Note: Descriptions are shown in the official language in which they were submitted.
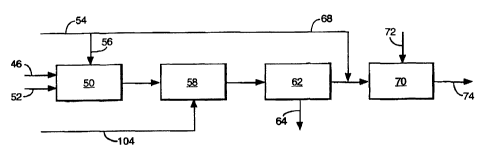
101520253035CA 02264611 2005-08-09HEAVY METAL CATALYST RECOVERY _This invention relates to the treatment of a catalyst-containing stream derivedfrom the liquid phase oxidation oi aromatic polycarboxylic acid precursors toproduce the polycarboxylic acid.Cobalt or manganese or a combination of cobalt and manganese. e.g. in theform of their acetates. together with a source of bromide ion provide catalysis forthe catalytic liquid phase oxidation of polycarboxylic acid precursors such asparaxylene to produce the polycarboxylic acid. e.g. terephthalic acid. The liquidphase oxidation is carried out using a lower monocarboxylic aliphatic acid such asacetic acid as a solvent in which the catalyst system is dissolved.The polycarboxylic acid produced by the oxidation process is withdrawn fromthe reactor as a slurry ol crystals in mother liquor comprising mainly the aliphaticcarboxylic acid together with water and dissolved catalyst components and organics(including the polycarboxylic acid. precursors thereof). Further precipitation of thepolycarboxylic acid is usually obtained by means of a crystallisation process beforeseparating the crystals from the mother liquor. The solids-liquid separation may becarried out by means of an integrated filtration and washing system as disclosed inEP-A-502628 and WO-A-93/24440.Alter separation of the aromatic acid product from the mother liquor from theslurry, conventional practice is to recycle a major part of the mother liquor and itscatalyst metal content to the oxidation reactor and to purge a minor part to avoidundue build-up of primarily organic contaminants within the reaction system. The» mother liquor purge is treated to recover said aliphatic carboxylic acid for recycle tothe oxidation reaction, leaving a high melting point and viscous residue whichcontains. inter alia, metal and bromine catalyst components and organic acidicmaterials.It has long been recognised that efficient utilisation oi catalyst and processeconomics call for the further processing of such residues to allow recovery ofcatalyst metal for reuse in the catalytic liquid oxidation process. The literature isreplete with methods for the recovery oi the catalyst metals. One commonly usedroute for recovery involves contacting the residue with water so as to extract thedesired metals. Usually the residue is contacted with water in such a way that themetal catalyst components dissolve while the organic contaminants remain largelyundissolved. Following separation ol the solution from the undissolved components.the solution is contacted with the alkali metal carbonate or bicarbonate to precipitatethe catalyst metals as carbonates or bicarbonates so that they can then berecovered for further treatment. il necessary. and recycled to the oxidation reactor.101520253035WO 98/08605CA 02264611 1999-02-26PCT/GB97/02158Such an approach is disclosed for instance in JPâB-81025195. JP-B-79037598âB.JP-B-71014339 and JP-A-51145486 and requires a solids-liquid separation step inorder to separate the solution containing dissolved catalyst metals from theundissolved material.The present invention is concerned with the treatment of catalyst containingresidues derived from the production of aromatic polycarboxyiic acids and seeks toprovide an improved process for the recovery of the catalyst metals.According to one aspect of the present invention there is provided a processfor the treatment of heavy metal cata|ystâcontaining residue from the production ofan aromatic polycarboxyiic acid having a solubility in water of less than 1% byweight at 25°C comprising dissolving substantially all of the residue in an aqueousmedium, precipitating the metal catalyst components by inclusion in the aqueousmedium of metal salt-forming anions and separating the precipitate from theaqueous medium.Conveniently the metal salt-forming anions comprise carbonate and/orbicarbonate ions.By dissolving substantially the whole of the residue. the catalyst metal yieldcan be increased since catalyst metals occluded. chemically or otherwise, with theorganics are taken into solution and can then be precipitated as salts, e.g.carbonates and/or bicarbonates.According to a second aspect of the present invention there is provided aprocess for the treatment of heavy metal catalyst-containing residue from theproduction of an aromatic polycarboxyiic acid. comprising dissolving substantially allof the residue in an aqueous medium, precipitating the metal catalyst componentsby inclusion in the aqueous medium of a carbonate and/or bicarbonate-containingliquor obtained following contacting a metal or ammonium hydroxide with a carbondioxide-containing offgas derived from the oxidation reaction in which saidpolycarboxyiic acid is produced.Typically the aromatic polycarboxyiic acid is one which has very low solubilityin water. viz. less than 1% by weight at 25°C.Preferably the aqueous medium comprises at least in part, for instance at least10% by weight (e.g. at least 20%), an organic material-containing mother liquorderived from the hydrogenation of an aqueous solution of the polycarboxyiic acid. Itis conceivable that a major part of the water employed in the dissolution step maybe contributed by the water content of said mother liquor. in contrast with priorcatalyst recovery schemes in which water is used to extract the desired metals fromthe residue, the process of the present invention involves the solubilisation ofsubstantially the whole of the residue before precipitating the catalyst metals. thus101520253035WO 98/08605CA 02264611 1999-02-26PCT/GB97/02158making it feasible to use an organics containing aqueous medium for dissolution ofthe metals and organic content of the residue.Soiubilisation of substantially all of the residue in the aqueous medium may beeffected by inclusion of an alkaline agent added to the aqueous medium. forinstance prior to and/or in the course of combining the residue with the aqueousmedium. The agent may comprise ammonium hydroxide or a metal hydroxide, suchas sodium hydroxide. Alternatively, pH may be increased initially by the inclusion ofsaid carbonate and/or bicarbonate reaction product in the aqueous medium either asthe sole alkaline agent or in combination with another alkaline agent or agents suchas ammonium hydroxide or a metal hydroxide. The inclusion of said carbonateand/or bicarbonate reaction product in the aqueous medium (by addition to theaqueous medium prior to or after contacting the same with the residue) is consideredadvantageous since the metal recovered in the form of carbonates and/orbicarbonates is of a higher quality, apparently because less oxide contaminationoccurs compared with use of hydroxide only in the initial soiubilisation of theresidue. For this reason. the process of the invention may be carried out using saidcarbonate and/or bicarbonate reaction product as the major or sole alkaline agent ineffecting initial soiubilisation of the residue.Typically the alkaline agent is introduced to raise the pH sufficiently,preferably to between 4.5 and 5.5 (more preferably 4.7 to 5.3), to dissolve theresidue and partially neutralise the acidic content of the residue (and, whereapplicable, that of the aqueous medium where the latter contains acidic componentsas in the case where it is constituted by the mother liquor derived from saidhydrogenation reaction). Subsequently the carbonate and/or bicarbonate reactionproduct is added to raise the pH further to precipitate the catalyst metals and securea pH compatible with downstream processing of the liquor remaining followingseparation of the solids. For instance, the pH is conveniently increased to about 6.5to about 9. preferably about 7 to 8. by addition of said carbonate and/or bicarbonatereaction product. Where the downstream processing includes biological treatment.e.g. anaerobic digestion. of the liquor to remove the COD content thereof, the pH ofthe liquor obtained following precipitation of the metals may be adjusted to 6.5 to 8,preferably about 7. for compatability with the biological treatment process. Suchadjustment may involve the addition of further mother liquor derived from thehydrogenation reaction and/or other acidic component such as a mineral acid (e.g.HCI) and/or alkaline component such as caustic soda. in order to secure a suitablepH for biological treatment.According to a further feature of the present invention there is provided aprocess for the treatment of heavy metal catalyst-containing residue from the1020253035W0 98l08605CA 02264611 1999-02-26PCT/GB97/02 158production of an aromatic polycarboxylic acid. comprising the steps of dissolvingsubstantially all of the residue in an aqueous medium. and precipitating the metalcatalyst components by inclusion in the aqueous medium of carbonate and/orbicarbonate ions in such a way that evolution of CO2 is substantially suppressedwhen the carbonate and/or bicarbonate ions are added.In one embodiment of the invention. the addition of the carbonate and/orbicarbonate ions is deferred until the pH of the aqueous medium has been increasedby addition of an alkaline agent other than a carbonate or bicarbonate to a levelsuch that evolution of CO2 is substantially suppressed when the carbonate and/orbicarbonate ions are added.Alternatively or additionally, the extent of dilution of the residue may becontrolled such that CO2 is suppressed upon addition of the carbonate and/orbicarbonate ions.CO2 suppression may also be effected by the application of overpressureduring the process.Suppression of carbon dioxide evolution is advantageous in order to avoidstripping volatiles such as acetic acid from the aqueous medium since the evolvedgases/vapours would then need to be treated before disposal. Also. suppression ofcarbon dioxide evolution avoids operating and/or design problems. e.g. foaming andlevel control. in dealing with such evolution during the addition of thecarbonate/bicarbonate ions.Following precipitation and separation of the catalyst metal carbonates and/orbicarbonates, the liquor is conveniently subjected to anaerobic treatment or wetoxidation, optionally followed by aerobic treatment.According to yet another aspect of the present invention there is provided aprocess for the disposal of the organics content of a heavy metal catalyst-containingresidue from the production of an aromatic polycarboxylic acid, comprisingbiologically digesting the organics after neutralising the acidity of said residue forcompatability with the biological treatment, the neutralisation step includingdissolving said residue in aqueous medium with the aid of an alkaline agent in sucha way as to precipitate the heavy metals as salts thereof, preferably as carbonateand/or bicarbonate salts.The heavy metals may in this way be separated from the liquor (e.g. forrecycle to the oxidation reactor) before the liquor is further processed by biologicaldigestion of its organic content thereby substantially eliminating the heavy metalsfrom the sludge produced by the biological digestion process. Thus. the metalsprecipitation step serves both to recover catalyst values from the residue while at10isâ203035CA 02264611 2005-08-09the same time serving as pre-treatment neutralisation of the residue feed to thebiological digestion system.Typically the process of the present invention involves producing thepolycarboxylic acid by the oxidation of a precursor thereof (such as p-xylene in thecase of terephthalic acid) in a solvent comprising a lower (C2-C6) aliphaticmonocarboxylic acid using an oxidising agent. usually air. oxygen enriched gas orsubstantially pure oxygen, in the presence of a dissolved catalyst system comprisingheavy metals such as cobalt and manganese and bromine ions. The polycarboxylicacid produced is extracted from theoxidation reactor in the form of a slurry ofcrystals in mother liquor mainly comprising the aliphatic carboxylic acid and.following separation of the crystals from the mother liquor (e.g. using one or moreintegrated solids-liquid separating and water washing units such as are described inEP~A-502628 and W0-A-93/24440), the mother liquor is divided into two fractionsfor recycle to the oxidation reactor and for purge respectively. The mother liquorpurge is concentrated by removal of the aliphatic carboxylic acid (e.g. byevaporation) and the residue is then contacted with said aqueous medium.in practice removal of any contaminant metals present in the residue. e.g. iron.copper and chromium. may be effected by suitably adapting the teaching of the priorart. for example see GB Patent Nos. 1413488 and 1319172 following the treatmentof the present invention.The invention will now be described by way of example with reference to theaccompanying drawings in which:Figure 1 is a flow sheet illustrating a process for the production of terephthalic acid;Figure 2 is a flow sheet illustrating a catalyst recovery system in accordance withthe process of the present invention: andFigure 3 is a schematic view of a scrubber unit for effecting scrubbing of off-gasfrom a reactor used in the production of terephthalic acid.In the process illustrated in Figure 1. terephthalic acid is produced in a reactor10 by reacting p-xylene (pX) with air (02) in acetic acid solvent containing somewater and a dissolved catalyst system comprising heavy metals. usually cobalt andmanganese. and bromine as a promoter. The p-xylene. acetic acid and catalyst maybe supplied to the reactor via a feed mix drum 12 in which these components aremixed with recycled mother liquor (M/L) from mother liquor drum 14. The oxygenlalris introduced separately into the reactor 10 via a feed line or lines (not shown).Further details of the reaction are given in our prior European Patent ApplicationsNos. 498591 and 502628.Typically the reaction is carried out at a temperature of 170-230°Cand a pressure of several kg/cmâ to 100 kg/cmâ. eg. 8-30 kg/cmâ.1015â-e20253035CA 02264611 2005-08-09The terephthalic acid is withdrawn lrom the reactor 10 in the form oi a slurry ofterephthalic acid crystals in mother liquor comprising acetic acid andsome water.The slurry is then subjected to crystallisation in one or more crystallisation vessels(not shown) by reducing pressure and temperature so as to precipitate furtherterephthalic acid. Following the crystallisation process. the slurry is typically at atemperature of the order of 70 to 200°C. The slurry next undergoes an integratedsolids-liquid separation process in which the crystals are separated from the motherliquor by filtration and are washed using water or acetic acid as the wash medium.The solids-liquid separation process is carried out in unit 18 under pressure using afilter medium across which a pressure differential is produced to effect displacementof the mother liquor and the wash liquor through the filter cake, comprisingterephthalic acid crystals. which develops on the filter medium. The pressuredifferential may be produced by pressurising the upstream side of the filter mediumwith a gas or vapour or by hydraulically pressurising the slurry and the wash liqour.The integrated filtration and washing process may be carried outusing for example abelt filter as disclosed in European Patent Application No. 502628 under conditionsdescribed therein or using a rotary suction filter or a pressure drum filter such as aBHSâFest drum filter or a centrifuge. In the illustrated embodiment. the filtration andwashing process is shown carried out in a rotary filter unit. if desired withcountercurrent washing of the filter cake with water. The iilter cake comprisingterephthalic acid crystals is removed from the unit 18 via line discharge outlet 20 forfurther processing. e.g. such further processing may comprise preparation for usedirectly in polyester production (without purification by hydrogenation) or it maycomprise purilication. lor instance by hydrogenation. to reduce the level ofimpurities in the terephthalic acid followed by subsequent use in the production orpolyesters, e.g. as disclosed in our prior international Patent Application No.WO93I24440.The mother liquor filtrate derived from the solidsâliquid separation unit 18 vialine 22 largely consists of acetic acid (typically 85 - 95% by weight) and water(typically 5 - 15°/o by weight). The mother liquor also contains solubleorganicby-products and intermediates produced in the reaction. reaction catalyst andresidual terephthalic acid. Also with this type of filter, the wash liquor often mixeswith the mother liquor stream. The recovered mother liquor is fed to a separator 24in which the liquor is separated from the gas used to provide the pressure differencefor the filtration and washing unit 18 (e.g. nitrogen). The gas is recovered via line 26and the mother liquor via line 28. The mother liquor is split into two fractions. one ofwhich is recycled via line 30 and mother liquor drum 14 back to the reactor and thesecond of which is purged from the process via line 32 in order to maintain the level101520253035WO 98108605CA 02264611 1999-02-26PCT/GB97/02 158of impurities in the system within acceptable limits. The mother liquor recyclefraction is typically in the range 0.7 to 0.99 (e.g. 0.7 to 0.95) and the purge fractionis correspondingly 0.3 to 0.01 (e.g. 0.3 to 0.05).The mother liquor purge is fed via line 32 to a stripper stillpot 38 in which asubstantial part of the solvent (acetic acid) is boiled off and fed to an acetic acidrecovery process (distillation column) via line 40. The residual liquor is fed toevaporator 42 for concentration. in evaporator 42, further acetic acid is driven offvia line 44 for feed to acetic acid recovery in such a way as to leave the evaporatorbottoms in a fluid state for supply of the resulting residue to a catalyst recoverywaste treatment system (see Figure 2) via line 46. The residue contains. interalia,cobalt, manganese and bromine catalyst components together with acidic organicmaterials.Referring to Figure 2, the residue obtained from the evaporator 42 is fed vialine 46 to a stirred tank 50 together with a 5% w/w caustic soda solution supplied vialine 52 and an aqueous medium supplied via lines 54, 56. Although in Figure 2. thevarious components are shown as being fed separately to the tank 50, the residuemay be slurried up in a portion of the aqueous medium in a slurry receiver upstreamof the tank 50. At least part. e.g. at least 10% by weight, of the aqueous medium isadvantageously constituted by mother liquor derived from plant for purifying crudeterephthalic acid by hydrogenation of an aqueous solution of the crude acid in thepresence of a noble metal catalyst such as platinum and/or palladium on an inert,e.g. carbon, support. Suitable plant for purifying crude terephthalic acid is describedin EP-A-498591, EP-A-502628 and WO-A-93/24440. As described in these priorpatent publications, following hydrogenation the solution is passed through acrystallisation train resulting in a slurry of purified terephthalic acid crystals inaqueous mother liquor and the slurry is filtered and washed. The mother liquorfiltrate (primary mother liquor) obtained may be used as the aqueous mediumsupplied to the tank 50. Alternatively. the primary mother liquor may be subjected tocooling or evaporation to precipitate further. but less pure. terephthalic acid crystalswhich, following separation from the secondary mother liquor, may be slurried inacetic acid for recycle to the oxidation reactor. The secondary mother liquor soobtained may then be used as the aqueous medium in the catalyst recovery system.If desired. the aqueous medium may comprise both primary and secondary motherliquor. The advantage of using the secondary mother liquor is that its organiccontent is reduced compared with the primary mother liquor. Typically the motherliquor supplied to the tank will comprise primarily water but will also contain smallamounts of acetic acid. benzoic acid. paratoluic acid. terephthalic acid andmanganese and cobalt acetates.101520253035CA 02264611 1999-02-26WO 98/08605 PCT/GB97/02158In the tank 50. at a temperature of about 60°C to about 80°C, 5% w/w causticsoda is added to raises the pH to for example about 5 and the metals and organicsare dissolved. The liquor obtained overflows into a precipitation tank 58 via abaffled outlet to prevent carryover of any solids still undergoing dissolution in tank50. Sodium carbonate and/or bicarbonate obtained from a scrubber as describedbelow is also supplied to the tank 58 via line 104, the rate of supply being such thatthe pH is raised to about 6.5 to about 9 leading to precipitation of the catalystmetals, primarily as carbonates and/or bicarbonates thereof. In practice, we havefound that significant precipitation of the catalyst metals is not initiated until a pH ofabout 6 has been attained. Beyond a pH of 6, precipitation increases rapidly and isvirtually complete when a pH of 8 is attained. Good metals recovery is achieved at apH of 7.5.As mentioned previously. some precipitation of the metals as oxides(especially manganese oxides and/or hydroxides) may also occur especially ifcaustic soda is used in tank 50. The oxides are considered to be contaminants andconsequently it may be preferred to substitute at least part of the caustic soda intank 50 with sodium carbonate and/or bicarbonate derived from the same source asthat supplied via line 104. However, we have found that as long as caustic soda isrestricted to raising the pH up to about 5.5 (preferably 4.7 to 5.3), the formation ofoxide/hydroxide contaminants is substantially avoided. Sodium carbonate and/orbicarbonate is then used to take the pH up to 6 and beyond. when pH increase ismanaged in this way, we find that the catalyst metal product recovered is in the formof freely flowing purple powder. If. on the other hand, caustic soda is used toincrease pH up to neutral pH, a material is obtained which is very different inappearance, both as a suspension and as a filtered solid, to the carbonate material.In this case, a fine black suspension is obtained which is difficult to filter and formsa brown or black filter cake on filtration which is believed to be attributable to thepresence of metal oxides/hydroxides.The contents of the precipitation tank 58 are passed to solids-liquid separatorunit 62 which may. for instance, comprise a clarifier producing a solids-containingunderflow and a liquor overflow. The underflow is pumped to a sludge buffer tank(not shown) and subsequently passed to a filter press to produce a relatively drycake containing the catalyst metal carbonates and/or bicarbonates. The catalystmetals recovered in this way may be recycled via line 64 to the oxidation reactor 10as their carbonates and/or bicarbonates or, alternatively, before recycle they may beconverted to for example acetates by reaction with acetic acid. The unit 62 mayalternatively comprise for example a centrifuge or a candle filter unit in which casethe filter press may be dispensed with.1001â S20253035CA 022646â11 2005-08-09The overï¬ow from the clarifier is mixed with additional mother liquorsupplied via lines 54. 68 and passes to a final neutralisation tank 70 where. ifnecessary. acid (e.g. a mineral acid such as HCl) or alkali (e.g. caustic soda) isadded via line 72 in order to adjust the pH of the liquor prior to feed to downstreamprocessing plant via line 74. The mother liquor supplied via line 54 typicallycorresponds to the amount which is to be purged from the purification plant tomaintain the levels of impurities within acceptable limits. especially when the motherliquor is to be recycled in the manner disclosed in EP-A-498591. EP-A-502628 andW0-A-93/24440. The purged mother liquor requires treatment before disposalbecause of its COD and such treatment will usually entail adjustment of its pH.it will be seen that the process described with reference to Figure 2 allows thepurge to be employed as a vehicle for use in recovery of catalyst metals despite theorganic content of the mother liquor purge. Rather than pass the entire amount ofmother liquor purge to the residue dissolution tank 50. it is preferably divided intotwo lractions. as indicated by lines 56 and 68. so that the equipment size and cost inthese stages can be reduced. Another factor that may influence the amount ofmother liquor employed in the dissolution stage (tank 50) is the evolution of CO, thatoccurs in the course of increasing pH in the dissolution stage. If the sodium (or otheralkali metal) carbonate andlor bicarbonate is added at low pH levels. for a givenamount of the liquor present. the amount of CO, that can remain in solution (andhence be available as carbonate ions at the precipitation stage) is reducedcompared with addition at higher pH levels.Consequently to avoid loss of CO, from solution on introduction of the sodiumcarbonate and/or bicarbonate. it may be desirable to secure conditions whichsuppress CO, evolution from the solution.This can be achieved by controlling pH(e.g. a pH of about 5 is suitable) and/or the level of dilution during the dissolution. process. Whilst equipment size and cost is a factor which implies minimising theamount of mother liquor used in the dissolution stage. it will be generally desirableto employ sufficient mother liquor consistent with suppressing CO, evolution if suchevolution is found to be a problem.The neutralisation carried out in tank 70 will usually involve adjustment of pHwithin the range 6.5 to 8. preferably 7. tor compatability with the downstreamprocessing of the liquor. Such downstream processing may take various forms suchas anaerobic treatment (e.g. using the UASB process - upflow anaerobic sludgeblanket) lollowed by aerobic treatment (e.g. activated sludge treatment). or wetF oxidation using for example the known ZIMPRO or LOPROX processes.As mentioned in relation to Figure 2. the carbonate used in the treatment ofthe residue may be derived from a scrubber. Figure 3 illustrates one form of1015..20253035CA 02264611 2005-08-09scrubbing unit for use in scrubbing eflluent gas from plant for the production ofterephthalic acid after treatment of the eliluent gas by catalytic oxidation underelevated pressure to convert methyl bromide in the effluent to bromine and/orhydrogen bromide. The elfluent gas stream is derived from the overheadscondensing system associated with a reactor for the production of terephthalic acidby liquid phase oxidation at p-xylene. for example by means of the processdisclosed in our prior EP-A-498591 and/or EP-A-502628. AThe ellluent gas stream is typically at apressure of the order oi 10 to 16 bare and a temperature of the order or 40°C andtypically contains. inter alia. volatile organics such as methyl bromide. acetic acidand benzene. together with nitrogen. water vapour. carbon monoxide. carbon dioxideand oxygen.The gas stream is preheated to a temperature of the order of 250 to 300°C.mixed with a combustion assistant and led to a catalytic combustion unit. Aconvenient combustion assistant is methyl acetate which is produced as aby-product in the terephthalic acid production process. Various other combustionassistants may be used instead or in addition. especially those which containoxygen. The amount of combustion assistant introduced is such that the temperatureof the combusted gas stream exiting the catalytic combustion unit is oi the order of400°C or greater. The catalyst employed in the catalytic combustion unit maycomprise any suitable oxidation catalyst to secure substantially total conversion ofmethyl bromide to bromine and HBr while also securing, in combination with thecombustion assistant (where needed). substantially total oxidation of other organicssuch as acetic acid and production ol heat to produce the desired exit temperature.Typically the catalyst employed comprises a noble metal catalyst such as platinumand/or palladium supported on an inert support. The support may be ceramic ormetallic in the form of a monolith or pellets. Suitable commercial catalysts areavailable from catalyst manufacturers such as Johnson Matthey. Engelhard andDegussa.Following catalytic combustion. the treated gas stream typically has atemperature of the order of 400 to 600°C and a pressure only marginally lower thanthe untreated gas stream. ie about 9.5 to 15.5 bare in the case where the untreatedgas stream has a pressure oi the order of 10 to 16 bara. The treated gas is thenpassed through an expander in which the energy content oi the gas stream isconverted into mechanical power which can be employed appropriately within theterephthalic acid production process. tor instance as power input lor an aircompressor for leading air under pressure to the oxidation reactor oi the productionprocess or tor generation ol electric power for distribution either within the plant or10101520253035WO 98/08605CA 02264611 1999-02-26PCT/GB97/02 158to other users. At the exit side of the expander, the gas stream temperature istypically of the order of 140 to 200°C (eg about 170°C) and its pressure is nearatmospheric, eg about 1.2 bara. The temperature and pressure conditions employedare such that the bromine and HBr derived from methyl bromide in the course of thecatalytic combustion remain in the gas phase thereby avoiding any risk of dew pointcorrosion. In this way, cost penalties otherwise incurred through the use of scrubbingplant upstream of the expander (with consequent reduction in energy available forextraction by means of the expander) or through the use of expensive materials ofconstruction for the expander, are avoided.Following energy recovery. the gas stream is processed to remove the brominecomponents so that any discharge to atmosphere is substantially free of suchcomponents. Such processing is effected by desuperheating the gas stream andcontacting the gas stream with a suitable aqueous scrubbing medium in thescrubbing unit of Figure 3 to remove the Brz and HBr so that the bromine content inthe discharged gas of less than 4 ppm, with 1 ppm being readily achievable. Thescrubbing unit comprises a vessel 80 having two packed sections 82 and 84. Thepackings employed may be conventional, e.g. Raschig rings. Pall rings etc. A liquidcollection tray 86 is located between the two sections 82. 84. The effluent gas(together with water employed to irrigate the pipeline), following treatment toremove HBr, is fed to an inlet 88 at the base of the vessel 80 where the gas andliquid entering the vessel impinge on a plate (not shown) within the vessel base toprevent the gas/liquid mixture impinging on that part of the vessel wall opposite theinlet 88. The gas rises through the vessel. traversing the packed sections 84, 82.and leaves the vessel via outlet 90 which may be a discharge to atmosphere.The scrubbing liquid employed may be any suitable liquid capable of removingbromine from the effluent gas. The scrubbing liquid is circulated around a loopincluding inlet line 96. the upper section 82, exit line 92, pump 94 and inlet line 96so that the liquid flows countercurrent to the direction of gas flow passing up throughthe vessel 80. A second recirculatory flow of scrubbing liquid is established in thelower part of the vessel 80. again in countercurrent relation to the gas flow, bymeans of outlet line 98. pump 100 and return line 102. Spent scrubbing liquid ispurged from the system via line 104 for supply to the precipitation tank 58 (Figure 2)and make-up liquid is supplied via line 106. The amount of scrubbing liquid pumpedthrough the vessel per unit time will generally be far in excess of that being purged.e.g. a ratio of at least 20:1, e.g. at least 30:1 (typically of the order of 40:1). A purgeline 108 interconnects the outlet of pump 94 and line 102 so that scrubbing liquidcollecting in the collection tray 86 is passed to the lower recirculatory liquid flowloop. A small amount of the scrubbing liquid is routed to the inlet 88 via line 110. for1110152025WO 98/08605CA 02264611 1999-02-26PCT/GB97/02158example from the pump 100, in order to prevent any risk of corrosion in the regionof the inlet.From the foregoing, it will be seen that the bromine containing gas is subjectedto a two stage scrubbing treatment allowing the bromine to be substantiallycompletely removed before the gas is discharged from the vessel. The scrubbingliquid is preferably caustic soda, which is converted to sodium carbonate andbicarbonate in the scrubbing vessel as a result of absorption into the hydroxide ofcarbon dioxide contained in the effluent gas. The sodium (bi)carbonate resultingfrom the scrubbing process is then used in the recovery of catalyst metals asdescribed above thereby making efficient use of the scrubbing liquor.EXAMPLESA. Effect of Dissolver pH variationsSamples of catalystâcontaining residue derived from a commercial scale plant forthe production of terephthalic acid were processed using a laboratory unitcomprising a dissolver vessel equipped with an overhead mechanical stirrer, aprecipitator vessel also equipped with an overhead mechanical stirrer, and arecovery filter in the form of a reduced pressure Buchner type filter using a vacuumfilter cloth.The composition of the plant residue is given in Table 1 below.Table 1Typical Unit Residue Feed Composition (all figures ppm wt)Component Concentration Component Concentration4CBAlc 3,425 TMA 12.292TA 39,347 BPTC 1 .9704CBA 1.115 p-TOL 3.258IPA 21.236 Co 1,400OPA 10,482 Mn 2.670BA 41 .408 Na 1.070Fe 33In Table 1, 4-CBAIc is 4-carboxybenzyl alcohol, TA is terephthalic acid, 4CBAis 4~carboxybenzaldehyde, IPA is isophthalic acid, CPA is orthophthalic acid. BA isbenzoic acid, TMA is trimellitic acid, BPTC is biphenyltricarboxylic acid, and p-TOLis paratoluic acid.The plant residue diluted 1:1 with water to render it for pumping and wassupplied to the dissolver vessel together with 5% w/w caustic soda to dissolvesubstantially all of the residue. Typically 4 parts of diluted residue are mixed in the12CA 02264611 1999-02-26WO 98/08605 PCT/GB97/02158dissolver vessel with 5 parts 2M caustic soda on a weight basis to effect dissolutionof the residue. The resulting solution was transferred to the precipitator vessel whereit was combined with a feed having a composition corresponding to the causticscrubber liquor recovered (line 104) from the offgas scrubber system described withreference to Figure 3, namely 3.2% w/w Na2CO3/4.8% w/w NaHCO3. The resultingprecipitate was then recovered by filtration. The laboratory unit was operated underdifferent temperature, pH and residence time conditions as set out below in Table 2.Table 2Conditions Used in TestsRun No. Dissoiver Precipitator Dissoiver pH Precipitator DissoiverTemp Temp pH ResidenceTime (min)1 68.2 34.7 4.8 7.4 73.32 67.8 45.2 5.1 8 84.63 78.1 49.4 6.1 8.1 494 76.9 47.7 5.9 8.2 43.25 50.6 47.7 4.5 8.1 104.26 79.1 42.1 4.5 8 87.4The filter cake recovered from filtration was dried in air at room temperatureand then analysed to determine the amounts of metals and organics present andparticle size measurements were also made using a Coulter LS 130 Laser diffractionand PIDS Particle Size Analyser fitted with the Fluid Module. as supplied by CoulterElectronics Limited of Northwell Drive, Luton. Bedfordshire. England, using slurry15 samples of the recovered catalyst in the process filtrate as the matrix. Metals contentswere determined using atomic absorption and organics were quantified using highpressure liquid chromatography. The volume mean particle size is the mean size ofthe particles in the sample examined on the basis of the volume of material accountedfor rather than the number of particles. The 5% quantile mentioned is a measure of20 the fines content of the sample. It is that particle size (in microns) below which 5% ofthe total sample lay in the particle size distribution measured. again on a volumerather than number basis. Thus a low 5% quantile figure indicates a higher finescontent. It is a more sensitive measure of the small particles in a sample than meanparticle size, and may vary widely for samples giving ostensibly similar mean particle25 size measurements. A lower mean particle size. and more especially a low 5%quantile figure. indicates a finer recovered catalyst material containing a higher13510152025WO 98/08605CA 02264611 1999-02-26PCT/GB97/02158proportion of fines which will tendfto consist of undesirable metal hydroxide particles.Samples with a high fines content will in general also be more difficult to filter.The following results (Table 3) were obtained for the different sets ofconditions specified above. where "Product" refers to the precipitate recovered byfiltration.Table 3Analytical Results under Table 2 ConditionsRun Product Product Product Product Product Volume 5% Co MnNo. Co °/o Mn °/o Na °/o Fe ppm Organic Mean Quantile Recovery RecoveryImpurities Particle Volume % %% Size Basis(micron) (micron)1 14.6 27.3 2.34 405 1.06 22.7 7.84 81.6 812 14.67 28.21 1.93 490 1.23 22.7 7.84 87.2 87.93 14.75 27.78 1.96 640 2.2 15.6 4.28 89.1 89.34 14.67 26.91 2.92 620 4.23 15.6 4.28 89.1 89.35 12.89 25.11 2.29 630 1 .46 27.7 6.45 65.4 69.26 13.86 26.2 2.49 715 1 .55 26.7 7.49 84.4 82.7All figures in Table 3 are weight %. Recovery refers to °/o metals recovered inthe filter cake as a % of those present in residue.From a comparison of Examples 1 and 2 with Examples 3 and 4. the data inTable 3 reveals that use of caustic soda to adjust the dissolver pH to 6 before theaddition of carbonate/bicarbonate produces a material containing significantly higherlevels of organic impurities. considerably lower particle size with a higher proportionof fines, and containing more iron. Comparison of Examples 1 and 2 with Examples5 and 6 reveals that use of less caustic soda to raise the dissolver pH to 4.5produces a material of equivalent particle size and organics content but the ironcontent is still higher.8. Comparison of Dissolution/Precipitation RegimesThe procedure described in Example A above was carried using the sameresidue composition using. in Run 1. 5% w/w NaOH in the dissolver vessel and 3.2%w/w Na2CO,,/4.8% w/w NaHCO3 (representative of the recovered scrubber liquorcomposition) in the precipitator vessel as in Example A and, in Run 2, using the3.2% w/w Na2CO_.,/4.8% w/w NaHCO, composition in both vessels. The conditionsemployed in each Run are given in Table 4. Tables 5 and 6 respectively give theanalytical results obtained from Runs 1 and 2 and the metals mass balance.14CA02264611 1999-02-26WO 98/08605 PCT/GB97/02158Table 4Dissolver/Precipitator ConditionsRun No. Dissolver Precipitator Dissolver pH Precipitator DissolverTemp Temp pH ResidenceTime min.1 69.58 48.88 5.05 8.05 752 69.73 48.6 5.06 8 71Table 5Analytical Results under Table 4 Conditions .Run No. Product Product Product Product Product Volume Mean 5% QuantileCo % Mn °/o Na % Fe ppm Organic Particle Size Volume BasisImpurities % (micron) (micron)1 14.7 25.5 2.2 1,020 0.67 16.47 4.282 13.1 25 3.1 930 1.33 18.9 4.285 All percentages in Table 5 are on a weight % basis.Table 6Metals Mass BalanceMn94.874.9C085.374.945 In Table 6. the Mn, Co and Fe metals recovery is expressed as a percentage ofthe metals fed as residue to the dissolver relative to the metals present in the10 recovered precipitate.From the results in Tables 5 and 6. it will be seen that the organic acid contentof the product is higher for the case using carbonate alone in the dissolution andprecipitation steps, and that the metals recovery is also lower in this case. The ironcontent of the two products is similar although the mass balance suggests that more15 is purged in the filtrate using carbonate alone. However. less significance isattached to this than to the Co/Mn mass balance information which is more accuratebecause of the higher concentrations involved.C. Comparison of Aqueous MediumIn Example A above. the residue sample was dissolved up using demineralised20 water in the dissolver vessel. Experimental runs, using the same batch of residuesample, were carried out using:Run 1 - demineralised water; and1510152025WO 98/08605CA 02264611 1999-02-26PCT/GB97/02158Run 2 - a sample of an aqueous mother liquor (PPML) typically derived as a purgefrom the hydrogenation stage of a commercially operating terephthalic acidproduction plant. in both runs, the amount of water added (either as demineralisedwater or in the form of aqueous mother liquor) were substantially the same.Both Runs were carried out in accordance with Example A in the laboratoryunit using caustic soda in the disssolver vessel and sodium carbonate/bicarbonate inthe precipitation vessel (see Table 7 for the conditions used in the laboratory unit).Table 7Laboratory unit conditionsDissolver pH Dissolver Dissolver Precipitator pH PrecipitatorTemp °C Residence Time Temp°C(minutes)5 70 90 7.5 60The results of HPLC (high pressure liquid chromatography) analysis showing theorganic contents of the recovered precipitate are given in Table 8.Table 8Organic Analysis of Recovered Precipitate (all figures ppm wt)4CBAlc TA 4CBA IPA OPADiluent Water 155 2.417 32 768 309Diluent PPML 147 1,156 N/D 391 184BA TMA BPTC p-TOL Total AcidsDiluent Water 1,633 434 83 126 0.73Diluent PPML 866 246 46 162 0.53All figures in Table 8 are ppm except the total organic acids content which isexpressed in wt %.Given the low levels of organics present. the differences in organiccontamination present in the recovered precipitate from each run are not consideredto be significant. i.e. for all practical purposes. the use of the aqueous mother liquorfrom the hydrogenation process is not considered to have a material affect on thequality of the recovered precipitate.D. Effect of precipitator temperatureThe procedure of Example A was carried out. using the same residue and withwater as the diluent. in order to illustrate the effect of temperature on theprecipitation stage, particularly in terms of the amount of iron contaminant presentin the Co/Mn product recovered. The results obtained are given in Table 9 below.1610CA 02264611 1999-02-26WO 98/08605 PCT/GB97/02158Table 9Effect of temperature in precipitation stageDissolver Dissolver pH Precipitator Precipitator pH Product IronTemp °C Temp °C ppm70 5 42 7.1 85070 5 58 7.5 94570 5 77 7.1 3,070From Table 9, it will be seen that the temperature at which precipitation iscarried out has a marked affect on the amount of iron contaminating the recoveredcatalyst product. For this reason, it is preferred to operate the precipitation stage ata temperature of no greater than 70°C, more preferably no greater than 65°C andtypically in the range of 20 to 60°C. The temperature of the precipitation stage maybe controlled by the temperature of the alkaline agent (e.g. scrubber liquor)introduced into the precipitation stage. If desired, the precipitation vessel may becooled during the precipitation process to maintain a temperature consistent withreduced iron recovery in the recovered catalyst product.17