Note: Descriptions are shown in the official language in which they were submitted.
CA 02271582 1999-OS-13
-1-
METHOD FOR ADMINISTRATION OF
THERAPEUTIC AGENTS, INCLUDING ANTISENSE, WITH REPEAT DOS1NG
FIELD OF THE INVENTION
This invention relates to a method for administration of therapeutic
compositions
that comprise a combination of a lipid and a therapeutic agent, and
particularly for administration
of lipid-nucleic acid compositions, for in vivo therapeutic use in a regimen
involving repeat
dosing. In these compositions the therapeutic agent is encapsulated and
protected from
degradation and clearance in serum.
BACKGROUND OF THE INVENTION
Therapeutic oligonucleotides, such as antisense oligonucleotides or ribozymes,
are
short segments of DNA that have been designed to hybridize to a sequence on a
specific mRNA.
The resulting complex can down-regulate protein production by several
mechanisms, including
inhibition of mRNA translation into protein and/or by enhancement of RNase H
degradation of
the mRNA transcripts. Consequently, therapeutic oligonucleotides have
tremendous potential
for specificity of action (i.e. the down-regulation of a specific disease-
related protein). To date,
1 S these compounds have shown promise in several in vitro and in vivo models,
including models of
inflammatory disease, cancer, and HIV (reviewed in Agrawal, Trends in Biotech.
14:376-387
(1996)). Antisense can also effect cellular activity by hybridizing
specifically with chromosomal
DNA. Advanced human clinical assessments of several antisense drugs are
currently underway.
Targets for these drugs include the genes or RNA products of c-myc, ICAM-1,
and infectious
disease organisms such as cytomegalovirus, and HIV-1.
One well known problem with the use of therapeutic oligonucleotides having a
phosphodiester internucleotide linkage is its very short half life in the
presence of serum or
within cells. (Zelphati, O et al. 1993. Inhibition of HIV-1 Replication in
Cultured Cells with
Antisense Oligonucleotides Encapsulated in Immunoliposomes. Antisense. Res.
Dev. 3:323-
338; and Thierry, AR et al. pp147-161 in Gene Regulation: Biology of Antisense
RNA and
DNA (Eds. Erickson, RP and Izant, JG) 1992. Raven Press, NY). No clinical
assessment
CA 02271582 1999-OS-13
-2-
currently employs the basic phosphodiester chemistry found in natural nucleic
acids, because of
these and other known problems.
This problem has been partially overcome by chemical modifications which
reduce serum or intracellular degradation. Modifications have been tested at
the internucleotide
phosphodiester bridge (i.e. using phosphorothioate, methylphosphonate or
phosphoramidate
linkages), at the nucleotide base (i.e.5-propynyl-pyrimidines), or at the
sugar (i.e. 2'-modified
sugars) (Uhlmann E., et al. 1997. Antisense: Chemical Modifications.
Encyclopedia of Cancer
Vol. X. pp 64-81 Academic Press Inc.). Others have attempted to improve
stability using 2'-5'
sugar linkages (see US Pat. No. 5,532,130). Other changes have been attempted.
However, none
of these solutions have proven entirely satisfactory, and in vivo free
antisense still has only
limited efficacy. Problems remain, such as in the limited ability of some
antisense to cross
cellular membranes (see, Vlassov, et al., Biochim. Biophys. Acta 1197:95-1082
(1994)) and in
the problems associated with systemic toxicity, such as complement-mediated
anaphylaxis,
altered coagulatory properties, and cytopenia (Galbraith, et al., Antisense
Nucl. Acid Drug Des.
4:201-206 (1994)). Further, as disclosed in US Pat. Appl. SN. 08/657,753 and
counterpart patent
application WO 97/46671, both incorporated herein by reference, modified
antisense is still
highly charged, and clearance from the circulation still takes place within
minutes.
To attempt to improve efficacy, investigators have also employed lipid-based
carrier systems to deliver chemically-modified or unmodified antisense. In
Zelphati, O and
Szoka, F.C. (1996) J. Contr. Rel. 41:99-119, the authors refer to the use of
anionic (conventional)
liposomes, pH sensitive liposomes, immunoliposomes, fusogenic liposomes and
cationic
lipid/antisense aggregates.
None of these compositions successfully deliver phosphodiester antisense for
in
vivo therapy. In another paper, Zelphati & Szoka note that antisense
phosphodiester
oligonucleotides associated with cationic lipids have not been active in cell
culture in vitro; and
that only one study has reported the activity of phosphodiester antisense
oligonucleotides
complexed to cationic lipids. The authors argue that these findings
"...necessitate[ ] the use [of -
sic] backbone-modified oligonucleotides that are relatively resistant to both
intracellular and
extracellular nucleases even if a carrier is used to deliver the
oligonucleotide into the target cell".
CA 02271582 1999-OS-13
-3-
(1997. J. Lip. Res. 7(1):31-49 at 34). This finding is corroborated by
Bennett, CF. (1995.
Intracellular Delivery of Oligonucleotides with Cationic Liposomes. Chp 14 CRC
Press) who
states at p. 224 that "In contrast, we have been unable to demonstrate
inhibition of gene
expression by uniform phosphodiester oligodeoxynucleotides directed towards a
number of
cellular targets in the presence of cationic lipids."
Prior art lipid formulations of modified antisense are also largely
ineffective in
vivo. They have poor encapsulation efficiency (15% or less for passive
encapsulation systems),
poor drug to lipid ratios (3% or less by weight), high susceptibility to serum
nucleases and rapid
clearance from circulation (particularly in the case of cationic
lipid/antisense aggregates made
from DOTMA, trade-name LIPOFECTINT"'), and/or large sized particles (greater
than 100 nm),
which make them unsuitable for systemic delivery to target sites. No
successful in vivo efficacy
studies of lipid-encapsulated (nuclease-resistant) modified antisense are
known in the prior art.
Two references to unique lipid-antisense compositions that may be
significantly
nuclease resistant bear consideration. Firstly, the anionic liposome (LPDII)
composition of Li, S.
and Huang, L (1997. J. Lip. Res. 7(1) 63-75), which encapsulates poly-lysine
coated antisense,
are said to have 60-70% encapsulation efficiency, but suffer from a large size
of around 200 nm
and a low drug to lipid ratio of 8% by weight. The effect of these particles
in vivo is unknown.
Secondly, the Minimal Volume Entrapment (MVE) technique for cardiolipin
(anionic) liposomes
results in the reasonably high encapsulation efficiency of 45-65% but again
the drug:lipid ratio
remains very small, approximately 6.5% by weight (see US Pat. No. 5,665,710 to
Rahman et al.;
Thierry AR, and Takle, GB. 1995, Liposomes as a Delivery System for Antisense
and Ribozyme
Compounds. in Delivery Strategies for Antisense Oligonucleotide Therapeutics,
S. Akhtar, ed,
CRC Press, Boca Raton, FL., pp. 199-221; Thierry, AR et al. pp147-161 in Gene
Regulation:
Biology of Antisense RNA and DNA (Eds. Erickson, RP and Izant, JG) 1992. Raven
Press,
NY). Note that US Pat. No. 5,665,710 also discloses encapsulation efficiencies
of 60-90% for
tiny, medically useless amounts of antisense (0.1 ug), where the drug to lipid
ratio must be very
low.
It is an observation of the inventors that a wide variety of prior art lipid
compositions used for conventional drugs could be tested for efficacy in the
antisense field, but
CA 02271582 1999-OS-13
-4-
the improvement (over free antisense) for in vivo efficacy is not known. In
this regard, it is noted
that although lipid compositions assertedly for use as drug carriers were
disclosed by Bailey and
Cullis (US Pat. 5552155; and (1994) Biochem. 33(42):12573-12580), they did not
disclose
formulations of any bioactive compounds with these lipids, and did not suggest
their utility for
high efficiency loading of polyanionic species.
What is needed in the art are improved lipid-therapeutic agent compositions
which are suitable for therapeutic use. Preferably these compositions would
encapsulate nucleic
acids or other therapeutic agents with high-efficiency, have high drug:lipid
ratios, be
encapsulated and protected from degradation and clearance in serum, and/or be
suitable for
systemic delivery. In addition, although not generally appreciated in the art,
at least some of the
known lipid compositions are unsuitable for therapeutic regimens involving
repeat dosing,
because the patient's system becomes primed by the first dose of the antisense
agent and rapidly
clears subsequent doses. It is an object of the present invention to provide a
method for lipid-
based therapy using therapeutic agents such an antisense oligonucleotides
which is not limited to
uses in regimens calling for single (or widely spaced) doses.
CA 02271582 1999-OS-13
-$-
SUMMARY OF THE INVENTION
In accordance with a preferred embodiment of the invention, there is provided
a
method for the treatment or prevention of a disease characterized by aberrant
expression of a
gene in a mammalian subject comprising,
preparing lipid-encapsulated therapeutic nucleic acid particles comprising a
therapeutic nucleic acid component that hybridizes specifically with the
aberrantly expressed
gene to reduce its expression in the subject encapsulated within a lipid
particle, said lipid particle
being formed from a lipid mixture including a steric-barrier lipid component
selected from
among lipids that prevent particle aggregation during lipid-nucleic acid
particle formation and
which exchange out of the lipid particle at a rate greater than PEG-CerC20;
and
administering a therapeutically effective or prophylactic amount of the
particles to
the mammalian subject in a plurality of separate doses separated in time by
intervals of up to
eight weeks.
The particles are preferably formulated using a PEG-modified or polyamide
oligomer-modified lipid as the first lipid component. To achieve high loading
levels, an
effective method for preparation of the lipid-encapsulated nucleic acid
particles involves the
use of a lipid mixture comprising at least the first lipid component and a
protonatable lipid
component selected from among lipids containing a protonatable group that has
a pKa such that
the lipid is in a cationic form at a pH below the pKa and a neutral form at
around physiological
pH.
The method the invention is particularly useful for therapy using lipid-
encapsulated antisense nucleic acids or ribozymes. In a more general sense,
however, the
method of the invention may also be used for delivery of non-cytotoxic lipid-
encapsulated
therapeutic agents generally in a repeat dosing regimen.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 illustrates a neutralization step which releases surface-bound
antisense
from the lipid-nucleic acid compositions according to the present invention.
CA 02271582 1999-OS-13
-6-
Figures ZA and 2B illustrate certain lipid components which are useful in the
present inventive methods. Figure 2A illustrates several groups of amino
lipids including the
chemical structure of DODAP. Figure 2B illustrates groups of PEG-modified
lipids.
Figure 3 illustrates the influence of ethanol on the encapsulation of
antisense
oligodeoxynucleotides. The liposomal antisense compositions were prepared as
described in the
Examples, with the final concentrations of antisense and lipids being 2 mg/mL
and 9.9 mg/mL,
respectively. The final ethanol concentration in the preparations was varied
between 0 and 60%,
vol/vol. Encapsulation was assessed either by analyzing the pre-column and
post-column ratios
of [3H]-antisense and ['~C]-lipid or by determining the total pre-column and
post-column
[3H]-antisense and ['''C]-lipid radioactivity.
Figure 4 illustrates the influence of ethanol on lipid and antisense loss
during
extrusion. The liposomal antisense compositions were prepared as described for
Figure 3. The
samples were extruded ten times through three 100 nm filters as described in
"Materials and
Methods". After extrusion, the filters were analyzed for [3H]-antisense and
['4C]-lipid
radioactivity by standard scintillation counting techniques. Results were
expressed as a percent
of the total initial radioactivity.
Figure 5 illustrates the influence of DODAP content on the encapsulation of
antisense oligodeoxynucleotides. A 0.6 mL aliquot of a [3H]-phosphorothioate
antisense
oligodeoxynucleotide (in 300 mM citrate buffer, pH 3.80) was mixed with 0.4 mL
of a 95%
ethanol solution of lipid (DSPC:CHOL:DODAP:PEG-CerCl4; 100-(55+X):45:X:10,
molar
ratio) at final concentrations of 2 mg/mL and 9.9 mg/mL, respectively. The
molar ratio of
DODAP was varied between 0 and 30%. The molar ratio of DSPC was adjusted to
compensate
for the changes in DODAP content. Encapsulation was assessed either by
analyzing the
pre-column and post-column ratios of [3H]-antisense and ['4C]-lipid or by
determining the total
pre-column and post-column [3H]-antisense and ['4C]-lipid radioactivity.
Figure 6 illustrates the influence of DODAP content on the encapsulation of
antisense oligodeoxynucleotides. Samples were identical to those prepared in
Figure 5. In this
instance, the amount of antisense associated with the lipid was assessed by a
solvent extraction
procedure as described in "Material and Methods". Antisense was extracted into
a
CA 02271582 1999-OS-13
7_
methanol:water aqueous phase, while the lipid was soluble in the organic
(chloroform) phase.
The aqueous phase was preserved and antisense concentration was determined by
measuring the
absorbance at 260 nm. This confirmed that the antisense was associated with
the lipid vesicles,
and that the [3H]-label on the antisense had not exchanged to the lipid.
Figure 7 illustrates the quasi-elastic light scattering analysis of
encapsulated
liposomal antisense. The size distribution of a liposomal preparation of
antisense was
determined by quasi-elastic light scattering (QELS) immediately after removal
of the free
antisense (A), and after storage of the preparation for 2 months at 4°C
(B), using a Nicomp
Model 370 sub-micron particle sizer.
Figure 8 illustrates the influence of the initial antisense concentration on
antisense loading in DODAP vesicles. Varying final concentrations of a 20mer
of
[3H]-phosphorothioate antisense oligodeoxynucleotide (in 300 mM citrate
buffer, pH 3.80) were
mixed with an ethanol solution of lipid (DSPC:CHOL:DODAP:PEG-CerCl4;
25:45:20:10,
molar ratio), 9.9 mg/mL (final concentration). Encapsulation was assessed
either by analyzing
the pre-column and post-column ratios of [3H]-antisense and ['4C]-lipid or by
determining the
total pre-column and post-column [3H]-antisense and ['4C]-lipid radioactivity.
EPC:CHOL
liposomes containing encapsulated antisense are included for comparison.
Figure 9 illustrates the plasma clearance of encapsulated antisense.
Encapsulated
liposomal antisense was prepared using the ethanol-citrate procedure as
described in "Material
and Methods". Initial lipid and antisense concentrations were 9.9 and 2 mg/mL,
respectively.
Liposomal formulations were composed of X:CHOL:DODAP:PEG-CerCl4 (25:45:20:10),
where X represents either distearoylphosphatidylcholine (DSPC), sphingomyelin
(SM), or
palmitoyloleoylphosphatidylcholine (POPC). The formulations contained a lipid
label
(['4C]-cholesterylhexadecylether) and [3H]-antisense and were injected (200
qL) intravenously
via the lateral tail vein of female (20-25 g) ICR mice at a lipid dose of 120
mg/kg. Blood was
recovered by cardiac puncture on anesthetized mice. Lipid and antisense
recoveries were
determined by standard scintillation counting techniques.
Figure 10 illustrates the biodistribution of encapsulated antisense.
Encapsulated
liposomal antisense was prepared using the ethanol-citrate procedure as
described in "Material
CA 02271582 1999-OS-13
_g_
and Methods". Initial lipid and antisense concentrations were 9.9 and 2 mg/mL,
respectively.
Liposomal formulations were composed of X:CHOL:DODAP:PEG-CerCl4 (25:45:20:10),
where X represents either distearoylphosphatidylcholine (DSPC), sphingomyelin
(SM), or
palmitoyloleoylphosphatidylcholine (POPC). The formulations contained a lipid
label
(['4C]-cholesterylhexadecylether) and [3H]-antisense and were injected (200
~L) intravenously
via the lateral tail vein of female (20-25 g) ICR mice at a lipid dose of 120
mg/kg. Mice were
terminated by cervical dislocation and the organs were recovered and processed
as described in
"Materials and Methods". Lipid and antisense recoveries were determined by
standard
scintillation counting techniques.
Figure 11 illustrates the differential release rates of antisense in plasma.
Encapsulated liposomal antisense was prepared using the ethanol-citrate
procedure as described
in "Material and Methods". Initial lipid and antisense concentrations were 9.9
and 2 mg/mL,
respectively. Liposomal formulations were composed of X:CHOL:DODAP:PEG-CerC 14
(25:45:20:10), where X represents either distearoylphosphatidylcholine (DSPC),
sphingomyelin
(SM), or palmitoyloleoylphosphatidylcholine (POPC). The formulations contained
a lipid label
(['4C]-cholesterylhexadecylether) and [3H]-antisense and were injected (200
~.L) intravenously
via the lateral tail vein of female (20-25 g) ICR mice at a lipid dose of 120
mg/kg. Blood was
recovered by cardiac puncture on anesthetized mice. Lipid and antisense
recoveries were
determined by standard scintillation counting techniques. Release rates were
determined by
measuring the [3H]/[14C] ratio over time.
Figure 12 illustrates the influence of PEG-acyl chain lengths on plasma
clearance
of encapsulated antisense. Encapsulated liposomal antisense was prepared using
the
ethanol-citrate procedure as described in "Material and Methods". Initial
lipid and antisense
concentrations were 9.9 and 2 mg/mL, respectively. Liposomal formulations were
composed of
DSPC:CHOL:DODAP:PEG-CerCl4 or C20 (25:45:20:10). The formulation contained a
lipid
label (['4C]-cholesterylhexadecylether) and [3H]-antisense and were injected
(200 ~L)
intravenously via the lateral tail vein of female (20-25 g) ICR mice at a
lipid dose of 120 mg/kg.
Blood was recovered by cardiac puncture on anesthetized mice. Lipid and
antisense recoveries
were determined by standard scintillation counting techniques.
CA 02271582 1999-OS-13
-9-
Figure 13 illustrates the enhanced efficacy of liposomal antisense containing
DODAP - ear swelling. Inflamed mice were treated at the time of ear challenge
with a 30 mg/kg
i.v. dose of either HBS (no oligo), EPC:CHOL liposomes with entrapped PS 3082
(identified as
AS 1000), POPC:CHOL:DODAP:PEG-CerCl4 with entrapped PS 3082 (identified as
4100), or
DSPC:CHOL:DODAP:PEG-CerCl4 with entrapped PS 3082 (identified as 4200). Ear
swelling
was measured at 24 hours after initiating inflammation using an engineer's
micrometer.
Figure 14 illustrates the enhanced efficacy of liposomal antisense containing
DODAP - cellular infiltration. Mice received 10 pCi of [3H]-methylthymidine,
i.p., 24 hours
before initiating inflammation. Inflamed mice were treated at the time of ear
challenge with a 30
mg/kg i.v. dose of either HBS (no oligo), EPC:CHOL liposomes with entrapped PS
3082
(identified as AS 1000), POPC:CHOL:DODAP:PEG-CerCl4 with entrapped PS 3082
(identified
as 4100), or DSPC:CHOL:DODAP:PEG-CerCl4 with entrapped PS 3082 (identified as
4200).
Cell infiltration was monitored by measuring the radioactivity in the
"challenged ear" versus the
non-treated ear. Results are expressed as the ratio of radioactivity in the
left (challenged ear)
versus right ear.
Figure 15 shows asymmetric loading of lipid-encapsulated-nucleic acid
particles
in accordance with the invention .
Figure 16 shows clearance of lipid-encapsulated antisense particles formulated
with several amino lipids at different levels.
Figure 17 shows blood levels of antisense-containing particles after repeat
dosages.
Figure 18 shows blood levels of antisense-containing particles after repeat
dosages.
Figure 19 illustrates results of a study on the in vivo efficacy of lipid-
encapsulated antisense particles in accordance with the invention in a mouse
tumor model.
Figure 20 shows encapsulation efficiency results for lipid-encapsulated
therapeutic agent particles in accordance with the invention.
Figure 21 shows results for studies on the use of murine ICAM1 in an ear
inflammation model.
CA 02271582 1999-OS-13
-10-
Figure 22 shows results for studies on the use of murine ICAM1 in an ear
inflammation model.
Figure 23 shows results for studies on the use of murine ICAM1 in an ear
inflammation model.
S
DETAILED DESCRIPTION OF THE INVENTION
CONTENTS
I. Glossary
II. General
III. Methods of Preparing Liposome/Nucleic Acid Complexes
IV. Pharmaceutical Preparations
V. Methods of Introducing the Lipid-Encapsulated Therapeutic Agents Into Cells
VI. Examples
VII. Conclusion
I. Glossary
Abbreviations and Definitions
The following abbreviations are used herein: ATTA, N-(cu-N'-acetoxy-
octal14'amino-3',6',9',12'-tetraoxatetradecanoyl)); CHE, cholesteryl-
hexadecylether; CHOL,
cholesterol; DODAP or AL-1, 1,2-dioleoyloxy-3-dimethylaminopropane (and its
protonated
ammonium form); DODMA, N-(1-(2,3-Dioleoyloxy) propyl)-N,N,-dimethyl ammonium
chloride; DSPC, distearoylphosphatidylcholine; EPC, egg phosphatidylcholine;
HBS,
HEPES-buffered saline; HEPES, N-2-hydroxyethylpiperazine-N'-2-ethanesulphonic
acid; MES,
2-(N-morpholino)ethane sulfonic acid; PS 3082, murine ICAM-1 phosphorothioate
oligodeoxynucleotide having the sequence: TGCATCCCCCAGGCCACCAT (SEQ ID No. 1);
NaCI, sodium chloride; OLIGREENTM, a dye that becomes fluorescent when
interacting with an
oligonucleotide; PEG-CerC20, polyethylene glycol coupled to a ceramide
derivative with 20
carbon acyl chain; POPC, palmitoyloleoylphophatidylcholine; SM, sphingomyelin.
CA 02271582 1999-OS-13
-11-
"Lipid-therapeutic agent particle" means a particle comprising lipids and a
charged (cationic or anionic) therapeutic agent. "Lipid-therapeutic nucleic
acid particle" means
a particle comprising a lipid and a therapeutic nucleic acid.
"Lipid-encapsulated therapeutic agent (nucleic acid) particle" means a lipid-
therapeutic agent particle wherein less than 50% and preferably less than 10%
of the therapeutic
agent (nucleic acid) is detectable on the external surface of the particle or
in the buffer external to
the particle. In the case of nucleic acids, the amount of encapsulated versus
unencapsulated
nucleic acid can be assayed by fluorescence assays or nuclease assays as
described herein.
Comparable assays can be used for other types of therapeutic agents.
"Therapeutically effective amount" means an amount which provides a
therapeutic benefit. For antisense oligonucleotide this means generally 0.5 to
50 mg/kg of body
weight, but when delivered in a lipid particle formulation, a below-toxic
amount of lipid must be
used.
"Lipid exchange out of particle" and the rate of this exchange is fully
explained in
US Pat. Apps. SN 08/486,214 and 08/485,608 and PCT Patent publications WO
96/10391 and
WO 96/10392, which are all incorporated herein by reference. Lipid exchange
into the
surrounding medium is possible for lipids which are reversibly associated with
the lipid particle
membrane. Each lipid has a characteristic rate at which it will exchange put
of a particle which
depends on a variety of factors including acyl chain length, saturation, head
group size, buffer
composition and membrane composition.
"Disease site" is the site in an organism which demonstrates or is the source
of a
pathology. The disease site may be focused, as in a site of neoplasm or
inflammation, or may be
diffuse as in the case of a non-solid tumor. "Administration at a site which
is distal to the disease
site" means that delivery to the disease site will require some kind of
systemic delivery, either by
blood or lymph circulation, or other fluid movement inside the organism.
The term "transfection" as used herein, refers to the introduction of
polyanionic
materials, particularly nucleic acids, into cells. The polyanionic materials
can be in the form of
DNA or RNA which is linked to expression vectors to facilitate gene expression
after entry into
the cell. Thus the polyanionic material or nucleic acids used in the present
invention is meant to
CA 02271582 1999-OS-13
-12-
include DNA having coding sequences for structural proteins, receptors and
hormones, as well as
transcriptional and translational regulatory elements (i.e., promoters,
enhancers, terminators and
signal sequences) and vector sequences. Methods of incorporating particular
nucleic acids into
expression vectors are well known to those of skill in the art, but are
described in detail in, for
example, Sambrook et al., Molecular Cloning: A Laboratory Manual (2nd ed.),
Vols. 1-3, Cold
Spring Harbor Laboratory, (1989) or Current Protocols in Molecular Biology, F.
Ausubel et al.,
ed. Greene Publishing and Wiley-Interscience, New York (1987), both of which
are incorporated
herein by reference.
The term "physiological pH" refers to pH levels conventionally encountered in
serum or blood. In general, this will be in the range of pH 7.2 to 7.5.
Preferred protonatable or
deprotonatable lipids have a pKa such that they are substantially neutral at
this pH, i.e., a pKa of
about 4 to 7 in the case of an amino lipid.
II. General
The present invention relates to therapeutic methods utilizing lipid-
encapsulated
therapeutic agent particles in which therapeutic agents such as antisense
nucleic acids or other
non-cytotoxic therapeutic materials are encapsulated within a lipid layer. The
application is
principally discussed in the specification and examples in terms of
therapeutic nucleic acids,
which are a preferred embodiment, but it will be appreciated that the same
techniques can be
applied to other therapeutic material, including proteins, peptides, cytokines
and heparins.
To evaluate the quality of a lipid/nucleic acid formulation the following
criteria,
among others, may be employed:
drug to lipid ratio;
encapsulation efficiency;
nuclease resistance/serum stability; and
particle size.
High drug to lipid ratios, high encapsulation efficiency, good nuclease
resistance and serum
stability and controllable particle size, generally less than 200 nm in
diameter are desirable In
addition, the nature of the nucleic acid polymer is of significance, since the
modification of
CA 02271582 1999-OS-13
-13-
nucleic acids in an effort to impart nuclease resistance adds to the cost of
therapeutics while in
many cases providing only limited resistance. The present invention provides
lipid-nucleic acid
particles and methods for preparing lipid-nucleic acid formulations which are
far superior to the
art according to these criteria.
Unless stated otherwise, these criteria are calculated in this specification
as
follows:
drug to lipid ratio: The amount of drug (therapeutic agent) in a defined
volume of
preparation divided by the amount of lipid in the same volume. This may be on
a mole per mole
basis or on a weight per weight basis, or on a weight per mole basis. For
final, administration-
ready formulations, the drug:lipid ratio is calculated after dialysis,
chromatography and/or
enzyme (e.g., nuclease) digestion has been employed to remove as much of the
external
therapeutic agent (e.g., nucleic acid) as possible. Drug:lipid ratio is a
measure of potency of the
formulation, although the highest possible drug:lipid ratio is not always the
most potent
formulation;
encapsulation efficiency: the drug to lipid ratio of the starting mixture
divided by
the drug to lipid ratio of the final, administration competent formulation.
This is a measure of
relative efficiency. For a measure of absolute efficiency, the total amount of
therapeutic agent
(nucleic acid) added to the starting mixture that ends up in the
administration competent
formulation, can also be calculated. The amount of lipid lost during the
formulation process may
also be calculated. Efficiency is a measure of the wastage and expense of the
formulation;
nuclease resistance/serum stability: the ability of the formulation to protect
the
nucleic acid therapeutic agents from nuclease digestion either in an in vitro
assay, or in
circulation. Several standard assays are detailed in this specification.
Encapsulated particles
have much greater nuclease resistance and serum stability than lipid-antisense
aggregates such as
DOTMA/DOPE (LIPOFECTINTM) formulations; and
size: the size of the particles formed. Size distribution may be determined
using
quasi-elastic light scattering (QELS) on a Nicomp Model 370 sub-micron
particle sizer. Particles
under 200 nm are preferred for distribution to neo-vascularized (leaky)
tissues, such as
neoplasms and sites of inflammation.
CA 02271582 1999-OS-13
- 14-
In addition, it has been surprisingly found that the composition of the lipid
particle has a substantial affect on the behaviors of the particle when used
in a therapeutic
regimen involving repeat dosing. In particular, in experiments using antisense
oligonucleotides
as the therapeutic agent, it was found that the acyl chain length of the lipid
derivatized to a steric
barrier (i.e. ATTA or PEG) moiety demonstrates a profound effect on clearance
rates. Repeat
dosages of PEG-CerC20, PEG-DSPE and ATTAB-DSPE formulations are rapidly
cleared from
the circulation compared to the first dosage, whereas the PEG-CerC 14
formulation is reasonably
consistent with the first dosage. (See Figs. 17 and 18).
Without intending to be bound by any particular theory of action, it is
believed
that lipids like the PEG-CerCl4 lipid, a lipid which exchanges out of the
outer monolayer of the
lipid particle with a T"Z on the order of minutes (i.e. up to 240 minutes,
preferably up to 60 mins)
in blood provides a tremendous benefit over lipids like PEG-CerC20, PEG-DSPE
and ATTAB-
DSPE which do not exchange out within any realistic time frame, where repeat
dosing of a lipid-
formulated compound, such as a therapeutic compound or diagnostic compound, is
required.
The mammalian blood clearance response may not recognize these as foreign
antigens if the
derivatized lipid is removed expeditiously from the lipid particle when in
circulation. However,
when the derivatized-lipid remains with the formulation for extended periods,
a clearance
response is invoked, which causes rapid clearance upon repeat dosing. This
data suggests that
any lipid derivatized with a steric barrier molecule that exchanges out of the
liposome membrane
faster than PEG-CerC20, PEG-DSPE or ATTA8-DSPE will be superior for use in
repeat dosing.
For example ATTAB-DMPE, or PEG-CerC8 to C18 all being exchangeable, will have
improved
circulation characteristics upon repeat administration.
Taken together, it will be evident to one skilled in the art, that on the
basis of
these teachings, any diagnostic or therapeutic agent that may be delivered in
a lipid formulation
comprising a steric-barrier derivatized lipid, such as a PEG-lipid or ATTA-
lipid, should be tested
with both a long and short acyl-chain anchors, in order to determine which
formulation is best for
repeat dosings.
Further, without intending to be bound by any theory of action, the invention
herein may prove to be particularly useful when the bioactive agent being
delivered is a non-
CA 02271582 1999-OS-13
-1$-
cytotoxic agent. Cytotoxic agents kill those cells which clear long
circulating (i.e. PEG-DSPE)
liposomes. This ensures that repeat dosings will not be rapidly cleared,
because the cells
responsible (usually macrophages) do not survive. In these situations, the
acyl-chain length may
not be significant. However, where the bioactive agent is non-cytotoxic, such
as in the case of
antisense drugs (regardless of chemistry or target), plasmids, proteins, etc.,
and many
conventional drugs, the invention will be useful for repeat dosing.
In general, the period of time between repeat dosings in the method of the
invention will be less than eight weeks, for example between one and eight
weeks, although
these periods of time are not critical. Lesser periods of time might result in
an accumulation of
the lipid-encapsulated therapeutic agent as a result of repeating the dose
prior to the clearance of
the prior dose. Longer periods of time would not be outside the scope of the
present invention,
although it is assumed that at some point the priming of the system relaxes
such that second
doses would not be subject to accelerated clearing.
The observation of the effect of chain length on clearance rate following
repeat
dosages can be used as a basis for providing repeat dosage formulations of any
type of lipid
particle which includes a lipid modified with a steric barrier moiety, such as
polyethylene glycol
or polyamide oligomers. In a preferred embodiment of the invention, however,
the method of
the invention makes use of certain lipids which can be present in both a
charged and an
uncharged form. These lipids, when used as part of the lipid mixture
containing lipids modified
with steric barrier moieties permit high efficiency loading of charged
therapeutic agents,
including without limitation nucleic acids having exclusively phosphodiester
internucleotide
linkages.
For example, amino lipids which are charged at a pH below the pKa of the amino
group and substantially neutral at a pH above the pKa can be used in a two-
step process. First,
lipid vesicles can be formed at the lower pH with (cationic) amino lipids and
other vesicle com-
ponents in the presence of nucleic acids. In this manner the vesicles will
encapsulate and entrap
the nucleic acids. Second, the surface charge of the newly formed vesicles can
be neutralized by
increasing the pH of the medium to a level above the pKa of the amino lipids
present, i.e., to
physiological pH or higher. Particularly advantageous aspects of this process
include both the
CA 02271582 1999-OS-13
-16-
facile removal of any surface adsorbed nucleic acid and a resultant nucleic
acid delivery vehicle
which has a neutral surface. Liposomes or lipid particles having a neutral
surface are expected to
avoid rapid clearance from circulation and to avoid certain toxicities which
are associated with
cationic liposome preparations.
It is further noted that the vesicles formed in this manner provide
formulations of
uniform vesicle size with high content of nucleic acids. Additionally, the
vesicles are not
aggregate complexes, but rather are large unilamellar vesicles having a size
range of from about
70 to about 200 nm, more preferably about 90 to about 130 nm.
Without intending to be bound by any particular theory, it is believed that
the very
high efficiency of nucleic acid encapsulation is a result of electrostatic
interaction at low pH.
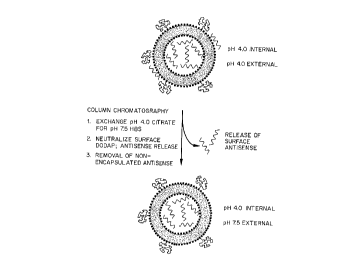
Figure 1 provides an illustration of the processes described herein. More
particularly, this figure
illustrates a lipid-nucleic acid composition of amino lipids and PEG-modified
lipids having
encapsulated antisense nucleic acid and surface-bound antisense nucleic acid.
At acidic pH
(shown as pH 4.0), the surface is charged and binds a portion of the antisense
through
electrostatic interactions. When the external acidic buffer is exchanged for a
more neutral (pH
7.5, HBS) buffer, the surface of the lipid particle or liposome is
neutralized, resulting in release
of the antisense nucleic acid.
Encapsulation efficiency results in Figs. 15 show a further unexpected benefit
of
the invention. As shown in the figure, for both phosphorothioate (PS-2302) and
phosphodiester
(PO-2302) formulations it is possible to obtain encapsulation efficiencies -
i.e., the amount of
nucleic acid that ends up on the inside of the particle - that are greater
than 50%.
Phosphodiesters achieve well over 60%, and phosphorothioates can be at least
up to 80%
encapsulated. The asymmetry of loading is surprising, given that in the
simplest model of
loading large unilamellar vesicles (LUV's) the therapeutic agent (nucleic
acid) would be equally
likely to associate with cationic charges on the inside and outside of the
particle. A 1:1
distribution (inside to outside) would suggest that the 50% on the outside
should be removed
upon neutralization of the outside surface charges, such that SO% efficiency
would be the
theoretical upper limit. Through some unclear mechanism, however, the
invention surprisingly
CA 02271582 1999-OS-13
17-
provides an active process whereby the majority of the therapeutic agent
(nucleic acid) ends up
protected on the inside of the particles.
III. Methods of Preparing Lipid/Therapeutic Agent (Nucleic Acid) Formulations
In view of the above, a preferred embodiment of the present invention provides
methods of preparing lipid/nucleic acid formulations. In the methods described
herein, a mixture
of lipids is combined with a buffered aqueous solution of nucleic acid to
produce an intermediate
mixture containing nucleic acid encapsulated in lipid particles wherein the
encapsulated nucleic
acids are present in a nucleic acid/lipid ratio of about 10 wt% to about 20
wt%. The intermediate
mixture may optionally be sized to obtain lipid-encapsulated nucleic acid
particles wherein the
lipid portions are large unilamellar vesicles, preferably having a diameter of
70 to 200 nm, more
preferably about 90 to 130 nm. The pH is then raised to neutralize at least a
portion of the
surface charges on the lipid-nucleic acid particles, thus providing an at
least partially
surface-neutralized lipid-encapsulated nucleic acid composition.
The mixture of lipids includes at least two lipid components: a protonatable
or
deprotonatable lipid component that is selected from among lipids which have a
pKa such that
the lipid is cationic at pH below the pKa and neutral at pH above the pKa, and
a steric-barrier
lipid component that is selected from among lipids that prevent particle
aggregation during lipid-
nucleic acid particle formation and which exchange out of the lipid particle
at a rate greater than
PEG-CerC20.
The protonatable or deprotonatable lipid component of is a lipid (or a mixture
of
lipid species with similar properties) which has at least one protonatable or
deprotonatable group,
such that the lipid is charged at a first pH (cationic or anionic, depending
on the nature and pKa
of the protonatable or deprotonatable group), and neutral at a second pH,
preferably near
physiological pH. It will of course be understood that the addition or removal
of protons as a
function of pH is an equilibrium process, and that the reference to a charged
or a neutral lipid
refers to the nature of the predominant species and does not require that all
of the lipid be present
in the charged or neutral form. Lipids which have more than one protonatable
or deprotonatable
group, or which are zwiterrionic are not excluded from use in the invention.
Protonatable lipids
CA 02271582 1999-OS-13
-1g-
are particularly useful as the first lipid component of the invention when the
pKa of the
protonatable group is in the range of about 4 to about 11. Most preferred is
pKa of about 4 to
about 7, because these lipids will be cationic at the lower pH formulation
stage, while particles
will be largely (though not completely) surface neutralized at physiological
pH around pH 7.5.
S One of the benefits of this pKa is that at least some antisense stuck to the
outside surface of the
particle will lose its electrostatic interaction at physiological pH and be
removed by simple
dialysis; thus greatly reducing the particle's susceptibility to clearance.
Preferred lipids with a protonatable group for use as the first lipid
component of
the lipid mixture are amino lipids. As used herein, the term "amino lipid" is
meant to include
those lipids having one or two fatty acid or fatty alkyl chains and an amino
head group (including
an alkylamino or dialkylamino group) which is protonated to form a cationic
lipid at
physiological pH (see Figure 2A). In one group of embodiments, the amino lipid
is a primary,
secondary or tertiary amine represented by the formula:
1
R2%N-R3
in which R' is a C,z to Cz4 alkyl group which is branched or unbranched, and
saturated or
unsaturated. RZ is hydrogen or a C, to Cz.~ alkyl group which is also branched
or unbranched, and
saturated or unsaturated (when three or more carbons are present). R3 is
hydrogen or a C~ to CG
alkyl group. Examples of these amino lipids include, for example,
stearylamine, oleylamine,
dioleylamine, N-methyl-N,N-dioleylamine, and N,N-dimethyloleylamine.
In another group of embodiments, the amino lipid is a lipid in which the amino
head group is attached to one or more fatty acid or fatty alkyl groups by a
scaffold such as, for
example, a glycerol or propanediol moiety. Illustrative of these amine lipids
is the formula:
8110
R13
8120 N~
wRl4
CA 02271582 1999-OS-13
-19-
wherein at least one and preferably both of R' ' and R'2 is a C'Z to C24 alkyl
or acyl group which is
branched or unbranched, saturated or unsaturated. In those embodiments in
which only one of
R" or R'2 is a long chain alkyl or acyl group, the other of R" or R'2 will be
a hydrogen or lower
alkyl or acyl group having from one to six carbon atoms. The remaining groups,
R'3 and R'4 are
typically hydrogen or C1 to C4 alkyl. In this group of embodiments, the amino
lipid can be
viewed as a derivative of 3-monoalkyl or dialkylamino-1,2-propanediol. An
example of a
suitable amino lipid is DODAP (1,2-dioleoyloxy-3-dimethylamino-propane, see
Figure 2A).
Other amino lipids would include those having alternative fatty acid groups
and other
dialkylamino groups, including those in which the alkyl substituents are
different (e.g., N-ethyl-
N-methylamino-, N-propyl-N-ethylamino- and the like). For those embodiments in
which R"
and R'Z are both long chain alkyl or acyl groups, they can be the same or
different. In general,
amino lipids having less saturated acyl chains are more easily sized,
particularly when the
complexes must be sized below about 0.3 microns, for purposes of filter
sterilization. Amino
lipids containing unsaturated fatty acids with carbon chain lengths in the
range of C,4 to Czz are
particularly preferred. Other scaffolds can also be used to separate the amino
group and the fatty
acid or fatty alkyl portion of the amino lipid. Suitable scaffolds are known
to those of skill in the
art.
Compounds that are related to DODAP that may be useful with this invention
include: 1-oleoyl-2-hydroxy-3-N,N-dimethylamino propane; 1,2-diacyl-3-N,N-
dimethylamino
propane; and 1,2-didecanoyl-1-N,N-dimethylamino propane. Further, it is
proposed that various
modifications of the DODAP or DODMA headgroup, or any compound of the general
formula:
can be modified to obtain a suitable pKa. Suitable headgroup modifications
that are useful in the
instant invention include:
CA 02271582 1999-OS-13
-20-
R' R2
1 H H
2 H CH3
3 CH3 CH3
4 H CHZCH3
CH3 CHZCH3
6 CHZCH3 CHZCH3
7 H CHZCHZOH
8 CH3 CHZCHZOH
9 CHZCH3 CHZCHZOH
CH2CHZOH CHZCHzOH
11* H CHzCH2NH2
12* CH3 CHzCH2NH2
13* CHzCH3 CHzCH2NH2
14* CHZCH20H CHZCHZNH2
15* CHZCHzNH2 CH2CHZNH2
In other embodiments, the amino lipid can be a derivative of a naturally
occurring
amino lipid, for example, sphingosine. Suitable derivatives of sphingosine
would include those
having additional fatty acid chains attached to either of the pendent hydroxyl
groups, as well as
alkyl groups, preferably lower alkyl groups, attached to the amino functional
group.
5 Other lipids which may be used as the first lipid component of the invention
include phosphine lipids (although toxicity issues may limit their utility),
and carboxylic acid
lipid derivative. These generally have a pKa of about S and are therefore
useful with cationic
therapeutic agents.
The second lipid component is selected to improve the formulation process by
10 reducing aggregation of the lipid particles during formation. This may
result from steric
stabilization of particles which prevents charge-induced aggregation during
formation. Examples
CA 02271582 1999-OS-13
-21 -
of suitable lipids for this purpose include polyethylene glycol (PEG)-modified
lipids,
monosialoganglioside Gm l, and polyamide oligomers ("PAO") such as ATTA
(disclosed in US
Pat. Appl. SN 60/073,852 and US Pat. Appl. SN 60/(not yet received TT&C
Attorney Docket
No.16303-005810 both assigned to the assignee of the instant invention and
incorporated herein
by reference). Other compounds with uncharged, hydrophilic, steric-barrier
moieties, that
prevent aggregation during formulation, like PEG, Gml or ATTA, can also be
coupled to lipids
for use as the second lipid component in the methods and compositions of the
invention.
Typically, the concentration of the second lipid component is about 1 to 15%
(by mole percent of
lipids).
Specific examples of PEG-modified lipids (or lipid-polyoxyethylene conjugates)
that are useful in the present invention can have a variety of "anchoring"
lipid portions to secure
the PEG portion to the surface of the lipid vesicle. Examples of suitable PEG-
modified lipids
include PEG-modified phosphatidylethanolamine and phosphatidic acid (see
Figure 2B,
structures A and B), PEG-modified diacylglycerols and dialkylglycerols (see
Figure 2B,
1 S structures C and D), PEG-modified dialkylamines (Figure 2B, structure E)
and PEG-modified
1,2-diacyloxypropan-3-amines (Figure 2B, structure F). Particularly preferred
are PEG-ceramide
conjugates (e.g., PEG-CerCl4) which are described in co-pending USSN
08/486,214,
incorporated herein by reference.
In embodiments where a sterically-large moiety such as PEG or ATTA are
conjugated to a lipid anchor, the selection of the lipid anchor depends on
what type of association
the conjugate is to have with the lipid particle. It is well known that
mePEG(mw2000)-
diastearoylphosphatidylethanolamine (PEG-DSPE) will remain associated with a
liposome until
the particle is cleared from the circulation, possibly a matter of days. Other
conjugates, such as
PEG-CerC20 have similar staying capacity, and thus these compounds are not
suitable as the
steric barrier lipids for this invention. PEG-CerC 14, however, rapidly
exchanges out of the
formulation upon exposure to serum, with a T"Z less than 60 mins. in some
assays. As illustrated
in US Pat. Application SN 08/486,214 at least three characteristics influence
the rate of
exchange: length of acyl chain, saturation of acyl chain, and size of the
steric-barrier head group.
CA 02271582 1999-OS-13
-22-
Compounds having propoerties comparable to PEG-CerC 14 are suitable for use in
the method of
the invention.
In addition to the protonatable or deprotonatable lipid component and the
steric
barrier lipid components, the lipid mixture of the preferred embodiment may
contain additional
lipid species. These additional lipids may be, for example, neutral lipids or
sterols.
Neutral lipids, when present in the lipid mixture, can be any of a number of
lipid
species which exist either in an uncharged or neutral zwitterionic form at
physiological pH. Such
lipids include, for example diacylphosphatidylcholine,
diacylphosphatidylethanolamine,
ceramide, sphingomyelin, cephalin, and cerebrosides. The selection of neutral
lipids for use in
the complexes herein is generally guided by consideration of, e.g., liposome
size and stability of
the liposomes in the bloodstream. Preferably, the neutral lipid component is a
lipid having two
acyl groups, (i.e., diacylphosphatidylcholine and
diacylphosphatidylethanolamine). Lipids
having a variety of acyl chain groups of varying chain length and degree of
saturation are
available or may be isolated or synthesized by well-known techniques. In one
group of
embodiments, lipids containing saturated fatty acids with carbon chain lengths
in the range of C1,~
to CZZ are preferred. In another group of embodiments, lipids with mono or
diunsaturated fatty
acids with carbon chain lengths in the range of C,4 to C22 are used.
Additionally, lipids having
mixtures of saturated and unsaturated fatty acid chains can be used.
Preferably, the neutral lipids
used in the present invention are DOPE, DSPC, POPC, or any related
phosphatidylcholine. The
neutral lipids useful in the present invention may also be composed of
sphingomyelin or
phospholipids with other head groups, such as serine and inositol.
The sterol component of the lipid mixture, when present, can be any of those
sterols conventionally used in the field of liposome, lipid vesicle or lipid
particle preparation. A
preferred sterol is cholesterol.
The mixture of lipids is typically a solution of lipids in an alcoholic
solvent.
Hydrophilic, low molecular weight water miscible alcohols with less than 10
carbon atoms,
preferably less than 6 carbon atoms are preferred. Typical alcohols used in
this invention are
ethanol, methanol, propanol, butanol, pentanol and ethylene glycol and
propylene glycol.
Particularly preferred is ethanol. In most embodiments, the alcohol is used in
the form in which
CA 02271582 1999-OS-13
- 23 -
it is commercially available. For example, ethanol can be used as absolute
ethanol (100%), or as
95% ethanol, the remainder being water.
In one exemplary embodiment, the mixture of lipids is a mixture of amino
lipids,
neutral lipids (other than an amino lipid), a sterol (e.g., cholesterol) and a
PEG-modified lipid
(e.g., a PEG-ceramide) in an alcohol solvent. In preferred embodiments, the
lipid mixture
consists essentially of an amino lipid, a neutral lipid, cholesterol and a PEG-
ceramide in alcohol,
more preferably ethanol. In further preferred embodiments, the first solution
consists of the
above lipid mixture in molar ratios of about 10-35% amino lipid:25-45% neutral
lipid:35-55%
cholestero1:0.5-15% PEG-ceramide. In still further preferred embodiments, the
first solution
consists essentially of DODAP, DSPC, Chol and PEG-CerC 14, more preferably in
a molar ratio
of about 10-35% DODAP:25-45% DSPC:35-55% Cho1:0.5-15% PEG-CerCl4. In another
group
of preferred embodiments, the neutral lipid in these compositions is replaced
with POPC or SM.
In accordance with a preferred embodiment of the invention, the lipid mixture
is
combined with a buffered aqueous solution of charged therapeutic agent,
preferably nucleic
acids. The buffered aqueous solution of therapeutic agents (nucleic acids)
which is combined
with the lipid mixture is typically a solution in which the buffer has a pH of
less than the pKa of
the protonatable lipid in the lipid mixture. As used herein, the term "nucleic
acid" is meant to
include any oligonucleotide or polynucleotide having from 10 to 100,000
nucleotide residues.
Antisense and ribozyme oligonucleotides are particularly preferred. The term
"antisense
oligonucleotide" or simply "antisense" is meant to include oligonucleotides
which are
complementary to a targeted nucleic acid and which contain from about 10 to
about 50
nucleotides, more preferably about 1 S to about 30 nucleotides. The term also
encompasses
antisense sequences which may not be exactly complementary to the desired
target gene. Thus,
the invention can be utilized in instances where non-target specific-
activities are found with
antisense, or where an antisense sequence containing one or more mismatches
with the target
sequence is the most preferred for a particular use.
The nucleic acid that is used in a lipid-nucleic acid particle according to
this
invention includes any form of nucleic acid that is known. Thus, the nucleic
acid may be a
modified nucleic acid of the type used previously to enhance nuclease
resistance and serum
CA 02271582 1999-OS-13
-24-
stability. Surprisingly, however, acceptable therapeutic products can also be
prepared using the
method of the invention to formulate lipid-nucleic acid particles from nucleic
acids which have
no modification to the phosphodiester linkages of natural nucleic acid
polymers, and the use of
unmodified phosphodiester nucleic acids (i.e., nucleic acids in which all of
the linkages are
phosphodiester linkages) is a preferred embodiment of the invention. Still
other nucleic acids
which are useful in the present invention include, synthetic or pre-formed
poly-RNA such as
poly(IC) IC.
The nucleic acids used herein can be single-stranded DNA or RNA, or double-
stranded DNA or DNA-RNA hybrids. Examples of double-stranded DNA include
structural
genes, genes including control and termination regions, and self replicating
systems such as
plasmid DNA. Single-stranded nucleic acids include antisense oligonucleotides
(discussed
above and complementary to DNA and RNA), ribozymes and triplex-forming
oligonucleotides.
In order to increase stability, some single-stranded nucleic acids may have
some
or all of the nucleotide linkages substituted with stable, non-phosphodiester
linkages, including,
for example, phosphorothioate, phosphorodithioate, phosphoroselenate,
boranophosphate,
methylphosphonate, or O-alkyl phosphotriester linkages. Phosphorothioate
nucleic acids (PS-
oligos) are those oligonucleotides or polynucleotides in which one of the non-
bridged oxygens of
the internucleotide linkage has been replaced with sulfur. These PS-oligos are
resistant to
nuclease degradation, yet retain sequence-specific activity. Similarly,
phosphorodithioate
nucleic acids are those oligonucleotides or polynucleotides in which each of
the non-bridged
oxygens of the internucleotide linkage have been replaced by a sulfur atom.
These
phosphorodithioate-oligos have also proven to be more nuclease resistant than
the natural
phosphodiester-linked form. Other useful nucleic acids derivatives include
those nucleic acids
molecules in which the bridging oxygen atoms (those forming the phosphoester
linkages) have
been replaced with -S-, -NH-, -CHz and the like. Preferably, the alterations
to the antisense or
other nucleic acids used will not completely affect the negative charges
associated with the
nucleic acids. Thus, the present invention contemplates the use of antisense
and other nucleic
acids in which a portion of the linkages are replaced with, for example, the
neutral methyl
CA 02271582 1999-OS-13
-25-
phosphonate or phosphoramidate linkages. When neutral linkages are used,
preferably less than
80% of the nucleic acid linkages are so substituted, more preferably less than
50%.
Those skilled in the art will realize that for in vivo utility, such as
therapeutic
efficacy, a reasonable rule of thumb is that if a thioated version of the
sequence works in the free
form, that encapsulated particles of the same sequence, of any chemistry, will
also be efficacious.
Encapsulated particles may also have a broader range of in vivo utilities,
showing efficacy in
conditions and models not known to be otherwise responsive to antisense
therapy. Those skilled
in the art know that applying this invention they may find old models which
now respond to
antisense therapy. Further, they may revisit discarded antisense sequences or
chemistries and
find efficacy by employing the invention.
Therapeutic antisense sequences (putatively target specific) known to work
with this
invention include the following:
Trivial Name: Gene Target, Chemistry and Sequence
PS-3082 murine ICAM-1 (Intracellular Adhesion Molecule-1)
(phosphorothioate)
TGCATCCCCCAGGCCACCAT
(SEQ ID. No 1)
PO-3082 murine ICAM-1 (phosphodiester)
TGCATCCCCCAGGCCACCAT
(SEQ ID. No 1)
PS-2302 human ICAM-1 (phosphorothioate)
GCCCAAGCTGGCATCCGTCA
(SEQ ID. No 2)
PO-2302 human ICAM-1 (phosphodiester)
GCCCAAGCTGGCATCCGTCA (SEQ ID. No 2)
PS-8997 human ICAM-1 (phosphorothioate)
GCCCAAGCTGGCATCCGTCA
(SEQ ID. No 2)
PO- 8997 human ICAM-1 (phosphodiester)
GCCCAAGCTGGCATCCGTCA
(SEQ ID. No 2)
US3 human erb-B-2 gene (phosphodiester or phosphorothioate)
GGT GCT CAC TGC GGC (SEQ ID. No 3)
CA 02271582 1999-OS-13
-26-
LR-3280 human c-myc gene (phosphorothioate)
AAC GTT GAG GGG CAT (SEQ ID. No
4)
Inx-6298 human c-myc gene (phosphodiester)
AAC GTT GAG GGG CAT (SEQ ID. No
4)
Inx-6295 human c-myc gene (phosphodiester or phosphorothioate)
T AAC GTT GAG GGG CAT (SEQ ID. No
5)
LR-3001 human c-myb gene (phosphodiester or phosphorothioate)
TAT GCT GTG CCG GGG TCT TCG GGC
(SEQ ID. No
6)
c-myb human c-myb gene (phosphodiester or phosphorothioate)
GTG CCG GGG TCT TCG GGC
(SEQ ID. No
7)
IGF-1R human IGF-1R (Insulin Growth Factor 1 - Receptor)
(phosphodiester or phosphorothioate)
GGA CCC TCC TCC GGA GCC (SEQ ID. No
8)
LR-42 human IGF-1 R (phosphodiester or phosphorothioate)
TCC TCC GGA GCC AGA CTT
(SEQ ID. No
9)
EGFR human EGFR (Epidermal Growth Factor Receptor)
(phosphodiester or phosphorothioate)
CCG TGG TCA TGC TCC (SEQ ID. No
10)
VEGF human VEGF (Vascular Endothelial Growth Factor)
gene
(phosphodiester or phosphorothioate)
CAG CCT GGC TCA CCG CCT TGG
(SEQ ID. No
11)
PS-4189 murine PKC-alpha (Phosphokinase C - alpha)
gene
(phosphodiester or phosphorothioate)
CAG CCA TGG TTC CCC CCA AC (SEQ ID. No
12)
PS-3521 human PKC-alpha
(phosphodiester or phosphorothioate)
GTT CTC GCT GGT GAG TTT CA (SEQ ID. No
13)
Bcl-2 human bcl-2 gene
(phosphodiester or phosphorothioate)
TCT CCC AgC gTg CgC CAT (SEQ ID. No
14)
CA 02271582 1999-OS-13
-2~-
ATG-AS human c-raf 1 protein kinase
(phosphodiester or phosphorothioate)
GTG CTC CAT TGA TGC
(SEQ ID. No 15 )
VEGF-R1 human VEGF-R -1 (Vascular Endothelial Growth Factor Receptor 1)
ribozyme
GAG UUG CUG AUG AGG CCG AAA GGC CGA AAG UCU G
(SEQ ID. No 16)
Using these sequences, the invention provides a method for the treatment of a
diseases, including tumors, characterized by aberrant expression of a gene in
a mammalian
subject. The method comprises the steps of preparing a lipid-encapsulated
therapeutic nucleic
acid particle according to the methods as described herein, where the
therapeutic nucleic acid
component hybridizes specifically with the aberrantly expressed gene; and
administering a
therapeutically effective amount of the resulting particle to the mammalian
subject. These
sequences are, of course, only representative of the possible therapeutic
oligonucleotide
compounds that can be delivered using the invention. It is well known that,
depending on the
target gene, antisense that hybridizes to any part of the target gene, such as
coding regions,
introns, the 5' untranslated region (5'UTR), start of translation, or 3'UTR
may have therapeutic
utility. Therefore, the sequences listed above are only exemplary of
antisense. Furthermore, all
the alternative chemistries that have been proposed (i.e. see Background) can
be tested with the
invention to determine efficacy along with all types of ribozymes. In short,
the compounds listed
above represent the broad class of therapeutic 5-50 mer oligonucleotides of
various chemistries
which are useful with this invention. Other oligonucleotides which are useful
include all those
which have previously demonstrated efficacy in the free form.
While the invention is generally described and exemplified with regard to
antisense oligonucleotides, other nucleic acids can be formulated and
administered to a subject
for the purpose of repairing or enhancing the expression of a cellular
protein. Accordingly, the
nucleic acid can be an expression vector, cloning vector or the like which is
often a plasmid
designed to be able to replicate in a chosen host cell. Expression vectors may
replicate
autonomously, or they may replicate by being inserted into the genome of the
host cell, by
CA 02271582 1999-OS-13
- 2g -
methods well known in the art. Vectors that replicate autonomously will have
an origin of
replication or autonomous replicating sequence (ARS) that is functional in the
chosen host
cell(s). Often, it is desirable for a vector to be usable in more than one
host cell, e.g., in E. coli
for cloning and construction, and in a mammalian cell for expression.
Additionally, the nucleic acid can carry a label (e.g., radioactive label,
fluorescent
label or colorimetric label) for the purpose of providing clinical diagnosis
relating to the presence
or absence of complementary nucleic acids. Accordingly, the nucleic acids, or
nucleotide
polymers, can be polymers of nucleic acids including genomic DNA, cDNA, mRNA
or
oligonucleotides containing nucleic acid analogs, for example, the antisense
derivatives
described in a review by Stein, et al., Science 261:1004-1011 (1993) and in
U.S. Patent Nos.
5,264,423 and 5,276,019, the disclosures of which are incorporated herein by
reference. Still
further, the nucleic acids may encode transcriptional and translational
regulatory sequences
including promoter sequences and enhancer sequences.
The nucleic acids used in the present invention will also include those
nucleic
acids in which modifications have been made in one or more sugar moieties
and/or in one or
more of the pyrimidine or purine bases. Examples of sugar modifications
include replacement of
one or more hydroxyl groups with halogens, alkyl groups, amines, azido groups
or functionalized
as ethers or esters. Additionally, the entire sugar may be replaced with
sterically and electron-
ically similar structures, including aza-sugars and carbocyclic sugar analogs.
Modifications in
the purine or pyrimidine base moiety include, for example, alkylated purines
and pyrimidines,
acylated purines or pyrimidines, or other heterocyclic substitutes known to
those of skill in the
art. As with the modifications to the phosphodiester linkages discussed above,
any modifications
to the sugar or the base moieties should also act to preserve at least a
portion of the negative
charge normally associated with the nucleic acid. In particular, modifications
will preferably
result in retention of at least 10% of the overall negative charge, more
preferably over 50% of the
negative charge and still more preferably over 80% of the negative charge
associated with the
nucleic acid.
The nucleic acids used in the present method can be isolated from natural
sources,
obtained from such sources as ATCC or GenBank libraries or prepared by
synthetic methods.
CA 02271582 1999-OS-13
-29-
Synthetic nucleic acids can be prepared by a variety of solution or solid
phase methods. Gener-
ally, solid phase synthesis is preferred. Detailed descriptions of the
procedures for solid phase
synthesis of nucleic acids by phosphite-triester, phosphotriester, and H-
phosphonate chemistries
are widely available. See, for example, Itakura, U.S. Pat. No. 4,401,796;
Caruthers, et al., U.S.
S Pat. Nos. 4,458,066 and 4,500,707; Beaucage, et al., Tetrahedron Lett.,
22:1859-1862 (1981);
Matteucci, et al., J. Am. Chem. Soc., 103:3185-3191 (1981); Caruthers, et al.,
Genetic Engin-
eering, 4:1-17 (1982); Jones, chapter 2, Atkinson, et al., chapter 3, and
Sproat, et al., chapter 4,
in Oligonucleotide Synthesis: A Practical Approach, Gait (ed.), IRL Press,
Washington D.C.
(1984); Froehler, et al., Tetrahedron Lett., 27:469-472 (1986); Froehler, et
al., Nucleic Acids
Res., 14:5399-5407 (1986); Sinha, et al. Tetrahedron Lett., 24:5843-5846
(1983); and Sinha, et
al., Nucl. Acids Res., 12:4539-4557 (1984) which are incorporated herein by
reference.
As noted above, the solution of therapeutic agent (nucleic acids) comprises an
aqueous buffer. Preferred buffers (in the case of anionic therapeutic agents)
are those which
provide a pH of less than the pKa of the first lipid component. Examples of
suitable buffers
include citrate, phosphate, acetate, and MES. A particularly preferred buffer
is citrate buffer.
Preferred buffers will be in the range of 1-1000 mM of the anion, depending on
the chemistry of
the oligonucleotide being encapsulated, and optimization of buffer
concentration may be
significant to achieving high loading levels (See. Figs 15 and 20).
Alternatively, pure water
acidified to pH 5-6 with chloride, sulfate or the like may be useful. In this
case, it may be
suitable to add S% glucose, or another non-ionic solute which will balance the
osmotic potential
across the particle membrane when the particles are dialyzed to remove
ethanol, increase the pH,
or mixed with a pharmaceutically acceptable carrier such as normal saline. The
amount of
therapeutic agent (nucleic acid) in buffer can vary, but will typically be
from about 0.01 mg/mL
to about 200 mg/mL, more preferably from about 0.5 mg/mL to about 50 mg/mL.
The mixture of lipids and the buffered aqueous solution of therapeutic agent
(nucleic acids) is combined to provide an intermediate mixture. The
intermediate mixture is
typically a mixture of lipid particles having encapsulated therapeutic agent
(nucleic acids).
Additionally, the intermediate mixture may also contain some portion of
therapeutic agent
(nucleic acids) which are attached to the surface of the lipid particles
(liposomes or lipid vesicles)
CA 02271582 1999-OS-13
-30-
due to the ionic attraction of the negatively-charged nucleic acids and
positively-charged lipids
on the lipid particle surface (the amino lipids or other lipid making up the
protonatable first lipid
component are positively charged in a buffer having a pH of less than the pKa
of the protonatable
group on the lipid). In one group of preferred embodiments, the mixture of
lipids is an alcohol
solution of lipids and the volumes of each of the solutions is adjusted so
that upon combination,
the resulting alcohol content is from about 20% by volume to about 45% by
volume. The
method of combining the mixtures can include any of a variety of processes,
often depending
upon the scale of formulation produced. For example, when the total volume is
about 10-20 mL
or less, the solutions can be combined in a test tube and stirred together
using a vortex mixer.
Large-scale processes can be carried out in suitable production scale
glassware.
Optionally, the lipid-encapsulated therapeutic agent (nucleic acid) complexes
which are produced by combining the lipid mixture and the buffered aqueous
solution of
therapeutic agents (nucleic acids) can be sized to achieve a desired size
range and relatively
narrow distribution of lipid particle sizes. Preferably, the compositions
provided herein will be
sized to a mean diameter of from about 70 to about 200 nm, more preferably
about 90 to about
130 nm. Several techniques are available for sizing liposomes to a desired
size. One sizing
method is described in U.S. Pat. No. 4,737,323, incorporated herein by
reference. Sonicating a
liposome suspension either by bath or probe sonication produces a progressive
size reduction
down to small unilamellar vesicles (SUVs) less than about 0.05 microns in
size.
Homogenization is another method which relies on shearing energy to fragment
large liposomes
into smaller ones. In a typical homogenization procedure, multilamellar
vesicles are recirculated
through a standard emulsion homogenizer until selected liposome sizes,
typically between about
0.1 and 0.5 microns, are observed. In both methods, the particle size
distribution can be
monitored by conventional laser-beam particle size determination. For the
methods herein,
extrusion is used to obtain a uniform vesicle size.
Extrusion of liposome compositions through a small-pore polycarbonate
membrane or an asymmetric ceramic membrane results in a relatively well-
defined size
distribution. Typically, the suspension is cycled through the membrane one or
more times until
the desired liposome complex size distribution is achieved. The liposomes may
be extruded
CA 02271582 1999-OS-13
-31-
through successively smaller-pore membranes, to achieve a gradual reduction in
liposome size.
In some instances, the lipid-nucleic acid compositions which are formed can be
used without any
sizing.
The present invention further comprises a step of neutralizing at least some
of the
surface charges on the lipid portions of the lipid-nucleic acid compositions.
By at least partially
neutralizing the surface charges, unencapsulated antisense or other nucleic
acid is freed from the
lipid particle surface and can be removed from the composition using
conventional techniques.
Preferably, unencapsulated and surface adsorbed nucleic acids is removed from
the resulting
compositions through exchange of buffer solutions. For example, replacement of
a citrate buffer
(pH about 4.0, used for forming the compositions) with a HEPES-buffered saline
(HBS pH about
7.5) solution, results in the neutralization of liposome surface and antisense
release from the
surface. The released antisense can then be removed via chromatography using
standard
methods, and then switched into a buffer with a pH above the pKa of the lipid
used.
In other aspects, the present invention provides lipid-encapsulated nucleic
acid
1 S compositions, preferably prepared by the methods recited above.
Accordingly, preferred
compositions are those having the lipid ratios and nucleic acid preferences
noted above.
In still other aspects, the present invention contemplates reversed-charge
methods
in which the lipid portion of the complex contains certain anionic lipids and
the component
which is encapsulated is a positively charged therapeutic agent. One example
of a positively
charged agent is a positively charged peptide or protein. In essentially an
identical manner,
liposome-encapsulated protein is formed at a pH above the pKa of the anionic
lipid, then the
surface is neutralized by exchanging the buffer with a buffer of lower pH
(which would also
release surface-bound peptide or protein).
IV. Pharmaceutical Preparations
The lipid-nucleic acid compositions prepared by the above methods can be
administered either alone or in mixture with a physiologically-acceptable
carrier (such as
CA 02271582 1999-OS-13
-32-
physiological saline or phosphate buffer) selected in accordance with the
route of administration
and standard pharmaceutical practice.
Pharmaceutical compositions comprising the lipid-nucleic acid compositions of
the invention are prepared according to standard techniques and further
comprise a
pharmaceutically acceptable carrier. Generally, normal saline will be employed
as the
pharmaceutically acceptable carrier. Other suitable carriers include, e.g.,
water, buffered water,
0.9% saline, 0.3% glycine, and the like, including glycoproteins for enhanced
stability, such as
albumin, lipoprotein, globulin, etc. In compositions comprising saline or
other salt containing
carriers, the carrier is preferably added following lipid particle formation.
Thus, after the lipid-
nucleic acid compositions are formed, the compositions can be diluted into
pharmaceutically
acceptable Garners such as normal saline. The resulting pharmaceutical
preparations may be
sterilized by conventional, well known sterilization techniques. The aqueous
solutions can then
be packaged for use or filtered under aseptic conditions and lyophilized, the
lyophilized prepara-
tion being combined with a sterile aqueous solution prior to administration.
The compositions
may contain pharmaceutically acceptable auxiliary substances as required to
approximate
physiological conditions, such as pH adjusting and buffering agents, tonicity
adjusting agents
and the like, for example, sodium acetate, sodium lactate, sodium chloride,
potassium chloride,
calcium chloride, etc. Additionally, the lipidic suspension may include lipid-
protective agents
which protect lipids against free-radical and lipid-peroxidative damages on
storage. Lipophilic
free-radical quenchers, such as a-tocopherol and water-soluble iron-specific
chelators, such as
ferrioxamine, are suitable.
The concentration of lipid-nucleic acid complexes in the pharmaceutical
formulations can vary widely, i. e., from less than about 0.01 %, usually at
or at least about 0.05-
5% to as much as 10 to 30% by weight and will be selected primarily by fluid
volumes,
viscosities, etc., in accordance with the particular mode of administration
selected. For example,
the concentration may be increased to lower the fluid load associated with
treatment. This may
be particularly desirable in patients having atherosclerosis-associated
congestive heart failure or
severe hypertension. Alternatively, complexes composed of irritating lipids
may be diluted to
low concentrations to lessen inflammation at the site of administration. In
one group of
CA 02271582 1999-OS-13
-33-
embodiments, the nucleic acid will have an attached label and will be used for
diagnosis (by
indicating the presence of complementary nucleic acid). In this instance, the
amount of
complexes administered will depend upon the particular label used, the disease
state being
diagnosed and the judgement of the clinician but will generally be between
about 0.01 and about
50 mg per kilogram of body weight, preferably between about 0.1 and about 5
mg/kg of body
weight.
As noted above, the lipid-therapeutic agent (nucleic acid) compositions of the
invention include polyethylene glycol (PEG)-modified phospholipids, PEG-
ceramide, or
ganglioside G~,-modified lipids or other lipids effective to prevent or limit
aggregation.
Addition of such components does not merely prevent complex aggregation,
however, it may
also provides a means for increasing circulation lifetime and increasing the
delivery of the lipid-
nucleic acid composition to the target tissues.
The present invention also provides lipid-nucleic acid compositions in kit
form.
The kit will typically be comprised of a container which is compartmentalized
for holding the
various elements of the kit. The kit will contain the compositions of the
present inventions,
preferably in dehydrated or concentrated form, with instructions for their
rehydration or dilution
and administration. In still other embodiments, the lipid-encapsulated-
therapeutic agent (nucleic
acid) particles will have a targeting moiety attached to the surface of the
lipid particle. Methods
of attaching targeting moieties (e.g., antibodies, proteins, small molecule
mimetics, vitamins,
oligosaccharides and hyaluronic acid) to lipids (such as those used in the
present compositions)
are known to those of skill in the art.
Dosage for the lipid-nucleic acid compositions will depend on the ratio of
nucleic
acid to lipid and the administrating physician's opinion based on age, weight,
and condition of
the patient.
V. Methods of Introducing Lipid-Encapsulated Therapeutic Agents Into Cells
The lipid-therapeutic agent compositions of the invention can be used for
introduction of those therapeutic agents into cells. In the case of nucleic
acid-containing
compositions, the composition of the invention are useful for the introduction
of nucleic acids,
CA 02271582 1999-OS-13
-34-
preferably plasmids, antisense and ribozymes into cells. Accordingly, the
present invention also
provides methods for introducing a therapeutic agent such as a nucleic acid
into a cell. The
methods are carried out in vitro or in vivo by first forming the compositions
as described above,
then contacting the compositions with the target cells for a period of time
sufficient for
transfection to occur.
The compositions of the present invention can be adsorbed to almost any cell
type. Once adsorbed, the complexes can either be endocytosed by a portion of
the cells,
exchange lipids with cell membranes, or fuse with the cells. Transfer or
incorporation of the
nucleic acid portion of the complex can take place via any one of these
pathways. In particular,
when fusion takes place, the liposome membrane is integrated into the cell
membrane and the
contents of the liposome combine with the intracellular fluid. Contact between
the cells and the
lipid-nucleic acid compositions, when carried out in vitro, will take place in
a biologically
compatible medium. The concentration of compositions can vary widely depending
on the
particular application, but is generally between about 1 pmol and about 10
mmol. Treatment of
the cells with the lipid-nucleic acid compositions will generally be carried
out at physiological
temperatures (about 37 ° C) for periods of time of from about 1 to 6
hours, preferably of from
about 2 to 4 hours. For in vitro applications, the delivery of nucleic acids
can be to any cell
grown in culture, whether of plant or animal origin, vertebrate or
invertebrate, and of any tissue
or type. In preferred embodiments, the cells will be animal cells, more
preferably mammalian
cells, and most preferably human cells.
In one group of preferred embodiments, a lipid-nucleic acid particle
suspension is
added to 60-80% confluent plated cells having a cell density of from about 103
to about 105
cells/mL, more preferably about 2 X 10° cells/mL. The concentration of
the suspension added to
the cells is preferably of from about 0.01 to 0.2 ~g/mL, more preferably about
0.1 pg/mL.
Typical applications include using well known transfection procedures to
provide
intracellular delivery of DNA or mRNA sequences which code for therapeutically
useful
polypeptides. In this manner, therapy is provided for genetic diseases by
supplying deficient or
absent gene products (i.e., for Duchenne's dystrophy, see Kunkel, et al.,
Brit. Med. Bull.
45(3):630-643 (1989), and for cystic fibrosis, see Goodfellow, Nature 341:102-
103 (1989)).
CA 02271582 1999-OS-13
-35-
Other uses for the compositions of the present invention include introduction
of antisense
oligonucleotides in cells (see, Bennett, et al., Mol. Pharm. 41:1023-1033
(1992)).
Alternatively, the compositions of the present invention can also be used for
the
transfection of cells in vivo, using methods which are known to those of skill
in the art. In
particular, Zhu, et al., Science 261:209-211 (1993), incorporated herein by
reference, describes
the intravenous delivery of cytomegalovirus (CMV)-chloramphenicol
acetyltransferase (CAT)
expression plasmid using DOTMA-DOPE complexes. Hyde, et al., Nature 362:250-
256 (1993),
incorporated herein by reference, describes the delivery of the cystic
fibrosis transmembrane
conductance regulator (CFTR) gene to epithelia of the airway and to alveoli in
the lung of mice,
using liposomes. Brigham, et al., Am. J. Med. Sci. 298:278-281 (1989),
incorporated herein by
reference, describes the in vivo transfection of lungs of mice with a
functioning prokaryotic gene
encoding the intracellular enzyme, chloramphenicol acetyltransferase (CAT).
Thus, the
compositions of the invention can be used in the treatment of infectious
diseases.
For in vivo administration, the pharmaceutical compositions are preferably
administered parenterally, i.e., intraarticularly, intravenously,
intraperitoneally, subcutaneously,
or intramuscularly. More preferably, the pharmaceutical compositions are
administered
intravenously or intraperitoneally by a bolus injection. For example, see
Stadler, et al., U.S.
Patent No. 5,286,634, which is incorporated herein by reference. Intracellular
nucleic acid
delivery has also been discussed in Straubringer, et al., METHODS ~N
ENZYMOLOGY, Academic
Press, New York. 101:512-527 (1983); Mannino, et al., Biotechniques 6:682-690
(1988);
Nicolau, et al., Crit. Rev. Ther. Drug Carrier Syst. 6:239-271 (1989), and
Behr, Acc. Chem. Res.
26:274-278 (1993). Still other methods of administering lipid-based
therapeutics are described
in, for example, Rahman et al., U.S. Patent No. 3,993,754; Sears, U.S. Patent
No. 4,145,410;
Papahadjopoulos et al., U.S. Patent No. 4,235,871; Schneider, U.S. Patent No.
4,224,179; Lenk
et al., U.S. Patent No. 4,522,803; and Fountain et al., U.S. Patent No.
4,588,578.
In other methods, the pharmaceutical preparations may be contacted with the
target tissue by direct application of the preparation to the tissue. The
application may be made
by topical, "open" or "closed" procedures. By "topical", it is meant the
direct application of the
pharmaceutical preparation to a tissue exposed to the environment, such as the
skin, oropharynx,
CA 02271582 1999-OS-13
-36-
external auditory canal, and the like. "Open" procedures are those procedures
which include
incising the skin of a patient and directly visualizing the underlying tissue
to which the
pharmaceutical preparations are applied. This is generally accomplished by a
surgical procedure,
such as a thoracotomy to access the lungs, abdominal laparotomy to access
abdominal viscera, or
other direct surgical approach to the target tissue. "Closed" procedures are
invasive procedures
in which the internal target tissues are not directly visualized, but accessed
via inserting
instruments through small wounds in the skin. For example, the preparations
may be
administered to the peritoneum by needle lavage. Likewise, the pharmaceutical
preparations
may be administered to the meninges or spinal cord by infusion during a lumbar
puncture
followed by appropriate positioning of the patient as commonly practiced for
spinal anesthesia or
metrazamide imaging of the spinal cord. Alternatively, the preparations may be
administered
through endoscopic devices.
The lipid-nucleic acid compositions can also be administered in an aerosol
inhaled
into the lungs (see, Brigham, et al., Am. J. Sci. 298(4):278-281 (1989)) or by
direct injection at
the site of disease (Culver, HUMAN GENE THERAPY, MaryAnn Liebert, Inc.,
Publishers, New
York. pp.70-71 (1994)).
The methods of the present invention may be practiced in a variety of hosts.
Preferred hosts include mammalian species, such as humans, non-human primates,
dogs, cats,
cattle, horses, sheep, and the like.
VI. Examples
Materials and Methods:
Lipids
Distearoylphosphatidylcholine (DSPC), sphingomyelin (SM), and
palmitoyloleoylphosphatidylcholine (POPC) were purchased from Northern Lipids
(Vancouver,
Canada). 1,2-dioleoyloxy-3-dimethylammoniumpropane (DODAP or AL-1) was
synthesized by
Dr. Steven Ansell (Inex Pharmaceuticals) or, alternatively, was purchased from
Avanti Polar
Lipids. Cholesterol was purchased from Sigma Chemical Company (St. Louis,
Missouri, USA).
PEG-ceramides were synthesized by Dr. Zhao Wang at Inex Pharmaceuticals Corp.
using
CA 02271582 1999-OS-13
-37-
procedures described in PCT WO 96140964, incorporated herein by reference.
[3H] or ['''C]-CHE
was purchased from NEN (Boston, Massachusetts, USA). All lipids were > 99%
pure.
Buffers and Solvents
Ethanol (95%), methanol, chloroform, citric acid, HEPES and NaCI were all
purchased from commercial suppliers.
Synthesis and Purification of Phosphorothioate Antisense
PS 3082, a 20mer phosphorothioate antisense oligodeoxynucleotide, was
synthesized, purified and donated by ISIS Pharmaceuticals (Carlsbad,
California, USA). The
sequence for this oligo is: TGCATCCCCCAGGCCACCAT. (Seq ID No 1) The details of
the
synthesis and purification can be found elsewhere (see, Stepkowski, et al., J.
Immunol.
153:5336-5346 (1994)).
Preparation of Liposomal Antisense
Lipid stock solutions were prepared in 95% ethanol at 20 mg/mL
(PEG-Ceramides were prepared at 50 mg/mL). DSPC, CHOL, DODAP, PEG-CerCl4
(25:45:20:10, molar ratio), 13 qmol total lipid, were added to a 13 x 100 mm
test tube containing
trace amounts of ['4C]-cholesterylhexadecylether. The final volume of the
lipid mixture was 0.4
mL. In some experiments, SM or POPC was substituted for DSPC. A 20mer
antisense
oligodeoxynucleotide, PS 3082 (2 mg), and trace amounts of [3H]-PS 3082 were
dissolved in 0.6
mL of 300 mM citric acid, pH 3.8 in a separate 13 x 100 mm test tube. The
antisense solution
was warmed to 65°C and the lipids (in ethanol) were slowly added,
mixing constantly. The
resulting volume of the mixture was 1.0 mL and contained 13 ~mol total lipid,
2 mg of antisense
oligodeoxynucleotide, and 38% ethanol, vol/vol. The antisense-lipid mixture
was subjected to 5
cycles of freezing (liquid nitrogen) and thawing (65 ° C), and
subsequently was passed l OX
through three stacked 100 nm filters (Poretics) using a pressurized extruder
apparatus with a
thermobarrel attachment (Lipex Biomembranes). The temperature and pressure
during extrusion
were 65 °C and 300-400 psi (nitrogen), respectively. The extruded
preparation was diluted with
CA 02271582 1999-OS-13
-38-
1.0 mL of 300 mM citric acid, pH 3.8, reducing the ethanol content to 20%. The
preparation was
immediately applied to a gel filtration column. Alternatively, the extruded
sample was dialyzed
(12 000-14 000 MW cutoff; SpectraPor) against several liters of 300 mM citrate
buffer, pH 3.8
for 3-4 hours to remove the excess ethanol. The sample was subsequently
dialyzed against HBS,
pH 7.5, for 12-18 hours to neutralize the DODAP and release any antisense that
was associated
with the surface of the vesicles. The free antisense was removed from the
encapsulated
liposomal antisense by gel exclusion chromatography as described below.
Gel Filtration Chromatography
A 20 x 2.5 cm glass column containing Biogel AlSm, 100-200 mesh, was
equilibrated in HEPES-buffered saline (HBS; 20 mM HEPES, 145 mM NaCI, pH 7.5).
The 2.0
mL liposomal antisense preparation was applied to the column and allowed to
drain into the gel
bed under gravity. The column was eluted with HBS at a flow rate of 50 mL/hr.
Column
fractions (1.0 mL) were collected and analyzed for radioactivity using
standard liquid
scintillation counting techniques. The fractions were pooled based on the
levels of ['4C]-CHE
present in the fraction. The size distribution of the pooled liposomal
antisense was determined
using a NICOMP Model 370 Sub-micron particle sizer and was typically 110 ~ 30
nm.
Ion Exchange Chromatography
As an alternative to gel filtration chromatography, samples were sometimes
dialyzed first in 300 mM citrate, pH 3.80, for 2-3 hours to remove residual
ethanol, followed by
at least a 12 hour dialysis in HBS, to exchange the external citrate for HBS
and remove residual
ethanol. The sample was applied to a 1.5 x 8 cm DEAE-Sepharose~ column
equilibrated in
HBS. Free oligonucleotide binds to the DEAE with very high affinity. The peak
containing the
lipid was pooled, concentrated, and analyzed for antisense content, as
described below.
CA 02271582 1999-OS-13
-39-
Assessment of Ahtisense Encapsulation
Antisense encapsulation was typically assessed by dual label ([3H]-antisense
and
['4C]-lipid) liquid scintillation counting after gel filtration chromatography
to separate the free
and encapsulated antisense. Antisense encapsulation was evaluated by summing
the total
[3H]-antisense radioactivity associated with the lipid peak and dividing by
the total [3H]-antisense
radioactivity. Alternatively, the [3H]/["C] ratio was determined before and
after (i.e., in the
pooled lipid peak) gel filtration chromatography. Antisense encapsulation was
also assessed by
measuring the absorbance of the sample at 260 nm, preceded by a Bligh and Dyer
extraction of
the antisense from the lipid, as described below.
Extraction of the Antisense
The antisense was extracted from the lipid according to the procedure outlined
by
Bligh and Dyer (Bligh, et al., Can. J. Biochem. Physiol. 37:911-917 (1959)).
Briefly, up to 250
~L of aqueous sample was added to a 13 x 100 mm glass test tube, followed by
the addition of
750 pL of chloroform:methanol (1:2.1, vol/vol), 250 pL of chloroform, and 250
pL of distilled
water. The sample was mixed after each addition. The sample was centrifuged
for 10 min. at
3000 rpm, resulting in a clear two-phase separation. The aqueous phase (top)
was removed into
a new 13 x 100 mm test tube. An aliquot (500 ~L) of this phase was diluted
with 500 ~L of
distilled water, mixed, and the absorbance at 260 nm was assessed using a
spectrophotometer. In
some instances, the organic phase (bottom) was washed with 250 pL of methanol,
centrifuged
for 10 min. at 3000 rpm, and the upper phase removed and discarded. This was
repeated 3 times.
The washed organic phase was assessed for phospholipid content according to
the method of
Fiske and Subbarrow (Fiske, et al., J. Biol. Chem. 66:375-400 (1925)).
OLIGREEN Assay
A fluorescent dye binding assay for quantifying single stranded
oligonucleotide in
aqueous solutions was established using a BioluminTM 960 fluorescent plate
reader (Molecular
Dynamics, Sunnyvale, California, USA). Briefly, aliquots of encapsulated
oligonucleotide were
diluted in HEPES buffered saline (HBS; 20mM HEPES, 145mM NaCI, pH 7.5) . A 10
~uL
CA 02271582 1999-OS-13
-40-
aliquot of the diluted sample was added to 100 ~L of a 1:200 dilution of
OligreenTM reagent, both
with and without 0.1% of Triton X-100 detergent. An oligo standard curve was
prepared with
and without 0.1 % Triton X-100 for quantification of encapsulated oligo.
Fluorescence of the
OLIGREENT"'-antisense complex was measured using excitation and emission
wavelengths of
485nm and 520nm, respectively. Surface associated antisense was determined by
comparing the
fluorescence measurements in the absence and presence of detergent.
Ear Inflammation Model and Efficacy Studies
Sensitization and Elicitation of Contact Sensitivity
Mice were sensitized by applying 25 pL of 0.5% 2,4-dinitro-1-fluorobenzene
(DNFB) in acetone:olive oil (4:1) to the shaved abdominal wall for two
consecutive days. Four
days after the second application, mice were challenged on the dorsal surface
of the left ear with
10 pL of 0.2% DNFB in acetone:olive oil (4:1). Mice received no treatment on
the contralateral
(right) ear. In some cases, control mice received 10 ~L of vehicle on the
dorsal surface of the left
ear.
Evaluation of Ear Swelling
Ear thickness was measured immediately prior to ear challenge, and at various
time intervals after DNFB challenge, using an engineer's micrometer (Mitutoyo,
Tokyo, Japan).
Increases in ear thickness measurements were determined by subtracting the pre-
challenge from
post-challenge measurements.
The progression of ear inflammation over a 3 day period for ICR (outbred) mice
is
indicated in Figures 12 and 13. Erythema was evident almost immediately after
ear challenge
and gradually declined in intensity over the remainder of the study. ICR mice
exhibited peak ear
thickness at 24 hours after the induction of ear inflammation. Maximal ear
thickness
measurements were found to be 170 x 10-4 inches, corresponding to a 70%
increase in ear
thickness. Although ear swelling gradually declines at 48 and 72 hours after
inflammation
initiation, ear measurements still have not returned to baseline thickness
levels (90-100 x 10-4
inches).
CA 02271582 1999-OS-13
-41 -
The mouse in vivo experimental systems in this specification were selected in
part
because of their high degree of correlation to human disease conditions. The
mouse ear
inflammation model , which can be treated using methods and compositions of
the invention, is
well known to be an excellent model for human allergic contact dermatitis and
other disease
conditions. The control therapeutic used in this model is a corticosteroid
which demonstrates
efficacy both in the mouse model and in related human disease conditions.
The mouse B 16 tumor model, a fast growing melanoma, which can be treated
using methods and compositions of the invention, is a standard, widely used
experimental
system. This tumor model can be successfully treated using vinca alkaloids,
such as vincristine
or vinblastine, which are known to be efficacious against human tumors as
well.
Treatments which demonstrate utility in the mouse models of this invention are
excellent candidates for testing against human disease conditions, at similar
dosages and
administration modalities.
1 S EXAMPLE 1
This example illustrates the effects of ethanol on the encapsulation of
antisense.
A 20mer of [3H]-phosphorothioate antisense oligodeoxynucleotide (in 300 mM
citrate buffer, pH 3.80) was mixed with an ethanol solution of lipid
(DSPC:CHOL:DODAP:
PEG-CerCl4; 25:45:20:10, molar ratio) at final concentrations of 2 mg/mL and
9.9 mg/mL,
respectively. The final ethanol concentration in the preparations was varied
between 0 and 60%,
vol/vol. The samples were extruded ten times through three 100 nm filters as
described in
"Materials and Methods". The samples were dialyzed for 2-3 hours in 300 mM
citrate buffer, pH
3.80, to remove a majority of the excess ethanol. The samples were switched to
HEPES-buffered
saline (HBS), pH 7.50, and dialyzed for a minimum of 12 hours to replace the
external citrate
buffer with HBS. This renders the majority of DODAP in the outer bilayer
neutral, and will
release any surface bound antisense. Non-encapsulated antisense was then
removed from the
liposomal antisense by DEAE-sepharose chromatography as described in "Material
and
Methods". Encapsulation was assessed either by analyzing the pre-column and
post-column
CA 02271582 1999-OS-13
- 42 -
ratios of [3H]-antisense and ['4C]-lipid or by determining the total pre-
column and post-column
[3H]-antisense and ['4C]-lipid radioactivity.
In another experiment, the formulations were prepared as described. After
extrusion, the filters were analyzed for [3H]-antisense and ['4C]-lipid
radioactivity by standard
scintillation counting techniques. Results were expressed as a percent of the
total initial
radioactivity.
Figure 3 demonstrates the effects of ethanol on the encapsulation of antisense
at
pH 3.8. The encapsulation efficiency of phosphorothioate antisense increases
in a near linear
manner up to a final ethanol concentration of 50%, vol/vol. At an ethanol
content greater than
50%, a large amount of aggregation/precipitation is observed. The effect of
ethanol on vesicle
formation can be further observed by monitoring both lipid and antisense loss
on the filters
during extrusion (Figure 4). At low ethanol contents, extrusion is slow and
the proportion of
lipid and antisense loss is the same, suggesting that the losses are due to
the formation of large
complexes which get trapped on the filter. At ethanol contents of 30 and 40%,
extrusion is very
quick and losses of both lipid and antisense are minimal. As the ethanol
content is increased
above 40%, the loss of antisense becomes disproportionally high relative to
the lipid. This can
be attributed to the insolubility of DNA in high concentrations of alcohol.
Furthermore, in the
presence of ethanol, PEG is required to prevent aggregation and fusion of the
vesicles (results not
shown).
EXAMPLE 2
This example illustrates the effects of DODAP on the encapsulation of
antisense,
and further illustrates the effect of initial antisense concentration on the
compositions.
Having demonstrated that ethanol can greatly facilitate the preparation of
lipid
vesicles containing entrapped antisense, the next step was to examine the
influence of DODAP
(AL-1) content on the encapsulation of antisense (Figure 5). Accordingly, a
4.6 mL aliquot of a
[3H]-phosphorothioate antisense oligodeoxynucleotide (in 300 mM citrate
buffer, pH 3.80) was
mixed with 0.4 mL of a 95% ethanol solution of lipid (DSPC:CHOL:DODAP:PEG-
CerCl4;
100-(55+X):45:X:10, molar ratio) at final concentrations of 2 mg/mL and 9.9
mg/mL,
CA 02271582 1999-OS-13
- 43 -
respectively. The molar ratio of DODAP was varied between 0 and 30%. The molar
ratio of
DSPC was adjusted to compensate for the changes in DODAP content. The samples
were
extruded ten times through three 100 nm filters as described in "Materials and
Methods", and
were dialyzed for 2-3 hours in 300 mM citrate buffer, pH 3.80, to remove a
majority of the
S excess ethanol. The samples were switched to HEPES-buffered saline (HBS), pH
7.50, and
dialyzed for a minimum of 12 hours to replace the external citrate buffer with
HBS.
Non-encapsulated antisense was then removed from the liposomal antisense by
DEAE-sepharose
chromatography as described in "Material and Methods". Encapsulation was
assessed either by
analyzing the pre-column and post-column ratios of [3H]-antisense and ['4C]-
lipid or by
determining the total pre-column and post-column [3H]-antisense and ['4C]-
lipid radioactivity.
As seen in Figure S, antisense encapsulation increased significantly between S-
20% DODAP. At
DODAP contents greater than 20-25%, extrusion of the vesicles became more
difficult
suggesting the formation of complexes. At DODAP concentration of 40 and SO%,
extrusion of
the lipid / antisense mixture took hours compared to minutes for a lipid
composition containing
20% DODAP. To verify that the antisense was indeed associated with the lipid
and that the
observed encapsulation was not due to exchange of the [3H]-label from the
antisense onto the
lipid, the antisense was extracted from the lipid using a Bligh and Dyer
extraction. Using this
technique, the antisense, which is soluble in the aqueous phase, was separated
from the lipid
(soluble in the organic phase) and quantified by measuring the absorbance at
260 nm (Figure 6).
While this method can underestimate the antisense concentration, the technique
substantiated that
the observed association of antisense with the lipid was not an artifact.
In yet another experiment, varying concentrations of a 20mer of
[3H]-phosphorothioate antisense oligodeoxynucleotide (in 300 mM citrate
buffer, pH 3.80) were
mixed with an ethanol solution of lipid (DSPC:CHOL:DODAP:PEG-CerCl4;
25:45:20:10,
molar ratio), 9.9 mg/mL (final concentration). The samples were extruded and
dialyzed twice as
described above. Non-encapsulated antisense was then removed from the
liposomal antisense by
DEAE-sepharose chromatography as described in "Material and Methods".
Encapsulation was
assessed either by analyzing the pre-column and post-column ratios of [3H]-
antisense and
['4C]-lipid or by determining the total pre-column and post-column [3H]-
antisense and ['~C]-lipid
CA 02271582 1999-OS-13
-44-
radioactivity. EPC:CH liposomes containing encapsulated antisense are included
for
comparison.
Optimization of the drug:lipid ratio was accomplished by increasing the
initial
antisense concentration that was mixed with 9.8 mg total lipid
(DSPC:CHOL:DODAP:PEG-CerCl4; 25:45:20:10) (Figure 8). Drug:lipid ratios of up
to 0.25,
w/w, were obtained using 10 mg/mL of antisense in the preparation. However,
the increased
drug:lipid ratio was accompanied by a decrease in encapsulation efficiency,
therefore a
compromise must be made between optimizing the drug:lipid ratio and
encapsulation efficiency.
In comparison, antisense encapsulated by hydration of a dry lipid film (i.e.
EPC:CHOL) in the
absence of cationic lipid typically yields low encapsulation efficiencies (<
12-15%) and
drug:lipid ratios (< 0.1, w/w). Consequently, significant quantities of
antisense are wasted
during the encapsulation procedure.
EXAMPLE 3
This example illustrates the properties of the liposomal antisense
formulations
provided in the Materials and Methods above.
The size distribution of a liposomal preparation of antisense was determined
by
quasi-elastic light scattering (QELS) immediately after removal of the free
antisense (A), and
after storage of the preparation for 2 months at 4°C (B), using a
Nicomp Model 370 sub-micron
particle sizer. A 0.6 mL aliquot of a [3H]-phosphorothioate antisense
oligodeoxynucleotide (in
300 mM citrate buffer, pH 3.80) was mixed with 0.4 mL of a 95% ethanol
solution of lipid
(DSPC:CHOL:DODAP:PEG-CerCl4; 25:45:20:10, molar ratio) at final concentrations
of 2
mg/mL and 9.9 mg/mL, respectively. The sample was extruded ten times through
three 100 nm
filters as described in "Materials and Methods", and dialyzed for 2-3 hours in
300 mM citrate
buffer, pH 3.80, to remove a majority of the excess ethanol. The sample was
switched to
HEPES-buffered saline (HBS), pH 7.50, and dialyzed for a minimum of 12 hours
to replace the
external citrate buffer with HBS. Non-encapsulated antisense was then removed
from the
liposomal antisense by DEAE-sepharose chromatography as described in "Material
and
Methods".
CA 02271582 1999-OS-13
- 45 -
The size distribution and storage stability of antisense preparations
described
herein is demonstrated in Figure 7. The size distribution of a standard
DSPC:CHOL:DODAP:
PEG-CerCl4 (25:45:20:10) preparation containing a 2 mg/mL initial antisense
concentration was
analyzed immediately after column chromatography to remove any free antisense.
A very
homogenous distribution is observed after preparation (119 ~ 32 nm). This size
distribution
remained stable for at least 2 months after storage at 4°C (119 ~ 32
nm).
EXAMPLE 4
This example illustrates the clearance pharmacokinetics, biodistribution and
biological activity of an encapsulated murine ICAM-1 phosphorothioate
antisense
oligodeoxynucleotide.
4.1 Plasma clearance
Encapsulated liposomal antisense was prepared using the ethanol-citrate
procedure as described in "Material and Methods". Initial lipid and antisense
concentrations
were 9.9 and 2 mg/mL, respectively. Liposomal formulations were composed of
X:CHOL:DODAP:PEG-CerCl4 (25:45:20:10), where X represents either
distearoylphospha-
tidylcholine (DSPC), sphingomyelin (SM), or palmitoyloleoylphosphatidylcholine
(POPC). The
formulations contained a lipid label (['4C]-cholesterylhexadecylether) and
[3H]-antisense and
were injected (200 pL) intravenously via the lateral tail vein of female (20-
25 g) ICR mice at a
lipid dose of 120 mg/kg. Blood was recovered by cardiac puncture on
anesthetized mice. Lipid
and antisense recoveries were determined by standard scintillation counting
techniques.
The plasma clearance of three formulations, DSPC:CHOL:DODAP:PEG-CerCl4,
SM:CHOL:DODAP:PEG-CerCl4, and POPC:CHOL:DODAP:PEG-CerCl4, of encapsulated
antisense were examined in inflamed ICR mice (Figure 9). The circulation time
was longest for
the DSPC version of the formulation.
4.2 Organ accumulation
CA 02271582 1999-OS-13
-46-
Liposomal antisense compositions were prepared and administered to mice as
outlined in the preceding section. Mice were terminated by cervical
dislocation and the organs
were recovered and processed as described in "Materials and Methods". Lipid
and antisense
recoveries were determined by standard scintillation counting techniques.
Organ accumulation of the various formulations was typical of previously
described liposome clearance patterns, with the RES organs, principally the
liver and spleen,
being responsible for the majority of clearance (Figure 10). One interesting
observation is that
the liver and spleen clearance account for only 40-45% of the total clearance
of the "DSPC"
formulation, suggesting that a significant population of vesicles is
accumulating in another organ
system or is being excreted.
4.3 Stability.
Liposomal antisense compositions were prepared and administered to mice as
outlined in the preceding section. Blood was recovered by cardiac puncture on
anesthetized
mice. Lipid and antisense recoveries were determined by standard scintillation
counting
techniques. Release rates were determined by measuring the [3H]/[14C] ratio
over time.
The stability of the formulations was also assessed by measuring the ratio of
antisense and lipid recovery in the blood at various times (Figure 11). A
ratio of 1.0 suggests
that the antisense and the lipid are staying together in the circulation. The
"DSPC" formulation
showed little deviation from a ratio of 1.0 over 24 h, suggesting that it is
very stable in the
circulation. The "POPC" formulation dropped to a ratio of 0.6 after 2 h, while
the ratio for the
"SM" formulation decreased more slowly, reaching 0.6 after 12 h in the
circulation. These
results indicate that it may be possible to deliberately alter the antisense
release rates by
modifying the lipid composition.
4.4 PEG-Acyl Influence on circulation half life of single dose of thioate
antisense
Encapsulated lipid-encapsulated antisense was prepared using the ethanol-
citrate
procedure as described in "Material and Methods". Initial lipid and antisense
concentrations
were 9.9 and 2 mg/mL, respectively. Liposomal formulations were composed of
CA 02271582 1999-OS-13
-47-
DSPC:CHOL:DODAP:PEG-CerCl4 or C20 (25:45:20:10). The formulation contained a
lipid
label (['4C]-cholesterylhexadecylether) and [3H]-antisense and were injected
(200 ~uL)
intravenously via the lateral tail vein of female (20-25 g) ICR mice at a
lipid dose of 120 mg/kg.
Blood was recovered by cardiac puncture on anesthetized mice. Lipid and
antisense recoveries
were determined by standard scintillation counting techniques.
The influence of PEG-acyl chain length on clearance rates of a
DSPC:CHOL:DODAP:PEG-Cer formulation was investigated using PEG-CerCl4 and
PEG-CerC20 (Figure 12). The inclusion of PEG-CerC20 in the formulation
resulted in
enhanced circulation times over the PEG-CerC 14. This corresponds to in vitro
data suggesting
that the C 14 version of the PEG is exchanged much more rapidly out of the
vesicle than the C20
version.
4.5 In vivo efficacy ofsingle dose of lipid encapsulated ICAM 1
(phosphorothioate)
antisense
The efficacy of PS- 3082 encapsulated in various lipid formulations containing
DODAP was tested in an ear inflammation model using ICR mice.
Inflamed mice were treated at the time of ear challenge with a 30 mg/kg i.v.
dose
of either HBS (no oligo), EPC:CHOL liposomes with entrapped PS- 3082
(identified as AS
1000), POPC:CHOL:DODAP:PEG-CerCl4 with entrapped PS- 3082 (identified as AS
4100), or
DSPC:CHOL:DODAP:PEG-CerCl4 with entrapped PS- 3082 (identified as AS 4200).
Ear
swelling was measured at 24 hours after initiating inflammation using an
engineer's micrometer.
Ear swelling measurements were made 24 hours after initiating inflammation in
mice treated i.v. at the time of ear challenge with either HBS (control), PS-
3082 encapsulated in
EPC:CHOL vesicles (30 mg/kg dose of oligo), PS- 3082 encapsulated in
POPC:CHOL:DODAP:PEG-CerCl4 vesicles (30 mg/kg dose of oligo), or PS- 3082
encapsulated in DSPC:CHOL:DODAP:PEG-CerCl4 vesicles (30 mg/kg dose of oligo)
(Figure
13). The "DSPC" formulation resulted in the greatest efficacy, exhibiting only
10% increase in
ear swelling over pre-challenge values. A similar trend was observed for
cellular infiltration into
the "challenged" ear versus the non-treated ear (Figure 14).
CA 02271582 1999-OS-13
-48-
In another evaluation, mice received 10 pCi of [3H]-methylthymidine, i.p., 24
hours before initiating inflammation. Inflamed mice were treated at the time
of ear challenge
with a 30 mg/kg i.v. dose of either HBS (no oligo), EPC:CHOL liposomes with
entrapped PS-
3082 (identified as AS 1000), POPC:CHOL:DODAP:PEG-CerCl4 with entrapped PS-
3082
(identified as AS 4100), or DSPC:CHOL:DODAP:PEG-CerCl4 with entrapped PS- 3082
(identified as AS 4200). Cell infiltration was monitored by measuring the
radioactivity in the
"challenged ear" versus the non-treated ear. Results are expressed as the
ratio of radioactivity in
the left (challenged ear) versus right ear.
4.6 In vivo efficacy of single dose of lipid encapsulated ICAM 1
(phosphodiester) antisense
This experiment demonstrates the in vivo efficacy of a phosphodiester
antisense
oligodeoxynucleotide encapsulated in lipid particles according to the
invention. In specific, the
phosphodiester was targeted to the ICAM-1 gene in an ear inflammation model.
1
i" .
G~ot~ i ~ ; , ,~~
~~ ~stx~a: ~~' ! ~ ~ r F,~.~~of
~ ' ~~~~ .
' ~ ~ ~ : ~.~
R~ ~ s ~~
~
~ 3
~ ~ :
. ii~"'~~. e i E i ~ k~,
~ ~. ' ~ n mr,..,~, i ~. (:~
r IO~~k z w i ir<.f~h'.a
~ ,.Y'.', '. r
~ yvrJ(i,,~, " 4Fz'~, ~' ' ~ i ~ .1
S, : r
.... wa"u ~ ,'-H~ag. ~
ai r~',t,,3 4~' ~1
~
~~'
" i~ii ~ i~;~~~~~~s i F
3' ~~ fy >C ~tb
rv' : : , t
i W. ., .. ~~
(i
~~
, p ~ , i i ~F
~ R i .. - ~ ,i:
~ 5~ ~ '. .!Fm i
LF ~~~4~f~~ .
~~ 'z
e ,.F irr~
3 ;~-~ t
.4 i A ~s
~
1 control 200 ~cl 24 hr
inflammation - HBS
2 corticosteroid 200 ,ul 24 hr
3 empty vesicles 200 ,ul 24 hr
4 PS-3082 200 ,ul 24 hr
5 PO-3082 200 ,ul 24 hr
Antisense Sample Preparation: Antisense was encapsulated using the standard
methods of
Examples 5-9, using the phosphodiester modification. The phosphodiester
formulation used 10-
50 mM citrate (preferably 20 mM citrate), pH 4.0 instead of 300 mM citrate, pH
4.0 preferred for
phosphorothioates. Empty vesicles consisted of lipid components only.
Corticosteroid (either
Halobetasol propionate 0.05% by weight (Westwood Squibb, Montreal) or
Dexamethasone (50
ug dissolved in 4:1 acetone:olive oil)) was applied topically in a thin film
to cover the surface of
the ear 15 minutes after ear challenge.
CA 02271582 1999-OS-13
-49-
Inflammation and Dosing: Mouse ear inflammation was induced using DNFB as
described
above in Materials and Methods. Female ICR mice (6-8 weeks old) received
intravenous tail
vein injections of antisense (200 ,ul). Antisense doses for the
phosphorothioate and
phosphodiester antisense were adjusted to be 20-30 mg/kg. 6 mice were tested
with each
formulation. Administration occurred 15 min. after the application of 0.2%
DNFB to the mouse
ear. Ear measurements were made on anaesthetized mice 24 hours after treatment
(unless shown
otherwise) and prior to termination. Mice are terminated by cervical
dislocation and the ears are
removed around the pinna. Ears are then weighted, digested (Solvable) and
analyzed for
radioactivity by liquid scintillation counting. Ears were analyzed for 1) Ear
edema - based on the
increase in ear thickness due to ear swelling. Calculated by subtracting pre-
ear thickness values
from post-ear thickness values Figure 21. 2) Cell infiltration - based on
radioactivity
accumulated in the inflamed (right) ear vs. the control (left) ear Figure 22;
and 3) Ear weights -
left ear versus right ear (measurement of edema) Figure 23.
Results: The controls consisting of buffer alone (HBS) or Empty Vesicles alone
demonstrated
no efficacity. Topical corticosteroid demonstrates its known excellent
efficacity by reducing
inflammation to below pre-challenge levels. Both the phosphorothioate and
phosphodiester
antisense show excellent efficacy through a systemic delivery administration,
reducing the
degree of inflammation by around 70% and 85%, respectively. Thus, it is
possible to administer
the compositions of the invention at a site where the disease site is distal
to the site of the
inj ection.
4.7 In vivo efficacy of US3 antisense (Tumor Window Model)
In this example, the anti-tumor activity of lipid encapsulated US3, an
antisense
oligonucleotide directed at the erb-B-2 gene, has been demonstrated in an in
vivo human breast
tumor model.
The human breast carcinoma line MDA-MB-453 was implanted in a mouse tumor
window model according to the method of Wu, N.Z., Da, D., Rudoll, T.L.,
Needham, D.,
Whorton, R. & Dewhirst, M.W. 1993. Increased microvascular permeability
contributes to
preferential accumulation of Stealth liposomes in tumor tissue. Cancer
Research 53: 3765-3770;
CA 02271582 1999-OS-13
-S0-
and Dewhirst, M.W., Tso, C.Y., Oliver, R., Gustafson, C.S., Secomb, T.W. &
Gross, J.F. 1989.
Morpholigic and hemodynamic comparison of tumor and healing normal tissue
microvasculature. Int. J. Radiat. Oncol. Biol. Phys. 17: 91-99. See also
Dewhirst, MW., and
Needham, D. 1995. Extravasation of Stealth Liposomes into Tumors: Direct
Measurement of
Accumulation and Vascular Permeability using a Skin Flap Window Chamber. In
Stealth
Liposomes (Eds. Lasic, D. and Martin, F.) CRC Press.
The lipid-antisense formulation consists of disteroylphosphatidylcholine
(DSPC,
25 mol%), cholesterol (Chol, 45 mol%), dioleoylphosphatidyldiaminopropane,
(DODAP, or
AL1, 20 mol%) and PEG-ceramide (C14 chain length, 10 mol%). For some
experiments
detailed below, proportions and constituents were altered, but the method of
preparation
remained the same. Lipids were dissolved in ethanol at 20 mg/ml (PEG-ceramide
at 50 mg/ml).
Routinely, 1 to 2 pCi'4C-cholesterylhexadecylether was added as a lipid
radiolabel. Lipids were
mixed in the correct proportions in ethanol to a final concentration of 10 mg
in 400 p,l. The lipid
mixture was then added dropwise to phosphorothioated antisense (US3: anti-
human erb-B-2
GGT GCT CAC TGC GGC (SEQ ID. No 3) dissolved in 300 mM citrate buffer pH 4.0
(600 pl
to make a final volume of 1 ml). The antisense was used at a variety of
concentrations, but the
optimum concentration for maximum encapsulation efficiency and drug:lipid
ratio was
determined to be 0.5 mg/ml final. During the addition, the solution becomes
opaque. The
DODAP is positively charged at pH 4.0 (pKa= 6.53) and so attracts the
negatively charged DNA
molecules. The mixture was subjected to five cycles of freezing in liquid N2
and thawing at 65
°C followed by extrusion through 100 nln filters ten times at 65
°C.
After extrusion, two methods can be used for removal of the external
antisense.
Firstly, the liposomes are diluted 2:1 with citrate (to reduce ethanol content
to 20%) then applied
to a Bio-Gel A18M 100-200 mesh column equilibrated with HBS. The column
profiles shown in
this report were generated in this manner. Alternatively, the liposomes are
dialysed 2h against
citrate to remove ethanol, the overnight against HBS to increase the external
pH. The resulting
mixture is then applied to a DEAE canon exchange column to remove external
oligo. This
method was the routine method used for sample preparation for in vivo studies.
Antisense
concentrations were routinely determined by A260 measurements. Lipid
concentrations were
CA 02271582 1999-OS-13
- S1 -
determined by scintillation counting after spiking initial mixture with a
known concentration of
3H or'''C cholesterylhexadecyl ether, or by HPLC. Encapsulation efficiency was
determined by
division of the final drug to lipid ratio by the initial drug to lipid ratio.
In vivo efficacy evaluation: When the tumor in the window has reached a
diameter
of 2-3 mm, treatment with free or TCS-encapsulated US3 oligonucleotide is
initiated. Treatment
consists of a 200 ul intravenous administration (tail vein) of either free US3
or TCS-encapsulated
US3 on a 3 administrations/week schedule and an antisense dose of 10
mg/kg/administration.
Tumor size is monitored 3 times per week by microscopy.
Results: The TCS-encapsulated US3 oligonucleotide was very effective at
preventing the
growth, or causing extensive size reduction, of the MDA-MB-453 human breast
carcinoma in the
window model. In contrast, unencapsulated oligonucleotide was ineffective at
inhibiting tumor
growth.
4.8 In vivo clearance of various formulations using alternative amino lipids:
DODAP or
1 S DODMA
Antisense particle formulations were prepared according to Example 2, with the
following modifications: In assay#1 and #2, 25% AL-1 (hydrochloride salt of
DODAP) and 25%
free base DODAP were employed, respectively, with a concomitant reduction in
the amount of
DSPC. Assay#3, 4 and 5 employed 30%, 25% and 20% DODMA (free base (prepared at
Inex
Pharmaceuticals Corp., Burnaby BC)), respectively, again with a concomitant
reduction of
DSPC.
Both the encapsulation efficiency and in vivo clearance of the formulations
were
studied. There was no significant difference between the encapsulation or
clearance of the free
base or HCI salt of DODAP. Decreasing DODMA concentration (30, 25, 20 %)
severely
decreased the encapsulation efficiency of PS-2302 (91%, 43%, 35%) and likewise
the
Drug/Lipid ratio of the resulting formulation.
In the clearance study outlined in Figure 16, DODMA formulations demonstrated
slightly higher rates of clearance than 25 % DODAP or AL-l, although all
formulations appear
to be retained in the circulation to a degree which is suitable for human
therapeutics.
CA 02271582 1999-OS-13
-52-
4.9 PEG-acyl influence on clearance rate of repeat doses of encapsulated EGF-R
phosphorothioate antisense
Lipid -encapsulated antisense was prepared using the ethanol-citrate procedure
as
described above, with changes to molar ratios of components as indicated.
Initial lipid and
antisense concentrations were about 9.9 and 2 mg/mL, respectively. DODAP
containing
formulations had drug:lipid ratios of 0.15 (+/-) 0.05. Passive encapsulation
systems had
drug:lipid ratios of 0.03. Nine different liposomal formulations were
prepared, using standard
techniques, in the following molar ratios:
Formu- DSPC Chol DODAP Steric Barrier Antisense
lation (mol%) (mol%) (mol%) Derivatized Lipid (EGF-R
(name: mol%) 2mg/ml)
1 55 45 Nil Nil Empty
2 50 45 Nil ATTAB-DSPE : Empty
5
3 50 45 Nil ATTA8-DSPE : AS
5
4 20 45 30 ATTA8-DSPE : AS
5
5 20 45 30 PEG-DSPE : 5 AS
6 25 45 25 PEG-CerC 14 : Empty
5
7 25 45 25 PEG-CerC14:5 AS
8 25 45 25 PEG-CerC20:5 Empty
9 25 45 25 PEG-CerC20:5 AS
Antisense ("AS") used was fully phosphorothioated EGFR (anti-human Epidermal
Growth
Factor Receptor) CCG TGG TCA TGC TCC (SEQ ID. No 10) (prepared by Hybridon,
Inc.)
PEG-CerCl4 is PEG(mw2000)-Ceramide with 14 carbon acyl chain.
PEG-CerC20 is PEG(mw2000)-Ceramide with 20 carbon acyl chain.
PEG-DSPE is PEG(mw2000)- 1,2-distearoyl-sn-glycero-3-phosphoethanolamine
ATTA8-DSPE is N-(c~-N'-acetoxy-octal14'amino-3',6',9',12'-
tetraoxatetradecanoyl))-1,2-
distearoyl-sn-glycero-3-phosphoethanolamine (molec weight about 2660).
Synthesis of ATTAB-
DSPE is fully disclosed in US Provisional Pat. Application Serial No.
60/073,852, filed 23 - Dec
- 1997 and US Provisional Pat. Application filed 2-Feb-1998 (Attorney Docket
No.: TT&C
CA 02271582 1999-OS-13
-53-
16303-005810) both assigned to the assignee of the instant invention and
incorporated herein by
reference.
Each formulation contained a lipid label (['4C]-cholesterylhexadecylether) and
[3H]-antisense, as described in Example 4.4, above. All samples were prepared
in 300 mM citrate
pH 4.0 containing 40% ethanol and extruded lOX through 100 nm filters.
Formulations
contained phosphorothioate antisense and lipid or empty lipid alone. Samples
were dialyzed in
HBS (20 mM Hepes, 145 mM NaCI, pH 7.45) to remove ethanol and citrate. Sample
lipid
concentrations were adjusted such that the injected lipid dose will be 1.8
,umol/mouse/week (5-
mg AS per kg mouse/week). Samples were filtered (0.22 ,um) prior to injection.
10 In this experiment female (20-25g) ICR mice (6-8 weeks old) were divided
into 9
groups of 6, plus other control groups. Each group received four injections of
the same
formulation. All injections were 200 ~L intravenous (via the lateral tail
vein) at a lipid dose of
120 mg/kg. Mice were dosed every week for 3 weeks (4 injections). At 4 weeks,
certain groups
(treated with lipid and antisense) were given an injection of empty lipid
carriers of varying
1 S composition to evaluate whether there is rapid clearance of the carrier in
the absence of antisense.
Blood (25 ,ul, pipettor) was collected 1 h post-injection each week for 3
weeks by tail nicks.
Mice were weighed each week to estimate blood volume (8.0 ml whole blood/100 g
body
weight). Blood was placed in a glass scintillation vial containing 200 ,ul of
5% EDTA. Solvable
(500 ,ul) was added and the blood was digested for 3 h at 65 °C.
Samples were decolorized by
the addition of 100 ~cl 70% hydrogen peroxide. Samples were analyzed for
radioactivity by
liquid scintillation counting. At the end of 4 weeks, mice were terminated by
COz inhalation or
cervical dislocation preceded by general anesthesia.
The results of this experiment are shown in Figure 17. For all formulations
not
containing antisense ("empty liposomes") repeat dosages demonstrated
circulation times
reasonably consistent with the first dosage. However, when antisense is used
in the formulation,
it was surprisingly found that the acyl chain length of the lipid derivatized
to the steric barrier
(i.e. ATTA or PEG) moiety demonstrates a profound effect on clearance rates.
Repeat dosages
of PEG-CerC20, PEG-DSPE and ATTAB-DSPE formulations are rapidly cleared from
the
CA 02271582 1999-OS-13
-54-
circulation compared to the first dosage, whereas the PEG-CerCl4 formulation
is reasonably
consistent with the first dosage.
Similar results are demonstrated in Figure 18. The formulations were identical
to
those of Figure 17, with the additional formulation of empty vesicles using
the same lipids as
formulations 4 and 5.
4.10 In vivo efficacy of repeat doses of encapsulated phosphorothioate c-myc
antisense in an
oncology model.
In vivo efficacy of repeat injections of using formulations of the invention
are shown
in a mouse tumor system in Figure 19. This experiment demonstrated efficacy of
the antisense
formulated according to the invention in a human oncology model, and showed
the importance of
PEG-acyl chain length on the efficacy of repeat dosings.
Lipid-antisense particle formulation: Formulations were prepared as described
in these Examples.
Formu-lation DSPC Chol DODAP Steric Barrier Antisense
(mol%) (mol%) (mol%) Derivatized (c-myc
Lipid
(name: mol%) 2mg/ml)
HBS Buffer Empty
AS4200 25 45 25 PEG-CerC14:5 LR-3280
(c-myc)
AS4204 25 45 25 PEG-CerC20:5 LR-3280
(c-myc)
AS4204 25 45 25 PEG-CerC20 : c-myc
5 SCR
(c-myc SCR)
AS4204 25 45 25 PEG-CerC20:5 PS-2302
(PS-2302)
AS4204 25 45 25 PEG-CerC20:5 PS-3208
(PS-3082)
c-myc LR-3280
c-myc SCR c-myc
SCR
PS-2302 PS-2302
CA 02271582 1999-OS-13
-55-
PS-3082 PS-3082
AS4200 (no 25 45 25 PEG-CerCl4 : 5 Empty
antisense)
AS4204 (no 25 45 25 PEG-CerC20 : 5 Empty
antisense)
Antisense used were:
LR-3280: human c-myc gene (phosphorothioate)
AAC GTT GAG GGG CAT
(SEQ ID. No 4)
c-myc SCR: GAA CGG AGA CGG TTT (SEQ ID. No 17)
PS-2302 human ICAM-1 (phosphorothioate)
GCCCAAGCTGGCATCCGTCA (SEQ ID. No 2)
PS-3082 murine ICAM-1 (Intracellular Adhesion Molecule-1)
(phosphorothioate)
TGCATCCCCCAGGCCACCAT
(SEQ ID. No 1)
Formulations were diluted in filtered HBS, pH 7.6 to achieve required
antisense dose
(i.e. lipid dose decreases as well). Samples were filtered (0.22 ,um) prior to
injection. External
buffer was HBS (20 mM Hepes, 145 mM NaCI, pH 7.6). Free antisense was
dissolved in HBS and
adjusted to the required dose by A260 (Extinction coefficients: active and
control c-myc = 30.6, PS
2302 = 32.8, PS-3082 = 33.6).
Tumour Inoculum: B 16/BL6 murine melanoma cells were maintained in culture in
MEM media
supplemented with 10% FBS. On day 0 of the study, 3 x105 cells were injected
sub-cutaneously
(s.c.) into the dorsal flank (injection volume: 50,u1) of female C57BL/6 mice
(20-23 g). Typically,
15% extra mice will be injected so non-spheroidal tumours or mice in which no
tumours are
observed can be excluded from the study. Tumours were allowed to grow for a
period of 5-7 days
until tumors reached 50-100 mm3 prior to initiating treatments with test
samples/controls.
Treatment: On the day of first treatment mice with acceptable tumours were
randomly grouped
with 5 animals per group. Treatment began when tumours were 50-100 mm3. Mice
were dosed
every other day for a total of 7 doses. Administrations were via intravenous
tail vein injections (200
ul). Initial drug: lipid ratio of formulation was 0.20 (w/w) and the final
drug:lipid ratio (0.14) was
CA 02271582 1999-OS-13
-56-
held constant; consequently, the lipid concentration varied as samples were
diluted to the desired
antisense concentration. The antisense dose was 10 mg/kg.
Endpoints: Primary tumour volume was measured using calipers. Length (mm) and
width (mm)
measurements were made every other day (on non-injection days) for the
duration of the study.
Tumour height measurements (mm) were made when feasible. Tumour volumes were
calculated
using the following formulas:
#1 Tumour Volume (mm3) _ (L x WZ)/2
#2 Tumour Volume (mm3) _ (L x W x H) x ~/6
Mice were euthanized when tumour volumes reach 10% of body weight or on the
first signs of
ulceration. Mouse weights were recorded every day during the dosing portion of
the study. On
termination, all tumours were excised, weighed, observed by FACS analysis or
by Northern/Western
analysis. Mice were euthanized by COZ inhalation or cervical dislocation
preceded by general
anesthesia.
Results: Figure 9 shows weights of tumors excised and weighed at day 18 for
all groups treated
1 S with antisense at 10 mg/kg/dose compared with empty lipid controls. Tumour
sizes for the
AS4200(c-myc) group exhibited the best efficacy and were very consistent with
only small ranges
in tumour volumes observed (285-451 mm3). The group treated with free c-myc
also resulted in
smaller tumours but exhibited more variability in tumour volume (156-838 mm3).
The encapsulated
c-myc controls (c-myc SCR/PS-2302/PS-3082), AS4204(c-myc), empty lipid
carriers, and free
antisense controls, however, showed no inhibitory effect on tumor volumes over
the 18 days when
compared to HBS controls.
c-myc expression in tumor tissue was also evaluated by FACS. A correlation
between tumour size and c-myc protein expression was detected (data not
shown).
To determine the importance of the stability of the PEG-polymers, PEG-acyl
chain
length was evaluated using formulations containing PEG-CerC 14 and PEG-CerC20.
Interestingly,
the formulation containing the PEG-CerC20 (AS4204) showed no apparent efficacy
at any of the
doses studied. The PEG-CerCl4 formulation (AS4200) showed a dose response. The
difference
observed between the PEG-CerC 14 and PEG-CerC20 formulations may reflect the
rapid clearance
phenomenon that has been observed in other models.
CA 02271582 1999-OS-13
-S7-
To establish the tolerability of free and encapsulated antisense, mouse
weights were
measured on a daily basis during the treatment phase of the study. No
significant changes in mouse
weights for either free or encapsulated formulations were apparent over the
course of the dosing
phase or throughout the study.
EXAMPLE 5
This example illustrates a high efficiency formulation according to Example 2,
but
instead of phosphorothioate antisense, employing 1) a phosphodiester antisense
compound having
exclusively phosphodiester internucleotide linkages (PO-2302 anti-human ICAM-1
GCCCAAGCTGGCATCCGTCA (SEQ ID. No 1 )) prepared by Inex Pharmaceuticals (USA),
Inc.,
Hayward CA) or 2) ribozyme molecule to VEGF-R-1 (human Vascular Endothelial
Growth Factor
Receptor 1) comprising a modified RNA sequence of GAG UUG CUG AUG AGG CCG AAA
GGC CGA AAG UCU G (SEQ ID. No 16).
A l5mer of [3H]-phosphodiester antisense oligodeoxynucleotide (PO-2302) in
citrate
buffer, pH 3.80 (experiments ranged from 10-1000 mM citrate) was mixed with an
ethanol solution
of lipid (DSPC:CHOL:DODAP:PEG-CerC 14; 25:45:20:10, molar ratio) at final
concentrations of
2 mg/mL and 9.9 mg/mL, respectively. The final ethanol concentration in the 1
ml preparation was
38% vol/vol. The sample was extruded ten times through three 100 nm filters as
described in
"Materials and Methods". The sample was dialyzed for 2-3 hours in citrate
buffer, pH 3.80 (same
molarity as experiment), to remove a majority of the excess ethanol. The
samples were switched to
HEPES-buffered saline (HBS), pH 7.50, and dialyzed for a minimum of 12 hours
to replace the
external citrate buffer with HBS. Non-encapsulated antisense was removed
either by this regular
dialysis, tangential flow dialysis, or chromatography. Encapsulation was
assessed either by
analyzing the pre-column and post-column ratios of [3H]-antisense and ['4C]-
lipid or by determining
the total pre-column and post-column [3H]-antisense and ['4C]-lipid
radioactivity.
Figure 15 illustrates results. Encapsulation efficiency was over 50% across
the 10-50
mM citrate range, and all final (administration ready) drug:lipid ratios were
greater than 10% by
weight. Parallel experiments varying citrate concentration were conducted with
phosphorothioate
CA 02271582 1999-OS-13
-58-
antisense PS-2302. Results are also above 50% encapsulation, and in fact show
a higher
encapsulation efficiency than phosphodiesters, particularly at higher citrate
concentrations.
This experiment was repeated using 20mM citrate instead of 300 mM citrate to
encapsulate the ribozyme molecule to VEGF-R-1 (human Vascular Endothelial
Growth Factor
Receptor 1 ) GAG UUG CUG AUG AGG CCG AAA GGC CGA AAG UCU G (SEQ ID. No 1~.
Figure 20 shows the encapsulation efficiency of the ribozyme at was over 50%,
approximately the
same as the phosphodiester.
EXAMPLE 6
This example illustrates a high efficiency formulation as in Example 5, but
replacing
DODAP with an alternative protonatable lipid. Typically, the preparation for
the alternative will be
X:DSPC:CHOL:PEG-CerCl4 at 20:25:45:10 molar ratio where X can be DODAC, OA,
DODMA
or any other lipid suitable for the invention.
Materials: distearoylphosphatidylcholine, DSPC; cholesterol, CHOL (both from
Northern Lipids, Vancouver, BC); N,N-dioleyl-N,N-dimethylammonium chloride,
DODAC;
Oleylamine, OA (prepared by Steve Ansell, Inex); N-(1-(2,3-Dioleoyloxy)
propyl)-N,N,-dimethyl
ammonium chloride, DODMA(Avanti Polar Lipids, Alabaster AB, chloride salt
prepared by Steve
Ansell, INEX); polyethylene glycol)2000 coupled to a ceramide derivative with
14 carbon acyl
chains, PEG-CerC 14 (Zhou Wang, INEX Pharmaceuticals); 13 x 100 mm glass tube;
filter sterilized
300 mM citrate buffer, pH 3.9 - 4.0 (use a 0.2 ~m filter). Fully thioated c-
myc antisense (1NEX
(USA), Hayward Ca), Anhydrous Ethanol (Commercial Alcohols, Toronto, On),
Citric acid,
Monobasic Sodium phosphate, Dibasic Sodium phosphate, Sodium hydroxide, HEPES
(BDH,
Mississauga On). Deionized water, Chloroform, Methanol, OligreenT"'
oligonucleotide reagent
(Molecular Probes, Eugene Or), Sodium chloride, Triton X-100, alcohol
dehydrogenase reagent kit,
(Sigma Chemical Co., St Louis Mo.),
Lipid stock solutions were made in 100 % ethanol with the working
concentrations
of the lipids which is as follows:
DSPC, 20 mg/ml; CHOL, 20 mg/ml (not very soluble above this concentration);
DODMA,
20 mg/ml; PEG-CerC 14; 50 mg/ml.
CA 02271582 1999-OS-13
-59-
To prepare stock solutions of antisense, the antisense molecules were
dissolved in the
filtered 300 mM citrate buffer at a concentration of 3.33 mg/ml. Lipids were
mixed in the desired
proportions in a 13 x 100 mm glass tube to achieve a final volume of 0.4 ml of
lipids using 100
ethanol as listed in table 1, below:
Table 1. Proportional mixture of lipids in a 13 x 100 mm glass test tube.
Lipid Mol % M. Wt. mg ~mol Stock Vol of
Stock
(mg/ml) (~1)
DODMA 20 652.6 1.69 2.60 20 84.5
DSPC 25 790 2.57 3.25 20 115
CHOL 45 386.7 2.26 5.85 20 113.1
PEG-CerCl4 10 2600 3.38 1.30 SO 67.6
100 9.9 13.00 380.2
In a separate 13 x 100 mm glass tube, 0.6 ml of antisense at 3.33 mg/ml was
added. The pH
of this solution should be 3.9 - 4Ø (NOTE: the antisense concentration is
NOT determined by
weight but rather by measuring absorbance at 260 nm). The lipid mixture
solution was warmed to
65 °C for about 2 minutes. The antisense tube was vortexed and during
this time, using a Pasteur
pipette, the lipids (in ethanol) were added slowly in a dropwise manner. The
mixture will get
"cloudy" and some bubbles may be observed due to the ethanol, but no
aggregates should be present.
The resulting volume of the antisense-lipid mixture was 1.0 ml with a 10 mg
(13 umol) total lipid
at 13 pmols, 2 mg of antisense, and 38 % ethanol, vol/vol. It can be expected
that the pH to rise to
about 4.4.
The antisense-lipid mixture was subjected to 5 (five) cycles of freezing in
liquid
nitrogen and thawing at 65°C in a waterbath. After each thaw, the
mixture was vortexed briefly.
Subsequently, the mixture was passed 10 times through three stacked 100 nm
polytcarbonate filters
(Poretics) or extruded using a pressurized extruder apparatus with a
thermobarrel attachment (Lipex
Biomembranes). The temperature and nitrogen pressure during extrusion were 65
°C and no more
CA 02271582 1999-OS-13
-60-
than 200 psi to 300 psi, respectively. Each pass should take no more than 2
minutes and is vortexed
after each pass.
After extrusion, the mixture was dialyzed in a dialysis tubing (3500 Mwt
cutoff;
SpectraPor) for 1 hour in 300 mM citrate at pH 3.9-4.0, removing the ethanol.
The mixture was
transferred into 5 L of HBS buffer at pH 7.5 and allowed to further dialyze to
a minimum of 12
hours, to neutralize the DODMA and release any surface bound antisense
associated with the
vesicles. Alternatively, tangential flow dialysis, ion exchange-chromatography
or gel filtration
chromatography can be used to process the extruded antisense-lipid mixture to
an administration
ready preparation.
EXAMPLE 7
This example illustrates a high efficiency formulation as in Example 5, but
replacing DSPC with SM to generate a preparation of DODAP:SM:CHOL:PEG-CerCl4
at
20:25:45:10 molar ratio. Antisense is processed with the formulation for a
standard 1.0 ml
volume, which can be scaled up proportionately as required.
Materials: Sphingomyelin SM; cholesterol, CHOL; dimethylaminopropane,
DODAP; polyethylene glycol coupled to a ceramide derivative with 14 carbon
acyl chains, PEG-
CerCl4; 13 x 100 mm glass tube; filter sterilized 300 mM citrate buffer, pH
3.9 - 4.0 (use a 0.2
~m filter).
Lipid stock solutions were made in 100 % ethanol with the working
concentrations of the lipids which is as follows:
SM, 20 mg/ml; CHOL, 20 mg/ml (not very soluble above this concentratnon);
DODAP, 20
mg/ml; PEG-CerCl4; 50 mg/ml.
To prepare stock solutions of antisense, the antisense molecules were
dissolved in the
filtered 300 mM citrate buffer at a concentration of 3.33 mg/ml. Lipids were
mixed in the desired
proportions in a 13 x 100 mm glass tube to achieve a final volume of 0.4 ml of
lipids using 100
ethanol as listed in Table 2, below:
Table 2. Proportional mixture of lipids in a 13 x 100 mm glass test tube.
CA 02271582 1999-OS-13
-61 -
Lipid Mol % M. Wt. mg ~mol Stock Vol of
(mg/ml) Stock (~1)
DODAP 20 684.5 1.78 2.60 20 89.0
SM 25 703 2.30 3.27 20 115
CHOL 45 386.7 2.26 5.85 20 113.1
PEG-CerCl4 10 2600 3.38 1.30 50 67.6
100 9.72 13.02 3 84.7
In a separate 13 x 100 mm glass tube, 0.6 ml of antisense at 3.33 mg/ml was
added. The pH
of this solution should be 3.9 - 4Ø (NOTE: the antisense concentration is
NOT determined by
weight but rather by measuring absorbance at 260nm). The lipid mixture
solution was warmed to
65 ° C for about 2 minutes. The antisense tube was vortexed and during
this time, using a Pasteur
pipette, the lipids (in ethanol) were added slowly in a dropwise manner. The
mixture will get
"cloudy" and some bubbles may be observed due to the ethanol, but no
aggregates should be present.
The resulting volume of the antisense-lipid mixture was 1.0 ml with a 10 mg
(13 umol) total lipid
at 13 ~mols, 2 mg of antisense, and 38 % ethanol, vollvol. It can be expected
that the pH to rise to
about 4.4.
The antisense-lipid mixture was subjected to 5 (five) cycles of freezing in
liquid
nitrogen and thawing at 65°C in a waterbath. After each thaw, the
mixture was vortexed briefly.
Subsequently, the mixture was passed 10 times through three stacked 100 nm
polytcarbonate filters
(Poretics) or extruded using a pressurized extruder apparatus with a
thermobarrel attachment (Lipex
Biomembranes). The temperature and nitrogen pressure during extrusion were 65
° C and no more
than 200 psi to 300 psi, respectively. Each pass should take no more than 2
minutes and is vortexed
after each pass.
After extrusion, the mixture was dialyzed in a dialysis tubing (3500 Mwt
cutoff;
SpectraPor) for 1 hour in 300 mM citrate at pH 3.9-4.0, removing the ethanol.
The mixture was
transferred into 5 L of HBS buffer at pH 7.5 and allowed to further dialyze to
a minimum of 12
hours, to neutralize the DODAP and release any surface bound antisense
associated with the vesicles.
Alternatively, tangential flow dialysis, ion exchange-chromatography or gel
filtration
CA 02271582 1999-OS-13
-62-
chromatography can be used to process the extruded antisense-lipid mixture to
an administration
ready preparation.
EXAMPLE 8
This example illustrates a high efficiency formulation as in Example 5, but
replacing PEG-CerCl4 with ATTAB-DSPE to prepare DODAP:DSPC:CHOL:ATTAB-DSPE at
40:10:45:5 molar ratio of antisense formulation.
Materials: distearoylphosphatidylcholine, DSPC; cholesterol, CHOL;
dimethylaminopropane, DODAP; N-(c~-N'-acetoxy-octa(14'amino-3',6',9',12'-
tetraoxatetradecanoyl))-1,2-distearoyl-sn-glycero-3-phosphoethanolamine, ATTAB-
DSPE; 13 x
100 mm glass tube; filter sterilized 300 mM citrate buffer, pH 3.9 - 4.0 (use
a 0.2 ~m filter).
Lipid stock solutions were made in 100 % ethanol with the working
concentrations of the lipids which is as follows:
DSPC, 20 mg/ml; CHOL, 20 mg/ml (not very soluble above this concentration);
DODAP,
20 mg/ml; ATTAB-DSPE; 50 mg/ml.
To prepare stock solutions of antisense, the antisense molecules were
dissolved in the
filtered 300 mM citrate buffer at a concentration of 3.33 mg/ml. Lipids were
mixed in the desired
proportions in a 13 x 100 mm glass tube to achieve a final volume of 0.4 ml of
lipids using 100
ethanol as listed in Table 3, below:
Table 3. Proportional mixture of lipids in a 13 x 100 mm glass test tube.
Lipid Mol % M. Wt. mg ~mol Stock Vol of Stock
(pl)
(mg/ml)
DODAP 40 684.5 4.16 6.08 20 208
DSPC 10 790 1.2 1.52 20 60
CHOL 45 386.7 2.6 6.72 20 130
ATTAB- S 2638 2.0 0.76 50 40
DSPE
100 10.26 15.1 438
In a separate 13 x 100 mm glass tube, 0.6 ml of antisense at 3.33 mg/ml was
added. The pH
of this solution should be 3.9 - 4Ø (NOTE: the antisense concentration is
NOT determined by
CA 02271582 1999-OS-13
-63-
weight but rather by measuring the absorbance at 260nm). The lipid mixture
solution was warmed
to 65 ° C for about 2 minutes. The antisense tube was vortexed and
during this time, using a Pasteur
pipette, the lipids (in ethanol) were added slowly in a dropwise manner. The
mixture will get
"cloudy" and some bubbles may be observed due to the ethanol, but no
aggregates should be present.
The resulting volume of the antisense-lipid mixture was 1.0 ml with a 10 mg
(13 umol) total lipid
at 13 qmols, 2 mg of antisense, and 38 % ethanol, vol/vol. It can be expected
that the pH to rise to
about 4.4.
The antisense-lipid mixture was subjected to 5 (five) cycles of freezing in
liquid
nitrogen and thawing at 65 ° C in a waterbath. After each thaw, the
mixture was vortexed briefly.
Subsequently, the mixture was passed 10 times through three stacked 100 nm
polytcarbonate filters
(Poretics) or extruded using a pressurized extruder apparatus with a
thermobarrel attachment (Lipex
Biomembranes). The temperature and nitrogen pressure during extrusion were 65
°C and no more
than 200 psi to 300 psi, respectively. Each pass should take no more than 2
minutes and is vortexed
after each pass.
After extrusion, the mixture was dialyzed in a dialysis tubing (3500 Mwt
cutoff;
SpectraPor) for 1 hour in 300 mM citrate at pH 3.9-4.0, removing the ethanol.
The mixture was
transferred into 5 L of HBS buffer at pH 7.5 and allowed to further dialyze to
a minimum of 12
hours, to neutralize the DODAP and release any surface bound antisense
associated with the vesicles.
Alternatively, tangential flow dialysis, ion exchange-chromatography or gel
filtration
chromatography can be used to process the extruded antisense-lipid mixture to
an administration
ready preparation.
EXAMPLE 9
This example illustrates use of tangential flow dialysis to clean up a large
scale (>50
ml) preparation of extruded antisense-lipid mixture to obtain an
administration ready preparation.
Tangential Flow Diafiltration has been shown to be useful in four functions in
the formulation
process 1) buffer exchange, 2) removal of ethanol, 3) removal of
unencapsulated antisense and 4)
concentration of the formulation. Using TF it is demonstrated that it is
possible to efficiently
CA 02271582 1999-OS-13
-64-
exchange these components using only 10-15 sample volumes with a single buffer
system at a very
significant reduction in the process time.
Materials for Tangential Flow Dialysis: Microcross SamplerT"~ Tangential Flow
column (Microgon, LagunaHills, Ca) MasterflexT"' console drive and EasyloadT"'
Pump head (Cole-
Parmer, Vernon Hills Ill.), Extruder (Lipex Biomembranes, Vancouver BC),
Polycarbonate
membranes, 100 pm, (AMD Manufacturing, Mississauga On).
Antisense (c-myc) is prepared by dissolving in 300 mM Na Citrate buffer to a
final
concentration of 4.17 mg/ml for c-myc as verified by absorbance at 260 nm. The
antisense stock
solution is typically warmed to 65°C for 2 minutes to dissolve and to
remove secondary structure.
AS4200 consists ofDODAP:DSPC:CHOL:PEG-CER-14 at the percent mol ratio
of25:20:45:10 and
the lipids are aliquoted from stock solutions to a total concentration of 10
mg/0.400 ml in anhydrous
ethanol. In this study 50 - 60 ml scale formulations were produced. Thus 20-24
ml of the ethanolic
lipid solution is added dropwise, at room temperature, using a peristaltic
pump at lml/nin into 30 -
36 ml of the AS solution which is stirring in a 100 ml round bottom flask with
a 2 cm magnet stir
bar (Stirrer setting 2-3). After mixing, the lipid/antisense suspension was
pipetted into a 100 ml
extruder prepared with 2-3, 100 ~m polycarbonate membranes and pre-
equilibrated at 65°C. The
suspension was extruded using ten passes at 300 psi. After extrusion the
formulation was processed
using tangential flow diafiltration.
Tangential Flow Ultrafiltration. A 230 cm2 Microcross tangential flow
cartridge
(50 kDa cut off) was attached to a Masterflex peristaltic pump, sample
reservoir and buffer reservoir
using Tygon tubing. The tubing length was adjusted so that the total circuit
of tubing, pump and TF
cartridge had a total dead volume of 30 ml. To this system a 60 ml sample
reservoir was attached.
The sample was loaded into the tubing and reservoir by running the peristaltic
pump at a low speed.
After loading, the system was closed and the pump speed gradually increased to
the pump maximum
(approx. 100 ml/min) until the initial TF cartridge inlet pressure was 12-15
psi and the outlet
pressure was 8-11 psi. When the system pressure stabilized, both the filtrate
outlet and the buffer
reservoir were opened. Opening these valves allowed filtrate to flow out of
the cartridge at ~ 10-15
ml/min while wash buffer (i.e. PBS, pH 7.5) was being collected. For a 50 - 60
ml formulation 700 -
900 ml of buffer was used to "wash" the sample. Fractions (10 ml) of the
filtrate were collected for
CA 02271582 1999-OS-13
- 65 -
analysis of ethanol removal, pH, and antisense. After diafiltration was
completed the wash buffer
reservoir was closed and with the pump continuing to run, filtrate was allowed
to flow, concentrating
the sample, typically reducing the preparation volume to the tubing dead
volume (30 - 35 ml). The
sample was collected from the system and the tubing and column were washed
with 15 ml wash
buffer to remove any remaining formulation.
Antisense Quantification. Antisense concentration was normally determined by
measuring absorbance at 260nm as outlined in the current protocol. Briefly,
antisense stock solutions
were quantified by diluting 1:500 in MilliQ water and measuring absorbance. TF
filtrate fractions
were diluted 1:10 in MilliQ water and absorbance was measured. Antisense in
suspension with lipids
was measured by adding 10 p l of the suspension to 250 ~1 MilliQ water. A
monophase was created
by adding 750 ~1 CHC13/MeOH (2.1:1) and 100 ~ul MeOH. Immediately after
vortexing the mixture
the absorbance was measured at 260 nm. In each case the extinction coefficient
for the given
antisense was multiplied by the dilution factor to determine the antisense
concentration.
Lipid Quantification. As outlined in the current protocol, 50 ql aliquots of
the
lipid/antisense suspension was diluted with 100 ~l MilliQ water and submitted
for analysis by
HPLC. The percent encapsulation efficiency of the formulation is determined by
dividing the
Drug/Lipid ratio of the finished product by the initial Drug/Lipid ratio
formed when the lipid and
antisense stock solutions are mixed.
Ethanol Assay. Ethanol in the TF filtrate was determined using an alcohol
dehydrogenase reagent kit supplied by Sigma Chemical Co.
DEAE Sephadex chromatography. A suspension ofthe processed formulation was
loaded onto a 1 X 10 cm column of DEAE sephadex equilibrated in 20 mM PBS, pH
7.5. After
eluting through the column the formulation was collected into a sterile falcon
tube. The volume,
antisense and lipid concentration were measured to determine recovery.
Particle Size. The particle size of the formulation was measured by QELS using
a
Nicomp Particle sizer, (Nicomp, Santa Barbara, CA.) and particle sizes are
reported in the particle
mode with volume weighing.
CA 02271582 1999-OS-13
-66-
Results
of Large
Scale Preparations:
Assay InitialInitial Final Final Initial Final Encaps.
Lipid AntisenseLipid AntisenseDrug:LipidDrug:LipidEffic.
ContentContent Content Content
(mg/ml)(mg/ml) (mg/ml) (mg/ml)
A 10.581 1.936 14.604 1.681 0.183 0.115 63%
B 8.727 2.284 7.926 1.008 0.262 0.127 48%
C 11.06 2.97 2.69 0.556 0.286 0.207 77%
EXAMPLE 10
Phosphodiester and phosphorothioate antisense oligonucleotides encapsulated
according to the methods in Example 2 and 5-9 were examined for their relative
susceptibility to
nuclease digestion by serum or S 1 nuclease. Protection of the phosphodiester-
linked
oligonucleotide was significantly higher in serum when encapsulated as opposed
to the free,
raising the T"Z of degradation from 10 mins to at least 8h. Free
phosphorothioate
oligodeoxynucleotide showed significant breakdown in serum within 30 minutes,
however
encapsulated phosphorothioate oligodeoxynucleotide did not show any sign of
degradation even
after 24h incubation in serum. In vivo data agrees with these findings,
showing no sign of
degradation of the encapsulated phosphorothioate antisense until 8h.
As a positive control, the free phosphodiester and phosphorothioate antisense
were subjected to very potent levels of S1 nuclease (100U/SO,ug) (lU of S1
nuclease will digest 1
ug DNA per minute at 37°C). The enzyme completely digested the free
phosphodiester and
phosphorothioate within seconds after its addition. The encapsulated
phosphodiester under the
same conditions was over 90% intact at 24h, and the encapsulated
phosphorothioate was fully
intact at 24h.
The experiments were conducted as described in the specification, or modified
as
follows.
SI Nuclease Digestion. SO,ug aliquots containing free, encapsulated, or
encapsulated + 0.5% Triton X100 were aliquoted into 1.5 ml eppendorf tubes. To
the tubes were
added 10 ~1 l OX S 1 nuclease buffer, dH20 (to make final volume 100 ,ul),
and, just prior to
CA 02271582 1999-OS-13
-67-
digestion, 1 OOU of S 1 nuclease to each eppendorf tube. The tubes were sealed
with parafilm and
incubated at 55 °C. A sample of the free, encapsulated, or encapsulated
+ 0.5% Triton X100 not
digested by nuclease (standard) was frozen in liquid nitrogen in an eppendorf
tube and stored at -
20°C. At each desired time point, an aliquot of each sample was
collected, added to GDP buffer
containing proteinase K (133 ~cg/ml) and immediately frozen in liquid nitrogen
in order to stop the
reaction. Once all of the time points were collected, the samples were
incubated at 55°C in a
waterbath to activate proteinase K enabling it to denature any remaining S 1
nuclease. Proteinase K
digested samples were applied to polyacrylamide gels, described below, to
assess levels of S 1
nuclease degradation
Normal MuriuelHuman Serum Digestion. 50i.cg of the free, encapsulated, or
encapsulated + 0.5% Triton X 100 was aliquoted into 1.5 ml eppendorf tubes. To
the tubes we added
45 ,ul normal murine/human serum, dH20 (to make final volume 50 ,ul), to each
eppendorf tube.
The tubes were sealed with parafilm and incubated at 37 °C. A sample of
the free, encapsulated, or
encapsulated + 0.5% Triton X100 not digested by nuclease (standard) was frozen
in liquid nitrogen
in an eppendorf tube and stored at -20 ° C. Aliquots were taken at
various time points, added to GDP
buffer containing proteinase K (133 ,ug/ml) and immediately frozen in liquid
nitrogen to stop the
reaction. Once all of the time points were collected, the samples were
incubated at 55*C in a
waterbath to activate proteinase K enabling it to denature any remaining
exonuclease. Proteinase
K digested samples were applied to polyacrylamide gels to assess levels of
exonuclease degradation
Micrococcal Nuclease. An alternative standard nuclease assay not employed in
the present
experiment is the assay disclosed by Rahman et al. US Pat. 5665710, wherein
nucleic acid/ lipid
particles are incubated for 30 mins at 37 °C in presence of an excess
of micrococcal nuclease in 1
mM CaClz.
Polyacrylamide Gel Electrophoresis (PAGE). Prepared 14 cm X 16 cm X 7.5mm
polyacrylamide (15% or 20% ) gels in 7M urea and TBE. Approximately 300 ng of
sample (at each
time point) and standard were aliquoted into eppendorf tubes. An equivalent
volume of 2X loading
buffer was added to each sample. The samples were then heated in a waterbath
to 90°C for 3 min
to reduce secondary structures and then applied to the gel. The loaded gel was
electrophoresed at
600V for 10 min (to sharpen the band) and then at 300V for the duration of the
gel. The gel was
CA 02271582 1999-OS-13
-6g-
incubated in 1X SyberGreen I stain in TBE for a minimum of 15 min and then
photographed while
illuminated under L1V light (3.5 sec exposure, 4.5 aperture).
VII. Conclusion
S As discussed above, the present invention provides methods of preparing
lipid-
encapsulated therapeutic agent (nucleic acid) compositions in which the
therapeutic agent (nucleic
acid) portion is encapsulated in large unilamellar vesicles at a very high
efficiency. Additionally,
the invention provides compositions prepared by the method, as well as methods
of introducing
therapeutic agents (nucleic acids) into cells. The compositions are
surprisingly efficient in
transfecting cells, both in vivo and in vitro.
All publications, patents and patent applications mentioned in this
specification are
herein incorporated by reference into the specification to the same extent as
if each individual
publication, patent or patent application was specifically and individually
indicated to be
incorporated herein by reference.
Although the foregoing invention has been described in some detail by way of
illustration and example for purposes of clarity of understanding, it will be
obvious that certain
changes and modifications may be practiced within the scope of the appended
claims.
CA 02271582 1999-07-26
68a
SEQUENCE LISTING
(1) GENERAL INFORMATION
(i) APPLICANTS: Semple, Sean C.; Klimuk, Sandra,Harasym,Troy;
K.;
Hope, Michael, J.~ Ansell, Steven, M.; Cullis, R.;Scherrer,
Pieter,
Peter; Mok, Wilson, W.K.
(ii) TITLE OF INVENTION: Method for AdministrationTherapeutic
of
Agents, including antisense with repeat dosing
(iii) NUMBER OF SEQUENCES: 17
(iv) CORRESPONDENCE ADDRESS:
(A) ADDRESSEE: Smart & Biggar
(B) STREET: Box 11560, Vancouver Centre, 2200-650W. Georgia
Street
(C) CITY: Vancouver
(D) STATE: British Columbia
(E) COUNTRY: Canada
(F) ZIP: V6B 4N8
(iv) COMPUTER-READABLE FORM
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
(C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: PatentIn Ver. 2.0
(v) CURRENT APPLICATION DATA
(A) APPLICATION NUMBER: CA 2,271,582
(B) FILING DATE: 13-MAY-1999
(vi) PRIOR APPLICATION DATA
(A) APPLICATION NUMBER: US 09/078,955
(B) FILING DATE: 14-MAY-1998
(vii) ATTORNEY/AGENT INFORMATION
(A) NAME: Smart & Biggar
(B) REFERENCE/DOCKET NUMBER: 80472-4
(2) INFORMATION FOR SEQ ID NO.: 1:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Murine
(ix) FEATURE
(D) OTHER INFORMATION: note= "therapeutic antisense sequence
for murine ICAM-1"
CA 02271582 1999-07-26
68b
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 1:
TGCATCCCCC AGGCCACCAT 20
(2) INFORMATION FOR SEQ ID NO.: 2:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo sapiens
(ix) FEATURE
(D) OTHER INFORMATION: /note= ~~therapeutic antisense sequence
for human ICAM-1"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 2:
GCCCAAGCTG GCATCCGTCA 20
(2) INFORMATION FOR SEQ ID NO.: 3:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 15 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo Sapiens
(ix) FEATURE
(D) OTHER INFORMATION: note= ~~therapeutic antisense sequence
for human erb-B-2"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 3:
GGTGCTCACT GCGGC 15
(2) INFORMATION FOR SEQ ID NO.: 4:
CA 02271582 1999-07-26
68c
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 15 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo sapiens
(ix) FEATURE
(D) OTHER INFORMATION: note= "therapeutic antisense sequence
for human c-myc"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 4:
AACGTTGAGG GGCAT 15
(2) INFORMATION FOR SEQ ID NO.: 5:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 16 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo sapiens
(ix) FEATURE
(D) OTHER INFORMATION: note= "therapeutic antisense sequence
for human c-myc"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 5:
TAACGTTGAG GGGCAT 16
(2) INFORMATION FOR SEQ ID NO.: 6:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 24 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo Sapiens
CA 02271582 1999-07-26
68d
(ix) FEATURE
(D) OTHER INFORMATION: note= "therapeutic antisense sequence
for human c-myb"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 6:
TATGCTGTGC CGGGGTCTTC GGGC 2Q
(2) INFORMATION FOR SEQ ID NO.: 7:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 18 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo sapiens
(ix) FEATURE
(D) OTHER INFORMATION: note= "therapeutic antisense sequence
for human c-myb gene"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 7:
GTGCCGGGGT CTTCGGGC 18
(2) INFORMATION FOR SEQ ID NO.: 8:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 18 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo Sapiens
(ix) FEATURE
(D) OTHER INFORMATION: note= "therapeutic antisense sequence
for human IGF-1R"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 8:
GGACCCTCCT CCGGAGCC 18
CA 02271582 1999-07-26
68e
(2) INFORMATION FOR SEQ ID NO.: 9:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 18 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo Sapiens
(ix) FEATURE
(D) OTHER INFORMATION: note= ~~therapeutic antisense sequence
for human IGF-1R"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 9:
TCCTCCGGAG CCAGACTT 18
(2) INFORMATION FOR SEQ ID NO.: 10:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 15 nucleic acids
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo Sapiens
(ix) FEATURE
(D) OTHER INFORMATION: note= ~~therapeutic antisense sequence
for human EGFR"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 10:
CCGTGGTCAT GCTCC 15
(2) INFORMATION FOR SEQ ID NO.: 11:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 21 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
CA 02271582 1999-07-26
68f
(A) ORGANISM: Homo Sapiens
(ix) FEATURE
(D) OTHER INFORMATION: note= "therapeutic antisense sequence
for human VEGF"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 11:
CAGCCTGGCT CACCGCCTTG G 21
(2) INFORMATION FOR SEQ ID NO.: 12:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Murine
(ix) FEATURE
(D) OTHER INFORMATION: note= "therapeutic antisense sequence
for murine PKC-alpha"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 12:
CAGCCATGGT TCCCCCCAAC 2C
(2) INFORMATION FOR SEQ ID NO.: 13:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo sapiens
(ix) FEATURE
(D) OTHER INFORMATION: note= "therapeutic antisense sequence
for human PKC-alpha"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 13:
CA 02271582 1999-07-26
68g
GTTCTCGCTG GTGAGTTTCA 20
(2) INFORMATION FOR SEQ ID NO.: 14:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 18 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo Sapiens
(ix) FEATURE
(D) OTHER INFORMATION: note= ~~therapeutic antisense sequence
for human bcl-2 gene"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 14:
TCTCCCAGCG TGCGCCAT 18
(2) INFORMATION FOR SEQ ID NO.: 15:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 15 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo Sapiens
(ix) FEATURE
(D) OTHER INFORMATION: note= ~~therapeutic antisense sequence
for human c-raf-1 protein kinase"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 15:
GTGCTCCATT GATGC 15
(2) INFORMATION FOR SEQ ID NO.: 16:
(i) SEQUENCE CHARACTERISTICS
CA 02271582 1999-07-26
68h
(A) LENGTH: 34 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: RNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo Sapiens
(ix) FEATURE
(D) OTHER INFORMATION: note= ~~therapeutic antisense sequence
for human VEGF-R-1"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 16:
GAGUUGCUGA UGAGGCCGAA AGGCCGAAAG UCUG 34
(2) INFORMATION FOR SEQ ID NO.: 17:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 15 base pairs
(B) TYPE: nucleic acid
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Homo Sapiens
(ix) FEATURE
(D) OTHER INFORMATION: note= ~~therapeutic antisense sequence
for human c-myc SCR"
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 17:
GAACGGAGAC GGTTT 15