Note: Descriptions are shown in the official language in which they were submitted.
CA 02267657 1999-03-30
HYDROXYPHENYL DER',IVATIVES WITH
HIV INTEGRASE INHIBITORY PROPERTIES
TECHNICAL FIELD OF THE INVENTION
The present invention relates to a hydroxyphenyl derivatives which have HIV
integrase
inhibitory properties that have been characterized by ;specific structural and
physicochemical
features. This inhibitory property may be advantageously used, for example, to
provide
medicinals (e.g. compositions) with antiviral properties against HIV viruses,
including the HIV-
1 and HIV-2 viruses, i.e. the hydroxyphenyl derivatives including
pharmaceutical compositions
thereof may be used to inhibit the activity of HIV intesgrase.
BACKGROUND OF THE INVENTION
The HIV (human immunodeficiency virus) retrovirus is the causative agent for
AIDS (acquired
immunodeficiency syndrome). Thus the HIV-1 retrovirus primarily uses the CD4
receptor (a 58 kDa
transmembrane protein) to gain entry into cells, through high-affinity
interactions between the viral
envelope glycoprotein (gp 120) and a specific region of the CD4 molecule found
in T-lymphocytes
and CD4 (+) T-helper cells (Lasky L. A. et al., Cell vol. 50, p. 975 - 985
(1987)). HIV infection is
characterized by a period immediately following infection called
"asymptomatic" which is devoid
of clinical manifestations in the patient. Progressive HTV-induced destruction
of the immune system
then leads to increased susceptibility to opportunistiic infections, which
eventually produces a
syndrom called AIDS-related complex (ARC) characterized by symptoms such as
persistent
generalized lymphadenopathy, fever, weight loss, followed itself by full blown
AIDS. After entry
of the retrovirus into a cell, viral RNA is converted into DNA, which is then
integrated into the host
cell DNA. The reverse transcriptase encoded by the virus genome catalyzes the
first of these
reactions (Haseltine W. A. FASEB J. vol 5, p. 2349 - 2360 (1991)). At least
three functions have
been attributed to the reverse transcriptase: RNA-dependent DNA polymerase
activity which
1
CA 02267657 1999-03-30
catalyzes the synthesis ofthe minus strand DNA from viral RNA, ribonuclease H
(RNase H) activity
which cleaves the RNA template from RNA-DNA hybrids and DNA-dependent DNA
polymerise
activity which catalyzes the synthesis of a second DNA strand from the minus
strand DNA template
(Guff S. P. J. Acq. Imm. Defic. Syndr. Vol 3, p. 81T - 831 (1990)). The double
stranded DNA
produced by reverse transcriptase, now called provirus, is then able to be
inserted into host genomic
DNA. At the end of reverse transcription, the viral ge:nome now in the form of
DNA is integrated
into host genomic DNA and serves as a template for viral gene expression by
the host transcription
system, which leads eventually to virus replication (Ruth et al.,1989). The
preintegration complex
consists of integrase, reverse transcriptase, pl7 and proviral DNA (Bukrinsky
M. L, Proc. Natn.
Acid. Sci. USA vol. 89 p.6580 - 6584 (1992)). The phosphorylated p17 protein
plays a key role in
targeting the preintegration complex into the nucleus of host cell (Gallay et
al., 1995).
The primary RNA transcripts made from the provilus are synthesized by the host
cell RNA
polymerise II which is modulated by two virus-encoded proteins called Tat and
Rev. The viral
proteins are formed as polyproteins.
Post-translational modifications of viral polyproteins include processing and
glycosylation of Env
(envelope) proteins, and myristylation of the N-terminal residue of the p17
protein in the Gag and
Gag-Pol polyproteins. The latter two precursors correspond to structural
proteins and viral enzymes.
The viral protease is involved in processing polyproteins Gag and Gag-Pol into
mature proteins, a
step essential for virus infectivity.
A number of synthetic antiviral agents have been desired to block various
stages in the
replication cycle of HIV. These agents include compounds which interfere with
viral binding to
CD4 T-lymphocytes (for example, soluble CD4), compounds which block viral
reverse
transcriptase (for example, didanosine and zidovudine (AZT)), budding of
virion from the cell
2
CA 02267657 1999-03-30
(interferon), or the viral protease (for example Ritonavir and Indinavir).
Some of these agents
proved ineffective in clinical tests. Others, targeting primarily early stages
of viral replication,
have no effect on the production of infectious virions in chronically infected
cells. Furthermore,
administration of many of these agents in effective therapeutic doses has led
to cell-toxicity and
unwanted side effects, such as anemia, neurotoxicity and bone marrow
suppression. Anti-
protease compounds in their present form are typically large and complex
molecules of peptidic
nature that tend to exhibit poor bioavailability and are not generally
consistent with oral
administration. These compounds often exhibit side effects such as nausea,
diarrhea, liver
abnormalities and kidney stones.
Accordingly, the need exists for compounds that can effectively inhibit the
action of the third
viral enzyme called integrase, for use as agents for treating HIV infections.
The terms HIV integrase and integrase as used herein are used interchangeably
and refer to the
integrase enzyme encoded by the human immunodefic;iency virus type 1 or 2. In
particular this
term includes the human immunodeficiency virus type: 1 integrase.
SUMMARY OF THE INVENTION
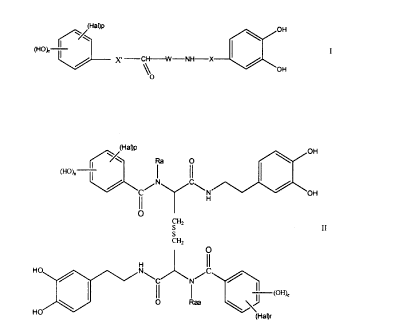
The present invention provides an hydroxyphenyl derivative selected from the
group consisting
of a compound of formula
3
CA 02267657 1999-03-30
~~(Ha~p
OH
(HOB, I
J~~ -'C~-NH-X
OH
or
OH
a O
~H~)"
N OH
H
O
CH,
II
S
CHz
H ~ N~
II
0
HO
and pharmaceutically acceptable derivatives thereof including where applicable
or appropriate
pharmaceutically acceptable salts thereof, wherein n :is 1, 2 or 3, a is l, 2
or 3, Hal represents a
halogen atom (e.g. Cl, Br, F or I), p is 0, 1 or 2, r is 0, 1 or 2, X and X'
each independently
represents a single bond, a saturated straight or branched hydrocarbon group
of 1 to 4 carbon
atoms or a straight or branched hydrocarbon group of 2 to 4 carbon atoms
comprising a carbon to
carbon double bond, Re represents H or -CH3, and R~, represents H or -CH3; W
represents an
amino acid residue or fragment (in particular alpha-amino acid residues) such
as for example a
4
CA 02267657 1999-03-30
residue based on tyrosine, DOPA, hydroxyproline, serine, threonine, histidine,
valine,
phenylalanine, lysine, alanine, glycine, glutamic acid, aspartic acid,
arginine, asparagine,
glutamine, leucine, lysine, isoleucine, proline, tryptophan, methionine,
cysteine, cystine,
thyroxine, meta-tyrosine, sarcosine, other alpha-methyl amino acids such as
alpha-methyl
DOPA, as well as other 3-substituted tyrosines, and the like.
W, for the above formulae I and II, may, for example, be derived from natural
or unnatural
alpha-amino acids. The term unnatural alpha-amino acid refers to alpha - amino
acids which do
not occur in nature but which can be derived from naturally occurring alpha -
amino acids or
other chemical reagents by methods known to those skilled in the art.
W may, for example, represent a group of formula
O O
II 11
-A
wherein k is 0 or 1, A and A' each independently represents a group of formula
~b
~b
or
N C'
N ~ ~~J
R Rc
Ra represents H or -CH3, Rti represents H or -CH3, F;~ represents H or OH, R
is selected from
the group consisting of H, CH3-, (CH3)ZCH-, (CH3)ZCHCH2-, CH3CHzCH(CH3)-,
C6HSCH2-,
CH3SCHzCHZ-, HOZCCHz-, HZNC(O)CHz-, HOZCCH,2CHz-, H2NC(O)CHzCH2-,
HzNCHzCH2CHzCHz-, HOCHz-, CH3CH(OH)-, HSCH2-
5
CA 02267657 1999-03-30
CH2_
HN ~ N
~CNHCH2CH2CH2-
/ N~ NH2 N CH2_
H H
HO I I
HO ~ ~CH2- HO CH2_ H ~ ~ ~ ~ C~-
( lal)f I i
and
(Haf)s\
CHr
HO
wherein Hal is as defined above and f is 0, 1 or 2 and g is 0, 1 or 2 and
provided that n and p
may not at the same time represent 3 and a and r may not at the same time
represent 3.
The group of structure
HO ~ ,--CH2
(~a~l)f
may in particular for example be a fluoride substituted structure of formula
6
CA 02267657 1999-03-30
F
HO \ / CHZ-
Similarly, the group of structure
(Hal)9~
C112-
HO
may in particular for example be a fluoride substituted structure of formula
F \ / C H2_
HO
As mentioned, the polyhydroxy derivatives include pharmaceutically acceptable
derivatives of
the compounds of formula I and II as defined above. As used herein the
expression
"pharmaceutically acceptable derivative" is to be understood as referring to
any pharmaceutically
acceptable salt, amide, ester, or salt of such ester, of a compound of this
invention.
The present invention provides, where appropriate, salts (e.g. derived from
appropriate bases or
acids) which include but are not limited to alkali metal (e.g., sodium) salts,
alkaline earth metal
(e.g., magnesium) salts, and ammonium salts such as acid addition salts of
amines (e.g.
7
CA 02267657 1999-03-30
ammonium chloride salts) as well as quaternary ammonium salts of for example N
- (R")4' type
wherein R" is an organic residue.
The pharmaceutically acceptable salts of the compounds of this invention
include those derived
from pharmaceutically acceptable inorganic and organic acids and bases.
Examples of such acid
salts include: acetate adipate, alginate aspartate benzoate, benzenesulfonate,
bisulfate, butyrate,
citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate,
dodecylhydrogen-
sulfate, dodecylsulfate, ethanesulfonate, formate, furnarate, glucoheptanoate,
glycerophosphate,
glycollate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide,
hydroiodide, 2-
hydroxyethanesulfonate, lactate, maleate, malonate, rnethanesulfonate, 2-
naphthylsulfonate,
nicotinate, nitrate, oxalate, pamoate, pectinate, perchlorate, persulfate, 3-
phenylpropionate,
phosphate, picrate, pivalate, propionate, salicylate, succinate, sulfate,
tartrate, thiocyanate,
tosylate, and undecanoate.
This invention also envisions the quaternization of any basic nitrogen
containing groups of the
compounds disclosed herein. The basic nitrogen can be quaternized with any
agents known to
those of ordinary skill in the art including, for example, lower alkyl
halides, such as methyl,
ethyl, propyl and butyl chloride, bromides and iodides; dialkyl sulfates
including dimethyl,
diethyl, dibutyl and diamyl sulfates; long chain halides such as decyl,
lauryl, myristyl and stearyl
chlorides, bromides and iodide; and arylalkyl halides including benzyl and
phenethyl bromides.
Water or oil-soluble or dispersible products may be obtained by such
quaternization.
This invention also envisions the presence of an ester groups) such as for
example on the acidic
end of an appropriate amino acid fragment(s), such as glutamic acid and
aspartic acid as having
some anti-integrase activity as such as acting as pro-drugs, i.e. capable of
hydrolysis of the ester
moiety to liberate in the systemic circulation the acid, also possessing anti-
integrase activity. For
example, the ether oxygen of an ester compound may be attached or linked to
benzyl, a lower
8
CA 02267657 1999-03-30
alkyl (e.g. C,-C6 alkyl) such as methyl, a lower cycloalkyl (e.g. C3-C~
cycloalkyl) such as
cyclohexyl, and the like. Alternatively, an esters) may be derived from a
carboxylic acids) and
one or more hydroxyl groups, such as for example are hydroxyl group on a
phenyl ring. A
carboxylic acid may, for example, comprise an acyl soup having from 2 to 8
carbon atoms; the
acyl group may for example comprise lower alkyl of 1 to 6 carbon atoms, lower
cycloalkyl of
from 3 to 7 carbon atoms, etc..
In addition, this invention also includes in its scope the possibility of
structures having an amide
functionality such as, for example, on the carboxylic end located on the side
chain of such acids.
These amides, such as simple primary, secondary or tertiary amides, possess
activity of their
1 S own. In addition, it is possible to couple such acids with dopamine to
yield compounds of
interest. The amino moiety of an amide compound may for example be -NH2, -
NH(C,-C6 alkyl),
or -N(C,-C6 alkyl)2, a pyrrolidine residue, a piperidine residue, a morpholine
residue and the like
In any event, it is also to be understood that for the present invention the
expression
"pharmaceutically acceptable derivative" is to be understood as refernng to
any other compound
having a structure such that, upon administration to a recipient, it is
capable of providing
(directly or indirectly) a compound of this invention ar an antivirally active
metabolite or residue
thereof. Thus the compounds of this invention may be modified by appending
appropriate
functionalities to enhance selective biological properties. Such modifications
are known in the art
and include those which increase biological penetration into a given
biological system (e.g.,
blood, lymphatic system, central nervous system), increase oral availability,
increase solubility to
allow administration by injection, alter metabolism and alter rate of
excretion.
The present invention in particular provides a dopamine derivative selected
from the group
consisting of a compound of formula Ia
9
CA 02267657 1999-03-30
OH
Ra O
~0)n I Ia
N C
c~ ~N~
II H OH
O R
and where applicable pharmaceutically acceptable salts, esters and amides
thereof wherein n, Ra
and R are as defined above.
The present invention also provides a dopamine derivative selected from the
group consisting of a
compound of formula Ib
H
O\C/N
O
~HO~n ~~ \ Ib
N
Rd
wherein n is as defined above (e.g. n may in particular be 1 or 2), and Ra is
selected from the
group consisting of H and OH.
The present invention further relates to dipeptide derivatives i.e. to
compounds of formula I
defined above wherein k is 1. The present invention in particular provides an
hydroxylphenyl
derivative wherein for the compound of general formula I above, W represents a
group of
formula
CA 02267657 1999-03-30
b b
- N C -C -N -- -C
- C -
R R
wherein n is as defined above (e.g. n may in particular be 1 or 2), p is as
defined above (p may in
particular be 0), each Ra is independently as defined above, each Rb is
independently as defined
above, and each R is independently as defined above:; more particularly, for
example, for each R, f
may be 0 or l and g may be 0 or 1.
The compounds of this invention contain one or more asymmetric carbon atoms
and thus may
occur as racemates and racemic mixtures, single enantiomer, diastereomeric
mixtures and
individual diastereoisomers. All such isomeric forms of these compounds are
expressly included
in the present invention. Each stereogenic carbon may be of the R or S
configuration.
The amino acid residues may, for example, in any event, be of L, D or DL form,
preferably of L
form; thus for example the amino acid residue (i.e. W) may be a L-a-amino
residue, a D-a-
amino residue, or a DL-a-amino residue.
Accordingly, the present invention father provides a dopamine derivative
selected from the group
consisting of a compound of formula Ic
11
CA 02267657 1999-03-30
OH
Ic
N ~. l
\N ~ OH
I I H
O R
and where applicable pharmaceutically acceptable salts, amides and esters
thereof,
wherein n is 1, or 2, Ra and R are as defined above (e.g. f and g may be 0 or
1 and the respective
group Hal thereof may be fluorine (F)).
The present invention furthermore provides a dopamine derivative selected from
the group
consisting of a compound of formula Id
(HO)~_ II R
N C ~ N
\N/~y/ Id
R Ra O
OH
OH
and where applicable pharmaceutically acceptable salts, amides and esters
thereof,
wherein n is 1; or 2, each Ra is independently as defined above, and each R is
independently as
defined above; more particularly, for example, for each R, f may be 0 or 1 and
g may be 0 or 1.
The compounits vfthapresent invention including where applicable their
pharmaceutically
acceptable derivatives have an affinity for integrase, in particular, HIV
integrase. Therefore, these
12
CA 02267657 1999-03-30
compounds are useful as inhibitors of such integrase, i.e. they are in
particular useful as HIV
integrase inhibitors. These compounds can be used alone or in combination with
other
therapeutic or prophylactic agents, such as antivirals, antibiotics,
immunomodulators or vaccines,
for the treatment or prophylaxis of viral infection.
According to the present invention, the compounds of this invention are
capable of inhibiting
HIV viral replication in human CD4+ T-cells, by inhibiting the ability of HIV
integrase to
integrate the double stranded DNA into host genomic; DNA for further virus
replication by the
host cell machinery (Sakai H., J. Virol. Vol. 67 p. 1169 - 1174 (1993)). These
novel compounds
can thus serve to reduce the production of infectious virions from acutely
infected cells, and can
1 S inhibit the initial or further infection of host cells. Accordingly, these
compounds are useful as
therapeutic and prophylactic agents to treat or prevent infection by HIV-I and
related viruses,
which may result in asymptomatic HIV-I infection, AIDS-related complex (ARC),
acquired
immunodeficiency syndrome (AIDS), AIDS-related dementia, or similar diseases
of the immune
system.
Thus the present invention also provides a pharmaceutical composition
comprising a
pharmaceutically acceptable carrier and a pharmaceutically effective amount of
at least one
hydroxyphenyl derivative as defined above. The pharmaceutical compositions may
be used to
inhibit integrase, including HIV integrase, thus providing protection against
HIV infection.
The expression " pharmaceutically effective amount" is to be understood herein
as referring to an
amount effective in treating HIV infection in a patient. The term
prophylactically effective
amount refers to an amount effective in preventing HIV infection in a patient.
As used herein, the
term patient refers to a mammal, including a human. The expressions
"pharmaceutically
acceptable carrier" (or adjuvant) and "physiologically acceptable vehicle" are
to be understood as
referring to a non-toxic carrier or adjuvant that may be administered to a
patient, together with a
13
CA 02267657 1999-03-30
compound of this invention, and which does not destroy the pharmacological
activity thereof.
These factors will be discussed in more detail below.
The compounds of this invention may be readily prepared using conventional
techniques from
commercially available and cheap starting materials. The relative ease of
synthesis of the
products described in this invention represents a marked advantage for the
large scale preparation
of these compounds. In general, the derivatives of the present invention may
be readily obtained
from amino acids through sequences recognized by those knowledgeable in the
art as
straightforward, requiring readily available reagents and easy techniques.
Using standard
techniques, amino acids may be transformed to the desired HIV integrase
inhibitors according to
approaches as shown in Scheme 1, Scheme 2 and Scheme 3 which are discussed
below. The
preparation of dipeptide derivatives may be accomplished by solid phase
peptide synthesis; this
type of process is generically illustrated in scheme 4 below (see example 42
below).
Scheme 1 illustrates example steps for the preparation of a derivative in
accordance with the
present invention:
Note:
a) For scheme l, PG and PG' may be any suitable (known) removable protecting
group for respectively protecting the amine functional group and the
carboxylic acid
functional group(s). PG may, for example, be Boc i.e. tert- butyloxycarbonyl
and
PG' may, for example, be tert-Butyl, 2,6-CI2Bz1 or Bzl, i.e. a functional
group of
the following formula
14
CA 02267657 1999-03-30
CH2_
b) For scheme 1 R, may, for example, be CH3-, BzIOOCCHZCH2-,
HZNC(O)CHzCH2-; R,a may, for example, be CH3-, HOOCCHZCHZ-,
HzNC(O)CHZCH2-
Step I )
0 0
'HN
// N
PG~
PG ( e.g. BOC) OH ~O-PG' (e.g. O-Bzl)
Ri R~
compound 1 compound 2
Step 2)
0 0
H
N NH z
PG~
O- P G' -O- P G'
R~ R~
compound 2 compound 3
Step 3)
Step 4)
CA 02267657 1999-03-30
0
NHz
O~G'
R~
compound 4 ~~d 3
(R2 = H a OH)
compound S
O
NH
O.?G'
R~
N
compound (5~
16
compound (6)
CA 02267657 1999-03-30
Step 5)
lO H
\ O \ OH
NH
R2
/ \OH H2N / OH
O Rya
compound 6 compound 7
-----~ H ~ \ O ~ \ OH
NH
R2 / ~N / OH
O Rya
compound 8
In accordance with Scheme l, illustrated above, different pharmacophores may
be attached to the
amino acid via the N terminal. Thus, for step 1 compound 1 is treated so as to
protect the
carboxylic acid functional group by means of a suitable protecting group PG';
for example
compound 1 may be a Boc-amino acid which is benzylated with benzyl bromide to
yield
compound 2 in the form of a benzyl ester using cesium carbonate in DMF
according to the
method of S.-S. Wang et al (J. Org. Chem. vol 49 p. 1286 (1977)). For step 2
the amino
protecting group PG is removed to provide compound 3 having a free amino
functional group; for
example the removal of the Boc group from compound 3 may be carried out by
stirring in a
mixture of TFA and methylene chloride (1:1 (v/v)). For step 3 the amino
compound 3 is then
coupled with an hydroxylated benzoic acid (compound 4) with EDC and HOBT in
DMF
providing the desired coupled product compound 5. F'or step 4 compound 5 is
treated to remove
17
CA 02267657 1999-03-30
the protecting group PG' to yield compound 6 having a free carboxylic acid
group; for example
the benzyl protecting group PG'may be removed by hydrogenolysis using Pd/C as
catalyst to
yield compound 6 having a free carboxylic acid group. Finally for step 5
compound 6 is coupled
with dopamine (compound 7) to provide the desired derivative, namely compound
8.
Scheme 2 (which is divided below into scheme 2a and scheme 2b) illustrates
example steps for an
alternate method for the preparation of a derivative in accordance with the
present invention:
Note:
a) For scheme 2a, PG, as mentioned above, may be any suitable (known)
removable protecting group for protecting the amine functional group. PG may,
for
example, be Boc i.e. tert-butyloxycarbonyl
b) For scheme 2a, R3 may, for example, be (CH3)zCHCHZ-, CH3SCHZCH2-, or a
functional group of the following formula
CH2-
N
H
18
CA 02267657 1999-03-30
Scheme 2a:
Step 1
O
H ~ OH
PG .g BOC) ~OH
R3 H2N / OH
compound 1 compound 9
O ~ OH
--
HN OH
R3
compound 10
Step 2:
O ~ OH
HN OH
R3
compound 10
19
CA 02267657 1999-03-30
p ~ .OH
NHz
~N / ~OH
R3
compound 11
Step 3:
H
O ~ OH
~z I / + R2 ( / OH
OH
Rs O
compound 11 compound 4
(R2 = H or OH)
H
OH
RZ / ~N / OH
O Rs
compound 12
15
CA 02267657 1999-03-30
Scheme 2b:
Stepl
PG o
OH
OH
H N~ I
2 OH
compound 1a
a~mpound 9a
C ~ OH
.~ N I
HN OH
Kd compound 10a
Step 2:
G p ~ OH
N I / -
htN OH
compound 10a
Rd
21
CA 02267657 1999-03-30
,OH
~OH
compound 11a
Step 3:
HO
O ~ OH \
+ ( / OH
N R
/ OH
O
compound 11a compound 4
(RZ = H or OH)
Rd
HO
N \ OH
R2
O N / OH
O
compound 12a
I 0 The second approach illustrated in scheme 2 above proceeds by the C-
terminal first with the
subsequent coupling taking place at a later stage after the removal of the
amino blocking group.
Thus, for step 1 of scheme 2a compound 1 (e.g. a Boc amino acid) is coupled
with dopamine
(compound 9) using EDC and HOBT as coupling reagents in DMF to obtain compound
10. For
step 2 of scheme 2a compound 10 is treated to remove the protecting or
blocking group PG to
obtain compound 11; for example, the removal of a Boc group may be performed
by stirring
compound la in a fiure of TFA aad methylene chloride at room temperature for a
short period
of time. For step 3 of scheme 2a compound 11 may then be coupled with the
appropriate
22
CA 02267657 1999-03-30
hydroxybenzoic acid (compound 4) using the EDC/HOBT coupling conditions to
obtain
compound 12. The desired product compound 12 may then be deprotected if needed
or
appropriate by hydrogenolysis using Pd/C as catalyst for those amino acid with
functionality on
the side chain. Scheme 2b may proceed in an analogous fashion.
Scheme 3 illustrates yet another example method for the preparation of a
derivative in accordance
with the present invention
Note:
a) For scheme 3, PG and PG" may be any suitable (known) independently
removable protecting group for respectively protecting different functional
groups
including a nitrogen atom. PG may, for example, be Boc i.e. tert-
butyloxycarbonyl
and PG" may, for example, be Fmoc, i.e. fluorenylmethyloxycarbonyl
b) For scheme 3, R4 may, for example, be -HNCHzCH2CHz- or a group of formula
N
and RS may, for example, be HzNCH2C:H2CH2- or a group of formula
N
23
CA 02267657 1999-03-30
Step 1
O ~ OH
OH
PG" R HZN /
(e.g. Fmoc) 4vPG OH
compound 13 compound 9
O ~ OH
NH
PG" ~ ~ /
N OH
R4
LPG
compound 14
Step 2
OH
O
PG"~ /
HN OH
R4
LPG oorrpourtd 14
N
H
24
a~mpomd 15
CA 02267657 1999-03-30
Step 3:
O ~ OH H
R2=HorOH
HN / OH R ~ / OH
2
\PG compound 15 ~P~nd 4
H
O ~ ~ OH
R~ / ~ /
OH
O ~~ compound 16
PG
Step 4:
Ho
O ~ OH
NH
R2 / N / OH
O ~ compound tE
PG
HO
O ~ OH
NH
R2 / N / OH
K5
compound 17
In scheme 3 illustrated above the starting amino acid (compound 13) is
provided with a pair of
independently removable protecting groups PG and PG"; the amino group may have
a protecting
group (PG") such as Fmoc for example. On the other hand if the group R4
includes a primary or
secondary amino component a protecting group PG may likewise be attached to
the nitrogen atom
I S of such an amino component; PG may, for example, be Boc or tert-
butyloxycarbonyl. Thus, for
CA 02267657 1999-03-30
step 1 of scheme 3 compound 13 is coupled with dopamine (compound 9) using EDC
and HOBT
as coupling reagents in DMF to obtain compound 14. For step 2 compound 14 is
treated to
remove the protecting or blocking group PG" to obtain compound 15. For step 3
compound 15
may then be coupled with the appropriate hydroxybenzoic acid (compound 4)
using the
EDC/HOBT coupling conditions to obtain compound 16 . For step 4 compound 15 is
treated to
remove the protecting group PG to yield compound 1.7.
Scheme 4 illustrates in a generic fashion an example method for the
preparation of a dipeptide
derivative in accordance with the present invention (see example 42 below for
a more specific
description of a process for making a dipeptide derivative):
Step 1
Ho ~ o
O ~ OH
NH
NHz
N / OH
I we Rx
O
compound 19
compound I S
26
CA 02267657 1999-03-30
O
O ~ OH
RZ
I NH ~ /
H / O ~'~1 OH
RX
compound 20
The compounds listed in Table 1 were prepared by following Scheme 1 or Scheme
2 (see
examples below); the numbers) in brackets after each root amino acid name is
the numer(s) of an
examples) below. Their activities are also listed in the same table
demonstrating their potential
usefulness.
Table 1. Anti-integrase activity f ICSO) of amino acid derivatives in
accordance with formula Ic above
Root Amino acid (i.e. Anti-integrase activity (ICso)
W for formula I is the 4-hydroxy derivative 3, 4-dihydroxy derivative
fragment thereof) ~M ,uM
Glycine (ex. 15) 100
L-Glutamic (ex. 21 & 22 - 64 11
step B )
L-Glutamic-4-O-benzyl (ex. 26
23 )
L-Tyrosine (ex. 11 & 12) 88 6
27
CA 02267657 1999-03-30
L-Tryptophan (ex. 29 & 30) 245 17
L-Proline (ex. 27 & 28) >200 gp
L-Leucine (ex. 25 & 26) >200 45
L-Phenylalanine (ex. 13 & >200 45
14)
L-Serine (ex. 18) 100
L-Methionine (ex. 31) 100
L-Dopa (ex. 16) g
D-Tyrosine (ex. 10) 67
D-Tyrosine-O-benzyl (ex. 10 42
- step D)
L-Alanine (ex. 19 & 20) 160 71
Dipeptide derivatives were also prepared and are listed in Table 2 ; the
numbers) in Table 2 with
respect to each product structure name therein indicated a number of an
example.
Table 2. Anti-integrase activity (ICSO) of dipeptide derivatives of I
Product X Y Anti-integraseExample
activity Number
(ICso)
X- Tyr-Tyr-Y 3,4-dihydroxybenzoylOH 177 36
X- Tyr-Tyr-Y 3,4-dihydroxybenzoyl3,4-dihydroxyphenethylamino17 52
X-Gly-Tyr-Y 3,4-dihydroxybenzoylOH > 200 39
28
CA 02267657 1999-03-30
X-Tyr- 3,4-dihydroxybenzoyl3,4-dihydroxyphenethylamino20 41
Asp(OBn)-Y
X-Tyr-Gly-Y 3,4-dihydroxybenzoylOH > 200 37
I O As can be appreciated by the skilled artisan, the above synthetic schemes
are not intended to
comprise a comprehensive list of all means by which the compounds described
and claimed in this
application may be synthesized. Further methods will be evident to those of
ordinary skill in the
art.
I S For the purposes of Table 1 (and Table 2) the HIV-1 integrase inhibition
assay was carried out as
follows. The 3'-processing and strand transfer reactions were performed with 1
picomole (pmole)
of duplex substrate consisting of S50-20 (5'-TGTGGAAAATCTCTAGCAGT-3')
radiolabeled at
the 5'-end with 3zP, SOS-20 (S'-ACTGCTAGAGATTTTCCACA-3'), and 400 ng of
purified HIV-I
integrase in 20 mM MOPS, pH 7.2, SO mM NaCI, 1 mM DTT, 0.1 mg/mL BSA, 10%
DMSO,
20 10% glycerol and 7.5 mM of either MnClz or MgClz in a total volume of 20
~L. For evaluating the
inhibitory potential of anti-integrase compounds, the enzyme was incubated in
the same conditions
in the presence of various concentrations of the compounds. The reactions were
carried out at 37°C
for 60 min and stopped by the addition of 1 volume of loading buffer
containing 95% formamide,
25 mM EDTA (pH 8.0), 0.05% bromophenol blue and 0.05% xylene cyanol.
The samples were heated at 95°C for 3 to 5 min just prior to gel
loading. Unless indicated
otherwise, 3 ~L of samples were loaded on 0.5 mm thick, 20% denaturing
polyacrylamide gels
containing 8 M urea, prepared in 89 mM Tris-HCI, pH 8.4, 89 mM boric acid, 1
mM EDTA
buffer (TBE). The gels were run at 200 V (20 mA) for approximately 90 min. At
the end of the
run, the gels were sealed in Saran Wrap plastic and directly exposed for
autoradiography at -70 °C
using Kodak X-GMAT LS films. Densitometric measurements were made using a Bio-
Rad model
29
CA 02267657 1999-03-30
GS-670 Imaging Densitometer apparatus with Molecular Analyst software version
1.3. The
inhibitory concentration (ICSOj of the anti-integrase compounds was determined
as the amount of
compound needed to decrease the activity of integrase by 50% compared to the
reaction carried out
in the absence of inhibitor.
The novel compounds of the present invention are excellent ligands for
integrase, particularly
HIV-1, and most likely HIV-2 and HTLV-1 integrase. Accordingly, these
compounds are capable
of targeting and inhibiting an early stage event in the replication, i.e. the
integration of viral DNA
into the human genome, thus preventing the replication of the virus.
In addition to their use in the prophylaxis or treatment of HIV infection, the
compounds according
to this invention may also be used as inhilrito~~ car interruptive agents for
other viruses which
depend on integrases, similar to HIV integrases, for obligatory events in
their life cycle. Such
compounds inhibit the viral replication cycle by inhibiting integrase. Because
integrase is essential
for the production of mature virions, inhibition of that process effectively
blocks the spread of
virus by inhibiting the production and reproduction of infectious virions,
particularly from acutely
infected cells. The compounds of this invention advantageously inhibit
enzymatic activity of
integrase and inhibit the ability of integrase to r.~a~talyze the integration
of the virus into the genome
of human cells.
The compounds of this invention may be employed in a conventional manner for
the treatment or
prevention of infection by HIV and other viruses which depend on integrases
for obligatory events
in their life cycle. Such methods of treatment, their dosage levels and
requirements may be
selected by those of ordinary skill in the art from available methods and
techniques. For example a
compound of this invention may be combined with a pharmaceutically acceptable
adjuvant for
administration to a virally infected patient in a pharmaceutically acceptable
manner and in an
amount effective to lessen the severity of the viral infection. Also, a
compound of this invention
CA 02267657 1999-03-30
may be combined with pharmaceutically acceptable adjuvants conventionally
employed in
vaccines and administered in prophylactically effective amounts to protect
individuals over an
extended period of time against viral infections, such as HIV infection. As
such, the novel
integrase inhibitors of this invention can be administered as agents for
treating or preventing viral
infections, including HIV infection, in a mammal. The compounds of this
invention may be
administered to a healthy or HIV-infected patient either as a single agent or
in combination with
other antiviral agents which interfere with the replication cycle of HIV. By
administering the
compounds of this invention with other antiviral agents which target different
events in the viral
replication cycle, the therapeutic effect of these compounds is potentiated.
For instance, the co-
administered antiviral agent can be one which targets early events in the life
cycle of the virus,
such as cell entry, reverse transcription and viral DNA integration into
cellular DNA. Antiviral
agents targeting such early life cycle events include, didanosine (ddI),
zalcitabine (ddC), stavudine
(d4T), zidovudine (AZT), polysulfated polysaccharides, sT4 (soluble CD4) --
which blocks
attachment or adsorption of the virus to host cells -- and other compounds
which block binding of
virus to CD4 receptors on CD4-bearing T-lymphocytes. Other retroviral reverse
transcriptase
inhibitors, such as derivatives of AZT, may also be co-administered with the
compounds of this
invention to provide therapeutic treatment for substantially reducing or
eliminating viral infectivity
and the symptoms associated therewith. i..acamples of other antiviral agents
include ganciclovir,
dideoxycytidine, trisodium phosphonoformiate, eflornithine, ribavirin,
acyclovir, alpha interferon
and trimenotrexate. Additionally, non-ribonucleoside inhibitors of reverse
transcriptase, such as
TIBO or nevirapine, may be used to potentiate the effect of the compounds of
this invention, as
may viral uncoating inhibitors, inhibitors of trans-activating proteins such
as tat or rev, or
inhibitors of the viral protease. These compounds may also be co-administered
with other
inhibitors of HIV integrase.
Combination therapies according to this invention exert a synergistic effect
in inhibiting HIV
replication bECause each component agent of the combination acts on a
different site of HIV
31
CA 02267657 1999-03-30
replication. The use of such combinations also advantageously reduces the
dosage of a given
conventional anti-retroviral agent that would be required for a desired
therapeutic or prophylactic
effect as compared to when that agent is administered as a monotherapy. These
combinations may
reduce or eliminate the side effects of conventional single anti-retroviral
agent therapies while not
interfering with the anti-retroviral activity of those agents. These
combinations reduce potential of
resistance to single agent therapies, while minimizing any associated
toxicity. These combinations
may also increase the efficacy of the conventional agent without increasing
the associated toxicity.
Preferred combination therapies include the administration of a compound of
this invention with
AZT, 3TC, ddI, ddC or d4T.
Alternatively, the compounds of this invention may also be co-administered
with other HN
protease inhibitors such as Ro 31-8959 (Roche), L-735,524 (Merck), XM 323
(Dupont Merck) and
A-80,987 (Abbott) to increase the effect of therapy or prophylaxis against
various viral mutants or
members of other HIV quasi species.
We prefer administering the compounds of this invention as single agents or in
combination with
retroviral reverse transcriptase inhibitors, such as derivatives of AZT or HIV
aspartyl protease
inhibitors. We believe that the co-administration of the compounds of this
invention with retroviral
reverse transcriptase inhibitors or HIV aspartyl protease inhibitors may exert
a substantial
synergistic effect, thereby preventing, substantially reducing, or completely
eliminating viral
infectivity and its associated symptoms.
The compounds of this invention can also be administered in combination with
immunomodulators (e.g., bropirimine, anti-human alpha interferon antibody, IL-
2, GM-CSF,
methionine enkephalin, interferon alpha, diethyldithiocarbante, tumor necrosis
factor, naltrexone
and rEPO); antibiotics (e.g., pentamidine isethionate) or vaccines to prevent
or combat infection
and disease associated with HIV infection, such as A)DS and ARC.
32
CA 02267657 1999-03-30
When the compounds of this invention are administered in combination therapies
with other
agents, they may be administered sequentially or concurrently to the patient.
Alternatively,
pharmaceutical or prophylactic compositions according to this invention may be
comprised of a
combination of an integrase inhibitor of this invention and another
therapeutic or prophylactic
agent.
Although this invention focuses on the use of the compounds disclosed herein
for preventing and
treating HIV infection, the compounds of this invention can also be used as
inhibitory agents for
other viruses that depend on similar integrases for obligatory events in their
life cycle. These
viruses include, but are not limited to, other diseases caused by
retroviruses, such as simian
immunodeficiency viruses, HTLV-I and HTLV-II.
Pharmaceutical compositions of this invention comprise any of the compounds of
the present
invention, and pharmaceutically acceptable salts thereof, with any
pharmacentically acceptable
carrier, adjuvant or vehicle. Pharmaceutically acceptable carriers, adjuvants
and vehicles that may
be used in the pharmaceutical compositions of this invention include, but are
not limited to ion
exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as
human serum albumin,
buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate,
partial glyceride
mixtures of saturated vegetable fatty acids, water, salts or electrolytes,
such as protamine sulfate,
disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride,
zinc salts,
colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-
based substances,
polyethyleneglycol, sodium carboxymethylcellulose, polyacrylates, waxes,
polyethylene-
polyoxypropylene-block polymers, polyethylene glycol and wool fat.
The pharmaceutical compositions of this invention may be administered orally,
parenterally by
inhalation spray, topically, rectally, nasally, buccally, vaginally or via an
implanted reservoir. We
33
CA 02267657 1999-03-30
prefer oral administration or administration by injection. The pharmaceutical
compositions of this
invention may contain any conventional non-toxic pharmaceutically acceptable
carriers, adjuvants
or vehicles. The term "parenteral" as used herein includes subcutaneous,
intracutaneous,
intravenous, intramuscular, infra-articular, intrasynovial, intrasternal,
intrathecal, intralesional and
intracranial injection or infusion techniques.
The phanmaceutical compositions may be in the form of a sterile injectable
preparation, for
example, as a sterile injectable aqueous or oleaginous suspension. This
suspension may be
formulated according to techniques known in the art using suitable dispersing
or wetting agents
(such as, for example, Tween 80) and suspending agents. The sterile injectable
preparation may
1 S also be a sterile injectable solution or suspension in a non-toxic
parenterally acceptable diluent or
solvent, for example, as a solution in 1,3-butanediol. Among the acceptable
vehicles and solvents
that may be employed are mannitol, water, Ringer's solution and isotonic
sodium chloride
solutions. In addition, sterile, fixed oils are conventionally employed as a
solvent or suspending
medium. For this purpose, any bland fixed oil may be employed including
synthetic mono- or
diglycerides. Fatty acids, such as oleic acid and its glyceride derivatives
are useful in the
preparation of injectables, as are natural pharmaceutically-acceptable oils,
such as olive oil or
castor oil, especially in their polyoxyethylated versions. These oil solutions
or suspensions may
also contain a long-chain alcohol diluent or dispersant, such as Ph. Helv. or
a similar alcohol.
The pharmaceutical compositions of this invention may be orally administered
in any orally
acceptable dosage form including, but not limited to, capsules, tablets, and
aqueous suspension and
solutions. In the case of tablets for oral and canters which are commonly used
include lactose and
corn starch. Lubricating agents, such as magnesium stearate, are also
typically added. For oral
administration in a capsule form, useful diluents include lactose and dried
corn starch. When
aqueous suspensions are administered orally, the active ingredient is combined
with emulsifying
and suspending agents. If desired, certain sweetening and/or flavoring and/or
coloring agents may
34
CA 02267657 1999-03-30
S be added.
The pharmaceutical compositions of this invention may also be administered in
the form of
suppositories for rectal administration. These compositions can be prepared by
mixing a
compound of this invention with a suitable non-irritating excipient which is
solid at room
temperature but liquid at the rectal temperature and therefore will melt in
the rectum to release the
active components. Such materials include, but are not limited to, cocoa
butter, beeswax, and
polyethylene glycols.
Topical administration of the pharmaceutical compositions of this invention is
especially useful
when the desired treatment involves areas or organs readily accessible by
topical application. For
application topically to the skin, the pharmaceutical composition should be
formulated with a
suitable ointment containing the active components suspended or dissolved in a
carrier. Carriers
for topical administration of the compounds of this invention include, but are
not limited to,
mineral oil, liquid petroleum, white petroleum, propylene glycol,
polyoxyethylene
polyoxypropylene compound, emulsifying wax and water. Alternatively, the
pharmaceutical
compositions can be formulated with a suitable lotion or cream containing the
active compound
suspended or dissolved in a car~ier. Suitable carriers include, but are not
limited to mineral oil,
sorbitan monostearate, polysorbate 60, cetyl esters wax cetearyl alcohol, 2-
octyldodecanol, benzyl
alcohol and water. The pharmaceutical compositions of this invention may also
be topically
applied to the lower intestinal tract by rectal suppository formulation or in
a suitable neat
fornulation. Topically-transdernal patches are also included in this
invention.
The pharmaceutical compositions of this invention may be administered by nasal
aerosol or
inhalation. Such compositions are prepared according to techniques well-known
in the art of
pharmaceutical formulation and may be prepared as solutions in saline
employing benzyl alcohol
or other suitable preservatives, absorption promoters to enhance
bioavailability, fluorocarbons,
CA 02267657 1999-03-30
and/or other solubilizing or dispersing agents known in the art.
Dosage levels of between about 0.01 and about 25 mg/kg body weight per day,
preferably between
about 0.5 and about 25 mg/kg body weight per day of the active ingredient
compound are useful in
the prevention and treatment of viral infection, including HIV infection.
Typically, the
pharmaceutical compositions of this invention will be administered from about
1 to about 5 times
per day or alternatively, as a continuous infusion. Such administration can be
used as a chronic or
acute therapy. The amount of active ingredient that may be combined with the
carrier materials to
produce a single dosage form will vary depending upon the patient treated and
the particular mode
of administration. A typical preparation will contain from about 5% to about
75% active
compound (w/w). Preferably, such preparations contain from about 20% to about
50% active
compound.
Upon improvement of a patient's condition, a maintenance dose of a compound,
composition or
combination of this invention may be administered if necessary. Subsequently,
the dosage or
frequency of administration, or both, may be reduced, as a function of the
symptoms, to a level at
which the improved condition is retained. When the symptoms have been
alleviated to the desired
level, treatment should cease, at least in principle. Patients may, however,
require intermittent
treatment on a long-term basis, upon any recurrence of disease symptoms,
especially for AIDS.
As the skilled artisan will appreciate, lower or higher doses than those
recited above may be
required. Specific dosage and treatment regimen for any particular patient
will depend upon a
variety of factors, including the activity of the specific compound employed,
the age, body weight,
general health status, sex, diet, time of administration, rate of excretion,
drug combination, the
severity and course of the infection, the patient's disposition to the
infection and the judgment of
the treating physician.
36
CA 02267657 1999-03-30
The compounds of this invention are also useful as commercial reagents which
effectively bind to
integrases, particularly HIV integrase. As commercial reagent, the compounds
of this invention,
and their derivatives, may be used to block integration of a target DNA
molecule by integrase, or
may be derivatized to bind to a stable resin as a tethered substrate for
affinity chromatography
applications. These and other uses which characterize commercial integrase
inhibitors will be
evident to those of ordinary skill in the art.
DETAILED DESCRIPTION OF THE INVENTION
In order that the invention herein described may be more fully understood, the
following
detailed description is set forth. In the description, the following
abbreviations are used:
Desi anon Reagent or Fragment
Et ethyl
Trityl triphenylmethyl
Ala DL, D - or L-alanine
Asn DL, D- or L-asparagine
Cys DL, D- or L-cysteine
Gly glycine
Gln DL, D- or L-glutamine
His DL, D- or L-histidine
Ile DL, D- or L-isoleucine
Leu DL, D- or L-leucine
Met DL, D- or L-methionine
Phe DL, D- or L-phenylalanine
Pro DL, D- or L-proline
Ser DL, D- or L - serine
37
CA 02267657 1999-03-30
Thr DL, D- or L-threonine
Trp DL, D- or L-tryptophan
Val DL, D- or L-valine
Boc tert-butyloxycarbonyl
EDC 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide
BOP benzotriazol-1-yloxytris(dimethylamino)phosphonium
hexafluorophosphate
TFA trifluoroacetic acid
EtOAC ethyl acetate
DMF dimethylformanlide
AZT zidovudine
IL-2 interleukin-2
rEPO recombinant erythropoietin
EtOH ethyl alcohol
MeOH methyl alcohol
THF tetrahydrofuran
CHzCl2 dichloromethane
Clz-Bzl 2,6-dichlorobenzyl
tert-Bu tert-butyl
Bzl benzyl
NMP N methylpyrrolidone
CHC13 chloroform
Combinations of substituents and variables envisioned by this invention are
only those that result
in the formation of stable compounds.
The term stable, as used herein, refers to compounds which possess stability
sufficient to allow
38
CA 02267657 1999-03-30
manufacture and administration to a mammal by methods known in the art.
Typically, such
compounds are stable at a temperature of 40 °C or less, in the absence
of moisture or other
chemically reactive conditions, for at least a week.
EXAMPLES
In order that this invention be more fully understood, the following examples
are set forth. These
examples are for the purpose of illustration only and a~-e not to be construed
as limiting the scope of
the invention in any way.
Materials and Methods
Analytical thin layer chromatography (TLC) was carried out with 0.25 mm silica
gel E. Merck 60 FZSa
plates and eluted with the indicated solvent systems. Preparative
chromatography was performed by
flash chromatography, using silica gel 60 (EM Science) with the indicated
solvent systems and a
positive nitrogen pressure to allow proper rate of elution. Detection of the
compounds was carried out
by exposing eluted plates (analytical or preparative) to llV light and/or
treating analytical plates with
a 2% solution ofp-anisaldehyde in ethanol containing 3% sulfuric acid and 1%
acetic acid followed
by heating.
Unless otherwise indicated, all starting materials were purchased from a
commercial source such as
Aldrich Co. or Sigma Co.
Nuclear magnetic resonance (NMR) spectra were recorded on a Broker AMX 500
equipped with a
reversed or QNP probe. Samples were dissolved in deuterochloroform (CDC13),
deuteroacetone
(acetone-db) or deuterodimethylsulfoxide (DMSO-db) for data acquisition using
tetramethylsilane as
internal standard. Chemical shifts are expressed in parts per million (ppm),
the coupling constants (J)
39
CA 02267657 1999-03-30
are expressed in hertz (Hz) whereas multiplicities are denoted as s for
singlet, d for doublet, dd for
doublet of doublets, t for triplet, q for quartet, m for multiplet, and br for
broad.
GENERAL PROCEDURES
Example 1. Preparation of N (tent-butyloxycarbonyl)amino acids
To a solution of amino acid (1 eq.) in water and dioxane were added at room
temperature
triethylamine (1.3-1.5 eq.) and Boc-ON (1.1 eq.) or di-tert-butyl-dicarbonate
(2 eq.). The mixture
was stirred at room temperature under argon for 3 to 5 h. The solution was
diluted with water and
extracted by ether at least six times. The aqueous layer was acidified to pH ~
2.5 with cold 1N HCl
to yield an oily layer. The mixture was extracted three times with methylene
chloride. The
combined organic extracts were washed with brine and dried over magnesium
sulfate. After
filtration, the filtrate was evaporated using a bath set at 30°C. The
residue was found to be of
sufficient purity for the next reaction step.
Example 2. Benzylation of N Boc amino acid
Three different solvent systems were used to achieve benzylation of acids or
hydroxyl groups.
a) methanol/water followed by DMF method
To a N Boc amino acid (1 eq.) in methanol was added cesium carbonate (1.4-2.0
eq.) as a 20%
solution in water, and then the solution was evaporated to dryness. The
residue was dissolved in
dimethylformamide (DMF) and benzyl bromide ( 1.1-1.5 eq.) was added. The
mixture was stirred
at room temperature under argon overnight. The mixture was diluted with water
and the organic
layer was extracted with ethyl acetate. The combined organic phases were
washed with brine and
CA 02267657 1999-03-30
dried over magnesium sulfate. The solids were filtered off and solvent was
evaporated under
vacuum yielding a residue that was purified by silica gel chromatography using
20% ethyl acetate
in hexane.
b) dimethylformamide method
To a N Boc amino acid ( 1 eq.) in dimethylformamide (DMF) were added cesium
carbonate ( 1.4-
2.0 eq.) and benzyl bromide (1.1-1.5 eq.). The reaction mixture was stirred at
room temperature
overnight under argon. A work-up and purification as previously described in
example 2a yielded
the desired product.
c) acetone method
To a N Boc amino acid (1 eq.) in acetone were added potassium carbonate (1.4-
2.0 eq.) and
benzylbromide (1.1-1.5 eq.). The reaction mixture was stirred at room
temperature for a period
of 3 - 5 h under argon. Work-up and purification as carried out in the
previous example 2a
afforded the desired product.
Example 3. Removal of the N tent-butyloxycarbonyl (Boc) group
A solution of N tert-butyloxycarbonyl amino acid (1 eq.) in a 1:1 mixture of
trifluoroacetic acid
(TFA) ( 10 eq.) and methylene chloride (CHZC12) was stirred at room
temperature for 15-30 min.
The solvent and excess acid were removed under vacuum to yield the desired
product that was
used without further purification.
41
CA 02267657 1999-03-30
Example 4. Coupling reaction of hydroxylated benzoic acid with the NH part of
an
amino acid
To a mixture of 3-hydroxy- or 3,4-dihydroxybenzoic acid (1.5 eq.),
hydroxybenzotriazole
hydrate (HOBT) (1.6 eq.), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide
(EDC) (1.6
eq.) in DMF was added a solution of product from example 3 (1 eq.) and
triethylamine or
diisopropylethylamine (1 eq.) in DMF. The mixture was stirred at room
temperature under
argon for either 6 h or overnight, monitoring the reaction by TLC. The
reaction mixture was
quenched by water and extracted three times with ethyl acetate. The organic
phases were
combined and washed with brine. After drying over magnesium sulfate, the
solution was
filtered and the solvent was evaporated under vacuum. The residue was purified
by silica gel
chromatography, eluting as indicated in each procedure.
Example 5. Cleavage of benzyl esters or benzyl ethers
The benzyl ester or benzyl ether of an amino acid derivative dissolved in
methanol was
hydrogenated over 10% Pd-C (less than 10% by weight of the weight the amino
acid benzyl
ester or ether) under 1 atmosphere of HZ for 1-2 h. The catalyst was filtered
off and the filtrate
was evaporated under vacuum to yield the desired product.
Example 6. Coupling reaction of dopamine with the COOH of a substituted amino
acid
To a solution of substituted carboxylic acid ( 1 eq.) prepared as in example
5, HOBT ( 1.5 eq.) and
EDC (1.5 eq.) in DMF at 0°C was added a solution of dopamine
hydrochloride (2 eq.) and
triethylamine or diisopropylethylamine (2 eq.) in DMF. The mixture was stirred
under argon for
0.5 h and the mixture was allowed to reach room temperature and stirred
overnight. The resulting
mixture was diluted with water and extracted three times with ethyl acetate.
The organic phases
42
CA 02267657 1999-03-30
were combined and washed with brine. After drying over magnesium sulfate, the
solution was
filtered and the solvent was evaporated under vacuum. The residue was purified
by silica gel
chromatography, eluting agent as indicated in each procedure.
Example 7. Removal of the N fluorenylmethyloxycarbonyl (Fmoc) group
A solution of N fluorenylmethyloxycarbonylamino acid (1 eq.) in 30%
diethylamine in
acetonitrile was stirred 15 min at room temperature. The solvent was removed
under vacuum to
yield the desired product that was used without further purifcation.
Example 8. Removal of the methyl ester group
Amino acid methyl ester (0.2 eq.) was dissolved in methanol at room
temperature 1N sodium
hydroxide (0.65 mL) was added, the mixture was stirred for 0.5 h and 1N HC1
(0.3 mL) was
added, maintaining the temperature at around 0°C. After removing the
methanol under vacuum,
a second portion of 1N HCl (0.3 mL) was added to adjust the pH at ~2.5. The
organic acid was
extracted with CHzCl2, dried over magnesium sulfate and concentrated in vacuo,
yielding the
desired product that was used for the next step without further purification.
Example 9. Coupling reaction of hydroxylated benzoic acid with the NH part of
an amino
acid using BOP reagent
The acid (O.1M in a 1-1 mixture of dioxan and dichloromethane) and BOP reagent
(1.0 eq.) was
stirred at room temperature under an inert atmosphere. The amine (1.2 eq.) was
directly added
followed by the base (triethylamine, 1.2 eq.). The reaction was stirred for 3
to 16 h. The
suspension was then poured in an extraction vessel containing ethyl acetate
and 1N hydrochloric
acid and the organic layer washed with 3 portions of water before drying over
magnesium sulfate.
43
CA 02267657 1999-03-30
The solution was filtered and concentrated in vacuo before purification by
flash chromatography.
Specific examples for the preparation of derivatives in accordance with the
present invention
Example 10. Preparation of the N [N'-(3',4'-dihydroxybenzoyl)-D-tyrosylJ-
dopamine
Step A. Preparation of N (tert-butyloxycarbonyl)-D-tyrosine
The title compound was prepared from D-tyrosine (543 mg, 3.0 mmol), by
following the
procedure described in example 1. The product was isolated as a colorless
syrup (740 mg, 88%
yield).
'H NMR (DMSO-db): 1.32 (s, 9H); 2.71 (dd, J = 3.3, 12.9, 1 H); 2.89 (dd, J =
3.8, J = 12.3,
1H); 4.00 (m, 1H); 6.64 (d, J = 8.6, 2H); 6.95 (d, J = 8.0, 1H); L03 (d, J =
8.6, 2H); 9.18 (br,
1 H); 12.46 (s, 1 H).
Step B. Preparation of N (tert-butyloxycarbonyl)-O-benzyl-D-tyrosine benzyl
ester
The title compound was prepared from the product obtained in step A of this
example (650
mg, 2.3 mmol) according to the indications of example 2a. The crude product
was purified by
silica gel column chromatography using 5% MeOH/CHC13 to yield the desired
product (650
mg, 61 %).
'H NMR (DMSO-db): 1.32 (s, 9H); 2.83 (dd, J = 9.8, 13.5, 1 H); 2.93 (dd, J =
5.5, 13.8, 1 H);
4.17 (q, J = 7.1, 8.3, 1H); 5.05 (s, 2H); 5.08 (s, 2H); 6.91 (d, J = 8.2, 2H);
7.13 (d, J = 8.2,
2H); 7.28-7.44 (m, lOH).
44
CA 02267657 1999-03-30
Step C. N (3',4'-dihydroxybenzoyl)-O-benzyl-D-tyrosine benzyl ester
The title compound was prepared from the product obtained in step B of this
example (110
mg, 0.24 mmol) by the removal of the Boc group following the indications of
example 3. The
resulting unblocked derivative was then coupled with 3,4-dihydroxybenzoic acid
according to
the indications of example 4. The crude product was purified by silica gel
column
chromatography using 5% methanol/chloroform to yield the desired product as a
white solid,
mp. 140°C (dec.), (88 mg, 74%).
'H NMR (CDC13): 3.15 (m, 2H); 4.98 (s, 2H); 5.01 (m, 1H); 5.18 (m, 2H); 6.63
(d, J = 7.4,
1H); 6.80 (d, J = 7.4, 2H); 6.85 (d, J = 8.9, 1H); 6.92 (d, J = 7.4, 2H); 7.08
(d, J = 7.4, 2H);
7.26 (s, 1H); 7.31-7.41 (m, lOH).
Step D. N [N'-(3',4'-dihydroxybenzoyl)-O-benzyl-D-tyrosyl]-dopamine
The title compound was prepared from the product of step C of this example
(200 mg, 0.39
mmol) by removing the benzyl ester group following the indications of example
5. The
resulting unblocked derivative was coupled with dopamine hydrochloride
according to the
indications of example 6. Purification by silica gel chromatography
(3%MeOH/EtOAc)
provided the desired product, (88 mg, 40%) as a white solid, mp. 131°C
(dec.).
'H NMR (DMSO-d6): 2.60 (m, 2H), 3.03 (dd, J = 7.9, 13.7, 1H); 3.16 (dd, J =
5.3, 14.2,
1 H); 3.36 (m, 2H); 4.79 (m, 1 H); 5.02 (s, 2H) 6.69 (d, J = 7.9, 1 H); 6.71
(s, 1 H); 6.85 (d, J =
7.9, 1H); 6.89 (d, J = 8.8, 2H); 7.19 (d, J = 6.1, 2H); 7.27 (t, J = 6.7, 1H);
7.30 (s, 1H); 7.34
(d, J = 7.4, 1H); 7.30-7.47 (m, SH); 7.58 (d, J = 7.7, 1F1); 8.04 (b, 4H).
Step E N [N'-(3',4'-dihydroxybenzoyl)-D-tyrosyl]-dopamine
CA 02267657 1999-03-30
The title compound was prepared from the product of step D of this example (60
mg, 0.11
mmol) by following the indications in example 5. Purification by silica gel
chromatography
(100% EtOAc) provided the desired product, (21 mg, 43%) as a white solid, mp.
178°C
(dec.).
'H NMR (DMSO-d6): 2.60 (m, 2H); 2,89 (dd, J = 8.0, 13.9, 1 H); 3.11 (dd, J =
5.5, 13.9, 1 H);
3.36 (m, 2H); 4.74 (m, 1H); 6.50 (d, J = 7.0, 1H); 6.68 (d, J = 8.1, 1H); 6.72
(d, J = 6.3, 1H);
6.73 (s, 1H); 6.83 (d, J = 7.5, 1H); 7.11 (d, J = 8.0, 2H); 7.38 (d, J = 7.4,
2H); 7.26 (t, J = 6.7,
1 H); 7.48 (d, J = 7.6, 1 H); 8.04 (b, SH).
Example 11. Preparation of N [N'-(p-hydroxybenzoyl)-L-tyrosyl]-dopamine
Step A. Np-hydroxybenzoyl-O-tert-butyl-L-tyrosine tert-butyl ester
The title compound was prepared from O-tert-butyl-L-tyrosine tert-butyl ester
(445 mg; 1.60
mmol) by following the indications of example 4. Purification by flash
chromatography
eluting with 20% ethyl acetate in hexane provided 291 mg (S 1 %) of the title
compound, mp.
106°C.
'H NMR: (CDC13) : 1.31 (s, 9H), 1.41 (s, 9H), 3.18 (d, J = 5.6, 2H), 4.91 (m,
1H), 6.68 (d, J =
7.3, 1H), 6.82 (d, J = 8.0, 2H), 6.92 (d, J = 8.3, 2H), 7.07 (d, J = 8.0, 2H),
7.56 (d, J = 8.3,
2H), 8.41 br, 1H).
Step )3. N (N'-(p-benzoyl)-L-tyrosyl]-dopamine
46
CA 02267657 1999-03-30
The tert-butyl protecting group was removed by treatment of the product of
step A of this
example (21 mg, 0.05 mmol) with trifluoroacetic acid according to the
conditions in example
3. The residue was treated without further purification with dopamine
hydrochloride
according to the procedure of example 6, providing the title product (16 mg,
53%), mp.
161°C.
'H NMR (acetone-db): 2.52 - 2.61 (m, 2H), 2.95 (dd, J = 14.4, 8.2, 1H), 3.10
(dd, J = 14.4,
8.2, 1H), 3.27 - 3.37 (m, 2H), 4.71 - 4.76 (m, 1H), 6.47 (d, J = 8.4, 1H),
6.63 - 6.70 (m, 4H),
6.82 (d, J = 6.9, 2H), 7.07 (m, 2H), 7.38 (d, J = 5.2, 1H), 7.54 (d, J = 8.0,
1H), 7.69 (d, J = 7.7,
2H), 7.4 - 7.9 (br, 2H), 8.12 (br, 1H).
Example 12. Preparation of N [N'-(3',4'-dihydroxybenzoyl)-L-tyrosyl]-dopamine
Step A. N 3',4'-dihydroxybenzoyl-O-tert-butyl-L-tyrosine tert-butyl ester
The title compound was prepared from O-tert-butyl-L-tyrosine tert-butyl ester
(500 mg; 1.8
mmol) by following the indications of example 4. Purification by flash
chromatography
eluting with 10% methanol in methylene chloride provided 511 mg (80%) of the
title
compound, mp. 122°C.
'H NMR: (CDC13) : 0.87 (s, 9H), 0.88 (s, 9H), 3.16 (m, 2H), 4.78 (d, J = 7.5,
1H), 6.85 (d, J =
9.1, 1H), 6.89 (d, J = 8.5, 2H), 7.21 (d, J = 8.5, 2H), 7.29 (d, J = 9.1, 2H),
7.42 (s, 1H), 7.49
(d, J = 7.5, 2H), 8.29 br, 1 H), 8.50 (br, 1 H).
Step B. N [N-(3',4'-dihydroxybenzoyl)-L-tyrosyl]-dopamine
47
CA 02267657 1999-03-30
The tert-butyl protecting group was removed by treatment of the product of
step A of this
example (184 mg, 0.42 mmol) with trifluoroacetic acid according to the
indications of
example 3. The residue was treated without further purification with dopamine
hydrochloride
according to the procedure of example 6, providing the title product (128 mg,
66%), mp.
205°C.
'H NMR (acetone-db): 2.56 (m, 2H), 2.99 (dd, J = 13.6, 7.9, 1H), 3.11 (dd, J =
13.6, 5.3, 1H);
3.36 (m, 2H), 4.78 (m, 1 H), 6.46 (d, J = 7.8, 1 H), 6.64 (d, J = 7.8, 1 H),
6.68 s, 1 H), 6.70 (d, J
= 8.3, 2H), 6.80 (d, J = 8.6, 1 H), 7.09 (d, J = 8.3, 2H), 7.24 (d, J = 8.6, 1
H), 7.39 (s, 1 H), 7.55
(m, 1 H), 7.65 (d, J = 7.6, 1 H), 7.5 - 8.4 (br, SH).
Example 13. Preparation of N [N'-(p-hydroxybenzoyl)-L-phenylalanyl]-dopamine
Step A. Np-hydroxybenzoyl-L-phenylalanine benzyl ester
The title compound was prepared from L-phenylalanine benzyl ester (400 mg;
1.58 mmol) by
following the indications of example 4. Purification by flash chromatography
eluting with
10% ethyl acetate in hexane containing 1% acetic acid provided 323 mg (92%) of
the title
compound, mp. 163°C.
'H NMR: (CDC13) : 3.16 (dd, J = 12.8, 8.9, 1 H), 3.24 ( dd, J = I2.8, 8.9, 1
H), 4.92 (m, 1 H),
5.12 (s, 2H), 6.83 (d, J = 6.6, 2H), 7.15 - 7.31 (m, lOH), 7.70 (m, 3H), 8.90
(br, 1H).
Step B. N [N'-(p-benzoyl)-L-phenylalanyl)-dopamine
The product obtained in step A of this example (287 mg, 0.76 mmol) following
the
indications of examples 5 and 6, provided, after flash chromatography eluting
with 50% ethyl
48
CA 02267657 1999-03-30
acetate in dichloromethane containing 1 % acetic acid, the desired product
(301 mg, 94%), mp.
196°C.
'H NMR (acetone-db): (two conformers) 2.48 (t, J = 7.6, 2H), 2.60 (t, J = 7.8,
2H), 2.93 (m,
1 H), 3.00 (dd, J = 13.3, 3.8, 1 H), 3.13 - 3.25 (m, 1 H), :3.32 (m, 1 H),
4.59 (m, 1 H), 6.41 (d, J =
7.1, 1H), 6.57 (m, 2H), 6.76 (m, 2H), 7.12 (m, 1H), 7.21 (d, J = 7.4, 2H),
7.26 (d, J = 7.5, 1H),
7.65 (d, J = 7.4, 2H), 8.00 (t, J = 5.7, 1 H), 8.21 (d, J = 8.3 ( 1 H), 8.59
(s, 1 H), 8.69 (s, 1 H),
9.92 (s, 1H).
Example 14. Preparation of N [N'-(3',4'-dihydroxybenzoyl)-L-phenylalanyl]-
dopamine
Step A. N 3,4-dihydroxybenzoyl-L-phenylalanine benzyl ester
The title compound was prepared from L-phenylalanine benzyl ester (400 mg;
1.57 mmol) by
following the indications of example 4. Purification by flash chromatography
eluting with
40% ethyl acetate in hexane containing 1% acetic acids provided 323 mg (60%)
of the title
compound, mp. 155°C.
'H NMR: (CDC13) : 3.24 (dd, J = 13.6, 8.5, 1H), 3.32 (dd, J = 13.6, 5.2, 1H),
4.92 (m, 1H),
5.03 (m, 1H), 5.17 (s, 2H), 6.92 (d, J = 8.0, 1H), 7.20 - 7.36 (m, 11H), 7.53
(s, 1H), 7.88 (d, J
= 7.3, 1H), 8.57 (br, 1H).
Step B. N [N'-(3',4'-benzoyl)-L-phenylalanyl]-dopamine
The product obtained in step A of this example (18 mg, 0.046 mmol) following
the
indications of examples 5 and 6, provided after flash chromatography eluting
with SO% ethyl
acetate containing 1% acetic acid, the desired product (19 mg, 73%), mp.
201°C.
49
CA 02267657 1999-03-30
'H NMR (acetone-db): 2.52 - 2.59 (m, 2H), 3.04 (dd, J = 13.7, 7.7, 1H), 3.18
(dd, J = 13.7,
7.7, 1 H), 3.32 (m, 2H), 4.77 (dt, J = 6.3, 7.6, 1 H), 6.45 (d, J = 7.1, 2H,
7.11 - 7.26 (m, 6H),
7.34 (s, 1H), 7.38 (m, 2H), 7.52 (d, J = 7.6, 1H), 7.98 (s, 4H).
Example 15. Preparation of N [N'-(3',4'-dihydroxybenzoyl)-glycyl]-dopamine
Step A. N (3,4-dihydroxybenzoyl)-glycine tert-butyl ester
The title compound was prepared from glycine tert-butyl ester (400 mg; 2.57
mmol) by
following the indications of example 4. Purification by flash chromatography
eluting with
40% ethyl acetate in hexane containing 1% acetic acid provided 337 mg (52%) of
the title
compound, mp. 139°C.
'H NMR: (acetone -db) : 1.52 (s, 9H), 4.09 (d, J = 5.5, 2H), 6.95 (d, J = 7.3,
1H), 7.42 (d, J =
7.3, 1 H), 7.55 (s, 1 H), 7.90 (br, 1 H), 8.34 (br, 1 H), 8.60 (br, 1 H).
Step B. N [lV'-(3',4'-benzoyl)-glycyl]-dopamine
The product obtained in step A of this example (277 mg, 1.03 mmol) following
the
indications of examples 3 and 6, provided, after flash chromatography eluting
with 30% ethyl
acetate in methylene chloride containing 1 % acetic acid, the desired product
( 163 mg, 45 %),
mp. 155°C.
'H NMR (acetone-d6): 2.58 (t, J = 7.1, 2H), 3.34 (dt, J = 7.1, 5.1, 2H), 3.99
(d, J = 5.3, 2H),
6.47 (d, J = 8.0, 1H), 6.64 (d, J = 8.0, 1H), 6.67 (s, 1H), 6.85 (d, J = 57.7,
1H), 7.31 (d, J = 7.7,
1H), 7.44 (s, 1H), 7.47 (t, J = 5.1, 1H), 7.94 (t, J = 5.0, 1H), 7.4 - 8.0
(br, 2H), 8.4 (br, 2H).
CA 02267657 1999-03-30
Example 16. Preparation of N [N'-(3',4'-dihydroxybenzoyl)-L-3,4-dihydroxy-
phenylalanyl]-dopamine
Step A. N (tert-butyloxycarbonyl)-O,O-dibenzyl-3,4-dihydroxy-phenyl-L-alanine
benzyl
ester
The title compound was prepared from L-3,4-dihydro~;yphenylalanine (DOPA) as
described
in examples 1 and 2b. In example 1, di-tert-butyl-dica~rbonate (960 mg, 4.4
mmol) was used
instead of Boc-ON to react with DOPA (790 mg, 4.0 rnmol) with triethylamine
(600 mg, 6.0
mmol) as base. The product was used for the next step without purification
following the
indications of example 2b, using 280 mg, (0.94 mmol). Purification by flash
chromatography
using 15% EtOAc/hexane provided the title compound as white crystals (180 mg,
34% ), mp.
106-108°C.
'H NMR (CDCl3): 1.41 (s, 9H); 3.00 (d, J = 4.9, 2H); 4.14 (s, 1H); 4.94 (d, J
= 7.4, 1H);
5.04-5.10 (m, 6H); 6.57 (d, J = 7.8, 1H); 6.78 (d, J = 8.4, 1H); 6.71 (s, 1H);
7.26-7.43 (m,
15H).
Step B. N (3',4'-dihydroxybenzoyl)-O,O'-(dibenzyl)-L-3,4-
dihydroxyphenylalanine benzyl
ester
The title compound was prepared by cleaving the laoc protecting group of the
product
prepared in step A of this example ( 130 mg, 0.23 nlmol) and coupling it with
3,4-
dihydroxybenzoic acid as described in example 3 and in example 4 respectively.
Purification by flash chromatography using S% Me;OH/CHC13 yielded the desired
product
as a white solid (86 mg, 60% ), mp. 215°C (dec.).
51
CA 02267657 1999-03-30
'H NMR (CDC13): 3.11 (m, J = 6.1, J = 14.1, 1H); 3.14 (dd, J= 6.7, 13.9, 1H);
5.00 (s,
1 H); 5.06-5.13 (m, 6H); 6.53 (d, J = 8.0, 1 H); 6.65 (d, J = 7.6, 1 H); 6.69
(s, 1 H); 6.76 (d, J
= 8.2, 1H); 6.82 (d, J = 8.1, 1H); 7,05 (d, J = 8.0, 11-i); 7.25-7.47 (m,
15H); 7.47 (s, 1H).
Step C. N (3',4'-dihydroxybenzoyl)-L-3,4-dihydroxyphenylalanine
The title compound was prepared by the reduction of the compound obtained in
step B of
this example (64 mg, 0.11 mmol) according to the indications found in example
5. The
product was purified by flash chromatography eluting with 10% MeOH/EtOAc,
affording
the desired product as a solid (27 mg, 73% yield), rnp. 121°C (dec.).
'H NMR (DMSO-db): 2.98 (m, 2H); 4.44 (m, 1H); 6.53 (d, J = 7.9, 1H); 6.59 (d,
J = 7.6,
1 H); 6.66 (s, 1 H); 6.75 (d, J = 8.2, 1 H); 7,17 (d, J =~ 7.9, 1 H); 7.24 (s,
1 H); 8.14 (d, J = 8.2,
1H); 8.69 (s, 2H), 9.11 (s, 2H); 12.50 (s, H).
Step D. N [N'-(3',4'-dihydroxybenzoyl)-L-3,4-dihydroxylphenylalanyl]-dopamine
The product from step C of this example was coupled with dopamine
hydrochloride
according to the procedure of example 6. Finally tile O-benzyl ether
protecting groups
were hydrogenolyzed by following the indications of example 5. Purification by
flash
chromatography using 5% MeOH/EtOAc yielded the desired product (13.8 mg, 28%
), mp.
142°C (dec.).
'H NMR (DMSO-d6): b 2.48 (m, 2H); 2.67 (m, 2H); 3.32 (m, 2H); 4.47 (m, J =
4.3, J = 9.1,
1 H); 6.42 (d, J = 7.0, 1 H); 6.51 (d, J = 7.4, 1 H); 6. _'> 8 (d, J = 7.5, 1
H); 6.59 (d, J = 8.4, 1 H);
52
CA 02267657 1999-03-30
6.66 (s, 1 H); 6.74 (d, J = 7.7, 1 H); 7,15 (s, 1 H); 7,17 (d, J = 7.6, 1 H);
7.22 (s, 1 H); 7.93 (t, J
= 5.7, 1 H); 7.95 (d, J = 8.5, 1 H); 8.67 (b, 6H, OH).
Example 17. Preparation of N (N'-(3',4'-dihydroxybenzoyl)-traps-4-
hydroxyprolyl]-
dopamine
Step A. N (tert-butyloxycarbonyl)-O-benzyl-traps-4-hydroxy-L-proline benzyl
ester
The title compound was prepared from traps-4-hyd~roxyproline as described in
example 1
1 S and 2b. The Boc derivative was prepared with the following quantities: di-
tert-butyl-
dicarbonate (960 mg, 4.4 mmol), traps-4-hydroxyproline (260 mg, 2.0 mmol),
triethylamine (300 mg, 3.0 mmol). The product wa:; used for the next step
without
purification. The benzylation was performed accordling to the indications of
example 2b.
Purification by flash chromatography using 20% EtOAc/hexane provided the title
compound as syrup (368 mg, 48%).
'H NMR (CDC13): 1.35 and 1.45 (s, 9H); 2.06 (m, 1H); 3.62 (m, 1H); 3.72 (m,
1H); 4.41
(t, J = 7.4, 1 H); 4.51 (m, 1 H); 5.1 S (s, 4H); 7.34-7.37 (m, 1 OH).
Step B. N (3,4-dihydroxybenzoyl)-O-benzyl-traps-4-hydroxy-L-proline benzyl
ester
The title compound was prepared by cleaving the Hoc group of the compound
obtained in
step A of this example (350 mg, 0.85 mmol) by following the indications of
example 3, and
coupling it with 3,4-dihydroxybenzoic acid (196 m,g, 1.27 mmol) according to
example 4.
Purification by flash chromatography using 5% MeOH/CHCl3, provided the desired
product as an oil (275 mg, 72.4% yield).
53
CA 02267657 1999-03-30
'H NMR (DMSO-db): 2.02 (m, 1H); 2.24 (m, 1H); 3.79 (d, J = 8.4,1H); 3.96 (d, J
= 10.1,
1 H); 4.18 (s, 1 H); 4.59 (t, J = 8.4, 1 H); 5.16 (s, 4EI); 6.77 (d, J = 7.9,
1 H); 6.87 (d, J = 7.7,
1 H); 6.98 (s, 1 H); 7.31-7.37 (m, IOH); 9.22 (b, 1 FI); 9.42 (b, 1 H).
Step C. N [N'-(3',4'-dihydroxybenzoyl)-t'~-benzylu-traps-4-hydroxyprolyl]-
dopamine
The title compound was prepared from the product obtained in step B of this
example
(420 mg, 1.0 mmol) by removal of the benzyl ester group as described in
example 5. The
resulting acid was then coupled with dopamine hydrochloride as described in
example 6.
Flash chromatography eluting with 1% M~OH/Et~OAc provided the desired product
as a
syrup ( 100 mg, 20%).
'H NMR (DMSO-db): 1.88 (m, 1H); 2.28 (m, 1H); 2.53 (m, 2H); 3.16 (m, 2H); 3.58
(d, J
= 11, 1H); 3.73 (d, J = 8.9, 1H); 4.11 (s, 1H) 4.35 (m, 2H); 4.48 (t, J = 7.7,
1H); 6.44 (d, J
= 7.7, 1 H); 6.5 8 (s, 1 H); 6.62 (d, J = 8.2, 1 H); 6. 77 (d, J = 8.2, 1 H);
6.90 (d, J = 7.9, 1 H);
7.00 (s, 1H); 7.21-7.30 (m, SH); 7.95 (t, J = 5.3, 1H); 8.62 (b, 1H); 8.72
(br, 4H); 9.16
(br, 1 H); 9.37 (br, 1 H).
Step D. N [N'-(3',4'-dihydroxybenzoyl)-traps-4-1'clydroxyprolyl]-dopamine
The title compound was prepared from the compound of step C of this example
(30 mg,
0.06 mmol) by following the indications of example 5. Purification by flash
column
chromatography with 3% MeOH/EtOAc afforded) the desired product (18 mg, 75%)
as
white crystals, mp. 59-62°C.
54
CA 02267657 1999-03-30
'H NMR (DMSO-db): 1.81 (m, 1H); 2.04 (m, 1H); 2.51 (m, 2H); 3.17 (m, 2H); 3.34
(d, J
= 6.6, 1 H); 3.69 (d, J = 8.3, 1 H); 4.21 (s, 1 H); 4.47 (t, J = 8.3, 1 H);
6.44 (d, J = 6.7, 1 H);
6.58 (s, 1 H); 6.61 (d, J = 7.7, 1 H); 6.76 (d, J = 8.2:, 1 H); 6.90 (d, J =
8.0, 1 H); 7.01 (s,
1 H); 8.90 (b, 4H); 7.90 (t, J = 5.3, 1 H).
Example 18. Preparation of N [N'-(3',4'-dihyfroxybenzoyl)-L-seryl]-dopamine
Step A. N (tert-butyloxycarbonyl)-O-benzyl-L-se;rine benzyl ester
The title compound was prepared from N (tert-butyloxycarbonyl)-O-benzyl-L-
serine,
(300 mg,l.0 mmol) as described in example 2b. Purification by flash
chromatography
eluting with 25% EtOAc/hexane provided the title compound as a syrup (370 mg,
96 %).
'H NMR (CDCl3): 1.38 (s, 9H); 3.68 (m, 2H); 4.:53 (q, J = 6.2, J = 6.0, 1H);
4.48 (m,
2H); 5.16 (m, 2H); 7.33 (m, lOH).
Step B. N (3,4-dihydroxybenzoyl)-O-benzyl-L-sf:rine benzyl ester
The title compound was prepared by cleaving the Boc group of the compound
obtained in
step A of this example (325 mg, 0.84 mmol) and coupling it with 3,4-
dihydroxybenzoic
acid as described in examples 3 and 4 respectively. Purification by flash
chromatography
eluting with 5% MeOH/CHCl3 provided the desired product (220 mg, 62 %).
'H NMR (DMSO-db): 3.83 (m, 2H); 4.52 (m, 2H); 4.74 (q, J = 6.3, J = 6.4, 1H);
5.16 (m,
2H); 6.78 (d, J = 8.4, 1H); 7.26 (d, J = 8.6, 1H); '1.28 (s, 1H); 7.29-7.32
(m; lOH); 8.42 (d,
J = 7.4, 1 H); 9.15 (b, 1 H); 9.50 (br, 1 H).
CA 02267657 1999-03-30
Step C. N [N'-(3',4'-dihydroxybenzoyl)-O-benzyl-L-Beryl]-dopamine
The title compound was prepared from the compound obtained in step B of this
example
(160 mg, 0.38 mmol) according to the procedures described in example 5 and 6
respectively. Purification by flash chromatography eluting with 2.5%
MeOH/EtOAc
provided the desired product (68 mg, 37%).
'H NMR (DMSO-db): 2.52 (m, 2H); 3.19 (m, 2H); 3.69 (d, J = 6.1, 2H); 4.49 (s,
2H);
4.64 (q, J = 5.6, J = 6.4, 1 H); 6.42 (d, J = 7.6, 1 H); 6.57 (s, 1 H); 6.60
(d, J = 8.2, 1 H);
6.77 (d, J = 8.3, 1H); 7.23 (d, J = 7.2, 1H); 7.25 (s, 1H); 7.29-7.327 (m,
SH); 8.01 (d, J =
7.9, 1 H); 8.02 (t, J = 5.4, 1 H); 8.62 (b, 1 H); 8.72 ( br, 1 H); 9.12 (br, l
H); 9.47 (br, l H).
Step D. N [N'-(3',4'-dihydroxybenzoyl)-L-serylJ-dopamine
The title compound was prepared from the compound obtained in step C of this
example
as described in example 5. The product was purified by flash chromatography
eluting
with 5% MeOH/EtOAc to provide the product (6ti%) as a solid, mp. 118°C
(dec.).
'H NMR (DMSO-db): 2.51 (m, 2H); 3.17 (m, 2H)~; 3.66 (d, J = 6.0, 2H); 4.39 (q,
J = 5.7, J
= 6.5, 1H); 6.43 (d, J = 7.9, 1H); 6.57 (s, 1H); 6.59 (d, J = 7.9, 1H); 6.79
(d, J = 8.5, 1H);
7.25 (d, J = 7.8, 1 H); 7.32 (s, 1 H); 7.86 (d, J = 7.8, 1 H); 7.95 (t, J =
5.4, 1 H); 9.00 (br,
SH).
Example 19. Preparation of N [N'-(p-hydroxyhenzoyl)-L-alanyl]-dopamine
Step A. N (tert-butyloxycarbonyl)-L-alanine bemzyl ester
56
CA 02267657 1999-03-30
The title compound was prepared from N (tert-butyloxycarbonyl)-L-alanine (567
mg, 3.0
mmol) as described in example 2a. Purification by flash chromatography eluting
with
20% EtOAc/hexane provided the desired product as a syrup (800 mg, 96 %).
'H NMR (CDCl3): 1.38 (d, J =7.3); 1.43 (s, 9H); 4.36 (s, 1H); 5.07 (s, 1H);
5.15 (m, 2H);
7.35 (s, SH).
Step B. N (p-hydroxybenzoyl)-L-alanine benzyl ester
The title compound was prepared from the product prepared in step A of this
example
(400 mg, 1.4 mmol) by removing the Boc group i:ollowing the indications of
example 3.
The resulting product was then coupled with p-hydroxybenzoic acid according to
example 4. Purification by flash chromatography eluting with 5% MeOH/CHC13
provided the desired product as white crystals (270 mg, 64%), mp. 82-
84°C.
'H NMR (DMSO-db): 1.40 (d, J = 7.1, 3H); 4.49 (m, 1H); 5.13 (s, 2H); 6.80 (d,
J = 8.2,
2H); 7.35 (s, 5H); 7.75 (d, J = 8.4, 2H); 8.53 (d, J = 6.8, IH); 9.99 (s, 1H).
Step C. N [N'-(p-hydroxybenzoyl)-L-alanyl]-dopamine
The title compound was prepared from the compound obtained in step B of this
example
(150 mg, 0.50 mmol) following the indications of examples 5 and 6 for the
cleavage of
the benzyl ester and the coupling with dopamine hydrochloride. Purification by
hash
chromatography eluting with 5% MeOH/EtOAc provided the desired product (147
mg,
85%), mp. 127°C (dec.).
57
CA 02267657 1999-03-30
'H NMR (acetone-db): 1.28 (d, J = 7.1, 3H); 2.51 (m, 2H); 3.17 (m, 2H), 4.40
(m, 1H);
6.43 (d, J = 7.8, 1H); 6.57 (s, 1H); 6.59 (d, J = 8.1), 1H); 6.80 (d, J = 8.0,
2H); 7.77 (d, J =
7.9, 2H); 7.86 (t, J = 5.1, 1H); 8.12 (d, J = 7.4, 1H:); 8.16 (s, 1H); 8.71
(s, 1H); 9.95 (s,
1 H).
Example 20. Preparation of N [N'-(3',4'-dibydroxybenzoyl)-L-alanyl]-dopamine
Step A. N (3,4-dihydroxybenzoyl)-L-alanine ben:~yl ester
The title compound was prepared from N (tert-butyloxycarbonyl)alanine benzyl
ester (400
mg, 1.4 mmol) by the removal of the Boc group following the indications of
example 3.
The resulting compound was then coupled with 3,4-dihydroxybenzoic acid
according to
indications of example 4. Purification by flash chromatography eluting with 5%
MeOH/CHC13 provided the desired product as a syrup (180 mg, 41%).
'H NMR (DMSO-db): 1.40 (d, J =7.1, 3H); 4.47 (m, 1H); 5.13 (s, 2H); 6.78 (d, J
= 8.2, 1H);
7.24 (d, J = 7.9, 1 H); 7.32 (s, 1 H); 7.3 S ( s, SH); 8.47 (d, J = 6.7, 1 H);
9.12 (s, 1 H), 9.48
(s, 1H).
Step B. N [N'-(3',4'-dihydroxybenzoyl)-L-alanyl=~-dopamine
The title compound was prepared from the compound of step A of this example
(115 mg,
0.36 mmol) according to indications of example 5 and example 6 for the
cleavage of the
benzyl group and the coupling reaction with dopamine hydrochloride.
Purification by flash
chromatography eluting with S% MeOH/EtOAc provided the desired product (56 mg,
15%), mp. 205°C (dec.).
58
CA 02267657 1999-03-30
'H NMR (DMSO-db): 1.40 (d, J = 7.1, 3H); 2.63 (t, J = 7.0, 2H); 3.38 (m, 2H);
4.58 (m,
1 H); 6.52 (d, J = 6.6, 1 H); 6.67 (d, J = 7.9, 1 H); 6.71 (s, 1 H); 6.87 (d,
J = 8.2, 1 H); 7.32 (d,
J = 7.9, 1 H); 7.39 (s, 1 H); 7.45 (s, 1 H), 7.60 (d, J = 6.9, 1 H); 8.04 (s,
4H).
Example 21. Preparation of N [N'-(p-hydroxytienzoyt)-L-glutamyl]-dopamine
Step A. N (tert-butyloxycarbonyl)-b-benzyloxy-1:..-glutamic acid benzyl ester
The title compound was prepared from N (tert-bu.tyloxycarbonyl)-b-benzyloxy-L-
glutamic
acid (169 mg, 0.50 mmol) as described in example 2a. Purification by flash
chromatography eluting eith 20% EtOAc/hexane provided the title compound as
white
crystals (186 mg, 87%), mp. 71.5-74°C.
'H NMR (CDC13): 1.42 (s, 9H); 1.99 (m, 1H); 2.2 2 (m, 1H); 2.44 (m, 2H); 4.39
(s, 1H);
5.09 (s, 4H); 7.33 (s, lOH).
Step B. N (p-hydroxybenzoyl)-8-benzyloxy-L-glutamic acid benzyl ester
The title compound was prepared from the product obtained in step A of this
example (350
mg, 0.80 mmol) by the removal of the Boc group following the indications of
example 3.
The resulting product was coupled with p-hydrox:ybenzoic acid according to the
indications found in example 4. Purification by clash chromatography eluting
with 15%
EtOAc/CHZC12 provided the desired product as a syrup (161 mg, 43%).
'H NMR (DMSO-db): 2.06 (d, J = 7.4, 1H); 2.15 (d, J = 5.8, 1H); 4.50 (m, 1H);
5.07 (s,
2H); 5.14 (s, 2H); 6.81 (d, J = 8.4, 2H); 7.34 (s, lOH); 7.77 ( d, J = 8.5,
2H); 8.51 (d, J =
7.5, 1 H); 10.02 (s, 1 H).
59
CA 02267657 1999-03-30
Step C. N ~N'-(p-hydroxybenzoyl)-L-glutamyl]-dopamine
The title compound was prepared from the compound prepared in step B of this
example
(116 mg, 0.26 mmol) according to indications focmd in examples 5 and 6 for the
cleavage
of the benzyl groups and the coupling reaction with dopamine hydrochloride.
Purification
by flash chromatography eluting with EtOAc provided the desired product (61.2
mg, 44%),
mp. 128°C (dec.).
'H NMR (DMSO-db): 1.89 (m, 1H); 1.'98 (m, 1H).; 2.14 (t, J = 7.6, 2H); 2.50
(t, J = 7.7,
4H~); 3.17 (m, 4H); 4.33 (m, 1H); 6.39-t:O.43 (m, 2H); 6.56-6.62 (m, 4H); 6.81
(d, J = 8.7,
2H)', 7.77 (d, J = 8.1, 2H); 7.85 (t, J = 5.4, 1H); a'.90 (t., J = 5.4, 1H);
8.16 (d, J = 7.8, 1H),
8.61 (s, 2H); 8.72 (s, 2H), 9.98 (s, 1H).
Example 22. Preparation of N (3,4-dihydroxylae~a~rl)-&-'b~enzyloxy-L-glutamic
acid
benzyl ester
Step A. N (3,4-dihydroxybenzoyl)-b-benzyloxy-~L-gluramic acid benzyl ester
The title compound was prepared from N (tert-butyloxycarbonyl)-b-benzyloxy-L-
glutamic
acid benzyl ester (350 mg 0.82 mmol) by the removal of the Boc group following
the
indications found in example 3. The resulting product was coupled with 3,4-
dihydroxybenzoic acid according to the indications found in example 4.
Purification by
flash chromatography eluting with 5% MeOH/EtOAc provided the desired product
as a
syrup (169 mg, 36%).
CA 02267657 1999-03-30
'H NMR (CDC13): 2.15 (m, 1H); 2.30 (m, 1H); 2.49 (m, 2H); 4.81 (m, 1H); 5.06
(s, 2H);
5.16 (s, 2H); 6.86 (d, J = 8.4, 2H); 7.12 ( d, J = 7.4, 1H); 7.19 ( d, J =
7.4, 1H); 7.26-7.31
(m, lOH); 7.46 (s, 1H).
Step B. N [N'-(3',4'-dihydroxybenzoyl)-L-glutamyl]-dopamine
The title compound was prepared from the compound obtained in step B of this
example
(130 mg, 0.28 mmol) according to the indications of example 5 and 6 for the
removal of
the benzyl groups and the coupling with dopamine hydrochloride. Purification
by flash
chromatography eluting with EtOAc provided the; desired product as a foam (23
mg,
15 %).
'H NMR (DMSO-db): 1.87 (m, 1H,); 1.96 (m, 1H); 2.12 (s, 2H); 2.48 (t, J = 7.6,
4H);
3.17 (m, 4H); 4.31 (m, 1H); 6.40 (d, J = 7.6, 1H); 6.42 (d, J = 7.3, 1H); 6.56
(s, 1H);
6.59 (d, J = 8.6, 1H); 6.60 (s, 1H); 6.62 (d, J = 7.7, 4H); 6.76 (d, J = 8.0,
1H); 7.23 ( d, J
= 9.6, 1H); 7.32 (s, 1H); 7.85 ( t, J = 5.3, 1H); 7.f~8 ( t, J = 5.9, 1H);
8.05 (d, J = 7.8, 1H),
8.60 (s, 2H); 8.71 (s, 2H), 9.10 (s, 1H); 9.45 (s, 1.H).
Example 23. Preparation of N [N'-(3',4'-dihyd~roxybenzoyl)-b-benzyloxy-L-
glutamyl]-
dopamine
Step A. N [N'-(tert-butyloxycarbonyl)-b-benzyloxy-L-glutamyl]-dopamine
The title product was prepared by the reaction of dopamine hydrochloride with
N (tert-
butyloxycarbonyl)-b-benzyloxy-L-glutamic acid (500 mg, 1.15 mmol) according to
the
indications of example 6. Purification by flash chromatography eluting with
25%
61
CA 02267657 1999-03-30
EtOAc/CHZC12 containing 1% acetic acid provided the desired product (400 mg,
65%) as a
solid, mp. 58°C.
'H NMR (acetone-db): 1.40 (s, 9H,); 1.95 (m, 1H); 2.14 (m, 1H); 2.46 (t, J =
7.3, 4H); 2.65
(t, J = 7.0, 2H), 3.38 (m, 2H); 4.19 (m, 1H); 5.11 (s, 2H), 6.19 (d, J = 7.4,
1H); 6.55 (d, J =
8.2, 1 H); 6.74 (m, 2H); 7.29 - 7.33 (m, 5H), 7.43 -(m, 1 H), 7.75 (br, 1 H),
7.83 (br, 1 H).
Step B. N [1V'-(3',4'-dihydroxybenzoyl)-b-benzyloxy-L-glutamyl]-dopamine
The title compound was prepared from the product of step A ~af this example
(400 mg, 0.84
mmol) by the removal of the Boc group following the indications of example 3.
The
resulting compound was then coupled with 3,4-dihydroxybenzoic acid according
to
indications found in exemple 4. Purification by flash chromatography eluting
with EtOAc
containing 1% acetic acid provided the desired product (124 3ng, 28%) as a
solid, mp
108°C.
'H NMR (acetone-db): 2.02 (m, 1H), 2.23 (m, 1H), 2.47 (m, ll~,); 2.50 (t, J =
7.0, 2H); 3.35
(m, 2H); 4.61 (m, 1 H); 5.05 (s, 2H), 6.48 (d, J = 7.6,1 H); 6.64 (d, J = 7.6,
1 H); 6.68 (s,
1 H); 6.82 (d, J = 8.8, 1 H); 6.90 - 7.03 (m, 1 H); 7.25 - 7.31 (rla, 5H),
7.44 (s, 1 H), 7.49 (m,
1 H), 7.66 (d, J = 7. 8, 1 H); 7.5 - 8. 8 (br, 4H)
Example 24. Preparation of N [N'-(3',4'-dihyd~roxybenzoyl~-L-glutaminyl)-
dopamine
Step A. N (tert-butyloxycarbonyl)-L-glutamine benzyl ester
The title compound was prepared from N (tert-butyloxycarbonyl)-L-glutamine
(250 mg,
1.0 mmol) as described in example 2b. Purificatiion by flash chromatography
eluting with
62
CA 02267657 1999-03-30
5% MeOH /EtOAc provided the desired product as crystals (320 mg, 96%), mp.
105.5-
107.5°C.
'H NMR (CDCl3): 1.43 (s, 9H); 1.94 (m, 1H); 2.19 ( m, IH); 2.27 (m, 1H); 4.36
(s, 1H);
5.15 (m, 2H); 5.37 (d, J = 7.3, 1H); 5.58 (s, 1H); 7.35 (m, SH).
Step B. N (3,4-dihydroxybenzoyl)-L-glutamine ben~y! -ester
The title compound was prepared from the product of step A of this example
(300 mg, 0.89
mmol) by the removal of the Boc group followin;; the indications of example 3.
The
resulting compound was then coupled with 3,4-di.hydra:~ybenzoic acid according
to
indications found in example 4. Purification by flash chrrc~matography eluting
with 5%
MeOH/EtOAc provided the desired product as a ;syrup ( 154 mg, 46%).
'H NMR (DMSO-db): 1.96 (m, 1 H); 2.06 (m, 1 H;); 2.21 (m, 2H); 4.40 (m, 1 H);
5.13 (s,
2H); 6.78 (d, J = 8.4, 1H); 6.82 (s, 1H); 7.24 ( d, J = 7.6, 1H); 7.31 ( s,
2H); 7.33-7.36 (m,
SH,); 8.52 (d, J = 6.9, 1H); 9.12 (s, 1H); 9.49 (s, 1H)
Step C. N [N'-(3',4'-dihydroxybenzeyl)-L-glutarnine]-dopamine
The title compound was prepared from the compound obtained in step B of this
example
(110 mg, 0.3 mmol) according to the indications found in example 5 and example
6 for
the removal of the benzyl group and the coupling; with dopamine hydrochloride.
Purification by flash chromatography eluting with 7.5% MeOH/EtOAc provided the
desired product as a solid (24.5 mg, 20%), mp. 154°C (dec.).
63
CA 02267657 1999-03-30
'H NMR (DMSO-db): 1.85 m, 1H); 1.95 m, 1H); 2.13 (s, 2H); 2.51 (s, 2H); 3.18
(m,
2H); 4.30 (m, 1H); 6.41 (d, J = 7.9, 1H); 6.59 (d, J = 7.8, 1H); 6.58 (s, 1H);
6.77 (d, J =
8.5, 1 H); 7.24 (d, J = 8.7, 1 H); 7.29 (s, 1 H); 7.31 (s, 2H); 7.89 (t, J =
5.3, 1 H); 8.12 (d, J =
7.6, 1 H), 8.61 (s, 1 H); 8.72 (s, 1 H), 9.10 (s, 1 H); 9.46 (s, 1 H).
Example 25. Preparation of N [N'-(p-hydroxybenzoyl)-L-leucyl]-dopamine
Step A. N (p-hydroxybenzoyl)-L-leucine methyl ester
The title compound was prepared from L-leucine methyl ester hydrochloride (182
mg, 1.0
mmol) as described in example 4. Purification by flash chromatography eluting
with 5%
MeOH/CHC13 provided the desired product as white crystals (138 mg, 52%), mp.
138-
140°C.
'H NMR (DMSO-db): 0.88 (d, J = 6.5, 3H); 0.93 (d, J = 6.6, 3H); 1.55 (m, 1H);
1.68 (m,
1H); 1.75 (m, 1H); 3.36 (s, 3H); 4.47 (m, 1H); 6.82 (d, J = 8.3, 2H); 7.77 (
d, J = 8.3, 2H);
8.42 (d, J = 7.7, 1 H); 9.99 (s, 1 H).
Step B. N [N'-(p-hydroxybenzoyl)-L-leucyl]-dopamine
The title compound was prepared from the compound obtained in step A of this
example
(96 mg, 0.40 mmol) by saponification of the methyl ester group according to
example 8.
The resulting acid was coupled with dopamine hydrochloride as in example 6.
Flash
chromatography eluting with 100% EtOAc provided the title compound (96 mg,
63%),
mp. 161°C (dec.).
64
CA 02267657 1999-03-30
'H NMR (DMSO-db): 0.84 {d, J = 5.5, 3H); 0.90 (d, J = 6.5, 3H); 1.47 (m, 1H);
1.60 (m,
2H); 2,52 (m, 2H); 3.17 (m, 2H); 4.43 (m, 1H); 6.42 (d, J = 8.0, 1H); 6.58 (d,
J = 6.1,
1H); 6.60 (s, 1H); 6.81 (d, J = 9.1, 2H); 7.78 (d, J = 8.4, 2H); 7.88 (t, J =
5.5, 1H); 8.09
(d, J = 8.5, 1 H); 8.61 (s, 1 H); 8.69 (s, 1 H); 9.95 (s, 1 H).
Example 26. Preparation of N [N'-(3',4'-dihydroxybenzoyl)-L-leucyl]-dopamine
Step A. N [N'(tert-butyloxycarbonyl)-L-leucyl]-d,opamine
The title compound was prepared from N (tert-bu.tyloxycarbonyl)-L-leucine (187
mg,
0.75 mmol) by coupling with dopamine hydrochloride as in example 6. Flash
column
chromatography eluting with 25% EtOAc/CHZCI., provided the title compound as a
syrup
( 195 mg, 71 %).
'H NMR (CDC13): 0.88 (t, J = 6.5, 6H); 1.42 (s, 9H); 1.59 (s, 2H); 2.64 (t, J
= 6.7, 2H);
3.44 (m, 2H); 4.13 (s, 1 H); 5.21 (s, 1 H); 6.54 (d, J =7.9, 1 H); 6.66 (s, 1
H); 6.61 (s, 1 H);
6.78 (d, J = 7.7, 1 H).
Step B. N [N'-(3',4'-dihydroxybenzoyl)-L-leucyl'~-dopamine
The title compound was prepared from the product obtained in step A of this
example
( 176 mg, 0.48 mmol) by removing the Boc group following the indications of
example 3
The resulting unblocked product was coupled wish 3,4-dihydroxybenzoic acid
according
to example 4. Purification by flash chromatography eluting with 5% MeOH/EtOAc
provided the desired product {42 mg, 22%), mp. 132°C (dec.),
'H NMR (DMSO-db): 0.85 (d, J = 5.6, 3H); 0.89 (d, J = 5.2, 3H); 1.45 (m, 1H);
1.60 (m,
2H); 2.50 (t, J = 7.2, 2H); 3.19 (m, 2H); 4.40 (m, 1H); 6.41 (d, J = 7.8, 1H);
6.57 (s, 1H);
CA 02267657 1999-03-30
6.59 (d, J = 7.6, 1 H); 6.75 (d, J = 7.5, 1 H); 7.24 (d, J = 10, 1 H); 7.31
(s, 1 H); 7.86 (t, J =
5.5, 1 H); 7.86 (d, J = 7.8, 1 H); 8.66 (s, 1 H); 9.08 (s, 1 H); 9.42 (s, 1
H).
Example 27. Preparation of N [N'-(p-hydroxyhenzoyl)-L-prolyl)-dopamine
Step A. N [N'(tert-butyloxycarbonyl)-L-prolyl]-dopamine
The title compound was prepared from N (tert-butyloxycarbonyl)-L-proline ( 108
mg,
0.50 mmol) in a coupling reaction with dopamine hydrochloride (189 mg, 1.0
mmol)
according to example 6. Purification by flash column chromatography (5%
MeOH/CHC13) provided the desired compound as a syrup (128 mg, 73%).
'H NMR (DMSO-db): 1.32 (s, 6H); 1.39 (s, 3H,); 1.72 (m, 2H); 1.76 (m, 1H);
2.06 (m,
1H); 2.50 (s 2H); 3.13 (m, 1H); 3.25 (m, 2H); 3.27 (m, 1H); 3.98 (m, 1H); 6.43
(d, J =
7.4, 1 H); 6.56 ( s, 1 H); 6.61 (d, J = 7.9, 1 H); 7.86 (s, 1 H); 8.31 (s, 1
H); 8.61 (s, 1 H); 8.71
(s, 1 H).
Step B. N [N'-(p-hydroxybenzoyl)-L-prolyl]-dopamine
The title compound was prepared from the product obtained in step A of this
example by
the removal of the Boc group following the indications found in example 3. The
resulting
unblocked derivative was then coupled with p-hydroxybenzoic acid according to
the
indications found in example 4. Purification by flash chromatography eluting
with
EtOAc afforded the desired product (23 mg, 31°/.), mp.
165°C (dec.).
'H NMR (DMSO-db): 1.74 (m, 2H); 1.85 (s, 1H); 2.10 (s, 1H); 2.50 (s 2H); 3.17
(s, 2H);
3.46 (s, 1H); 3.57 (s, 1H); 4.09 (s, 1H); 6.43 (d, J = 6.8, 1H); 6.58 (s, 1H);
6.61 (d, J =
66
CA 02267657 1999-03-30
7.9, 1 H); 6.78 (d, J = 7.2, 2H); 7.43 (d, J = 7.0, 2Hf); 7.83 (s, 1 H); 8.63
(s, 1 H); 8.73 (s,
1 H); 9.88 (s, 1 H).
Example 28. Preparation of N [N'-(3',4'-dihydroxybenzoyl)-L-prolyl]-dopamine
The title compound was prepared from N (tert-butyloxycarbonyl)-L-prolyl-
dopamine
( 150 mg, 0.43 mmol) by the removal of the Boc group following the indications
found in
example 3. The resulting unblocked derivative was then coupled with 3,4-
dihydroxybenzoic acid as in example 4. Purification by flash chromatography
eluting
with EtOAc afforded the desired product (54 mg, 32%) as a solid, mp.
131°C (dec.).
'H NMR (DMSO-db): 1.73 (s, 2H); 1.85 (s, 1H); :?.09 (s, 1H); 2.50 (s 2H); 3.17
(s, 2H);
3.48 (s, 1 H); 3.56 (s, 1 H); 4.09 (s, 1 H); 6.43 (d, J = 6.7, 1 H); 6.5 8 (s,
1 H); 6.61 (d, J =
78.4, 1 H); 6.76 (d, J = 7.3, 1 H); 6.90 (s, 1 H); 7.0(1 (s, 1 H); 7.84 ( s, 1
H); 8.62 (s, 1 H);
8.72 (s, 1H); 9.13 (s, 1H); 9.33 (s, 1H).
Example 29. Preparation of N [N'(p-hydroxybenzoyl)-L-tryptophanyl]-dopamine
Step A. N [N'(tert-butyloxycarbonyl)-L-tryptophamyl]-dopamine
The title compound was prepared from N (tert-b~utyloxycarbonyl)-L-tryptophan
(204 mg,
0.65 mmol) by coupling with dopamine hydrochloride according to the procedure
of
example 6. Purification by f<ash chromatography eluting with 2.5% MeOH/EtOAc
provided the title compound as a syrup (215 mg, 75%).
'H NMR (DMSO-db): 1.31 (s, 9H); 2.46 (t, J = 7.4, 2H); 3.02 (m, 2H); 3.14 (m,
1H);
3.22(s, 1H); 4.15 (m, 1H); 6.43 (d, J = 7.6, 1H); fi.58 (s, 1H); 6.62 (d, J =
7.5, 1H); 6.66
(d, J = 8.1, 1 H); 6.97 (t, J = 7.5, 1 H); 7.05 (t, J = 7.3, 1 H); 7.10 (s, 1
H); 7.31 (d, J = 7. 7,
67
CA 02267657 1999-03-30
1 H); 7.58 (d, J = 7.7, 1 H); 7.86 (t, J = 4.7, 1 H); 8.62 (s, 1 H); 8.71 (s,
1 H); 10.77 (s, 1 H).
Step B. N [N'(p-hydroxybenzoyl)-L-tryptophanyl]-dopamine
The title compound was prepared from the product obtained in step A of this
example
(175 mg, 0.40 mmol) by removing the Boc group following the indications of
example 3.
The resulting unblocked derivative was then coupled with p-hydroxybenzoic acid
according to the indications of example 4. Purification by flash
chromatography eluting
with 5% MeOH/EtOAc afforded the desired product (125 mg, 68%) as a foam.
'H NMR (DMSO-db): 2.50 (t, J = 7.6, 2H); 3.11(~m, 1H, CHz ~2~); 3.18 (m, 2H)
3.24 (m,
1H); 4.65 (m, 1H); 6.43 (d, J = 8.1, 1H); 6.59 (s, 1H,); 6.61 (d, J = 8.1,
1H); 6.76 (d, J =
8.6, 2H); 6.98 (t, J = 7.4, 1H); 7.05 (t, J = 7.6, 1Hf); 7.17 (s, 1H); 7.30
(d, J = 8.1, 1H);
7.65 (d, J = 8.0, 1H); 7.67 (d, J = 8.3, ZH); 8.03 (t, J = 5.3, 1H); 8.12 (d,
J = 8.2, 1H);
8.69 (s, 2H); 9.95 (s, 1 H); 10.74 (s, 1 H).
Example 30. Preparation of N [N'-(3',4'-dihydlroxybenzoyl)-L-tryptophyl]-
dopamine
The title compound was prepared from N [(N'-(t~ert-butyloxycarbonyl)-L-
tryptophyl]-
dopamine (44 mg, 0.10 mmol) by the removal of the Boc group following the
indications
of example 3. The resulting unblocked derivative; was then coupled with 3,4-
dihydroxybenzoic acid according to the conditions found in example 4.
Purification by
flash chromatography eluting with EtOAc afforded the desired product (25 mg,
53%) as
a yellow solid, mp. 119°C (dec.)
'H NMR (DMSO-db): 2.47 (m, 2H); 3.13 (m, 1H:); 3.17 (m, 2H); 3.22 (m, 1H,);
4.62 (m,
68
CA 02267657 1999-03-30
1 H); 6.43 ( d, J = 7.4, 1 H); 6.59 ( s, 1 H); 6.61 (d, ;f = 7.9, 1 H); 6.73
(d, J = 8.0, 1 H); 6.97
(t, J = 7.7, 1 H); 7.04 (t, J = 7.3, 1 H); 7.15 ( s, 1 H): ?.29 (d, J = 8.1, 1
H); 7.24 (s, 1 H); 7.29
(d, J = 6.1, 1 H); 7.64 (d, J = 8.0, 1 H); 7.66 (t, J = Ei.6, 1 H); 8.00 (d, J
= 7.00, 1 H); 8.61 (s,
1 H); 8.72 (s, 1 H); 9.07 (s, 1 H); 9.44 (s, 1 H); 10.74 (s, 1 H);.
Example 31. Preparation of N [N'-(3',4'-dihydroxybenzoyl)-L-methionyl]-
dopamine
Step A. N [N'(tert-butyloxycarbonyl)-L-methion,yl]-dopamine
The title compound was prepared from N-(tert-butyloxycarbonyl)-L-methionine
(250 mg,
1.0 mmol) by coupling with dopamine (380 mg, 2.0 mmol) according to example 6.
Purification by flash chromatography eluting with. 30% EtOAc/CHZCIz yielded
the title
compound (230 mg, 60%).
'H NMR (DMSO-db): 1.38 (s, 9H); 1.73 (m, 1H); 1.80 (m, 1H); 2.02 (s, 3H); 2.40
(m,
2H); 2.51 (t, J = 7.6, 2H); 3.24 (m, 2H); 3.96 (m, 1H); 6.43 (d, J = 7.6,
1H,); 6.57 (s, 1H);
6.61 (d, J = 8.4, 1 H); 6.87 (d, J = 7.9, 1 H); 7.78 (t., J = 5.0, 1 H); 8.62
(s, 1 H); 8.70 (s, 1 H).
Step B. N [N'-(3',4'-dihydroxybenzoyl)-L-methionine]-dopamine
The title compound was prepared from the product prepared in step A of this
example
(150 mg, 0.40 mmol) by the removal of the Boc group following the indications
found in
example 3. The resulting unblocked derivative w~~s coupled with 3,4-
dihydroxybenzoic
acid according to the indications of example 4. Purification by flash
chromatography
eluting with 5% MeOH/EtOAc afforded the desired product (40 mg, 24%) as a
solid, mp.
126°C (dec.).
69
CA 02267657 1999-03-30
S 'H NMR (DMSO-db): 1.93 (m, 2H); 2.05 (s, 3H); 2.42 (m, 1H); 2.47 (m, 1H);
2.53 (t, J =
7.8, 2H); 3.17 (m, 2H); 4.43 (m, 1H); 6.43 (d, J = l3.0, 1H); 6.57 (s, 1H,);
6.59 (d, J = 8.0,
1 H); 6.77 (d, J = 8.1, 1 H); 7.25 (d, J = 8.1, 1 H); 7.:32 (s, 1 H); 7.89 (t,
J = 5.6, 1 H); 8.08 (d,
J = 7.9, 1 H); 8.67 (s, 4H).
Example 32. Preparation of N (N'-(3',4'-dihydroxybenzoyl)-L-lysyl]-dopamine
Step A. N [Na'-(9-fluorenylmethoxycarbonyl)-NE"-(tert-butyloxycarbonyl)-L-
lysyl]-
dopamine
The title compound was prepared from Na (fluorenylmethyloxycarbonyl)--NE'-
(tert-
butyloxycarbonyl)-L-lysine (230 mg, 0.50 mmol) by coupling with dopamine
hydrochloride as described in example 6. Purification by flash chromatography
eluting
with 40% EtOAc/CHzCl2 provided the desired product as white crystals, mp. 58-
61°C
(280 mg, 93%).
'H NMR(DMSO-db): 1.20 (m, 1H); 1.23 (m, 1H); 1.33 (me, 2H); 1.48 (m, 1H); 1.55
(m,
1H); 2.52 (m, 2H); 2.89 (s, 2H); 3.15 (m, 1H); 3.22 (m, 1H); 3.89 (q, J~ =
8.5, J = 6.8,
1 H); 4.21 (t, J = 6.1, 1 H); 4.22 (d, J = 7.4, 2H); 6.43 (d, J = 7.9, 1 H);
6.75 (s, 1 H); 6.61
(d, J = 7.6, 1H); 6.74 (s, 1H); 7.37 (d, J = 8.0, 1H); 7.32 (t, J = 7.4, 2H,);
7.42 (t, J = 7.4,
2H); 7.72 (d, J = 6.6, 2H); 7.86 (t, J = 5.7, 1H); 7.138 (d, J = 7.5, 2H);
8.62 (b, 1H); 8.71
(b, 1 H).
Step B. N [N'-(3',4'-dihydroxybenzoyl)-L-lysyl]-dopamine
The title compound was prepared from the compound prepared in step A of this
example
(140 mg, 0.23 mmol) by the removal of the Fmoc group following the indications
found in
CA 02267657 1999-03-30
example 7. The resulting unblocked derivative was coupled with 3,4-
dihydroxybenzoic acid
according to the indications of example 4. Purification by flash
chromatography using
EtOAc provided the desired product (85 mg, 41 %). The cleaving of the Boc
group of the side
chain of the coupled product (20 mg, 0.04 mmol) 'was achieved via an acid
hydrolysis
as described in example 3. The solvent and acid were removed under vacuum to
afford the
desired product (14.4 mg, 86%), mp.93.5°C (dec.).
'H NMR(DMSO-db): 1.17 (m, 2H); 1.52 (m, 2H); 1.68 (m, 2H,; 2.53 (m, 2H); 2.76
(s,
2H); 3. I 8 (m, 2H), 4.34 (q, J = 6.7, J = 8.2, 1 H); 6.43 (d, J = 8.0, 1 H);
6.5 7 (s, 1 H); 6.61
(d, J = 8.0, 1 H); 6.78 ( d, J = 8.2, 1 H); 7.24 ( d, J == 8.2, 1 H); 7.33 (s,
1 H); 7.91 (t, J = 4.7,
1 H); 7.97 (d, J = 7.9, 1 H); 8.68 (s, 4H).
Example 33. Preparation of N [N'-3',4'-di~~droxybenzoyl)-L-histidyl]-dopamine
Step A. N [Na'-(fluorenylmethoxycarbonyl)-N;~"~-(trityl)-L-histidyl]-dopamine
The title compound was prepared from Na (fluorenylmethoxycarbonyl)-N';m-trityl-
histidine
(619 mg, 1.0 mmol) according to the indications found in example 6 with
dopamine
hydrochloride. Purification by flash chromato;~aphy eluting with 40%
EtOAc/CHZC12
provided the desired product (390 mg, 52% yield).
'H NMR(DMSO-db): 3.16 (m, 1H); 4.I4 (m, 1H); 4.18 (d, J = 7.4, 2H); 4.20 (t, J
= 5.4,
1 H); 6.39 (d, J = 7.6, 1 H); 6.56 (s, 1 H); 6.60 (d, J = 7.8, I H); 7.03 (s,
6H); 7.07 (t, J = 7.4,
1H); 7.23 (s, 1H); 7.27 (m, 2H); 7.31 (s, 9H); 7.40 (m, 2H); 7.68 (m, 2H);
7.85 (d, J = 7.8,
1 H); 7.90 (d, J = 7.7, 2H); 7.95 (s, 1 H); 8.63 (b, I 1~); 8.72 (b, I H).
Step B. N [N'(3',4'-dihydroxybenzoyl)-L-histidyl]-dopamine
71
CA 02267657 1999-03-30
The title compound was prepared from the compound prepared in step A of this
example (320
mg, 0.42 mmol) by the removal of the Fmoc group as described in example 7. The
resulting
unblocked derivative was coupled with 3,4-dihydroxybenzoic acid as in example
4.
Purification by flash chromatography eluting with 5% MeOH/EtOAc provided the
desired
compound (85 mg, 41%). For the removal of the trityl group located on the side
chain, the
product (60 mg, 0.089 mmol) was hydrogenolyzed following the conditions found
in example
5 affording the desired product (30 mg, 79%).
'H NMR(DMSO-db): 2.47 (t, J = 7.6, 2H); 2.96 (rn, 2H); 3.17 (m, 2H); 4.57 (q,
J = 7.8, J~,H
= 6.7, 1 H); 6.41 (d, J = 9.1, 1 H); 6.57 (s, 1 H); 6.60 (d, 3 = 8.2, 1 H);
6.78 ( d, J = 8.4, 1 H);
6.87 (s, 1H); 7.20 (d, J = 7.8, 1H); 7.28 (s, 1H); 7.78 (s, 1H); 7.88 (t, J =
5.7, 1H); 8.23 (d, J
= 7.9, 1 H); 8.65 (s, 2H); 9.15 (s, 1 H). 9.55 (s, 1 H).
Example 34. Preparation of N [lV'-(3',4'-dihydroxybenzoyl)-L-aspartyl]-
dopamine
Step A. N tent-butyloxycarbonyl-y-cyclohexyl-L-aspartic acid benzyl ester
The title compound was prepared from N tert-but'yloxycarbonyl-L-aspartic acid
y-
cyclohexyl ester (1.0 g, 3.2 mmol) by an alkylation with benzyl bromide
following the
indication of example 2c. The resulting ester was obtained in 98% yield after
purification
by flash chromatography eluting with 15% EtOAc;/hexane.
Step B. N (3,4-dihydroxybenzoyl)-y-cyclohexyloxy-L-aspartic acid benzyl ester
The title compound was prepared from the compound prepared in step A of this
example
by the removal of the Boc group according to the indications found in example
3 and
coupling with 3,4-dihydroxybenzoic acid as indicated in example 4.
Purification by flash
72
CA 02267657 1999-03-30
chromatography eluting with 20% etltyt ~~tate/CIKZCI2 provided the title
compound (260
mg, 52%).
Step C. N [N'-(3',4'-dihydroxybenzoyl)-y-cyclohexyloxy-L-aspartyl]-dopamine
The title compound was prepared from the compound prepared in step B of this
example
(259 mg, 0.59 mmol) by hydrogenolysis of the benzyl ester following the
conditions
outlined in example 5. The resulting product was then subjected to coupling
with
dopamine hydrochloride according to example 6. Purification by flash
chromatography
eluting with ethyl acetate afforded the title compound in 49% yield.
Example 35. Preparation of N [lV'-(3',4'-dibydroxybenzoyl)-sarcosyl]-dopamine
Step A. N-tent-butyloxycarbonyl-sarcosine benzyl ester
The title compound was prepared from N tert-butyloxycarbonyl-sarcosine (2.0 g,
I0.6
mmol) by an alkylation with benzyl bromide following the indication of example
2c. The
resulting ester (2.89 g; 98%) was obtained after purification by flash
chromatography
eluting with 15% EtOAc/hexane.
'H NMR (CDCl3): 1.43 (d, 9H), 2.94 (d, 3H), 3.9 7 (d, 2H), 7.36 (s, SH)
Step B. N'-(3,4-dihydroxybenzoyl)-sarcosine ben:~yl ester
The title compound was prepared from the compound prepared in step A of this
example
by the removal of the Boc group according to the indications found in example
3 and
coupling with 3,4-dihydroxybenzoic acid as indicated in example 4.
Purification by flash
73
CA 02267657 1999-03-30
chromatography eluting with 80% ethyl acetate/ClEI2Clz provided the title
compound (433
mg, 43%).
'H NMR (CDC13): 3.1 (d, 3H), 3.5 (s, 2H), 5.2 (d, 2H), 6.9 (m, 3H), 7.40 (m,
SH), 9.40
(br, 2H).
Step C. N [N'-(3',4'-dihydroxybenzoyl)-sarcosinyll]-dopamine
The title compound was prepared from the compound prepared in step B of this
example
(278 mg, 0.88 mmol) by hydrogenolysis of the benzyl ester following the
conditions
outlined in example 5. The resulting product was then subjected to coupling
with
dopamine hydrochloride according to example 6. Purification by flash
chromatography
eluting with 5% MeOH/CHZCIz/1% AcOH afforded the title compound in 50% yield.
'H NMR (CDC13): 2.60 (t, 2H), 2.9 (d, 3H), 3.2 (q,, 2H), 3.3 (t, 2H), 6.4 -
7.0 (m, 6H), 9.5
(br, 4H).
Example 36. Preparation of N [N'-(3',4'-dihyd.roxybenzoyl)-L-tyrosyl]-L-
tyrosine
The dipeptide was prepared using the peptide synthesizer (ABI 430A) utilising
Wang
resin (0.5 mmol).
Step A. HZN-L-tyrosyl-L-tyrosyl-resin
The N Fmoc-L-tyrosine(t-Bu) - OH (1 mmol was activated over a period of 45 min
with
HOBT (1.0 eq.) and DCC (1.0 eq.) in 5 mL of N ~methylpyrrolidone (NMP). At the
same
time, in a separate flask, the Fmoc protecting group on the tyrosine amino
group bound to
74
CA 02267657 1999-03-30
the polymer was removed by two successive treatments of 15 min with a solution
of 30%
piperidine in N methylpyrrolidone, followed by a series of washes with NMP.
The
activated ester was then filtered and added to the resin. The suspension was
stirred for 2
h. The Fmoc blocking group was then removed as previously described and the
resin was
successively washed with N methylpyrrolidone and CHZC12.
Step B. N [N'-(3',4'-dihydroxybenzoyl)-L-tyrosyl;~-L-tyrosyl-resin
To the dityrosyl moiety bound on the resin (1 g) and 3,4-dihydroxybenzoic acid
(85 mg;
app. 3 eq.) in DMF (5 mL) and CHzCl2 (2 mL) we:re added BOP (benzotriazol-1-
yloxy-
tris-dimethylamino)phosphonium hexafluorophosphate) (240 mg, app. 3 eq.) and
diisopropylethylamine (125 ~L; 4 eq.). The flask was stirred under nitrogen
for a period
of 16 h. The resin was filtered off, washed successively with DMF, MeOH and
CHZC12,
yielding 518 mg of crude resin.
Step C. N [N (3',4'-dihydroxybenzoyl)-L-tyrosyl]~-L-tyrosine
A mixture of 0.5 mL water in 4.5 mL of trifluoroacetic acid was cooled to
0°C and was
added to the crude resin. The resulting suspension was stirred, allowing the
mixture to
reach room temperature over a period of 2 h. The mixture was then filtered and
the resin
washed with 5 mL of acetic acid. The filtrate was evaporated to dryness in
vacuo and the
residue was purified by preparative HPCL, using a Supelcosil C-18 column, flow
rate: 18
mL/min; gradient: 0.1%TFA from 0-30% acetonitrile over 40 min. Retention time:
21
min. 5 mg (4%) of the title compound was recovered (4% yield).
CA 02267657 1999-03-30
Example 37. Preparation of N [N (3',4'-dihydroxybenzoyl)-L-tyrosyl]-glycine
Step A. N (3,4-dihydroxybenzoyl)-O-tent-butyltyrosine tert-butyl ester
The hydrochloric salt of L-O-tert-butyltyrosine tent-butyl ester (800 mg, 2.4
mmol) was
coupled with 3,4-dihydroxybenzoic acid by follovring the directions found in
example 9.
Purification by flash chromatography eluting with 40% ethyl acetate in hexane
afforded
the desired product (385 mg, 37%).
'H NMR (CDC13): 1.31 (s, 9H), 1.42 (s, 9H), 3.13 (dd, J = 5.8, 14.3, 1H), 3.17
(dd, J =
6.3, 14.3, 1H), 4.85 (ddd, J = 5.8, 6.3, 4.0, 1H), 6.77 (d, J = 7.4, 1H), 6.5-
6.9 (br, 0.5H)
and 7.26 (s, 0.5H), 6.80 (d, J = 8.2,), 6.91 (d, J = 8.0, 2H,), 7.03 (d, J =
8.2, 1H), 7.08 (d, J
= 8.0, 2H), 7.54 (s, 1 H), 7.8-8.4 (br, 1 H).
Step B. N [lV'-(3',4'-dihydroxybenzoyl)-L-O-tent-butyltyrosyl]-glycine
The product of step A of this example (375 mg) was stirred with TFA (10 mL)
for a
period of 90 min. The mixture was evaporated to dryness in vacuo and several h
with a
mechanical pump providing a quantitative yield (200 mg) of the free acid. The
acid was
then condensed using the BOP procedure as found in example 9 with glycine tent-
butyl
ester hydrochloride salt (80 mg,l.5 eq.) and trieth:ylamine (132 ~L, 3.0 eq.)
for a period
of 15 h. Purification by flash chromatography eluting with 80% EtOAc in hexane
afforded the desired product (120 mg, 89%) as the; tert-butyl ester. The ester
was
hydrolysed using 0.5 mL of water and 7 mL of TF~A with stirring for 1 h. The
mixture
was then evaporated in vacuo and purified by flash chromatography eluting with
5%
methanol in dichloromethane containing 1% acetic acid to provide the desired
product
(35 mg, 34%).
76
CA 02267657 1999-03-30
'H NMR (DMSO-db): 2.86 (dd, J = 10.8, 13.7,1H), 2.98 (dd, J = 1.4, 13.7, 1H),
3.66-3.92
(m, 2H), 6.61 (d, J = 8.2, 2H), 6.73 (d, J = 8.4, 1H), 7.09 (d, J = 8.2, 1H),
7.16 (d, J = 8.4,
1H), 7.23 (s, 1H), 8.09 (d, J = 8.5, 1H), 8.26 and E~.38 (2t, J = 5.6, 2x
O.SH).
Example 38. N [N'-[N"-(3",4"-dihydroxyben~;oyl)-L-tyrosyl]-glycyl]-dopamine
Step A. N Boc-glycyl-dopamine
The title compound was prepared by coupling dopamine hydrochloride with tert-
butyloxycarbonylglycine (200 mg, 1.1 mmol) according to the procedure of
example 9.
Purification by flash chromatography afforded the; title compound (214 mg,
64%).
'H NMR (DMSO-db): 1.38 (s, 9H), 2.5-2.58 (m, 2H), 3.16-3.20 (m, 2H), 3.49 (d,
J = 5.7,
2H), 6.42 (d, J = 7.9, 1H), 6.57 (s, 1H), 6.62 (d, J = 7.9), 6.86 (d, J = 5.2,
1H), 7.71-7.77
(m, 1 H), 8.62 (s, 1 H), 8.72 (s, 1 H).
Step B. N [N'-[N"-(3",4"-dihydroxybenzoyl)-L-tyrosy]-glycyl]-dopamine
The product obtained in step A of this example was hydrolyzed under the
conditions of
example 3 providing an intermediate that was coupled with N (3,4-
dihydroxybenzoyl)-L-
tyrosine under the conditions outlined in example 9. Purification by flash
chromatography
eluting with S% methanol in ethyl acetate containing 1% acetic acid provided
the title
compound (30 mg, 19%).
'H NMR (DMSO-d6): 2.52 (t, J = 7.8, 2H), 2.88 (<ld, J = 9.9, 13.8, 1H), 2.97
(dd, J = 4.3,
13.8, 1H), 3.16-3.21 (m, 2H), 3.61 (dd, J = 5.2, 16.5, 1H), 3.70 (dd, J = 5.6,
16.5, 1H),
4.46-4.5 3 (m, 1 H), 6.44 (dd, J = 1.5, 8.2, 1 H), 6.5 6 (d, J = 1.5, 1 H),
6.62 (d, J = 8.2, 1 H),
77
CA 02267657 1999-03-30
6.63 (d, J = 8.5, 2H), 6.74 (d, J = 7.9, 1H), 7.09 (d, J = 8.5, 2H), 7.18 (dd,
J = 1.7, 7.9, 1H),
7.25 (d, J = 1.7, 1H), 7.76 (t, J = 5.4, 1H), 8.19-8.23 (m, 2H), 8.1-9.7 (br,
SH).
Example 39. Preparation of N [1V'-(3',4'-dihydroxybenzoyl)-glycyl]-L-tyrosine
Step A. N 3,4-dihydroxybenzoyl-glycine
Coupling of the hydrochloride salt of glycine tent-butyl ester with 3,4-
dihydroxybenzoic
acid was obtained by following the indications of example 9. Purification by
flash
chromatography afforded 288 mg (81 %) of the title compound after eluting with
4%
methanol in methylene chloride.
'H NMR (DMSO-db): 1.41 (s, 9H), 3.82 (d, J = 5.6, 2H), 6.76 (d, J = 8.0, 1H),
7.20 (d, J =
8.0, 1H), 7.28 (s, 1H), 8.45 (t, J = 5.6, 1H),9.13 (s, 1H), 9.47 (s, 1H).
Step B. N [N'-(3',4'-dihydroxybenzoyl)-glycyl]-L-tyrosine
The product obtained in step A of this example was hydrolyzed by stirnng in
TFA (10 mL)
for a period of 90 min. Evaporation of the acid in vacuo afforded a product
that was then
reacted with O-tert-butyl-L-tyrosyl-dopamine using the conditions outlined in
example 9.
Purification by flash chromatography eluting with. 70% ethyl acetate in hexane
provided
230 mg (67%) of the O-tert-butyl derivative of the; title compound.
'H NMR (CDC13) d 1.28 (s, 9H), 1.30 (s, 9H), 3.00 (d, J = 5.4, 2H), 3.89-4.08
(m, 2H),
4.60-4.67 (m, 1 H), 6.64 (d, J = 7.3, 1 H), 6.83 (d, J~ = 8.0, 2H), 6.97-7.05
(m, 1 H), 7.04 (d, J
= 8.1, 2H), 7.09-7.18 (m, 2H), 7.32-7.41 (m, 1H).
78
CA 02267657 1999-03-30
Treatment of the tert-butyl derivative with TFA for a period of 2 h provided a
quantitative
yield of the title product.
'H NMR (DMSO-d6): 2.75-2.93 (m, 2H), 3.73-3.87 (m, 2H), 4.35-4.44 (m, 1H),
6.62 (d, J
= 8.1, 2H), 6.76 (d, J = 8.5, 1H), 6.98 (d, J = 8.1, ;?H), 7.20 (dd, J = 1.6,
J = 8.5, 1H), 7.30
(d, J = 1.6, 1H), 7.96 (d, J = 8.1, 0.5H) 8.18 (d, J == 7.7, O.SH), 8.29-8.36
(m, 1H), 9.00-9.60
(br, 4H).
Example 40. N [N'-[N"-(3",4"-dihydroxybenzoyl)-glycylJ-L-tyrosylJ-dopamine
Step A. N L-tyrosine-dopamine
The title compound was prepared from N tert-but:yloxycarbonyl-O-2,6-
dichlorobenzyl-L-
tyrosine (2.50 g, 5.7 mmol) according to the indications found in example 6
with dopamine
hydrochloride. Purification by flash chromatography eluting with 50%
EtOAc/hexane
provided the desired product (3.03 g, 93%).
'H NMR (DMSO-db): 1.31 (s, 9H), 2.45-2.55 (m, 2H), 2.67 (dd, J = 10.2, 13.4,
1H), 2.86
(dd, J = 4.1, 13.4, 1H), 3.10-3.18 and 3.20-3.29 (2,m, 2H), 4.07 (ddd, J =
4.1, 10.2, 8.5, 1H),
5.17 (s, 2H), 6.43 (d, J = 7.8, 1H), 6.59 (s, 1H), 6.64 (d, J = 7.8, 1H), 6.75
(d, J = 8.5, 1H),
6.94 (d, J = 8.2, 2H), 7.15 (d, J = 8.2, 2H), 7.45 (t., J = 8.1, 1H), 7.53 (d,
J = 8.1, 2H), 7.87
(t, J = 4.9, 1 H), 8.00-9.15 (br, 2H).
Step B. N-[N'-[N"-(3",4"-dihydroxybenzoyl)-gl:ycyl]-L-tyrosyl]-dopamine
The product obtained in step A of this example was hydrolyzed by stirnng in
TFA (10 mL)
for a period of 30 min. Evaporation of the acid in vacuo afforded a product,
part of which
79
CA 02267657 1999-03-30
(148 mg, 0.70 mmol) was then reacted with N 3,4-dihydroxybenzoyl-glycine using
the
conditions outlined in example 9. Purification by flash chromatography eluting
with 5%
methanol in ethyl acetate provided 115 mg (25%) of the O-(2,6-dichlorobenzyl
derivative
of the title compound. The latter protected dipeptide derivative (115 mg, 0.17
mmol) in 4
mL of methanol was then hydrogenolyzed according to the procedure found in
example 5.
Purification by flash chromatography eluting with 2% methanol in ethyl acetate
provided
60 mg (68%) of the desired product.
'H NMR (DMSO-db): 2.47 (t, J = 7.7, 2H), 2.65 (dd, J = 8.9, J = 13.6, 1H),
2.83 (dd, J =
4.7, 13.6, 1 H), 3.08-3.23 (m, 2H), 3.71 (dd, J = 5.6, J = 16.2, 1 H), 3.83
(dd, J = 5.8, 16.2,
1H), 4.35 (ddd, J = 4.7, 8.9, 8.4, 1H), 6.42 (d, J = 7.7, 1H), 6.57 (s, 1H),
6.59 (d, J = 8.3,
2H), 6.62 (d, J = 7.7, 1H), 6.76 (d, J = 8.0, 1H), 6.95 (d, J = 8.3, 2H), 7.21
(dd, J = 1.7,8.0,
1 H), 7.3 0 (d, J = 1.7, 1 H), 7.94 (d, J = 8.4, 1 H), 7. 96 (t, J = 5.5, 1
H), 8.3 7 (t, J = 5.7, 1 H),
8.50-8.85 (br, 2H), 9.05-9.22 (br, 2H), 9.20-9.60 Ibr, 1H).
Example 41. N [1V'-[1V"-(3",4"-dihydroxybenzoyl)-L-tyrosyl]-L-y-O-benzyl-
aspartyl]-
dopamine
Step A. N Boc-L-aspartyl-dopamine y-benzyl ester trifluoroacetate
Coupling of N Boc-L-aspartic acid 'y-benzyl ester (1.50 g, 4.63 mmol) dopamine
hydrochloride by following the indications of example 6. Purification by flash
chromatography eluting with 50% ethyl acetate in hexane afforded 2.06 g (93%)
of the title
compound after eluting with 40% ethyl acetate in hexane.
'H NMR (DMSO-d6): 1.37 (s, 9H), 2.45-2.52 (m, 2H) 2.57 (dd, J = 8.6, 15.9,
1H), 2.70-
2.78 (m, 1H), 3.10 - 3.25 (m, 2H), 4.28 - 4.35 (m, 1H), 5.08 (s, 2H), 6.41 (d,
J = 7.9, 1H),
CA 02267657 1999-03-30
6.56 (s, 1H), 6.62 (d, J - 7.9, 1H), 7.04 (d, J = 7.8., 1H), 7.29 - 7,40 (m,
SH), 7.80 (s, 1H),
8.62 (s, 1H), 8.71 (s, 1H).
Step B. N [N'-[N'-(3",4"-dihydroxybenzoyl)-L-tyrosyl]-L-y-O-benzyl-aspartyl]-
dopamine
The product obtained in step A of this example (500 mg, 1.00 mmol) was
hydrolyzed by
following the indications of example 3. The resulting product was then coupled
with N
3,4-dihydroxybenzoyl-L-tyrosine as indicated in example 6. Purification by
flash
chromatography eluting with 5% methanol in methylene chloride containing 1%
acetic acid
afforded 69 mg (35%) of the desired title compound.
'H NMR (DMSO-d6): 2 signal sets 2.45-2.67 (m, 3H), 2.75-2.98 (m, 3H), 3.10-
3.25 (m,
2H), 4.39-4.52 (m, 1H), 4.57-4.69 (m, 1H), 5.04-_'i.06 (m, 2H), 6.42 (d,
J=7.6, 1H), 6.57-
6.76 (m, 4H), 7.05-7.35 (m, 7H), 7.71 (t, J= 5.5, 1 H), 7.93 (t, J=5.5, 1 H),
8.12 (d, J - 7.6,
1 H), 8.26 (d, J = 7.7, 1 H), 8u.63 (s, 1 H), 8.73 (s, l~ H), 9.10 (s, 1 H),
9.20 (s, 1 H), 9.51 (s,
1 H).
Example 42. N [N'-[N"-(3",4"-dihydroxybenzoyl)-L-tyrosyl]-glycyl]-dopamine
Step A. N glycyl-dopamine
Boc-L-tyrosine (200 mg, 1.1 mmol) was coupled with dopamine hydrochloride
following
the indications of example 4. Purification by flaslh chromatography eluting
with 75% ethyl
acetate in hexane afforded 214 mg (64%) of the pure amide.
'H NMR (DMSO-db): 1.38 (s, 9H), 2.5-2.58 (m, f,H), 3.16-3.20 (m, 2H), 3.49 (d,
J = 5.7,
2H), 6.42 (d, J = 7.9, 1H), 6.57 (s, 1H), 6.62 (d, J = 7.9, 1H), 6.86 (d, J =
5.2, 1H), 7.71-
81
CA 02267657 1999-03-30
7.77 (m, 1 H), 8.62 (s, 1 H), 8.72 (s, 1 H).
Step B. N [N'-[N"-(3",4"-dihydroxybenzoyl)-L-tyrosyl]-glycyl]-dopamine
The product obtained in step A of this example (214 mg, 0.70 mmol) was
hydrolyzed by
stirring in TFA (10 mL) for a period of 90 min. Evaporation of the acid in
vacuo afforded a
product that was then reacted with N (3,4-dihydroxybenzoyl)-L-tyrosine (80 mg,
0.24
mmol) using the conditions outlined in example 9. Purification by flash
chromatography
eluting with 5% methanol in ethyl acetate containing 1% acetic acid provided
30 mg (19%)
of the pure title compound.
'H NMR (DMSO-db): 2.52 (t, J = 7.8, 2H), 2.88 (dd, J = 9.9, J = 13.8, 1H),
2.97 (dd, J =
4.3, 13.8, 1H), 3.16-3.21 (m, 2H), 3.61 (dd, J = 5.2, J = 16.5, 1H), 3.70 (dd,
J = 5.6, 16.5,
1H), 4.46-4.53 (m, 1H), 6.44 (dd, J = 1.5, 8.2, 1H), 6.56 (d, J = 1.5, 1H),
6.62 (d, J = 8.2,
1 H), 6.63 (d, J = 8. S, 2H), 6.74 (d, J = 7.9, 1 H), 7.09 (d, J = 8.5, 2H),
7.18 (dd, J = 1.7, 7.9,
1H), 7.25 (d, J = 1.7, 1H), 7.76 (t, J = 5.4, 1H), 8.19-8.23 (m, 2H), 8.1-9.7
(br, SH).
Example 43. Preparation of N [N'-(3',4'-dihydroxyhydrocinnamoyl)-L-tyrosyl]-L-
tyrosine
Step A. N 3,4-dihydroxyhydrocinnamoyl-O-benzyloxy-L-tyrosine
O-benzyloxy-L-tyrosine benzyl esterp-toluenesulfonate salt (1.01 g. 1.9 mmol)
was
coupled with 3,4-dihydroxyhydrocinnamoic acid :following the indications of
example 4.
Purification by flash chromatography eluting with 50% ethyl acetate in hexane
afforded
855 mg (84%) of the pure amide.
82
CA 02267657 1999-03-30
'H NMR (DMSO-db): 2.29 (t, J = 7.8, 2H), 2.55 (t, J = 7.8, 2H), 2.85 (dd, J =
8.5, 7.6, 1H),
2.94 (dd, J = 6.1, 13.8, 1H), 4.45 (ddd, J = 6.1, 8.~~, 7.6, 1H), 5.03 - 5.09
(m, 4H), 6.39 (d, J
= 8.0, 1H), 6.56 (s, 1H), 6.61 (s, 1H), 6.88 (d, J = 8.2, 2H), 7.07 (d, J =
8.2, 2H), 7.27 (d, J
= 7.0, 2H), 7.30 - 7.40 (m, 8H), 7.43 (d, J = 7.5, 1 H), 8.29 (d, J = 7.6, 1
H), 8.61 (s, 1 H),
8.70 (s, 1H).
Step B. N 3,4-dihydroxyhydrocinnamoyl-L-tyrosine
The deprotection of the amide was carned out using the conditions outlined in
example 5.
Purification by flash chromatography eluting with 10% methanol in
dichloromethane
afforded 190 mg (73%) of the title compound.
'H NMR (DMSO-db): 2.27 (t, J = 7.9, 2H), 2.53 (t, J = 7.9, 2H), 2.72 (dd, J =
9.4, 13.7,
1H), 2.89 (dd, J = 5.0, 13.7, 1H), 4.33 (ddd, J = 5.0, 9.4, 8.2, 1H), 6.39 (d,
J = 7.3, 1H),
6.55 (s, 1H), 6.59 (d, J = 7.3, 1H), 6.64 (d, J = 8.1, 2H), 6.97 (d, J = 8.1,
2H), 8.04 (d, J =
8.2, 1H), 8.4 - 8.9 (br, 2H), 12 - 13 (br, 1H).
Step C. N [N'-(3',4'-dihydroxyhydrocinnamoyl)-I,-tyrosyl]-L-tyrosine
The product obtained in step B of this example (48 mg, 0.14 mmol) was coupled
with O-
benzyloxy-L-tyrosine benzyl esterp-toluenesulfonate salt following the
indications of
example 4. Purification by flash chromatography eluting with 4% methanol in
ethyl acetate
containing 1% acetic acid afforded 68 mg (100%)~ of the pure title compound.
'H NMR (DMSO-db): 2.21 (t, J = 8.4, 2H), 2.47 It, J = 8.4, 2H), 2.57 (dd, J =
9.9, 14.2,
1H), 2.80 (dd, J = 7.9, 14.0, 1H), 2.86 (dd, J = 3.3, 14.2, 1H), 2.93 (dd, J =
4.9, 14.0, 1H),
83
CA 02267657 1999-03-30
4.33 (ddd, J = 4.9, 7.9, 7.6, 1 H), 4.44 (ddd, J = 3..., 9.9, 8.2, 1 H), 6.36
(d, J = 7.9, 1 H), 6.54
(s, 1H), 6.59 (d, J = 7.9, 1H), 6.61 (d, J = 8.1, 2H), 6.65 (d, J = 7.9, 2H),
6.98 (d, J = 8.1,
2H), 7.00 (d, J = 7.9, 2H), 7.90 (d, J = 8.2, 1 H), 8.05 (d, J = 7.6, 1 H),
8.57 (s, 1 H), 8.60 -
8.80 (br, 1H), 9.11 (s, 1H), 9.18 (s, 1H), 12.1 - 12.9 (br, 1H).
Example 44. Preparation of N (N'-[N"-(3",4"~-dihydroxyhydrocinnamoyl)-L-
tyrosyl]-L-tyrosyl]-dopamine
The product obtained in step A of example 43 (100 mg, 0.29 mmol) was coupled
with O-
2,6-dichlorobenzyloxy-L-tyrosyl-dopamine salt following the indications of
example 4.
The crude material (315 mg) was used as isolated and subjected to
hydrogenolysis using
the conditions of example 5. Purification by flash chromatography eluting with
7%
methanol in ethyl acetate containing 1% acetic acid afforded 100 mg (54%) of
the pure title
compound.
'H NMR (DMSO-db): 2.42 - 2.50 (m, 2H), 2.58 - 2.65 (m, 2H), 2.69 - 2.76 (m,
2H), 2.79
(dd, J = 8.3, 14.0, 1H), 2.85 (dd, J = 8.0, 13.8, 1H), 2.96 (dd, J = 6.3,
14.0, 1H), 3.03 (dd, J
= 6.2, 13.9, 1H), 3.29 - 3.42 (m, 2H), 4.47 - 4.53 (m, 1H), 4.53 - 4.59 (m,
1H), 6.52 - 6.59
(m, 2H), 6.69 - 6.80 (m, 8H), 7.02 (d, J = 8.2, 2H), 7.06 (d, 8.5, 2H),
Example 45. Preparation of N'-(3',4'-dihydroxyhydrocinnamoyl)-L-3,4-
dihydroxyphenylalanine
Step A. N'-(3',4'-dihydroxyhydrocinnamoyl)-L-3.,4-O-dibenzyloxyphenylalanine
benzyl
ester
84
CA 02267657 1999-03-30
The title compound was prepared by cleaving the Boc protecting group of the
product
prepared in step B of the example 16 (310 mg, 0.80 mmol) and coupling it with
3,4-
dihydroxyhydrocinnamic acid as described in ex~unple 3 and in example 4
respectively.
Purification by flash chromatography using 5% MeOH/CHZCIz containing 1% acetic
acid
yielded 220 mg (61%) of the desired product.
'H NMR (DMSO-db): 2.29 (t, J = 8.0, 2H), 2.55 (t, J = 8.0, 2H), 2.73 (dd, J =
8.5, 13.8,
1H), 2.82 (dd, 6.4, 13.8, 1H), 4.41 ddd, J = 6.4, 8.5, 7.5, 1H), 5.03 (d, J =
12.6, 1H), 6.56 (s,
1H), 6.60 (s, 1H), 6.60 (d, J = 8.5, 1H), 6.61 (d, J = 7.7, 1H), 7.22 - 7.38
(m, SH), 8.26 (d, J
1 S = 7.5, 1 H), 8.4 - 9.2 (br, 4H).
Step B. N'-(3',4'-dihydroxyhydrocinnamoyl)-L-3,4-dihydroxyphenylalanine
The title compound was prepared by the reduction of the compound obtained in
step A of
this example (135 mg, 0.30 mmol) according to the indications found in example
S. The
product (90 mg, 83%) was obtained by filtering off the catalyst and
evaporating the filtrate
to dryness.
'H NMR (DMSO-db): 2.27 (t, J = 8.0, 2H), 2.55 ( t, J = 8.0, 2H), 2.65 (dd, J =
8.6, 13.5,
1 H), 2.66 (dd, 3.5, 13 .5, 1 H), 4.26 ddd, J = 3.5, 8, 6, 7.5, 1 H), 6.39 (d,
J = 8.3, 1 H), 6.41 (d,
J = 8.5, 1H), 6.56 - 6.60 (m, 4H), 7.82 (d, J = 5.9, 1H), 8.0 - 9.7 (br, 4H).
Example 46. Preparation of N'-[N"-(3",4"-dihydroxyhydrocinnamoyl)-L-3',4'-
dihydroxyphenylalanyl]-L-3,4-dihydroxyphenylalanine
Boc-L-3,4-di-O-benzyloxyphenylalanine benzyl ester (325 mg, 0.68 mmol) was
deprotected by treatment with TFA as indicated in example 3, providing the
desired L-di0-
CA 02267657 1999-03-30
benzyloxyphenylalanine benzyl ester that was coupled with Boc-L-
dihydroxyphenylalanine
following the indications of example 6. Purification by flash chromatography
eluting with
2% methanol in methylene chloride afforded 275 mg (62%) of the dipeptide
intermediate.
Subsequent removal of the Boc group, again following the indications of
example 3,
provided the intermediate that was coupled with 3,4-dihydroxyhydrocinnamic
acid as
indicated in example 4. The coupled product was hydrogenolyzed as isolated
according to
the indications of example 5. Purification by flash chromatography eluting
with 7%
methanol in ethyl acetate containing 1% acetic acid provided 75 mg (35%) of
the title
compound.
'H NMR (DMSO-db): 2.22 (t, J = 8.0, 2H), 2.46 - 2.53 (m, 3H), 2.74 (dd, J =
8.1, 13.5,
1H), 2.80 - 2.85 (m, 1H), 2.86 (dd, 5.3, 13.5, 1H), 4.31 ddd, J = 8.1, 5.3,
7.2, 1H), 4.38 -
4.46 (m, 1H), 6.37 (d, J = 7.8, 1H), 6.43 - 6.49 (m, 2H), 6.54 (s, 1H), 6.56 -
6.63 (m, 4H),
6.64 (s, 1 H), 7.93 (d, J = 8.3, 1 H), 8.01 (d, J = 7.2, 1 H), 8.5 - 8.9 (br,
6H), 12.2 - 12.8 (br,
1 H).
Example 47. Preparation of N [N'-[3',4'-dihydlroxyhydrocinnamoyl)-L-3,4-
dihydroxyphenylalanyl]-dopamine
Step A. N Boc-3,4-dihydroxyphenylalanyl-dopamine
N Boc-L-3,4-dihydroxyphenylalanine (1.00 g, 3.16 mmol) was coupled with
dopamine
hydrochloride according to the indications of example 6. Purification by flash
chromatography eluting with 5% methanol in me~.hylene chloride containing 1%
acetic acid
provided 1.17 g (83%) of the pure coupled product.
'H NMR (DMSO-db): 1.32 (s, 9H), 2.44 - 2.52 (rn, 2H), 2.53 (dd, J = 9.7, 13.5,
1H), 2.71
86
CA 02267657 1999-03-30
(dd, J = 4.3, 13.5, 1H), 3.07 - 3.28 (m, 2H), 3.98 (~idd, J = 4.3, 9.7, 8.3,
1H), 6.42 (d, J =
7.8, 1 H), 6.45 (d, J = 8.0, 1 H), 6.57 - 6.63 (m, 4H), 6.64 (d, J = 8.3, 1
H), 7.80 (t, J = 5.0,
1H), 8.0 - 9.8 (br, 4H).
Step B. N [N'-[N"-[3",4"-dihydroxyhydrocinnam.oyl)-L-3',4'-
dihydroxyphenylalanyl]-L-
3,4-dihydroxyphenylalanyl]-dopamine
The product of step A of this example (1.17 g, 2.79 mmol) was treated with TFA
as in
example 3 to remove the Boc protecting group. A portion of the product thus
obtained
(260 mg, 0.60 mmol) was then coupled with 3,4-dihydroxyhydrocinnamic acid
using the
conditions as in example 4. Purification by flash chromatography eluting with
10%
methanol in ethyl acetate containing 1% acetic acid provided 113 mg (38%) of
the desired
product.
'H NMR (DMSO-db): 2.26 (t, J = 7.7, 2H), 2.42 - 2.48 (m, 2H), 2.52 (t, J =7.7,
2H), 2.54
(dd, J = 8.0, 13.6, 1H), 2.73 (dd, J = 5.0, 13.6, 1H), 3.05 - 3.24 (m, 2H),
4.31 (ddd, J = 5.0,
9.0, 8.3, 1H), 6.37 (d, J = 8.0, 1H), 6.39 - 6.43 (m, 2H), 6.55 - 6.63 (m,
6H), 7.79 (t, 4.9,
1H), 7.91 (d, J = 8.3, 1H), 7.4 - 10.0 (br, 6H).
Example 48. Preparation of N (N'-[N"-(3",4"-~dihydroxyhydrocinnamoyl)-L-3',4'-
dihydroxyphenylalanyl]-L-3,4-dihydroxyphenylalanyl]-dopamine
The product obtained in step A of example 47 (355 mg, 0.58 mmol) was
deprotected by
treating with TFA according to the indications of example 3. The product thus
obtained
was coupled with 3,4-dihydroxyhydrocinnamic acid according to the conditions
of example
4. Purification by flash chromatography eluting with 5% methanol in ethyl
acetate
containing 1% acetic acid provided 70 mg (18%) of the title compound.
87
CA 02267657 1999-03-30
'H NMR (DMSO-db): 2.22 - 2.28 (m, 2H), 2.45 (t, J = 6.8, 2H), 2.46 - 2.57 (m,
3H), 2.62
(dd, J = 8.3, 13.6, 1H), 2.71 - 2.80 (m, 2H), 3.06 - 3.21 (m, 2H), 4.25 - 4.32
(m, 1H), 4.32 -
4.40 (, 1 H), 6.33 - 6.46 (m, 4H), 6.54 - 6.67 (m, 8:H), 7.74 (t, J = 5.0, 1
H), 7.83 (d, J = 8.0,
1 H), 8.00 (d, J = 8.0, 1 H), 2.8 - 4.7 (br, 8H).
Example 49. Preparation of N [N'-(3',4'-dihydroxybenzoyl)-DL-3-
fluorotyrosinyl]-
dopamine
Step A. N'-3',4'-dihydroxybenzoyl-O-benzyl-DL-3-fluorotyrosine benzyl ester
N Boc-O-benzyl-DL-3-fluorotyrosine benzyl ester (524 mg, 1.09 mmol) was
deprotected
by treating with TFA as indicated in example 3, providing the crude
intermediate that was
coupled with 3,4-dihydroxybenzoic acid as indicated in example 4. Purification
of the
product eluting with 15% ethyl acetate in methyle,ne chloride provided 205 mg
(34%) of
the desired amide product.
'H NMR (acetone-db): 3.17 (dd, J = 5.6, 5.0, 2H), 5.13 (m, 1H), 5.13 (d, J =
12, 2H), 5.23
(d, J = 7.2, 2H), 6.58 - 7.40 (m, 16H).
Step B. N'-[(3',4'-dihydroxybenzoyl)-DL-3-fluorotyrosyl]-dopamine
The product obtained in step A of this example (179 mg, 0.32 mmol) was
unblocked by
hydrogenolysis according to example 5, providing; a product that was then
reacted with
dopamine hydrochloride according to the indicati~~ns of example 6.
Purification by flash
chromatography eluting with 10% methanol in ethyl acetate containing 1 %
acetic acid
provided 118 mg (71%) of the title compound.
88
CA 02267657 1999-03-30
'H NMR (acetone-db): 2.62 (d, J = 6.0, 2H), 3.25 (dd, J = 5.3, 6.8, 2H), 3.38
(q, J = 6.2, 2H)
4.75 (q, J = 6, 1H), 6.50 (d, J = 7.7, 2H), 6.60 (m, 2H), 6.83 (d, J = 8.3,
1H), 6.89 (d, J =
8.9, 1 H), 6.90 (s, 1 H), 7.05 (d, J = 11.7, 1 H), 7.25 (d, J = 8.2, 1 H),
7.37 (s, 1 H).
Example 50. Preparation of N (3,4-dihydroxybenzoyl)-8-N'-(3',4'-
dihydroxyphenethyl)-L-glutamine a-benzyl ester
Step A. N Boc-b-N'-(3,4-dihydroxyphenethyl)-L-glutamine benzyl ester
N Boc-L-glutamic acid a-benzyl ester (800 mg, 2.37 mmol) was coupled with
dopamine
hydrochloride according to example 6. Purification by flash chromatography
eluting with
5% methanol in ethyl acetate containing 1% acetic; acid yielded 896 mg (80%)
of white
crystals, mp. °C.
'H NMR (acetone-db): 1.07 - 2.15 (m, 2H), 2.27 (t, J = 6.7, 2H), 2.63 (t, 7.2
, 2H), 3.35 (q,
T = 6.4, 2H), 4.20 (m, 1H), 5.16 (q, J = 14.0, 2H), 6.45 (d, J = 5.8, 1H),
6.53 (d, J = 7.8,
1H), 6.70 (s, 1H), 6.72 (d, J = 7.6, 1H), 7.07 (s, 1>=~), 7.30 - 7.40 (m, SH),
7.62 - 7.71 (s,
2H).
Step B. N (3,4-dihydroxybenzoyl)-b-N'-(3,4-dihydroxyphenethyl)-L-glutamine a-
benzyl
ester
The product prepared in step A of this example (800 mg, 2.37 mmol) was
deblocked with
TFA as described in example 3. The product thus obtained was then coupled with
3,4-
dihydroxybenzoic acid according to the indications of example 4. Purification
by flash
chromatography eluting with S% methanol in ethyl acetate containing 1% acetic
acid
yielded 896 mg (80%) of the title compound.
89
CA 02267657 1999-03-30
'H NMR (acetone-db): 2.15 - 2.26 (m, 2H), 2.39 (d, J = 4.7, 2H), 2.62 (t, 7.0,
2H), 3.35 (m,
2H), 4.62 (s, 1H), 5.16 (q, J = 5.2, 2H), 6.50 (d, J == 7.8, 1H), 6.70 (s,
1H), 6.90 (d, J = 7.9,
1H), 7.03 (dd, J = 8.3, 8.7, 1H), 7.31 (dd, J = 6.4, :8.9, 1H), 7.35 - 7.41
(m, 6H), 7.68 (q, J =
9.5, 1 H), 8.2 (d, J = 6.6, 1 H).
Example 51. Preparation of N [N'-[(3',4'-dihydroxybenzoyl)-DL-m-tyrosyl]-
dopamine
Step A. Preparation of N (tert-butyloxycarbonyl)-DL-m-tyrosine
The title compound was prepared from DL-m-tyrosine (1.0 g, 5.5 mmol), by
following the
procedure described in example 1. The product was isolated as a colorless
syrup ( 1.18 g, 76%).
'H NMR (DMSO-db): 1.30 (s, 9H); 3.15 (dd, J = 3.1, 13.0, 1H); 4.10 (q, J =
7.2, 1H); 6.20
(d, J = 8.0, 1H); 6.50 - 6.80 (m, 4H), 7.2 (s, 1H), 10.0 (br, 1H).
Step B. Preparation of N (tert-butyloxycarbonyl)-~D-benzyl-DL-m-tyrosine
benzyl ester
The title compound was prepared from the product obtained in step A of this
example (1.0
g, 3.56 mmol) according to the indications of example 2c. The crude product
was purified
by silica gel column chromatography using 5% MeOH/CHZC12 to yield the desired
product
(1.06 g, 65%).
'H NMR (CDC13): 1.45 (s, 9H); 3.15 (d, J = 3.3, 2H); 4.15 (q, J = 7.2, 1H);
4.70 (J = 5.7,
1H), 5.20 (d, J = 11, 4H), 6.71 (d, J = 6.8, 1H); 6.f> (s, 1H), 6.90 (d, 8.5,
1H), 7.2 (t, J = 7.6,
1H), 7.3 - 7.5 (m, 1H).
CA 02267657 1999-03-30
Step C. N (3,4-dihydroxybenzoyl)-O-benzyl-DL-;m-tyrosine benzyl ester
The title compound was prepared from the product obtained in step B of this
example (520
mg, 1.09 mmol) by the removal of the Boc group following the indications of
example 3.
The resulting unblocked derivative was then coupled with 3,4-dihydroxybenzoic
acid
according to the indications of example 4. The cnzde product was purified by
silica gel
column chromatography using 10% ethyl acetate in methylene chloride to yield
205 mg
(34%) of the desired product.
'H NMR (DMSO-db): 3.15 (d, J = 3.1, 2H); 4.20 m, 1H), 5.20 (m, 4H); 6.70 -
7.80 (m,
17H), 8.6 (d, J = 7.2, 2H).
Step D. N [N'-(3',4'-dihydroxybenzoyl)-DL-m-tyrosyl]-dopamine
The title compound was prepared from the product of step C of this example
(208 mg, 0.56
mmol) by removing the benzyl ester group followiing the indications of example
5. The
resulting unblocked derivative was coupled with dopamine hydrochloride
according to the
indications of example 6. Purification by silica gel chromatography with 10%
MeOH in
ethyl acetate containing 1% acetic acid provided the desired product, (26 mg,
14%).
'H NMR (DMSO-db): 2.62 (t, J = 5.9, 2H), 3.20 (m, 2H); 4.7 (s, 1H); 6.70 - 7.5
(m, 12H),
8.5 (s, 1H), 9.3 (br, 4H).
Example 52. Preparation of N ~N'-~N" -(3",4'''-dihydroxybenzoyl -L-tyrosyl]-L-
tyrosyl]-dopamine
91
CA 02267657 1999-03-30
N [N'-(3',4'-dihydroxybenzoyl)-L-tyrosyl]-tyrosine (prepared as described in
example 36,
step C) (30 mg, 0.06 mmol) was coupled with dopamine hydrochloride according
to the
indications of example 6. Purification by flash chromatography eluting with 5%
methanol
in ethyl acetate containing 1 % acetic acid provided 5 mg (14%) of the title
compound.
1H NMR (acetone-db): 2.57 (t, J=7.0, 2H), 2.84 (d~d, J=7.8, 13.6, 1H), 2.87 -
303 (m, 2H),
3.08 (dd, J= 5.3, 13.9, 1H), 3.23 - 3.36 (m, 2H), 4.45 - 4.52 (m, 1H), 4.58 -
4.66 (m, 1H),
6.50 (dd, J - 2.0, 7.8, 1H), 6.65 (d, J=8.3, 2H), 6.6!~ (d, J - 7.8 1H), 6.70
(d, J= 2.0, 1H),
7.07 -7.11 (m, 1H), 7.10 (d, J- 7.9, 2H), 7.21 (dd, :f =1.9, 8.2,qH), 7.39 (d,
J=1.9, 1H), 7.40
- 7.46 (m, 1H), 7.55 (d, J= 7.2, 1H).
92