m/z): 1161 (M+ - C1), 899 (M+ -
CH2CH2C6F13)-
CA 02259183 1998-12-22
WO 98/00376 5 3 PCT/US97/i 1215
14. Representative Example of a Preparative Stille Coupling:
A sealed tube under nitrogen was charged with tin reactant la
(2.97 g, 2.40 mmol), 1-bromo-4-nitrobenzene (904 mg, 2.00
mmol), lithium chloride (259 mg, 6.00 mmol),
dichlorobis(triphenylphosphine)palladium(II) (28.1 mg, 0.04
mmol), dry DMF (5 mL), and dry THF (5 mL). The mixture was
heated to 80°C and a homogeneous solution resulted. The
mixture was stirred at 80°C for 22 h. After azeotropic
evaporation with toluene at 75°C (to remove THF and some of
the DMF), the resulting residue was partitioned between water
(40 mL), dichloromethane (60 mL), and FC-72 (40 mL). The
three phases were separated. Evaporation of the FC-72 phase
provided 2.31 g (80.60 from la) of tin chloride 8 as a
colorless oil. The dichloromethane phase was washed three more
times with water (90 mL) and FC-72 (40 mL). Evaporation of
the combined FC-72 phases (including the first phase) provided
2.85 g (99.40 from la) of tin chloride 8. The final
dichloromethane phase was dried over anhydrous MgS04 and
evaporated to give yellow crystals free of fluorous reactant
la and fluorous tin halides. The crude organic product was
further purified by column chromatography on silica gel to
provide the cross-coupled product, 9-nitrobiphenyl (337 mg,
85%) as yellow crystals, and the homo-coupled product,
biphenyl (17 mg, 50), as white crystals.
15. Representative Example of Recycling of Tin Reactants: The
tin chloride 8 (2.85 g) isolated by evaporation of FC-72 phase
after the above Stille coupling was treated with a 3M solution
of phenyl magnesium bromide in ether (1.04 mL, 3.12 mmol ) in
dry ether (25 mL) under stirring at 25°C for 6 h. The reaction
mixture was hydrolyzed with NH4C1 solution and diluted with
ether, and the organic phase was washed with deionized water
then dried over anhydrous MgS04. The solvent was evaporated to
dryness. Column chromatography on neutral alumina with hexane
CA 02259183 1998-12-22
WO 98/00376 5 4 PCT/US97/11215
yielded pure compound la (2.85 g, 96% overall from la in the
preceding section) as a colorless oil.
16. Bis [iris (3,3,4,4,5,5,6,6,7,7,8,8,8-
Tridecafluorooctyl)tin Oxide, (Bis [iris (2-
Perfluorohexyl)ethyltin]oxide [(C6F13CH2CH2)3Sn]20; an
Alternative Procedure for the Preparation of Tin Hydride 3:
Sodium hydroxide (254 mg, 6.33 mmol) in 8.4 mL of water was
added to a solution of tris (2-perfluorohexyl)ethyltin bromide
(5.23g, 4.22 mmol) in acetone (55 mL). The mixture was heated
at reflux for 12 h. The solvents mixture was evaporated. To
the residue was added IO ml of anhydrous toluene, and the
resulting solution heated in a reflux apparatus equipped with
a Dean-Stark type water trap for 12 h. The toluene solution
was evaporated and the residue was dried over P205 in a vacuum
desiccator for 12 h. The residue was extracted with dried
hexane. The organic fraction collected was concentrated
yielding the bis [iris (2-perfluorohexyl)ethyltin] oxide (3g,
610) as a viscous yellow oil.
1H NMR (CDC13) d 2.45 (m, 12H); 1.55 (t, J - 8.3 Hz, 2J
(119Sn-H) - 53.4 Hz, 12 H); 119Sn NMR (CDC13} 165.59 ppm.
A mixture of bis [iris (2-perfluorohexyl)ethyltin]
oxide (3 g, 1.28 mmol) and polymethylhydrosiloxane (191 mL;
3.22 mmol) was stirred at 25 °C for 12 h. After addition of
ether, the presence of the tin hydride was shown by TLC
comparison with an authentic sample.
17. Preparation of Iris (3,3,4,4,5,5,6,6,7,7,8,8,8-
Tridecafluorooctyl)allyltin, (iris (2-
perfluorohexylethyl)allyltin), [(C6F13CHZCH2)3SnCH2CHCH2]:
Allylmagnesium bromide (0.10 ml, 0.10 mmol) 1M in ether was
CA 02259183 1998-12-22
WO 98/00376 5 5 PCT1US97/11215
added to a solution of 2 ( 100 mg, 0 . 08 mmol ) in ether ( 4 mL) .
The mixture was heated at reflux for 2 h with stirring. To the
reaction cooled to 0 °C, and a saturated solution of aqueous
ammonium chloride (3 ml) and ether (5 ml) were added. The two
phases were separated and the aqueous phase was extracted
twice with ether (2 x 10 ml) . The ether phase was dried over
MgS04 yielding, after evaporation, the allyl derivative (62
mg, 640) as a white oil.
1H NMR (CDC13) d 5.95 (m, 1H); 5.0 - 4.8 (m, 2H); 2.30 (m,
6H) ; 1. 95 (d, 2H, J--- 9 Hz) ; 1.20 (t, J= 8. 3 Hz, 6H) .
18. Bromo tris(3,3,4,4,5,5,6,6,7,7,8,8,8-
tridecafluorooctyl)silane (Bromo tris(2-
(perfluorohexyl)ethyl]silane) 9: Tris(3,3,4,4,5,5,6,6,7,7,
8,8,8-tridecafluorooctyl)silane (1.00 g, 0.94 mmol; prepared
as described in J. Fluorine Chem. 1993, 60, 211) was dissolved
in FC-72 (9 mL) at 25 °C under argon. Bromine (0.08 mL, 1.91
mmol) was slowly added and the resulting solution was stirred
for 8 h at 25 °C. The reaction mixture was washed twice with
CH2C12. Evaporation of the fluorous layer yielded the
bromosilane 9 as a colorless oil (1.08 g, 990): IR (neat)
2952, 1443, 1362, 1317, 1240, 1209, 1145, 1122, 1073, 905,
812, 737, 707 cm-1; 1H NMR (300 MHz, CDC13) d 1.24-1.30 (m, 6
H), 2.11-2.28 (m, 6 H); lyF NMR (970MHz, CDC13) d (rel. to
FCC13) -126.82, -123.88, -123.53, -122.55, -116.32, -81.43.
19. Allyloxy-Iris [2- (perfluorohexyl) ethyl] silane (11) : A11y1
alcohol 10 (0.06 mL, 0.91 mmol) and triethylamine (0.13 mL,
0.91 mmol) were dissolved in dry THF (2 mL) under argon. A
mixture of bromo Iris[2-(perfluorohexyl)ethyl]silane 9 (260
mg, 0.23 mmol) in THF (2 mL) was slowly added to the above
mixture at 25 °C. The resulting mixture was stirred at 25 °C
for 3 h. After removal of the solvent, the residue was
CA 02259183 1998-12-22
WO 98/00376 5 6 PCT/US97/11215
purified by 3-phase extraction with FC-72 (10 mL), CH2C12 {10
mL), and H20 (10 mL). The organic-aqueous biphase was
additionally extracted twice with FC-72 (10 mL). After
evaporation of the combined fluorous extracts, the residue was
further purified by flash-chromatography (hexanes-Et20, 50:1),
yielding allyloxy-tris[2-(perfluorohexyl)ethyl]silane 11 as a
colorless oil (115 mg, 450): IR (neat) 2949, 2910, 1362,
1317, 1234, 1211, 1196, 1149, 1121, 1075, 1040, 904, 845, 812,
736, 707 cm-1; 1H NMR (300 MHz, CDC13) d 0. 93-0.99 (m, 6 H) ,
2.03-2.20 (m, 6 H) , 4. 21 (d, J = 5. 0 Hz, 2 H) , 5. 18 (dd, Jl -
10. 4 Hz, J2 = 1. 5 Hz, 1 H) , 5.26 (dd, JI - 17. 1 Hz, J2 - 1. 5
Hz, 1 H), 5.84-5.97 (m, 1 H); 13C NMR (75 MHz, CD3COCD3) d
3.44, 25.56 (t, J = 23.3 Hz), 64.93, 108.21-123.06 {m, CF2,
CF3), 115.93, 137.85; 19F NMR (470 MHz, CDC13) d (rel. to
CFC13) -126.89 (t, J = 4.7 Hz), -129.06, -123.60, -122.64, -
116.96 (t, J = 18.8 Hz), -81.57 (t, J = 9.4 Hz); MS m/z 1126
(M+) , 451, 349, 309, 239, 195.
20. tris[2-(Perfluorohexyl)ethyl](2-methylallyloxy)silaae:
2-Methyl-2-propen-1-of (0.97 mL, 5.59 mmol) and triethylamine
(0.79 mL, 5.59 mmoI) were dissolved in dry THF (10 mL) under
argon. A mixture of bromo tris[2-(perfluorohexyl)ethyl]silane
9 (1.60 g, 1.39 mmol) in THF (5 mL) was slowly added to the
above solution at 25 °C. The resulting mixture was stirred at
°C for 2 h. After removal of the solvent, the residue was
25 purified by 3-phase extraction with FC-72 (20 mL) , CH2C12 (20
mL), and H20 (20 mL). The organic-aqueous biphase was
additionally extracted twice with FC-72 (20 mL). After
evaporation of the combined fluorous extracts, the residue was
further purified by flash-chromatography (hexanes-Et20, 50:1),
yielding tris[2-(perfluorohexyl)ethyl]-(2-methyl-
allyloxy)silane as a colorless oil (602 mg, 96%): IR (neat)
2949, 2916, 1362, 1317, 1237, 1206, 1195, 1121, 1074, 905,
736, 707 cm-1; 1H NMR (300 MHz, CDC13) d 0.99-0.99 (m, 6 H),
..._ ._.._ _ . . T
CA 02259183 1998-12-22
WO 98/00376 5 7 PCT/US97/11215
1.72 (s, 3 H), 2.03-2.21 (m, 6 H), 4.08 (s, 3 H), 4.89 (s, 1
H), 9.94 (s, 1 H); 13C NMR (75 MHz, CD3COCD3) d 3.48, 18.96,
25.68 (t, J - 22.5 Hz), 67.65, 105.87-129.09 (m, CF2, CF3),
111.91, 145.11; 19F NMR (470 MHz, CDC13) d (rel. to CFC13) -
- 5 126.95 (q, J = 9.7 Hz), -124.13, -123.64, -122.68, -117.01 (t,
J = 14. 1 Hz) , -81. 64 (t, J = 9. 4 Hz) ; MS m/z 1190 (M+) , 239,
137.
21. tris[2-(Perfluorohexyl)ethyl](prop-2-ynyloxy)silane:
Propargyl alcohol (0.10 mL, 1.79 mmol) and triethylamine (0.26
mL, 1.74 mmol) were dissolved in dry THF (10 mL) under argon.
A mixture of bromo tris[2-(perfluorohexyl)ethyl]silane 9 (1.00
g, 0.87 mmol) in THF (2 mL) was slowly added to the above
solution at 25 °C. The resulting suspension was stirred at 25
°C for 3 h. After removal of the solvent, the residue was
purified by 3-phase extraction with FC-72 (15 mL), CH2C12 (15
mL), and H20 (15 mL). The organic-aqueous biphase was
additionally extracted twice with FC-72 (15 mL). Evaporation
of the combined fluorous extracts yielded a mixture of Iris[2-
(perfluorohexyl)ethyl](prop-2-ynyloxy)silane and silanol in a
ratio of 87 to 13 as a colorless oil (960 mg, 98 0): IR
(neat) 3317, 2950, 2911, 1443, 1362, 1317, 1249, 1234, 1221,
1198, 1144, 1120, 1076, 905, 812, 736, 708 cm-1; 1H NMR (300
MHz, CDC13) d 0.98-1.09 (m, 6 H), 2.07-2.29 (m, 6 H), 2.50 (t,
J = 2. 5 Hz, 1 H) , 4. 38 (d, J = 2. 6, 2 H) ; 13C NMR (75 MHz,
CD3COCD3) d 3.72, 25.71 (t, J = 23.3 Hz), 52.50, 75.85, 82.46,
105.94-124.18 (m, CF2, CF3); MS m/z 778 ((M+ + 1) -
CH2CH2 (CF2) 5CF3) , 374, 293, 226, 184.
22. General Procedure for the Preparation of Isoxazol(in)es
by Mukaiyama's Method (General Procedure 1): To a solution of
the silyl ether (0.10 mmol) in BTF (4 mL) were added the nitro
alkane (0.99 mmol), phenyl isocyanate (0.22 mL, 1.98 mmol),
and two drops of triethylamine. The reaction mixture was
CA 02259183 1998-12-22
WO 98/00376 5 8 PCT/iJS97/11215
stirred at 25 °C for 3 days. After removal of the solvent,
the residue was purified by 3-phase extraction with FC-72 (20
mL), H20 (20 mL), and benzene (20 mL). The organic-aqueous
biphase was additionally extracted twice with FC-72 (20 mL).
The combined fluorous extracts were evaporated to yield the
desired isoxazol(in)e. (For small scale reactions the crude
reaction mixture was diluted with benzene and extracted three
times with FC-72. The combined fluorous extracts were
filtered and evaporated.)
23. General Procedure for the Preparation of Isoxazol(in)es
by Huisgen's Method (General Procedure 2): The silyl ether
(0.09 mmol) and the oxime (0.36 mmol) were placed at 25 °C in
CH2C12 (6 mL), triethylamine (0.36 mmol) was added and the
reaction mixture was stirred at 25 °C for 24 h. After removal
of the solvent, the residue was purified by three-phase
extraction with FC-72 (15 mL), H20 (15 mL), and benzene (15
mL). The organic-aqueous biphase was additionally extracted
twice with FC-72 (15 mL). The combined fluorous extracts were
evaporated to yield the desired isoxazol(in)e. (For small
scale reactions the crude reaction mixture was suspended in
benzene and extracted three times with FC-72.)
24 . 3-Methyl-5-Iris [2- (perfluorohexyl) ethyl] silanyl-
oxymethyl-4,5-dihydroisoxazole: Prepared according to general
procedure 1 with allyl silyl ether 11 (0.050 g, 0.044 mmol),
BTF (2 mL), nitroethane (0.03 mL, 0.44 mmol), and phenyl
isocyanate (0.10 mL, 0.88 mmol) to afford the isoxazoline (53
mg, 990): IR (neat) 2947, 2932, 1441, 1361, 1352, 1317, 1236,
1206, 1149, 1122, 1071, 1022, 905, 845, 811, 737, 707 cm-1; 1H
NMR (300 MHz, CDC13) d 0.91-1.00 (m, 6 H), 1.96 (s, 3 H),
2.00-2.22 (m, 6 H), 2.75 (dd, J1 = 17.1 Hz, J2 = 7.3 Hz, 1 H),
2.98 (dd, JI - 17.0 Hz, J2 - 10.8 Hz, 1 H), 3.68 (dd, J1 -
11.4 Hz, J2 = 9.5 Hz, 1 H), 3.80 (dd, J1 - 11.4 Hz, J2 - 3.1
_,_..._...~~_ T..
CA 02259183 1998-12-22
WO 98/00376 5 9 PCT/US97/11215
Hz, 1 H) , 9.57-4.66 (m, 1 H) ; 13C NMR (750 MHz, CDC13) d 2. 94,
12.74, 29.68 (t, J = 23.3 Hz), 39.85, 64.73, 79.51, 104.61-
121.39 (m, CF2, CF3) , 155.11; 19F NMR (970 MHz, CDC13) d (rel.
to CFC13) -126.74 (t, J = 4.7 Hz), -123.86, -123.46, -122.51,
-116.72 (t, J = 14.1 Hz), -81.37 (t, J = 9.4 Hz); MS m/z 1164
(M+ - F), 936, 836, 508, 309, 239, 195; HRMS calcd. for
C21H16N~2F26Si (M+ - (CH2) 2 (CF2) 5CF3) m/z 836.0535, found
836.0514.
25. 3-Phenyl-5-tris[2-(perfluorohexyl)ethyl]silanyl-
oxymethyl-4,5-dihydroisoxazole: Prepared according to
general procedure 2 with the allyl silyl ether 11 (0.069
g,
0.061 mmol), phenyl hydroximic acid chloride (38.0 mg, 0.25
mmol), and triethylamine (0.037 mL, 0.25 mmol) in CH2C12
(4
mL) to afford the isoxazoline (73 mg, 960) : IR (neat) 2945,
2912, 1942, 1359, 1316, 1240, 1205, 1144, 1122, 10 72, 1018,
904, 745, 737, 707 cm-l; 1H NMR (300 MHz, CDC13) d 0.91-1.00
(m, 6 H), 2.01-2.19 (m, 6 H), 3.20 (dd, J1 - 16.6 Hz, J2 = 8.1
Hz, 1 H), 3.90 (dd, Jl - 16.6 Hz, J2 - 10.9 Hz, 1 H), 3.78
(dd, JI - 11.5 Hz, J2 = 4. 4 Hz, 1 H) , 3. 91 (dd, Jl - 11. 5
Hz,
J2 - 2.9 Hz, 1 H), 4.79-4.87 (m, 1 H), 7.35-7.40 (m, 3
H),
7.63-7.66 (m, 2 H) ; 13C NMR (75 MHz, CDC13) d 2. 95, 24.66
(t,
J - 23.3 Hz), 36.13, 64.85, 80.47, 109.61-119.13 (m, CF2,
CF3), 126.55, 128.79, 129.06, 130.28, 156.51; 19F NMR (470
MHz, CDC13) d (rel. to CFC13) -126.72 (t, J - 9. 7 Hz),
-
123.86, -123.45, -122.52, -116.72 (t, J = 14.1 Hz), - 81.34
(t,
J - 9.4 Hz); MS m/z 1295 (M+), 1226 (M+ - F), 898 (M+ -
(CH2) 2 (CF2) 5CF3) , 378, 309, 239, 195.
26. 3-tert-Butyl-5-tris[2-(perfluorohexyl)ethyl]silanyl-
oxymethyl-4,5-dihydroisoxazole: Prepared according to general
procedure 2 with the allyl silyl ether 11 (0.054 g, 0.048
CA 02259183 1998-12-22
WO 98/00376 6 0 PCT/US97/11215
mmol), tert-butyl hydroximic acid chloride (46.0 mg, 0.34
mmol), and triethylamine (0.05 mL, 0.34 mmol) in CH2C12 (4 mL)
to afford the isoxazoline (59 mg, 99%): IR (neat) 2363, 1442,
1363, 1317, 1240, 1199, 1194, 1121, 1072, 905, 811, 749, 737,
707 cm-l; 1H NMR (300 MHz, CDC13) d 0.91-1.00 (m, 6 H), 1.10
(s, 9 H), 2.04-2.22 (m, 6 H), 2.78 (dd, Jl = 16.9 Hz, J2 = 7.2
Hz, 1 H), 3.00 (dd, JI - 16.9 Hz, J2 - 10.6 Hz, 1 H), 3.66
(dd, Jl - 11. 4 Hz, J2 = 4 . 9 Hz, 1 H) , 3. 76 (dd, Jl - 11.2 Hz,
J2 = 3.3 Hz, 1 H), 4.57-4.65 (m, 1 H); 13C NMR (75 MHz, CDC13)
d 2.78, 24.61 (t, J = 23.3 Hz), 27.71, 32.86, 35.42, 64.59,
79.47, 104.54-122.85 (m, CF2, CF3), 165.81; 19F NMR (470 MHz,
CDC13) d (rel. to CFC13) -126.78 (t, J = 4.7 Hz), -123.87, -
123.50, -122.54, -116.72 (t, J = 14.1 Hz), -81.41 (t, J = 14.1
Hz) ; MS m/z 1206 (M+ - F) , 309, 239, 195, 126.
27. 3-Propyl-5-tris[2-(perfluorohexyl)ethyl]silanyl-
oxymethyl-4,5-dihydroisoxazole: Prepared according to general
procedure 1 with the allyl silyl ether 11 (0.111 g, 0.099
mmol), BTF (4 mL), nitrobutane (0.10 mL, 0.99 mmol), and
phenyl isocyanate (0.22 mL, 1.98 mmol) to afford the
isoxazoline (125 mg, 990): IR (neat) 2971, 2994, 2914, 2881,
1362, 1317, 1239, 1207, 1144, 1121, 1071, 906, 745, 736, 707
cm-l; 1H NMR (300 MHz, CDC13) d 0.89-1.00 (m, 9 H), 1.51-1.63
(m, 2 H) , 2. 03-2.21 (m, 6 H) , 2.29 (t, J = 7 . 4 Hz, 2 H) , 2. 73
(dd, J1 - 17. 0 Hz, J2 = 7. 3 Hz, 1 H) , 2. 96 (dd, J1 - 17. 0 Hz,
J2 = 10.8 Hz, 1 H), 3.67 (dd, JI = 11.4 Hz, J2 = 4.7 Hz, 1 H),
3.78 (dd, JI = 11.3 Hz, J2 = 3.1 Hz, 1 H), 4.57-4.65 (m, 1 H);
13C NMR (75 MHz, CD3COCD3) d 3.48, 13.98, 20.44, 25.47 (t, J =
23.3 Hz), 38.85, 65.81, 80.38, 109.32-123.54 (m, CF2, CF3),
159.19; MS m/z 1211 (M+), 1192 (M+ - F), 848, 803, 293, 226,
157.
____... . ... ._ _ _ T
CA 02259183 1998-12-22
WO 98/00376 61 PCT/US97/11215
28. 3,5-Dimethyl-5-tris[2-(perfluorohexyl)ethyl]silanyl-
oxymethyl-4,5-dihydroisoxazole: Prepared according to general
procedure 1 with the allyl silyl ether (0.107 g, 0.094 mmol),
BTF (4 mL), nitroethane (0.07 mL, 0.94 mmol), and phenyl
isocyanate (0.20 mL, 1.88 mmol) to afford the isoxazoline (112
mg, 99%) : IR (neat) 2935, 1442, 1362, 1352, 1336, 1316, 1238,
1206, 1144, 1120, 1072, 906, 811, 745, 736, 707 cm-1; 1H NMR
(300 MHz, CDC13) d 0. 93-0. 99 (m, 6 H) , 1. 30 (s, 3 H) , 1. 91 (s,
3 H), 2.02-2.19 (m, 6 H), 2.58 (d, J = 17.1 Hz, 1 H), 2.91 (d,
J = 17.1 Hz, 1 H), 3.57 (d, J = 11.0 Hz, 1 H), 3.63 (d, J =
10.9 Hz, 1 H); 13C NMR (75 MHz, CDC13) d 2.87, 12.92, 22.26,
24 . 68 (t, J = 24.0 Hz) , 45. 68, 68. 06, 85.35, 105. 16-122. 96 (m,
CF2, CF3), 155.05; MS mlz 1178 (M+ - F), 928, 850 (M+ -
(CH2) 2 (CF2) 5CF3) , 795, 309, 239.
29. 3-tert-Butyl-5-methyl-5-Iris[2-(perfluorohexyl)ethyl]-
silanyloxymethyl-4,5-dihydroisoxazole: Prepared according to
general procedure 2 with the allyl silyl ether (0.104 g, 0.091
mmol), tert-butyl hydroximic acid chloride (50.0 mg, 0.36
mmol), and triethylamine (0.054 mL, 0.364 mmol) in CH2C12 (9
mL) to afford the isoxazoline (115 mg, 990): IR (neat) 2976,
2939, 2913, 2875, 2361, 2343, 2331, 1365, 1316, 1239, 1208,
1144, 1120, 1073, 899, 745, 736, 707 cm-l; 1H NMR (300 MHz,
CDC13) d 0.94-0.99 (m, 6 H), 1.16 (s, 9 H), 1.31 (s, 3 H),
2.10-2.21 (m, 6 H), 2.61 (d, J = 16.7 Hz, 1 H), 2.94 (d, J =
16.7 Hz, 1 H), 3.57 (d, J = 10.9 Hz, 1 H), 3.62 (d, J = 11.0
Hz, 1 H); 13C NMR (75 MHz, CDC13} d 2.89, 22.15, 29.73 (t, J =
24.0 Hz), 27.73, 33.03, 41.32, 67.80, 85.22, 105.16-122.96 (m,
CF2, CF3) , 165. 95; 19F NMR (470 MHz, CDC13) d (rel. to CFC13)
-127.06 (t, J - 4.7 Hz), -124.10, -123.72, -122.74, -116.96
(t, J = 14 . 1 Hz) , -81. 79 (t, J = 14 . 1 Hz) ; MS m/z 1220 (M+ -
F), 1126, 795, 475, 309, 239, 195, 140.
CA 02259183 1998-12-22
WO 98/00376 62 PCT/US97/11215
30. 5-Methyl-3-propyl-5-tris[2-(perfluorohexyl)ethyl]-
silanyloxymethyl-4,5-dihydroisoxazole: Prepared according to
general procedure 1 with the allyl silyl ether (0.100 g, 0.088
mmol), BTF (4 mL), nitrobutane (0.09 mL, 0.88 mmol), and
phenyl isocyanate (0.19 mL, 1.76 mmol) to afford the
isoxazoline and starting silane (ratio 94:6, 106 mg): IR
(neat) 2974, 2943, 2914, 2881, 1442, 1362, 1317, 1236, 1207,
1144, 1121, 1072, 907, 811, 736, 707 cm-l; 1H NMR (300 MHz,
CDC13) d 0.89-0.99 (m, 9 H), 1.31 (s, 3 H), 1.49-1.62 (m, 2
H) , 2. 03-2. 19 (m, 6 H) , 2.26 (t, J = 7 . 4 Hz, 2 H) , 2. 56 (d, J
- 16.7 Hz, 1 H) , 2. 90 (d, J = 16. 7 Hz, 1 H) , 3. 58 (d, J = 10. 9
Hz, 1 H), 3.63 (d, J - 10.9 Hz, 1 H); 13C NMR (75 MHz,
CD3COCD3) d 3.49, 13. 96, 20. 44, 22. 65, 25. 50 (t, J = 23.2 Hz) ,
44.73, 68.86, 85.92, 105.76-124.42 (m, CF2, CF3), 159.13; MS
m/z 1226 (M+ + 1) , 1206 (M+ - F) , 878 (M+ - (CH2) 2 (CF2) 5CF3) ,
795, 309, 239, 195.
31. 5-Methyl-3-phenyl-5-tris[2-(perfluorohexyl)ethyl]-
silanyloxymethyl-4,5-dihydroisoxazole: Prepared according to
general procedure 2 with the allyl silyl ether (0.098 g, 0.086
mmol), phenyl hydroximic acid chloride (53.0 mg, 0.34 mmol),
and triethylamine (0.051 mL, 0.34 mmol) in CH2C12 (4 mL} to
afford the isoxazoline and starting silane (ratio 97:3): IR
(neat) 2934, 2915, 2363, 1443, 1361, 1316, 1238, 1206, 1144,
1121, 1079, 907, 736, 707 cm-l; 1H NMR (300 MHz, CDC13) d
0.93-0.99 (m, 6 H), 1.43 (s, 3 H), 2.02-2.16 (m, 6 H), 3.03
(d, J = 16. 6 Hz, 1 H) , 3. 37 (d, J = 16. 6 Hz, 1 H) , 3. 69 (d, J
- 11.1 Hz, 1 H), 3.74 (d, J = 11.0 Hz, 1 H), 7.37-7.42 (m, 3
H), 7.60-7.63 (m, 2 H); 13C NMR (75 MHz, CD3COCD3) d 3.47,
22.66, 25.45 (t, J - 22.5 Hz), 42.57, 68.98, 87.86, 105.75-
123.50 (m, CF2, CF3), 127.26, 129.50, 130.58, 131.39, 157.25;
19F NMR (470 MHz, CDC13) d (rel. to CFC13) -126.78 (t, J = 4.7
Hz), -123.73, -123.51, -122.57, -116.78 (t, J - 14.1 Hz), -
___ ._ __.
CA 02259183 1998-12-22
WO 98/00376 6 3 PCT/US97/I1215
81.43 (t, J = 9.4 Hz); MS m/z 1240 (M+ - F), 990, 912 (M+ -
(CH2) 2 (CF2) 5CF3) , 795, 309, 239, 160.
32. 3-Methyl-5-tris[2-(perfluorohexyl)ethyl]silanyl-
oxymethylisoxazole: Prepared according to general procedure 1
with propargyl silyl ether (0.100 g, 0.089 mmol), BTF (6 mL),
nitroethane (0.064 mL, 0.89 mmol), and phenyl isocyanate (0.18
mL, 1.78 mmol) to afford the isoxazoline (105 mg, 99%): IR
(neat) 2947, 2910, 1443, 1364, 1317, 1295, 1249, 1197, 1144,
1121, 1075, 904, 895, 812, 795, 736, 707 cm-l; 1H NMR (300
MHz, CDC13) d 0. 95-1.01 (m, 6 H) , 1. 99-2. 17 (m, 6 H) , 2.28 (s,
3 H), 4.77 (s, 2 H), 6.05 (s, 1 H); 13C NMR (75 MHz, CD3COCD3)
d 3.37, 11.17, 25.41 (t, J = 23.3 Hz), 57.61, 103.98, 105.27-
124.41 (m, CF2, CF3), 160.53, 171.10; MS m/z 1162 (M+ - F),
910, 828, 506, 309, 239.
33. 3-tert-Butyl-5-Iris[2-(perfluorohexyl)ethyl]silanyl-
oxymethylisoxazole: Prepared according to general procedure 2
with propargyl silyl ether (0.100 g, 0.089 mmol), tert-butyl
hydroximic acid chloride (97.0 mg, 0.72 mmol), and
triethylamine (0.11 mL, 0.72 mmol) in CH2C12 (6 mL) to afford
the isoxazoline (108 mg, 990): IR (neat) 2973, 2950, 2912,
2360, 2392, 1367, 1317, 1237, 1206, 1144, 1121, 1076, 904,
812, 745, 736, 707 cm-1; 1H NMR (300 MHz, CDC13) d 0.95-1.01
(m, 6 H), 1.31 (s, 9 H), 2.00-2.17 (m, 6 H), 4.77 (s, 2 H),
6. 11 (s, 1 H) ; 13C NMR (75 MHz, CD3COCD3) d 3. 92, 25.46 (t, J
- 23.3 Hz), 32.76, 57.81, 101.19, 105.78-124.00 (m, CF2, CF3),
170.96, 172.74; MS m/z 1209 (M+ - 14), 1204 (M+ - F), 918,
871, 813, 962, 310.
34. 3-Propyl-5-tris[2-(perfluorohexyl)ethyl]silanyl-
oxymethylisoxazole: Prepared according to general procedure 1
with propargyl silyl ether (0.100 g, 0.089 mmol) , BTF (6 mL) ,
CA 02259183 1998-12-22
WO 98/00376 6 4 PCT/US97/11215
nitrobutane (0.09 mL, 0.89 mmol), and phenyl isocyanate (0.18
mL, 1.78 mmol) to afford the isoxazoline (107 mg, 990): IR
(neat) 2972, 2945, 2912, 2883, 1443, 1362, 1317, 1294, 1237,
1209, 1144, 1121, 1075, 905, 812, 745, 736, 707 cm-l; 1H NMR
(300 MHz, CDC13) d 0.94-1.02 (m, 9 H), 1.64-1.71 (m, 2 H),
2. 00-2. 17 (m, 6 H) , 2. 62 (t, J = 7. 4 Hz, 2 H) , 4. 77 (s, 2 H) ,
6.06 (s, 1 H); 13C NMR (75 MHz, CD3COCD3) d 3.39, 13.91,
22.29, 25.43 (t, J = 23.3 Hz), 28.54, 57.74, 102.90, 105.75-
123.47 (m, CF2, CF3), 169.63, 171.06; MS m/z 1209 (M+), 1190
(M+ - F) , 857, 309, 239, 195.
35. 3-Phenyl-5-tris[2-(perfluorohexyl)ethyl]silanyl-
oxymethylisoxazole: Prepared according to general procedure 2
with propargyl silyl ether (0.100 g, 0.089 mmol), phenyl
hydroximic acid chloride (110 mg, 0.71 mmol), and
triethylamine (0.11 mL, 0.71 mmol) in CH2C12 (6 mL) to afford
the isoxazoline and silanol (40 o silanol as determined by 1H-
NMR) . 1H NMR (300 MHz, CDC13) d 1. 00-1.06 (m, 6 H) , 2.04-2.21
(m, 6 H), 4.87 (s, 2 H), 6.53 (s, 1 H), 7.95-7.47 (m, 3 H),
7.76-7.80 (m, 2 H); 13C NMR (75 MHz, CD3COCD3) d 3.35, 25.37
{t, J = 23.3 Hz), 57.71, 101.37, 107.77-123.44 (m, CF2, CF3).
127.55, 129.87, 130.10, 131.01, 163.23, 172.26; MS m/z 1293
(M+) , 1224 (M+ - F) , 893, 568, 309, 290, 158 .
36. General Procedure for Cleavage of the Silyl Group
(General Procedure 3): The silylated isoxazol(in)e (0.079
mmol) was dissolved in Et20 (THF) (3 mL) at 25 °C.
HF~pyridine (0.1 mL) was added and the solution was stirred
for 1 h at 25 °C. After removal of the solvent the residue
was dissolved in CH2C12 (20 mL). Sat. aq. NHqCl (10 mL) was
added and the organic-aqueous biphase was washed twice with
FC-72 (10 mL). After separation of the layers, the aqueous
phase was extracted twice with CH2C12. The combined organic
_ .... .
CA 02259183 1998-12-22
WO 98/00376 6 5 PCT/US97/11215
phases ~ were dried (MgS04) and evaporated to yield the
deprotected isoxazol(in)e. The purity was determined by GC-
analysis.
37. (3-Phenyl-4,5-dihydroisoxazol-5-yl)methanol: Prepared
according to general procedure 3 with isoxazoline (0.097 g,
0.078 mmol) and HF~pyridine (0.1 mL) in THF (3 mL) to afford
after extraction the isoxazoline (13.6 mg, 990) with 950
purity. The physical data are in agreement with those
reported in literature.
38. (3-Methyl-4,5-dihydroisoxazol-5-yl)methanol: Prepared
according to general procedure 3 with the silyl isoxazoline
(0.300 g, 0.254 mmol) and HF~pyridine (0.2 mL) in Et20 (6 mL)
to afford after extraction the isoxazoline (8.5 mg, 290) with
93o purity. The physical data are in agreement with those
reported in literature.
39. (3-tert-Butyl-4,5-dihydroisoxazol-5-yl)methanol (13a):
Prepared according to general procedure 3 with the silyl
isoxazoline (0.085 g, 0.069 mmol) and HF~pyridine (0.1 mL) in
THF (3 mL) to afford after extraction the isoxazoline (10.6
mg, 99%) with 91o purity. The physical data are in agreement
with those reported in literature.
40. (3-Propyl-4,5-dihydroisoxazol-5-yl)methanol (13b):
Prepared according to general procedure 3 with the silyl
isoxazoline (0.096 g, 0.079 mmol) and HF~pyridine (0.1 mL) in
THF (3 mL) to afford after extraction the isoxazoline (5.9 mg,
48%) with 94o purity: IR (neat) 3700-3100 br., 2962, 2935,
2875, 1462, 1435, 1383, 1361, 1334, 1313, 1096, 1074, 1049,
908, 874, 849 cm-l; 1H NMR (300 MHz, CDC13) d 0.98 (t, J = 7.9
Hz, 3 H), 1.54-1.66 (m, 2 H), 1.92 (s, br 1 H), 2.32 (t, J =
7 . 4 Hz, 2 H) , 2. 82 (dd, J1 - 17 . 0 Hz, J2 = 7 . 6 Hz, 1 H) , 2 . 96
CA 02259183 1998-12-22
WO 98/00376 6 PCT/US97/11215
6
(dd, J1 = 17.0 Hz, J2 = 10.7 Hz, 1 H), 3.56(dd, J1 - 12.1 Hz,
J2 = ~l . 6 Hz, H) , 3. 77 J1 - 12. Hz, J2 = 3. 1 Hz, 1
1 (dd, 1 H) ,
4.62-9.70 (m, 1 H); 13C NMR (75 MHz, CDC13) d 13.79, 19.78,
29.61, 38.50, 63.68, 6; m/z 143 (M~), 115, 112,
80.00, 159.6 MS
84; HRMS calcd. for C7H13N02 m/z 143.0 946,found 143.0943.
41. (3,5-Dimethyl-4,5-dihydroisoxazol-5-yl)methanol (13d):
Prepared according to general procedure 3 with the silyl
isoxazoline (O.I20 g, 0.100 mmol) and HF~pyridine (0.1 mL) in
THF (3 mL) to afford after extraction the isoxazoline (4 mg,
310) with 99o purity: IR (CHC13) 3588, 2976, 2954, 2925,
2874, 1431, 1388, 1353, 1334, 1291, 1222, 1178, 1055 cm-1; 1H
NMR (300 MHz, CDC13) d 1.31 (s, 3 H), 1.95 (s, 3 H), 2.55 (s,
br 1 H), 2.58 (d, J = 17.0 Hz, 1 H), 3.09 (dd, J1 - I7.0 Hz,
J2 - 0.9 Hz, 1 H), 3.44 (d, J = 12.0 Hz, 1 H), 3.61 (d, J =
11.9 Hz, 1 H); 13C NMR (75 MHz, CDC13) d 13.59, 22.73, 45.92,
67.46, 86.36, 156.29; MS m/z 129 (M+), 98, 74, 59; HRMS calcd.
for C6H11N02 m/z 129.0790, found 129.0790.
42. (3-tert-Butyl-5-methyl-4,5-dihydroisoxazol-5-yl)methanol:
Prepared according to general procedure 3 with the silyl
isoxazoline (0.113 g, 0.091 mmol) and HF~pyridine (0.1 mL) in
Et20 (3 mL) to afford after extraction the isoxazoline (15.6
mg, 990) with 99o purity: IR (neat) 3700-3100 br., 2968,
2931, 2871, 2361, 2392, 1979, 1462, 1434, 1395, 1367, 1340,
1260, 1244, 1205, 1126, 1056, 895, 797 cm-1; 1H NMR (300 MHz,
CDC13) d 1.19 (s, 9 H), 1.31 (s, 3 H), 1.98 (s, br 1 H), 2.62
(d, J = 16.7 Hz, 1 H), 3.08 (d, J = 16.7 Hz, 1 H), 3.46 (d, J
- 12. 0 Hz, 1 H) , 3. 62 (d, J = 11.8 Hz, 1 H) ; 13C NMR (75 MHz,
CDC13) d 22.56, 28.21, 33.36, 41.59, 67.49, 86.20, 167.03; MS
m/z 171 (M+), 140, 98, 82; HRMS calcd. for CgH17N02 m/z
171.1259, found 171.1253.
CA 02259183 1998-12-22
WO 98/00376
PCT/US97/11215
67
43. (5-Methyl-3-propyl-4,5-dihydroisoxazol-5-yl)methanol:
Prepared according to general procedure 3 with the silyl
isoxazoline (0.062 g, 0.051 mmol) and HFpyridine (0.1 mL)
in
Et20 (3 mL) to afford after extraction the isoxazoline (8
mg,
99} with 99o purity: IR (neat) 3700-3100 br., 2963, 2929,
2874, 2855, 1457, 1934, 1378, 1363, 1336, 1317, 1236, 1054,
909 cm-1; 1H NMR (300 MHz, CDC13) d 0.95 (t, J = 7.4 Hz, 3 H),
1.32 (s, 3 H), 1.58 (q, J = 7.5 Hz, 2 H), 1.97 (s, br 1 H),
2.29 (t, J = 7.6 Hz, 2 H), 2.58 (d, J = 17.0 Hz, 1 H) , 3.04
(d, J = 17.0 Hz, 1 H) , 3. 46 (d, J = 11. 9 Hz, 1 H) , 3. (d,
63 J
- 11.9 Hz, 1 H); 13C NMR (75 MHz, CDC13) d 13.92, 19.94,
22.79, 30.03, 44.27, 67.59, 85.93, 159.97; MS m/z 157 (M+),
126, 97; HRMS calcd. for C8H15N02 m/z 157.1103, found
157.1099.
44. (5-Methyl-3-phenyl-4,5-dihydroisoxazol-5-yl)methanol:
Prepared according to general procedure 3 with the silyl
isoxazoline (0.080 g, 0.064 mmol) and HF~pyridine (0.1 mL) in
THF (3 mL) to afford after extraction the isoxazoline (11.5
mg, 95o) with 98o purity. The physical data are in agreement
with those reported in literature.
45. (3-tert-Butylisoxazol-5-yl)methanol: Prepared according
to general procedure 3 with the silyl isoxazole (0.099 g,
0.073 mmol, around 100 of silanol) and HF~pyridine (0.1 mL) in
Et20 (3 mL} to afford after extraction the isoxazole (11.4 mg,
990) with 99% purity: IR (neat) 3700-3100 br., 2966, 2935,
2910, 2872, 1607, 1488, 1465, 1409, 1367, 1211, 1193, 1068,
1042, 998 cm-l; 1H NMR (300 MHz, CDC13) d 1.31 (s, 9 H}, 2.57
(s, br 1 H), 9.71 (s, 2 H), 6.15 (s, 1 H); 13C NMR (75 MHz,
CDC13) d 29.63, 32.22, 56.67, 100.03, 171.09, 172.36; MS m/z
155 (M+), 140, 124, 94, 68, 57; HRMS calcd. for C8H13N02 m/z
155.0946, found 155.0939.
CA 02259183 1998-12-22
WO 98/00376 6 8 PCT/US97/11215
46. (3-Propylisoxazol-5-yl)methanol (13c): Prepared
according to general procedure 3 with the silyl isoxazole
(0.102 g, 0.076 mmol, around l00 of silanol) and HF~pyridine
(0.1 mL) in Et20 (3 mL) to afford after extraction the
isoxazole (10,7 mg, 990) with 97o purity. The physical data
are in agreement with those reported in literature.
47. (3-Methylisoxazol-5-yl)methanol: Prepared according to
general procedure 3 with the silyl isoxazole (0.098 g, 0.075
mmol, around l00 of silanol) and HF~pyridine (0.1 mL) in Et20
(3 mL) to afford after extraction the isoxazole (8.5 mg, 990)
with 99o purity. The physical data are in agreement with
those reported in literature.
48. (3-Phenylisoxazol-5-yl)methanol: Prepared according to
general procedure 3 with the silyl isoxazole (0.05 g, 0.04
mmol) and HF~pyridine (0.1 mL) in Et20 (3 mL) to afford after
extraction the isoxazole (6.8 mg, 99°) with 98° purity. The
physical data are in agreement with those reported in
literature.
49. Simulated Combinatorial Synthesis without Characterization
of Intermediates:
50. (3-tent-Butyl-4,5-dihydroisoxazol-5-yl)methanol (13a):
The allyl silyl ether 11 was prepared as described before with
0.65 mmol of bromo Iris[2-(perfluorohexyl)ethyl]silane 9.
Cycloaddition according to general procedure 2 with tert-butyl
hydroxamic acid chloride (440 mg, 3.20 mmol), and
triethylamine (0.48 mL, 3.20 mmol) in CH2C12 (20 mL) afforded
the isoxazoline. Silyl cleavage according to general
procedure 3 with HF~pyridine (0.5 mL) in Et20 (20 mL) afforded
after extraction the isoxazoline (68 mg, 660) with 99o purity.
____
CA 02259183 1998-12-22
WO 98/00376 6 g PCTIUS97/11215
51. (3-Propyl-4,5-dihydroisoxazol-5-yl)methanol (13b): The
allyl silyl ether was prepared as described before with 0.38
mmol of bromo tris[2-(perfluorohexyl)ethyl]silane 9.
Cycloaddition according to general procedure 1 with
nitrobutane (0.38 mL, 3.80 mmol) and phenyl isocyanate (0.73
mL, 7.20 mmol) in BTF (15 mL) afforded the isoxazoline. Silyl
cleavage according to general procedure 3 with HF~pyridine
(0.4 mL) in Et20 (12 mL) afforded after extraction the
isoxazoline (39.5 mg, 730) with 94% purity.
52. (3,5-Dimethyl-4,5-dihydroisoxazol-5-yl)methanol (13d):
The allyl silyl ether was prepared as described before with
0.38 mmol of bromo tris[2-(perfluorohexyl)ethyl]silane 9.
Cycloaddition according to general procedure 1 with nitro
ethane (0.27 mL, 3.80 mmol) and phenyl isocyanate (0.73 mL,
7.20 mmol) in BTF (15 mL) afforded the isoxazoline. Silyl
cleavage according to general procedure 3 with HF~pyridine
(0.4 mL) in Et20 (12 mL) afforded after extraction the
isoxazoline (15.5 mg, 32%) with 99o purity.
53. (3-Propylisoxazol-5-yl)methanol (13c): Silyl propargyl
ether was prepared as described before with 0.31 mmol of bromo
tris[2-(perfluorohexyl)ethyl]silane 9. Cycloaddition
according to general procedure 1 with nitrobutane (0.31 mL,
3.10 mmol) and phenyl isocyanate (0.63 mL, 6.20 mmol) in BTF
(15 mL) afforded the isoxazole. Silyl cleavage according to
general procedure 3 with HF~pyridine {0.5 mL) in Et20 (10 mL)
afforded after extraction the isoxazoline (37.3 mg, 830) with
97o purity.
54. erythro/threo-a-Methyl-3-phenyl-2-isoxazoline-5-methanol
(13e): rac-3-Buten-2-of (0.083 mL, 0.960 mmol) and
triethylamine (0.14 mL, 0.96 mmol) were dissolved in dry THF
(4 mL) under argon. A mixture of bromo tris[2-
CA 02259183 1998-12-22
WO 98/00376 7 0 PCT/LTS97/11215
{perfluorohexyl)ethyl]silane 9 (275 mg, 0.24 mmol) in THF (2
mL) was slowly added to the above solution at 25 °C. The
resulting suspension was stirred at 25 °C for 3 h. Workup as
described for the preparation of the allyl silyl ether
afforded crude rac-tris[2-(perfluorohexyl)ethyl]-{1-methyl-
allyloxy)silane. Cycloaddition according to general procedure
2 with rac-tris[2-(perfluorohexyl)ethyl]-(1-
methylallyloxy)silane (100 mg) and phenyl hydroximic acid
chloride (53 mg, 0.35 mmol), and triethylamine (0.055 mL, 0.37
mmol) in CH2C12 (6 mL) afforded the isoxazoline. Silyl
cleavage according to general procedure 3 with HF~pyridine
(0.5 mL) in Et20 (20 mL) afforded after extraction
erythro/threo-a-methyl-3-phenyl-2-isoxazoline-5-methanol (10.4
mg, 620) as a 70:30 diastereoisomer mixture with 97o purity.
The diastereoisomer ratio was determined by 1H NMR analysis.
w55. Fluorous Phase Switch. Representative Experimental
Procedure for Grignard Reaction/Silylation: The Grignard
reagent (3M in ether, 0.75 mmol, 0.25 mL) was added at 0° C to
a solution of the carbonyl derivative (0.5 mmol) in dry THF (5
mL) . The mixture was stirred 15 min at 0° C and then 30 min
at 25 °C. Bromo tris[2-(perfluorohexyl)ethyl]silane (9) (1
mmol, containing about 10-15~ C6F13(CH2)4C6F13) in FC-72 (6
mL) was added at 25 °C to the mixture. After 14 h, the
residue obtained by evaporation was extracted with water (20
mL), chloroform (20 mL) and FC-72 (1 x 15 mL and 2 x 10 mL) in
a triphasic extraction. After separation of the chloroform
extract, the remaining aqueous phase was washed with
chloroform (1 x 10 mL). The combined organic phases (CHC13)
were dried (MgS04) and evaporated. The resulting residue was
taken up in acetonitrile (15 mL) and washed with FC-72 (1 x 10
mL then 2 x 5 mL) in a biphasic extraction. The fluorous
phases were combined (from triphasic and biphasic extraction)
and evaporated, and the residue was diluted with 1:1
j
CA 02259183 1998-12-22
WO 98/00376 ~ 1 PCT/US97/11215
THF:benzotrifluoride (10 mL). KHC03 (100 mg) and cesium
fluoride (2 mmol, 2 mL in MeOH 1M) were added at 25 °C, and
the mixture was stirred for 2 h. The residue obtained after
evaporation was extracted with water (20 mL), FC-72 (20 mL)
and chloroform (3 x 15 mL). The fluorous phase was washed
with acetonitrile (1 x 10 mL). The combined organic
(chloroform and acetonitrile) phases were dried (MgS04) and
evaporated. The residue was diluted with acetonitrile (15 mL)
and this was washed with FC-72 (1 x 10 mL then 2 x 5 mL). The
acetonitrile solution was evaporated and the corresponding
alcohols 15 were obtained. All the alcohols are known
compounds.
56. Fluorous Tagging of Biproducts. General Procedure for
Radical Addition and Hydrostannylation: A suspension of 1-
iodoadamantane (26.2 mg, 0.1 mmmol), benzyl acrylate (81.1 mg,
0.5 mmol), tris(2-perfluorohexylethyl)tin hydride (3) ('11.6
mg, 0.01 mmol), sodium cyanoborohydride (9.6 mg, 0.13 mmol),
and cat. AIBN in BTF (0.5 mL) and t-butanol (0.5 mL) was
heated at reflux under nitrogen for 12 h. After cooling, a
mixture of tris(2-perfluorohexylethyl)tin hydride (696 mg, 0.6
mmol ) and cat . AIBN in BTF ( 0 . 2 mL) was added to the reaction
mixture. Then, the mixture was heated at 90°C under nitrogen
for 24 h. After cooling, the reaction mixture was dissolved
in chloroform (10 mL) and extracted with FC-72 (10 mL) three
times. The organic layer was filtered through neutral alumina
and evaporated under reduced pressure to give the product (246
mg, 820) free of alkene.
5?. General Procedure for 1,3-bipolar Cycloaddition and
Hydrostannylation: To a solution of styrene (31.3 mg, 0.3
mmol) and triethylamine (12.1 mg, 0.13 mmol) in
dichloromethane (1 mL) was added dropwise tert-butyl
hydroximic acid chloride (13.6 mg, 0.1 mmol) diluted with
dichloromethane (1 mL) at 25 °C. The mixture was stirred for
CA 02259183 1998-12-22
WO 98/00376 7 2 PCT/US97/11215
12 h. After evaporating the solvent, tris(2-
perfluorohexylethyl)tin hydride 3 (348 mg, 0.3 mmol) and cat.
AIBN dissolved in BTF (1 mL) were added to the crude residue
and the mixture was heated at 90°C under nitrogen for 24 h.
After cooling, the reaction mixture was diluted with
dichloromethane (10 mL) and this was extracted with FC-72 (10
mL) three times. The organic layer was evaporated under
reduced pressure to give the product (20 mg, 99a) free of
alkene.
58 . Tris (2- (perfluorohexyl) ethyl) tin azide (16) ; Tris
(3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)tin azide: To a
solution of Iris(2-(perfluorohexyl)ethyl)tin bromide (2) (10
g, 8.06 mmol) in ether (14 mL) was added a solution of sodium
azide (629 mg, 9.67 mmol) in water (2 mL) and the resulting
biphasic mixture was stirred at 25 °C for 12 h. Ether (20 mL)
and water (20 mL) were added to the reaction mixture. The two
layers were separated and the ethereal phase was washed with
water (3 x 20 mL), and dried over anhydrous MgS04. The
solvent was evaporated to dryness to yield the tin azide ( 9. 4
g, 97o yield) as a colorless oil: IR (thin film) 2080, 1360,
734 cm-1; 1H NMR (CDC13) d 2.48 (m, 6 H) , 1.51 (m. 6 H) ; 119Sn
NMR (BTF-C6D6) d 11.53
59 Representative Experimental Procedure for the Preparation
of 5-Substituted Tetrazoles 19: A solution of tris (2-
(perfluorohexyl)ethyl)tin azide 16 (0.5 g, 0.416 mmol) and p-
tolunitrile (97.5 mg, 0.832 mmol) in benzotrifluoride (BTF,
0.84 mL) was heated in a sealed tube at 80 °C for 12 h. The
BTF was evaporated and the crude product was partitioned
between benzene and FC-72 (10 mL each). After separation of
the 2 layers, the benzene layer was washed twice with FC-72
(10 mL). Evaporation of the benzene phase yielded the
unreacted p-tolunitrile. The fluorinated phase was evaporated
T
CA 02259183 1998-12-22
WO 98/00376 7 3 PCT/US97/11215
and a saturated solution of HC1 in ether (10 mL) was added to
the residue. The mixture was stirred for 12 h at 25 °C.
After evaporation of the ether, the residue was dissolved in
FC-72 and acetonitrile (10 mL of each). After separation of
the two layers, the organic phase was washed with FC-72 (3 x
mL). Evaporation of the acetonitrile phase yielded the 5-
p-tolyltetrazole (41 mg, 61o yield), which was identified by
comparing physical and spectral data with those of the
authentic sample. {The other tetrazoles prepared are also
10 known compounds.) The FC-72 phase was evaporated as well
yielding tris (2-(perfluorohexyl)ethyl)tin chloride 8 (449 mg,
900) as a colorless oil.
60.Iris[2-(Perfluorodecyl)ethyl]silane: Magnesium powder
(0.45 g, 18.5 mmol) was suspended in dry Et20 (20 mL) and 1-
iodo-1H, 1H, 2H, 2H-perfluorododecane (0. 50 g, 0. 77 mmol) was
added. The resulting suspension was sonicated for 30 minutes.
A solution of 1-iodo-1H,1H,2H,2H-perfluorododecane (9.50 g,
14.7 mmol) in Et20 (70 mL) was slowly added. The mixture was
heated at reflux for 2 h. Trichlorosilane (0.40 mL, 3.87
mmol) was slowly added and the reaction mixture was stirred
under reflux for 16 h. After cooling to 25 °C, sat. aq. NH4C1
and CH2C12 were added. The cloudy biphase was extracted 5
times with FC-72. Evaporation of the combined fluorous
extracts yielded the crude product as a white solid. Removal
of the impurity (dimer, Wurtz coupling product) by bulb-to-
bulb distillation (0.5 Torr, 210 °C) yielded Iris[2-
(perfluorodecyl)ethyl]silane as a white solid (9.7 g, 760):
mp 76-78 °C; IR (FC-72) 2977, 2950, 2919, 2873, 2136, 1444,
1426 cm-1; 1H NMR (300 MHz, FC-72 with benzene as internal
lock) d 1.15-1.22 (m, 6 H), 2.24-2.41 (m, 6 H), 4.14 (s, 1 H).
61. Bromo tris[2-(perfluorodecyl)ethyl]silane: Iris[2-
(Perfluorodecyl)ethyl]silane (0.56 g, 0.34 mmol) was dissolved
CA 02259183 1998-12-22
WO 98/00376 7 4 PCT/US97/11215
under argon in FC-72 (10 mL). Bromine (0.03 mL, 0.50 mmol)
was added and the mixture was stirred at 25 °C for 12 h. FC-
72 (40 mL) was added and the fluorous phase was washed with
CH2C12. Evaporation of the fluorous layer yielded bromo
tris[2-(perfluorodecyl)ethyl]silane as a white solid (586 mg,
990): mp 80-81 °C; IR (FC-72) 2980, 2953, 2907, 2847, 1444,
1423 cm-1; 1H NMR (300 MHz, FC-72 with benzene as internal
lock) d 1.41-1.55 (m, 6 H), 2.33-2.99 (m, 6 H).
62. Tripropyl 4-Bromoorthothiobenzoate: 4-Bromobenzoic acid
(2.00 g, 9.95 mmol) was suspended in thionyl chloride (3 mL,
41.2 mmol) and heated at reflux for 60 min. Removal of the
excess thionyl chloride and vacuum drying provided 4-
bromobenzoyl chloride as a colorless solid. Propanethiol (10
mL, 109 mmol) was slowly added to a mixture of 4-bromobenzoyl
chloride and anhydrous A1C13 (5.30 g, 39.7 mmol). The mixture
was heated at 60 °C for 48 h, cooled, and poured slowly with
stirring into ice-cooled 4N aqueous NaOH (75 mL). Extraction
with ether and washing of the organic phase with brine
afforded after drying (MgSOq ) the crude product as a red oil .
Purification by flash-column chromatography (Si02, hexanes
containing to of NEt3) provided the orthothiobenzoate as a
colorless oil (1.78 g, 450): IR (neat) 2962, 2929, 2871,
1582, 1483, 1456, 1391, 1378, 1337, 1291, 1237, 1076, 1008 cm-
1~ 1H NMR (300 MHz, CDC13) d 0. 93 (t, J = 7.3 Hz, 9 H) , 1.48-
1.55 (m, 6 H), 2.54 (t, J = 7.3 Hz, 6 H), 7.43 (d, J = 8.7 Hz,
2 H), 7.73 (d, J = 8.7 Hz, 2 H); 13C NMR (75 MHz, CDC13) d
13.80, 21.80, 33.82, 72.87, 121.53, 129.68, 130.99, 141.33. MS
m/z 351 (M+ - propyl), 399, 319 (M+ - S(CH2)2CH3), 317, 201,
109; HRMS calcd. for C13H1gS281Br (M+ - S(CH2)2CH3) m/z
319.0013, found 319.0000.
_ ._ T
CA 02259183 1998-12-22
WO 98/00376 7 5 PCT/US97/11215
63. 4-Iris[2-(Perfluorodecyl)ethyl]silyl-thiobenzoic S-Propyl
Ester 22: Tripropyl 4-bromoorthothiobenzoate (250 mg, 0.66
mmol) was dissolved in Et20 (7.5 mL) and cooled to -78 °C
under argon. t-BuLi (1.7 molar in pentane, 0.82 mL, 1.39
mmol) was slowly added and the resulting yellow solution was
stirred at that temperature for 45 min. The yellow
aryllithium solution was then transferred via canula to a 25
°C mixture of bromo Iris[2-(perfluorodecyl)ethyl]silane (500
mg, 0.29 mmol) in BTF (15 mL) and FC-72 (2.5 mL). The
reaction mixture was stirred at 25 °C for 30 min. After
addition of H20, the reaction mixture was extracted three
times with CH2C12. The combined organic layers were dried
(MgS04) and evaporated to afford an oil which was taken up
into FC-72 and washed with benzene. The benzene layer was
additionally extracted twice with FC-72. The combined
fluorous layers were evaporated to yield the crude
or.thothioester which was dissolved in BTF (7.5 mL), THF (7.5
mL), acetone (5 mL), and H20 (0.5 mL) at 25 °C. AgN03 (135
mg, 0.80 mmol) was added and the resulting suspension was
stirred at 25 °C for 12 h. After filtration and evaporation
of the filtrate, the crude product was purified by flash-
column chromatography (Si02, Et20/hexanes; 1/40) to afford 4-
tris[2-(perfluorodecyl)ethyl]silylthiobenzoic S-propyl ester
as a colorless solid (319 mg, 60%): mp 69-71 °C; IR (CHC13)
2970, 2935, 1668, 1296, 1193, 1157; 1H NMR (300 MHz, CDC13) d
1.04 (t, J = 7.3 Hz, 3 H), 1.15-1.21 (m, 6 H), 1.68-1.75 (m, 2
H) , 1 . 99-2. 06 (m, 6 H) , 3. 08 (t, J = 7. 1 Hz, 2 H) , 7. 54 (d, J
- 8.0 Hz, 2 H) , 8.03 (d, J = 8. 0 Hz, 2 H) ; 13C NMR (125 MHz,
CDC13, 30 °C) d 1.60, 13.93, 23.00, 25.60 (t, J = 23.0 Hz),
31.19, 108.95-118.40 (m, CF2, CF3), 127.23, 133.99, 137.79,
139.27, 191.85.
64. 4-tris[2-(Perfluorodecyl)ethyl]silyl-benzoic acid 20: 9-
tris[2-(Perfluorodecyl)ethyl]-silyl-thiobenzoic S-propyl ester
CA 02259183 1998-12-22
WO 98/00376 ,/ 6 PCT/US97/11215
(210 mg, 0.11 mmol) was dissolved in FC-72 (15 mL}. Bromine
(0.05 mL, 0.83 mmol) was added at 25 °C and the mixture was
stirred for 3 h. After addition of FC-72 (15 mL) and washing
with CH2C12, the fluorous layer was evaporated to afford 4-
tris[2-(perfluorodecyl)ethyl]silylbenzoic acid bromide as a
colorless solid. The acid bromide was dissolved in THF (12
mL) and BTF (3 mL). H20 (1.5 mL) was added and the solution
was stirred at 25 °C for 12 h. Evaporation of the solvents
afforded 4-tris[2-(perfluorodecyl)ethyl]silylbenzoic acid as a
colorless solid (196 mg, 970). mp 134-136 °C; 1H NMR (300
MHz, TFA-d) d 1.35-1.39 (m, 6 H), 2.10-2.35 (m, 6 H), 7.78 (d,
J = 8. 1 Hz, 2 H) , 8.27 (d, J = 7. 9 Hz, 2 H) ; 13C NMR (75 MHz,
TFA-d) d 2.95, 27.42, 105-120 (m, CF2, CF3), 131.78, 132.05,
136.15, 142.95, 175.38. MS (EI} m/z 1243 (M+ -
CH2CH2(CF2)9CF3), 706, 601, 474, 423, 378, 175.
65. General Procedure for the Ugi-Four-Component-Condensation:
4-Iris[2-(Perfluorodecyl)ethyl]silylbenzoic acid (20) (26.2
mg, 0.015 mmol), the amine (0.25 mmol), the aldehyde (0.25
mmol), and the isocyanide (0.25 mmol) were added to a sealed
tube with CF3CH20H (0.3 mL). (For some examples, the
preformed imine was used.) The suspension was heated under
argon to 90 °C for 98 h. After removal of the solvent, the
residue was dissolved in FC-72 (15 mL) and washed with benzene
(15 mL). The benzene layer was additionally washed twice with
FC-72 (15 mL). The combined fluorous phases were evaporated
to yield the perfluorosilylated amino acid amide. For
desilylation the amino acid amide was dissolved at 25 °C in
THF (2 mL), TBAF (1 molar in THF, 0.022 mL, 0.022 mmol) was
added and the resulting solution was stirred at 25 °C for 30
min. After removal of the solvent, the residue was taken up
into benzene (30 mL} and washed twice with FC-72 (15 mL).
Et20 (30 mL) was added to the organic layer which was washed
with 0.1N HCl, sat. aq. Na2C03, and brine (15 mL each). The
CA 02259183 1998-12-22
WO 98/00376 7 7 PCT/US97/11215
organic phase was dried (MgS04) and evaporated to yield the
benzoylated amino acid amide. The purity was checked by GC-
analysis.
66. N-Benzoyl-N-benzyl-phenylglycine-tert-butylamicle:
Prepared according to the general procedure with the acid (20)
(26.2 mg, 0.015 mmol), benzyl benzylidene amine (51 mg, 0.25
mmol), and tert-butyl isocyanide (30 mL, 0.25 mmol) to afford
the silylated amino acid: IR (CHC13) 1684, 1631, 1518, 1240,
1218, 1157 cm-1; 1H NMR (300 MHz, CDC13) d 1, 14 (s, br. 6 H) ,
1.34 (s, 9 H), 1.90-2.20 (br. 6 H), 4.35-4.50 (br. 1 H), 4.71
(d, ~l = 17. 6, 1 H) , 5. 52 (s, 1 H) , 5. 63 (s, 1 H) , 6. 91 (s, br.
2 H) , 7.26 (s, br. 9 H) , 7.30-7. 49 (m, 8 H) ; 13C NMR (75 MHz,
CDC13) d 1.50, 25.54, 28.75, 51.93, 105-125 (m, CF2, CF3),
127.08, 127.15, 127.24, 128.45, 128.95, 129.15, 129.25,
129.98, 137.63, 138.89, 168.42, 172.70. Desilylation as
described in the general procedure in THF (2 mL) with TBAF
(0.022 mL, 0.022 mmol) afforded N-benzoyl-N-benzyl-
phenylglycine-tert-butylamide (5.0 mg, 83 0) with 85o purity:
IR (CHC13) 3424, 3066, 3032, 2969, 2934, 2907, 2296, 1681,
1635, 1514, 1496, 1453, 1430,1409 cm-1; 1H NMR (300 MHz,
CDC13) d 1.30 (s, 9 H), 9.47 (s, br. 1 H), 4.73 (d, J = 16.4
Hz, 1 H), 5.46 (s, 1 H), 5.40-5.75 (s, br. 1 H), 7.03 (s, br.
2 H) , 7. 13-7. 16 (m, 4 H) , 7. 26-7. 49 (m, 9 H) ; 13C NMR (75 MHz,
CDC13) d 28.67, 51.74, 126.79, 127.02, 128.41, 128.66, 128.92,
129.73, 129.90, 135.33, 136.46, 168.53, 173.38; MS m/z 328 (M+
- NHC(CH3)3), 300 (M+ - CONHC(CH3)3), 210, 191, 105, 91, 77;
HRMS calcd. for C21H1gN0 (M+ - CONHC(CH3)3) m/z 300.1388,
found 300.1389.
67. N-Benzoyl-N-benzyl-4-methoxyphenylglycine-tert-butylamide:
Prepared according to the general procedure with the acid 20
(26.1 mg, 0.015 mmol), benzyl amine (27 mL, 0.25 mmol),
CA 02259183 1998-12-22
WO 98/00376 PCT/US97/11215
78
anisaldehyde (30 mL, 0.25 mmol), and tert-butyl isocyanide (30
mL, 0.25 mmol) to afford after desilylation N-benzoyl-N-
benzyl.-4-methoxyphenylglycine-tert-butylamide (5.1 mg, 810)
with 87o purity: IR (CHC13) 3423, 3066, 3031, 2968, 2937,
2911, 2246, 1681, 1633, 1581, 1512, 1455, 1412, 1395, 1366,
1339, 1306, 1252, 1224, 1179, 1033 cm-1; 1H NMR (300 MHz,
CDC13) d 1.30 (s, 9 H) , 3.77 (s, 3 H) , 4.43 (s, br. 1 H) , 4.72
(d, J = 16. 4 Hz, 1 H) , 5. 43 (s, 1 H) , 5.40-5. 70 (s, br. 1 H) ,
6.79 (d, J = 8.3 Hz, 2 H), 7.02-7.47 (m, 12 H); 13C NMR (75
MHz, CDC13) d 28.70, 51.67, 55.42, 114.24, 126.77, 126.92,
127.25, 128.35, 128.58, 129.79, 131.18, 136.60, 159.76,
168. 84, 173.28; MS m/z 930 (M+) , 358 (M+ - NHC (CH3) 3) , 330 (M+
- CONHC(CH3)3), 240, 224, 105, 91, 77; HRMS calcd. for
C22H20N02 (M+ - CONHC(CH3)3) m/z 330.1994, found 330.1498.
68. N-Benzoyl-N-benzylcyclohexylglycine-tert-butylamide:
Prepared according to the general procedure with the acid (20)
(24.0 mg, 0.013 mmol), benzyl amine (27 mL, 0.25 mmol),
cyclohexane carboxaldehyde (30 mL, 0.25 mmol), and tert-butyl
isocyanide (30 mL, 0.25 mmol) to afford after desilylation N-
benzoyl-N-benzyl-cyclohexylglycine-tert-butylamide (1.7 mg,
320) with 89° purity: mp 140-141 °C; IR (CHC13) 2964, 2935,
2858, 2249, 1674, 1618, 1510, 1452, 1363 cm-1; 1H NMR (300
MHz, CDC13) d 0.90-2.00 (m, 10 H), 1.31 (s, 9 H), 2.38 (m, 1
H), 4.14 (d, J = 10.5 Hz, 1 H), 4.44 (d, J = 16.1 Hz, 1 H),
4.70 (d, J = 16.1 Hz, 1 H), 5.30 (s, br. 1 H), 6.90-7.50 (m,
10 H) ; 1~C NMR (75 MHz, CDC13) d 25.06, 25.88, 26.49, 28. 80,
29.77, 30.32, 36.50, 51.19, 52.65, 126.72, 127.25, 127.64,
128.31, 128.53, 129.74, 136.99, 137.41, 169.42, 173.99; MS m/z
406 (M+), 306 (M+ - CONHC(CH3)3), 216, 197, 105, 91, 77; HRMS
calcd. for C26H34N202 m/z 406.2620, found 406.2635.
__...__..__._
CA 02259183 1998-12-22
WO 98/00376 7 9 PCT/US97/11215
69. N-Benzoyl-N-propyl-cyclohexylglycine-cyclohexylamide:
Prepared according to the general procedure with the acid (20)
(26.4 mg, 0.015 mmol), propyl amine (21 mL, 0.25 mmol),
cyclohexane carboxaldehyde (30 mL, 0.25 mmol), and cyclohexyl
isocyanide (31 mL, 0.25 mmol) to afford after desilylation Id-
benzoyl-N-propyl-cyclohexylglycine-cyclohexylamide (5.7 mg,
990) with > 95% purity: mp I10-112 °C; IR (CHC13) 2934, 2856,
2244, 1660, 1613, 1578, 1529, 1450, 1419, 1352 cm-1; 1H NMR
(300 MHz, CDC13) d 0.61 (t, J = 7.3 Hz, 3 H) , 0.85-1.84 (m, 22
H) , 2.39 (m, 1 H) , 3.20 (m, 2 H) , 3. 75-3.78 (m, 1 H) , 4. 00 (s,
br. 1 H), 7.34-7.43 (m, 5 H); 13C NMR (75 MHz, CDC13) d 11.21,
22.59, 24.71, 25.79, 26.50, 29.69, 30.47, 32.79, 32.97, 35.53,
47.75, 126.64, 128.61, 129.76, 136.96, 170.27, 173.70; MS m/z
384 (M+), 258 (M+ - CONHC6H11), 223, 105, 77; HRMS calcd. for
C24H36N2O2 m/z 389.2777, found 384.2781.
70. N-Benzoyl-N-benzylphenylglycine-cyclohexylamide: Prepared
according to the general procedure with the acid (20) (27.1
mg, 0.015 mmol), benzyl benzylidene amine (51 mg, 0.25 mmol),
and cyclohexyl isocyanide (31 mL, 0.25 mmol) to afford after
desilylation N-benzoyl-N-benzyl-phenylglycine-cyclohexylamide
(5.2 mg, 920) with 80o purity: IR (CHC13) 3422, 3066, 3031,
2935, 2857, 2246, 1673, 1634, 1603, 1515, 1497, 1452, 1431,
1411, 1349, 1313, 1253 cm-1; 1H NMR (300 MHz, CDC13) d 1.07-
2.20 (m, 10 H) , 3. 83 (m, br. 1 H) , 4.47 (s, br. 1 H) , 4.70 (s,
br. 1 H), 5.47 (s, 1 H), 5.67 (s, br. 1 H), 7.12-7.49 (m, 15
H); 13C NMR (75 MHz, CDC13) d 24.91, 25.60, 32.89, 48.71,
126.83, 127.15, 128.51, 128.73, 128.99, 129.73, 129.96,
135.24, 136.34, 168.39, 173.36; MS m/z 321 (M+ - PhCO), 300
(M+ - CONHCHH11), 217, 210, 105, 91, 77; HRMS calcd. for
C21H18N0 (M+ - CONHC6H11) 300.1388, found 300.1398.
CA 02259183 1998-12-22
WO 98/00376 8 0 PCT/US97/11215
71. N-Benzoyl-N-propylvaline-cyclohexylamide: Prepared
according to the general procedure with the acid 20 (25.8 mg,
0.014 mmol), propyl amine (21 mL, 0.25 mmol), isobutyraldehyde
(23 mL, 0.25 mmol), and cyclohexyl isocyanide (31 mL, 0.25
mmol) to afford after desilylation N-benzoyl-N-propylvaline-
cyclohexylamide (3.5 mg, 710) with > 95o purity. The physical
data are in agreement with those reported in literature.
72. N-Benzoyl-N-benzylvaline-cyclohexylamide: Prepared
according to the general procedure with the acid (20) (26.1
mg, 0.015 mmol), benzyl amine (27 mL, 0.25 mmol),
isobutyraldehyde (23 mL, 0.25 mmol), and cyclohexyl isocyanide
(31 mL, 0.25 mmol) to afford after desilylation N-benzoyl-N-
benzylvaline-cyclohexylamide (3.5 mg, 610) with > 95~ purity:
mp 131-133 °C; IR (CHC13) 3423, 3303, 3064, 3029, 2970, 2931,
2860, 2242, 1665, 1617, 1518, 1950, 1340, 1309 cm-l; 1H NMR
(300 MHz, CDC13) d 0.78-1.37 (m, 11 H), 1.51-1.79 (m, 5 H),
2. 69 (m, br. 1 H) , 3. 63 (m, br. 1 H) , 4 . 16 (d, J = 10. 6 Hz, 1
H), 4.44 (d, J = 15.8 Hz, 1 H), 4.67 (d, J = 15.9 Hz, 1 H),
6. 93 (s, br. 2 H) , 7. 11-7.26 (m, 7 H) ; 13C NMR (75 MHz, CDC13)
d 19.39, 19.91, 24.63, 25.53, 27.29, 32.58, 32.87, 47.76,
52.54, 68.17, 126.57, 127.20, 127.49, 128.17, 128.42, 129.64,
136.79, 137.08, 169.27, 173.92; MS m/z 287 (M+ - PhCO), 266
(M+ - CONHC6H11), 211, 183, 105, 91, 77; HRMS calcd. for
C1gH27N20 (M+ - PhCO) m/z 287.2123, found 287.2128.
73. N-Benzoyl-N-benzyl-cyclohexylglycine-cyclohexylamide:
Prepared according to the general procedure with the acid (20)
(26.2 mg, 0.015 mmol), benzyl amine (27 mL, 0.25 mmol),
cyclohexane carboxaldehyde (30 mL, 0.25 mmol), and cyclohexyl
isocyanide (31 mL, 0.25 mmol) to afford after desilylation N-
benzoyl-N-benzylcyclohexylglycine-cyclohexylamide (5.3 mg,
84%) with > 95% purity: mp 181-182 °C; IR (CHC13) 2933, 2856,
1
CA 02259183 1998-12-22
WO 98/00376 81 PCT/US97/11215
2246, 1671, 1620, 1515, 1451, 1415, 1351, 1329 cm-1; 1H NMR
(300 MHz, CDC13) d 0.85-1.84 (m, 20 H), 2.41 (m, 1 H), 3.65-
3.71 (m, 1 H), 4.15 (d, J = 11.0 Hz, 1 H), 4.45 (d, J = 15.8
Hz, 1 H), 4.67 (d, J = 15.9 Hz, 1 H), 9.92 (s, br. 1H), 6.96
(s, br. 2 H), 7.15-7.43 (m, 8 H); 13C NMR (75 MHz, CDC13) d
24.72, 25.62, 25.77, 26.41, 29.73, 30.32, 32.69, 32.99, 36.31,
47.85, 126.67, 127.25, 127.50, 128.25, 128.47, 129.70, 136.89,
137.24, 169.26, 174.01; MS m/z 932 (M+), 327 (M+ - PhCO), 306
(M+ - CONHC6H11), 223, 105, 92, 77; HRMS calcd. for C2gH36N202
(M+) m/z 432.2793, found 432.2778.
74. General Procedure for Conventional Biginelli reactions: A
solution of 174 mg (0.89 mmol) benzoyloxyethylurea in THF (5
mL) was treated with 3 equiv of f3-keto ester, 3 equiv of
aldehyde and of concentrated HC1 (25 uL). The solution was
stirred until completion (TLC), concentrated in vacuo and
purified by chromatography on Si02 (ethyl acetate/hexane.s
l:l).
75. 1-(Benzoyloxyethyl)-6-methyl-2-oxo-4-phenyl-1,2,3,4-
tetrahydropyrimidine-5-carboxylic acid ethyl ester: mp 129
°C; IR (CHC13) 3425, 3016, 1711, 1684, 1623, 1450, 1392, 1270,
1178; 1H NMR (CDC13) d 7. 96 (d, 2 H, J = 7. 8 Hz) , 7. 60 - 7. 10
(m, 8 H), 5.39 (bs, 2 H), 4.50 - 4.40 (m, 3 H), 4.15 - 3.90
(m, 3 H), 2.60 (s, 3 H), 1.18 (t, 3H, J = 7.1 Hz); 13C NMR
(CDC13) d 166.3, 165.9, 153.4, 148.0, 143.0, 133.1, 129.6,
128.6, 128.4, 127.7, 126.0, 105.3, 63.4, 60.2, 54.0, 40.9,
16.4, 14.1; MS (El) m/z (relative intensity) 408 (M+, 9) , 393
(16), 331 (16), 303 (8), 286 (6), 259 (20), 209 (10), 149
(100), 105 (99), 77 (59); HRMS (E1) calculated for C23H24N205
408.1672, found 408.1685.
CA 02259183 1998-12-22
WO 98/00376 8 2 PCT/ITS97/11215
76. I-(Benzoyloxyethyl)-6-methyl-2-oxo-4-(2-naphthyl)-1,2,3,4-
tetrahydropyrimidine-5-carboxylic acid ethyl ester: IR (neat)
3342, 2977, 1714, 1679, 1621, 1450, 1390, 1269, 1219, 1182,
1108, 1070; 1H NMR (CDC13) d 7. 95 - 7.25 (m, 12 H) , 5. 58 (d, 1
H, J = 2. 8 Hz) , 5. 49 (d, 1 H, J = 2.8 Hz) , 4.60 - 4. 35 (m, 3
H) , 4 . 15 - 4. 00 (m, 3 H) , 2. 63 (s, 3 H) , 1. 18 (t, 3H, J = 7. 1
Hz); 13C NMR (CDC13) d 166.4, 166.2, 153.7, 148.4, 140.5,
133.2, 132.9, 129.7, 128.8, 128.5, 128.0, 127.6, 126.3, 126.0,
125.0, 124.4, 105.3, 63.6, 60.4, 54.3, 91.2, 16.6, 14.3; MS
(E1) m/z (relative intensity) 458 (M+, 26), 443 (22), 912
(9), 385 (11), 353 (11), 331 (29), 309 (30), 263 (6), 209
(12), 149 (100), 105 (87), 77 (36); HRMS (E1) calculated for
C27H26N205 458.1842, found 958.1842.
77. 1-(Benzoyloxyethyl)-6-methyl-2-oxo-4-(4-methoxyphenyl)-
1,2,3,4-tetrahydropyrimidine-5-carboxylic acid ethyl ester:
mp 75°C; IR (CHC13) 3425, 3025, 1709, 1677, 1621, 1514, 1454,
1392, 1270, 1179, 1114, 1069; 1H NMR (CDC13) d 7.96 (d, 2 H, J
- 7.1 Hz), 7.56 (t, 1 H, J = 7.6 Hz), 7.41 (t, 2 H, J = 7.6
Hz), 7.14 (d, 2 H, J = 8.7 Hz), 6.62 (d, 2 H, J = 8.7 Hz),
5.39 (bs, 2 H), 4.50 - 4.40 (m, 3 H), 4.15 - 3.95 (m, 3 H),
3.66 (s, 3H), 2.60 (s, 3 H), 1.19 (t, 3H, J = 7.1 Hz); 13C NMR
(CDC13) d 166.4, 166.2, 159.0, 153.9, 197.9, 135.5, 133.2,
129.7, 128.5, 127.4, 113.9, 105.8, 63.6, 60.3, 55.1, 53.3,
40.9, 16.5, 19.3; MS (El) m/z (relative intensity) 938 (M+,
5), 423 (27), 365 (13), 331 (7), 316 (6), 289 (30), 243 (6),
209 (5), 149 (92), 105 (100), 77 (50); HRMS (E1) calculated
for C24H26N206 438.1787, found 438.1791.
78. 1-(Benzoyloxyethyl)-6-methyl-2-oxo-4-(2-naphthyl)-1,2,3,4-
tetrahydropyrimidine-5-carboxylic acid methyl ester: mp 116°C;
IR (CHC13) 3425, 3012, 1708, 1683, 1623, 1454, 1392, 1275,
1188, 1115, 1076; 1H NMR (CDC13) d 7. 88 (d, 2 H, J = 8.0 Hz) ,
_. .. _
CA 02259183 1998-12-22
WO 98/00376 8 3 PCT/US97/11215
7.70 - 7.30 (m, 10 H), 5.57 (d, 1 H, J = 2.6 Hz), 5.52 (bs, 1
H), 4.55 - 4.45 (m, 3 H), 4.10 - 3.95 (m, 1 H), 3.66 (s, 3 H),
2.63 (s, 3 H); 13C NMR (CDC13) d 166.5, 166.3, 153.9, 148.7,
140.3, 133..1, 132.8, 129.5, 128.7, 128.3, 128.0, 127.5, 126.1,
125.9, 124.7, 124.2, 104.9, 63.5, 53.8, 51.5, 41.0, 16.5; MS
(E1) m/z (relative intensity) 494 (M+, 10), 429 (10), 339
(6), 317 (12), 295 (15), 149 (81), 105 (100), 77 (35); HRMS
(E1) calculated for C26H24N2~5 444.1686, found 444.1685.
79. 1-(Benzoyloxyethyl)-6-ethyl-2-oxo-4-(2-naphthyl)-1,2,3,4-
tetrahydropyrimidine-5-carboxylic acid ethyl ester: IR (neat)
3390, 2981, 1709, 1681, 1614, 1454, 1382, 1269, 1182, 1108,
1067; 1H NMR (CDC13) d 7.85 (d, 2 H, J = 7. 1 Hz) , 7.70 - 7.25
(m, 10 H) , 5.55 (d, 1 H, J = 3. 3 Hz) , 5. 52 (bs, 1 H) , 4.55 -
4.95 (m, 3 H), 4.20 - 9.05 (m, 2 H), 4.05 - 3.85 (m, 1 H),
3.55 - 3. 35 (m, 1 H) , 2. 90 - 2. 75 (m, 1 H) , 1.25 (t, 3H, J =
7.2 Hz), 1.20 (t, 3H, J = 7.1 Hz); 13C NMR (CDC13) d 166.4,
165.7, 159.4, 154.1, 140.4, 133.2, 133.0, 129.7, 129.5, 128.9,
128.9, 128.1, 127.7, 126.3, 126.1, 125.0, 124.3, 104.5, 63.7,
60.5, 53.9, 40.8, 22.2, 14.3, 13.1; MS (El) m/z (relative
intensity) 472 (M+, 14), 443 (25), 350 (11), 323 (14), 277
(7), 223 (13), 149 (100), 105 (95), 77 (41); HRMS (E1)
calculated for C2gH2gN205 472.1980, found 972.1998.
80. 1-(Benzoyloxyethyl)-6-ethyl-2-oxo-4-phenyl-1,2,3,4-
tetrahydropyrimidine-5-carboxylic acid ethyl ester: mp 111
°C; IR (neat) 3340, 2981, 1714, 1687, 1616, 1450, 1383, 1269,
1209, 1172, 1108, 1079; 1H NMR (CDC13) d 7.94 (d, 2 H, J = 7.2
Hz), 7.55 - 7.05 (m, 8 H), 5.41 (bs, 1 H), 5.36 (d, 1 H, J =
3.2 Hz), 4.55 - 4.40 (m, 3 H), 4.20 - 4.00 (m, 2 H), 4.00 -
3.85 (m, 1 H), 3.50 - 3.30 (m, 1 H), 2.90 - 2.70 (m, 1 H),
1.23 (t, 3 H, J = 7.3 Hz), 1.19 (t, 3 H, J = 7.1 Hz); 13C NMR
(CDC13) d 166.4, 165.7, 159.2, 154.1, 143.2, 133.2, 129.7,
CA 02259183 1998-12-22
WO 98/00376 8 4 PCT/US97/11215
128.7, 128.5, 127.8, 126.2, 104.6, 63.7, 60.4, 53.7, 40.7,
22.0, 14.2, 13.0; MS (E1) m1z (relative intensity) 422 (M+,
5}, 393 (95), 377 (10), 345 (31), 317 (19), 273 (35), 293 (8),
223 (36), 195 (11), 149 (100), 105 (92), 77 (52); HRMS (El)
calculated for C24H26N205 422.1827, found 422.1842.
81. 4-(tris(Perfluorodecylethyl)silyl)benzoyloxyethylurea 23.
A solution of 4-(tris(perfluorodecylethyl)silyl)benzoic acid
propyl thioester 22, 88 mg, 47.6 umol} in FC-72 (6 mL) was
treated at 25 °C with bromine (30 uL, 0.58 mmol). After 5 h,
the mixture was extracted with dichloromethane (10 mL). The
dichloromethane phase was extracted with FC-72 (3 x 10 mL).
The combined fluorous phases were evaporated. The resulting
acid bromide (88 mg) was diluted with BTF (1 mL) and added to
a suspension of hydroxyethylurea (27 mg, 0.26 mmol),
triethylamine (36 ~L, 0.26 mmol) and 9-dimethylaminopyridine
(3 mg, 25 umol) in dry dioxane (0.50 mL) at 35 °C. After
stirring for 22 h at 35°C, the volatiles were removed in vacuo
and FC-72 (20 mL ), water ( 10 mL) and toluene ( 5 mL) were
added. The combined water/toluene phases were washed with FC-
72 (5 x 10 mL). The combined fluorous phases were filtered
and concentrated to give 23 as a white solid (79 mg , 890): 1H
NMR ( acetone-d6 ) d 8 . 10 ( d, 2 H, J = 8 . 1 Hz ) , 7 . 8 6 ( d, 2 H, J
- 8.1 Hz), 5.96 (bs, 1 H), 5.13 (bs, 2 H), 4.35 (t, 2 H, J =
5.5 Hz) , 3.53 (q, 2 H, J = 5. 5 Hz) , 2. 45 - 2.20 (m, 6 H) , 1.50
- 1.40 (m, 6 H).
82. General Procedure for Biginelli reactions. A solution of
23 (18 mg, 9.6 umol) in THF/BTF (2/1, 0.75 mL) was treated at
25°C with 10 equiv of f3-keto ester, 10 equiv of aldehyde and
concentrated HC1 (1 uL). After 3 days at 50°C, volatiles were
removed in vacuo and FC-84 and toluene (10 mL each) were
added. The toluene phase was extracted with FC-84 (5 x 5 mL).
The combined fluorous phases were filtered and concentrated.
_. . __.. . T.
CA 02259183 1998-12-22
WO 98/00376 8 5 PCTlUS97/11215
The resulting white solid 24 was diluted with THF/BTF (1:1,
0.50 mL) and treated dropwise with a 1 M tributylammonium
fluoride (TBAF) solution in THF (10 uL, 10 umol). After
stirring for 0.5 h at 25°C, volatiles were removed in vacuo
and FC-84 and toluene were added (10 mL each). The fluorous
phase was extracted with toluene (3 x 5 mL). The combined
toluene phases were extracted with sat. aqueous NaHC03
solution (3 x 10 mL) and brine (3 x 10 mL), dried (Na2S04),
filtered and concentrated. The resulting products 25 (Figure
14) were spectroscopically (1H NMR) identical to those
prepared by the conventional procedure.
Although the present invention has been described in
detail in connection with the above examples, it is to be
understood that such detail is solely for that purpose and
that variations can be made by those skilled in the art
without departing from the spirit of the invention except as
it may be limited by the following claims.