Note: Descriptions are shown in the official language in which they were submitted.
CA 02276284 1999-06-28
WO 98/24480 PCT/US96/19352
RADIOIODINATED PHOSPHOLIPID ETHER ANALOGS
AND METHODS OF USING THE SAME
FIELD OF THE INVENTION
The present invention relates generally to the field of radiopharmaceuticals
and
biological probes. and more specifically to radiolabelled analogs of
phospholipid ethers useful
in cancer diagnosis and treatment.
BACKGROUND OF THE INVENTION
The early detection of cancer has been one of the primary goals of modern
imaging
technology, since the identification of a suspected tumor in a localized stage
significantly
improves the chances for successful treatment and elimination of the cancerous
tissue. A
large number of imaging strategies have therefore been designed. using a
variety of techniques
and modalities. to aid the physician in making an accurate diagnosis as early
as possible.
Unfortunately. conventional imaging techniques such as computerized tomography
(CT) and MRI (magnetic resonance imaging) are limited in their ability to
afford a conclusive
diagnosis of a suspected lesion, since they are only capable of observing
differences in the
density or morphology of tissues. A more invasive and costly biopsy procedure
is often
necessary to provide a definitive diagnosis. In contrast, nuclear medicine
techniques such as
positron emission tomography (PET) and single photon emission tomography
(SPELT) can
provide functional or biochemical information about a particular organ or area
of interest.
However. the success of these nuclear imaging techniques depends in large part
on the
selective uptake and detection of appropriate radiopharmaceuticals. Selective
uptake. in turn,
depends upon the development of radiopharmaceuticals with a high degree of
specificity for
the target tissue. Unfortunately, the tumor-localizing agents developed thus
far for
oncological applications have had only limited application.
For example. one of these prior art compounds, 6'Ga gallium citrate. was
originally
identified for its ability to accumulate in tumor tissue. Unfortunately, 6'Ga
gallium citrate is
taken up by a variety of other non-tumorous lesions as well, including
inflammatory lesions,
and unacceptable amounts of radioactivity can also accumulate in liver and
spleen tissue. The
rapid buildup of a radiopharmaceutical in these organs can seriously interfere
with the
imaging of nearby lesions, and also negatively impacts the dosage that can
safely be given to
a patient.
CA 02276284 2002-10-25
74667-117
An alternative approach has been to develop
radiolabelled monoclonal antibodies (blabs) directed to
tumor-specific antigens. However, these monoclonal
antibodies are specific only to the particular tumor tissue
for which they have been produced and therefore will not
localize generally in neoplastic tissue. Moreover, the use
of blabs for diagnostic imaging has lead to additional
problems, including varying degrees of antigen expression,
low tumor uptake, non-specific binding and adverse
immunogenic reactions.
In an attempt to address these problems, the
present inventors have recently identified and developed a
series of novel compounds demonstrating useful tumor
specificity. See, e.g., U.S. Patent Nos. 4,925,649;
4,965,391; 5,087,721; and 5,347,030. It is believed that
these radioiodinated phospholipid ether analogs take
advantage of a unique biochemical characteristic of
malignant tumor cells: i.e. the large concentration of
naturally-occurring ether lipids in the cell membranes
relative to corresponding normal tissues. Although the
precise mechanism of action is not fully understood, the
prevailing hypothesis is that the phospholipid ether analogs
become entrapped in tumor membranes. Accordingly, these
compounds localize in tumor tissue and remain in place for
diagnostic and/or therapeutic applications.
The selective retention of the radiolabelled
phospholipid ether analogs described in the above patents
has been demonstrated in a variety of rodent and animal
tumors. Unfortunately, the data obtained from these studies
has also demonstrated a relatively rapid clearance of the
radiopharmaceutical compound from the blood, and an
undesirable accumulation by non-target tissues. As noted
above, non-target tissue uptake can decrease the efficacy of
-2
CA 02276284 2005-10-05
74667-117
radiodiagnostic imaging by creating high background
activity, or by causing excessive exposure of radiosensitive
tissues to the injected radioactivity.
Accordingly, there remains a significant need in
the art for radiopharmaceuticals which exhibit a rapid
clearance from non-target tissues as well as an extended
half-life in the blood plasma, while still retaining its
specificity and avidity for neoplastic tissue. Such an
agent should not only assist in the non-invasive imaging of
primary tumors and metastases, but should also provide a
potential cytotoxic agent for site-specific eradication of
the tumor tissue.
SU1~ARY OF THE INVENTION
The present invention provides a compound of the
general formula:
0 Y X
Z-CH2CH20 i 0 CH2CHCH20 (CH2) n
m
where X is selected from the group consisting of radioactive
isotopes of iodine; m is equal to 1 or 0; n is an integer
between 16 and 30; Y is selected from the group
O
II
consisting of H, OH, COOH, OCR, and OR, and Z is selected
from the group consisting of NH2, NR2 and NR3+, wherein R is
an alkyl or aralkyl substituent.
In accordance with a further aspect of the present
invention, there are provided compounds having the general
Formula I:
-3-
CA 02276284 2002-10-25
74667-117
X
CHZO(CH2)n
Y-C H 0
CHZOPOCHzCHz-Z
0-
where X is a radioactive isotope and n is an integer between
16 and 30. In a preferred embodiment of the present
invention, X is a radioactive isotope of iodine, preferably
selected from the group comprising 1231, lzSl ~ and 1311. It is
further contemplated that X can be substituted at the ortho,
meta or para position on the aromatic ring. In a preferred
embodiment, the radioactive isotope is substituted at the
para position.
Y is selected from the group comprising H, OH,
0
COOH, o~~R , and OR, and Z is selected from the group
comprising NH2, NRz, and NR3, wherein R is an alkyl or
aralkyl substituent.
In accordance with a specific illustrative
embodiment of the invention, the improved compound is
1-O-[18-(p-iodophenyl)octadecyl]-1,3-propanediol-3-
phosphocholine.
CH20 (CHz)
H-C H 0 18
CHzOPOCH2CHzN+(CH3) 3
0-
In accordance with another specific illustrative
embodiment, the improved compound is 1-O-[18-(p-
Iodophenyl)octadecyl]-2-O-methyl-rac-glycero-3-
phosphocholine:
-3a-
CA 02276284 2002-10-25
74667-117
CHzO (CH2)18 \ / I
CH30-CH 0
CHZOPOCHZCHzN+(CH3)3
0-
In a further embodiment of the invention, the
radiolabelled aralkyl side chain may be substituted directly
onto the alkyl phosphocholine moiety in accordance with
general Formula II:
X 0
I I
~ / ~CH2)nOPOCHZCH2-Y
0-
where X is a radioactive isotope substituted at the ortho,
meta or para position, preferably of iodine, n is an integer
from 16 to 30, and Y is selected from the group comprising
NH2, NR2, and NR3, wherein R is an alkyl or aralkyl
substituent.
-3b-
CA 02276284 1999-06-28
WO 98/24480 PCT/US96/19352
In accordance with a specific illustrative embodiment of the invention, the
improved
compound is 18-(p-iodophenyl)octadecyl phosphocholine.
O
(CH2)~$OPOCH2CH2N(CH3)3
O_
BRIEF DESCRIPTION OF THE FIGURES
Figure 1 is an illustrative preparatory scheme for 18-(p-
iodophenyi)octadecanol;
Figure 2 is an illustrative preparatory scheme for 18-(p-iodophenvl)octadecyl
phosphocholine;
Figure 3 is an illustrative preparatory scheme for 1-O-[18-(p-
iodophenyl)octadecylJ-
1.3-propanediol-3-phosphocholine;
Figure 4 is an illustrative preparatory scheme for 1-O-[18-(p-
iodophenyl)octadecyl]-2-
O-methyl-rcrc-glycero-3-phosphocholine;
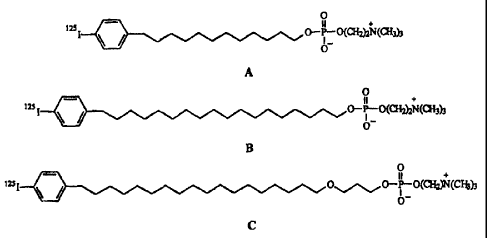
Figures SA-SC provide the chemical structures of the alkyl chain len~~th
analogs 12-(p-
iodophenyl)dodecyl phosphocholine (Figure ~A), 1~-(p-iodophenyl)pentadecvi
phosphocholine
(Figure ~B), and 18-(p-iodophenyl)octadecyl phosphocholine (Figure ~C) used in
biodistribution studies with rats bearing Walker-256 tumor;
Figure 6 provides a line graph illustrating the blood clearance profile of 12-
(p-
iodophenyl)dodecyl phosphocholine (C-12), 15-(n-iodophenyl)pentadecyl
phosphocholine (C-
15) and 18-(p-iodophenyl)dodecylphosphocholine (C-18) in rats bearing Walker-
256 tumors;
Figures 7A-7C provide the chemical structures of the alkyl chain length
analogs 12-(p-
iodophenyl)dodecyl phosphocholine (Figure 7A), 18-(p-iodophenyl)octadecyl
phosphocholine
(Figure 7B), and 1-O-[18-(p-iodophenyl)octadecylJ-1,3-propanediol-3-
phosphocholine used in
biodistribution studies with rats bearing Dunning (MATLyLu) prostate tumors;
Figures 8A-bC are depictions of whole-body gamma-camera scintigraphy scans of
Dunning (MATLyLu) prostate tumor-bearing using 18-(p-iodophenyl joctadecyl
phosphocholine (Figure 8A), 12-(p-iodophenyl)dodecyl phosphocholine (Figure
8B) and 1-O-
[18-(p-iodophenyl)octadecvl]-1,3-propanediol-3-phosphocholine (Figure 8C).
GENERAL DESCRIPTION OF THE INVENTION
The present invention represents a significant improvement of the
radiolabelled
phospholipid ether analogs previously described in the prior art, providing a
series of
radiopharmaceutical compounds exhibiting greatly increased plasma half life
and a
-4-
CA 02276284 1999-06-28
WO 98/24480 PCT/US96/19352
significantly lower accumulation in non-target organs. Thus. these improved
radiopharmaceuticals provide superior tumor imaging capabilities by reducing
the amount of
background radiation from non-target tissues, and the rapid clearance of the
compounds from
non-target organs also allows for an increase in the radiation dosimetry of
the
radiopharmaceutical. for further enhancement of tumor imaging capabilities and
cytotoxic
cancer therapy.
Surprisingly, the nature of the phospholipid ether compounds which exhibit
these
enhanced capabilities are compounds having an extension of the carbon chain
bearing the
radiolabelled phenyl group. Previous studies with related alkyl phosphocholine
analogs had
demonstrated that blood levels actually decreased with increasing chain
length. while tumor
levels increased. See, e.g., Kotting et al., "Alkylphosphocholines: influence
of structural
variation on biodistribution at antineoplastically active concentrations."
Cancer Chemother.
Pharna. 30:105-1 12 ( 1992). In contrast, the improved compounds of the
present invention
have displayed a propensity to remain in the circulation much longer than the
original shorter
1 ~ chain analogs, and the delayed clearance from the blood plasma
advantageously results in
additional opportunities for uptake of the radiopharmaceuticals by tumor
tissue as they
continuously circulate through the vasculature.
Although an understanding of the underlying mechanism is not essential to the
beneficial use of the improved compounds provided by the present invention,
the inventors
believe that the uptake and transport of the radiolabelled analogs by plasma
components may
be an important factor relating to the tumor retention of the compounds.
Certainly, increasing
the length of the carbon chain results in a corresponding increase in the
lipophilicity of the
analogs, and greater lipophilicity would presumably increase the affinity of
these compounds
for the cell membrane. There is also evidence (not shown) to indicate that the
longer carbon
chain alters the binding affinity of these compounds for plasma components
such as plasma
albumin.
The differences in the clearance and quantity of radioactivity in non-target
tissues
observed with the improved compounds of the present invention significantly
enhances the
chances for the imaging of tumors in human patients. Moreover, it should also
be noted that
the improved phospholipid ether analogs of the present invention are cytotoxic
to tumor cells,
even without the presence of a radioactive isotope in the compound. Therefore,
the inclusion
of a long-lived radioactive isotope of iodine, for example, yields tumor-
specific
radiopharmaceuticals which are tissue-destructive by more than one mode.
-5-
*rB
CA 02276284 1999-06-28
WO 98/24480 PCT/US96/19352
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
The following examples relate to specific embodiments and methods of using the
improved radiolabelled phospholipid analogs of the present invention, and
include illustrative
methods for synthesizing the analogs. All starting materials and reagents were
obtained from
Aldrich Chemical Company, Milwaukee, Wisconsin.
In the experimental disclosure which follows, the following abbreviations
apply: eq
(equivalents); M (Molar); ~M (micromolar); N (Normal); mol (moles); mmol
(millimoles);
Pmol (micromoles): nmol (nanomoles); g (grams); mg (milligrams); kg
(kilograms); ug
(micrograms); L (liters); ml (milliliters); ~l (microliters); em
(centimeters); mm (millimeters);
~m (micrometers); nm (nanometers); °C (degrees Centigrade); h (hours):
min (minutes); sec
(seconds); msec (milliseconds); Ci (Curies) mCi (milliCuries); ~Ci
(microCuries); TLC (thin
layer achromatography); Ts (tosyl); Bn (benzyl); Ph (phenyl); Ms (mesyl); Et
(ethyl). Me
(methyl).
EXAMPLE 1
The synthesis of 18-(p-Iodophenyl)octadecanol (Compound VI), which is a
preliminary
compound for the illustrative preparatory schemes for the phospholipid ether
analogs
discussed in detail in Examples 2 through 4 below, was accomplished from
commercially-
available 1 S-(p-iodophenyl) pentadecanoic acid (Compound I) in accordance
with the
illustrative preparatory scheme shown in Figure I.
In general terms, aliphatic chain elongation from C-1~ to C-18 was achieved by
Li,CuCl,-catalyzed cross-coupling between Grignard reagent (Compound IV)
prepared from
benzyl 3-bromopropyl ether and I S-(p-iodophenyl) pentadecyl tosylate
(Compound III). See,
e. g., Fouquet and Schlosser. "Improved carbon-carbon linking by controlled
copper catalysis,"
Angew. Chem. Int. Ed. 13:82-83 ( 1974); Schlosser and Bossert, "The 'two-fold
reaction'
benchmark applied to the copper catalyzed assembling of I,w-difunctional
hydrocarbon
chains," Tetrahedron 47:6287-92 ( I 991 ). Cleavage of the benzyl protective
group in
Compound V by anhydrous aluminum chloride-anisole (see, e.g., Akiyama et al.,
"AICl3 -
N,N-dimethylaniline: a novel benzyl and allyl ether cleavage reagent." Bull.
Chem. Soc. Jpn.
65:1932-38 { 1992)) afforded alcohol VI with 18 carbon atoms in aliphatic
chain.
Specifically, to a solution of 1 S-(p-iodophenyl)dodecanoic acid (Compound I)
(210
mg; 0.47 mmol) in 3 ml of tetrahydrofuran was added BH3 ~THF complex ( 1 ml of
1.0 M
solution; 1 mmol) dropwise at 0°C. The reaction mixture was then
stirred at room temperature
-6-
CA 02276284 1999-06-28
WO 98!24480 PCT/US96/19352
under anhydrous conditions for 10 h. It was again cooled to 0°C and
quenched with water.
Ethyl acetate and additional water were added. The organic layer was
separated. washed with
water and dried (Na,SOa). Evaporation of solvent gave the white solid Compound
II, 15-(p-
iodophenyl)pentadecanol (204 mg, 100 %). Compound II was used in the next step
without
purification.
A mixture of 15-(p-iodophenyl)pentadecanol (Compound II) (200 mg; 0.47 mmol),
tosyl chloride ( 102 mg; 0.53 mmol) and 4-dimethylaminopyridine (66 mg; 0.53
mmol) in
dichloromethane (3 ml) was stirred at room temperature for 12 h. The reaction
mixture was
then partitioned between chloroform (40 ml), methanol (40 ml) and 0.1 N
hydrochloric acid
(36 ml). The chloroform layer was separated and extraction was repeated (2 x
40 ml of
chloroform). Extracts were combined, dried (Na,SOa) and evaporated. The
residue was
chromatographed on silica gel in hexane-chloroform (from 95:5 to 40:60) to
give Compound
III. 15-(p-Iodophenyl)pentadecyl tosylate (259 mg; 95 %).
To a solution of 3-benzyioxypropanol (5.28 g; 31.8 mmoi) and carbon
tetrabromide
(13.1 g; 39.6 mmol) in dichloromethane (50 ml) was slowly added
triphenylphosphine (10.4
g; 39.7 mmol) at 0°C. The reaction mixture was stirred at 0°C
for 30 min and at room
temperature for I h. By that time TLC in chloroform showed completion of the
reaction.
Most of the solvent was evaporated, the residue was diluted with hexane (200
ml) and the
precipitate of triphenylphosphinoxide was removed by filtration. Evaporation
of the filtrate
gave an oily residue which was purified by chromatography in 1 % ether in
hexane. This
afforded Compound IV, benzyl 3-bromopropyl ether (5.61 g; 77 %).
To a suspension of magnesium powder (60 mg: 2.5 mmol) in tetrahydrofuran ( 1.5
ml)
was added dibromoethane (0.02 ml) for activation. After the reaction with
dibromoethane had
ceased, the solution was removed by syringe and replaced with fresh
tetrahydrofuran (2.5 ml).
Then. benzyl 3-bromopropyl ether (Compound IV) (0.12 ml; 0.67 mmol) was added
dropwise
to the stirred suspension of magnesium for 30 min. When all the halide had
been added,
stirring was continued for an additional 2 h at room temperature. The green-
gray solution of
Grignard reagent was carefully withdrawn by syringe and transferred to 25 ml
flask, which
was then cooled in a dry ice bath to -78°C. A solution of Li,CuCl4
(0.0067 mmol/ml) in
tetrahydrofuran (0.5 ml; 0.0034 mmol) was added to the Grignard reagent under
stirring,
followed by Compound III, I S-(p-iodophenyl)pentadecyl tosylate (260 mg; 0.44
mmol) in 3
ml of tetrahydrofuran.
-7-
CA 02276284 1999-06-28
WO 98/24480 PCT/US96/19352
The reaction mixture was allowed to warm to room temperature during a 2 h
period,
and stirring was continued for an additional 12 h. The reaction was quenched
by ammonium
chloride solution and extracted with ethyl acetate. The extract was washed
with water, dried
{Na~SO,) and evaporated. Silica gel chromatography with 2% ether in hexane
provided
Compound V. 1-Benzyloxy-18-(p-iodophenyl)octadecane (190 mg; 76 %).
Finally, to a solution of 1-benzyloxy-18-(p-iodophenyl)octadecane (Compound V)
( 185
mg; 0.33 mmol) and anisole (0.1 S ml; 1.32 mmol) in dichloromethane (3 ml) was
added
anhydrous aluminum chloride ( 135 mg; 1 mmol) at the room temperature.
Stirring was
continued for 2 h. The reaction was quenched by diluted hydrochloric acid and
extracted by
chloroform. Extract was dried {Na~SOa) and evaporated. The residue was
chromatographed in
hexane - ethyl acetate (gradient from 95:5 to 85:15) to give 18-(p-iodophenyl)
octadecanol,
Compound VI ( 123 mg; 77 %).
EXAMPLE 2
Further conversion of 18-(p-iodophenyl) octadecanol into the desired
phosphocholines
was performed as described in detail for the C-12 analogs in Rampy et al.,
"Synthesis and
biological evaluation of radio-iodinated phospholipid ether analogs," Nucl.
Med. Biol. 22, 505-
512 ( 1990. In one preferred embodiment, the improved phospholipid ether
analog
contemplated by the present invention is a simple straight chain alkyl
phospholipid, 18-(p-
iodophenyl)octadecyl phosphocholine (Compound XVI). The synthesis of Compound
XVI
was accomplished according to the illustrative preparatory scheme shown in
Figure 2.
As illustrated in Figure 2. 2-chloro-2-oxo-1,3.2-dioxaphospholane (0.02 ml;
0.27
mmol) was added to the stirred solution of 18-(p-iodophenyl)octadecanol
(Compound VI)
( 115 mg; 0.24 mmol) in dry benzene (3 ml) containing triethylamine (0.042 ml;
0.29 mmol).
Stirring was continued overnight. The precipitated triethylamine hydrochloride
was filtered off
and the solvent was removed in vacuo. The residue was transferred into a
pressure bottle. A
solution of trimethyiamine in acetonitrile ( 5 ml; 25 % w/v) was added. The
bottle was
sealed and heated at 75°C for 24 h. The acetonitrile was then
evaporated and the residue was
chromatographed on silica gel with chloroform-methanol (gradient from 10:0 to
5:5), followed
by final elution with chloroform-methanol-water (65:25:4). After evaporation
of the solvent,
the product was precipitated by addition of acetone to give a white solid (130
mg; 84 %).
_g_
CA 02276284 1999-06-28
WO 98/24480 PCT/IJS96119352
EXAMPLE 3
In another preferred embodiment, the improved phospholipid ether analogs
contemplated by the present invention are constructed using a propanediol
backbone. In this
example. the synthesis of 1-O-[ 18-(p-Iodophenyl)octadecylJ-1,3-propanediol-3-
phosphocholine
(Compound XIV) was accomplished according to the illustrative preparatory
scheme shown in
Figure 3.
To a solution of 18-(p-iodophenyl)octadecanol {Compound VI) ( 150 mg; 0.317
mmol)
and triethylamine (0.07 ml; 0.475 mmol) in dry methylene chloride (2 ml) was
added methane
sulfonyl chloride (0.03 ml; 0.38 mmol) at 0°C. Stirring was continued
for 40 min and the
reaction was quenched by addition of water. The reaction mixture was diluted
with
chloroform and washed several times with water. The chloroform Layer was dried
(Na,S04)
and evaporated. The residue was chromatographed in hexane - ethyl acetate (9:1
). This
afforded pure Compound VII. 18-(p-Iodophenyl)octadecyl methanesulfonate ( 142
mg; 82
yield).
1 ~ To a solution of 3-benzyloxy propanol (Compound VIII) (0.03 ml; 0.18 mmol)
and
18-(p-iodophenyl) octadecyl methanesulfonate (Compound VII) (66 mg; 0.12 mmol)
in dry
dimethylformamide (3 ml) was added sodium hydride (8 mg of 60% suspension in
oil; 0.2
mmol) at the room temperature. The reaction mixture was stirred for 12 hr,
quenched by
water and extracted with ethyl acetate. The extract was washed with brine,
dried and
evaporated. Column chromatography in hexane-ethyl acetate (gradient from 95:~
to 85:15)
afforded Compound X, 1-O-(18-(p-Iodophenyl) octadecylJ-3-O-benzyl-1.3-
propanediol (60
mg; 81 % yield).
Compound X was debenzyiated by AICh-anisole as described for the svnthesis of
Compound VI, to produce Compound XII, 1-O-[18-(p-Iodophenyl)octadecylJ-1,3-
propanediol
(50 mg; 0.08 mmol). (42 mg, 99 % yield). Finally, the desired phosphocholine
XIV was
synthesized above from the alcohol XII (42 mg; 79 mmol) in an analogous manner
to that
described in Example 2 for Compound XVI. (45 mg, SS % yield).
EXA1MPLE 4
This example illustrates yet another preferred embodiment of the improved
phospholipid ether analogs contemplated by the present invention, wherein the
hydrogen
located at the 2-position of Compound XIV is replaced with an O-methyl group.
The
synthesis of 1-O-(18-(p-Iodophenyl)octadecylJ-2-O-methyl-rac-glycero-3-
phosphocholine
-9-
CA 02276284 1999-06-28
WO 98/24480 PCT/US96/19352
{Compound XV) was accomplished according to the illustrative preparatory
scheme shown in
Figure 4.
Compound XI, l-O-[ 18-(p-IodophenyI)octadecyl]-2-O-methyl-3-O-benzyl-rac-
glycerol,
was synthesized as described above in Example 3 above for Compound X, from 18-
(p-
iodophenyl) octadecyl methanesulfonate (Compound VII) (67 mg) and 1-O-benzyl-2-
O-
methyl-rac-glycerol (Compound IX) (36 mg). (62 mg, 78% yield). The synthesis
of
Compound IX is known in the art and is described in detail in Pinchuk er al.,
Chem. Phys.
Lipids, 59:263-65 (1991). Briefly, to 2-O-methyl-1,?-O,O-benzylideneglycerol
(2.4 g, 12.4
mmol) cooled in an ice bath was added 18 ml of 1.0 M borane-THF solution ( I 8
ml, 18
IO mmol) with stirring. After 10 minutes the mixture was left at room
temperature for I? h,
then for 48 h at 40-45°C until TLC (hexane/ethyl acetate 6:4) showed no
starting material
remained. The mixture was cooled and water was added drop-wise to destroy the
borane
excess. The solvent was evaportated. the residue was dissolved in water.
extracted with ether
(3 x l00 nil), extracts were washed with water, dried (Na,SO,), and evaporated
to give 2.32 g
I ~ (96%) of a slightly yellow oil. The compound was used for the following
step without any
further purification.
Compound XIII. 1-O-[I8-(p-Iodophenyl)octadecyl]-2-O-methyl-rac-glycerol, was
synthesized from the benzyl ether (Compound XI ) (58 mg, 0.09 mmol) by the
procedure
described for Compound VI in Example 1. (40 mg, 80% yield). The desired
phosphocholine
20 XV was synthesized from the alcohol XIII (33 mg, 0.06 mmol) by the
procedure described in
Example 3 above for Compound XVI. (32 mg 75% yield).
EXAMPLE 5
Radioiodination of Phosphotipid Ether Analogs
25 For certain uses, such as scintigraphy or experimental evaluation of tissue
distribution,
it is desirable to create radioactive compounds. Radioiodination of the
iodinated phospholipid
ether analogues disclosed herein, or one of the intermediates in the synthesis
pathway, can be
accomplished by a variety of techniques. some of which are known in the art.
For example,
aromatic compounds with electron donating groups (such as anilines) can be
radiolabelled by
30 electrophilic iodination in the presence of radioiodine, iodine
monochloride, chloramine-T,
iodogen, etc. Unactivated aromatic rings can be radioiodinated by exchange of
a leaving
group, such as aryl boronic acids, aryl thallium trifluoroacetates, triazenes
or metallated arenes
with radioiodine. Direct electrophilic radioiodination of a phenyl ring is yet
another
- 10-
CA 02276284 1999-06-28
WO 98/24480 PCT/US96/19352
alternative, but may produce isomeric mixtures which are difficult to
separate. Iodine
exhange of aryl iodides with radioiodine may be a preferable approach insofar
as no complex
separation techniques are necessary, since the substrate and radioiodinated
product are
chemically identical.
In a preferred embodiment of the invention, an isotope exchange-type technique
is
utilized wherein the substrate and radioiodine are reacted at an elevated
temperature in a
"melt." The molten reaction medium possesses a sufficiently high dielectric
constant to
solubilize both the substrate and radioiodide. Examples of reaction media
currently in use are
benzoic acid (mp 122°C., by 249°C.) and acetamide (mp
182°C., by 221°C.). In a specific
preferred embodiment. an acidic exchange medium comprising pivalic acid, a
homolog of
acetic acid, also known as trimethyl acetic acid, can be used. Pivalic acid
has a melting point
of 33°C. and a boiling point of 164°C.
The phospholipid ether analogs discussed herein were made via isotope exchange
in
pivalic acid. This technique is described in detail in Weichert et al..
"Radioiodination via
isotope exchange in pivalic acid," Appl. Radiat. Isot. 37:907-13 (1986).
Briefly, the unlabeled
compound (0.5 mg) was placed in a 300-~l V-vial (Wheaton. Millville. NJ)
fitted with teflon
faced seal and screw cap. Absolute ethanol (20 ~l) was added via a microliter
syringe,
followed by aqueous Na''-51 (0.5-3.0 mCi, 2-10 ul) (no-carrier-added in
reductant-free 0.1 N
NaOH from Amersham Radiochemicals). The vial was gently swirled to dissolve
the contents
and ensure homogeneity.
Inlet and outlet cannuli were inserted and a gentle stream of nitrogen was
applied to
remove the solvents. Two successive in-line charcoal traps were placed on the
outlet side in
order to trap any volatile radioiodine present in the reaction vial. Once dry,
solid pivalic acid
(5 mg), previously dried by azeotropic removal of water with toluene and
distilled under
nitrogen. was added. The vial was sealed and heated at 160°C in a
preheated single well
aluminum heating block containing 1 cm of sand in the bottom of the well.
After 1 hr., the
reaction vial was removed from the heating block and allowed to cool to room
temperature.
Absolute ethanol (70 ~l) was added through a micro syringe followed by gentle
agitation and
subsequent removal of a TLC sample (I-2 pl).
The entire contents of the reaction vial were then injected directly onto a
silica gel
HPLC column eluted with hexane/isopropanol/water (40:52:8) at 0.8 ml/min.
Peaks were
analyzed by both UV (230 and 254 nm) and radiodetection. After pooling
appropriate
fractions, the radiochemical purity of the final product was monitored by TLC
(gamma and
CA 02276284 1999-06-28
WO 98/24480 PCT/US96/I9352
UV detection) and by HPLC (UV at 230254 nm and radiochemical detector).
Fractions were
combined and the solvent was removed with a gentle stream of nitrogen. HPLC
analysis of
the final compound confirmed both chemical (UV at 230/254 nm) and
radiochemical
(radioactivity) purity.
Of course, any isotope of iodine such as the clinically used isotopes, ''-'-I.
''-'I, ''-SI and
'''I can be used. ''-SI is preferred for in vitro work in the laboratory due
to its relatively long
half life. For radiodiagnostic purposes in humans, ''-3I or '3'I are preferred
due to their shorter
half lives and favorable imaging energies. Thus, the radioiodination procedure
described
above may be modified, as known by those of skill in the art, to compensate
for the
difference in half life.
It is further contemplated that the radioiodinated phospholipid ether analogs
of the
present invention may be solubilized in a suitable transport agent or carrier
vehicle, and
administered to mammalian subjects as radio.logic agents by any known manner.
preferably
parentally such as intravenously or intraperitonally. It is not intended that
the present
I S invention be limited by the particular nature of the therapeutic
preparation. For example,
such compositions can be provided together with physiologically tolerable
liquid. gel or solid
carriers. diluents. adjuvants and excipients.
These therapeutic preparations can be administered to mammals for veterinary
use,
such as with domestic animals. and clinical use in humans in a manner similar
to other
therapeutic agents. In general, the dosage required for therapeutic efficacy
will varv
according to the type of use and mode of administration. as well as the
particularized
requirements of individual hosts.
Such compositions are typically prepared as liquid solutions or suspensions,
or in solid
forms. Oral formulations for cancer usually will include such normally
employed additives
such as binders, fillers. carriers, preservatives, stabilizing agents,
emulsifiers. buffers and
excipients as, for example, pharmaceutical grades of mannitol, lactose,
starch, magnesium
stearate, sodium saccharin, cellulose. magnesium carbonate, and the like.
These compositions
take the form of solutions, suspensions, tablets, pills, capsules, sustained
release formulations,
or powders. and typically contain I%-95% of active ingredient, preferably 2%-
70%.
The compositions are also prepared as injectables, either as liquid solutions
or
suspensions: solid forms suitable for solution in, or suspension in, liquid
prior to injection
may also be prepared. The radioiodinated compounds of the present invention
are often
mixed with diluents or excipients which are physiologically tolerable and
compatible.
- 12-
rB
CA 02276284 1999-06-28
WO 98/24480 PCTIUS96/19352
Suitable diluents and excipients are, for example, water, saline, dextrose,
glycerol. or the like,
and combinations thereof. In addition. if desired the compositions may contain
minor
amounts of auxiliary substances such as wetting or emulsifying agents,
stabilizing or pH
buffering agents.
Additional formulations which are suitable for other modes of administration,
such as
topical administration. include salves, tinctures, creams, lotions, and, in
some cases,
suppositories. For salves and creams, traditional binders. carriers and
excipients may include,
for example, poiyalkylene glycols or triglycerides.
EXAMPLE 6
Biodistribution Studies With Walker-256 Carcinosarcoma
The three radiolabelled compounds illustrated in Figure ~, including the
improved C-
18 analog 18-{p-iodophenyl)octadecyl phosphocholine (Compound XVI) described
in Example
2, were prepared and administered to two sets of female Sprague Dawley rats
(Charles River,
1 ~ Portage. Michigan). One normal set was used as a control, and one set was
inoculated with
Walker-2~6 carcinosarcoma cells (~ x 106 cells) in 0.2 ml saline in the right
hindlimb. The
Walker carcinoma cell line was provided by Dr. James Varani of the Department
of Pathology
at the University of Michigan, and is an accepted cell line representative of
effect in humans.
See, e.k. Tayck et al., "Influence of the Walker 256 Carcinosarcoma on Muscle.
Tumor, and
Whole-Body Protein Synthesis and Growth Rate in the Cancer-Bearing Rat, Cancer
Res.
46:5649-5~4 ( 1986).
The animals were used 6-8 days later when the tumor weight averaged 10 g. The
radiolabelled compounds were dissolved in absolute ethanol (50-500 pl) and
Tween-20 (0.1
ml/mg of compound) was added to the solution. Ethanol was removed by
evaporation under
a stream of nitrogen. Physiological saline or sterile water was added, to give
a 2-3% Tween-
20 solution which was subsequently mixed by vortex. The solubilized
radiolabelled
compounds (5-10 ~Ci, 0.3 ml) were administered intravenously via the tail vein
to tumor
bearing rats, and the animals were sacrificed by exsanguination while under
ether anesthesia at
the various time points. Blood samples were collected through cardiac puncture
and selected
tissues (liver, kidney, etc.) were removed, trimmed, blotted to remove excess
blood and
weighed. Large organs were thoroughly minced with scissors to obtain random
representative
tissue samples.
-13-
CA 02276284 1999-06-28
WO 98/24480 PCT/US96/19352
For the biodistribution experiments with 12-(p-iodophenyl)dodecyl
phosphocholine,
weighed tissue samples were counted with a well scintillation counter (85%
counting
efficiency). The concentration of radioactivity in each tissue was expressed
as a percentage of
administered dose per gram of tissue.
S Tissue distribution was assessed at various times following administration
of the
radioiodinated chain length isomers. An illustration of the data for each
analog is set forth in
Table 1 below. The results are expressed as the mean % administered dose per
gram ~ SEM
(% Dose/g ~ SEM).
C-12 Analog C-15 C-1$
Analog Analog
Tissue 24hr 48hr 24hr 48hr 24hr 48hr
Adrenal 0.33 0.29 0.73 0.49 0.85 0.93
0.01 0.01 0.08 0.05 0.04 0.06
BIOOd 0.2710.040.270.030.160.010.14O.Oi0.960.000.640.07
Duodenum1.35 0.77 1.13 1.38 0.69 0.68
0.13 0.06 0.13 0.24 0.05 0.04
Kidney 4.10 3.44 1.14 0.91 0.65 0.59
0.44 0.08 0.11 0.04 0.00 0.04
Liver 1.620.17 I.370.011.290.100.830.040.550.040.590.05
Lung 1.06 0.53 0.97 0.81 0.88 0.76
0.04 0.01 0.03 0.04 0.06 0.07
Plasma 0.390.05 0.390.050.160.010.150.011.470.030.950.11
Tumor 2.99 1.84 1.47 1.65 0.98 I .14
0.34 0.09 0.10 0.23 0.07 0.0I
As the above data clearly demonstrates, the clearance of the C18 analog from
non-
target tissues was much more rapid than from tumor, and the quantities of
radioactivity
detected in the liver. kidney and duodenum were significantly lower following
administration
of the C 18 analog, as compared to the same organs in the C I S and C I 2
analog studies. In
addition. the C 18 analog was retained in the circulation to a much greater
extent than the
other chain length isomers surveyed. As Table I illustrates, the plasma level
for the C-18
analog was significantly higher than the plasma levels for both the C-12 and
the C-1 ~ analog.
Moreover. even at 12.0 hours, blood levels for the C18 analog were 0.60 ~ 0.01
(% Dose/g ~
SEM), as compared to levels of 0.07 ~ 0.01 and 0.22 ~ 0.03 for the C 1 S and C
12 analogs,
- 14-
CA 02276284 1999-06-28
WO 98/24480 PCT/US96/19352
respectively. This effect is illustrated in Figure 6, which shows the blood
clearance profile of
all three analogs up to 120 hours post-injection.
While the rapid decline in radiation levels in non-target tissue was
accompanied by a
much more subtle reduction in the radioactivity present in the tumor, the C18
analog still
retained a beneficial degree of tumor specificity in this tumor model. In
fact, the level of
C18 in the tumor increased significantly over a longer time period, most
likely as a result of
the longer plasma half life.
EXAMPLE 7
Biodistribution Studies With Dunning Prostate Tumor
The three radiolabelled compounds illustrated in Figures 7A-7C, including the
improved C-18 analogs 18-p-iodophenyl)octadecyl phosphocholine {Compound XVI -
Figure
7B) and I-()-(18-(p-iodophenyljoctadecyl)-1,3-propanediol-3-phosphocholine
(Compound XIV
- Figure 7C) described in Examples 2 and 3, respectively, were prepared and
administered to
1 ~ two sets of male Copenhagen rats (Harlan Sprague-Dawley, Indianapolis,
IN). One group of
normal animals was used as a control, and another group was inoculated with
Dunning
prostate tumor cells ( 1 x 1 O6 MAT-LyLu cells) in 0.2 ml saline in the right
hindlirnb. The
MAT-LyLu subline of the Dunning (R-3327) adenocarcinoma prostate tumor was
provided by
Dr. Ken Pienta of the Department of Urology at the University of Michigan, and
is an
accepted cell line representative of effect in humans. See, e.g., Pienta et
al., "Inhibition of
Spontaneous Metastasis in a Rat Prostrate Cancer Model by Oral Administration
of Modified
Citrus Pectin, J. Natl. Cancer Inst. 87:348-53 (I995).
The animals were maintained with free access to food and water until day 8-10.
The
radioiodinated phospholipid ether analog formulated in 2% Tween 20 - sterile
water (5-10
pCi, 0.3-0.4 ml, described above in Example 6) was administered intravenously
into the tail
vein of the tumor-bearing animals under metofane anesthesia. The animals were
euthanized at
selected time points after injection (n=3 per time point) and samples of
blood, plasma and a
variety of tissues including tumor and prostate are collected and analyzed for
radioiodine
content. Biodistribution values are calculated as the mean of the individual %
administered
dose per gram of tissue and % dose per organ values. An illustration of the
data for each
analog is set forth in Table 2 below. These results are also expressed as the
mean
administered dose per gram ~ SEM (% Doselg ~ SEM).
- IS -
CA 02276284 1999-06-28
WO 98/24480 PCT/US96/19352
C-12 Compound Compound
Analog XIV XVI
i
Tissue 24hr 48hr 24hr 48hr 24hr 48hr
Adrenal 0.29 0.23 1.37 1.04 0,88 0.76
0.41 0.01 0.03 0.07 0.02 0.03
Blood 0.23 0.22 t 0.41 0.29 0.88 0.64
0.02 0.01 t 0.01 0.01 0.04 0.05
J Kldnev 4.45 3.38 0.73 0.65 0.59 0.51
0.27 0.43 0.02 0.01 0.02 0.03
Liver 1.270.021.050.03 0.570.020.360.010.570.020.400.03
Lung 0.48 0.33 0.89 0.78 0.84 0.69
0.03 0.02 0.07 0.02 0.01 0.05
Plasma 0.32 0.32 0.58 0.42 1.43 0.99
0.03 0.01 0.01 0.03 0.06 0.11
Tumor 0.37 0.30 0.43 0.39 0.75 0.81
0.03 0.01 0.05 0.01 0.04 0.04
As with the Walker-256 tumor in Example 6, the C-18 analogs again demonstrated
kidney and liver levels several times lower than the C-12 analog in the
prostate tumor model.
Importantly, however, the longer chain analogs also demonstrated a superior
tumor avidity,
with tumor levels of Compound XVI more than twice as high as the shorter chain
analog.
I S Thus. the improved compounds of the present invention may find
particularly advantageous
use in the imaging and/or treatment of prostate tumors.
EXAMPLE 8
Gamma Camera Scintigraphy Studies With Dunning 83327 Prostate Tumor
Comparative in vivo scintigaphic studies were also conducted using the C-12
analog
12-(p-iodophenyl)dodecyl phosphocholine and the C-18 analogs 18-(p-
iodophenyl)octadecyl
phosphocholine (Compound XVI} and 1-D-[18-(p-iodophenyl) octadecyl]-1,3-
propanediol-3-
phosphocholine (Compound XIV) utilized in Example 7. The imaging studies were
performed on Dunning 83327 prostate tumor-bearing animals injected with > 40
pCi of the
radioiodinated phospholipid ether formulations described above. The
anesthetized animals
were imaged at selected times after injection on a gamma camera set to the ''-
'I window and
outfitted with a low energy collimator. Static fifteen-minute images were
obtained for each
animal at the specified time. The results are illustrated in Figures 8A-8C.
- 16-
CA 02276284 1999-06-28
WO 98/24480 PCT/US96/19352
As illustrated in Figures 8A-8C for 18-(p-iodophenyl)octadecyl phosphocholine
(Compound XVI), 12-(p-iodophenyl)dodecyl phosphocholine and 1-O-[18-(p-
iodophenyl)
octadecylJ-1,3-propanediol-3-phosphocholine (Compound XIV), respectively, the
mass
designated as T corresponds to the tumor tissue. Administration of the C-18
compounds
resulted in excellent tumor localization in comparison with the C-12 analog,
wherein the
tumor was barely visible.
In addition to the foregoing specifically mentioned uses of the inventive
compounds,
the compounds of the present invention may find applicability as carrier
molecules for
radiosensitizers. Radiosensitizers are agents administered to sensitize tumor
tissue to the
effects of externally applied radiation. Well known radiosensitizers, such as
misonidazole and
metronidazole are substituted nitroimidazoles. Substitution of an electron-
capturing moiety,
such as nitroimidazole. for the iodophenyl moiety in the phospholipid ether
analogues of the
present invention would permit tumor-localized sensitization for radiation
therapy.
1 ~ 1n yet another proposed use. the phospholipid analogues of the present
invention could
incorporate boron containing substituents for use as boron-neutron activation
therapeutic
agents. These therapeutic agents are administered using the stable isotope of
the electron-
capturing boron. External radiation activates the boron to create tissue
destructive activity.
Although the invention has been described in terms of specific embodiments and
applications, persons skilled in this art can, in light of this teaching,
generate additional
embodiments without exceeding the scope or departing from the spirit of the
claimed
invention. In particular, the methods of synthesis are merely illustrative and
can be modified
by those of skill in the art for the production of various substituted
phospholipid ether
analogues in accordance with the invention. Moreover, other techniques of
radio-tagging the
analogues may be employed. Of course, the invention contemplates any one of
the ortho-,
meta- and para-isomers of iodobenzyl as the iodine-bearing moiety.
In addition, while certain preferred embodiments of the improved phosphilipid
ether
analogs described herein incorporate an 18-carbon alkyl chain into the
compound, both longer
and shorter chains are also contemplated to be within the scope of the present
invention. In
their association with the plasma membrane of cells, the long aliphatic chain
of the
phospholipid ether analogs of the present invention presumably become buried
in the
hydrophobic phosphotipid bilayer. Both NMR and x-ray diffraction studies have
shown that
the thickness of this hydrophobic phase of the lipid bilayer is between 3~ and
40 Angstroms
- 17-
F rB
CA 02276284 1999-06-28
WO 98/24480 PCT/US96/19352
depending on the type of phospholipid making up the membrane. See Yeagle, P.,
Ed., "The
Structure of Biological Membranes," page 345, CRC Press, Boca Raton. FL ( 1991
).
Accordingly, based on the expectation that hydrophobic chains as long as 40
Angstroms can
be expected to intercalate into such Lipid bilayers, the present invention
contemplates that the
extended alkyl chain can be up to 30 carbons in length.
Accordingly, it is to be understood that the drawings and descriptions in this
disclosure
are proffered to facilitate the comprehension of the invention and should not
be construed to
limit the scope thereof. Other improvements and modifications which become
apparent to
persons of ordinary skill in the art only after reading this disclosure, the
drawings and the
following claims are deemed within the spirit and scope of the present
invention.
-18-