Note: Descriptions are shown in the official language in which they were submitted.
CA 02270478 1999-04-29
1 '
I NZ 0 I IV S
TI~EIR PREPARATION AND THEIR THERAPEUTIC APPL~IC~rTION
The present invention relates to N-
(imidazolylbutyl)benzenesulphonamide derivatives, to
their preparation and to their therapeutic application.
Patent applications EP 718,307 and EP S65,396
describe 1-oxo-2-(phenylsulphonylamino)pentylpiperidine
derivatives and 1-(2-(arylsulphonylamino)-1-
oxoethyl)piperidine derivatives, respectively, these
compounds having antithrombotic activity. Patent
application EP 713,865 describes compounds which are
useful as intermediates in the synthesis of compounds
with antithrombotic activity.
The compounds of the invention correspond to
formula (I)
S 0=NFi
R3~ /
(I)
A R
~W1 R i
x
in which
R1 and R'1 represent, independently of each other,
either a hydrogen atom or a halogen atom or a (C1-
CA 02270478 1999-04-29
A.
C,) alkyl group,
Rz represents either a 1-piperidyl group optionally
substituted in position 4 with one or more groups
chosen from hydroxyl, straight or branched (C1-C,) alkyl,
hydroxy (Cl-C,) alkyl, (C,-C,) alkoxy (Cl-C,) alkyl,
C,) alkoxy, (Cl-C,) alkylthio, nitrile, monofluoromethyl,
difluoromethyl, trifluoromethyl, 2-fluoroethoxy, 2,2,2-
trifluoroethoxy, (C3-C6)cycloalkyl, -COOR' and -CONR'R~
(R' being a~
CA 02270478 1999-04-29
a
(C1-C,)alkyl group and R" being a hydrogen atom or a
(C1-C,) alkyl group) or by a =CYZ group [Y and Z being
chosen. independently of oae another, from hydrogen and
halogen atoms and (Ci-C,)alkyl (optionally substituted
by one or more halogen atoms) cyano and -COOR' groups,
R1 being defined as above or by a group (r
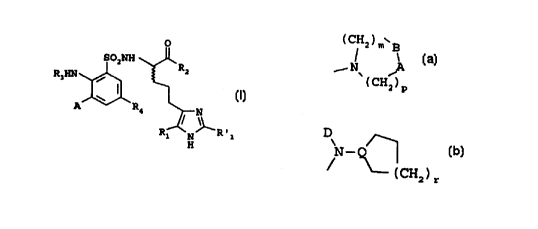
= 1 to 3 ) or by an aNOCH~ group; or a spiro [ (C,-
C~)cyclo-alkane-1,4'-piperidinl-1-yl group; or a
1.a,3.6-tetra-hydropyridin-1-yl group optionally
substituted is the 4 position by a straight or branched
(C,-C,)alkyl group (optionally substituted by one or
more halogen atoms) or a (C3-C,) cycloalkyl group; or a
hexahydro-lE-azepin-1-yl group optionally substituted
in the 4 position by a trifluoromethyl or =CPS group; or
a heptahydroazocin-1-yl group; or an octahydro-lA-
azonia-1-yl group: or a
(~)' H ~ 0.
group ~ N ~ (a-B being a group -CONR", rn = 1 to
.(~)a
a and p = 1 to Z) ; or a DN- ~ group (Q being
1
(CH )
i r
a carbon or nitrogen atom. D a (C,-C,) alkyl or -CH~CF,
2 0 group and r = 1 t0 3 ) ;
R~ represents either a straight or branched (Cl-Cs) alkyl
group; or a -CORs group where RS is a (Cl-C,) alkyl, the
latter being straight or breached, - (C8=) sOCH~, -
CH=O (CzH,O),CH~. - (CH=) ,CF3 Or - (CH=) o0H (a = 1 t0 4 )
group; or an -SO~R~ group; or a -CONHR, group; or an
CA 02270478 1999-04-29
3
-SO,N (R6) s group where R6 is a straight or branched (C1-
C,) alkyl group,
R, represents either a hydrogen atom or a halogen atom
and
A represents either a phenyl group optionally
substituted by one or more substituents chosen from
halogen atoms and straight or branched (C1-C,)alkyl,
straight or branched (C1-C,)alkoxy, trifluoromethyl,
trifluoromethoxy, formyl, -CHsORlo, -CHZOCORlo,
-CHzOCONRloRll, -COORlo, -CONRloRll, nitro. -NRloRil, -NHCORio
and -NH (CHs) QORlo groups with Rlo and Rll each being,
independently of one another, a hydrogen atom or a
straight or branched (C1-C~)alkyl group and q between 0
and 6; or a heterocycle chosen from the pyridinyl,
thienyl, furyl, pyrimidyl and thiazolyl groups, it
being possible for the said groups to be substituted as
above; or a cyclo (CS-Ce) alkyl group.
According to the invention, the preferred
compounds are the compounds of formula (I) in which
Rl and R'1 each represent, independently of one another,
either a hydrogen atom or a halogen atom or a (C1
Cs) alkyl group,
Rs represents either a piperidin-1-yl group optionally
substituted in the 4 position by one or more groups
chosen from (Cl-C4)alkyl, the latter being straight or
branched, hydroxy (C~-C,) alkyl, (C1-C,) alkoxy (C1-C,) alkyl,
(CI-C,) alkoxy, (Cl-C,) alkylthio, nitrile, difluoromethyl,
trifluoromethyl, 2,2,2-trifluoroethoxy and (C,-C6)cyclo-
1
CA 02270478 1999-04-29
4
alkyl groups or by a =CYZ group (Y and Z being chosen,
independently of one another, from hydrogen and halogen
atoms and (Ci-C,) alkyl groups) or by an =NOCH, group; or
a spiro[(Cl-C~)cycloalkane-1,4~-piperidial-1-yl group;
or a 1.Z.3.6-tetrahydropyridin-1-yl group optionally
substituted in the ~ position by a straight or branched
(C1-C,)alkyl group (optionally substituted by one or
more halogen atoms): or a hexahydro-iX-azepin-1-yl
group optionally substituted in the 4 position by a =CF=
group; or as octahydro-1X-azonia-1-yl group; or a
(C'H.,): B jai
group N' ~ (a-B being a group -CONR", m - 1 to 2
(~) a D
and p = 1 to 2): or a N - ~ group (Q being a
( CH )
Z
nitrogen or carbon atom, D a (Cl-C,) alkyl or -C&~CFs
group, and r = 1 to 3).
R, represents either a straight or branched (C,-CS)alkyl
group: or a -CORS group where Rs is a (C1-C,) alkyl, the
latter being straight or branched, -CH,O(C=H,O),CH,,
- (C8~)nOH or - (C8=)nOCH, group; or a -CONBR, group and
A represents either a phenyl group optionally
substituted by one or more substitueats chosen from
halogen atoms and straight or branched (C1-C,)alkyl,
straight or breached (C1-C,)alkoxy, trifluoroaethyl,
trifluoromethoxy and -NR1oR11 groups with Rla and Ri, each
being, independently of one another, a hydrogen atom or
a straight or branched (C1-C,) alkyl group; or a
CA 02270478 1999-04-29
pyridinyl or thienyl group which can be substituted as
above; or a cyclo (Cs-Ce) alkyl group.
Among these compounds, the compounds of
choice are those in which R1 represents a (C1-C,) alkyl
5 group and R~l a hydrogen atom, Rz represents a
piperidin-1-yl group substituted in the 4 position by a
straight or branched (C1-C,,)alkyl group or by a =CFs
group, R, represents a -CORS group where RS is a
straight or branched (Cl-C,,)alkyl group and A represents
a thienyl group optionally substituted as above or a
cyclo (CS-Ce) alkyl group.
The preferred configuration of the central
amino acid part
0
so2x~~
is tS] .
The compounds of the invention can exist in
the form of racemates or of pure enantiomers or of a
mixture of enantiomers. They can also exist in the form
of the free acid or the free base or of
pharmaceutically acceptable addition salts.
All these forms form part of the invention.
In the following schemes, the -CPh3 group
represents the triphenylmethyl group.
According to the invention, the compounds of
formula (Ia), in which R~ represents a -CORS group where
CA 02270478 1999-04-29
6
Rs is a straight or branched (Ci-C,) alkyl group or a
- (CH,)nCF, group (n = 1 to 4) , can be synthesized
according to Schema 1. A compouad~ of formula (II), in
which R1 and R'i each represent either a hydrogen atom
or a (C1-C,) alkyl group and R= is as def iaed above, is
reacted with a compound of formula (III), in which A
represents either a phenyl group optionally substituted
as above or a heterocycle chosen from the pyridinyl,
thienyl, furyl, pyrimidyl and thiazolyl groups, ft
being possible for the said groups to be substituted as
above, or a cyclo (CS-C,) alkyl
group, and R, and RS are as defined above, in an aprotic
l0 solvent such as dichloromethane in the presence of a
base such as triethylamine, and a compound of formula
(IV) is obtained which is treated with an acetic
acid/ethanol, acetic acid/water or acetic
acid/tetrahydrofuran/water mixture at the reflux ,
temperature.
CA 02270478 1999-04-29
7
Scheme 1
O
HC1. H2N
Rz
(II)
Ri N R , i
i
CPh3
RSOC SOZC1
i
RSOC~N i I (III)
A Ra
O
RsOC1 S02NH
Rz
RsOC ~ N
I (IV)
A R4 N
/
Ri N R y
i
CPh3
O
S02NH
R~
RSOCHN
i
I (Ia)
R' N
Ri N R ~ i
H
CA 02270478 1999-04-29
8
In an alternative form of the invention
illustrated by Scheme 2, for the preparation of the
compounds of formula (Ia) in which RS is a (C1-C~) alkyl,
the latter being straight or branched, -(CH~)nOCH,, -
CHaO ( CzH~O ) aCH3 , - ( CH, ) aCF3 Or - ( CHI ) nOH group (n = 1 t0
4), a compound of formula (II) can be reacted with a
compound of formula (V), in which A represents either a
phenyl group substituted as above, or a heterocycle
chosen from the pyridinyl, thienyl, furyl, pyrimidyl
and thiazolyl groups, it being possible for the said
groups to be substituted as above, or a cyclo(CS-
C8)alkyl group, and R, and RS are as defined above, in
an aprotic solvent, such as dichloromethane, in the
presence of a base, such as triethylamine, and a
compound of formula (VI) is obtained which is treated
with an acetic acid:ethanol, acetic acid:water or
acetic acid:tetrahydrofuran:water mixture at the reflux
temperature.
CA 02270478 1999-04-29
9
Scheme 2
0
HC1. HzN
RZ
(II)
N
,
Ai N R i
i
CPh3
SO~C1
Rs N / (V)
C1 w
A R~
R
s
i
A ~ S02 O
N~N A (VI)
'~' s
Rs
f ,
Rl N R i
I CPh~
O
SOiNH
Ri
RSOCHN
(Ia)
A R~ /
Ri N A, i
H
CA 02270478 1999-04-29
In an alternative form of the invention, it is also
possible to use the process illustrated in Scheme 3. A
compound of formula (VII), in which Rl and R'1 each
represent either a hydrogen atom or a (C1-C~)alkyl group
5 and Rz, R, and R5 are as defined above, is reacted with
a compound of formula (VIII), in which R represents a
(C1-C~)alkyl group and A is as defined above, in a
solvent, such as dimethylformamide, in the presence of
a catalyst, such as tetrakis(triphenylphosphine)-
10 palladium(0), in order to form a compound of formula
(IX) which is heated at the reflux temperature in
acidic medium, for example in an acetic acid: water
mixture.
If it is desired to obtain a compound of
formula (Ia) in which R, is a hydrogen atom, then the
corresponding compound of formula (Ia) in which R, is a
halogen atom can be hydrogenolysed. When it is desired
to obtain a compound of formula (Ia) in which R1 and/or
R'1 represent a halogen atom, then the corresponding
compound of formula (Ia) in which R1 and/or R'1
represent a hydrogen atom is treated with a
halogenating agent, such as, for example, N-bromo-
succinimide or N-chlorosuccinimide, in a solvent, such
as di.methylformamide.
CA 02270478 1999-04-29
11
Scheme 3
0
SOiNH
RSOCHN Rz
Ra N ('JII)
Ri N R ~ i
CPh3
ASn(R)~ :VIII)
O
SOzNH
RSOCHN R~
A Ra N iIX)
/
Ri N Ry
CPh3
O
SOzNH
RSOCHN RZ
i
:a)
A~ Ra N
Ri N Ry
H
CA 02270478 1999-04-29
12
Alternatively, in order to prepare the
compounds of formula (Ib) in which R3 represents either
a -CORS group, or an -SO=R6 group, or a -CONHR6 group, or
an -SO1N (R6) s group, where RS is a (C1-C,) alkyl, the
latter being straight or branched, -(CHz)nOCH3,
-CHsO (C,H,,O) nCH3, - (CH=) aCF3 or - (CH,) aOP group, P is a
protecting group (n = 1 to 4) and R6 is as defined
above, the process illustrated in Scheme 4 is used. A
compound of formula (II) is reacted with a compound of
formula (X), in which A and R,, are as defined above, and
a compound of formula (XI) is obtained which is reacted
with an acid chloride of formula RSCOC1 or an alkyl
isocyanate of formula R6NC0 or a sulphonyl chloride of
formula R6SO,C1 or a sulphamoyl chloride of formula
(R6) 2NSOzCI and a compound of formula (XII) is obtained
which is treated with an acetic acid: ethanol, acetic
acid: water or acetic acid:tetrahydrofuran:water
mixture at the reflux temperature.
When it is desired to obtain a compound of
formula (Ib) in which R1 and/or R'1 represent a halogen
atom, then the corresponding compound of formula (Ib)
in which Rl and/or R', represent a hydrogen atom is
treated with a halogenating agent, such as, for
example, N-bromosuccinimide or N-chlorosuccinimide, in
a solvent, such as dimethylformamide.
If it is desired to obtain a compound of
formula (Ib) in which R, is a hydrogen atom, then the
corresponding compound of formula (Ib) in which R, is a
CA 02270478 1999-04-29
13
Scheme 4
0
HC1. HEN
R:
(II)
R~~R'
N
CPh~
SOiCl
H=N / IX1
A ~ R,
O
SO~NH
R=
HzN
A ~ R~ N (!( I )
Ri N R,i
CPh~
O
SO~NH
R HN
(XII)
A R~
R~ N Ry
CPh~
0
SO~NH
R=
R~HN
(:bi
A R, N
/
R1 N R 1
H
CA 02270478 1999-04-29
14
halogen atom is hydrogenolysed.
In order to prepare the compounds of formula
(Ic) in which Rl~ corresponds to R, when R3 represents a
straight or branched (Cl-C5)alkyl group, the process
illustrated in Scheme 5 is used. A compound of formula
(II) is reacted with a compound of formula (XIV) in
which Rls represents a straight or branched (C1-CS) alkyl
group and A and R, are as defined above and a compound
of formula (XV) is obtained which is treated with an
acetic acid: ethanol, acetic acid: water or acetic
acid:tetrahydrofuran:water mixture at the reflux
temperature.
When it is desired to obtain a compound of
formula (Ic) in which Rl and/or R'1 represent a halogen
atom, then the corresponding compound of formula (Ic)
in which R1 and/or R'1 represent a hydrogen atom is
treated with a halogenating agent, such as, for
example, N-bromosuccinimide or N-chlorosuccinimide, in
a solvent, such as dimethylformamide.
If it is desired to obtain a compound of
formula (Ic) in which R~ is a hydrogen atom, then the
corresponding compound of formula (Ic) in which R, is a
halogen atom is hydrogenolysed.
The starting compounds are commercially
available or are described in the literature or can be
prepared according to methods which are described
therein or which are known to a person skilled in the
art.
CA 02270478 1999-04-29
15
Scheme 5
O
HC1. HzN
Rz
(I.)
N
Ri N R ~ i
C Phi
SOzCl
Rl~~ i XIVI
A R,
O
SOZNH
Rz
R:ZHN
A R, N (xv>
Ri N Ry
CPh~
O
SOzNH
Rz
R1~~ ~ ( Ic)
A ~~ Rv N
Ri N R y
H
CA 02270478 1999-04-29
16
Thus, the compounds of formula (II) are prepared
according to a method analogous to that described in
European Patent Application EP 0,643,047.
Some compounds of formula (III) are described in
European Patent Application EP 0,718,307.
The compounds of formula (VII) are prepared according
to a method analogous to that described in European
Patent Application EP 0,718,307.
The compounds of formula (X) and (XIV) are described in
European Patent Application EP Q,713,865.
5-Ethyl-1X-imidazole is prepared according to the
method described by Horne D.A., (l994), Heterocycles,
39, No. 1, 139.
The preparation of 4-cyclopropylpyridine is described
by Eisch J.J., (l974), J. Org. Chem., 39, No. 21, 5l10.
4-Difluoromethylenepiperidine is prepared according to
a method analogous to that described by Schmidt W. et
al., (1995), Liebigs Ann., l319-1326.
The preparation of N-cyclopentylformamide is described
by Bossio R. et al., (1993). Synthesis, 8, 783-785.
The following Examples 1 to 11 illustrate the
preparation of some compounds of formula (II); Examples
12 to 2? illustrate the preparation of some compounds
of formula (I) according to the invention.
The microanalyses and the IR and NMR spectra
confirm the structure of the compounds obtained.
The numbers of the compounds exemplified refer to those
in the table given later, which illustrates the
CA 02270478 1999-04-29
17
chemical structures and the physical properties of some
compounds according to the invention.
The ratios between brackets represent the (base: acid)
ratio.
Example 1
(S)-5-Ethyl-a-[(4-ethylpiperidin-1-yl)carbonyl]-1-(tri-
phenylmethyl)-1H-imidazole-4-butanamine hydrochloride
(1:1)
1.1. 5-Ethyl-4-iodo-1H-imidazole
22.7 g (89 mmol) of iodine, in solution in
800 ml of chloroform, are added dropwise with stirring
at 0~C over 3 hours to a solution of 8.6 g (89 mmol) of
5-ethyl-1H-imidazole in 600 ml of a 2N aqueous sodium
hydroxide solution. Stirring is continued for 4 hours
at this temperature and then the chloroform is
evaporated under reduced pressure. The aqueous phase is
cooled to 0~C, neutralization is carried out using a
12N aqueous hydrochloric acid solution and extraction
is carried out with 3 times 1 1 of ethyl acetate. The
organic phases are combined, washed with l00 ml of a
saturated sodium chloride solution, dried over sodium
sulphate and concentrated under reduced pressure. The
residue obtained is purified by chromatography on a
column of silica gel, elution being carried out with a
methanol:dichloromethane (1.5:98.5) mixture. 10.5 g of
product are obtained in the form of a white powder.
Yield = 53%
CA 02270478 1999-04-29
18
Melting point = 155~C
1.2. 5-Ethyl-4-iodo-1-[(4-methylphenyl)sulphonyl]-
1H-imidazole
0.91 g (22.7 mmol) of 60% sodium hydride in
oil is added, portionwise, with stirring at 0~C under
nitrogen to a solution of 4.8 g (21.6 a~ol) of 5-ethyl-
4-iodo-1H-imidazole in 25 ml of anhydrous dimethyl-
formamide. Stirring is continued for 0.5 hour at 0~C
and 4.35 g (22.7 mmol) of 4-(methylphenyl)sulphonyl
chloride are added. Stirring is maintained for one hour
at 0~C, the temperature of the mixture is allowed to
return to room temperature, stirring is continued for
one hour and then the reaction mixture is concentrated
under reduced pressure. The residue is taken up in
400 ml of ethyl acetate and washing is carried out
successively with 100 ml of a 0.5N aqueous hydrochloric
acid solution, l00 ml of water and 100 ml of a
saturated sodium chloride solution. Finally, the
solution is dried over sodium sulphate and concentrated
under reduced pressure.
6.1 g of product are obtained in the form of a white
solid after precipitation from an ethyl acetate: pentane
mixture.
Yield = 75%
Melting point = 95~C
CA 02270478 1999-04-29
19
1.3 Methyl (S)-2-[[(l,l-dimethylethoxy)carbonyl]-
amino]-5-[5-ethyl-1-[(4-methylphenyl)-
sulphonyl]-1H-imidazol-4-yl]pent-4-ynoate
1.3.1. Methyl (S)-2-[[(1,1-dimethylethoxy)carbonyll-
amino]pent-4-ynoate
a) (S)-2-[[(1,1-Dimethylethoxy)carbonyl]amino]pent-
4-ynoic acid
11.5 g (77 m~aol) of (S)-2-aminopent-4-ynoic
acid hydrochloride, 100 ml of dioxane, 50 ml of water
and 80 ml of 2N sodium hydroxide solution are
introduced into a 250 ml round-bottomed flask under a
nitrogen atmosphere. 17.9 g (82 mmol) of tert-butyl
dicarbonate are added to the solution and stirring is
carried out for 3 hours at room temperature. 200 ml of
ethyl acetate are added and acidification is carried
out to pH 2 by addition of a 2N hydrochloric acid
solution. The phases are separated and the aqueous
phase is extracted with 50 ml of ethyl acetate. Drying
is carried out over magnesium sulphate and evaporation
is carried out to dryness.
18.78 g of product are obtained in the form of a
colourless oil which is used as is in the following
stage.
b) Methyl (S)-2-[[(l.l-dimethylethoxy)carbonyl]-
amino]pent-4-ynoate
13 g (154 mmol) of sodium hydrogencarbonate
CA 02270478 1999-04-29
are added, in a 250 ml round-bottomed flask under a
nitrogen atmosphere, to a solution of 18.78 g (77 mmol)
of (S) -2- [ [ (1, 1-dimethylethoxy) carbonyl] amino] pent-
4-ynoic acid in 150 ml of dimethylformamide. 20 ml
5 (318 mmol) of methyl iodide are added and the mixture
is stirred for 18 hours at room temperature. The
mixture is poured into water and extraction is carried
out with ethyl acetate. The organic phase is washed
with water and then dried over magnesium sulphate.
10 Evaporation is carried out to dryness. l5.85 g of
product are obtained in the form of a yellow oil which
is used as is in the following stage.
1.3.2. Methyl (S)-2-[[(l,l-dimethylethoxy)carbonyl]-
amino]-5-[5-ethyl-1-[(4-methylphenyl)-
15 sulphonyl]-1H-imidazol-4-yl]pent-4-ynoate
A mixture of 9.87 g (26.3 mmol) of 5-ethyl-
4-iodo-1-[(4-methylphenyl)sulphonyl]-1H-imidazole,
8.94 g (39.4 mmol) of methyl (S)-2-[[(l,l-dimethyl-
ethoxy)carbonyl]amino]pent-4-ynoate, 0.25 g (1.3 mmol)
20 of copper iodide, 10.84 ml (105 mmol) of diethylamine
and 0.92 g (1.3 mmol) of dichlorobis(triphenyl-
phosphine)palladium in 26 ml of dimethylformamide is
heated for 8 hours at 50~C under argon. The reaction
mixture is concentrated under reduced pressure and the
residue obtained in taken up in 300 ml of ethyl acetate
and washed successively with 3 times 100 ml of water
and 100 ml of a saturated sodium chloride solution.
Finally, the solution is dried over sodium sulphate and
CA 02270478 1999-04-29
21
concentrated under reduced pressure. The residue is
purified by chromatography on a column of silica gel,
elution being carried out with an ethyl acetate: hexane
(3:7) mixture.
11 g of product are obtained in the form of a viscous
oil.
Yield = 88%
1.4. Methyl (S)-2-[[(1,1-dimethylethoxy)carbonyl]-
amino] -5- [5-ethyl-1- [ (4-methylphenyl) -
sulphonyl]-1H-imidazol-4-yl]pentanoate
A mixture of 13.5 g (28.4 mmol) of methyl
(S) -2- [ [ (1,1-dimethylethoxy) carbonyl] amino] -5- [5-ethyl-
1-[(4-methylphenyl)sulphonyl]-1X-imidazol-4-yl]pent-4-
ynoate in the presence of 1.8 g of 10% palladium-on-
charcoal in 50 ml of methanol is hydrogenated for
10 hours at room temperature under a pressure of
0.35 MPa (50 psi). The reaction mixture is filtered
through celite and the filtrate is concentrated under
reduced pressure. The residue is purified by
chromatography on a column of silica gel, elution being
carried out with a cyclohexane:ethyl acetate (7:3)
mixture.
10.5 g of product are obtained in the form of a viscous
oil.
Yield = 77%
CA 02270478 1999-04-29
22
1.5. Methyl (S)-2-[[(1,1-dimethylethoxy)carbonyl]-
amino]-5-(5-ethyl-1H-imidazol-4-yl)pentanoate
A mixture of 10.4 g (21.6 mmol) of methyl
(S) -2- [ [ (1. 1-dimethylethoxy) carbonyl] amino] -5- [5-ethyl-
1-[(4-methylphenyl)sulphonyl]-1H-imidazol-4-yl]-
pentanoate and 8.79 g (65.2 mmol) of 1-hydroxybenzo-
triazole hydrate in 150 ml of methanol is stirred at
room temperature for 4 hours. The reaction mixture is
concentrated under reduced pressure and the residue is
taken up in l00 ml of ether and Washed with 450 m1 of a
0.7N aqueous hydrochloric acid solution. The phases are
separated, the pH of the aqueous phase is adjusted to
8-9 with a sodium hydrogencarbonate solution and
extraction is carried out with 2 times 500 ml of ethyl
acetate. The organic phases are combined, dried over
sodium sulphate and concentrated under reduced
pressure.
8.79 g of compound are obtained in the form of a
viscous oil used as is in the following stage.
Yield = 88%
1.6. Methyl (S)-2-[[(1,1-dimethylethoxy)carbonyl]-
amino]-5-[5-ethyl-1-(triphenylmethyl)-
1H-imidazol-4-yl]pentanoate
2.9 ml (20.3 mmol) of triethylamine and
5.77 g (20.7 mmol) of triphenylmethyl chloride are
successively added at 0~C to a solution of 5.95 g
(18.3 mmol) of methyl (S)-2-[[(1,1-dimethylethoxy)-
CA 02270478 1999-04-29
23
carbonyl]amino]-5-(5-ethyl-1H-imidazol-4-yl)pentanoate
in 70 ml of dichloromethane. The temperature of the
mixture is allowed to return to room temperature,
stirring is continued for 18 hours at this temperature
and then the reaction mixture is concentrated under
reduced pressure. The residue is taken up in 300 ml of
ethyl acetate and washed successively with 200 ml of a
0.1N aqueous hydrochloric acid solution, 200 ml of a
saturated sodium hydrogencarbonate solution and 100 ml
of a saturated sodium chloride solution. The solution
is dried over magnesium sulphate and concentrated under
reduced pressure. The residue obtained is purified by
chromatography on a column of silica gel, elution being
carried out with a dichloromethane:methanol (99:1)
mixture.
9.4 g of product are obtained in the form of a viscous
oil.
Yield = 90.6%
1.7 . (S) -a- [ [ (1,1-Dimethylethoxy) carbonyl] amino] -
5-ethyl-1-(triphenylmethyl)-1H-imidazole-4-
pentanoic acid
0.83 g (19.8 mmol) of lithium hydroxide
monohydrate is added with stirring at 0~C to 9.4 g
(16.6 mmol) of methyl (S)-2-[[(l,l-dimethylethoxy)-
carbonyl]amino]-5-[5-ethyl-1-(triphenylmethyl)-
1H-imidazol-4-yl]pentanoate in a mixture of 48 m1 of
methanol and 16 ml of water. The temperature of the
CA 02270478 1999-04-29
24
mixture is allowed to return to room temperature and
stirring is continued at this temperature for 24 hours.
Evaporation is carried out under reduced pressure and
the aqueous phase is acidified to pH 2 at 0~C with a 1N
aqueous hydrochloric acid solution before extracting
with 2 times 300 ml of dichloromethane. The organic
phases are combined, washed with 100 ml of a saturated
sodium chloride solution, dried over magnesium sulphate
and concentrated under reduced pressure. The residue is
triturated in ether, filtered off and dried under
reduced pressure.
8.87 g of product are obtained in the form of a white
powder.
Yield = 96.7%
Melting point = 141~C
1.8. 1,1-Dimethylethyl (S)-[1-[(4-ethylpiperidin-
1-yl)carbonyl]-4-[5-ethyl-1-(triphenyl-
methyl)-1H-imidazol-4-yl]butyl]carbamate
1.8.1. 4-Ethylpiperidine hydrochloride
a) 1,1-Dimethylethyl 4-ethylpiperidine-1-carboxylate
20 g (190 mmol) of 4-ethenylpyridine are
hydrogenated for 4 hours at 50~C, under an atmosphere
of 0.42 MPa (60 psi), in the presence of 2 g of
platinum(IV) oxide, the reaction mixture is filtered
through celite and the filtrate is concentrated under
reduced pressure. The residue is taken up in l50 ml of
water, the pFI is adjusted to 8 with a saturated aqueous
CA 02270478 1999-04-29
sodium carbonate solution and 44 g (190 mmol) of
bis(1,1-dimethylethyl) dicarbonate, in solution in
100 ml of tetrahydrofuran, are added dropwise. The
temperature of the reaction mixture is allowed to
5 return to room temperature and stirring is maintained
for 18 hours at this temperature. Evaporation is
carried out under reduced pressure and the aqueous
phase is extracted with 2 times 300 ml of ethyl
acetate. The organic phases are combined and washed
10 with 100 m1 of a saturated sodium chloride solution.
The combined organic phases are dried over sodium
sulphate and concentrated under reduced pressure. The
residue thus obtained is purified by chromatography on
a column of silica gel, elution being carried out with
15 a cyclohexane:ethyl acetate (9:1) mixture.
13.8 g of product are obtained in the form of an oil.
Yield = 34%
b) 4-Ethylpiperidine hydrochloride
A solution of l3.8 g (64.8 mmol) of
20 1,1-dimethylethyl 4-ethylpiperidiae-1-carboxylate in
200 ml of ether is treated with a stream of gaseous
hydrochloric acid for 1 hour at 0~C. The temperature of
the mixture is allowed to return to room temperature,
stirring is continued at this temperature for 18 hours
25 and the mixture is concentrated under reduced pressure.
The residue thus obtained is triturated in ether,
filtered off and dried under reduced pressure.
CA 02270478 1999-04-29
26
6.62 g of product are obtained in the form of a white
powder which is used as is in the following stage.
Yield = 70%
Melting point = 138~C
l.8.2. l,l-Dimethylethyl (S)-[1-[(4-ethylpiperidin-
1-yl)carbonyl]-4-[5-ethyl-1-(triphenyl-
methyl)-1H-imidazol-4-yl]butyl]carbamate
0.37 g (2.9 mmol) of diisopropylethylamine
and 0.46 g (1.2 mmol) of [(benzotriazol-1-
yl)oxy]tris(dimethylamino)phosphonium
hexafluorophosphate are successively added at 0~C,
under nitrogen and with stirring, to a mixture of 0.6 g
(1.08 mmol) of (S)-a-[[(1,1-dimethylethoxy)carbonyl]-
amino]-5-ethyl-1-(triphenylmethyl)-1H-imidazole-
4-pentanoic acid and 0.18 g (1.2 mmol) of 4-ethyl-
piperidine hydrochloride in 8 ml of dichloromethane.
The temperature of the mixture is allowed to return to
room temperature, stirring is continued at this
temperature for 18 hours and the reaction mixture is
concentrated under reduced pressure. The residue is
taken up in l00 ml of ethyl acetate, washed
successively with 80 ml of a 1N aqueous hydrochloric
acid solution, 50 ml of a saturated sodium hydrogen-
carbonate solution and 50 ml of a saturated sodium
chloride solution, dried over magnesium sulphate and
concentrated under reduced pressure. The residue is
purified by chromatography on a column of silica gel,
CA 02270478 1999-04-29
27
elution being carried out with a methanol:dichloro-
methane f2:98) mixture.
0.7 g of product is obtained in the form of a viscous
oil.
Yield = 98%
1.9. (S)-5-Ethyl-a-[(4-ethylpiperidin-
1-yl)carbonyl]-1-(triphenylmethyl)-
1H-imidazole-4-butanamine hydrochloride (l: l)
A solution of 0.7 g (1.07 mmol) of
1, 1-dimethylethyl (S) - [1- [ (4-ethylpiperidin-1-yl) -
carbonyl]-4-[5-ethyl-1-(triphenylmethyl)-1H-imidazol-4-
yl]butyl]carbamate in 50 ml of benzene is treated with
a stream of gaseous hydrochloric acid for 15 minutes at
0~C. The temperature of the mixture is allowed to
return to room temperature, stirring is continued at
this temperature for 1.5 hours and the mixture is
concentrated under reduced pressure. The residue thus
obtained is triturated in ether, filtered off and dried
under reduced pressure.
0.61 g of product is obtained in the form of a white
powder which is used as is in the following stage.
Yield = 97%
Example 2
(S)-5-Methyl-a-[[4-(trifluoromethyl)-piperidin-1-
yl]carbonyl]-1-(triphenylmethyl)-1H-imidazole-4-
butanamine hydrochloride (1:1)
CA 02270478 1999-04-29
2a
2.1. 1.1-Dimethylethyl (S)-[4-[5-methyl-1-(tri-
phenylmethyl)-1H-imidazol-4-yl]-1-[[4-(tri-
fluoromethyl)piperidin-1-yl]carbonyl]butyl]-
carbamate
2.2.1. (S)-a-[[(l,l-Dimethylethoxy)carbonyl]amino]-
5-methyl-1-(triphenylmethyl)-1H-imidazole-
4-pentanoic acid
It is prepared according to the method
described in Example 1.7, from methyl
(S)-2-[[(1,1-dimethylethoxy)carbonyl]amino]-
5-[5-methyl-1-(triphenylmethyl)-1H-imidazol-4-yl]-
pentanoate.
2.2.2. 1,1-Dimethylethyl (S)-[4-[5-methyl-1-(tri-
phenylmethyl)-1H-imidazol-4-yl]-1-[[4-(tri-
fluoromethyl)piperidin-1-yl]carbonyl]butyl]-
carbamate
0.834 g (2.2 mmol) of [(benzotriazol-
1-yl)oxy]tris(dimethylamino)phosphonium hexafluoro-
phosphate is added portionwise at 0~C, under argon and
with stirring, to a mixture of 1.08 g (2 mmol) of
(S)-a-[[(1,1-dimethylethoxy)carbonyl]amino]-5-methyl-
1-(triphenylmethyl)-1H-imidazole-4-pentanoic acid,
0.306 g (2 mmol) of 4-(trifluoromethyl)piperidine and
1.04 ml (6 a~aaol) of diisopropylethylamine in 25 ml of
dichloromethane. The temperature of the mixture is
allowed to return to room temperature, stirring is
continued at this temperature for 18 hours and the
reaction mixture is concentrated under reduced
CA 02270478 1999-04-29
29
pressure. The residue is taken up in 100 ml of ethyl
acetate, washed successively with 50 ml of a 1N aqueous
hydrochloric acid solution, 50 ml of a saturated sodium
hydrogencarbonate solution and 50 ml of a saturated
sodium chloride solution, dried over sodium sulphate
and concentrated under reduced pressure. The residue is
purified by chromatography on a column of silica gel,
elution being carried out with a methanol:dichloro-
methane (2:98) mixture.
1.2 g of product are obtained in the form of a viscous
oil.
Yield = 89%
2.2. (S)-S-Methyl-a-[[4-(trifluoromethyl)-
piperidin-1-yllcarbonyl]-1-(triphenylmethyl)-
1H-imidazole-4-butanamine hydrochloride (1:1)
A solution of 1.2 g (1.78 mmol) of
1,1-dimethylethyl (S)-[4-[5-methyl-1-(triphenyl-
methyl)-1H-imidazol-4-yl]-1-[[4-(trifluoromethyl)-
piperidin-1-yl]carbonyl]butyl]carbamate in 100 ml of
benzene is treated with a stream of gaseous
hydrochloric acid for 20 minutes at 0~C. The reaction
mixture is left stirring at this temperature for 1 hour
and concentrated under reduced pressure. The residue
thus obtained is triturated in ether, filtered off and
dried under reduced pressure.
1.05 g of product are obtained in the form of a white
powder which is used as is in the following stage.
Yield: 97%
CA 02270478 1999-04-29
Melting point = 78~C
Example 3
(S)-a-[(4-Methoxypiperidin-1-yl)carbonyl]-5-methyl-
1-(triphenylmethyl)-1H-imidazole-4-butanamine
5 hydrochloride (1:1)
3.1. 4-Methoxypiperidine hydrochloride
3.l.1. 1,1-Dimethylethyl 4-hydroxypiperidine-
1-carboxylate
12 g (55 mmol) of bis(1,1-dimethylethyl)
10 dicarbonate, in solution of 50 ml of methanol, are
added dropwise at room temperature to a solution of
5.06 g (50 amnol) of piperidin-4-of in 50 ml of
methanol. The reaction mixture is left stirring at this
temperature for two hours and concentrated under
15 reduced pressure. The residue thus obtained is purified
by chromatography on a column of silica gel, elution
being carried out with a dichloromethane:methanol
(95:5) mixture.
9.74 g of product are obtained in the form of an oil.
20 Yield = 97%
3.l.2. 1,1-Dimethylethyl 4-methoxypiperidine-
1-carboxylate
l.59 g (39.8 mmol) of 60% sodium hydride in
oil are added portionwise at 0~C, under argon and with
25 stirring, to a mixture of 8 g (39.8 mmol) of
1,1-dimethylethyl 4-hydroxypiperidine-1-carboxylate and
of 4.95 ml (79.5 mmol) of iodomethane in solution of
CA 02270478 1999-04-29
31
40 ml of dimethylformamide and stirring is continued at
this temperature for 2 hours. The reaction mixture is
poured into l00 ml of a saturated ammonium chloride
solution and extracted with 2 times 200 ml of ethyl
acetate. The organic phases are combined, washed
successively with l00 ml of water and l00 ml of a
saturated sodium chloride solution, dried over
magnesium sulphate and concentrated under reduced
pressure. The residue is purified by chromatography on
a column of silica gel, elution being carried out with
an ethyl acetate:cyclohexane (2:8) mixture.
7.1 g of product are obtained in the form of an oil.
Yield = 85%
3.1.3. 4-Methoxypiperidine hydrochloride
A solution of 7 g (32.5 mmol) of l,l-
dimethylethyl 4-methoxypiperidine-1-carboxylate in 100
ml of tetrahydrofuran is treated with a stream of
gaseous hydrochloric acid for 30 minutes at 0~C. The
temperature of the mixture is allowed to return to room
temperature and stirring is continued for 18 hours at
this temperature. The reaction mixture is concentrated
under reduced pressure and the residue is triturated in
ether, filtered off and dried under reduced pressure.
4.2 g of product are obtained in the form of a white
powder which is used as is in the following stage.
Yield = 86%
Melting point = l32~C
3 . 2 . 1,1-Dimethylethyl (S) - [1- [ (4-methoxy-
CA 02270478 1999-04-29
32
piperidin-1-yl)carbonyl]-4-[5-methyl-
1-(triphenylmethyl)-1H-imidazol-4-yl]-
butyl]carbamate
0.71 g (1.87 mmol) of [(benzotriazol-I-yl)-
oxy]tris(dimethylamino)phosphonium hexafluorophosphate
is added portionwise at 0~C, under nitrogen and with
stirring, to a mixture of 0.9l8 g (1.7 mmol) of
(S)-a-[[(1,1-dimethylethoxy)carbonyl]amino]-5-methyl-
1-(triphenylmethyl)-1H-imidazole-4-pentanoic acid,
0.28l g (1.87 mmol) of 4-methoxypiperidine hydro-
chloride and 0.63 ml (3.57 mmol) of diisopropylethyl-
amine in 12 ml of dichloromethane. The temperature of
the mixture is allowed to return to room temperature,
stirring is continued at this temperature for 18 hours
and the reaction mixture is concentrated under reduced
pressure. The residue is taken up in 100 mI of ethyl
acetate, washed successively with 50 ml of a 0.5N
aqueous hydrochloric acid solution, 50 ml of a
saturated sodium hydrogencarbonate solution and 50 m1
of a saturated sodium chloride solution, dried over
sodium sulphate and concentrated wader reduced
pressure. The residue is purified by chromatography on
a column of silica gel, elution being carried out with
a methanol:dichloromethane (2:98) mixture.
1.05 g of product are obtained in the form of a viscous
oil.
Yield = 97'k
3.3. (S)-a-[(4-Methoxypiperidin-1-yl)carbonyl]-
CA 02270478 1999-04-29
33
5-methyl-1-(triphenylmethyl)-1H-imidazole-
4-butanamine hydrochloride (1:1)
A solution of 1.05 g (1.65 mmol) of
1,1-dimethylethyl (S) - [1- [ (4-methoxypiperidin-1-yl) -
carbonyl]-4-[5-methyl-1-(triphenylmethyl)-1H-imidazol-
4-yl]butyl]carbamate in 50 ml of benzene is treated
with a stream of gaseous hydrochloric acid for 20
minutes at 0~C. The mixture is left stirring at this
temperature for 30 minutes and concentrated under
reduced pressure. The residue thus obtained is
triturated in ether, filtered off and dried under
reduced pressure.
0.93 g of product is obtained in the form of a white
powder which is used as is in the following stage.
Yield = 98%
Melting point = 112~C
Example 4
(S)-5-Methyl-a-((4-methylenepiperidin-1-yl)-carbonyl]-
1-(triphenylmethyl)-1H-imidazole-4-butanamine
hydrochloride (1:1)
4.1. 4-Methylenepiperidine hydrochloride
4.1.1. 1,1-Dimethylethyl 4-methylenepiperidine-
1-carboxylate
17 ml of a 1.6M solution of a-butyllithium in
hexane are added at room temperature, under a nitrogen
atmosphere, to a mixture of 9.81 g (27.5 mmol) of
methyltriphenylphosphonium bromide in 60 ml of
CA 02270478 1999-04-29
34
anhydrous tetrahydrofuran. The mixture is left stirring
for 4 hours at room temperature and a solution of 5 g
(25 mmol) of 1,1-dimethylethyl 4-oxopiperidine-
1-carboxylate in 20 ml of anhydrous tetrahydrofuran ie
rapidly added. The reaction mixture is heated for
hours at the reflux temperature, poured into 400 ml
of a saturated ammonium chloride solution and extracted
with 2 times 300 ml of ether. The organic phases are
combined, dried over sodium sulphate and concentrated
10 under reduced pressure. The residue is purified by
chromatography on a column of silica gel, elution being
carried out with an ethyl acetate:n-hexane (5:95)
mixture.
2.8 g of product are obtained in the form of a glassy
I5 oil.
Yield = 57$
4.1.2. 4-Methylenepiperidine hydrochloride
This compound is obtained, from 1,1-dimethyl-
ethyl 4-methylenepiperidine-1-carboxylate, according to
the method described in Example 3.1.3.
4.2. (S)-5-Methyl-a-[(4-methylenepiperidin-1-yl)-
carbonyl]-1-(triphenylmethyl)-1H-imidazole-
4-butanamine hydrochloride (1:1)
(S)-a-[[(1,1-Dimethylethoxy)carbonyl]amino]-
5-methyl-1-(triphenylmethyl)-1H-imidazole-4-pentanoic
acid is reacted with 4-methylenepiperidine hydro-
chloride according to the method described in Example
3.2. and 1,1-dimethylethyl (S)-[4-[5-methyl-1-(tri-
CA 02270478 1999-04-29
phenylmethyl)-1H-imidazol-4-yl]-1-[(4-methylene-
piperidin-1-yl)carbonyl]butyl]carbamate, an amorphous
product, is obtained.
Melting point = 85~C
5 This product is treated with a stream of gaseous
hydrochloric acid according to the method described in
Example 3.3.
0.74 g of product are obtained.
Yield = 100%
10 Melting point = 148~C
Examvle 5
(S) -cx- [ (4-Cyclopropylpiperidin-1-yl) -carbonyl] -5-
methyl-1-(triphenylmethyl)-1H-imidazole-4-butanamine
hydrochloride (l: l)
15 5.1. 4-Cyclopropylpiperidine hydrochloride
5.1.1. 4-Cyclopropylpiperidine
13 g (109 mmol) of 4-cyclopropylpyridine, in
solution in 150 ml of acetic acid, are hydrogenated at
50~C in a Parr apparatus uader a pressure of 0.35 MPa
20 (50 psi) in the presence of 0.7 g of platinum(IV)
oxide. The reaction mixture is filtered through celite
and the filtrate is concentrated under reduced
pressure.
l2.85 g of product are obtained, which product is used
25 as is in the following stage.
Yield = 94%
5.l.2. 1,1-Dimethylethyl 4-cyclopropylpiperidine-
CA 02270478 1999-04-29
36
1-carboxylate
g (40 mmol) of 4-cyclopropylpiperidine are
dissolved in 40 ml of dichloromethane, the mixture is
cooled to 0~C and 6.98 g (32 mmol) of
5 bis (1,1-dimethylethyl) Bicarbonate and 4.85 g (48 amnol)
of triethylamine are added dropwise. The reaction
mixture is concentrated and the residue is purified by
chromatography on a column of silica gel, elution being
carried out with a dichloromethane:methanol (99:1)
mixture.
4 g of product are obtained.
Yield = 44%
5.1.3. 4-Cyclopropylpiperidine hydrochloride
A stirred solution of 6.5 g (28.8 mmol) of
1,1-dimethylethyl 4-cyclopropylpiperidine-1-carboxylate
in l00 ml of benzene is treated with a stream of
gaseous hydrochloric acid for 30 minutes at 0~C. The
temperature of the reaction mixture is allowed to
return to room temperature, stirring is maintained for
4 hours at this temperature and the reaction mixture is
concentrated under reduced pressure. The residue thus
obtained is triturated in ether, filtered off and dried
under reduced pressure.
4.1 g of product are obtained in the form of a white
powder which is used as is in the following stage.
Yield = 88%
Melting point = 186~C
5.2. 1,1-Dimethylethyl (S)-[1-[(4-cyclopropyl-
CA 02270478 1999-04-29
37
piperidin-1-yl)carbonyl]-4-[5-methyl-1-(tri-
phenylmethyl)-1X-imidazol-4-yl]butyl]-
carbamate
A mixture of 6 g (11 mmol) of (S)-a-
[[(l,l-dimethylethoxy)carbonyl]amino]-5-methyl-1-(tri-
phenylmethyl)-1X-imidazole-4-pentanoic acid, l.79 g (11
mmol) of 4-cyclopropylpiperidine hydrochloride and 9.6
ml (55.5 mmol) of diisopropylethylamine in l00 ml of
dichloromethane is stirred and 4.62 g (12.2 mmol) of
[(benzotriazol-1-yl)oxy]tris(dimethylamino)phosphonium
hexafluorophosphate are added portionwise at 0~C, under
argon and with stirring. The temperature of the mixture
is allowed to return to room temperature, stirring is
continued at this temperature for 4 hours and the
reaction mixture is concentrated under reduced
pressure. The residue is taken up in 300 ml of ethyl
acetate, washed successively with l00 ml of a 1N
aqueous hydrochloric acid solution, l00 ml of a
saturated sodium hydrogencarbonate solution and 100 ml
of a saturated sodium chloride solution, dried over
sodium sulphate and concentrated under reduced
pressure. The residue is purified by chromatography on
a column of silica gel, elution being carried out with
a methanol:dichloromethane (1:99) mixture.
5 g of product are obtained.
Yield = 70$
5.3. (S)-a-[(4-Cyclopropylpiperidin-1-yl)-
carbonyl]-5-methyl-1-(triphenylmethyl)-
CA 02270478 1999-04-29
38
1H-imidazole-4-butanamine hydrochloride (1:1)
A stirred solution of 5.3 g (8 mmol) of
1,1-dimethylethyl (S)-[1-[(4-cyclopropylpiperidin-
1-yl)carbonyl]-4-[5-methyl-1-(triphenylmethyl)-
1H-imidazol-4-yl]butyl]carbamate in 200 ml of benzene
is treated with a stream of gaseous hydrochloric acid
for 30 minutes at 0~C. The temperature of the reaction
mixture is allowed to return to room temperature,
stirring is maintained for 3 hours and the reaction
mixture is concentrated under reduced pressure. The
residue thus obtained is taken up in two times 280 m1
of dichloromethane and dried under reduced pressure.
4.7 g of product are obtained in the form of a white
powder which is used as is in the following stage.
Yield = 100%
Melting point = 124~C
Example 6
(S)-5-Methyl-a-[[4-(difluoromethylene)-piperidin-1-
yl]carbonyl]-1-(triphenylmethyl)-1H-imidazole-4-
butanamine hydrochloride (l: l)
6.1. 4-(Difluoromethylene)piperidine hydrochloride
6.1.l. 1,1-Dimethylethyl 4-(difluoromethylene)-
piperidine-1-carboxylate
45.6 ml (252 mmol) of hexamethylphosphor-
amide, in solution in 30 ml of triglyme, are added
dropwise at 0~C under an argon atmosphere to 12 ml
(120 mmol) of difluorobromomethane in solution in
CA 02270478 1999-04-29
39
180 ml of triglyme, which is kept stirring. The
temperature of the reaction mixture is allowed to
return to room temperature, stirring is maintained for
30 minutes at this temperature and the reaction mixture
is again cooled to 0~C. 1l.94 g (60 mmol) of
1,1-dimethylethyl 4-oxopiperidine-1-carboxylate, in
solution in 30 ml of triglyme, are then added, the
temperature of the mixture is allowed to return to room
temperature and the reaction mixture is stirred for 30
minutes at this temperature. The reaction mixture is
heated for 2 hours at 80~C, cooled, poured into 1 litre
of water and extracted with 3 times 400 ml of pentane.
Washing is carried out with water, drying is carried
out over sodium sulphate and evaporation is carried
out. The residue is purified by chromatography on a
column of silica gel, elution being carried out with a
cyclohexane:ethyl acetate (97:3) mixture.
8.5 g of product are obtained.
Yield = 61%
6.1.2. 4-(Difluoromethylene)piperidine hydrochloride
This compound is obtained in the form of a
white powder, from 1,1-dimethylethyl 4-(difluoro-
methylene)piperidine-1-carboxylate, according to the
method described in Example 3.l.3.
Yield = 100%
Melting point = 196~C
6.2. (S)-5-Methyl-a-[[4-(difluoromethylene)-
piperidin-1-yl]carbonyl]-1-(triphenylmethyl)-
CA 02270478 1999-04-29
1H-imidazole-4-butanamine hydrochloride (l: l)
(S)-a-[[(1,1-Dimethylethoxy)carbonyl]amino]-
5-methyl-1-(triphenylmethyl)-1H-imidazole-4-pentanoic
acid is reacted with 4-(difluoromethylene)piperidine
5 hydrochloride according to the method described in
Example 3.2 and l,l-dimethylethyl (S)-[4-[5-methyl-
1-(triphenylmethyl)-1H-imidazol-4-yl]-1-[[4-(difluoro-
methylene)piperidin-1-yl]carbonyl]butyl]carbamate is
obtained in the form of a glassy solid.
10 Yield = 75%
Melting point = 86~C
This product is treated with a stream of gaseous
hydrochloric acid according to the method described in
Example 3.3.
15 The product is obtained in the form of a white powder.
Yield = 99%
Melting point = 117~C
Example 7
(S)-5-Methyl-a-[[4-(methylthio)piperidin-
20 1-yl]carbonyl]-1-(triphenylmethyl)-1H-imidazole-
4-butanamine hydrochloride (1:1)
7.1. 4-(Methylthio)piperidine hydrochloride
7.1.l. 1,1-Dimethylethyl 4-[(methylsulphonyl)oxy]-
piperidine-1-carboxylate
25 5.6 ml (72 mmol) of methanesulphonyl chloride
are added dropwise at 0~C under nitrogen to a solution
of 13.9 g (69 mmol) of 1,1-dimethylethyl 4-hydroxy-
CA 02270478 1999-04-29
41
piperidine-1-carboxylate and of 5.6 ml (76 mmol) of
triethylamine in 80 ml of dichloromethane. The reaction
mixture is left stirring for 6 hours at this
temperature and is concentrated under reduced pressure.
The residue is taken up in 200 ml of ethyl acetate and
washed successively with 2 times l00 ml of a 1N aqueous
hydrochloric acid solution, 100 ml of water and 100 ml
of a saturated sodium chloride solution. The organic
layer is dried over sodium sulphate and concentrated
under reduced pressure. The residue is purified by
chromatography on a column of silica gel, elution being
carried out with ethyl acetate.
16.2 g of product are obtained in the form of white
crystals.
Yield = 95%
Melting point = 93.9~C
7.1.2. 1,1-Dimethylethyl 4-(methylthio)piperidine-
1-carboxylate
A mixture of 2.47 g (10 mmol) of
1,1-dimethylethyl 4-[(methylsulphonyl)oxy]piperidine-
1-carboxylate, 0.7l g (10.1 mmol) of sodium thio-
methoxide and 0.37 g (1 mmol) of tetrabutylammonium
iodide in 10 ml of tetrahydrofuran is stirred for 72
hours at room temperature and then the mixture is
concentrated under reduced pressure. The residue is
purified by chromatography on a column of silica gel,
elution being carried out with an n-hexane: ethyl
acetate (9:1) mixture.
CA 02270478 1999-04-29
42
1.5 g of product are obtained in the form of a viscous
oil.
Yield = 63%
7.l.3 4-(Methylthio)piperidine hydrochloride
This compound is obtained, from l,l-dimethyl-
ethyl 4-(methylthio)piperidine-1-carboxylate, according
to the method described in Example 3.l.3.
Yield = 100%
Melting point = 156.5~C
7.2. (S)-5-Methyl-a-[[4-(methylthio)piperidin-
1-yl]carbonyl]-1-(triphenylmethyl)-
1H-imidazole-4-butanamine hydrochloride (1:1)
(S)-a-[[(1,1-Dimethylethoxy)carbonyl]amino]-
5-methyl-1-(triphenylmethyl)-1H-imidazole-4-pentanoic
acid is reacted with 4-(methylthio)piperidine
hydrochloride according to the method described in
Example 3.2 and 1,1-dimethylethyl (S)-[4-[5-methyl-
1-(triphenylmethyl)-1H-imidazol-4-yl]-1-[[4-(methyl-
thin)piperidin-1-yl]carbonyl]butyl]carbamate is
obtained in the form of an amorphous powder.
Yield = 93%
Melting point = l01.2~C
This product is treated with a stream of gaseous
hydrochloric acid according to the method described in
Example 3.3.
The product is obtained in the form of an amorphous
powder.
Yield = 100%
CA 02270478 1999-04-29
43
Melting point = 127.7~C
Example 8
(S)-5-Methyl-a-[(4-methyl-1,2,3,6-tetrahydro-pyridin-1-
yl)carbonyl]-1-(triphenylmethyl)-1H-imidazole-4-
butanamine hydrochloride (1:1)
8.1. 4-Methyl-1,2,3,6-tetrahydropyridine
hydrochloride
8.l.1. 1,1-Dimethylethyl 4-methyl-
1,2,3,6-tetrahydropyridine-1-carboxylate
A solution of 18 ml (28.8 mmol) of
methyllithium in ether is added at 0~C under nitrogen
to a solution of 4.95 g (25 mmol) of 1,1-dimethylethyl
4-oxopiperidine-1-carboxylate in 30 ml of anhydrous
tetrahydrofuran and stirring is continued for 2 hours
at this temperature. 3 ml (38 mmol) of methanesulphonyl
chloride are then added dropwise, stirring is continued
for 4 hours at 0~C and then the reaction mixture is
concentrated under reduced pressure. The residue is
taken up in 200 ml of ethyl acetate and washed
successively with 2 times 100 ml of a O.1N aqueous
hydrochloric acid solution, 100 ml of water and 100 ml
of a saturated sodium chloride solution. The organic
layer is dried over sodium sulphate and concentrated
under reduced pressure. The residue is taken up in
100 ml of toluene and 15 ml of triethylamine, heated
for 18 hours at the reflux temperature and concentrated
under reduced pressure. The residue is purified by
CA 02270478 1999-04-29
44
chromatography on a column of silica gel, elution being
carried out with an n-hexane: ether (95:5) mixture.
0.9 g of product is obtained in the form of a viscous
oil.
Yield = 18%
8.l.2. 4-Methyl-1,2,3,6-tetrahydropyridine
hydrochloride
This compound is obtained, from
1,1-dimethylethyl 4-methyl-l,2,3,6-tetrahydropyridine-
1-carboxylate, according to the method described in
Example 3.1.3.
Yield = 100%
8.2. (S)-5-Methyl-a-[(4-methyl-l,2,3,6-tetrahydro-
pyridin-1-yl)carbonyl]-1-(triphenylmethyl)-
1H-imidazole-4-butanamine hydrochloride (1:1)
(S)-a-[[(l,l-Dimethylethoxy)carbonyl]amino]-
5-methyl-1-(triphenylmethyl)-1H-imidazole-4-pentanoic
acid is reacted with 4-methyl-1,2,3,6-tetrahydro-
pyridine hydrochloride according to the method
described in Example 3.2 and 1,1-dimethylethyl
(S)-[4-[5-methyl-1-(triphenylmethyl)-1H-imidazol-4-yl]-
1-[(4-methyl-1,2,3,6-tetrahydropyridin-1-yl)carbonyl]-
butyl]carbamate is obtained in the form of an amorphous
powder.
Yield = 90%
Melting point = 90.7~C
This product is treated with a stream of gaseous
hydrochloric acid according to the method described in
CA 02270478 1999-04-29
Example 3.3.
The product is obtained in the form of a white powder.
Yield = 100%
Melting point = 1l8~C
5 Example 9
1-(2-Amino-5-(5-methyl-1-(triphenylmethyl)-1H-imidazol-
4-yl)-1-oxopentyl]-hexahydro-5H-1,4-diazepin-5-one
hydrochloride
9.1. Hexahydro-5H-1,4-diazepin-5-one hydrochloride
10 9.l.1. 1-(Phenylmethyl)-hexahydro-5H-1,4-diazepin-
5-one
A solution of 9.86 g (87.18 amnol) of
hydroxylamine-O-sulphonic acid in acetic acid is added
over 10 minutes to a solution of 11 g (58.12 Col) of
15 1-phenylmethylpiperidin-3-one in 60 ml of formic acid.
The reaction mixture is heated for 4 hours at the
reflux temperature. The mixture is allowed to cool and
is poured into an ice: water mixture and then
neutralized with a 5% aqueous sodium hydroxide
20 solution. Extraction is carried out with chloroform and
the organic phase is recovered, dried and evaporated to
dryness. The residue is purified by chromatography on a
column of silica gel, elution being carried out with a
dichloromethane:methanol (2:98) mixture.
25 6.94 g of product are obtained.
Yield = 58.5%
9.1.2. Hexahydro-5H-1,4-diazepin-5-one hydrochloride
CA 02270478 1999-04-29
46
5.5 g (26.2 mmol) of 1-(phenylmethyl)-
hexahydro-5H-1,4-diazepin-5-one are dissolved in 100 ml
of methanol, 0.7 g of 10% palladium-on-charcoal is
added and the reaction mixture is heated for 3 hours at
45~C under a pressure of 0.29 MPa (42 psi). The
reaction mixture is filtered, the solvents are
evaporated and the residue is taken up in 30 ml of
ethanol. Heatiag is carried out, the insoluble material
is filtered off and rinsed with ether and the solvent
is evaporated.
2.44 g of product are obtained in the form of an off-
White powder which is used as is in the following
stage.
9.2. 1-[2-Amino-5-[5-methyl-1-(triphenylmethyl)-
1H-imidazol-4-yl]-1-oxopentyl]-hexahydro-
5H-1,4-diazepin-5-one hydrochloride
9.2.1. 1,1-Dimethylethyl (S)-[4-[5-methyl-1-(tri-
phenylmethyl)-1H-imidazol-4-yl]-1-[(5-oxo-
hexahydro-5H-1,4-diazepin-1-yl)carbonyl]-
butyl]carbamate
2 .15 g (4 m~mol) of (S) -a- [ [ (1, 1-dimethyl-
ethoxy)carbonyl]amino]-5-methyl-1-(triphenylmethyl)-1H-
imidazole-4-pentanoic acid, followed in succession by
2.8 ml (16 mmol) of N,N-diisopropylethylamine and by
1. 5 g (4 mmol) of O- (benzotriazol-1-yl) -
N,N,N',N'-tetramethyluronium hexafluorophosphate, are
added at 0~C to a solution of 0.6 g (4 Col) of
hexahydro-5H-1,4-diazepin-5-one hydrochloride in 40 ml
CA 02270478 1999-04-29
47
of dichloromethane. The temperature of the reaction
mixture is allowed to return to room temperature,
stirring is continued overnight at this temperature and
the reaction mixture is concentrated under vacuum. The
residue is taken up in 200 ml of ethyl acetate and
washed successively with 3 times 30 ml of a 1N aqueous
hydrochloric acid solution, 2 times 20 ml of a
saturated sodium hydrogencarbonate solution and then
20 ml of a saturated sodium chloride solution. The
organic layer is dried over magnesium sulphate and
concentrated under reduced pressure. The residue is
purified by chromatography on a column of silica gel,
elution being carried out with a dichloro-
methane: methanol (98:2 to 97:3) gradient.
1.87 g of product are obtained in the form of an off-
white foam.
Yield = 74%
9.2.2. 1-[2-Amino-5-[5-methyl-1-(triphenylmethyl)-
1H-imidazol--4-yl]-1-oxopentyl]-hexahydro-
5H-1,4-diazepin-5-one hydrochloride
A solution of 1.87 g (2.94 armol) of
l, 1-dimethylethyl (S) - [4- [5-methyl-1- (triphenyl-
methyl)-1H-imidazol-4-yl]-1-[(5-oxohexahydro-
5H-1,4-diazepin-1-yl)carbonyl]butyl]carbamate in 200 ml
of toluene is treated with a stream of gaseous
hydrochloric acid for 10 seconds at 0~C. The
temperature of the mixture is allowed to return to room
temperature and then the reaction mixture is
CA 02270478 1999-04-29
48
concentrated under reduced pressure. The residue is
dissolved in a minimum volume of dichloromethane and
200 ml of ether are added. The mixture is triturated,
filtered and dried.
1.64 g of product are obtained, which product is used
as is in the following stage.
Yield = 97%
Example 10
(S)-a-Amino-N-cyclopentyl-N,5-dimethyl-
1-(triphenylmethyl)-1H-imidazole-4-pentanamide
hydrochloride
10.l. N-Methylcyclopentanamine hydrochloride
10.1.1. N-Cyclopentylformamide
A mixture of 10 g (117 mmol) of cyclopentan-
amine and of 10.8 ml (140 mmol) of ethyl formate is
heated for 4 hours at the reflux temperature and then
the reaction mixture is concentrated under reduced
pressure. The residue is purified by chromatography on
a column of silica gel, elution being carried out with
an ethyl acetate:cyclohexane (1:9 to 6:4) gradient.
10 g of product are obtained in the form of an oil.
Yield = 75%
10.1.2. l,l-Dimethylethyl cyclopentylmethylcarbamate
50 ml (50 mmol) of a 1M solution of lithium
aluminium hydride in tetrahydrofuran are added dropwise
at 0~C under nitrogen to a solution of 4.37 g (38 a~ol)
of N-cyclopentylformamide in 20 ml of anhydrous
CA 02270478 1999-04-29
49
tetrahydrofuran. The temperature of the mixture is
allowed to return to room temperature and the reaction
mixture is heated at the reflux temperature for 8
hours. The reaction mixture is cooled to 0~C and
acidified to pH 2 with a 1N aqueous hydrochloric acid
solution and the pH is adjusted to 8 with potassium
carbonate. 8.6 g (40 mmol) of bis(l,l-dimethylethyl)
dicarbonate, in solution in 40 ml of methanol, are then
added dropwise. The temperature of the mixture is
allowed to return to room temperature and stirring is
continued for 15 hours at this temperature. The
reaction mixture is extracted with 2 times 300 ml of
ether and the organic phases are combined. They are
washed with 2 times 200 ml of a 1N aqueous hydrochloric
acid solution and then with 200 ml of a saturated
sodium chloride solution. The combined organic phases
are dried over sodium sulphate, filtered and
concentrated under reduced pressure. The residue is
purified by chromatography on a column of silica gel,
elution being carried out with a cyclohexane:ether
(95:5) mixture.
2.91 g of product are obtained in the form of an oil.
Yield = 38~
10.1.3. N-Methylcyclopentanamine hydrochloride
A solution of 2.9 g (14.5 mmol) of
1,1-dimethylethyl cyclopentylmethylcarbamate is treated
with a stream of gaseous hydrochloric acid for 5
minutes at 0~C. The mixture is left stirring for
CA 02270478 1999-04-29
4 hours at this temperature and is then concentrated
under reduced pressure.
1.96 g of product are obtained in the form of a white
hygroscopic powder.
5 Yield = 100%
Melting point = l23-l26~C
10.2. (S)-a-Amino-N-cyclopentyl-N,5-dimethyl-
1-(triphenylmethyl)-1H-imidazole-
4-pentanamide hydrochloride
10 10.2.l. 1,1-Dimethylethyl (S)-[1-[(cyclopentylmethyl-
amino)carbonyl]-4-[5-methyl-1-(triphenyl-
methyl)-1X-imidazol-4-yl]butyl]carbamate
0.68 g (5 mmol) of N-methylcyclopentanamine
hydrochloride, 2.l5 ml (12.3 mmol) of N,N-diisopropyl-
15 ethylamine and 1.98 g (5.24 mmol) of O-(benzotriazol-
1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate
are successively added, at 0~C under nitrogen, to a
solution of 2.57 g (4.76 mmol) of (S) -a-
[[(l,l-dimethylethoxy)carbonyl]amino]-5-methyl-1-(tri-
20 phenylmethyl)-1H-imidazole-4-pentanoic acid in 15 ml of
dichloromethane. The temperature of the reaction
mixture is allowed to return to room temperature,
stirring is continued for 15 hours at this temperature
and the reaction mixture is concentrated under vacuum.
25 The residue is taken up in l50 ml of ethyl acetate and
washed successively with 100 ml of a 1N aqueous
hydrochloric acid solution, 100 ml of a saturated
sodium hydrogencarbonate solution and then l00 ml of a
CA 02270478 1999-04-29
51
saturated sodium chloride solution. The organic layer
is dried over sodium sulphate and concentrated under
reduced pressure. The residue is purified by
chromatography on a column of silica gel, elution being
carried out with an ethyl acetate:cyclohexane (3:7 to
8:2) gradient.
2,26 g of product are obtained in the form of an
amorphous solid.
Yield = 77~
Melting point = 86-90~C
10.2.2. (S)-a-Amino-N-cyclopentyl-N,5-dimethyl-
1-(triphenylmethyl)-1X-imidazole-
4-pentanamide hydrochloride
A solution of 2.2 g (3.5 mmol) of
l,l-dimethylethyl (S)-[1-[(cyclopentylmethylamino)-
carbonyl]-4-[5-methyl-1-(triphenylmethyl)-1H-imidazol-
4-yl]butyllcarbamate is treated with a stream of
gaseous hydrochloric acid for 5 minutes at 0~C. The
mixture is left stirring for 5 hours at this
temperature and is then concentrated under reduced
pressure.
2 g of product are obtained, which product is used as
is in the following stage.
Yield = 100
Melting point = l38-142~C
Example 11
(S)-a-Amino-N.5-dimethyl-N-pyrrolidin-1-yl-
CA 02270478 1999-04-29
' 52
1-(triphenylmethyl)-1H-imidazole-4-pentan-amide
hydrochloride
11.1. N-Methylpyrrolidin-1-amine hydrochloride
1l.1.1. 1,1-Dimethylethyl pyrrolidin-1-ylcarbamate
1.13 ml (8.15 mmol) of triethylamine are
added dropwise to a solution of 1 g (8.15 mmol) of
pyrrolidin-1-amine hydrochloride and of 1.62 g
(7.4 mmol) of bis(1,1-dimethylethyl) dicarbonate in
8 ml of dichloromethane. The mixture is left stirring
for 15 hours and is concentrated under reduced
pressure. The residue is taken up in 100 ml of ether
and washed successively with 10 ml of water and 100 ml
of a saturated aqueous sodium chloride solution. The
organic layer is dried over sodium sulphate, filtered
through silica and concentrated under reduced pressure.
1 g of product is obtained.
Yield = 67~
Melting point = 108~C
11.1.2. 1,1-Dimethylethyl methylpyrrolidin-1-yl-
carbamate
7.7 ml (7.7 mmol) of a 1M lithium bis-
(trimethylsilyl)amide solution in tetrahydrofuran are
added dropwise at -78~C under nitrogen to a solution of
1.34 g (7 mmol) of 1,1-dimethylethyl pyrrolidin-1-yl-
carbamate and of 1.75 ml (28 mmol) of methyl iodide in
3 ml of anhydrous tetrahydrofuran. The temperature of
the mixture is allowed to return to room temperature
and stirring is continued for 30 minutes at this
CA 02270478 1999-04-29
53
temperature. 150 ml of ether are added and washing is
carried out successively with l00 mI of water and
100 ml of a saturated sodium chloride solution. The
organic layer is dried over sodium sulphate. filtered
and concentrated under reduced pressure. The residue is
purified by chromatography on a column of silica gel,
elution being carried out with a cyclohexane:ethyl
acetate (9:1) mixture.
0.75 g of product is obtained in the form of an oil.
Yield = 55%
11.l.3. N-Methylpyrrolidin-1-amine hydrochloride
This product is prepared, from 0.75 g
(3.7 mmol) of l,l-dimethylethyl methylpyrrolidin-1-yl-
carbamate, according to the method described in 10.l.3.
0.5 g of product is obtained in the form of a viscous
oil.
Yield = 100%
11.2. (S)-a-Amino-N,5-dimethyl-N-pyrrolidin-1-yl-
1-(triphenylmethyl)-1X-imidazole-4-pentan-
amide hydrochloride
l1.2.1. 1,1-Dimethylethyl (S)-[1-[(methylpyrrolidin-
1-ylamino)carbonyl]-4-[5-methyl-1-(triphenyl-
methyl)-1H-imidazol-4-yl]butyl]carbamate
The product is prepared according to the
procedure described in 10.2.l., from 1.8 g (3.3 mmol)
of (S)-a-[[(1.1-dimethylethoxy)carbonyl]amino]-
5-methyl-1-(triphenylmethyl)-1X-imidazole-4-pentanoic
acid and from 0.48 g (3.5 mmol) of N-methylpyrrolidin-
CA 02270478 1999-04-29
54
1-amine hydrochloride.
1.8 g of product are obtained in the form of an
amorphous solid.
Yield = 88%
Melting point = 70-75~C
l1.2.2. (S)-a-Amino-N,5-dimethyl-N-pyrrolidin-1-yl-
1-(triphenylmethyl)-1H-imidazole-4-pentan-
amide hydrochloride
The product is prepared according to the
procedure described in 10.2.2., from 1.8 g (2.8 manol)
of 1,1-dimethylethyl (S)-[1-[(methylpyrrolidin-1-yl-
amino)carbonyl]-4-[5-methyl-1-(triphenylmethyl)-
1H-imidazol-4-yl]butyl]carbamate.
1.65 g of product are obtained in the form of an
amorphous solid.
Yield = l00%
Melting point = 130-135~C
Example 12 (Compound No. 67)
(S) -N- [3- [ [ [4- (5-Ethyl-1H-imidazol-4-yl) -1- [ (4-
ZO ethylpiperidin-1-yl)carbonyl]butyl]-
amino] sulphonyl] [1,1' -biphenyl] -2-yl] propan-amide
hydrochloride (1:1)
12 .1. (S) -N- [3- [ [ [1- [ (4-Ethylpiperidin-1-yl) -
carbonyl]-4-[5-ethyl-1-(triphenylmethyl)-
1X-imidazol-4-yl]butyl]amino]-
sulphonyl][1,1'-biphenyl]-2-yl]-N-(1-oxo-
propyl)propanamide
CA 02270478 1999-04-29
0.48 ml (3.4 mmol) of triethylamine is added
dropwise at 0~C under nitrogen to a mixture of 0.435 g
(1.25 amnol) of 2- [bis (1-oxopropyl) amino] -
[1,1'-biphenyl]-3-sulphonyl chloride and of 0.61 g
5 (1.04 Col) of (S) -5-ethyl-a- [ (4-ethylpiperidin-1-yl) -
carbonyl]-1-(triphenylmethyl)-1H-imidazole-4-butan-
amine hydrochloride in 8 ml of dichloromethane. The
mixture is left stirring for 4 hours and is
concentrated under reduced pressure. The residue is
10 taken up in 100 ml of ethyl acetate, washed
successively with 50 ml of a 1N aqueous hydrochloric
acid solution, 50 ml of a saturated sodium hydrogen-
carbonate solution and 50 ml of a saturated sodium
chloride solution, dried over magnesium sulphate and
15 concentrated under reduced pressure.
0.85 g of product is obtained in the form of a viscous
oil which is used as is in the following stage.
Yield = 95%
12 .2 . (S) -N- [3- [ [ [4- (5-Ethyl-1H-imidazol-4-yl) -
20 1-[(4-ethylpiperidin-1-yl)carbonyl]butyl]-
amino] sulphonyl] [1,1' -biphenyl] -2-yl]propan-
amide hydrochloride (1:1)
0.85 g (0.95 mmol) of (S) -N- [3- [ [ [1-
[(4-ethylpiperidin-1-yl)carbonyl]-4-[5-ethyl-1-(tri-
25 phenylmethyl)-1H-imidazol-4-yl]butyl]amino]-
sulphonyl] [1,1' -biphenyl] -2-yl] -N- (1-oxopropyl) -
propanamide, in solution in a mixture of 30 ml of
acetic acid and 10 ml of water, is heated for 16 hours
CA 02270478 1999-04-29
56
at the reflux temperature and the reaction mixture is
concentrated under reduced pressure. The residue is
taken up in 150 ml of ethyl acetate, washed
successively with 50 ml of a saturated sodium hydrogen-
carbonate solution and with 50 ml of a saturated sodium
chloride solution, dried over sodium sulphate and
concentrated under reduced pressure. The residue is
purified by chromatography on a column of silica gel,
elution being carried out with a methanol:dichloro-
methane (4:96) mixture.
0.44 g of product is obtained in the form of a base
which is taken up in 10 ml of a O.1N solution of
hydrochloric acid in isopropanol and concentrated under
reduced pressure. The residue is purified by
chromatography on an RP 18 column, elution being
carried out with an acetonitrile:water (3:7) mixture.
After lyophilization, 0.42 g of product is obtained in
the form of a white powder.
Yield = 69%
Melting point = 132~C
[a]D = +l12~: (c = 0.2, methanol)
Example 13 (Compound No. 40)
(S) -N- [3- ( [ [4- (5-Methyl-1H-imidazol-4-yl) -1- [ [4
(trifluoromethyl)piperidin-1-yl]carbonyl]butyl]
amino] sulphonyl] [1,1' -biphenyl] -2-yl] propanamide
hydrochloride (1:1)
CA 02270478 1999-04-29
57
13.1. (S)-N-[3-[[[4-[5-Methyl-1-(triphenylmethyl)-
1H-imidazol-4-yl]-1-[[4-(trifluoromethyl)-
piperidin-1-yl]carbonyl]butyl]amino]-
sulphonyl][l,l'-biphenyl]-2-yl]-N-(1-oxo-
propyl)propanamide
0.79 ml (5.7 mmol) of triethylamine is added
dropwise at 0~C under argon to a mixture of 0.65 g
(1.72 mmol) of 2-[bis(1-oxopropyl)amino]-
[1,1'-biphenyl]-3-sulphonyl chloride and of l.05 g
(1.72 amnol) of (S) -5-methyl-a- [ [4- (trifluoromethyl) -
piperidin-1-yl]carbonyl]-1-(triphenylmethyl)-
1H-imidazole-4-butanamine hydrochloride in 20 ml of
dichloromethane. The temperature of the mixture is
allowed to return to room temperature, stirring is
continued for 18 hours at this temperature and the
reaction mixture is concentrated under reduced
pressure. The residue is taken up in 100 ml of ethyl
acetate, washed successively with 50 ml of a 1N aqueous
hydrochloric acid solution, 50 ml of a saturated sodium
hydrogencarbonate solution and 50 ml of a saturated
sodium chloride solution, dried over sodium sulphate
and concentrated under reduced pressure. The residue is
purified by chromatography on a column of silica gel,
elution being carried out with a
methanol:dichloromethane (2:98) mixture.
1.04 g of product are obtained in the form of a viscous
Oll.
Yield = 70%
CA 02270478 1999-04-29
58
13.2. (S)-N-[3-[[[4-(5-Methyl-1H-imidazol-4-yl)-
1-[[4-(trifluoromethyl)piperidin-1-yl]-
carbonyl] butyl] amino] sulphonyl] -
[1,1'-biphenyl]-2-yl]propanamide
S hydrochloride (I:1)
1.02 g (l.l amtol) of (S) -N- [3- [ [ [4- [5-methyl-
1-(triphenylmethyl)-1H-imidazol-4-yl]-1-[[4-(trifluoro-
methyl) piperidin-1-yl] carbonyl] butyl] amino] sulphonyl] -
[1,1'-biphenyl]-2-yl]-N-(1-oxopropyl)propanamide, in
ZO solution in a mixture of 25 ml of acetic acid and 25 ml
of water, are heated for 10 hours at the reflex
temperature and the reaction mixture is concentrated
under reduced pressure. The residue is taken up in
100 ml of ethyl acetate, washed with 50 ml of a
15 saturated sodium hydrogencarbonate solution, dried over
sodium sulphate and concentrated under reduced
pressure. The residue is purified by chromatography on
a column of silica gel, elution being carried out with
a methanol:dichloromethane (5:95) mixture. 0.42 g of
20 product is obtained in the form of a base which is
taken up in 12 ml of a O.1N solution of hydrochloric
acid in isopropanol and concentrated under reduced
pressure. The residue is purified by chromatography on
an RP Z8 column, elution being carried out with an
25 acetonitrile:water (3:7) mixture.
After lyophilization, 0.33 g of product is obtained.
Yield = 46%
Melting point = 146-1S0~C
CA 02270478 1999-04-29
59
[a] p~ _ +80~ (c = 0.2, methanol)
Example 14 (Compound No.34)
(S) -N- [2- [ [ [1- [ (4-Methoxypiperidin-1-yl) -carbonyl] -4
(5-methyl-1X-imidazol-4-yl)-butyl]amino]sulphonyl]-6
thien-2-ylphenyl]-propanamide hydrochloride (1:1)
l4.1. (S)-1-[2-(3-Ethyl-1,1-dioxo-5-thien-2-yl-
2H-1,2.4-benzothiadiazin-2-yl)-5-[5-methyl-
1-(triphenylmethyl)-1H-imidazol-4-yl]-1-oxo-
pentyl]-4-methoxypiperidine
14.1.1. 2-[(1-Chloropropylidene)amino]-3-thien-2-yl-
benzenesulphonyl chloride
2.86 ml (33 mmol) of propionyl chloride are
added dropwise at 0~C to a mixture of 3.8 g (15 mmol)
of 2-amino-3-(thien-2-yl)benzenesulphonic acid and of
4 ml (49.5 mmol) of pyridine in 30 ml of dichloro-
methane. The reaction mixture is left stirring for
5 hours at this temperature and is then concentrated
under reduced pressure. The residue is taken up in
40 ml of dichloromethane, 7.8 g (37.5 mmol) of
phosphorous pentachloride are added portioawise and the
mixture is left stirring for 1 hour at 0~C and then for
2 hours at room temperature. 200 ml of ether are added
to the reaction mixture, filtration is carried out and
the filtrate is washed successively with 2 times 200 ml
of ice-cold water and 50 ml of a saturated sodium
chloride solution, dried over sodium sulphate and
concentrated under reduced pressure. The residue is
CA 02270478 1999-04-29
purified by chromatography on Florisil', elution being
carried out rapidly with ether.
3.18 g of product are obtained after crystallization
from pentane.
5 Yield = 61%
Melting point = 74~C
14.l.2. (S)-1-[2-(3-Ethyl-1.1-dioxo-5-thien-2-yl-
2H-1,2,4-benzothiadiazin-2-yl)-5-[5-methyl-
1-(triphenylmethyl)-1H-imidazol-4-yl]-1-oxo-
10 pentyl]-4-methoxypiperidine
0.55 ml (3.96 mmol) of triethylamine is added
dropwise at 0~C to a mixture of 0.42 g (1.2 mmol) of 2-
[(1-chloropropylidene)amigo]-3-thien-2-
ylbenzenesulphonyl chloride and of 0.69 g (1.2 mmol) of
15 (S)-a-[(4-methoxypiperidin-1-yl)carbonyl]-5-methyl
1-(triphenylmethyl)-1H-imidazole-4-butanamine
hydrochloride in 15 ml of dichloromethane. The
temperature of the mixture is allowed to return to room
temperature, stirring is continued for 18 hours at this
20 temperature and the reaction mixture is concentrated
under reduced pressure. The residue is taken up in
l00 ml of ethyl acetate. washed successively with 50 ml
of a 1N aqueous hydrochloric acid solution, 50 ml of a
saturated sodium hydrogencarbonate solution and 50 ml
25 of a saturated sodium chloride solution, dried over
sodium sulphate and concentrated under reduced
pressure.
l.07 g of product are obtained in the form of a viscous
CA 02270478 1999-04-29
61
oil which is used as is in the following stage.
Yield = 100%
l4.2 . (S) -N- [2- [ [ [1- [ (4-Methoxypiperidin-1-yl) -
carbonyl]-4-(5-methyl-1H-imidazol-4-yl)-
butyl ] amino] sulphonyl ] - 6 - thi en- 2 -ylphenyl ] -
propanamide hydrochloride (l: l)
1.07 g (1.2 mmol) of (S) -1- [2- (3-ethyl-
1,1-dioxo-5-thien-2-yl-2H-1,2,4-benzothiadiazin-2-yl)-
5-[5-methyl-1-(triphenylmethyl)-1H-imidazol-4-yl]-
1-oxopentyl]-4-methoxypiperidine, in solution in a
mixture of 50 ml of acetic acid and 50 ml of water, are
heated for 6 hours at the reflux temperature and the
reaction mixture i.s concentrated under reduced
pressure. The residue is taken up in 150 ml of ethyl
acetate, washed successively with 50 ml of a saturated
sodium hydrogencarbonate solution and 50 ml of a
saturated sodium chloride solution, dried over sodium
sulphate and concentrated under reduced pressure. The
residue is purified by chromatography on a column of
silica gel, elution being carried out with a
methanol:dichloromethane (5:95) mixture.
0.43 g of product is obtained in the form of a base
which is taken up in 12 ml of a 0.1N solution of
hydrochloric acid in isopropanol and concentrated under
reduced pressure. The residue is purified by
chromatography on an RP 18 column, elution being
carried out with an acetonitrile:water (3:7) mixture.
After lyophilization, 0.34 g of product is obtained in
CA 02270478 1999-04-29
62
the form of a white powder.
Yield = 45%
Melting point = 126~C
[a]D = +87~ (c = 0.2, methanol)
Exarnnple 15 (Compound No. 47)
(S)-N-[2-[[I4-(5-Methyl-1H-imidazol-4-yl)-1-[(4-
methylenepiperidin-1-yl)carbonyl]-
butyl]amino]sulphonyl]-6-thien-2-ylphenyl]-propanamide
hydrochloride (1:1)
This compound is prepared according to the
method described in Example 14, from (S)-5-methyl-
a-[(4-methylenepiperidin-1-yl)carbonyl]-1-(triphenyl-
methyl)-1H-imidazole-4-butanamine hydrochloride and
2-[(1-chloropropylidene)amino]-3-thien-2-ylbenzene-
sulphonyl chloride.
Melting point = 1l5-l20~C
[a] D~ _ +54 ~ (c = 0 .2, methanol)
Example 16 (Compound No. 45)
(S) -N- [2- [ [ [1- [ (4-Cyclopropylpiperidin-1-yl) -carbonyl]
4-(5-methyl-1H-imidazol-4-yl)-butyl]amino]sulphonyl]-6
thien-2-ylphenyl]-propanamide hydrochloride (l: l)
16.1. (S)-4-Cyclopropyl-1-I2-(3-ethyl-l,l-dioxo-
5-thien-2-yl-2H-l,2,4-benzothiadiazin-2-yl)-
5-[5-methyl-1-(triphenylmethyl)-1H-imidazol-
4-yl]-1-oxopentyl]piperidine
0.5 ml (3.63 mmol) of triethylamine is added
CA 02270478 1999-04-29
63
dropwise at 0~C to a mixture of 0.38 g (1.1 mmol) of
2-[(1-chloropropylidene)amino]-3-thien-2-ylbenzene-
sulphonyl chloride and of 0.64 g (1.1 a~ol) of
(S)-a-[(4-cyclopropylpiperidin-1-yl)carbonyl]-5-methyl-
1-(triphenylmethyl)-1H-imidazole-4-butanamine
hydrochloride in 20 ml of dichloromethane. The
temperature of the mixture is allowed to return to room
temperature, stirring is continued for 2 hours at this
temperature and the reaction mixture is concentrated
under reduced pressure. The residue is taken up in
100 ml of ethyl acetate, washed successively With 50 ml
of a 1N aqueous hydrochloric acid solution, 50 ml of a
saturated sodium hydrogencarbonate solution and 50 ml
of a saturated sodium chloride solution, dried over
sodium sulphate and concentrated under reduced
pressure.
1.1 g of product are obtained in the form of a viscous
oil which is used as is in the following stage.
Yield = l00%
l6.2 . (S) -N- [2- [ [ [1- [ (4-Cyclopropylpiperidin-1-yl) -
carbonyl]-4-(5-methyl-1H-imidazol-4-yl)-
butyl] amino] sulphonyl] -6-thien-2-ylphenyl] -
propanamide hydrochloride (1:1)
1.1 g (l.l mmol) of (S)-4-cyclopropyl-
1-[2-(3-ethyl-1,1-dioxo-5-thien-2-yl-2H-1,2,4-benzo-
thiadiazin-2-yl)-5-[5-methyl-1-(triphenylmethyl)-
1H-imidazol-4-yl]-1-oxopentyl]piperidine, in solution
in a mixture of 25 ml of acetic acid and 25 m1 of
CA 02270478 1999-04-29
64
water, are heated for 4 hours at the reflux temperature
and the reaction mixture is concentrated under reduced
pressure. The residue is taken up in l50 ml of ethyl
acetate, washed successively with 50 ml of a saturated
sodium hydrogencarbonate solution and 50 ml of a
saturated sodium chloride solution, dried over sodium
sulphate and concentrated under reduced pressure. The
residue is purified by chromatography on a column of
silica gel, elution being carried out with a
methanol:dichloromethane (2:98 to 8:92) mixture.
0.528 g of product is obtained in the form of the base.
Yield = 80%
0.528 g of base is taken up in 10 m1 of a 0.1N solution
of hydrochloric acid in isopropanol and concentrated
under reduced pressure. The residue is purified by
chromatography on an RP 18 column, elution being
carried out with an acetonitrile:water (3:7) mixture.
After lyophilization, 0.27 g of product is obtained in
the form of a white powder.
Yield = 39%
Melting point = l08-l10~C
[a]D - +98~ (c = 0.2, methanol)
Example 17 (Compound No. 20)
(S) -N- [3' - (Ethylamino) -3- [ [ [1- [ (4-ethyl-piperidin-1-
yl)carbonyl]-4-(5-methyl-1H-imidazol-4-
yl)butyl] amino] sulphonyl] - [1,1' -biphenyl] -2-
yl]propanamide hydrochloride (1:2)
CA 02270478 1999-04-29
17 .1. (S) -1- [2- [7-Bromo-3-ethyl-5- (3-nitrophenyl) -
l,l-dioxo-2H-l,2,4-benzothiadiazin-2-yl]-
5-[5-methyl-1-(triphenylmethyl)-1H-imidazol-
4-yl]-1-oxopentyl]-4-ethylpiperidine
5 17.l.1. 5-Bromo-2-[(1-chloropropylidene)amino]-
3'-vitro[1,1'-biphenyl]-3-sulphonyl chloride
a) Pyridine salt of 5-bromo-3'-vitro-2-[(1-oxo-
propyl)amino][1,1'-biphenyl]-3-sulphonic acid
1.62 ml (18.6 mmol) of propionyl chloride are
10 added dropwise at 0~C under a nitrogen atmosphere to a
solution of 3.15 g (8.45 mmol) of 2-amino-5-bromo-3'-
vitro[l,l'-biphenyl]-3-sulphonic acid and 2.4 ml
(29.6 mmol) of pyridine in 10 ml of dichloromethane.
The temperature of the mixture is allowed to return to
15 room temperature and stirring is continued for 18
hours. The reaction mixture is concentrated under
reduced pressure.
The.residue is used as is in the following stage.
b) 5-Bromo-2-[(1-chloropropylidene)amino]
20 3'-vitro[l, l'-biphenyl]-3-sulphonyl chloride
The residue obtained above is dissolved in
20 ml of dichloromethane and 4.6 g (21.2 mmol) of
phosphorous pentachloride are added at 0~C under a
nitrogen atmosphere. The temperature of the mixture is
25 allowed to return to room temperature and the mixture
is kept stirring for 5 hours at this temperature. The
reaction mixture is concentrated under reduced pressure
and the residue is taken up in 150 ml of ether and
CA 02270478 1999-04-29
66
filtered through sintered glass. The filtrate is washed
with 2 times 100 ml of water and then 100 ml of a
saturated sodium chloride solution, dried over
magnesium sulphate and concentrated under reduced
pressure.
3 g of product are obtained in the form of a glassy oil
which is used as is in the following stage.
Yield = ??%
1? .1.2. (S) -1- (2- (?-Bromo-3-ethyl-5- (3-nitrophenyl) -
1,1-dioxo-2H-1,2,4-benzothiadiazin-2-yl]-
5-(5-methyl-1-(triphenylmethyl)-1X-imidazol-
4-yl]-1-oxopentyl]-4-ethylpiperidine
0.42 ml (3.5 mmol) of triethylamine is added
dropwise at 0~C to a mixture of 0.53 g (1.15 mmol) of
5-bromo-2-((1-chloropropylidene)amino]-
3'-vitro(1,1'-biphenyl]-3-sulphonyl chloride and of
0 . 58 g (I.02 mmol) of (S) -a- ( (4-ethylpiperidin-1-yl) -
carbonyl]-5-methyl-1-(triphenylmethyl)-1H-imidazole-
4-butanamine hydrochloride in 5 ml of dichloromethane.
The temperature of the mixture is allowed to return to
room temperature, stirring is continued for 18 hours at
this temperature and the reaction mixture is
concentrated under reduced pressure. The residue is
taken up in 100 ml of ethyl acetate, washed
successively with 50 ml of a 1N aqueous hydrochloric
acid solution, 50 ml of a saturated sodium hydrogen-
carbonate solution and 50 ml of a saturated sodium
chloride solution, dried over sodium sulphate and
CA 02270478 1999-04-29
67
concentrated under reduced pressure.
1 g of product is obtained in the form of a viscous oil
which is used as is in the following stage.
Yield = 100%
17.2. (S)-N-[5-Bromo-3-[[[1-[(4-ethylpiperidin-
1-yl)carbonyl]-4-(5-methyl-1H-imidazol-4-yl)-
butyl]smino]sulphonyl]-
3'-nitro(1,1'-biphenyl]-2-yllpropanamide
1 g (1 mmol) of (S) -1- [2- [7-bromo-3-ethyl-
5-(3-nitrophenyl)-l,l-dioxo-2H-1,2,4-benzothiadiazin-
2-yl]-5-E5-methyl-1-(triphenylmethyl)-1X-imidazol-
4-yl]-1-oxopentyl]-4-ethylpiperidine, in solution in a
mixture of 30 ml of acetic acid and 20 ml of water, is
heated for 8 hours at the reflux temperature and the
reaction mixture is concentrated under reduced
pressure. The residue is taken up in l00 ml of ethyl
acetate, washed with 50 ml of a saturated sodium
hydrogencarbonate solution, dried over sodium sulphate
and concentrated under reduced pressure. The residue is
purified by chromatography on a column of silica gel,
elution being carried out with a methanol:dichloro-
methane (5:95) mixture.
0.42 g of product is obtained in the form of a white
solid.
Yield = 60%
Melting point = 2l5~C
17.3. (S)-N-[3'-Amino-3-[[[1-[(4-ethylpiperidin-
1-yl)carbonyl]-4-(5-methyl-1H-imidazol-
CA 02270478 1999-04-29
68
4-yl) butyl] amino] sulphonyl] [1,1' -biphenyl] -
2-yl] propanamide
0.4 g (0.56 mmol) of (S) -N- [5-bromo-
3-[[[1-[(4-ethylpiperidin-1-yl)carbonyl]-4-(5-methyl-
1H-imidazol-4-yl)butyl]aminolsulphonyl]-
3' -nitro [l, l' -biphenyl] -2-yl] propanamide in 20 ml of
ethanol is hydrogenated for 10 hours at room
temperature at 0.35 MPa (50 psi) in the presence of
0.1 g of 10% palladium-on-charcoal. The reaction
mixture is filtered through celite and the filtrate is
concentrated under reduced pressure. The residue is
taken up in 100 ml of ethyl acetate, washed with 50 ml
of a saturated sodium hydrogencarbonate solution, dried
over sodium sulphate and concentrated under reduced
pressure.
0.33 g of product is obtained in the form of a white
solid.
Yield = 100%
Melting point = 160~C
17.4. (S)-N-[3'-(Sthylamino)-3-[[[1-[(4-ethyl-
piperidin-1-yl)carbonyl]-4-(5-methyl-
1H-imidazol-4-yl) butyl] amino] sulghonyl] -
[1,1'-biphenyl]-2-yl]propanamide
hydrochloride (1:2)
0.043 ml (0.78 mmol) of acetaldehyde and
70 mg of 10% palladium-on-charcoal are added to 0.33 g
(0.56 mmol) of (S) -N- [3' -amino-3- [ [ [1- [ (4-ethyl-
piperidin-1-yl)carbonyl]-4-(5-methyl-1H-imidazol-4-yl)-
CA 02270478 1999-04-29
69
butyl] amino] sulphonyl] [1,1' -biphenyl] -2-yl] propanamide
in 10 ml of ethanol and the mixture is stirred for
8 hours at room temperature under a pressure of
0.35 MPa (50 psi). The reaction mixture is filtered
through celite, 4 ml of a O.1N solution of hydrochloric
acid in isopropanol are added to the filtrate and the
filtrate is concentrated under reduced pressure. The
residue is purified by chromatography on an RP18
column. elution being carried out with an aceto-
nitrile:water (3:7) mixture.
After lyophilization, 0.2 g of product is obtained in
the form of a white powder.
Yield = 53%
Melting point = 163~C
[a]D = +l30~ (c = 0.2, methanol)
Exaa~le 18 (Compound No. 29)
(S) -N- [3- [ [ [1- [ (4-Ethylpiperidin-1-yl) -carbonyl] -4- (5-
methyl-1H-imidazol-4-yl)-butyl]amino]sulphonyl)[1,1'-
biphenyl]-2-yl]-2-(2-methoxyethoxy)acetamide
hydrochloride (l: l)
l8.1. (S) -2-Amino-N- [1- [ (4-ethylpiperidin-1-yl) -
carbonyl]-4-(5-methyl-1-(triphenylmethyl)-
1H-imidazol-4-yl]butyl][1,1'-biphenyl]-
3-sulphonamide
0.365 ml (2.61 mmol) of triethylamine is
added dropwise at 0~C under an argon atmosphere to a
mixture of 0.5 g (0.87 mmol) of (S)-5-ethyl-
CA 02270478 1999-04-29
a-[(4-ethylpiperidin-1-yl)carbonyl]-1-(triphenyl-
methyl)-1H-imidazole-4-butanamine hydrochloride and of
0.281 g (1.05 mmol) of 2-amino[1,1'-biphenyl]-3-
sulphonyl chloride in solution in 10 ml of
5 dichloromethane. The mixture is left stirring for
1 hour at this temperature and is then concentrated
under reduced pressure. The residue is taken up in
50 ml of ethyl acetate, washed successively with 20 ml
of a 1N hydrochloric acid solution, 20 ml of a
10 saturated sodium hydrogenearbonate solution and 20 ml
of a saturated sodium chloride solution and then dried
over magnesium sulphate. Finally, the organic phase is
filtered and concentrated to dryness. The residue is
purified by chromatography on a column of silica gel,
15 elution being carried out with a methanol:dichloro-
methane (1:99 then 3:97) mixture.
0.53 g of product is obtained in the form of a white
solid.
Yield = 79%
20 18.2. (S)-N-[3-[[[1-[(4-Ethylpiperidin-1-yl)-
carbonyl]-4-(5-methyl-1H-imidazol-4-yl)-
butyl] amino] sulphonyl] [l, l' -biphenyl] -2-yl] -
2-(2-methoxyethoxy)acetamide hydrochloride
(1:1)
25 2 g (13 amnol) of 2- (2-methoxyethoxy) acetyl
chloride are added at room temperature under argon to
1 g (1.3 mmol) of (S)-2-amino-N-[1-[(4-ethylpiperidin-
1-yl)carbonyl]-4-[5-methyl-1-(triphenylmethyl)-
CA 02270478 1999-04-29
71
1H-imidazol-4-yl]butyl][l,l'-biphenyl]-3-sulphonamide
in solution in 10 ml of dimethylacetamide. The reaction
mixture is left stirring at this temperature for
0.5 hour and is then cooled with an ice bath. l00 ml of
ethyl acetate and l00 ml of a 1N aqueous hydrochloric
acid solution are added and the organic phase is
recovered. It is washed successively with 50 ml of a
saturated sodium hydrogencarbonate solution and 50 ml
of a saturated sodium chloride solution and is then
concentrated under vacuum. The residue is taken up in
an acetic acid: water:tetrahydrofuran (2:1:1) mixture,
the mixture is heated for 3 hours at 80~C and is
concentrated under vacuum. The residue is taken up in
l00 ml of ethyl acetate, washed successively with 50 ml
of a 1N hydrochloric acid solution, 50 m1 of a
saturated sodium hydrogencarbonate solution and 50 ml
of a saturated sodium chloride solution and is then
dried over magnesium sulphate. Finally, the organic
phase is filtered and concentrated under vacuum. The
residue is purified by chromatography on a column of
silica gel, elution being carried out with a
dichloromethane:methanol (98:2 then 90:l0) mixture.
0.54 g of product is obtained in the form of the base.
The hydrochloride is prepared in 10 ml of a O.1N
solution of hydrochloric acid in isopropanol.
After lyophilization, 0.57 g of product is obtained.
Yield = 64.6%
Melting point = 98~C
CA 02270478 1999-04-29
72
[a]D - +55.5~ (c = 0.2, methanol)
Example 19 (Compound No. 30)
(S)-N-[2-Cyclopentyl-6-[[[1-[(4-ethyl-piperidin-1-
yl)carbonyl]-4-(5-methyl-1H-imidazol-4-
yl) butyl] amino] sulphonyl] -phenyl] -N' -ethylurea
hydrochloride (l: l)
19.1. (S)-2-amino-3-cyclopentyl-N-[1-[(4-ethyl-
piperidin-1-yl)carbonyl]-4-[5-methyl-1-(tri-
phenylmethyl)-1H-imidazol-4-yl]butyl]benzene-
sulphonamide
0.95 ml (6.75 mmol) of triethylamine is added
dropwise at 0~C to a solution of 0.70 g (2.7 mmol) of
2-amino-3-cyclopentylbenzenesulphonyl chloride and of
1.54 g (2.7 mmol) of (S)-5-methyl-a-[(4-ethylpiperidin-
1-yl)carbonyl]-1-(triphenylmethyl)-1H-imidazole-
4-butanamine hydrochloride in 6 ml of dichloromethane.
The reaction mixture is left stirring for 5 hours at
this temperature and is then concentrated under reduced
pressure. The residue is taken up in l00 ml of ethyl
acetate, washed successively with 50 ml of a 1N
hydrochloric acid solution, 50 ml of a saturated sodium
hydrogencarbonate solution and 50 ml of a saturated
sodium chloride solution, dried over magnesium sulphate
and concentrated under reduced pressure. The residue is
purified by chromatography on a column of silica gel,
elution being carried out with a
dichloromethane:methanol (98:2) mixture.
CA 02270478 1999-04-29
73
1,8 g of product are obtained in the form of a viscous
oil.
Yield = 88%
19 . 2 . (S) -N- [2-Cyclopentyl-6- [ [ [1- [ (4-ethyl-
piperidin-1-yl)carbonyl]-4-[5-methyl-1-(tri-
phenylmethyl)-1H-imidazol-4-yl]butyl]amino]-
sulphonyl]phenyl]-N'-ethylurea
A solution of 0.7S g (1 mmol) of (S)-2-amino-
3-cyclopentyl-N-[1-[(4-ethylpiperidin-1-yl)carbonyl]-
4-[5-methyl-1-(triphenylmethyl)-1H-imidazol-4-yl]-
butyl]benzenesulphonamide and of 0.32 ml (4 mmol) of
ethyl isocyanate in dimethylformamide is heated for
38 hours at 50~C and then the reaction mixture is
concentrated under reduced pressure. The residue
obtained is purified by chromatography on a column of
silica gel, elution being carried out with a
methanol:dichloromethane (2:98) mixture.
0.53 g of product is obtained in the form of a white
solid.
Yield = 65%
Melting point = 1l5~C
19 . 3 . (S) -N- [2-Cyclopeatyl-6- [ [ [1- [ (4-ethyl-
piperidin-1-yl)carbonyl]-4-(5-methyl-
1H-imidazol-4-yl) butyl] amino] sulphonyl] -
phenyl]-N'-ethylurea hydrochloride (1:1)
A mixture of 0.53 g (0.64 mmol) of
(S)-N-[2-cyclopentyl-6-[[[1-[(4-ethylpiperidin-1-yl)-
carbonyl]-4-[5-methyl-1-(triphenylmethyl)-1H-imidazol-
CA 02270478 1999-04-29
74
4-yi]butyl]aminolsulphonyl]phenyl]-N'-ethylurea and of
0.2 g of 10% palladium-on-charcoal in 8 ml of a 0.1N
solution of hydrochloric acid in isopropanol is stirred
for 40 hours at room temperature under a pressure of
0.35 MPa (50 psi). The mixture is filtered through
celite and the filtrate is concentrated under reduced
pressure. The residue thus obtained is purified on an
RP 18 column, elution being carried out with an
acetonitrile:water (4:6) mixture.
After lyophilization, 0.30 g of product is obtained.
Yield = 77%
Melting point = 147~C
[a]D~ _ +86~ (c = 0.2, methanol)
Example 20 (Compound No. 70)
(S)-N-[3-[[[4-(5-Chloro-1H-imidazol-4-yl)-1-[(4-
ethylpiperidin-1-yl)carbonyl]butyl]-
amino] sulphonyl] [1,1' -biphenyl] -2-yl] -propanamide
hydrochloride (l:l)
20.1. (S) -N- [3- [ [ [1- [ (4-Ethylpiperidin-1-yl) -
carbonyl] -4- (1H-imidazol-4-yl)butyl] amino] -
sulphonyl ] [ l , l' -bipheayl ] - 2 -yl ] propanamide
20.1.1. 2-[(1-Chloropropylideae)-
amino][1,I'-biphenyl]-3-sulphonyl chloride
It is prepared according to the method
described in Example 14.1.1., from 2-amino-
[1,1'-biphenyl]-3-sulphonic acid.
The product is obtained in the form of a viscous oil
CA 02270478 1999-04-29
which is used as is in the following stage.
20.l.2. (S)-N-I3-[[[1-[(4-Ethylpiperidin-1-yl)-
carbonyl] -4- (1H-imidazol-4-yl) butyl] amino] -
sulphonyl][1.1'-biphenyl]-2-yllpropanamide
5 2.2 ml (Z5.84 mmol) of triethylamine are
added dropwise at 0~C under nitrogen to a mixture of
1.8 g (5.3 mmol) of 2-[(1-chloropropylidene)-
amino][1,1'-biphenyl]-3-sulphonyl chloride and of 2.7 g
(4.8 mmol) of (S)-a-[(4-ethylpiperidin-1-yl)carbonyl]-
10 1-(triphenylmethyl)-1H-imidazole-4-butanamine
hydrochloride in 30 ml of dichloromethane. The
temperature of the mixture is allowed to return to room
temperature, stirring is continued for 18 hours at this
temperature and the reaction mixture is concentrated
15 under reduced pressure. The residue is taken up in
150 ml of ethyl acetate, washed successively with
100 ml of a 1N aqueous hydrochloric acid solution,
50 ml of a saturated sodium hydrogencarbonate solution
and 50 ml of a saturated sodium chloride solution,
20 dried over sodium sulphate and concentrated under
reduced pressure.
The residue is taken up in a mixture of 60 ml of acetic
acid and of 40 ml of water, the mixture is heated for
6 hours at the reflux temperature and the reaction
25 mixture is then concentrated under reduced pressure.
The residue is takes up is 200 ml of ethyl acetate,
washed successively with 50 ml of a saturated sodium
hydrogencarbonate solution and SO ml of a saturated
CA 02270478 1999-04-29
76
sodium chloride solution, dried over sodium sulphate
and concentrated under reduced pressure. The residue is
purified by chromatography on a column of silica gel,
elution being carried out with a methanol:dichloro-
methane (5:95) mixture.
2.2 g of product are obtained in the form of a viscous
oil.
Yield = 81%
20.2. (S)-N-[3-[[[4-(5-Chloro-1H-imidazol-4-yl)-
1-[(4-ethylpiperidin-1-yl)carbonyl]butyl]-
amino] sulphonyl] [l, l' -biphenyl] -2-yl] -
propanamide hydrochloride (1:1)
A solution of 0.56 g (1 mmol) of
(S) -N- [3- [ [ [1- [ (4-ethylpiperidin-1-yl) carbonyl] -
4-(1H-imidazol-4-yl)butyl]amino]-
sulphonyl] [1,1' -biphenyl] -2-yl] propanamide and of
0.l16 g (1.1 mmol) of N-chlorosuccinimide in 2 ml of
dimethylformamide is stirred for 5 hours at 0~C and the
reaction mixture is then concentrated under reduced
pressure. The residue is purified by chromatography on
a column of silica gel, elution being carried out with
a methanol:dichloromethane (2:98) mixture.
0.3 g of product is obtained in the form of the base.
Yield = 50%
The base is taken up in 15 ml of a 0.1N solution of
hydrochloric acid is isopropanol and concentration is
carried out under reduced pressure. The residue thus
obtained is purified on an RP 18 column, elution being
CA 02270478 1999-04-29
77
carried out with an acetonitrile:water (6:4) mixture.
After lyophilization, 0.30 g of product is obtained.
Yield = 94%
Melting point = l00~C
(a]p~ _ +l02~ (c = 0.2, methanol)
Example 21 (Compound No. 23)
(S) -N- (2- ( [ [1- [ (4-Ethylpiperidin-1-yl) -carbonyl] -4- (5-
methyl-1H-imidazol-4-yl)-butyl]amino]sulphonyl]-6-
pyridin-2-ylphenyl]-propanamide hydrochloride (1:2)
21.l. (S) -N- I4-Bromo-2- [ ( [1- [ (4-ethylpiperidin-
1-yl)carbonyl]-4-[5-methyl-1-(triphenyl-
methyl)-1H-imidazol-4-yl]butyl]amino]-
sulphonyl]-6-iodophenyl]propanamide
2.24 g (4.4 mmol) of 2-[bis(1-oxopropyl)-
amino]-5-bromo-3-iodobenzenesulphonyl chloride and
then, dropwise, 1.84 ml (13.2 mmol) of triethylamine
are successively added at 0~C to a solution of 2.28 g
(4 mmol) of (S)-5-methyl-a-[(4-ethylpiperidin-1-yl)-
carbonyl]-1-(triphenylmethyl)-1H-imidazole-4-butanamine
hydrochloride in 25 ml of dichloromethane. The mixture
is left stirring for 6 hours at this temperature and is
concentrated under reduced pressure. The residue is
taken up in 200 ml of ethyl acetate, washed
successively with 100 ml of a 1N aqueous hydrochloric
acid solution, 100 ml of a saturated sodium
hydrogencarbonate solution and 100 ml of a saturated
sodium chloride solution, dried over sodium sulphate
CA 02270478 1999-04-29
78
and concentrated under reduced pressure. The residue is
taken up in l50 ml of tetrahydrofuran, a stream of
gaseous ammonia is passed through the solution at 0~C
for 2 hours and the mixture is concentrated under
reduced pressure. The residue is purified by
chromatography on a column of silica gel, elution being
carried out with a dichloromethane:methanol (98:2)
mixture.
2.64 g of product are obtained in the form of a glassy
oil.
Yield = 70%
21.2. (S)-N-[4-Bromo-2-[[[1-[(4-ethylpiperidin-
1-yl)carbonyl]-4-[5-methyl-1-(triphenyl-
methyl)-1H-imidazol-4-yl)butyl]amino]-
sulphonyl]-6-pyridin-2-ylphenyl]propanamide
A mixture of 1.9 g (2 mmol) of
(S)-N-[4-bromo-2-[[[1-[(4-ethylpiperidin-1-yl)-
carbonyl]-4-[5-methyl-1-(triphenylmethyl)-1H-imidazol-
4-yl] butyl] amino] sulphonyl] -6-iodophenyl] propanamide,
0.883 g (2.4 mmol) of 2-(tributylstannyl)pyridine,
0.1 g (0.17 mmol) of bis(dibenzylideneacetone)-
palladium(0), 0.033 g (0.17 mmol) of copper iodide and
0.98 g (0.34 a~ol) of triphenylarsine in 4 ml of
dimethylformamide is heated at 80~C under argon for
5 hours. The reaction mixture is taken up in l50 ml of
ethyl acetate and washed with two times 100 m1 of a 10%
aqueous ammonia solution and then with 50 ml of a
saturated sodium chloride solution. The organic layer
CA 02270478 1999-04-29
79
is dried over sodium sulphate and concentrated under
reduced pressure. The residue is purified by
chromatography on a column of silica gel, elution being
carried out with a dichloromethane:methanol (98:2)
mixture.
1.15 g of product are obtained in the form of a viscous
oil.
Yield = 64%
21.3. (S)-N-[4-Bromo-2-[[[1-[(4-ethylpiperidin
1-yl)carbonyl]-4-(5-methyl-1H-imidazol-4-yl)
butyl]amino]sulphonyl]-6-pyridin-2-ylphenyl]-
propanamide
1.14 g (1.26 mmol) of (S)-N-[4-bromo-
2-[[[1-[(4-ethylpiperidin-1-yI)carbonyl]-4-[5-methyl-
1-(triphenylmethyl)-1H-imidazol-4-yl)butyl]amino]-
sulphonyl]-6-pyridin-2-ylphenyl]propanamide in a
mixture of 30 ml of acetic acid and 15 ml of water are
heated for 1 hour at the reflux temperature. The
reaction mixture is concentrated under reduced pressure
and the residue is taken up in 150 ml of ethyl acetate.
The organic layer is washed successively with 50 ml of
a saturated sodium hydrogencarbonate solution and 50 m1
of a saturated sodium chloride solution, dried over
sodium sulphate and concentrated under reduced
pressure.
The residue is purified by chromatography on a column
of silica gel, elution being carried out With a
methanol:dichloromethane (5:95) mixture.
CA 02270478 1999-04-29
0.66 g of product is obtained in the form of a glassy
oil.
Yield = 79.5%
21.4 . (S) -N- [2- [ [ [1- [ (4-Ethylpiperidin-1-yl) -
5 carbonyl]-4-(5-methyl-1H-imidazol-4-yl)-
butyl]amino]sulphonyl]-6-pyridin-2-ylphenyl]-
propanamide hydrochloride (1:2)
A mixture of 0.65 g (0.98 mmol) of
(S) -N- [4-bromo-2- [ [ [1- [ (4-ethylpiperidin-1-yl) -
10 carbonyl]-4-(5-methyl-1H-imidazol-4-yl)butyl]amino]-
sulphonyl]-6-pyridin-2-ylphenyl]propanamide, l.24 g
(20 mmol) of ammonium formate and 0.065 g of palladium-
on-charcoal in 10 ml of methanol containing 0.2 ml of
acetic acid is heated for 3 hours at the reflex
15 temperature and then the reaction mixture is
concentrated under reduced pressure. The residue is
taken up in l00 ml of ethyl acetate, washed
successively with 50 ml of a saturated sodium hydrogen-
carbonate solution and 50 ml of a saturated sodium
20 chloride solution, dried over sodium sulphate and
concentrated under reduced pressure. The residue is
purified by chromatography on a column of silica gel,
elution being carried out with a methanol:dichloro-
methane (5:95) mixture.
25 0.55 g of base is obtained in the form of a viscous
oil.
Yield = 96%
The base is taken up in 25 ml of a O.1N solution of
CA 02270478 1999-04-29
81
hydrochloric acid in isopropanol and concentrated uader
reduced pressure. The residue thus obtained is purified
on an RP 18 column, elution being carried out with an
acetonitrile:water (6:4) mixture.
0.44 g of product is obtained.
Yield = 68%
Melting point = 138-l44~C
[a]D~ _ +l21~ (c = 0.2, methanol)
Example 22 (Compound No. 22)
(S) -N- [2- [ [ [1- [ (4-Ethylpiperidin-1-yl) -carbonyl) -4- (5-
methyl-1H-imidazol-4-yl)-butyl]amino]sulphonyl]-4-
fluoro-6-thien-2-yI-phenyl]propanamide hydrochloride
(1:1)
22.1. 2-[Bis(1-oxopropyl)amino]-5-fluoro-3-thien-2-
ylbenzenesulphonyl chloride
22.1.1. N,N-Diethylethanamine salt of 2-[bis(1-oxo-
propyl)amino]-5-fluoro-3-thien-2-ylbenzene-
sulphonic acid
A mixture of 10.92 g (40 mmol) of 2-amino-5-
fluoro-3-thien-2-ylbenzenesulphonic acid aad of 5.6 ml
(40 mmol) of triethylamine in 77 ml (600 mmol) of
propionic anhydride is heated for 24 hours at the
reflex temperature and then the reaction mixture is
concentrated under reduced pressure. The residue is
crystallized from an ethyl acetate: ether mixture.
16.9 g of product are obtained.
Yield = 90%
CA 02270478 1999-04-29
82
Melting point = 239~C
22.1.2. 2-[His(1-oxopropyl)amino]-5-fluoro-3-thien-
2-ylbenzenesulphonyl chloride
14.22 g (68.2 mmol) of phosphorous
pentachloride are added at 0~C under nitrogen to a
solution of 1.60 g (34.1 amnol) of N,N-diethylethanamine
salt of 2-Ibis(1-oxopropyl)amino]-5-fluoro-3-thien-
2-ylbenzenesulphonic acid in 60 ml of dichloromethane.
The mixture is kept stirring for 5 hours at this
temperature, the temperature is allowed to rise to room
temperature and stirring is continued for 1 hour at
this temperature. 200 ml of ether are added, the
mixture is filtered and the filtrate is concentrated
under reduced pressure. The residue is taken up in
800 ml of ether, filtered and concentrated under
reduced pressure. The residue is purified by
chromatography on a Florisil' column, elution being
carried out with an ether: pentane (1:9 then l:l)
mixture.
6.4 g of product are obtained in the form of a viscous
oil.
Yield = 55%
22 .2 . (S) -N- [2- [ [ [1- [ (4-Ethylpiperidin-1-yl) -
carbonyl)-4-(5-methyl-1H-imidazol-4-yl)-
butyl]amino]sulphonyl]-4-fluoro-6-thien-2-yl-
phenyl]propanamide hydrochloride (1:1)
6.16 g (18.1 mmol) of 2-[bis(1-oxopropyl)-
amino]-5-fluoro-3-thien-2-ylbenzenesulphonyl chloride
CA 02270478 1999-04-29
83
and then 5.5 mmol of triethylamine are added under
nitrogen at 0~C to a solution of 9.83 g (17.2 mmol) of
(S)-a-[(4-ethylpiperidin-1-yl)carbonyl]-5-methyl-
1-(triphenylmethyl)-1H-imidazole-4-butanamine
hydrochloride in 60 ml of dichloromethane. The
temperature of the reaction mixture is allowed to
slowly return to room temperature, stirring is
continued at this temperature for 15 hours and the
reaction mixture is concentrated under reduced
pressure. The residue is taken up in 300 ml of ethyl
acetate and washed successively with 300 ml of a 1N
aqueous hydrochloric acid solution, 300 ml of a
saturated sodium hydrogencarbonate solution and then
300 ml of a saturated sodium chloride solution. The
organic layer is dried over sodium sulphate, filtered
and concentrated under reduced pressure.
The residue is heated for 12 hours in 225 ml of an
acetic acid: water (2:1) mixture and concentrated under
reduced pressure. The residue is taken up in 400 ml of
dichloromethane and washed successively with 200 ml of
a saturated sodium hydrogencarbonate solution and then
200 ml of a saturated sodium chloride solution. The
organic layer is dried over sodium sulphate, filtered
and concentrated under reduced pressure. The residue
thus obtained is purified by chromatography on a column
of silica gel, elution being carried out with a
dichloromethane:methanol (96:4) mixture.
7.7 g of product are obtained in the form of the base.
CA 02270478 1999-04-29
84
Yield = 74%
Melting point = 145-150~C
The hydrochloride is prepared by adding 45.l ml of a
0.1N solution of hydrochloric acid is isopropanol to
2.5 g (4.5 mmol) of base.
2.7 g of product are obtained in the form of the
hydrochloride.
Melting point = 145~C
[a] D~ - +112 ~ (c = 0 .2, methanol)
Example 23 (Compound No. 53)
(S) -N- [2- [ [ [1- [ [4- (Difluoromethylene) -piperidin-1-
yl] carbonyl] -4- (5-methyl-1H-imidazol-4-yl)butyl] amino] -
sulphonyl]-6-thien-2-ylphenyl]propanamide hydrochloride
(1:1)
This product is prepared according to the
procedure described is Example 19, from 0.8l g
(1.36 mmol) of (S)-5-methyl-a-[[4-(difluoromethylene)-
piperidin-1-yl]carbonyl]-1-(triphenylmethyl)-
1H-imidazole-4-butanamine hydrochloride and from 0.47 g
(1.36 mmol) of 2-[(1-chloropropylidene)amino]-3-thien-
2-ylbenzenesulphonyl chloride.
0.58 g of product is obtained in the form of a white
powder.
Yield = 67%
Melting point = 144-l45~C
[a]D~ - +l07.9~ (c = 0.2, methanol)
CA 02270478 1999-04-29
Example 24 (Compound No. 25)
(S)-N-[6-Cyclopentyl-2-[[[1-[(4-ethyl-piperidin-1-
yl)carbonyl]-4-(5-methyl-1H-imidazol-4-yl)butyl]-
amino]sulphonyl]-phenyl]propanamide hydrochloride (l: l)
5 24.l. 3-Cyclopentyl-2-(diacetylamino)benzene-
sulphonyl chloride
24.l.1. N,N-Diethylethanamine salt of 3-cyclopentyl-
2-(diacetylamino)benzenesulphonic acid
6.5 g (19 mmol) of N,N-diethylethanamine salt
10 of 2-amino-3-cyclopentylbenzenesulphonic acid, in
solution in acetyl chloride, are heated for 48 hours at
the reflex temperature and then the reaction mixture is
concentrated under reduced pressure.
8.22 g of product are obtained, which product is used
15 as is in the following stage.
Yield = 95%
Melting point = l86~C
24.1.2. 3-Cyclopentyl-2-(diacetylamino)benzene-
sulphonyl chloride
20 This compound is prepared according to the
procedure described in Example 22.l, from 8.2 g
(18.1 mmol) of N,N-diethylethanamine salt of 3-cyclo-
pentyl-2-(diacetylamino)benzenesulphonic acid.
3.81 g of product are obtained in the form of a viscous
25 oil which is used as is in the following stage.
Yield = 57%
24.2. (S) -N- [6-Cyclopentyl-2- [ [ [1- [ (4-ethyl-
piperidin-1-yl)carbonyl]-4-(5-methyl-
CA 02270478 1999-04-29
86
1X-imidazol-4-yl)butyl]amino]sulphonyl]-
phenyllpropanamide hydrochloride (l: l)
This product is prepared according to the
procedure described in Example 22.2, from 0.743 g
(2 mmol) of 3-cyclopentyl-2-(diacetylamino)benzene-
sulphonyl chloride and from 1.14 g (2 mmol) of
(S)-a-[(4-ethylpiperidin-1-yl)carbonyl]-5-methyl-
1-(triphenylmethyl)-1H-imidazole-4-butanamine
hydrochloride.
0.73 g of product is obtained.
Yield = 60%
Melting point = 124~C
[a]D~ _ +89~ (c = 0.2, methanol)
Exan~le 25 (Compound No. 64)
(S) -N- [2- [ [ [4- (5-Methyl-1H-imidazol-4-yl) -1- [ (5-
oxohexahydro-1H-1,4-diazepin-1-yl)carbonyl]butyl]-
amino]sulphonyl]-6-thien-2-yl-phenyl]propanamide
hydrochloride (1:1)
25.1. 1-[2-[[(2-Amino-3-thien-2-ylphenyl)-
sulphonyl]amino)-5-[5-methyl-1-(triphenyl-
methyl)-1X-imidazol-4-yl]-1-oxopentyll-hexa-
hydro-5X-1,4-diazepin-5-one
0.6l8 g (2.26 mmol) of 2-amino-3-thien-2-
ylbenzenesulphonyl chloride in 80 ml of dichloromethane
is placed wader argon and 1.29 mg (2.26 mmol) of 1-[2-
amino-5-[5-methyl-1-(triphenylmethyl)-1H-imidazol-4-
yl]-1-oxopentyl]-hexahydro-5H-1,4-diazepin-5-one
CA 02270478 1999-04-29
87
hydrochloride are added. The mixture is cooled to 0~C
with an ice bath and 0.94 ml (6.74 mmol) of
triethylamine is slowly added. The temperature of the
reaction mixture is allowed to return to room
temperature and stirring is continued overnight at this
temperature. 10 ml of a 0.5N aqueous hydrochloric acid
solution are then added and the organic phase is washed
with a 0.5N aqueous hydrochloric acid solution and then
with a saturated sodium chloride solution and dried
over magnesium sulphate. The residue is purified by
chromatography on a column of silica gel, elution being
carried out with a dichloromethane:methanol (97.5:2.5)
mixture.
l.33 g of product are obtained.
Yield = 76%
25.2. (S)-N-[2-[[[4-[5-Methyl-1-(triphenylmethyl)-
1H-imidazol-4-yl]-1-[(5-oxohexahydro-
1H-1,4-diazepin-1-yl)carbonyl]butyl]amino]-
sulphoayl]-6-thien-2-ylphenyl]propanamide
1.33 g (1.72 a:mol) of 1- [2- [ [ (2-amino-
3-thien-2-ylphenyl)sulphoayl]amino]-5-[5-methyl-1-(tri-
phenylmethyl)-1H-imidazol-4-yl]-1-oxopeatyl]-hexahydro-
5H-1,4-diazepin-5-one are placed in 1.3 ml of dimethyl-
acetamide and 0.3 ml (3.44 mmol) of propionyl chloride
is added. The mixture is left stirring for 2 hours and
then ethyl acetate is added. Evaporation is carried out
to dryness and the residue is taken up in
dichloromethane. The organic layer is washed with 2
CA 02270478 1999-04-29
88
times 20 ml of a 1N aqueous hydrochloric acid solution,
dried over magnesium sulphate and evaporated.
1.4 g of product are obtained, which product is used as
is in the following stage.
Yield = l00%
25.3. (S)-N-[2-[[[4-(5-Methyl-1H-imidazol-4-yl)-
1-I(5-oxohexahydro-1H-1,4-diazepin-1-yl)-
carbonyl]butyl]amino]sulphonyl]-6-thien-2-yl-
phenyl]propanamide hydrochloride (l: l)
0.25 g (0.31 mmol) of (S) -N- [2- [ [ I4- [5-
methyl-1-(triphenylmethyl)-1H-imidazol-4-yl]-1-[(5-
oxohexa-hydro-1H-1,4-diazepin-1-
yl) carbonyl] butyl] amino] -sulphonyl] -6-thien-2-
ylphenyl]propanamide are placed in 4 ml of
tetrahydrofuran, and 2 ml of acetic acid and 2 ml of
water are added. The mixture is heated overnight at
50~C, evaporated to dryness and the residue is taken up
in 100 ml of dichloromethane. The solution is washed
with an aqueous sodium hydrogencarbonate solution and
dried over magaesium sulphate. It is filtered aad the
residue is purified by chromatography on a column of
silica gel, elution being carried out with a dichloro-
methane: methanol (90:10) mixture.
The base is dissolved in a O.1N solution of
hydrochloric acid in isopropanol and the product is
purified by chromatography on an RP 18 silica column,
CA 02270478 1999-04-29
x
89
elution being carried out with an acetonitrile/water
mixture.
0.026 g of product are obtained in the form of the
hydrochloride is obtained.
Yield = 14~
Melting point = 155~C
[a]D~ - 80~ (c = 0.1, methanol)
Example 26 (Compound No. 65)
(S)-N-Cyclopentyl-N,5-dimethyl-a-[[L2-[(1-oxopropyl)-
amino]-3-thien-2-yl-phenyl]sulphonyl]amino]-1H-
imidazole-4-pentanamide hydrochloride (l: l)
26.1. (S)-a-[[(2-Amino-3-thien-2-ylphenyl)-
sulphonyl]amino]-N-cyclopentyl-N,5-dimethyl-
1-(triphenylmethyl)-1H-imidazole-4-pentan-
amide
1 g (3.66 mmol) of 2-amino-3-thien-2-
ylbenzenesulphonyl chloride and then l.12 ml
(7.85 amnol) of triethylamine in 5 ml of dichloromethane
are added successively at 0~C under nitrogen to a
solution of l.95 g (3.5 mmol) of (S)-a-amino-N-cyclo-
pentyl-N,5-dimethyl-1-(triphenylmethyl)-1H-imidazole-
4-pentanamide hydrochloride in 15 ml of dichloro-
methane. The temperature of the reaction mixture is
allowed to return to room temperature and stirring is
continued for 15 hours at this temperature. The
reaction mixture is concentrated under reduced
pressure. The residue is taken up in 150 ml of ethyl
acetate and washed successively with 50 ml of a 1N
CA 02270478 1999-04-29
aqueous hydrochloric acid solution, with 50 ml of a
saturated sodium hydrogencarbonate solution and then
with 50 ml of a saturated sodium chloride solution. The
organic layer is dried over sodium sulphate. The
5 residue is purified by chromatography on a column of
silica gel, elution being carried out with a dichloro-
methane: methanol (97:3) mixture.
2.35 g of product are obtained in the form of an
amorphous solid.
10 Yield = 87%
Melting point = 102-107~C
26.2. (S)-N-Cyclopentyl-N,5-dimethyl-
a-[[[2-[(1-oxopropyl)amino]-3-thien-2-yl-
phenyl]sulphonyl]amino]-1H-imidazole-4-
15 pentanamide hydrochloride (l: l)
0.5l ml (5.9 mmol) of propionyl chloride is
added dropwise at room temperature to a solution of
2.23 g (2.94 mmol) of (S) -a- [ [ (2-amino-3-thien-2-yl-
phenyl)sulphonyl]amino]-N-cyclopentyl-N,5-dimethyl-
20 1-(triphenylmethyl)-1H-imidazole-4-pentanamide in
1.5 ml of dimethylacetamide. The mixture is left
stirring for 10 hours and then 150 ml of ethyl acetate
are added. The organic layer is washed with l50 ml of
water and then with 100 ml of a saturated sodium
25 chloride solution. The organic layer is dried over
sodium sulphate, filtered and concentrated under
reduced pressure. The residue is taken up in 60 ml of
ethyl acetate and 30 ml of water and is then heated at
CA 02270478 1999-04-29
91
the reflex temperature for 2 hours. l50 ml of
dichloromethane are then added and the organic layer is
washed successively with l00 ml of a saturated sodium
hydrogencarbonate solution and 100 ml of a saturated
sodium chloride solution. The organic layer is dried
over sodium sulphate, filtered and concentrated under
reduced pressure. The residue thus obtained is taken up
in 40 ml of a 0.1N solution of hydrochloric acid in
isopropanol and concentrated under reduced pressure.
The residue is purified by chromatography on a column
of silica gel, elution being carried out with an aceto-
nitrile:water (2:8) mixture.
1.l5 g of product are obtained.
Yield = 64%
Melting point = 140-l45~C
[a] D~ - + 103 ~ (c = 0 .2, methanol)
Example 27 (Compound No. 66)
(S) -N, 5-Dimethyl-a- [ [ [2- [ (1-oxopropyl) amino] -3-thien-2-
ylphenyl]sulphonyl]amino]-N-pyrrolidin-1-yl-1X-
imidazole-4-pentanamide hydrochloride (l: l)
27.1. (S)-a-[I(2-Amino-3-thien-2-ylphenyl)-
sulphonyl]amino]-N,5-dimethyl-N-pyrrolidin-
1-yl-1-(tripheaylmethyl)-1H-imidazole-
4-pentanamide
This compound is prepared according to the
procedure described in 26.l, from 0.8 g (2.94 u~mol) of
2-amino-3-thien-2-ylbenzenesulphonyl chloride and
CA 02270478 1999-04-29
92
1.56 g (2.8 mmol) of (S)-a-amino-N,5-dimethyl-
N-pyrrolidin-1-yl-1-(triphenylmethyl)-1H-imidazole-
4-pentanamide hydrochloride.
1.84 g of product are obtained in the form of an
amorphous solid.
Yield = 83%
Melting point = 102-106~C
27 .2 . (S) -N, 5-Dimethyl-a- [ [ [2- [ (1-oxopropyl) amino]
3-thien-2-ylpheayl]sulphonyl]amino]
N-pyrrolidin-1-yl-1H-imidazole-4-pentanamide
hydrochloride (l: l)
This compound is prepared according to the
procedure described in 26.2, from 1.8 g (2.37 mmol) of
(S) -a- [ [ (2-amino-3-thien-2-ylphenyl) sulphonyl] amino] -
N,5-dimethyl-N-pyrrolidin-1-yl-1-(triphenylmethyl)-
1F1-imidazole-4-pentanamide.
1 g of product is obtained.
Yield = 69%
Melting point = l54-160~C
[a]D~ - +1l9~ (c = 0.2, methanol)
Key to the table:
is the "salt" Column,
"HC1" corresponds to a hydrochloride and the ratio
between brackets is the (base: acid) ratio,
is the " [a] w~" column,
c = 0.2, methanol, except for the compound 1 (c =
CA 02270478 1999-04-29
93
0.22, methanol), for the compound 6 (c = 0.265,
methanol), for the compound 12 (c = 0.25,
methanol) and for the compounds 51, 54 and 56 (c =
0.4, methanol)
S'6L ; L'4ii ('(JH) ., ' H_ EH'OE(tHJIOtH~- EH'--~N- H_ t~' b
t6 ~ TT( (LJH) ~ H ~HJLHJOJ- ~HJ ~N-- H- ~HJ~ E
60T ~ b9i-09I ~iJH? ( H_ EHJtHJOJ- tFiJ-~N- H_ E~_ Z
';' 9L + ZZL-8ii (jJH? ( H' EHJOt(tHJIOtHJOJ- ~N - H- tHJ_ i
o _
o, 01 a ~ ~ ~ ~U~OfI ;~B$ H EM t8 t~l t ~ ~t t~l 'ON
0L ~ ~UI~~BW
r -~._.
0
r H
N
N 1W N
(Tl
/ NH~2!
HN~OS
0
ajqes
Melting
(a~
po
No R R Ry Ry R~ A Saltpoint
~
1
. ~ (
C~
HCl
5 -CN, -H '-'N~~ -CHzCH~CH3 -H ~ ~ (1:1)62-66
+ 135
CH,
~ HCl 114.3 0
H H '-N\ ' \ -COCHy -H
i1:1)13A
f, y -
-C
CH,
0
~ HCl
- N\~ -COCHZCHy -H ~ (1 144
1) + 105.5
-H ~ ' :
CH,
0
8 -CH3 -H - N'~ -COCHZCHZOH -H \ ( HC1 l50
i1:1)+ 68
cH,
_H - N -COCHZCH~ -H \ ~ (iCl)132
+ 10S
CH,
F
10 -CH3 -H "-N\ / \ -COCHzCH3 -H ~ '' (iCi) 130 + 113
CH,
F
'H~ 'xa~ ~
H_ 'HJ' St
(i'T) H- EHJ~HJOJ- ~N -
TZT + Z6T ZJH ~ ~ ~
i
'Ha 'H~
(W H- 'HJtHaOJ- H' cHJ- 6T
T) -
N
+ ZET TJH ~
tb
av (I cH~ 'HJtHJOJ- 'x~ (,I._
~ ~..~ H' 'HJ- ET
T~ H-
N ett + 9Et IJH I
0
o, ~ 3
'H~ ZT
00 ~ 'HJZHJOJ- _ H-
'Ha_
( N .-
) H
0 66 a 8ZT IJN ' 1
r
N
0 3
c
U H_
t~_ Lt
(I't1 ~ 'HJLHJOJ- H~
H- ~~~N _
+ ~6'C TJH ~ f
Sot /
to ~ , it ~a 'o
(aof
d ' ~
'b
,wod ups
of ip~ ~ ~ ~ _
6un18W
'H~
vT) H- 'HJrHJOJ. H_ 'HJ- 6T
~ f1
y U (T ~
iJH S
'x~ ~ H_ EHJ- et
(T ~ H- 'HJtHJOJ- ~~--~H'
tl
99 4 9ZT t~H ~ ~/
N
d'
O
IT'T) \ H- EHJiHJO~- N
H- 'HJ- LT
OET + ObT t~H I
/
o ~HJ _
r _
N
o 'HJ 'H~
d T'TI 'HJtHJOJ- ~'~ ~
H- 'HJ- 9T
H- H
9TT + 9ZT I \
i~H
LH t,~ tb 'ON
I'o) ~a f~
iulod~IeB
f 1') ~
o sulilBW
(t;T' 'Ha ~
H- EHOtHaoa- \~N - H- EW 5z
'
se i dzT ~
i~
H
EHa
~z't) ~ H- ~HJtHJOJ- \~M._. H_ ~~_ bL
SET t 9bT
TJH ~ N
~H~
~ H_ E~_ Z
~ ~ H- EHJiHaOJ- ~N--
~
~
t'H
TZT + bbT-AEt
N _N.
~H~
{T~T) ~ ~ (~-~HJiHJOJ- ~~N - H- EHJ- ZZ
Ztt + Sbt
ZJH
tHJ t TZ
tW I) J ~ H- tHJtHJOJ ~~N - H- HJ-
bTt 8Zt
' ZJH
o
v
_
~Ha
t ~ H- EHJtHJOJ- ~~N - H_ Ha' OZ
'
0I + E9t jJH i c
HN~J H ,
~Jo~ ~ y ~2!
i,21tM 'oN
a
~u~od Heg
d
z ~ p i BuisiaW
~
laNinn 7A
__ ~ A Salt
0
Na.A' R
'- N -COCHyCH; ~ (HC1) 145 + 85
~~ -F
26 -CH; -H
H
C 3 y
0
HCl N
- N\~~ COCHtCH3l; I l32 95
H (1:1) o
CH3 H
CHI
F
/ HC1
r N~ -COCH~OCH3 I Q 1;1)106 + 10l
~~ -H
2g -CH -H \
3
CH
N
~ N )yOCH3 -H ~ (lCl) 98 + SS.S '
~~ O(CH
-COCH
-CH; -N Z
Z
CH
a
30 -CH; -H - N~ -CONHCHzCH; -H ~ tlC1?147 + 86
~~
CH
CH3 / HCl
H N -COCHZCH3 -H ~ (1a1)114-120+ 96
~
-~
31 -CH; - '
~ H
C
Melting
No.R~ R'1 Rt R H p I
~ ' a
~
20
Salt
Point
(C)
S
32 -CH -H '- N -COCHZCH1 -H ' ~ ( icy)
1 198-150
OH +
9A
33 CH3 -H 'N COCH~CH) -H 'S~
12G
130
84
0
(lel)
N
N
J
O
34 -CH -H -N O~ -COCH~CH3 -H ~S ~
h~
CHI (r
(lCl)
126
+
8'T
o
0
i HC1
35 -CH -H - N S~ -COCHZCH3 -H ~ '
CHI (1:1)
190
+
149
N
S
36 -CH3 -H -N_, J-C-t3 -COCH~CH3 -H ~ ~ (lCl)168-1~2~ 99
F S
3~ -CH1 -H -" H~~F -COCHZCH3 H 1 ! ' (1~1)194-146t 103
Melting
~Q
~
00
p Salt
R point
(
)
No. Rl R'i RZ ~
(C)
R3
F
H ~ N -COCH2CH; -H
(1C11
~~ 194-14B
1
71
38 -CH3 -
F n
0
N
N
HC1 0
~ -COCH; -H I 154
39 -CH; -H IN~CF~ '
+ aaa
~ 90
(1:1i
H' o
D ~o
0
HCl
80
-H ~ -COCHyCH; -H I
(1:1) '
0 -CH -N~CF~ '~
146-150
+
;
4
HCl
121
41 -CH; -H -!3 ~ CF; -COCHZCH3 -H ' I
+
105
(1:1)
HC1
42 -CH; -H ---N'~ -COCHZCH; -H ' I
(1:1i 140 + 108
CH
3
i
Melting
R~ A Salt t ( o )
R 'on
NO. R1 R' R~ ~ C
1 )
43 -CHI -H
~ // -COCHyCH~ ~ 4 (lCl) 148 101.5
- N\ -H +
,~ F n
0
/ N
N
HC1
44 -CH3 -H -f1\~-~ -COCHZCH3 ~ (1:1) 133 + 122
-H
CHI a
N
_
S HC1
- -COCHzCH3 ~ ~ l1:1) 108-l10+ 98
-H
45 -CH -H N
7
HC1
-CH -H - N ~ -COCHZCH3 -H ~ ~ (1:1) 159-163+ 84
~'~
46 3 OH
~ HCl
47 -CH -H -N -COCHyCH3 -H ~ ~ (1:1) 115-120+ 54
J'-CH
3 _ .
I
CA 02270478 1999-04-29
l03
m (~ ..
o
rv o... ~ ~ ~ ~
.- o _
t a + +
p P N
a
ra
d ~ ~ N
r1
_ ~, a .. a ..
v~ ~ ~-~ x ~' _ '~ x ~
x
U
i
cn rs N \ / O
a x x s x x
x z x
a x r
z n U x U U
U
O ~-' ~ ' U
U G V
n
x w w w w w
x U
V
x
x~ x
x zJ
x x x x x
a ,
x x x
.. x
V U V V
N
Z a~ C ~n ~n ~n
CA 02270478 1999-04-29
104
a
n 0... I~ N m m ~ ni
d O N m ri
t t
t 1
f
O U1 N IV
C ~ ~ r ~ N
r ~ a r ? 1f1
V' O ~ m r~t rl
p Q t~1
,H -w rl
ri ~-1 .~ .m-i .a e-i n-1 n-1 .i ri ~-1
H x:.. s.~ x.. x', s'. x::
E
x s m m x x
r r r r r
x
x V Z x x x
n = n n a n
a n ~ ~ x x
x
U p U
.J ~ r r r
w w m w w w x
c1. a
I I I I ~~o
I I i ~J
5e x x x x x
v r r r
n n n n
.z a a a U
r r r
d r1 a~ m to e~ ao
Z 1I7 N M I!1 1f1 1I1
CA 02270478 1999-04-29
10S
~ r~) v r, r, a~ o
s 01 m 1f~ ~ P CD
t ~ t t t
_. N _. _... m P
m _ a~ o e~ rv
o -~ a .a
c U Q, ~ ,
~. ~~ o P P ~ ,-I
v ao e~~ .r
-a .r .a
r.y .1 r.~ H ., .~ .~ r~1 'r
. a '~
v i :: i :: s :: s :: s ::
x..
a
m c~ cn t~ t~
x x x x x x
~ ~ ~ ~
U U U U V V
eV n ~ v x n
U
V ~ V U V V
U U ~'' O
y zJ Z ~z
z z / ~z~/
i
x x s x x s
' ~ ' '
a a
0 0~ o ., n ",
z u, ~o ~o ~o ~o ~o
Melting~ (y
~ po
R4 p Saltpoint ( a
)
No. R2 ; (~C)
R1
R'1
HC1
CH -H ~ ( 1 190-145+ 103
-COCH 1(
65-CH; -H I ; ' _
2
CHI y
o
S
N-N - HC1
-COCH~CH3 -H ' il;l)154-i60~ 119
\ / 0
66-CH -H J
3
CH3
CH -H / I iCi 132 + 112
LOCH ~ '
67-CH=CH;-H
3 (
; )
-
H
I
C N
_ S HCi
- CH -H 120 + 88
~~ -COCH
68-CHZCH;-H N ; ~ l (1:11
Z
CHI
HC1
- CH -H 142 + 78
\~ -COCH
69-CH; -CH3N ; 1 ~ (1;1)
Z
CH3
-N -COCHZCH; -H / I (lCl
70-C1 -H \~ 100 + 102
y )
3
CA 02270478 1999-04-29
i
l07
CA 02270478 1999-04-29
4
l08
The compounds of the invention have formed
the subject of pharmacological studies which have
demonstrated their antithrombotic properties and their
advantage as substances possessing therapeutic
S activity.
1. Determination of the inhibition constants (Ri with
respect to thrombin
The following are deposited in each well of a
96-well microplate: 25 ~.l of a solution of compound to
be tested (7 concentrations are studied), 50 ~C1 of a
solution of chromogenic substrate (2 concentrations are
studied; S2238 Chromogenix") in solution in Tris buffer
at pH 7.5 (50 mM Tris, 100 mM NaCl and 0.1% HSA) and
finally 25 ~.1 of a 300 U/ml thrombin solution. The
release of 4-nitroaniline is monitored at 405 nm using
a plate reader.
The Ri is determined by the Dixon method.
The compounds of the invention are inhibitors of
thrombin and their Ri is between 0.001 and l00 ACM.
2. Clotting of rat plasma by human thrombin ex vivo
Male CD rats weighing 150 to 200 g are
treated with the compound to be tested or with the
vehicle by the i.v., oral or subcutaneous route. The
animals are then anaesthetized with Nembutal" (60 mg/kg;
0.1 ml/kg), the blood is withdrawn onto 3.8% trisodium
citrate (1 vol/9 vol of blood) from the retro-orbital
sinus and the plasma is prepared by centrifuging at
3600 g for 15 minutes at room temperature. 200 ~,1 of
CA 02270478 1999-04-29
l09
plasma are then incubated at 37~C with 200 ~l of a
solution of human thrombin, the final concentration of
human thrombin being 0.75 NIH uaits/ml, and the
clotting time is recorded. The anticoagulant effect is
expressed by the dose which increases the clotting time
by 100%.
They inhibit the clotting of rat plasma at doses of
0.0l to 5 mg/kg i.v. They are also active by the oral
and subcutaneous routes.
The compounds of the invention can be used in
a11 clinical indications related to thrombosis or in
those where thrombotic complications could occur.
To this end, they can be presented in any
form appropriate for oral, parenteral or intravenous
administration, such as tablets. dragees, capsules,
including hard gelatin capsules, suspensions or
solutions to be taken orally or to be injected, and the
like, in combination with suitable excipients. All
these forms contain doses which make possible an
administration of 1 to 1000 mg per day and per patient.
in one or more doses.