Note: Descriptions are shown in the official language in which they were submitted.
CA 02266847 1999-03-22
WO 98113527 PCTAUS97/17413
COMPOSITIONS AND METHODS FOR
ENHANCING HYBRIDIZATION SPECIFICITY
TECHNICAL FIELD
The present invention relates generally to compositions and methods for
hybridization of oligonucleotides, and more specifically to certain solutions and/or
oligonucleotide analogues which may increase hybridization specificity.
BACKGROUND OF THE INVENTION
The detection of diseases is increasingly important in prevention and
treatments. While multifactorial diseases are difficult to devise genetic tests for, more
than 200 known human disorders are caused by a defect in a single gene, often a change
of a single arnino acid residue (Olsen, Biotechnology: An industry comes of age~National Academic Press, 1986). Many of these mutations result in an altered amino
acid that causes a disease state.
Sensitive mutation detection techniques offer extraordinary possibilities
for mutation screening. For example, analyses may be performed even before the
implantation of a fertilized egg (Holding and Monk, Lancet 3:532, 1989). Increasingly
efficient genetic tests may also enable screening for oncogenic mutations in cells
exfoliated from the respiratory tract or the bladder in connection with health checkups
(Sidransky et al., Science 252:706, 1991). Also, when an unknown gene causes a
genetic disease, methods to monitor DNA sequence variants are useful to study the
inheritance of disease through genetic linkage analysis. However. detecting and
diagnosing mutations in individual genes poses technological and economic challenges.
Several dirr~.e~ll approaches have been pursued, but none are both efficient andinexpensive enough for truly widescale application.
Mutations involving a single nucleotide can be identified in a sample by
physical, chemical, or enzymatic means. Generally, methods for mutation detection
may be divided into sc~nning techniques, which are suitable to identify previously
unknown mutations, and techniques designed to detect, distinguish, or quantitate known
sequence variants.
Several sc~nning techniques for detection of mutations have been
developed on the observation that heteroduplexes of mi~m~t~hed complementary, DNA
strands exhibit an abnormal behavior, especially when denatured. This phenomenon is
exploited in denaturing and temperature gradient gel electrophoresis (DGGE and
TGGE, respectively) methods. Duplexes mi~m~tched in even a single nucleotide
CA 02266847 1999-03-22
W O 98/13527 PCTAUS97/17413
position can partially denature, resulting in retarded migration, when electrophoresed in
an increasingly denaturing gradient gel (Myers etal., Nature 313:495, 1985; Abrams
et al., Genomics 7:463, 1990; Henco et al., Nucl. Acids ~es. 18:6733, 1990). Although
mutations may be detected, no information is obtained regarding the precise location of
a mutation. Mutant forms must be further isolated and subjected to DNA sequence
analysls.
Alternatively, a heteroduplex of an RNA probe and a target strand may
be cleaved by RNase A at a position where the two strands are not properly paired. The
site of cleavage can then be determined by electrophoresis of the denatured probe.
10 However, some mutations may escape detection because not all mi~m~tches are
efficiently cleaved by RNase A.
Mism~tched bases in a duplex are also susceptible to chemical
modification. Such modification can render the strands susceptible to cleavage at the
site of the mi~m~tch or cause a polymerase to stop in a subsequent extension reaction.
15 The chemical cleavage technique allows identification of a mutation in target sequences
of up to 2 kb and it provides information on the approximate location of mi~m~tched
nucleotide(s) (Cotton et al., PNAS US~ 85:4397, 1988; Ganguly et al., Nucl. Acids ~es.
18:3933, 1991). However, this technique is labor intensive and may not identify the
precise location of the mutation.
An alternative strategy for detecting a mutation in a DNA strand is by
substituting (during synthesis) one of the normal nucleotides with a modified
nucleotide, thus altering the molecular weight or other physical parameter of the
product. A strand with an increased or decreased number of this modified nucleotide
relative to the wild-type sequence exhibits altered electrophoretic mobility (Naylor et
25 al., Lancet 337:635, 1991). This technique detects the presence of a mutation, but does
not provide the location.
Two other strategies visualize mutations in a DNA segment by altered
gel migration. In the single-strand conformation polymorphism technique (SSCP),
mutations cause denatured strands to adopt different secondary structures, thereby
30 influencing mobility during native gel electrophoresis. Heteroduplex DNA molecules,
cont~ining internal mi~m~tches, can also be separated from correctly m~tched molecules
by eleckophoresis (Orita, Genomics 5:874, 1989; Keen, Trends Genet. 7:5, 1991). As
~vith the techniques discussed above, the presence of a mutation may be determined but
not the location. As well, many of these tech~iques do not distinguish bet~,veen a single
35 and multiple mutations.
CA 02266847 1999-03-22
W O 98/13527 PCT~US97/17413
All of the above-mentioned techniques indicate the presence of a
mutation in a limited segment of DNA and some of them allow approximate
localization within the segment. However, sequence analysis is still required to unravel
the effect of the mutation on the coding potential of the segment. Sequence analysis is a
5 powerful tool, allowing, for exarnple, screening for the same mutation in individuals of
an affected family, monitoring disease progression in the case of m~ n~nt disease, or
for detecting residual malignant cells in bone marrow before autologous transplantation.
Despite these advantages, the procedure is unlikely to be adopted as a routine diagnostic
method because of the high expense involved.
A large number of other techniques have been developed to analyze
known sequence variants. Automation and economy are very important considerations
for implementation of these types of analyses. In this regard, none of the alternative
techniques discussed below combine economy and automation with the required
specificity.
A number of strategies for nucleotide sequence distinction all depend on
enzymes, some very costly, to identify sequence differences (Saiki, PNAS USA
86:6230, 1989;Zhang,Nucl.AcidsRes. l9:3929, 1991).
For example, restriction enzymes recognize sequences of about 4-8
nucleotides. Based on an average G+C content, approximately half of the nucleotide
positions in a DNA segment can be monitored with a panel of 100 restriction enzymes.
As an alternative, artificial restriction enzyme recognition sequences may be created
around a variable position by using partially mi.cm~tched PCR primers. With thistechnique, either the mutant or the wild-type sequence alone may be recognized and
cleaved by a restriction enzyme after amplification (Chen et al., Anal. Biochem. 195:51,
1991; Levi et al., Cancer Res. 51:3497, 1991).
Another method exploits the property that an oligonucleotide primer that
is mi~m~tçlled to a target sequence at the 3' penultimate position exhibits a reduced
capacity to serve as a primer in PCR. However, some 3' mi~m~tçh~s, notably G-T, are
less inhibitory than others, thus limiting its usefulness. In attempts to improve this
technique, additional mi~m~tshes are incorporated into the primer at the third position
from the 3' end. This results in two mi~m~tched positions in the three 3' nucleotides of
the primer hybridizing with one allelic variant, and one mi~m~tch in the third position in
from the 3' end when the primer hybridizes to the other allelic variant (Newton et al.,
- Nucl. Acids Res. 17:2503, 1989). For this technique to be successful, it is necessary to
define amplification conditions that significantly favor amplification of a 1 bpmi~m~tçh .
CA 02266847 1999-03-22
W O 98/13527 PCT~US97/17413
DNA polymerases have also been used to distinguish allelic sequence
variants by determining which nucleotide is added to an oligonucleotide primer
immediately uy~L,eal~ of a variable position in the target strand. Based on thisapproach, a ligation assay has been developed. In this method, two oligonucleotide
5 probes hybridizing in immediate juxtaposition on a target strand are joined by a DNA
ligase. Ligation is inhibited if there is a mi~m~tch where the two oligonucleotide probes
abut.
Mutations may be identified via their destabilizing effects on the
hybridization of short oligonucleotide probes to a target sequence (see Wetmur, Crit.
Rev. Biochem. Mol. Biol. 26:227, 1991). Generally, this technique, allele-specific
oligonucleotide hybridization, involves amplification of target sequences and
subsequent hybridization with short oligonucleotide probes. An amplified product can
be scanned for many possible sequence variants by determining its hybridization pattern
to an array of immobilized oligonucleotide probes. Many of these techniques~
15 especially allele-specific oligonucleotide hybridization~ require establishing conditions
that favor the hybridization of an exact match over a mismatch. As is well known, such
conditions are difficult to achieve. One approach to improving hybridization is the
addition of a chaotrope.
Chaotropes decrease the melting temperature of an oligonucleotide duplex
20 (see Van Ness and Chen, Nucleic Acids Research 19:5143, 1 99 l ). Oligonucleotide probes
( 12-50 mers) possess some functional properties that are not shared by long DNA probes.
These parameters include different rates of duplex formation as a function of (a) the
difference between the hybridization t~nll~l~ure and the Tm~ (b) stringency requirements
for m~ims~l selectivity/specificity of hybridization, and (c) sequence-specific anomalous
25 behavior.
Chaotropes are useful in DNA probe-based diagnostic assays, as they
can simultaneously Iyse the cells of org~ni~m~ of interest, inhibit nucleases and
proteases, and provide adequate hybridization stringency without chemically altering
the target analyte. Chaotropic lysis and hybridization solutions elimin~te the need to
30 isolate nucleic acid prior to conducting the DNA probe assay, and facilitate the
development of rapid and simple assay formats (see Van Ness and Chen, Nucleic Aci~s
Research 19:5143, 1991, for review). However, the commonly used chaotropes do not
substantially increase the differential hybridization of matchedlmi~m~tched sequences.
In addition, special problems arise when hybridization methods are
35 employed that involve the use of mixed pools of oligonucleotide probes (12- to
50-mers) having differing base sequences and G+C content. Many applications utilize
CA 02266847 1999-03-22
WO 98/13527 PCT/USg7/17413
mixed pools of oligonucleotides and are frustrated by a host of problems. For example~
many gene isolation strategies involve the reverse translation of a known polypeptide
sequence into a set of all possible DNA sequences that can encode that protein (Jaye
etal., Nucl. Acids Res~ 2325-2335, 1983). A pool of oligonucleotide probes,
5 homologous to the set of possible protein encoding DNA sequences, are then used to
screen a genomic or cDNA library from the relevant organism or cell type in order to
identify the desired gene sequence. While the length of all of the oligonucleotide
probes is the sarne, the G+C content of each probe may vary significantly, making the
selection of hybridization conditions that are suitable and specific for each
10 oligonucleotide problematic. As a result, often many false positive clones will be
selected when screening highly complex libraries for genes of low abundance.
This problem of simultaneously and accurately hybridizing many
differing oligonucleotides of differing G+C content is even greater for sequenceanalysis of a specific region of DNA or identifying single base change mutations using
15 large arrays of oligonucleotides (which may vary from 100% A+T to 100% G+C)
bound to a fixed surface (Southern etal., Genomics 13:1008-1017, 1992; Maskos and
Southern, Nucl. Acids Res. 20:1675-1678, 1992). These methods, while theoretically
powerful, have been sorely limited by the inability to identify hybridization conditions
that will facilitate accurate hybridization (i.e., no mi~m~tch hybrid duplexes formed)
20 and allow all possible perfect hybrids to be stably forrsled.
One attempted solution has been to use a class of salts composed of
small alkylammonium ions (most commonly tetramethylammonium (TMA+) and tetra-
ethylammonium (TEA+)), that can greatly decrease the effect of base composition on
DNA melting (Marky etal., Biochemistry 20:1427-1431, 1981; De Murcia etal..
25 Biophys. Chem. 8:377-383, 1978; Melchior and Von Hippel, Proc. Nat. Acad. Sci. USA
70:298-302, 1973). Of the tetraalkylammonium salts, only TMA+ and TEA+ are smallenough to fit into the major groove of the B-form DNA double helix where they bind to
the A+T base pairs of DNA (perhaps to the 0-2 of thymine) (see De Murcia et al.,Biophysical Chemistry 8:377 1978). The overall effect on stability is two-fold with the
30 first being that the tetraalkylammonium salts increase the non-polar character of the
solvent which destabilizes the base stacking interactions in native DNA (see Rees et al..
Biochemistry 32:137, 1993). The second effect is that the A+T base pairs are stabilized.
Specifically, TMACI prevents DNA premelting by decreasing the transient openingsbetween the base pairs from occurring below the melting te~ t~ re (see De Murcia et
35 al., Biophysical Chemistry 8:377 1978; Marky et al., Biochemistry 20:1427, 1981). The
exact nature of TEACI stabilization is not known. Overall, the A+T pairing is stabilized
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
resulting in a rise in the melting temperature for the A+T pairs (see Marky et al.,
Biochemistry 20:1427 1981; De Murcia et al., Biophysical Chemistry 8:377 1978). For
100% A+T oligonucleotide duplexes, the Tm in TMACI is actually 6~C higher than that
found in a sodium solution (see Marky et al., Biochemistry 20:1427, 1981).
When genomic DNA is melted in TMACI or TEACl at the specific
concentrations of 3M and 2.4M, respectively, identical melting t~m~ ul~s are
exhibited for A+T and G+C pairs (see Melchior et al., Proc. Natl. Acad. Sci. USA70:298, 1973). Usually what is observed is that synthetic DNA duplex stability in
concentrated TMACl and TEACI stability is somewhat ~imini.ched and has little base
compositional dependence (see Wood et al., Proc. Natl. Acad. Sci. USA 82:1585 1985;
Marky et al., Biochemistry 20:1427 1981; Jacobs et al., Nucleic Acids Res. 16:4637,
1988). For example, a series of 19-mers ranging from 26% G+C to 79% G+C content
had melting temperatures over a range of 18~C in 2X SSC, while in 3M TMACI the
range narrowed to 5~C and in 2.4M TEACI, the temperatures were virtually unchanged
negating all influence from G+C content (see Jacobs et al., Nucleic Acids Res. 16:4637,
1988). TEACI had the added benefit of reducing the melting temperature
approximately 22~C over TMACI and SSC (see Jacobs et al., Nucleic Acids Res.
16:4637, 1988). When various lengths of hybridization probes are measured and the
corresponding melting temperatures plotted versus length, the plot is a smooth curve
even though the G+C content varied from 31-66% (see Wood et al., Proc Natl. Acad.
Sci. US,4 82:1585 1985). In addition, the width of the melting curve, or the HCT, for
natural DNA fragments is significantly reduced in TMACI (1~C) than in sodium
solutions (5-10~C) (see Wood et al., Proc. Natl. Acad. Sci. USA 82:1585 1985).
Narrowing of the HCT is indicative of the stabilization of the A+T pairing since the
A+T pairs normally melt at a lower temperature than the G+C pairs creating broadmelting curves.
In the context of gene isolation from complex libraries, the number of
false positive clones isolated using a 17-mer mixed oligo pool (G+C range of 47% to
71%) was reduced 100-fold when performed in 3 M TMACl rather than using a NaCl
hybridization solution (Wood etal., Proc. Nat. Acad. Sci. USA ~2:1585-1588, 1985).
However, even when using TMACl to elimin~te the base composition effect on Tm~ asignificant number of false positive clones are still isolated due to formation of
mi~m~tched hybrids.
Using deoxyinosine at the third codon position (Honoré et al., J.
Biochem. Biophys. Methods 27:39-48, 1993) of highly degenerate oligonucleotide pools
from backtranslated protein sequences allows the oligonucleotide pool size to be
CA 02266847 1999-03-22
W O 98/13527 PCT~US97tl7413
significantly reduced. However, when screening a more complex genomic library for
clones, the isolation of false positive clones may still be a significant problem (Jacobs
et al., Nucl. Acids Res. 16:4637-4650, 1988). While the presence of
tetramethylammonium and tetraethylarnmoniurn salts made oligonucleotide melting
5 independent of base composition, there was no or little effect of mi~m~tches on the
thermal melting of oligonucleotides. That is, duplexes cont~ining a mi.cm~tch had a
similar Tm to duplexes which were perfectly base-paired.
Another method used to enhance specificity in hybridization reactions
creates base mi~m~tc.hes using base analogs to replace any of the A, G, C, or T
10 nucleotides. Research has shown that some primers containing a base pair mi~m~tch
have increased specificity when the mi~m~tch is placed in precise locations (seeWenham et al., Clinical Chemistry 37:241, 1991; Newton et al., Nucleic Acids Research
17:2503, 1989; Ishikawa et al., Human ~mmunology 42:315, 1995). However,
differences of as little as 0.5~C in the melting temperatures are equally comrnon
15 between perfectly matched hybrids and the same hybrid with a single base mi~m~tch
introduced ~see Tibanyenda et al.. European lournal of Biochemistry 139:19, 1984;
Werntges et al., Nucleic Acids Research 14:3773, 1986). Even better specificity has
been noted between one and two base mi.~m~tched duplexes than has been observed
between a perfectly matched duplex and the same duplex with a single mi.~m~tch (see
Guo et al., Nature Biotechnology 15:331, 1997). Guo et al. found a (Tm Of 4~C between
zero and one mi.~m~tches and a ~Tm of 13~C between one and two adjacent mi~m~tches
for a 20-mer duplex. However, even with two mi~m~tches, often there is still little
destabilization of the duplex. This inability to consistently discriminate mi~m~tl~.hes
lends to the lack of specificity in PCR.
The use of more than one base pair mi~m~tch per hybridization
employing at least one nucleotide analog has been evaluated (see Guo et al., Nature
Biotechnology 15:331, 1997). In this case, the analog compound consists of 3-
nitropyrrole repl~cf~n~ent of the purine or pyrimidine bases. 3-Nitropyrrole has the
ability to minim~lly hydrogen bond with all four bases ~see Nichols et al., Nature
369:492, 1994; Be~ onl et al., Journal of the American Chemical Society 117:1201,
1995). By introducing an artificial mi~m~tch, large differences in the duplex melting
~el~lpel~lules occur ranging from approximately 5~C to 15~C with the largest difference
occurring when the mi.cm~teh is located at the center of the 15-mer hybridizing oligo.
Significant differences in ~T" occur when an artificial nucleotide is introduced into a
duplex that already contains a base mi.~m~tch creating a two-mi.~m~trh duplex. The
degree of destabilization depends upon the type of base mi~m~tch (e.g, G/T) and the
CA 02266847 1999-03-22
wO 98/13527 PCT/USg7/17413
separation between the two mi.~m~tches. In experimental ex~min~tion, the base analog
nucleotide ranged from 1 to 7 bases to the 3' side of the base mism~tch, which was held
in the center of the 1 5-mer. Differences in ~Tm for the three different base mi~m~tched
15-mers ranged from a 2~C stabilization (in the C/T mi~m~tch case only and when the
5 mi~m~t~hes are adjacent) to a 7~C further destabilization with the maximum
destabilization consistently occurring at a 3 or 4 base mi.~m~tch separation (see Guo et
al., Nature Biotechnology 15:331, 1997).
When two artificial mism~tches are introduced, the proximity of the
artificial bases greatly influences the degree of destabilization. The two artificial
10 mism~t~hes were centered on the middle of a 21-mer duplex beginning with a
separation of 6 bp. The destabilization, or ~Tm, is minim~lly 12~C when compared to
the perfectly matched duplex. The greatest difference of over 20~C occurs when the
two artificial mi~m~tches are 10 base pairs apart. This difference corresponds to one
helical turn and indicates that some kind of interaction occurs between the two artificial
15 bases that decreases the stability of the duplex.
Experimentally, when the PCR primer utilized contained one or two
artificial mi~m~t~hes between the primer and the DNA sample, the PCR gave results as
would be expected for a perfectly matched primer (see Guo et al., Nature Bio~ec~nology
15:331, 1997). However, when the primer contained both a true and an artificial
20 mi.~m~tch, the PCR failed to produce any measurable results; while PCR with perfectly
matched and true mi.cm~tf~hes all produced measurable amounts of PCR product. The
same study found similar results when using hybridization probes: those with perfect
matches, true mism~tchçs and artificial mismatches annealed while the probes
cont~ining artificial and true mism~tches did not. These studies indicate greater
25 specificity is created when artificial base mism~tches are incorporated in hybridization
reactions such that when naturally occurring mi~m~tches occur, they are
thermodynamically less stable than a perfectly matched hybridization reaction and thus
less likely to produce a false positive in an assay or PCR. Interestingly, however, the
difference in thermodynamic stability noted above for duplexes cont~ining only
30 artificial mi~m~lches is not manifested in the experimental situation.
A further means of effecting hybridization discrimination is through
differences in the stability between hybridization duplexes that contain nicks and gaps.
In these reactions, duplexes are formed from tandemly stacked short oligomers
hybridized to a longer strand that either align contiguously or non-contiguously leaving
35 a few base pair gap. Hybridizations that result in a nick are subject to "stacking
hybridization" where another DNA strand hybridizes across the nick site. Stacking
CA 02266847 1999-03-22
W O 98113S27 PCT~US97/17413
hybridization does not occur where gaps are present in the non-contiguous oligomers.
The stacking has the effect of increased discrimination as evidenced by decreased
dissociation rates and greater thermodynamic stability than the non-contiguous
co~ ~L~ (see Lane et al., Nucleic ,4cids Res. 25:611, 1997). Thermodynamic
5 measurements show differences between the hybridization stacked duplexes standard
free energy change (~G) and the gapped duplexes is 1.4 to 2.4 kcal/mol. Therefore,
discrimination in hybridization can be afforded through the use of multiple short probes.
Most of the base mimics in current use are the result of the pursuit for a
universal base. Many utilize nitroazole base analogues and have demonstrated reduced
10 discrimination in base pairing. A series of nitroazole nucleobase analogues have been
studied in ~Ue,llpl~ to gain additional insight into the significance of electronic structure
and heterocyclic size in base pairing for the development of more effective universal
bases (see Bergstrom et al., NucleicAcids Res. 25:1935, 1997). In this work, thethermodynamic ~.ol)~,lies of the deoxyribonucleosides of 3-nitropyrrole, 4-
15 nitropyrazole, 4-nitroimidazole, and 5-nitroindole were measured. For comparison,
thermodynamic measurements were also made on the deoxyribonucleosides of
hypo~n~hine and pyrazole as well an abasic spacer, 1,2-dideoxyribose. Four
oligonucleotides were synthesized for each modified nucleoside in order to obtain
duplexes in which each of the four natural bases was placed opposite the base mimic.
20 All of the base mimics analyzed proved to be far less stable than the natural base
pairings (A+T: Tm = 65.7~C, C+G: T", = 70.5~C) with the T",s ranging from 35-46~C for
5-nitroindole to 18-29~C for the other nitroazole bases analyzed. The only exception
was 4-nitroimidazole paired with dGTP where the Tm was 40.9~C. In analyzing the free
energy for the duplex melting, the 3-nitropyrrole base mimic was found to have the
25 least discrimination when pairing with any of the four naturally occurring bases with an
overall AG of 0.4kcal/mol. The next least discrimin~fing was 5-nitroindole with a ~G
of 0.8 kcal/mol. Both of these values are less than the ~G of 1.1 kcal/mol foundbetween the natural base pairings of A+T and G+C. 4-Nitropyrazole showed a slight
preference for pairing with A with a ~G of I kcal/mol more stable than C. G, and T free
30 energies. Finally, 4-nitroimidazole showed a high selectivity for pairing to G (as was
evidenced by its high T~" value) due to the ability of the imid~ole N3 to hydrogen bond
with the deoxyguanosine N1. It should be noted, however, that the above values are
dependent upon the nearest base neighbors to the mimic. Further studies altered the
nearest neighbors and found that 3-nitropyrrole and 5-nitroindole are quite non-
35 discrimin~ting base pairing partners.
CA 02266847 1999-03-22
W O98/13527 PCTrUS97/17413
Of interest, the enthalpy and entropy changes were found to track one
another (i.e., a large enthalpy change correlates to a large entropy change) regardless of
the base mimic utilized implying that the correlation between ~S and AH is independent
of the mode of association of the bases. What was observed was that small enthalpy
5 and entropy changes were found in the non-hydrogen bonding base mimics. The low
values for entropy change reflect the greater degree of freedom of movement possible
for bases that are not locked into the duplex by hydrogen bonding interactions. The
small enthalpy changes reflect alterations in hydrogen bonding interactions as a result of
the loss of hydrogen bonding interactions for the base opposite the base mimic. If a
10 natural base remains stacked in the helix without an opposing hydrogen bonding partner
then it has lost hydrogen bonding interactions with water without regaining a new
donor/acceptor partner.
A similar study involved ex~mining acyclic nucleoside analogues with
carboxamido- or nitro-substituted heterocyclic bases (see Aerschot et al., Nucleic Acids
15 Res. 23:4363, l99S). Utilization of acyclic nucleosides endows the constructs with
enough flexibility to allow good base stacking as well as allow the base mimics to
obtain an orientation to best base-pair with the corresponding base. The heterocyclic
bases examined included: 4,5-imidazoledicarboxamide, 4-nitroimidazole, and 5-
nitroindazole. These complexes were referenced against acyclic hypoxanthine, 1-(2(-
20 deoxy-(-D-ribofuranosyl)-3-nitropyrrole, S-nitroindole, and 2-deoxyinosine. All the
new acyclic complexes had melting temperatures 7-20~C less than those observed for
the natural bases. S-Nitroindazole when paired against each of the four natural bases
had the least spread in ~Tm of only 2.2~C while the 4-nitroimid~ole had a spread of
8.0~C with dG being significantly out of line with the other three bases as had similarly
25 been observed above. Of the reference compounds, deoxyinosine had a aTn, of 5.6~C,
S-nitroindoles ~Tm was 1.0~C, 1-(2(-deoxy-(-D-ribofuranosyl)-3-nitropyrrole had a ~Tm
of 5.1~C, and the ~Tn~ of acyclic hypoxanthine was 4.8~C. However, all base mimics
showed about the sarne destabilization (~Tm of 4-5~C) when placed in an oligo
consisting almost exclusively of adenosines with exception of 4-nitroimid~ole and
30 acyclic deoxyinosine that had ~Tms of 7.0~C and 8.9~C, respectively.
Aerschot and co-workers also examined the effect of incorporation of
multiple base mimics into an oligo (see Aerschot et al., Nucleic Acids ~es. 23:4363,
1995). Overall, melting temperatures dropped but most markedly with the
incorporation of three base mimics. The nitroindoles, however, showed the least
35 amount of temperature differential.
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
11
Another base mimic, 1-(2(-deoxy-(-D-ribofuranosyl) imidazole-4-
carboxamide (Nucleoside 1), mimics preferentially dA as well as dC nucleosides (see
Johnson et al., Nucleic Acids Res. 25:559, 1997). The ability to substitute for both dA
and dC results from rotation about the carboxamide/imidazole bond as well as the bond
between the imidazole and furanose ring. When the imidazole is anti to the furanose
and the carboxamide group is anti to the imidazole, the lone pair on the oxygen and one
of the amide NH hydrogens is in a position that mimics the NH2 and N-l of adenosine.
Imidazole rotation about the glycosidic bond to the syn orientation places the amide
group in a position that approximately matches the positions of the NH2 and N-3 of
cytosine.
When Nucleoside 1 is substituted for any naturally occurring nucleoside,
the enthalpy increases with the greatest increase for a dG substitution for the 1-C
pairing (from ~H = 74.7 (kcal/mol)/~G = -16.5 (kcal/mol) for the GtC pairing to ~H =
-45.5 (kcal/mol)/~G = -5.8 (kcal/mol)). The smallest enthalpy change occurs for a dA
substitution (AH = -72.9 (kcal/mol)/~G = -15.4 (kcal/mol) for A/T pairing to ~H =
-66.7 (kcal/mol)/~G = -11.7 (kcal/mol) for the l-T pairing). Correspondingly, Tmsignificantly decreases from 65.7~C and 70.5~C for the A-T and C-G couples,
respectively, to 46.6~C for the l-T pairing, 43.4~C for l-G, 27.6~C for l-A, and 14.6~C
for l-C.
When used in a PCR reaction, Nucleoside I and its N-propyl derivative
are pl~r~rell~ially incorporated as dATP analogues (see Sala et al., Nucleic Acids Res.
24:3302, 1996). However, once incorporated into a DNA template, their ambiguous
hydrogen bonding potential gave rise to misincorporation of any of the naturallyoccurring bases at frequencies of 3 x 10-2 per base per amplification. Most of the
substitutions (primarily consisting of G) were a result of rotation about the carboxamide
bond when part of the template. Between 11-15% of the substitutions were due to
rotation of the imidazole moiety about the glycosidic bond. As part of a DNA template,
the N-propyl derivative behaved in the same way as Nucleoside 1 despite its propyl
moiety. This study indicates that while Nucleoside 1 preferentially behaves as dATP, it
has the ability in a PCR type environment to behave as all four naturally occurring
nucleotides as well. From this and the above studies, it is evident that a wide range of
duplex stability can be obtained through variations in base mimics and their placement
within an oligonucleotide.
- Petrruska et al., Proc. Natl. Acad Sci. USA 85:6252-6256. 1988 have
35 reported on the correlation between the thermodynamic stability of mi~m~tched primers
and DNA polymerase fidelity. By analyzing the melting profiles of a perfectly based
~ . ......
CA 02266847 1999-03-22
W O 98/13527 PCT~US97/17413
12
paired primer with a A/T correct match at the 3'-end compared to primers that had either
the incorrect base pair G/T, C/T, or T/T it was noted that there was a shift in free energy
changes upon dissociation (~G~) of 0.2, 0.3 and 0.4 kcal/mole for the terminal A/T
compared to the G/T, C/T, or T/T mi~m~tches. Interestingly, the A/T mi~m~tch was5 extended (Drosphilia DNA polymerase) about 200 times faster than the G/T mi~m~tch
and about 1400 and 2500 times faster than the C/T and T/T mi~m~tched respectively.
The authors hypothesized that the binding cleft of the polymers excludes water and
amplifies by amplifying free energy differences by increasing enthalpy differences in
mi~m~tched primers.
Livshits et al., J. Bimolecular Structure and Dynamics 11:783-795,
Adenine Press, ISSN 0739-1102, has presented interesting data and theoretical
considerations of dissociation curve of oligonucleotides immobilized in gel-based
arrays. The gel introduces a complicating factor of having to account for the effect of
diffusion on the dissociation process. However, there was agreement between the
15 calculated and experimental dissociation temperatures due in part to enthalpylentropy
compensation.
Many DNA hybridization-based diagnostic tests are being developed to
identify persons who might be suffering from (or be predisposed to) specific genetic
diseases (see for example, Norari et al., Gene 43:23-28, 1986) or to determine a genetic
20 histocompatibility profile, which is useful for tissue matching between donor and
patient (e.g., for a bone marrow transplant) (Sorg et al., Eur. J. lmmunogen 19:391-401.
1992). However, significant problems are encountered when trying to develop simple
and reliable hybridization methods using allele-specific oligonucleotide probes that
differ in sequence at one nucleotide position. Norari etal. solved the mi.~m~t~h25 hybridization problem by the addition of 10-times more unlabeled complementary
oligonucleotide than the mi~m~tched labeled oligonucleotide. However, this is animpractical solution when multiplex hybridization methods are being used.
Diagnostic tests that rely on the polymerase chain reaction (PCR)
technique also experience problems associated with the hybridization of
30 oligonucleotides. Rychlik (BioTechniques 18:84-90, 1995) examined the effects on
PCR of varying the G+C content of primers at either the 5' or 3' end of a priming
oligonucleotide. Using standard PCR buffers and conditions, oligonucleotides having a
high G+C content at the 3' end (the end used to extend DNA synthesis during PCR)results in high priming efficiency, but also promotes false priming due to greater
35 tolerance for mi~m~tches at the 5' end. Moreover, the effects of mi~m:~t~.hes in PCR are
variable; mi~m~tches located in the middle of a primer-template duplex do not
CA 02266847 1999-03-22
WO 98/13527 PCT/US97117413
13
significantly affect the efficiency of PCR arnplification, while 3'-terminal base
mi~m~tçhes sometimes strongly affects PCR product yield. As a fu~ther complication,
the strength of the effect that the various base pair mism~tches have on PCR
~ amplification is not the same as that observed for oligonucleotide hybrid formation and
5 stability (Ikuta et al., Nucl. Acids. Res. 15:797-811, 1987; Jacobs et al., Nucl. Acids Res.
16:4637-4650, 1988).
The present invention provides methods and compositions for detecting
base changes by improving the specificity and accuracy of hybridization of an
oligonucleotide with a target DNA sequence, and further provides other related
10 advantages.
SUMMARY OF T~IE INVENTION
This invention generally provides compositions and methods to increase
the specificity of hybridization of nucleic acids.
In one aspect, the invention provides a composition comprising a nucleic
15 acid and a salt, the salt comprising an anion and a cation, the anion selected from
halogenated acetate, propionate and halogenated propionate, the cation selected from
primary, secondary and tertiary ammonium comprising 1-36 carbon atoms, and
quaternary ammonium comprising 4-48 carbon atoms.
In another aspect, the invention provides a composition which is non-
20 flowing comprising a oligonucleotide of 6- l 00 nucleotides and a salt, the salt
comprising an anion and a cation, the anion selected from acetate, halogenated acetate,
propionate, and halogenated propionate, the cation selected from primary, secondary
and tertiary ammonium comprising 1-36 carbon atoms, and quaternary ammonium
comprising 4-48 carbon atoms. A "non-flowing" composition does not flow, as
25 solutions flow during chromatography.
In another aspect, the invention provides a composition which is free
from organic solvent, comprising a oligonucleotide of 6-100 nucleotides and a salt, the
salt comprising an anion and a cation, the anion selected from acetate, halogenated
acetate, propionate, and halogenated propionate, the cation selected from primary,
30 secondary and tertiary ammonium comprising 1-36 carbon atoms, and quaternary
amrnonium comprising 4-48 carbon atoms.
In another aspect, the invention provides a composition which includes a
nucleic acid and a salt, where the nucleic acid is immobilized on a solid support, and the
salt is formed from an anion and a cation, the anion selected from acetate, halogenated
35 acetate, propionate and halogenated propionate, the cation selected from primary,
-
CA 02266847 1999-03-22
WO 98113S27 rCTtUS97117413
14
secondary and tertiary ammonium comprising 1-36 carbon atoms, and quaternary
ammonium comprising 4-48 carbon atoms.
In another aspect, the invention provides a salt selected from the group:
(a) an acetate salt of a cation of the formula HN(CH3)2Ra wherein Ra
is a C4-C7hydrocarbyl;
(b) a halogenated acetate salt of a cation of the formula HN(CH3)2Rb
wherein Rb is a C,-C,2hydrocarbyl;
(c) acetate and halogenated acetate salts of a cation of the formula
H2N(C5-C7cycloalkyl)Rc where Rc is a Cl-C~2hydrocarbyl; and
(d) acetate and halogenated acetate salts of N-substituted piperdine,
wherein the nitrogen atom of piperidine is substituted with C,-C,2hydrocarbyl. Such
salts may be prel)aled by combining the respective acid and bases.
In another aspect, the invention provides an oligonucleotide in solution,
where an oligonucleotides if formed, at least in part, from a plurality of fragments, each
fragment shown schematically by structure (1)
B I B2 B3 B5
wherem,
~1 1 1
Bl B2 B3 represents a sequence of at least three nucleotides as
found in wild-type DNA, where "B" represents a base independently selected at each
location;
~ represents a series of covalent chemical bonds termed a
"specificity spacer," which separates and connects two bases B3 and B5;
the specificity spacer having steric and chemical properties such that
(a) it does not prevent hybridization between a fragment of structure
(1) and an oligonucleotide fragment having a complementary base sequence, as shown
schematically as structure (2)
(I) ~1 1 1 1~
B l B2 B3 B5
B ' B ' B ' B ' B5'
CA 02266847 1999-03-22
W O 98/13527 PCTAUS97/17413
In addition, the specificity spacer has at least one of the following
properties: it cannot enter into hydrogen bonding with a base positioned opposite itself
in a hybridized complementary base sequence of structure (2); it can enter into
hydrogen bonding with a base positioned opposite itself in a hybridized complementary
S base sequence of structure (2), however it does not hydrogen-bond through any of
adenine, guanine, cytosine, thymine or uracil according to standard Watson-Crickhydrogen bonding.
In another aspect, the invention provides an array, where the array
includes a plurality of oligonucleotides immobilized in an array format to a solid
10 support. Each oligonucleotide of the plurality is forrned, at least in part, from a
plurality of fragments, each fragment shown schematically by structure ( 1 )
Bl B2 B3 B5
1 5 wherein,
~1 1 1
Bl B2 B3 represents a sequence of at least three nucleotides as
found in wild-type DNA, where "B" represents a base independently selected at each
location;
~ represents a series of covalent chemical bonds terrned a
20 "specificity spacer," which separates and connects two bases B, and B5;
the specificity spacer having steric and chemical properties such that
(a) it does not prevent hybridization between a fragment of structure
(1) and an oligonucleotide fragment having a complementary base sequence, as shown
schematically as structure (2)
(1) ~1 1 1 1~
B~ B2 B3 Bs
B ' B ' B ' B ' B '
In addition, the specificity spacer has at least one of the following
properties: it cannot enter into hydrogen bonding with a base positioned opposite itself
30 in a hybridized complementary base sequence of structure (2); it can enter into
hydrogen bonding with a base positioned opposite itself in a hybridized complementary
CA 02266847 1999-03-22
W O 98/13527 PCTrUS97117413
16
base sequence of structure (2), however it does not hydrogen-bond through any ofadenine, guanine, cytosine, thymine or uracil according to standard Watson-Crickhydrogen bonding.
In another aspect, the invention provides a composition which includes
an oligonucleotide and a salt in solution, the oligonucleoditde being formed, at least in
part of a plurality of fragments, each fragment shown schematically by structure (1)
(1) ~ I I I I ~
Bl B2 B3 B5
wherein,
~1 1 1
Bl B2 B3 represents a sequence of at least three nucleotides as
found in wild-type DNA, where "B" represents a base independently selected at each
location;
represents a series of covalent chemical bonds termed a
"specificity spacer," which separates and connects two bases B3 and B5;
the specificity spacer having steric and chemical properties such that
(a) it does not prevent hybridization between a fragment of structure
(1) and an oligonucleotide fragment having a complementary base sequence, as shown
schematically as structure (2)
(1) ~ I I I I ~
Bl B2 B3 Bs
B ' B ' B ' B ' B '
at least one of (b) and (c) where
(b) the specificity spacer cannot enter into hydrogen bonding with a
base positioned opposite itself in a hybridized complementary base sequence of
structure (2);
(c) the specificity spacer can enter into hydrogen bonding with a
base positioned opposite itself in a hybridized comlementary sequence of structure (2)
but the specificity spacer does not provide any base selected from adenine, guanine,
thymine, uracil or cytosine for the hydrogen bonding;
and the salt is a hybotrope.
CA 02266847 1999-03-22
W O98/13527 PCTrUS97/17413
17
In another aspect, the invention provides an array composition formed, at
least in part of a plurality of oligonucleotides immobilized in an array format to a solid
support, each oligonucleotide of the plurality formed, at least in part of a plurality of
fragments, each fragment shown schematically by structure ( I )
(1) ~1 1 1 1~
Bl B2 B3 Bs
wherein,
~1 1 1
Bl B2 B3 represents a sequence of at least three nucleotides as
found in wild-type DNA, where "B" represents a base independently selected at each
1 0 location;
~ represents a series of covalent chemical bonds termed a
'ispecificity spacer," which separates and connects two bases Bl and B5;
the specificity spacer having steric and chemical properties such that
(a) it does not prevent hybridization between a fragment of structure
15 (1) and an oligonucleotide fragment having a complementary base sequence. as shown
schematically as structure ~2)
(1) ~1 1 1 1~
Bl B2 B3 B5
B, IB2 IB3 IB4 IBs
, and
20 at least one of (b) and (c) where
(b) the specificity spacer cannot enter into hydrogen bonding with a
base positioned opposite itself in a hybridized complementary base sequence of
structure (2);
(c) the specificity spacer can enter into hydrogen bonding with a
25 base positioned opposite itself in a hybridized comlementary sequence of structure (2)
but the specificity spacer does not provide any base selected from adenine, guanine.
thymine, uracil or cytosine for the hydrogen bonding; and the nucleic acid of formula
( l ) being in contact with a hybotrope.
In yet other aspects, methods are provided for distinguishing between
30 hybridization of a complementary nucleic acid target and a nucleic acid probe in which
CA 02266847 l999-03-22
W O 98/13527 PCTrUS97/17413
18
the probe and target are either perfectly complementary or have one or more basemismAt~hes, comprising the steps: (a)mixing the target and probe in a solution
comprising a hybotrope; and (b)hybridizing at a discrimin~ting temperature, and
detecting the arnount of probe hybridized to the target, thereby de~rmining whether the
5 duplex is perfectly complementary or mi.cm~t~hed In preferred embodiments, theprobe or target is from 6 to 40 bases. In another preferred embodiment, the probe is
labeled. In a related aspect, the probe has one or more abasic residues and the solution
does not contain a hybotrope. In a preferred embodiment, the hybridization reaction
mixture comprises a hybotrope.
In yet another aspect, methods are provided for increasing discrimination
in a nucleic acid synthesis procedure, such as polymerase chain reaction. In these
methods, a single stranded nucleic acid target is mixed with an oligonucleotide primer
or a solution comprising a hybotrope and a polymerase, the primer is annealed to the
target at a discrimin~ting temperature, and a complementary strand to the target is
synthesized. The arnount of duplex formed for a mismatched primer and target is less
than for a perfectly matched primer and target.
These and other aspects of the present invention will become evident
upon reference to the following detailed description and attached drawings. In addition,
various references are identified below and are incorporated by reference in their
entirety.
BRIEF DESCRIPTION OF THE DRAWINGS
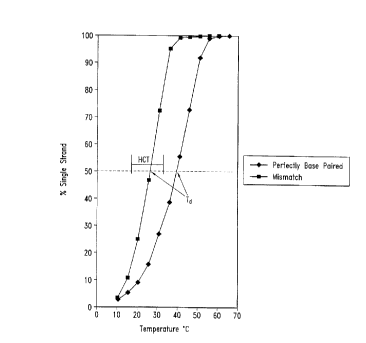
Figure I is a graph illustrating therrnal melt profiles of oligonucleotide
duplexes. Percentage single strand DNA (a, y-axis) is plotted versus temperature (x-
axis). The Td Of the duplex is defined as the temperature at which 50% of the strands
are in single strand forrn. The helical coil transition (HCT) is defined as the
temperature difference between an a of 0.2 (or 20%) and 0.8 (or 80%). The melting
curve denoted by the squares represents the behavior of a duplex in contact with a
hybotrope (e.g, LiTCA) and the melting curve denoted by the diamonds represents the
behavior of an oligonucleotide duplex in a NaCI-based hybridization solution.
Figure 2 is a graph illustrating the relationship of the Td ~f an oligo
duplex and salt concentration in hybridization solutions (LiTCA, GuSCN, NaSCN~
NaClO4, KI, NaCI, GuCI, CsTFA). The Td in degrees C is plotted versus molarity of
the salt.
Figure 3 is a graph showing the difference in Td between two duplexes,
one that is perfectly based-paired and the other that contains a single mism:~tçh. The
CA 02266847 1999-03-22
WO 98/13527 PCT/US97117413
19
temperature difference between any two Tds at a = 0.5 is defined as the ~Td. Thepercentage of single strand DNA (y-axis) is plotted versus temperature (~C; x-axis).
Figure 4 is a graph displaying melting profiles for an 1 8-mer
- oligonucleotide duplex that is perfectly based paired (diamonds) and the same
S oligonucleotide duplex that contains a central mism~tch (squares A/A, position 9). The
~Td is 6~C. The melting profiles were determined in 2.0 M LiTCA. The percentage
single strand (y-axis) is plotted versus temperature (~C; x-axis).
Figure 5 is a graph illustrating melting profiles for an 1 8-mer
oligonucleotide duplex that is perfectly based-paired (diamonds) and the same
10 oligonucleotide duplex that contains a central mism~tch (squares; A/A, position 9). The
melting curves are determined in QY low stringency hybridization buffer (Promega,
Madison, Wl). The percentage single strand (y-axis) is plotted versus temperature (~C;
x-axis).
Figure 6 is a graph showing melting profiles for a set of 1 9-mer
15 oligonucleotides duplexes that vary in G+C composition from 26% to 73%. All of the
duplexes are perfectly based paired. The ATd is 5~C across the entire G+C range. The
melting profiles are determined in 3M TMATCA. The ~/0 single strand (y-axis) is
plotted versus temperature (~C; x-axis).
Figure 7 is a graph displaying melting profiles for a set of l9-mer
20 oligonucleotides duplexes that vary in G+C composition from 26% to 73%. All of the
duplexes are perfectly based paired. The ~Td is 4~C across the entire G+C range. The
melting profiles are determined in 3M TEATCA. The % single strand (y-axis) is
plotted versus temperature (~C; x-axis).
Figure 8 is a graph illustrating melting profiles for a set of 19-mer
25 oligonucleotide duplexes that vary in G+C composition from 26% to 73%. All of the
duplexes are perfectly base-paired. The ~~Tm is 1 6~C across the entire G+C range. The
melting profiles are determined in 0.165M NaCI. The % single strand (y-axis) is
plotted versus telllpeldlule (~C; x-axis).
Figure 9 is a graph illustrating melting profiles for an 1 8-mer
30 oligonucleotide duplex that is perfectly based paired and the same oligonucleotide
duplex that contains either a central mi~m~tch (A/A) or abasic substitution at position 9.
The melting profiles are determined in GuSCN. The % single strand (y-axis) is plotted
versus temperature (~C; x-axis).
Figure 10 is a graph showing the relationship between molarity and Td of
35 the data obtained from the melting curves described in Figure 9. The Td on the y-axis is
plotted versus the molarity of GuSCN on the x-axis.
.. . .
CA 02266847 1999-03-22
W O 98/13527 PCTrUS97/17413
Figure 11 is a graph illustrating melting profiles for an 1 8-mer
oligonucleotide duplex that is perfectly based paired in I x PCR buffer or LiTCA over a
concentration range of 0.05 M to 0.4 M. The % single strand (y-axis) is plotted versus
temperature (x-axis).
S Figure 12 is a photograph of a 2% agarose gel that shows the presence or
absence of an amplicon 381 bp in length. "m", marker; and Hl 7, H14, H11, AB1, dN1,
dN2, dN3 and dN6 are the 5' primers used in amplification.
Figure 13 is the text scan of a set of arrayed oligonucleotides that when
duplexed with probe contain the mi.~m~tch indicated in the top row. "C" indicates
control probe, "6S" indicates the 6S abasic substituted probe and "8S" indicates the 8S
abasic substituted probe. The figure is a compilation of 3 separate filters.
Figure 14 is a graph showing the difference in Td between three
duplexes, that vary in G+C content from 27% to 83%. The percentage of single strand
DNA (y-axis) is plotted versus temperature (~C; x-axis). The melting solution was 100
mM 2-methoxyethylamine trifluoroacetate.
Figure 15 is a graph showing the difference in Td between three
duplexes, that vary in G+C content from 27% to 83%. The percentage of single strand
DNA (y-axis) is plotted versus temperature (~C; x-axis). The melting solution was l O0
mM diisobutylamine acetate.
Figure 16 is a graph showing the difference in Td between three
duplexes, that vary in G+C content from 27% to 83%. The percentage of single strand
DNA (y-axis) is plotted versus temperature (~C; x-axis). The melting solution was 2 M
Guanidinium thiocyanate.
Figure 17 is a graph showing the difference in Td between three
duplexes, that vary in G+C content from 27% to 83%. The percentage of single strand
DNA (y-axis) is plotted versus temperature (~C; x-axis). The melting solution was 1x
PCR buffer.
Figure 18 is a graph showing the difference in Td between three
duplexes, that vary in G+C content from 27% to 83%. The percentage of single strand
DNA (y-axis) is plotted versus temperature (~C; x-axis). The melting solution was lx
SSC.
Figure 19 is a graph showing the difference in Td between three
duplexes, that vary in G+C content from 27% to 83%. The percentage of single strand
DNA (y-axis) is plotted versus temperature (~C; x-axis). The melting solution was 20%
formamide, 10 mM Tris pH 7.6, and 5 mM EDTA with 0.1 % sarkosyl.
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
21
Figure 20 is a graph showing the difference in Td between three
duplexes, that vary in G+C content from 27% to 83%. The percentage of single strand
DNA (y-axis) is plotted versus temperature (~C; x-axis). The melting solution was I M
dicyclohexylammonium acetate.
Figure 21 is a graph showing the difference in Td between three duplexes,
that vary in G+C content from 27% to 83%. The percentage of single strand DNA (y-
axis) is plotted versus temperature (~C; x-axis). The melting solution was 500 mM n-
ethylbutyl~mmmonium acetate.
Figure 22 is a graph showing the difference in Td between three
duplexes, one that is perfectly based-paired and the other two that contains a mism~tch
or a deoxynebularine substitution. The percentage of single strand DNA (y-axis) is
plotted versus temperature (~C; x-axis). DMO-2060: 5'-hexylamine-
GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3' (oligonucleotide
immobilized on the nylon bead.; DMO-2055: 5'-TEXAS RED-
TGT/GGA/TCA/GCA/AGC/AGG/AGT/ATG-3 ' (perfect complement); DMO-2058;
5'-TEXAS RED- TGT/GGAITCA/GGA/AGC/AGG/AGT/ATG-3' (mism~tch
complement); and DMO-2058-dN: 5'-TEXAS RED-
TGT/GGA/TCA/G(deoxynebularine)A/AGC/AGG/AGT/ATG-3' (deoxynebularine
mism~tch complement). The melting solution was I M diisopropylamine acetate. Themaximum difference between the 3 melting curves in the Td or Tm is 6 C. The helical
coil transition (HCT) of the true mi.sm:~tch was 14 C; the HCT for the deoxynebularine
mi.sm~tch duplex was 14 C and the HCT for the perfectly based paired duplex was 16
C.
Figure 23 is a graph showing the difference in Td between three
duplexes, one that is perfectly based-paired and the other two that contains a mism~tch
or a deoxynebularine substitution. The t~ peldlLIre difference between any two Tds at
a = 0.5 is defined as the ~Td. The percentage of single strand DNA (y-axis) is plotted
versus temperature (~C; x-axis). DMO-2060: 5'-hexylamine-
GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3' (oligonucleotide
immobilized on the nylon bead.; DMO-2055: 5'-TEXAS RED-
TGT/GGA/TCA/GCA/AGC/AGG/AGT/ATG-3' (perfect complement); DMO-2058;
5'-TEXAS RED- TGT/GGA/TCA/GGA/AGC/AGG/AGT/ATG-3' (mi.sm~tch
complement); and DMO-2058-dN: 5'-TEXAS RED-
TGT/GGA/TCA/G(deoxynebularine)A/AGC/AGG/AGT/ATG-3' (deoxynebularine
mi~m~tch complement). The melting solution was 1 M n,n-dicyclohexylamine acetate.
The maximum difference between the 3 melting curves in the Td was 4 C. The helical
....
CA 02266847 1999-03-22
WO 98113527 PCT/US97/17413
22
coil transition (HCT) of the true mi~m~tc,h was 15 C; the HCT for the deoxynebularine
mi~m~tc,h duplex was 15 C and the HCT for the perfectly based paired duplex was 15
C.
Figure 24 is a graph showing the difference in Td between three
duplexes, one that is perfectly based-paired and the other two that contains a mism~ch
or a deoxynebularine substitution. The temperature difference between any two Tds at
a = 0.5 is defined as the ~Td. The percentage of single strand DNA (y-axis) is plotted
versus temperature (~C; x-axis). DMO-2060: 5'-hexylamine-
GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3' (oligonucleotide
immobilized on the nylon bead.; DMO-2055: 5'-TEXAS RED-
TGTIGGAITCA/GCAIAGC/AGG/AGT/ATG-3' (perfect complement); DMO-2058;
5'-TEXAS R~D- TGT/GGAITCA/GGAIAGCIAGGIAGTIATG-3' (mism~tch
complement); and DMO-2058-dN: 5'-TEXAS RED-
TGT/GGA/TCA/G(deoxynebularine)A/AGC/AGG/AGT/ATG-3' (deoxynebularine
mi~m~ch complement). The melting solution was 1 M n,n-dicyclohexylamine acetate.The maximum difference between the 3 melting curves in the Td was 4 C. The helical
coil transition (HCT) of the true mi.~m~tch was 17 C; the HCT for the deoxynebularine
mismatch duplex was 17 C and the HCT for the perfectly based paired duplex was 15
C.
2Q Figure 25 is a graph showing the difference in Td between three
duplexes, one that is perfectly based-paired and the other two that contains a mism~tch
or a deoxynebularine substitution. The te",pelal~lre difference between any two Tds at
a = 0.5 is defined as the ~-Td. The percentage of single strand DNA (y-axis) is plotted
versus temperature (~C; x-axis). DMO-2060: 5'-hexylamine-
GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3' (oligonucleotide
immobilized on the nylon bead.; DMO-2055: 5'-TEXAS RED-
TGT/GGAITCAIGCAIAGC/AGG/AGT/ATG-3' (perfect complement); DMO-2058;
S'-TEXAS RED- TGT/GGA/TCA/GGA/AGC/AGG/AGT/ATG-3' (mism~tch
complement); and DMO-2058-dN: S'-TEXAS RED-
TGT/GGA/TCAlG(deoxynebularine)A/AGC/AGG/AGT/ATG-3'
(deoxynebularine mi~m~tch complement). The melting solution was 100 mM n,n-
dimethylhexylamine acetate. The maximum difference between the 3 melting curves in
the Td was 9 C. The helical coil transition (HCT) of the true mi~m~tch was 15 C; the
HCT for the deoxynebularine mism~tch duplex was 15 C and the HCT for the perfectly
based paired duplex was 15 C.
CA 02266847 1999-03-22
W O 98113527 PCTAUS97/17413
23
Figure 26 explains a convention used herein to denote oligonucleotides
having a specificity spacer.
DETAILED DESCRIPTION OF Tl~E INVENTION
Prior to setting forth the invention, it may be helpful to an underst~n-ling
S thereof to define certain terms used herein.
As used herein, "hybotrope" refers to any chemical or any mixture of a
chemical in an aqueous or organic environment with buffers, chelators, salts and/or
detergents that changes the enthalpy of a nucleic acid dup}ex by at least 20% when
referenced to a standard salt solution (0.165 M NaCl, 0.01 M Tris pH 7.2, 5 mM EDTA
and 0.1% SDS). That is, the energy content of the nucleic acid duples is decreased.
The reference oligonucleotide is 5'-
GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3' as the immobilized
oligonucleotide and 5'-TGT/GGA/TCA/GCA/AGC/AGG/AGTIATG-3' as the solution
nucleotide which is typically labeled at the 5'-end with a fluorochrome such as Texas
Red. The oligonucleotide duplex (24 nucleotides in length) has a helical to coiltransition (HCT) of 25~C or less. The HCT is the difference between the temperatures
at which 80% and 20% of the duplex is single stranded. The average minimum slopefor a solution to be defined as a hybotrope is the first derivative of the HCT and is equal
to 2.4 in units of 1/temperature in degrees C ((80% single strand - 20% single-
strand)/25~C).
As used herein, "stringency" is the percentage of mi~m~tched base pairs
that are tolerated for hybridization under a given condition.
As used herein, "discrimination" is the difference in Td between a
perfectly base-paired duplex and a duplex cont:linin~ a mi~m~t~.h.
As used herein, a "discrimination temperature" is a temperature at which
a hybridization reaction is performed that allows detectable discrimination between a
mi~m~tched duplex and a perfectly matched duplex. As shown herein, a range of
temperatures satisfy criteria of a discrimin~tion temperature.
As used herein, an "abasic" residue in an oligonucleotide refers to a
compound that approximates the length of a ribofuranose sugar, is covalently attached
to neighboring bases (e.g, via phosphodiester or equivalent linkages), and is substituted
at the beta anomeric position with a group that does not interact with the base on the
opposite strand of a duplex. An abasic residue may be an apurine or apyrimidine
structure, an anucleoside structure, or an analogue of a phosphate backbone. The abasic
substitution may also consist of a backbone of N-(2-aminoethyl)-glycine linked.
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
24
As used herein, a "base analog" in an oligonucleotide refers to a
compound that has a ribofuranase sugar and is substituted at the beta anomeric position
with a group that has a similar 3-D shape as an A, C, G, T, or U base, but does not
hydrogen bond to the base on the opposite strand of a duplex.
As used herein, "deoxyNebularine" refers to a 2'-deoxynubularine,
which is 9-(beta-D-2'-deoxyribofuranosyl) purine (Eritja et al., Nucl. Acids Res.
14:8135, 1986). The molecular formula is C,oH,2N4O4.
As used herein, "nucleic acid" or "nucleic acid molecule" refers to any of
deoxyribonucleic acids (DNAs), ribonucleic acids (RNAs), oligonucleotides, fragments
generated by the polymerase chain reaction, and fragments generated by any of ligation,
scission, endonuclease action, and exonuclease action. Nucleic acids can be composed
of naturally occurring bases and analogs of naturally occurring bases, or a combination
of both. Nucleic acids can be either single stranded or double stranded.
As used herein, "Tm" is the temperature at which half the molecules of a
nucleic acid duplex are single stranded. Tm is measured in solution, while Td ismeasured for the duplex affixed to a solid support, both terms indicate the temperature
at which half of a duplex are single stranded.
A. HYBOTROPES
As noted above, the present invention provides compositions, including
hybotropes, that can change the enthalpy of a nucleic acid duplex (i.e., that can decrease
the energy content of the oligonucleotide duplex, so that the cooperativity of the
melting processes is increased, as discussed in more detail below). Generally, enthalpy
of a duplex in a solution cont~ining a hybotrope is increased at least 20%, and
preferably, 30-100% over a duplex in a reference solution comprising 0.165M NaCl.
Several consequences flow from increased enthalpy. Importantly, the
temperature range over which a duplex melts is decreased, likely due to increased
cooperativity of melting. The difference between a hybrotropic solution and a
hybridization solution used in most molecular biology protocols is illustrated in Figure
4 and Figure 5. In Figure 4, the difference in Td between a duplex containing a
mi~m~tch and duplex which is perfectly base-paired is about 5~C and is clearly
distinguished. The hybotrope in Figure 4 is LiTCA. In Figure 5 the difference in Td
between a duplex cont~ining a mi~m~tch and duplex which is perfectly base-paired is
less than 2~C and is not distinct. Also, the HCT of the hybotrope in Figure 4 is less
than 25~C and the HCT of the SSC-based solution is greater than 25~C.
CA 02266847 1999-03-22
W Og8/13527 PCTrUS97/17413
Because the temperature range of a melt is smaller in a hybotropic
solution, there is a greater difference in the Tm of a perfectly complementary duplex and
a duplex cont:~ining one or more mi~m~tchçd base pairs (e.g, base pairing other than
A:T, G:C, A:U). This property is illustrated in Figure 3 in which an 18 mer duplex
perfectly complement or cont~ining a 1 bp mi~m~teh is melted in a solution comprising
a hybotrope. As shown, the difference in Td between the two duplexes is substantial. In
general, a hybotrope causes an increase in "Td of 22~C (e.g., > 2~C, > 2.5~C, > 3~C~
> 3.5~C, > 4~C) over the ATd of the matched and mi~m~tched duplexes in a reference
solution (e.g, 0.18M Na+). For a 6 to 18 base pair duplex (50% G+C) a hybotrope
induces a ~Td of > 2~C (e.g., > 2~C, > 2.5~C, > 3~C, > 3.5~C, > 4~C, > 4.5~C, > 5~C).
for a 19 to 24 base pair duplex, a hybotrope induces a ~Td of ~ 1~C (e.g., > 1~C.
> 1 5~C,>2~C~25~C~3~C~3 5~C~>4oc~>4~5oc~>5oc)andfora25to36base
pair duplex, a hybotrope induces a ~Td of ~ 0.5~C (e.g., > 0.5~C, > 1 ~C, > 1.5~C, > 2~C.
>2.5oc~>3oc~>3.5oc~>4oc~>4.5oc~>5oc)~
The melting of a duplex causes a transition from a helical state (duplex~
to a coil state (single stranded). The transition, called HCT (helical to coil transition) is
readily measured and is expressed in units of temperature. As used herein, HCT is the
temperature difference between which a duplex is 80% (a = 0.8) and 20% (a = 0.2)single-stranded.
A hybotrope may be identified as any chemical or any mixture of a
chemical in an aqueous or organic environment with buffers, chelators, salts and/or
detergents that decreases the enthalpy of a nucleic acid duplex by 20% when referenced
to a standard salt solution (0.165 M NaCI, 0.01 M Tris pH 7.2, 5 mM EDTA and 0.1%
SDS). The reference oligonucleotide is 5-
GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3' as the immobilized
oligonucleotide and 5'-TGT/GGA/TCA/GCA/AGC/AGG/AGT/ATG-3' as the solution
nucleotide which is typically labeled at the 5'-end with a fluorochrome such as Texas
Red. The oligonucleotide duplex (24 nucleotides in length) has a helical to coiltransition (HCT) of 25~C or less. The HCT is the difference between the temperatures
at which 80% and 20% of the duplex is single stranded.
1. Relationship of H~botrope to HCT
In Figure 1, the characteristic parameters of a thermal melting profile
(helical coil transition) of an oligonucleotide duplex in two different hybridization
solutions are presented. The squares represent the melting profile of an oligonucleotide
duplex in NaCI based hybridization solution (e.g., SSPE, SSC). 20xSSPE is 173.~g
CA 02266847 l999-03-22
W O 9U13527 PCTrUS97/17413
26
NaCl, 27.6g NaHPO4, and 7.4g EDTA at pH7.4 in lL water. 20xSSC is 175.3g NaCI,
88.2g NaCitrate at pH 7 in lL water. The diamonds represent the melting profile of the
same oligonucleotide duplex in a hybotrope-based hybridization solution, in this case
LiTCA (lithium trichloroacetate~. Td is the temperature (~C) at which half of the
5 molecules in a population are single-strand and half of the molecules are double-
stranded. The HCT (helical coil transition) is the width of the melting curve from a
value of 20% single-strand to 80% single-strand and possesses the unit of temperature
(e.g., ~C, ~K). The stringency factor is the value of the slope (partial derivative) of the
helical coil transition at the Td. Either stringency factor or HCT may be used to identify
10 a hybotrope.
In Table 1, the slope (k) of the linear equation that relates concentration
of solute to Td, the helical coil transition, and the ~Td for 9 different hybotropic and
hybridization solutions is presented. An 18 bp oligonucleotide duplex was melted in
the respective solutions and the values are obtained as described in the exarnples.
Table I
Hybridization Solution Type Slope (k)HCT(~C) Stringency Factor
LiTCA 19 8 7.5
GuSCN 13 10 6.0
NaSCN 8.5 11 5.4
NaClO4 7 12 5.0
KI 5 15 4.0
NaCI 4-5 17 5 3-4
GuCI 3.5 18 3.3
CsTFA 2.5 18 3.3
30% formamide ND 20 3.0
* = not determined
Thus, from these data, HCT is inversely proportional to the stringency
factor for a given hybridization solution type; the lower the value of HCT, the higher
the stringency factor. The HCT increases as the slope of the linear function that relates
20 salt concentration to Td decreases ((Td[salt] = Td[0] - k[Cx 3), where Td[0] is the
extrapolated Td at zero salt concentration, k is the salt specific constant and Cx~ is the
concentration of the salt or hybotrope; see Figure 2).
CA 02266847 1999-03-22
W O98/13S27 PCT~US97/17413
27
A hybotrope may be identified as any chemical or any mixture of a
chemical in an aqueous or organic environment with buffers, chelators, salts and/or
detergents that decreases the enthalpy of a nucleic acid duplex by 20% when referenced
to a standard salt solution (0.165 M NaCI, 0.01 M Tris pH 7.2, 5 mM EDTA and 0.1%
5 SDS). The reference oligonucleotide is 5'-
GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3' as the immobilized
oligonucleotide and 5'-TGT/GGA/TCA/GCA/AGC/AGG/AGT/ATG-3' as the solution
nucleotide which is typically labelled at the S'-end with a fluorochrome such as Texas
Red. The oligonucleotide duplex (24 nucleotides in length) has a helical to coil10 transition (HCT) of 25~C or less..
2. Relationship of HCT to Discrimination
Either stringency factor or HCT is related directly to another readily
measurable parameter of oligonucleotide duplexes. This parameter, ~Td, is the
15 temperature difference between the Td of an oligonucleotide duplex that is perfectly
base paired and the Td of the same oligonucleotide duplex that contains a mi~m~tch at
some position in the duplex (see Figure 3). As shown herein, the temperature difference
between a perfectly base paired duplex and a duplex cont~ining a mi.~m~tch is a
function of the stringency factor (or HCT) of a given hybridization solution or
20 hybotrope. The relationship is expressed as: ~Td increases as the stringency factor of a
solution increases. In Table 2, this relationship is presented for 18 bp oligonucleotide
duplexes. The duplex is melted in the respective hybridization solution and HCT and
ATd is determined as described herein.
CA 02266847 1999-03-22
W O 98/13527 PCTrUS97/17413
28
Table 2
Hybridization Solution TypeSlope (k)HCT (~C) ~Td (~C)
LiTCA 19 8 7.5
GuSCN 13 10 6.0
NaSCN 8.5 11 5.5
NaClO4 7 12 4.5
KI 5 15 3.0
NaCl 4.5 17.5 1.5
GuCl 3.5 18 1.2
CsTFA 2.5 18 l.2
30% forrnarnide ND* 20 1.5
= not determined
The data presented in Table 2 show that HCT is inversely proportional to
the ATd between a perfectly base paired duplex and a duplex cont~ining a mism~t~h.
That is, either stringency factor or HCT predicts the ability of given hybridization
solution to discriminate mi~m~tched duplexes. This aspect of hybotrope-based
hybridization is further illustrated in Figures 4 and 5. Figure 4 is a graph showing
melting profiles in 2.0 M LiTCA for an 1 8-mer oligonucleotide duplex that is perfectly
based paired (diamonds) and the sarne oligonucleotide duplex that contains a central
10 mism~tch (A/A, position 9). The ~Td is 6~C. FigureS is a graph showing melting
profiles for an 18-mer oligonucleotide duplex in QY low stringency hybridizationbuffer (Promega, Madison, WI) that is perfectly based paired (squares) and the same
oligonucleotide duplex that contains a central mism~tch (A/A, position 9). The ~Td is
0~C. Therefore, the ~Td value relates to the ability of a chemical to discriminate
15 between perfectly base paired duplexes and duplexes that contain a mi~m~tch The
practical utility of this result is discussed below.
In addition, transition enthalpies between a fully base-paired and base
stacked double helix to two unpaired and unstacked single strands can be calculated.
(Breslauer, K.J., Chapter 15, "Methods for Obtaining Therrnodynamic Data on
20 Oligonucleotide Transitions," in Thermodynamic Data for Biochemistry and
Biotechnology, ed. H. Hinz, Academic Press, New York, NY, 19~6.) The difference
between a non-cooperative and cooperative transition is expressed in terrns of AHVIl
(van't Hoff enthalpy). In a cooperative transition, the value of (da/dT)Td is high, and
therefore, the ~HVH is also high. In a non-cooperative transition, the value of (da/dT)Td
CA 02266847 1999-03-22
W O98/13527 PCTAUS97117413 29
is low, and therefore, the /~HVH is also low. (The term (datdT)Td is the derivative of the
slope of the melting curve at the Td, a is defined as the % single strand on the ordinate
axis.)
In this regard, thermodynamic parameters for two different sets of
5 oligonucleotides (42% G+C; 63% G+C) in three types of hybridization solution are
shown in Table 3. The data show that the enthalpy values are inversely related to the
values obtained for the temperature range of the thermal coil transition of the duplex
(HCT).
Table 3
AHvH
Solution Type % G+C Length (nt)Td(~C) HCT (~C) (kcal/mol)
2 M LiTCA 42 19 nt 35.5 12 -52.8
2 M TMATCA 42 19 55.4 18 -47.0
3 M TMATCA 42 19 43.0 11.5 -60.7
3 M TMACl 42 19 60.0 15.5 -46.2
2 M LiTCA 63 l9 42.0 15 -42.0
2 M TMATCA 63 19 48.0 19.5 -38.6
3 M TMATCA 63 19 47.0 13 -61.8
3 M TMACl 63 19 59.0 17.5 -39.7
3. Characterization of a Hybotrope
a. Characteristics of a hybotrope.
As noted herein, a hybotrope is useful within the context of the present
invention if it is a solution or is miscible from about 0.05 M to about lO M in water.
15 other protic, or aprotic solvent. In certain preferred embodiments, the hybotrope does
not inactivate polymerases. In other preferred embodiments, the anion part of a
hybotrope has a pK, of less than 2.2.
The chaotrope is a chemical that increases the enthalpy of an
oligonucleotide or nucleic acid duplex by at least 20% when referenced to a standard
20 salt solution (i e., 0.165 M NaCl). Enthalpy is measured by plotting the slope of the
therrnal transition, a, versus temperature (see Figure l) and applying the following:
The van't Hoff enthalpy can be obtained from the dirrelellliated
- equilibrium melting curve (Marky and Breslauer, 1987) by plotting da versus
CA 02266847 1999-03-22
WO 98/13S27 PCT/US97/17413
temperature. Briefly, thermodynamic data provide a basis for predicting the stability
(~G ) and t~ lul~- dependent melting behavior (also described here as the helical
coil transition (HCT), (~H~)) from the primary sequence of bases in the duplex. We use
a thermally induced helical coil transition (from double strand to single strand) to obtain
5 values for the ~HVH. The analysis of the shape of the helical coil transition is used to
calculate the van't Hoff transition enthalpy. As described by Marky and Breslauer,
(1987), a is equal to the fraction of single strands in the duplex state. If a is plotted
versus temperature the temperature at which a takes the value of 0.5 is defined as the
Td. The equilibrium constant K for any transition can be expressed in the form of a, the
10 van't Hoff enthalpy can be expressed as:
~-HVH = RT2[dlnK/dT] or ~-HVH= -R[dlnK/d(l/T)]
To solve the general expression when a takes the value of 0.5 in terms
of a the foregoing equation is differentiated and solved for a at the Td:
~ -HVH = (2 + 2n)RT2(a(x/~T)T Td which can also be written:
~-HVH = (2 ~ 2n)R(~a/~(l /T)T-Td
In this series of experiments it is assumed that a bi-molecularity exists
where n=2 for the preceding equations and therefore the corresponding coefficient is
equal to 6. Another assumption employed is that there is no dependence of Td on
concentration since at every temperature increment the concentration of single strands is
zero (recall that all unhybridized material is washed away from the solid support prior
to the melting process and that at each 5~C temperature increment, the solid support is
placed in a fresh solution). For any process at equilibrium, ~G = -RT(lnKeq) and ~G =
~H - T~S it is possible to write -RT (In K) = ~H - TAS.
As has been shown by Gralla and Crothers (Gralla, J., and Crothers,
D.M., J. Mol. Biol. 73:497-511, 1973) for bimolecular transitions, the full width or half-
width of a differen~i~ted melt curve at the half-height is inversely proportional to the
van't Hoff transition enthalpy. As suggested, for an equilibrium of the form nA ~ An
the general forms of the van't Hoff equation are:
~-HVH = B/((1/T,)-(1/T2) (for the full width at half-height)
~-HV~, = B'/((l/Tma~L)-(1/T2) (for the upper half-width at half-height)
where Tm~ is the temperature at the maximum, and T, and T2 correspond to the upper
and lower temperatures at which value the change in the plotted temperature is equal to
one-half of [(~a/~3(1/T)ma,J. For a molecularity of 2, -B = 10.14 and -B = 4.38. The
detailed derivations are given in Marky and Breslauer, (1987). This approach of
measuring the van't Hoff enthalpies is particularily amenable to melting duplexes off
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
31
solid supports as all problems associated with baselines and background are completely
elimin~te.l
The equilibriurn constant K for a helical transition of a molecularity of 2
can be expressed as the extent of a (the fraction of single strand molecules in a duplex).
5 The value of K is usually determined at the Tm of the helical coil transition where a =
0.5. This value of the Tm is then extrapolated to some reference temperature (e.g,
298K) using the empirically determined Tm (or Td) and the calculated van't Hoff
enthalpy (assumed to be temperature independent) and the integrated form of the van't
Hoff equation:
ln[K(Tm)/K(Tref)] = ~H~IR(I/T-1/Tm)
From the empirically determined value of K(Tref), it is possible to
determine ~G~ for the helical coil transition using the relation /~G~ = ~H~ - T~S~. Since
the melting curves described here are concentration independent7 the ln(KTm) = 0 since
K = I at the rI'm. Therefore the van't Hoff equation reduces to:
-In K(T) = ~H~/R(l/T-l/Tm),
which upon multiplying both sides by RT, provides
-RT lnK(T) = ~H~(l -T/Tm) = ~G~
This expression can be used to calculate the transition free energy ~G~ at
any temperature of interest (T) from the experimentally measured values of T~n and
~HVII. The corresponding ~S~ can be calculated from relation ~G~= ~H~- T~S".
As a result of reducing the HCT, a hybotrope increases the stringency
factor of a hybridization solution or solvent, where the stringency factor is the value of
the slope (partial derivative) of the helical coil transition at the value of the T"l. As
discussed above, the stringency factor can be used to identify a hybotrope.
A hybotrope is generally soluble or miscible in water, polar, apolar or
organic solvent from about 0.05 to 10 M, or a hybotrope can be composed solely of a
polar, apolar or organic solvent.
b. Structure of hybotropes.
The term "hybotrope" refers to any chemical or any mixture of a
chemical in an aqueous or organic environment with buffers, chelators, salts and/or
detergents that changes the enthalpy of a nucleic acid duplex by at least 20% when
referenced to a standard salt solution (0.165 M NaCI, 0.01 M Tris pH 7.2, 5 mM EDTA
and 0.1% SDS). That is, the energy content of the nucleic acid duples is decreased.
The reference oligonucleotide is 5'-
GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3' as the immobilized
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
32
oligonucleotide and 5'-TGT/GGA/TCA/GCAIAGC/AGG/AGT/ATG-3' as the solution
nucleotide which is typically labeled at the 5'-end with a fluorochrome such as Texas
Red. The oligonucleotide duplex (24 nucleotides in length) has a helical to coiltransition (HCT) of 25~C or less. The HCT is the difference between the temperatures
5 at which 80% and 20% of the duplex is single stranded. The average minimum slope
for a solution to be defined as a hybokope is the first derivative of the HCT and is equal
to 2.4 in units of I /temperature in degrees C ((80% single strand - 20% single-strand)/25~C).
The hybotrope may be a salt selected from LiTCA, RbTCA, GuSCN,
10 NaSCN, NaCI04, EU, TMATCA TEATCA, TMATBA, TMTCA, TMTBA, TBATCA
or TBATBA.
Preferred hybotropes are a salt formed of an anion and a cation, where
the anion is selected from acetate, propionate and halogenated versions thereof. The
halogen of the halogenated anion is selected from fluorine, chlorine, bromine and
15 iodine, but is preferably fluorine and/or chlorine. The halogenated anion may contain
as few as one and as many as three halogen atoms for halogenated acetate. The
halogenated propionate may contain as few as one or as many as five halogen atoms.
Trichloroacetate and trifluoroacetate are two prefered anions.
The cation is preferably an ammonium ion, not including NH4. Thus, the
20 cation is a primary, secondary or tertiary arnmonium comprising 1-36 carbon atoms, or
a quaternary ammonium comprising 4-48 carbon atoms. Preferably, the cation is
formed from atoms selected from 2-20 carbon atoms, 0-5 oxygen atoms and 1-5
nitrogen atoms. Thus, the cation substituents, where the groups bonded to the central
nitrogen of the ammonium ion are called the "cation substituents" may contain ester~
25 ether, hydroxyl, amine and amide functionality. Preferably, the cation substiuents are
hydrocarbyl groups, i.e., groups formed entirely of carbon and hydrogen? where
hydrocarbyl groups may be saturated or unsaturated, and the carbon atoms of a
hydrocarbyl group may be linear, branched or arranged in a cyclic fashion.
A preferred ammonium ion is a quaternary ion of the structure N(R)4
30 wherein R is a C,-CI2hydrocarbyl and any two R groups may join together to form a
cyclic structure with the nitrogen atom. The phrase "any two R groups may join
together to form a cyclic structure with the nitrogen atom" means that the arnmoniurn
ion may be heterocyclic in that the central nitrogen atom is part of a cyclic structure.
For example, the central nitrogen atom may be the nitrogen atom in piperidine, where
35 this nitrogen atom is also bonded to other R groups. Preferred R groups for the
CA 02266847 1999-03-22
WO 98113527 PCT/US97/17413
33
quaternary ammonium ion are independently selected from C,-C,2alkyl, C3-
C,2cycloa}kyl and C7-CI2arylalkyl.
Another pfer~ ed ammonium ion is a tertiary ion of the structure
HN(R)3 wherein R is a C,-CI2hydrocarbyl and any two R groups may join together to
5 form a cyclic structure with the nitrogen atom. Again, preferred R groups for the
tertiary amrnonim are are independently selected from C,-C,2alkyl, C3-C,2cycloalkyl
and C7-C,2arylalkyl.
Yet another preferred ammonium ion is a secondary ion of the structure
N(H)2(R)2 wherein R is a C,-C,2hydrocarbyl and the two R groups may join together to
10 form a cyclic structure with the nitrogen atom. Again, preferred R groups for the
tertiary ammonim are are independently selected from C,-CI2alkyl, C3-C,2cycloalkyl
and C7-C,2arylalkyl.
Suitable salts include, without limitation, those cont~ining an ammonium
cations selected from ethylbutylammonium, I-methylimidizole, 1-methylpiperidine, ]-
15 methylpyrrolidine, 3-methoxypropylamine, triethylamine, bis(2-methoxyethyl)amine,
diallylamine, dibutylamine, diisobutylamine, N,N-dimethylaminobutane, N,N-
dimethylclyclohexylamine, N,N-dimethylheptylamine, N,N-dimethylhexylamine,
triethanolamine, l-ethylpiperidine, dicyclohexylamine, diisopropylamine,
dipropylamine, N,N-dimethylisopropylamine, N-ethylbutylamine? tetraethylarnonium,
20 tripropylamine, 2-methoxyethylamine, and N,N-dimethyloctylamine, and the anion is
selected from acetate, trichloroacetate and trifluoroacetate.
As used herein, the following terms have the indicated meanings.
Alkyl refers to an aliphatic hydrocarbon radical,--(CH2)DCH3, either
branched or unbranched such as methyl, ethyl, N-propyl, iso-propyl, N-butyl, iso-butyl.
25 sec-butyl, tert-butyl, dodecyl or the like.
Aryl refers to a radical derived from an aromatic hydrocarbon by
removal of one hydrogen atom such as phenyl, o~-naphthyl, ,B-naphthyl, biphenyl,anthryl and the like.
Arylalkyl, ~CN2)n--Ar, refers to an alkyl radical as defined above
30 joined to an aryl radical.
Hydroxyalkyl refers to a radical--(CH2)nOH.
Cycloalkane or cycloalkyl refers to a radical of a saturated hydrocarbon
in a ring structure such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl,
cycloheptyl, ~ m~ntyl and the like.
Unless otherwise stated, all number ranges are inclusive of the stated
range (e.g, 1 to 5 carbons, includes to and 5 carbons).
CA 02266847 l999-03-22
W O98/13527 PCT~US97/17413
34
Halogen refers to chlorine, bromine, iodine or fluorine.
c. Novel hybotropes.
Some of the hybotropes disclosed herein form novel hybridization
5 solutions that improve the specificity o~ oligonucleotide probes. For example, tetramethylammonium trichloroacetate (TMATCA) and tetraethylammonium
trichloroacetate (TEATCA) confer a high level of hybridization stringency. Moreover,
these hybotropes neutralize G+C content influence Td. In the Examples, random
oligonucleotide probes (all 19-mers) differing in G+C content from 25% to 73% are
10 shown to possess a Td within 5~C of each other in the presence of TMATCA (seeFigure 6); the average Td in 3 M TMATCA was 45~C. Similar results are obtained with
TEATCA (see ~igure 7). As a control, the Tds of these 19 mers were determined in 3 M
TEACI. The resulting differences in Td was 6~C and the average Td of the 6
oligonucleotides was about 62~C. Furthermore, in 30% formamide, the 6
15 oligonucleotide probes differed in Td by 15~C, in 0.165 M NaCI, the range in Td values
was 15~C (see Figure 8); and in 2 M LiTCA, the difference in Td was about 10~C. Most
signi~lcantly, however, the HCT in TMATCA ranges from 8~C for the 25% G+C
content oli~o to 14~C for the 73% G+C oligonucleotide. In contrast, the HCT in
TMACI ranges from 12.5~C for the 25% G+C content oligo to 17.5~C for the 73% G+C20 oligonucleotide. This 4~C to 5~C shift in the HCT of the oligos in the presence of
TMATCA results in a significant improvement in the stringency factor of TMATCA
compared to TMACI. Thus, TMATCA is a significantly better salt than any previousiy
described solution for conducting oligonucleotide-based assays.
Novel hybridization solutions have also been identified which neutralize
25 the effects of G+C content on the melting behaviour of nucleic acid duplexes. These
solutions are in some cases hybotropes and in other cases can be used as PCR buffers or
as hubridization solutions which minimi7~ the effects of G+C content on nucleic acid
duplexes. These new hybridization solutions, their properties, and their preparation are
described in Example 12. Figure 14 is a graph showing the difference in Td between
30 three duplexes, that vary in G+C content from 27% to 83%. The capture
oligonucleotide is a 36-mer (DMO-GC36cap: 5'- hexylamine-
GCA/GCC/TCG/CGG/AGG/CGG/ATG/ATC/GTC/ATT/AGT/ATT-3 ' ) and three
complementary oligos which are labelled with the fluorochrome are DMO-83GC: 5'-
Texas Red- CCG/CCT/CCG/CGA/GGC/TGC-3'; DMO-50GC: 5~-Texas Red-
35 AAT/GAC/GAT/CAT/CCG/CCT-3'; DMO-27GC: -Texas Red-
AAT/ACT/AAT/GAC/GAT/CAT-3'. The temperature difference between any two Tds
CA 02266847 1999-03-22
W O 98113527 PCTAUS97/17413
at a = 0.5 is defined as the ~Td. The pelcelltdge of single strand DNA (y-axis) is
plotted versus te,~ dl~lre (~C; x-axis). The melting solution was 100 mM 2-
methoxyethylamine trifluoroacetate. The maximum difference between the 3 melting- curves in the Td was 6 C. The helical coil transition of the 27% G+C content was 21 C,
5 50% G+C was 33 C and for the 83% G+C duplex was 29 C. Note that the helical coil
- transitions (HCTs) of the 3 different G+C content oligonucleotides is different. This is
in contrast to the case with diisobutylamine as shown in Figure 15. Figure 15 is a graph
showing the dirrerence in Td between three duplexes, that vary in G+C content from
27% to 83% (the same system as described in Figure 14. The temperature difference
10 between any two Tds at a = 0.5 is defined as the ~Td. The percentage of single strand
DNA (y-axis) is plotted versus t~ pGld~ (~C; x-axis). The melting solution was 100
mM diisobutylamine acetate. The maximum difference between the 3 melting curves in
the Td was 5 C. The helical coil transition of the 27% G+C content was 22 C, 50%G+C was 26 C and for the 83% G+C duplex was 25 C. The helical coil transitions for
15 the three oligonucletide duplexes are very similar. This is the behaviour that is
preferred for use in array hybridizations or polymerase chain reactions.
In Figure 16 the inability of GuSCN to neutralize G+C content is shown.
Figure 16 is a graph showing the difference in Td between three duplexes, that vary in
G+C content from 27% to 83% (the same capture and probe oligonucleotides as
20 described in figure 14). The temperature difference between any two Tds at a = 0.5 is
defined as the ~Td. The percentage of single strand DNA (y-axis) is plotted versus
temperature (~C; x-axis). The melting solution was 2 M Guanidinium thiocyanate. The
maximum difference between the 3 melting curves in the Td was 16 C. The helical coil
transition of the 27% G+C content was 28 C, for the 50% G+C duplex was 30 C and for
25 the 83% G+C duplex was 32 C. Similar results were obtained with lx PCR buffer(Figure 17) and lx SSC buffer (Figure 18). There was also no neutralization of G+C
content with 20% formamide (Figure 19).
In contrast to the situation in Figures 17, 18 and 19, Figure 20 shows the
melting behaviour of the 3 different G+C oligonucleotide duplexes in I M
30 dicyclohexylamine acetate. Figure 20 is a graph showing the difference in Td between
three duplexes, that vary in G+C content from 27% to 83% (same duplexes as described
in Figure 14). The tt;ml~eldlule difference between any two Tds at a = 0.5 is defined as
the ~Td. The percentage of single strand DNA (y-axis) is plotted versus temperature
(~C; x-axis). The melting solution was 1 M dicyclohexylamine acetate. The maximum
35 difference between the 3 melting curves in the Td or Tm is 3 C. The helical coil
transition of the 27% G+C content was 13 C, for the 50% G+C duplex was 17 C and for
CA 02266847 1999-03-22
WO g8/13527 rCT/US97/17413
36
the 83% G+C duplex was 19 C. This is an ideal profile for a hybrotrope. In contrast the
the narrow helical coil transition observed in Figure 20, a much wider HCT is observed
with 500 mM n-ethylbutylamine acetate. Figure 21 is a graph showing the difference in
Td between three duplexes, that vary in G+C content from 27% to 83% (the identical
duplex system as described in Figure 14). The temperature difference between any two
Tds at a = 0.5 is defined as the ~Td. The percentage of single strand DNA (y-axis) is
plotted versus te~ a~ule (~C; x-axis). The melting solution was 500 mM n-
ethylbutylamine acetate. The maximum difference between the 3 melting curves in the
Td is 1 C. The helical coil transition of the 27% G+C content was 22 C, for the 50%
G+C duplex was 22 C and for the 83% G+C duplex was 26 C.
The ability of some of the G+C neutralizing buffer to act as hybrotropes
is illustrated in Figure 22. Figure 22 is a graph showing the difference in Td between
three duplexes, one that is perfectly based-paired and the other two that contains a
mi~m~tch or a deoxynebularine substitution. The temperature difference between any
two TdS at a = 0.5 is defined as the ~Td. The percentage of single strand DNA (y-axis)
is plotted versus temperature (~C; x-axis). DMO-2060: 5'-hexylamine-
GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3' (oligonucleotide
immobilized on the nylon bead.; DMO-2055: S'-TEXAS RED-
TGT/GGA/TCA/GCA/AGC/AGG/AGT/ATG-3' (perfect complement); DMO-2058;
S'-TEXAS RED- TGT/GGA/TCA/GGA/AGC/AGG/AGT/ATG-3' (mism~tch
complement); and DMO-2058-dN: S'-TEXAS RED-
TGT/GGA/TCA/G(deoxynebularine)A/AGC/AGG/AGT/ATG-3' (deoxynebularine
mi~m~tch complement). The melting solution was 1 M diisopropylamine acetate. Themaximum difference between the 3 melting curves in the Td is 6 C. The helical coil
transition (HCT) of the true mi~m~trh was 14 C; the HCT for the deoxynebularine
mi~m~tch duplex was 14 C and the HCT for the perfectly based paired duplex was 16
C. The same situation was observed for 1 M diisopropylamine acetate (Figure 22), 1 M
n,n-dimethylcyclohexylarnine acetate (Figure 23) and I M dicyclohexylamine acetate
(Figure 24) and N,N-dimethylhexylamine acetate (Figure 25).
Preferred hybotropes of the present invention include, without limitation,
bis(2-methoxyethyl)amine acetate, l-ethylpiperidine acetate, l-ethylpiperidine
trichloroacetate, l-ethylpiperidine trifluoroacetate, 1-methylimidizole acetate, 1-
methylpiperidine acetate, I-methylpiperidine trichloroacetate, l-methylpyrrolidine
acetate, l-methylpyrrolidine trichloroacetate, l-methylpyrrolidine trifluoroacetate, 2-
methoxyethylamine acetate, N,N-dimethylcyclohexylamine acetate, N?N-
dimethylcyclohexylamine trifluoroacetate, N,N-dimethylcyclohexylamine, N,N-
CA 02266847 1999-03-22
WO 98/13527 PCT/USg7/17413
37
dimethylheptylamine acetate, N,N-dimethylheptylarnine acetate, N,N-
dimethylhexylarnine acetate, N,N-dimethylhexylamine acetate, N,N-
dimethylisopropylamine acetate, N-ethylbutylamine acetate, N-ethylbutylamine
trifluoro~et~te, N,N-dimethylaminobutane trichloroacetate, N,N-
5 dimethylisopropylarnine trichloroacetate, triethanolamine acetate, triethylamine acetate,triethylamine trichloroacetate, tripropylamine acetate, tetraethylammoniurn acetate.
These compounds or chemicals can be combined with buffers, chelating agents and/or
d~lelge~
d. Effect of hybotrope concentration on discrimination.
As shown herein, the discrimination between mi.~m~tched
oligonucleotides ~mutant abbreviated as "mt") and perfectly based-paired
oligonucleotides (abbreviated as "wt") is not a function of concentration of a particular
hybotrope but rather a function of hybotrope type. Surprisingly, the HCT for thehybotropes LiTCA, GuSCN, GuHCl, and NaClO4 does not change over about the range
of about 0.5 M to about 6.0 M. Moreover, the slope of the mt duplex is always
observed to be greater than for wt duplexes (see Figure 9). Furthermore, the difference
between the Tm of the wt duplex and the mutant duplex (~Td) is not affected by the
concentration of the hybotrope. However, the T,~ is directly propor~ional to
concentration (see Figure 10). Because ~Td does not change over a wide concentration
range for the hybotropic solutions, a wide temperature range can be employed forconducting oligonucleotide-based assays (e.g, 20~C to ~0~C). Second, relatively low
concentrations (e.g, 0.5 M) of hybotrope may be employed in hybridization assays,
including polymerase catalyzed reactions.
The approximate concentration range at which a solution of a compound
(such as LiTCA) exhibits the characteristics of a hybotropic solution is approximately
0.2 to 0.4 M (Figure 11; Examples).
e. Effect of length of duplex.
The length of an oligonucleotide probe (i.e., resultant duplex) has the
effect of increasing the T", as length increases. Due to this relationship, discrimination
using a hybotrope is effectively limited to hybridization lengths of 6-40 bases and
preferably 6-30 bases.
CA 02266847 1999-03-22
W O 98/13527 PCTrUS97/17413
38
f. Assays for determining if a compound is hybotropic.
As discussed above, a hybotrope is a chemical that can increase the
enthalpy of a nucleic acid duplex by 20% or more when referenced to a standard salt
solution. A convenient assay for measuring this increased enthalpy is a thermal
5 transition assay. A hybotrope may be identified as any chemical or any mixture of a
chemical in an aqueous or organic environment with buffers, chelators, salts and/or
detergents that decrease the enthalpy of a nucleic acid duplex by 20% when referenced
to a standard salt solution (0.165 M NaCl, 0.01 M Tris pH 7.2, 5 mM EDTA and 0.1%
SDS). The reference oligonucleotide is 5'-
10 GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3 ' as the immobilized
oligonucleotide and 5'-TGT/GGA/TCA/GCA/AGC/AGG/AGT/ATG-3' as the solution
nucleotide which is typically labelled at the S'-end with a fluorochrome such as Texas
Red. The oligonucleotide duplex (24 nucleotides in length) has a helical to coiltransition (HCT) of 25~C or less." Moreover, a suitable hybotrope is soluble in water,
15 other protic solvent or aprotic solvent. Although not required, a hybotrope preferably
does not inactivate polymerases when in they are with polymerases and the like in PCR
reactions (and the like). Assays for these properties are briefly discussed below.
HCT of an 18-24 mer with a 50% G+C content are readily measured for
a given solution. Briefly, an 18-24 mer oligonucleotide and its complement with a 50%
20 G+C are synthesized. The oligonucleotides are dissolved to 2 ~M in the candidate
hybotrope solution. The mixture is heated to 85~C (at 0.5~C/min) and then cooled to
10-15~C to allow hybridization. Absorbance versus time is recorded at 260 nrn by a
UV-VIS spectrophotometer equipped with a thermal programmer. The HCT is
determined from a plot of normalized absorbance values (fully annealed= 0% single
25 strand; fully denatured= 100% single strand) versus temperature. A solution in which
the temperature difference between 80% and 20% single stranded (HCT) is < 25~C is a
suitable hybotropic solution within the context of this invention.
Solubility may be measured by making a saturated solution with the
respective salt, filtering off undissolved salt, removing the liquid or aqueous material
30 and then determining the weight of the rem:~ining salt.
pK values are measured using standard titration methods.
Polymerase activity in a hybotropic solution may be measured according
to the use of the polymerase. For example, in amplification reactions, duplicatereactions with and without the hybotrope are run. The hybotrope does not inactivate the
35 enzyme if 10% of activity is retained.
CA 02266847 1999-03-22
WO 98/13S27 PCT/USg7/17413
39
B. ABASIC, ANUCLEOSIDIC ANALOGS AND DEOXY NEBULARINE RESIDUEC .
AS described herein, an increase in specificity of priming or probing
when using synthetic oligonucleotides is accomplished by minimi7:in~ the helical coil
transition of the respective primer duplex, thereby increasing the stringency factor of the
S respective sequence. An increased stringency factor of an oligonucleotide decreases the
stability of a mi~m~tch and therefore promotes a high fidelity hybridization.
Furthermore, increasing the stringency factor or decreasing HCT may also result in an
increase in the specificity of priming. One way to increase stringency is to introduce
one or more abasic anucleosidic or deoxynebularine residues into one strand of a10 duplex. Thus, introducing one of these residues leads to a "base pair" that is not
hydrogen bonded. In effect, this is analogous to a mi.~m~trh and will decrease the Td
and HCT of the respective derived oligonucleotide compared to a perfectly base-paired
oligonucleotide that has the same sequence. Although, for the sake of simplicity the
oligonucleotides in the examples below incorporate only one type of these residues at a
15 time, combinations such as an abasic and an anucleosidic residue may be utilized.
As noted above, an abasic residue is a compound that approximates the
length of a ribofuranose sugar, is covalently attached to neighboring bases and is
substituted at the beta anomeric with a group that does not interact [i. e., hydrogen bond]
with the base on the opposite strand of a duplex. Abasic residues in oligonucleotides
20 can be introduced by the chemical or enzymatic hydrolysis of the glycosidic bond. The
resulting structure is apurinic or apyrimidinic, lacks coding information, and fails to
base pair. One abasic residue, the CE phosphoramidite of the tetrahydrofuran
derivative, is commercially available (dSPACER, Glenn Research, Sterling, Virginia)
along with other "spacer" phosphoramidites (Glenn Research, Sterling, Virginia).25 Alternatively, an abasic substitution may comprise a backbone of N-(2-aminoethyl)-
glycine linked by amide bonds. Unlike native DNA or RNA backbone, this structurehas no deoxyribose or phosphate groups.
The typical placement of the abasic site is approximately in the middle
of the oligonucleotide. A typical primer has the following configuration: 5'-N,0-spacer-
30 Nlo-3'. However, multiple abasic sites may be placed in the oligonucleotide(s) at
regular or irregular intervals, depending on the value of HCT to be achieved. Generally,
a primer ranges in length from 6 to 40 or from 16 to 30 nt in length and contains from 1
to 5 abasic sites. Thus, abasic sites can be incorporated at a spacing of 3~ 4, 5, 6, or 8
nucleotides or incorporated in any combination of nucleotides (or analogues) that base-
35 pair with abasic sites. For example, a 6-mer may have one 1 abasic site, an 1 8-mer, 2
abasic sites, a 24-mer, 3 basic sites, etc. As a general guideline when an
CA 02266847 1999-03-22
W O 98/13527 PCTrUS97/17413
oligonucleotide is used to detect a mutation, the abasic site is preferably not located at
the site of the mutation. However, abasic sites may be placed at the site of mutations
that are not of interest (e.g., a polymorphism that does not result in a phenotype).
As shown in the table below, introduction of an abasic residue into 5'-
5 hexylarnine-TGTGGATCAGCA-spacer-GCAGGAGTATG-3', wherein the spacer is
either the C3-spacer or dSPACER from Glenn Research (Sterling, VA, where these two
chemicals have the same effect but are chemically distinct), lowers the HCT from 2.5~C
to 6~C compared to a normal oligonucleotide, and depending on the solution used.
Table 4
Buffer Type FactorOligo Type I~CT (~C) Td (~C) Stringency
factor
lX PCR buffer norrnal 18
1 X PCR bufferabasic (dSPACER) 12
lX PCR bufferabasic (C3 spacer) 12
0.5 M TMATCA normal 14
0.5 M TMATCAabasic (dSPACER) 8
0.5 M TMATCAabasic (C3 spacer) 8
2.0 M LiTCA normal 12.5 44.5 4.97
2.0 M LiTCAabasic (dSPACER) 10 39 6.37
2.0 M LiTCAabasic (C3 spacer) 10 39 6.25
3.0 M GuSCN norrnal 16 35.5 3.85
3.0 M GuSCNabasic (dSPACER) 12.5 32 5.24
3.0 M GuSCNabasic (C3 spacer) 12.5 31 5.31
DeoxyNebularine (dN) can also be used to increase the enthalpy of an
oligonucleotide duplex. Preferably, deoxyNebularine replaces a G, C, or T base in a
probe or primer. Multiple deoxyNebularine sites may be placed in the
oligonucleotide(s) at regular or irregular intervals, depending on the value of HCT to be
15 achieved. Generally, a primer ranges in length from 6 to 40, preferably from 16 to 30
bases and contains from I to 5 deoxyNebularine sites. A typical primer has the
following configuration: 5~-N~o~ deoxyNebularine -N~o~3l. As shown in the table
below, introduction of a deoxyNebularine residue into 5'-hexylarnine-
CA 02266847 1999-03-22
W O 98/13S27 PCTAJS97117413 41
TGTGGATCAGCA-dN-GCAGGAGTATG-3' lowers the HCT from 2.5~C to 6~C,
depending on the hybridization solution or hybotrope used.
Table 4B
Stringency
- Buffer Type Oligo Type HCT Td(~C) Factor
IXPCRbuffer normal 18
lX PCR buffer deoxyNebularine 12
lX PCR buffer deoxyNebularine 12
0.5 M TMATCA normal 14
0.5 M TMATCA deoxyNebularine 8
0.5 M TMATCA deoxyNebularine 8
2.0 M LiTCA norrnal 12 44 5.0
2.0 M LiTCA deoxyNebularine 10 39 6.3
2.0 M LiTCA deoxyNebularine 10 39 6.3
3.0 M GuSCN normal 16 35 3.9
3.0 M GuSCN deoxyNebularine 12.5 32 5.2
3.0 M GuSCN deoxyNebularine 12.5 31 5.3
The invention thus provides an oligonucleotide comprising a plurality of
fragments, each fragment shown schematically by structure ( 1 )
(I) ~1 1 1 1~
Bl B2 B3 B5
I 0 wherein,
~1 1 1
Bl B2 B3 represents a sequence of at least three nucleotides (and
preferably 4-12) as found in wild-type DNA, where "B" represents a base independently
selected at each location;
~ represents a series of covalent chemical bonds termed a
15 "specificity spacer," which separates and connects two bases B3 and Bs.
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
42
The meaning of strucuture (1) can be understood by reference to Figure
26, where it can be seen that structure (1) corresponds to a sequence of nucleotides
wherein at least the base and perhaps more of the nucleotide is mis~ing in the region
termed the "specificity spacer". Each specificity spacer occupies no more that a single
5 nucleotide site.
The specificity spacer has steric and chemical properties such that it does
not prevent hybridization between a fragment of structure (1) and an oligonucleotide
fragment having a complementary base sequence, as shown schematically as structure
(2)
(1) ~ I I I I ~
Bl B2 B3 B5
I l IB2 1 3 ~ 4 1 5
Thus, in the structure above, it can be seen that the specificity spacer
occupies a single nucleotide site, and does not prevent the "wild-type" nucleotides, i. e.,
15 the nucleotides having the standard phosphate-sugar-base group found in naturally
occuring oligonucleotides (e.g., DNA, cDNA, RNA) from base-pairing. The wild-type
nucleotides are represented by the straight lines termin~ting in a "B", where "B"
represents a standard base selected from adenine, guanine, cytosine, uracil and thymine.
The specificity spacer may or may not hydrogen bond with the
20 nucleotide in the complementary chain (2) with which the chain having structure (1)
forms a duplex. Preferably, the specificity spacer cannot hydrogen bond with anything.
However, in another preferred embodiment, the specificity spacer can hydrogen bond
with the "opposite base" (shown as ~'B4- n the duplex of (1) and (2)), but not in the
conventional Watson-Crick manner. In fact, if the specificity spacer can hydrogen bond
25 with the base in the complementary chain, then that hydrogen bonding is preferably
much weaker than the hydrogen bonding that would occur if the specificity spacer were
to bond to the opposite base by standard Watson-Crick base pairing.
A preferred specificity spacer has the formula
Y
--~--~~ SSC ~o_ 1_
Y Y
wherein
CA 02266847 1999-03-22
WO 98/13527 PCTtUSg7/l7413
43
Y is selected from oxygen, sulfur, methyl and amino when X is oxygen,
or Y is selected from oxygen and sulfur when X is sulfur; and
SSC represents a specificity spacer component having a chain of 2-5
carbon atoms shown in the formula
~C--(C)n--C~
wherein n is 0, 1, 2 or 3. The SSC should not have less than two carbon atoms because
that would cause the nucleotides which neighbor the specificity spacer to be too close
10 together to effective hydrogen bond with a complentary oligonucleotide. Likewise, the
SSC should not have more than 5 carbons because again that would disrupt the ability
of nucleotides in the specificity spacer-cont~ining sequence to hydrogen bond with a
complementary sequence. Preferably, the specificity spacer component has a total of 3
or 4 carbons directly sepald~ g the two fl~nking -O-P groups.
The 2-5 carbon atoms of the SSC may be substituted with essentially any
atoms, so long as the arrangement of those atoms is not such that the specificity spacer
completely stops a complementary oligonucleotide chain from hybridizing with thespecificity spacer-containing oligonucleotide. Preferred SSCs are either unsubstituted
(i.e., are alkylene chains) or are alkylene chains substituted with sterically non-
20 dem~n~ing substituents such as halogen, C 1 -C I Ohydrocarboxyloxy (a hydrocarbyl
group joined to the "2-5 carbon atoms'~ through an ether oxygen atom), hydroxyl, C1-
CShydrocarbyl and like-sized or smaller groups.
The specificity spacer component may contain a five- or six-
memembered carbocyclic or heterocyclic ring. For instance, the SSC may be a ribose
25 or deoxyribose group as found in a standard nucleotide, however this ribose or
deoxyribose is "abasic" in that the purine or pyrimidine base is absent, and is preferably
replaced with a hydrogen. A specificity spacer component of this structure may be
represented by the formula (2)
~C--/C_C~
X~ )
1-2 (2)
wherein n is 1 and X is selected from carbon, oxygen and sulfur, such that any carbon
shown in formula (2), including X when it is carbon, may be substituted with hydrogen.
.. ......
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
44
C,-C5hydrocarbyl, C,-Cshydrocarbyloxy, a non-hydrogen bonding purine base analog or
a non-hydrogen bonding pyrimidine base.
The invention provides oligonucleotides having specificity spacers
which may be in solution or may be bound to a solid support. ~specially when bound
5 to a solid support, the invention provides compositions in an array form, having a
plurality of oligonucleotide sequences, each having specificity spacers.
Each oligonucleotide cont~ining a specificity spacer contains a plurality
of such spacers. Thus, specificity spacers preferably constitute 15-60% of the
nucleotide positions in an oligonucleotide. However, the specificity spacers are not
10 adjacent to one another, i.e, there is at least one "wild-type" nucleotide located between
any two specificity spacers. In fact, all of the specificity spacers in an oligonucleotide
preferably are separated by 4-12 "wild-type" nucleotides, and are more preferably
separated by 5-8 or 8-12 wild-type nucleotides. The specificity spacers are preferably
arranged in a repeating pattern, such that there would be 5 wild-type nucleotides
15 followed by a specificity spacer, followed by 5 more wild-type nucleotides, followed by
a specificity spacer, followed by 5 more wild-type nucleotides, etc. At each occurrence
in an oligonucleotide chain, the chemical structure of the specificity spacer isindependently selected.
Methods of synthesizing oligonucleotides are known to those of skill in
20 the art. In paticular, methods of synth~si7ing oligonucleotides with natural and non-
nucleotidic monomers are found in: Oligonucleotide Synthesis: A Practical Approach,
Gait, ed., IRlL Press, Oxford (1984). The 5'-Dimethoxytrityl-2'-deoxynucleoside-3'-
(N,N-diisopropyl-2-cyanoethyl)phosphoramidites for the synthesis of standard
oligonucleotides are available from Glen Research (Herndon, VA); Beckman
25 Instruments (Brea, CA) or Applied Biosystems (Foster City, CA).
The specificity spacers suitable for direct incorporation into
oligonucleotides for use in this invention are commercially as cynanoethyl
phosphoramidites from m~nuf~turers like Glen Research, Midland Certified Reagents
(Midland,TX), and Clonetech (Palo Alto, CA). Alternatively, other specificity spacers
30 can be prepared from compounds that contain the required diol by a three stage process
f~mili~r to those skilled in the art and described in Gait. Stage 1 involves protecting
any amine as benzamides or other suitable protecting group. Stage 2 involves
protecting one of the hydroxyls, preferably a primary hydroxyl, as a dimethoxytrityl
ether using dimethoxytrityl chloride in pyridine. In the third stage the second hydroxyl
35 is converted into a N,N-diisopropyl-2-cyanoethyl)phosphoramidite by phosphitylation
with N,N,N,N-tetraisopropylphosphoramidite and diisopropylarnmonium tetrazolide.
CA 02266847 1999-03-22
WO 9Ut3S27 PCT/US97/17413
These phosphoramidites can be used on automated DNA synthesi7~rs availible from
Beckman, ABI or Perseptive Biosystems.
C. METHODS OF USING HYBOTROPES AND OLIGONUCLEOTIDES CONTAINING AN
ABASIC OR ANUCLEOSIDIC RESIDUE
As noted herein, a hybotrope may be used in essentially any reaction
involving hybridization of a duplex in which the annealed region is from about 6 to
about 40 base pairs long. Such reactions include screening for one or few base changes
(e.g., genetic screen), DNA sequence analysis by random oligonucleotide hybridization,
amplification reactions, RTase polymerization, such as synthesis of cDNA, differential
1 0 amplification.
AS used herein, a "discrimination temperature" is a temperature at which
a hybridization reaction is performed that allows discrimination between a mism~tched
duplex and a perfectly matched duplex. As shown herein, a range of temperatures
satisfy criteria of a discrimination temperature. The discrimination temperature ranges
from the temperature at which an a value (fraction of single stranded nucleic acid) is
0.2 for a given oligonucleotide duplex (or nucleic acid duplex) containing a mi.~m~tch at
any place in the duplex, to the temperature at which a value for (x equals 0.8 for the
same given oligonucleotide duplex (or nucleic acid duplex), but which does not contain
a mi.~m~tch at any place in the duplex. An o~ value is the fraction of single stranded
nucleic acid at any given temperature generated during the thermal transition of a DNA
strand from a double-stranded to a single stranded form. In deterrnining o~, themi~m~tch can be due to any type of modified nucleotide, nucleoside~ or derivative
thereof. A discrimination temperature is applicable to any given duplex 6 nt to 250 nt
in length, of any given G+C content, cont~ining modified or substituted nucleotides or
nucleosides, and in which the duplex is composed of deoxyribonucleotides.
ribonucleotides, or mixtures of different types of strands. As an example, for an
oligonucleotide duplex of 18 nucleotides in length, the critical discrimination
temperature (range) would be from 10 to 15~C. The lowest temperature of the
discrimination temperature range is dependent on the concentration and type of
hybotrope used and can range from 0 to 80~C, preferably from 20 to 50~C.
1. Detection of a Mutation.
Mutations are a single-base pair change in genomic DNA. Within the
context of this invention, most such changes are readily detected by hybridization with
oligonucleotides that are complementary to the sequence in question. In the system
.
CA 02266847 1999-03-22
W O 98/13527 PCTrUS97/17413
46
described here, two oligonucleotides are employed to detect a mutation. One
oligonucleotide possesses the wild-type sequence and the other oligonucleotide
possesses the mutant sequence. When the two oligonucleotides are used as probes on a
wild-type target genomic sequence, the wild-type oligonucleotide will form a perfectly
based paired structure and the mutant oligonucleotide sequence will form a duplex with
a single base pair mi~m~tch. As shown herein, the resulting two types of duplexes
(wild-type and mutant) have different Tds as a result of a single base pair mism~tch in
one of the duplexes. For example, a 6 to 7~C difference between the Td of a wild-type
(wt) duplex (perfectly based paired) and duplex cont~ining a mi.~m~tch (described above
10 as ~Td) is obtained in a hybotrope (LiTCA), but not in standard salt-based hybridi~ation
solutions (e.g., SSC). The ~Td value for a 30-mer was 6~C, 6.4~C for a 24-mer, and
7~C for a 18-mer.
A ~'long" oligonucleotide probe (> 18nt) may thus be used for mutation
detection in a long polynucleotide target nucleic acid. For this application, a probe that
15 hybridizes only to a single copy portion of the human genome is preferable. A 30-mer
can be used to search a collection of 1.15 x 10l8 30 mers (complexity= 3.45 x 10l9
nucleotides), a 24-mer can be used to search a collection of 2.81 x 1024 24 mers(complexity = 6.75 x 10'5 nucleotides) and a 18-mer can be used to search a collection
of 6.87 x 10'~ 10-mers (complexity= 1.23 x 10'' nucleotides). In addition, from a
20 empirical point of view, oligonucleotides work best as probes or primers of eucaryotic
DNA or RNA when greater than 23 nt in length.
As discussed above, a 6 to 7~C difference in the Td of a wild type versus
mi~m~tçlled duplex permits the ready identification or discrimination of the two types
of duplexes. To effect this discrimination, hybridization is performed at the Td of the
25 mi.~m~tched duplex in the respective hybotropic solution. The extent of hybridization is
then measured for the set of oligonucleotide probes. When the ratio of the extent of
hybridization of the wild-type probe to the mi~m~tched probe is measured, a value to
10/1 to greater than 20/l is obtained. These types of results permit the development of
robust assays for mutation detection.
For exemplary purposes, one assay format for mutation detection utilizes
target nucleic acid (e.g, genomic DNA) and oligonucleotide probes that span the area
of interest. The oligonucleotide probes are greater or equal to 24 nt in length (with a
maximum of about 36 nt) and labeled with a fluorochrome at the 3' or 5' end of the
oligonucleotide probe. The target nucleic acid is obtained via the lysis of tissue culture
35 cells, tissues, or~ni~m~, etc., in the respective hybridization solution. The lysed
solution is then heated to a temperature which denatures the target nucleic acid (15-
CA 02266847 1999-03-22
W O 98/13527 PCT~US97/17413
47
25~C above the Td of the target nucleic acid duplex). The oligonucleotide probes are
added at the denaturation temperature, and hybridization is conducted at the Td of the
mi~m~tclled duplex for 0.5 to 24 hours. The genomic DNA is then collected and bypassage through a GF/C (GF/B, and the like) glass fiber filter. The filter is then washed
5 with the respective hybridization solution to remove any non-hybridized
oligonucleotide probes (RNA, short oligos and nucleic acid does not bind to glass fiber
f1lters under these conditions). The hybridization oligo probe can then be thermally
eluted from the target DNA and measured (by fluorescence for example). For assays
requiring very high levels of sensitivity, the probes are concentrated and measured.
Other highly sensitive hybridization protocols may be used. The
methods of the present invention enable one to readily assay for a nucleic acid
conl~ining a mutation suspected of being present in cells, samples, etc., i.e., a target
nucleic acid. The "target nucleic acid" contains the nucleotide sequence of
deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) whose presence is of interest,
and whose presence or absence is to be detected for in the hybridization assay. The
hybridization methods of the present invention may also be applied to a complex
biological mixture of nucleic acid (RNA and/or DNA). Such a complex biological
mixture includes a wide range of eucaryotic and procaryotic cells, including protoplasts;
and/or other biological materials which harbor polynucleotide nucleic acid. The method
is thus applicable to tissue culture cells, animal cells, animal tissue, blood cells (e.g.,
reticulocytes, lymphocytes), plant cells, bacteria, yeasts, viruses, mycoplasmas,
protozoa, fungi and the like. By detecting a specific hybridization between nucleic acid
probes of a known source, the specific presence of a target nucleic acid can be
established.
A typical hybridization assay protocol for detecting a target nucleic acid
in a complex population of nucleic acids is described as follows: Target nucleic acids
are separated by size on a gel matrix (electrophoresis), cloned and isolated, sub-divided
into pools, or left as a complex population. The target nucleic acids are transferred,
spotted, or immobilized onto a solid support such as a nylon membrane or nitrocellulose
membrane. (This "immobilization" is also referred to as "arraying"). The immobilized
nucleic acids are then subjected to a heating step or UV radiation, which irreversibly
immobilizes the nucleic acid. The membranes are then immersed in "blocking agents"
which include Dendhart's reagent (Dendhart, Biochem. Biophys. Res. Comm. 23:641,1966), heparin (Singh and Jones, Nucleic Acids Res. 12:5627, 1984), and non-fat dried
milk (Jones et al., Gene Anal. Tech. 1:3, 1984). Blocking agents are generally included
in both the prehybridization step and hybridization steps when nitrocellulose is used.
CA 02266847 1999-03-22
wO 98/13527 PCT/US97/17413
48
The target nucleic acids are then probed with labeled oligonucleotide probes under
conditions described above in hybotrope-based solutions. Probes may be detected by a
conjugated enzyme. Unbound enzyme is then washed away and the membrane is
immersed in a substrate solution. Signal is then detected by colorimetric means, by
5 fluorescence or by chemiluminescence, depending on substrate type. Alternatively, the
probe is directly labeled (e.g., radioactive isotope, fluorescent molecule, mass-
spectrometry tags; see U.S. Application No. 08/589,250, filed January 23, 1996,
chemiluminescent tags and the like).
2. DNA sequence analysis.
DNA sequence analysis is conventionally performed by hybridizing a
primer to target DNA and performing chain extensions using a polymerase. Specific
stops are controlled by the inclusion of a dideoxynucleotide. The specificity of priming
in this type of analysis can be increased by including a hybotrope in the annealing
15 buffer and/or incorporating an abasic residue in the primer and annealing at a
discrimin~ting temperature.
Other sequence analysis methods involve hybridization of the target with
an assortment of random, short oligonucleotides. The sequence is constructed by
overlap hybridization analysis. In this technique, precise hybridization is essential. Use
20 of hybotropes or abasic residues and :lnnP~Iing at a discrimin~ting temperature is
beneficial for this technique to reduce or elimin~te mism~tched hybridization. The goal
is to develop automated hybridization methods in order to probe large arrays of
oligonucleotide probes or large arrays of nucleic acid samples. Application of such
technologies include gene mapping, clone characterization, medical genetics and gene
25 discovery, DNA sequence analysis by hybridization, and finally, sequencing
verification.
Many parameters must be controlled in order to automate or multiplex
oligonucleotide probes. The stability of the respective probes must be similar, the
degree of mi~m~tch with the target nucleic acid, the t~m~ e, ionic strength, the30 A+T content of the probe (or target), as well as other parameters when the probe is short
(i.e., 6 to 50 nucleotides) should be similar. Usually, the conditions of the ~el;l-lent
and the se~uence of the probe are adjusted until the formation of the perfectly based
paired probe is therrnodynamically favored over the any duplex which contains a
mismatch. Very large scale applications of probes such as sequencing by hybridization
35 (SBH), or testing highly polymorphic loci such as the cystic fibrosis trans-membrane
protein locus re~uire a more stringent level of control of multiplexed probes. William
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
49
Bains (GATA 11:49, 1994), has ascertained that the ability to use multiplexed
oligonucleotide probes is generally much more difficult to implement than is suggested
by theory. Hybotropes and abasic residues will essentially overcome the limitations in
the use of multiplexed probes as presented by Bains.
The actual length of an oligonucleotide probe to uniquely prime any
natural nucleic acid target is far longer than is predicted by theory. In general, the
probability that a given probe is unique is related to the length. Theoretically, the
length is 12 to 15 nucleotides when the target is 520 kilobases in length. However, it is
shown that a probe needs to be 24 nucleotides in order to possess a 90% probability of
being unique. Therefore, using longer "short" probes (i.e., 24-36 nucleotide lengths) in
hybridization assays that need to be specific is highly desirable. The methods and
compositions presented here substantially aid in the use of long oligonucleotide probes
(i.e., 24-36 nucleotide lengths) in terms of discrimination.
3. Amplification reactions.
The observation that ~Td does not change as a function of concentration
of hybotrope has substantial utility for use in DNA, RNA or nucleic acid amplifications
based on primer extension by a polymerase (e.g., polymerase chain reaction, see U.S.
Patent Nos. 4,683,195; 4,683,202; and 4,800,159, cycling probe technology, NASBA),
ligation (LCR, ligation chain reaction), and RNA amplification (see Lizardi etal.,
Bio/Technology 6:1197, 1988, Kramer et al., Nature 339:401, 1989; Lomeli et al., Clin.
Chem. 35:1826, 1989; U.S. Patent No. 3,786,600). The observation that wt and mt 30-
mer oligonucleotides can be distinguished on the basis of thermal melting in 0.5 M
LiTCA permits the possibility of a substantial improvement in priming efficiency in
PCR. In its current configuration, the PCR buffer is optimized for the polymerase
rather for specific priming. That is, conditions have evolved since the introduction of
the technique that favor performance of the polymerase over the performance of
specificity of priming with oligonucleotides. Thus, PCR buffer as currently
commercially available does not provide or support a high level of stringency ofhybridization of PCR primers.
Commercially available PCR buffers are examined with respect to the
melting behavior of 24-mer oligonucleotides in both the wild-type (wt) and mutant (mt)
forms. In Table 5, the level of discrimination achieved in PCR buffer versus a low
molarity concentration of hybotrope is shown.
.. . .
CA 02266847 1999-03-22
W O 98/13S27 PCTrUS97J17413
Table 5 ~Td for PCR buffers and low molaritY hybotropes
Solution Conc.Oligo Length HCT' ~Td~ Td,
PCR buffer lx 24-mer wt 15 61
PCR buffer lx 24-mer mt 14 1 60
LiTCA 0.1 M24-merwt 12 65.5
LiTCA 0.1 M24-mermt 8 4 61.5
* =~C
As shown, the HCT for standard PCR buffer is about 15~C, whereas the
HCT for 0.1 M LiTCA is about 12~C. The ~Td for the lx PCR buffer is only 1~C for5 the 24-mer, whereas the ~Td for 0.1 M LiTCA is 4~C. Therefore, priming specificity is
significantly improved in 0.1 M LiTCA versus IX PCR buffer. Higher concentrations
of hybridization solutions may also be used (0.1 M LiTCA to 3.0 M LiTCA or 0.1 to
3.0 TMATCA).
Alternatively, priming is performed in a hybotrope solution and chain
10 extension is performed in a separate buffer that supports the polymerase. For example,
a solid phase PCR could be employed where the solid phase is moved through two
solutions. Priming would occur in some apl)lopl;ate concentration of LiTCA or
TMATCA and then the polymerase chain reaction would take place in a different PCR
buffer cont~ining the polymerase. It is also possible to conduct the first few rounds in
15 the amplification in a hybotrope based hybridization solution and conducting the
rem~ining rounds on normal PCR buffer (generally, only the first few rounds are
important for specificity).
The use of deoxyNebularine modified oligonucleotides also increases the
specificity of priming in the PCR. One deoxyNebularine substitution incorporated into
20 an oligonucleotide reduces the HCT by 2.5~C. Two oligonucleotides probes cont~inin~;
3 deoxyNebularine sites per 24-mer decrease HCT by 8~C relative to the unsubstituted
control. This decrease in the HCT dramatically increases the level of specificity of
priming in an amplification reaction (e.g, polymerase chain reaction). This is likely
due to the reduction of false or mis-priming during the first few (e.g., 10) cycles of
25 PCR. That is, the enthalpy of the deoxyNebularine substituted oligonucleotideincreases relative to the unsubstituted primer, thus increasing the specificity of priming.
Within the context of this invention, the primer is preferably 6 to 36 bases in length and
contains I to 6 deoxyNebularine sites. The sites are preferably separated by 4, 5, 6, 7 or
8 nucleotides and may be separated by up to 12 to 24 nucleotides. The substitutions are
CA 02266847 1999-03-22
W O 98/13527 PCT~US97117413
51
also preferably clustered at the 3' end of the primer to ensure specificity of primer
extension by nucleic acid polymerases, which may be, for example, DNA or RNA
primers. Moreover, the temperature range over which priming occurs is dramatically
reduced when deoxyNebularine-substituted primers are used.
As shown in the examples (see Example 8), the temperature range in
which amplifications are observed is decreased from about 25~C - 65~C to about 25~C -
35~C. In addition, this decrease is observed for two different DNA polymerases.
The results also indicate that the dSpacer substitution prevents the
polymerase from "reading through" the abasic site. That is, when the polymerase
10 encounters an abasic residue, chain extension is termin~te(l However, unlike abasic
residues, a deoxyNebularine residue does not terminate chain extension. Although, as
noted above7 the tclllpc~dlllre range over which the amplification range is much reduced
compared to non-substituted oligonucleotides. Therefore, deoxyNebularine substituted
primers can substantially increase the specificity of a DNA polymerase chain reaction.
Furthermore, the combination of an deoxyNebularine site in an
amplification PCR primer and a hybotrope salt solution, which promotes a high
enthalpy value for the primer duplex, ~i~nific~ntly lowers the HCT of the primerduplex. As discussed above, when the HCT decreases, the stringency factor increases
and high-discrimination priming of the polymerase chain reaction can take place. These
20 are conditions required for favorable multiplexing PCRs. The term multiplexing refers
to the ability to use more than one set of primers in a PCR reaction and generate
multiple products or the ability to use more than one target nucleic acid per set of PCR
primers.
The use of the hybotrope tetramethylammonium trichloroacetate is of
25 particular utility because the dependence of G+C content on Td (stability) is neutralized.
However, other hybotropes of the present invention which may be used in the
polymerase chain reaction include, without limitation, bis(2-methoxyethyl)amine
acetate, 1-ethylpiperidine acetate, l-ethylpiperidine trichloroacetate, l-ethylpiperidine
trifluoro~cet~te, I-methylimidizole acetate, 1-methylpiperidine acetate, 1-
30 methylpiperidine trichloroacetate, I-methylpyrrolidine acetate, 1-methylpyrrolidine
trichloroacetate, 1-methylpyrrolidine trifluoroacetate, 2-methoxyethylamine acetate,
N,N-dimethylcyclohexylamine acetate, N,N-dimethylcyclohexylamine trifluoroacetate,
N,N-dimethylcyclohexylamine, N,N-dimethylheptylamine acetate, N,N-
dimethylheptylamine acetate, N,N-dimethylhexylamine acetate, N,N-
35 dimethylhexylamine acetate, N,N-dimethylisopropylamine acetate, N-ethylbutylamine
acetate, N-ethylbutylamine trifluoro~cet:~te, N,N-dimethylaminobutane trichloroacetate,
CA 02266847 1999-03-22
W O98/13527 PCT~US97/17413 52
N,N-dimethylisopropylamine trichloroacetate, triethanolarnine acetate, triethylamine
acetate, triethylamine trichloro~çet~t~, tripropylamine acetate, tetraethylarnrnonium
acetate. These compounds or chemicals can be combined in amplification reaction with
divalent cations such as Mg++, buffers, detergents, co-factors, nucleotides and their
5 analogs, polymerases and/or ligases. The compounds listed above can be used inconcentration ranging from 5 mM to 6 M, preferably from 100 mM to 2.5 M.
D. HYBOTROPES AND ABASIC NUCLEIC ACIDS IN ARRAYS
In the fields of molecular biology and microbiology, it has long been
common to employ solid supports having biomolecules imnlobilized thereon.
10 Immobilization provides various advantages, such as, allowing for multiplexing of
samples and ready measurements of tags employed in a large number of signal systems.
Recently, intense attention has focused on creating arrays of
biomolecules, and particularly polynucleotides, on a flat solid support. The following
publications (and the references cited therein), which are exemplary only, provide
15 general and specific overviews of various utilities for these biomolecular arrays, as well
as methods of E,le~)a~ g such arrays: M.D. Eggers et al., Advances in DNA Sequencing
Technology, SPIE 1891:113-126, 1993; A.B. Chetverin et al., Bio/Technology 12:1093-
1099, 1994; E.M. Southern, Nucleic Acids Research 22: 1368-1373, 1994; R.J. Lipshutz
et al., BioTechniques 19:442-447, 1995; M~ Schena, BioEssays 18:427-431~ 1996; A.P.
20 Blanchard et al., Biosensors & Bioelectronics I 1 :687-690, 1996; M.J. O'Donnell-
Maloney et al., Genetic Analysis Biomolecular Engineering 13:151-157, 1996; A
Regalado, Start-Up 24-30, Oct. 1996; and D. Stipp, ~ortune 30-41, March 31, 1997.
The advent of large scale genomic projects and the increasing medical
use of molecular diagnostics, has prompted the development of large volume
25 throughput methods for screening recombinant DNA libraries representing entire
genomes, the performance of large scale DNA sequencing projects, and executing
replicative immunological assays, nucleic acid hybridization assays, or polymerase
chain reaction assays. The following publications (and the references cited therein),
which are exemplary only, provide general and specific overviews of large throughput
30 methods that rely on biomolecular arrays, as well as methods of preparing such arrays:
M.D. Eggers et al., Advances in DNA Sequencing Technology, SPIE 1891:113-126,
1993; A.B. Chetverin et al., Bio/Technology 12:1093-1099, 1994; E.M. Southern,
Nucleic Acids Research 22:1368-1373, 1994; R.J. Lipshutz et al., BioTechniques
19:442-447, 1995; M. Schena, BioEssays 18:427-431, 1996; A.P. Blanchard et al.,
35 Biosensors & Bioelectronics 11:6X7-690, 1996; M.J. O'Donnell-Maloney et al., Genetic
CA 02266847 1999-03-22
WO 98/13527 PCTrusg7/l74l3
53
Analysis. Biomolecular Engineering 13:151-157, 1996; A. Regalado, Start-Up 24-30.
Oct. 1996; and D. Stipp, Fortune 30-41, March 31, 1997.
The need for high throughput methodology has led, in some cases, to a
~ change from a 96-well microtiter dish format, to a 384-well (Maier et al., J.
Biotechnology 35:191, 1994) or 864-well (Drmanac et al., Electrophoresis 13:120,1992) format, which can also be used in conjunction with robotic devises (see, e.g.,
Belgrader et al., BioTechniques 19.426, 1995; Wilke et al., Diagnostic Microbiology
and Infect. Disease 21: 181, 1995). However, all of these automated techniques require
the use of a robotic pin-tool devise that is capable of reproducibly transferring equal
volumes of liquid from one arrayed configuration (i.e., 96-well microtiter plate) to
another (i. e., 96-spot array on a hybridization filter membrane).
Recently, methods have also been developed to synthesize large arrays
of short oligodeoxynucleotides (ODNs) bound to a glass surface that represent all, or a
subset of all, possible nucleotide sequences (Maskos and Southern, Nucl. Acids Res.
20:1675, 1992). Once such an ODN array has been made may be used to perform DNA
sequencing by hybridization (Southern et al., Genomics 13.1008, 1992; Drmanac et al.~
Science 260:1649, 1993). The utility of this method of DNA sequencing would be
greatly improved if better methods existed for the transfer and arraying of the precise
amounts of the biochemical reagents required for the synthesis of large sets ODNs
bound to hybridizable surfaces. This would enable greater equality of ODN yield at
each position within the array and also increase the nucleotide chain length it is possible
to synthesize.
The polymerase chain reaction (PCR) has found wide application to
many different biological problems. Two major limitations to the commercial
utilization of PCR are the high cost of the reagents and the inability to automate the
performance of the process. Reagent costs can be lowered if the total volume of each
reaction can be decreased~ allowing a concomitant decrease in DNA polymerase andnucleotides. An accurate and reliable means to array small volumes of reagents using a
robotically controlled pin tool could help solve both of these PCR problems.
The combination of hybotropes and modified oligonucleotide probes
described in this application will permit the useful multiplexing of probes and 'capture '
oligonucleotides in the array format. Hybotropes that neutralize the G+C content effect
on Tm or Td are especially useful in the application and use of array technology. In
traditional hybridization solutions the difference in Tm or Td when the G+C content is
varied from 20% to 80% is generally 12 to 16~C. Therefore is it impossible to maintain
the ideal hybridization temperature which is I to 8 degrees below the Tm of the
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
54
respective oligonucleotide duplex as the G+C content is varied. Solutions (hybotropes)
which neutralize the effect of G+C on T", or Td perrnit the useful multiplexing of probes.
Hybotropes such as bis(2-methoxyethyl)amine acetate, 1-ethylpiperidine acetate, 1-
ethylpiperidine trichloroacetate, 1-ethylpiperidine trifluoroacetate, 1-methylimidizole
acetate, 1-methylpiperidine acetate, I-methylpiperidine trichloroacetate, l-
methylpyrrolidine acetate, I-methylpyrrolidine trichloroacetate, 1-methylpyrrolidine
trifluoro~cet~te, 2-methoxyethylamine acetate, N,N-dimethylcyclohexylamine acetate,
N,N-dimethylcyclohexylamine trifluoroacetate, N,N-dimethylcyclohexylamine, N,N-
dimethylheptylamine acetate, N,N-dimethylheptylamine acetate, N,N-
dimethylhexylamine acetate, N,N-dimethylhexylamine acetate, N,N-
dimethylisopropylamine acetate, N-ethylbutylamine acetate, N-ethylbutylamine
trifluoroacetate, N,N-dimethylaminobutane trichloroacetate, N,N-
dimethylisopropylamine trichloroacet~tç, triethanolamine acetate, triethylamine acetate,
triethylamine trichloro~ret~te~ tripropylamine acetate, tetraethylammonium acetate are
useful in array formats.
A number of genetic diseases are caused by single, or a limited set, of
mutations due to founder effects or advantages to heterozygous carriers. There is an
increasing clinical interest in monitoring sequence variants associated with, for
example, altered metabolism of drugs or serving as genetic markers in forensic
medicine, and in the diagnosis of in~ectious disease, identification of drug-resistant
variant strains may require distinction between similar sequence variants.
The solutions described herein are used to increase the specificity of
priming in the PCR. There are several options in terms of a mechanism in which the
specificity of the priming step can be improved. The first is a through the use of a solid
support to which one of the PCR primers is (covalently) attached. The solid support
can take many forms such as beads, membranes, etc. The priming step can take place in
the hybotrope and then the solid support can be washed and moved into a solution that
supports the polymerase chain extension. The solid support is then moved back into the
nesstrope for the priming reaction and the cycle is repeated. The cycling of the solid
support between the two dirl~lel~t solutions only has to occur to a limited number of
times (1-15 cycles) after which time the traditional amplification cycle in a standardized
PCR buffer can be allowed proceed. Alternatively, the target nucleic acids of interest
are moved between the priming solution and the polymerase extension reaction solution
using electric fields (i.e., electrophoresis).
The use of hybotropes andlor abasic or anucleosidic oligonucleotide
probes can be used to increase the specificity and efficiency of isothermal applications
CA 02266847 1999-03-22
W O 98/13527 PCT~US97/17413
of polymerases to the arnplification of nucleic acid sequences. Applications of
isothermal conditions for using nucleic acid polymerases include nucleic acid
sequencing, genotyping, mutation detection, oligonucleotide ligation assays, mutation
detection, and the like.
s
The following examples are offered by way of illustration, and not by
way of limitation.
CA 02266847 l999-03-22
W O98/13527 PCTAUS97/17413
56
EXAMPLES
EXAMPLE 1
PREPA~ATION, PROPERTIES, AND USES OF NOVEL HYBOTROPES
A novel hybotrope is synthesized which demonstrates properties not
previously described for a salt solution. Tetramethyl ammonium- and tetraethyl
ammonium-trichloroacetate are synthesized by neutralizing tetramethyl ammonium-
and tetraethyl ammonium-hydroxide with trichloroacetate to pH 7.0 to pH 8.5,
10 depending upon the application. The resulting salt solution is then dried under vacuum
to complete dryness and the mass is determined. The salt is then dissolved in water to a
final concentration of 0.5 to 3.0 M. The resulting salt solution is then buffered with a
buffer such as Tris-HCI, pH 7.0-8.5, and detergents, such as sarkosyl, are added to
about 0.1%, and optionally EDTA is added to 0.5 to 5 mM.
These hybotropes possess the property of neutralizing the differences in
G+C and A+T base-pairing strength while simultaneously lowering the Td and ~Td,
increasing ~Td. In the table below the characteristics of the novel TEATCA and
TMATCA hybotropes.
20 25% G+C content: 5'-AAATAATTCAGGGTCAAAA-3'
36% G+C content: 5'-CTGTCGTAGGTAAATAACT-3'
42% G+C content: 5'-AAAAAGTGGGGAAGTGAGT-3'
57% G+C content: 5'-GTGTTAACTTCCGCTCCTC-3'
63% G+C content: 5'-GGCGTAGGTCTGTCGTGCT-3'
25 73% G+C content: 5'-GGTGTGGGTCCGTCGTGCC-3'
CA 02266847 1999-03-22
W O 98113527 PCTAUS97/17413
57
The following Tds are obtained in the hybridizations described below:
Table '
Solution Type Length of Probe G+C Content Td HCT
3 M TEATCA 19-mer 25% 38.0 7.5
3 M TEATCA 19-mer 36% 38.5 10
3 M TEATCA 19-mer 42% 39 11.5
3 M TEATCA 19-mer 57% 40 12
3 M TEATCA 19-mer 63% 41 13
3 M TEATCA 19-mer 73% 42 14
3 M TMACI l9-mer 25% 61 12.5
3 M TMACI 19-mer 36% 62 14
3 M TMACI 19-mer 42% 60 15.5
3 M TMACI l9-mer 57% 65 17
3 M TMACI 19-mer 63% 59 17.5
3 M TMACI 19-mer 73% 59 17.5
3 M TMATCA 19-mer 25% 44.5 8
3 M TMATCA 19-mer 36% 45.5 10
3 M TMATCA 19-mer 42% 43 11.5
3 M TMATCA l9-mer 57% 48.5 12
3 M TMATCA l 9-mer 63% 47 13
3 M TMATCA 19-mer 73% 48.5 14
2 M TMATCA 19-mer 25% 43 15
2 M TMATCA 19-mer 36% 44.5 17
2 M TMATCA l9-mer 42% 44.5 18
2 MTMATCA l9-mer 57% 53 19.5
2 MTMATCA l9-mer 63% 48 19.5
2 M TMATCA l9-mer 73% 52 19
30% i;olmalllide 19-mer 25% 25 20
30% formamide l9-mer 36% 27.5 20
30% formamide l 9-mer 42% 29 20
30% formamide l9-mer 57% 40 21
30% formamide 19-mer 63% 37.5 22
30% formamide l9-mer 73% 40 23
CA 02266847 1999-03-22
W O 98/13527 PCT~US97/17413
58
The data in Table 6 clearly indicate a decrease in the helical coil
transition in solutions cont~ining 3 M TMATCA or 3 M TEACl compared to the
control solution which is TMACL. An average decrease of 3.5~C is observed for
5 solutions cont~inin~ 3 M TMATCA or 3 M TEACI compared to the control solution
which is TMACL. Also, formamide has a surprisingly high value for the helical coil
transition, which ranges from 20 to 23~C depending on the G+C value. Also shown is
the concentration dependence of the ability of a TMATCA solution to neutralize G+C
content. At 2 M, TMATCA is neither able to neutralize G+C content or reduce the
10 HCT. This property is unique as no other hybotrope displays a concentration
dependence.
EXAMPLE 2
DETERMINATION OF THE MELTING TEMPERATURE OF OLIGONUCLEOTIDE DUPLEXES IN
VARIOUS HYBOTROPE AND NoN-HYsoTRoPE BASED HYBRIDIZATION SOLUTIONS.
This example describes the determination of the Td of wild type and
mutant oligonucleotides when hybridized to a target nucleic acid. It is shown that
hybotrope based hybridization solutions allow the detection of single base pair
20 mutations in a nucleic acid target with a probe up to a 30 nucleotides in length.
Solutions and Rea~ents
Filter wash (FW) is 0.09 M NaCI, 540 mM Tris pH 7.6, 25 mM EDTA.
SDS/FW is FW with 0.1% sodium dodecyl sulfate (SDS). Hybridization solutions
25 contain the text specified concentration of hybotrope 2% N-lauroylsarcosine (sarcosyl),
50 mM Tris pH 7.6 and 25 mM EDTA. Formamide hybridization solution contains
30% formamide, 0.09 M NaCI, 40 mM Tris-HCI pH 7.6, 5 mM EDTA and 0.1% SDS.
GuSCN is purchased from Kodak (Rochester, NY). GuCI, lithium hydroxide,
trichloroacetic acid, NaSCN, NaCI04 and KI, are purchased from Sigma (St. Louis,30 MO). Rubidium hydroxide is purchased from CFS Chemicals (Columbus, OH).
CsTFA is purchased from Pharmacia (Piscataway, NJ).
Plel)alalion of LiTCA~ TMATCA and TEATCA
LiTCA and TMATCA, and TEATCA are prepared by the dropwise
titration of a 3 N solution of LiOH, TEAOH and TMAOH respectively, with
CA 02266847 1999-03-22
W~ 98/13527 PCT/US97/17413
59
trichloracetic acid (100% w/v, 6.1 N) to pH 7.0 on ice with constant stirring. The salt is
evaporated to dryness under vacuum, washed once with ether and dried.
- Oli~onucleotide SYnthesis
Oligonucleotides are synthPsi7~d on a commercial synthesizer using
5 standard cyanoethyl-N,N-diisorpropylamino-phosphoramidite (CED-phosphoramidite)
chemistry. Amine tails are incorporated onto the S'-end using the commercially
available N-monomethoxytritylaminohex-6-yloxy-CED-phosphoramidite.
Alternatively, oligonucleotides are commercially purchased.
Preparation of Nylon Bead Supports (ODN-Bead)
ODN (oligonucleotide)-beads (3/32nd inch diameter) are prepared as
previously described (Van Ness et al., Nucl. Acids Res. 19:3345, 1991). The ODN-beads contain 0.01 to 1.2 mg/bead of covalently immobilized ODN.
Determination of Td and T~r~ Values Using ODN-Beads in Various HYbridization
Solution Containin~ H~botropic Salts
To label the probe oligonucleotides, amine ODNs are reacted with
amine-reactive fluorochromes. The derived ODN plep~dtion is divided into 3 portions
and each portion is reacted with either (a) 20-fold molar excess of Texas Red sulfonyl
chloride (Molecular Probes, Eugene, OR), with (b) 20-fold molar excess of Lissamine
sulfonyl chloride (Molecular Probes, Eugene, OR), or (c) 20-fold molar excess of20 fluorescein isothiocyanate. The final reaction conditions consist of 0.15 M sodium
borate at pH 8.3 for 1 hour at room tt;~ Lule. The unreacted fluorochromes are
removed by size exclusion chromatography on a G-50 Sephadex column.
For the determination of ODN/ODN Td from the ODN-bead,
fluorescently-labeled ODN is incubated in various hybridization solutions with a25 complementary ODN immobilized on ODN-beads. From 5 to 5000 ng of ODN are
hybridized in 300-400 ~11 volumes at various temperatures (19-65~C) for 5-30 minutes
with constant agitation. The beads are washed with 3 x 1 ml of the respective
hybridization solution, and then once with the respective melting solution at the starting
temperature of the melting process. The beads in 300-400 ~1 of the respective melting
30 solution are then placed in a 0-15~C water bath. At 5 minute intervals, the temperature
is raised 5~C, the solution dec~ntPd into a well of a microtiter plate, and fresh solution
(5~C below the next increment) is added to the beads. The "melting" or duplex
CA 02266847 1999-03-22
W O 98/13527 PCT~US97/17413
dissociation is conducted over a temperature range of 15~C to 95~C. Fluorescence is
measured with a commercial fluorescence plate reader.
To calculate the Td, cumulative counts eluted at each temperature are
plotted against temperature. The temperature at which 50% of the material is
5 dissociated from the bead is the Td.
For the determination of RNA/ODN or DNA/ODN Td mems from nylon
membranes (Schleicher & Schuell, Keene, N.H.), 32P-labeled ODN (3'-labeled with
t~rrnin~l transferase) is incubated with 0.5 cm2 pieces of membrane, in text-specified
hybridization solutions. For the (non-covalent) immobilization of genomic DNA onto
10 nylon membranes, purified DNA is denatured in 0.3 M NaOH at 20~C for 10 minutes.
An equal volume of 2 M ammoniurn acetate is added and the sample was applied to
Nytran membranes assembled in a slot blot al,pa,dlus. RNA was denatured in 4.6 Mformaldehyde-6 x SSC (0.9 M NaCl, 90 mM sodium citrate) for 15 min. at 60~C and
applied to the membranes as above. After immobilization of the nucleic acids, the
15 filters were baked at 80~C for 2 hours, then stored dry at arnbient temperature. The
hybridizations and dissociations were then performed as described above for the nylon
bead solid supports.
To determine the TnptODN (the temperature at which the maximum rate
of hybridization of target nucleic acid to ODNs occurs, under near stringent to stringent
20 conditions (-20 to -5~C below the Td)), complementary 32P-labeled ODN is hybridized
(to the Cot,") to either covalently immobilized ODN sequences on the ODN-bead asdescribed above, or in a sandwich assay forrnat when RNA is used as the target nucleic
acid. The hybridizations are performed over a 40~C range (+ or -20~C around the Td of
the respective duplex in 5~C increments). The extent of hybridization is then measured
25 as a function of temperature at the Cot,,, of the respective hybridization.
Spectroscopic Thermal Denaturation
Thermal transitions determined in solution (T",) are recorded at 260 nm
using a Gilford System 2600 UV-VIS spectrophotometer equipped with a Gilford 2527
Thermo-programmer. ODNs (2 mM/strand) are dissolved in the respective
30 hybridization or melting solutions. The ODN mixtures were heated to 85~C, then
cooled to 10-15~C to allow hybridization. The samples were slowly heated to X5~Cemploying a temperature increase of 0.5~C/min. Absorbance versus time is recorded,
and the first derivative is computed automatically. The Tm values are determined using
the first derivative maxima.
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
61
Oli~onucleotides
The following oligonucleotides are used to measure the difference in Td
between a wild type oligonucleotide and a mutant oligonucleotide. The wild type
oligonucleotide represents fully and perfectly base-paired duplex and a mutant
5 oligonucleotide represents a single base pair mism~tch (generally in the middle of the
oligonucleotide).
The sequence of the "capture" oligonucleotide is 5'-
GTCATACTCCTGCTTGCTGATCCACATCTG-3'. The sequence of the wild type 30-
mer is 5'-CAGATGGGTATCAGCAAGCAGGAGTATGAC-3', the sequence for the
10 wild type 24-mer 5'-ATGGGTATCAGCAAGCAGGAGTAT-3', the sequence for the
wild type 18-mer 5'-GGTATCAGCAAGCAGGAG-3'. The sequence of the mutant 30-
mer is 5'-CAGATGGGTATCAGGAAGCAGGAGTATGAC-3', the sequence for the
mutant 24-mer 5'-ATGGGTATCAGGAAGCAGGAGTAT-3', the sequence for the
mutant 18-mer 5'-GGTATCAGGAAGCAGGAG-3'.
The helical coil transition of an oligonucleotide or nucleic acid duplex
can be measured by essentia}ly an adaptation of methods previously described by
Martinson (Biochemistry 12:145-165, 1973) for the thermal elution of DNA or RNA
duplexes or hybrids from hydroxylapatite. For the determination of the helical coil
transition from a solid support, fluorescently-labeled oligonucleotide (ODN) was20 incubated in various hybridization solutions with a complementary ODN immobilized
on ODN-beads. From 5 to 5000 ng of ODN were hybridized in 300-400 ~11 volumes atvarious temperatures (19-65~C) for 5-30 minutes with constant agitation. The beads
were washed with 3 x 1 ml of the respective hybridization solution, and then once with
the respective melting solution at the starting t~lllpeldlure of the melting process. The
25 beads in 300-400 ~11 of the respective melting solution were then placed in a 0-15~C
water bath. At 5 minute intervals, the temperature was raised 5~C, the solution decanted
into a well of a microtiter plate, and fresh solution (5~C below the next increment) was
added to the beads. The "melting" or duplex dissociation was conducted over a
temperature range of 15~C to 95~C. Fluorescence was measured with a commercial
30 fluorescence plate reader. To calculate the Td, cumulative counts eluted at each
temperature were plotted against temperature. The telllpel~Lule at which 50% of the
material had been dissociated from the bead was taken as the Td. The helical coil
transition is defined as the temperature at which a value of a equals 0.2 for a given
oligonucleotide duplex (or nucleic acid duplex, cont~ining or not containing a mism~tch
35 at any place in the duplex) to the tempeld~uie at which a value for a equals 0.8 for the
same given oligonucleotide duplex (or nucleic acid duplex).
CA 02266847 1999-03-22
W O 98/13527 PCTr~S97/17413
62
The following Tds are obtained in the hybridizations described below:
Table 7
Td Td
Length of(Mutant)(Wild Type) ~Td HCT
SolutionProbe (~C) (~C) (~C) (~C)
Type
2.5 m 30-mer27 33 6 13/14
LiTCA
2.5 m 24-mer25.5 326.5 13/14.5
LiTCA
2.5m18-mer 24 31 7 9/14
LiTCA
2.0 m 30-mer42 47 5 13.5/16
LiTCA
2.0 m 24-mer38 44 6 14/15
LiTCA
2.0 m 18-mer37 43 6 14.5/16.5
LiTCA
3.0 m 30-mer37 42.5 5.5 13.5/17.5
GuSCN
3.0 m 24-mer34.5 416.6 12.5/17
GuSCN
3.0 m 18-mer33.5 40.5 7 14.5/15
GuSCN
3.0 m 30-mer55.5 604.5 16/21
GuHCI
3.0 m 24-mer52.5 585.5 15/20
GuHCI
3.0 m 18-mer50 57 7 18/20
GuHCI
Rapid Hybe 30-mer 8080 0 na*
Rapid Hybe 24-mer 8080 0 na
Rapid Hybe 18-mer 6870 2 18/23
5x SSC 30-mer72.5 72.5 0 18/18
5x SSC 24-mer69 70 1 18/18
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
63
Td Td
Length of (Mutant)(Wild Type) ~Td HCT
Solution Probe (~C) (~C)(~C) (~C)
Type
5x SSC 18-mer 67 72 5 16/18
Promega QY30-mer80 80 0 na
Promega QY24-mer80 80 0 na
Promega QY18-mer62 65 3 20/23
* na indicates not applicable or too large to
accurately deterrnine.
The data indicate that the hybotropic solutions (LiTCA. GuSCN and
GuHCI) permit the detection of a single base-pair mi~mAtch in a 24-mer and 30-mer
probe whereas the detection of a single base-pair mi~mAt~h in standard hybridization
5 solutions (Rapid Hybe, Promega QY or 5x SSC) is not possible.
A similar experiment is performed for the 24-mers described above in a
series of hybridization solutions.
Table 8
Hybridization Solution Type Slope ([..], k) HCT ~Td
LiTCA,3M l9 8C 7.5 C
GuSCN,3M 13 10 6.0
NaSCN,3 M 8.5 ll 5.5
NaClO4, 3 M 7 12 4.5
KI,3M 5 15 3.0
NaCl, 0.165 M 4.5 17.5 1.5
GuCI,3M 3.5 18 1.2
CsTFA, 2M 2.5 18 1.2
30% formamide ND 20 1.5
Td(wt) is the Td of a perfectly base-paired oligonucleotide duplex and
Td(mt) is the Td of a oligonucleotide duplex Cont:~ining a single mi~mAtch. The values
are for a 24-mer duplex of sequence described in Example l. From the data presented
CA 02266847 1999-03-22
W O 98/13527 rcTrusg7/17413
64
in the table above, the stringency factor is directly proportional to the difference
between a perfectly base paired duplex and a duplex cont~inin~ a mism~tch. That is,
the stringency factor predicts the ability of given hybridization solution to discriminate
mi~m~tched duplexes.
EXAMPLE 3
EFFECT OF CONCENTRATION OF SALT OR HYBOTROPE ON HCT AND TD
The discrimination between mi~m~tched oligonucleotides (mutant
10 abbreviated as "mt") and perfected based-paired oligonucleotides (abbreviated as "wt")
is not a function of concentration of a particular hybotrope but rather a function of
hybotrope type. HCT is defined as the temperature range over which a duplex melts
during a melting process under defined conditions. To calculate HCT, the temperature
at which 80% of the duplexes are melted is subtracted from the temperature at which
15 20% melting is observed. Surprisingly, for the hybotropes LiTCA, GUSCN, GUHC1,
NaClO4 the HCT does not change over about the range of 0.5 M to about 6.0 M. Theslope of the mt duplex is always observed to be greater than for wt duplexes (see
Table 8). Another parameter which does not change as a function of concentration is
the difference between the Td ~f the wt duplex and the mutant duplex (~Td). The Td of
20 the mt and wt duplexes is observed to be strictly dependent on concentration in a
precisely linear relation. In Table 9, the HCT and Td for mt and wt 30-mer duplexes
and mt and wt 18-mers are presented.
The effect of concentration and hybotrope type on HCT, ~Td, and Td.
Table 9
Oligo
Hybotrope Conc.Length HCT ~Td Td
GUSCN 0.5 M24-mer wt 12.5 C 70 C
GUSCN 0.5 M24-mer mt 10.0 C 4.0 C 74
GUSCN 1.0 M24-mer wt 12.5 C 65
GuSCN 1.0 M24-mt mt l 0.0 C3.5 C 68.5
GUSCN 2.0 M24-mer wt 12.5 C 52
GUSCN 2.0 M24-mer mt 10.0 C 4.0 C 56
GuSCN 2.5 M24-mer wt 12.5 C 46.5
GUSCN 2.5 M24-mer mt 12.5 C 3.5 C 50
CA 02266847 1999-03-22
W O 98/13527 PCT~US97/17413
The data from Table 9 is graphically represented in Figure 9.
Because ~Td does not change over a wide concentration range for the
hybotropic solutions described above, a wide temperature range can be employed for
5 conducting oligonucleotide-based assays (i.e., 20 to 80~C). In addition, relatively low
concentrations (e.g, 0.5 M) of oligonucleotide can be employed in assays and
polymerase based assays.
EXAMPLE 4
DETECTION OF A SINGLE BASE-PAIR MISMATCH ON A SOLID PHASE.
This example describes the detection of a single-base pair mi~m:~tch in
an immobilized probe using complementary fluorescently labeled oligonucleotides.The set of probe oligonucleotides consists of one probe which forms perfect base-
15 pairing and one oligonucleotide which contains the mi~m~tch when hybridized. Thetwo oligonucleotides are labeled with different fluorochromes, and after hybridization is
allowed to occur at the Td of the mi.cm~tch, the ratio of hybridized fluorochromes is
determined.
A "target" oligonucleotide (DMO50 1: 5'-
20 TTGATTCCCAATTATGCGAAGGAG-3') was immobilized on a set of solid supports.
ODN-beads (3/32nd inch diameter) were prepared as previously described (Van Nesset al., Nucl. Acids Res. 19:3345, 1991). The ODN-beads contained 0.01 to 1.2 mg/bead
of covalently immobilized ODN. DMO578 is the complement to DMO501 (perfect
complement). DMO1969 is the complement to DMO501 with a G--->T change at
25 position 11. DMO1971 is the complement to DMO501 with a A--->T change at
position 12. Each probe oligonucleotide was labeled with either BODIPY, TAMRA orTexas Red. Hybridization reactions were assembled in 3 M GuSCN, 0.01 M Tris pH
7.6, 5 mM EDTA at 50 ng/ml respective probe. Equal molar ratios of each probe type
were used in each hybridization in the presence of 3 solid supports per tube.
30 Hybridizations are performed at 42~C for 30 minutes with constant agitation. The
beads were washed twice with 3 M GuSCN at 42~C and then with SDS/FW 5 times.
To denature the probe oligonucleotide, the solid supports are placed in
200 ~11 TE (TE is 0.01 M Tris, pH 7.0, 5 mM EDTA). The mixture is incubated for 10
mimltes at 100~C. Fluorescence is measured in a black microtiter plate. The solution is
35 removed from the incubation tubes (200 microliters) and placed in a black microtiter
plate (Dynatek Laboratories, Chantilly~ VA). The plates are then read directly using a
CA 02266847 1999-03-22
W Og8/13527 PCTrUS97tl7413
66
Fluoroskan II fluorometer (Flow Laboratories, McLean, VA) using an excitation
wavelength of 495 nm and monitoring emission at 520 nm for fluorescein, using anexcitation wavelength of 591 nm and monitoring emission at 612 nm for Texas Red,and using an excitation wavelength of 570 nm and monitoring emission at 590 nm for
5 li~mine or TAMRA.
The results are as follows:
Table 10
Fluorochrome ratio in Fluorochrome ratio after
Probe Mixhybridizationmix denaturing
578TR/578BD 1.9/1 1.9/1
57~TR/1969BD 2.0/1 25/1
578TR/1971TA 0.025/1 0.58/1
578BD/1971TA 0.014/1 0.48/1
The results indicate that there is no effect of the fluorochrome on the
10 hybridization as indicated in line I that Texas Red (TR) 578 oligonucleotide and 578-
BD (BODIPY) competed evenly for hybridization to the immobilized target since the
ratio of labels did not change after hybridization. There is an average of a 20-fold
enrichment of perfectly based probes over the mi.~m~tçlled probes in GuSCN allowing
certain detection of base-pair mi~m~tçhes
EXAMPLE 5
DETERMINATION OF THE HELICAL COIL TRANSITIONS IN PCR
AND LOW MOLARITY HYBOTROPE SOLUTIONS.
The observation that ~Td does not change as a function of concentration
of hybotrope has substantial utility for uses in DNA, RNA or nucleic acid
amplifications based on primer extension by polymerases (i.e., polymerase chain
reaction). The observation that mi~m:lt~l~ed probes as long as 30-mer oligonucleotides
can be distinguished on the basis of thermal melting in 0.5 M LiTCA, perrnits the
possibility of a substantial improvement in priming efficiency in the PCRS. In its
current configuration, the PCR buffer is optimized for the polymerase rather for specific
priming.
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
67
Commercially available PCR buffers were exarnined with respect to the
melting behavior of 18-mers, 24-mers and 30-mers in both the wild-type (wt) and
mutant (mt) forms. In Table 11, the level of discnmin~tion achieved in PCR buffer
versus a low molarity concentration of hybokope is presented.
Table 11: ~Td for PCR Buffers and Low Molarit~ H~botropes
HybotropeConc.Oligo Length HCT ~Td Td
PCRbuffer lx 24-merwt 15 C 61 C
PCR buffer1 x 24-mer mt 14 C 1 C60 C
LiTCA 0.1 M 24-mer wt12 C 65.5
LiTCA0.lM 24-merwt 8C 4C 61.5
Note that the HCT for standard PCR buffer is about l 5~C whereas the
HCT for 0.1 M LiTCA is about 12~C. The ~Td for the lx PCR buffer is 1~C for the 24-
mer whereas the ~Td in 0.1 M LiTCA is 4~C. Thus, priming specificity is significantly
improved in a 0.1 M LiTCA versus lX PCR buffer.
EXAMPLE 6
INTRODUCTION OF AN ABASIC SITE INTO AN OLIGONUCLEOTIDE TO INCREASE THE HCT
OF THE OLIGONUCLEOTIDE AND IMPROVE PRIMING SPECIFICITY
It is shown in Example 3 that the introduction of an abasic site or
mi~m~tched site into an oligonucleotide primer will decrease the Td and HCT of the
respective derived primer compared to a perfected based pair "sister" primer. Abasic
sites in polynucleotides of oligonucleotides can be introduced by the chemical or
enzymatic hydrolysis of the glycosidic bond. The resulting structure is apurinic or
apyrimidinic which lacks the coding information and fails to base pair. The CE
phosphoramidite of the tetrahydroduran derivative is commercially available
(dSPACER, Glenn Research, Sterling, Virginia) as well as other spacer
phosphoramidites (Glenn Research, Sterling, Virginia).
Effect of abasic substitution on the HCT of a set of oligonucleotides is
shown in the Table below.
CA 02266847 1999-03-22
W O 98/13527 PCT~US97/17413
68
Table 12
Stringency Factor
BufferType Oligo Type HCT' Td'
1 X PCR buffer norrnal 18
lX PCR buffer abasic (dSPACER) 12
IX PCR buffer abasic (C3 spacer) 12
0.5 M TMATCA normal 14
0.5 M TMATCA abasic (dSPACER) 8
0.5 M TMATCA abasic (C3 spacer) 8
2.0 M LiTCA normal 12.5 44.5 4.97
2.0 M LiTCA abasic (dSPACl~R)10 39 6.37
2.0 M LiTCA abasic (C3 spacer) 10 39 6.25
3.0 M GuSCN normal 16 35.5 3.85
3.0 M GuSCN abasic (dSPACER)12.5 32 5.24
3.0 M GuSCN abasic (C3 spacer) 12.5 31 5.31
* = ~C
The oligonucleotide is a 24-mer with the following sequence: 5'-
hexylamine-TGTGGATCAGCA-spacer-GCAGGAGTATG-3' where the spacer is
5 either the C3-spacer or dSPACER from Glenn Research (Sterling, VA).
EXAMPLE 7
DETECTION OFA SINGLE BASE-PAIR MISMATCH ON A SOLID
PHASE USING ABASIC SUBSTITUTED OLIGONUCLEOTIDES
This example describes the hybridization of an oligonucleotide
containing an abasic site to an irnmobilized oligonucleotide using fluorescent tags. The
set of probe oligonucleotides consists of one probe which forrns perfect base-pairing
15 and one oligonucleotide which contains the an abasic site when hybridized. The two
oligonucleotides are labeled with different fluorochromes, and after hybridization at the
Td of the mi~m~tch, the ratio of hybridized fluorochromes is determined.
A "target" oligonucleotide (DMO501: 5'-
TTGATTCCCAATTATGCGAAGGAG-3') was immobilized on a set of solid
CA 02266847 1999-03-22
W O 98/13527 PCTrUS97/17413
69
supports. ODN-beads (3/32nd inch diameter) were prepared as previously described(Van Ness et al., Nuc. Acids Res. 19:3345, 1991). The ODN-beads contained 0.01 to
1.2 mg/bead of covalently immobilized ODN. DMO578 is the complement to
- DMO501 (perfect complement). DMO1969 is the complement to DMO501 with an
5 abasic site at position 11. DMO1971 is the complement to DMO501 with an abasic site
at position 12. Each probe oligonucleotide is labeled with either BODIPY, TAMRA or
Texas Red. Hybridization reactions were assembled in 3 M GuSCN, 0.01 M Tris p~
7.6, 5 mM EDTA at 50 ng/ml respective probe. Equal molar ratios of each probe type
were used in each hybridization in the presence of 3 solid supports per tube.
10 Hybridizations were at 42~C for 30 minutes with constant agitation. The beads were
washed twice with 3 M GuSCN at 42~C and then with SDS/FW 5 times.
To denature the probe oligonucleotide, the solid supports were placed in
200 ,Lll TE (TE is 0.01 M Tris, pH 7.0, 5 mM EDTA). The mixture is incubated for 10
minutes at 100~C. Fluorescence is measured in a black microtiter plate. The solution is
15 removed from the incubation tubes (200 microliters) and placed in a black microtiter
plate (Dynatek Laboratories, Chantilly, VA). The plates are then read directly using a
Fluoroskan Il fluorometer (Flow Laboratories, McLean, VA) using an excitation
wavelength of 495 nrn and monitoring emission at 520 nm for fluorescein, using an
excitation wavelength of 591 nm and monitoring emission at 612 nrn for Texas Red,
20 and using an excitation wavelength of 570 nm and monitoring emission at 590 nm for
li~s~n~ine or TAMRA.
The results are as follows:
Table 13
Fluorochrome ratio in Fluorochrome ratio after
Probe Mix hybridization mixdenaturing
578TR/578BD 2.1/1 2.1/1
578TR/1969BD 1.8/1 25/1
578TR/1971TA 0.024/1 0.6/1
578BD/1971TA 0.015/1 0.36/1
The results indicate that there is no effect of the fluorochrome on the
hybridization as indicated in line 1 that Texas Red (TR) 578 oligonucleotide and 578-
BD (BODIPY) competed evenly for hybridization to the imrnobilized target since the
ratio of labels did not change after hybridization. There is an average of a 20-fold
-
CA 02266847 1999-03-22
W O 98/13527 PCTrUS97117413
enrichment of perfectly based probes over the abasic modified in GuSCN; allowingmuch higher levels of discrimination in hybridization reactions.
EXAMPLE 8
EFFECTS OF INTRODUCING A DEoxYNEsuLARINE RESIDUE INTO AN
OLIGONUCLEOTIDE PRIMER uSED IN AMPLIFICATION
This example describes the use of oligonucleotide primers substituted
with either abasic or deoxyNebularine residues to increase the specificity of priming in
10 amplification reactions.
The primers used in this experiment are described by Rychlik (Rychlik,
Biotechniques, 18:84-907 1995). Primers may be synthesized or obtained as gel-
filtration grade primers from Midland Certified Reagent Company (Midland Texas).Amplification reactions are either Ta~ DNA polymerase-based (10 mM
15 Tris-HCI pH 8.3, 1.5 mM MgCI2, 50 mM KCI), or Pfu DNA polymerase based (20 mMTris-HCI pH 8.75, 2.0 mM MgCI2, 10 mM KCI, 10 mM (NH4)2SO4, 0.1% Triton X-100,
0.1 mg/ml bovine serum albumin (BSA)). The total deoxynucleoside triphosphate
(dNTPs) concentration in the reactions is 0.8 mM, the primer concentration is 200 nM
(unless otherwise stated) and the template amount is 0.25 ng of bacteriophage lambda
20 DNA per 25 ~11 reaction. The amplification cycles consist of a denaturation step at 94~C
for 5 minutes followed by 30 cycles of: 94~C for 45 seconds, 52~C for 45 seconds, at
72~C for 30 seconds, followed by a single step of 72~C for 5 minutes. Amplified DNA
fragments are electrophoresed along with DNA standards through a 2% agarose gel in
0.5 X TBE buffer (45 mM Tris-borate, pH 8.0, 0.1 mM EDTA) and visualized after
25 staining with ethidium bromide. DNA is quantitated by densitometry. Each experiment
is performed twice.
Two regions in the bacteriophage lambda DNA sequence (GenBank
Accession #J02459) are chosen as the priming sites for amplification. The 5' primer has
a stable GC-rich 3' end; the 3' primer is chosen so that a 381 bp product will result from
30 the amplification. The primers used in this example are as follows:
Forward (5') primers:
H17: 5'-GAACGAAAACCCCCCGC-3'
35 H14: 5'-CTTCGAAAACCCCCCGC-3'
CA 02266847 1999-03-22
W O98/13527 PCTAJS97/17413
71
Hl 1: 5'-CTTGCTAAACCCCCCGC-3'
ABI: S'-GAACGA(dS)AACCCC(dS)CGC-3'
AB2: S'-GAACGA(dS)AACCC(dS)CCGC-3'
AB3: 5'-GAACGA(dS)AACCCCCCG(dS)C-3'
5 DN1: S'-GAACGA(dS)AACCCC(dN)CGC-3'
DN2: S'-GAACGA(dNAACCC(dN)CCGC-3'
DN3: 5'-GAACGA(dN)AACCCCCCG(dN)C-3'
DN4: 5'-GAACG(dN)AAACCC(dN)CCGC-3'
DN5: 5'-GAACG(dN)AAACC(dN)CCCGC-3'
10 DN6: 5'-CTTCGAAAACCC(dN)CCGC-3'
Reverse (3') primer:
reverse: 5'-GATCGCCCCCAAAACACATA-3'
l S (dS) represents "dSPACER" residue and (dN) represents deoxyNebularine residue.
The forward primers are designed with their 5' ends variably
mi~m~trhed to the target DNA. The H17 primer is a perfect match to the intended
target, whereas the primer H14 is complementary only for the 14 nucleotides at the 3'
end (the 3 nucleotides at the S' end are mi~m~tched). All of the primer pairs are used in
20 separate amplification reactions, and the annealing temperature is varied from 25~C to
65~C. A set of typical results are presented in the following table. Similar results are
obtained for both Taq and Pfu polymerases.
CA 02266847 1999-03-22
W O 98/13527 PCT~USg7/17413
72
Table 14
Primer Name Number Substitutions Temp. Range (~C) that
mi~m~tçhes ~ amplifications observed
position in primer
H17 none none 25 ---> 65
H14 3 (~ 5' none 25 ---> 65
H11 6 @, 5 none 25 ---> 50
AB1 2, ~ 7, 14 dSpacer no amplification
AB2 2, ~7, 13 dSpacer noamplification
AB3 2, ~ 7, 16 dSpacer noamplification
DN1 2,~7, 14 dNeb. 25 --->35
DN2 2, ~ 7? 13 dNeb. 25 ---> 35
DN3 2, ~7, 16 dNeb. 25 ---> 30
DN4 2, ~6, 13 dNeb. 25 --->35
DN5 2, ~ 6, 12 dNeb. 25 ---> 35
DN6 2,3~5', 13 dNeb. 25--->30
These results indicate that the dSpacer substitution prevents the Taq or
Pfu DNA polymerase from "reading through" the abasic site. That is, when the
S polymerase encounters an abasic residue, chain extension is termin~te~l Therefore, the
priming site is not conserved during the second strand synthesis, and amplification of
the target nucleic acid is not achieved. However, the polymerases can read through
deoxyNebularine residues present in the oligonucleotide primers. Most likely, but not
verified, deoxythymidine is inserted as the complementary base to deoxyNebularine.
10 However, the tem~ dLulc range over which amplification is achieved is reducedcompared to the temperature range for amplification using the H17 primer (from 25~C -
65~C down to 25~C to approximately 35~C). It is therefore a~p~cllt that the
deoxyNebularine substituted primers can substantially increase the specificity of the
PCR reaction. Priming was improved which led to the amplification of a specific
15 amplicon.
In a second series of experiments, the primer pairs are used in separate
amplification reactions lltili~ing an ~nn~ling t~lllpCldllllC of 42~C. The results are
presented in Figure 12. Similar results are obtained for both the Taq and Pfu
polymerases. As expected, the H17, H14 and Hll primers all give rise to a 381 bp20 amplicon, despite the 3 base mi.~m~tc~.h~s at the 5' end for the H14 primer and the 6 base
mi~m~tches at the 5' end for the H11 primer. As above, no amplification is observed
using the ABl primer Cont~ining abasic residues. In contrast, the DNl, DN2, and DN3
primers all give rise to a 381-bp amplicon, although no amplification is observed using
CA 02266847 1999-03-22
W O 98/13S27 PCTrUS97117413
73
the DN6 primer, probably due to the micm~tçh of 3 bases at the S'-end of the primer
and the deoxyNebularine substitution at the 3' end of the primer. Thus,
deoxyNebularine substituted primer can greatly increase the specificity of priming in
the polymerase chain reaction.
EXAMPLE 9
EFFECTS OF INTRODUCING A DEoxYNEsuLARINE RESIDUE INTO AN
OLIGONUCLEOTIDE ON THE HCT OF THE OLIGONUCLEOTIDE
In Example 3, the introduction of an abasic site or mi.cm~tc.hed site into
an oligonucleotide primer decreases the Td and HCT of the modified primer as
compared to a perfectly based pair "sister" primer. The effect of deoxyNebularine
substitutions on the HCT is also investigated.
DeoxyNebularine modified oligonucleotides can be synthesized by
15 standard methods utiii7.in~ phosphoramidites. The CE phosphoramidite of the
tetrahydroduran derivative, as well as other spacer phosphoramidites are cGmmercially
available (deoxyNebularine, Glenn Research, Sterling, Virginia). The oligonucleotide
for the following ex~ nents is synthPci7~d as a 24-mer having the following
sequence: 5'-hexylamine-TGTGGATCAGCA(dN)GCAGGAGTATG-3'.
The effect of the deoxyNebularine (dN) substitution on the HCT of a set
of oligonucleotides is shown in the Table below.
CA 02266847 1999-03-22
W O 98/13527 PCT~US97/17413
74
Table 15
Stringency Factor
Buffer Type Oligo Type ~HCT Td
1 X PCR buffer normal 18
lX PCR buffer abasic 12
lX PCR buffer deoxyNebularine 12
0.5 M TMATCA normal 14
0.5 MTMATCA abasic 8
0.5 M TMATCA deoxyNebularine 8
2.0 M LiTCA normal 12 44 5.0
2.0 M LiTCA abasic 10 39 6.3
2.0 M LiTCA deoxyNebularine 10 39 6.3
3.0 M GuSCN normal 16 35 3.9
3.0 M GuSCN abasic 12.5 32 5.2
3.0 M GuSCN deoxyNebularine 12.5 31 5.3
~ = ~C
The deoxynebularine substituted oligonucleotide showed the same
decrease in the HCT as the abasic substituted oligonucleotide.
EXAMPLE 10
DETECTION OF A SINGLE BASE-PAIR MISMATCH ON A SOLID
PHASE USING DEoxYNEsuLARINE SUBSTITUTED OLIGONUCLEOTIDES
This example describes the hybridization of an oligonucleotide
cont~ining a deoxyNebularine site to an immobilized oligonucleotide (target). The set
of probe oligonucleotides consists of one probe that is perfectly complementary to the
target, and a second oligonucleotide that contains a deoxyNebularine site. The probe
oligonucleotides are labeled with fluorescent tags to aid in detection of hybridization.
15 For this data, the two oligonucleotides are labeled with different fluorochromes, and
after hybridization at the Td of the mi~m~tch, the ratio of hybridized fluorochromes is
deterrnined.
A target oligonucleotide, 5'-TTGATTCCCAATTATGCGAAGGAG-3'
(DMO501), is irnmobilized on a solid support. Oligonucleotide co~ .in;~g beads
CA 02266847 1999-03-22
WO 98/13527 PCT/US97/17413
(ODN-beads) that are 3t32nd inch diameter are prepared as previously described (Van
Ness et al., Nuc. Acids Res. 19:3345, 1991). The ODN-beads contain from 0.01 to 1.2
mg/bead of covalently immobilized ODN. Probe oligonucleotides include DMO578,
- which is the perfect complement to DMO501. DMO1969, which is the complement to
5 DMO501 but has a deoxyNebularine residue at position 11, DMO1971, which is thecomplement to DMO501 but has a deoxyNebularine site at position 12. Each probe
oligonucleotide is labeled with either BODIPY, TAMRA or Texas Red. Hybridizationreactions contain 50 ng/ml of each probe in a solution comprising 3 M GuSCN, 0.01 M
Tris plI 7.6, and 5 mM EDTA. Equal molar ratios of each probe are used for each
10 hybridization to 3 solid supports contained in a tube. Hybridizations are carried out at
42~C for 30 mimltes with constant agitation. The beads are washed twice with 3 MGuSCN at 42~C followed by five washes of SDS/FW.
To denature the probe/target duplexes, the solid supports are placed in
200 1ll TE (0.01 M Tris, pH 7.0, 5 mM EDTA) and incubated for 10 minutes at 100~C.
15 The solution (200 ~11) is removed from the incubation tubes and placed in a black
microtiter plate (Dynatek Laboratories, Chantilly, VA) for measurement of
fluorescence. The plates are then read directly in a Fluoroskan Il fluorometer (l;low
Laboratories, McLean, VA) using an excitation wavelength of 495 nrn and monitoring
emission at 520 nm for fluorescein, using an excitation wavelength of 591 nm and20 monitoring emission at 612 nm for Texas Red, and using an excitation wavelength of
570 nm and monitoring emission at 590 nm for li~s~min~ or TAMRA.
The results are presented in the following table:
Table 16
Fluorochrome ratio in Fluorochrome ratio after
Probe Mix hybridizationmixdenaturing
578TR/578BD 5.6/1 5.6/1
578TR/1969BD 2.0/1 36/1
578TR/1971TA 0.018/1 0.7/1
578BD/1971TA 0.022/1 0.48/1
The results indicate an average of 20-fold enrichment of perfectly based
probes over the deoxyNebularine modified probes in GuSCN; allowing much higher
levels of ~ rimin~tion in hybridization reactions. This enri~hment is not due to the
presence of the fluorochrome, as the fluorochrome has no measurable effect on the
- hybridization. As indicated in line 1, Texas Red (TR) 578 oligonucleotide and 578-BD
.,
CA 02266847 1999-03-22
W O 98/13527 PCTAJS97/17413
76
(BODIPY) competed equivalently for hybridization to the immobilized target as
evidenced by the same ratio of labels before and after hybridization.
EXAMPLE 1 1
S DETECTION OFA SINGLE BASE-PAIR MISMATCH ON A SOLID PHASE
USING ABASIC SUBSTITUTED OLIGONUCLEOTIDES
This example describes the use of abasic substituted oligonucleotide
probes to detect single base pair mi~m~tches As shown herein, an increase in
10 efficiency is observed in detecting single base-pair mi~m~tches using abasic substituted
oligonucleotide probes as compared to standard probes.
Target oligonucleotides are covalently attached to membrane filters
(Magna Graph nylon membrane filters, Micron Separations, Westboro, MA) (Van Nesset al., Nuc. Acids Res. 19:3345, 1991). The target oligonucleotides are based on the
15 sequence: 5'-TGTGGATCAGCAAGCAGGAGTATC-3' and contain either a G~A,
T~C, T~T, G~T, or T~G mt~m~tch at positions 13 or 14 in the target
oligonucleotides. After attachment of the oligonucleotides to the membrane, the sheet is
blocked for l0 min with gentle mixing in a succinnic anhydride solution (2.5 g of
succinnic anhydride dissolved in 25 ml m-pyrol mixed with 125 ml 0.1 M NaBorate pH
20 8.5). The sheets are then washed 5 times with a solution of 10 mM Tris, 5 mM EDTA
(TE). The sheets are additionally blocked for 30 min with gentle mixing with a solution
of 1% bovine serum albumin (Fraction 5, Sigma) and cont~ining 100 llg/ml fragmented,
single strand herring sperm DNA. The sheets were then washed 5 times in TE.
The following biotinylated probes
control probe: 5'-ACACCTAGTCGTTCGTCCTCATAC-3',
8S abasic probe: 5'-ACACCT(dS)GTCGTTCGTCCTCATAC-3', and
6S abasic probe: 5'-ACACCT(dS)GTCGTTCGTCCTC(DS)TAC-3'
are added to the sheet at a final concentration of 10 ng/ml in 1 ml of 3 M
GuSCN, and the sheets are incubated at 28~C for 30 minutes. The sheets are then rinsed
30 four times in lxSSC/0.1% SDS for 1 minute each wash, followed by two rinses in Wash
Solution (0.01 M Tris pH 7.2, 0.1 M NaCl, 0.005 M EDTA, 0.1% Tween 20).
The streptavidin/alkaline phosphatase conjugate (Vector, Burlingame,
CA) is diluted 1 :10,000 in wash solution. The sheets are then incubated in this solution
for 1 hour at room temperature with shaking. The sheets are subsequently rinsed four
35 times with wash solution and once with detection buffer (0.1 M NaCl, 0.01 M Tris pH
8.5, 0.05 M MgCl2) for 5 minutes. The alkaline phosphatase substrate is prepared by
CA 02266847 1999-03-22
WO 98113527 PCT/US97/17413
77
dissolving a BCIP/NBT tablet (Schleicher and Schuell, part #78349, Keene, NH) in30ml dH20. The reaction is carried out for 0.5 to 4 hours at room t~ dLule. The
sheets are then rinsed with water and dried. A text scanner is used to detect signal.
-As shown in Figure 13, hybridization to the control probe is observed for
S each target oligonucleotide, even for those that are micm~tche~ However, the 6S
abasic-modified probe hybridized nearly exclusively to the perfect match target
oligonucleotide. The 8S abasic-modified probe also hybridized plel~lelllially to the
perfect match target oligonucleotide. The density for each target is presented in the
following table in relative intensity units:
Table 17
Oligo None G/A C/T T/T G/T T/G
Control 90 82 26 90 91 45
6S 52 0 0 0 10 3
8S 76 35 2 35 45 30
The following table presents the ratio of mi.cm~trh density to control
density.
Table 18
Oligo None G/A C/T T/T G/T T/G
Control I 0.91 0.28 1 1.01 0.5
6S I 0 0 0 0.19 0.06
8S 1 0.46 0.03 0.46 0.59 0.39
EXAMPLE 12
HIGH THROUGHPUT ANALYSIS OF HELICAL COIL TRANSITIONS
OF OLIGONUCLEOTIDES.
A capture oligonucleotide (36-mer) was covalently linked to nylon bead
via a C6-amine tail as previously described (Van Ness et al., Nuc. Acids Res. 19:3345,
1991). Oligonucleotides (of various lengths as described in the text) were labeled via a
25 C6 amine spacer with Texas Red (fluorescein, li.cs~mine or TAMRA can also be used)
CA 02266847 1999-03-22
wo 98/13S27 PCT/USg7/17413
78
and were hybridized to the capture oligonucleotide in a 1.5M guanidinium thiocyanate
solution (other hybridization solutions as described in the text can also be used).
Specifically, the "signal" oligonucleotide was synthesized by Midland
Certified Reagent Company (Midland, Texas) at I~M scale. The oligo was diluted to
5 250 ~lL in TE buffer which was used as a stock solution. The signal oligo was further
diluted for hybridization by removing 25 ~lL of the stock solution and mixing it into
975uL of 1.5M guanidinium thiocyanate solution (other hybridization solutions asdescribed in the text can also be used). This working stock was aliquoted into a Cetus
tube (lOOuL/tube). A nylon pin was immersed in the solution for 15 minutes at ambient
10 temperature to allow the signal oligo to hybridize to the immobilized capture oligo. The
beads were then washed to remove unhybridized signal oligonucleotide lx with 0.01 M
Tris pH 7.0, 5 mM EDTA, and 0.1 M NaCI; 2x with 0.01 M Tris pH 7.0, 5 mM 3~DTA,
0.1 M NaCl, and 0.1% SDS; lx with 0.01 M Tris pH 7.0, 5 mM EDTA, and 0.1 M
NaCI (TEN: O.OlM Tris pH 7.5, lmM EDTA, lOOmM NaCl; TENS: O.OIM Tris pH
15 7.5, lmM EDTA. lOOmM NaCl, 0.1% SDS).
Test solutions were aliquoted into wells of a polycarbonate thermowell
plate (Corning Costar Corp., Cambridge, MA) and the plate placed in an MJ thermal
cycler (MJ Research Company, Watertown, MA). The beads were serially transferredbetween the wells of the plate; every 2.5 to 5 minutes the tempeld~LIre increases by 5~C
20 starting at 10~C and reaching 85 to 100~C at the fmal point. After the melting process
was completed, the liquid in the polycarbonate thermowell plates was transferred to a
black 96 well microtiter plate (Dynatek Laboratories, Chantilly, VA). The plates were
then read directly using a Fluoroskan II fluorometer (Flow Laboratories, McLean, VA)
using an excitation wavelength of 495 nm and monitoring emission at 520 nm for
25 fluorescein, using an excitation wavelength of 591 nm and monitoring emission at 612
nm for Texas Red, and using an excitation wavelength of 570 nm and monitoring
emission at 590 nm for li~ min~ or TAMRA. The level of fluorescence correlates with
the amount of signal oligonucleotide that has melted from the capture oligo.
To calculate the Td, cumulative counts eluted at each temperature were
30 plotted against temperature. The teml)eldl~lre at which 50% of the material dissociates
from the bead is the Td. The data was exported into a spre~h-oet and melt curves were
generated for each solution. From these melt curves, Td, ~HCT, and ~Td were
calculated.
CA 02266847 1999-03-22
WO 98/13527 rcT/usg7/l74l3
79
EXAMPLE 13
IDENTIFICAT1ON OF HYBRIDIZATION SOLUTIONS WHICH EFFECTIVELY NEUTRALlZE THE
G+C CONTENT OF NUCLEIC ACID DUPLEXES.
-
This example describes the identification and use of novel compounds
that reduce or elimin~te the effects of G+C content on the melting behaviour of nucleic
acid duplexes.. Also, as shown herein, an increase in efficiency is observed in detecting
single base-pair mi.sm~tches using modified oligonucleotide probes as compared to
standard probes.
10 Solutions and Rea~eents
Filter wash (FW) is 0.09 M NaCI, 540 mM Tris pH 7.6, 25 mM EDTA.
SDS/FW is FW with 0.1% sodium dodecyl sulfate (SDS). Hybridization solutions
contain the text specified concentration of hybotrope of G+C neutralizing compound,
0.1 to 2% N-lauroylsarcosine (sarcosyl), 50 mM Tris pH 7.6 (in some cases) and 0.5 to
15 25 mM EDTA. Formamide hybridization solution contains 30% formamide, 0.09 M
NaCl, 40 mM Tris-HCl pH 7.6, 5 mM EDTA and 0.1% SDS. GuSCN is purchased
from Kodak (Rochester, NY). GuCl, lithium hydroxide, trichloroacetic acid, NaSCN,
NaCl04 and KI, are purchased from Sigma (St. Louis, MO). CsTFA is purchased fromPharmacia (Piscataway, NJ). The amine based compounds were purchased from Sigma
20 (St. Louis, MO), Aldrich (Milwaukee, WI) or from Fluka (Ronkonkoma, NY)
P~pal ation of LiTCA~ TMATCA and TEATCA and other Amine-based TCA~ TFA and
acetate salts.
LiTCA and TMATCA, and TEATCA are pr~ared by the dropwise
titration of a 3 N solution of LiOH, TEAOH and TMAOH respectively, with
25 trichloracetic acid (100% w/v, 6.1 N) to pH 7.0 on ice with constant stirring. The salt is
evaporated to dryness under vacuurn, washed once with ether and dried. The acetate,
trichoroacetate, or trifluoroacetate salts of the arnine col-t~ compounds were
synthesized by neutralizing the respective arnines with acetic acid, trichloroacetic acid
or with trifluoroacetate to pH 6.0 to pH 8.5, depending upon the application. The
30 resulting salt solution was then diluted to the concentration desired as stated in the
figures or tables in this exarnple. In some cases the salt was then dissolved in water to a
final concentration of 0.1 to 3.0 M. The resulting salt solution was in some cases then
buffered with Tris-HCl, pH 7.0-8.5, and detergents, such as sarkosyl, are added to about
0.1%, and optionally EDTA is added to 0.5 to 5 mM. The oligonucleotide that was
. . .~.
CA 02266847 1999-03-22
W O 98/13527 PCTrUS97/17413
tethered to the bead was DMO-2060:5'-hexylamine-
GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3' (oligonucleotide
imrnobilized on the nylon bead); abd the probe oligonucleotides were: DMO-2055: 5'-
TEXAS RED- TGT/GGA/TCA/GCA/AGC/AGG/AGT/ATG-3' (perfect complement);
5 DMO-2058; 5'-TEXAS RED- TGT/GGA/TCA/GGA/AGC/AGG/AGT/ATG-3'
(mi~mslt~ll complement); and DMO-2058-dN: 5'-TEXAS RED-
TGT/GGA/TCA/G(deoxynebularine)A/AGC/AGG/AGT/ATG-3' (deoxynebularine
mi.~m~tch complement).
I 0 Table 1 9
1 M 500mM 100mM
Stock Solution Stock pH ~Tm (27-83 GC) ~Tm (27-83 GC) ~Tm (27-83 GC
-e:~y l . ,e acetate 7 0
-e ly ~ ,e ., u hloluacetdle . - 0
'-e-lV~, 3' ,e-rfluoroacetate .-~ - o
-me ly .: - ~ acetate ~. 7 1
-me ly, -r c "e acetnte ~ ~ 14
-me lypperc le lli1h oact:ldle 8.4 3
-me ly pyrro c ne ace-ate 7 - - ~
-me ly pyrro c ne ., . hlc uac~lal~ 8.~ ~
-me-nypyr o c n~-rfluoruac~:latt, 7. ~. 4
~ _ loxy~.. hy.. l, ,eacetate ~ -
_-me loxy~.l,ydr, letrifluoloacet.t~ ~.~ . 1 1 .
v-",r ,o,-y~,rupylamine acetate ~.
~-~t ne in 1X TE ~.~ 1v
s :-methoxyethy)amine acetate
s~2-methoxyethy )amine trifluo(oac~tah ~. 3
~i 'yl~." ,eaceta-e . .
~yldr" ,e trifluoruac~ldl~
ci ~utylc mine acetate 8
~ cyclolexylamine Acetate .
c sobu .yk mir e acetate
- u~,y am ne ac~tate ~' 2
~ ~,crupy am ne trif uoluact:t.. tc . ~ -
c propylamine aceta-P 2
1~ N \I N' te-,detllylel ,yl_nedid,~, le acetate - 3
n n-c memy ar" lo~u-ane cetate -v 2
n n-c memy a,ll ~o .u .ane .I .;h ruac~ldle .2
n n-c memy .A., . ,o ~u-ane -r fluorc,ac~:l. tu
n n-~ "t: . ,y . utylamine ace-ate ~ ~ 0
n n-c memy cyulohexyla." ,e acetate ~.
n n-c me-ly cy~ lohexyld", le 3 -5
trifluoruact:t le
n n-d me . ly cyclohe,-vld" l ,e/TE/Sark
n n-~ "~: . ,y l~,lylam ne acetate
n n-c me-ly ,e~ Iyb ., ,e ac~t--te ~.~
n n-c me . ly lexylam ne ace~a~e ~ ;~ v v
n n-c memy lexylam ne ace a.e ~. ~ 1
n n-c me.ly ~ op,u~,l all, ,e acetate . r~
n n-c memy sc)pr-,pyamne l,i- l, ruac~:tale . ~ _ 1.
n n- ~"e ,y oclylamine trifluoroacetate . -~
CA 02266847 1999-03-22
W O 98/13527 rCTAUS97/17413
81
1 M 500mM 100mM
Stock olution Stock pH ~Tm (27-83 GC ~Tm (27-83 GC) ~Tm (27-83 GC'
n-e- lyl ~utylamine acetate
-e . lyl ~utylamine tr flu~ acetdt~ 6.
,~.,anol ". ,eace-ate 6. - L 1-.
-r e- ly am ne acetate
." ,e l,i~ r~a
upJl.A.Il ,eacetate 6.5 - ~ ~
ldlll~ n acet te
-e-ra e~hyla,~on ~rn ace-ate 3M -~
Ul Illd-I ' ~ %ITE/Sarc
X PCR Bu er
X SSC
As shown in Table 19 numerous amine-based hybridization solutions (in
the 100 mM concentration range, 500 mM concentration range and 1000 mM
concentration range) have been identified which give rise to a ~Td of 9 ~C or less
5 between oligonucleotide duplexes of G+C content of 27% to 83% Novel hybridization
solutions were prepared which demonstrate properties not previously described for a
hybridization solution
These hybidization solutions possess the ~ulopelly of neutralizing the
differences in G+C and A+T base-pairing strength. Some of the solutions (most
10 tripropylamine acetate, bis(2-methoxyethyl)amine trifluoroacetate, disopropylamine
trifluoroacetate, n,n dimethylaminobutane trifluoroacetate at 100 mM; triethanolamine
acetate, noteably n,n dimethylcyclohexylamine trifluoroacetate, n,n
dimethylheptylarnine acetate at 500 mM; noteably n,n dimethylcyclohexylamine
trifluoroacetate, tripropylarnine acetate, dibutylamine acetate, n,n dimethylheptylamine
15 acetate, dimethylhexylamine acetate, dicyclohexylamine acetate at 1000 mM)
simultaneously lowers the Td and ~Td, Others such as increase ~Td (I-ethylpiperidine
acetate, etc.) In the table below the characteristics of the novel hybridization solutions
and hybotropes are described. The following ~Tds as a function of G+C content were
obtained from the melt curves described below: Novel hybridization solutions have
20 also been identified which neutralize the effects of G+C content on the melting
behaviour of nucleic acid duplexes. These solutions are in some cases hybotropes and
in other cases can be used as PCR buffers or as hubridization solutions which minimi7~-
the effects of G+C content on nucleic acid duplexes. These new hybridization
solutions, their l)lo~llies, and their ~rel)~dlion are described in Example 12. Figure 14
25 is a graph showing the difference in Td between three duplexes. that vary in G+C
content from 27% to 83%. The capture oligonucleotide is a 36-mer (DMO-GC36cap:
5 ' -hexylamine-GCA/GCC/TCG/CGG/AGG/CGG/ATG/ATC/GTC/ATT/AGT/ATT-3 ' )
and three complt-.rnent~ry oligos which are labelled with the fluorochrome are DMO-
. . .
CA 02266847 1999-03-22
W O98/13527 . PCTAUS97/17413
82
83GC: 5'-Texas Red- CCG/CCT/CCG/CGA/GGC/TGC-3'; DMO-50GC: 5'-Texas
Red- AAT/GAC/GAT/CAT/CCG/CCT-3'; DMO-27GC: -Texas Red-
AAT/ACT/AAT/GAC/GAT/CAT-3'. The temperature difference between any two Tds
at ~ = 0.5 is defined as the ~Td. The percentage of single strand DNA (y-axis) is
plotted versus temperature (~C; x-axis). The melting solution was 100 mM 2-
methoxyethylamine trifluoroacetate. The maximum difference between the 3 meltingcurves in the Td was 6 C. The helical coil transition of the 27% G+C content was 21 C,
50% G+C was 33 C and for the 83% G+C duplex was 29 C. Note that the helical coiltransitions (HCTs) of the 3 different G+C content oligonucleotides is different. This is
in contrast to the case with diisobutylamine as shown in Figure 15. Figure 15 is a graph
showing the difference in Td between three duplexes, that vary in G+C content from
27% to 83% (the same system as described in Figure 14. The temperature difference
between any two Tds at o~ = 0.5 is defined as the ~Td. The percentage of single strand
DNA (y-axis) is plotted versus temperature (~C; x-axis). The melting solution was 100
mM diisobutylamine acetate. The maximum difference between the 3 melting curves in
the Td was 5 C. The helical coil transition of the 27% G+C content was 22 C, 50%G+C was 26 C and for the 83% G+C duplex was 25 C. The helical coil transitions for
the three oligonucletide duplexes are very similar. This is the behaviour that is
preferred for use in array hybridizations or polymerase chain reactions.
In Figure 16 the inability of GuSCN to neutralize G+C content is shown.
Figure 16 is a graph showing the difference in Td between three duplexes, that vary in
G+C content from 27% to 83% (the same capture and probe oligonucleotides as
described in figure 14). The temperature difference between any two Tds at a = 0.5 is
defined as the ~Td. The percentage of single strand DNA (y-axis) is plotted versus
temperature (~C; x-axis). The melting solution was 2 M Guanidinium thiocyanate. The
maximum difference between the 3 melting curves in the Td or Tm is 16 C. The helical
coil transition of the 27% G+C content was 28 C, for the 50% G+C duplex was 30 Cand for the 83% G+C duplex was 32 C. Similar results were obtained with lx PCR
buffer (figure 17) and lx SSC buffer (Figure 18). There was also no neutralization of
G+C content with 20% formamide (Figure 19). Figure 17 is a graph showing the
difference in Td between three duplexes, that vary in G+C content from 27% to 83%
(the same duplex system as described in Figure 14). The temperature difference
between any two Tds at a = 0.5 is defined as the ~Td. The percentage of single strand
DNA (y-axis) is plotted versus temperature (~C; x-axis). The melting solution was lx
PCR buffer. The maximum difference between the 3 melting curves in the Td was 14 C.
The helical coil transition of the 27% G+C content was 17 C, for the 50% G+C duplex
CA 02266847 1999-03-22
WO 98/13527 PCTtUSg7/17413
83
was 22 C and for the 83% G+C duplex was 23 C. Figure 18 is a graph showing the
difference in Td between three duplexes, that vary in G+C content from 27% to 83%.
The tt;lllpeldLule difference between any two Tds at a = O.S is defined as the ~Td. The
percentage of single strand DNA (y-axis) is plotted versus te-llpc~l~Lule (~C; x-axis).
S The melting solution was lx SSC. The maximum difference between the 3 melting
curves in the Td iS 13 C. The helical coil transition of the 27% G+C content was 20 C,
for the 50% G+C duplex was 22 C and for the 83% G+C duplex was 23 C. Figure 19 is
a graph showing the difference in Td between three duplexes, that vary in G+C content
from 27% to 83%. The temperature difference between any two Tds at a = O.S is
10 defined as the ~Td. The percentage of single strand DNA (y-axis) is plotted versus
temperature (~C; x-axis). The melting solution was 20% formamide, 10 mM Tris pH
7.6, and S mM EDTA with 0.1 % sarkosyl. The maximum difference between the 3
melting curves in the Td is 14 C. The helical coil transition of the 27% G+C content
was 15 C, for the 50% G+C duplex was 16 C and for the 83% G+C duplex was 20 C.
In contrast to the situation in Figures 17, 18 and 19, Figure 20 shows the
melting behaviour of the 3 different G+C oligonucleotide duplexes in 1 M
dicyclohexylamine acetate. Figure 20 is a graph showing the difference in Td between
three duplexes, that vary in G+C content from 27% to 83%. The temperature difference
between any two Tds at a = 0.5 is defined as the ATd. The percentage of single strand
20 DNA (y-axis) is plotted versus t~lllpc;,d~lre (~C; x-axis). The melting solution was 1 M
dicyclohexylamine acetate. The maximum difference between the 3 melting curves in
the Td was 3 C. The helical coil transition of the 27% G+C content was 13 C, for the
50% G+C duplex was 17 C and for the 83% G+C duplex was 19 C. This is an ideal
profile for a hybotrope. In contrast the the narrow helical coil transition observed in
25 Figure 20, a much wider HCT is observed with SOO mM n-ethylbutylamine acetate.
Figure 21 is a graph showing the difference in Td between three duplexes, that vary in
G+C content from 27% to 83%. The capture oligonucleotide is a 36-mer (DMO-
GC36cap: 5'- hexylamine-GCA/GCC/TCG/CGG/AGG/CGG/ATG/ATC/GTC/ATT/
AGT/ATT-3') and three complementary oligos which are labelled with the
30 fluorochrome are DMO-83GC: 5'-Texas Red- CCG/CCT/CCG/CGA/GGC/TGC-3';
DMO-SOGC: S'-Texas Red- AAT/GAC/GAT/CAT/CCG/CCT-3'; DMO-27GC: -Texas
Red-AAT/ACT/AAT/GAC/GAT/CAT-3'. The temperature difference between any
two Tds at a = O.S is defined as the ~Td. The percentage of single strand DNA (y-axis)
is plotted versus temperature (~C; x-axis). The melting solution was 500 mM n-
35 ethylbutylamine acetate. The maximum difference between the 3 melting curves in the
CA 02266847 1999-03-22
W O 98/13527 PCT~US97/17413
84
Td was 1 C. The helical coil transition of the 27% G+C content was 22 C, for the 50%
G+C duplex was 22 C and for the 83% G+C duplex was 26 C.
The ability of some of the G+C neutralizing buffer to act as hybrotropes
is illustrated in Figure 22. Figure 22 is a graph showing the difference in Td between
three duplexes, one that is perfectly based-paired and the other two that contains a
mi~m~tch or a deoxynebularine substitution. The temperature difference between any
two Tds at a = 0.5 is defined as the ~Td. The percentage of single strand DNA (y-axis~
is plotted versus t~lllp~.dlure (~C; x-axis). DMO-2060: 5'-hexylamine-
GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3' (oligonucleotide
10 immobilized on the nylon bead.; DMO-2055: 5'-TEXAS RED-
TGTIGGA/TCAIGCAIAGCIAGGIAGTIATG-3' (perfect complement); DMO-2058;
5'-TEXAS RED- TGTIGGAITCAIGGA/AGC/AGGIAGTIATG-3' (mi~m~tch
complement); and DMO-2058-dN: 5'-TEXAS RED-
TGT/GGA/TCA/G(deoxynebularine)A/AGC/AGG/AGT/ATG-3' (deoxynebularine
15 mi~m~tch complement). The melting solution was I M diisopropylamine acetate. The
maximum difference between the 3 melting curves in the Td was 6 C. The helical coil
transition (HCT) of the true mi~m~tch was 14 C; the HCT for the deoxynebularine
mi~m~tc.h duplex was 14 C and the HCT for the perfectly based paired duplex was 16
C.
The same situation was observed for 1 M diisopropylamine acetate
(Figure 22), I M n,n-dimethylcyclohexylamine acetate (Figure 23) and I M
dicyclohexylamine acetate (Figure 24) and n,n-dimethylhexylamine acetate (Figure 25).
Figure 23 is a graph showing the difference in Td between three duplexes. one that is
perfectly based-paired and the other two that contains a mi~m~tch or a deoxynebularine
substitution. The temperature difference between any two Tds at a = 0.5 is defined as
the ATd. The percentage of single strand DNA (y-axis) is plotted versus temperature
(~C; x-axis). DMO-2060: 5'-hexylamine-
GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3' (oligonucleotide
immobilized on the nylon bead., DMO-2055: 5'-TEXAS RED-
TGT/GGA/TCAIGCAIAGC/AGGIAGT/ATG-3' (perfect complement); DMO-2058;
5'-TEXAS RED- TGTIGGAITCAIGGA/AGC/AGG/AGT/ATG-3' (mismatch
complement); and DMO-2058-dN: 5'-TEXAS RED-
TGT/GGA/TCA/G(deoxynebularine)A/AGC/AGG/AGT/ATG-3' (deoxynebularine
mi~m~tch complement). The melting solution was 1 M n,n-dicyclohexylamine acetate.
The maximum difference between the 3 melting curves in the Td or Tm is 4 C. The
helical coil transition (HCT) of the true mi~m~tch was 15 C; the HCT for the
CA 02266847 1999-03-22
WO 98/13~27 PCT/USg7/17413
deoxynebularine mi~m~tçh duplex was 15 C and the HCT for the perfectly based paired
duplex was 15 C.
Figure 24 is a graph showing the difference in Td between three
duplexes, one that is perfectly based-paired and the other two that contains a mi.~m~trh
5 or a deoxynebularine sllbstit-~tion. The temperature difference between any two Tds at
a = 0.5 is defined as the ~Td. The percentage of single strand DNA (y-axis) is plotted
versus temperature (~C; x-axis). DMO-2060: 5'-hexylamine-
GTC/ATA/CTC/CTG/CTT/GCT/GAT/CCA/CAT/CTG-3' (oligonucleotide
immobilized on the nylon bead.; DMO-2055: S'-TEXAS RED-
10 TGT/GGA/TCA/GCA/AGC/AGG/AGT/ATG-3' (perfect complement); DMO-2058;
5'-TEXAS RED- TGT/GGA/TCA/GGA/AGC/AGG/AGT/ATG-3' (mi.cm~tch
complement); and DMO-2058-dN: 5'-TEXAS RED-
TGT/GGA/TCA/G(deoxynebularine)A/AGC/AGG/AGT/ATG-3' (deoxynebularine
mi~m~tçh complement). The melting solution was I M n,n-dicyclohexylarnine acetate.
15 The maximum difference between the 3 melting curves in the Td iS 4 C. The helical coil
transition (HCT) of the true mi~m~tch was 17 C; the HCT for the deoxynebularine
mi~m~tch duplex was 17 C and the HCT for the perfectly based paired duplex was
15 C.
Figure 25 is a graph showing the difference in Td between three
20 duplexes, one that is perfectly based-paired and the other two that contains a mi~m~tch
or a deoxynebularine substitution. The temperature difference between any two Tds at
a = 0.5 is defined as the ~Td. The percentage of single strand DNA (y-axis) is plotted
versus temperature (~C; x-axis). The melting solution was l O0 mM n,n-
dimethylhexylamine acetate. The maximum difference between the 3 melting curves in
25 the Td iS 9 C. The helical coil transition (HCT) of the true mi~cm~tçh was 15 C; the HCT
for the deoxynebularine mi~m~3tçh duplex was 15 C and the HCT for the perfectly based
paired duplex was 15 C.
It will be appreciated that, although specific embodiments of the
invention have been described herein for purposes of illustration, various modifications
30 may be made without departing from the spirit and scope of the invention.
Accordingly, the invention is not limited except as by the appended claims.