Note: Descriptions are shown in the official language in which they were submitted.
CA 02277105 1999-07-06
WO 98/30541 PCTIUS98100142
OF MATRIX METALLOPROTEINASES AND TNFA
C-TERMINAL KETONE HYDROXAMIC ACID INHIBITORS
SECRETION
Technical Field
This invention relates to compounds having activity to inhibit matrix
metalloproteinases and
TNFa secretion, to pharmaceutical compositions comprising these compounds) and
to a medical
method of treatment. More particularly) this invention concerns C-terminal
ketone compounds
which inhibit matrix metalloproteinases and TNFa secretion, pharmaceutical
compositions
comprising these compounds and a method of inhibiting matrix
metalloproteinases and TNFa
secretion.
Background of the Inve_n_r;n.,
The matrix metalloproteinases (MMP's) are a class of extracellular enzymes
including
collagenase, stromelysin, and gelatinase which are believed to be involved in
the tissue destruction
which accompanies a large number of disease states varying from arthritis to
cancer.
Typical connective tissue cells are embedded within an extracellular matrix of
high
molecular weight proteins and glycoproteins. In healthy tissue, there is a
continual and delicateiy-
balanced series of processes which include cell division) matrix synthesis)
and matrix degradation.
In certain pathological conditions, an imbalance of these three processes can
lead to improper
tissue restructuring. For example) in arthritis, joint mobility can be lost
when there is improper
remodelling of load-bearing joint cartilage. In the case of cancer, lack of
coordination of cell
division and the two processes of matrix synthesis and degradation can lead to
conversion of
transformed cells to invasive phenotypes in which increased matrix turnover
permits tumor cells to
penetrate basement membranes surrounding capillaries leading to subsequent
metastasis.
There has been hightened interest in discovering therapeutic agents which bind
to and
inhibit MMP's. The discovery of new therapeutic agents possessing this
activity will lead to new
drugs having a novel mechanism of action for combatting disease states
involving tissue
degenerative processes including, for example, rheumatoid arthritis,
osteoarthritis) osteopeniax
such as osteoporosis, periodontitis, gingivitis) corneal) epidermal or gastric
ulceration, and tumor
growth and metastasis or invasion.
Tumor Necrosis Factor a (TNFa) is a potent proinflammatory mediator which has
been
implicated in inflammatory conditions including arthritis) asthma) septic
shock, non-insulin
dependent diabetes mellitus and inflammatory bowel disease. TNFa is originally
expressed as a
membrane-bound protein of about 26 kD, which is proteolytically cleaved to
release a soluble 17
kD fragment (TNFa processing) which combines with two other secreted TNFa
molecules to
- 35 form a circulating 51 kD homotrimer. Recently, several MMP inhibitors
were found to inhibit
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
2
TNFa processing (see Mohler, et al., Nature) 1994, 370, 218; Gearing, et al.,
Nature, 1994,
370, 555; and McGeehan, et al., Nature, 1994) 370, 558), leading to the
hypothesis that TNFa
processing is caused by an as yet uncharacterized metalloproteinase residing
in the plasma
membrane of cells producing TNFa. Inhibitors of this metalloproteinase would
therefore be
useful as therapeutics to treat disease states involving TNFa secretion.
Transforming growth factor alpha (TGFa) is a potent mitogen which ellicites
its biological
activity by binding to cell surface receptors, in particular epidermal growth
factor (EGF) receptor.
It is known to promote angiogenesis and to stimulate epithelial cell migration
and therefore has
been implicated in a number of malignant disorders such as breast cancer and
ovarian carcinoma.
TGFa is produced by proteolytic cleavage of a 160 amino acid membrane bound
precursor.
Several cleavage sites have been identified including A1a38-Va139, similar to
the cleavage site of
proTNFa (Ala-76-Va177). This common cleavage site suggests that inhibitors of
TNFa
processing may also block the cleavage of proTGFa and therefore would be
therapeutically useful
in diseases mediated by TGFa.
Summarv of the Invention
The present invention provides a novel class of C-terminal ketone inhibitors
of matrix
metalloproteinases and/or TNFa secretion.
In its principle embodiment, the present invention provides a macrocyclic
compound of
formula I
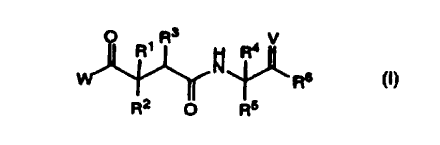
3
O R ~ R H Ra V
N
Rs
R2 O RS
or a pharmaceutically acceptable salt, ester or prodrug thereof wherein
W is NHOH or -OH.
R1 and R4 are independently selected at each occurrence from hydrogen or alkyl
of one to
four carbon atoms.
V is O or NOR ~ .
R2 is selected from the group consisting of
(a) hydrogen,
(b) hydroxy,
(c) alkoxy of one to six carbon atoms,
CA 02277105 1999-07-06
WO 98/30541 PCT/US98I00142
3
(d) alkyl of one to six carbon atoms,
(e) alkyl of one to six carbon atoms substituted with
( I ) halogen,
(2) hydroxy,
(3) alkoxy of one to six carbon atoms,
(4) cycloalkyl of three to eight carbon atoms)
(5) alkanoyloxy wherein the alkyl portion is of one to four carbon atoms,
(6) pyridyl,
(7) pyridyl substituted with alkyl of one to four carbon atoms,
(8) phenoxy wherein the phenyl ring is unsubstituted or substitued with I, 2
or 3 substituents
independently selected from (8a) alkyl of one to four carbon atoms,
(8b)hydroxy, (8c) alkoxy of
one to four carbon atoms, (8d) halogen) (8e) haloalkyl of one to four carbon
atoms, (8f) cyano,
(8g) cyanoallcyl, (8h) -C02R~ wherein R~ is hydrogen or alkyl of one to four
carbon atoms) (8i) -
CONR~R~ wherein R~ is defined above and R~ is selected from hydrogen) alkyl of
one to four
1 S carbon atoms, alkanoyl of one to four carbon atoms, phenyl, and phenyl
substituted with 1, 2, or 3
substutuents independently selected from alkyl of one to four carbon atoms)
hydroxy, alkoxy of
one to four carbon atoms) halogen, haloalkyl of one to four carbon atoms,
cyano, cyanoalkyl) -
CONR'~R ) ~ wherein Ry and R ) ~ are independently selected from hydrogen and
alkyl of one to four
carbon atoms) and -C02R9,
R'
1
~ O
~ 1 O~O
..( (9)
( 10 ) -S(O)AR > > wherein n is 0, 1 or 2 and R > > is selected from ( 1 Ua)
alkyl of one to six carbon
atoms, ( l Ob) phenyl. ( 1 Oc) phenyl substituted with 1, 2 ar 3 substituents
independently selected
from alkyl of one to four carbon atoms) hydroxy) alkoxy of one to four carbon
atoms) halogen)
haloalkyl of one to four carbon atoms, cyano, cyanoalkyl, -C02R~, -COIVR~RH) (
l Od) thienyl,
25 ( l0e) thienyl substituted with alkyl of one to four carbon atoms, ( 1 Uf)
phenylalkyl wherein the
alkyl portion is of one to four carbon atoms, ( 1 Og) phenylalkyl wherein the
alkyl portion is of one
to four carbon atoms, and the phenyl ring is substituted with l ) 2 or 3
substituents independently
selected from alkyl of one to four carbon atoms, hydroxy, alkoxy of one to
four carbon atoms,
halogen, haloalkyl of one to four carbon atoms, cyano) cyanoalkyl) -C02R~, and
-CONR~Rg)
( 30 ( 1 Oh ) thienylalkyl wherein the alkyl portion is of one to four carbon
atoms, and ( 1 Oi ) thienylalkyl
wherein the alkyl portion is of one to four carbon atoms and the thienyl ring
is substituted with
alkyl of one to four carbon atoms, and
(11) -NR12R13 wherein R12 is hydrogen or alkyl of one to four carbon atoms and
R13 is selected
from ( I I a) hydrogen, ( I 1 b) alkyl of one to four carbon atoms, ( 11 c) -
C02R 14 wherein R ~ 4 is
35 independently selected at each occurrence from (i) alkyl of one to four
carbon atoms, (ii) haloalkyl
CA 02277105 1999-07-06
WO 98/30541 PCT/US98I00142
4
of one to four carbon atoms, (iii) phenyl, (iv) phenyl substituted with 1, 2,
or 3 substituents
independently selected from alkyl of one to four carbon atoms, allcoxy of one
to four carbon
atoms, halogen, haloallcyl of one to four carbon atoms, nitro, cyano,
cyanoalkyl, -S02NH2, -
COZR~, and -CONR~Rg, (v) phenylalkyl wherein the alkylene portion is of one to
four carbon
atoms, (vi) phenylalkyI wherein the alkylene portion is of one to four carbon
atoms, and the phenyl
ring is substituted with 1, 2, or 3 substituents independently selected from
alkyl of one to four
carbon atoms, alkoxy of one to four carbon atoms, halogen, haloalkyl of one to
four carbon atoms)
cyano, cyanoalkyl, -S02NH2, -C02R~, and -CONR~Rg, (vii) heteroarylalkyl
wherein the alkylene
portion is of one to four carbon atoms, and the heteroaryl group is selected
from furyl, pyridyl,
thienyl, benzimidazolyl, imidazolyl, thiazolyl, and benzothiazolyl wherein the
heteroaryl group is
unsubstituted or substituted with alkyl of one to four carbon atoms) and ( 11
d) -S02R 14,
or R12 and R13, together with the N atoms to which they are attached define a
heterocycle selected
from morpholinyl) thiomorpholinyl, thiomorpholinyl sulfone, pyrrolidinyl)
piperazinyl,
piperidinyl, succinimidyl, maleimidyl, glutarimidyl, phthalimidyl,
naphthalimidyl,
0 0 0 0
H3C~ N"\ H3C~ N'\ H3C~ N' \ O' \
- H C~N~ ~3C~ - H3C
N i N- N N
0 3 ° ~c o -~cI ~o
, , , ,
0
O ~ ~ O O N/
N ~ ~ N- N_ ' O
/ I
O O H O, and ~H3 '
,
(f) alkenyl of two to six carbon atoms,
(g) alkenyl of two to six carbon atoms substituted with
2(1 ( I ) halogen)
(2) hydroxy)
(3) alkoxy of one to six carbon atoms,
(4) cycloalkyl of three to eight carbon atoms)
(5) alkanoyloxy wherein the alkyl portion is of one to four carbon atoms,
(6) pyridyl)
(7) pyridyl substituted with alkyl of one to four carbon atoms,
(8) phenoxy wherein the phenyl ring is unsubstituted or substitued with 1, 2
or 3 substituents
independently selected from (8a) alkyl of one to four carbon atoms, (8b)
hydroxy, (8c) alkoxy of
one to four carbon atoms) (8d) halogen, (8e) haloallcyl of one to four carbon
atoms, (8f) cyano,
(8g) cyanoalkyl, (8h) -C02R~, (8i) -CONR~Rg, (8j) phenyl, and (8k) phenyl
substituted with 1,
2, or 3 substutuents independently selected from alkyl of one to four carbon
atoms, hydroxy,
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
alkoxy of one to four carbon atoms, halogen, haloalkyl of one to four carbon
atoms, cyano,
cyanoallcyl) -C02R9) and -CONR9R10,
R'
i~ ( .~ O
1
O
(9) ,
( I0) -S(O)nR i i and
5 (11) -NR12Ri3~
R3 is selected from the group consisting of
(a) alkyl of one to ten carbon atoms,
(b) alkenyl of two to ten carbon atoms,
(c) cycloallcyl of three to eight carbon atoms)
(d) (cycloalkyl)alkyl wherein the cycloalkyl portion is of three to eight
carbon atoms, and the
alkylene portion is of one to six carbon atoms,
(e) cycloalkylene of five to eight carbon atoms,
(f) (cycloalkylene)alkyl wherein the cycloalkylene portion is of three to
eight carbon atoms)
and the alklene portion is of one to six carbon atoms)
IS (g) phenyl wherein the phenyl ring is unsubstituted or substituted with 1,
2 or 3 subsdtuents
independently selected from (g 1 ) alkyl of one to four carbon atoms) (g2)
allcoxy of one to four
carbon atoms) (g3) halogen, (g4) haloalkyl of one to four carbon atoms) (g5)
cyano, (g6)
cyanoalkyl) (g7) -C02R~, (g8) -C02NR~Rg, (g9)) alkoxyalkyloxy and (gl0) phenyl
substituted
with 1. 2, or 3 substutuents independently selected from alkyl of one to four
carbon atoms)
2U hydroxy, alkoxy of one to four carbon atoms, halogen) haloalkyl of one to
four carbon atoms,
cyano, cyanoalkyl, -CO~R'~, and -CONR'~R i~)
(h 1 phenylalkyl wherein the alkylene portion is of one to six carbon atoms,
and the phenyl ring
is unsubstituted or substituted with 1, 2 or 3 substituents independently
selected from (h 1 ) alkyl of
one to four carbon atoms, (h2) alkoxy of one to four carbon atoms, (h3)
halogen) (h4) haloalkyl of
~5 one to four carbon atoms, (h5) cyano, (h6) cyanoall:yl) (h7) -C02R~) (h8) -
C02NR~R~, (h9)
phenyl, and (h10) phenyl substituted with 1) 2) or 3 substutuents
independently selected from
alkyl of one to four carbon atoms, alkoxy of one to four carbon atoms,
halogen, haloalkyl of one
to four carbon atoms, cyano, cyanoalkyl, -C02R~ and -C02NR~R8)
(i) -(CH2)m-T-(CH2)~-RiS wherein m and n are independently 0, 1, 2, 3 or 4) T
is O or S,
- 30 and R 15 is selected from the group consisting of (i 1 ) alkyl of one to
four carbon atoms, (i2)
phenyl, and (i3) phenyl substituted with 1, 2) or 3 substituents selected from
(i) alkyl of one to
four carbon atoms) (ii) hydroxy, (iii) allcoxy of one to four carbon atoms)
(iv) halogen) (v)
haloalkyl of one to four carbon atoms, (vi) cyano) (vii) cyanoalkyl, (viii) -
C02R~, (ix) -
CONR~Rg, (x) phenyl, and (xi) phenyl substituted with 1, 2, or 3 substutuents
independently
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
6
selected from alkyl of one to four carbon atoms, hydroxy, alkoxy of one to
four carbon atoms,
halogen, haloalkyl of one to four carbon atoms, cyano, cyanoalkyl) -C02R~, and
-CONR~RR) and
(j) fluorenylalkyl wherein the alkylene portion is of one to four carbon
atoms, and
RS is selected from the group consisting of
(a) alkyl of one to six carbon atoms,
(b) alkyl of one to six carbon atoms substituted with (b 1 ) cycloallcyl of
three to eight carbon
atoms, (b2) hydroxy, (b3) alkoxy, (b4) -SRS) (b5) -NR~Rg, (b6) -C02R~, (b7) -
CONR~R~, (b8)
guanidyl, (b9) phenyl, (b10) phenyl substituted with 1, 2, or 3 substituents
independently selected
from alkyl of one to four carbon atoms, hydroxy, alkoxy of one to four carbon
atoms, halogen,
haloalkyl of one to four carbon atoms, vitro, cyano) cyanoalkyl,
carboxyalkyloxy, -S(O)"R»
wherein n is 0, 1 or 2 and R16 is alkyl of one to four carbon atoms, -SOZN~-
I2, -C02R~, and
CONR~Rg, and (b 11 ) phenyl substituted with 1, 2, or 3 substituents
independently selected from
alkyl of one to four carbon atoms, hydroxy) alkoxy of one to four carbon
atoms, halogen, and
haloalkyl of one to four carbon atoms, (b 10) naphthyl, (b 11 ) naphthyl
substituted with 1, 2, or 3
I S substituents independently selected from alkyl of one to four carbon
atoms, hydroxy, alkoxy of
one to four carbon atoms, halogen) haloalkyl of one to four carbon atoms,
(b12) indolyl,
(b13) indolyl substituted with alkyl of one to four carbon atoms, hydroxy)
alkoxy of one to four
carbon atoms) halogen, haloalkyl of one to four carbon atoms, -S02R1~, -
S02NH2, -C02R~ and -
CONR~RR) (b 14) pyridyl) (b 1 S) pyridyl substituted with alkyl of one to four
carbon atoms, (b 16)
pyrazolyl, (b17) pyrazolyl substituted with alkyl of one to four carbon atoms,
(b18) 5-oxadiazolyl,
(b 19) imidazolyl) and (b-20) imidazolyl substituted with alkyl of one to four
carbon atoms)
(c ) phenyl and
(d) phenyl substituted with 1, 2) or 3 substituents independently selected
from alkyl of one to
four carbon atoms) hydroxy, alkoxy of one to four carbon atoms, halogen, and
haloalkyl of one to
2_5 four carbon atoms.
R~ is selected from the group consisting of
(a) alkyl of one to six carbon atoms,
(b) alkyl of one to six carbon atoms substituted with hydroxy, alkoxy,
halogen, and -C02R
wherein R 1~ is selected from hydrogen, alkyl of one to four carbon atoms and
alkenyl of two to
3(1 four carbon atoms)
(c) phenyl,
(d) phenyl substituted with 1, 2, or 3 substituents selected from (d 1 ) alkyl
of one to four
carbon atoms, (d2) halogen, (d3) hydroxy, (d4) hydroxyalkyl of one to four
carbon atoms,
(d5) haloalkyl of one to four carbon atoms, (d6) allcoxy of one to four carbon
atoms)
35 (d7) cyano, (d8) -NR~Rg, (d9) -S02NR~Rg, (d 10) -S02R ~ 6, (d 11 ) -CH2NR 1
gR 19, wherein R 1 g
and R19 are independently selected at each occurrence from hydrogen and alkyl
of one to four
carbon atoms, or R1g and R19 together with the N atom to which they are
attached define a a 5-or
CA 02277105 1999-07-06
WO 98/30541
7
PCT/US98/00142
6-membered heterocyclic ring selected from morpholinyl, thiomorpholinyl,
thiompholinyl sulfone,
pyrrolidinyl, piperazinyl, 3-ketopiperazinyl and piperidinyl, (d 12) -CONR~Rg,
(d 13) -COZR~)
and (d I 4) phenyl, wherein the phenyl ring may be substituted with 1, 2, or 3
substituents selected
from alkyl of one to four carbon atoms, halogen) and haloalkyl of one to four
carbon atoms,
(e) 1,3-benzodioxole,
{f) indolyl,
(g) indolyl substituted with (gl) alkyl of one to four carbon atoms, (g2)
halogen, (g3)
haloalkyl of one to four carbon atoms) {g4) alkoxy of one to four carbon
atoms) (g5) -S02NR~RR,
(g6) -C02R~) (g7) alkylsulfonyl of one to four carbon atoms, and (g8) phenyl,
wherein the phenyl
ring may be substituted with l, 2, or 3 substituents selected from alkyl of
one to four carbon
atoms, halogen, haloalkyl of one to four carbon atoms) and alkoxy of one to
four carbon atoms,
(h) Pyrrolyl,
(i) pyrroIyl substituted with alkyl of one to four carbon atom
(j) imidazolyl,
I S (k) imidazolyl substituted with alkyl of one to four carbon atoms,
(1) benzimidazolyl)
(m) benzimidazolyl substituted with 1, 2 or 3 substituents independently
selected from alkyl of
one to four carbon atoms) halogen and haloalkyl of one to four carbon atoms)
provided that in (f)-(m) above, when the heterocycle is attached at a carbon
atom, the N atom may
2l1 bear a substituent selected from the group consisting of alkyl of one to
six carbon atoms) -
CONR~RB, -S02NR~Rg and -S02Rt4,
(n) pyridyl,
(o) pyridyl substituted with alkyl of one to four carbon atoms,
(p) thienyl,
='S (q) thienyl substituted with halogen, alkyl of one to four carbon atoms,
and haloalkyl of one to
four carbon atoms,
(r) thiazolyl)
(s) thiazolyl substituted with halogen, alkyl of one to four carbon atoms, and
haloalkyl of one
to four carbon atoms)
30 (t) oxazolyl,
(u) oxazolyl substituted with halogen, alkyl of one to four carbon atoms, and
haloallcyl of one
to four carbon atoms,
(v) furyl,
(w) furyl substituted with halogen, alkyl of one to four carbon atoms, and
haloalkyl of one to
35 four carbon atoms,
(x) benzofuryl,
CA 02277105 1999-07-06
WO 98130541 PCT/US98/00142
8
(y) benzofuryl substituted with 1, 2, or 3 substituents selected from alkyl of
one to four carbon
atoms, halogen, and haloalkyl of one to four carbon atoms,
(z) benzothiazolyl, and
(aa) benzothiazolyl substituted with 1, 2, or 3 substituents selected from
alkyl of one to four
carbon atoms, halogen, and haloallcyl of one to four carbon atoms.
In another aspect, the present invention provides pharmaceutical compositions
which
comprise a therapeutically effective amount of compound of formula I in
combination with a
pharmaceutically acceptable carrier.
In yet another aspect, the present invention provides a method of inhibiting
matrix
metalloproteinases and/or TNFa secretion in a host mammal in need of such
treatment comprising
administering to a mammal in need of such treatment a therapeutically
effective amount of a
compound of formula 1.
Detailed Description
As used throughout this specification and the appended claims, the following
terms have
the meanings specified.
The term alkyl refers to a monovalent group derived from a straight or
branched chain
saturated hydrocarbon by the removal of a single hydrogen atom. Alkyl groups
are exemplified by
methyl, ethyl, n- and iso-propyl, n-, sec-, iso- and tort-butyl) and the Like.
2U The term allcylsulfonyl represents an alkyl group, as defined above,
attached to the parent
molecular group through a S02 group.
The term "alkanoyl" represents an alkyl group, as defined above, attached to
the parent
molecular moiety through a carbonyl group. Alkanoyl groups are exemplified by
fonmyl, acetyl,
propionyl, butanoyl and the like.
The terms allcoxy and alkoxyl denote an alkyl group, as defined above, at
ached to the
parent molecular moiety through an oxygen atom. Representative alkoxy groups
include methoxy,
ethoxy, propoxy, butoxy) and the like.
The term "alkoxycarbonyl" represents an ester group; i.e. an all:oxy group)
attached to the
parent molecular moiety through a carbonyl group such as methoxycarbonyl,
ethoxycarbonyl) and
3() the like.
The term alkenyl as used herein refer to monovalent straight or branched chain
groups of 2
to 6 carbon atoms containing a carbon-carbon double bond, derived from an
alkene by the removal
of one hydrogen atom and include) but are not limited to groups such as
ethenyl, 1-propenyl, 2-
propenyl, 2-methyl-1-propenyl, 1-butenyl, 2-butenyl and the like.
The term alkylene denotes a saturated divalent hydrocarbon group derived from
a straight
or branched chain saturated hydrocarbon containing by the removal of two
hydrogen atoms, for
example -CH2-, -CH2CH2-, -CH(CH3)CH2- and the like.
CA 02277105 1999-07-06
WO 98/30541 PCT/LTS98/00142
9
The term alkenylene denotes a divalent group derived from a straight or
branched chain
hydrocarbon containing at least one carbon-carbon double bond. Examples of
alkenylene include -
CH=CH-, -CH2CH=CH-, -C(CH3)=CH-) -CH2CH=CHCHZ-, and the like.
The terms alkynylene refers to a divalent group derived by the removal of two
hydrogen
atoms from a straight or branched chain acyclic hydrocarbon group containing
at least one carbon-
carbon triple bond. Examples of alkynylene include -CH_--CH-, -CH-_-_-C-CH2-, -
CH-CH-
CH(CHg)- and the like.
The term cycloalkyl as used herein refer to a monovalent saturated cyclic
hydrocarbon
group. Representative cycloalkyl groups include cyclopropyl, cyclobutyl,
cyclopentyl)
1(1 cyclohexyl) cycloheptyl, bicyclo(2.2.I]heptane and the like.
Cycloalkylene denotes a divalent radical derived from a cycloalkane by the
removal of two
hydrogen atoms.
The terms "(cycIoallcyl)alkyl" and "(cycloalkenylene)alkyl" refer,
respectively, to a
cycloalkyl group or cycloalkenylene group as defined above attached to the
parent molecular
1 S moiety through an alkylene group.
The term cyanoalkyl denotes an alkyl group) as defined above, substituted by a
cyano
group and includes, for example, cyanomethyl, cyanoethyl, cyanopropyl and the
like.
The term haloalkyl denotes an alkyl group, as defined above, having one, two)
or three
halogen atoms attached thereto and is exemplified by such groups as
chloromethyl, bromoethyl,
2(I trifluoromethyl, and the like.
The term "hydroxyalkyl" represents an alkyl group) as defined above)
substituted by one to
three hydroxyl groups with the proviso that no more than one hydroxy group may
be attached to a
single carbon atom of the alkyl group.
The term "phenoxy" refers to a phenyl group attached to the parent molecular
moiety
'_'S through an oxygen atom.
By pharmaceutically acceptable salt is meant those salts which are) within the
scope of
sound medical judgement, suitable for use in contact with the tissues of
humans and lower animals
without undue toxicity, irritation, allergic response and the like, and are
commensurate with a
reasonable beneflt/risk ratio. Pharmaceutically acceptable salts are well
known in the art . For
3U example, S. M Berge, et al. describe pharmaceutically acceptable salts in
detail in J.
Pharmaceutical Sciences, 1977, 66:1 - 19 . The salts can be prepared in situ
during the final
isolation and purification of the compounds of the invention, or separately by
reacting the free base
function with a suitable organic acid. Representative acid addition salts
include acetate, adipate,
alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate,
butyrate, camphorate,
35 camphersulfonate, citrate, cyclopentanepropionate, digluconate,
dodecylsulfate, ethanesulfonate,
fumarate, glucoheptonate, glycerophosphate, hemisulfate, heptonate, hexanoate)
hydrobromide,
hydrochloride, hydroiodide) 2-hydroxy-ethanesulfonate, lactobionate, lactate,
laurate, lauryl
CA 02277105 1999-07-06
WO 98130541 PCT/US98/00142
sulfate, malate, maleate, malonate) methanesulfonate, 2-naphthalenesulfonate,
nicotinate, nitrate,
oleate, oxalate) palirutate, pamoate, pectinate, persulfate, 3-
phenylpropionate, phosphate, picrate,
pivalate, propionate, stearate, succinate, sulfate, tarnate, thiocyanate,
toluenesulfonate,
undecanoate, valerate salts, and the like. Representative alkali or alkaline
earth metal salts include
5 sodium, lithium, potassium, calcium, magnesium, and the like, as well as
nontoxic ammonium,
quaternary ammonium, and amine canons) including, but not limited to ammonium,
tetramethylammonium, tetraethylammoruum, methylamine, dimethylamine,
trimethylamine,
triethylamine) ethylamine, and the like.
As used herein, the term "pharmaceutically acceptable ester" refers to esters
which
10 hydrolyze in vivo and include those that break down readily in the human
body to leave the parent
compound or a salt thereof. Suitable ester groups include, for example, those
derived from
pharmaceutically acceptable aliphatic carboxylic acids) particularly alkanoic)
alkenoic,
cycloalkanoic and alkanedioic acids, in which each alkyl or alkenyl moiety
advantageously has not
more than 6 carbon atoms. Examples of particular esters includes formates,
acetates, propionates,
butyates, acrylates and ethylsuccinates.
The term "pharmaceutically acceptable prodrugs" as used herein refers to those
prodrugs of
the compounds of the present invention which are, within the scope of sound
medical judgement,
suitable for use in contact with with the tissues of humans and lower animals
with undue toxicity,
irritation, allergic response, and the like) commensurate with a reasonable
benefitJrisk ratio) and
2f effective for their intended use, as well as the zwitterionic forms, where
possible, of the
compounds of the invention. The term "prodrug" refers to compounds that are
rapidly transformed
in vivo to yield the parent compound of the above formula, for example by
hydrolysis in blood. A
thorough discussion is provided in T. Higuchi and V. Stella) Pro-drugs as
Novel Delivery
S sv terns, Vol. 14 of the A.C.S. Symposium Series, and in Edward B. Roche,
ed., Bioreversible
2_5 Carriers in Drult Desien) American Pharmaceutical Association and Pergamon
Press) 1987) both
of which are incorporated herein by reference.
Asymmetric centers may exist in the compounds of the present invention. The
present
invention contemplates the various stereoisomers and mixtures thereof.
Individual stereoisomers
of compounds of the present invention are made by synthesis from starting
materials containing the
30 chiral centers or by preparation of mixtures of enantiomeric products
follwed by separation as, for
example) by conversion to a mixture of diastereomers followed by separation by
recrystallization
or chromatographic techniques) or by direct separation of the optical
enantiomers on chiral
chromatographic columns. Starting compounds of particular stereochemistry are
either
commercially available or are made by the methods detailed below and resolved
by techniques well
35 known in the organic chemical arts.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
Il
Preferred Embodiments
Preferred compounds of the present invention have formula I wherein R6 is
defined
therein; R1 and R4 are hydrogen; R2 is selected from the group consisting of
(a) hydrogen)
(b) hydroxy,
(c) alkoxy of one to six carbon atoms,
(d) alkyl of one to six carbon atoms,
(e) alkyl of one to six carbon atoms substituted with
R'
~ O
0
(1) ,
1(l (2) -S(O)~RI~ wherein n is 0, 1 or 2 and R11 is selected from (2a) phenyl)
(2b) phenyl substituted
with 1, 2 or 3 substituents independently selected from alkyl of one to four
carbon atoms)
hydroxy, alkoxy of one to four carbon atoms) halogen, haloalkyl of one to four
carbon atoms,
cyano) cyanoalkyl) -C02R~, and -CONR~R~, (2c) thienyl and (2d) thienyl
substituted with alkyl
of one to four carbon atoms and
(3) -NR 12R 1 ~ wherein R 12 and R 13 are independently selected from hydrogen
and alkyl of one to
four carbon atoms or R 1 Z and R 13, together with the N atoms to which they
are attached define a
0
H3C~ N' \
N
heterocycle of formula ~c o , and
(f) alkenyl of two to six carbon atoms: R-~ is selected from the group
consisting of
(a) alkyl of one to ten carbon atoms,
2U (b) cycloalkyl of three to eight carbon atoms, and
(c) phenylalkyl wherein the alkylene portion is of one to six carbon atoms,
and the phenyl ring
is unsubstituted or substituted with 1) 2 or 3 substituents independently
selected from (cl ) alkyl of
one to four carbon atoms, (c2) alkoxy of one to four carbon atoms) (c3)
halogen, (c4) haloalkyl of
one to four carbon atoms) (c5) cyano, (c6) cyanoalkyl) (c7) -C02R~) (c$) -
C02NR~RR) (c9)
phenyl, and (c10) phenyl substituted with 1, 2, or 3 substutuents
independently selected from alkyl
of one to four carbon atoms, alkoxy of one to four carbon atoms, halogen,
haloalkyl of one to four
carbon atoms, cyano, cyanoalkyI, -C02R~ and -C02NR~Rg; and RS is selected from
(a) alkyl of one to six carbon atoms,
(b) alkyl of one to six carbon atoms substituted with (b 1 ) cycloalkyl of
three to eight carbon
atoms, (b2) -C02R~, (b3) -SRS, (b4) phenyl, and (b5) phenyl substituted with
1, 2, or 3
substituents independently selected from alkyl of one to four carbon atoms,
hydroxy, alkoxy of
one to four carbon atoms, halogen, haloalkyl of one to four carbon atoms,
nitro, cyano,
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
12
cyanoallryl, -S(O)nRl6 wherein n is 0, 1 or 2 and R16 is alkyl of one to four
carbon atoms, -
S02NH2, -C02R~, and -CONK?Rg.
More prefenred compounds have the structure immediately above wherein W is -
NHOH
and V is O.
Still more preferred compounds have the structure immediately above wherein R2
is
selected from the group consisting of hydrogen, hydroxy and alkenyl of two to
six carbon atoms;
R3 is selected from the group consisting of isobutyl) cyclohexyl, 3-
phenylpropyl, 3-(4-
toIyl)propyl and biphenyloxy; RS is selected from the group consisting of
alkyl of one to six
carbon atoms, and alkyl of one to six carbon atoms substituted with
cycloallcyl of three to eight
1 O carbon atoms, carboxy, phenyl, and hydroxyphenyl; and R~ is selected from
(a) alkyl of one to six carbon atoms,
(b) alkyl of one to six carbon atoms substituted with -C02R1~,
(c) phenyl,
(d) phenyl substituted with 1, 2, or 3 substituents selected from alkyl of one
to four carbon
atoms, halogen, hydroxy) hydroxyalkyl of one to four carbon atoms) haloalkyl
of one to four
carbon atoms, alkoxy of one to four carbon atoms, -NR~Rg) cyano, -S02NR~RR, -
S02R~6,~-
CH2NR(8R19) -CONR~RA and -C02R~)
(e) indolyl,
(f) indolyl substituted with alkyl of one to four carbon atoms, halogen,
haloalkyl of one to
2U four carbon atoms, alkoxy of one to four carbon atoms and phenyl, wherein
the phenyl ring may
be substituted with 1, 2, or 3 substituents selected from alkyl of one to four
carbon atoms,
halogen, haloallcyl of one to four carbon atoms, and alkoxy of one to four
carbon atoms,
(g) PYrTOIyI.
(h) pyrrolyl substituted with alkyl of one to four carbon atoms,
?_5 (i ) benzimidazolyl,
(j ) benzimidazolyl substituted with 1, 2 or 3 substituents independently
selected from alkyl of
one to four carbon atoms, halogen and haloalkyl of one to four carbon atoms)
provided that in (e)-(j) above, when the heterocycle is attached at a carbon
atom, the N atom may
bear a substituent selected from the group consisting of alkyl of one to six
carbon atoms, -
30 CONR~Rg and -S02NR~Rg,
(k) thienyl,
(1) thienyl substituted with halogen, alkyl of one to four carbon atoms, and
haloalkyl of one to
four carbon atoms,
(m) thiazolyl,
35 (n) thiazolyl substituted with halogen, alkyl of one to four carbon atoms,
and haloalkyl of one
to four carbon atoms,
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/U0142
13
(o) oxazolyl and
(p) oxazolyl substituted with halogen, alkyl of one to four carbon atoms, and
haloalkyl of one
to four carbon atoms.
Still yet more preferred compounds have the structure immediately above
wherein R6 is
selected from the group consisting of
(a) phenyl,
(b) phenyl substituted with l, 2, or 3 substituents selected from alkyl of one
to four carbon
atoms, halogen, hydroxy) hydroxyalkyl of one to four carbon atoms) haloalkyl
of one to four
carbon atoms, alkoxy of one to four carbon atoms) -NR~RB, cyano) -S02NR~Rg, -
S02R 1~ -
1 () CH2NR I gR 19, -CONR~Rg and -C02R~,
(c) indolyl,
(d) indolyl substituted with alkyl of one to four carbon atoms, halogen,
haloalkyl of one to
four carbon atoms, alkoxy of one to four carbon atoms and phenyl, wherein the
phenyl ring may
be substituted with 1, 2, or 3 substituents selected from alkyl of one to four
carbon atoms,
halogen, haloalkyl of one to four carbon atoms, and alkoxy of one to four
carbon atoms,
(e) pyrrolyl)
(f) pyrrolyl substituted with alkyl of one to four carbon atoms)
(g) benzimidazolyl,
(h) benzimidazolyl substituted with 1) 2 or 3 substituents independently
selected from alkyl of
2() one to four carbon atoms) halogen and haloalkyl of one to four carbon
atoms,
provided that in (c)-(h) above, when the heterocycle is attached at a carbon
atom, the N atom may
bear a substituent selected from the group consisting of alkyl of one to six
carbon atoms, -
CONR15R» and -S02NR~SR~6)
(i) thienyl,
(j) thienyl substituted with halogen, alkyl of one to four carbon atoms, and
haloalkyl of one to
four carbon atoms)
(k ) thiazolyl,
(1) thiazolyl substituted with halogen) alkyl of one to four carbon atoms, and
haloalkyl of one
to four carbon atoms)
(m) oxazolyl and
(n) oxazolyl substituted with halogen, alkyl of one to four carbon atoms, and
haloallcyl of one
to four carbon atoms.
The most preferred compounds of this invention have the structure immediately
above
. wherein R6 is selected from the group consisting of phenyl and phenyl
substituted with 1, 2, or 3
substituents selected from alkyl of one to four carbon atoms, halogen,
hydroxy, hydroxyallcyl of
one to four carbon atoms, haloalkyl of one to four carbon atoms, alkoxy of one
to four carbon
atoms, -NR~Rg, cyano, -S02NR~Rg, -S02R~6, -CH2NR~gRl9, -CONR~Rg, -C02R~, and
CA 02277105 1999-07-06
WO 98130541 PCT/US98/00142
14
phenyl, wherein the phenyl ring may be substituted with 1, 2, or 3
substituents selected from alkyl
of one to four carbon atoms, halogen, and haloallcyl of one to four carbon
atoms.
Determination of Stromelvsin Inhibition
The efficacy of the compounds of this invention as matrix metalloproteinase
inhibitors was
determined by measuring the inhibition of stromelysin. The inhibition of
stromelysin by the
compounds of this invention was determined as follows: Recombinant truncated
stromelysin
(human sequence) produced in E. coli was prepared by expression and
purification of the protein
as described by Ye et al., Biochemistry , 1992, 31, 11231- I I 235. The enzyme
was assayed by
its cleavage of the thiopeptide ester substrate Ac-Pro-Leu-Gly-[2-mercapto-4-
methyl-pentanoyl)-
Leu-Gly-OEt described by Weingarten and Feder, Anal. Biochem. . 1985,147) 437-
440 ( 1985),
as a substrate of vertebrate collagenase. The reported conditions were
modified to allow assays to
be carried out in a microtiter plate. Upon hydrolysis of the thioester bond.
the released thiol group
reacts rapidly with 5,5'-dithio-bis(2-nitrobenzoic acid) (DTNB)) producing a
yellow color which is
measured by a microtiter plate reader set at 405 nm. The rates of cleavage of
the substrate by
stromelysin in the presence or absence of inhibitors are measured in a 30 min
assay at ambient
temperature. Solutions of the compounds in DMSO are prepared, and these are
diluted at various
concentrations into the assay buffer (50 mM MES/NaOH pH 6.5 with 10 mM CaCl2
and 0.2%
Pluronic F-68), which is also used for dilution of the enzyme and substrate.
The potency of the
2~ compounds [ICSp] are calculated from the inhibition/inhibitor concentration
data. The compounds
of this invention inhibit stromelysin as shown by the data for representative
examples in Table 1.
Table 1
Inhibitory Potencies against Stromelysin of Representative Compounds
Exam le ICS (nM)
1 36
1 G 2.3
1 H 88
3.6
5.6
6 12
7 8.0
8 8.6
9 1.2
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
10 7100
~
_
11 93000
12 7.8
13 99
14 1.2
15 27
16 36
17 16
18 4.5
19 1.5
2() 7.3
21 1.7
22 10
23 6.6
24 3.2
1.8
26 220
27 2.7
28 320
320
3 740
I
33 110
Pharmaceutical Compositions
The present invention also provides pharmaceutical compositions which comprise
compounds of the present invention formulated together with one or more non-
toxic
5 pharmaceutically acceptable carriers. The pharmaceutical compositions may be
specially
formulated for oral administration in solid or liquid form, for parenteral
injection, or for rectal
administration.
The pharmaceutical compositions of this invention can be administered to
humans and
other animals orally, rectally, parenterally , intracisternally,
intravaginally, intraperitoneally)
10 topically (as by powders, ointments, or drops), bucally, or as an oral or
nasal spray. The term
"parenteral" administration as used herein refers to modes of administration
which include
CA 02277105 1999-07-06
WO 98/30541 PCT/LTS98/00142
16
intravenous, intramuscular) intraperitoneal, intrasternal, subcutaneous and
intraarticular injection
and infusion.
Pharmaceutical compositions of this invention for parenteral injection
comprise
pharmaceutically acceptable sterile aqueous or nonaqueous solutions,
dispersions, suspensions or
emulsions as well as sterile powders for reconstitution into sterile
injectabIe solutions or
dispersions just prior to use. Examples of suitable aqueous and nonaqueous
carriers, diluents,
solvents or vehicles include water) ethanol, polyols (such as glycerol,
propylene glycol,
polyethylene glycol, and the like), and suitable mixtures thereof) vegetable
oils (such as olive oil),
and injectable organic esters such as ethyl oleate. Proper fluidity can be
maintained> for example)
by the use of coating materials such as lecithin, by the maintenance of the
required particle size in
the case of dispersions, and by the use of surfactants.
These compositions may also contain adjuvants such as preservative, wetting
agents)
emulsifying agents, and dispersing agents. Prevention of the action of
microorganisms may be
ensured by the inclusion of various antibacterial and antifungal agents, for
example, paraben,
chlorobutanol, phenol sorbic acid, and the like. It may also be desirable to
include isotonic agents
such as sugars, sodium chloride, and the like, Prolonged absorption of the
injectabie
pharmaceutical form may be brought about by the inclusion of agents which
delay absorption such
as aluminum monostearate and gelatin.
in some cases, in order to prolong the effect of the drug, it is desirable to
slow the
2() absorption of the drug from subcutaneous or intramuscular injection. This
may be accomplished
by the use of a liquid suspension of crystalline or amorphous material with
poor water solubilit)~.
The rate of absorption of the drug then depends upon its rate of dissolution
which, in turn, may
depend upon crystal size and crystalline form. Alternatively, delayed
absorption of a parenterally
administered drug form is accomplished by dissolving or suspending the drug in
an oil vehicle.
Injectable depot forms are made by forming microencapsule matrices of the drug
in
biodegradable polymers such as polylactide-polyglycolide. Depending upon the
ratio of drug to
polymer and the nature of the particular polymer employed, the rate of drug
release can be
controlled. Examples of other biodegradable polymers include poly(orthoesters)
and
poly(anhydrides) Depot injectable formulations are also prepared by entrapping
the drug in
Iiposomes or microemulsions which are compatible with body tissues.
The injectable fotrnulations can be sterilized) for example, by filtration
through a bacterial-
retaining filter, or by incorporating sterilizing agents in the form of
sterile solid compositions
which can be dissolved or dispersed in sterile water or other sterile
injectable medium just prior to
use.
Solid dosage forms for oral administration include capsules, tablets, pills,
powders, and
granules. In such solid dosage forms, the active compound is mixed with at
least one inert,
pharmaceutically acceptable excipient or carrier such as sodium citrate or
dicalcium phosphate
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
17
and/or a) fillers or extenders such as starches, lactose, sucrose) glucose,
mannitol, and silicic acid)
b) binders such as, for example, carboxymethylcellulose, alginates, gelatin)
polyvinylpyrrolidone,
sucrose, and acacia, c) humectants such as glycerol) d) disintegrating agents
such as agar-agar,
calcium carbonate, potato or tapioca starch, alginic acid, certain silicates,
and sodium carbonate, e)
solution retarding agents such as paraffin, f) absorption accelerators such as
quaternary ammonium
compounds, g) wetting agents such as, for example) cetyl alcohol and glycerol
monostearate, h)
absorbents such as kaolin and bentonite clay) and i) lubricants such as talc,
calcium stearate)
magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate, and
mixtwes thereof. In
the case of capsules, tablets and pills, the dosage form may also comprise
buffering agents.
Solid compositions of a similar type may also be employed as fillers in soft
and hard-filled
gelatin capsules using such excipients as lactose or milk sugar as well as
high molecular weight
polyethylene glycols and the like.
The solid dosage forms of tablets, dragees, capsules, pills, and granules can
be prepared
with coatings and shells such as enteric coatings and other coatings well
known in the
pharmaceutical formulating art. They may optionally contain opacifying agents
and can also be of
a composition that they release the active ingredients) only) or
preferentially, in a certain part of
the intestinal tract, optionally, in a delayed manner. Examples of embedding
compositions which
can be used include polymeric substances and waxes.
The active compounds can also be in micro-encapsulated form, if appropriate)
with one or
2(1 more of the above-mentioned excipients.
Liquid dosage forms far oral administration include pharmaceutically
acceptable emulsions,
solutions) suspensions, syrups and elixirs. In addition to the active
compounds, the liquid dosage
forms may contain inert diluents commonly used in the art such as, for
example, water or other
solvents, solubilizing agents and emulsifiers such as ethyl alcohol, isopropyl
alcohol, ethyl
carbonate, ethyl acetate, benzyl alcohol) benzyl benzoate, propylene glycol,
1,3-butylene glycol,
dimethyl formamide) oils (in particular, cottonseed, groundnut, corn) germ)
olive) castor, and
sesame oils)) glycerol) tetrahydrofurfuryl alcohol, polyethylene glycols and
fatty acid esters of
sorbitan, and mixtures thereof.
Besides inert diluents, the oral compositions can also include adjuvants such
as wetting
agents, emulsifying and suspending agents) sweetening, flavoring) and
perfuming agents.
Suspensions, in addition to the active compounds, may contain suspending
agents as) for
example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and
sorbitan esters,
microcrystalline cellulose, aluminum metahydroxide, bentonite) agar-agar, and
tragacanth, and
mixtwes thereof.
Compositions for rectal or vaginal administration are preferably suppositories
which can be
prepared by mixing the compounds of this invention with suitable non-
irritating excipients or
carriers such as cocoa butter, polyethylene glycol or a suppository wax which
are solid at room
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
IS
temperature but liquid at body temperature and therefore melt in the rectum or
vaginal cavity and
release the active compound.
Compounds of the present invention can also be administered in the form of
liposomes.
As is known in the art, liposomes are generally derived from phospholipids or
other lipid
substances. Liposomes are formed by mono- or mufti-lamellar hydrated liquid
crystals that are
dispersed in an aqueous medium. Any non-toxic, physiologically acceptable and
metabolizable
lipid capable of forming liposomes can be used. The present compositions in
liposome form can
contain, in addition to a compound of the present invention, stabilizers)
preservatives, excipients)
and the like. The preferred lipids are the phospholipids and the phosphatidyl
cholines (lecithins),
both natural and synthetic.
Methods to form liposomes are known in the art. See, for example) Prescott,
Ed.,
Methods in Cell Biolo~v) Volume XIV, Academic Press, New York, N.Y. (1976), p.
33 et seq.
Dosage forms for topical administration of a compound of this invention
include powders)
sprays, ointments and inhalants. The active compound is mixed under sterile
conditions with a
I S pharmaceutically acceptable carrier and any needed preservatives) buffers)
or propellants which
may be required. Opthalmic formulations) eye ointments) powders and solutions
are also
contemplated as being within the scope of this invention.
Actual dosage levels of active ingredients in the pharmaceutical compositions
of this
invention may be varied so as to obtain an amount of the active compounds)
that is effective to
2() achieve the desired therapeutic response for a particular patient,
compositions, and mode of
administration. The selected dosage level will depend upon the activity of the
particular
compound, the route of administration, the severity of the condition being
treated, and the
condition and prior medical history of the patient being treated. However, it
is within the skill of
the art to start doses of the compound at levels lower than required for to
achieve the desired
2S therapeutic effect and to gradually increase the dosage until the desired
effect is achieved.
Generally dosage levels of about 1 to about 50) more preferably of about 5 to
about 20 mg
of active compound per kilogram of body weight per day are administered orally
to a mammalian
patient. If desired, the effective daily dose may be divided into multiple
doses for purposes of
administration, e.g. two to four separate doses per day.
Preparation of Compounds of this Invention
The compounds of this invention may be prepared by a variety of synthetic
routes.
Representative procedures are outlined in the following Schemes I-3.
Abbreviations which have been used in the descriptions of the schemes and the
examples
that follow are: THF for tetrahydrofuran; DMF for N,N-dimethylformamide; ETOAc
for ethyl
acetate; Et20 for diethyl ether, IPA for isopropanol; ETOH for ethanol; MeOH
for methanol; AcOH
for acetic acid; HOBT for 1-hydroxybenzotriazole hydrdate; EDC for 1-(3-
dimethylaminopropyl)-
CA 02277105 1999-07-06
WO 98/30541 PCT/US98100142
19
3-ethylcarbodiimide hydrochloride; NMM for N-methylmorpholine; Bu3P for
tributylphosphine;
ADDP for 1,1'-(azodicarbonyl)dipiperidine; and DMPU for 1,3-dimethyl-3,4,5,6-
tetrahydro-
2( 1H)-pyrimidinone.
The preparation of representative compounds of the invention, wherein Ri-R6
and W are
defined above, is outlined in Scheme 1. Coupling of succinic acid derivative 1
with keto amine _2
in the presence of an tertiary amine base) hydroxybenzotriazole (HOBt), and a
suitable coupling
agent such as 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
(EDCI~HCl) gives 3.
Conversion of 3 to the corresponding carboxylic acid 4 is accomplished by
acidic removal of the
tert-butyl ester using, for example, trifluoroacetic acid or hydrogen chloride
in dioxane. Treatment
of this acid with hydroxylamine or a hydroxylamine equivalent such as O-tert-
butyldimethylsilylhydroxylamine in the presence of a suitable coupling agent
such as EDCI~HCl
gives hydroxamate 5. O-Benzylhydroxylamine can also be employed in this
coupling reaction.
The resulting0-benzylhydroxamate can then be treated with hydrogen and a
palladium catalyst such
I S as 10% palladium on carbon to produce hydroxamate 5.
Scheme 1
3
O R R H R O
O R~ R3 R40 ~ a
t-BuO~~OH + H2N~R6 t-BuO~~~N~Rs
R2 O R5 ~ R2 O R5
3_
3
O R' R N R40 4: W = -OH
Rs
R2 O R5 ~~ W = -NHOH
2U
Preparation of keto amine 2 is accomplished as shown in Scheme 2. Conversion
of the
protected amino acid 6_ to the methyl ester or N,O-dimethylamide is
accomplished by known
methods. Reaction of 7 with R6MgX wherein X is Br) Cl or I, or R6Li generates
ketone 8_. Acidic
removal of the tert-butyl protecting groups gives amino ketone 2.
Alternatively, 6 can be treated
25 with a carbon anion such as phenyllithium which gives 8 directly.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
Scheme 2
O 40
t-BuOuN R4 t-BuO~N
~O H I I T P _
O R5 O RS
$ Z
P = -OCH3 or -NCH30CH3
4~
t-BuO~N~Rs H2N R4 s
' -R
O R5 R5
$ 2
The preparation of the succiruc acid derivative _l is shown in Scheme 3.
Treatment of
5 oxazolidinone ~ with a suitable base such as lithium diisopropylamide
followed by addition of tert-
butyl bromoacetate and basic hydrolysis gives carboxylic acid 1_Q. This acid
is treated with at least
two equivalents of a strong base such as lithium diisopropylamide followed by
an alkylating agent
R2X wherein X is Br, Cl or I. The resulting dialkyl succinate 11 is again
treated with a strong
base such as lithium diisopropylamide followed by either methanol (R t = H) or
an alkyl halide (R I
1 (1 = alkyl) such as methyl iodide to give substituted succinate _1.
Scheme 3
R
R3 N O R3 O H O R3 O ~ R3
t-BuO~ -" t-Bu0~0 H -''t-gu0~0 H
O O R2 O R2 O
Ph
$ ~ t~ t
I5 The foregoing may be better understood by reference to the following
examples which are
presented for illustration and are not intended to Iimit the scope of the
invention as defined in the
appended claims.
CA 02277105 1999-07-06
WO 98/30541 PCTILTS98/00142
21
ation of Succinate E ter I
0
t-Bu0 OH
O
1_
Step 1
O O
OH SOCK
CI
t
A mixture of 4-methylvaleric acid (50.7 g) 0.43 mmol) and thionyl chloride (40
mL, 65.2
g, 0.54 mole) was stirred at ambient temperature for 18 hours. The mixture was
heated to distill
the excess reagent through a 10 cm Vigreux column. The acid chloride was then
distilled to give i
(48.43 g, 84 %), by 135-138 °C.
Step 2:
0 0 0 0
NHVO + n-BuLi + ~ CI --~.. ~ ~ p
! L ~"'/ _
I 5 To a -78 °C solution of 4S-benzyl-2-oxazoIidinone (62.2 g) U.35
mole) in THF (600 mL)
was addedn-butyllithi um ( 140 mL, 2.5 M in hexane j over 1 hour. After 30
minutes i (0.359 mole )
was added over 10 minutes during which time the temperature rose to -60
°C. After 1 hour the
bath was removed and the reaction mixture was warmed to 0 °C. The
reaction was quenched with
saturated ammonium chloride) the mixture was allowed to settle) and the
supernatant was decanted
and concentrated. The combined residues were partitioned between water and
ethyl acetate. The
organic layer was washed with water, 1 M sodium bicarbonate) water and brine)
dried over sodium
sulfate) filtered and concentrated. .The residue was distilled discarding a
small forerun to give Z
(92.9 g) 96%), by 154-156 °C / 0.15 mm.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
22
Step 3:
0 o
0 0
° + NaN(TMS )2 + °'' .-~ ~ °
B~~ °-t Bu t-Bu0 U
\ / ~ o
\ /
To a mechanically-stirred -78 °C solution of ii (92.9 g, 0.337 mole) in
THF ( 1 L) was
added sodium bis(trimethylsilyl)amide (375 mL, I M in THF) over 40 minutes.
The reaction
mixture was stirred for 30 minutes and t-butyl bromoacetate (55 mL, 72.6 g,
0.372 mole)was
added over 30 minutes. The reaction mixture was stirred for 30 minutes and
then the cold bath
was removed and the mixture was warmed to 0 °C. The reaction was
quenched with saturated
ammonium chloride. After mixing well, the mixture was allowed to settle and
the supernatant was
1 () decanted, concentrated, and recombined with the residue. This mixture was
partitioned between
water and ethyl acetate. The organic layer was washed with water, 1 M sodium
bicarbonate, water
and brine) dried over sodium sulfate and concentrated by distillation to about
250 mL. After
dilution with 750 mL hexane and cooling in an ice bath the resulting crystals
were collected and
washed with hexane to provide iii ( 104.6 g) mp 101-102 °C. The mother
liquors were
1 S concentrated and the residue was purified by chromatography on silica gel
(S - 10°lo ethyl acetate
hexane) and the product fraction crystallized to yield 7.6 g more for a total
of 112.2 g (85 %).
Step 4:
°
° °
t-Bu0 ° + LiOH + H~O~ -~ ° + N LJ
a t-B~o °"
i~ ° iv ° \ /
\ /
To a 0 °C solution of iii ( I I2.2 g, ().288 mole) in THF ( 1.2 L) was
added water ( 100 mL)
and 30 % hydrogen peroxide ( 110 mL, 36.6 g, 1.08 mole). A solution of lithium
hydroxide
monohydrate ( 17.8 g, 0.424 mole) in water (400 mL) was added in portions over
25 minutes and
the resulting solution was stirred for 1 hour. The mixture was concentrated
under a slow nitrogen
stream to about 800 mL. After seeding with the chiral oxazolidinone the
mixture was chilled and
filtered removing a portion of the auxiliary which was washed well with water.
The fil~ate was
extracted with dichloromethane (3x) to remove the balance of the chiral
oxazolidinone. The
combined organic extracts were washed with aqueous 0.5 N sodium hydroxide. The
base layers
were acidified with 1 M sulfuric acid to pH 3 and extracted with ethyl
acetate. After washing with
CA 02277105 1999-07-06
WO 98/30541 PCT/US98100142
23
water and brine) drying over sodium sulfate, and evaporation of solvents the
residue amounted to
64.9 g (98%) of R-2-(i-butyl)-succinic acid-4-t-butyl ester.
Ste~S
0 0
t-Bu0 OH + LDA + wl ~ OH
t-Bu0
iv O O
/
To a -78 °C solution of lithium diisopropylamide) prepared by the
addition of n-
butyllithium ( I 1.4 ml, 28.4 mmol, 2.SM in hexanes) to a solution of
diisopropylamine (3.7 ml,
28.4 mmol) in 60 ml THF at -78 °C, was added a solution of iv (2.7 g,
11.8 mmol) in THF (20
mL) at -78 °C by cannula in a stream. The resulting clear, yellow
solution was stirred at -78 °C for
I hour and then butenyl iodide (2.58 g, 14.2 mmol) was added by syringe. This
mixture was
allowed to warm to ambient temperature and stir overnight. The reaction
mixture was poured into
l :l ether-water and the separated aqueous layer was extracted with ether
(2x). The combined
organic layers were washed with aq 1 M NaHS04 and brine, dried with MgSOa,
filtered and
concentrated. Flash chromatography (2%-5% isopropanol-hexane) gave epimeric
succinates _v
(2.30 g, >9: I Syn/anti) as a clear liquid.
t 6
0 0
t-Bu0 OH ~) LDA OH
2) MeOH t-Bu0
O .O
_v ~ v~
2()
To a -78 °C solution of lithium diisopropylamide, prepared by the
addition of n-
butyllithium (7.8 ml, 19.5 mmol, 2.SM in hexanes) to a solution of
diisopropylamine (2.6 ml)
19.5 mmol) in 30 ml THF at -78 °C) was added a solution of epimeric
isobutyl succinate _v (2.3 g,
8. I mmol) in THF ( 10 mL) at -78 °C by cannula in a stream. The
resulting clear, yellow solution
was stirred at -78 °C for I hour, warmed to 0 °C and recooled to
-78 °C. Methanol (1 ml) was
added and the solution was warmed to 0 °C. The reaction mixture was
poured into 1:1 ether-water
. and the separated aqueous layer was extracted with ether (2x). The combined
organic layers were
washed with aq 1 M NaHS04 and brine, dried with MgS04, filtered and
concentrated to give an
epimeric mixture (2:1 anti/syn) of succinates vi which could be separated by
flash chromatography
(10-50% ethyl acetate-hexanes). .
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
24
Prev~tiQn of~uccinate Ester 2
0
t-Bu0 OH
~ O
The desired compound was prepared according to the method used to prepare
succinate
ester 1) except substituting allyl bromide for 4-bromo-1-butene.
Preparation of Succinate Ester
O
t-Bu0 OH
O
The desired compound was prepared according to the method used to prepare
succinate
ester 1, except substituting 5-bromo-1-pentene for 4-bromo-1-butene.
Preparation of Succinate Ester 4
/ CH3
O
t-Bu0 OH
O
4
SteR 1
Br O O
H C~ + ~ OH ~ ( OH
H3(~, Vil
mizture of isomers
A mixture under nitrogen of 4-bromotolucne (36.9 mL, 51.3 g, 0.3 mole), 4-
pentenoic
acid (30.6 mL, 30.0 g, 0.3 mole), acetonitrile (500 mL)) >Tiethylamine ( 126
mL, 91.5 g, 0.90
mole), palladium acetate ( 1.35 g, 6 mmole) and tri-(o-tolyl)phosphine (4.65
g, 15 mmole) was
heated slowly to a gentle reflux. (A mild exotherm was observed as reflux
begins.) After 18
hours at reflux, the mixture was cooled in an ice bath and the solid was
removed by filtration and
rinsed well with ethyl acetate. The filtrate was concentrated to a small
volume and the residue was
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
partitioned between aqueous I M sodium carbonate and ether. The aqueous phase
was extracted
with ether. The combined ether layers were extracted with aqueous 1 M sodium
carbonate. The
basic solution was treated with charcoal and filtered. The filtrate was
acidified with 3 M
hydrochloric acid. After cooling in an ice bath, the soft solid was filtered)
washed with ice water,
5 and dried over sodium hydroxide to give vii (45 g) as a mixture of isomers
which was used
without further purification.
SteR 2
O O
OH I ~ OH
H3C ~ vii H3C
The mixtureof isomers vii was hydrogenated in 600 mL TIC over 9 g of 10%
palladium
on carbon at 4 atmospheres of hydrogen for 18 hours. After filtration and
concentration of the
solution, the residue was crystallized from hexane to yield 5-(4-
tolyl)pentanoic acid viii, 33 g) mp
77-78 °C).
Step 3
O o
OH ~ ~ cl
HaC ~ ~' H3C ~ 1.~
A mixture of ~0ø ( 11.02 g) 57 mmole) and l2 mL thionyl chloride was stirred
at 24 °C for
2(~ 18 hours and then heated to distill most of the excess thionyl chloride.
Short path distillation gave
1 1.74 g (97 °lo) of 5-(4-tolyl)pentanoyl chloride (~, by - 110
°C at 0.35 mm).
Step 4
o O O o
N VO + n-BuLi + I ~ CI -----~-
H3C'v j~ H3C~ x V
To a -78 °C solution of 4S-benzyl-2-oxazolidinone ( 10.36 g, 58 mmole)
in THF ( 1 SO mL)
was added ra-butyllithium (23.5 mL 2.5 M) over 25 minutes. After 30 minutes, ~
(55.7 mmole)
was added quickly, during which time the reaction temperature rose to -45
°C. The reaction
mixture was warmed to 0 °C and the reaction was quenched with saturated
aqueous ammonium
chloride. The mixture was allowed to settle and the supernatant was decanted
and concentrated.
CA 02277105 1999-07-06
WO 98130541 PCT/US98/00142
26
The residue was partitioned between water and ethyl acetate. The organic layer
was washed with
water, aqueous 1 M sodium bicarbonate, water and brine. After drying over
sodium sulfate the
solution was concentrated and the residue was chromatographed ( 10-20 % ethyl
acetate-hexane) to
give ,~ ( 17.83 g, 89%).
Sten 5
/ CH3
O O
O
v v -N O
H ~ I / ~ ~--~ _ t-Bu0 OH
O
4
The desired compound was prepared using Step 4 of the preparation of succinate
ester 1,
except substituting x_ for "~i.
Preparation of Succinate E ter 5
/ CH3
O
t-Bu0 OH
O
S
The desired compound was prepared using steps 5 and 6 of the preparation of
succinate
ester 1, except substituting succinate ester 4 for iv.
Preparation of Succinate Ester 6
O F F
t-Bu0
O / F
F
To a cold (0°) solution of succinate ester 2_ (0.79g, 3mmol) in 10 mL
methylene chloride
was added pentaflurophenol (0.65g, 3.Smmo1) and EDCI (0.698, 3.Smmol). The
resulting
solution was stirred for 16 hours while warming to ambient temperature. The
reaction mixture was
Quenched with 2N Na2C03. The organic layer was washed with 2N HCl and brine,
dried (sodium
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
27
sulfate) and concentrated to give succinate ester _8 (0.8 g) as a crude yellow
oil, which was used
without further purification.
Preparation of succinate E ter 7
O
t_Bu.O H
7
The desired compound was prepared using steps 1-4 of the preparation of
succinate ester 1,
except substituting 4- -pentenoic acid for 4-methyl valeric acid in step 1.
Preparation of succinate E ter 8
8
The desired compound was prepared from the succinate ester 7 using the Suzuki
coupling
conditions described in Example 41B.
CA 02277105 1999-07-06
WO 98/30541 PCT/ITS98/00142
28
Preparation of succinate Ester 9
II y
The desired compound was prepared from succinate ester _8) using step 5 of the
preparation
of succinate ester I , except substituting allyl iodide for butenyl iodide.
Preparation of succinate Ester 10
O
O
t-Bu.O OH
O I ()
The desired compound was prepared using steps 1-4 of the preparation of
succinate ester 1, except
substituting 6-benzyloxyhexanoic acid for 4-methyl valeric acid in step I.
Preparation of succinate ester 1 I
f
OH
O 11
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
29
~telL
OH OH
(H3Cy~S
xi
S Prepared as described for 4-(trimethylsilyl)-3-butyn-1-of in Organic
Syntheses 1993,
Volume VIII, p. 609.
IH NMR (300 MHz, CDCl3) 8 3.68 (t, 2H)) 2.27 (t, 2H), 1.68-1.62 (m, 4H), 0.14
(s, 9H).
Ste~2
OH OH
(H3C
~3$~ (HaC)aSl
O
X1V
Prepared as described in Tetrahedron Letters 1979, p. 399.
1H NMR (300 MHz, CDCl3) 8 2.50 (t, 2H)) 2.32 (t) 2H), 1.84 (t, 2H)) 0.14 (s,
9H).
st
O
OH
O
O
The desired compound was prepared using steps 1-4 of the preparation of
succinate ester 1, except
substituting 6-(trimethylsilyl)-5-hexynoic acid for 4-methyl valeric acid in
step 1.
~ H NMR (300 MHz) CDC13 ) 8 3.03-2.94 (m, 1 H)) 2.64 (dd, 1 H), 2.47 (dd, 1
H)) 2.31 (td) 2H))
2(t 2.00 (t) 1H), 1.99-1.90 (m) 1H), 1.81-1.69 (m, IH), 1.45 (s) 9H).
MS (DCI/NH3) m/e 227 (M+I )+.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
Exam In a 1
O
H
HOHN N
O
E~lhe l A
O
MeO~N
II OH
O
5
To a 0° C solution of L-phenylalanine (25 g) 151 mmol) in aqueous 1 N
NaOH ( I 75 mL)
was added methyl chloroformate ( I S g, 159 mmol ) via syringe over several
minutes. The pH was
adjusted to 14 with 1 N NaOH and the resulting clear solution was stirred for
1 hour. The basic
1 () solution was extracted with ether (3x) and the organics were discarded.
The pH was adjusted to 3
with a cold phosphoric acid (~ 1 N) and the acidic solution was extracted with
methylene chloride
(3x). The combined organics were washed with brine, dried (Na?S04) and
concentrated in vacuo
to give 1a (32g) as an extremely viscous oil which was carried on without
further purification.
15 Example 1 B
H O H O i
MeO~N~OH MeO~N
'' II \ T
O O
/ ~ ~ /
To a 0 °C solution of la (4.9 g, 21 mmol) in anhydrous diethyl ether
(250- mL) was added
PCIS (5.25g) 25.Smmo1) over several minutes. The resulting suspension was
allowed to stir for 1
20 hour during which time it slowly became a pale yellow solution. Solvent was
removed in vacuo
and the resulting acid chloride was dried under high vacuum for 1 hour. The
crude acid chloride
was then dissolved in methylene chloride (250 mL)) cooled to 0 °C, and
indole (2.9 g, 25.2 mmol)
was added over 10 minutes. AlCl3 (S.Sg, SOmmol) was then added over a period
of 5 minutes,
during which time the solution became a blood-red color, and the reaction
mixture was allowed to
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
31
warm to ambient temperature and stir for 16 hours. The reaction mixture was
poured into cold
water and extracted with methylene chloride. The combined organics were washed
with brine,
dried (Na2S04) and concentrated in vacuo to give 7.Sg of a crude red solid.
Flash
chromatography (hexane-ethyl acetate) gradient elution 3:1 to 1:1 ) gave 2.6 g
of a product 1 b
containing ~60% of the desired acylation product which was carried on without
further
purification.
Example 1
O
MeO~N ~ ~ H2
O 1 NT
H
lU
To a solution of 1 b (2.4 g) in 3:1 MeOH/water (40 mL) was added KOH (2.1 g,
37.3
mmol). The resulting solution was heated at reflux for 18 hours) cooled and
acidifed with 1 N
phosphoric acid. The acidic aqueous Iayer was extracted with ethyl acetate
(3x) and the organic
layer discarded. The aqueous layer was made basic with aqueous 3N NaOH
solution and extracted
with methylene chloride (3x). The combined organics were washed with brine)
dried (Na2S04)
and concentrated in vacuo to give the desired compound i cc (482mg) as a
racemic mixture.
Example 1 D
O F F O
O ~ H2 \ J O H O
t-Bu0 ~ / F + ~ N ~ t-6u0 N \ /
O H O
f / \ ~ H
1~ /
2()
To a solution of ~ (0.48 g, 1.8 mmol) in DMF was added succinate ester f (0.8
g, 1.8
mmol). The reaction was allowed to stir at ambient temperature for 16 hours)
then was warmed to
45 °C for 3 days. The reaction mixture was diluted with ethyl acetate
and the organic layer washed
with 1 N NaOH, water (4x), dried (Na2S04), filtered and concentrated to give 1
g of a tan foam.
Flash chromatography (hexane-ethyl acetate 5:1 ) gave 1 d (510 mg) as a 1:1
mixture of epimers at
the Phe center.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
32
Example 1 E
O H O
t-Bu0 HO
O L. N
H
Ester 1 d (0.5 g, 1.0 mmoi) was dissolved in cold (0°)- TFA and stirred
for 5 hours while
warming to ambient temperature. Solvent was removed under a stream of nitrogen
and the residue
was azeotroped with methylene chloride and dried on high vacuum for 16 hours
to give 1 a (250
mg) as a 1:1 mixture of epimers at the Phe center.
Example 1 F
H
To a cold (0°) solution of le (0.25g, 0.54mmo1) in DMF was added- NMM
(0.08 g, 0.81
mmol, 0.09 mL), HOBT (0.08 g, 0.59 mmol) and EDCI (0.11 g) 0.59 mmol). The
resulting
solution was stirred for 5 minutes and tertr-butyltrimethylsilylhydroxylamine
(0.09 g) 0.59 mmol )
was added in one portion. The resulting solution was warmed to ambient
temperature and allowed
to stand for 97 hours. The reaction mixture was diluted with EtOAc, washed
with water (3x ) and
brine. dried (Na2S04), filtered and concentrated in vacuo . Flash
chromatography ( 1-3~7c
methanol-methylene chloride) gave the desired compound (80mg) as a tan solid
which was a 1:1
mixture of epimers at the Phe center. mp 18()-210° (dec). ~H NMR (3(>n
MHz) DMSO-d6) 8
2U 12.02 (s, 1H)) I().4 (s) 1H), 8.64-8.17 (m, 3H), 8.22-8.17 (m, 2H), 7.48-
7.13 (m, 7H), 5.42-
5.39 (m, 2H), 4.82-4.52 (m) 3H), 3.12-30.7 (m, 2H), 2.97-2.91 (m) 2H), 2.41-
2.38 (m, 2H),
2.0-1.94 (m, 3H), 1.30-1.25 (m, 2H), 1.10-1.01 (m, 2H), 0.8-0.56 (m, 8H). MS
(DCI/IVH3)
m/e 476 (M+H)+. Anal calcd for C2gH33N304: C, 70.71; H, 6.99; N) 8.83. Found:
C, 70.50;
H) 6.99; N, 8.60.
CA 02277105 1999-07-06
WO 98/30541 PCTlUS98/00142
33
Example 1 G
O H O
HOHN N
O N
\ H
Separation of the diastereomers prepared in Example 1 F by HPLC gave the
compound of
Example 1G. mp 190-210 °C (dec). 1H NMR (300 MHz, DMSO-d6) 8 11.95 (s,
1H)) 10.36 (s,
1H)) 8.68 (s, 1H), 8.51-8.46 (m) 2H), 7.48-7.46 (m, 1H), 7.40-7.37 (d, 2H, J =
7.1 Hz), 7.25-
7. I 0 (m, 4H), 5.44-5.33 (m, 2H), 4.82-4.67 (m, 2H)) 3.14-3.12 (dd, I H, J =
4.1, 9.4 Hz))
2.97-2.89 (m, 1H), 2.39-2.35 (m, 1H), 1.98-1.83 (m, 2H), 1.30-1.25 (m) 2H))
1.21-0.99 (m,
1 H), 0.85-0.77 (m, I H)) 0.67-0.65 (d, 3H, J = 6.4 Hz), 0.55-0.53 (d, 3H, J =
6.8 Hz). MS
(DCI/NH3) m/e 476 (M+N)+. Anal calcd for C2gH33N304~0.75 H20: C, 68.76; H,
7.11; N)
8.59. Found: C, 68.77; H, 6.66; N, 8.52. [a]d = -4.19° (c) 0.31 ) DMF).
Exam I» a 1 H
O
H
HOHN N
O
IS
The desired compound was isolated in the chromatography of Example 1 G. mp I70-
210
°C (dec). IH NMR (DMSO) 8 12.03 (s, 1 H), 10.39 (s) I H), 8.71 (s, 1
H)) 8.64-8.61 (d) I H) J =
7.8 Hz)) 8.55 (s) 1 H)) 8.22-8.19 (d, I H, J = 6.4 Hz)) 7.49-7.47 (d) 1 H, J =
6.4 Hz)) 7.38-7.35
(d) 2H) J = 7.8 Hz)) 7.27-7.17 (m) SH), 5.41-5.40 (m, 2H)) 4.72-4.68 (d) 1H) J
= 7.8 Hz))
4.56-4.50 (d, 1 H) J = 14.9 Hz)) 3.12-3.07 (m) 1 H), 2.95-2.91 (m, 1 H)) 2.42-
2.41 (m, 1 H),
2.00-1.99 (m, 1 H ), 1.76-1.72 (m) 1 H ), I .26-1.25 ( m, 1 H ), 0.80-0.72 (m,
1 H ), U.62-0.5 6 (m,
6H). MS (DCI/IVH3) m/e 476 (M+H)+. Anal calcd for C2gH33N304~5/4 H20: C,
67.52; H,
7.18; N, 8.44. Found: C) 67.54; H, 6.94; N, 8.42. [a]d = 36.67° (c,
0.24, DMF).
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
34
_~xam~le 2
H
I
OH
a O
/ \ H
i '
Example 2A
0
/D" . dH
o
/ \ ~~/ H ~ ~
s
To a solution of methyl carbamate 2a (63.32 g, 280 mmol) in ether ( 1 L) was
added PBr3
( 10.8 mL, 110 mmol) via syringe at ambient temperature. The solution was
allowed to stir
overnight. The solvent was removed in vacuo and the resulting N-
carboxyanhydride ~ø (s4.2 g,
10010) was dried on under high vacuum for 2 hours. The product was carried
forward without
further purification.
Example 2B_
0
-- H
H ~ / \
To a solution of 2b (28.3s g) 148 mmol) was added indole (139.12 g, 740 mmol)
in one
portion. The reaction mixture was cooled to 0 °C and A1C13 (59.22 g,
445 mmol) was added
slowly via solid addition funnel. Upon complete addition of AICl3, the cold
bath was removed
and the solution was allowed to stir for 4 hours while warming to ambient
temperature. The
reaction was quenched by pouring onto 250 mL ice. The pH was adjusted to 12 by
the dropwise
addition of NI~OH. The aqueous layer was extracted twice with CH2C12. The
combined organic
layers were washed with brine, dried (MgS04)) filtered, and concentrated in
vacuo to a brown oil.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
~5
Flash chromatography (1:2:97 to 1:3:96 NH40H-MeOH-CH2C12) gave 2c (5 g, 16%)
as a tan
solid.
H
.H
\ N
H
r
A solution of acid succinate ester 4 (1.107 g, 3.7 mmol) in 20 mL DMF was
cooled to U
°C. NMM (975 mg, 8.9 mmol) was added via syringe, followed by HOBT (602
mg, 4.5 mmol))
EDCI (856 mg, 4.5 mmol), and 2c ( 1.18 g, 4.5 mmol). The solution was allowed
to stir
overnight while warming to ambient temperature. The reaction was quenched with
water and
extracted twice with EtOAc. The combined organic layers were washed with
brine, dried
(MgS04}, filtered, and concentrated in vacuo. The resulting orange foam was
chromatographed
( 1 % MeOH-CH2C12) to give 2sl ( I .607 g, 80%).
Ex
O H O _
t-Bu0 ~ ~ ~ HOH
r
O N O
S
I \ H
a
r
The desired compound was prepared according to the method of Examples 1 E and
F,
except substituting ~ for ~. mp I 88 °C (dec). 1 H NMR (300 MHz, DMSO-
d6) b I I .99 (d, 1 H,
J = 3.0 Hz)) 10.41 (d, 1 H) J = 1.5 Hz}, 8.74 (d) 1 H, J = 1.8 Hz), 8.56-8.44
(m, 2H), 8.20-8.17
(m, IH), 7.49-7.16 (m, 8H), 5.74-5.58 (m, 1H), 5.49-5.35 (m, 1H), 4.95-4.86
(m, 2H)) 3.18-
2.93 (m, 2H), 2.42-2.30 (m) 1H), 2.21-2.11 (m, IH), 1.96-1.56 (m) 3H), 1.47-
0.72 (m) 6H),
0.6I (dd, 6H, J = 36.4, 6.7 Hz). MS (APCI) m/e 504 (M+1 ). Anal calcd for
C3pH37N304:C,
71.54; H) 7.40; N, 8.34. Found: C, 70.72; H) 7.05; N, 7.10. [a]d = -
35°.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
36
Example 3
HOH
O
Example 3A
Bn O
H~ C02H "~ Bri f ~ O' gn
3~ .
i i
To a solution of phenylalanine ( 10 g, 61 mmol) in 500 mL of H20 was added
K2C03
(27.6 g, 200 mmol) and benzyl bromide (24 mL) 200 mmol). The reaction mixture
was heated at
reflux for 2.5 days and then was quenched with I M HCl and extracted with
CH2C12. The organic
layer was washed with H20) 1 M NaOH and brine. The organic layer was dried
(MgS04),
filtered) and concentrated in vacuo. Flash chromatography (20% EtOAc-hexane)
gave 3a (48%).
I 5 Example 3B
Bn 8n
Bri p' Bn Bpi ~ COpH
w ~ ~ w
i U
To a solutiong of 3a (5.05 g, 1.16 x 10-2 mol) in 2:1 dioxane/water (300 mL)
was added
KOH (0.65 g, 1.16 x 10'2 mol) and the reaction mixture was stirred for 2 days.
The reaction
mixture was acidified with 1 M HCl and extracted with EtOAc. The organic layer
was dried
(MgS04)) filtered, and concentrated in vacuo. Flash chromatography (40% EtOAc-
hexane) gave
fib,.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
37
Examl 1~
Bn Bn O
Bri ~C02H Bn~~~OMe
\ ~ ~ \
/ /
To a 0 °C solution of ,~, (2.0 g, 5.8 x 10'3 mol) in 30 mL CH2Cl2 was
added EDCI ( I .22
g, 6.36.x 10-3 mol), N,O-dimethylhydroxylamine hydrochloride (0.62 g, 6.36 x
10-3 mol), and
NMM (0.764 mL) 6.96 x 10-3 mol) and the reaction mixture was stirred at 0
°C for 10 minutes.
The cold bath was removed and the reaction was allowed to warm to ambient
temperature and stir
overnight. The reaction was quenched with H20 and exuacted with CH2Cl2. The
organic layer
was washed with citric acid) saturated aqueous NaHC03, pH 7 buffer and brine.
The organic
layer was dried (MgS04)) filtered) and concentrated in vacuo. Flash
chromatography (20%
EtOAc-hexane) gave ,~c (48% yield).
Exam lie 3D
Bn Bn
B~ -'OMe
en
3E ~ \ ~ ~ \
/ /
IS
To a 0 °C solution in THF (20 mL) of Weinreb amide ~ (588 mg, 1.5 mmol)
was added
ethylmagnesium bromide (I mL, 3 mmol) via syringe. The ice bath was removed
and the reaction
mixuture was allowed to warm to ambient temperature and stir for 3.5 hours.
The reaction was
quenched with H20 and extracted with EtOAc. The organic layer was washed with
brine, dried
2U (MgS04), filtered, and concentrated in vacuo. Flash chromatography
(5°!o EtOAc-hexane) gave
the desired product ~ in 36% yield.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
38
Exam lp a 3E
Bri Bn : H2N~
_ _e
i
To a solution of ketone ~j, (554 mg) 1.55 mmol) in 50 mL MeOH was added 20%
Pd(OH)2/C and the reaction was stirred for 24 hours. The reaction was filtered
to remove the
catalyst and concentrated in vacuo to give the desired product in ~g 91 %
yield.
'H\~
H HOHN N
w o \
The desired compound was prepared according to the method of Examples 1 D-F,
except
substituting 3e for Ic. mp 145 °C (dec). IH NMR (300 MHz) DMSO-d6) 8
10.36 (d. 1H, J =
1.7 Hz), 8.69 (d, 1 H, J = 1.7 Hz), 8.20 (d) 1 H) J = 8.8 Hz)) 7.09-7.30 (m,
SH)) 5.23-5.39 (m)
I H ), 4.6U-5.00 (m, 2H ), 4.46-4.57 (m, 1 H)) 2.56-2.94 (m) 2H )) 2.29-2.39
(m, 1 H)) 1.64-1.94
(m, 3H), 1.26-1.50 (m) 2H), 0.98-1.08 (m, 1H), 0.92 (t) 3H) J = 7.4 Hz), 0.84-
0.96 (m) 2H),
0.7R (dd, 6H) J = 6.4, 16.6 Hz). [a]d = +30.4°
x 1 4
i
H l
H
s
O
/ \ H
The desired compound was prepared by coupling of ~ andR-2-(i-butyl)-succinic
acid-4-t-
butyl ester according to the method of Example 2C, followed by hydrolysis of
the ten-butyl ester
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
39
using the method of Example lE. mp 110 °C (dec). IH NMR (300 MHz, DMSO-
d6) 8 12.08 (s,
1 H), 11.92 (d, 1 H, J = 2.7 Hz), 8.43 (d, 1 H, J = 8.5 Hz), 8.30 (d, 1 H, J =
3.1 Hz), 8.16-8.19
(m, 1 H), 7.44-7.47 (m, 1 H), 7.1 I -7.30 (m, 8H), 5.32-5.42 (m, 1 H), 2. 87-
3.19 (m, 2H)) 2.61-
2.74 (m, 1 H), 2.04-2.25 (m, 2H)) 1.18-1.40 (m, 2H), 0.97-1.08 (m) I H), 0.69
(dd, 6H, J =
6.5, 16.3 Hz). MS (APCI) m/e 421 (M+1 ), 438 (M+18). Anal calcd for C25H28N204
O.SH20: C, 69.90; H) 6.80; N) 6.52. Found: C, 69.83; H) 6.80; N, 6.49. [a]d =
+7.2°.
Example 5
o
HOH
O
Example SA
i O ~ O i
H H \ I ~ ~ H \ I
O ~ 1 N OBn O 1 N
~H ~ ~ ~ ~H
A pH 4 solution in 1.5:1 THF-H20 of the compound of Example 4 (50 mg) 1.0 x 10-
1
mmol) and O-benzylhydroxylamine hydrochloride (26 mg) 1.6 x 10-1 mmol) was
cooled to 0 °C
and EDCI (63 mg) 3.3 x 10-1 mmol ) was added. The reaction stirred at 0
°C for 1 hour. The ice
bath was removed and the reaction mixture was warmed to ambient temperature
and stirred
overnight. The THF was removed in vacuo and the remaining liquid was partioned
between
EtOAc and citric acid. The organic layer was washed with brine, dried (MgS04),
filtered and
concentrated in vacuo. Flash chromatography (1 % MeOH-CH2C12) gave the desired
product ~ in
46% yield.
CA 02277105 1999-07-06
WO 98/30541 PCT/LTS98/00142
Example SB
Hog
Oen O O
A mixture of benzyl hydroxamate Sa (0.266 g, 5.1 x 10-4 mol) and Pd/C (0.133
g) in 10
5 mL of THF was stirred overnight under I atm of H2. The reaction mixture was
gravity filtered
through a plug of celite. Solvent was removed in vacuo to give the desired
product in 82% yield as
a tan solid. mp 120 °C. I H NMR (300 MHz, DMSO-d6) 8 11.94 (s) 1 H),
10.33 (s) 1 H), 8.71
(s) 1H), 8.47 (d, IH, J = 8.5 Hz), 8.33 (s, IH), 8.16-8.20 (m) 1H)) 7.44-7.48
(m, 1H)) 7.11-
7.31 (m, 7H)) 5.30-5.39 (m, 1 H)) 2.89-3.16 (m, 2H), 2.64-2.77 (m) I H), 1.83
(d, 2H) J = 7.4
10 Hz)) I .29-1.40 (m) 1 H), I .14-1.26 (m, I H ) ) 0.86-0.97 (m, 1 H )) 0.66
(dd, 6H) J = 6.6) 10.3
Hz). MS (CI) m/e 436 (M+1 ). Anal calcd for C25H29N304 ~ 1.00 H20: C) 66.20;
H, 6.88; N)
9.26. Found: C) 66.34; H) 6.74; N) 8.99. [a]d = -1 I.5°.
I5 Exam a 6
H
HOH
O 1 N
\ ~H
The desired compound was prepared by coupling of 2c and succinate ester 5
according to
the method of Example 2C, followed by hydrolysis of the tert-butyl ester using
the method of
20 Example 1 E, and conversion of the acid to the hydroxamate according to the
method of Example 5.
mp 185 °C. 1 H NMR (300 MHz, DMSO-d6) 8 I 1.94 (d, 1 H, J = 2.6 Hz))
10.35 (s, I H), 8.70
(s, 1 H)) 8.53 (d, 1 H, J = 8.8 Hz), 8.35 (d, 1 H, J = 3.4 Hz), 8.20-8.23 (m,
1 H), 7.46-7.51 (m,
1 H), 7.10-7.31 (m, 7H), 6.70 (dd, 4H, J = 8.1, 25.7 Hz), 5.33-5.43 (m, 1 H),
2.87-3.19 (m,
CA 02277105 1999-07-06
WO 98/30541 PCTIUS98/00142
41
2H), 2.61-2.75 (m, 1H), 2.14-2.4.4 (m) 2H), 2.14 (s, 3H), 1.88 (d, 2H, J = 7.3
Hz), 1.11-1.42
(m, 4H). MS (CI) m/e 512 (M+1 ). Anal calcd for C31 H33N304 ~ 0.75 H20: C,
70.90; H) 6.62;
N, 8.00. Found: C, 71.20; H, 6.63; N, 7.72. [oc]d = -5.8°,
Ex 7
o i
\ H
r
The desired compound was prepared by coupling of 2c and succinate ester 6
according to
the method of Example 2C, followed by hydrolysis of the tert-butyl ester using
the method of
1 () Example 1 E. I H NMR (300 MHz, DMSO-d6) 8 12.01 (d, I H, J = 3.0 Hz))
8.63 (d) 1 H) J = 9.2
Hz), 8.49 (d, I H, J = 3.3 Hz)) 8.25-8.22 (m, 1 H)) 7.52-7.48 (m, 1 H), 7.37-
7.13 (m) 7H)) 6.68
(dd, 4H, J = 34.2, 8.1 Hz), 5.7405.61 (m, 1 H), 5.52-5.45 (m, I H), 4.97-4.87
(m, 2H), 3.91
(br, 1H), 3.16-2.92 (m) 2H), 2.40-2.10 (m, 4H)) 2.15 (s, 3H)) 1.87-1.63 (m)
2H), 1.44-0.96
(m, 7H)) O.S4-0.7U (m, 1H). MS (CI) m/e 565 (M+1). Anal calcd for C36H4004N2 ~
I.5 H20:
C, 73.07; H) 7.32; N, 4.73. Found: C, 73.13; H, 6.97; N, 4.98.
CA 02277105 1999-07-06
WO 98/30541 PCT/L1S98/00142
Ex~ arn~lg~
42
o i
H
HOH i
o ~ N
H
The desired compound was prepared according to the method of Example 5, except
substituting the compound of Example 7 for the compound of Example 4. mp 220
°C. 1 H NMR
(300 MHz) DMSO-d6) & 12.02 (s, 1H), 10.48 (s, IH), 8.77 (s) IH), 8.60 (d) 1H)
J = 9.2 Hz))
8.53 (s) 1 H )) 8.29-8.26 (m, 1 H), 7.56-7.15 (m) 8H), 6.70 (dd) 4H, J = 34.6)
7.8 Hz), 5.80-
5.64 (m, 1 H), 5.55-5.44 (m) 1 H), 5.02-4.90 (m) 2H), 3.23-2.90 (m, 2H), 2.44-
2.28 (m) 3H ),
2.19 (s, 3H)) 2.08-1.96 (m, IH), 1.91-1.66 (m, 2H)) 1.48-0.96 (m) 7H), 0.76-
0.63 (m, IH).
1 () MS (ESI) m/e 58U (M+1 ), 602 (M+Na). Anal calcd for C36H41 N304 ~ I.25
H20: C, 71.79; H,
7.28; N) 6.97. Found: C) 71.59; H, 7.05; N) 7.06. [a]d = -20.4°.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
43
x 9
0 0
H I
HOH ~ \
O
H
Ex~ar thehe 9A
H2 \ I
1 N
~ H
The desired compound was prepared according to the method of Examples 1 A-C,
except
substituting L-ten-leucine for L-phenylalanine.
I
H \ H 1
----~ HOH
O ~ N
H ~ ~H
The desired compound was prepared according to the method of Examples 28 and
C,
except substituting Q~ for ~ and substituting succinate ester ~ for succinate
ester _4. mp 218 °C
I 5 (dec). I H NMR (300 MHz. DMSO-d6) 8 I 1.92 (s, 1 H)) 1 U.45 (s, 1 H)) 8.73
(s) I H)) 8.36 (d)
1 H) J = 3.1 Hz), 8.17-8.21 (m, 1 H), 8. I 2 (d, 1 H, J = 8.8 Hz), 7.44-7.47
(m, 1 H), 7.13-7.25
(m) 2H), 5.54-5.70 (m, 1 H), 5.1 U (d, 1 H, J = 8.8 Hz), 4.87-4.97 (m) 2H),
2.73 (dt) 1 H, J =
2.7, 10.9 Hz), 2.23-2.37 (m, 1H), 1.98-2.20 (m) 2H), 1.28-1.39 (m) 1H), 0.96-
1.16 (m, 1H),
1.00 (s) 9H)) 0.82-0.94 (m) 1 H), 0.62 (dd) 6H, J = 6.1, 40.3 Hz). MS (DCI)
m/e 442 (M+H)+.
20 Anal calcd for C25H35N304 ~ 0.5 H20: C) 66.64; H) 8.05; N, 9.32. Found: C)
66.59; H, 8.01;
N, 9.10. [a]d = -55.9°.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
44
Exam I;Re 10
O H O / I
N
HO
OH O \ NH
O OPFP H O / I
O + O N
~O O ~~ ~NH
ls~ l~ I \
Pentafluorophenol ester ~ (0.605 g) I .53 mmol), prepared as described in
W094/U2446,
and ~ (0.448 g) 1.70 mmol) were combined in dry DMF (6 mL). The solution was
heated at 30
°C for 24 hours, then reduced in volume by rotary evaporation under
high vacuum. The residue
was diluted with ethyl acetate) then washed succesively with brine) pH3
buffer) aqueous Na2C03,
pH7 buffer and brine. The organics were dried over Na2S04 and evaporated to
give II Ob (0.764
g) as a tan solid which was carried forward without purification.
Exam Ip a l OB
O H O / I O H O / I
N ~ N
O \ ~ HO
O O ~NH OH O \ NH
\ \
IS
To a 0 °C solution in THF ( 12 mL) of dioxolanone ,~ (0.728 g, 1.5
mmol) was added 2.1
M HCl ( 12 mL) and the solution allowed to warm to ambient temperature over 18
hours. The
solution was evaporated to drynessto give a tan foam (0.70 g). The crude
material was purified by
reverse phase HPLC to give the desired compound (0.223 g) as a white foam. 1 H
NMR (DMSO-
d6) 0.65 (d, 3H) J = 6.4 Hz), 0.72 (d, 3H, J = 6.5 Hz), 1.02 (m, 1 H), 1.23
(m, 1 H), 1.41 (m,
1H), 2.62 (m, 1H), 2.94 (dd, 1H, J = 7.4, 13.9 Hz), 3.17 (dd, 1H, J = 6.5,
13.9 Hz), 3.91 (d,
1 H, J = 7.1 H z)) 5.38 (m, 1 H ), 7. 22 (m, 6H), 7.45 (dd, 1 H, J = 2.3, 6.4
Hz)) 8.16 (dd, 1 H, J =
2.3, 6.1 Hz), 8.27 (d, 1 H, J = 3.0 Hz)) 8.41 (d, 1 H, J = 3.0 Hz), 8.58 (bds,
1 H), 11.91 (d, 1 H,
CA 02277105 1999-07-06
WO 98/30541 PCT/(JS98/00142
J = 2.7 Hz). MS (DCI/IVH3) M/e 454 (M+NH4)+, 437 (M+H)+, 419, 265. Anal. Calcd
for C25
H28 N2 OS ~.75 H20: C, 66.72; H) 6.61; N, 6.22. Found: C, 66.71; H, 6.30; N,
5.90.
Exam lp a I 1
o H ~ ~ L
N \
HO
OH O ~NH
The desired compound was the slower eluting species in the chromatography
described in
Example I . I H NMR (DMSO-d6) 0.63 (d, 3H) J = 5.5 Hz)) 0.71 (d) 3H) J = 5.5
Hz), 0.87
10 (m, 2H), 1.27 (m, 1 H), 2.59 (m, 1 H)) 2.89 (dd, 1 H, J = 10.0, 13.6 Hz))
3.09 (dd, 1 H) J = 4.7,
13.6 Hz), 3.83 (d, 1H, J = 7.8 Hz), 5.43 (m, 1H), 7.21 (m) 4H)) 7.32 (d, 2H, J
= 6.9 Hz), 7.46
(dd, 1 H) J = 1.8) 5.5 Hz), 8.21 (dd) 1 H, J = 2.6, 5.5 Hz), 8.47 (d) 1 H) J =
3.3 Hz), 8.64 (d)
1 H, J = 8.9 Hz), I I .98 (d) 1 H, J = 2.6 Hz). MS (DCI/NH3 ) m/e 454 (M+NI-I4
)+, 437 ( M+H )+)
265. Anal calcd for C25H28N205: C, 68.79; H, 6.47; N, 6.42. Found: C) 67.87;
H, 7.14; N,
IS 5.54.
Example 12
O H O
HO N \
N _
NH
H OH O
The desired compound was prepared according to the method of Example 5) except
substituting the compound of Example 10 for the compound of Example 4. mp I22-
125 °C. 1 H
NMR (DMSO-d6) 0.63 (d, 3H, J = 6.8 Hz), 0.68 (d, 3H, J = 6.4 Hz), 0.85 (m)
1H), 1.08 (d,
1 H)) I .37 (m, 1 H), 2.63 (m, I H), 2.94 (dd, 1 H, J = 6.8, 13.9 Hz), 3.18
(dd) 1 H, J = 7.2, 13.9
Hz), 3.77 (dd) 1 H, J = 6.8, 8.5 Hz), 5.22 (d, 1 H, J = 6.8 Hz), 5.37 {dd, 1
H, J = 6.8, 7.2 Hz),
7.19 (m, 6H), 7.45 (dd) 1H, J = 0.7, 7.8 Hz)) 8.13 (dd, 1H, J = 2.0, 5.7 Hz),
8.25 (d, 1H, J =
2.0 Hz), 8.37 (d, 1H, J = 8.1 Hz), 8.83 (s, 1H), 10.59 (s, 1H), 11.92 {s, 1H).
MS (DCI/NH3)
CA 02277105 1999-07-06
WO 98/30541 PCTIUS98/00142
46
m/e 469 (M+NHd)+, 452 (M+H)+. Anal calcd for C25H29N305~0.33 H20: C, 65.64; H,
6.53;
N, 9.19. Found: C, 65.65; H, 6.54; N, 8.20. [ot]d = +12 ° (C = 0.95)
CH3OH).
Exam l
O H O / I
H O~N N \
I _ ~
H OH O ~NH
The desired compound was prepared according to the method of Example 5) except
substituting the compound of Example 11 for the compound of Example 4. mp 172-
175 °C. 1 H
NMR (DMSO-d6) 0.62 (d, 3H) J = 6.4 Hz), 0.69 (d) 3H, J = 6.1 Hz), 0.79 (m) 1
H)) 0.88 (m,
1 H ), 1.23 (m, 1 H ), 2.62 (m) 1 H)) 2.90 (dd, 1 H, J = 9.5, 13.9 Hz)) 3.09
(dd, 1 H, J = 4.7, 13.9
Hz)) 3.68 (dd, 1 H, J = 6.5, 8.8 Hz)) S.U 1 (d(OH), 1H, J = 6.1 Hz), 5.41 (m,
1 H), 7.22 (m)
SH), 7.30 (s) 1 H), 7.31 (dd, 1 H) J = 1.7, 7.0 Hz)) 7.46 (dd) 1 H, J = 1.3,
8.5 Hz), 8.20 (dd)
1 H) J = 2.6, 8.5 Hz)) 8.44 (d, 1 H, J = 9.5 Hz)) 8.46 (s) 1 H), 8.75 (s, 1
H), 10.51 (s, 1 H), 11.92
(s) 1H). MS (DCI/NH3) m/e 469 (M+MH4)+, 452 (M+H)+) 391. Anal calcd for
C25H2gN305~0.33 H20: C, 65.64; H, 6.53; N) 9.19. Found: C, 65.63; H, 6.74; N)
8.31. [
- -9.1 ° (C = 1.1, CH30H).
2« Example 14
O H O _
H O~N N
- = t Y
H OH O ~ N
H
The desired compound was prepared according to the method of Examples 10 and
12,
except substituting 9a for Ic. mp 144-146 °C. 1H NMR (DMSO-d6) 0.65 (d,
3H, J = 6.8 Hz),
0.69 (d, 3H, J = 6.5 Hz), 0.89 (m, 1 H), 0.98 (s, 9H), 1.20 (m, 1 H), 1.41 (m,
1 H), 2.77 (m,
1 H)) 3.73 (t) 1 H, J = 8.2 Hz}, 5.12 (d, 1 H, J = 9.5 Hz), 5.24 (d, 1 H, J =
8.1 Hz)) 7.12 (m,
2H), 7.46 (d, 1H, J = 6.8 Hz), 7.78 (d, 1H, J = 9.4 Hz), 8.21 (dd, 1H, J =
1.7, 6.1 Hz), 8.36
(d, 1H, J = 2.7 Hz), 8.84 (bds, 1H), 10.59 (bds, 1H), 11.96 (bds, 1H). 13C NMR
(DMSO-d6)
21.58, 23.49, 25.12, 27.05) 34.42, 37.22, 47.57, 60.23, 71.47, 112.07, 116.61,
121.45,
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
47
121.68, 122.81, 125.49) 134.45) 136.59, 168.77, 172.44, 193.57 . MS (DCI/NH3)
m/e 435
(M+NH4)+, 418 (M+H)+. Anal calcd for C22H31 N305'0.5 H20: C, 61.95; H, 7.56;
N, 9.95.
Found: C) 61.64; H, 7.69; N, 9.67. [a]d = -57 ° (C = 1.2, CH30H).
Fpm- I?~ a 15
O H O
HO~ N
H OH O
Example 15A
0
HpN
IU ~ /
The desired compound was prepared according to the method of Examples 3A-E,
except
substituting phenyllithium for ethylmagnesium bromide.
Example I SB
0
0 0
2N H 0~ H
N N
H OH O \
/
The desired compound was prepared according to the method of Examples 10 and
12)
except substituting ~ for lc. IH NMR (CD30D) $ 0.78 (d, 3H, J = 6.5 Hz), 0.80
(d) 3H, J =
6.5 Hz), I .10 (m, 1 H), I .30 (m) 1 H), 1.54 (m) 1 H), 2.77 (m) 1 H)) 3.01
(dd, 1 H, J = 6.5) 13.6
Hz), 3.20 (dd, 1 H, J = 6.8) 13.9 Hz), 3.98 (d, 1 H, J = 7.1 Hz), 5.73 (t, 1
H, J = 6.8 Hz), 7.18
(m, 5H)) 7.45 (t) 2H, J = 7.8 Hz), 7.58 (m, IH)) 7.93 (d, 2H, J = 7.1 Hz). MS
(DCI1NH3) m/e
430 (M+NH4)+, 413 (M+H)+. Anal calcd for C23H28N205~O.SH20: C, 65.54; H, 6.93;
N,
6.65. Found: C, 65.57; H, 6.99; N, 6.52.
CA 02277105 1999-07-06
WO 98130541 PCTILJS98/OOI42
48
Example 16
O
H
HOHN N
O
The desired compound was prepared according to the method of Example 1, except
substituting 1-methylindole for indole. ~H NMR (300 MHz, DMSO-d6) 8 10.36 (s,
1H), 8.68-
8.46 (m, 3H), 8.32-8.18 (m, 1 H)) 7.56-7.18 (m, 7H), 5.43-5.33 (m, 2H), 4.82-
4.6 (m, 2H),
3.88 (s, 3H), 3.12-3.07 (m) 1H), 2.95-2.90 (m, 1H)) 2.40-2.39 (m, 1H), 2.03-
1.88 (m, 2H),
1.34-1.26 (m, 2H), 0.81-0.56 (m, 7H). MS (DCI/NH3) m/e 490 (M+H)+. Anal calcd
for
C29H35N3o4'0-25 H20: C, 70.49; H, 7.24; N, 8.42. Found: C, 70.39; H, 7.37; N,
8.35.
O H
HOHN N
O
Example 17A
O
~O~N~
'IO
To a -78 °C solution of N-Boc-phenylalanine (2.69 g) 10 mmol) in 10 mL
THF was added
MeLi (22.9mL, 32mmoL, 1.4M in ether) via addition funnel over 10 minutes. The
cold bath
removed and the solution allowed to warm to ambient temperature and stirred 2
hours. The
reaction was quenched with 25 mL of 2N HCl solution, stirred for 10 minutes,
and the aqueous
layer was extracted with ether (3x). The combined organics were washed with
brine) dried
CA 02277105 1999-07-06
WO 98130541 PCT/US98100142
49
(Na2S04) and concentrated in vacuo. Flash chromatography (hexane-ethyl acetate
S: I ) gave 17a
( 1.31 g) as a waxy solid.
Example 17B
O
HCI~H2N~
A 0 °C solution of 17~ ( 1.31 g, 4.83 mmol) in 4N HCI/dioxane was
stirred for 2 hours and
then was diluted with diethyl ether. The residual solid was filtered and dried
under high vacuum to
give 17ø (U.9 g) as the HCl salt.
O
O
HC!~H2N~ O N
HOHN
O
The desired compound was prepared according to the method of Examples 1 D-F,
except
I S substituting 17b for 1 c. mp I 80 °C (dec). 1 H NMR (DMSO-D6) 8 1
U.37 (S, I H), 8.71 (S, 1 H))
8.47-8.38 (D, IH) J = 8.1 Hz), 7.30-7.13 (M, SH)) 5.36-5.30 (M) 1H)) 4.80-4.U6
(M) 3H))
3.10-3.()4 (M, 1 H), 2.72-2.64 (M, 1 H)) 2.41-2.39 (M, l H), 2.11 (S, 3H),
1.92-1.90 (M, I H))
1.7y-1.70 (M, 1 H > I .39-1.35 (M) 2H )) 1.23-1.20 (M. 2H)) U.84-U.75 ( M)
7H). MS (DCUNH3 )
m/e 375 ( M+H ). Anal calcd for C2 ~ H 3pN~ 04C: C, 67.35; H, 8.U 1; N ) 7.48.
Found: C, 66.95;
2() H) 8.01; N) 7.31.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98100I42
Examplg 18
O H O
HOHN N \
O
Examine 18A
H O
Ou N \
'OI
5
To a cold 0 °C solution of bromobenzene (5 g, 32 mmol) in THF was added
nBuLi ( 12.8
mL, 32 mmol, 2.SM in diethyl ether) over the course of 5 minutes. The
resulting yellow solution
was stirred at 0 °C for 25 minutes and then was added to a -78
°C solution of N-BOC-1-
10 phenylalanine (2.69 g, 10 mmol) over 25 minutes. The resulting yellow
solution was allowed to
warm to room temperature overnight ( 16 hours) and then was quenched with 1 N
HCl solution.
The aqueous layer was extracted with ether (3x) and the combined organics were
washed with 1 N
NaHC03 and brine, dried (MgS04) and concentrated in vacuv.. Flash
chromatography (hexane-
ethyl acetate 6:1 ) gave 1 a (0.25 g) as a waxy solid.
Example 18B
O N O O H O
HOHN N I \
O /
1$.a
The desired compound was prepared according to the method of Examples 17B and
C,
except substituting 18a for ~ 7a. mp 209-211 °. 1 H NMR (DMSO-d6) 8
10.39 (s, 1 H)) 8.72 (s,
1 H ) ) 8.59-8.57 (d, 1 H, J=8.5 Hz), 8/06-8.02 (d, 2H, J=8. I Hz), 7.69-7.64
(t, 1 H, J=7.1 Hz),
7.55-7.52 (t, 2H, J=8.1 Hz), 7.40-7.38 (d, 2H, J=7.4 Hz)) 7.30-7.28 (m, 2H),
7.20-7.18 (m,
1 H), 5.66-5.62 (m, 1 H), 5.44-5.35 (m, 1 H), 4.86-4.70 (m, 2H), 3.19-3.13
(dd, 1 H, J=7.9,4.1
Hz)) 2.94-2.86 (m, 1H), 2.38-2.34 (m, 1H), 1.96-1.81 (m, 2H), 1.33-1.26 (m,
2H), 0.88-0.81
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
51
(m) 2H), 0.68-0.59 (m, 6H). MS (DCI/NH4) 437 (M+1 ). Anal. Calcd for:
C26H32N204: C,
70.57; H, 7.44; N, 6.33. Found: C, 70.60; H) 7.13; N, 6.42.
Examgle 19
O H O
HOHN N
O I/
OH
Example 19A
H O
t-BuO~ N
1~
Ot-8u
The desired compound was prepared according to the method of Example 18A)
except
substituting N-Boc-O-tBu-L-tyrosine for N-BOC-1-phenylalanine.
Exam~he 19B
O
HCI~HpN
1~ ~ \
I OH
A cold 0 °C solution of 19a ( 1.8 g, 4.7 mmol) in trifluoroacetic acid
was stirred for 30
minutes. The excess TFA was removed in vacuo and the residue was taken up in 1
N HCl in Et20,
allowed to stir for 30 minutes, diluted with diethyl ether, and the residual
solid was filtered. The
extremely hygroscopic solid was dried in a vacuum oven for several hours and
then was dried
under high vacuum for 16 hours to give 19b (0.48 g) as a hygroscopic) white
HCI salt.
CA 02277105 1999-07-06
WO 98130541 PCT/US98/00142
52
Exam In a 19C
HCI~HpN
t-Bu0'~ N
O /
OH ~ ' OH
To a 0 °C solution of succinate ester ~ (1.5 g, Smmol) in 20 mL of
methylene chloride was
added HOBT (0.81 g) 6 mmol) and EDCI ( 1.17 g, 6 mmol). The suspension became
a clear
solution after 10 minutes and was allowed to stir for 4 hours total. The
solution was diluted with
methylene chloride and the organics were washed with water (3x) and brine and
concentrated in
vacuo. The crude HOBT ester was dissolved in DMF ( l OmL) and added to a
solution of 19b ( 1.7
g, 6 mmol) and NMM (1.2 g, 10 mmol) in 10 mL of DMF. The reaction stirred for
3 days and
1 () then was diluted with ethyl acetate and the organic layer was washed with
water (3x) and brine)
dried (Na2S04) and concentrated iia vacuo. Flash chromatography (gradient
elution: methanol-
methylene chloride 0-2%) gave 19c (2.45 g) as a while solid.
Example 19D
O H O O H O
t-Bu0 O N I \ HOHN O N
1~
OH ~ ' OH
The desired compound was prepared according to the method of Examples I E and
F)
except substituting ~ for _l~. mp 208-210 °C (dec). tH NMR (3(?0 MHz,
DMSO-d6) 8 10.42 (s,
1 H ), 9. I 5 (s, 1 H), 8.71 (s) 1 H ) ) 8.49-8.47 (d, 1 H, J = 7.8 Hz), 7.99-
7.97 (d, 2H, J = 7.4 Hz),
7.63-7.60 (t) 1 H, J = 7.2 Hz), 7.53-7.48 (t, 2H, J = 7.8 Hz), 7.13-7.10 (d,
ZH, J = 8.2 Hz),
6.65-6.62 (d, 2H, J = 8.5 Hz), 5.73-5.64 (m, 1 H), 5.49-5.48 (m) 1 H), 4.95-
4.86 (m, 2H),
3.01-2.95 (m) 1H), 2.77-2.69 (m, 1H), 2.36-2.34 (m, 1H), 1.89-1.68 (m, 3H),
1.28-I.18 (m,
SH), 0.82-0.58 (m, 7H). MS (DCI/NH3) m/e 481 (M+H)+.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
53
Exanr~le 20
O H O
HOHN N I
O
The desired compound was prepared according to the method of Example 19,
except
substituting N-BOC-alpha-cyclohexyl alanine for N-Boc-O-tBu-L-tyrosine. mp 209-
210 °C. 1H
NMR (300 MHz) DMSO-d6) b 10.47 (s, 1 H)) 8.76 (s, 1 H), 8.54-8.52 (d, I H, J =
7.5 Hz))
7.92-7.89 (d, 2H, J = 8. I Hz), 7.66-7.60 (t, I H, J = 8.5 Hz), 7.55-7.49 (t,
2H, J = 7.8 Hz))
5.62-5.50 (m, 1 H), 5.33-5.29 (m, 1 H), 4.93-4.86 (m, 2H)) 2.20-2.08 (m, 2H),
1.91-1.87 (m,
2H), 1.61-1.32 (m, 7H)) 1.13-1.10 (m) 4H)) 0.9-0.80 (m, 3H)) 0.76-0.74 (d, 3H,
J = 6.4 Hz),
1 () 0.67-0.65 (d) 3H) J = 6.8 Hz). MS (DCI/NH3) m/e 443 (M+H)+. Anal calcd
for C2~,H3gN2p4:
C, 70.55; H, 8.65; N, 6.32. Found: C, ?0.21; H, 8.65; N) 6.32.
E~caml 1~ a 21
O ~ O
H
HOHN N /
O Phi H
Example 21 A
H O
BOCN /
Phi H
21a
To a -40 °C solution under nitrogen of methylmagnesium bromide (9.54
ml, 3.0 M in
Et20, 28.6 mmol) in dry toluene (20 ml) was added pyrrole (3.2 ml, 46.5 mmol)
dropwise and the
resulting solution was stirred at -10 °C for 10 minutes. The Grignard
reagent was cannulated into a
solution of BOC-L-phenylalanine-methyl ester (1.0 g, 3.58 mmol) in dry toluene
(lOml) at -65 °C,
the temperature was allowed to warm up to 0 °C over 4 hours, and the
reaction was quenched by
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
54
addition of sat. NH4C1 solution, extracted with CH2C12 (3x), dried over
Na2S04, filtered and
concentrated in vacuo to give 1.9 g of a crude mixture which was purified by
flash
chromatography(15% ethyl acetate-hexane) followed by recrystallization from
Et20-hexanes to
give 21 ~ (677 mg) as a white solid.
Example 21 B
H O O
BOCN / HpN
j NJ = NJ
Ph H Ph H
21a ?~
A solution of 21 a (600 mg, 1.91 mmol) in 4M HCl/dioxane (8 ml) was stirred at
room
temperature for 50 minutes. The solvent was evaporated to give 21 b (507 mg)
as a purple solid
which was used in the next step without further purification.
Example 21 C
O O O
H2N ~ N
t-Bu0 ~/~
Phi H O phi H /
21b
To a 0 °C solution in DMF (20 mL) of 21 b (6(>D mg, 1.91 mmol) was
added HOBT (258
mg, 1.91 mmol)) NMM(630 ltl, 1.91 mmol), succinate ester 2 (515.7 mg) 1.91
mmol) and EDC
(366 mg) 1.91 mmol) and the reaction mixture was stirred for 20 minutes at 0
°C and for 15 hours
at ambient temperature. The reaction mixture was diluted with ethyl acetate
and washed with
2() brine-H20 (1:1 ). The aqueous wash was extracted with ethyl acetate (3x)
and the combined
organic extracts were washed with brine-H20 ( 1:1 ), dried over Na2S04,
filtered and concentrated
in vacuo to give a brown foam. Purification by chromatography on silica gel
(20% ethyl acetate-
hexanes to give 21c (797 mg) as a yellow foam.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
Exam lp a 21 D
O H O O H O
t-Bu0 N / ----~ HO N /
O Phi H O Phi H
21~
A solution of ~ (781 mg, 1.67 mmol) in trifluoroacetic acid (8ml) was stirred
at ambient
temperature for 50 minutes. The ssolvent was evaporated to give 2~ (808 mg) as
a yellow foam.
Exam 1e~21~
O ~ O
H O O
HO N / / HO. N N /
O Phi H H O = N
Ph H
10 To a 0 °C solution in DMF (15 mL) under nitrogen of ~Il (753 mg)
1.84 mmol} was added
HOBT (273 mg) 2.02 mmol), NMM(443 ~tl) 4.04 mmol)) TBDMSONH2 (298 mg, 2.02
mmol)
and EDC (387 mg. 2.02 mmol). The reaction mixture was stirred at 0 °C
for 1 hour and at ambient
temperature for 17 hours. The reaction mixture was diluted with ethyl acetate
and washed with
brine-H20 ( I :1 ). The aqueous wash was extracted with ethyl acetate (3x) and
the combined
15 organic extracts were washed with brine-H20 ( 1:1 ), dried over Na2S04)
filtered and concentrated
in vacuo to give a yellow solid. Purification by chromatography on silica gel
( 10°lo MeOH-
CH2Cl2) gave the desired compound (448 mg) as a white solid. mp 2(>4-205
°C (dec). IH NMR
(300 MHz) DMSO-d6) 8 U.65 (d, 3H, J = 3 Hz), 0.77 (d) 3H) J = 3 Hz),0.83 (m, I
H), 1.13-
1.38 (m) 3H), 1.77-1.98 (m) 2H), 2.41 (dt) 1 H) J = 3) 12 Hz), 2.88 (dd, 1 H)
J = 3, 1 U.SHz))
20 3.07 (dd) 1 H, J = 4.5) 15 Hz), 4.66-4.84 (m, 2H), 5.23-5.46 (m, 2H), 6.22
(m, 1 H), 7.09-7.40
(m, 7H), 8.44 (d, 1H) J = 9 Hz), 8.685 (s, 1H), 10.36 (s, 1H)) 11.88 (s, 1H).
MS (DCI/NH3)
m/z 426 (M+H)+. [a]d =+19.69°(EtOH).
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
56
Example 22
O ~ O
H
HOHN N I
OPh ~NJ
Examha a 22A
H O
BOCN
Ph ~N~
To a -78 °C solution under nitrogen of n-butyl lithium (2.SM in
hexanes, 21.5 ml, 53.8
mmol) in ether ( I 80m1) was added 3-bromopyridine (5.18 ml, 53.8 mmol)
dropwise and the
reaction mixture was stirred for I hour. A solution of BOC-L-phenylalanine
methyl ester (6.U g)
21.5 mmol) in ether (25 ml) was added and the reaction mixture was stirred at -
78 °C for 3 hours
and 0 °C for two hours. The reaction mixture was poured onto water,
extracted with CH2C12
(3x)) dried over Na2S04) filtered and concentrated in vacuo to give an orange
oil which was
purified by flash chromatography (3U°lo ethyl acetate-hexanes) to give
2a (1.2 g) as a yellow oil.
Example 22B
H O O O
BOCN H
HOHN N
Ph N OPh ~N~
The desired compound was prepared according to the method of Examples 21 B-E)
except
substituting ~ for 1 a. mp 196.3- I 97.7 °C. 1 H-NMR (300 MHz) DMSO-d6)
8 0.49-0.62 (m,
6H), 0.69-0.82 (m, 2H )) 1.04-1.28 (m, 2H), 1.66- I .80 (m, 1 H)) 1.825-1.97
(m, 1 H), 2.23-
2.35 (m, 1 H ), 2.84-2.98 (m, 1 H ), 3.08-3.21 (m, 1 H ), 4.62-4.84 (m, 2H),
5.24-5.63 (m, 2H),
7.09-7.38 (m, 6H), 7.49-7.57 (m, 1H), 8.26-8.37 (1H), 8.61-8.78 (3H), 9.0?-
9.I5 (1H). MS
(DCI/NH3) m/z 438 (M+H)+. a]d =-18.86° (EtOH).
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
57
Example 23
O H O
HOHN N
O Ph
Example 23A
Br
N
TI PS
~3a
To a -78 °C solution under nitrogen of 1-triisopropylsilyIpyrrole (2.8
g, 12.6 mmol ) in
THF (30 ml) was added NBS (2.23 g) 12.6 mmol) via a solid addition funnel. The
reaction
mixture was stirred at -78 °C for 1 hour and then was warmed to ambient
temperature over 1 hour.
The reaction mixture was concentrated) carbon tetrachloride was added to
precipitate the
succinimide and the solid was filtered and washed with carbon tetrachloride.
The filtrate was
concentrated, and the crude product was purified by flash chromatography
(hexanes) to afford 3-
bromo-I-triisopropylsilylpyrrole (3.18 g) as a colorless oil.
I5 Example 23B
H O
Br BOCN
N Ph ~N~
TIPS TIPS
To a -78 °C solution under nitrogen of 3-bromo-I-(triisopropylsilyl-
pyrrole (3.18 g, 10.5
mmol) in dry THF (50 ml) was added n-BuLi ( 1.6 M, 6.56 ml, 10.5 mmol) and the
reaction
mixture was stirred for 0.5 hours. A solution of BOC-L-phenylalanine methyl
ester (1.25 g, 4.2
mmol) in dry THF (2 ml) was then added and the resulting mixture was stirred
at -78 °C for 1.5
hours. The reaction mixture was poured onto water) extracted with CH2C12 (3x),
dried over
Na2S04) filtered, and concentrated in vacuo. Chromatography on silica gel (10%
ethyl acetate-
hexanes) provided ~ (268 mg) as a light yellow oil.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
58
Example 23C
H O p H O
80CN HOHN N
Ph N ~ O Ph
TIPS
The desired compound was prepared according to the method of Examples 21B-E)
except
substituting 23b for 21a. ~H NMR (300 MHz, DMSO-d6) 8 0.53-0.88 (7H), 1.06-
1.36 (m, 2H))
1.73-2.13 (m, 2H)) 2.32-2.46 (m, 1 H), 2.70-2.91 (m, 1 H), 2.98-3.08 (m, 1 H
)) 4.65-4.85 (m,
2H), 5.18-5.54 (m, 2H)) 6.54 ( I H), 6.85 ( 1 H), 7.09-7.40 (m, SH), 7.75 ( 1
H), 8.34-8.54 ( 1 H ))
8.70 (s, I H). MS (DCI/NH3) m/z 426 (M+H)+.
Example 24
O ~ O
H
H2N N /
O Ph S
Exams lei 24A
H O
BOCN /
1
S
Ph
24a
To a -78 °C solution under nitrogen of BOC-L- phenylalanine methyl
ester (2.0 g, 7.16
mmol) in dry THF (80 ml) was added 2-thienyllithium ( 17.9 ml, 17.9 mmoI) and
the reaction
mixture stirred for 1 hour. The reaction mixture was poured onto water,
extracted with CH2C12
(3x), dried over Na2S04, filtered and concentrated in vacuo to give an orange
oil. Purification by
chromatography on silica gel (0.5°lo acetone-CH2C12) gave 24a (882 mg)
as a yellow solid.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
59
Ex~, lp a 24B
H O O
BOCN / H O
HOHN N /
Ph S O S
Ph
The desired compound was prepared according to the method of Examples 21 B-E
except
substituting ~ for ~. 1 H NMR(300 MHz, DMSO-d6) 8 0.54-0.87 (m, 7H), 0.95-1.35
(m)
2H ),1.68-2.11 (m, 2.SH), 2.32-2.47 (m, O.SH), 2.83-3.15 (m, 2H)) 4.63-4.85
(m, 2H), 5.29-
5.52 (m) 2H)) 7.11-7.40 (m, 6H), 8.02-8.20 (2H), 8.58-8.75 (1H), 8.73 (s, 1H).
MS
(DCI/NH3)) m/z 443 (M+H)+.
Example 25
O ~ O
HOHN N N
O p
Ph
Example 25A
H OH
BOCN
Ph O
~Sa
To a -70 °C solution under nitrogen of oxazole (3.36 g, 48.8 mmol) in
THF (80 ml) at was
added n-BuLi (30.5 ml, 48.8 mmol) and the mixture was stirred at -7U °C
for 20 minutes. A
solution of N-BOC-phenylalaninal (4.86 g, 19.5 mmol) in THF (20 ml) was then
added and the
mixture was stirred at -50- -70 °C for 6 hours. The reaction was
quenched with H20, extracted
with CH2CI2 (3x), dried over Na2S04, filtered and concentrated in vacuo.
Chromatography on
silica gel (40% ethyl acetate-hexanes) provided ~5 ( 1.12 g).
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
Example 25B
H OH H O
BOCN N BOCN
o~ \o~
Ph Ph
To a 0 °C solution of ~ (969 mg, 3.05 mmol) in CH2C12 (60m1) was added
KBr/H20
5 (36.3 mg, 613p1) and 2,2,6,6-tetramethyl-1-piperidinyloxy, free radical
(4.76 mg, 0.0305 mmol).
In another vial, NaOCI solution (10.9 ml) was adjusted to pH 8 with saturated
aqueous NaHC03
solution, and the resulting solution was added to the 25a solution and the
reaction mixture was
stirred at 0 °C for S hours. The reaction mixture was poured into H20-
brine, extracted with
CH2C12 (3x), dried over Na2S04) filtered and concentrated in vacuo.
Chromatography on silica
10 gel (30%-40% ethyl acetate-hexanes) gave 25b (731 mg).
Example 25C
H O O H O
BOCN N
HOHN N
Ph O Ph O
25b
15 The desired compound was prepared according to the method of Examples 21 B-
E except
substituting 25b for 21 a. 1 H NMR (300 MHz, DMSO-d6) 8 0.54-0.90 (m) 7H)) I
.14-1.39 (m)
3H ), 1.75-2.06 (m, 2H)) 2.71-2.82 (m, 1H), 3.11-3.22 (m, 1H), 4.64-4.91 (m,
2H)) 5.29-5.42
(m) 2H)) 7.12-7.41 (m, SH), 8.53 (d, 1 H, J = 7.5 Hz), 8.60-8.65 ( 1 H), 8.74
(s, 1 H). 9.02-9.08
(1H)) 10.36-10.48 (1H). MS (DCI/NH3) m/z 428 (M+H)+.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
61
Examlhe 26
O H O
HOHN N
O Ph S
Example 26A
H O
BOCN
S
Ph
26a
To a -78 °C solution under nitrogen of thiazole ( 1.1 ml) 15.8 mmol) in
THF (8() ml) was
added n-butyl lithium (1.6M in hexanes, 9.88 ml) 15.8 mmol) and the reaction
mixture was stirred
for 0.5 hours. A solution of BOC-L-phenylalanine methyl ester (2.0g) 7.17
mmol) in THF (5 ml)
was added and the mixture was stirred at -78 °C for 30 minutes. The
reaction mixture was poured
into water) extracted with CH2C12 (3x)) dried over Na2S04) filtered) and
concentrated in vacuo.
Chromatography on silica gel (20% ethyl acetate-hexanes) gave 26a ( 1.95g) as
a yellow solid.
Example 26B
BOCN O N HO' O H O
N N N/
Ph S H O S
~ ~ Ph
The desired compound was prepared according to the method of Examples 2l B-E
except
substituting 2~(a for 21a. 1H NMR (300 MHz) DMSO-d6) 8 0.55-0.92 (m, 7H)) 1.18-
1.40 (m,
2H),1.80-2.29 (m) 3H), 2.75-2.90 (m, 1H), 4.66-4.95 (m) 2H), 5.30-5.76 (m,
2H)) 7.13-7.38
(m, SH)) 8.22-8.30 (2H)) 8.58-8.75 (2H). MS (DCI/NH3) m/z 444 (M+H)+.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
62
Example 27
O H O _
HOHN~N
O H IOI
\ /
Example 27A
I' o
H~OH O
O' ~ ~ O
OH O
Malic acid (53.2 g, .397 mol) was dissolved in 400 mL of HCI saturated 2-
propanol and
the solution was heated at reflux for 22 hours. The solution was reduced in
volume by rotary
evaporation, diluted with EtOAc ( I L), and extracted twice with saturated
aqueous Na2C03. The
1 () organics were dried over MgS04 and concentrated to give diiso-propyl
malate 27a (67g).
Example 27B
i
- o'/
~o~° °
OH O OH TO
I S Diiso-propyl malate 27a, 25.6 g) 117 mmol) was added slowly to a 1 M
solution of LDA
in THF (235 mL) 235 mmol) at -78 °C. The solution was allowed to slowly
warm to -50 °C over 2
hours, and then was recooled to -78 °C. Cinnamyl bromide (25.Ug) 127
mmol ) in THF (SU mL)
was added dropwise, and the solution was stirred at -78 °C for 15
hours. The dry ice bath was
removed and the reaction was quenched with 1 M HCI. The solution was diluted
with ether and
20 extracted twice with 1 M HCI. After drying (Na2S04) and solvent removal,
the crude material was
chromatographed on silica gel ( I S°lo ether-hexanes) to give 27b as a
10:1 mixture of diastereomers
(9.5 g).
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
63
Exam 1e~27r
o _
OH O
A mixture of 27~ (9.29g) and 10% Pd/C (0.45 g) and were placed in a Parr
shaker
S containing 150 mL methanol, and exposed to 4 atm pressure of hydrogen for 18
hours. Filtration
and solvent removal provided 27c as a yellow liquid (9.39 g).
Example 27D
i
I
0 0
O _ O OH
HO
OH O ~ OH O
A solution of 27c (9.39 g) 27.9 mmol) in 20 mL dioxane was treated with 3 M
KOH (30
mL) and stirred overnight at 90 °C. The solution was poured over ice
and acidified to pH 3 with
concentrated HCI. The reaction mixture was extracted twice with EtOAc, and the
organics were
dried over MgS04. Solvent removal gave 27~ as a yellow liquid.
Example 27E
H O
To a solution of 27~c (7.U g, 28 mmol) DMF (50 mL) and 2,2-dimethoxypropane (
I 80 mL)
was added Dowex-50 resin and the mixture was stirred at ambient temp for 3
days. The resin was
filtered off and the solution concentrated to give a DMF solution of 27e which
was used as is in the
next step.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/OOI42
64
Example 27F
0 0
\ 7rZg ~ \ ?Zf
Crude acid 27e (theoretical yield 28 mmol) with residual DMF was diluted with
CH2C12
( 1 I 0 mL) and cooled to 0 °C. Pentafluorophenol (8.33 g, 45 mmol) was
added, followed by
EDCI (6.49 g) 33.8 mmol). The mixture was stirred at 0 °C for 2.5
hours, then extracted
succesively with saturated aqueous Na2C03 and brine. The organic phase was
reduced in volume
in vacuo, diluted with ethyl acetate, washed three times with brine, dried
over Na2S04) filtered,
and concentrated in vacuo. Chromatography on silica gel (gradient elution 10-
15-20% ether-
I U hexanes) gave ~ (8.88 g) as a 7:4 mixture of diastereomers.
Exam,~]e 27G
O O H O _
O OPFP HOHN ~ I
~O O O H O
/ \\
The desired compound was prepared according to the method of Example 1 (),
except
substituting 27f for 10a. mp 111- I I 3 °C. ~ H NMR (30() MHz, DMSO-d6)
8 1.23 ( m, 3H ). 1.37
(m, 1H). 2.25 (m) 1H)) 2.38 (m) 1H), 2.62 (m) IH), 2.90 (dd) 1H, J = 6.6)
14.() Hz), 3.21 (dd,
I H, J =7.3) I 3.6 Hz)) 3.84 (dd, 1 H) J = 6.6, 8.5 Hz)) 5.24 (d, 1 H, J = 6.6
Hz), 5.44 (q, 1 H, J
= 8.1 Hz), 6.77 (dd, 2H, J = 1.5, 8.2 Hz), 6.96 (m, 3H), 7.15 (m) 3H), 7.22
(m, 6H), 7.47 (dd)
2l ) 1 H ) J = 1.9, 5.9 H z), 8.19 (dd) I H, J = 2.2, 5.9 H z), 8.32 (d, I H,
J = 3.0 H z), 8.52 (d, 1 H, J =
8.4 Hz), 8.83 (bds, 1 H), 10.63 (bds, 1 H), 11.94 (d, I H, J = 2.6 Hz). 13C
NMR (CD30D) 8
30.03, 30.11, 36.55, 39.80, 50.99, 57.05, 73.04, 112.98, 1 I 6.67, 122.89,
123.37, 124.49,
126.50, 127.15, 127.50) 129.12, 129.20, 129.28, 130.45, 135.53, 138.43,
138.69, 143.14,
171.52) 175.07, 194.40. MS (APCI) m/e 514 (M+H)+, 453. Anal calcd for C3pH31
N3O5~0.8
H20: C, 68.24; H, 6.22; N, 7.96. Found: C, 68.40; H, 6.27; N, 7.56. [a]d = -
129 (CH30H, c =
0.02).
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
Ex~l~8_
o H o _
HOHN~N
O H OII ~ N\
H
The desired compound was isolated in the purification of the compound of
Example 27.
5 mp 116-118 °C. 1 H NMR (300 MHz, DMSO-d6) 8 1.18 (m, 2H), 1.28 (m, 1
H)) 1.53 (m, 1 H ),
2.0 I (m, 2H), 2.70 (m, 1 H)) 2.89 (dd, I H, J = 4. I , 13.3 Hz), 3.12 (dd, I
H, J = 4.8, I 3.3 Hz),
3.97 (t, 1 H, J = 4.9 Hz)) 5.21 (d, 1 H, J = 5.2 Hz), 5.38 (m, I H), 7.04 (d)
2H, J = 7.0 Hz). 7.1-
7.3 (m, IOH)) 7.47 (m, 1H)) 8.20 (dd, 1H, J =2.3, 5.6 Hz)) 8.41 (d) 1H) J =
2.9 Hz), 8.51 (d.
1 H) J = 8.5 Hz)) 8.63 (bds) 1 H), 10.37 (bds, I H), 1 I .97 {bds, I H). MS
(APCI) m/e 514
10 (M+H)+) 453 . Anal calcd for C3pH3~N305~U.5 H20: C, 68.95; H) 6.17; N,
8.(14. Found: C,
69.11; H, 6.17; N, 7.92. [aJd = -22.3 (CH~OH) c = 0.01 ).
Example 29
O H O
HOHN N
O N
Ph
Exam 1"~ 2yA
OH OH H NHp
BocHN~ BocHN ~ N
COOH ---
~ Ph ~ Ph
29b
To a solution of 29a (0.32g, I.08 mmol) in DMF (6.0 mL) was added EDC (0.23 g,
1.I9
mmol), HOBT (0.16 g, 1.19 mmol), NMM (0.13 mL, 1,19 mmol) and 1,2-
phenethyldiamine
(0.12 g) 1.13 mmoI) and the reaction mixture was stirred for 6 hours. the
reaction mixture was
partitioned between ethyl acetate and brine. The aqueous layer was separated
and extracted twice
with ethyl acetate. The ethyl acetate extracts were combined, washed with
brine, dried over
CA 02277105 1999-07-06
WO 98130541 PCT/US98/00142
66
MgS04, filtered and evaporated to dryness. The crude 29b was used for next
reaction without
purification.
Exam In a 29B
OH NH2 OH
BocHN ~ ~ BocHN ~N
_ O - N
~ Ph ~ Ph
A mixture of ~ø and camphorsulfonic acid (12 mg, mmol) in toluene (20 mL) was
heated
at 80 °C for 4 hours. The reaction mixture was evaporated to a small
volume and partitioned
between CH2C12, brine and saturated aqueous NaHC03. The aqueous layer was
extracted with
CH2C12. The CH2C12 layers were combined) dried (MgS04), filtered and
evaporated to dryness.
Flash chromatography (40%-80% ethyl acetate-hexanes) gave (~ (261 mg) as white
crystals.
Exam 1e~29r.
OH
BacHN ~ N . OH H
H2N
- N_~(~ '_' ~(N
~ Ph
~ Ph
A mixture of 29c (255 mg) 0.69 mmol ) and trifuloroacetic acid ( 1.5 mL) was
stirred for 30
minutes, and then was evaporated to dryness. Residual trifluoroacetic was
removed by azeotropic
evaporation with toluene to give 29d as brownish crystals which was used
without further
purification.
Example 29D
OH H
H2N~~ O ~ OH
'' '(~ ~ ~ t-Bu0
N~ O t
Ph N
Ph
To a solution of succinate ester 2 (U.828 mmol) in DMF (2 mL) was added EDC (
159 mg,
0.828 mmol), HOBT( 112 mg, 0.828 mmol) and NMM (0.19 mL, 1.73 mmol). The
reaction
mixture was stirred at room temperature for 10 minutes and a solution of ~
(0.69 mmol) in DMF
(2 mL) was added. The reaction mixture was stirred for 10 hours and then was
partitioned
CA 02277105 1999-07-06
WO 98/30541 PCT/US98100I42
67
between ethyl acetate and brine. The aqueous layer was separated and extracted
twice with ethyl
acetate. The combined ethyl acetate extracts were washed with brine) dried
(MgS04), filtered and
evaporated to dryness. Chromatography on silica gel (20-40 % ethyl acetate-
hexanes) gave 29e
( 156 mg) as yellow crystals.
S
Example 29E
O ~ OH ~ O
t-Bu0 , \ "'.~' t_gu0 - ~ o N
O ~ ~ O
N N
Ph ~ Ph
The desired compound was prepared according to the method of Example 25B,
except
substituting ,fig for ~.
Fxamnle 29F
0 0
t-Buo O ~ ~ ~ O
---~ HOHN -
O ' N O
~ Ph ~ Ph
The desired compound was prepared according to the method of Examples 21 D and
E,
except substituting 29f for 21c. Mixture of two stereoisomers: mp: 159.5-161.0
°C (dec). iH
NMR (3(?0 MHz, DMSO-d6) 8 0.58 (d, 3H, J = 5.6 Hz)) 0.65 (d, 3H, J = 5.6 Hz))
0.72 (d, 3H,
J = 6.2 Hz), 0.87 (d, 3H, J = 6.2 Hz), U.79-U.89 (m) I H), 0.70-0.89 (m, 1 H),
1.20-1.30 (m)
2H ), 1.3U-1.42 (m, 2H), 1.83-2.17 (m. 3H)) 1.83-2.17 (m) 3H)) 2.78-2.87 (m) I
H)) 2.78-2.87
2() (m) 1 H)) 3.4U (m) 2H), 3.45 (m, 2H), 4.7U (dd, 1 H, J = 16.5, 1.6 Hz))
4.81 (dd) I H, J = 1 U)
1.6 Hz)) 4.88 (d) I H, J = 10.5 Hz), 4.89 (d, I H, J = I 5.8 Hz)) 5.38 (m) I
H)) 5.55 (m) I H),
5.79 (m) IH)) 5.84 (m, IH), 7.21 (m, 2H), 7.26-7.32 (m) SH)) 7.41 (m, 2H))
7.36-7.43 (m,
SH), 7.58 (d, 2H) J = 8.4 Hz), 7.89 (d, 2H, J = 8.7 Hz), 8.61 (d, 1H) J = 8.4
Hz)) 8.70 (s, IH,
J = 8.2 Hz), 8.72 (s, IH), 8.72 (s, 1H), 10.40 (d, 1H, J = 1.2 Hz), 10.42 (d,
IH, J = 1.2 Hz))
13.52 (s, 1H), 13.52 (s, 1H). MS (DCl-NH3) m/e 477 (M+H)+, 433. Anal calcd for
C27H32N404: C, 66.78; H, 6.84; N, 11.53. Found: C, 66.6; H, 6.69; N, I 1.25.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
68
Example 30
~,O
H
H~N N
OH O
I
Example 30A
I
H i H N~ i
N \ ' t-Bu0 N I
t-Bu0 ~
O L--N O ~N
I ~ ,H ~ I ~ ,H
'
To a solution of ~ (2.45 g, S.15 x 10-3 mol)) prepared by coupling of 2c andR-
2-(i-
butyl)-succinic acid-4-t-butyl ester according to the method of Example 2C) in
pyridine (50 mL)
was added O-methylhydroxylamine hydrochloride {0.80 g, 1.00 x 10-2 mol) in one
portion and
1 U the mixture was heated at 80 °C for 3 days. The pyridine was
removed in vacuo. The residue was
taken up in CH2C12 and the solvent was removed in vacuo. Flash chromatography
(CH2C12)
gave ~(_)b as a mixture of oxime diastereomers.
Example 30B
(
H ~~O i H ~p i
t-Bu0 N \ ' HOHN N \
1 T
O N O N
I \ H I \ H
' ''
The desired compound was prepared according to the method of Examples SA and
B,
except substituting 30b for the compound of Example 4. 1 H NMR (300 MHz, DMSO-
db) 8
11.43 (s, 1 H), 10.37 (s, 1 H), 8.73 (s, 1 H), 8.36 (d, 1 H, J = 8.1 Hz)) 8.08
(d, 1 H, J = 7.7 Hz),
7.92 (d, 1H, J = 2.6 Hz), 7.41-6.98 (m, 8H}, 5.60-5.49 (m, 1H), 3.90 (s, 3H))
3.20-2.98 (m,
2H), 2.81-2.69 (m, 1 H), 1.87-1.77 (m, 2H), 1.29-1.17 (m, 1 H), 1.02-0.74 (m,
2H), 0.58-0.49
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
69
(m, 6H). MS (DCI-NH3) m/e 465 (M+H)+. Anal calcd for C26H32N404~U.25 H20: C)
66.57;
H, 6.98; N, 11.94. Found: C, 66.72; H, 7.11; N, 11.85.
Examl 1~ a 31
O H O i
HOHN
O ø ~ N
\C02H \H
Example 31 A
H O
HN~ H
d
1
O8n O
OBn H
O
To a suspension in THF (25 mL) of OBn-Asp ( 1 g, 4.48 mmol) and activated
charcoal (25
mg) was added diphospgene (0.416 mL) 3.45 mmol) via syringe at ambient
temperature and the
reaction mixture was heated at 55 °C for 1.5 hours. The solution was
then filtered through celite)
the filter cake was washed with EtOAc, and the solvent was removed in vacuo.
The resulting solid
I S was recrysnrllized (EtOAc-Hexane) to give the desired product in
55°lo yield.
Example 31 B
0
O O H O i
,,,~ HO
;' -O
OBn H O
OBn N
31a ~ ~ H
0
-2U The desired compound was prepared according to the method of Examples 2A
and B,
except substituting ~ for ~ and substituting for R-2-(i-butyl)-succinic acid-4-
t-butyl
estersuccinate ester 4.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
Exam lp a 31 C
O H O ~ H
I
HO ~ 1 ~ HOH
O ~ OBn ~ O
H \C02H ~H
The desired compound was prepared according to the method of Example 5, except
5 substituting ~1 for the compound of Example 4.
Example 32
O H O
N
HO
O
HO
Example 32A
0
N~O
'O H
Cyclohexylacetic acid (25 g, 0.176 mol ) was dissolved in 5() mL thionyl
chloride, and the
solution was heated at reflux for 1 hour. The solution was concentrated in
vacuo and placed under
vacuum for 1 hour. The acid chloride was then added to a -78 °C
solution in THF (450 mL) of 1-
lithio 2-(S)-benzyloxazolidinone (0.158 mol). After 10 minutes, the dry ice
bath was removed,
and after a further 30 minutes) the mixture was quenched with aqueous NH4CI
solution. The
solution was extracted with 1 M NaOH and washed with pH 7 buffer solution. The
organic phase
was dried over MgS04) filtered and concentrated in vacuo to give 2a as a white
solid (42 g)
which was used without further purification.
CA 02277105 1999-07-06
WO 98/30541 PCT/IJS98/00142
71
Exam~he 32B
0
N~O
t-Bu0
O
To a -78 °C solution in THF (420 mL) of acyl oxazolidinone ~ (42 g, 140
mmol) was
added sodium hexamethyldisilazide (140 mL of a 1 M soultion in THF, 140 mmol)
dropwise over
40 minutes. After 30 minutes, a solution often-butyl bromoacetate (23 mL, 156
mmol) in 70 mL
THF was added dropwise over 30 minutes. One hour after the addition was begun,
the dry ice
bath was removed and replaced with an ice bath. After 2 hours at 0 °C)
the reaction was quenched
with aquesou NHqCI. The solution was concentrated, diluted with EtOAc and
extracted twice with
aqueous NH4Cl. After drying (Na2S04) and solvent removal, the crude material
was
rec,-rystallized from 3:I hexanes-EtOAc to give ~2b (31.8 g) as white needles)
mp 141-142 °C.
Flash chromatography of the mother liquors provided a further 2.60 g product.
Example 32
0
0
U
f-Bu0 N O O
OH
O t-Bu0
O
To a 0 °C solution of acyloxazolidinone ~ (34.4 g, 83 mmol) in 360 mL
THF was added
30 mL water and 33 ml 3U~lo hydrogen peroxide) followed by a solution of LiOH
(5.28 g) 126
mmol) in 120 mL water. After 6.5 hours) the peroxides were quenched with
NaHS03 (300
2() mmol), then KOH (300 mmol) was added. The solution volume was reduced in
vacuo and the pH
adjusted to 9 with 50% aqueous NaOH. The solution was extracted twice with
methylene
chloride, then acidified to pH3 with concnetrated HCI. The aqueous phase was
extracted with
ethyl acetate. The organic phase was dried over Na2S04) filtered and
concentrated in vacuo to give
32c as a slightly yellow oil ( 10.4 g).
CA 02277105 1999-07-06
WO 98130541 PCT/US98100142
72
Exa~r le 32D
O H O
O t-Bu0 N ~ \
t-Bu0 O H O /
O
HO
The desired compound was prepared according to the method of Example 19C,
except
substituting 32c for succinate ester 3.
Example 32E
O H O
N O H O
t-Bu0 \ H O N \
O ~ / O
HO
HO
1 () A solution of ~ ( 1.37 g, 2.86 mmol) in 30 mL HCl saturated acetic acid
was stirred for 4
hours at ambient temperature. The reaction mixture was concentrated in vacuo
and the residue was
azeotroped twice with toluene. Vacuum drying provided the desired compound as
a white foam.
tH NMR (300 MHz, CD30D) 8 0.87 (m) 2H)) 1.08 (m) 2H), 1.42 (m) 3H)) 1.60 (m.
4H), 2.5
(m) 3H), 2.87 (dd, 1 H, J = 6.7, 13.9 Hz), 3.07 (dd, 1 H, J = 6.8) 13.9 Hz),
5.62 (t) 1 H, J = 6.8
Hz), 6.61 (d. 2H, J = 8.4 Hz), 6.98 (d, 2H) J = 8.5 Hz)) 7.42 (t) 2H) J = 7.4
Hz), 7.55 (m, 1 H ))
7.89 (d) 2H, J = 7.2 Hz) . t3C NMR (CD~OD) 8 27.40, 31.29, 31.66, 34.72)
37.81, 41.63)
49.11, 56.37) 116.15, 129.07, 129.61, 129.70, 131.45, 134.45) 137.18, 157.08,
176.06,
176.35, 200.32. MS (DCI/NH3) m/e 424 (M+H)+) 195.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
73
Exa~33
. O H O
N
HOHN
O
HO
The desired compound was prepared according to the method of Example 5, except
substituting the compound of Example 33 for the compound of Example 4. mp 144-
145 °C. ~ H
NMR (300 MHz, DMSO-d6) 8 0.7-1.0 (bdm, SH), 1.2-1.6 (bdm, 6H), 2.01 (m, 2H),
2.58 (m)
I H), 2.76 (dd) 1 H, J = 7.4, 13.9 Hz), 2.97 (dd) 1 H, J = 6.8, 13.9 Hz), 5.39
(q) 1 H, J = 7.1
Hz)) 6.59 (d) 2H) J = 8.5 Hz)) 7.01 (d, 2H, J = 8.4 Hz), 7.46 (t) 2H, J = 7.8
Hz), 7.62 (m) 1 H ),
7.88 (d, 2H) J = 7.1 Hz), 8.65 (s, IH)) 9.14 (s, 1H), 10.32 (s) 1H). MS
(DCI/NH3) m/e 456
(M+NH4)+, 439 (M+H)+. Anal calcd for C~SH3~N205~.75 HBO: C) 66.43; H, 7.02; N,
6.20.
Found: C) 66.53; H, 6.79; N) 6.22. [a]d = -12° (CH~OH, c = .013
g/mL).
Example 34
o l ~ o
H O
O
IS o
Example 'i4A
o
HCI~H
i
OH
The desired compound was prepared by adding 4-bromo-tert-butylbenzene to a 0
°C
solution of nBuLi in diethyl ether . The resulting 4-tert-butylphenyllithium
solution was added to a
-78 °C solution of N-BOC-tBu(OH) tyrosine in diethyl ether. The
solution was stirred at -78 °C
for 30 minutes, warmed to 0° over 1 hour and quenched with an aqueous
solution of NH4C1. The
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
74
aqueous layer was extracted twice with diether ether and the combined organics
were washed with
brine, dried (Na2S04), filtered and concentrated in vacuo. Flash
chromatography gave the BOC-
protected compound which was immediately taken up in 4N HCl-dioxane and
stirred for 30
minutes. The resulting slurry was diluted with diethyl ether, filtered and
dried for 16 hours under
high vacuum, to give ~.
Exam lp a 34B
O O H O
HCI'H2N
HO N \
o I
off
HO
The desired compound was prepared according to the method of Examples 19C and
32E,
except substituting 34a for 19b, and substituting succinate ester 1 for
succinate ester 3. 1H NMR
(300 MHz) CD~OD) 8 0.67 (d) 3H, J = 6.5 Hz), 0.75 (d, 3H, J = 6.4 Hz), 0.95
(m, 3H), 1.16
(m) 1H), 1.35 (s, 9H), 1.52 (m, IH), 1.82 (m. IH)) 1.94 (m) IH)) 2.34 (m, 1H),
2.46 (m, 1H),
2.8 I (dd, 1 H, J = 6.2, 14.2 Hz)) 3. I 1 (dd, 1 H) J = 4.7, 14.2 Hz)) 4.89
(bds) 1 H), 4.94 (m,
1 H), 5.59 (m) 1 H), 5.73 (m, 1 H)) 6.68 (d) 2H, J = 8.5 Hz), 7.1 I (d, 2H, J
= 8.4 Hz), 7.55 (d,
2H, J = 6.7 Hz), 7.97 (d) 2H, J = 6.8 Hz), 8.59 (d) 1 H, J = 5.6 Hz). MS
(DCl/NH3) m/e 508
( M+H )+.
2O Example 35
O H O
HOHN N I \
O
HO
The desired compound was prepared according to the method of Example 1 F,
except
substuting the compound of Example 34 for I e. ~ H NMR (300 MHz) CD30D) 8 0.67
(d, 3H) J =
6.4 Hz), 0.74 (d, 3H, J = 6.4 Hz), 0.86 (m, 2H)) 0.95 (m, 1H), 1.14 (m) 1H),
1.34 (s, 9H),
1.42 (m, 1 H), 1.77 (m) 1 H), 2.03 (m) 1 H), 2.43 (m) I H), 2.83 (dd, 1 H, J =
9.8, 13.9 Hz), 3.10
(dd) 1H, J = 4.4, 13.9 Hz), 4.80 (m, 1H), 4.92 (m, 1H), 5.53 (m, 1H)) 5.77 (m,
1H), 6.69 (d,
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
2H, J = 7.6 Hz), 7.13 (d, 2H, J = 8.5 Hz), 7.54 (d, 2H, J = 8.5 Hz), 7.97 (d,
2H) J = 7.6 Hz).
13C NMR (CD30D) 8 21.46, 24.45, 26.49, 30.55, 31.45, 32.29, 36.00) 37.37,
41.63, 47.08,
49.84) 56.31, 115.63, 116.28, 126.74) 129.24, 129.70) 131.31, 134.12, 138.71,
157.17,
158.54, 172.87, 176.02, 199.57. MS (DCI/NH3) m/e 523 (M+H)+. Anal calcd for
5 C3~H42N205~0.5 H20: C, 70.03; H, 8.15; N, 5.27. Found: C, 69.92; H) 8.15; N,
5.21.
Example 36
0 0
HOHN
0 0
Example 36A
O
O
O
H O~~O
p ~ O
A suspension of succinic anhydride (4 g) 40 mmol )) allyl alcohol (2.7 mL) 40
mmol ) and
1 S DMAP (5.9 g) 48 mmol) in 200 mL toluene was refluxed for 4 hours and then
cooled to ambient
temperature and concentrated. The residue waspartitioned between EtOAc and
brine. The aqueow
layer was extracted with EtOAc and then acidified to pH 2 with 6M HCI. The
acidic aqueous
phase was extracted with EtOAc (3x) to give an organic layer which was washed
with brine (2x))
dried (MgS04)) filtered and concentrated to afford ~ (5.96 g) as a clear
liquid which was used
2U without further purification.
Example ~6B
O O
H O~~ O ~ O
O 2 O
25 To a solution of carboxylic acid 3~a (3 g, 19 mmoI) in 95 mL CHZC12 was
added EDC
( 1.8 g, 9.5 mmol). The reaction mixture was stirred for 3 hours and then
poured into a separatory
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
76
funnel containing 20 mL of ice water. The organic layer was washed with ice-
cold water,
saturated aqueous NaHC03 and brine) dried with MgS04, filtered and
concentrated in vacuo to
afford 36b (2.8 g) which was used without purification.
Example 36C
° 0 0
"ZN~ a I'
c.B~o °"+ ° ~ ~ t.e~o
o _ ~ o
\ / HCt -
\ /
To a solution of R-2-(i-butyl)-succinic acid-4-t-butyl ester ( 1 g) 4.35
mmol), O-
benzylphenylalanine hydrochloride salt ( 1.52 g, 5.22 mmol, Aldrich), HOBT
(704 mg, 5.22
1 () mmol) and NMM ( 1.4 mL) 13 mmol) in 22 mL DMF at 0 °C was added
EDC ( 1 g, 5.22 mmol) in
a single portion. The resulting solution was allowed to slowly warm to ambient
temperature and
then was stirred for 3 days. The reaction mixture was partitioned between
water and ethyl acetate.
The aqueous layer was extracted with EtOAc (3x) and the combined organic
layers were washed
with I M NaHS04) I M NaHC03 and brine, dried with MgS04) filtered and
concentrated in vacuo.
15 The residue was purified by flash clu-omatography (CH2Cl2 then 2% MeOH-
CH~C12) to give 36c
( 1.96 g) as a yellow oil.
Example 36D
O o
O ~ O
t.e~o a.~o ~ t_e~o
0
Hydrogenation of benzyl ester ~r ( 1.96 g, 4.2 mmol; 20U mg 10% Pd/C;
methanol; 1 atm
hydrogen) gave 36d (1.57 g) as a thick oil which was used without further
purification.
CA 02277105 1999-07-06
WO 98/30541 PCTlUS98/00142
77
Example ~~F
O O~ O H O
t-Bu0 v 'O H 2 O t-Bu0 N O ~
O O ~ O \
\ / ~ / 36e
To neat anhydride ~Cb. (2.7 g, 9 mmol) was added a solution of carboxylic acid
,3,~d ( 1.57
g, 4.16 mmol) in 10 mL CH2CI2, Et3N (864 uL, 6.24 mmol) and DMAP (21 mg, 9
mmol). The
resulting yellow solution was refluxed in an oil bath at 50 °C for 3
hours, cooled, concentrated and
then stirred in the presence of 50 mL 5% NaHC03 for 30 minutes. This mixture
was diluted with
EtOAc and the separated aqueous layer was extracted with EtOAc. The combined
organic layers
were washed with 1 M NaHS04 and brine, dried with MgS04, filtered and
concentrated. The
residue was purified by flash chromatographed (IS% ethyl acetate-hexane then
35% ethyl acetate
hexane) to give 36e (952 mg) as a yellow foam.
Example 36E
0 0 0 0
t-Bu0 ~ O~ ----~. HOHN ~ O
O O O O
/ \ /
The desired compound was prepared according to the method of Examples 1 E and
F)
except substituting 36e for I d. mp 126- I 29 °C. I H NMR (300 MHz)
DMSO-d6) 8 0.6-1.0 (m)
8H ), 1.1-1.4 (m) 2H )) 1.8-2.2 (m, 2H)) 2.6-3.2 (m, 6H)) 4.2-4.6 (m, 3H)) 5.2-
5.4 (m, 2H))
5.8-6.0 (m) IH), 7.1-7.3 (m) SH)) 8.4-8.6 (m) 1H)) 8.45-8.55 (m) IH)) 8.70 and
8.73 (two s,
IH), 10.36 and 10.40 (two s, IH). MS (DCI/NH3) m/e 433 (M+H)+, 450 (M+NH4)+.
CA 02277105 1999-07-06
WO 98130541 PCT/US98/00142
78
Exam 1R a 37
0 0
HOHN ~ OH
O O
To a solution of the compound of Example 36 (329 mg, 0.76 mmol) in 6 mL THF
was
S added palladium(0) bis(dibenzylideneacetone) (44 mg, 0.08 mmol),
triphenylphosphine (42 mg)
0.16 mmol) and morpholine (662 uL, 7.6 mmol). The resulting clear) yellow
solution was stirred
for I hour and then concentrated. The residue was partitioned between CHZCl2
and HBO and the
separated aqueous layer was extracted with CH2C12 (2x). The aqueous layer was
concentrated,
redissolved in H20, filtered and the desired compound ( 135 mg) was isolated
by reverse-phase
HPLC (0-30% CH3CN/H20; 2 I mm Dynamax 60A C 18 column; 12 mL/minutes). mp 120-
121
°C. 1H NMR (300 MHz, DMSO-d6) b 0.60 (d, 3H, J = 5.4 Hz), 0.67 (d, 3H,
J = 5.7 Hz), 0.8-
1.0 (m, 2H), 1.1-1.3 (m, 1H), 1.91 (dd) 1H) J = 14.4,7.8 Hz)) 2.10 (dd) IH) J
= 14.4, 6.6 Hz),
2.36 (t) 2H, J = 6.3 Hz)) 2.6-2.9 (m, SH), 2.70 (t) 4H, J = 4.8 Hz)) 3.05 (dd,
1 H, J = 14.1, 4.2
Hz)) 3.52 (t, 4H, J = 4.8 Hz)) 4.4-4.5 (m) IH)) 7.1-7.3 (m) SH), 8.47 (d, 1H,
J = 8.4 Hz). MS
(DCI/NH3) m/e 393 (M+H)+. Anal calcd for C2pH2gN20~~1.0 H20: C, 57.93; H,
7.90; N, 8.44.
Found: C, 57.91; H) 7.55; N, 8.76. [oc] _ +62 ° (c 0.3, MeOH).
Example 38
o H O
HOHN N
O
HO
The desired compound was prepared according to the method of Example 6
substituting
succinate ester 4 for 5 and ketone 19b for 2c. ~ H NMR (300MHz, DMSO-d6) d 9.
I 8 (s, 1 H),
8.76 (s, 1H), 8.5-8.4 (m, 1H), 7.92-7.88 (d, 2H) J=7.1 Hz), 7.58-7.57 (m, IH),
7.48-7.45 (m,
IH)) 7.03-6.98 (m, 3H), 6.85-6.82 (d, 2H, J=7.4 Hz), 6.61-6.58 (d, 2H, J=8.2
Hz), 5.45-5.40
(m, I H), 3.10-2.87 (m, 2H), 2.8-2.6 (m) 3H), 2.23 (s, 3H), I .88-. I 87 (m, I
H), 1.2-1.15 (m,
2H).
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
79
MS (ESI) m/e 487 (M-H)-. Anal. Calcd for: C29H32N2Og~0.75H20: C, 69.37; H,
6.72; N,
5.57. Found: C, 69.37; H, 6.74; N, 5.88.
Jrxample 39
H
HOHN - N I \
OH 0 ~ NH
The desired compound was prepared according to the method of Examples 10A, l
OB and 5
except substituting 27f for l0a and 9a for lc. 1H NMR (300MHz, DMSO-d6) d
11.98 (s, 1H),
I 0.63 (s, I H), 8.83 (s) I H), 8.41 (s, 1 H), 8.27-8.2 (m, 1 H), 8.01-7.98
(d) I H) J=9.5 Hz))
7.50-7.48 (m, 1 H), 7.27-7.18 (m) 2H)) 6.98-6.85 (m, 3H), 6.76-6.74 (d, 2H,
J=8.4 Hz)) 5.27-
5.22 (d, 1 H, J=8.4 Hz), 5. I 8-5.1 S (d, 1 H, J=9.6 Hz)) 3.82-3.77 (t, 1 H>
J=9.5, 7.7 Hz)) 2.80-
2.75 (m, 1 H)) 2.40-2.33 (m) I H)) 2.25-2.21 (m, 1 H), 1.41-1.30 (m, I H), I
.25-1.2U (m, 3H))
1.00 (s) 9H). MS (ESI) m/e 480 (M+H)+) 478 (M-H)-. Anal. Caicd for:
C27H33N3O4~O.SOH20: C) 66.37; H. 7.01; N) 8.68. Found: C) 66.39; H) 6.96; N)
8.45.
Example 4p
H
HOHN N
O
The desired compound was prepared according to the method of Example 6
substituting
succinate ester _4 for ~ and ketone ~ for ~. tH NMR (300MHz, DMSO-d6) d 10.31
(s, 1H),
,20 8.61 (m, IH), 8.55-8.52 (d) 1H) J=8.1 Hz), 7.94-7.91 (d, 2H, J=8.5 Hz),
7.58-7.55 (m, 1H),
7.48-7.45 (m) 2H), 7.25-7.14 (m) SH), 7.00-6.98 (d, 2H, J=7.8 Hz), 6.85-6.82
(d, 2H, J=7.9
Hz), 5.51-5.49 (m, 1 H), 3.14-3.08 (m, 1 H ), 2.91-2.83 (m, 1 H), 2.71-2.59
(m, 1 H), 2.33-2.26
(m, 2H), 1.83-1.80 (m, 2H), 1.23-1.18 (m, 3H). MS (ESI) m/e 471 (M-H)-. Anal.
Calcd for:
C29H32N2O4~O.SOH20: C, 72.32; H, 6.90; N, 5.81. Found: C, 72.57; H, 6.88; N,
5.80.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
0
I
-n N
H
Example 41 A
0
o--<
0
S ~ OH O
41a
The desired compounds was prepared according to the method of Example 27B,
except
substituting allyl bromide for cinnamyl bromide.
Example 41 b
OCH3
OCH3
1~) OH O
A solution of 4~(S.0 g, 19.4 mmol) in THF (60 mL) at 0°C was treated
with 9-BBN)
stirred at ambient temperature for 1.S hours, treated sequentially with DMF, [
1) 1'-
bis(diphenylphosphino)-ferrocene)dichloropalladium (790 mg) 0.97 mmol), 3,4,5-
trimethoxybromobenzene (14.4 g) 58.3 mmol) and cesium carbonate (38.6 g, 118.5
mmol),
1 S stirred at 60°C for S.S hours, cooled to room temperature and
diluted with water, extracted with
ethyl acetate, and the combined organic layers were washed with water and
brine, dried (Na2S04)
and concentrated to an oil. The oil was purified on silica gel with 10% to 30%
ethyl
acetate/hexane to provide 4.95 g (59.9%) of ~1 as a yellow oil.
MS (APCI) m/e 427 (M+H)+.
20 Exam In a 41 C
Exam lr a 41
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
81
OCH3
OCH3
The desired compound was prepared according to the method of Example27D and
27E, except
substituting 41 j2 for ~. MS (ESI) m/e 383 (M+H)+ .
Exarrrole 41 D
' H _41d
A solution of 41~ (755 mg, 1.97- mtnol) in DMF (15 mL) at 0 °C was
treated sequentially
with HOBT (294 mg, 2.17 mmol), NMM (477 mL) 438.8 mg, 4.35 mmol), EDC (417 mg,
2.17
mmol ) and indoleketone-ten-leucine) 1 Oa (500 mg, 2.17 mmol) , stirred at
room temperature for
1 ( i 17 hours, and diluted with water, extracted with ethyl acetate. The
combined organic layers were
washed with water and brine, dried (Na2S04), and concentrated. The residue was
purified on
silica gel with 50% ethyl acetate/hexane to provide 402 mg (34%) of the title
compound as a pale
yellow foam. MS (APCI) m/e 595 (M+H)+.
Example 41 E
_41e
A solution of 4l~lc (400 mg, 0.6734 mtnol) in trifluoroacetic acid (3 mL) was
stirred at room
temperature for 4 hours, and concentrated. The residue was purified on silica
gel with 0.1 % acetic
CA 02277105 1999-07-06
WO 98130541 PCT/US98/00142
82
acid in 10% MeOH/CH2C12 to provide 353.4 mg (94.7%) of title compound as a
white solid. MS
(ESI) m/e 555 (M+H)+.
Example 41F
o~
-o
I
0 0
H
HOHN _ N
OH O
A solution of ~1 (326 mg) 0.588 mmol) in DMF (IS mL) at 0 °C was
treated sequentially
with HOBT ( 103.4 mg, 0.765 mmol), NMM ( 168 mL, 154.6 mg, 1.53 mmoi), EDC (
146.6 mg,
0.765 mmol) and O-(tert-butyldimethyl-silyl)hydroxyamine (112.6 mg) 0.765
mmol)) stirred at
room temperature for 17 hours, and diluted with water) extracted with ethyl
acetate. The combined
organic layers were washed with water and brine) dried (Na2S04)) and
concentrated. The residue
was purified on silica gel with 7% MeOH/CH2Cl~ to provide 32 mg (9.56%) of the
title compound
as a pale pink solid. 1 H NMR (300 MHz, DMSO-d6) 8 0.979 (s) 9H)) 1.130-2.000
(m, 4H),
2.272-2.392 (m) 2H), 2.700 (m, IH). 3.546 (s, 3H)) 3.568 (s, 6H), 3.783 (t.
1H)) 5.146 (d,
1 H), 5.255 (d) 1 H)) 6.282 (s, 2H), 7.138-7.237 (m, 2H)) 7.457 (d, 1 H))
7.909 (d, 1 H)) 8. I 82
(d. IH), 8.412 (s) IH), 8.849 (s, IH)) 10.633 (s, 1H), 11.969 (s) 1H). MS
(APCI) m/e 570
(M+H)+. High resolution MS (FAB) m!e calcd for (M+H)+ : C3~H4pN30R: 570.2815.
Found
570.2822.
Example 42
o~
-o
I
0 0
H
HOHN N
OH O /
N
H
The desired compound was prepared according to the methods of Example 41,
except substituting
2c for 1l 0a in Example 41 D. 1 H NMR (300 MHz, DMSO-d6) 8 1.236-1.377 (m,
4H}, 2.274-
2.441 (m, 2H), 2.918-2.988 (dd, 1 H), 3.111-3.183 (dd, I H), 3.570 (s, 3H),
3.638 (s, 6H),
3.826 (t, 1 H), 5.222 (d, 1 H), 5.387 (q, 1 H), 6.343 (s, 2H), 7.118-7.276 (m,
9H), 7.439 (d,
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
83
1 H)) 8.125 (d, 1 H), 8.297 (s, 1 H), 8.435 (d, 1 H). MS (APCI) m/e 604
(M+H)+. Anal. calcd
for C33H3~N3Og~HOAc: C, 63.33; H, 6.22; N, 6.33. Found: C, 63.10; H, 6.05; N,
6.05.
~x;3mnle 43
o'
'o
o H o I o~
HORN - N
OH O ~ /
~xamnle 43A
0 0'
BocHN
_43a
A solution of Boc-tert-leucine-N-methoxyl-N'-methylamide ( I .15 g) 4.2 mmol )
in ethyl
ether (70 mL) at -78°C was treated with 2-lithioanisole (prepared by
addition of 2-bromoanisole
( 1.57 mL, 2.36 g, 12.6 mmol) to a solution of n-butyllithium (2.SM/hexane,
5.04 mL) 12.6
mmol ) in ethyl ether ( I S mL) at 0°C)) stirred at -30°C to -
45°C for I hour and poured onto I :1
Et~O: U.1 M HCI. The aqueous layer was separated, and extracted with ether)
the combined
organic layers were washed with brine) dried (Na2S04), and concentrated. The
residue was
purified on silica get with 2%-5%-10% ethyl acetate/hexane to provide 788 mg
(58%) of the title
compound as a colorless oil. MS (DCI/NH3) m/e 322 (M+H)+.
Example 43B
0 0'
HCI HzN
/
A solution of 43~ (786 mg, 2.45 mmol) in HCI/dioxane (4M, 6 mL) was stirred at
room
temperature for I hour, diluted with ether, filtered. The filtrate was washed
with ether, and dried
under high vacuum to provide 550.6 mg (87.3%) of the title compound as a white
solid. MS
(APC1) m/e 222 (M-HCl+H)+.
Exam In a 43C
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
84
o~
'o
o H o I o~
HOHN . N \
OH O
The desired compound was prepared according to the method of Examples 41 D-F,
except
substituting ketone 43b for II Oa. I H NMR (300 MHz) DMSO-d6) 8 0.883 (s, 9H),
1.171-1.399
(m, 4H), 2.291-2.515 (m, 2H), 2.728-2.833 (m, 1H), 3.575 (s, 3H), 3.635 (s,
6H), 3.797 (s,
3H),3.772-3.813 (m, IH), 5.199 (d, 1H), 5.304 (d) 1H), 6.388 (s, 2H), 6.960-
7.009 (t, 1H),
7.104 (d, 1 H), 7.449-7.501 (t, 1 H), 7.550-7.588 (dd, 1 H), 7.929 (d) 1 H),
8.852 (s, 1 H))
10.645 (s, 1H). MS (ES1) m/e 561 (M+H)+. Anal. calcd for C29H4pN209: C) 62.12;
H, 7.19;
N, 4.99. Found: C, 62.20; H, 7.22; N, 4.71.
Example 44
o~
'o
I
O H O
HOHN , N
OH O
Example 44A
0
HCI HZN \ O~
/
44a
The desired compound was prepared following the methods of Examples 43A and
43B,
except substituting 3-lithioanisole for 2-lithio anisole. MS (ESI) m/e 222 (M-
HCl+H)+.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
Example 44B
o~
'o
I
O H O
HOHN . N ! \
OH O
The desired compound was prepared following the methods of Example 43C, except
5 substituting 44a for 43a. IH NMR (300 MHz, DMSO-d6) 8 0.933 (s, 9H)) 1.138-
1.328 (m)
4H), 2.244-2.428 (m, 2H), 2.794-2.826 (m) 1H), 3.584 (s) 3H)) 3.666 (s, 6H),
3.793 (s,
3H),3.774-3.812 (m, 1 H)) 5.223 (d, IH), 5.298 (d, 1 H), 6.339 (s) 2H)) 7.151-
7. I 87 (dd, I H),
7.388-7.441 (m, 2H)) 7.571 (d, 1H), 8.137 (d) IH), 8.$59 (s, 1H), 10.655 (s,
1H). MS (ES1)
m/e 561 (M+H)+. Anal. calcd for C29H4pN209~0.25H20: C) 61.63; H, 7.22; N,
4.95. Found:
1 (1 C, 61.78; H, 7.48; N) 4.58.
i
~l
1
N
~H
Example 45
CA 02277105 1999-07-06
WO 98130541 PCTlUS98/00142
86
Exacrlple 45A
o i
H
N
45a
The desired compound was prepared according to the method of Example 2C,
except
coupling succinate 7 instead of 4 with ketone 2c. MS (ESI) m/e 461 (M + N)+.
i
'~ N
H
The desired compound was prepared according to the method of Example 41 B,
except using 45~
instead of 4_j,~.
Example 45B
CA 02277105 1999-07-06
WO 98/3054I PCT/US98/00142
87
i
H°~
\ T
N
The desired compound was prepared according to the method of Examples 1 E and
5A-B
except substituting ~5 from above for _l~. mp 104 °C. I H NMR (300 MHz,
DMSO-d~,) 11.92
(s, 1 H)) 10.35 (d) 1 H) J~.7 Hz), 8.70 (d, 1 H, J=1.4 Hz), 8.47 (d) 1 H)
J=8.5 Hz), 8.36 (d,
IH, J=3.0 Hz), 8.18-8.14 (m) 1H)) 7.47-7.42 (m) 1H)) 7.32-7.11 (m) 7H), 6.33
(s) 2H), 5.41-
5.31 (m, 1 H)) 3.63 (s, 6H), 3.56 (s) 3H), 3. I 6-3.07 (m, 1 H), 2.98-2.88 (m)
1 H)) 2.77-2.65 (m)
1H)) 2.77-2.65 (m, 3H)) 1.93-1.R7 (m, 1H), 1.44-1.18 (m) 4H). MS (ESI) m/e 588
(M + H)+.
i
"°~ \
o : \ T
\H
The desired compound was prepared according to the method of Example 45A-45C,
except
substituting ketone Q~ for ~ in Example 45A. mp 126 °C. t H NMR (300
MHz, DMSO-d6) I 1.96
I 5 (d, 1 H, J=2.2 Hz), 10.37 (d, 1 H, J=1.4 H z), 8.69 (d, 1 H, J= I .5 Hz),
8.39 (d, I H ) J=2.9 Hz),
8.18 (d, 1 H, J=7.4 Hz)) 8.05 (d, 1 H, J=9.2 Hz), 7.48-7.43 (m) 1 H), 7.24-
7.13 (m, 2H), 6.32
(s, 2H)) 5.09 (d, 1 H, J=8.8 Hz), 3.60 (s) 6H)) 3.56 (s, 3H), 2.96-2.87 (m, 1
H), 2.47-2.27 (m)
E~ple 45~
Example 46
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
88
2H), 2.22-1.98 (m, 2H)) 1.47-1.24 (m, 4H), 0.97 (s, 9H).13C NMR (300 MHz, DMSO-
d6)
193.9, 174.0, 1 S2.S, 137.7, 136.6, 135.4, 134.4, 12S.S, 122.9, 121.7, 121.4,
116.9, 112.1,
lOS.3, 60.3, 59.9) 41.0, 35.3, 34.2, 31.5, 28.2, 27.1. MS (APCI) m/e SS4 (M +
H)+.
S
Example 47
o
Example 47A
O
HpN
The desired compound was prepared according to the method of Example 43A and
43B, except
using phenyl lithium in place of 3-lithioanisole.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
89
Examlhe 47B
o
The desired compound was prepared according to the method of Example 45A-45C,
except
substituting ketone 47a for 2c in Example 45A. mp 146 °C. ~H NMR (300
MHz) DMSO-d6)
10.36 (s, 1 H), 8.67 (s) 1 H), 8.22 (d) 1 H) J=8.0 Hz)) 7.97-7.92 (m) 2H),
7.63-7.45 (m) 3H))
6.38 (s, 2H), 5.27 (d, 1H) J=8.1 Hz), 3.68 (s) 6H), 3.59 (s, 3H), 2.98-2.87
(m, 1H)) 2.46-2.26
(m, 2H), 2.22-1.96 (m, 2H)) 1.44-1.30 (m, 4H)) 0.92 (s) 9H).13C NMR (300 MHz,
DMSO-d~)
1 U 200.3) 174.4 , 167.6, 164.8) 152.6, 138.2, 137.8) 135.4) 133.0, 128.6)
128.0, 105.3, 59.9,
59.5) 55.7) 4U.6) 35.3, 35.2, 34.0) 31.6, 28.0) 26.9.
H
The desired compound was prepared according to the method of Example 2C and
2D, except
coupling succinate _9 instead or 4 with ketone 9a instead of 2c.
x 48
CA 02277105 1999-07-06
WO 98130541 PCT/US98/00142
1H NMR (300 MHz) DMSO-d6) 11.93 (s, 1H), 10.48 (d, 1H, J=1.7 Hz), 8.74 (d> 1H,
J=1.7
Hz), 8.42 (d) 1 H, J=3.1 Hz)) 8.16 (t) 2H, 3=8.1 Hz), 7.47-7.43 (m, 1 H), 7.23-
7.11 (m, 2H))
7.23-7.11 (m, 2H), 6.28 (s, 2H), 5.76-5.56 (m, 1H), 5.14 (d, 1H, J=8.5 Hz),
4.96-4.90 (m,
2H), 3.58 (s, 6H), 3.56 (s, 3H), 2.80-2.70 (m, 1H), 2.46-2.33 (m, 1H), 2.33-
2.13 (m, 3H))
5 2.10-1.98 (m, 1H), 1.38-1.18 (m, 4H), 1.00 (s, 9H). MS (ESI) m/e 594 (M +
H)+~ Anal. Calcd
for: C33 H43 N3 O7~0.50H20: C) 65.76; H) 7.35; N, 6.97. Found: C, 65.70; H,
7.46; N, 6.98.
Example 49
O \ /
O H O
HO.N N
H O
The desired compound was prepared according to the method of Example 2C and
2D, except
coupling succinate _1Q instead of 4 with ketone ~ instead of _2~.
~ H NMR (DMSO-d6) 8 0.93 (s) 9H), 1.0-1.43 (m, 6H), 1.95-2.02 (m, 1 H), 2.10-
2.21 (m, 1 H ))
2.78-2.90 (m, 1H), 3.21 (t, 2H, J=9 Hz), 4.34 (s, 2H), 7.28 (d) 1H, J=9 Hz).
7.21-7.36 (m)
5H)) 7.44-7.50 (m, 2H), 7.53-7.62 (m) 1 H). 7.94 (d, 2H) J=8 Hz), 8.24 (d. 1
H, J=9 Hz), 8.68
(s, 1 H ), 10.38 (s, 1 H ). MS (DCI/NH3 ) m/e 469 (M+H)+. Anal. calcd for:
C27H36N205: C,
69.20; H, 7.74; N, 5.99. Found: C) 69.35; H, 7.70; N, 6.02.
Example 50
O \
O H O
HO.N N
H O /
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
91
Example SOA
O
H2
The desired compound was prepared according to the methods of Examples I 8A
and B, except
substituting N-Boc-alpha-cyclohexyl alanine for N-Boc-phenylalanine.
Example SOB
O
O H O
HO.N N
H O /
The desired compound was prepared according to the method of Example 2C and
2D) except
coupling succinate 1 () instead or 4 with ketone ()a instead of 2c. ~ H NMR
(300 MHz, DMSO-d6) 8
0.78-1.0 (m, 2H), 1.03-1.68 (m, 16H), 1.78-1.90 (m, 1 H), I .91-2.13 (m, 2H))
2.60-2,74 (m)
1l~ 1H), 3.21-3.29 (m, 2H), 4.4 (s) 2H), 5.20-5.37 (m, IH), 7.20-7.39 (m) SH),
7.41-7.52 (m) 2H),
7.55-7.63 (m, 1 H)) 7.90 (d, 2H) J=8 Hz)) 8.38 (d) 1 H, J=8 Hz), 8.70 (s, 1
H)) 10.38 (s) I H); MS
(DCI/NH3) m/e 509 (M+H)+. Anal. calcd for: C3oH40N2OS: C) 70.83; H, 7.92; N,
5.50. Found:
C. 70.63: H, 8.13; N, 5.63.
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
92
Example 51
HO~
N
H
H
~O H ~O N ,
O O
H
The desired compound was prepared according to the method of Example 2C except
substituting succinate ester 1 1 for 4 and ketone ~ for 2c.
~H NMR (300 MHz, DMSO-d6) 8 8.39 (s) 1H)) 8.21 (d, 1H), 8.06 (d, 1H), 7.47 (d,
1H), 7.24-
7.18 t m) 2H), 5.10 (d, 1 H), 2.89-.287 (m, 1 H), 2.69 )t) 1 H), 2.45 (dd, 1
H), 2.29 (dd) 1 H),
2.07-1.91 (m, 2H)) 1.58-1.42 (m, 2H)) 1.37 )s, 9H), 0.97 (s, 9H). MS (DCI/NH3)
m/e 439
(M+1 )+.
Example 51 A
CA 02277105 1999-07-06
WO 98130541 PCT/US98/00142
93
Example 1 B
O~
,O
O H O O H O
O N , / '-~ N
O ~ ~NY O O I ~ /
H ~ N
Sla H
A solution of the alkyne I a (2 I 1 mg) 0.48 mmol) and 1-bromo-3,4,5-
trimethoxybenzene
(131 mg, 0.528 mmol) in 2:1 triethylamine/acetonitrile (4.8 mL) was degassed
with N2 for 20
minutes, treated with 10% palladium on activated carbon (20 mg, 0.0192 mmol)
and copper iodide
(5 mg, 0.024 mmol)) heated at reflux for 24 hours) cooled to 23 °C,
filtered through Celite, and
concentrated to a residue. The residue was purified on silica gel with 50%
ethyl acetate/hexane to
provided 13U mg of 51 b as a white solid. ~H NMR (300 MHz) DMSO-d6) b 8.41 (s)
1 H), 8.22 (d,
1 H), 8.08 (d) I H), 7.47 (d) 1 H), 7.22-7.18 (m, 2H), 6.63 (s) 2H)) 5.14 (d,
1'H), 3.73 (s, 6H),
3.63 (s, 3H), 2.96-.290 (m, 1H)) 2.37-2.10 (m, 4H), 1.65-I.55 (m, 2H), 1.38
(s) 9H)) 0.98 (s,
9H). MS (DCI/NN3) m/e 605 (M+I)+,
Example 5 i c;
A solution of the alkyne ~ ( 115 mg) 0.19 mmol) in 1: I methanol/ethyl acetate
(4 mL}
was treated with 10% palladium on activated carbon (20 mg, 0.019 mmol) under
an atmosphere of
hydrogen (H2 balloon) for 16 hours, filtered through Celite) and concentrated
to provide 115 mg
of Sue. 1H NMR (300 MHz, DMSO-d6) 8 8.35 (s) 1H}) 8.24 (d, 1H), 8.11 (d, 1H),
7.48 (d,
CA 02277105 1999-07-06
WO 98/30541 PCT/US98100142
94
1H)) 7.24-7.14 (m, 2H), 6.29 (s, 2H), 5.12 (d) 1H), 3.69 (s, 6H), 3.57 (s,
3H), 2.82-2.81 (m,
1 H), 2.42 (dd, 1 H ), 2.20 (dd, 1 H), 2.13 (t, 1 H ) ) 1.37 ( s, 9H), 1.33-
1.24 (m, 6H ), 0.98 ( s, 9H ).
MS (ESI) m/e 607 (M-1)+.
Exarr~le 51 D
\ /
H
The ester 1 c was convereted to the desired compound following the procedures
described
1 () in Examples 1 E, SA and SB. ~ H NMR (300 MHz) DMSO-d~) 8 8.68 (s) 1 H),
8.36 (d, 1 H}, 8.24
(d) 1 H), 8.07 (d, 1 H)) 7.48 (d) 1 H), 7.24-7. I 4 (m) 2H)) 6.29 (s, 2H))
5.10 (d, 1 H)) 3.69 (s,
6H), 3.57 (s, 3H), 2.82 (m, 1H), 2.21-1.98 (m, 4H), 1.40-1.18 (m, 6H)) 0.99
(s) 9H). MS
(DCl/NH3) m/e 568 (M+1)+.
Anal. calcd for C3 ~ H4 ~ N30~~H20: C, 63.57; H, 7.40: N, 7.17. Found: C)
63.52; H) 7.15; N)
1 S f~.67.
Exam lp a 52
0 0
H
HON N
H OH O '
H
Example 52A
Br
0
o o~
20 off o
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
9S
The desired compound was prepared according to the method of Example 27B,
except substituting
1,3-dibromo-1-propene for cinnamyl bromide.
Example S2B
NHAc
O
S ~ OH O
A solution of Example 2a (3.0 g, 8.9 mmol) in DMF (100 mL) at room temperature
was
treated with [ 1,1 '-bis(diphenylphosphino)-ferrocene] dichloropalladium (363
mg, 0.445 mmol )) 3-
acetimidobenzenoboronic acid (2.39 g, 13.35 mmol) and cesium carbonate (8.7 g,
26.7 mmol ),
stirred at 60°C for 7 hours, cooled to room temperature and diluted
with water, extracted with
ethyl acetate, and the combined organic layers were washed with water and
brine) dried (Na2S04)
and concentrated to an oil. The oil was purified on silica gel with SO% ethyl
acetate/hexane to
provide 716.9 mg (20%) of S2b as a yellow oil. MS (ESI) m/e 392 (M+H)+.
Example S2C
NHAc
r0
off o ~
The olefin 5~ was converted to the desired compound ~ following the procedure
of
Example S 1 C .
NHAc
HO~
N
H
OH O
CA 02277105 1999-07-06
WO 98/30541 PCT/US98I00142
96
The desired compound was prepared according to the methods of Example 41 C-F,
except
substituting 52c for 41b. IH NMR (DMSO-d6) 8 0.989 (s, 9H), 1.23-1.39 (m, 4H),
1.98 (s,
3H), 2.19-2.36 (m, 2H), 2.72-2.76 (m, 1 H), 3.77-3.83 (t, 1 H, J=8.1 Hz)) 5.12-
5.16 (d, 1 H,
J=6 Hz), 5.26-5.29 (d, 1 H, J=7.5 Hz), 6.37-6.39 (d, 1 H, J=7.8 Hz), 6.72-6.77
{t, I H, J=7.8
Hz), 7.16-7.25 (m, 4H), 7.45-7.48 ( 1 H), 7.94-7.97 (d, 1 H, J=9.6 Hz), 8.2 I -
8.24 ( I H), 8.38 (s,
1 H ), 8.80 (s, 1 H), 9.71 (s, 1 H), 10.6 {s, 1 H), 11.94 (s, 1 H). MS (ESI)
537 (M+H)+, 559
(M+Na)+.
Example 53
OMe
H O
N
OH O , I N
H
The desired compound was prepared according to the method of Example 52B-D
except
substituting 3-methoxybenzenoboronic acid for 3-acetimidobenzenoboronic acid.
1 H NMR
(DMSO-d6 ) b U.99 (s, 9H), 1.22- I .27 (m) 4H)) 2.27-2.39 (m, 2H)) 2.74 (dt, 1
H), 3.59 (s, 3H),
3.76-3.8 I (t) 1 H) J=8.4 Hz), 5.13-5.16 (d, 1 H, J=9.6 Hz), 5.25-5.27 (d, I
H) J=7.2 Hz)) 6.31-
6.33 (d) 1 H, J=7.2 Hz), 6.55-6.68 (2H)) 6.75-6.81 (t) I H, J=7.R Hz), 7.18-
7.23 (m, 2H), 7.45-
7.48 (d. 1 H. J=8.7 Hz), 7.94-?.98 (d, I H, J=9.3 Hz), 8.21-8.24 (d) 1 H,
J=9.6 Hz), 8.40 (s,
1 H ), 8. R4 ( s, 1 H ), 10.63 ( s, 1 H ), 11.97 ( s, 1 H ). M S (ES 1 )
510(M+H )+, 532 (M+Na)+.
2() Example 54
'o
I
H O
N
OH O
N
I
S02CH3
CA 02277105 1999-07-06
WO 98/30541 PCT/US98/00142
97
Exam lie 54A
/ ../
/ ~ w / o\
0
i ~ -o
0 o i
0 0
o \- o b _
0 0 ~~ o ~ , v ~
H ~ N
SOzMe
A solution of 4 ~,~ (249 mg) 0.419 mmol) in dichloromethane (5 mL) at room
temperature
was treated with methanesulfonyl chloride ( 144.2 mg) 97.4 ml, 1.26 mmol), and
triethylamine
( 127 mg, 175 ml, 1.26 mmol), stirred for 6 hours, and quenched with water)
extracted with
dichloromethane, dried (Na2S04) and concentrated . The residue was purified on
silica gel with
40% to 60% ethyl acetate/hexane to provide 239.4 mg (85%) of S~ as a white
foam. MS (ESI)
m/e 673 (M+H)+.
Example 54B
o'
o~
~ I o
I
O H O
N -._
HO
OH O
N
SOZMe
A solution of 4a (237 mg, 0.353 mmol) in THF (4.5 mL) at 0°C was
treated with I N HCl (4.5
mL), stirred at room temperature for I7 hours, and concentrated. The residue
was extracted with
dichloromethane, dried (Na2S04), and concentrated. The residue was purified on
silica gel with
0.1 % acetic acid in 10% MeOH/CH2Cl2 to provide 177 mg (79%) of 5~ as a white
solid. MS
(ESI) m/e 631 (M-H)-.
CA 02277105 1999-07-06
WO 98/30541 PCT/LTS98/00142
98
Exam 1
-o
I
H
N
OH O
I~ N
1
SOpCH3
The desired compound was prepared according to the method of Examples 5A-B,
except
substituting S~ø for 4_. 1 H NMR (DMSO-d6) 8 1.004 (s, 9H), 1.247-1.347 (m,
4H), 2.269-
2.418 (m, 2H), 2.800-2.833 (m) 1H), 3.554 (s, 3H), 3.593 (s, 6H)) 3.649 (s)
3H) , 3.768-3.822
( 1 H), 5.100-5.127 (d, 1 H, J=8.1 Hz), 5.232-5.256 (d, 1H, J=7.2 Hz), 6.301
(s, 2H), 7.403-
7.464 (m, 2H), 7.874-7.900 (d, 1 H, J=7.8 Hz), 8.136-8.165 (d, 1 H) J=8.7 Hz),
8.215-8.240
(d, 1 H) J=7.5 Hz), 8.634 (s, 1 H)) 8.855 (s, 1 H)) 10.652 (s) 1 H). MS (ES1 )
648 (M+H)+, 665
(M+NH4)+.