Note: Descriptions are shown in the official language in which they were submitted.
CA 02261808 1999-01-2~
1-Azoniabicyclo[2.2.1]heptane derivatives and pharmaceutical compositions
containin~ them
The present invention relates to novel substituted heterocyclic compounds,
to a method of preparing them and to the pharmaceutical compositions containing
5 them as the active principle.
More particularly, the present invention relates to a novel class of
substituted heterocyclic compounds for therapeutic use in pathological phenomenainvolving the tachykinin system, such as: pain (D. Regoli et al., Life Sciences,1987, 40, 109-117), allergy and inflammation (J.E. Morlay et al., Life Sciences,1987, 41, 527-544), circulatory insuff1ciency (J. Losay et al., 1977, Substance P,
Von Euler, I.S. and Pemow ed., 287-293, Raven Press, New York), gastrointestinaldisorders (D. Regoli et al., Trends Pharmacol. Sci., 1985, 6, 481-484), respiratory
disorders (J. Mizrahi et al., Pharmacology, 1982, 25, 39-50), neurological disorders
and neuropsychiatric disorders (C.A. Maggi et al., J. Autonomic Pharmacol., 1993,
13, 23-93), these examples being neither limiting nor exclusive.
In recent years, numerous research studies have been carried out on
tachykinins and their receptors. Tachykinins are distributed throughout both thecentral nervous system and the peripheral nervous system. The tachykinin
receptors have been recognized and are classifled into three types: NKl, NK2, NK3.
Substance P (SP) is the endogenous ligand of the NKl receptors, neurokinin A
(NKA) that of the NK2 receptors and neurokinin B (NKB) that of the NK3 receptors.
The NKl, NK2 and NK3 receptors have been identified in different species.
A review by C.A. Maggi et al. looks at the tachykinin receptors and their
antagonists and gives an account of the pharmacological studies and the
applications in human therapeutics (J. Autonomic Pharmacol., 1993, 13, 23-93).
The following non-peptide compounds may be mentioned among the
antagonists specif1c for the NKl receptor: CP-96345 (J. Med. Chem., 1992, 35,
2591-2600), RP-68651 (Proc. Natl. Acad. Sci. USA, 1991, 88, 10208-10212),
SR 140333 (Curr. J. Pharmacol., 1993, 250, 403-413).
For the NK2 receptor, a non-peptide selective antagonist, SR 48968, has
been described in detail (Life Sci., 1992, 50, PL101-PL106).
As far as the human NK3 receptor is concerned, the selective non-peptide
antagonist (+)-N-[1-[3-[1-benzoyl-3-(3,4-dichlorophenyl)piperid-3-yl]propyl]-4-
phenylpiperid-4-yl]-N-methylacetamide hydrochloride, or SR 142801, has been
described (Peptides and their antagonists in tissue injury, Montreal, Canada, 1994,
CA 02261808 1999-01-2~
July 31 - August 3. C~n~di~n J. Physiol. Pharmacol., 1994, 72 (suppl. 2), 25,
Abst. III. 0. 9.; Life Sci., 1994, 56 (1), 27-32; British Pharmacol. Society,
Canterbury, 1995, April 6 - 8; Eur. J. Pharmacol., 1995, 278 (1), 17-25; 1st Eur.
Congress Pharmacol., Milan, 1995, June 16- 19).
Patent application EP-A-336230 describes peptide derivatives which are
substance P and neurokinin A antagonists useful for the treatment and preventionof asthma.
International patent applications WO 90/05525, WO 90/05729, WO
91/09844 and WO 91/18899 and European patent applications EP-A-0436334, EP-
10 A-0429466 and EP-A-0430771 describe substance P antagonists.
European patent applications EP-A-0428434, EP-A-0474561, EP-A-
512901, EP-A-515240, EP-A-559538, EP-A-591040, EP-A-0625509 and EP-A-
0630887 and international patent applications WO 94/10146, WO 94/29309,
WO 94/26735, WO 95/05377, WO 95/12577, WO 95/16682, WO 95/28389,
15 WO 96/06094 and WO 96/05193 also relate to neurokinin receptor antagonists.
Novel substituted heterocyclic compounds have now been found which are
neurokinin receptor antagonists.
Thus, according to one of its features, the present invention relates to
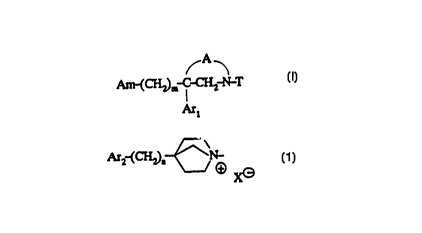
compounds of the formula
~A
Am-(CH2)n,-C-CH,-N-T (I)
Ar~
in which:
- A is a divalent radical selected from:
A ~ ) -O-CO-
A2) -CH2-O-CO-
A3)-O-CH2-CO-
A4) -O-CH2-CH2-
As) -N(Rl )-CO-
A6) -N(RI )-CO-CO-
A7) -N(RI )-CH2-CH2-
A8)-O-CH2-
in which R~ is a hydrogen or a (C~-C4)alkyl;
- m is or 3;
- Arl is a phenyl which is unsubstituted or monosubstituted or polysubstituted by
CA 02261808 1999-01-2~
a substituent selected from a halogen atom, a hydroxyl, a (C~-C4)alkoxy, a
(C,-C4)alkyl, a trifluoromethyl and a methylenedioxy, said substituents being
identical or different; a thienyl which is unsubstituted or substituted by a
halogen atom; a benzothienyl which is unsubstituted or substituted by a
halogen atom; a naphthyl which is unsubstituted or substituted by a halogen
atom; an indolyl which is unsubstituted or N-substituted by a (Cl-C4)alkyl or a
benzyl; an imidazolyl which is unsubstituted or substituted by a halogen atom;
a pyridyl which is unsubstituted or substituted by a halogen atom; or a
biphenyl;
10 - T is a group selected from CH,-Z, -CH(C6H5)2 and -C(C6H5)3; T can also be
the group -CO-B-Z if A is a divalent radical selected from -O-CH,-CH,-,
-N(R~)- CH2-CH2- and -O-CH2-;
- B is a direct bond or a methylene;
- Z is an optionally substituted mono-, di- or tri-cyclic aromatic or
heteroaromatic group; and
- Am is a group of the formula
Ar2-(CH2)"~N-
~3 X(~)
in which:
- Ar, is a pyridyl; a phenyl which is unsubstituted or monosubstituted or
polysubstituted by a substituent selected from a halogen atom, a hydroxyl, a
(C~-C4)alkoxy, a (C~-C4)alkyl, a trifluoromethyl, a nitro and a methylenedioxy,
said substituents being identical or different; a thienyl; a pyrimidyl; or an
imidazolyl which is unsubstituted or substituted by a (C,-C4)alkyl;
- n is zero or one; and
_5 - X Q is an anion;
and the salts thereof, where appropriate, with mineral or organic acids.
The compounds of formula (I) according to the invention include the
racemates as well as the optically pure isomers.
More particularly, the radical Z can be a phenyl group which can be
30 unsubstituted or may contain one or more substituents.
If Z is a phenyl group, it can be monosubstituted or disubstituted, especially
in the 2,4-position but also, for example, in the 2,3-, 4,5-, 3,4- or 3,5-position; it
can also be trisubstituted, especially in the 2,4,6-position but also, for example, in
CA 02261808 1999-01-2j
the 2~3,4-, 2,3,5-, 2,4,5- or 3,4,5-position; tetrasubstituted, for example in the
2,3,4,5-position; or pentasubstituted.
The radical Z can also be a bicyclic aromatic group such as 1- or 2-
naphthyl; or 1-, 2-, 3-, 4-, 5-, 6- or 7-indenyl in which one or more bonds can be
hydrogenated, it being possible for said groups to be unsubstituted or optionally to
contain one or more substituents such as the alkyl, phenyl, cyano, hydroxyalkyl,hydroxyl, oxo, alkylcarbonylamino, alkoxycarbonyl, thioalkyl, halogen, alkoxy ortrifluoromethyl group, in which the alkyl and alkoxy groups are Cl-C4.
The radical Z can also be a pyridyl, thi~di~701yl, indolyl, indazolyl,
10 imidazolyl, benzimidazolyl, benzotriazolyl, benzofuranyl, benzothienyl, benzo-
thiazolyl, benzisothiazolyl, quinolyl, isoquinolyl, benzoxazolyl, benzisoxazolyl,
benzoxazinyl, benzodioxinyl, isoxazolyl, benzopyranyl, thiazolyl, thienyl, furyl,
pyranyl, chromenyl, isobenzofuranyl, pyrrolyl, pyrazolyl, pyrazinyl, pyrimidinyl,
pyridazinyl, indolizinyl, phth~l~7inyl, quinazolinyl, acridinyl, isothiazolyl, iso-
15 chromanyl or chromanyl group, in which one or more double bonds can be
hydrogenated, it being possible for said groups to be unsubstituted or optionally to
contain one or more substituents such as the alkyl, phenyl, cyano, hydroxyalkyl,hydroxyl, alkylcarbonylamino, alkoxycarbonyl or thioalkyl group, in which the
alkyl and alkoxy groups are C~-C~.
In particular, the invention relates to compounds of formula (I) in which:
- Z is Z' and is:
- a phenyl which is unsubstituted or monosubstituted or polysubstituted by a
substituent selected from a halogen atom; a trifluoromethyl; a cyano; a
hydroxyl; a nitro; an amino which is unsubstituted or monosubstituted or
disubstituted by a (Cl-C~)alkyl; a benzylamino; a carboxyl; a (C~-C~0)alkyl; a
(C3-C8)cycloalkyl which is unsubstituted or monosubstituted or polysubstituted
by a methyl; a (C~-C~0)alkoxy; a (C3-C8)cycloalkoxy which is unsubstituted or
monosubstituted or polysubstituted by a methyl; a mercapto; a (C,-
C~0)alkylthio; a formyloxy; a (C~-C6)alkylcarbonyloxy; a formylamino; a (C~-
C6)alkylcarbonylamino; a benzoylamino; a (C~-C4)alkoxycarbonyl; a (C3-
C7)cycloalkoxycarbonyl; a carbamoyl which is unsubstituted or
monosubstituted or disubstituted by a (C ~-C4)alkyl; a ureido which is
unsubstituted or monosubstituted or disubstituted in the 3-position by a (C~-
C~)alkyl or a (C3-C7)cycloalkyl; a phenyl which is unsubstituted or
monosubstituted or polysubstituted by a halogen, a trifluoromethyl, a (C~-
CA 02261808 1999-01-2~
C4)alkyl, a hydroxyl or a (C,-C4)alkoxy, said substituents being identical or
different; and a (pyrrolidin- 1 -yl)carbonylamino, said substituents being
identical or different;
- a naphthyl which is unsubstituted or monosubstituted or polysubstituted by a
halogen, a trifluoromethyl, a (C,-C4)alkyl, a hydroxyl or a (C~-C4)alkoxy; or
- a pyridyl, a thienyl, an indolyl, a quinolyl, a benzothienyl, an imidazolyl or a
furyl.
It is possible to form salts of the compounds of formula (I) other than the
quaternary ammonium salts. These salts include those with mineral and organic
10 acids which permit a suitable separation or crystallization of the compounds of
formula (I), such as picric acid, oxalic acid or an optically active acid, for example
a mandelic or camphosulfonic acid, and those which form pharmaceutically
acceptable salts such as the hydrochloride, hydrobromide, sulfate, hydrogensulfate,
dihydrogenphosphate, methanesulfonate, methylsulfate, maleate, fumarate,
15 naphthalene-2-sulfonate, benzenesulfonate, gluconate, citrate, isethionate or p-
toluenesulfonate .
The anions X Q are those normally used to salify quaternary ammonium ions
and are preferably chloride, bromide, iodide, acetate, hydrogensulfate, methane-sulfonate, paratoluenesulfonate and benzenesulfonate ions.
O It is preferable to use the pharmaceutically acceptable anions, for example
chloride, methanesulfonate or benzenesulfonate.
In the present description, the alkyl groups or alkoxy groups are linear or
branched; halogen atom is understood as meaning a chlorine, bromine, fluorine oriodine atom.
In the substituents of the group Z = phenyl, (C,-C,O)alkyl is understood as
meaning for example a methyl, an ethyl, an n-propyl, an isopropyl, an n-butyl, an
isobutyl, a sec-butyl, a tert-butyl, a pentyl or n-pentyl, a hexyl or n-hexyl, a heptyl
or n-heptyl, an octyl or n-octyl, a nonyl or n-nonyl or a decyl or n-decyl;
(C3-C8)cycloalkyl optionally substituted by a methyl is understood as meaning for
30 example a cyclopropyl~ a cyclobutyl, a cyclopentyl, a 1-, 2- or 3-methylcyclopentyl,
a cyclohexyl, a 1-, 2-, 3- or 4-methylcyclohexyl, a cycloheptyl or a cyclooctyl;(Cl-C~u)alkoxy is understood as meaning for example a methoxy, an ethoxy, an n-
propoxy, an isopropoxy, an n-butoxy, an isobutoxy, a sec-butoxy, a tert-butoxy, a
pentoxy, a hexyloxy, a heptyloxy, a nonyloxy or a decyloxy; (C3-C8)cycloalkoxy
35 optionally substituted by a methyl is understood as meaning for example a
CA 02261808 1999-01-2~
cyclopropoxy, a cyclohexyloxy, a 1-, 2-, 3- or 4-methylcyclohexyloxy, a cyclo-
heptyloxy or a cyclooctyloxy; (C~-C~0)alkylthio is understood as meaning for
example a methylthio, an ethylthio, an n-propylthio, an isopropylthio, an n-
butylthio, an isobutylthio, a sec-butylthio, a tert-butylthio, a pentylthio, a hexylthio,
S a heptylthio, an octylthio, a nonylthio or a decylthio; (C~-C6)alkylcarbonyloxy is
understood as meaning for example an acetoxy, a propionyloxy, a butyryloxy, a
valeryloxy, a caproyloxy or a heptanoyloxy; (C ~ -C6)alkylcarbonylamino is
understood as meaning for example an acetylamino, a propionylamino, a butyryl-
amino, an isobutyrylamino, a valerylamino, a caproylamino or a heptanoylamino;
10 (C~-C4)alkoxycarbonyl is understood as meaning for example a methoxycarbonyl,an ethoxycarbonyl, an n-propoxycarbonyl, an isopropoxycarbonyl, an n-butoxy-
carbonyl, an isobutoxycarbonyl, a sec-butoxycarbonyl or a tert-butoxycarbonyl;
and (C3-C7)cycloalkoxycarbonyl is understood as meaning for example a cyclo-
propoxycarbonyl, a cyclobutoxycarbonyl, a cyclopentoxycarbonyl, a
15 cyclohexyloxycarbonyl or a cycloheptyloxycarbonyl.
Advantageously, the radical Z is a phenyl which is unsubstituted or
monosubstituted or polysubstituted by a halogen atom, more particularly a
chlorine, fluorine or iodine atom, a trifluoromethyl7 a (C~-C~)alkyl, a hydroxyl or a
(C,-C~)alkoxy; a naphthyl which is unsubstituted or monosubstituted or poly-
20 substituted by a halogen, a trifluoromethyl, a (C~-C4)alkyl, a hydroxyl or a
(C,-C~)alkoxy; a pyridyl; a thienyl; an indolyl; a quinolyl; a benzothienyl; or an
imidazolyl.
One group of preferred compounds according to the present invention
consists of those of the formula
,Aa~
Am-CH2-CH,-C-CH2-N-CH2-Za (la)
Arla
in which:
- Aa is a divalent radical selected from: -O-CO-, -CH2-O-CO-,
-O-CH2-CO,-N(R~)-CO- and -N(R~)-CO-CO-,
in which R~ is a hydrogen or a (C,-C4)alkyl;
30 - Am is a group of the formula
~ C
Ar2 (CH,)n~--N--
X~)
CA 02261808 1999-01-2~
- nisOor 1;
- X(~) is a pharmaceutically acceptable anion;
- Ar2 is as defined above for a compound of formula (I);
- Arla is a phenyl which is unsubstituted or monosubstituted or polysubstituted
by a substituent selected from a halogen atom, a hydroxyl, a (C~-C4)alkoxy, a
(C I -C4)alkyl and a trifluoromethyl, said substituents being identical or
different; and
- Za is a phenyl which is unsubstituted or monosubstituted or polysubstituted by
a substituent selected from a halogen atom, a trifluoromethyl, a (C~-C~O)alkyl, a
(Cl-CIO)alkoxy and a hydroxyl, said substituents being identical or different.
Among these compounds, those of the formula
, Aa
Am-CH2-CH,-C-CH2-N-CH2-Z'a (I'a)
Ar'la
in which:
- Aa is as defined above for a compound of formula (Ia);
15 - Ama is a group of the formula
~ ,3
- xQ is a pharmaceutically acceptable anion;
- Ar'la is a 3,4-dichlorophenyl or a 3,4-difluorophenyl; and
- Z'a is a 3,5-bis(trifluoromethyl)phenyl, a 3,5-dimethylphenyl or a 274-bis-
(trifluoromethyl)phenyl;
are particularly preferred.
Another group of preferred compounds according to the invention consists
of those of the formula
,Ab ~
Am-CH2-CH,-C- CH2-N-CO-B-Za (Ib)
Arla
25 in which:
- Ab is the divalent radical -O-CH2-CH2-, -N(RI)-CH2-CH2- or -O-CH2-,
in which R~ is a hydrogen or a (C~-C4)alkyl;
- Am is a group of the formula
CA 02261808 1999-01-2~
Ar, (CH~)n~ N--
X¢
- nisOor l;
- B is a direct bond or a methylene;
- X~ is a pharmaceutically acceptable anion;
5 - Ar, is as defined above for a compound of formula (I); and
- Arla and Za are as defined above for a compound of formula (Ia).
Among these compounds, those of the formula
Al~
Ama-CH7-CH,-C-CH2-N-CO-B-Z"a (I~b)
Ar'~a
in which:
10 - B is a direct bond or a methylene;
- Ama is as defined above for a compound of formula (I'a);
- Ab is as defined above for a compound of formula (Ib);
- Ar'la is as defined above for a compound of formula (I'a); and
- Z"a is a phenyl substituted in the 3-position by a halogen or a (C~-C~O)alkoxygroup if B is a methylene, or Z"a is a 3,5-bis(trifluoromethyl)phenyl, a 3,5-
dimethylphenyl or a 2,4-bis(trifluoromethyl)phenyl if B is a direct bond;
are particularly preferred.
According to another of its features, the present invention relates to a
method of preparing the compounds of formula (I) and the salts thereof,
20 characterized in that:
I ) a compound of the formula
A
E-O-(CH2)m-C-CH~-NH (II)
Arl and A are as defined above for a compound of
formula (I) and E is hydrogen or an O-protecting group, is treated
- either with a functional derivative of an acid of the formula
HOCO-B-Z (III)
in which B and Z are as defined above for (I), if it is intended to prepare a
compound of formula (I) in which T is -CO-B-Z,
- or with a halogenated derivative of the formula
CA 02261808 1999-01-2~
Hal-CH,-Z (IV)
in which Z is as defined above and Hal is a halogen, preferably bromine or
chlorine, if it is intended to prepare a compound of formula (I) in which T is -CH2-
Z,
- or with a halogenated derivative of the formula
Hal-CH(C6Hs)2 (V)
if it is intended to prepare a compound of formula (I) in which T is a group
-CH(C6Hs)2,
- or with a halogenated derivative of the formula
Hal-C-(C6Hs)3 (VI)
if it is intended to prepare a compound of formula (I) in which T is a group
-C(C6Hs)3~
to give a compound of the formula
, A~
E-O-(CH~)",-C-CH,-N-T (VII)
Arl
2) the O-protecting group is removed, if apl)lopliate, by reaction with an
acid or a base to give the alcohol of the formula
~A ~
HO-(CH2)m-C-CH2-N-T (VIII)
Arl
3) the alcohol (VIII) is treated with a compound of the formula
-Cl (IX)
20 in which Y is a methyl, phenyl, tolyl or trifluoromethyl group, to give a compound
of the forrnula
, A
Y-SO2-O-(CH2)",-C-CH2- N-T (X)
Ar,
4) the compound (X) is reacted with a cyclic tertiary amine of the formula
Ar2-(CH2) ~ ' N (XI)
25 in which Ar2 and n are as defined for a compound of formula (I); and
5) the resulting product is isolated in the forrn of a sulfonate and, if
CA 02261808 1999-01-2~
appropriate, a sulfonic acid salt, or optionally the anion and, if apl)ropliate, the
resulting acid salt are exchanged with another anion and, if appropriate, another
salt with a pharmaceutically acceptable mineral or organic acid.
If E is an O-protecting group, this is selected from the conventional O-
protecting groups well known to those skilled in the art~ such as, for example,
tetrahydropyran-2-yl, benzoyl or a (Cl-C4)alkylcarbonyl.
In step I ), the functional derivative of the acid (III) used is the acid itself or
alternatively one of the functional derivatives which react with amines, for example
an anhydride, a mixed anhydride, the acid chloride or an activated ester such as the
10 paranitrophenyl ester.
If the acid of formula (III) itself is used, the reaction is carried out in the
presence of a coupling agent used in peptide chemistry, such
as 1,3- dicyclohexylcarbodiimide or benzotriazol-l-yloxytris(dimethylamino)-
phosphonium hexafluorophosphate, in the presence of a base such as triethylamine15 or N,N-diisopropylethylamine, in an inert solvent such as dichloromethane or N,N-
dimethylformamide, at a temperature between 0~C and room temperature.
If an acid chloride is used, the reaction is carried out in an inert solvent such
as dichloromethane or benzene, in the presence of a base such as triethylamine or
N-methylmorpholine, at a temperature between -60~C and room temperature.
~0 If a halogenated derivative of formula (IV), (V) or (Vl) is used~ the reaction
is carried out in an inert solvent such as tetrahydrofuran, N,N-dimethylfonnamide
or dimethyl sulfoxide, in the presence of a base such as potassium te)t-butylate,
sodium hydride or lithium diisopropylamide, at a temperature between 0~C and
80~C.
The resulting compound of formula (VII) is deprotected in step 2), if
appropriate, by the methods known to those skilled in the art. For example, if E is
a tetrahydropyran-2-yl group, the deprotection is effected by acid hydrolysis using
hydrochloric acid in a solvent such as ether or methanol or a mixture of these
solvents, or using pyridinium p-toluenesulfonate in a solvent such as methanol, or
30 using an Amberlyst~ resin in a solvent such as methanol. The reaction is carried
out at a temperature between room temperature and the reflux temperature of the
solvent. If E is a benzoyl group or a (Cl-C4)alkylcarbonyl group, the deprotection
is effected by hydrolysis in an alkaline medium using for example an alkali metal
hydroxide such as sodium hydroxide, potassium hydroxide or lithium hydroxide, in35 an inert solvent such as water, methanol, ethanol or dioxane or a mixture of these
CA 02261808 1999-01-2~
solvents, at a temperature between 0~C and the reflux temperature of the solvent.
In step 3), the reaction of the alcohol of formula (VIII) with a sulfonyl
chloride of formula (IX) is carried out in the presence of a base such as
triethylamine, pyridine, N,N-diisopropylethylamine or N-methylmorpholine, in an
inert solvent such as dichloromethane, benzene or toluene, at a temperature
between -20~C and the reflux temperature of the solvent.
In step 4), if a compound of formula (X) is reacted with a compound of
formula (XI), the reaction is carried out in an inert solvent such as N,N-
dimethylformamide, acetonitrile, methylene chloride, toluene or propan-2-ol, in the
10 presence or absence of a base. If a base is used, it is selected from organic bases
such as triethylamine, N,N-diisopropylethylamine or N-methylmorpholine, or from
alkali metal carbonates or bicarbonates such as potassium carbonate, sodium
carbonate or sodium bicarbonate. In the absence of a base, the reaction is carried
out using an excess of the compound of formula (XI), in the presence of an alkali
15 metal iodide such as potassium iodide or sodium iodide. If -A- in the compound of
formula (X) is the divalent radical -0-C0- or -CH2-0-C0-, the reaction is carried
out at a temperature between room temperature and 80~C. If -A- in the compound
of formula (X) is the divalent radical -0-CH2-C0-, -0-CH2-CH~-, -N(R~)-C0-C0-,
-N(R~)-CH,-CH~-? -N(R~)-C0- or -0-CH2-, the reaction is carried out at a
0 temperature between room temperature and 1 00~C.
The resulting products of forrnula (I) are isolated in the form of a sulfonate
(YS030 ) or alternatively the sulfonate anion of the resulting quaternary salt is
optionally exchanged with another pharmaceutically acceptable anion.
The sulfonate anion YS03 Q origin~ting from the reaction between the
~5 compound of formula (XI) and the compound of formula (X) can be exchanged, insitu or after isolation of the compound of formula (I) n which X(3 is the ion YS03~),
with another ion xQ by the conventional methods, for example by exchange in
solution with saturated sodium chloride solution or with hydrochloric acid solution
if X (~ is a chloride anion, or by exchange of the anion via elution of the compound
(I) on an ion exchange resin, for example Amberlite IRA68~ or Duolite A375~.
The compounds of formula (II) are prepared by a variety of procedures.
The cornpounds of fomlula (lI) in which -A- is the divalent radical ~H2~C0- and E is
hyd~gen or an O~otecbng group are p~ed acw[ding to SCHEME 1 below, in which m and Ar~
are as defined for a compound of fonnula (~) and Pr is an O~tecbng g[~up as defir~l above for E.
.
CA 02261808 1999-01-25
SCHEME 1
Pr-O-(CH2)m-Br + Arl-CH2-CN
(XII) I (XIII)
al
Pr-O-(CH~)m-CH-CN (XIV)
Ar~
bl
CH,-OH
Pr-O-(CH2)m-f-CN (XV)
Ar~
c
.
CH~-OH
Pr-O-(CH, ),ll-C- CH~-NH2 (XVI)
Ar
dl
/o\ ~/o
f H2 C II: -A- = -CH2-O-CO-,
Pr-o-(cH2)m-c\~cH ~NH ~ E = -Pr
Ar,
el
~\ C '~O II: -A- = -CH2-O-CO-,
HO-(CH~)n,-C\cH ~NH ~ E = H
Ar,
.
CA 02261808 1999-01-2~
In step _ of SCHEME 1, a compound of formula (XII) is reacted with a
compound of formula (XIII) by the method described in patent applications EP-A-
0428434 and EP-A-0474561.
The resulting compound (XIV) is reacted in step b 1 with aqueous
5 formaldehyde solution in the presence of a base such as 1,8-diazabicyclo[5.4.0]-
- undec-7-ene, in a solvent such as 1,2-dimethoxyethane, at a temperature between
room temperature and the reflux temperature of the solvent.
The nitrile derivative of formula (XV) is reduced in step cl to give the
primary amine of formula (XVI). This reduction can be effected by means of
hydrogen in the presence of a catalyst such as Raney~ nickel, platinum oxide or
palladium-on-charcoal, in an inert solvent such as an alcohol, for example ethanol,
by itself or mixed with aqueous ammonia, or by means of a reducing agent such aslithium aluminum hydride, diisobutylaluminum hydride or borane in THF, in a
solvent such as toluene, hexane, petroleum ether, xylene or tetrahydrofuran. Thereaction is carried out at a temperature between 0~C and 70~C.
In step ~, the compound (XVI) is reacted with a reactive derivative of
carbonic acid, such as phosgene, in solution in toluene, or 1, I '-
carbonyldiimidazole, in the presence of a base such as triethylamine, N,N-
diisopropylethylamine or N-methylmorpholine, in a chlorinated solvent such as
dichloromethane or 1,2- dichloroethane, or an ether such as tetrahydrofuran, at a
temperature between -70~C and room temperature, to give a compound of the
expected formula (II) in which E is an O-protecting group.
Using the methods described above, the O-protecting group is removed by
hydrolysis (step e 1) to give the compound of formula (II) in which E is hydrogen.
The compounds of formula (II) in which -A- is the divalent radical -O-CH,-
CO- and E is hydrogen or an O-protecting group are prepared according to
SCHEME 2 below, in which m and Arl are as defined for a compound of formula
(I). Prl and Pr~ are the O-protecting group Pr as defined above for E; more
particularly, Prl is an O-protecting group hydrolyzable in an acid medium and Pr2
is an O-protecting group hydrolyzable in a basic medium.
CA 02261808 1999-01-25
14
SCHEME 2
H~ ~ O (XVII)
C'
Arl
a2
.
HO-CH-CN (XVIII)
Ar~
b2
.
Pr~-O-CH-CN (XIX)
Ar,
c~
O-Pr~
Pr2-O-(CH7)m-C-CN (XX)
Ar~
d2
.
O-Prl
Pr2~0~(CH2)m~C~CH2~NH2 (XXI)
Ar,
j2 1 e2
~ OH O-Pr~ O
Pr2-o-(cH2)m-c-cH2-NH2Pr2-o-(cH2)m-f-cH2-NH-c-cH2
Ar~ Ar,
(XXIV) (XXII)
k2 f2
.
CA 02261808 1999-01-2~
OH O
Pr2-O-(CH2)m-~-CH2~NH~ C-CH2-Hal (XXIII)
Arl
g2 h2
o,CH~\C~~ o~CH2\C~~
Pr2-O-(CH2)m~c\cH ,NH HO-(CH2) -C\ NH
Arl Arl
II:-A-=-O-CH2-CO- II:-A-=-O-CH2-CO-
E=Pr2 ,E=H
li2
o/CH2\ C~~
Prl-o-(cH~)m-c\cH ~NH
Ar,
II: -A- = -O-CH2-CO-
E = Prl
In step a2 of SCHEME 2, the synthesis of a cyanohydrin of formula (XVIII)
from an aldehyde of formula (XVII) is effected by the methods well known to
those skilled in the art, such as, for example, the one described in Organic
Syntheses; Wiley, New York, 1932; Collect. vol. 1, p. 336, or by an adaptation of
this method utilizing the action of sodium metabisulfite and potassium cyanide in
aqueous solution.
In step b2, the hydroxyl group of the compound of fonnula (XVIII) is
protected by the methods known to those skilled in the art.
The resulting compound of formula (XIX) is treated in step c2 with a strong
base~ such as lithium diisopropylamide, potassium tert-butylate or sodium hydride,
to give a carbanion, which is reacted with a compound of the formula
CA 02261808 1999-01-2~
16
Hal-(CH2)m-O-Pr2, in which Hal is a halogen, preferably bromine or chlorine, to
give the compound of formula (XX). The reaction is carried out in an inert solvent
such as an ether (for example tetrahydrofuran, diethyl ether or 1,2-dimethoxy-
ethane), an amide (for example N,N-dimethylformamide) or an aromatic hydro-
S carbon (for example toluene or xylene), at a temperature between -70~C and
+60~C.
The nitrile derivative of formula (XX) is reduced in step d2 by the methods
described above to give the primary amine of formula (XXI).
In step e2, the compound of fonnula (XXI) is reacted with a compoulld of
10 the formula Hal-CO-CH2-Hal, in which Hal is a halogen, preferably chlorine orbromine, in the presence of a base such as a tertiary amine (for example
triethylamine, N-methylmorpholine or pyridine), to give a compound of formula
(XXII). The reaction is carried out in an inert solvent such as a chlorinated solvent
(for example dichloromethane, dichloroethane or chloroform), an ether (for
15 example tetrahydrofuran or dioxane) or an amide (for example N,N-dimethyl-
formamide), at a temperature between -70~C and room temperature.
In step f2, the O-protecting group Pr~ is removed from the compound of
formula (XXII) by acid hydrolysis using the methods described above.
Alternatively, the O-protecting group Pr~ is removed from the compound of
20 formula (XXI) by acid hydrolysis in step j2, after which the resulting compound
(XXIV) is reacted in step k2 with a compound of the formula Hal-CO-CH2-Hal by
the methods described above in step e2.
The resulting compound of formula (XXIII) is cyclized in the presence of a
base to give the compound of the expected fonnula (II). If it is desired to obtain a
25 compound of formula (II) in which E is a protecting group Pr2, a base such as an
alkali metal carbonate (for example potassium carbonate), an alkali metal hydride
(for example sodium hydride) or potassium tert-butylate is used in an inert solvent
such as an aromatic hydrocarbon (for example xylene or toluene), an amide (for
example N,N-dimethylformamide) or an ether (for example tetrahydrofuran), at a
30 temperature between -30~C and the reflux temperature of the solvent (step 2). If
it is desired to obtain a compound of fonnula (II) in which E is hydrogen, a base
such as an alkali metal hydroxide (for example sodium hydroxide or potassium
hydroxide) in concentrated aqueous solution is used in a solvent such as an alkanol
(for example propan-2-ol) or an amide (for example N,N-dimethylformamide) or a
35 mixture of these solvents, at a temperature between room temperature and the
CA 02261808 1999-01-2~
reflux temperature of the solvent (step h2).
If appropliate, a compound of formula (II) in which E is an O-protecting
group Prl is prepared in step i2 by the methods known to those skilled in the art.
- The compounds of formula (II) in which -A- is the divalent radical -O-CH2-
5 CH,- and E is hydrogen or an O-protecting group are prepared according to
SCHEME 3 below, in which m and Ar~ are as defined for a compound of formula
(I) and Prl and Pr2 are as defined in SCHEME 2 abo~/e.
SCHEME 3
, CH,, C~~ (II): -A- = O-CH2-CO-
E-O-(CH?),l,-C, ,NHE = H, Prl or Pr2
CH,
Arl
a3
O, CH2, CH (II): -A- = O-CH2-CH2-
E-O-(CH~)m-C--CH / NH~ E = H~ Prl or Pr2
Ar~
In step a3 of SCHEME 3, a compound of formula (II) in which -A- is the
divalent radical -O-CH2-CO- and E is hydrogen or an O-protecting group, obtainedaccording to SCHEME 2, is reduced. The reduction is effected by means of a
15 reducing agent such as lithium aluminum hydride, diisobutylaluminum hydride,
sodium borohydride or borane in THF, in an inert solvent such as tetrahydrofuran,
diethyl ether, 1,2-dimethoxyethane or toluene, at a temperature between room
temperature and the reflux temperature of the solvent.
The compounds of formula (II) in which -A- is the divalent radical -O-CO-
20 and E is hydrogen or an O-protecting group are prepa ed according to SCHEME 4below, in which m and Arl are as defined for a compound of formula (I) and Pr
and Pr, are as defined in SCHEME 2 above.
CA 02261808 1999-01-2
18
SCHEME 4
O-Prl
Pr-O-(CH2)m-C-CH2-NH2 (XXI)
Ar~
a4
OH
Pr-O-(CH,)m-C-CH2-NH (XXIV)
Ar~
b4
.
o C II:-A-=-O-CO-
Pr2-O-(CH2)~ll~c\cH ~N ' E = Pr2
Ar~
c4
o C ~ II:-A-=-O-CO---
HO-(CH2) -C\ NH ~ E =H
Ar~
In step a4 of SCHEME 4 the O-protecting group Prl of the compound of
5 fonnula (XXI) obtained in step d2 of SCHEME 2 is removed by acid hydrolysis
using the methods described above.
The resulting compound of formula (XXIV) is reacted in step b4 with a
reactive derivative of carbonic acid such as l l -carbonyldiimidazole phosgene in
toluene or p-nitrophenyl chloroformate in the presence of a base such as
10 triethylamine NN-diisopropylethylamine or N-methylmorpholine to give a
compound of the expected formula (II) in which E is an O-protecting group. The
reaction is carried out in an inert solvent such as a chlorinated solvent (for example
CA 02261808 1999-01-2
19
1,2-dichloroethane or dichloromethane), an ether such as tetrahydrofuran, an amide
such as N,N-dimethylformamide or an aromatic solvent such as toluene, at a
temperature between -60~C and room temperature.
Using the methods described above, the O-protecting group Pr2 is removed
5 by basic hydrolysis (step c4) to give the compound of formula (II) in which E is
hydrogen.
The compounds of formula (II) in which -A- is the divalent radical -N(RI)-
CO-CO- and E is hydrogen or an O-protecting group are prepared according to
SCHEME 5 below, in which m, Arl and R~ are as defined for a compound of
10 formula (I) and Pr~ is as defined above.
SCHEME 5
H ~ ~ (XVII)
Arl
a5
.
Rl-NH-CH-CN (XXV)
Arl
b5
.
R~
Boc-N-CH-CN (XXVI)
Ar,
c5
Boc-N-Rl
Prl-O-(CH,)",-C-CN (XXVII)
Arl
CA 02261808 1999-01-25
dS
Boc-N-R~
Prl-O-(CH2)m-C-cH2~NH2 (XXVIII)
Ar,
le
NH-R~
HO-(CH2)m-f-CH2-NH2 (XX )
f5
o
N/ C\ c~~
HO-(CH2)m-f \CH / NH
Ar~
(II): -A- = -N(RI )-CO-CO-
E=H
gS
o
R~ \ ~ C c~ ~
Prl-o-(cH2)m-c\cH ~N
Ar~
(II): -A- = -N(R~)-CO-CO-
E = Prl
In step aS of SCHEME S an a-amino nitrile compound of formula (XXV)
is prepared from an aldehyde of formula (XVII) by the method described in
Tetrahedron Letters 1984 25 (41) 4583-4586 using an amine of the formula
CA 02261808 1999-01-2~
21
H~N-R~ .
The amino group of the compound of forrnula (XXV) is protected in step
bS by an N-protecting group such as tert-butoxycarbonyl (Boc) or benzyloxy-
carbonyl, for example, using the methods known to those skilled in the art. The
5 tert-butoxycarbonyl group is illustrated in SCHEME 5 above.
The resulting compound of formula (XXVI) is treated in step c5 with a
strong base to form a carbanion, which is reacted with a compound of the formulaHal-(CH2)m-O-Prl to give a compound of formula (XXVII). The reaction is carried
out by the method described in step c2 of SCHEME 2.
The nitrile derivative of formula (XXVII) is reduced in step dS by the
methods described above to give the primary amine of formula (XXVIII).
In step e5, the O-protecting group and the N-protecting group are removed
from the compound of formula (XXVIII) by acid hydrolysis with hydrochloric acid
or trifluoroacetic acid, for example, in a solvent such as an alcohol (for example
methanol), an ether (for example diethyl ether, dioxane or tetrahydrofuran) or achlorinated solvent (for example dichloromethane), at a temperature between 0~C
and the reflux temperature of the reaction mixture.
In step f5, the compound of the expected formula (II) is prepared by
application or adaptation of the method described by R. Granger, H. Orzalesi andY. Robbe in Trav. Soc. Pharm. Montpellier, 1965, 25, Fasc. 4, 313-317, using thereaction of a compound of formula (XXIX) with diethyl oxalate in an alcoholic
solvent such as ethanol, or an aromatic solvent such as toluene, or a mixture ofthese solvents, at a temperature between room temperature and the reflux
temperature of the reaction mixture.
~5 If appropriate, a compound of formula (II) in which E is an O-protecting
group Prl is prepared in step ~5 by the methods known to those skilled in the art.
The compounds of formula (II) in which -A- is the divalent radical -N(RI)-
CH,-CH,- and E is hydrogen or an O-protecting group are prepared according to
SCHEME 6 below, in which m, R~ and Arl are as defined for a compound of
fonnula (I) and Pr~ is an O-protecting group as defined above for E.
CA 02261808 1999-01-2~
SCHEME 6
R,\ /C c~O
(II):-A-=-N(R~)-CO-CO-
Pr~ o-(CH )n,-C~CH ' ~ E = Pr~
Arl
a6
.
R~ \ /CH ~ CH
, (II):-A-=-N(Rl)
Prl-o-(cH~)nl-c\cH, NH l E = Pr~
Arl
b6
.
R~\N~' ?~CH? ~
(II): -A- = -N(R~ )-CH~-CH?-
HO-(CH2),,,-C~cH ~ ~ E = H
Arl
In step a6 of SCHEME 6, a compound of formula (II) in which -A- is the
5 divalent radical -N(R~)-CO-CO- and E is an O-protecting group, obtained in step
~5 of SCHEME 5, is reduced. The reduction is effected by means of a reducing
agent such as lithium aluminum hydride, in an inert solvent such as an ether (for
example tetrahydrofuran, 1,2-dimethoxyethane or diethyl ether) or an aromatic
solvent such as toluene, at a temperature between room temperature and the reflux
10 temperature of the solvent.
If appropriate, the O-protecting group is r moved in step b6 by acid
hydrolysis using the methods described above to give the compound of formula (II)
in which E is hydrogen.
The compounds of formula (II) in which -A- is the divalent radical -N(R~)-
15 CO- and E is hydrogen or an O-protecting group are prepared according to
SCHEME 7 below, in which m, R~ and Ar~ are as defined for a compound of
CA 02261808 1999-01-2~
formula (I) and Pr~ is as defined in SCHEME 2 above.
SCHEME 7
NH-R~
HO-(CH2)m-C-CH2-NH2 (XXIX)
Arl
I a7
NH-RI
Prl-O-(CH2)m-C-cH2~NH2 (XXX)
Arl
b7
.
R~ ~ ~~ ~
N C II: -A- = -N(R~ )-CO-
Pr~-O-(CH~ ,-C~CH -NH ~ E=Pr
Ar,
c7
.
R~ "0
N C II:-A-=-N(R,)-CO-
HO-(CH~)n~-c\cH "NH ' E = H
Ar,
In step a7, the hydroxyl of the compound of formula (XXIX), obtained in
step e5 of SCHEME 5, is protected by the methods known to those skilled in the
art.
In step b7, the resulting compound of formula (XXX) is reacted with a
10 reactive derivative of carbonic acid, such as l,l'-carbonyldiimidazole, phosgene in
toluene, or p-nitrophenyl chloroformate, in the presence of a base such as
triethylamine, N,N-diisopropylethylamine or N-methylmorpholine, to give an
expected compound of forimula (II) in which E is an O-protecting group. The
CA 02261808 1999-01-2~
24
reaction is carried out in an inert solvent such as a chlorinated solvent (for example
1,2-dichloroethane or dichloromethane), an ether (for example tetrahydrofuran), an
amide (for example N,N-dimethylformamide) or an aromatic solvent (for example
toluene), at a temperature between -60~C and 60~C.
If applopliate, a compound of formula (II) in which E is hydrogen is
prepared in step c7 by the methods known to those skilled in the art.
The compounds of formula (II) in which -A- is the divalent radical -O-CH2-
and E is hydrogen or an O-protecting group are prepared according to SCHEME 8
below, in which m and Ar~ are as defined for a compound of formula (I) and Pr, is
10 as defined in SCHEME 2 above.
SCHEME 8
OH
Pr2-O-(CH2)n,-C-cH2~NH2 (XXIV)
Ar~
a8
.
O CH, Il:-A-=-O-CH2-
Pr,-O-(CH2)m-C\cH ,N ~ E = Pr2
Arl
!b8
O CH2 II:-A-=-O-CH2-
HO-(CH2) -C NH E = H ,1
In step a8 of SCHEME 8, a compound of formula (XXIV) is reacted with
aqueous formaldehyde solution in an inert solvent such as tetrahydrofuran, at a
temperature between room temperature and the reflux temperature of the solvent,
to give a compound of the expected formula (II) in which E is an O-protecting
group.
CA 02261808 1999-01-2~
Using the methods described above, the O-protecting group Pr2 is removed
by basic hydrolysis (step b8) to give the compound of formula (n) in which E is
hydrogen.
The compounds of formula (XI) are prepared by known methods such as
5 those described in the following publications:
- J. Chem. Soc., 1937, 1523-1526;
- J. Chem. Soc., 1938, 400.
The compounds of formula (XI) can also be prepared according to
SCHEME 9 below, in which n and Ar2 are as defined for a compound of formula
10 (I) and Bz is the benzyl radical.
CA 02261808 1999-01-2
26
SCHEME 9
Ar2-(cH2)n /--\
~ NH
HOOC/ \J
~ (XXXI) ~
Ar~-(cH2)n / \ Ar~-(cH2)n /--\
HOOC>~/N-Bz HO-CH>~/NH
(XXXII) \ / (XXXIV)
b9~ ~d9
Ar~-(cH2)n
~( N-Bz
HO-CH2 --/
(XXXIII)
Ar,-(CH,~N-Bz, CH3SO3
(XXXV)
Ar2-(CH2)~N
(XI)
In step a9 of SCHEME 9, the nitrogen atom of the piperidine of fonnula
5 (XXXI) is protected by a beuzyl group using the methods known to those skilled in
the art.
The carboxyl group in the 4-position of the resulting piperidine of formula
(XXXII) is reduced in step b9 to give the compound of formula (XXXIII)
substituted in the 4-position by a hydroxymethyl group. The reduction is effected
10 by means of a reducing agent such as borane in THF or the borane/dimethyl sulfide
CA 02261808 1999-01-2~
complex, in an inert solvent such as tetrahydrofuran, diethyl ether, 1,2-
dimethoxyethane or dichloromethane, at a temperature between room temperature
and the reflux temperature of the solvent.
The compound of formula (XXXIII) can also be obtained from the
5 compound of formula (XXXI) by reduction of the carboxyl group (step c9),
followed by protection of the nitrogen of the resulting piperidine of formula
(XXXIV) (step d9) by the methods mentioned above.
In step e9, the compound (XXXIII) is reacted with methanesulfonyl
chloride, in the presence of a base such as triethylamine, to give the cyclized
10 compound of formula (XXXV) in quaternary ammonium form. The reaction is
carried out in an inert solvent such as dichloromethane or toluene, at a temperature
between -20~C and the reflux temperature of the solvent.
The compound (XXXV) is deprotected in step f9 by the methods known to
those skilled in the art. This gives the expected compounds of forrnula (XI).
lS The piperidines of formula (XXXI) are known or are prepared by known
methods such as those described in international patent application WO 94/26735.In particular, a compound of formula (XXXI) in which n = 1 can be
prepared by the acid hydrolysis of a corresponding piperidine substituted in the 4-
position by a cyano group, which itself is obtained by reacting a 4-cyanopiperidine
20 with a halide of the formula Ar~-CH,-Hal in the presence of a base such as sodium
diisopropylamide.
The 4-cyanopiperidine is obtained by reacting isonipecotamide with
phosphorus oxychloride.
The enantiomers of the compounds according to the invention, of the
25 formula
~A
Am-(cH2)nl-c*-cH~-N-T (I*)
Ar
in which:
- ' *" means that the carbon atom carrying this label has the determined (+) or (-)
absolute configuration; and
30 A and T are as defined for the compounds of formula (I);
and the salts thereof, where appropriate, with mineral or organic acids, are novel
compounds which form part of the invention.
Resolution of the racemic mixtures of the compounds of formula (I) makes
CA 02261808 1999-01-2
28
it possible to isolate the enantiomers of formula (I*). It is preferable however to
resolve the racemic mixtures from a compound of formula (II) or an intermediate
which is useful for the preparation of a compound of formula (II).
Thus if it is desired to prepare the enantiomers (I*) of the compounds of
S formula (I) in which -A- is the divalent radical -CH -O-CO- the racemic mixture
of an intermediate of the formula
CH2-OH
HO-(CH2),l,-C-CH.-NH (XXXVI)
Ar~ are as defined for a compound of formula
(I) which is obtained by removal of the O-protecting group Pr from a compound offormula (XVI) by the methods described above is resolved.
If it is desired to prepare the enantiomers (I*) of the compounds of formula
(I) in which -A- is the divalent radical -O-CO- -O-CH2-CO- -O-CH2-CH2- or -O-
CH - the racemic mixture of an intermediate of the formula
OH
Pr2-o-(cH2)m-c-cH~-NH2 (XXIV)
Ar,
in which m and Ar~ are as defined for a compound of formula (I) and Pr2 is as
15 defined in SCHEME 2 above is resolved.
If it is desired to prepare the enantiomers (I*) of the compounds of formula
(I) in which -A- is the divalent radical -O-CH2-CH2- it is also possible to resolve
the racemic mixture of a compound of the formula
CH \CH I (II): -A- = -O-CH2-CH2-
HO-(CH2) -C\ NH l E = H
Arl
20 in which m and R~ are as defined for a compound of formula (I).
If it is desired to prepare the enantiomers (I*) of the compounds of formula
(I) in which -A- is the divalent radical -N(R~)-CO- -N(R~)-CO-CO- or -N(R~)-
CH,-CH,- the racemic mixture of an intermediate of the formula
NH-R~
HO-(CH2)m-C-cH2-NH2 (XXIX)
Ar~
CA 02261808 1999-01-2~
29
in which m, Ar~ and Rl are as defined for a compound of formula (I), is resolved.
If resolution of the racemates is effected on the intermediates of formula
(XXXVI), (XXIV), (XXIX) or (II) [-A- = -O-CH2-CH2- and E = H], this can be
done by known methods involving the formation of a salt with optically active
5 acids, for example with (+)- or (-)-tartaric acid. The diastereoisomers are then
separated by conventional methods such as crystallization or chromatography, after
which the optically pure enantiomers are obtained by hydrolysis.
The compounds of formula (I) above also include those in which one or
more hydrogen, carbon or iodine atoms have been replaced with their radioactive
isotope, for example tritium, carbon-14 or iodine-125. Such labeled compounds
are useful in research, metabolic or pharmacokinetic studies and in biochemical
tests as receptor ligands.
The affinity of the compounds for the tachykinin receptors was evaluated in
vitro by several biochemical tests using radioligands:
1) The binding of [1-5I]BH-SP (substance P labeled with iodine-125 using
Bolton-Hunter's reagent) to the NK, receptors of human Iymphoblasts.
2) The binding of [l25I]His-NKA to the NK2 receptors of the rat duodenum
or bladder.
3) The binding of [~~5I]His[MePhe7]NKB to the NK3 receptors of the rat
0 cerebral cortex, the guinea-pig cerebral cortex and the gerbil cerebral cortex, and to
the human NK3 cloned receptors expressed by CHO cells (Buell et al., FEBS
Letters, 1992, 299, 90-95).
The tests were performed according to X. Emonds-Alt et al. (Eur. J.
Pharmacol., 1993, 250, 403- 413).
The compounds according to the invention generally have an affinity for the
above-mentioned tachykinin receptors, with an inhibition constant Ki preferably
below 10-S M.
In particular, the compounds of the present invention are active principles
of pharmaceutical compositions, the toxicity of which is compatible with their use
as drugs.
The compounds of the present invention are generally atlmini~tered in
dosage units. Said dosage units are preferably formulated in pharmaceutical
compositions in which the active principle is mixed with a pharmaceutical
excipient.
Thus, according to another of its features, the present invention relates to
CA 02261808 1999-01-2
pharmaceutical compositions in which a compound of formula (I) or a
phannaceutically acceptable salt thereof is present as the active principle.
The compounds of formula (I) above and the pharmaceutically acceptable
salts thereof can be used in daily doses of 0.01 to 100 mg per kilogram of body
weight of the m~mm~l to be treated, preferably in daily doses of 0.1 to 50 mg/kg.
In humans, the dose can preferably vary from 0.5 to 4000 mg per day, more
particularly from 2.5 to 1000 mg, depending on the age of the subject to be treated
or the type of treatment: prophylactic or curative.
In the pharmaceutical compositions of the present invention for oral,
10 sublingual, inhalational, subcutaneous, intramuscular, intravenous, transdermal,
local or rectal a-lmini~tration, the active principles can be a-lmini~tered to animals
and humans in unit forms of a(lmini~tration, mixed with conventional
pharmaceutical carriers. The appropriate unit forms of a~mini~tration include oral
forrns such as tablets, gelatin capsules, powders, granules and solutions or
15 suspensions to be taken orally, sublingual and buccal fonns of a~lmini~tratioll,
aerosols, implants, subcutaneous, intramuscular, intravenous, intranasal or
intraocular forms of a~lminictration and rectal forms of administration.
When a solid composition in the fonn of tablets is prepared, the main active
principle is mixed with a phannaceutical vehicle such as silica, gelatin, starch,
20 lactose, magnesium stearate, talc, gum arabic or the like. The tablets can be coated
with sucrose~ various polymers or other appropriate substances or else they can be
treated so as to have a prolonged or delayed activity and so as to release a
predetennined amount of active principle continuously.
A preparation in the form of gelatin capsules is obtained by mixing the
25 active principle with a diluent such as a glycol or a glycerol ester, and
incorporating the mixture obtained into soft or hard gelatin capsules.
A preparation in the form of a syrup or elixir can contain the active
principle together with a sweetener, which is preferably calorie-free, methylparaben
and propylparaben as antiseptics, a flavoring and an appropriate color.
The water-dispersible granules or powders can contain the active principle
mixed with dispersants or wetting agents or with suspending agents such as
polyvinylpyrrolidone, as well as with sweeteners or taste correctors.
Rectal a~mini~tration is effected using suppositories, which are prepared
with binders melting at the rectal temperature, for example cocoa butter or
35 polyethylene glycols.
CA 02261808 1999-01-2~
Parenteral, intranasal or intraocular admini~tration is effected using aqueous
suspensions, isotonic saline solutions or sterile and injectable solutions whichcontain pharmacologically compatible dispersants and/or wetting agents, for
example propylene glycol or butylene glycol.
Administration by inhalation is effected using an aerosol which contains for
example sorbitan trioleate or oleic acid, as well as trichlorofluoromethane,
dichlorofluoromethane, dichlorotetrafluoroethane or any other biologically
compatible propellant gas; it is also possible to use a system containing the active
principle in powder form, by itself or associated with an excipient.
The active principle can also be formulated as microcapsules, with one or
more carriers or additives if appropriate.
In each dosage unit, the active principle of formula (I) is present in the
amounts appropriate to the daily doses envisaged. In general, each dosage unit is
suitably adjusted according to the dosage and the intended type of a~mini~tration,
l S for example tablets, gelatin capsules and the like, sachets, ampoules, syrups and the
like, and drops, so that such a dosage unit contains from 0.5 to 1000 mg of active
principle, preferably from 2.5 to 250 mg, to be a~lmini~tered one to four times a
day.
The above-mentioned compositions can also contain other active products
such as, for example, bronchodilators, antitussives, antihistamines, anti-
inflamm~tories, antiemetics and chemotherapeutic agents.
According to another of its features, the present invention relates to the use
of the products of formula (I) for the preparation of drugs intended for the
treatment of physiological disorders associated with an excess of tachykinins, and
all neurokinin-dependent pathological conditions of the respiratory,
gastrointestinal, urinary, immune, cardiovascular and central nervous systems, as
well as pain and migraine.
Non-limiting examples are:
- acute and chronic pain associated for example with migraine, with pains
experienced by cancer and angina patients, and with chronic inflamm~tory
processes such as osteoarthritis and rheumatoid arthritis,
- infl~mm:~tions such as neurogenic inflamm~tions, chronic inflamm~tory diseases,
for example obstructive chronic respiratory diseases, asthma, allergies, rhinitis,
coughs, bronchitis, hypersensitivity, for example to pollen and mites, rheumatoid
arthritis, fibrositis, osteoarthritis, psoriasis, ulcerative colitis, Crohn's disease,
CA 02261808 1999-01-2~
inflamm:~tion of the intestines (irritable colon), prostatitis, nervous bladder,incontinence, cystitis, urethritis and nephritis, ophthalmic diseases such as
conjunctivitis and vitreoretinopathy, and skin diseases such as contact dermatitis,
atopical dermatitis, urticaria, eczema, pruritus and burns, especially sunburn,
- diseases of the immune system associated with suppression or stimulation of the
functions of the immune cells, for example rheumatoid arthritis, psoriasis, .Crohn's
disease, diabetes, lupus and rejection reactions following transplantation,
- small-cell lung cancers and demyelination diseases such as multiple sclerosis or
amyotrophic lateral sclerosis,
- diseases of the central nervous system of the neuropsychiatric or neurologicaltype, such as anxiety, vigilance disorders, mood disorders, depression, psychosis,
schizophrenia, mania, dementia, epilepsy, Parkinson's disease, Alzheimer's
disease, drug dependence, alcoholism, Down's syndrome and Huntington's chorea,
as well as neurodegenerative diseases and stress-related somatic disorders,
- diseases of the gastrointestinal system, such as nausea, vomiting of any origin,
irritable colon, gastric and duodenal ulcers, esophageal ulcers, diarrhea and
hypersecretions,
- diseases of the cardiovascular system, such as hypertension, the vascular aspects
of migraine, edema, thrombosis, angina pectoris, vascular spasms, circulatory
diseases due to vasodilation, Reynauld's diseases, fibrosis and collagen diseases,
and
- heart rate and rhythm disorders, in particular those caused by pain or stress.The present invention also includes a method of treating said complaints at
the doses indicated above.
2 5 The following abbreviations are used in the Preparations and in the
Examples:
Me. Ome: methyl, methoxy
Et, Oet: ethyl, ethoxy
EtOH: ethanol
30 MeOH: methanol
Ether: diethyl ether
Iso ether: diisopropyl ether
DMF: dimethylformamide
DMSO: dimethyl sulfoxide
35 DCM: dichloromethane
CA 02261808 1999-01-2~
THF: tetrahydrofuran
AcOEt: ethyl acetate
Na2CO3 sodium carbonate
NaHCO3 sodium hydrogencarbonate
NaCI: sodium chloride
Na2SO4 sodium sulfate
MgSO4 magnesium sulfate
NaOH: sodium hydroxide
HCI: hydrochloric acid
10 TFA: trifluoroacetic acid
Hydrochloric ether: saturated solution of hydrochloric acid in ether
KCN: potassium cyanide
Na2S,Os sodium metabisulfite
DBU: 178-diazabicyclo[5.4.0]undec-7-ene
15 NH~CI: ammonium chloride
M.p.: melting point
RT: room temperature
Silica H: silica gel 60H, marketed by Merck (DARMSTADT)
NMR: nuclear magnetic resonance
20 ~: chemical shift
s: singlet
bs: broad singlet
d: doublet
t: triplet
25 qd: quadruplet
mt: multiplet
u: unresolved signals
PREPARATIONS
Preparation 1.1
5-(3 ,4-Dichlorophenyl)-5 -[2-(tetrahydropyran-2-yloxy)ethyl]tetrahydro-2H-
1,3-oxazin-2-one
A) 2-(3,4-Dichlorophenyl)-4-(tetrahydropyran-2-yloxy)butanenitrile
A suspension of 17.75 g of sodium hydride (80% dispersion in oil) in 750
35 ml of THF is cooled in an ice bath, a solution of 100 g of 3,4-dichlorophenyl-
CA 02261808 1999-01-2~
34
acetonitrile in 250 ml of THF is added dropwise and the reaction mixture is stirred
for two hours at RT. It is cooled to -20~C, a solution of 112.36 g of 1-bromo-2-(tetrahydropyran-2-yloxy)ethane in 120 ml of THF is added dropwise and the
reaction mixture is stirred for 2 hours at RT. It is concentrated under vacuum, the
residue is taken up with water and extracted with ether, the organic phase is
washed twice with a buffer solution of pH 4, with a buffer solution of pH 7 and
twice with saturated NaCI solution and dried over Na2SO4 and the solvent is
evaporated off under vacuum. The residue is chromatographed on silica using
toluene and then a toluene/AcOEt mixture (100/3; v/v) as the eluent to give 113.5 g
10 of the expected product, which is used as such.
B) 2-(3,4-Dichlorophenyl)-2-(hydroxymethyl)-4-(tetrahydropyran-2-yloxy)-
butanenitrile
A mixture of 12.56 g of the compound obtained in the previous step, 9.6 g
of 37% aqueous formaldehyde solution and 0.3 g of DBU in 25 ml of 1,2-
15 dimethoxyethane is refluxed for 1 hour. The reaction mixture is concentrated
under vacuum, the residue is extracted with ether, the organic phase is washed
twice with water, twice with a buffer solution of pH 4, twice with water and twice
with saturated NaCI solution and dried over Na2SO~ and the solvent is evaporatedoff under vacuum to give 17 g of the expected product, which is used as such.
20 C) 2-(3,4-Dichlorophenyl)-2-(hydroxymethyl)-4-(tetrahydropyran-2-yloxy)-
butylamine
A mixture of 17 g of the compound obtained in the previous step and 6 g of
Raney~ nickel in 300 ml of EtOH and 40 ml of 20% aqueous ammonia solution is
hydrogenated for 5 hours at 40~C and at atmospheric pressure. The catalyst is
25 filtered off and the filtrate is concentrated under vacuum. The residue is taken up
with DCM, the organic phase is washed with water and with saturated NaCI
solution and dried over MgSO~ and the solvent is evaporated off under vacuum to
give 16.5 g of the expected product in the form of an oil, which is used as such.
D) 5-(3,4-Dichlorophenyl)-5-[2-(tetrahydropyran-2-yloxy)ethyl]tetrahydro-2H-1,3- oxazin-2-oDe
24.6 g of a 20% solution of phosgene in toluene, diluted in l50 ml of DCM,
are cooled to -70~C, a solution of 16.5 g of the compound obtained in the previous
step and 5.7 g of triethylamine in 100 ml of DCM is added dropwise and the
reaction mixture is stirred while the temperature is allowed to rise to RT. It is
35 concentrated under vacuum, the residue is taken up with a water/AcOEt mixture
CA 02261808 1999-01-2S
- 35
and the product which crystallizes at the interphase is filtered off to give a first
crop of the expected product. After dec~nt~tion of the filtrate, the organic phase is
washed with water, with a buffer solution of pH 4 and with saturated NaCI solution
and dried over MgSO4 and the solvent is evaporated off under vacuum. The
5 residue is taken up with AcOEt and the crystalline product formed is filtered off to
give the second crop of the product. A total of 4.5 g of the expected product isobtained.
Preparation 1.2
5-(3,4-Dichlorophenyl)-5-[3-(tetrahydropyran-2-yloxy)propyl]tetrahydro-
10 2H- 1,3-oxazin-2-one
A) 2-(3,4-Dichlorophenyl)-5-(tetrahydropyran-2-yloxy)pentanenitrile
A solution of 50.8 g of 3,4-dichlorophenylacetonitrile in 250 ml of THF is
added dropwise at a temperature below 20~C to a suspension of 12 g of sodium
hydride (55% dispersion in oil) in 175 ml of THF and the reaction mixture is
stirred for 2 hours at RT. It is cooled to -20~C, a solution of 62.5 g of 1-bromo-3-
(tetrahydropyran-2-yloxy)propane in 60 ml of THF is added dropwise and the
reaction mixture is stirred while the temperature is allowed to rise to RT. It is
poured into a solution of 31 g of ammonium chloride in 1.4 liters of water and
extracted with ether, the combined organic phases are washed with saturated NaClsolution and dried over MgSO~ and the solvents are evaporated off under vacuum.
The residue is chromatographed on silica using toluene and then a toluene/AcOEt
mixture (95/5; vlv) as the eluent to give 64 g of the expected product, which isused as such.
B) 2-(3,4-Dichlorophenyl)-2-(hydroxymethyl)-5-(tetrahydropyran-2-yloxy)-
pentanenitrile
A mixture of 15 g of the compound obtained in the previous step. 11.2 g of
37% aqueous formaldehyde solution and 0.35 g of DBU in 30 ml of 1,2-
dimethoxyethane is refluxed for 1 hour. The reaction mixture is concentrated
under vacuum, the residue is extracted with ether, the organic phase is washed with
water, twice with a buffer solution of pH 4, twice with water and twice with
saturated NaCI solution and dried over Na,SO4 and the solvent is evaporated off
under vacuum. The residue is chromatographed on silica using toluene and then a
toluene/AcOEt mixture (80/20; v/v) as the eluent to give 15.5 g of the expected
product, which is used as such.
C) 2-(3,4-Dichlorophenyl)-2-(hydroxymethyl)-5-(tetrahydropyran-2-yloxy)-
CA 02261808 1999-01-2~
36
pentylamine
A mixture of 15.5 g of the compound obtained in the previous step and 5 g
of Raney~ nickel in 200 ml of EtOH and 40 ml of 20% aqueous ammonia solution
is hydrogenated for S hours at 30~C and at atmospheric pressure. The catalyst is5 filtered off and the filtrate is concentrated under vacuum. The residue is taken up
with DCM, the organic phase is washed with water and with saturated NaCI
solution and dried over MgSO4 and the solvent is evaporated off under vacuum to
give 14.9 g of the expected product in the form of an oil, which is used as such.
D) 5-(3,4-Dichlorophenyl)-5-[3-(tetrahydropyran-2-yloxy)propyl]tetrahydro-2H-
1,3-oxazin-2-one
21.4 g of a 20% solution of phosgene in toluene~ diluted in 120 ml of DCM,
are cooled to -70~C, a solution of 14.9 g of the compound obtained in the previous
step and 4.98 g of triethylamine in 80 ml of DCM is added dropwise and the
reaction mixture is stirred while the temperature is allowed to rise to RT. It is
concentrated under vacuum, the residue is taken up with water and extracted withether, the organic phase is washed with water and with saturated NaCI solution and
dried over MgSO4 and the solvent is evaporated off under vacuum to give 12.5 g of
the expected product, which is used as such.
Preparation 1.3
6-(3,4-Dichlorophenyl)-6-[2-(tetrahydropyran-2-yloxy)ethyl]morpholin-3-
one
A) 2-(3,4-Dichlorophenyl)-2-hydroxyacetonitrile
A mixture of 70 g of 3,4-dichlorobenzaldehyde and 90 g of Na2S2O5 in 300
ml of water is stirred overnight at RT. The reaction mixture is cooled to 0~C, a'5 solution of 52 g of KCN in 100 ml of water is added dropwise and the reaction
mixture is stirred while the temperature is allowed to rise to RT. It is extracted
with ether, the organic phase is washed with water and dried over Na2SO4 and thesolvent is evaporated off under vacuum to give 76 g of the expected product, which
is used as such.
B) 2-(3,4-Dichlorophenyl)-2-(tetrahydropyran-2-yloxy)acetonitrile
A solution of 76 g of the compound obtained in the previous step and 0.25
g of p-toluenesulfonic acid monohydrate in 300 ml of DCM is cooled to 0~C, a
solution of 39 g of 3,4-dihydro-2H-pyran in 50 ml of DCM is added dropwise and
the reaction mixture is stirred while the temperature is allowed to rise to RT. It is
washed with saturated NaHCO3 solution and with water, the organic phase is dried
CA 02261808 1999-01-2~
over Na2SO~, and the solvent is evaporated off under vacuum to give 33 g of the
expected product after cryst~lli7~tion at 0~C from pentane. M.p. = 61~C.
C) 4-(Benzoyloxy)-2-(3,4-dichlorophenyl)-2-(tetrahydropyran-2-yloxy)-
butanenitrile
56 ml of a 2 M solution of lithium diisopropylamide in THF are cooled to
-60~C, a solution of 32 g of the compound obtained in the previous step in 50 ml of
THF is added dropwise and the mixture is stirred for I hour at -60~C. A solutionof 25.4 g of2-bromoethyl benzoate in 50 ml of THF is then added dropwise at -
60~C and the reaction mixture is stirred while the temperature is allowed to rise to
10 RT. It is concentrated under vacuum, the residue is extracted with ether, theorganic phase is washed with water and with a buffer solution of pH 4 and dried
over Na2SO4 and the solvent is evaporated off under vacuum. The residue is
chromatographed on silica using a toluene/AcOEt mixture (100/5; v/v) as the
eluent to give 34 g of the expected product, which is used as such.
15 D) 4-(Benzoyloxy)-2-(3,4-dichlorophenyl)-2-(tetrahydropyran-2-yloxy)butylamine
A mixture of 34 g of the compound obtained in the previous step and 10 g
of Raney~ nickel in 400 ml of EtOH and 40 ml of concentrated aqueous ammonia
solution is hydrogenated at RT and at atmospheric pressure. The catalyst is filtered
off and the filtrate is concentrated under vacuum. The residue is taken up with
water and extracted with ether, the organic phase is washed with saturated NaCI
solution and dried over Na,SO4 and the solvent is evaporated off under vacuum.
The residue is chromatographed on silica H using a gradient of a DCM/MeOH
mixture (from 100/1; v/v to 100/3; v/v) as the eluent to give 16 g of the expected
product, which is used as such.
E) N-(2-Bromoacetyl)-4-(benzoyloxy)-2-(3,4-dichlorophenyl)-2-hydroxy-
butylamine
A solution of 16 g of the compound obtained in the previous step and 4.8 g
of triethylamine in 100 ml of DCM is cooled to -60~C, a solution of 5.68 g of
bromoacetyl chloride in 20 ml of DCM is added dropwise and the reaction mixture
is stirred for 30 minutes. It is concentrated under vacuum, the residue is extracted
with ether, the organic phase is washed with water and with a buffer solution of pH
4 and dried over Na2SO4 and the solvent is evaporated off under vacuum. The
product obtained is dissolved in the minimum amount of MeOH and acidified to
pH I by the addition of a saturated solution of gaseous HCI in ether, and the
solvents are evaporated off under vacuum. The residue is taken up with water and
CA 02261808 1999-01-2~
38
extracted with AcOEt, the organic phase is washed with saturated NaHCO3
solution and with water and dried over Na2SO~ and the solvent is evaporated off
under vacuum to give 16 g of the expected product, which is used as such.
F) 6-(3,4-Dichlorophenyl)-6-(2-hydroxyethyl)morpholin-3-one
A mixture of 16 g of the compound obtained in the previous step, 50 ml of
propan-2-ol, 15 ml of 10 N NaOH solution and 10 ml of DMF is stirred for 4
hours. The reaction mixture is concentrated under vacuum, the residue is extracted
with AcOEt, the organic phase is washed with water and dried over Na2SO4 and
the solvent is evaporated off under vacuum. The residue is chromatographed on
10 silica H using a gradient of a DCM/MeOH mixture (from 100/3; v/v to 100/5; v/v)
as the eluent to give 6.1 g of the expected product.
Proton NMR spectrum at 200 MHz in DMSO-d6
~: 2.05 ppm: mt: 2H
3.0 to 4.4 ppm: u: 6H
4.5 ppm: t: lH
7.3to8.3ppm:u:4H
G) 6-(3,4-Dichlorophenyl)-6-[2-(tetrahydropyran-2-yloxy)ethyl]morpholin-3-one
A solution of 1.7 g of the compound obtained in the previous step and
0.003 g of p-toluenesulfonic acid monohydrate in 50 ml of DCM is cooled to 0~C,
20 0.588 g of 3,4-dihydro-2H-pyran in 10 ml of DCM is added dropwise and the
reaction mixture is stirred for I hour at RT. It is concentrated under vacuum, the
residue is extracted with ether, the organic phase is washed witll saturated NaHCO3
solution and with water and dried over Na2SO4 and the solvent is evaporated off
under vacuum. The residue is chromatographed on silica H using a DCM/MeOH
25 mixture (100/2; v/v) as the eluent to give 1.8 g of the expected product, which is
used as such.
Preparation 1.4
6-[2-(Benzoyloxy)ethyl]-6-(3,4-dichlorophenyl)morpholin-3-one
A mixture of 4.8 g of the compound obtained in step E of Preparation 1.3
30 and 1.4 g of K2CO3 in 100 ml of xylene is heated at 130~C overnight. After
cooling to RT, the reaction mixture is filtered and the filtrate is concentrated under
vacuum. The residue is extracted with ether, the organic phase is washed with a
buft'er solution of pH 2 and with water and dried over MgSO4 and the solvent is
evaporated off under vacuum. The residue is chromatographed on silica H using a
35 DCM/MeOH mixture (100/1; v/v) as the eluent to give 1.4 g of the expected
, . . .
CA 02261808 1999-01-2~
39
product.
Proton NMR spectrum at 200 MHz in DMSO-d6
~: 2.45 ppm: mt: 2H
3.75 ppm: AB system: 2H
3.9 to 4.5 ppm: u: 4H
7.4 to 7.9 ppm: u: 8H
8.25 ppm: bs: lH
This compound can also be obtained by following the three steps of the
method described below.
A') 4-(Benzoyloxy)-2-(3,4-dichlorophenyl)-2-hydroxybutylamine hydrochloride
This compound is described in step A of Preparation 1.7.
B') N-(2-Chloroacetyl)-4-(benzoyloxy)-2-(3,4-dichlorophenyl)-2-hydroxy-
butylamine
A solution of 20 g of the compound obtained in the previous step and 10.3
g of triethylamine in 100 ml of DCM is cooled to 0~C, 5.8 g of chloroacetyl
chloride are added dropwise and the reaction mixture is stirred for 30 minutes. It is
concentrated under vacuum, the residue is extracted with AcOEt, the organic phase
is washed with water, with a buffer solution of pH 2 and with water and dried over
MgSO4 and the solvent is evaporated off under vacuum to give 22 g of the
expected product, which is used as such.
C') 6-[2-(Benzoyloxy)ethyl]-6-(3,4-dichlorophenyl)morpholin-3-one
A solution of 22 g of the compound obtained in the previous step in 600 ml
of THF is cooled to -10~C, 11.42 g of potassium tert-butylate are added and the
reaction mixture is stirred until dissolution is complete. It is concentrated under
vacuum, the residue is extracted with AcOEt, the organic phase is washed with a
buffer solution of pH 2 and with water and dried over MgSO4 and the solvent is
evaporated off under vacuum to give 12.8 g of the expected product after
crystallization from ether.
Preparation 1.5
2-(3,4-Dichlorophenyl)-2-(2-hydroxyethyl)morpholine
A suspension of 1.6 g of lithium aluminum hydride in 25 ml of THF is
heated to 60~C, a solution of 4 g of the compound obtained in step F of Preparation
1.3 in 20 ml of THF is added dropwise and the mixture is stirred for 30 minutes
under reflux. After cooling, 1.5 ml of water, 1.5 ml of 4 N NaOH and then 4.5 mlof water are added. The mineral salts are filtered off on Célite~, the filtrate is
CA 02261808 1999-01-2~
decanted and the organic phase is evaporated under vacuum. The residue is taken
up with ether and dried over Na,SO4 and the solvent is evaporated off under
vacuum to give 3.6 g of the expected product.
Preparation 1.6
2-(3,4-Dichlorophenyl)-2-(3-hydroxypropyl)morpholine
A) S-(Benzoyloxy)-2-(3,4-dichlorophenyl)-2-(tetrahydropyran-2-yloxy)-
pentanenitrile
47 ml of a 1.5 M solution of lithium diisopropylamide in THF are
cooled to -60~C, a solution of 19.3 g of the compound obtained in step B of
10 Preparation 1.3 in 100 ml of THF is added dropwise and the mixture is stirred for
30 minutes at -60~C. 17 g of 3-bromopropyl benzoate are then added dropwise at
-60~C and the reaction mixture is stirred while the temperature is allowed to rise to
RT. It is concentrated under vacuum, the residue is extracted with ether, the
organic phase is washed with water and with saturated NaCI solution and dried
15 over Na,SO4 and the solvent is evaporated off under vacuum to give 21 g of the
expected product after crystallization from hexane.
B) S-(Benzoyloxy)-2-(3,4-dichlorophenyl)-2-(tetrahydropyran-2-
yloxy)pentylamine
A mixture of 20 g of the compound obtained in the previous step and 7 g of
20 Raney~ nickel in 300 ml of MeOH is hydrogenated at RT and at atmospheric
pressure. The catalyst is filtered off and the filtrate is concentrated under vacuum.
The residue is taken up with water and extracted with ether, the organic phase is
washed with water and dried over Na2SO4 and the solvent is evaporated off under
vacuum to give 20 g of the expected product, which is used as such.
25 C) N-(2-Chloroacetyl)-S-(benzoyloxy)-2-(3,4-dichlorophenyl)-2-(tetrahydropyran-
2-yloxy)pentylamine
A solution of 9 g of the compound obtained in the previous step and 2.4 g
of triethylamine in 100 ml of DCM is cooled to 0~C, a solution of 2.23 g of
chloroacetyl chloride in 20 ml of DCM is added dropwise and the reaction mixture30 is stirred for 30 minutes. It is concentrated under vacuum, the residue is extracted
with ether, the organic phase is washed with water and dried over Na2SO4 and thesolvent is evaporated off under vacuum to give 9.5 g of the expected product,
which is used as such.
D) N-(2-Chloroacetyl)-S-(benzoyloxy)-2-(3,4-dichlorophenyl)-2-hydroxy-
pentylamine
CA 02261808 1999-01-2
~ 41
A saturated solution of gaseous HCI in ether is added to a solution of 9 g of
the compound obtained in the previous step in 50 ml of DCM and 50 ml of MeOH
until the pH is 1, and the mixture is stirred for 30 minutes at RT. It is concentrated
under vacuum, the residue is extracted with ether, the organic phase is washed with
5 water and with saturated NaHCO3 solution and dried over Na2SO4 and the solventis evaporated off under vacuum. The residue is chromatographed on silica H usinga DCM/MeOH mixture (100/3; v/v) as the eluent to give 4.7 g of the expected'
product, which is used as such.
E) 6-(3,4-Dichlorophenyl)-6-(3-hydroxypropyl)morpholin-3-one
A mixture of 2.95 g of the compound obtained in the previous step, 40 ml
of propan-2-ol and 3 ml of 10 N NaOH solution is stirred for 2 hours at RT. The
reaction mixture is concentrated under vacuum, the residue is extracted with DCM,
the organic phase is washed with water and dried over Na2SO4 and the solvent is
evaporated off under vacuum. The residue is chromatographed on silica H using a
gradient of a DCM/MeOH mixture (100/2; v/v to 100/5; v/v) as the eluent to give
0.5 g of the expected product. M.p. = 130- 132~C.
F) 2-(3,4-Dichlorophenyl)-2-(3-hydroxypropyl)morpholine
A suspension of 0.82 g of lithium aluminum hydride in 10 ml of THF is
heated to 60~C, a solution of 2 g of the compound obtained in the previous step in
20 ml of THF is added dropwise and the mixture is stirred for 30 minutes under
reflux. After cooling, I ml of water, 1 ml of 4 N NaOH and then 3 ml of water are
added. The mineral salts are filtered off on Célite~, the filtrate is decanted and the
organic phase is evaporated under vacuum. The residue is taken up with ether anddried over Na2SO4 and the solvent is evaporated off under vacuum to give 2 g of
the expected product.
Proton NMR spectrum at 200 MHz in DMSO-d6
~: 0.8 to 2.2 ppm: u: 4H
2.5 to 3.7 ppm: u: 8H
4.3 ppm: t: lH
7.2 to 7.7 ppm: u: 3H
CA 02261808 1999-01-2~
42
Preparation 1.7
5-[2-(Benzoyloxy)ethyl]-5-(3,4-dichlorophenyl)oxazolidin-2-one
A) 4-(Benzoyloxy)-2-(3,4-dichlorophenyl)-2-hydroxybutylamine hydrochloride
A saturated solution of gaseous HCI in ether is added at RT to a solution of
12 g of the compound obtained in step D of Preparation 1.3 in 50 ml of MeOH
until the pH is 1, and the reaction mixture is stirred for I hour at RT. It is
concentrated under vacuum, the residue is taken up with DCM and the precipitate
formed is filtered off and washed with ether to give 3.4 g of the expected product
after recrystallization from propan-2-ol. M.p. = 200-204~C.
10 B) 5-[2-(Benzoyloxy)ethyl]-5-(3,4-dichlorophenyl)oxazolidin-2-one
1.4 g of 1,1 '-carbonyldiimidazole are added at RT to a solution of 3 g of the
compound obtained in the previous step and 0.85 g of triethylamine in 30 ml of
1,2-dichloroethane and the reaction mixture is stirred for 30 minutes at RT and
then heated at 50~C for 2 hours. It is concentrated under vacuum, the residue is15 taken up with water and extracted with DCM, the organic phase is washed with a
buffer solution of pH 2 and with water and dried over Na,SO4 and the solvent is
evaporated off under vacuum to give 3 g of the expected product.
Proton NMR spectrum at 200 MHz in DMSO-d6
~: 6 ppm: mt: 2H
0 3.75 ppm: AB system: 2H
4.35 ppm: mt: 2H
7.4 to 7.8 ppm: u: 8H
7.9 ppm: s: IH
Preparation 1.8
'5 6-(3,4-Dichlorophenyl)- I -methyl-6-[2-(tetrahydropyran-2-yloxy)ethyl]- piperazine-2,3 -dione
A) 2-(3,4-Dichlorophenyl)-2-(methylamino)acetonitrile hydrochloride
A mixture of 10 g of 3,4-dichlorobenzaldehyde and 9.5 ml of cyano-
trimethylsilane is cooled in an ice bath, 10 mg of zinc iodide are added and themixture is stirred for 30 minutes at RT. 20 ml of a 33% solution of methylamine in
EtOH are then added and the mixture is heated at 40~C for 2 hours. The solvent is
concentrated under vacuum, the residue is extracted with ether and the organic
phase is dried over MgSO4 and filtered. A saturated solution of gaseous HCI in
ether is added to the filtrate until the pH is 1, and acetone is then added until the
product precipitates. The precipitate formed is filtered off, washed with ether and
.
CA 02261808 1999-01-2~
43
dried to give 12.8 g ofthe expected product. M.p. = 172~C.
B) 2-(tert-Butoxycarbonyl-N-methylamino)-2-(3,4-dichlorophenyl)acetonitrile
Concentrated NaOH solution is added to an aqueous suspension of 12.8 g
of the compound obtained in the previous step until the pH is 13, the mixture is5 extracted with ether, the organic phase is dried over MgSO4 and the solvent isevaporated off under vacuum. The residue is taken up with 20 ml of 1,4-dioxane,
12.5 g of di-tert-butyl dicarbonate are added and the reaction mixture is heated at
60CC for 2 hours. It is concentrated under vacuum, the residue is extracted withether, the organic phase is washed with a buffer solution of pH 2, with saturated
10 NaCI solution and with 10% Na,CO3 solution and dried over MgSO4 and the
solvent is evaporated off under vacuum. The residue is chromatographed on silicausing heptane and then a heptane/AcOEt mixture (96/4; v/v) as the eluent to give12.7 g of the expected product, which is used as such.
C) 2-(tert-Butoxycarbonyl-N-methylamino)-2-(3,4-dichlorophenyl)-4-(tetrahydro-
pyran-2-yloxy)butanenitrile
A solution of 11.1 g of the compound obtained in the previous step in 60 ml
of DMF is added dropwise to a suspension of 1.5 g of sodium hydride (60%
dispersion in oil) in 50 ml of DMF, the temperature being maintained at 25~C, and
the mixture is stirred for I hour at RT. A solution of 8.1 g of 1-bromo-2-
(tetrahydropyran-2-yloxy)ethane in 20 ml of DMF is then added and the reaction
mixture is heated at 60~C for 4 hours. At'ter cooling to RT, it is poured into amixture of ice and a buffer of pH 2 and extracted with ether, the organic phase is
washed twice with water and dried over MgSO~ and the solvent is evaporated off
under vacuum. The residue is chromatographed on silica using heptane and then a
heptane/AcOEt mixture (75/25; vlv) as the eluent to give 13.2 g of the expected
product, which is used as such.
D) 2-(tert-Butoxycarbonyl-N-methylamino)-2-(3,4-dichlorophenyl)-4-(tetrahydro-
pyran-2-yloxy)butylamine
A mixture of 13.2 g of the compound obtained in the previous step and 4 g
of Raney~ nickel in 150 ml of EtOH and 50 ml of 20% aqueous ammonia solution
is hydrogenated at 30~C and at atmospheric pressure. After 5 hours, the catalyst is
filtered off and the filtrate is concentrated under vacuum. The residue is extracted
with ether, the organic phase is washed twice with water and dried over MgSO4
and the solvent is evaporated off under vacuum to give 12.6 g of the expected
product, which is used as such.
CA 02261808 1999-01-2~
E) 2-(3,4-Dichlorophenyl)-4-hydroxy-2-(methylamino)butylamine hydrochloride
A mixture of 4.6 g of the compound obtained in the previous step and 10 ml
of concentrated HCI solution in 40 ml of MeOH is heated at 70~C for 1 hour. The
solvent is then concentrated under vacuum, the residue is taken up with acetone
and the precipitate formed is filtered off, washed with ether and dried to give 2.79
g of the expected product. M.p. = 240~C (dec.).
F) 6-(3,4-Dichlorophenyl)-6-(2-hydroxyethyl)- 1 -methylpiperazine-2,3-dione
Concentrated NaOH solution is added to an aqueous suspension of 5.3 g of
the compow~d obtained in the previous step until the pH is 13, the mixture is
10 extracted with ether, the organic phase is dried over MgSO4 and the solvent is
evaporated off under vacuum. The product obtained (4 g) is taken up with 50 ml of
EtOH, 2.57 g of diethyl oxalate are added and the reaction mixture is stirred for 1
hour at RT. It is concentrated under vacuum and the residue is taken up with 60 ml
of toluene and refluxed for 70 hours. It is concentrated under vacuum to give 2.8 g
15 of the expected product after crystallization from DCM. M.p. = 260~C.
G) 6-(3,4-Dichlorophenyl)-l-methyl-6-[2-(tetrahydropyran-2-yloxy)ethyl]-
piperazine-2,3 -dione
0.1 g of p-toluenesulfonic acid monohydrate and then 1.26 ml of 3,4-
dihydro-2H-pyran are added to a suspension of 2.8 g of the compound obtained in
20 the previous step in 50 ml of DCM and the reaction mixture is stirred overnight at
RT. It is washed with 10% Na,CO3 solution and with water, the organic phase is
dried over MgSO~ and the solvent is evaporated off under vacuum. The residue is
chromatographed on silica using AcOEt and then an AcOEt/MeOH mixture (93/7;
v/v) as the eluent to give 3.1 g of the expected product, which is used as such. Preparation 1.9
2-(3,4-Dichlorophenyl)- I -methyl-2-[2-(tetrahydropyran-2-yloxy)ethyl]-
piperazine
A solution of 2 g of the compound obtained in Preparation 1.8 in 20 ml of
THF is added dropwise to a suspension of 1.2 g of lithium aluminum hydride in 2030 ml of THF and the mixture is refluxed for 1 hour. After cooling, 5 ml of water are
added, the mineral salts are filtered off, the filtrate is concentrated under vacuum,
the residue is taken up with ether, the organic phase is dried over MgSO4 and the
solvent is evaporated off under vacuum to give 1.9 g of the expected product in the
form of an oil.
CA 02261808 1999-01-2~
Preparation 1.10
6-[2-(Benzoyloxy)ethyl]-6-(3,4-difluorophenyl)morpholin-3-one
A) 2-(3,4-Difluorophenyl)-2-hydroxyacetonitrile
A solution of 80.2 g of Na2S2Os in 250 ml of water is heated to 50~C, 50 g
5 of 3,4-difluorobenzaldehyde are added and the reaction mixture is stirred for 1 hour
at 50~C and left to stand overnight at RT. It is cooled to 0~C, a solution of 77.7 g
of KCN in 100 ml of water is added dropwise, the mixture is stirred while the
temperature is allowed to rise to RT, and stirring is then continued for 1 hour at
RT. The reaction mixture is extracted with ether, the organic phase is washed with
10 water and dried over MgSO4 and the solvent is evaporated off under vacuum to
give 48 g of the expected product, which is used as such.
B) 2-(3,4-Difluorophenyl)-2-(tetrahydropyran-2-yloxy)acetonitrile
A solution of 48 g of the compound obtained in the previous step and 0.2 g
of p-toluenesulfonic acid monohydrate in 500 ml of DCM is cooled to 0~C, a
solution of 28.6 g of 3,4-dihydro-2H-pyran in 50 ml of DCM is added dropwise,
the mixture is stirred while the temperature is allowed to rise to RT, and stirring is
then continued overnight at RT. The reaction mixture is concentrated under
vacuum, the residue is extracted with DCM, the organic phase is washed with 10%
Na2CO3 solution and with water and dried over Na,SO4 and the solvent is
evaporated off under vacuum. The residue is chromatographed on silica using a
toluene/AcOEt mixture (100/15; v/v) as the eluent to give 43 g of the expected
product, which is used as such.
C) 4-(Benzoyloxy)-2-(3,4-difluorophenyl)-2-(tetrahydropyran-2-
yloxy)butanenitrile
133 ml of a 1.5 M solution of lithium diisopropylamide in THF are cooled
to -60~C, a solution of 43 g of the compound obtained in the previous step in 250
ml of THF is added dropwise and the mixture is stirred for 30 minutes at -60~C. A
solution of 45.8 g of 2-bromoethyl benzoate in 100 ml of THF is then added
dropwise at -60~C and the reaction mixture is stirred while the temperature is
allowed to rise to RT. It is concentrated under vacuum, the residue is extractedwith ether, the organic phase is washed with water and with a buffer solution of pH
4 and dried over Na2SO4 and the solvent is evaporated off under vacuum. The
residue is chromatographed on silica using a toluene/AcOEt mixture (100/5; v/v) as
the eluent to give 47 g of the expected product, which is used as such.
D) 4-(Benzoyloxy)-2-(3,4-difluorophenyl)-2-(tetrahydropyran-2-yloxy)butylamine
CA 02261808 1999-01-2
46
A mixture of 47 g of the compound obtained in the previous step and 10 g
of Raney~ nickel in 400 ml of EtOH is hydrogenated at RT and at atmospheric
pressure. The catalyst is filtered off and the filtrate is concentrated under vacuum.
The residue is taken up with water and extracted with ether, the organic phase is
5 washed with water and dried over Na,SO4 and the solvent is evaporated off under
vacuum to give 45 g of the expected product, which is used as such.
E) 4-(Benzoyloxy)-2-(3,4-difluorophenyl)-2-hydroxybutylamine hydrochloride
A saturated solution of gaseous HCI in ether is added at RT to a solution of
45 g of the compound obtained in the previous step in 250 ml of MeOH until the
10 pH is 1, and the reaction mixture is stirred for 30 minutes at RT. It is concentrated
under vacuum, the residue is taken up with ether and the precipitate formed is
filtered off and washed with ether to give 15 g of the expected product after
recrystallization from propan-2-ol. M.p. = 202-204~C.
F) N-(2-Chloroacetyl)-4-(benzoyloxy)-2-(3,4-difluorophenyl)-2-hydroxy-
butylamine
A solution of 12.2 g of the compound obtained in the previous step and
7.88 g of triethylamine in 100 ml of DCM is cooled to 0~C, a solution of 3.85 g of
chloroacetyl chloride in 100 ml of DCM is added dropwise and the reaction
mixture is stirred for 30 minutes. It is concentrated under vacuum, the residue is
extracted with an ether/AcOEt mixture (50/50; v/v), the organic phase is washed
with water, with a buffer solution of pH 4 and with water and dried over Na2SO4
and the solvent is evaporated off under vacuum to give 13.5 g of the expected
product, which is used as such.
G) 6-[2-(Benzoyloxy)ethyl]-6-(3,4-difluorophenyl)morpholin-3-one
A mixture of 13.5 g of the compound obtained in the previous step and 20.7
g of' K,C03 in 100 ml of toluene is refluxed overnight. The reaction mixture is
concentrated under vacuum, the residue is taken up with ether and the mixture isfiltered. The filtrate is washed with water, with a buffer solution of pH 2 and with
water and dried over MgSO4 and the solvent is evaporated off under vacuum. The
residue is chromatographed on silica H using a DCM/MeOH mixture (100/1; v/v)
as the eluent to give 4.9 g of the expected product.
Proton NMR spectrum at 200 MHz in DMSO-d6
~: 2.35 ppm: mt: 2H
3.65 ppm: AB system: 2H
3.8 to 4.35 ppm: u: 4H
CA 02261808 1999-01-2~
47
7.1 to 7.8 ppm: u: 8H
8.2 ppm: bs: lH
Preparation 1.11
- 2-(3,4-Difluorophenyl)-2-(2-hydroxyethyl)morpholine
A suspension of 1.8 g of lithium aluminum hydride in 20 ml of THF is
added dropwise at RT to a solution of 2.8 g of the compound obtained in
Preparation 1.10 in 20 ml of THF and the mixture is then refluxed for 5 hours.
After cooling, 2 ml of water, 2 ml of 4 N NaOH and then 6 ml of water are added.The mineral salts are filtered off and the filtrate is concentrated under vacuum.
10 The residue is dissolved in DCM, acidified to pH I by the addition of a saturated
solution of gaseous HCI in ether, and extracted with water, the aqueous phase iswashed with ether, rendered alkaline to pH 8 by the addition of concentrated NaOH
solution, and extracted with DCM, the organic phase is washed with water and
dried over MgSO4 and the solvent is evaporated off under vacuum to give 0.77 g of
15 the expected product.
Preparation 1.12
5-[2-(Benzoyloxy)ethyl]-5-(3,4-difluorophenyl)oxazolidin-2-one
I g of 1 ?1 '-carbonyldiimidazole is added at RT to a solution of 2.1 g of the
compound obtained in step E of Preparation 1.10 and 0.63 g of triethylamine in 50
20 ml of 1,2-dichloroethane and the reaction mixture is then stirred for I hour at RT
and heated for 2 hours at 50~C. It is concentrated under vacuum, the residue is
extracted with DCM, the organic phase is washed with water, with a buffer
solution of pH 2 and with water and dried over MgSO~ and the solvent is
evaporated off under vacuum to give 1.5 g of the expected product.
Preparation 1.13
5-(3,4-Dichlorophenyl)- 1 -methyl-5-[2-(tetrahydropyran-2-yloxy)ethyl]-
imidazolidin-2-one
A) 2-(3,4-Dichlorophenyl)-2-(methylamino)-4-(tetrahydropyran-2-yloxy)-
butylamine
A mixture of 3.4 g of the compound obtained in step E of Preparation 1.8,
0.05 g of p-toluenesulfonic acid monohydrate and 2.1 ml of 3,4-dihydro-2H-pyran
in 30 ml of DMF is heated at 60~C for 45 minutes. The reaction mixture is pouredonto ice, rendered alkaline by the addition of concentrated NaOH and extracted
with ether, the organic phase is washed with water and dried over MgSO4 and the
35 solvent is evaporated off under vacuum to give 4.3 g of the expected product,
CA 02261808 1999-01-2~
48
which is used as such.
B) 5-(3,4-Dichlorophenyl)- I-methyl-5-[2-(tetrahydropyran-2-yloxy)ethyl]-
imidazolidin-2-one
1.6 g of l,1'-carbonyldiimidazole are added to a solution of 3.2 g of the
5 compound obtained in the previous step in 100 ml of 1,2-dichloroethane and thereaction mixture is stirred for 30 minutes at RT. It is then heated at 60~C for 2
hours and concentrated under vacuum. The residue is extracted with AcOEt, the
organic phase is washed with a buffer solution of pH 2, with saturated NaCl
solution and with 10% Na,CO3 solution and dried over MgSO4 and the solvent is
10 evaporated off under vacuum. The residue is chromatographed on silica H usingDCM and then a DCM/MeOH mixture (98/2; vlv) as the eluent to give 2.4 g of the
expected product in the form of an oil.
Preparation 1.14
6-[3 -(Benzoyloxy)propyl] -6-(3,4-dichlorophenyl)morpholin-3-one
l 5 A mixture of 7.5 g of the compound obtained in step D of Preparation 1.6,
9.3 g of K,CO3 and 3.75 g of sodium iodide in 100 ml of methyl ethyl ketone is
refluxed overnight. After cooling, the reaction mixture is concentrated under
vacuum, the residue is taken up with ether, the mineral salts are filtered off, the
filtrate is washed with a buffer solution of pH 2 and with water, the organic phase
20 is dried over MgSO, and the solvent is evaporated off under vacuum. The residue
is chromatographed on silica H using DCM and then a gradient of a DCM/MeOH
mixture (from 99/1; v/v to 9812; vlv) as the eluent to ~ive 1.3 g of the expected
product.
Proton NMR spectrum at 200 MHz in DMSO-d6
~: 1.1 to2.3ppm:u:4H
3.65 ppm: AB system: 2H
3.8 to 4.4 ppm: u: 4H
7.2 to 8.05 ppm: u: 8H
8.2ppm:s:1H
Preparation 1.15
6-[2-(Benzoyloxy)ethyl]-6-(3,4-difluorophenyl)morpholin-3-one, (-) isomer
A) 4-(Benzoyloxy)-2-(3,4-difluorophenyl)-2-hydroxybutylallline, (+) isomer
A solution of 41 g of L-(+)-tartaric acid in 1200 ml of MeOH is heated to
the reflux temperature, a solution of 81.4 g of the compound obtained in step E of
Preparation 1.10, in the form of the free base, in 200 ml of MeOH is then added all
CA 02261808 1999-01-2
49
at once and the mixture is left to crystallize for 48 hours while the temperature is
allowed to return to RT. The crystals formed are filtered off and washed with ether
to give 42.5 g of the tartaric acid salt.
[a]D20 = +36.2~ (c = l; DMF)
The resulting salt is recrystallized from 1450 ml of 70~ EtOH to give 35 g
of the tartaric acid salt after the crystals formed have been filtered off and washed
with ether.
[a]D20 = +38.9o (c = 1; DMF)
The resulting salt is taken up with 2000 ml of AcOEt, 40 ml of 10%
10 Na2CO3 solution are added and then, after stirring and decantation, the organic
phase is washed with water and dried over MgSO4 and the solvent is evaporated
off under vacuum to give 23.5 g of the expected product after cryst~lli7~tion from
an iso ether/pentane mixture. M.p. = 100.5-100.6~C.
[a]D20 = +42.5o (c = 1; MeOH)
15 B) N-(2-Chloroacetyl)-4-(benzoyloxy)-2-(3,4-difluorophenyl)-2-hydroxy-
butylamine, (+) isomer
A solution of 23.5 g of the compound obtained in the previous step and 8.1
g of triethylamine in 300 ml of DCM is cooled to 0~C, a solution of 8.3 g of
chloroacetyl chloride in 50 ml of DCM is added dropwise and the reaction mixtureis stirred for 30 minutes at 0~C. It is concentrated under vacuum, the residue is
extracted with an AcOEt/ether mixture (50/50; v/v), the organic phase is washed
with a buffer solution of pH 2 and dried over MgSO4 and the solvent is evaporated
off under vacuum to give 24.8 g of the expected product after crystallization from
an iso ether/pentane mixture.
~5 [a]D~~ = +26.1~ (c = l; MeOH)
C) 6-[2-(Benzoyloxy)ethyl]-6-(3,4-difluorophenyl)morpholin-3-one, (-) isomer
A solution of 23.6 g of the compound obtained in the previous step in 750
ml of THF is cooled to 0~C, 13.7 g of potassium te7t-butylate are added and the
reaction mixture is stirred for 15 minutes. It is concentrated under vacuum, theresidue is extracted with AcOEt, the organic phase is washed with a buffer solution
of pH 2 and with water and dried over MgSO4 and the solvent is evaporated off
under vacuum. The residue is chromatographed on silica H using a DCM/MeOH
mixture (from 100/1; v/v to 100/1.5; v/v) as the eluent to give 14.5 g of the
expected product after crystallization from pentane.
[a]D20= -7.1~ (c = l; MeOH)
CA 02261808 1999-01-2
Preparation 1.16
6-[2-(Benzoyloxy)ethyl]-6-(3,4-difluorophenyl)morpholin-3-one, (+)
isomer
A) 4-(Benzoyloxy)-2-(3,4-difluorophenyl)-2-hydroxybutylamine, (-) isomer
The filtration and wash liquors obtained from the crystallization and then
recrystallization of the tartaric acid salt prepared in step A of Preparation 1.15 are
concentrated under vacuum. The residue is treated with 10% Na2CO3 solution and
extracted with AcOEt, the organic phase is dried over MgSO4 and the solvent is
evaporated off under vacuum to give 49 g of the amino alcohol in the form of a
mixture of isomers. The amino alcohol is dissolved in 120 ml of MeOH and this
solution is added all at once to a refluxing solution of 24.1 g of D-(-)-tartaric acid
in 730 ml of MeOH. The mixture is left to crystallize for 48 hours while the
temperature is allowed to return to RT. The crystals formed are filtered off andwashed with ether to give 40.7 g of the tartaric acid salt.
The resulting salt is recrystallized from 1445 ml of 70~ EtOH to give 35 g
of the tartaric acid salt after the crystals fonned have been filtered off and washed
with ether. The resulting salt is taken up with 2000 ml of AcOEt, 10% Na.CO3
solution is added and then, after stirring and decantation, the organic phase iswashed with water and dried over MgSO~ and the solvent is evaporated off under
vacuum to give 27 g of the expected product. M.p. = 102~C.
[a]D20 = -40.5~ (c = l; MeOH)
B) N-(2-Chloroacetyl)-4-(benzoyloxy)-2-(3,4-difluorophenyl)-2-hydroxy-
butylamine, (-) isomer
A solution of 27 g of the compound obtained in the previous step and 13 ml
of triethylamine in 500 ml of DCM is cooled to -30~C, 5.8 g of chloroacetyl
chloride are added dropwise and the reaction mixture is stirred for 15 minutes. It is
then washed with water, with 1 N HCI solution and with 10% Na2CO3 solution, the
organic phase is dried over MgSO4 and the solvent is evaporated off under vacuumto give 28.6 g of the expected product after crystallization from an iso
ether/pentane mixture. M.p. = 63~C.
[a]D~~=-31.5~ (c = I; MeOH)
C) 6-[2-(Benzoyloxy)ethyl]-6-(3,4-difluorophenyl)morpholin-3-one, (+) isomer
A solution of 28.5 g of the compound obtained in the previous step in 250
ml of THF is cooled to -30~C, 18.5 g of potassium tert-butylate are added all atonce and the reaction mixture is stirred for 45 minutes. It is poured into 1000 ml of
CA 02261808 1999-01-2~
a buffer solution of pH 2 and extracted with ether, the organic phase is dried over
MgSO4 and the solvent is evaporated off under vacuum to give 20.5 g of the
expected product after crystallization from an ether/iso ether mixture. M.p. =
9~~C.
[a]D20 = +8.2o (c = 1; MeOH)
Preparation 1.17
2-[2-(Benzoyloxy)ethyl]-2-(3,4-difluorophenyl)morpholine, (-) isomer
A solution of 12 g of the compound obtained in Preparation 1.15 ((-)
isomer) in 75 ml of THF is added dropwise at RT to 100 ml of a 1 M solution of
borane in THF and the reaction mixture is stirred for 30 minutes at RT. It is then
refluxed for 3 hours, 40 ml of a I M solution of borane in THF are added and
reflux is continued for 30 minutes. 80 ml of boiling MeOH are added and reflux is
continued for 30 minutes. The reaction mixture is cooled in an ice bath, 30 ml of a
saturated solution of gaseous HCI in ether are added and the reaction mixture isstirred overnight at RT. It is concentrated under vacuum, the residue is taken up
with 10% Na CO3 solution and extracted with ether, the organic phase is washed
with water and with saturated NaCI solution and dried over MgSO4 and the solventis evaporated off under vacuum to give 11.2 g of the expected product in the form
of an oil.
0 Preparation 1.18
2-[2-(Benzoyloxy)ethyl]-2-(3,4-difluorophenyl)morpholine, (+) isomer
A mixture of 19.9 g of the compound obtained in Preparation 1.16 ((+)
isomer) and 300 ml of a I M solution of borane in THF is heated at 60~C for 2
hours. 60 ml of boiling MeOH are added and reflux is continued for 30 minutes.
25 The reaction mixture is cooled to 10~C, 50 ml of a solution of gaseous HCI in ether
are added and the reaction mixture is left to stand overnight at RT. It is
concentrated under vacuum, the residue is taken up with 300 ml of 10% Na,CO3
solution,300 ml of ether are added and the mixture is stirred for 30 minutes. After
decantation, the organic phase is dried over MgSO4 and the solvent is evaporated30 off under vacuum to give 20 g of the expected product in the form of an oil.
Preparation 1.19
2-[2-(Benzoyloxy)ethyl]-2-(3,4-dichlorophenyl)morpholine
A solution of 6 g of the compound obtained in Preparation 1.4 in 30 ml of
THF is added dropwise at RT to 76 ml of a 1 M solution of borane in THF and the
35 mixture is then refluxed for 4 hours. 30 ml of MeOH are added dropwise and
... . .
CA 02261808 1999-01-2~
reflux is continued for 30 minlltç~. The reaction mixture is cooled to 0~C,30 ml of
a saturated solution of gaseous HCI in ether are added and the reaction mixture is
stirred overnight at RT. It is concentrated under vacuum, the residue is taken up
with 10% Na2CO3 solution and extracted with ether, the organic phase is washed
with 10% Na2CO3 solution and with water and dried over MgSO4 and the solvent
is evaporated off under vacuum to give 5 g of the expected product.
Preparation 1.20
5-[2-(Benzoyloxy)ethyl]-5-(3,4-difluorophenyl)oxazolidin-2-one, (-) isomer
A mixture of 4 g of the compound obtained in step A of Preparation 1.16
((-) isomer) and 2.2 g of l,l'-carbonyldiimidazole in 40 ml of DCM is stirred for
30 minutes at RT and then refluxed for I hour. After cooling to RT, the reactionmixture is washed twice with I N HCI solution, the organic phase is dried over
MgSO4 and the solvent is evaporated off under vacuum to give 4.18 g of the
expected product after crystallization from iso ether. M.p. = 147~C.
[a]D20 = -62.2~ (c = l; DMF)
Preparation 1.21
2-[2-(Benzoyloxy)ethyl]-2-(3,4-difluorophenyl)morpholine
48 g of the compound obtained in Preparation I .10 are added in portions at
RT to 700 ml of a 1 M solution of borane in THF and the mixture is then heated at
60~C for 2 hours. 150 ml of MeOH are added dropwise and heating is continued
for 30 minutes. The reaction mixture is cooled to 10~C, 120 ml of hydrochloric
ether are added and the reaction mixture is left to stand overnight at RT. It isconcentrated under vacuum, the residue is taken up with 600 ml of saturated
Na,CO3 solution and 500 ml of ether and the mixture is stirred for 1 hour at RT.After decantation, the organic phase is washed with water and dried over MgSO4
and the solvent is evaporated off under vacuum. The residue is taken up with 400ml of propan-2-ol, 15 g of fumaric acid are added, the mixture is stirred for 30minutes and the precipitate formed is filtered off. It is taken up with 400 ml of
10~/o Na2CO3 solution and extracted with ether, the organic phase is dried over
MgSO4 and the solvent is evaporated off under vacuum to give 22 g of the
expected product in the form of an oil.
Preparation 1.22
5-[2-(Benzoyloxy)ethyl]-5-(3,4-dichlorophenyl)oxazolidine
4 g of 37% aqueous formaldehyde solution are added to a solution of 9 g of
the compound obtained in step A of Preparation 1.7 (in the form of the free base) in
.
CA 02261808 1999-01-2~
- 53
100 ml of THF and the mixture is then refluxed for 30 minutes and stirred
overnight at RT. It is concentrated under vacuum, the residue is extracted with
ether, the organic phase is washed with water and dried over Na2SO4 and the
solvent is evaporated off under vacuum to give 9 g of the expected product.
Proton NMR spectrum at 200 MHz in DMSO-d6
~: 2.4 ppm: mt: 2H
3.2 ppm: mt: 2H
3.8 to 5.0 ppm: u: 4H
7.0 to 8.0 ppm: u: 8H
Preparation 1.23
5-[2-(Benzoyloxy)ethyl]-5-(3,4-difluorophenyl)oxazolidin-2-one, (+)
isomer
This compound is prepared by the procedure described in Preparation 1.20
from the compound obtained in step A of Preparation 1.15 ((+) isomer).
[a]D20 = +61~ (c = 1; DMF)
Preparation 2.1
4-Phenyl- I -azabicyclo[2.2.1]heptane methanesulfonate
A) 1-Benzyl-4-carboxy-4-phenylpiperidine
106 ml of 30% NaOH solution are added to a suspension of 100 g of 4-
carboxy-4-phenylpiperidine p-toluenesulfonate in 600 ml of water, the resulting
solution is then cooled to 5~C, a solution of 47.6 g of benzyl bromide in 100 ml of
acetone is added dropwise and the mixture is stirred for 1 hour while the
temperature is allowed to rise to RT. The acetone is evaporated off under vacuumand the pH of the remaining aqueous phase is brought to 9.5 by the addition of
concentrated HCI solution, and then adjusted to 8.5 by the addition of 2 N HCI
solution. The precipitate formed is filtered off and washed with water and then
with acetone to give 70 g of the expected product. M.p. = 286~C.
B ) I -Benzyl-4-(hydroxymethyl)-4-phenylpiperidine
A suspension of 70 g of the compound obtained in the previous step in 150
ml of THF is cooled to 5~C, 237 ml of a 1 M solution of borane in THF are added
rapidly and the mixture is then refluxed for I hour. A further 474 ml of a 1 M
solution of borane in THF are then added and reflux is continued for 3 hours.
While the mixture is hot, 100 ml of MeOH are added over 30 minutes and then 150
ml of concentrated HCI solution are added over 30 minutes. After cooling to RT,
the reaction mixture is diluted with water, rendered alkaline by the addition of 30%
CA 02261808 1999-01-2~
NaOH solution and extracted with an ether/THF mixture, the organic phase is
washed with water and dried over Na,SO~ and the solvents are evaporated off
under vacuum to give 16 g of the expected product after two cryst~lli7~tions from
cyclohexane. M.p. = 127~C .
This compound can also be obtained by following the two steps of the
method described below.
A') 4-(Hydroxymethyl)-4-phenylpiperidine hydrochloride
A mixture of 10.25 g of 4-carboxy-4-phenylpiperidine p-toluenesulfonate
and 150 ml of a 1 M solution of borane in THF is refluxed for 1 hour. While the
10 mixture is hot, 30 ml of MeOH are added over 20 minutes; after cooling to RT, a
saturated solution of gaseous HCI in ether is added until the pH is 1, and the
mixture is stirred overnight at RT. The solvents are concelltrated under vacuum
and the residue is taken up with hot acetone and left to crystallize. The crystals
fomled are filtered off and washed with acetone and then with ether to give 11.1 g
15 of the expected product. M.p. = 263~C.
B') l-Benzyl-4-(hydroxymethyl)-4-phenylpiperidine
A solution of 20.5 g of the compound obtained in the previous step in 100
ml of water and 200 ml of acetone is heated to 40~C, 27 ml of 30% NaOH solution
are added, a solution of 17 g of benzyl bromide in 50 ml of acetone is then added
20 dropwise and the mixture is stirred for 1 hour. The acetone is evaporated off, the
residue is diluted with water and the precipitate formed is filtered off, washed with
water and dried. The precipitate is taken up with 800 ml of hot cyclohexane, an
insoluble material is filtered off and the filtrate is left to crystallize at RT to give
19.3 g of the expected product.
25 C) 4-Phenyl- 1 -benzyl- 1 -azoniabicyclo[2.2. l ]heptane methanesulfonate
A mixture of 14.05 g of the compound obtained in step B and 5.55 g of
triethylamine in 200 ml of DCM is cooled to 5~C, a solution of 6.01 g of methane-
sulfonyl chloride in 15 ml of DCM is added dropwise and the mixture is stirred for
15 minutes at RT. It is concentrated under vacuum at 30~C, the residue is extracted
30 with 500 ml of AcOEt, the organic phase is washed with water and dried over
Na,SO~ and the solvent is evaporated off under vacuum to give 19.1 g of an oil,
which is dissolved in 180 ml of n-butanol and refluxed for 2 hours. It is
concentrated under vacuum, the residue is taken up with 150 ml of hot acetone and
the mixture is stirred at RT. The crystalline product obtained is filtered off and
washed with ether to give 16.85 g of the expected product. M.p. = 193~C.
CA 02261808 1999-01-2~
D) 4-Phenyl- 1 -azabicyclo[2.2.1]heptane methanesulfonate
A mixture of 15 g of the compound obtained in the previous step and 1.5 g
of 10% palladium-on-charcoal in 150 ml of 95~ EtOH is hydrogenated for 4 hours
at 40~C and at atmospheric pressure. The catalyst is filtered off on Célite~ and5 washed with EtOH and the filtrate is concentrated under vacuum. The residue istaken up with hot AcOEt and left to crystallize to give 10.1 g of the expected
product after filtration. M.p. = 130~C.
EXAMPLE 1
5-(3,4-Difluorophenyl)-5-[2-[4-phenyl- 1 -azoniabicyclo[2.2.1]hept- 1 -yl]-
10 ethyl]-3-[3,5-bis(trifluoromethyl)benzyl]oxazolidin-2-one chloride monohydrate,
(+) isomer
A) 5-[2-(Benzoyloxy)ethyl]-5-(3,4-difluorophenyl)-3-[3,5-bis(trifluoromethyl)-
benzyl]oxazolidin-2-one, (+) isomer
1.2 g of potassium tert-butylate are added at RT to a solution of 3.5 g of the
compound obtained in Preparation 1.23 ((+) isomer) in a mixture of 50 ml of THF
and 10 ml of DMF and the mixture is stirred for 30 minutes at RT. 2.7 g of 3,5-
bis(trifluoromethyl)benzyl chloride are then added and the reaction mixture is
heated at 60~C for 6 hours. It is poured into 200 ml of buffer of pH 2 and extracted
with ether, the organic phase is washed with water and dried over MgSO4 and the
solvent is evaporated off under vacuum. The residue is chromatographed on silicaH using DCM as the eluent to give 3.6 g of the expected product.
B) 5-(3,4-Difluorophenyl)-5-(2-hydroxyethyl)-3-[3,5-bis(trifluoromethyl)benzyl]- oxazolidin-2-one, (+) isomer
A mixture of 3.6 g of the compound obtained in the previous step and 0.32
g of lithium hydroxide monohydrate in 50 ml of MeOH and 5 ml of water is stirredfor 2 hours at RT. The reaction mixture is concentrated under vacuum, the residue
is taken up with water and extracted with ether, the organic phase is washed twice
with water and dried over MgSO4 and the solvent is evaporated off under vacuum.
The residue is chromatographed on silica H using DCM and then a DCM/MeOH
mixture (98/2; vlv) as the eluent to give 2.6 g of the expected product.
C) 5-(3,4-Difluorophenyl)-5-[2-methanesulfonyloxyethyl]-3-[3,5-bis-
(trifluoromethyl)benzyl]oxazolidin-2-one, (+) isomer
A solution of 2.6 g of the compound obtained in the previous step in 50 ml
of DCM is cooled to 0~C, 1.14 ml of triethylamine and then 0.62 ml of methane-
sulfonyl chloride are added and the reaction mixture is stirred for 10 minutes. It is
CA 02261808 1999-01-2~
56
concentrated under vacuum, the residue is extracted with ether, the organic phase is
washed twice witll water and dried over MgSO4 and the solvent is evaporated off
under vacuum to give 3 g of the expected product, which is used as such.
D) 5-(3,4-Difluorophenyl)-5-[2-[4-phenyl- 1 -azoniabicyclo[2.2.1]hept- 1 -yl]ethyl]-
3-[3,5-bis(trifluoromethyl)benzyl]oxazolidin-2-one chloride monohydrate, (+)
isomer
A mixture of 2.4 g of the compound obtained in the previous step, 1.6 g of
4-phenyl-1-azabicyclo[2.2.1]heptane methanesulfonate and 1.7 g of K,C03 in 2 ml
of DMF is heated at 80~C for 5 hours. After cooling to RT, the reaction mixture is
10 poured into water and extracted with DCM, the organic phase is washed with 10%
HCI solution and with saturated NaCI solution and dried over Na~SO4 and the
solvent is evaporated off under vacuum. The residue is chromatographed on silicaH using a DCM/MeOH mixture (100/5; v/v) as the eluent to give 1.46 g of the
expected product.
[a]D~~ = +25.8~ (c = l; MeOH)
Proton NMR spectrum at 200 MHz in DMSO-d6
~: 2.0 to 2.4 ppm: u: 4H
2.5 ppm: t: 2H
3.1 to 4.0 ppm: u: 10H
4.6 ppm: AB system: 2H
7.0to8.1 ppm:u: llH
EXAMPLE 2
6-(3,4-Dichlorophenyl)-6-[2-[4-phenyl- 1 -azoniabicyclo[2.2.1]hept- 1 -yl]-
ethyl]-4-[3,5-bis(trifluoromethyl)benzyl]morpholin-3-one chloride
A) 6-[2-(Benzoyloxy)ethyl]-6-(3,4-dichlorophenyl)-4-[3,5-bis(trifluoromethyl)-
benzyl]morpholin-3 -one
A mixture of 2.1 g of the compound obtained in Preparation 1.4 and 0.616
g of potassium tert-butylate in 50 ml of THF is stirred for 30 minutes at RT, 1.44 g
of 3,5-bis(trifluoromethyl)benzyl chloride are added and the reaction mixture isrefluxed for 1 hour. It is concentrated under vacuum, the residue is taken up with a
buffer solution of pH 4 and extracted with ether, the organic phase is washed with
water and dried over Na,SO4 and the solvent is evaporated off under vacuum. The
residue is chromatographed on silica H using a DCM/MeOH mixture (100/0.5; v/v)
as the eluent to give 1.8 g of the expected product.
CA 02261808 1999-01-2~
Proton NMR spectrum at 200 MHz in DMSO-d6
~: 2.4 ppm: mt: 2H
4.05 ppm: AB system: 2H
4.15to4.6ppm:u:4H
4.75 ppm: AB system: 2H
7.3to8.2ppm:u: llH
B) 6-(3,4-Dichlorophenyl)-6-(2-hydroxyethyl)-4-[3,5-bis(trifluoromethyl)benzyl]- morpholin-3-one
A mixture of 1.8 g of the compound obtained in the previous step and 4 ml
of concentrated NaOH solution in 30 ml of MeOH is stirred for 30 minutes at 0~C
and stirring is then continued for 1 hour at RT. The reaction mixture is
concentrated under vacuum, the residue is extracted with ether, the organic phase is
washed with water and dried over Na2SO4 and the solvent is evaporated off under
vacuum to give 1.1 g of the expected product.
Proton NMR spectrum at 200 MHz in DMSO-d6
~: 2.0ppm:t:2H
2.9to3.45ppm:mt:2H
3.9 ppm: AB system: 2H
4.2 ppm: AB system: 2H
4.45ppm:t: lH
4.7 ppm: AB system: 2H
7.1to8.2ppm:u:6H
C) 6-(3,4-Dichlorophenyl)-6-[2-(methanesulfonyloxy)ethyl]-4-[3,5-bis(trifluoro-
methyl)benzyl]morpholin-3-one
0.25 g of methanesulfonyl chloride is added at RT to a solution of 1.1 g of
the compound obtained in the previous step and 0.22 g of triethylamine in 30 ml of
DCM and the reaction mixture is stirred for 1 hour. It is concentrated under
vacuum, the residue is extracted with AcOEt, the organic phase is washed with
water and dried over Na2SOil and the solvent is evaporated off under vacuum to
give 1.2 g of the expected product, which is used as such.
D) 6-(3,4-Dichlorophenyl)-6-[2-[4-phenyl- 1 -azoniabicyclo[2.2.1]hept- 1 -yl]ethyl]-
4-[3,5-bis(trifluoromethyl)benzyl]morpholin-3-one chloride
A mixture of 1.5 g of the compound obtained in the previous step, 0.67 g of
4-phenyl-1-azabicyclo[2.2.1]heptane methanesulfonate and 1 g of K2CO3 in 2 ml
of DMF is heated at 80-100~C for 3 hours. After cooling to RT, the reaction
CA 02261808 1999-01-2~
mixture is poured into water and extracted with DCM, the organic phase is washedwith 10% HCI solution, with saturated NaCI solution and with water and dried over
Na2SO4 and the solvent is evaporated off under vacuum. The residue is
chromatographed on silica H using a DCM/MeOH mixture (from 100/5; v/v to
5 100/7.5; v/v) as the eluent. The product obtained is dissolved in hot DCM and
poured into pentane and the precipitate formed is filtered off to give 0.4 g of the
expected product.
Proton NMR spectrum at 200 MHz in DMSO-d6
~: 2.0to2.7ppm:u:6H
3.0to4.05ppm:u: 10H
4.2to5.0ppm:u:4H
7.2to8.2ppm:u: llH