Note: Descriptions are shown in the official language in which they were submitted.
. CA 02263671 1999-02-18
WO 98/07449 1 PCT/EP97/0404:~
~ ., _ ... , . . . ~
Description
Polymeric bile acid absorption inhibitors having simultaneous bile acid
5 adsorber action
The invention relates to polymers having bile acid absorption inhibitor and
simultaneous bile acid adsorber action, a process for their preparation and
10 the use of these polymers as pharmaceuticals.
Bile acids and their salts are natural detergents and have an important
physiological function in fat digestion and fat absorption. As the end
products of cholesterol metabolism, they are synthesized in the liver,
15 stored in the gall bladder and released from there as a constituent of the
bile into the intestine, where they display their physiological action. The
largest part (about 85-90%) of the secreted bile acids (about 1 6g/day) is
absorbed again from the intestinal wall via the enterohepatic circulation,
chiefly in the terminal ileum, and transported back to the liver, i.e. recycled.20 Only 10-15% of the bile acids are excreted with the feces. In the liver, a
reduction in the amount of bile acid can be compensated for up to a certain
degree via a control loop system by de novo synthesis of bile acids from
cholesterol. A reduction in the liver cholesterol level leads to the increase
of the absorption of cholesterol from the blood serum and thus lowers the
25 cholesterol level in the blood serum. Finally, by suppression of bile acid
reabsorption by means of suitable inhibitors or bile acid adsorbers, the
enterohepatic circulation can thus be interrupted in the intestine and as a
result the serum cholesterol level in the blood lowered. Too high a serum
cholesterol level is recognized in medicine as serious, since it leads to
30 atherosclerosis and thus increases the risk of cardiac infarct. There are
therefore many therapeutic approaches for the treatment of
hypercholesterolemia. One of these approaches is the interruption of the
enterohepatic circulation. Using this approach, it is furthermore possible to
treat all diseases in which an inhibition of bile acid reabsorption in the small
~ CA 02263671 1999-02-18
- intestine appears to be desirable.
The following have been described in the prior art:
a) Polymeric bile acid adsorbers:
Nonabsorbable polymers have been used therapeutically for some time for
the binding of bile acids. In particular, insoluble, usually crosslinked
polymers, which contain quaternized nitrogen centers and act like anion
10 exchangers, are employed for this purpose. These polymers bind some of
the bile acid anions occurring in the intestine by means of mainly ionic
interactions and transport them from the intestine. Commercial products of
this type contain, for example, the active compounds cholestyramine and
colestipol. They are employed, for example, for the therapy of
1 5 hypercholesterolemia.
b) Bile acid absorption inhibitors (receptor blockers):
In addition to the polymeric bile acid adsorbers, the active bile acid
absorption inhibition approach has also been pursued. The bile acid
20 receptor sites in the terminal ileum are blocked here by molecules which,
analogously to the bile acids, can interact with the receptors, but unlike the
bile acids are not absorbed. As a result of this receptor blockade, the bile
acids can no longer be absorbed and are then excreted with the feces.
Examples of polymeric bile acid receptor blockers are found in EP 0 549
25 967. Bile acid polymers and oligomers are described therein in which bile
acid molecules are linked laterally to a polymer backbone.
- CA 02263671 1999-02-18
. ~
The compounds described in the prior art have the following
disadvantages.
a) Polymeric bile acid adsorbers:
1. The disadvantage of all polymeric bile acid adsorbers on the market to
date is the high dosage (10-30 g/day; recommended dose in the case of
cholestyramine, for example, 12 g/day). In the case of the polymers known
to date, the high daily dose is to be attributed to a low binding rate or to a
10 partial rerelease of the adsorbed bile acids in the isotonic intestinal
medium.
2. Low compliance in patients, on account of the fishy odor and
unpleasant, sandy taste and the sandy consistency of the powder of the
adsorber (e.g. cholestyramine). The present administration form is
15 problematic, since the adsorber powder does not dissolve in water but can
only be suspended. To improve compliance, in some cases more than
50% of taste- and odor-improving additives must be added, such that as a
result the daily dose of adsorber medicament is further increased.
3. The adsorbers known to date do not act selectively enough and also
20 bind vitamins (e.g. vitamin K) and other physiologically important
substances, so deficiency symptoms (e.g. avitaminosis) can occur.
4. A damping action on the cholesterol metabolism of the intestinal bacteria
is lacking.
b) Bile acid absorption inhibitors:
1. With all low molecular weight absorption inhibitors known to date, there
is the danger of cytotoxic side effects due to absorption in the intestine.
30 Thus pinocytosis and other transport mechanisms for the absorption of
these low molecular weight inhibitors cannot be excluded. A nonsystemic
action cannot be guaranteed.
2. An unpleasant side effect which can occur with the bile acid absorption
inhibitors known to date, because of the increase in the bile acid
- CA 02263671 1999-02-18
. . .
concentration in the intestine caused by the receptor blockade, is diarrhea.
Objective of the present invention:
5 It was the object of the present invention to prepare a nonsystemically
acting polymeric active compound to interrupt the enterohepatic circulation,
which no longer has the abovementioned disadvantages.
The object is achieved by binding bile acid molecules or low molecular
10 weight bile acid absorption inhibitor molecules firmly to a polymer molecule
covalently or via a spacer group, such that they are no longer absorbable
themselves, but their absorption-inhibiting action is still retained. In this
way, the systemic cytotoxic side effects of the low molecular weight
absorption inhibitors which occur in some cases and can be caused by
15 their absorption, are avoided. The polymer, for its part, is too large to be
absorbed. The polymer additionally contains bile acid adsorber centers,
e.g. quaternized nitrogen centers, in the molecule. These reduce the bile
acid concentration in the intestine, which is increased by the receptor
blockade, by binding and adsorbing bile acid anions.
20 Polymers of this type thus have a dual action. On the one hand, they act as
polymeric bile acid absorption inhibitors due to the covalently firmly bonded
receptor blocker units and, on the other hand, as bile acid adsorbers.
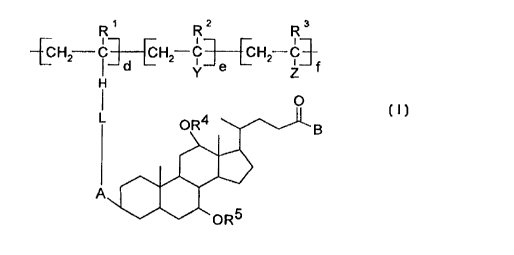
The invention therefore relates to vinyl copolymers consisting of units of
25 the formula I
- CA 02263671 1999-02-18
~CH2 C3~CCH2--C3~ECH2--c3
L oR4 ~ B
~\1 1
~~oR5
and their physiologically tolerable salts, in which:
R1, R2, R3 are hydrogen or CH3;
R4, R5 are hydrogen, (C1-C6)-alkyl, (C1-C6)-acyl;
d is 0.01 to 1.00;
e is 0 to 0.99;
f is 0 to 0.99;
where d + e + f must be equal to 1;
L is a bond, -NH-, -N(CH3)-, -+NH2CI--, -+NH(CH3)CI--, -+N(CH3)2CI--,
NH-CO-, -NH-(CH2)n-, ~NH-[(cH2)n-o-]m-(cH2)p-~ ~NH~(CH2)n~
-NH-co-(cH2)p-~-NH-(cH2)n-co-NH(cH2)m-N-(cH3)2 Cl~(CH2)m~~
-NH-[CH2-CH(CH3)-O-]m-CH2-CH(CH3)-,
-NH-(cH2)m-N(cH3)2 Cl~(CH2)n~~
-O-(CH2)n-, ~~~(CH2)n~C~~~
-CO-, -CO-NH-, -CO-N(CH3)-,
-CO-NH-CO-, -CO-NH-(CH2)n-, -CO-NH-[(CH2)n-O-]m-(CH2)p-,
-CO-NH-(CH2)n-CO-, -CO-NH-CO-(CH2)n -,
-co-NH-[cH2-cH(cH3)-o-]m- CH2-CH(CH3)-'
-CO-NH-(CH2)m-+N(CH3)2CI ~(CHZ)n~~
-CO-(CH2)n-O-(CH2)p-CO-,
-Ar-, -Ar-CO-, -Ar-CH2-, -Ar-CH2-+N(CH3)2CI ~(CH2)n~~ -CO-Ar-CO-,
-(C1 -C1 2)-alkylene-,
CA 02263671 1999-02-18
CH3
I
-NH-CH2-Ar-CH2-N ~(CH2)n~~
I
CH3 Cl
-NH-Ar-CO-, -NH-CH2-Ar-CH2-,
-NH-CO-Ar-CO-;
H is a bond, -CH2-, -Ar-, -Ar-CH2-,
where Ar is phenylene, naphthylene;
m is 1 to 18;
n is 1 to 18;
p is 1 to 18;
A is -O-, -NH-, a bond;
B js-oH~-oNal-oK7-NH2~-NH-cH3~-N(cH3)2~-NH-cH2-cH2-
SO3Na, -NH-CH2-COONa, -NH-CH2-CH2-+N(CH3)3CI-, -~-(C1 -
C18)-alkyl~-NH-(c1-c6)-alkyl~ NH-(C1-C6)-alkYIene-
HO
~ ~ ~R7
~A3H OH
R7 is-OH,-O-(C1-C6)-alkYl~-NH2;
Y is -NH2, -+NH3CI--, -NH-R9, -+NH2R9 Cl--, -NR9R10, -+NR9R10R11
Cl~,-(C1-C18)-alkylene-NH2-,-(C1-C18)-alkylene-+NH3CI~-,-(C1-C18)-
alkylene-NHR9, -(C1-C18)-alkylene-NR9R1~, -(C1-C18)-alkylene-
+NR9R10R1 1CI-, -NH-CO-(C1-C18)-alkyl, -NH-CO-(C1-C12)-alkylene-
N R9R1 ~, -N H-CO-(C1 -C1 2)-alkylene-+N R9R1 0R11 Cl-, -COR9, -CO-
, CO NH-(C1-C18)-alkylene-+NR9R1~R11CI~ ph
- CA 02263671 1999-02-18
,
-phenylene-(CO-C6)- alkylene-NH2, -phenylene-(CO-C6)-alkylene-
NH-R9, -phenylene-(C0-C6)-alkylene-NR9R1 ~, -phenylene-
(C0-C6)-alkylene- +NR9R10R11CI-, -CO-NH-R9, -NH-(C1-C18)-
alkylene-NHR9, -NH-(C1 -C18)-alkylene-NR9R1~, -NH-(C1 -C18)-
alkylene-+NR9R10R1 1 Cl-, -COOH, -O-R9, -CONH2, -O-CO-R9, -CO-
(Cl-Cl2)-alkyl, -O-CO-(Cl-Cl2)-alkylene-NR9Rl~, -O-CO-(Cl-C12)-
alkylene-+NR9R10Rll Cl-,
N~N +) F ~ --N/~ ~ --N~3 Cl
N~ Cl- , - C - O - C H 2 - C H - C H 2-~ H
-~-NH-(CH2)m-NR9R1 ~
-c-o-(c1-c18)-alkylene-+N-(c1-c18-alkyl)3 Cl-
1~ 9 10
-C-O-(C1-C18)-alkylene-NR R
Z is -NH2, -+NH3CI--, -NH-R9, -+NH2R9 Cl--, -NR9R10, -+NR9R10R11
Cl-, -(C1-C18)-alkylene-NH2-, -(C1-C18)-alkylene-+NH3CI -, -(C1-C1g)-
alkylene-NHR9,-(C1-C18)-alkylene-NR9R1~,-(C1-C18)-alkylene-
+NR9Rl0Rl lCI-, -NH-CO-(Cl -Cl8)-alkyl, -NH-CO-(Cl-Cl2)-alkylene-
- CA 02263671 1999-02-18
.
NR9R1~, -NH-CO-(C1-C12)-alkylene-+NR9R1~R1 1CI~, -COR9,
-CO-OR9, -CO-NH-(C1-C18)-alkylene-+NR9R1~R1 1CI-, -phenyl,
-p h e nyle n e-(C O-C 6)-alkyle n e-N H 2, -p h e nyle n e-(C O-C 6)-alkyle n e-N H-
R9, -phenylene-(C0-C6)-alkylene-N R9R1 ~,
-phenylene-(C0-C6)-alkylene- +NR9R10R11CI-, -CO-NH-(C1-C12)-
alkyl, -NH-(C1-C18)-alkylene-NHR9, -NH-(C1-C18)-alkylene-NR9R1~,
-NH-(C1-C18)-alkylene-+NR9R1~R11 Cl-,-COOH,
-O-R9, -CONH2, -O-CO-R9, -CO-(C1 -C12)-alkyl,
-O-CO-(C1 -C12)-alkylene-NR9R1~, -O-CO-(C1 -C12)-alkylene-
+NR9R10R11 Cl-
--N/~ Cl- /~ + fi~ Cl-
\~N(+) ~ --N ' --N~
N~ Cl- , -C -O -C H 2-l H -C H 2-~ H
O
-~-NH-(CH2)m-NR9R1 ~
-C-O-(C,-C,8)-alkylene-N+-(C1-C,8-alkyl)3 Cl,
or a crosslinker selected from the group consisting of:
CH--CH2
-C~X-(C2-C6)-alkylene-X-CC CR1-CH2-
' CA 02263671 1999-02-18
. ~
-(~-(C1 ~c3)~alkYlene)1 -1 8-0-CH-CH2 ,
-NH-(C1 -C1 2)-alkylene-NH-CH-CH2-
-NH-cH2-cH(oH)-cH2-NH-TH-cH2-,
-NH-CHOH-(C,-C12)-alkylene- CHOH-NH-CH-CH2-
X is 0, -NH-;
R9, R10 are (C1-C18)-alkyl-, -phenyl, -CH2-phenyl;
R1 1 is H, (C1 -C1 8)-alkyl-, -phenyl, -CH2-Phenyl;
where at least one of the radicals L, Y and Z must contain an ammonium
center.
20 Preferred compounds of the formula I and their physiologically tolerable
salts are those in which:
R1, R2, R3 are hydrogen or CH3;
R4, R5 are hydrogen;
d is 0.01 to 1.00;
e isOtoO.99;
f isOtoO.99;
where d+e+f must be equal to 1;
30 L is-NH-,-NH-(C1-C18)-alkylene-,
-NH-((C1 -C3)-alkylene-0-)1 -1 8-(C1 -C3)-alkylene-,
-CO-NH-, -CO-NH-(C1 -C18)-alkylene-,
,
- CA 0226367l l999-02-l8
- a~3 a-
-CaNH-(C1 -C1 8)-alkYIene- ~-(C1 -Cl g)-alkylen~,
CH
o
Il
-CO-, -NH-(C1 -C6)-alkylene-C-NH-;
H is a bond, -CH2-;
A is -O-, a bond, -NH-;
15 B is-OH,-ONa,-OCH3,-NH-CH2-CH2-OCH3,
HO \,/~\~
~O~OH
~'
~A~N~J\OH
Y is-NH2,-NHR9,-NR9Rl~,-+NR9R1~R1lCI~,-NH-(C1-C18)-alkylene-
+NR9Rl0RllCI-,-CH2-NH2,-CH2-NH-R9,-CH2-NH-(Cl-Cl8)-
alkylene-NR9R1~,-CH2-NH-(C1-C18)-alkylene-+NR9R1~R11CI~,-NH-
CO-R9, -CO-NH-R9, -CO-NH-propylene-+NR9R10R1 1CI-,
~ +
\~ N
N~
-CO-O-(Cl-Cl8)-alkylene-NR9R1~;
CA 02263671 1999-02-18
1 1
- Z is-NH2,-NHR9,-NR9R1~,-NH-(C1-C18)-alkylene-+N-(CH3)3CI~,
-CH2-NH2, -CH2-NH-R9, -CH2-NR9R1~, -CH2-NH-(C1 -C18)-alkylene-
+NR9R10R11CI-, or a crosslinker selected from the group consisting
of:
-NH(C1 -C1 2)-alkylene-NH-CH-CH2-
-CO-X-(C2-C18)-alkylene-X-C~CR1-CH2-
-NH-CH2-CH(OH)-CH2-NH-CH-CH2-;
I
X is-O-,-NH-;
R9, R10, R11 are (C1-C6)-alkyl-, -phenyl, -CH2-phenyl,
15 where at least one of the radicals L, Y and Z must contain an ammonium
center.
Particularly preferred compounds of the formula I and their physiologically
tolerable salts are those in which:
R1, R2, R3 are hydrogen;
R4, R5 are hydrogen;
d is 0.01 to 1.00;
e isOtoO.99;
f is O to 0.99;
where d+e+f must be equal to 1;
L is-NH-cH2-cH2-o-cH2-cH2-o-cH2-cH2
o
-C-NH-(C1 -C1 8)-alkylene-,
-NH-(C1 -C1 8)-alkylene-,
-CO-, -NH-,
.
CA 0226367l l999-02-l8
12
CH3CI-
-C-NH-(C1 -C6)alkYIene-~ -(C1 -C1 8)-alkYIene
O H3
-(C1 -C6)-alkylene-NH-(C~ -C6)-alkylene-;
H is a bond, -CH2-;
A is -O-, -NH-, a bond;
B is-OH,-ONa,-NH-CH2-CH2-OCH3;
Y is-NH2,-NHR9,-NH-(C1-C18)-alkylene-+N(CH3)3CI~,-CO-NH-(C1-
C10)-alkylene-+N(CH3)3CI~, -CO-NH-(C1 -C6)-alkylene-N(CH3), -CH2-
NH2,-CH2-NHR9;
Z is-NH2,-NHR9,-CH2-NH2,-CH2NHR9,-NH-(C1-C18)-alkylene-+N-
(CH3)3CI~,-CH2-NH-(C1-C18)-alkylene-+N-(CH3)3CI~,-CO-NH-
propylene-+N(CH3)3CI~;
where at least one of the radicals L, Y and Z must contain an ammonium
center.
25 Physiologically tolerable acid addition salts is understood as meaning
readily water-soluble, soluble or sparingly soluble compounds according to
the definition in the "German Pharmacopeia" (9th Edition 1986, official
edition, Deutscher Apotheker-Verlag Stuttgart), page 19. The
hydrochlorides and sulfates of the compounds are preferred.
An ammonium center is understood as meaning a positively charged
nitrogen atom (quaternized).
The invention furthermore relates to a process for the preparation of the
' CA 02263671 1999-02-18
. _ .
13
- polymers consisting of units of the formula 1.
General description of the preparation process of the polymers:
5 The synthesis of the cholate-containing monomers was carried out as
described in the examples.
Method 1:
Bonding of a linker to cholic acid and subsequent polymer-analogous
10 reaction with a polymer containing amino groups
Cholic acid is first mesylated with methanesulfonyl chloride in basic
medium. The mesyl group is a good leaving group and makes possible the
addition of side chains, e.g. of a triethylene glycol unit, by nucleophilic
15 substitution. The free hydroxyl group of the triethylene glycol unit is then
selectively activated by reaction with tosyl chloride. The tosyl leaving group
thus formed makes possible the polymer-analogous reaction with polymers
containing amino groups, e.g. polyallylamine or polyvinylamine (Example
1 d). The degree of substitution can be adjusted by changing the
20 polyamine: cholic acid derivative ratio.
Method 2:
Preparation of cholic acid amido ethers and subsequent bonding of a linker
and polymer-analogous reaction with polymers containing amino groups
Cholic acid is first converted into the active ester using p-nitrophenol. The
amido ether is then obtained by reaction with 2-methoxyethylamine
(Example 3b). This is linked via a linker to a polymer containing amino
groups as described in Method 1.
Method 3:
Free radical copolymerization of acrylic-substituted cholic acid derivatives
with vinylic monomers
CA 0226367l l999-02-l8
14
- An acrylic-substituted cholic acid derivative is reacted with a vinylic
(preferably acrylic) monomer by free radical copolymerization (see
Example 5). Ester groups which may possibly still be present in the cholate
radical can be selectively hydrolyzed under basic conditions.
Method 4:
Polymer-analogous reaction of mesyl-substituted cholic acid with polymers
containing amino groups
Cholic acid mesylate can be directly reacted with polymers containing
amino groups, e.g. polyamines, in a polymer-analogous reaction at
pH = 8-10. Substituted polyamines can also be employed here (Examples
6, 17).
Method 5
Introduction of a linker group by reaction of cholic acid with
dibromoalkanes and subsequent polymer-analogous reaction with
polymers containing amino groups
In this method, a direct reaction of cholic acids with a dibromoalkane, e.g.
dibromohexane, is achieved in THF in basic medium (Examples 7a, 15). A
large excess of dibromoalkane is important here. The omega-bromoalkoxy
cholic acid obtained is then converted into the polymer according to the
invention in a polymer-analogous reaction with a polymer containing amino
groups, e.g. a polyamine (Examples 7b, 12, 13, 15).
Method 6
Synthesis of a cholic acid derivative with a cationic center in the linker
group and subsequent homo- or copolymerization of the monomer
30 obtained
3-(N,N-Dimethylaminopropyl)methacrylamide is reacted with a bromo- or
mesyl-substituted cholic acid derivative in a Menschutkin reaction. The
3-amino group is quaternized here and a highly water-soluble acrylate-
- CA 02263671 1999-02-18
- substituted cholic acid derivative is produced (Examples 8, 9a). This can
be homopolymerized under free radical conditions or copolymerized with
other vinylic comonomers (Example 9).
5 Method 7
Free radical copolymerization of acrylic-substituted cholic acid with
allylamine (hydrochloride) or another vinylic polymerizable amine
Allylamine (hydrochloride) and other vinylic amines which can be
10 polymerized under free radical conditions can be directly polymerized with
acrylic-substituted cholic acid derivatives (Examples 11, 16).
Method 8
Polymer-analogous Michael addition of acrylic-substituted cholate to
15 polymers containing amino groups
In this reaction, an acrylic-substituted cholic acid derivative is reacted with
a polymer containing amino groups, e.g. a polyamine, in alcoholic solution
at pH = 9-10 in a Michael addition.
The present invention also relates to pharmaceutical preparations which
comprise one or more of the active compounds according to the invention
and, if appropriate, further hypolipidemic agents.
25 The active compounds according to the invention are suitable for use as
hypolipidemic medicaments.
The active compounds according to the invention are used, for example,
as pharmaceutical preparations, foodstuffs additives, formulation
30 auxiliaries, detergents, medicaments for influencing the enterohepatic
circulation of the bile acids, medicaments for influencing lipid absorption,
medicaments for influencing the serum cholesterol level, medicaments for
the concentration-dependent inhibition of bile acid absorption in the
gastrointestinal tract or as medicaments for the prevention of
' CA 02263671 1999-02-18
16
- arteriosclerotic symptoms.
Experimental section:
Example 1:
a) Example 1a: Cholic acid mesylate:
O O
,~OH ,~, J OH
~ ~l ~
HO OH H3C-S-O OH
O
1a
9.2 ml (117 mmol) of methanesulfonyl chloride were added dropwise at
0~C to 40.9 g (100 mmol) of cholic acid in 200 ml of pyridine in the course
of 20 min. The mixture was first stirred at 0~C for 15 min and then at room
temperature for 5 h. After standing overnight, the mixture was poured onto
1000 ml of ice water/200 ml of conc. sulfuric acid and stirred for a further
10 min. The resulting precipitate was filtered off with suction and washed
with water. The precipitate was then dissolved in methylene chloride and
the solution was extracted with water. The organic phase was dried using
sodium sulfate and evaporated. The product (Example 1 a) was obtained
quantitatively and employed for the subsequent step without further
purification.
1H NMR: (CDCI3) o = 0.69 ppm (s, 3H, CH3); 0.91 (s, 3H, CH3); 0.99
(d, J=6.0 Hz, CH3CH), 0.9-2.6 (m, 24 H, aliphat. CH), 2.99 (s, 3H,
CH3SO3); 3.87 (br. s, 1 H, CHOH); 4.01 (br. s, 1 H, CHOH); 4.50 (m; 1 H,
CHOS02)
' CA 02263671 1999-02-18
17
Example 1b:
Adduct of triethylene glycol to cholic acid:
OH ,~OH
~~ ~X~ ~HO ~
H3C-S-O OH ~O ~ ~OH
O 3
la lb
6.1 9 (12.5 mmol) of Example 1a were suspended in 25 ml (150 mmol) of
triethylene glycol and dissolved by briefly heating to 100~C. 4.0 9
(100 mmol) of magnesium oxide were added and the mixture was stirred at
100~C for 5 h. After standing overnight,100 ml of methylene chloride were
added and the resulting precipitate was filtered off with suction and washed
with methylene chloride. The organic phase was extracted with 200 ml of
2N aq. hydrochloric acid and then concentrated. Crude yield: 7.2 9. The
crude material was purified by chromatography on silica gel (gradient ethyl
acetate ~ ethyl acetate: methanol = 1: 1). The product fraction was
precipitated with methylene chloride with stirring under ultrasound and the
precipitate was filtered off. The filtrate contains pure Example 1 b. Yield
after concentrating: 1.7 9 (25%) of Example 1 b.
1H NMR: (CDCI3) o = 0.69 ppm (s, 3H, CH3); 0.91 (s, 3H, CH3); 0.99
(d, J=6.0 Hz, CH3CH), 0.8-2.4 (m, 24 H, aliphat. CH); 3.5-3.75 (m,13H,
O-CH2-CH2-O and CHR2OCH2); 3.85 (br. s,1 H, CHOH); 3.97 (br. s,1 H,
CHOH) .
CA 02263671 1999-02-18
18
Example 1c:
~OH ~OH
~ ~ ~
~O~OH TsO~E o~ 'OH
1b 1c
248 mg (1.3 mmol) of tosyl chloride, dissolved in 20 ml of dichloromethane,
were added in portions at 0~C in the course of 30 min to 600 mg
(1.1 mmol) of Example 1b and 140 mg (2.5 mmol) of potassium hydroxide
powder in 30 ml of dichloromethane. Since TLC checking still did not show
complete reaction, a further 100 mg (1.8 mmol) of potassium hydroxide
powder were added. The mixture was stirred at room temperature for a
further 1 h. The resulting precipitate was filtered off. The filtrate was
concentrated. Crude yield: 1.3 g. The crude product was dissolved in a little
dichloromethane and purified by means of column chromatography on
silica gel (ethyl acetate: methanol = 9: 1). Yield: 540 mg (70%).
1H NMR: (CDCI3) o = 0.68 ppm (s, 3H, CH3); 0.90 (s, 3H, CH3); 0.98
(d, J=6.0 Hz, CH3CH), 0.6-2.5 (m, 24 H, aliphat. CH); 2.44 (s, 3H, Ts-CH3);
3.5-3.75 (m,12H, O-CH2-CH2-O); 3.84 (br. s,1H, CHOH); 3.97 (br. s,1H,
CHOH); 4.15 (m, 1H, CHR2OCH2); 7.34 (d, J=9Hz, 2H, aryl H),
7.79 (J=9Hz, 2H, aryl H).
~ , .
- CA 0226367l l999-02-l8
19
Example 1d:
NH2 NH NH
2 I m:n=0.99:0.01
x HCI xHCI
;~ I 011 (~ 1~ 0 Na
TsO~ OH
1c 1d
200 mg of polyvinylamine were dissolved in 20 ml of water and pH=10 was
set. 66 mg (2 mol%) of 1 c, dissolved in 20 ml of ethanol, were added and
pH=10 was again set. The mixture was stirred at 50~C for 6 h, the pH
changing to pH = 8. The pH was again adjusted to pH = 10 and the mixture
10 was allowed to stand ovemight at room temperature. The product 1 d was
purified by ultrafiltration (membrane 5000A) using ethanol:water = 1:1 and
isolated by freeze-drying of the retentate. Yield: 180 mg. 1 H NMR analysis
showed a degree of substitution on the polyvinylamine of n = 1%.
1H NMR (D2O): o = 0.57 ppm (s, 3H, cholate CH3); 0.79 (br. m, 6H,
cholate-CH3 and CH3CH); 1.0-1.6 (m, aliphat. cholate CH); 1.6-2.5 (m,
CH2-CHNH and aliphat. cholate CH); 3.3-3.5 (O-CH2-CH2-O); 3.5-4.0 (br.
s, CHNH-CH2); 4.0-4.4 (m, CHOH and CHR2OCH2).
CA 02263671 1999-02-18
- Example 2:
NH2 NH2 NH m: n = 0.95: 0.05
xHa xHa
+ ~ ~o Na
T90'E--o~/
1c 2
0.7 ml of 2N sodium hydroxide solution was added to a solution of 100 mg
of polyvinylamine in 20 ml of ethanol and the mixture was warmed to 40-
50~C. 496 mg of Example 1 c were added in portions in the course of 6 h.
10 The pH was kept at pH = 9 and the mixture was allowed to stand at room
temp. for 3 days. The product 2 was purified by ultrafiltration (5000A
membrane) using ethanol:water = 1:1 and isolated by freeze-drying of the
retentate. Yield: 170 mg, 1H NMR analysis showed a degree of substitution
on the polyvinylamine of n = 5%.
1H NMR: as Example 1d.
CA 02263671 1999-02-18
- Example 3:
Example 3a:
O O
~J~OH ~ J1'O~3NO2
5 HO--/~OH HO--~OH
3a
20 g (49 mmol) of cholic acid were dissolved in 300 ml of THF and 10.2 g
(73 mmol) of p-nitrophenol and a spatula tipful of p-N,N-dimethylamino-
pyridine were added. The mixture was cooled to 0~C and 13.2 g (64 mmol)
of dicyclohexylcarbodiimide, dissolved in 50 ml of THF, were then added.
The mixture was stirred at 0~C for 30 min and then at room temp. for 4 h, a
precipitate forming. This was filtered off. The filtrate was concentrated to a
half of the volume and treated with pentane until it began to get cloudy.
The mixture was stirred for a further 1 h and the resulting precipitate was
filtered off. The combined precipitates were washed with toluene and dried.
Yield: 11.5 g. The combined filtrates were evaporated to dryness and the
residue was dissolved in 100 ml of THF. The solution was again treated
with pentane until it began to get cloudy and stirred for a further 1 h. The
resulting precipitate was again filtered off. Yield: 4.5 g.
Total yield of Example 3a: 16.0 g (62%).
1H NMR: (CDCI3) o = 0.70 ppm (s, 3H, CH3); 0.90 (s, 3H, CH3); 1.06
(d, J=6.0 Hz, CH3CH), 0.9-2.8 (m, 24 H, aliphat. CH); 3.47 (br. s,1 H,
CHOH); 3.85 (br. s,1 H, CHOH); 3.99 (br. s, 1 H, CHOH); 7.28 (d, J=9Hz,
2H, aryl H), 8.26 (d, J=9Hz, 2H, aryl H).
CA 02263671 1999-02-18
22
- Example 3b:
O O
~o~3No2 ~NH~oCH3
HO~OH HO--~OH
3a 3b
750 mg (10 mmol) of 2-methoxyethylamine were initially introduced into a
mixture of 100 ml of ethanol and 100 ml of water and 5.9 g (11 mmol) of
Example 3a were added in portions, the pH being kept at 10-11 by addition
of sodium hydroxide solution. The cloudy mixture was stirred at 40-50~C for
4 h and then allowed to stand at room temp. for 3 days. The mixture was
filtered and the filtrate was concentrated to a volume of 50 ml. By addition
of hydrochloric acid, the pH was adjusted to pH = 1. The mixture was
extracted twice with diethyl ether and then twice with dichloromethane. The
ether extracts were discarded. The combined dichloromethane phases
were evaporated after drying over sodium sulfate. Yield: 4.4 g (94%) of
Example 3b.
1H NMR: (CDCI3) o = 0.68 ppm (s, 3H, CH3); 0.99 (s, 3H, CH3); 1.00
(d, J=6.0 Hz, CH3CH), 0.9-2.4 (m, 24 H, aliphat. CH); 3.36 (s, 3H, CH30);
3.45 (m, 5H, CHOH and NH-CH2-CH2-O); 3.84 (br. s,1 H, CHOH); 3.96 (br.
s,1 H, CHOH); 6.20 (br. s, 1 H, NH) .
CA 02263671 1999-02-18
23
~ Example 3c:
~ , ~ ~NH~oCH3
~ ~ ~
HO OH H3C-S-O~OH
3b 3c
4.1 g (9.9 mmol) of Example 3b were dissolved in 20 ml of pyridine.
0.92 ml of methanesulfonyl chloride were added at 0~C and the mixture
was stirred at 0~C for 30 min and at room temp. for 1 h. The mixture was
treated with ice. 20 ml of conc. sulfuric acid were then slowly stirred in. The
mixture was stirred for a further 5 min. The resulting precipitate was filtered
off, washed with water and then taken up in dichloromethane. This solution
was extracted with water. The organic phase was evaporated after drying
over sodium sulfate.
Crude yield: 4.7 g. The crude product was purified by column
chromatography on silica gel (ethyl acetate). Yield: 2.9g (54%) of Example
3c.
1H NMR: (CDCI3) o = 0.69 ppm (s, 3H, CH3); 0.91 (s, 3H, CH3); 1.00
(d, J=6.0 Hz, CH3CH), 0.9-2.4 (m, 24 H, aliphat. CH); 2.99 (s, 3H,
CH3SO3); 3.36 (s, 3H, CH30); 3.45 (m, 4H, NH-CH2-CH2-O); 3.86 (br. s,
1 H, CHOH); 3.99 (br. s,1 H, CHOH); 4.50 (br. s,1 H, CHOSO2); 6.05 (br. s,
1H, NH).
.. , ~.. .. .
- CA 02263671 1999-02-18
24
- Example 3d:
O O
--~NH~OCH3 ~ NH~OCH3
~l ~X-~ ~ ~/~/
H3C-S-O OH ~O~----OH
O 3
3c 3d
1 ml of triethylamine was added to a solution of 2.9 9 (5.3 mmol) of
Example 3c and 12.0 9 (80 mmol) of triethylene glycol in 5 ml of
dichloromethane. The methylene chloride was distilled off and the mixture
was then heated to 100~C. 2 ml of triethylamine were added and the
mixture was stirred at 100~C for 5 h.1 ml of triethylamine was then added
again and the mixture was stirred at 100~C for a further 8 h. The mixture
was allowed to stand at room temp. for 1 week and then dissolved in
dichloromethane. The solution was poured into 50 ml of ice water/conc.
sulfuric acid (1:1). This mixture was extracted 3 times with
dichloromethane. The organic phase was washed with water, dried over
sodium sulfate and evaporated. Crude yield: 2.5 9. The crude material was
purified by chromatography on silica gel (gradient ethyl acetate ---> ethyl
acetate: methanol = 9:1). Yield: 0.9 9 (30%) of Example 3d.
1H NMR: (CD30D) ~ = 0.70 ppm (s, 3H, CH3); 0.91 (s, 3H, CH3); 1.02
(d, J=6.0 Hz, CH3CH), 0.8-2.5 (m, 24 H, aliphat. CH); 3.25-3.35 (m, 2H,
NH-CH2-CH2-O); 3.34 (s, 3H, CH30); 3.41-3.46 (m, 2H, NH-CH2-CH2-O);
3.50-3.67 (m,13H, O-CH2-CH2-O and CHR2OCH2); 3.79 (br. s, 1H,
CHOH); 3.95 (br. s,1 H, CHOH).
- CA 02263671 1999-02-18
- Example 3e:
~NH~OCH3 ~,I~NH~~CH3
~O~ OH TsO~O~OH
3d 3e
900 mg (1.5 mmol) of Example 1d were dissolved in 30 ml of
dichloromethane. 343 mg (1.8 mmol) of p-toluenesulfonyl chloride and
140 mg (2.5 mmol) of potassium hydroxide powder were added to this
solution at 0~C. The mixture was warmed to room temp. with stirring. After
stirring at room temp. for30 min,100 mg (1.8 mmol) of potassium
hydroxide powder were again added and the mixture was again stirred for
2 h. The precipitate was filtered off. The filtrate was evaporated. Crude
yield: 1.2 g. Pure Example 3e was obtained by column chromatography on
silica gel (gradient dichloromethane ---> dichloromethane: methanol = 95:
5). Yield: 870 mg (77%) of 3e.
1H NMR: (CDCI3) o = 0.69 ppm (s, 3H, CH3); 0.90 (s, 3H, CH3); 0.98
(d, J=6.0 Hz, CH3CH),1.0-2.4 (m, 24 H, aliphat. CH); 2.45 (s, 3H, Ts-CH3);
3.36 (s, 3H, CH30); 3.4-3.65 (m,13H, O-CH2-CH2-O, NH-CH2-CH2-O and
CHR2OCH2); 3.70 (t, J= 6Hz, 2H, Ts-O-CH2-CH2); 3.84 (br. s,1 H, CHOH);
3.97 (br. s,1H, CHOH); 4.16 (t, J= 6Hz, 2H, O-CH2-CH2); [lacuna]
1 H, CHR2OCH2); 7.35 (d, J=9Hz, 2H, aryl H), 7.80 (d, J=9Hz, 2H, aryl H).
CA 02263671 1999-02-18
26
- Example 3f:
NH2 NH NH
2 I m:n=0.99:0.01
x HCI x HCI
--Jl'hH ~NH
~/~/ OCH3 <~)0 ~~/~/ OCH3
TsO~o~J'OH
5 150 mg of polyvinylamine were dissolved in a mixture of 20 ml of water and
20 ml of ethanol. pH = 10 was set.131 mg (5 mol%) of Example 3e were
added in portions at 40-50~C. The solution was stirred at room temp. for
4 h and then heated under reflux for 3 h. After allowing to stand for a
further 3 days, Example 3e was still detectable. The mixture was adjusted
10 to pH = 12 and then heated under reflux for a further 7 h. After cooling, thepolymeric product was isolated by ultrafiltration (5000A membrane;
ethanol/water = 1 :1) and subsequent freeze-drying. Yield: 180 mg of
Example 3f. The 1H NMR analysis showed a degree of substitution of
about n = 1 %.
1H NMR (D2O): o = 0.73 ppm (s, 3H, cholate CH3); 0.96 (br. m, 6H, cholate
CH3); 0.99 (d, J= 6.0Hz, 3H, CH3CH); 1.0-2.4 (m, aliphat. cholate CH); 1.3-
1.8 (br. m, 2H, CHNH-CH2); 2.94-3.16 (br. m,1H, CHNH-CH2); 3.38 (s, 3H,
CH30); 3.4-3.9 (O-CH2, N-CH2, O-CH); 3.91 (br. s,1 H, CHOH); 4.08 (br. s,
20 1 H, CHOH).
~ CA 02263671 1999-02-18
27
Example 4:
5NH2 NH2 NH m: n= 0.95: 0.05
XHa XHa
nH (~ H
~/ / OCH3 <)~~~ / O~H3
Tso'~O~OH
100 mg of polyvinylamine were dissolved in a mixture of 20 ml of water and
20 ml of ethanol. pH = 10 was set. 350 mg (20 mol%) of Example 3e were
added in portions at 40-50~C. The solution was stirred at room temp. for
4 h and then heated under reflux for 3 h. After allowing to stand for a
further 3 days, Example 3e was still detectable. The mixture was adjusted
to pH = 12 and then heated under reflux for a further 7 h. After cooling, the
polymer product was isolated by ultrafiltration (5000A membrane; ethanol/
water = 1 :1) and subsequent freeze-drying. Yield: 170 mg of Example 3f.
The 1H NMR analysis showed a degree of substitution of about n = 5%.
1H NMR: (D2O) d = 0.73 ppm (s, 3H, cholate CH3); 0.96 (br. m, 6H, cholate
CH3); 0.99 (d, J= 6.0Hz, 3H, CH3CH); 1.0-2.4 (m, aliphat. cholate CH); 1.3-
1.8 (br. m, 2H, CHNH-CH2); 2.94-3.16 (br. m,1H, CHNH-CH2); 3.38 (s, 3H,
CH30); 3.5-3.85 (O-CH2, N-CH2, O-CH); 3.89 (br. s,1 H, CHOH); 4.05 (br.
s,1 H, CHOH).
CA 02263671 1999-02-18
28
- Example 5:
+
O NH O NH o
OCH3
Cl~
o~OH
I
= / m: n= 0.57: 0.43
~ rr-- ~--n
O NH O NH 1~l
~~'~0 Na
lCI-
o~OH
132 mg of 3-methacryloylamidopropyltrimethylammonium chloride were
added to a solution of 344 mg of comonomer I (synthesis as described in
Pat. EP 548793) in 2 ml of ethanol. Nitrogen was passed into the mixture
for 45 min. 662 ,ug of Trigonox 62 (t-butylperoxydiethyl acetate; AKZO
Chemicals) or dibenzoyl peroxide were then added under a nitrogen
atmosphere. The mixture was stirred at 75~C for 27 h with exclusion of air.
As starting material was still detectable, 687 ,ug of VA-044 (2,2 -azobis-
[2-(2 -cyanovaleric acid)]; 75% strength solution in water; Wako
Chemicals) were added under a nitrogen atmosphere. The mixture was
stirred at 45-50~C for 20 h. 5 ml of 20% strength aq. sodium hydroxide
solution were then added and the mixture was stirred at 45~C for 18 h.
150 ml of water were then added. pH = 7 was set by addition of dilute
hydrochloric acid. Example 5 was then isolated by ultrafiltration (5000A
~ CA 02263671 1999-02-18
29
- membrane; methanol/water = 1 :2).
1H NMR: (D2O) o= 0.73 ppm (s, 3H, cholate CH3); 0.96 (br. m, 6H, cholate
CH3); 0.99 (d, J= 6.0Hz, 3H, CH3CH); 1.0-2.4 (m, aliphat. cholate CH); 1.3-
1.8 (br. m, 2H, CHNH-CH2); 2.94-3.16 (br. m,1H, CHNH-CH2); 3.38 (s, 3H,
CH30); 3.5-3.85 (O-CH2, N-CH2, O-CH); 3.89 (br. s, 1 H, CHOH); 4.05 (br.
s,1 H, CHOH).
Ratio: 57:43
Example 6:
OH ~ ~ OH
NH2 NH +
(CH2),2 ~ >
--IN--_ OH
0
~ ~OH
NH2NH NH I >
(CH2) 12 ~'
--N-- OH
I Cl
l: m: n = 0.84: 0.15: 0.01
600 mg of trimethylammoniumdodecyl-substituted polyvinylamine (degree
30 of substitution 20%) were dissolved in a mixture of 10 ml of water and
10 ml of methanol. The mixture was warmed to 45-50~C. 239 mg
(10 mol%) of 3-mesylcholic acid (Example 1 a) were then added, the pH
being kept at 8-10 by addition of dilute sodium hydroxide solution. The
clear solution was stirred at 45-50~C for 15 h. The product was purified by
. . ,
CA 02263671 1999-02-18
ultrafiltration (5000A membrane) in methanol/water = 1 :1 and isolated by
freeze-drying. Yield: 550 mg.
1H NMR: (D2O) o = 0.8-2.2 (m), 2.25-4.2 (m), 3.04 (s, N-CH3). Degree of
substitution as indicated above.
Example 7:
Example 7a:
O O
~OH ; 1~OH
+ Br-(CH2)6~r ~ ,~
HO OH Br--(CH2)6--O OH
12.3 9 (30 mmol) of cholic acid were dissolved in 200 ml of THF. 73.2 9
(300 mmol) of 1,6-dibromohexane were then added and the mixture was
heated under reflux.10.2 9 (180 mmol) of potassium hydroxide powder
were then added in portions in the course of 6 h. The mixture was then
stirred for a further 1 h. After cooling, the resulting precipitate was filteredoff with suction and washed with THF. The filtrate was concentrated.
Excess dibromohexane was distilled off in vacuo. The viscous residue was
purified by column chromatography (ethyl acetate -> ethyl
acetate:methanol = 9:1). Yield: 6 9
1H NMR: (CDCI3) ~ = 0.68 ppm (s, 3H, cholate CH3); 0.89 (br. m, 6H,
cholate CH3); 0.99 (d, J= 6.0Hz, 3H, CH3CH); 1.0-2.4 (m, aliphat. cholate
CH); 1.3-1.8 (br. m, 2H, CHNH-CH2); 2.6-2.9 (br. m, 3H); 3.42 (d,
J= 6.0Hz, 2H); 3.84 (br. s,1 H, CHOH); 3.96 (br. s,1 H, CHOH); 4.06 (d,
J= 6.0Hz, 2H).
MS: Cl (ammonia): m/e[%]= 590 (M+NH4 of 31Br isotope, 95); 588 (M+NH4
CA 02263671 1999-02-18
31
- von 79Br isotope, 100).
Example 7b:
O
~ OH I OH
NH2 1 >
X HCI ~
Br--(CH2)6--O~OH O
OH ~OH
NH2 NH--(CH2)6--~ OH
x HCI
m: n= 0.98: 0.02
10 9 of polyallylamine hydrochloride were dissolved in 100 ml of water and
the pH was adjusted to pH = 10 using dil. sodium hydroxide solution. 3.1 9
of Example 7a were added at 50-60~C in the course of 3 h, a temporary
cloudiness occurring each time. The pH was kept at pH = 9.5-10 by
addition of dil. sodium hydroxide solution. The mixture was stirred at 60~C
for 4 h. The product was purified by ultrafiltration in methanol/water = 1:1
and subsequent freeze-drying. Yield: 7.4 9.
1H NMR: (D2O) o = 0.72 (br. s, 3H, cholate CH3), 0.92 (br. s, 3H, cholate
CH3), 0.95-2.2 (m, aliphat. CH), 2.72 (br. s, 2H, CH2-NH2), 2.9-4.2 (m,
CHOH, CH2O inter alia). Degree of substitution of cholate: 2%.
. CA 02263671 1999-02-18
32
Example 8:
OH \~OH
~NH N~
Meso~ O OH O
~ OH
~/
MesO l l
~ NH I ~ o~~OH
O
233 mg (0.44 mmol) of 3-(2-mesyl)ethoxycholic acid were dissolved in 1 ml
of methanol and 75 mg (0.44 mmol) of 3-(N,N-dimethylaminopropyl)-
methacrylamide were then added. After 20 min, a precipitate resulted. The
mixture was stirred at 50~C for 14 days. The solvent was stripped off and
the residue was chromatographed 3 times on silica gel (methanol ->
methanol/water/acetic acid = 10:0.5:0.05). 50 mg of product were obtained.
1H NMR: (CD30D) ~ = 0.71 (s, 3H, cholate CH3), 0.94 (s, 3H, cholate
CH3),1.01 (d, J= 7 Hz, 3H, cholate CH3CH),1.0-2.5 (m, aliphat. CH), 2.80
(s, 3H, mesylate anion), 3.0-4.0 (several m, CHOH, CH2O inter alia), 3.16
(s, 6H, N-CH3), 5.40 (br. s, 1 H, vinyl-H), 5.74 (br. s, 1 H, vinyl-H).
MS: m/e[%]= 605 (M+).
CA 02263671 1999-02-18
- Example 9:
Example 9a:
o
OH \~OH
NH~N~ + ~/
Br--(CH2)6--O~OH
OH
,~,J
O N--(CH2)6--O~OH
251 mg (0.44 mmol) of 3-(6-bromohexyloxy)cholate were dissolved in 2 ml
of methanol and 75 mg (0.44 mmol) of 3-(N,N-dimethylaminopropyl)-
methacrylamide were then added. The mixture was heated under reflux for
6 h and then allowed to stand overnight. The solvent was stripped off and
the residue was chromatographed on silica gel (methanol -> methanol/
water/acetic acid = 99:0.5:0.5).160 mg of crude product were obtained,
which was further purified by means of a weakly acidic ion exchanger.
Yield: 80 mg.
1H NMR: (CDCI3) o= 0.67 (s, 3H, cholate CH3), 0.87 (s, 3H, cholate CH3),
0.99 (d, J= 7 Hz, 3H, cholate CH3CH), 1.0-2.4 (m, aliphat. CH), 3.1-4.1
(several m, CHOH, CH2O inter alia), 3.15 (s, 6H, N-CH3), 3.86 (s), 5.35
(br. s,1 H, vinyl-H), 5.85 (br. s,1 H, vinyl-H).
~ CA 02263671 1999-02-18
34
Example 9b:
o
d~NH~ I + + ~OH
O I )
/\~\
~ (CH2)6--~ ./ OH
~ m:n= 0.70:0.30
n O
O NH O NH OH ~O Na
(CH2)6--O~
0.52 mg of free radical initiator VA 044 (Wako) was added to a solution of
55 mg (77 I~mol) of Example 9a and 17 mg (77 ,umol) of trimethyl-
20 ammoniumpropyl methacrylate chloride in 3 ml of water. The mixture was
deaerated and then stirred at 45~C for 70 h. The mixture was evaporated
and the residue was dissolved in 10 ml of water and purified by
ultrafiltration (5000A membrane). After freeze-drying, 66 mg of Example 9b
were obtained.
1H NMR: (CDCI3) ~i = 0.67 (s, 3H, cholate CH3), 0.87 (s, 3H, cholate CH3),
0.99 (d, J= 7 Hz, 3H, cholate CH3CH),1.0-2.4 (m, aiiphat. CH), 3.1-4.1
(several m, CHOH, CH2O inter alia), 3.15 (s, 6H, N-CH3), 3.86 (s), 5.35 (br.
s,1 H, vinyl H), 5.85 (br. s,1 H, vinyl H).
. CA 02263671 1999-02-18
Example 9c:
OH ~OH
~~\
~NH----N--(CH2)6--O~OH
-- ~--n O
O NH OH~O Na
~ Cl J '
(CH2)6--o OH
Nitrogen was passed through a solution of 741 mg (1.0 mmol) of Example
9a in 3.5 ml of methanol for 30 min. The solution was then warmed to
60~C. 10 mg of free radical initiator VA 044 (Wako) were added. The
mixture was then stirred at 60~C under a nitrogen atmosphere for 4 h. It
was then diluted with water and purified by ultrafiltration (5000 A
membrane). For the exchange of the counterion Br~ - Cl- it was then
washed twice with dilute aqueous NaCI solution and then twice with water.
After freeze-drying, 456 mg were obtained.
Elemental analysis:
Calculated: C 66.4 % H 10.4 % N 14.3 % Cl 5.3 %
Found: C 66.2 % H 10.5 % N 14.2 % Cl 5.2 %
~ CA 02263671 1999-02-18
36
- Example 10:
+
ONH O NH o
~J\OCH3
~N~
O~OH
m: n = 0.50: 0.50
~,~--n~ n
O NH O NH O
î~ X ' ~ Na
x HCI
O~OH
38 mg (0.35 mmol) of N-(3-N,N-dimethylaminopropyl)methacrylamide and
200 mg (0.35 mmol) of comonomer I were dissolved in 1.39 ml of ethanol.
5 Nitrogen was passed into the mixture with stirring for 45 min. 0.66 mg of
VA 044 initiator was then added. The mixture was stirred at 45-50~C for 27
h. After cooling, the resulting copolymer was isolated by ultrafiltration in
water (5000A membrane) and subsequent freeze-drying. Yield: 196 mg.
.. . .. .. . ..
. CA 02263671 1999-02-18
37
Example 11:
~--~OCH3
NH2 ~ NH~
l m:n= 0.95 005
OH ~\~O Na
_- - ~
~Hi'-- ~H
x HCI
181 mg (3.19 mmol) of allylamine were added at room temp. to a solution
of 80 mg (0.17 mmol) of I in 3 ml of ethanol. Nitrogen was passed into the
mixture for 1 h. 1.14 mg (1 mol%) of VA 044 initiator were then added. The
mixture was stirred at 50~C for 15 h. The mixture was concentrated and the
residue was stirred at 70~C for 4 h in 5 ml of 20% strength aq. sodium
hydroxide solution. The solution was adjusted to pH = 7. The product was
isolated by ultrafiltration in water (5000A membrane) and subsequent
freeze-drying. Yield: 106 mg.
... .. . . .
CA 02263671 1999-02-18
.
38
- Example 12:
~ m:n= 0.8:0.2
5CH2 CIH2
NH2 NH + 1~l
x HCI (CH2)10 OH\~OH
--N+ _
I Cl ~/
Br--(CH2)6--O~----'OH
~ ~ OH : I~OH
CH2 CH2 Cl H2 , ~, >
NH2 NH NH ~ T
(CH2) 10 (CH2)6 ~ OH
--N--
I Cl-
I: m: n = 0.79: 0.20: 0.01
1.74 9 of sodium hydroxide powder were added at room temp. to a solution
of 5.00 9 (43 mmol) of I in 100 ml of water. A solution of 1.98 9 of ll in
60 ml of methanol was then added. The mixture was stirred at 60~C for 6 h.
The mixture was diluted to a volume of 21 with water. The pH was adjusted
to pH = 7. The product was isolated by ultrafiltration in water (5000A
membrane) and subsequent freeze-drying.
Yield: 5.63 9.
1H NMR (D2O): I: m: n = 0.79: 0.20: 0.01
. CA 02263671 1999-02-18
39
- Example 13:
~ m: n= 0.8: 0.2
5 NH2 NH + ~
x Ha (CH2)l0 OH \r~ OH
--N+ ~
Cl~
10Br--(CH2)6--O'~ 'OH
OH ~OH
(CHz)lo (CH2)6 ~ OH
I Cl-
I: m: n =0.79: 0.20: 0.01
2.22 9 (55.6 mmol) of sodium hydroxide powder were added at room temp.
to a solution of 5.00 9 (55.6 mmol) of I in 100 ml of water. A solution of
2.54 9 of ll in 75 ml of methanol was then added. The mixture was stirred
25 at 60~C for 8 h. The mixture was diluted to a volume of 2 I with water. The
pH was adjusted to pH = 7. The product was isolated by ultrafiltration in
water (5000A membrane) and subsequent freeze-drying.
Yield: 5.50 9.
1H NMR (D2O): I: m: n = 0.79: 0.20: 0.01
CA 02263671 1999-02-18
- 40
- Example 14:
o
,~OCH3
~" + ~,
xNHHcl ~ NHJ--J'OH
_ _ _ I
~;~ ~ ~ O Na
x HCI <> ~~
O NH~OH
m: n = 0.99: 0.01
274 mg (0.50 mmol) of I were added at room temp. to a solution of 107 mg
(2.5 mmol) of polyvinylamine in 5 ml of ethanol. The pH of the solution was
adjusted to pH = 9-10 and the mixture was stirred at room temp. for 1
week. The mixture was concentrated to a quarter of the volume and
treated with 10 ml of 10% strength aqueous sodium hydroxide solution. It
was stirred at room temp. for 2 days and the pH was then adjusted to
pH = 7.
The product was isolated by ultrafiltration in water (5000A membrane) and
subsequent freeze-drying.
Yield: 90 mg.
1H NMR (D2O): m: n = 0.99: 0.01
CA 02263671 1999-02-18
41
- Example 15:
Example 15a:
O O
~X ~OH ~OH
~~ + Br-(CH2)~0-Br
HO~OH Br--(CH2)l0 O~OH
4.1 g (10 mmol) of cholic acid were dissolved in 100 ml of THF. 9.0 g
(30 mmol) of 1,10-dibromodecane were then added and the mixture was
15 heated under reflux. 3.4 g (60 mmol) of potassium hydroxide powder were
then added in portions in the course of 5 h. The mixture was then stirred
for a further 1 h. After cooling, the resulting precipitate was filtered off with
suction and washed with THF. The filtrate was concentrated. Excess
dibromodecane was distilled off in vacuo. The viscous residue was purified
20 by column chromatography (ethyl acetate). Yield: 0.81 g
. CA 02263671 1999-02-18
42
Example 15b:
+ ~OH
NH2 1 >
XHa ~
Br--(CH2),0 O~OH O
OH ~OH
NH2 NH--(CH2),0 O OH
x HCI
m: n = 0.99: 0.01
1.5 g of polyallylamine hydrochloride were dissolved in 15 ml of water and
10 ml of methanol and the pH was adjusted to pH = 10 using dil. sodium
hydroxide solution. 0.5 g (5 mol%) of Example 1 5a were added at 50-60~C
in the course of 2 h, a temporary cloudiness occurring. The pH was kept at
pH = 9.5-10 by addition of dil. sodium hydroxide solution. The mixture was
stirred at 60~C for 5 h. The product was purified by ultrafiltration (3000A
membrane) in ethanol/water = 1:1 and then in water and subsequent
freeze-drying. Yield: 0.95 g.
1H NMR: (D2O) degree of substitution of cholate: 1%.
. CA 02263671 1999-02-18
43
- Example 16:
OH ''f--I~OCH3
~N O O~X/
l O
OH \~O Na
_- -
O
x HCI m: n = 0.97: 0.03
Nitrogen was passed for 30 min through a solution of 63 mg (0.14 mmol) of
methyl 3-acryloylcholate in 2 ml of ethanol. 250 mg (2.60 mmol) of
N-vinylimidazole (Polyscience) and 8.9 mg (0.027 mmol) of VA 044 initiator
were then added. The mixture was warmed to 45-48~C under nitrogen and
stirred at this temperature for 2 days. A viscous material was obtained.
This was dissolved in 10 ml of methanol. 3 ml of 20% strength aqueous
sodium hydroxide solution were then added and the mixture was stirred at
50~C for 12 h. The pH was adjusted to pH = 7. The product was isolated by
ultrafiltration (5000A membrane) in water and subsequent freeze-drying.
Yield: 200 mg.
1H NMR: (D2O) degree of substitution of cholate: 3%.
. CA 02263671 1999-02-18
44
Example 17:
OH \~\/ OH
S ,~ +
MesO OH
l O
~k ~OH
X HCI --/
~1~ OH
m: n= 0.99: 0.01
500 mg of polyvinylamine were dissolved in a mixture of 10 ml of water and
10 ml of ethanol. 57 mg of 3-mesylcholic acid were added in portions at 40-
50~C. The pH was kept between 9 and 10. The mixture was stirred at 40-
50~C for 9 h. 200 ml of ethanol:water = 1 :1 were then added. The product
was isolated by ultrafiltration (5000A membrane) in water and subsequent
freeze-drying.
Yield: 500 mg.
1H NMR: (D2O) degree of substitution of cholate: 1%.
,
CA 02263671 1999-02-18
- Example 18:
+
O NH O NH o
O~OH
m: n= 0.50: 0.50
--rr-- ~--n
O NH O NH O
~ ~ ~ O Na
x HCI
O--/~OH
139 mg (0.63 mmol) of 3-methacrylamidopropyltrimethylammonium
chloride and 4.1 mg (0.013 mmol) of VA 044 initiator were added to a
deaerated solution of 300 mg (0.63 mmol) of methyl 3-acryloylcholate in
2.1 ml of ethanol. The mixture was stirred at 45-48~C for 2 days under
nitrogen. Monomers were no longer detectable on TLC checking. The pH
was adjusted to pH = 7 and 50 ml of water were added. The product was
isolated by ultrafiltration (5000A membrane) in water and subsequent
freeze-drying. Yield: 370 mg.10 ml of THF and 1 ml of 20% strength
sodium hydroxide solution were added to this product. The mixture was
warmed at 50~C. Since a homogeneous solution was not obtained, the
THF was distilled off in vacuo.100 ml of water were added and the mixture
was stirred at room temp. ovemight. The pH was adjusted to pH = 7.
Example 18 was then isolated by ultrafiltration (5000A membrane;
CA 02263671 1999-02-18
46
- methanol/water = 1 :1 ) and subsequent freeze-drying.
1H NMR: (D2O): ratio:70:30.
In the bovine bile assay adsorption test, the ability of the polymer to adsorb
bile acids is measured.
Bovine bile assay adsorption test:
The samples were prepared as follows:
An aqueous solution is first prepared which contains the following salts in
the concentrations indicated below:
NaCI 90 mmol/l
KCI 6 mmol/l
CaCI2 3 mmol/l
NaHCO3 10 mmol/l
Sodium taurocholate1.38 g/l
Sodium glycocholate2.49 g/l
The pH of the solution is adjusted to pH = 7.0 + 0.2.
25 10 ml of the above solution are taken and added to a sample vessel.
20 mg of the polymer (Examples 1 to 18) are then added. The mixture is
slowly stirred for 2 h and then centrifuged (5000 rpm). A sample of 30 ,ul is
taken from the supernatant for analysis and analyzed as described in the
following.
CA 02263671 1999-02-18
47
- HPLC with FLUORESCENCE DETECTiON
Equipment: HPLC unit from Kontron, consisting of three pumps
and mixing chamber, autosampler, UV detector and
analysis unit with MT2 software.
Fluorescence detector from Merck-Hitachi
As the samples are light- and temperature-sensitive,
the autosampler is cooled to about 5~C.
Mobile phase: Eluent A: ~'Millipore water (own unit)
Eluent B: acetonitrile/methanol 60:30
Column: ~'LiChrospher 100 RP-18, 25 mm, 5 ,um from Merck
Precolumn: LiChrospher 60 RP-select B, 4 mm, 5,um from Merck
Flow rate: 1.3 ml/min
Detection: Excitation: 340 nm
Emission: 410 nm
Gradient: 0.00 min 66 % B
7.50 min 66 % B
8.00 min 76 % B
12.50 min 76 % B
13.00 min 83 % B
25.00 min 83 % B
25.50 min 91 % B
40.00 min 91 % B
~ CA 02263671 1999-02-18
48
Enzymatic determination of the total bile acid
- 900 ~l each of the following mixture are added to Eppendorf
vessels:
6 ml of tetrasodium diphosphate buffer 0.1 M, pH 8.9,
2 ml of NAD solution (4 mg/ml of water),
20 ml of Millipore water
- 30 ,ul of the sample (concentration: 2 mg of polymer per 1 ml of
water) and 30 ,ul of enzyme solution are pipetted in.
10 - Enzyme solution: 3-alpha-hydroxysteroid dehydrogenase
0.5 units/ml
- The batches are mixed and incubated at room temperature for 2 h.
- Subsequent transfer to 1 ml disposable cuvettes and measurement
in a photometer at 340 nm.
Results of the enzyme test
Substance Adsorption
Ex. 7b 53 %
Ex. 14 52 %
Cholestyramine 33 %
Substance from EP 5 %
0549 967, Example 15
CA 02263671 1999-02-18
49
HPLC with UV DETECTION
Equipment: HPLC unit from Kontron, consisting of three pumps
and mixing chamber, autosampler, UV detector and
analysis unit with MT2 software.
Mobile phase: Eluent A: ammonium carbamate buffer 0.019 M,
adjusted to pH 4.0 with phosphoric acid.
Eluent B: acetonitrile
Column: LiChrospher 100 RP-8, 25 mm, 5 ,um from Merck
Precolumn: LiChrospher 60 RP-select B, 4 mm, 5,um from Merck
Flow rate: Gradient: 0.00 min 0.8 ml/min
20.00 min 0.8 ml/min
23.00 min 1.3 ml/min
51.00 min 1.3 ml/min
Detection: 200 nm (for preparations additionally at 254 nm)
G radient: 0.00 min 32 % B
8.00min 35%B
17.00 min 38 % B
20.00 min 40 % B
24.00 min 40 % B
30.00min 50%B
45.00min 60%B
. CA 02263671 1999-02-18
. ~ ....
Binding of the bile acids [%]
CholestyramineExample 6 Example 17
Taurocholate (TC) 52 57 57
Glycocholate (GC) 34 50 48
Taurodeoxycholate (TDC) 86 93 89
Glycodeoxycholate (GDC) 74 90 88
Taurochenodeoxycholate (TCDC) 100 100 100
Glycochenodeoxycholate (GCDC) 77 89 84
The "in vivo perfused rat intestine" test investigates the ability of the
polymers to block the bile acid reabsorption in the region of the ileum.
In vivo perfused rat intestine:
The in vivo investigation was carried out as described in F.G.J. Poelma et
al. (J. Pharm. Sci. 78 (4), 285-89,1989) - modifications of the test are
indicated.
Taurocholate and taurocholic acid, and cholate and cholic acid,
respectively, are used synonymously in the investigations.
Cannulation of the bile duct
The bile duct is exposed and a catheter is tied in (PE 50, Intramedic~). An
adaptor to accept 100 ~I disposal pipette tips (Brandt) is attached to its
end. The bile is collected in these pipettes and filled into weighed
Eppendorf reaction vessels at specific time intervals. At the end of the
experiments, the bile, as well as the medium samples, are weighed and
aliquots are measured in a scintillation counter. To this end,10 ~l of
sample are pipetted into a Sarstedt sample vessel, 58 x 22 mm, with 10 ml
of Quickszint 212 (Zinsser GmbH, Frankfurt am Main, Germany) and
counted in a Beckman 2800 t3-counter after a 30 min decay time.
1. The compounds according to the invention, Examples 1 to 18, were
CA 0226367l l999-02-l8
51
instilled into the intestinal segment together with 10 mM taurocholate using
3H-taurocholate or 14C-taurocholate as tracer and the perfusion solution
was circulated for 2 h with the aid of a peristaltic pump. The decrease in
the tracer in the intestine (medium) or the appearance of the tracer in the
bile fluid (bile) was determined with the aid of scintillation measurements
and HPLC.10 mM taurocholate with the tracer without compound
according to the invention was installed as control and the change in the
intestine and in the bile fluid was determined.
2. In vivo perfused intestine
The experimental animals used are Wistar rats bred in-house (Hoechst
animal husbandry) with an average body weight of 230-290 9. The
experimental animals are not fasted before anesthesia (urethane 1 g/kg
i.p.). After the onset of anesthesia, the animals are fixed to a temperature-
controlled (constant 37~C) operating table (Medax), shaved on the ventral
side and then the abdominal wall of the animals is opened using an
incision about 7 cm long. A Luer adaptor (Hoechst precision mechanics) is
then tied into the lower intestine about 8 cm from the ileocecal flap and the
continuing small intestine is tied off here. Further tying-in and tying-off of
the small intestine is then carried out 13-14 cm from the beginning of the
small intestine. The contents of this intestinal segment are carefully rinsed
out with warm isotonic saline solution at 37~C. The experimental solution is
later instilled into this segment, the end part of the jejunum/beginning of the
ileum.
The pump tubing is first filled from the 2 ml of installation solution (10 mM
taurocholate, polymer (Example 1 to18) in a concentration of 0.1 mmol/l in
0.9% strength phosphate-buffered saline solution, tracer: 3.5 ,uCi [3H(G)]-
taurocholic acid, NET-322, Lot 2533-081, DuPont de Nemour GmbH,
Dreieich, Germany, dissolved in phosphate-buffered isotonic saline
solution (silicone tubing A, 0.5 mm Desaga, Heidelberg, Germany, order
No.132020). The pump tubing is then attached to the intestinal segment
using two Luer adaptors and the residual solution is instilled via a three-
CA 02263671 1999-02-18
52
way tap (Pharmaseal K 75a) and a 2 ml disposable syringe (Chirana).
Immediately afterwards, the peristaltic pump (LKB Multiperpex 2115) is
switched on, and the medium is recirculated at 0.25 ml/min. A sample for
measurement of the activity (decrease in radioactivity in the intestine =
5 absorption rate) is taken at regular intervals by means of a Hamilton
syringe and a cannula (Termo 0.4 x 20) from an infusion tube integrated
into the circuit.
To demonstrate the prolonged action of the oligomeric or polymeric bile
10 acids, in this special experimental design the outflow (intestine) and the
filling (bile) of the radioactive tracer is tested with inhibitor during the first
instillation (Inst. l) and without the inhibitor during the second instillation
(Inst. ll).
Cholestyramine: 25% inhibition of absorption
Example 6: 36% inhibition of absorption
Example 7b: 50% inhibition of absorption
Example 15b: 60% inhibition of absorption
20 Evaluation of the tests
The bovine bile assay adsorption test shows that the polymers according to
the invention have a distinctly greater ability to adsorb bile acids than the
substance of Example 15 from EP 0 549 967. The ability of the polymers
25 according to the invention to adsorb bile acids is similar to that of
cholestyramine.
In the test system "in vivo perfused rat intestine", the polymers according to
the invention show an absorption-inhibiting action of 36% to 60%. In
contrast, cholestyramine shows a smaller absorption-inhibiting action of
30 25%. The polymers according to the invention are thus superior even to
cholestyramine in their action, as in addition to a great ability to adsorb bileacids, they are themselves readily bound to the bile acid receptor, and thus
show an absorption-inhibiting action.