Note: Descriptions are shown in the official language in which they were submitted.
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
STREPTOCOCCUS UBERIS LACTOFERRIN-BINDING PROTEIN
Technical Field
The present invention relates generally to
bacterial antigens. More particularly, the present
invention pertains to the characterization and
recombinant production of a bovine lactoferrin-binding
protein from Streptococcus uberis (S. uberis) and the
use of the same. The invention also relates to the
characterization of a regulatory region, mga, located
upstream of the lactoferrin-binding protein gene.
Backcrround
Mastitis, an infection of the mammary gland,
causes major economic losses to the dairy industry
yearly. Streptococcus uberis (S. uberis) is an
environmental pathogen responsible for a high
proportion of cases of mastitis in lactating cows and
is the predominant organism isolated from mammary
glands during the nonlactating period (Bramley, A.J.
(1984) Br. Vet. J. 140:328-335; Bramley and Dodd
(1984) J. Dairy Res. 51:481-512; Oliver, S.P. (1988)
Am. J. Vet. Res. 49:1789-1793). Mastitis resulting
from infection with S. uberis is commonly subclinical,
characterized by apparently normal milk with an
increase in somatic cell counts due to the influx of
leukocytes. The chemical composition of milk is
changed due to suppression of secretion with the
. transfer of sodium chloride and bicarbonate from blood
to milk, causing a shift of pH to a more alkaline
level. S. uberis mastitis may also take the form of
-1-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
an acute clinical condition, with obvious signs of
disease such as clots or discoloration of the milk and
swelling or hardness of the mammary gland. Some cases
of the clinical disease can be severe and pyrexia may
be present. For a review of the clinical
manifestations of S. uberis mastitis, see, Bramley
(1991) Mastitis: physiology or pathology, p. 3-9. In
C. Burvenich, G. Vandeputte-van Messom, and A. w. Hill
(ed. ) , New insights into the pathogenesis of mastitis.
Rijksuniversiteit Gent) Belgium; and Schalm et al.
(1971) The mastitis complex-A brief summary. p. 1-3.
In Bovine Mastitis. Lea & Febiger, Philadelphia.
The pathogenesis of S. uberis infection is
poorly understood. Furthermore, the influence of S.
uberis virulence factors on host defense mechanisms
and mammary gland physiology is not well defined.
Known virulence factors associated with S. uberis
include a hyaluronic acid capsule, hyaluronidase, R-
like protein, plasminogen activator and CAMP factor.
However, very little is known of their roles in
pathogenicity.
_ Lactoferrin (Lf) is a mammalian iron-binding
glycoprotein secreted by polymorphonuclear leukocytes
(PMNs) and various exocrine glands (Baggiolini et al.
(1970) J. Exp. Med. 131:559-570; Masson et al. {1966)
Clin. Chim. Acta 14:735-739). This protein is found
at high concentrations in milk and at mucosal surfaces
(Masson et al., supra; Reiter and Oram (1967) Nature
-- - 216:328-330). For example, bovine lactoferrin (bLf)
concentrations in lacteal secretions can increase up
to 30-fold during acute bovine mastitis, depending on
the severity of infection (Harmon et al. (1976)
Infect. Immun. 13:533-542).
A regulatory function for Lf in various
physiological pathways, including the adhesion of PMNs
to the endothelial surface, feedback inhibition of the
-2-
CA 02270404 1999-04-30
WO_98/21231 PCT/CA97/00867
granulocyte-monocyte colony-stimulating factor, and
the regulation of antibody production, has been
suggested. Specific interaction of Lf with certain
mammalian cells seems to be involved in the above
pathways and specific receptors for Lf have been
identified on macrophages, monocytes, B lymphocytes,
PMNs, activated T lymphocytes, and hepatocytes (Bennet
and Davis (1981) J. Immuno~. 127:1211-1216; Dehanne et
al. (1985) Am. J. Physiol. 248:463-469; Maneva et al.
(1983) Int. J. Biochem. 15:981-984; Rochard et al.
(1989) FEBS Lett. 255:201-204; and van Snick and
Masson (1976) J. Exp. Med. 144:1568-1580).
Lf inhibits the growth of E. coli and
certain other microorganisms in vitro (Bullen et al.
(1972) Br. Med. J. 1:69-75). This Lf-mediated
antimicrobial action has mainly been attributed to its
iron deprivation capacity with bacteria (Arnold et al.
(1977) Science 197:263-265; Law and Reiter (1977) J.
Dairy Res. 44:595-599; Oram and Reiter (1968) Biochim.
Biophys. Acta 170:351-365). In this regard, it is
well known that with few exceptions, iron is essential
for microbial growth (Weinberg, E.D. (1978) Microbiol.
Rev. 42:45-66. Even though iron is abundant within
mammalian tissues, virtually all iron within the
mammalian body is held intracellularly as ferritin or
as heme compounds, pools which are generally
inaccessible to invading microorganisms.
Additionally, the small amount of iron present in
extracellular spaces is effectively chelated by high-
affinity iron-binding host glycoproteins such as
transferrin (Tf), present in serum and lymph, and Lf,
present in secretory fluids and milk (Otto et al.
(1992) Crit. Rev. Microbiol. 18:217-233).
To overcome this deficiency, bacterial
pathogens have developed specific iron uptake
mechanisms. In many bacterial species, these
-3-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
mechanisms involve the synthesis and secretion of
small compounds called s-iderophores which display high
affinity for ferric iron (FeIII). Siderophores are
capable of removing TF- or Lf-bound iron to form
ferrisiderophore complexes which in turn are
recognized by specific iron-repressible membrane
receptors and internalized into the bacterium where
the iron is released (Crosa, J.H. (1989) Microbiol.
Rev. 53:517-530}. This iron uptake mechanism has been
described for many gram-negative bacterial species.
Some gram-negative bacteria do not secrete detectable
siderophores when grown in an iron-deficient
environment but produce outer membrane proteins that
bind directly and specifically to Tf or Lf, thereby
allowing iron transport into the bacterial cell.
Tf binding appears to be mediated by the
activity of two proteins present in bacterial outer
membranes, transferrin-binding protein 1 and 2 (Tbpl
and Tbp2) (Gonzalez et al. (1990) Mol. Microbiol.
4:1173-1179; Ogunnariwo and Schryvers (1990) Infect.
Imrnun. 58:2091-2097; Schryvers, A.B. (1989) J. Med.
Microbiol. 29:121-130); Schryvers and Lee (1989) Can.
J. Microbiol. 35:409-415; Schryvers and Morris (1988)
Mol. Microbiol. 2:467-472). Transferrin binding
proteins tend to be highly specific for the
transferrin of their natural host.
However, the mechanism of iron uptake from
Lf has not been well characterized. A putative 105
kDa receptor for Lf utilization, Lbpl, has been
identified in gonococcus by affinity isolation
(Schryvers and Lee, supra; Cornelissen et al. (1992)
J. Bacteriol. 174:5788-5797; Lee and Bryan (1989) J.
Med. Microbiol. 28:199-204). The structural gene for
Lbpl, termed lbpA, has been isolated (Biswas and
Sparling (1995} Infect. Immun. 63:2958-2967). The
genes for meningococcal lactoferrin receptors have
-4-
CA 02270404 1999-04-30_
WO 98/21231 PCT/CA97/00867
also been characterized (Petterson et al. (1993)
Infect. Immun. 61:4724-4733; Petterson et al. (1994)
J. Bacteriol. 176:1764-1766; Petterson et al. (1994)
' Microb. Pathog. 17:395-408). The DNA sequence of lbpA
and the predicted amino acid sequence of Lbpl in
' gonococcus and the meningococcus are highly conserved
(94% identity). Lbp1 has been shown to be 46%
identical to Tbpl (Cornelissen et al. (1992) J.
Bacteriol. 174:5788-5797) of the same gonococcal
strain but only 18% identical to Tbp2 (Anderson et al.
(1994) J. Bacteriol. 176:3162-3170). Both gonococcal
and meningococcal genes contain relatively well-
conserved Fur boxes and the proteins are homologous to
the Tong-dependent family of receptors, as is true for
Tbp1 (Cornelissen et al., supra) but not for Tbp2
(Anderson et al., supra). The strong similarity
between the Lf receptor protein, Lbpl, and the Tf
receptor protein, Tbpl, suggests that binding of Lf to
bacterial cells might be similar to Tf binding.
Consistent-with this hypothesis is the fact that the
putative protein encoded by lbpB, the open reading
frame upstream of lbpA, shows extensive homology to
Tbp2 (Pettersson et al., {1994) Microb. Pathog.
17:395-408), suggesting that iron-acquisition from Lf,
as from Tf, requires two specific proteins in the
outer membrane.
In contrast to the knowledge of the iron
uptake systems of Gram-negative bacteria, there is
-- comparatively little information concerning the
mechanisms by which Gram-positive pathogens acquire
iron during growth in extracellular body fluids. Both
S. aureus and the coagulase-negative staphylococci
have been reported to produce siderophores
(Konetschny-Rapp et al. (1991) Eur. J. Biochem.
191:65-74; Meiwes et al. (1990) FEMS Microbiol. Lett.
67:201-206). S. aureus appears capable of binding
-5-
CA 02270404 1999-04-30
WO 98!21231 PCT/CA97/00867
both human and bovine Lfs and human Tf (Naidu et al.
(1991) J. Med. Micrbiol. 34:323-328; Naidu et al.
(1991) J. Dairy Sci. 74:1218-1226; Naidu et al. (1992)
J. Med. Microbiol. 36:177-183; Modun et al. (1994)
Infect. Immun. 62:3850-3858). The interaction of Lf
with a bovine S. agalactiae strain has also been
reported (Rainard, P. (1992) FEMS Microbiol. Lett.
98:235-240). However, the iron acquisition functions
of these Tf- or Lf-binding proteins have not been ___
studied.
The group A streptococcal M protein is
considered to be one of the major virulence factors of
this organism by virtue of its ability to impede
attack by human phagocytes (Lancefield, R.C. (1962) J.
Immunol. 89:307-313). The bacteria persist in the
infected tissue until antibodies are produced against
the M molecule. Type-specific antibodies to the M
protein are able to reverse the antiphagocytic effect
of the molecule and allow efficient clearance of the
invading organism. For example, M proteins are one of
the key virulence factors of S. pyogenes, due to their
involvement in mediating resistance to phagocytosis
(Kehoe, M.A. (1991) Vaccine 9:797-806) and their
ability to induce potentially harmful host immune
responses via their superantigenicity and their
capacity to induce host-cross-reactive antibody
responses (Hisno, A.L. (1991) New Engl. J. Med.
325:783-793; Froude et al. (1989) Curr. Top.
Microbiol. Immunol. 145:5-26; Stollerman, G.H. (1991)
Clin. Immunol. Immunopathol. 61:131-142).
In group A streptococci (GAS), the genes for
M protein (emm) as well as a peptidase (scpA) and, if
present, genes encoding M protein-related IgG- and
IgA-binding proteins (fcrA and enn, respectively) are
clustered on the chromosome (Haanes et al. (1992) J.
Bacteriol. 174:4967-4976; Hollingshead et al. (1993)
-6-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97100867
Mol. Microbiol. 8:707-717; Podbielski, A. (1993) Mol.
Gen. Genet. 237:-287-300). Expression of these
virulence-associated surface proteins is co-regulated
at the level of transcription by the protein Mga
(which stands for multigene regulator of group A
' Streptococcus), formerly called Mry or VirR (Caparon
and Scott (1987) Proc. Natl. Acad. Sci. USA 84:8677-
8681; Chen et al. (1993) Mol. Gen. Genet. 241:685-693;
Haanes and Cleary (1989) J. Bacteriol. 171:6397-6408;
McIver et al. (1995) J. Bacteriol. 177:6619-6624;
Perez-Casal et al. (1991) J. Bacteriol. 173:2617-2624;
Podbielski et al. (1995) Infect. Immun. 63:9-20;
Podbielski, A. (1992) Med. Microbiol. Immunol.
181:227-240; Robbins et al. (1987) J. Bacteriol.
169:5633-5640). It is thought that Mga is a part of a
crucial regulatory system in GAS, possibly functioning
as a second component in a two-component regulatory
system.
Vaccination is one approach to enhance
resistance of the mammary gland to new infection and
reduce clinical severity of the disease. Previous
studies have shown that primary infection with S.
uberis can considerably reduce the rate of infection
following a second challenge with the same strain
(Hill, A.W. (1988) Res.Vet. Sci. 44:386-387}. Local
vaccination with killed S. uberis protects the bovine
mammary gland against intramammary challenge with the
homologous strain (Finch et al. (1994) Infect. Immun.
62:3599-3603). Similarly, subcutaneous vaccination
with live S. uberis has been shown to cause a dramatic
modification of the pathogenesis of mastitis with the
same strain (Hill et al. (1994) FEMS Immunol. Med.
Microbiol. 8:109-118}. Animals vaccinated in this way
shed fewer bacteria in their milk and many quarters
remain free of infection.
_7_
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
However, vaccination with live or attenuated
bacteria can pose risks to the recipient. It would
therefore be desirable to provide a subunit vaccine
composition for use against S. uberis. Until now, the
S. uberis lactoferrin-binding protein has not been
characterized and it use in vaccine compositions_has
not been described.
Disclosure of the Invention
The present invention is based on the
discovery of a bovine lactoferrin (bLF) binding
protein (bLbp) from S. uberis, and the
characterization thereof. The gene coding for bLF-
binding protein, lbp, as well as an upstream regulator
of the gene, mga, have been cloned. bLF-binding
protein, immunogenic fragments and analogs thereof,
and/or chimeric proteins including the same, can be
used, either alone or in combination with other
antigens, in novel subunit vaccines to provide
protection from bacterial infection in mammalian
subjects.
Accordingly, in one embodiment, the subject
invention is directed to an isolated, immunogenic S.
uberis bLF-binding protein, as well as a nucleic acid
molecule comprising a coding sequence for an
immunogenic S. uberis bLF-binding protein. In
additional embodiments, the invention is directed to
recombinant vectors including the same, host cells
_ transformed with these vectors and methods of
recombinantly producing S. uberis bLF-binding
proteins.
In still further embodiments, the subject
invention is directed to vaccine compositions
comprising a pharmaceutically acceptable vehicle and
an immunogenic S. uberis bLF-binding protein.
_g_
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
In yet other embodiments, the present
invention is directed to methods of treating or
preventing S. uberis infections, such as mastitis, in
a mammalian subject. The method comprises
administering to the subject a therapeutically
' effective amount of the above vaccine compositions.
In additional embodiments, the invention
pertains to methods of producing vaccine compositions
comprising (a) providing an immunogenic S. uberis bLF-
binding protein; and (b) combining the protein with a
pharmaceutically acceptable vehicle.
In further embodiments, the invention is
directed to antibodies against the S. uberis bLF-
binding proteins.
In additional embodiments, the invention is
directed to methods of detecting S. uberis antibodies
in a biological sample comprising:
(a) providing a biological sample;
(b) reacting the biological sample with a S.
uberis bLF-binding protein under conditions which
allow S. uberis antibodies, when present in the
biological sample, to bind to the S. uberis bLF-
binding protein to form an antibody/antigen complex;
and
(c) detecting the presence or absence of the
complex,
thereby detecting the presence or absence of
S. uberis antibodies in the sample.
In yet further embodiments, the invention is
_ 30 directed to an immunodiagnostic test kit for detecting
S. uberis infection. The test kit comprises a S.
uberis bLF-binding protein and instructions for
conducting the immunodiagnostic test.
In further embodiments, the invention is
directed to an isolated S. uberis Mga protein, as well
_g_
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
as a nucleic acid molecule comprising a coding
sequence for the same_
These and other embodiments of the present
invention will readily occur to those of ordinary
skill in the art in view of the disclosure herein.
Brief Description of the Fi uqures
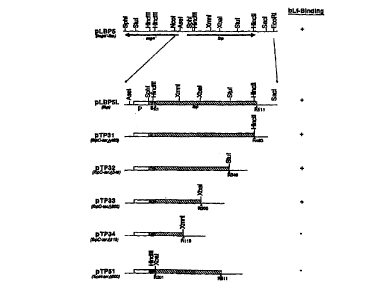
Figure 1 is a restriction enzyme map of Ibp
and shows progressive deletions and a summary of bLf
binding data. Open boxes (p region) represent 5~-
sequences containing promoter and ribosome binding
sites, shaded boxes (s region) represent lbp sequences
encoding the signal peptides, and hatched boxes
represent the Ibp sequences coding for the mature or
truncated proteins. R1 and 8201 (R represents
residue) indicate the first codon of the mature
protein in pLBP5 and the truncated protein in pTP5l,
respectively. Other numbers indicate the last codon
of each protein. Restriction sites are present on the
top of the Ibp. The bLf binding ability of each clone
is shown on the right.
Figures 2A-2C (SEQ ID NOS:1-2) depict the
nucleotide sequence and deduced amino acid sequence of
S. uberis bovine Lbp. Nucleotides and amino acids are
numbered on the right of the sequences. The deduced
amino acid sequence is shown in the single-letter code
below the nucleotide sequence. Two possible ATG start
codons at positions 232 and 262, and the TAA stop
codon at 1915, are shown in bold. Two putative -35
and -10 promoter sequences and Shine-Dalgarno
sequences (SD) are indicated. A putative rho-
independent transcription terminator (T) is
underlined. The double underline shows the presence
of a putative signal peptide at the N-terminus of the
ORF. The C-terminal hydrophobic trans-membrane domain
is indicated by italics and the nearby surface anchor
-10-
SUBSTITUTE SHEET (RULE 26)
CA 02270404 1999-04-30 -
WO 98/21231 PCT/CA97/00$67
motif is shown by italics and double underline. The
central repeated amino acid sequences are indicated by
the letters A, B, and C.
Figure 3 shows profiles of the secondary
structures, charged residues and hydrophobicity of
Lbp. The deduced amino acid sequence of Lbp was
analyzed with the Novotny-Auffray algorithm. Plots
marked Turn, Beta, and Alpha indicate the potential
for beta turn-random coil, beta sheet, and alpha helix
formation, respectively. The +/- plot shows regions
of the molecule with net positive (upper) and negative
(lower) charges. The hydrophobicity (Hydro) plot
shows the hydrophobic regions of the protein. The
- positions of the amino acids are shown on the
horizontal axis.
Figure 4 depicts the construction of pMGAI4F
from pLBPSi and pMGAl4. Plasmid pMGAI4F was generated
by inserting the 1.5 kb SphI-NheI fragment of pLBPSi
into the SphI and NheI sites of pMGAl4. Lines
indicate S. uberis DNA, while the box represents the
multiple cloning sites of vector pTZl8R. The probe
fragments used for Southern or Northern blot
experiments are indicated by the hatched bars. The
arrows indicate the locations of the open reading
frames of lbp, mga' and mga.
Figures 5A-5D (SEQ ID NOS:3-12) show the
nucleotide sequence of mga and deduced amino acid
sequence of Mga, as well as the ORFs downstream of
mga. Nucleotides and amino acids are numbered on the
right of the sequences. The deduced amino acid
sequence is shown in the single-letter code below the
nucleotide sequence. Possible ATG start codons are
shown in bold and the stop codons are indicated by
"*". A putative -35 and -10 promoter sequence and
Shine-Dalgarno sequence (SD) are indicated.
-11-
SUBSTITUTE SHEET (RULE 26)
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
Figure 6 shows the probes used in
hybridization analysis, as well as the structure of
Lbp (on the top), constructed from the DNA sequence
analysis. The signal peptide, proline rich region and
transmembrane domain are indicated by S, Pro and TM
respectively. The A, B and C repeat regions are also
shown. Probes used in hybridization are indicated by
the hatched bars below the map.
Figure 7 shows the time course of lzsl-bLf
binding to S. uberis. 109 bacteria were incubated with
6.9 nm lzsl_bLf in 0.2 ml of PBS-1% BSA. At time
intervals indicated, bacteria were pelleted and the
amount of cell associated lzsI_bLf was determined.
Figure 8 depicts the results of a
competition binding assay using bLf (33% iron-
saturated) as radiolabelled ligand and competitor.
Percentage binding values were calculated as
percentage of lzsI-bLf binding in the presence of
increasing amounts of unlabelled bLf to bacteria
suspended in PHS-1% BSA in the absence of unlabelled
bLf. Inset: Scatchard plot and affinity (Kd) of the
binding of lzsl_bLf to S. uberis. The line represents
the best fit as determined by a linear regression
analysis. A concentration of 270 nM of unlabelled bLf
caused 50% displacement of lzsl-bLf binding (indicated
by dotted lines).
Figure 9 shows the influence of iron
chelators on the expression of lactoferrin-binding by
S. uberis. Cells grown in THB-YE with or without
EDDA, dipyridyl or desferrioxamine mesylate were
incubated with 6.9 nM lzsl_bLf in 0.2 ml of PBS-1% BSA
at room temperature for 2 h. After three washes,
cell-bound radioactivity was determined.
Figure 10 shows the physical map of the
recombinant plasmid used to express the Lbp in Example
3. The plasmid pLBPS contains 3.7 kb of S. uberis
-12-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
derived kDNA, in the vector pTZl8R. The plasmid pGH-
LBP was constructed by subcloning an SphI-RsaI
fragment from pLBP5 into vector pGH433. Ptac indicates
the location of the tac promoter. The Lbp gene is
shown by the arrows labelled as Ibp.
Figure 11 shows inhibition of recombinant
bLf-binding protein on l2sl-labelled bLf binding to S.
uberis. Increasing amounts of a mixture of supernatant
(sup.) and whole cell lysate (with equal volume) of E.
coli pLBPS (~) or E. coli pTZl8R (o) were mixed with
109 cells and incubated with 0.69 nM l2sI-bLf in 0.2 ml
volumes. Inhibition values were calculated as relative
percentage of bLf binding to bacteria suspended in
PBS-1% BSA in the absence of any E. coli samples.
Detailed Description
The practice of the present invention will
employ, unless otherwise indicated, conventional
techniques of molecular biology, microbiology,
recombinant DNA technolo
gy, and immunology, which are
within the skill of the art. Such techniques are
explained fully in the literature. See, e.g.,
Sambrook, Fritsch & Maniatis, Molecular Cloning: A
Laboratory Manual, Vols. I, II and III, Second Edition
(lggg); DNA Cloning, Vols. I and II (D.N. Glover ed.
1985) ; Oligonucleotide Synthesis (M.J. Gait ed.
1984); Nucleic Acid Hybridization (B. D. Hames & S.J.
Higgins eds. 1984); Animal Cell Culture (R. K.
--- Freshney ed. 1986); Immobilized Cells and Enzymes
(IRL press, 1986); Perbal, B., A Practical Guide to
Molecular Cloning (1984); the series, Methods In
Enzymology (S. Colowick and N. Kaplan eds., Academic
Press, Inc.); and Handbook of Experimental Immunology,
Vols. I-IV (D. M. Weir and C.C. Blackwell eds.,
1986, Blackwell Scientific Publications).
-13-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
A. Definitions
In describing the present invention, the
following terms will be employed, and are intended to
be defined as indicated below.
It must be noted that, as used in this
specification and the appended claims, the singular
forms "a", "an" and "the" include plural referents
unless the content clearly dictates otherwise. Thus,
for example, reference to "an Lbp" includes a mixture
of two or more Lbps, and the like.
The terms "lactoferrin-binding protein",
"LF-binding protein" and "Lbp" (used interchangeably
herein) or a nucleotide sequence encoding the same,
intends a protein or a nucleotide sequence,
respectively, which is derived from an S. uberis 1bp
gene. The nucleotide sequence of a representative S.
uberis lbp gene, and the corresponding amino acid
sequence of an LF-binding protein encoded by this
gene, are depicted in Figures 2A-2C (SEQ ID NOS:l-2).
However, an LF-binding protein as defined herein is
not limited to the depicted sequence as several
subtypes of S. uberis are known and variations in LF-
binding proteins will occur between strains of S.
uberi s .
Furthermore, the derived protein or
nucleotide sequences need not be physically derived
from the gene described above, but may be generated in
any manner, including for example, chemical synthesis,
isolation (e. g., from S. uberis) or by recombinant
production, based on the information provided herein.
Additionally, the term intends proteins having amino
acid sequences substantially homologous (as defined
below) to contiguous amino acid sequences encoded by
the genes, which display immunological and/or
lactoferrin-binding activity.
-14-
SUBSTITUTE SHEET (RULE 26)
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
Thus, the terms intend full-length, as well
as immunogenic, truncated and partial sequences, and
active analogs and precursor forms of the proteins.
Also included in the term are nucleotide fragments of
the gene that include at least about 8 contiguous base
pairs, more preferably at least about 10-20 contiguous
base pairs, and most preferably at least about 25 to
50 or more contiguous base pairs of the gene. Such
fragments are useful as probes and in diagnostic
methods, discussed more fully below.
The terms also include those forms
possessing, as well as lacking, the signal sequence,
as well as the nucleic acid sequences coding therefor.
Additionally, the term intends forms of LF-binding
protein which lack the membrane anchor region, and
nucleic acid sequences encoding such deletions. Such
deletions may be desirable in systems that do not
provide for secretion of the protein. Furthermore, an
LF-binding domain, found within about the N-terminal
200 codons, may or may not be present. Thus, for
example, if the Lf binding protein will be used to
purify LF, the LF-binding domain will generally be
retained. If the protein is to be used in vaccine
compositions, immunogenic epitopes which may or may
not include the LF-binding domain, will be present.
The terms also include proteins in neutral
form or in the form of basic or acid addition salts
depending on the mode of preparation. Such acid
addition salts may involve free amino groups and basic
salts may be formed with free carboxyls.
Pharmaceutically acceptable basic and acid addition
salts are discussed further below. In addition, the
proteins may be modified by combination with other
biological materials such as lipids (both those
occurring naturally with the molecule or other lipids
that do not destroy immunological activity) and
-15-
CA 02270404 1999-04-30
WO 98121231 PCT/CA97/00867
saccharides, or by side chain modification, such as
acetylation of amino groups, phosphorylation of
hydroxyl side chains, oxidation of sulfhydryl groups,
glycosylation of amino acid residues, as well as other
modifications of the encoded primary sequence.
The term therefore intends deletions,
additions and substitutions to the sequence, so long
as the polypeptide functions to produce an
immunological response as defined herein. In this
regard, particularly preferred substitutions will
generally be conservative in nature, i.e., those
substitutions that take place within a family of amino
acids. For example, amino acids are generally divided
into four families: (1) acidic -- aspartate and
glutamate; (2) basic -- lysine, arginine, histidine;
(3) non-polar -- alanine, valine, leucine, isoleucine,
proline, phenylalanine, methionine, tryptophan; and
(4) uncharged polar -- glycine, asparagine, glutamine,
cystine, serine threonine, tyrosine. Phenylalanine,
tryptophan, and tyrosine are sometimes classified as
aromatic amino acids. For example, it is reasonably
predictable that an isolated replacement of leucine
with isoleucine or valine, or vice versa; an aspartate
with a glutamate or vice versa; a threonine with a
serine or vice versa; or a similar conservative
replacement of an amino acid with a structurally
related amino acid, will not have a major effect on
the biological activity. Proteins having
substantially the same amino acid sequence as the
reference molecule, but possessing minor amino acid
substitutions that do not substantially affect the
immunogenicity of the protein, are therefore within
the definition of the reference polypeptide.
By "mastitis" is meant an inflammation of
the mammary gland in mammals, including in cows, ewes,
goats, sows, mares, and the like, caused by the
-16-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
presence of S. uberis. The infection manifests itself
by the infiltration of phagocytic cells in the gland.
Generally, 4 clinical types of mastitis are
recognized: (1) peracute, associated with swelling,
heat, pain, and abnormal secretion in the gland and
accompanied by fever and other signs of systemic
disturbance, such as marked depression, rapid weak
pulse, sunken eyes, weakness and complete anorexia;
(2) acute, with changes in the gland similar to those
above but where fever, anorexia and depression are
slight to moderate; (3) subacute, where no systemic
changes are displayed and the changes in the gland and
its secretion are less marked: and (4) subclinical,
where the inflammatory reaction is detectable only by
standard tests for mastitis.
Standard tests for the detection of mastitis
include but are not limited to, the California
Mastitis Test, the Wisconsin Mastitis Test, the Nagase
test, the electronic cell count and somatic cell
counts used to detect a persistently high white blood
cell content in milk. In general, a somatic cell
count of about 300,000 to about 500,000 cells per ml
or higher, in milk will indicate the presence of
infection. Thus, a vaccine is considered effective in
the treatment and/or prevention of mastitis when, for
example, the somatic cell count in milk is retained
below about 500,000 cells per ml. For a discussion of
mastitis and the diagnosis thereof, see, e.g., The
- Merck Veterinary Manual. A Handbook of Diagnosis,
Therapy, and Disease Prevention and Control for the
Veterinarian, Merck and Co., Rahway, New Jersey, 1991.
An "isolated" nucleic acid molecule is a
nucleic acid molecule separate and discrete from the
whole organism with which the molecule is found in
nature; or a nucleic acid molecule devoid, in whole or
part, of sequences normally associated with it in
-17-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
nature; or a sequence, as it exists in nature, but
having heterologous sequences (as defined below) in
association therewith.
Hy "subunit vaccine composition" is meant a
composition containing at least one immunogenic
polypeptide, but not all antigens, derived from or
homologous to an antigen from a pathogen of interest.
Such a composition is substantially free of intact
pathogen cells or particles, or the lysate of such
cells or particles. Thus, a "subunit antigen
composition" is prepared from at least partially
purified (preferably substantially purified)
immunogenic polypeptides from the pathogen, or
recombinant analogs thereof. A subunit antigen
composition can comprise the subunit antigen or
antigens of interest substantially free of other
antigens or polypeptides from the pathogen.
The term "epitope" refers to the site on an
antigen or hapten to which specific B cells and/or T
cells respond. The term is also used interchangeably
with "antigenic determinant" or "antigenic determinant
site." Antibodies that recognize the same epitope can
be identified in a simple immunoassay showing the
ability of one antibody to block the binding of
another antibody to a target antigen.
An "immunological response" to a composition
or vaccine is the development in the host of a
cellular and/ or antibody-mediated immune response to
the composition or vaccine of interest. Usually, an
"immunological response" includes but is not limited
to one or more of the following effects: the
production of antibodies, B cells, helper T cells,
suppressor T cells, and/or cytotoxic T cells and/or yb
T cells, directed specifically to an antigen or
antigens included in the composition or vaccine of
interest. Preferably, the host will display either a
-18-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
therapeutic or protective immunological response such
that resistance_of the mammary gland to new infection
will be enhanced and/or the clinical severity of the
disease reduced. Such protection will be demonstrated
by either a reduction or lack of symptoms normally
displayed by an infected host, a quicker recovery time
and/or a lowered somatic cell count in milk from the
infected quarter.
The terms "immunogenic" protein or
polypeptide refer to an amino acid sequence which
elicits an immunological response as described above.
An "immunogenic" protein or polypeptide, as used
herein, includes the full-length sequence of the LF-
binding protein, with or without the signal sequence,
membrane anchor domain and/or LF-binding domain,
analogs thereof, or immunogenic fragments thereof. By
"immunogenic fragment" is meant a fragment of an LF-
binding protein which includes one or more epitopes
and thus elicits the immunological response described
above. Such fragments can be identified using any
number of epitope mapping techniques, well known in
the art. See, e.g., Epitope Mapping Protocols in
Methods in Molecular Biology, Vol. 66 (Glenn E.
Morris, Ed., 1996) Humana Press, Totowa, New Jersey.
For example, linear epitopes may be determined by
e.g., concurrently synthesizing large numbers of
peptides on solid supports, the peptides corresponding
to portions of the protein molecule, and reacting the
peptides with antibodies while the peptides are still
attached to the supports. Such techniques are known
in the art and described in, e.g., U.S. Patent No.
4,708,871; Geysen et al. (1984) Proc. Natl. Acad.
Sci. USA 81:3998-4002; Geysen et al. (1986) Molec.
Immunol. 23:709-715. Similarly, conformational
epitopes are readily_identified by determining spatial
conformation of amino acids such as by, e.g., x-ray
-19-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
crystallography and 2-dimensional nuclear magnetic
resonance. See, e.g., Epitope Mapping Protocols,
s upra .
Immunogenic fragments, for purposes of the
present invention, will usually include at least about
3 amino acids, preferably at least about 5 amino_
acids, more preferably at least about 10-15 amino
acids, and most preferably 25 or more amino acids, of
the Lbp molecule. There is no critical upper limit to
the length of the fragment, which could comprise
nearly the full-length of the protein sequence, or
even a fusion protein comprising two or more epitopes
o f Lbp .
"Native" proteins or polypeptides refer to
proteins or polypeptides isolated from the source in
which the proteins naturally occur. "Recombinant"
polypeptides refer to polypeptides produced by
recombinant DNA techniques; i.e., produced from cells
transformed by an exogenous DNA construct encoding the
desired polypeptide. "Synthetic" polypeptides are
those prepared by chemical synthesis.
A "vector" is a replicon, such as a plasmid,
phage, or cosmid, to which another DNA segment may be
attached so as to bring about the replication of the
attached segment.
A DNA "coding sequence" or a "nucleotide
sequence encoding" a particular protein, is a DNA
sequence which is transcribed and translated into a
- polypeptide in vitro or in vivo when placed under the
control of appropriate regulatory elements. The
boundaries of the coding sequence are determined by a
start codon at the 5' (amino) terminus and a
translation stop codon at the 3' (carboxy) terminus.
A coding sequence can include, but is not limited to,
procaryotic sequences, cDNA from eucaryotic mRNA,
genomic DNA sequences from eucaryotic (e. g.,
-20-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
mammalian) DNA, and even synthetic DNA sequences. A
transcription termination sequence will usually be
located 3' to the coding sequence.
DNA "control elements" refers collectively
to promoters, ribosome binding sites, polyadenylation
signals, transcription termination sequences, upstream
regulatory domains, enhancers, and the like, which
collectively provide for the transcription and
translation of a coding sequence in a host cell. Not
all of these control sequences need always be present
in a recombinant vector so long as the desired gene is
capable of being transcribed and translated.
"Operably linked" refers to an arrangement
of elements wherein the components so described are
configured so as to perform their usual function.
Thus, control elements operably linked to a coding
sequence are capable of effecting the expression of
the coding sequence. The control elements need not be
contiguous with the coding sequence, so long as they
function to direct the expression thereof. Thus, for
example, intervening untranslated yet transcribed
sequences can be present between a promoter and the
coding sequence and the promoter can still be
considered "operably linked" to the coding sequence.
_ A control element, such as a promoter,
"directs the transcription" of a coding sequence in a
cell when-RNA polymerase will bind the promoter and
transcribe the coding sequence into mRNA, which is
then translated into the polypeptide encoded by the
coding sequence.
A "host cell" is a cell which has been
transformed, or is capable of transformation, by an
exogenous nucleic acid molecule.
A cell has been "transformed" by exogenous
DNA when such exogenous DNA has been introduced inside
the cell membrane. Exogenous DNA may or may not be
-21-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
integrated (covalently linked) into chromosomal DNA
making up the genome of-the cell. In procaryotes and
yeasts, for example, the exogenous DNA may be
maintained on an episomal element, such as a plasmid.
With respect to eucaryotic cells, a stably transformed
cell is one in which the exogenous DNA has became
integrated into the chromosome so that it is inherited
by daughter cells through chromosome replication.
This stability is demonstrated by the ability of the
eucaryotic cell to establish cell lines or clones
comprised of a population of daughter cells containing
the exogenous DNA.
"Homology" refers to the percent identity
between two polynucleotide or two polypeptide
moieties. The correspondence between the sequence
from one moiety to another can be determined by
techniques known in the art. For example, homology
can be determined by a direct comparison of the
sequence information between two polypeptide molecules
by aligning the sequence information and using readily
available computer programs such as ALIGN, Dayhoff,
M.O. (1978) in Atlas of Protein Sequence and
Structure 5:Suppl. 3, National biomedical Research
Foundation, Washington, DC.
Alternatively, homology can be determined by
hybridization of polynucleotides under conditions
which form stable duplexes between homologous regions,
followed by digestion with single-stranded-specific
nuclease(s), and size determination of the digested
fragments. Two DNA, or two polypeptide sequences are
"substantially homologous" to each other when the
sequences exhibit at least about 80~-855, preferably
at least about 90%, and most preferably at least about
95~-98~ sequence identity over a defined length of the
molecules, as determined using the methods above. As
used herein, substantially homologous also refers to
-22-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
sequences showing complete identity to the specified
DNA or polypeptide sequence. DNA sequences that are
substantially homologous can be identified in a
Southern hybridization experiment under, for example,
stringent conditions, as defined for that particular
system. Defining appropriate hybridization conditions
is within the skill of the art. See, e.g., Sambrook
et al . , supra; DNA Cloning, supra; Nucleic Acid
Hybridization, supra.
The term "functionally equivalent" intends
that the amino acid sequence of an LF-binding protein
is one that will elicit a substantially equivalent or
enhanced immunological response, as defined above, as
compared to the response elicited by an LF-binding
protein having identity with the reference LF-binding
protein, or an immunogenic portion thereof.
A "heterologous" region of a DNA construct
is an identifiable segment of DNA within or attached
to another DNA molecule that is not found in
association with the other molecule in nature. Thus,
when the heterologous region encodes a bacterial gene,
the gene will usually be flanked by DNA that does not
flank the bacterial gene in the genome of the source
bacteria. Another example of the heterologous coding
sequence is a construct where the coding sequence
itself is not found in nature (e. g., synthetic
sequences having codons different from the native
gene). Allelic variation or naturally occurring
mutational events do not give rise to a heterologous
region of DNA, as zlsed herein.
The term "treatment" as used herein refers
to either (i) the prevention of infection or
reinfection (prophylaxis), or (ii) the reduction or
elimination of symptoms of the disease of interest
(therapy) .
-23-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
As used herein, a "biological sample" refers
to a sample of tissue or fluid isolated from a
subject, including but not limited to, for example,
blood, plasma, serum, fecal matter, urine, bone
marrow, bile, spinal fluid, lymph fluid, samples of
the skin, external secretions of the skin,
respiratory, intestinal, and genitourinary tracts,
tears, saliva, milk, blood cells, organs, biopsies and
also samples of in vitro cell culture constituents
including but not limited to conditioned media
resulting from the growth of cells and tissues in
culture medium, e.g., recombinant cells, and cell
components.
As used herein, the terms "label" and
"detectable label" refer to a molecule capable of
detection, including, but not limited to, radioactive
isotopes, fluorescers, chemiluminescers, enzymes,
enzyme substrates, enzyme cofactors, enzyme
inhibitors, chromophores, dyes, metal ions, metal
sols, ligands (e. g., biotin or haptens) and the like.
The term "fluoresces" refers to a substance or a
portion thereof which is capable of exhibiting
fluorescence in the detectable range. Particular
examples of labels which may be used under the
invention include fluorescein, rhodamine, dansyl, -
umbelliferone, Texas red, luminol, NADPH and a-/3-
galactosidase.
B. General Methods
Central to the present invention is the
discovery of a bovine LF-binding protein in S. uberis.
The gene for the S. uberis bLF-binding protein ("Ibp")
has been isolated and characterized, and the protein
encoded thereby sequenced. The complete DNA and amino
acid sequences of S. uberis bLF-binding protein are
shown in Figures 2A-2C (SEQ ID NOS:l-2). In
-24-
SUBSTITUTE SHEET (RULE 26~
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
particular, as described in the examples, a single ORF
of 1683 bp, depicted as residues 232-1914, inclusive
of Figures 2A-2C {SEQ ID NOS:1-2), encoding 561 amino
acid residues, gives rise to two protein species able
to bind bovine lactoferrin, having molecular weights
of 76 kDa and~165 kDa, respectively. The 165 kDa
protein is likely a dimer of the 76 kDa protein since
urea treatment results in a single band and Northern
blot analysis shows only one major transcript in S.
uberis, as well as in recombinant E. coli transformed
with a construct encoding the LF-binding protein.
S. uberis bovine LF-binding protein includes
a putative N-terminal signal peptide of about 50 amino
acids (if translation starts at the first ATG codon
shown in Figure 2A). Thus, the full-length bovine LF-
binding protein depicted, including the signal
sequence, is found at amino acid positions 1-561,
inclusive, (encoded by nucleotide positions 232-1914,
inclusive) of Figures 2A-2C (SEQ ID NOS:1-2). The
mature protein, lacking the signal peptide, is found
at amino acid positions 52-561, inclusive, (nucleotide
positions 445-1914, inclusive) of Figures 2A-2C. A
membrane anchor motif at the C-terminus is also
present, as indicated in Figures 2A-2C (SEQ ID NOS:1-
2). A bovine Lf binding domain is present in a 200
codon N-terminal region of the molecule. The protein
appears to lack disulfide bridges.
As shown in the examples, the binding of
~zsl_bLf to S. uberis was time-dependent and
displaceable by unlabelled bLf. Apo-bLf inhibits lzsl-
bLf binding as effectively as iron-saturated bLf.
Bovine transferrin, human lactoferrin and human
transferrin do not interfere with bLf binding. The
Scatchard plot is linear and approximately 7800
binding sites are expressed by each bacterial cell,
with an affinity of 1.0 x 10-' M. Reduced iron
-25-
SUBSTITUTE SHEET (RULE 26)
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
availability does not significantly modify the
saturation of S. uberis by bLf. The bLf binding
protein described herein is lactoferrin species-
specific, in that human Lf does not appear to
effectively block the binding of bovine Lf.
The Lf binding protein of S. uberis differs
from the transferrin receptors of Haemophilus and
Neisseria spp., which consist of two distinct
transferrin-binding proteins, termed Tbpl and Tbp2,
which range in molecular weight from 68 to 105 kDa
depending on the strain. Similarly, the bovine Lf
receptor of S. aureus consists of two distinct bLf
binding proteins with estimated molecular weights of
92 and 67 kDa (Naidu et al. (1991) J. Dairy Sci.
74:1218-1226) and therefore appears to be different
from the receptor described herein. Also, the
streptococcal LF-binding protein described herein
appears to be different from the S. aureus human Lf
binding protein, an approximately 450 kDa protein
which, under reducing SDS-PAGE gel conditions,
resolves into two components of 67 and 62 kDa.
Analysis of the primary and secondary
structure of the S. uberis bLf binding protein
suggests that it is an M-like protein. In particular,
a gene homologous to the group A streptococcal mga, a
positive regulator of M and M-like proteins, has been
found in the upstream adjacent region of lbp.
Southern blot analysis reveals that mga is present in
all S. uberis strains tested that contained the lbp.
The sequence of S. uberis mga and the
protein product therefrom is presented in Figures 5A-
5D (SEQ ID NOS:3-12). Starting at the ATG initiation
codon at nucleotides 361-363 and terminating at a TAA
codon at nucleotides 1858-1860, the deduced gene
product, Mga, is comprised of 499 amino acid residues
with a calculated molecular weight of 58,454 Da. The
-26-
SUBSTITUTE SHEET (RULE 26)
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
N-terminus of Mga lacks the features of a signal
peptide, suggesting that it is a cytoplasmic protein.
Preceding the start codon of mga is a putative
ribosome binding site AGGAGA. Sequences resembling
the -35 and -10 promoter motifs have also been
identified, as shown in Figures 5A-5D (SEQ ID
NOS:3-12).
S. uberis LF-binding protein, immunogenic
fragments thereof or chimeric proteins including the
same, can be provided in subunit vaccine compositions
to treat or prevent bacterial infections caused by S.
uberis, including mastitis in mammals, such as in
bovine, equine, ovine and goat species. In addition
to use in vaccine compositions, the proteins and
fragments thereof, antibodies thereto, and genes
coding therefor, can be used as diagnostic reagents to
detect the presence of infection in a mammalian
subject. Similarly, the genes encoding the proteins
can be cloned and used to design probes to detect and
isolate homologous genes in other bacterial strains.
For example, fragments comprising at least about 15-20
nucleotides, more preferably at least about 20-50
nucleotides, and most preferably about 60-100 or more
nucleotides, will find use in these embodiments. The
S. uberis LF-binding proteins also find use in
purifying bovine LFs from streptococcal species and
from recombinant host cells expressing the same.
S. uberis Lf binding proteins can be used in
vaccine compositions either alone or in combination
with other bacterial, fungal, viral or protozoal
antigens. These antigens can be provided separately
or even as fusion proteins comprising one or more
epitopes of an LF-binding protein fused to one or more
of these antigens. For example, other immunogenic
proteins from S. uberis, such as the CAMP factor,
hyaluronic acid capsule, hyaluronidase, R-like protein
-27-
SUBSTITUTE SHEET (RULE 26)
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
and plasminogen activator, can be administered with
the LF-binding protein. Additionally, immunogenic
proteins from other organisms involved in mastitis,
such as from the genera Staphylococcus,
Corynebacterium, Pseudomonas, Nocardia, Clostridium,
Mycobacterium, Mycoplasma, Pasteurella, Prototheca,
other streptococci, coliform bacteria, as well as
yeast, can be administered along with the bLF-binding
proteins described herein to provide a broad spectrum
of protection. Thus, for example, immunogenic
proteins from Staphylococcus aureus, Str. agalactiae,
Str. dysgalactiae, Str. zooepidemicus, Corynebacterium
pyogenes, Pseudomonas aeruginosa, Nocardia asteroides,
Clostridium perfringens, Escherichia coli,
Enterobacter aerogenes and Klebsiella spp. can be
provided along with the bLF-binding proteins of the
present invention.
Production of LF-Binding Protein
The above described LF-binding proteins and
active fragments, analogs and chimeric proteins
derived from the same, can be produced by a variety of
methods. Specifically, LF-binding proteins can be
isolated directly from bacteria which express the
same. This is generally accomplished by first
preparing a crude extract which lacks cellular
components and several extraneous proteins. The
desired proteins can then be further purified i.e. by
column chromatography, HPLC, immunoadsorbent
techniques or other conventional methods well known in
the art.
Alternatively, the proteins can be
recombinantly produced as described herein. As
explained above, these recombinant products can take
the form of partial protein sequences, full-length
sequences, precursor forms that include signal
-28-
CA 02270404 1999-04-30 -
WO 98/21231 PCT/CA97100867
sequences, mature forms without signals, or even
fusion proteins (e.g., with an appropriate leader for
the recombinant host, or with another subunit antigen
sequence for Streptococcus or another pathogen).
The 1bp genes of the present invention can
be isolated based on the ability of the protein
products to bind LF, using LF-binding assays as
described below. Thus, gene libraries can be
constructed and the resulting clones used to transform
an appropriate host cell. Colonies can be pooled and
screened for clones having LF-binding activity.
Colonies can also be screened using polyclonal serum
or monoclonal antibodies to the LF-binding protein.
Alternatively, once the amino acid sequences
are determined, oligonucleotide probes which contain
the codons for a portion of the determined amino acid
sequences can be prepared and used to screen genomic
or cDNA libraries for genes encoding the subject
proteins. The basic strategies for preparing
oligonucleotide probes and DNA libraries, as well as
their screening by nucleic acid hybridization, are
well known to those of ordinary skill in the art.
See, e.g., DNA Cloning: Vol. I, supra; Nucleic Acid
Hybridization, supra; 0ligonucleotide Synthesis,
supra; Sambrook et al., supra. Once a clone-from the
screened library has been identified by positive
hybridization, it can be confirmed by restriction
enzyme analysis and DNA sequencing that the particular
library insert contains an LF-binding protein gene or
a homolog thereof. The genes can then be further
isolated using standard techniques and, if desired,
PCR approaches or restriction enzymes employed to
delete portions of the full-length sequence.
Similarly, genes can be isolated directly
from bacteria using known techniques, such as phenol
extraction and the sequence further manipulated to
-29-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
produce any desired alterations. See, e.g., Sambrook
et al., supra, for a description of techniques used to
obtain and isolate DNA.
Alternatively, DNA sequences encoding the
proteins of interest can be prepared synthetically
rather than cloned. The DNA sequences can be designed
with the appropriate codons for the particular amino
acid sequence. In general, one will select preferred
codons for the intended host if the sequence will be
used for expression. The complete sequence is
assembled from overlapping oligonucleotides prepared
by standard methods and assembled into a complete
coding sequence. See, e.g., Edge (1981) Nature
292:756; Nambair et al. (1984) Science 223:1299; Jay
et al. (1984) J. Biol. Chem. 259:6311.
Once coding sequences for the desired
proteins have been prepared or isolated, they can be
cloned into any suitable vector or replicon. Numerous
cloning vectors are known to those of skill in the
art, and the selection of an appropriate cloning
vector is a matter of choice. Examples of recombinant
DNA vectors for cloning and host cells which they can
transform include the bacteriophage ~ (E. coli),
pBR322 (E. coli), pACYC177 (E. coli), pKT230
(gram-negative bacteria), pGV1106 (gram-negative
bacteria), pLAFRl (gram-negative bacteria), pME290
(non-E. coli gram-negative bacteria), pHVl4 (E. coli
and Bacillus subtilis), pHD9 (Bacillus), pIJ61
(Streptomyces), pUC6 (Streptomyces), YIp5
(Saccharomyces), YCpl9 (Saccharomyces) and bovine
papilloma virus (mammalian cells). See, Sambrook et
al . , supra; DNA Cloning, supra; B . Perbal , supra .
The gene can be placed under the control of
a promoter, ribosome binding site (for bacterial
expression) and, optionally, an operator (collectively
referred to herein as "control" elements), so that the
-30-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
DNA sequence encoding the desired protein is
transcribed into RNA in the host cell transformed by a
vector containing this expression construction. The
coding sequence may or may not contain a signal
peptide or leader sequence. If a signal sequence is
included, it can either be the native, homologous
sequence, or a heterologous sequence. For example,
the signal sequence for S. uberis LF-binding protein
(shown in Figure 2A), can be used for secretion
thereof, as can a number of other signal sequences,
well known in the art. Leader sequences can be
removed by the host in post-translational processing.
See, e.g., U.S. Patent Nos. 4,431,739; 4,425,437;
4,338,397.
Other regulatory sequences may also be
desirable which allow for regulation of expression of
the protein sequences relative to the growth of the
host cell. Regulatory sequences are known to those of
skill in the art, and examples include those which
cause the expression of a gene to be turned on or off
in response to a chemical or physical stimulus,
including the presence of a regulatory compound.
Other types of regulatory elements may also be present
in the vector, for example, enhancer sequences.
The control sequences and other regulatory
sequences may be ligated to the coding sequence prior
to insertion into a vector, such as the cloning
vectors described above. Alternatively, the coding
sequence can be cloned directly into. an expression
vector which already contains the control sequences
and an appropriate restriction site.
In some cases it may be necessary to modify
the coding sequence so that it may be attached to the
control sequences with the appropriate orientation;
i.e., to maintain the proper reading frame. It may
also be desirable to produce mutants or analogs of the
-31-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97l00867
LF-binding protein. Mutants or analogs may be
prepared by the deletion of a portion of the sequence
encoding the protein, by insertion of a sequence,
and/or by substitution of one or more nucleotides
within the sequence. Techniques for modifying
nucleotide sequences, such as site-directed
mutagenesis, are described in, e.g., Sambrook et al.,
supra; DNA Cloning, supra; Nucleic Acid Hybridization,
supra .
The expression vector is then used to
transform an appropriate host cell. A number of
mammalian cell lines are known in the art and include
immortalized cell lines available from the American
Type Culture Collection (ATCC), such as, but not
limited to, Chinese hamster ovary (CHO) cells, HeLa
cells, baby hamster kidney (BHK) cells, monkey kidney
cells (COS), human hepatocellular carcinoma cells
(e.g., Hep G2), Madin-Darby bovine kidney ("MDBK")
cells, as well as others. Similarly, bacterial hosts
such as E. coli, Bacillus subtilis, and Streptococcus
spp., will find use with the present expression
constructs. Yeast hosts useful in the present
invention include inter alia, Saccharomyces
cerevisiae, Candida albicans, Candida maltosa,
Hansenula polymorpha, Kluyveromyces fragili~,
Kluyveromyces lactis, Pichia guillerimondii, Pichia
pastoris, Schizosaccharomyces pombe and Yarrowia
lipolytica. Insect cells for use with baculovirus
expression vectors include, inter alia, Aedes aegypti,
Autographa californica, Bombyx mori, Drosophila
melanogaster, Spodoptera frugiperda, and Trichoplusia
ni.
Depending on the expression system and host
selected, the proteins of the present invention are
produced by culturing host cells transformed by an
expression vector described above under conditions
-32-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
whereby the protein of interest is expressed. The
protein is then isolated from the host cells and puri-
fied. If the expression system secretes the protein
into the growth media, the protein can be purified
directly from the media. If the protein is not
secreted, it is isolated from cell lysates. The
selection of the appropriate growth conditions and
recovery methods are within the skill of the art.
The proteins of the present invention may
also be produced by chemical synthesis such as solid
phase peptide synthesis, using known amino acid
sequences or amino acid sequences derived from the DNA
sequence of the genes of interest. Such methods are
known to those skilled in the art. See, e.g., J. M.
Stewart and J. D. Young, Solid Phase Peptide
Synthesis, 2nd Ed., Pierce Chemical Co., Rockford, IL
(1984) and G. Barany and R. B. Merrifield, The
Peptides: Analysis, Synthesis, Biology, editors E.
Gross and J. Meienhofer, Vol. 2, Academic Press, New
York, (1980), pp. 3-254, for solid phase peptide
synthesis techniques; and M. Bodansky, Principles of
Peptide Synthesis, Springer-Verlag, Berlin (1984) and
E. Gross and J. Meienhofer, Eds., The Peptides:
Analysis, Synthesis, Biology, supra, Vol. 1, for
classical solution synthesis. Chemical synthesis of
peptides may be preferable if a small fragment of the
antigen in question is capable of raising an
immunological response in the subject of interest.
The LF-binding proteins of the present
invention, or their fragments, can be used to produce
antibodies, both polyclonal and monoclonal. If
polyclonal antibodies are desired, a selected mammal,
(e. g., mouse, rabbit, goat, horse, etc.) is immunized
with an antigen of the present invention, or its
fragment, or a mutated antigen. Serum from the
immunized animal is collected and treated according to
-33-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
known procedures. See, e.g., Jurgens et al. (1985) J.
Chrom. 348:363-370. I~ serum containing polyclonal
antibodies is used, the polyclonal antibodies can be
purified by immunoaffinity chromatography, using known
procedures.
Monoclonal antibodies to the LF-binding
proteins and to the fragments thereof, can also be
readily produced by one skilled in the art. The
general methodology for making monoclonal antibodies
by using hybridoma technology is well known. Immortal
antibody-producing cell lines can be created by cell
fusion, and also by other techniques such as direct
transformation of H lymphocytes with oncogenic DNA, or
transfection with Epstein-Barr virus. See, e.g., M.
Schreier et al., Hybridoma Techniques (1980);
Hammerling et al., Monoclonal Antibodies and T-cell
Hybridomas (1981); Kennett et al., Monoclonal
Antibodies (1980); see also U.S. Patent Nos.
4,341,761; 4,399,121; 4,427,783; 4,444,887; 4,452,570;
4,466,917; 4,472,500, 4,491,632; and 4,493,890.
Panels of monoclonal antibodies produced against the
LF-binding protein, or fragment thereof, can be
screened for various properties; i.e., for isotype,
epitope, affinity, etc. Monoclonal antibodies are
useful in purification, using immunoaffinity
techniques, of the individual antigens which they are
directed against. Both polyclonal and monoclonal
antibodies can also be used for passive immunization
or can be combined with subunit vaccine preparations
to enhance the immune response. Polyclonal and
monoclonal antibodies are also useful for diagnostic
purposes.
Vaccine Formulations and Administration
The LF-binding proteins of the present
invention can be formulated into vaccine compositions,
-34-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
either alone or in combination with other antigens,
for use in immunizing subjects as described below.
Methods of preparing such formulations are described
in, e.g., Remington's Pharmaceutical Sciences, Mack
Publishing Company, Easton, Pennsylvania, 18 Edition,
1990. Typically, the vaccines of the present
invention are prepared as injectables, either as
liquid solutions or suspensions. Solid forms suitable
for solution in or suspension in liquid vehicles prior
to injection may also be prepared. The preparation
may also be emulsified or the active ingredient
encapsulated in liposome vehicles. The active
immunogenic ingredient is generally mixed with a
compatible pharmaceutical vehicle, such as, for
example, water, saline, dextrose, glycerol, ethanol,
or the like, and combinations thereof. In addition,
if desired, the vehicle may contain minor amounts of
auxiliary substances such as wetting or emulsifying
agents and pH buffering agents.
Adjuvants which enhance the effectiveness of
the vaccine may also be added to the formulation.
Adjuvants may include for example, muramyl dipeptides,
avridine, aluminum hydroxide, dimethyldioctadecyl
ammonium bromide (DDA), oils, oil-in-water emulsions,
saponins, cytokines, and other substances known in the
art.
The Lf binding protein may be linked to a
carrier in order to increase the immunogenicity
thereof. Suitable carriers include large, slowly
metabolized macromalecules such as proteins, including
serum albumins, keyhole limpet hemocyanin,
immunoglobulin molecules, thyroglobulin, ovalbumin,
and other proteins well known to those skilled in the
art; polysaccharides, such as sepharose, agarose,
cellulose, cellulose beads and the like; polymeric
amino acids such as polyglutamic acid, polylysine, and
-35-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
the like; amino acid copolymers; and inactive virus
particles.
The LF-binding proteins may be used in their
native form or their functional group content may be
modified by, for example, succinylation of lysine
residues or reaction with Cys-thiolactone. A
sulfhydryl group may also be incorporated into the
carrier (or antigen) by, for example, reaction of
amino functions with 2-iminothiolane or the
N-hydroxysuccinimide ester of 3-(4-dithiopyridyl
propionate. Suitable carriers may also be modified to
incorporate spacer arms (such as hexamethylene diamine
or other bifunctional molecules of similar size) for
attachment of peptides.
Other suitable carriers for the LF-binding
proteins of the present invention include VP6
polypeptides of rotaviruses, or functional fragments
thereof, as disclosed in U.S. Patent No. 5,071,651.
Also useful is a fusion product of a viral protein and
the subject immunogens made by methods disclosed in
U.S. Patent No. 4,722,840. Still other suitable
carriers include cells, such as lymphocytes, since
presentation in this form mimics the natural mode of
presentation in the subject, which gives rise to the
immunized state. Alternatively, the proteins of the
present invention may be coupled to erythrocytes,
preferably the subject's own erythrocytes. Methods of
coupling peptides to proteins or cells are known to
those of skill in the art.
Furthermore, the LF-binding proteins (or
complexes thereof) may be formulated into vaccine
compositions in either neutral or salt forms.
Pharmaceutically acceptable salts include the acid
addition salts (formed with the free amino groups of
the active polypeptides) and which are formed with in-
organic acids such as, for example, hydrochloric or
-36-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
phosphoric acids, or such organic acids as acetic,
oxalic, tartaric, mandelic, and the like. Salts
formed from free carboxyl groups may also be derived
from inorganic bases such as, for example, sodium,
potassium, ammonium, calcium,- or ferric hydroxides,
and such organic bases as isopropylamine,
trimethylamine, 2-ethylamino ethanol, histidine,
procaine, and the like.
Vaccine formulations will contain a
"therapeutically effective amount" of the active
ingredient, that is, an amount capable of eliciting an
immune response in a subject to which the composition
is administered. In the treatment and prevention of
mastitis, for example, a "therapeutically effective
amount" would preferably be an amount that enhances
resistance of the mammary gland to new infection
and/or reduces the clinical severity of the disease.
Such protection will be demonstrated by either a
reduction or lack of symptoms normally displayed by an
infected host, a quicker recovery time and/or a
lowered somatic cell count in milk from the infected
quarter. For example, the ability of the composition
to retain or bring the somatic cell count (SCC) in
milk below about 500,000 cells per ml, the threshold
value set by the International Dairy Federation, above
which, animals are considered to have clinical
mastitis, will be indicative of a therapeutic effect.
The exact amount is readily determined by
one skilled in the art using standard tests. The LF-
binding protein concentration will typically range
from about 1% to about 95% (w/w) of the composition,
or even higher or lower if appropriate. With the
present vaccine formulations, 20 to 500 ~g of active
ingredient per ml of injected solution should be
adequate to raise animmunological response when a
dose of 1 to 3 ml per animal is administered.
-37-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/0(1867
To immunize a subject, the vaccine is
generally administered parenterally, usually by
intramuscular injection. Other modes of
administration, however, such as subcutaneous,
intraperitoneal and intravenous injection, are also
acceptable. The quantity to be administered depends
on the animal to be treated, the capacity of the
animal's immune system to synthesize antibodies, and
the degree of protection desired. Effective dosages
can be readily established by one of ordinary skill in
the art through routine trials establishing dose
response curves. The subject is immunized by
administration of the vaccine in at least one dose,
and preferably two doses. Moreover, the animal may be
administered as many doses as is required to maintain
a state of immunity to infection.
Additional vaccine formulations which are
suitable for other modes of administration include
suppositories and, in some cases, aerosol, intranasal,
oral formulations, and sustained release formulations.
For suppositories, the vehicle composition will
include traditional binders and carriers, such as,
polyalkaline glycols, or triglycerides. Such
suppositories may be formed from mixtures containing
the active ingredient in the range of about 0.5% to
about 10% (w/w), preferably about 1% to about 2%.
Oral vehicles include such normally employed
excipients as, for example, pharmaceutical grades of
mannitol, lactose, starch, magnesium stearate, sodium
saccharin cellulose, magnesium carbonate, and the
like. These oral vaccine compositions may be taken in
the form of solutions, suspensions, tablets, pills,
capsules, sustained release formulations, or powders,
and contain from about 10% to about 95% of the active
ingredient, preferably about 25% to about 70%.
-38-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
Intranasal formulations will usually include
vehicles that neither cause irritation to the nasal
mucosa nor significantly disturb ciliary function.
Diluents such as water, aqueous saline or other known
substances can be employed with the subject invention.
The nasal formulations may also contain preservatives
such as, but not limited to, chlorobutanol and
benzalkonium chloride. A surfactant may be present to
enhance absorption of the subject proteins by the
nasal mucosa.
Controlled or sustained release formulations
are made by incorporating the protein into carriers or
vehicles such as liposomes, nonresorbable impermeable
polymers such as ethylenevinyl acetate copolymers and
Hytrel° copolymers, swellable polymers such as
hydrogels, or resorbable polymers such as collagen and
certain polyacids or polyesters such as those used to
make resorbable sutures. The LF-binding proteins can
also be delivered using implanted mini-pumps, well
known in the art.
The LF-binding proteins of the instant
invention can also be administered via a carrier virus
which expresses the same. Carrier viruses which will
find use with the instant invention include but are
not limited to the vaccinia and other pox viruses,
adenovirus, and herpes virus. By way of example,
vaccinia virus recombinants expressing the novel
proteins can be constructed as follows. The DNA
.. encoding the particular protein is first inserted into
an appropriate vector so that it is adjacent to a
vaccinia promoter and flanking vaccinia DNA sequences,
such as the sequence encoding thymidine kinase (TK).
This vector is then used to transfect cells which are
simultaneously infected with vaccinia. Homologous
recombination serves to insert the vaccinia promoter
plus the gene encoding the instant protein into the
-39-
CA 02270404 1999-04-30
WO 98/21231 PCTlCA97/00867
viral genome. The resulting TK~recombinant can be
selected by culturing the cells in the presence of 5-
bromodeoxyuridine and picking viral plaques resistant
thereto.
An alternative route of administration
involves gene therapy or nucleic acid immunization.
Thus, nucleotide sequences (and accompanying
regulatory elements) encoding the subject LF-binding
proteins can be administered directly to a subject for
in vivo translation thereof. Alternatively, gene
transfer can be accomplished by transfecting the
subject's cells or tissues ex vivo and reintroducing
the transformed material into the host. DNA can be
directly introduced into the host organism, i.e., by
injection (see International Publication No.
WO/90/11092; and Wolff et al. (1990) Science 247:1465-
1468). Liposome-mediated gene transfer can also be
accomplished using known methods. See, e.g., Hazinski
et al. (1991) Am. J. Respir. Cell MoI. Biol. 4:206-
209; Brigham et al. (1989) Am. J. Med. Sci. 298:278-
281; Canonico et al. (1991) Clin. Res. 39:219A; and
Nabel et al. (1990) Science 249:1285-1288. Targeting
agents, such as antibodies directed against surface
antigens expressed on specific cell types, can be
covalently conjugated to the liposomal surface so that
the nucleic acid can be delivered to specific tissues
and cells susceptible to infection.
Diacrnostic Assays
As explained above, the LF-binding proteins
of the present invention may also be used as
diagnostics to detect the presence of reactive
antibodies of S. uberis in a biological sample in
order to determine the presence of S. uberis
infection. For example, the presence of antibodies
reactive with LF-binding proteins can be detected
-40-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
using standard electrophoretic and immunodiagnostic
techniques, including immunoassays such as
competition, direct reaction, or sandwich type assays.
Such assays include, but are not limited to, Western
blots; agglutination tests; enzyme-labeled and
mediated immunoassays, such as ELISAs; biotin/avidin
type assays; radioimmunoassays; immunoelectrophoresis;
immunoprecipitation, etc. The reactions generally
include revealing labels such as fluorescent,
chemiluminescent, radioactive, enzymatic labels or dye
molecules, or other methods for detecting the
formation of a complex between the antigen and the
antibody or antibodies reacted therewith.
The aforementioned assays generally involve
separation of unbound antibody in a liquid phase from
a solid phase support to which antigen-antibody
complexes are bound. Solid supports which can be used
in the practice of the invention include substrates
such as nitrocellulose (e.g., in membrane or
microtiter well form); polyvinylchloride (e. g., sheets
or microtiter wells); polystyrene latex (e. g., beads
or microtiter plates); polyvinylidine fluoride;
diazotized paper; nylon membranes; activated beads,
magnetically responsive beads, and the like.
Typically, a solid support is first reacted
with a solid phase component (e.g., one or more LF-
binding proteins) under suitable binding conditions
such that the component is sufficiently immobilized to
the support. Sometimes, immobilization of the antigen
to the support can be enhanced by first coupling the
antigen to a protein with better binding properties.
Suitable coupling proteins include, but are not
limited to, macromolecules such as serum albumins
including bovine serum albumin (BSA), keyhole limpet
hemocyanin, immunoglobulin molecules, thyroglobulin,
ovalbumin, and other proteins well known to those
-41-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
skilled in the art. Other molecules that can be used
to bind the antigens to the support include
polysaccharides, polylactic acids, polyglycolic acids,
polymeric amino acids, amino acid copolymers, and the
like. Such molecules and methods of coupling these
molecules to the antigens, are well known to those of
ordinary skill in the art. See, e.g., Brinkley, M.A.
Bioconjugate Chem. (1992) 3:2-13; Hashida et al., J.
Appl. Biochem. (1984) 6:56-63; and Anjaneyulu and
Staros, International J. of Peptide and Protein Res.
(1987) 30:117-124.
After reacting the solid support with the
solid phase component, any non-immobilized solid-phase
components are removed from the support by washing,
and the support-bound component is then contacted with
a biological sample suspected of containing ligand
moieties (e. g., antibodies toward the immobilized
antigens) under suitable binding conditions. After
washing to remove any non-bound ligand, a secondary
binder moiety is added under suitable binding
conditions, wherein the secondary binder is capable of
associating selectively with the bound ligand. The
presence of the secondary binder can then be detected
using techniques well known in the art.
More particularly, an ELISA method can be
used, wherein the wells of a microtiter plate are
coated with an LF-binding protein. A biological
sample containing or suspected of containing anti-LF-
binding protein immunoglobulin molecules is then added
to the coated wells. After a period of incubation
sufficient to allow antibody binding to the
immobilized antigen, the plates) can be washed to
remove unbound moieties and a detectably labeled
secondary binding molecule added. The secondary
binding molecule is allowed to react with any captured
sample antibodies, the plate washed and the presence
-42-
CA 02270404 1999-04-30
WO 98121231 PCT/CA97/00867
of the secondary binding molecule detected using
methods well known in the art.
Thus, in one particular embodiment, the
presence of bound anti-LF-binding antigen ligands from
a biological sample can be readily detected using a
secondary binder comprising an antibody directed
against the antibody ligands. A number of anti-bovine
immunoglobulin (Ig) molecules are known in the art
which can be readily conjugated to a detectable enzyme
label, such as horseradish peroxidase, alkaline
phosphatase or urease, using methods known to those of
skill in the art. An appropriate enzyme substrate is
then used to generate a detectable signal. In other
related embodiments, competitive-type ELISA techniques
can be practiced using methods known to those skilled
in the art.
Assays can also be conducted in solution,
such that the LF-binding proteins and antibodies
specific for those proteins form complexes under
precipitating conditions. In one particular
embodiment, LF-binding proteins can be attached to a
solid phase particle (e.g., an agarose bead or the
like) using coupling techniques known in the art, such
as by direct chemical or indirect coupling. The
antigen-coated particle is then contacted under
suitable binding conditions with a biological sample
suspected of containing antibodies for the LF-binding
proteins. Cross-linking between bound antibodies
-- causes the formation of particle-ant-igen-antibody
complex aggregates which can be precipitated and
separated from the sample using washing and/or
centrifugation. The reaction mixture can be analyzed
to determine the presence or absence of antibody-
antigen complexes using any of a number of standard
methods, such as those immunodiagnostic methods
described above.
-43-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
In yet a further embodiment, an
immunoaffinity matrix can be provided, wherein a
polyclonal population of antibodies from a biological
sample suspected of containing anti-LF-binding
molecules is immobilized to a substrate. In this
regard, an initial affinity purification of the sample
can be carried out using immobilized antigens. The
resultant sample preparation will thus only contain
anti-S. uberis moieties, avoiding potential ___
nonspecific binding properties in the affinity
support. A number of methods of immobilizing
immunoglobulins (either intact or in specific
fragments) at high yield and good retention of antigen
binding activity are known in the art. Not being
limited by any particular method, immobilized protein
A or protein G can be used to immobilize
immunoglobulins.
Accordingly, once the immunoglobulin
molecules have been immobilized to provide an
immunoaffinity matrix, labeled LF-binding proteins are
contacted with the bound antibodies under suitable
binding conditions. After any non-specifically bound
antigen has been washed from the immunoaffinity
support, the presence of bound antigen can be
determined by assaying for label using methods known
in the art.
Additionally, antibodies raised to the LF-
binding proteins, rather than the LF-binding proteins
themselves, can be used in the above-described assays
in order to detect the presence of antibodies to the
proteins in a given sample. These assays are
performed essentially as described above and are well
known to those of skill in the art.
The above-described assay reagents,
including the LF-binding proteins, or antibodies
thereto, can be provided in kits, with suitable
-44-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
instructions and other necessary reagents, in order to
conduct immunoassays as=described above. The kit can
also contain, depending on the particular immunoassay
used, suitable labels and other packaged reagents and
materials (i.e. wash buffers and the like). Standard
immunoassays, such as those described above, can be
conducted using these kits.
Below are examples of specific embodiments
for carrying out the present invention. The examples
are offered for illustrative purposes only, and are
not intended to limit the scope. of the present
invention in any way.
C. Experimental
Example 1
Identification of a Bovine Lactoferrin-Bindin
Protein in S. Uberis
Materials and Methods
Bacterial strains and cultural conditions.
S. uberis strain (su-1) from the American
Type Culture Collection (ATCC 9927) was used for
study. Bacteria were grown on base #2 sheep blood agar
plates (PML Microbiologicals) at 37°C for 18 h. Iron-
restricted conditions were achieved in Todd-Hewitt
broth supplemented with 0.35 yeast extract (THB-YE) by
the addition of 800 ~M EDDA, 800 ~,M dipyridyl or 100
~cM desferrioxamine mesylate. All of the iron
chelators were obtained from Sigma.
Preparation of bacterial cell wall
Cell wall components of S. uberis were
extracted as described by Baker et al. (1976) J. Exp.
Med. 143:258-270. Twenty base #2 blood agar plates
were inoculated with S. uberis and incubated for 18 h
-45-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
at 37°C. Bacterial cells were collected, washed once
with 200 ml 0.85% saline and resuspended in 50 ml of
extraction buffer (0.05 M Na2HP04, 0.15 M NaCl, 0.01 M
EDTA, pH 7.4). Cell walls were extracted by shaking
with glass beads (4-mm diameter) for 20 h at 37°C.
After centrifugation at 48,300 g for 20 min,
supernatant (cell wall extract) was collected,
filtered (0.22 ~.M Nalgene filter), dialysed against
distilled water, lyophilized and resuspended in 1 ml
of distilled water.
Preparation of iron-binding proteins.
All iron-binding proteins, including bLf
(from bovine milk), bovine transferrin (bTf), human
lactoferrin (hLf) and human transferrin (hTf), were
purchased fram Sigma in the most iron-free form
available. Iron-saturated proteins and the
apoproteins were prepared by methods described
previously (Mazurier and Spik (1980) Biochim. Biophys.
Acta 629:399-408).
Preparation of l2sl-labelled bLF.
Bovine Lf was iodinated by the
lactoperoxidase method of Thorell and Johansson (1971)
Biochim. Biophys. Acta 251:363-368. Approximately 70
of bLf (33% iron-saturated) was used for
iodination; Izsl_labelled protein was separated from
free Nal2sl by chromatography on a Sephadex G-25
column. The labelled protein was aliquoted and stored
at -70°C until use. Lactoperoxidase was purchased
from Boehringer-Mannheim; and NalzSI from Amersham.
Lactoferrin-binding assays.
The binding assays were performed as
described (Naidu et al. (1990) J. Clin. Microbiol.
28:2312-2319; Naidu et al. (1991) J. Med. Micrbiol.
-46-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
34:323-328; Naidu et al. {1991) J. Dairy Sci. 74:1218-
1226; Naidu et al. {1992) J. Med. Microbiol. _36:177-
183). Bacterial cells were harvested from culture
media, washed once in 0.1 M phosphate-buffered saline
(PBS), pH 7.2, and resuspended in PBS containing 1%
bovine serum albumin (PBS-1% BSA) to a density of 101°
bacteria/ml (c. ODsoo=1.5). To determine saturation
time, 109 bacteria {in 0.1 ml of PBS-1% BSA) were mixed
and incubated with 0.1 ml of lzSI-bLf solution (6.9 nM
in PBS-1 % BSA) for periods of 5, 10, 15, 20, 25, 30,
60, 90, 120, 150 min at room temperature. Bacteria
were pelleted and washed three times with 1 ml of ice-
cold PBS containing 0.1% Tween 20. Radioactivity
bound to the bacterial pellet was measured in a 'y-
counter. In competitive binding experiments, 109
bacteria were mixed with 2 x 105 cpm lzSI-bLf in the
presence of serially-diluted unlabelled bLf and
incubated at room temperature for 2 h. Total input,
cell bound and free proteins were calculated and
subjected to Scatchard analysis (Scatchard, G. (1949)
Ann. N. Y. Acad. Sci. 51:660-672). When evaluating
the inhibitory effect of bLf, apo-bLf, bTf, hLF and
hTf on 1251-bLf binding, the unlabelled proteins were
used at concentration of 5.5 ~,M. All samples were
tested in triplicate, and each experiment was repeated
at least twice. The data presented are the means of
two independent experiments {unless otherwise stated).
Proteolvtic and heat treatment of S uberis
Bacteria (1 ml containing 101° cells) were
treated with proteases at 37°C for 2 h. Trypsin
{Sigma) hydrolysis was performed in 100 mM Tris-HCl
(pH 8.0), with a final enzyme concentration of 2,500
u/ml, and the reaction was stopped by addition of
phenylmethylsulfonyl fluoride (500 ~.g/ml). Pepsin
(Sigma) digestion was performed in 100 mM sodium
-47-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
acetate buffer (pH 4.5), with an enzyme concentration
of 1,000 u/ml, and the pH of the reaction mixture was
raised to 7.4 to stop the hydrolysis. Proteinase K
(Boehringer mannheim) treatment was carried out in 40
mM potassium phosphate buffer (pH 7.5), and the
digestion was inhibited by the addition of
phenylmethylsulfonyl fluoride (500 ~.g/ml). For heat
treatment, the bacterial suspension (101° cell/ml) was
incubated in a water bath for 1 h at each of the
following temperatures: 50°C, 80°C and 100°C. Both
enzyme and heat-treated cells were washed once in PBS
and resuspended in PBS-1% BSA prior to the binding
experiments.
PAGE and Western blottin
SDS-polyacrylamide gel electrophoresis
(PAGE) of proteins was performed using the method
described by Laemmli (Laemmli, U.K. (1970) Nature
227:680-685). Samples were solubilized in sample
buffer at 37°C for 30 min in the absence of 2-
mercaptoethanol (non-reducing conditions) or at 100°C
for 5 min in the presence of 1% 2-mercaptoethanol
(reducing conditions). Proteins were
electrophoretically transferred to nitrocellulose
membranes as recommended by the supplier (Bio-Rad) and
blocked with TBS-1% BSA.
To identify the putative bLf-binding
protein, Western blots were probed with l2sI_bLf as
follows. 125I-bLf was added to the membrane to a final
concentration of 80 ng/ml in TBS-1% BSA and incubated
at room temperature for 2 h. After three washes with
TBS containing 0.05% Tween 20, the membrane was
exposed to X-ray film for 24 h at room temperature.
To compete the lzsI-bLf binding, the transferred
membrane was incubated with 35 ~g/ml of unlabelled bLf
for 2 h before incubation with lzsl_bLf .
-48-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
Results
Time-dependent bindincr of bLF to S. uberis.
S. uberis strain su-1 was tested for bLf
binding in alzsI-labelled protein binding assay. To
study the kinetics of lzsI-bLf binding with S. uberis,
binding was measured at different time intervals
{Figure 7). The time course showed thatlzSI-bLf could
bind to S. uberis in a time-dependent manner, with a
requirement of approximately 90 min for 100%
saturation. This binding saturation time was the
basis for determining the incubation time in later
binding experiments.
BLf receptor saturability, affinity and copy number.
A competitive binding experiment using bLf
(33% iron-saturated) as both radioligand and
competitor was performed, and the specificity of bLf
binding by S. uberis was demonstrated {Figure 8).
Unlabelled bLf effectively displaced the binding of
lzSI-bLf to S. uberis in a dose-dependent manner. A
concentration of approximately 270 nM of unlabelled
bLf caused 50% blocking of lzsl_bLf uptake {indicated
by dotted lines). Scatchard plot analysis showed
linearity, thus the demonstration of one bLf binding
component is expected. The number of bLf molecules
bound per S. uberis cell calculated from Scatchard
plot was approximately 7800, with an affinity (Kd) of
1.0 x 10-'M.
To determine whether bLf iron saturation
could influence receptor binding, apo-bLf was used as
a competitor in the lzsI-bLf binding assay. The
results showed that apo-bLf could inhibit lzSI-bLF-
binding as effectively as the iron-saturated bLf
(Table d), indicating that both apo-bLf and iron-
saturated bLf had the same binding receptor on the S.
uberis cell.
-49-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
To examine further the specificity of
binding, the abilities of bTf, hLf and hTf to inhibit
the binding of lzsl_bLf to S. uberis cells were also
evaluated (Table 1). None of these proteins
interfered with the binding of lzSI-bLf to S. uberis,
suggesting that the binding was bLf-specific.
Influence of iron-restricted conditions on bLf
binding.
In an attempt to determine if the bLf-
binding property of S. uberis was mediated by an iron-
regulated bacterial component, EDDA, dipyridyl or
desferrioxamine mesylate were incorporated in THB-YE
broth to reduce the availability of iron. Cells from
these iron-restricted conditions did not show higher
~zsl_bLf binding than those from normal cultural
condition (Figure 9), indicating that iron-restricted
condition did not notably modify the saturation of S.
uberis by bLf.
25
35
-50-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97I00867
Table 1. Inhibitory effects of unlabelled proteins and
enzyme or heat treatment of bacteria lzsI-bLf
on
binding to S. uberis.
- Inhibitor or Percentage inhibition)
or
cell treatment decreasez in binding
mean SD
Inhibitor
bLf 94.6 0.1
apo-bLf 94.7 0.2
bTf 2.5 1.4
hLf -14.0 2.4
hTf 1.9 1.3
Proteases
Pepsin 85.7 3.7
Trypsin 89.1 4.4
Proteinase K 92.7 2.4
Heating
50°C -4.3 6.0
80°C 29.6 2.1
100°C 65.6 4.8
Inhibition values were calculated as relative
percentage of bLf binding to bacteria suspended in PBS
in the absence of any inhibitor.
2 Decrease in binding were calculated as relative
percentage of bLf binding to bacteria without any
treatment.
Sensitivity of the bLf binding component to protease
h-ydrolysis and heat treatment.
Pepsin, trypsin and proteinase k treatment
of S. uberis cells could abolish bLf-binding (Table
-51-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
1), indicating the involvement of a surface-exposed
cell wall proteins) in the binding. This
proteinaceous component was susceptible to
temperature, since heat treatment of bacteria reduced
binding to a certain degree Table 1).
Identification of a cell wall bLf-binding protein.
The presence of the functionally active bLf-
binding protein (Lbp) in the cell wall preparation of
S. uberis was detected by a Western blot probed with
~zsl-bLf. Under non-reducing conditions, two
components with apparent molecular weights of I65 and
76 kDa, respectively, were identified as bLf-binding
proteins of S. uberis. Proteins under reducing
conditions lost the bLf binding activity to a great
extent. The protein bands were demonstrated to be
from specific binding to 125I-bLf, since the presence
of unlabelled bLf effectively blocked the binding.
Discussion
To demonstrate whether S. uberis was able to
express a specific receptor for bLf, the inventors
herein sought to determine whether the major
prerequisites for a biological receptor could be
fulfilled, namely, ligand specificity and
concentration-dependent saturability. Bovine Lf
binding to S. uberis was time and concentration
dependent {demonstrated by the ability of the
unlabelled ligand to compete for binding with 1251-
bLf), indicating the existence of a limited number of
binding receptors on the cell surface. Scatchard plot
analysis estimated that there were 7800 bLf-binding
sites/cell. The affinity of the streptococcal
receptor for bLf (1.0 x 10-' M) is slightly lower than
that described by Naidu et al. (1991) J. Dairy Sci.
-52-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
74:1218-1226 for the bLf receptor of S. aureus (7.1 x
10-a M) .
S. uberis did not distinguish between bovine
. apo-Lf and iron-saturated Lf, since both forms were
equally effective at blocking lzsl-bLf binding in the
competitive binding assay. This finding is similar to
that described for the N. meningitides Lf receptor
(Schryvers and Morris (1988) Infect. Immun. 56:1144-
1149) but contrasts with that for the Tf receptors.
A common feature which has emerged from
studies of the transferrin-binding proteins of Gram-
negative bacteria is their remarkable specificity for
the transferrin of their natural host. In competition
binding assays, the streptococcal receptor described
here also demonstrated some lactoferrin species
specificity in that human Lf could not effectively
block the binding of bovine Lf. Interestingly, S.
aureus Lf receptors have been shown to bind
lactoferrins from both human and bovine sources (Naidu
et al. (1990) J. Clin. Microbiol. 28:2312-2319; Naidu
et al. (1991) J. Med. Micrbiol. 34:323-328; Naidu et
al. (1991) J. Dairy Sci. 74:1218-1226; Naidu et al.
(1992) J. Med. Microbiol. 36:177-183). In this
context, given that S. uberis is exclusively a bovine
pathogen, while S. aureus causes infection in both
humans and bovine, it is possible that the specificity
for lactoferrins may also contribute to the host
specificity of these bacteria: In addition, neither
bovine nor human transferrin was able to block binding
of lzsl_labelled bovine lactoferrin to the lactoferrin
receptor of S. uberis. This observation is similar to
that reported by Naidu et al. (1991) J. Dairy Sci.
74:1218-1226; Naidu et al. (1992) J. Med. Microbiol.
36:177-183, in which human or bovine lactoferrin-
binding to lactoferrin receptors of S, aureus could
not be blocked by human or bovine transferrin.
-53-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
Although the expression of most of the
transferrin and lactoferrin receptors of Gram-negative
bacteria such as N. meningitidis and H. influenza is
iron regulated, S. uberis receptor activity was not
regulated by growth medium iron availability. The
results are consistent with the findings of Modun et
al. (1994) Infect. Immun. 62:3850-3858 and Rainard, P.
(1992) FEMS Microbiol. Lett. 98:235-240 who showed
that the binding of S. aureus to Tf and S. agalactiae
to Lf was not regulated by iron.
Having demonstrated the existence of bovine
lactoferrin-receptor on S. uberis, the inventors
herein sought to identify the bacterial cell wall
components) involved. Treatment of S. uberis cells
with heat and proteolytic enzymes abolished.
lactoferrin-binding, indicating the probable
involvement of a proteins) on the cell surface. A
cell wall extract of S, uberis was prepared and
solubilized in SDS-PAGE sample buffer under non-
reducing condition. Two lactoferrin-binding proteins
with molecular weights of 165 kDa and 76 kDa were
identified by 125I_bLf binding after SDS-PAGE and
transfer to nitrocellulose. Since Scatchard plot
analysis indicated that S. uberis bears only one bLf
binding_component, the 165 kDa protein is likely the
dimer form of the 76 kDa.
The lactoferrin-binding protein of S. uberis
differs from the transferrin receptors of Haemophilus
and Neisseria spp., which consist of two distinct
transferrin-binding proteins, termed Tbpl and Tbp2,
which range in molecular weight from 68 to 105 kDa
depending on the strain. The S. uberis Lbp is also
different from the lactoferrin receptors of the
mentioned two species. To date, only one lactoferrin-
binding protein with a molecular weight of 98 to 105
kDa has been identified from those species above and
-54-
CA 02270404 1999-04-30 -
WO 98/21231 PCT/CA97/00867
the gonococcal lactoferrin protein (Lbpl) has been
shown to share features with the functionally related
gonococcal Tbpl (Biswas and Sparling, 1995). The
bovine lactoferrin receptor of S. aureus consists of
two distinct bLf-binding proteins with estimated
molecular weights of 92 and 67 kDa (Naidu et al.
(1991) J. Dairy Sci. 74:1218-1226) and therefore
appears to be different than the receptor described
herein. Also, the streptococcal Lbp described here
appears to be different from the S. aureus human
lactoferrin-binding protein which is as an
approximately 450 kDa protein which under reducing
SDS-PAGE gel conditions resolves into two components
of 67 and 62 kDa.
Example 2
Clonincr and Characterization of S.- uberis Lbp Gene
and its Upstream Mcta
Materials and Methods
Bacterial strains, plasmids and media.
S. uberis strains used are listed in Table 2
below. Bacteria were grown on base #2 sheep blood
agar plates (PML Microbiologicals) at 37°C for 18 h,
or in Todd-Hewitt broth supplemented with 0:3% yeast
extract (THB-YE) at 37°C overnight. E. coli cells
were grown in Luria broth or on Luria broth-agar
plates. Ampicillin was used at 50 ~.g/ml for the
growth of E. coli strains containing recombinant
plasmids. The cloning vector used was pTZl8R (Mead et
al. (1986) Protein Eng. 1:67-74).
Pret~aration of bacterial cell wall cell membranes
periplasma, whole cell lvsate and culture supernatant
Cell wall components of S. uberis were
extracted as described in Example 1. Outer and inner
-55-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
membranes were isolated from E. coli cells by sucrose
density gradient centrifugation. E. coli periplasmic
proteins were prepared by the cold osmotic shock
method.
To prepare whole cell lysates of E. coli
transformants, bacteria were grown overnight,
collected, washed once in 0.1 M phosphate-buffered
saline (PBS), pH 7.2, resuspended in water, exposed to
one freeze-thaw cycle and sonicated for 2 min. After
centrifugation at 6000 x g for 20 min, the supernatant
(whole cell lysate) was collected, lyophilized and
resuspended in 1/100 culture volume of water.
The 100 fold-concentrated culture
supernatant of the recombinant E. coli was obtained by
precipitation with 10% trichloroacetic acid (TCA).
Preparation of iron-saturated bLf and lzSl_labelled
bLf .
33% iron-saturated bovine lactoferrin was
prepared and iodinated as described in Example 1.
Lactoferrin-bindincr assay.
Bacterial cells were harvested from culture
media, washed once in 0.1 M PBS (pH 7.2), and
resuspended in PBS containing 1% bovine serum albumin
(PBS-1% BSA) to a density of 101° bacteria/ml (c.
ODsoo=1.5). To assess the bLf-binding ability of the
recombinant protein, increasing amounts of E. coli
cell lysate plus supernatant were mixed with
approximately 1Q9-S. uberis and incubated with 1.0 x
10' cpm 125I-bLf in a total volume of 200 ~,1. After 2
hours of incubation at room temperature, bacterial
cells were pelleted and washed three times with 1 ml
of ice-cold PBS containing 0.1 % Tween 20.
Radioactivity present in the bacterial pellet was
measured in a gamma-counter. All samples were tested
-56-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
in triplicate, and the experiment was repeated twice.
The data presented are=the mean of two independent
experiments t standard deviation.
Preparation of antiserum.
Serum against the recombinant Lbp of S.
uberis was raised in rabbits by subcutaneous injection
of 0.5 ml of TCA-precipitated culture supernatant of
recombinant E. coli in complete Freund's adjuvant.
Two subcutaneous boosts with the same amount of sample
in incomplete Freund's adjuvant were given to each
animal.
PAGE and Western blotting.
SDS-polyacrylamide gel electrophoresis
(PAGE) of proteins was performed as described by
Laemmli (Laemmli, U.K. (1970) Nature 227:680-685).
Samples under reducing and non-reducing conditions
were prepared as described in Example 1. Samples
dissolved in sample buffer with 3 M urea were boiled
for 30 min prior to loading. l2sl-bLf probed-Western
blotting was performed as in Example 1.
Immunoblotting probed with rabbit antiserum against
the recombinant Lbp was performed as follows.
proteins were electroblotted onto nitrocellulose
membranes as recommended by the supplier (Bio-Rad).
Nonspecific binding was blocked by incubation in TBS
(10 mM Tris-HC1, pH 7.5, 140 mM NaCl)-1% bovine serum
albumin (BSA). Blots were incubated with antibody
diluted 1:200 in TBS-1% BSA at room temperature for 1
h. After three washes in TBS containing 0.05% Tween
20, seroreactive proteins were detected with goat
anti-mouse (or rabbit) IgG coupled to alkaline
phosphatase (Kirkegaard & Perry Laboratories, Inc.) at
1:5,000 in TBS-1% BSA. Alkaline phosphatase activity
was detected using the Nitro Blue Tetrazolium-5-bromo-
-57-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
4-chloro-3-indolylphosphate toluidinium system as
described by the supplier (Promega).
Recombinant DNA techniques.
Plasmid DNA was purified as described by
Sambrook et al., supra. When required, DNA fragments
were isolated from agarose gels using a Gene Clean kit
(Bio/can Scientific).
To construct a gene library for S. uberis,
chromosomal DNA was prepared as previously described
(Caparon and Scott (1987) Proc. Natl. Acad. Sci. USA
84:8677-8681) and partially digested with Sau3AI.
Fragments of 2,000 to 5,000 by were recovered
following sucrose density gradient centrifugation
(Sambrook et al., supra). The ends of these fragments
were partially filled in with dGTP and dATP and
ligated into pTZl8R which was cut with SalI and
partially filled in with dTTP and dCTP.
Transformation of E. coli DF-I5a competent cells was
carried out as recommended by the supplier (GIBCO BRL,
Gaithersburg, Md). To identify Lf-binding clones,
transformants were replica-plated onto nitrocellulose
discs (Schleicher & Schuell, Keene; NH) and lysed in
chloroform vapor. Nonspecific binding was blocked by
incubation with TBS-1~ BSA. Membranes were further
incubated with l2sI-bLf as described in Example 1.
Restriction endonucleases, T4 DNA ligase,
DNA polymerase I Klenow fragment and calf intestinal
alkaline phosphatase were utilized according to the
manufacturer's directions (Pharmacia Canada Ltd.,
Quebe c , Canada ) .
DNA sequences were determined by the
dideoxy-chain termination method of Sanger et al.
(1977) Proc. Natl. Acad. Sci. USA 74:5463-5467 on
double-stranded plasmid templates by using a T7
Sequencing kit (Pharmacia Canada Ltd.).
-58-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
RNA analyses.
RNA from E~ coli strains was isolated as
described previously (Lloubes et al. (1986) Nucleic
Acid Res. 14:2621-2636) with an additional RNase-free
DNase I digestion.
RNA from S. uberis was prepared as follows.
The cell pellet from a 10 ml culture (OD6oo=0.6) was
resuspended in 250 ~C1 of TE buffer (pH 8.0) containing
500 a of mutanolysin (Sigma) and incubated at 37°C for
30 min. Lysis buffer (250 ~1) (60 mM Tris-HC1 pH 7.4,
200 mM NaCl, 10 mM EDTA, 2% SDS) and 100 ~.g/ml (final
concentration) of proteinase K was added and the
incubation continued for 1 h. The sample was
extracted once with 65°C phenol (water saturated, pH
4.0) and twice with room temperature phenol. RNA was
recovered by ethanol precipitation and treated with
DNase I (Pharmacia Canada Ltd.).
Northern blot analysis was carried out as
described by Sambrook et al., supra.
Results
Clonina and expression of the 1bp gene.
A gene library was constructed in pTZl8R
with chromosomal DNA from S. uberis (su-1). About
S000 transformants were initially screened for
expression of bLf binding protein (Lbp) by colony
blotting with lzsl_bLf. One colony with the strongest
signal and six with weaker signals were selected and
used to make whole cell lysates, which were further
tested for their ability to bind l2sI-bLF under non-
reducing conditions. The clone with the strongest
signal, E. coli pLBPS, generated three major bands;
two of them had molecular weights of 165 and 76 kDa
which are quite close in size to those of S. uberis.
A band slightly larger than 165 kDa was also observed
which could be the precursor form with the uncleaved
-59-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
signal peptide. No corresponding band was found from
the whole cell lysate of E. coli pTZl8R, the host
strain control. The recombinant Lbp was also detected
from the periplasm and supernatant of E. coli pLBP5,
but not from outer or inner membranes, indicating that
the Lbp expressed in E. coli DHSa was not membrane-
localized, but instead could be secreted from the
cell.
The presence of free and functionally active
recombinant Lbp in the cell lysate and supernatant of
E. coli pLBP5 was also detected by performing
competitive inhibition assays. The E. cold pLBP5 cell
lysate and supernatant mixture effectively inhibited
1251-bLf binding to S. uberis cells in a dose-dependent
manner, while samples from E. coli pTZlBR did not
(Figure 11).
To demonstrate whether the protein with a
molecular weight of 165 kDa was a dimer of the 76 kDa
molecule, samples treated with 2-mercaptoethanol or
urea were analyzed by Western blotting using rabbit
antiserum against the supernatant of E. coli pLBP5.
Mercaptoethanol treatment was shown to have no effect
on the electrophoretic mobility of Lbp from either S.
uberis or recombinant E. coli while urea-treatment
resulted in disappearance of the 165 and 76 kDa bands
and appearance of a new band with an apparent
molecular weight of 105 kDa. This data indicates that
the 165 kDa protein is likely a dimer of the 76 kDa
subunit. The dimer could have been disassociated by
urea to monomers which could be further denatured and
unfolded, resulting in the apparent molecular weight
increase from 76 kDa to 105 kDa. A significant
increase in the apparent molecular weight of Tbp2
following urea-treatment has been observed in
Neisseria meningitides by other researchers (yonder
haar et al. (1994) J. Bacteriol. 176:6207-6213). The
-60-
CA 02270404 1999-04-30_
WO 98/21231 PCT/CA97/00867
above data also indicates that disulfide bonds are not
essential for the formation of the oligomer.
Nucleotide seauence determination and analysis
To determine the nucleotide sequence of the
Lbp gene, each of the HincII, HindIII, Sacl, SphI and
XbaI fragments of pLBPS (Figure 1) was individually
cloned into the corresponding site of pTZlBR. By
using universal and reverse primers, each fragment was
sequenced in both orientations. Subsequent primers
were synthesized on the basis of sequence information
thereby obtained.
Two open reading frames were found from
pLBP5 (Figure 1). One was the Lbp-encoding gene lbp,
whose presence in subclone pLBPSL resulted in a bLf-
binding phenotype. The other ORF on the complementary
DNA strand was incomplete. A GenBank database search
showed that the presumed ORF gene product had
significant homology to the VirR and Mry positive
regulators in group A streptococci. Thus, this ORF
was named mga' (Figure 1). The cloning and sequencing
of the complete mga gene is discussed further below.
The 1bp sequence contained two potential
translation start codons (ATG) at positions 232 and
262 of the DNA sequence. Hoth are associated with
putative Shine-Dalgarno sequences (Figures 2A-2C).
These start points would give proteins with predicted
sizes of 62.857 and 61.454, respectively. The
predicted sizes are comparable to 76 KDa molecular
weight protein. The reason for the discrepancy
between the observed and calculated molecular weights
of this protein is not clear. Posttranslational
modification, such as lipid modification might have
occurred and increased the apparent molecular mass in
the SDS-PAGE determination. A similar difference
observed in Gram-negative bacteria has been
-61-
CA 02270404 1999-04-30 -
WO 98/21231 PCT/CA97/00867
demonstrated to be caused by protein lipid
modification (Theism et al. (1992) Infect. Immun.
60:826-831; Theisen et al. (1993) Infect. Immun.
61:1793-1798).
The DNA sequence shows two putative -10 and
-35 promoter regions present at -88 and -I02 from the
first ATG. Downstream of lbp there is a potential
rho-independent transcription terminator (Figures 2A-
2C) .
Analysis of the N-terminus of the predicted
sequence of Lbp showed amino acids characteristic of
signal sequences (Simonen and Palva (1993) Microbiol.
Rev. 57:109-137) (Figures 2A-2C). The features of the
sequence are a positively charged N-terminus, rich in
K and R residues, followed by a hydrophobic domain
from amino acids 25 to 48 (Figure 3) and the signal
peptidase cleaving site, VKA, at positions 49 to 50,
where cleavage occurs after the A residue. The
presence of this putative signal sequence indicates
that the Lbp is exported across the cytoplasmic
membrane of S. uberis.
A search of the GenBank database revealed
that the C-terminus of the Lbp was highly homologous
to the C- terminal ends of streptococcal M proteins,
plasminogen binding protein, fibrinogen and-IgG-
binding proteins. It shows all the general features
well established for surface proteins of Gram-positive
cocci (Fischetti et al. (1991) Common characteristics
of the surface proteins from Gram-positive cocci, p.
290-294. In G. M. Dunny, P. P. Cleary, and L. L. McKay
(ed), Genetics and molecular biology of streptococci,
lactococci, and enterococci. American Society for
Microbiology, Washington, D. C.). These features
include a small cluster of four charged amino acids at
the C-terminus followed by a hydrophobic domain of 21
amino acids (Figures 2A-2C and Figure 3). Adjoining
-62-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867 .
the hydrophobic domain is the consensus membrane
anchor motif LPSTGD. The next region of 50 amino
acids is the cell wall-associated region characterized
by a high proportion of proline and glycine (12%).
GenBank database searches also showed that Lbp is
homologous to mammalian myosin heavy chain and kinesin
heavy chain; it has 48% and 46% overall sequence
homology with these two fibrillar proteins,
respectively.
The region beyond the membrane anchor motif
and proline/glycine rich region contains three blocks
of amino acids which were found to have internal
homologies (Figures 2A-2C). The Al block contains 52
amino acids homologous to the A2 block; the B1 block
contains I3 amino acids homologous to the B2 block;
and the 59 amino acid C1 block is homologous to the C2
block.
Analysis of the secondary structure of the
translated protein showed an extensive a-helix region
stretching from the proline/glycine-rich region to the
~i-sheet and turn region near the cleavage site of the
signal sequence (Figure 3).
Localization of the Lf-binding domain of Lbp
To localize the Lf-binding domains of Lbp,
gene deletions were constructed (Figure 1). 3'-
deletions, pTP3l, pTP32, pTP33 and pTP34 were
generated at the restriction enzyme sites HincII,
StuI, XbaI and XmnI of pLBPSL. A 5'-deletion pTP51
was constructed by removing the 643 by HindIII-XbaI
fragment of pLBPSL. All these deletions contained the
lbp promoter. The resulting truncated Lbps were
produced in E. coli and were visualized after SDS-
PAGE, transfer to nitrocellulose, and reaction with
specific rabbit antiserum or lzsI-bLf. All the samples
were tested under non-reducing conditions in these
-63-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
experiments. The bLf binding results are summarized
in Figure 1. Like full-length Lbp, truncated proteins
from E. coli pTP3l, pTP32 and pTP33 could bind bLf in
both monomeric and dimeric forms. Some degradation
products from these clones also possessed binding
ability. No band was found from E. coli pTP34 or
pTP5l. These data indicate that the primary domain of
Lbp involved in the binding of bLf resides in a 22 kDa
N-terminal fragment.
Cloning and sectuencing of the complete mQa Qene
As described above, the plasmid pLBP5
contained an incomplete gene, mga', in addition to
lbp. The pLBP5 molecule was inverted to pLBPSi
(Figure 4) for convenience of description.
To obtain the complete mga gene, a 991 by
StyI fragment from mga' was radioactively (32P)
labelled as a probe (VP1 in Figure 4) and used to
screen an S. uberis gene library. A positive E. coli
DHSa clone was obtained and the restriction enzyme map
of the plasmid pMGAl4 is shown in Figure 4. To
determine the nucleotide sequence of pMGAl4, each of
the BamFiI, HincII, HindIII and KpnI restriction
fragments of pMGAl4 (Figure 4) was individually cloned
into the corresponding site of pTZl8R and sequenced
from both orientations using universal and reverse
primers. Subsequent primers were synthesized on the
basis of sequence information thereby obtained.
Plasmid pMGAl4 contained the complet-a ORF of Mga.
However, the associated promoter region was not
present. Since the 1.5 kb SphI-NheI fragment of pLBPSi
contained the majority of the 5' region of the mga'
and the complete promoter region, it was inserted into
the SphI and NheI sites of pMGAl4 to generate pMGAI4F
which then contained the complete mga gene including
the promoter (Figure 4).
-64-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
The sequence of a 3558 by DNA fragment of
pMGAI4F is presented in Figures 5A-5D. Starting at
the ATG initiation codon at nucleotides 361-363 and
terminating at a TAA codon at nucleotides 1858-1860,
the deduced gene product, Mga, is comprised of 499
amino acid residues with a calculated molecular weight
of 58,454 Da. The N- terminus of Mga lacks the
features of signal peptides as described (Simonen and
Palva (1993) Microbiol. Rev. 57:109-137), suggesting
that it is a cytoplasmic protein. Preceding the start
codon of mga is a putative ribosome binding site
AGGAGA. Sequences resembling the -35 and -10 promoter
motifs have also been identified.
A search of the GenBank database revealed
that the deduced protein of mga has 34% overall
sequence identity to the VirR and Mry proteins (Chen
et al. (1993) Mol. Gen. Genet. 241:685-693); Perez-
Casal et al. (1991) J. Bacteriol. 173:2617-2624).
Studies have shown that these positive regulators of
group A streptococcal M proteins contain helix-turn-
helix DNA binding domains, and it is believed that via
these domains, the regulators may interact directly
with specific DNA sequences to influence
transcriptional activity (Chen et al., supra; Perez-
Casal et al., supra). Attempts to identify a similar
DNA-binding domain with visual inspection and analysis
with PCGENE software failed to detect such a motif in
Mga of S. uberis. However, a region at amino acid
residues 106 to 125 (Figures SA-5D) showed 90%
identity to the sequence of the DNA-binding domain of
VirR49 (Podbielski et al. (1995) Infect. Immun. _63:9-
20). Following mga, there are four more ORFs
with 181, 85, 78 and 128 amino acid residues,
respectively (Figures 5A-5D). No significant sequence
alignments were found from GenBank.
-65-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
Northern blot analysis of the 1bp and mcra transcri~~ts
To analyze the lbp and mga transcripts, RNA
was prepared from S. uberis (su-1), E. coli
DHSa(pLBPS), E. coli DHSa(pLBPSL), E. coli
DHSa(pMGAI4F) and E. coli DHSa(pTZl8R), and used for
two Northern blots. One blot was probed with the 1.5
kb HindIII-HpaI internal fragment of 1bp (LP1 in
Figure 6). A 2.0 kb band was seen in lanes containing
RNA from S. uberis, E, coli DHSa(pLBP5) and E. coli
DH5~(pLBPSL), but was absent in the sample which
contained RNA from E. coli DHSa(pTZl8R). This data
indicates that only one major transcript was generated
by Ibp in recombinant E. coli as well as in native S.
uberis. The second blot was probed with the 1.0 kb
NcoI-NheI internal fragment of mga (VP2 in-Fig. 4). A
1.8 kb band was seen in samples which contained RNA
from E. coli DH5a(pLBPS) and E. coli DHSa(pMGAI4F),
respectively. No band was found from S. uberis and E.
coli DHScx(pTZl8R) RNA samples. The mga and mga' genes
were transcribed in recombinant E. coli, and the stop
codon of the vector must have been utilized during the
transcription of mga' gene. However, it is unclear
whether the absence of a visible hybridization band
from S. uberis was due to the low quantity of the gene
transcript or the inactivity of the mga gene.
Southern blotanalvsis of the lbp and mqa distribution
in S. uberis strains.
A collection of S. uberis including five
ATCC strains and 42 field isolates (Table 2) was used
in Southern hybridization experiments. Chromosomal
DNA was prepared and digested with the restriction
endonuclease HindIII and separated on agarose gels.
To analyze the lbp gene distribution, a 1.5 kb
HindIII-HpaI fragment which contained the most DNA
sequence from the region encoding Lbp (LP1 in Figure
-66-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
6) was used as a probe. This probe hybridized with DNA
from 42/47 strains (Table 2). This result meant
either that 1bp shared a region of homology with most
of the S. uberis strains or that among these strains,
there were different regions of the Ibp gene
homologous to the Ibp probe. To locate the homologous
regions) more specifically, this Ibp probe was
subdivided into smaller segments (Figure 6). Two
probes, the 643 by HindIII-XbaI fragment (LP2) and the
437 by XbaI-StuI fragment (LP3) covering the N-
terminal and central portion of the coding region in
the Ibp gene, respectively, hybridized only with su-1,
the strain from which the 1bp gene was originally
- cloned, whereas the 409 by StuI-HapI fragment from the
C-terminal region (LP4) hybridized with all the
strains that were hybridized by probe LP1 (Table 2).
It appears to be clear that the region of the lbp gene
encompassing the coding sequences for part of the C
repeat, the proposed wall attachment region and the
membrane anchor, is conserved among S. uberis strains,
while the region encoding the N-terminal portion
varies greatly. The diversity in sizes of chromosomal
restriction fragments from the different strains that
hybridized with the lbp probes indicates some degree
of restriction site heterogeneity (a different
location for HindIII site) among these strains.
35
-67-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
Table 2. S. uberis strains and DNA hybridization with LP
and VP probes
Probe
Strain Sources
LP1 LP2 LP3 LP4 VP3
su-1 ATCC9927 3.40b 3.40 3.40 3.40 0.87
su-2 ATCC13386 - - _ _ -
su-3 ATCC13387 3.60 - - 3.60 3.60
su-4 ATCC19436 2.70 - - 2.70 1.70
su-5 Chirino 93-1869 - - - _ _
su-6 Chirino 93-2017 - - -
_ _
su-7 Chirino 93-8678-2 4.40 - - 4.40 4.40
su-8 Chirino-S. uberis 5.00 - - 5.00 0.91
su-9 ATCC27958 3.80 - - 3.80 0.87
su-10 Greenfield 93-4997 3.20 - - 3.20 1.50
su-11 Greenfield 93-4997 3.20 - - 3.20 1.50
20
30 -
-68-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
Table 2. (continued)
Probe
Strain Sources
LP1 LP2 LP3 LP4 VP3
su-12 Leduc- 3.80 - - 3.80 0.87
su-13 Leduc 3.80 - - 3.80 0.87
su-14 Alix 5.00 - - 5.00 5.00
su-15 Magrath 4.10 - - 4.10 0.90
su-16 Ohaton 5.70 - - 5.70 0.90
su-17 Winfield 2.90 - - 2.90 1.40
su-18 Not known 4.10 - - 4.10 1.00
su-19 Gibbons 3.60 - - 3.60 1.20
su-20 Ardrossan 4.40 - - 4.40 4.40
su-21 Not known 3.40 - - 3.40 1.00
su-22 Dapp 2.90 - - 2.90 1.42
su-23 Thorsby 3.20 - - 3.20 1.40
su-24 Ponoka 3.80 - - 3.80 0.87
su-26 Warburg 4.40 - - 4.40 0.90
su-27 Leduc - - - - _
su-28 Fort MacLeod 3.00 - - 3.00 1.80
su-29 Sherwood Park 5.00 - - 5.00 5.00
su-30 Barrhead 5.00 - - 5.00 5.00
su-31 Wainwright 5.00 - - 5.00 5.00
su-32 Tofield 3.60 - - 3.60 1.03
su-33 Barrhead 5.00 - - 5.00 5.00
su-34 Barrhead 5.00 - - 5.00 5.00
su-35 Medicine Hat 5.00 - - 5.00 5.00
su-36 Ponoka 5.00 - - 5.00 5.00
su-37 Ponoka 5.00 - - 5.00 5.00
.su-38 Ponoka 5.00 - - 5.00 5.00
su-39 Ponoka 5.00 - - 5.00 5.00
su-40 Millet 3.60 - - 3.60 1.03
su-41 Lacombe 3.00 - - 3.00 1.40
su-42 Daysland 5.00 - - 5.00 5.00
su-43 Ohaton 5.30 - - 5.30 0.97
su-44 Ohaton 5.00 - - 5.00 5.00
su-45 Didsbury - - - _ _
su-46 Didsbury 4.70 - - 4.70 4.70
su-47 Didsbury 5.00 - - 5.00 5.00
su-48 Didsbury 4.70 - - 4.70 4.70
a All isolates except five ATCC
strains
are
field
(American
Type
Culture
Collection)
strains.
Size of hybridizing
fragment (in kilobase).
Lack of hybridization.
In order to study whether there was an mga-
related gene in other S. uberis strains, the previous
blot was stripped and reprobed with a 572 by HindIII
-69-
CA 02270404 1999-04-30
WO 98/21231 PCTlCA97/00867
fragment that encompassed the 5'-region of the mga
gene (VP3 in Figure 4). Hybridization was detectable
in all S. uberis strains that showed homology with the
lbp gene (Table 2). The specific band which reacted
with the mga probe in each strain is listed in Table
2.
Discussion
The Lbp gene of S. uberis was cloned in E.
coli after screening a gene library by colony blotting
with lzsl_bLf. E. coli transformants produced
functionally active bLf-binding proteins with
molecular weights of 76 kDa and 165 kDa, similar to
the native proteins produced by S. uberis.
Considering the one-affinity binding phenomenon of bLf
with S. uberis (Example 1), we wondered whether the
165 kDa protein was a dimer of the 76 kDa molecule.
Treatment with the reducing reagent (3-mercaptoethanol
did not have any effect on the mobilities of these
proteins, indicating the absence of disulfide bridges
in the protein structure. This was confirmed after
the sequencing data was available; there are no
appropriate cysteine residues within this protein.
After boiling for 30 min in the presence of 3 M urea,
proteins were completely denatured. This treatment
resulted in a significant molecular weight decrease of
the 165 kDa protein, probably resulting from the
dissociation of the dimer, and an increase in
molecular weight of the 76 kDa resulting from protein
unfolding. The dimer structure seems to be very
stable, since treatment with lower concentration of
urea or shorter boiling time could not alter the
mobility of the 165 kDa band. The presence of both
monomeric and dimeric Lbp in S. uberis cell wall
preparations may imply the existence of both forms on
the bacterial surface. However, it is possible that
-70-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
only the dimeric form exists on the bacterial cell;
the monomer might come from the cytoplasm due to
partial cell lysis caused by glass bead treatment.
The dissociation of the dimer to a monomer during
sample preparation seems unlikely because of the
stability of the dimer.
The complex nature of the S. uberis Lbp was
further confirmed from the nucleotide sequence
analysis. As expected, the two Lbps were encoded by a
single ORF of 1,683 bp. It is quite possible that two
copies of the translated product interact with each
other to form a homodimer. Interactions between
subunits could be extensive and extend throughout the
length of the molecule due to the existence of the
high a-helical content. Like M proteins (Fischetti,
V.A. (1989) Clin. Microbiol. Rev. 2:285-314), Lbp
shares significant sequence homology with a number of
a-helical coiled structure-containing mammalian
fibrillar proteins such as human myosin heavy chain
and kinesin heavy chain.
The deduced amino acid sequence of Lbp
indicated the existence of a signal peptide of 50
amino acids (if translation started at the first ATG
start codon) at the N-terminus, as expected for a
protein that appears on the outside of a bacterial
cell. The structure of this signal peptide is
comparable to the consensus structures for signal
peptides in proteins from Gram positive bacteria
described previously (Simonen and Palva (1993)
Microbiol. Rev. 57:109-137; Goward et al. (1993)
Trends Biochem. Sci. 18:136-140; Perlman and Halvorson
(1983) J. Mol. Biol. 167:391-409; von-Heijne, G.
(1983) Eur. J. Biochem. 133:17-21; van-Heijne, G.
(1986) Nucleic Acids Res. 14:4683-4690).
Analysis of the C-terminus of Lbp revealed
the presence of an amino acid sequence typical of
-71-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
membrane anchor motifs found in many other proteins
from Gram- positive bacteria (Fischetti et al. (1991)
Common characteristics of the surface proteins from
Gram-positive cocci, p. 290-294. In G. M. Dunny, P. P.
Cleary, and L. L. McKay (ed), Genetics and
molecular biology of streptococci, lactococci, and
enterococci. American Society for Microbiology,
Washington, D. C.). Such regions have been shown to
play a role in the anchoring of proteins in the cell
wall (Pancholi and Fischetti (1988) J. Bacteriol.
170:2618-2624; Schneewind et al. (1992) Cell 70:267-
281; Schneewind et al. (1993) Embo. J. 12:4803-4811).
It is probably via the C-terminal amino acids that Lbp
becomes anchored on the cell surface. The membrane
anchor motifs described here appear to have no
function in E. coli since the Lbp expressed in E. coli
was mainly secreted, similar to the streptococcal M6.1
protein which was found predominantly in the periplasm
(Fischetti et al., 1984).
5'- and progressive 3'-deletions to the lbp
gene allowed the definition of a large domain (about
200 codons) at the N-terminus which could bind bLf. A
comparison of this region with the transferrin binding
domain of Tbp2 from Neisseria (yonder Haar et al.
(1994) J. Bacteriol. 176:6207-6213) showed no
significant homology. Also, the bLf binding region
reported here did not contain any of the domains with
transferrin binding activity found in the transferrin-
binding protein (TfbA) of Actinobacillus
pleuropneumoniae (Strutzberg et al. (1995) Infect.
Immun. 63:3846-3850). This is not surprising since,
as shown in Example 1, bovine or human transferrin
could not block the binding of bovine lactoferrin to
S. uberis. This indicates a difference between the
bLf-binding domain and the Tf-binding domain.
A second ORF, mga, was found adjacent to the Ibp gene
-72-
CA 02270404 1999-04-30_
WO 98/21231 PCT/CA97/00867
region. The putative product of this gene (containing
499 amino acid residues) has comparable molecular size
and sequence to VirRl2 (499 residues) and Mry (530
residues), the positive regulators of the M protein
genes of group A streptococci (Chen et al. (1993) Mol.
Gen. Genet. 241:685-693); Perez-Casal et al. (1991) J.
Bacteriol. 173:2617-2624). However, Mry and VirRl2
had 98% homology (Chen et al., supra), whereas Mga
showed only 34% homology with them. This may be
attributed to the difference of species. Similarly,
VirR49 of an OF* GAS showed less homology (76%) to Mry
or VirR of OF- GAS (Podbielski et al. (1995) Infect.
Immun. 63:9-20).
The cytoplasmic location of Mga was
suggested by the absence of a signal peptide at the N-
terminus of the deduced protein. A potential -10 and
-35 promoter was found.
Studies have shown that mry is autoregulated
and environmentally regulated in response to the level
of COZ (Okada et al. (1993) Mol. Microbiol. 7:893-903).
Expression of mry was stimulated by increased
concentrations of CO2. In our experiments, S. uberis
cells were cultured under conditions without an
additional supplementation of C02. The absence of a
stimulating environment could have resulted in a very
low level of mga expression. This could be the reason
that Northern blot analysis did not detect any mga
transcript from S. uberis. It would be expected that
_in recombinant E. coli, iriga would not likely be
regulated by environmental signals due to the absence
of other regulatory components such as the sensing
protein.
It would be interesting to know whether mga
regulates the expression of lbp. No potential VirR-
binding boxes that are present in the promoters of M
and M-like protein genes (Podbielski et al. (1992)
-73-
CA 02270404 1999-04-30 -
WO 98121231 PCT/CA97/00867
Mol. Microbiol. 6:2253-2265; Podbielski et al. (1995)
Infect. Immun. 63:9-20) were found in the immediate
upstream portion of the -35 region of lbp by homology
comparisons. 47 S. uberis strains were blotted with
mga and 1bp specific probes in order to determine the
distribution of mga and its relation to Ibp in this
streptococcal group. Results showed that all strains
that were Ibp positive contained mga.
Southern blot analysis of Ibp in S. uberis
strains using subgenomic probes revealed that the C-
terminal sequence is conserved among strains, whereas
the N-terminal region shows greater variation. This
phenomenon resembles that described for M protein
genes in group A streptococci. By using DNA
hybridization with probes from the structural gene for
the M6 protein, Scott et al. (1986) Infect. Immun.
52:609-612 showed that the C-terminal region is highly
conserved among strains of different M serotypes and
the N-terminal region is highly variable. Consistent
with this, detailed amino acid sequence comparisons
and antibody reactivities revealed limited differences
within the conserved C-terminal regions and much more
extensive variability at the N- termini (Bessen and
Fischetti (1990) J. Exp. Med. 172:1757-1764; Bessen et
al. (1989) J. Exp. Med. 169:269-283; Kehoe,-(1991)
Vaccine 9:797-806). In the case of Lf binding
proteins of S. uberis, however, limited sequence and
serological data are available for comparisons. In M
proteins, the N-terminal region is distal to the
streptococcal cell surface, and thus would be expected
to be the region of the molecule most exposed to
immunological selective pressure. Therefore it is not
surprising that the N-terminal region varies in
sequence among different serological types of M
protein. In contrast, the sequence of the C-terminal
region of the molecule should be evolutionarily
-74-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
conserved to assure attachment to the streptococcal
surface. Since Lbp has a structure similar to that of
M protein, it is not surprising to find that its C-
terminal sequence is conserved among strains, whereas
the N-terminal region shows variation.
Since the N-terminal part of the Lbp is
variable among S. uberis strains, our finding that
this part of the molecule is responsible for Lf
binding seems surprising. Similar observations have
been reported in Actinobacillus pleuropneumoniae.
Three isoforms of transferrin binding proteins (Tbp2)
from different A. pleuropneumoniae serotypes contain a
variable N-terminal half and a conserved C-terminal
half (Bunka et al. (1995) Cloning and sequencing of
the transferrin-binding protein genes of
Actinobacillus pleuropneumoniae, biotype 1-serotype 5
and biotype 2-serotype 2. Unpublished; GenBank
accession no. 246774; Gerlach et al. (1992) Infect.
Immun. 60:3253-3261; Gerlach et al. (1992) Infect.
Immun. 60:892-898); the N-terminal half of the
molecule is responsible for transferrin binding
(Strutzberg et al. (1995) Infect. Immun. 63:3846-
3850) .
Example 3
Evaluation of the Protective Capacity of
Recombinant Lactoferrin-Bindinct Protein against
Challenge by S. U&ERIS
Materials and Methods
Bacterial strains, vectors and media.
S. uberis strain su-1 (ATCC 9927) was
obtained from a clinical case of bovine mastitis.
Bacterial cells were grown in tryptic soy broth,
aliquoted and stored_at -70°C on blood beads until
needed. E. coli strain DHSa was used in all
-75-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
transformation experiments. E. coli cells were
cultured in Luria medium, and the medium for the
growth of transformants was supplemented with 50 ~.g/ml
of ampicillin. The plasmid pGH433 (Anderson et al.
(1991.) Infect. Immun. 59:4110-4116) was used to
express the recombinant proteins under the control of
an IPTG (isopropyl ~i-D-thiogalacto-pyranoside)-
inducible promoter.
PAGE and Western blotting.
Lbp inclusion bodies were dissolved in
sample buffer in the presence of 4 M urea and run on
an SDS-PAGE gel containing 4 M urea. Western blotting
was done using convalescent serum from an su-1-
infected cow using a 1:75 dilution of cow convalescent
serum and a 1:5000 dilution of goat anti-bovine IgG
coupled to alkaline phosphatase.
Protein purification.
A culture of E. coli transformants (1 L) was
grown to an absorbance at 660 nm of 0.5 and induced
with 2 mM IPTG. After 2 h of continuous, vigorous
shaking at 37°C, the cells were harvested and the
protein inclusion bodies were prepared as described by
Gerlach et al. (1992) Infect. Immun. 60:892-898. A
mid-log phase broth culture (1 L) grown at 30°C was
cultured at 42°C for 2 h with vigorous shaking for
protein induction. Cells were harvested by
centrifugation, resuspended in 5 ml of 25% sucrose-50
mM Tris-HC1 buffer (pH 8.-0), and frozen at -70°C.
Lysis was achieved by the addition of 1 mg of lysozyme
in 250 mM Tris-HC1 buffer (pH 8.0), 10 min of
incubation on ice, addition of 25 ml of a detergent
mix (5 parts of 20 mM Tris-HC1 buffer, pH 7.4, 300 mM
NaCl, 2% deoxycholic acid, 2% Nonidet P-40 and 4 parts
of 100 mM Tris-HC1 buffer, pH 8.0, 50 mM EDTA, 2%
Triton X-100), and sonication. Inclusion bodies were
-76-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
harvested by centrifugation for 30 min at 15,000 x g
and resuspended in H20-to a concentration of 5 to 10
mg/ml. Antigens for ELISA were purified from SDS-PAGE
gels or 4 M urea SDS-PAGE gels by elution.
Determination of protein purity and concentration.
Protein purity was estimated by SDS-PAGE and
subsequent Coomassie blue staining. The protein
concentration was determined using Bio-Rad DC protein
assay as described by the supplier. Bovine serum
albumin (Pierce Chemical Co., Rockford, IL) was used
as a standard. The amount of target protein vs. total
protein was determined after scanning the Coomassie
blue stained
SDS-PAGE with a Bio-Rad 620 Densitometer.
Vaccine preparation and vaccination.
The vaccines consisted of proteins
emulsified in the adjuvant VSA3 which had been diluted
with 0.1 M PHS, pH 7.2. Each 2 ml dose of vaccine
contained VSA (0.67 ml), PBS (1.33 ml), and 50 ~,g of
protein antigen dissolved in 5-10 ~,1 of 4 M guanidine
hydrochloride. Fifteen healthy lactating dairy cows
from the Pennsylvania State University Mastitis
Research Herd were vaccinated intramuscularly at
drying off and again 28 days later. Groups of five
cows were given Lbp or adjuvant only.
Challenge.
The bacterial challenge culture was prepared
by rolling the stock bead cultures onto esculin blood
agar plates containing 5~ whole blood. After 24 hours
incubation at 37°C, a single colony was used to
inoculate 100 ml of Ultra High Temperature pasteurized
(UHT) milk and incubated for 12 hours at 37°C. The 24
hour culture was mixed well and a 100 ~.1 aliquot was
_77_
CA 02270404 1999-04-30
WO 98121231 PCT/CA97/00867
removed to inoculate a second 100 ml of UHT milk.
After a second 9 hour incubation at 37°C, the culture
was serially diluted in 10-fold increments using
sterile saline. The colony forming units (CFU) per ml
of each dilution was determined by absorbance on a
spectrophotometer and confirmed by plating onto blood
agar. Animals were challenged by intramammary
infusion of 200 CFU of S. uberis in 1 ml of saline in
the teat canal of one quarter on day four of
lactation.
Sampling.
Milk and blood samples were obtained at the
times outlined in Table 3.
Table 3. Immunization, challenge and Sampling
schedule..
TIME SAMPLE
dry off, D-0 serum, milk, immunization
14 days dry, D+14 serum
28 days dry, D+28 serum, immunization
52 days dry, D+52 serum
calving, C-0 serum, milk
4 days lactation, CH-0 serum, milk, challenge
7 days lactation, CH+3 serum, milk
14 days lactation, CH+10 serum, milk
21 days lactation, CH+17 serum, milk
Antibody Titers.
Total Ig titers for the Lbp antigen were
determined by an indirect ELISA. Nunc Immunlon-2
plates were coated with antigen in carbonate buffer.
Prior to use, the plates were blocked with TBST (100
mM Tris-C1, pH 8.0; 150 mM NaCl; 0.05% Tween-20) and
3% BSA for 1 hour. After blocking, the plates were
_78_
CA 02270404 1999-04-30
WO 98!21231 PCT/CA97/00867
washed with distilled water. Serum and milk samples
were serially diluted in 3-fold increments using TBST
containing 1% BSA. Rabbit antisera for the Lbp
antigen was also diluted and served as a positive
control. Negative control samples contained TBST with
1% BSA. The diluted samples and controls were
transferred to the coated plates and were incubated
for 1 hour at room temperature. The plates were washed
thoroughly with distilled water and all wells were
incubated with a horse radish peroxidase conjugate of
goat anti-IgG diluted 1:2000 in TBST containing 1%
BSA. Following a 1 hour incubation at room
temperature, the plates were washed with distilled
water. The amount of antibody present in samples was
visualized using ABTS substrate. The titers of each
sample were based on the absorbance reading at 405 nm
with a reference wavelength of 495 nm. A positive
reading for samples was one in which the absorbance
was two times the absorbance of the blank (negative
control). Titers were determined by taking the
reciprocal of the last dilution giving a positive
reading. Consistency among assay plates was monitored
by the absorbance reading of positive controls.
Milk compositional anal~rsis.
Milk pH values, total somatic cell counts,
fat, protein, and lactose were determined using a
Fossomatic Cell and Milk Analyzer (A/S Foss Electric,
Hillerrad, Denmark) .
- 30
DNA manipulations.
All molecular techniques were as recommend
by the supplier (Pharmacia Canada Ltd.) or Sambrook et
al. Molecular Cloning: A Laboratory Manual, Second
Edition (1989). When required, plasmid DNA fragments
-79-
CA 02270404 1999-04-30
WO._98/21231 PCT/CA97/00867
were isolated from agarose gels using a Gene Clean kit
(Bio/can Scientific).
Results
Expression of Lbp under the control of the tac
Qromoter.
Plasmid pGH-LBP was constructed by inserting
the 1.8 kb SphI-RsaI fragment from pLBPS into the
BamHI site of pGH433 (Fig. 10) which provides a 12
amino acid leader peptide and an IPTG-inducible tac
promoter. Before ligation, the insert was treated
with mung bean nuclease to remove the 3' overhang from
the end generated by SphI, and the BamHI-cut vector
was filled in by the Klenow fragment to produce blunt
ends. The pGH-LBP contained the carboxy-terminal 96°s
of the Lbp gene (lbp), which was preceded by a 12
amino acid leader peptide and the tac promoter
provided by vector pGH433. Analysis of the nucleotide
sequence at the fusion site revealed identity to the
sequence of the Ibp presented in Figure 1.
Purification of recombinant proteins.
Lbp of S. uberis was expressed in E. coli DHSa
using pGH-LBP. In this system, expression of the
recombinant protein was repressed under normal growth
conditions. Upon IPTG-induction, the recombinant
protein was produced in an aggregated form. The 82
and 90 kD Lbp made up 36% of the total protein.
Isolated protein aggregates were dissolved in 4 M
guanidine hydrochloride and used for vaccine
formulation. Lbp was purified from a 4 M urea SDS-
PAGE gel and used as the antigen for ELISA. Both
aggregated and purified Lbp were demonstrated to be
antigenically active by Western blotting using
convalescent serum from S. uberis infected cow.
-80-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
Somatic cell counts in milk following experimental
bacterial challenge.
The mean somatic cell count in milk from
challenged quarters of nonvaccinated control animals
increased up to 3,000,000 cell/ml 3 days following
challenge. The challenged quarters of animals
vaccinated with Lbp showed a rapid increase in the
mean SCC which was close to that in the control group.
Effect of vaccination of Lbp-specific antibody titers.
The mean levels of antibodies specific to
Lbp in the serum and milk of the control animals were
low prior to experimental challenge. In one trial,
Lbp-specific antibody levels in the serum and milk of
animals vaccinated with Lbp were not significantly
increased following vaccination when compared with
prevaccination levels or levels in the control
animals. In a subsequent trial, an antibody response
was seen.
Chancres of pH value and main constituent of milk after
challenge.
The mean pH values of milk from animals
vaccinated with placebo and Lbp changed in a similar
pattern after challenge. The percentage of fat,
lactose and protein in milk from immunized and
unimmunized animals also changed in a similar pattern
after challenge. No obvious decrease of these values
- occurred.
Discussion
The surface exposure of S. uberis Lbp makes
it a potential vaccine candidate by increasing the
levels of opsonic antibody in milk and potentiating
the speed of PMN recruitment into the mammary gland.
Furthermore, the above example shows that Lbp is
-81-
CA 02270404 1999-04-30
WO 98!21231 PCT/CA97/00867
recognized by bovine serum from an animal which
recovered from an S. uberis infection, indicating that
the protein is expressed in vivo and cows respond to
it immunologically. Based on these considerations,
the protective capacity of recombinant Lbp was tested
in lactating dairy cows against challenge by S.
uberis.
Lbp was produced as inclusion bodies in the
expression system used, facilitating the purification
of large quantities of pure recombinant proteins. In
one study, Lbp did not induce high levels of specific
antibodies in the immunized cows, likely due to the
formulation parameters. In these animals, bacterial
challenge resulted in high somatic cell counts,
indicative of mastitis. Therefore, vaccination with
Lbp did not prevent the occurrence of mastitis.
However, the serological response to vaccination was
very poor and therefore it is not possible to draw any
conclusions regarding the protective capacity of Lbp
from this study.
In a subsequent study, Lbp elicited an
antibody response in immunized cows. Protection
studies are on-going and preliminary results indicate
that Lbp is effective in protecting subjects from
mastitis.
The potential involvement of mammary gland
lymphocytes in the protection against S. uberis
mastitis following vaccination with killed S. uberis
via the intramammary or subcutaneous route has been
suggested elsewhere (Finch et al. (1994) Infect.
Immun. 62:3599-3603). Recent observations have
indicated that protection against mastitis caused by
S. uberis does not appear to be related to levels of
specific antibody. Intramammary or subcutaneous
administration of S.--uberis at drying off has been
shown to dramatically reduce both the incidence of
-82-
CA 02270404 1999-04-30
WO 98/21231 PCT/CA97/00867
clinical mastitis and numbers of bacteria recovered
from the milk following-challenge with the same strain
during the next lactation (Finch et al., supra; 1994;
Hill et al. (1994) FEMS Immunol. Med. Microbiol.
8:109-118). Although there was a significant increase
in the levels of S. uberis-specific immunoglobulin
following vaccination, there was no increase in the
opsonic activity of serum and milk. Furthermore, it
has been demonstrated that S. uberis can resist the
bactericidal activity of neutrophils despite the
presence of Ig bound to the surface of the bacteria
(Leigh and Field (1994) Infect. Immun. 62:1854-1859).
Therefore, assessment of cellular immune response
should be of equal importance to that of antibody
response when evaluating a vaccination regime against
S. uberis mastitis.
During our vaccine trial, no obvious
clinical signs of mastitis were observed after
challenge in immunized or unimmunized animals.
Therefore, it is not surprising that the pH value and
main constituent of milk were not influenced.
Thus, S. uberis lactoferrin-binding proteins
are disclosed, as are methods of making and using the
same. Although preferred embodiments of the subject
invention have been described in some detail, it is
understood that obvious variations can be made without
departing from the spirit and the scope of the
invention as defined by the appended claims.
-83-