Note: Descriptions are shown in the official language in which they were submitted.
CA 02263846 1999-02-19
S017-052.200
New Pyranoside Derivatives
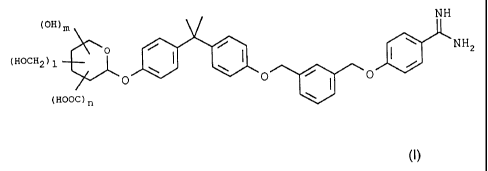
The present invention relates to new pyranoside derivatives, processes for
preparing them and their use in pharmaceutical compositions. The new
pyranoside derivatives correspond to general formula I
NH
(OH)m
o / I \ I \ _NHZ
(HOCH2)1 I
\ /
O O / I _O
(HOOC)n
wherein
I, m and n independently of one another denote an integer chosen from 0, 1,
2, 3 and 4 and I+m+n<_4
- in the form of their racemates, in enantiomerically pure or concentrated
form, optionally as pairs of diastereomers and are obtained in the form of
free
bases or salts, preferably with physiologically acceptable acids.
The compounds according to the invention are potent LTB4-antagonists. In
particular the compound of Example 1 is obtained in vivo as a metabolite of
an LTB4-antagonistic compound and in the receptor binding test it has a Ki-
value of 1.0 nM.
As has been found, the compounds of formula I are characterised by their
versatility of use in the therapeutic field. Particular mention should be made
of --
those applications in which the LT84-receptor-antagonistic properties play a
part. Examples include, in particular:
arthritis, asthma, chronic obstructive lung diseases, such as chronic
bronchitis, psoriasis, ulcerative colitis, gastropathy or enteropathy induced
by
nonsteroidal antiinflammatories, cystic fibrosis, Alzheimer's disease, shock,
reperiusion damage/ischaemia, atherosclerosis and multiple sclerosis.
CA 02263846 2005-08-25
27855-79
2
The new compounds may also be used to treat diseases or conditions
in which the passage of cells from the blood through the vascular endothelium
into the tissues is of importance (such as metastasis) or diseases and
conditions in which the combination of LTB4 or another molecule (such as 12-
HETE) with the LTB4-receptor influences cell proliferation (such as chronic
myeloid leukaemia).
The new compounds may also be used in combination with other active
substances, e.g. those which are used for the same indications, or for
example with antiallergics, secretolytics, f32-adrenergics, inhaled steroids;
antihistamines and/or PAF-antagonists. They may be administered by topical,
oral, transdermal, nasal or parenteral route or by inhalation.
The activity ratios may be investigated pharmacologically and biochemically
using tests such as those described in WO 93/16036, pp. 15 to 17.
The therapeutic or prophylactic dose depends not only on the potency of the
individual compounds and the body weight of the patient but also on the
nature and gravity of the illness. For oral administration the dose is between
and 500 mg, preferably between 20 and 250 mg. For inhalation the
patient is given between about 0,5 and 25 mg, preferably between about 2
and 20 mg of active substance.
Inhalable solutions generally contain between about 0.5 and 5 % of active
substance. The new compounds may be administered in conventional
preparations, e.g. as plain or coated tablets, capsules, lozenges, powders,
granules, solutions, emulsions, syrups, inhalable aerosols, ointments or
suppositories.
The Examples which follow show some possible ways of formulating the
preparations:
CA 02263846 1999-02-19
3
Formulation examples
1. Tablets
Composition:
Active substance according to invention 20 parts by weight
Stearic acid 6 parts by weight
Glucose 474 parts by weight
The ingredients are processed in the usual way to form tablets weighing 500
mg. If desired the content of active substance may be increased or reduced
and the quantity of glucose reduced or increased accordingly.
2. Suppositories
Composition:
Active substance according to invention 100 parts by weight
Lactose, powdered 45 parts by weight
Cocoa butter 1555 parts by weight
The ingredients are processed in the usual way to form suppositories
weighing 1.7 g.
3. Powder for Inhalation _
Micronised active substance powder (compound of formula I; particle
size about 0.5 to 7 p.m) is packed into hard gelatin capsules in a quantity
of 5 mg, optionally with the addition of micronised lactose. The powder
is inhaled from conventional inhalers, e.g. according to DE-A 33 45 722,
which is referred to herein.
The compounds according to the invention are prepared by methods which
are known ep ~ se from the prior art. Thus, the compounds of general
formula I
CA 02263846 1999-02-19
4
NH
(OH)m
(HOCH2) 1 '~ / I / I I ~ 'NHZ
p ~ ~ p / p ~
I
(HOOC)n
wherein I, m and n are as hereinbefore defined, are prepared by reacting 4-
[[3-[[4-[1-(4-hydroxyphenyl)-1-methylethyl]phenoxy]methyl]phenyl]methoxy]-
benzenecarboximidamide with a glucose derivative of general formula II
(a3)m
v -o
(~2 ) 1
x
(HOOC) n
wherein when n>0 the carboxyl group is optionally protected in the form of a
C1-4-alkyl ester and the hydroxy groups are protected in the form of acyl
groups of an aliphatic or aromatic carboxylic acid and X denotes a leaving
group which may be displaced by a phenoxide oxygen, converted from a
phenoxide and optionally the ester groups are saponified.
The compounds according to the invention may also be prepared from an
optionally protected glucose derivative (II) and the abovementioned phenol
using basic heavy metal compounds such as Ag20 or CdC03 in inert
solvents such as toluene or dichloromethane. The product is optionally freed
of the protecting groups by saponification.
The compounds (I) may also be prepared from derivatives of formula (II) and
the abovementioned phenol using Lewis acids such as, for example, BFg,
AICIg, ZnCl2, SnCl4, TiCl4, or from alkoxide derivatives of these Lewis acids
in inert solvents such as toluene, dichloromethane, etc.
CA 02263846 1999-02-19
Furthermore, the compounds according to the invention may be prepared
from an optionally protected derivative (II) wherein X=OH and the
abovementioned phenol using acid catalysts such as e.g. methanesulphonic
acid or tetrafluoroboric acid or using Lewis acids such as for example BFg,
AICIg, ZnCl2, SnCl4, TiCl4, or from alkoxide derivatives of these Lewis acids
in inert solvents such as aliphatic, aromatic, alkylsubstituted aromatics or
in a
halogenated hydrocarbon - preferably in toluene or in dichloromethane.
C1-4-alkyl in the preparation processes described above generally represents
a branched or unbranched hydrocarbon group having 1 to 4 carbon atoms,
which may optionally be substituted with one or more halogen atoms -
preferably fluorine -, which may be identical to or different from one
another.
Examples include the following hydrocarbon groups: methyl, ethyl, propyl, 1-
methylethyl (isopropyl), n-butyl, 1-methylpropyl, 2-methylpropyl and 1,1-
dimethylethyl.
The compounds according to the invention may be prepared starting from
compounds which are known from the prior art, using infer alia the methods
described in the following Examples. Various other embodiments of the
processes will become apparent to the skilled person from the description
provided. However, it is expressly pointed out that these Examples and the
associated description are provided solely for illustration and must not be
regarded as limiting the invention.
Examples
Example 1
HO O
NH
HO,,. O ~ , ~ NH _
HO _ O \ \ O ~ I O
OH
4-[[3-[[4-[1-(4-hydroxyphenyl)-1-methylethyl]phenoxy]methyl]phenyl]methoxy]-
benzenecarboximidamide-O-glucuronide
CA 02263846 1999-02-19
To a solution of 37.8 g of 4-[[3-[[4-[1-(4-hydroxyphenyl)-1-
methylethyl]phenoxy]methyl]phenyl]methoxy]-benzenecarboximidamide in
1000 ml dimethoxyethane is added a 30% solution of sodium methoxide in
methanol and the mixture is stirred for 10 min. After cooling to -10oC, 32.4 g
of methyl acetobromo-a-D-glucuronate are added and the mixture is stirred
for 48 h at ambient temperature. After the addition of a solution of 3.5 g of
LiOH in 100 ml of water the mixture is stirred for a further 48 h and the
solvent
is distilled off at ambient temperature in vacuo. The residue is dissolved in
200 ml of eluant H1) and the aqueous phase precipitated is separated off.
The substance is chromatographed using preparative thin-layer plates,
isolated after extraction with methanol and crystallised with water. Yield:
5.3 g
(Mp. 205oC (decomp.)).
1 ) 36 parts of n-butanol, 15 parts of formic acid, 15 parts of water, 15
parts of
acetone, 5 parts of chloroform are combined, shaken thoroughly, and after 3
days the aqueous phase precipitated is separated off.
Example 2
HO
NH
HO ,,. NH2
-O i i
I
HO O \ ~ O ~ O
OH w I
4-[[3-[[4-[1-(4-hydroxyphenyl)-1-methylethyl]phenoxy]methyl]phenyl]methoxy]-
benzenecarboximidamide-O-glucose
The compound is prepared according to the method of Example 1 from 4-([3-
[[4-[1-(4-hydroxyphenyl)-1-methylethyl]phenoxy]methyl]phenyl]methoxy]-
benzenecarboximidamide and tetraacetylbromoglucose.