Note: Descriptions are shown in the official language in which they were submitted.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
Macrocyclic Inhibitors of Matrix Metalloproteinases and TNFa Secretion
Technical Field
This invention relates to compounds having activity to inhibit matrix
metalloproteinases and
TNFa secretion, to pharmaceutical compositions comprising these compounds, and
to a medical
method of treatment. More particularly, this invention concerns macrocyclic
compounds which
inhibit matrix metalloproteinases and TNFa secretion, to pharmaceutical
compositions comprising
these compounds and to a method of inhibiting matrix metalloproteinases and
TNFa secretion.
Background of the Invention
The matrix metalloproteinases (MMP's) are a class of extracellular enzymes
including
collagenase, stromelysin, and gelatinase which are believed to be involved in
the tissue destruction
which accompanies a large number of disease states varying from arthritis to
cancer.
Typical connective tissue cells are embedded within an extracellular matrix of
high
I S molecular weight proteins and glycoproteins. In healthy tissue, there is a
continual and delicately-
balanced series of processes which include cell division, matrix synthesis,
and matrix degradation.
In certain pathological conditions, an imbalance of these three processes can
lead to improper
tissue restructuring. For example, in arthritis, joint mobility can be lost
when there is improper
remodelling of load-bearing joint cartilage. In the case of cancer, lack of
coordination of cell
division and the two processes of matrix synthesis and degradation can lead to
conversion of
transformed cells to invasive phenotypes in which increased matrix turnover
permits tumor cells to
penetrate basement membranes surrounding capillaries leading to subsequent
metastasis.
There has been hightened interest in discovering therapeutic agents which bind
to and
inhibit MMP's. The discovery of new therapeutic agents possessing this
activity will lead to new
drugs having a novel mechanism of action for combatting disease states
involving tissue
degenerative processes including, for example, rheumatoid arthritis,
osteoarthritis, osteopenias
such as osteoporosis, periodontitis, gingivitis, corneal, epidermal or gastric
ulceration, and tumor
growth and metastasis or invasion.
Tumor Necrosis Factor a (TNFa) is a potent proinflammatory mediator which has
been
implicated in inflammatory conditions including arthritis, asthma, septic
shock, non-insulin
dependent diabetes mellitus and inflammatory bowel disease. TNFa is originally
expressed as a
membrane-bound protein of about 26 kD, which is proteolytically cleaved to
release a soluble 17
kD fragment (TNFa processing) which combines with two other secreted TNFa
molecules to
form a circulating 51 kD homotrimer. Recently, several MMP inhibitors were
found to inhibit
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
2
TNFa processing (see Mohler, et al.) Nature, 1994, 370, 218; Gearing, et al.,
Nature, 1994,
370, 555; and McGeehan, et al., Nature, 1994, 370, 558), leading to the
hypothesis that TNFa
processing is caused by an as yet uncharacterized metalloproteinase residing
in the plasma
membrane of cells producing TNFa. Inhibitors of this metalloproteinase would
therefore be
useful as therapeutics to treat disease states involving TNFa secretion.
Transforming growth factor alpha (TGFa) is a potent mitogen which ellicites
its biological
activity by binding to cell surface receptors, in particular epidermal growth
factor (EGF) receptor.
It is known to promote angiogenesis and to stimulate epithelial cell migration
and therefore has
been implicated in a number of malignant disorders such as breast cancer and
ovarian carcinoma.
TGFa is produced by proteolytic cleavage of a 160 amino acid membrane bound
precursor.
Several cleavage sites have been identified including A1a38-Va139, similar to
the cleavage site of
proTNFa (Ala-76-Va177). This common cleavage site suggests that inhibitors of
TNFa
processing may also block the cleavage of proTGFa and therefore would be
therapeutically useful
in diseases mediated by TGFa.
Summary of the Invention
The present invention provides a novel class of macrocyclic inhibitors of
matrix
metalloproteinases and/or TNFa secretion.
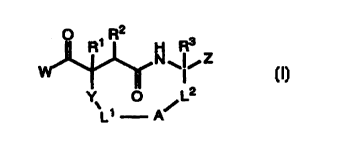
In its principle embodiment, the present invention provides a macrocyclic
compound of
formula I
2
H Rs
N~~~Z
I
Y' O ~ L2
L1 A
or a pharmaceutically acceptable salt, ester or prodrug thereof wherein
W is NHOH or OH.
R1 and R3 are independently selected from hydrogen or alkyl of one to four
carbon atoms.
R2 is selected from the group consisting of
(a) alkyl of one to ten carbon atoms,
(b) allcenyl of two to ten carbon atoms,
(c) cyeloalkyl of three to eight carbon atoms,
(d) (cycloalkyl)alkyl wherein the cycloalkyl portion is of three to eight
carbon atoms, and the
alklene portion is of one to six carbon atoms)
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
3
(e) cycloalkenylene of five to eight carbon atoms.
(f) (cycloalkenylene)alkyl wherein the cycloalkenylene portion is of five to
eight carbon atoms,
and the alklene portion is of one to six carbon atoms,
(g) phenyl)
(h) phenyl substituted with I ) 2, or 3 substutuents independently selected
from alkyl of one to
four carbon atoms, alkoxy of one to four carbon atoms, halogen, haloalkyl of
one to four carbon
atoms, cyano, cyanoaIkyl, -C02R4 wherein R4 is independently selected at each
occurrence from
hydrogen and alkyl of one to four carbon atoms, and -CONR4R5 wherein R4 is
defined above and
and RS is independently selected at each occurrence from hydrogen and alkyl of
one to four carbon
atoms,
(i) phenylalkyl wherein the alkylene portion is of one to six carbon atoms,
(j) phenylalkyl wherein the alkylene portion is of one to six carbon atoms and
the phenyl ring
is substituted with 1, 2, or 3 substituents independently selected from
alkoxyalkyloxy, alkyl of one
to four carbon atoms, alkoxy of one to four carbon atoms, halogen, haloalkyl
of one to four carbon
atoms, cyano, cyanoalkyl, -COZR4, -CONR4R5, phenyl, and phenyl substituted
with 1, 2, or 3
substutuents independently selected from alkyl of one to four carbon atoms,
hydroxy, alkoxy of
one to four carbon atoms, halogen, haloalkyl of one to four carbon atoms,
cyano, cyanoalkyl, -
C02R4, and -CONR4R5,
(k) -(CH2)m-T-(CH2)~-R6 wherein m and n are independently 0, 1, 2, 3 or 4, T
is O or S ) and
R6 is selected from the group consisting of alkyl of one to four carbon atoms,
phenyl, and phenyl
substituted with I, 2, or 3 substituents selected from alkyl of one to four
carbon atoms, hydroxy,
alkoxy of one to four carbon atoms, halogen, haloalkyl of one to four carbon
atoms, cyano,
cyanoalkyl, -C02R4, -CONR4R5, phenyl, and phenyl substituted with 1, 2, or 3
substutuents
independently selected from alkyl of one to four carbon atoms, alkoxy of one
to four carbon
atoms, halogen, haloalkyl of one to four carbon atoms, cyano, cyanoalkyl, -
C02R4, and -
CONR4R5, and
(1) fluorenylalkyl wherein the alkylene portion is of one to four carbon
atoms.
Y is absent or -O-.
Ll is alkylene of two to six carbon atoms.
L2 is selected from the group consisting of
(a) alkylene of one to six carbon atoms, and
(b)
a
Rr\..~i b 3
c~ D J L
R wherein D is CH or N, L3 is absent or is alkylene of one to four
carbon atoms, and Ra) Rb and R~ are independently selected from hydrogen,
alkyl of one to four
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
4
carbon atoms, hydroxy, alkoxy of one to four carbon atoms, halogen, haloalkyl
of one to four
carbon atoms, cyano, -S02R6 wherein R6 is alkyl of one to four carbon atoms, -
S02NH2, -
C02R4) 2-tetrazolyl, and -CONR~R8 wherein R~ and Rg are independently selected
at each
occurrence from hydrogen and alkyl of one to four carbon atoms, or R~ and Rg
together with the N
atom to which they are attached define a a 5-or 6-membered heterocyclic ring
selected from the
group consisting of morpholinyl) thiomorpholinyl, thiomorphoiinyl sulfone,
pyrrolidinyl,
piperazinyl, piperidinyl, and 3-ketopiperazinyl.
A is absent or is selected from the group consisting of
(a) -O-,
(b) -NR9- wherein R9 is selected from the group consisting of
( 1 ) hydrogen,
(2) alkyl of one to four carbon atoms,
(3) -C02R 1 o wherein R 1 o is independently selected at each occurrence from
the group consisting
of alkyl of one to four carbon atoms, haloalkyl of one to four carbon atoms,
phenyl, phenyl
substituted with 1, 2, or 3 substituents independently selected from alkyl of
one to four carbon
atoms, alkoxy of one to four carbon atoms, halogen, haloalkyl of one to four
carbon atoms, nitro,
cyano, cyanoalkyl, -S02NH2, -C02R4, and -CONR4R5, phenylalkyl wherein the
alkylene portion
is of one to four carbon atoms, phenylalkyl wherein the alkylene portion is of
one to four carbon
atoms, and the phenyl ring is substituted with 1, 2, or 3 substituents
independently selected from
alkyl of one to four carbon atoms, aIkoxy of one to four carbon atoms,
halogen, haloalkyl of one
to four carbon atoms, cyano, cyanoalkyl, -S02NH2, -COZR4, and -CONR4R5,
heteroarylalkyl
wherein the alkylene portion is of one to four carbon atoms, and the
heteroaryl group is selected
from furyl, pyridyl, thienyl, benzimidazolyl, imidazolyl, thiazolyl, and
benzothiazolyl wherein the
heteroaryl group is unsubstituted or substituted with alkyl of one to four
carbon atoms,
(4} -CONR~Rg,
(5) -CORIO, and
(6) -S02RIO,
(c) -S(O)S- wherein n is 0, 1, or 2,
(d) -S-S
(e) -CH=CH-,
(g)
V
wherein V is O or NOR4,
O
\ d~ wherein J is O or NR4,
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
(h)
O
J~
- (i)
O
5 \~~K~ wherein J is defined above an 4
d K is selected from O and NR , provided
that J and K are nat simultaneously O,
V
~N~N~
Ra R5
(k)
O
4
~N~L W /
R° O wherein L4 is alkylene of two to six carbon atoms,
(1)
(m)
(n)
O
~N~L5 ,O~
R4 , wherein.Ls is alkylene of one to three carbon atoms,
O
rO~L~N/
R4
Ra
5
rN i~ W
1 R,2
O wherein R4 is defined above and R12 is selected from hydrogen, alkyl
of one to four carbon atoms, -COR1~, -C02R1~, and -S02R10,
(o)
O
~ N
~~N~
R4
' 20 (P)
R4 O
N 5 'I
L wN~LS /O~
Ra
O
CA 02277121 1999-07-06
WO 98130551 PCT/US98/00144
6
(Q)
Ra Ra _
/N~ L ~N\
'O1 IO'
,
(r)
O
4
\O/L\N
Ra
(s)
O
R~2
O Ls /N\
,
(t)
R12 O
/N\ Ls/ \O/
,
(u)
O
/O~Ls~
,
(v)
0
~Ls i0\
,
(w) -J'-L4-K'- wherein J' and K' are independently selected from O and NR12,
(x) -NR4S02-,
(Y) -SOZNR4-,
(z) -NR4S02NR5_)
(aa)
-T-~ L3 -V-
Ra wherein T and V are independently selected from O and S and
Ra is defined above,
(bb)
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
7
.r N
R
\J
b
_ R R wherein Ra, Rb, and R~ are defined above,
(cc)
\ _
~Z N
tR
R~~.\Rb
(dd)
Ra
i
~N
~N
O ( ! R
\J
R~~' Rb
Ra
r-NY N
. Re , wherein Rd and Re are independently selected from hydrogen and alkyl of
one to four carbon atoms, and
Ra
l'l1i
.~-N~\~
Re
provided that when A is selected from (aa), (bb), (cc), (dd) and (ff) above,
L2 is alkylene, and
further provided that when both Y and A are absent, Ll is alkylene of three to
six carbon atoms.
Z is absent or is selected from the group consisting of
(a) -C02H,
IS (b) -C02R10,
(c)
O
~NR~3R~4 wherein R13 is h dro en or ally 1 of
y g y one to six carbon atoms, and
R14 is selected from the group consisting of
( 1 ) hydrogen,
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/OOI44
8
(2) alkyl of one to six carbon atoms,
(3) cycloalkyl of three to eight carbon atoms,
(4) (cycloalkyl)alkyl wherein the cycloalkyl portion is of three to eight
carbon atoms and the alkyl
portion is of one to four carbon atoms)
(5) cycloalkenyl of five to eight carbon atoms,
(6) (cycloalkenyl)alkyl wherein the cycloalkenyl portion is of five to eight
carbon atoms and the
alkyl portion is of one to four carbon atoms,
(7) -S02R»,
(8) -CH2CHZM(L3M)p-R4 wherein p is 1, 2 or 3, L3 is alkylene of from one to
four carbon atoms
and M is selected at each occurrence from O and S,
(9) -L4-(NR4L4)g-NR~Rg wherein q is 0, 1 or 2,
(10) _L,4_(1~TR4L,4)q_j~TR4S02NR7R8,
(11) aryl wherein the aryl group is selected from (a) phenyl, (b) phenyl
substituted with 1, 2, or 3
substituents selected from alkyl of one to four carbon atoms, halogen,
haloalkyl of one to four
carbon atoms, hydroxy, alkoxy of one to four carbon atoms, cyano, -C(O)R4, -
NR4R5, -C02R4, -
SOZR4, -S02NR4R5, (c) naphthyl, (d) naphthyl substituted with l, 2 or 3
substituents
independently selected from alkyl of one to four carbon atoms, halogen,
haloalkyl of one to four
carbon atoms, hydroxy, allcoxy of one to four carbon atoms, cyano, -C(O)R4, -
NR4R5, -C02R4, -
S02R4, -S02NR4R5,
(12) heteroaryl selected from the group consisting of (a) pyridyl, (b)
thiazolyl, (c) furyl, (d)
thienyl) (e) pyrrolyl, (fj tetrahydrofuryl, (g) imidazolyl, (h)
phenylthiazolyl, (i) benzothiazolyl, (j)
benzimidazolyl, (k) pyrazinyl, (1) pyrimidyl, (m) quinolyl, (n) piperazinyl,
and (o) indolyl wherein
the heteroaryl group is unsubstituted or substituted with alkyl of one to four
carbon atoms,
( 13) arylalkyl wherein the alkylene portion is of one to four carbon atoms
and the aryl group is
defined above,
(14) heteroarylalkyl wherein the alkylene portion is of one to four carbon
atoms and the heteroaryl
group is defined above,
(15)
\ La ~ N
O
(16)
CA 02277121 1999-07-06
WO 98/30551 PCT/LTS98/00144
9
Ris
O
N ~
wherein R15 is selected from hydrogen, hydroxy, alkyl of one to four
carbon atoms, alkoxy of one to four carbon atoms, and alkoxyalkyl of one to
four carbon atoms,
and
(17)
O
NR4R5
R" wherein Rz is the side chain of a naturally occurring amino acid,
(18)
OH
N H O
O
HN _ OH
~ OH
H3C' \O _
or R 13 and R 14, together with the N atom to which they are attached define a
5-or 6-membered
heterocyclic ring selected the group consisting of
(1) morpholinyl,
(2) thiomorpholinyl,
(3) thiomorpholinyl sulfone,
(4) pyrrolidinyl,
(5) piperazinyl,
(6) piperidinyl,
(7) 3-ketopiperazinyl, and
(8)
~ N
CO
2Rts wherein R~6 is hydrogen or benzyl, and
(d)
V
Rl~ wherein V is defined abov 17 '
a a~ R is selected from the group
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
consisting of
(1 ) alkyl of one to six carbon atoms,
{2) carboxyalkyl wherein the alkylene portion is of two to six carbon atoms,
(3) phenyl,
5 (4) phenyl substituted with 1, 2, or 3 substituents selected from alkyl of
one to four carbon atoms,
halogen, hydroxy, hydroxyalkyl of one to four carbon atoms, haloalkyl of one
to four carbon
atoms, alkoxy of one to four carbon atoms, amino. cyano, -NR4R5, -S02NR4R5. -
SO2R4,
-CH2NR~Rg, -CONR~Rg, -C02R4, and phenyl, wherein the phenyl ring may be
substituted with
1, 2, or 3 substituents independently selected from alkyl of one to four
carbon atoms, halogen, and
10 haloalkyl of one to four carbon atoms,
(5) 1,3-benzodioxole,
(6) indolyl,
(7) indolyl substituted with alkyl of one to four carbon atoms, halogen,
haloallryl of one to four
carbon atoms, alkoxy of one to four carbon atoms, -S02NR4R5, -C02RIO, and
phenyl, wherein
the phenyl ring may be substituted with 1, 2, or 3 substituents independently
selected from alkyl of
one to four carbon atoms, halogen, haloalkyl of one to four carbon atoms, and
alkoxy of one to
four carbon atoms,
(8) pyrrolyl,
(9) pyrrolyl substituted with alkyl of one to four carbon atoms,
( 10) imidazolyl,
( 11 ) imidazolyl substituted with alkyl of one to four carbon atoms, provided
that in (6)-( 11 ) above,
when the heterocycle is attached at a carbon atom, the N atom may bear a
substituent selected from
the group consisting of alkyl of one to six carbon atoms -CONR~Rg, -SOZNR~Rg
and -S02R10,
( 12) pyridyl,
(13) pyridyl substituted with alkyl of one to four carbon atoms,
( 14) thienyl,
(15) thienyl substituted with halogen, alkyl of one to four carbon atoms, and
haloalkyl of one to
four carbon atoms,
( 16) thiazolyl,
( 17) thiazolyl substituted with halogen, alkyl of one to four carbon atoms,
and haloallcyl of one to
four carbon atoms,
(18) oxazolyl,
( 19) oxazolyl substituted with halogen, alkyl of one to four carbon atoms,
and haloalkyl of one to
four carbon atoms,
(20) furyl,
(21 ) furyl substituted with halogen, alkyl of one to four carbon atoms, and
haloalkyl of one to four
carbon atoms,
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
11
(22) benzofuryl,
(23) benzofuryl substituted with 1, 2, or 3 substituents selected from alkyl
of one to four carbon
atoms, halogen, and haloalkyl of one to four carbon atoms,
(24) benzothiazolyl,
S (25) benzothiazolyl substituted with I, 2, or 3 substituents selected from
alkyl of one to four
carbon atoms, halogen, and haloallcyl of one to four carbon atoms,
(26) benzimidazolyl and
(27) benzimidazolyl substituted with I, 2 or 3 substituents independently
selected from alkyl of
one to four carbon atoms, halogen, and haloalkyl of one to four carbon atoms,
1 U (e)
Rlg
N
N R19
14
R wherein R1g and R19 are independently selected from the group
consisting of
( 1 ) alkyl of one to four carbon atoms)
(2) halogen,
15 (3) haloaIkyl of one to four carbon atoms,
(4) alkoxyalkyl wherein the alkoxy and alkylene portions are independently of
one to six carbon
atoms,
(5) alkanoyl of one to six carbon atoms)
(6) -CH(OH)R4,
20 (7) -CONR4R5,
(8) -C02R4,
{9) phenyl,
(10) phenyl substituted with 1, 2, or 3 substituents selected from alkyl of
one to four carbon
atoms, halogen, hydroxy, haloalkyl of one to four carbon atoms, alkoxy of one
to four carbon
25 atoms, amino, cyano, -S02NR4RS, -S02R4, -CH2NR4R5, -CONR4R5, and -C02R4 or
R1g and
R 19 together with the carbon atoms to which they are attached define a fused
5-7 membered
carbocyclic aryl or heterocyclic aryl ring wherein the ring may be substituted
with alkyl of one to
four carbon atoms or haloalkyl of one to four carbon atoms.
In another aspect, the present invention provides pharmaceutical compositions
which
30 comprise a therapeutically effective amount of compound of formula I in
combination with a
pharmaceutically acceptable carrier.
In yet another aspect, the present invention provides a method of inhibiting
matrix
metalloproteinases and/or TNFa secretion in a host mammal in need of such
treatment comprising
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
12
administering to a mammal in need of such treatment a therapeutically
effective amount of a
compound of formula I.
Detailed Description
As used throughout this specification and the appended claims, the following
terms have
the meanings specified.
The term alkyl refers to a monovalent group derived from a straight or
branched chain
saturated hydrocarbon by the removal of a single hydrogen atom. Alkyl groups
are exemplified by
methyl, ethyl, n.- and iso-propyl, n.-, sec-, iso- and ter-r-butyl, and the
like.
The term "alkanoyl" represents an alkyl group, as defined above, attached to
the parent
molecular moiety through a carbonyl group. Alkanoyl groups are exemplified by
formyl, acetyl,
propionyl, butanoyl and the like.
The terms alkoxy and alkoxyl denote an alkyl group. as defined above, attached
to the
parent molecular moiety through an oxygen atom. Representative alkoxy groups
include methoxy,
ethoxy, propoxy, butoxy, and the like.
The term "allcoxycarbonyl" represents an ester group; i.e. an alkoxy group,
attached to the
parent molecular moiety through a carbonyl group such as methoxycarbonyl,
ethoxycarbonyl, and
the like.
The term alkenyl as used herein refer to monovalent straight or branched chain
groups of 2
to 6 carbon atoms containing a carbon-carbon double bond, derived from an
alkene by the removal
of one hydrogen atom and include, but are not limited to groups such as
ethenyl, 1-propenyl, 2-
propenyl, 2-methyl-1-propenyl, 1-butenyl, 2-butenyl and the like.
The term alkylene denotes a saturated divalent hydrocarbon group derived from
a straight
or branched chain saturated hydrocarbon containing by the removal of two
hydrogen atoms) for
example -CH2-, -CH2CH2-, -CH(CH3)CH2- and the like.
The term alkenylene denotes a divalent group derived from a straight or
branched chain
hydrocarbon containing at least one carbon-carbon double bond. Examples of
alkenylene include
CH=CH-, -CH2CH=CH-, -C(CHg)=CH-, -CH2CH=CHCH2-, and the like.
The terms alkynylene refers to a divalent group derived by the removal of two
hydrogen
atoms from a straight or branched chain acyclic hydrocarbon group containing
at least one carbon-
carbon triple bond. Examples of alkynylene include -CH~H-, -CH--_C-CH2-, -CH-
CH-
CH(CH3)- and the like.
The term "benzyloxy" as used herein refers to -O-(CH2)-phenyl.
The term cycloalkyl as used herein refers to a monovalent saturated cyclic
hydrocarbon
group. Representative cycloalkyl groups include cyclopropyl, cyclobutyl,
cyclopentyl,
cyclohexyl, cycloheptyl, bicyclo[2.2.1 ]heptane and the like.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
13
Cycloalkylene denotes a divalent radical derived from a cycloalkane by the
removal of two
hydrogen atoms.
The terms "(cycloalkyl)alkyl" and "(cycloalkenylene)alkyl" refer,
respectively, to a
cycloalkyl group or cycloalkenylene group as defined above attached to the
parent molecular
moiety through an alkylene group.
The term cyanoatkyl denotes an alkyl group, as defined above, substituted by a
cyano
group and includes, for example, cyanomethyl, cyanoethyl, cyanopropyl and the
like.
The term haloallcyl denotes an alkyl group, as defined above, having one, two,
or three
halogen atoms attached thereto and is exemplified by such groups as
chloromethyl, bromoethyl,
trifluoromethyl, and the like.
The term "hydroxyalkyl" represents an alkyl group, as defined above)
substituted by one to
three hydroxyl groups with the proviso that no more than one hydroxy group may
be attached to a
single carbon atom of the alkyl group.
The term "phenoxy" refers to a phenyl group attached to the parent molecular
moiety
through an oxygen atom.
By pharmaceutically acceptable salt is meant those salts which are, within the
scope of
sound medical judgement, suitable for use in contact with the tissues of
humans and lower animals
without undue toxicity, irritation, allergic response and the like, and are
commensurate with a
reasonable benefit/risk ratio. Pharmaceutically acceptable salts are well
known in the art . For
example, S. M Berge, et al. describe pharmaceutically acceptable salts in
detail in J.
Pharmaceutical Sciences,1977, 66:1 - 19 . The salts can be prepared in situ
during the final
isolation and purification of the compounds of the invention) or separately by
reacting the free base
function with a suitable organic acid. Representative acid addition salts
include acetate, adipate,
alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate,
butyrate, camphorate,
camphersulfonate, citrate, cyclopentanepropionate, digluconate,
dodecylsulfate, ethanesulfonate,
fumarate, glucoheptonate, glycerophosphate, hemisulfate, heptonate, hexanoate,
hydrobromide,
hydrochloride, hydroiodide, 2-hydroxy-ethanesulfonate, lactobionate, lactate,
laurate, lauryl
sulfate, malate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate,
nicotinate, nitrate,
oleate, oxalate, palmitate, pamoate, pectinate, persulfate, 3-
phenylpropionate, phosphate, picrate,
pivalate, propionate, stearate, succinate, sulfate, tartrate, thiocyanate,
toluenesulfonate,
undecanoate, valerate salts, and the like. Representative alkali or alkaline
earth metal salts include
sodium, lithium, potassium, calcium, magnesium, and the Like, as well as
nontoxic ammonium,
quaternary ammonium, and amine cations, including, but not limited to
ammonium)
tetramethylammonium, tetraethylammonium, methylamine, dimethylamine)
trimethylamine)
triethylamine, ethylamine, and the like.
As used herein, the term "pharmaceutically acceptable ester" refers to esters
which
hydrolyze in vivo and include those that break down readily in the human body
to leave the parent
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
14
compound or a salt thereof. Suitable ester groups include, for example, those
derived from
pharmaceutically acceptable aliphatic carboxylic acids, particularly alkanoic,
alkenoic,
cycloalkanoic and alkanedioic acids, in which each alkyl or alkenyl moiety
advantageously has not
more than 6 carbon atoms. Examples of particular esters includes formates)
acetates, propionates,
butyates, acrylates and ethylsuccinates.
The term "pharmaceutically acceptable prodrugs" as used herein refers to those
prodrugs of
the compounds of the present invention which are, within the scope of sound
medical judgement,
suitable for use in contact with with the tissues of humans and lower animals
with undue toxicity,
irritation, allergic response, and the like, commensurate with a reasonable.
benefit/risk ratio, and
effective for their intended use, as well as the zwitterionic forms, where
possible, of the
compounds of the invention. The term "prodrug" refers to compounds that are
rapidly transformed
in vivo to yield the parent compound of the above formula, for example by
hydrolysis in blood. A
thorough discussion is provided in T. Higuchi and V. Stella, Pro-druQS as
Novel Delivery
Systems, Vol. 14 of the A.C.S. Symposium Series) and in Edward B. Roche, ed.,
Bioreversible
Carriers in Dru Design, American Pharmaceutical Association and Pergamon
Press, 1987, both
of which are incorporated herein by reference.
Asymmetric centers may exist in the compounds of the present invention. The
present
invention contemplates the various stereoisomers and mixtures thereof.
Individual stereoisomers
of compounds of the present invention are made by synthesis from starting
materials containing the
chirai centers or by preparation of mixtures of enantiomeric products followed
by separation as,
for example, by conversion to a mixture of diastereomers followed by
separation by
recrystallization or chromatographic techniques, or by direct separation of
the optical enantiomers
on chiral chromatographic columns. Starting compounds of particular
stereochemistry are either
commercially available or are made by the methods detailed below and resolved
by techniques well
known in the organic chemical arts.
Preferred Embodiments
Preferred compounds of the present invention have formula II
O R'R2 Rs
~~,~~ N ~~Z
Y O L2
~'-A
II
wherein W and L2 are defined above,
Y is absent or -O-;
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
RI and R3 are H;
L1 is alkylene of two to six carbon atoms;
. A is selected from the group consisting of
(a) -O-,
5 (b) -NR9- wherein R9 is selected from the group consisting of
( 1 ) hydrogen,
(2) alkyl of one to four carbon atoms,
(3) -C02RI~ wherein Rl~ is independently selected at each occurrence from the
group consisting
of alkyl of one to four carbon atoms, phenyl, phenyl substituted with 1, 2, or
3 substituents
10 independently selected from alkyl of one to four carbon atoms, alkoxy of
one to four carbon
atoms, halogen, haloalkyl of one to four carbon atoms, vitro, cyann,
cyanoalkyl, -S02NH2, -
COZR4, and -CONR4R5, phenylalkyl wherein the alkylene portion is of one to
four carbon atoms,
phenylalkyl wherein the alkylene portion is of one to four carbon atoms, and
the phenyl ring is
substituted with 1, 2, or 3 substituents independently selected from alkyl of
one to four carbon
15 atoms, alkoxy of one to four carbon atoms, halogen, haloalkyl of one to
four carbon atoms) cyano.
cyanoalkyl, -S02NH2, -COZR4, and -CONR4R5, and
(4) -S02R10,
(c) -CH=CH-)
(d)
O
~N~
Ra
(e)
O
~N~N~
R4 RS , and
(fj
Ra
~'N~N
a
R , wherein Rd and Re are independently selected from hydrogen and alkyl of
one to four carbon atoms, provided that when A is (f) above, L2 is allrylene;
CA 02277121 1999-07-06
WO 98/30551 PCTlUS98l00144
16
R2 is selected from the group consisting of isobutyl, cyclohexyl,
cyclopentylmethyl
phenyl, 3-(4-tolyl)propyl) 3-(4-chlorophenyl)propyl, 2-(4-propylphenyl)ethyl,
3-
benzyloxypropyl, 4-phenoxybutyl) 4-(4-butylphenoxy)butyl, 4-biphenyloxy, and _
2-{4-(4'cyano)biphenyloxy)ethyl; and
Z is absent or is selected from the group consisting of
(a) -CO~H,
(b) -C02R ~ ~,
(c)
O
~NR~3R~° wherein R13 is h dro en or ally 1 of one to six carbon at m
Y g Y o s, and
R14 is selected from the group consisting of
(1) hydrogen,
(2) alkyl of one to four carbon atoms,
(3) 2-phenylethyl,
(4) 2-{4-aminosulfonyl)phenylethyl,
(S) cyclopropyl,
(6) phenyl,
(7) phenylsulfonyl,
(8) 2-thiomethylethyl,
(9) 2-dimethylaminoethyl,
(10) -(CH~)~OCH20(CH2)20CH3)
( 11 ) 2-morpholinylethyl)
(12) 4-pyridinylethyl,
(13) 2-furylmethyl,
(14) 2-pyridyl)
(15) 2-thiazolyl,
Rls
O
(16) ~ wherein R15 is hydrogen, and
O
NHCH3
( 1 ~) CH3
or R 13 and R 14, together with the N atom to which they are attached define a
5-or 6-membered
heterocyclic ring selected the group consisting of morpholinyl, pyrrolidinyl,
piperidinyl, and
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
17
~N
' C02R16 "',herein R~6 is hydrogen or benzyl,
(d)
- V
R 1~ wherein V is defined above and R ~ ~ i
s selected from the group consisting of
( 1 } phenyl)
(2) phenyl substituted with alkyl of one to four carbon atoms, methanesulfonyl
or
dimethylaminomethyl,
(3) 3-indolyl,
(4) 2-pyrrolyl, and
(5) 1-dimethylaminocarbamoylindol-3-yl,
(e)
N
N
H and
N
N
H
More preferred compounds of the present invention have formula II wherein W is
-NHOH.
Still more preferred compounds have formula II wherein Y, R~, R3, Ll and A are
defined
above; R2 is selected from isobutyl, 3-(4-tolyl)propyl, 2-(4-
propylphenyI)ethyl; and Z is absent or
is selected from the group consisting of -C02H, -C02CH3, -C02benzyl, -CONHCH3,
-
CON(CH3)2,
S02NH2 ~ ( O
O
N ~ N~ ~ ~ SCH3 N CH
H H H ~ N~ ( s)2
H
> >
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
18
O
~ \ I ~ ~ ~N'1
N N N N S \ 'O
H ~ H ~ H , H V
O .N O O O
I\ I\ I\
C02Rts / ~S02CH' / t-Bu
, , , , ,
/ I
O O \ l \ \
N I ~ N "'t/N I / ~/ I
N
H , H , H , and H
Still yet more preferred compounds have the formula II wherein W is -NHOH and
Z is -
CONHCH3.
The most preferred compounds of the present invention have formula III
R2
Ot' / H
~,~~N~~Z
L~-~ IIO ~a
\
III
wherein
W is NHOH;
L1 is alkylene of two to six carbon atoms;
L3 is absent or methylene;
A is selected from the group consisting of
(a) -O-,
(b) -NR9- wherein R9 is selected from hydrogen, -C02benzyl,-S02CH3, -SOZ-(4-
tolyl),
(c) -CH=CH-, and
(d) -C(O)NH-;
R2 is selected from isobutyl) 3-(4-tolyl)propyl, 2-(4-propylphenyl)ethyl; and
.
Z is is selected from the group consisting of -CONHCH3, -CON(CH3)2,
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
19
O
0
~\
and
Determination of Stromelvsin Inhihitinn
The efficacy of the compounds of this invention as matrix metalloproteinase
inhibitors was
determined by measuring the inhibition of stromelysin. The inhibition of
stromelysin by the
compounds of this invention was determined as follows: Recombinant truncated
stromelysin
(human sequence) produced in E. coli was prepared by expression and
purification of the protein
as described by Ye et al., Biochemistry , 1992, 3J, 11231-11235. The enzyme
was assayed by
its cleavage of the thiopeptide ester substrate Ac-Pro-Leu-Gly-[2-mercapto-4-
methyl-pentanoyl]-
Leu-Gly-OEt described by Weingarten and Feeler, Anal. Biochem. , 1985,147, 437-
440 ( 1985),
as a substrate of vertebrate collagenase. The reported conditions were
modified to allow assays to
be earned out in a microtiter plate. Upon hydrolysis of the thioester bond)
the released thiol group
reacts rapidly with 5,5'-dithio-bis(2-nitrobenzoic acid) (DTNB), producing a
yellow color which is
measured by a microtiter plate reader set at 405 nm. The rates of cleavage of
the substrate by
stromelysin in the presence or absence of inhibitors are measured in a 30 min
assay at ambient
temperature. Solutions of the compounds in DMSO are prepared, and these are
diluted at various
concentrations into the assay buffer (50 mM MES/NaOH pH 6.5 with 10 mM CaCl2
and 0.2%
Pluronic F-68), which is also used for dilution of the enzyme and substrate.
The potency of the
compounds [ICSp] are calculated from the inhibition/inhibitor concentration
data. The compounds
2U of this invention inhibit stromelysin as shown by the data for
representative examples in Table 1.
Table 1
Inhibitory Potencies against Stromelysin of Representative Compounds
Exam le ICsp (nM)
6.9
2 7.2
5.8
4 2.4
9.3
6 2.6
3.6
8 s n 570
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
8 anti 3.9
9 2100
10 7.8
11 6.1
12 12
13 2.4
14 2.7
15 7.2
16 23
18 0.61
19 20
20 92
21 39
23 8.1
24 g.g
44
26 27
20
31 2.9
32 6.7
34 1.5
38 0.56
41 26
Pharmaceutical Compositions
The present invention also provides pharmaceutical compositions which comprise
compounds of the present invention formulated together with one or more non-
toxic
5 pharmaceutically acceptable carriers. The pharmaceutical compositions may be
specially
formulated for oral administration in solid or liquid form, for parenteral
injection, or for rectal
administration.
The pharmaceutical compositions of this invention can be administered to
humans and
other animals orally, rectally, parenterally , intracisternally,
intravaginally, intraperitoneally,
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
21
topically (as by powders) ointments, or drops), bucally, or as an oral or
nasal spray. The term
"parenteraI" administration as used herein refers to modes of administration
which include
intravenous, intramuscular, intraperitoneal, intrasternal, subcutaneous and
intraarticular injection
and infusion.
Pharmaceutical compositions of this invention for parenteral injection
comprise
pharmaceutically acceptable sterile aqueous or nonaqueous solutions)
dispersions, suspensions or
emulsions as well as sterile powders for reconstitution into sterile
injectable solutions or
dispersions just prior to use. Examples of suitable aqueous and nonaqueous
carriers, diluents,
solvents or vehicles include water, ethanol, polyols (such as glycerol,
propylene glycol,
polyethylene glycol, and the like), and suitable mixtures thereof, vegetable
oils (such as olive oil),
and injectable organic esters such as ethyl oleate. Proper fluidity can be
maintained, for example,
by the use of coating materials such as lecithin, by the maintenance of the
required particle size in
the case of dispersions, and by the use of surfactants.
These compositions may also contain adjuvants such as preservative, wetting
agents,
l5 emulsifying agents, and dispersing agents. Prevention of the action of
microorganisms may be
ensured by the inclusion of various antibacterial and antifungal agents, for
example, paraben,
chlorobutanol, phenol sorbic acid, and the like. It may also be desirable to
include isotonic agents
such as sugars, sodium chloride, and the like, Prolonged absorption of the
injectable
pharmaceutical form may be brought about by the inclusion of agents which
delay absorption such
as aluminum monostearate and gelatin.
In some cases, in order to prolong the effect of the drug, it is desirable to
slow the
absorption of the drug from subcutaneous or intramuscular injection. This may
be accomplished
by the use of a liquid suspension of crystalline or amorphous material with
poor water solubility.
The rate of absorption of the drug then depends upon its rate of dissolution
which, in turn, may
depend upon crystal size and crystalline form. Alternatively, delayed
absorption of a parenterally
administered drug form is accomplished by dissolving or suspending the drug in
an oil vehicle.
Injectable depot forms are made by fornvng microencapsule matrices of the drug
in
biodegradable polymers such as polylactide-polyglycolide. Depending upon the
ratio of drug to
polymer and the nature of the particular polymer employed, the rate of drug
release can be
controlled. Examples of other biodegradable polymers include poly(orthoesters)
and
poly(anhydrides) Depot injectable formulations are also prepared by entrapping
the drug in
' liposomes or microemulsions which are compatible with body tissues.
The injectable formulations can be sterilized, for example, by filtration
through a bacterial-
retaining filter, or by incorporating sterilizing agents in the form of
sterile solid compositions
which can be dissolved or dispersed in sterile water or other sterile
injectable medium just prior to
use.
CA 02277121 1999-07-06
WO 98130551 PCT/US98100144
22
Solid dosage forms for oral administration include capsules, tablets, pills,
powders, and
granules. In such solid dosage forms, the active compound is mixed with at
least one inert,
pharmaceutically acceptable excipient or carrier such as sodium citrate or
dicalcium phosphate
and/or a) fillers or extenders such as starches, lactose, sucrose, glucose,
mannitol, and silicic acid,
S b) binders such as, for example, carboxymethylcellulose, alginates, gelatin,
polyvinylpyrrolidone,
sucrose, and acacia, c) humectants such as glycerol, d) disintegrating agents
such as agar-agar,
calcium carbonate, potato or tapioca starch, alginic acid, certain silicates,
and sodium carbonate, e)
solution retarding agents such as paraffin, f) absorption accelerators such as
quaternary ammonium
compounds, g) wetting agents such as, for example, cetyl alcohol and glycerol
monostearate, h)
l0 absorbents such as kaolin and bentonite clay, and i) lubricants such as
talc, calcium stearate,
magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate, and
mixtures thereof. In
the case of capsules, tablets and pills, the dosage form may also comprise
buffering agents.
Solid compositions of a similar type may also be employed as fillers in soft
and hard-filled
gelatin capsules using such excipients as lactose or milk sugar as well as
high molecular weight
15 polyethylene glycols and the like.
The solid dosage forms of tablets, dragees) capsules, pills, and granules can
be prepared
with coatings and shells such as enteric coatings and other coatings well
known in the
pharmaceutical formulating art. They may optionally contain opacifying agents
and can also be of
a composition that they release the active ingredients) only, or
preferentially, in a certain part of
20 the intestinal tract, optionally, in a delayed manner. Examples of
embedding compositions which
can be used include polymeric substances and waxes.
The active compounds can also be in micro-encapsulated form, if appropriate,
with one or
more of the above-mentioned excipients.
Liquid dosage forms for oral administration include pharmaceutically
acceptable emulsions,
25 solutions, suspensions, syrups and elixirs. In addition to the active
compounds, the liquid dosage
forms may contain inert diluents commonly used in the art such as, for
example, water or other
solvents) solubilizing agents and emulsifiers such as ethyl alcohol, isopropyl
alcohol, ethyl
carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, I
,3-butylene glycol,
dimethyl formamide, oils (in particular, cottonseed, groundnut, corn, germ,
olive, castor, and
30 sesame oils)) glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols
and fatty acid esters of
sorbitan, and mixtures thereof.
Besides inert diluents, the oral compositions can also include adjuvants such
as wetting
agents, emulsifying and suspending agents, sweetening, flavoring, and
perfuming agents.
Suspensions, in addition to the active compounds, may contain suspending
agents as, for
35 example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and
sorbita.n esters,
microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar) and
tragacanth, and
mixtures thereof.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
23
Compositions for rectal or vaginal administration are preferably suppositories
which can be
prepared by mixing the compounds of this invention with suitable non-
irritating excipients or
carriers such as cocoa butter, polyethylene glycol or a suppository wax which
are solid at room
temperature but liquid at body temperature and therefore melt in the rectum or
vaginal cavity and
release the active compound.
Compounds of the present invention can also be administered in the form of
liposomes.
As is known in the art, liposomes are generally derived from phospholipids or
other lipid
substances. Liposomes are formed by mono- or mufti-lamellar hydrated liquid
crystals that are
dispersed in an aqueous medium. Any non-toxic, physiologically acceptable and
metabolizable
lipid capable of forming liposomes can be used. The present compositions in
liposome form can
contain, in addition to a compound of the present invention, stabilizers,
preservatives, excipients,
and the like. The preferred lipids are the phospholipids and the phosphatidyl
cholines (lecithins),
both natural and synthetic.
Methods to form liposomes are known in the art. See, for example, Prescott)
Ed.,
I S Methods in Cell Biolo~v_, Volume XIV, Academic Press, New York, N.Y.
(1976), p. 33 et seq.
Dosage forms for topical administration of a compound of this invention
include powders,
sprays, ointments and inhalants. The active compound is mixed under sterile
conditions with a
pharmaceutically acceptable carrier and any needed preservatives, buffers, or
propellants which
may be required. Opthalmic formulations, eye ointments, powders and solutions
are also
contemplated as being within the scope of this invention.
Actual dosage levels of active ingredients in the pharmaceutical compositions
of this
invention may be varied so as to obtain an amount of the active compounds)
that is effective to
achieve the desired therapeutic response for a particular patient,
compositions, and mode of
administration. The selected dosage level will depend upon the activity of the
particular
compound) the route of administration, the severity of the condition being
treated, and the
condition and prior medical history of the patient being treated. However, it
is within the skill of
the art to start doses of the compound at levels lower than required for to
achieve the desired
therapeutic effect and to gradually increase the dosage until the desired
effect is achieved.
Generally dosage levels of about 1 to about 50, more preferably of about 5 to
about 20 mg
of active compound per kilogram of body weight per day are administered orally
to a mammalian
patient. If desired, the effective daily dose may be divided into multiple
doses for purposes of
administration, e.g. two to four separate doses per day.
Preparation of Cornnounds of this L vention
The compounds of this invention may be prepared by a variety of synthetic
routes.
Representative procedures are outlined in the following Schemes 1-10. It is
understood that while
the following schemes describe the preparation of macrocycles predominately
derived from
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
24
tyrosine, the substitution of any of a number of both natural and unnatural
amino acids will result
in the formation of the desired macrocycles.
Abbreviations which have been used in the descriptions of the schemes and the
examples
that follow are: THF for tetrahydrofuran; DMF for N.N-dimethylformamide; ETOAc
for ethyl
acetate; Et20 for diethyl ether, IPA for isopropanol; ETON for ethanol; MeOH
for methanol; AcOH
for acetic acid; HOBT for 1-hydroxybenzotriazole hydrdate; EDC for 1-(3-
dimethylamin opropyl)-
3-ethylcarbodiimide hydrochloride; NMM for N-methyhnorpholine; Bu3P for
tributylphosphine;
ADDP for 1,1'-(azodicarbonyl)dipiperidine; and DMPU for 1,3-dimethyl-3,4,5,6-
tetrahydro-
2( 1H)-pyrimidinone.
The preparation of compounds of formula _6, wherein W is -OH and 7, wherein W
is -
NHOH and R 1, R2, R3, R 13 and R 14 are defined above is described in Schemes
1 a and b.
According to Scheme Ia, coupling of acid 1 with amino amide _2 in the presence
of an tertiary
amine base, hydroxybenzotriazole (HOBt), and a suitable coupling agent such as
1-(3-
dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI~HCl) provides
amide 3_.
Hydroboration of 3 using, for example, a tetrahydrofuran solution of borane
followed by work up
with aqueous hydrogen peroxide gives alcohol 4. Cyclization of 4 can be
achieved using
Mitsunobu conditions (Mitsunobu et al., J. Am. Chem. Soc., 1972, 94, 679}. For
example
addition of 4 to a solution of triphenylphosphine and diethylazodicarboxylate
gives macrocycle S_.
Conversion of 5_ to the corresponding earboxylic acid C is accomplished by
acidic removal of the
tent-butyl ester with, for example, trifluoroacetic acid or hydrogen chloride
in dioxane. Treatment
of this acid with hydroxylamine or a hydroxylamine equivalent such as D-tert-
butyldimethylsilylhydroxylamine in the presence of a suitable coupling agent
such as EDCI~HCl
gives hydroxamate 7. O-Benzylhydroxylamine can also be employed in this
coupling reaction.
The resulting0-benzylhydroxamate can then be treated with hydrogen and a
palladium catalyst such
as 10% palladium on carbon to produce hydroxamate Z.
CA 02277121 1999-07-06
WO 98130551 PC"T/US98100144
Scheme la
O R~ R2 Ra0
O OH + HpN NRi3R~a
O
/ HO ~
1_
O R~R2 H Ra0 ~ OR~R2 H Ra0
O NR'aR,a ~ N NR~aR~4
O O
/
HO
HO HO
$ 4
~ O R~ 2 H Ra0 O R~ R2 H R30
N NR~aR~a N NR~aR~a
--~ O
O
O
$ ~ I't: W = -OH
-~ Z: W = -NHOH
5 An alternative route to hydroxamate 7, as outlined in Scheme lb, involves
reaction of acid
1 with the benzyl ester analog of 2 under the conditions used to prepare ~.
The resulting amide 8 is
subjected to the cyclization conditions described above followed by
hydrogenolytic removal of the
benzyl ester giving carboxylic acid ~. Treatment of _9 with EDCI~HCI, HOBt, N-
methylmorpholine, and a primary or secondary amine of the formula HNR13R14
gives amide 5
10 which can be converted to hydroxamate 7 as described above.
Scheme lb
z
O R~ R N Ra0 ~ R~ R2 H Ra0
O~ Ph
O ----... O
HO ~ O I i
$ $
15 Preparation of intermediate _2 is accomplished by treating commercially
available acid 10
with the requisite amine of general formula HNR13R~4 using) for example,
EDCI~HCI) HOBt, and
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
26
N-methylmorpholine as shown in Scheme 2. The resulting amide 11 is subjected
to acidic removal
of the N-t-butoxycarbonyl nitrogen protecting group using trifluoroacetic acid
or hydrogen chloride
in dioaxane giving amide 2_. Amino ketones of the general formula 15 are
prepared by treating acid
12 with ethereal diazomethane to produce methyl ester 13. This compound is
subsequently reacted
with a anion such as R 1 ~MgX wherein X is Br, Cl or I, or R 1 ~Li to generate
ketone 14. Acidic
removal of the tert-butyl protecting groups gives amino ketone 15.
Alternatively, carboxylic acid
12 can be treated with a carbon anion such as phenyllithium which gives 14
directly. Amino
ketone 15 can be used in place of amino amide 2 in Scheme 1 for the
preparation of macrocyclic
compounds where "Z" _ -COR1~.
Scheme 2
H Rs0 H Rs0
~~N OH + HNR~3R~a------ ~O~N NR~3R'" . 2_
O O
HO
HO
H R30 H 30
~O~ N R Oi
1OI .-.. IOI ---.
1~
O N R30 R30
R » H2N R
O
HO
~4 1~
The preparation of intermediate 1 is shown in Scheme 3. Treatment of
oxazolidinone ~
with a suitable base such as lithium diisopropylamide followed by addition of
tort-butyl
bromoacetate and basic hydrolysis gives carboxylic acid ~, 7. This acid is
treated with at least two
equivalents of a strong base such as lithium diisopropylamide followed by an
alkenyl halide such
as 4-bromo-1-butene. The resulting dialkyl succinate 1_$ is again treated with
a strong base such as
lithium diisopropylamide followed by either methanol (R1 = H) or an alkyl
halide (R1= alkyl) such
as methyl iodide giving substituted succinate ~.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
27
Scheme 3
0,'
R2 ~O -~ ~ O R2 ~ O R2 O ~R2
N O~ ~ ~ O OH ---. ~ O R OH
O ~ fOI O
Ph O
~ 1$ i 1
Preparation of compounds of this invention where "Y" is -O- is shown in Scheme
4. The
known acetonide 19 (British Biotechnology PCT application WO 94/02446) is
first treated with a
base such as potassium carbonate then with an alkenyl halide such as allyl
bromide. Acidic
removal of the acetonide group using, for instance, aqueous hydrogen chloride
gives the
corresponding hydroxy acid which is subjected to treatrnent with a base such
as potassium
carbonate and benzyl bromide giving alcohol ZQ. O-Alkylation of 2Q using
sodium hydride and
allyl bromide followed by palladium catalyzed ester deprotection using, for
instance,
tetrakis(triphenylphosphine)palladium (0) gives allyl ether 21. Coupling of 21
with 2, followed by
hybroboration and cyclization as described in Scheme 1 above provides
macrocycle 22.
Hydrogenolytic removed of the benzyl ester using, for instance, hydrogen and
10% palladium on
carbon gives acid _2~ which can be converted to macrocyclic hydroxamate 24 as
described above.
Scheme 4
O R2 O R2 O R2
O~ ~ ~ Phi O~~ ~ ----~ Phi O%~ OH
~O O OH O O O
O R, R2 H Ra0 O R~R2 H Ra0
_.~. Ph~C1'~~N NR~3Rta ~ V1T~ NR~3R~a
O O O O
O ~ ~ i ~ 2,~: W - -OH
--~ ~ø: W = -NHOH
Benzimidazole-containing macrocycles are prepared according to Scheme 5. o-
Amino
amide 2~, prepared as described in Scheme 1 wherein HNR13R14 is 1,2-
phenylenediamine, is
heated with an acid such as camphor sulfonic acid to generate benzimidazole
2~. Conversion of
this compound to the corresponding carboxylic acid 27 and hydroxamate ?$ is
accomplished by
analogy with the sequence shown in Scheme 1.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
28
Scheme 5
H2N
O R~ R2 N R30 \ I O R~ 2 N R3 ' w
O _H O ~H
,2~: W = -Ot butyl
(\
O i O i ~ ?'Z: W = -OH
~,$: W = -NHOH
Macrocyclic olefins such as ,3Q are prepared by treating aryl iodide 29 with a
suitable
palladium catalyst, for instance tetrakis(triphenylphosphine)pallaium (0), and
an amine base such
as triethylamine and heating in a solvent such as acetorutrile. Olefln ,~0 is
converted to the
corresponding acid 31 and hydroxamate ~ according to the sequence outlined in
Scheme 1.
Scheme 6
2 2
O RJR H R3Z O RiR H R3Z
O
O O
I ~ I ~ ~ $Q: W = -Ot butyl
,~: W = -OH
~2: W = -NHOH
Tryptophan-derived macrocycles are prepared according to Scheme 7. Alcohol ,~
is
converted to a suitable leaving group, for example by reaction with p-
toluenesulfonyl chloride in
the presence of a tertiary base such as pyridine to give tosylate ~_4. This
compound is subjected to
phase-transfer alkylation conditions using) for example, potassium hydroxide
and benzyltrimethyl
ammonium chloride in a mixture of water and methylene chloride. The resulting
macrocyclic ester
~ is converted to the corresponding acid ~ø and hydroxamate ~ according to the
sequence
outlined in Scheme 1.
Scheme 7
2 z
O R~ N RaZ O R~ N RsZ
O O
N ~ ~: W = -Ot butyl
,~: X = -OH - ~ ~: W = -OH
H
,fig: X = -OTs
~: W = -NHOH
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
29
p-Aminophenylalanine-derived compounds of this invention are prepared as
outlined in
Scheme 8. Alcohol 38 is first converted to its mesylate using methanesulfonyl
chloride and a
tertiary amine base such as triethylamine. Hydrogenation of this material
using 10% palladium on
' carbon and triethylamine in a solvent such as iso-propanol generates
macrocyclic ester ~9 directly.
S Conversion to acid ~ and hydroxamate 41 is accomplished by the reaction
sequence shown in
Scheme 1. Alcohol 38 can also be oxidized to acid 42 using, for example)
chromic acid in sulfuric
acid. Hydrogenation of the aromatic nitro group is achieved using hydrogen
over a palladium
catalyst. Lactam formation is completed by treatment with a coupling agent
such as bis(2-oxo-3-
oxazolidinyl)phosphinic chloride (BOP-CI) in the presence of a tertiary amine
base such as
triethylamine giving 4~ which is converted to the corresponding acid 44 and
hydroxamate 45 as
outlined in Scheme 1.
Scheme 8
v R'rc H R3Z O R~ z H R3
O ~ --~) Z
O O
HO - _ ~ ~Q: W = -Ot butyl
\ / \ / 4Q: W = -OH
~,: W = -NHOH
z
O R~ R H Rs Z O R~ R2 H R3
O ~ N Z
0 0 ~ 4~: W = -Or-butyl
HO - - ~ ~: W = -OH
O
02N N \ / 4~: W = -NHOH
H
Carbamate and urea-derived macrocycles can be prepared according to Scheme 9.
Alcohol
~ is treated with bromoacetyl bromide in the presence of sodium carbonate. The
resulting ester
is subjected to hydrogenation conditions using, for example 10% palladium on
carbon in the
presence of a tertiary amine base such as triethylamine which gives macrocycle
4_$. The tert-butyl
ester group of ~, can be converted to acid 4_~ and to hydroxamate S,Q under
the conditions
illustrated in Scheme 1. Alcohol 4~ is converted to the corresponding
methanesulfonate ,5~ by
reaction with methanesulfonyl chloride in the presence of triethylamine.
Mesylate ,5~ is reacted
with trimethylsilyl azide in the presence of tri-n-butylammonium fluoride to
produce azide ,5,~.
This compound can be subjected to hydrogen over a palladium catalyst and the
resulting diamine
treated with a phosgene derivative such as carbonyldiimidazoIe to generate
urea 5,~,~. Conversion of
CA 02277121 1999-07-06
WO 98/30551 PC'T/US98J00144
this ester to the corresponding acid ~4 and hydroxamate 55 is accomplished as
described in
Scheme 1.
Scheme 9
R, rs_ H R3 R, ' H Rs
O N Z N Z
X l O '--~ O
'~~..JJ( ~ ~$: W = -Ot-butyl
O O H
~: X = -OH Ph~O~ N O~/ ~: W ° -OH
~: X = -OCOCH2Br
~Q: W = -NHOH
2
O R' N R3 Z R~ 2 H Rs
p ~ N Z
X O ---~ O
O ~ ;~: W = -Ot butyl
~,: X = -OMS Ph~O~ N
;L4: W = -OH
N
;2~,: X = -N3 H N H : W =
-NHOH
5
The preparation of macrocyclic lactones is shown in Scheme 10. Hydrogenolysis
of the
benzyl ester of 55 is achieved using hydrogen over a palladium catalyst. The
resulting hydroxy
acid is treated with 1,1'-(azodicarbonyl)dipiperdine and tributylphosphine in
a suitable solvent
such as tetrahydrofuran to produce 56. This ester can be converted to the
corresponding acid ~7
10 and hydroxamate 58 as described in Scheme 1.
Scheme 10
z
O RJR H RaZ O R,R2 H Rs
Z
O ~ o ~ ~: W = -Ot-butyl
~: W = -OH
~Ph
O
5,~: W = -NHOH
The foregoing may be better understood by reference to the following examples
which are
presented for illustration and are not intended to limit the scope of the
invention as defined in the
appended claims.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
31
Preparation of Succina P Ester I
0
t-Bu0 OH
O
Steg 1
O O
OH SOCK
~ CI
A mixture of 4-methylvaleric acid (50.7 g, 0.43 mmol) and thionyl chloride (40
mL, 65.2
g, 0.54 mole) was stirred at ambient temperature for 18 hours. The mixture was
heated to distill
the excess reagent through a IO cm Vigreux column. The acid chloride was then
distilled to give i
IO (48.43 g, 84 %), by 135-138 °C.
to 2:
O o 0 0
N ~O + n-BuLi + ~ CI ----,.
! ll
/ ~ /
To a -78 °C solution of 4S-benzyl-2-oxazolidinone (62.2 g, 0.35 mole)
in THF (600 mL)
was addedn-butyllithium (140 mL, 2.5 M in hexane) over 1 hour. After 30
minutes ~ (0.359 mole)
was added over 10 minutes during which time the temperature rose to -60
°C. After 1 hour the
bath was removed and the reaction mixture was warmed to 0 °C. The
reaction was Quenched with
saturated ammonium chloride, the mixture was allowed to settle, and the
supernatant was decanted
and concentrated. The combined residues were partitioned between water and
ethyl acetate. The
organic layer was washed with water, 1 M sodium bicarbonate, water and brine,
dried over sodium
sulfate, filtered and concentrated. The residue was distilled discarding a
small forerun to give g
(92.9 g, 96%), by 154-156 °C / 0.15 mm.
0 o
o
O + NaN(TMS)2 + ~ JO( O
ii B~~ O-t Bu i t-Bu0
~ / ~,i o
. ~
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
32
To a mechanically-stirred -78 °C solution of ii (92.9 g, 0.337 mole) in
THF ( I L} was
added sodium bis(trimethylsilyl)amide (375 mL, 1 M in THF) over 40 minutes.
The reaction
mixture was stirred for 30 minutes and t-butyl bromoacetate (55 mL, 72.6 g,
0.372 mole)was
added over 30 minutes. The reaction mixture was stirred for 30 minutes and
then the cold bath
was removed and the mixture was warmed to 0 °C. The reaction was
quenched with saturated
ammonium chloride. After mixing well, the mixture was allowed to settle and
the supernatant was
decanted, concenuated, and recombined with the residue. This mixture was
partitioned between
water and ethyl acetate. The organic layer was washed with water, 1 M sodium
bicarbonate, water
and brine, dried over sodium sulfate and concentrated by distillation to about
250 mL. After
dilution with 750 mL hexane and cooling in an ice bath the resulting crystals
were collected and
washed with hexane to provide iii ( 104.6 g) mp I 0 I -102 °C. The
mother liquors were
concentrated and the residue was purified by chromatography on silica gel (5 -
10% ethyl acetate -
hexane) and the product fraction crystallized to yield 7.6 g more for a total
of 112.2 g (85 %).
Step 4:
0
0 0 ''
O NH~ O
+ LiOH + H202 -..~. ~/
t-Bu0 OH + _
t-Bu0
O iv O
To a 0 °C solution of iii ( 1 I 2.2 g, 0.288 mole) in THF ( I .2 L) was
added water ( 100 mL)
and 30 % hydrogen peroxide (I 10 mL, 36.6 g, 1.08 mole). A solution of lithium
hydroxide
monohydrate ( 17.8 g, 0.424 mole) in water (400 mL) was added in portions over
25 minutes and
the resulting solution was stirred for 1 hour. The mixture was concentrated
under a slow nitrogen
stream to about 800 mL. After seeding with the chiral oxazolidinone the
mixture was chilled and
filtered removing a portion of the auxiliary which was washed well with water.
The filtrate was
extracted with dichloromethane (3x) to remove the balance of the chiral
oxazolidinone. The
combined organic extracts were washed with aqueous 0.5 N sodium hydroxide. The
base layers
were acidified with 1M sulfuric acid to pH 3 and extracted with ethyl acetate.
After washing with
water and brine, drying over sodium sulfate, and evaporation of solvents the
residue amounted to
64.9 g (98%) of R-2-,(i-butyl)-succinic acid-4-t-butyl ester.
CA 02277121 1999-07-06
WO 98130551 PCT/US98/00144
33
Sten 5
0 0
t-Bu0 OH + LDA + wi ~ OH
' t-Bu0
iv O
To a -78 °C solution of lithium diisopropylamide, prepared by the
addition of n-
butyllithium (11.4 ml, 28.4 mmol, 2.SM in hexanes) to a solution of
diisopropylamine (3.7 ml,
28.4 mmol) in 60 ml THF at -78 °C, was added a solution of iv (2.7 g,
11.8 mmol) in THF (20
mL) at -78 °C by cannula in a stream. The resulting clear, yellow
solution was stirred at -78 °C for
1 hour and then butenyl iodide (2.58 g, 14.2 mmol) was added by syringe. This
mixture was
allowed to warm to ambient temperature and stir overnight. The reaction
mixture was poured into
1:1 ether-water and the separated aqueous Iayer was extracted with ether (2x).
The combined
organic layers were washed with aq 1M NaHS04 and brine, dried with MgS04,
filtered and
concentrated. Flash chromatography (2%-5% isopropanol-hexane) gave epimeric
succinates v_
(2.30 g, >9:1 syn/anti) as a clear liquid.
Step
0 0
OH 1) LDA OH
t-Bu0 1( 2) MeOH i t-Bu0
O .O
_v ~ Yl
/ /
To a -78 °C solution of lithium diisopropylamide, prepared by the
addition of n
butyllithium (7.8 mI, 19.5 mmol, 2.SM in hexanes) to a solution of
diisopropylamine (2.6 ml,
19.5 mmol) in 30 ml THF at -78 °C, was added a solution of epimeric
isobutyl succinate v_ (2.3 g,
8.1 mmol) in THF ( 10 mL) at -78 °C by cannula in a stream. The
resulting clear, yellow solution
was stirred at -78 °C for 1 hour, warmed to 0 °C and recooled to
-78 °C. Methanol (1 ml) was
added and the solution was warmed to 0 °C. The reaction mixture was
poured into 1:1 ether-water
and the separated aqueous layer was extracted with ether (2x). The combined
organic layers were
washed with aq 1M NaHS04 and brine, dried with MgS04, filtered and
concentrated to give an
epimeric mixture (2: I anxi/syn) of succinates vi which could be separated by
flash chromatography
(10-50% ethyl acetate-hexanes).
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
34
Preparation of Succinate Ester 2
0
t-Bu0 OH
2O
The desired compound was prepared according to the method used to prepare
succinate
ester 1, except substituting allyl bromide for 4-bromo-1-butene.
Preparation of SuccinateEster
0
t-Bu0 OH
O
The desired compound was prepared according to the method used to prepare
succinate
ester 1, except substituting 5-bromo-1-pentene for 4-bromo-1-butene.
Preparation of Succinate Ester 4
CH3
O
t-Bu0 OH
O
4_
IS t 1
Br O O
H C ' ~ + ~ OH ~ J / OH
H3C Yll
mixture of isomers
A mixture under nitrogen of 4-bromotoluene (36.9 mL, 51.3 g, 0.3 mole), 4-
pentenoic
acid (30.6 mL,, 30.0 g, 0.3 mole), acetonitrile (500 mL), triethylamine ( 126
mL, 91.5 g, 0.90
mole), palladium acetate (1.35 g, 6 mmole) and tri-(o-tolyl)phosphine (4.65 g,
15 mmole) was
heated slowly to a gentle reflux. (A mild exotherm was observed as reflux
begins.) After 18
hours at reflux, the mixture was cooled in an ice bath and the solid was
removed by filtration and
rinsed well with ethyl acetate. The filtrate was concentrated to a small
volume and the residue was
CA 02277121 1999-07-06
WO 98!30551 PCT/US98/00144
partitioned between aqueous 1 M sodium carbonate and ether. The aqueous phase
was extracted
with ether. The combined ether layers were extracted with aqueous 1 M sodium
carbonate. The
basic solution was treated with charcoal and filtered. The filtrate was
acidified with 3 M
hydrochloric acid. After cooling in an ice bath, the soft solid was filtered,
washed with ice water,
5 and dried over sodium hydroxide to give vii (45 g) as a mixture of isomers
which was used
without further purification.
Step 2
0 0
OH ' ~ OH
H3C ~ H3C
The mixtureof isomers vu was hydrogenated in 600 mL THF over 9 g of 10%
palladium
on carbon at 4 atmospheres of hydrogen for 18 hours. After filtration and
concentration of the
solution, the residue was crystallized from hexane to yield 5-(4-
tolyl)pentanoic acid (viii, 33 g, mp
77-78 °C).
Step 3
0 0
OH ,--~ I ~ CI
HsC ~ ~ H3C ~ lg
A mixture of 3,,Qb (11.02 g, 57 mmole) and 12 mL thionyl chloride was stirred
at 24 °C for
18 hours and then heated to distill most of the excess thionyl chloride. Short
path distillation gave
11.74 g (97 %) of 5-(4-tolyl)pentanoyl chloride (~, by ~ 110 °C at 0.35
mm).
Step 4
0 0 0 0
N v0 + n-BuLi + I CI ---~
,- ,- - o
H3C'v j~ H3C ~ ~ LJ
\ / \ /
To a -78 °C solution of 4S-benzyl-2-oxazolidinone (10.36 g, 58 mmole)
in THF (150 mL)
was added n-butyllithium (23.5 mL 2.5 M) over 25 minutes. After 30 minutes) ~l
(55.7 mmole)
was added quickly, during which time the reaction temperature rose to -45
°C. The reaction
mixture was warmed to 0 °C and the reaction was quenched with saturated
aqueous ammonium
chloride. The mixture was allowed to settle and the supernatant was decanted
and concentrated.
CA 02277121 1999-07-06
WO 98130551 PCT/LTS98100144
36
The residue was partitioned between water and ethyl acetate. The organic layer
was washed with
water, aqueous 1 M sodium bicarbonate, water and brine. After drying over
sodium sulfate the
solution was concentrated and the residue was chromatographed ( 10-20 % ethyl
acetate-hexane) to
give 3~d_ (17.83 g, 89%).
Sten 55
CH3
O O
N~ O O
t-Bu0 OH
H3C ~ ~ ~ O
4_
The desired compound was prepared using Steps 4, 5 and 6 of the preparation of
succinate
ester 1, except substituting x_ for iii and substituting 5-bromo-1-pentene for
4-bromo-1-butene.
Pret~aration of Succinate Ester 5
CH3
O
t-Bu0 OH
O _5
OTBDMS
The desired compound was prepared using step 5 of the procedure for the
preparation of
succinate ester 4, except substituting TBDMSO(CH2),4I ( 10.8g, 34.Smmo1),
prepared as described
by Helquist et al., Tetrahedron Lett., 1985, 26, 5393, for 5-bromo-1-pentene.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
37
Preparation of Succinate Ester 6
i ( a
0
off
t-Buo
0
OTBDMS
Step 1
O ~ O
OCH3
OC H3
To a 0 °C solution in anhydrous THF (100 mL) of methyl 3-butenoate
(97%, 10.0 g, 96.9
mmol)was added 9-BBN (0.5 M in THF, 194 mL, 97 mmol) dropwise via dropping
funnel. After
the addition was completed, the reaction mixture was stirred at ambient
temperature for 5 hours.
To the resulting solution was added anhydrous THF (300 mL),
tetrakis(triphenylphosphine)palladium(0) (3.03 g, 2.62 mmol), 1-bromo-4-
propylbenzene (98%,
13.8 mL, 87 mmol), and powdered sodium methoxide (95%, 7.72 g) 136 mmol). The
reaction
mixture was stirred at reflux for 16 hours. The reaction mixture was cooled to
ambient temperature
and ported into saturated aqueous ammonium chloride. The mixture was extracted
twice with
ether. The combined organic extracts were washed with brine, dried with MgS04
and concentrated
to afford crude product as a light brown oil. Flash chromatography (CH2Cl2-
hexane, 1:4) afforded
Vii' (6.4 g).
i ~ a
i I o
OCH3 O
t-Bu0
O
OTBDMS
The desired compound was prepared by hydroysis of ester '~'u, followed by
conversion of
the carboxylic acid to succinate ester ~ according to steps 3, 4 and 5 of the
preparation of succinate
ester 4_, substituting TBDMSO(CH2)4I, for 5-bromo-1-pentene.
CA 02277121 1999-07-06
WO 98/30551 PG"T/US98/00144
38
Preparation of Succinate Ester 7
o w
OH
t-Bu0
O Z
OTBDMS
The desired compound was prepared according to the procedure used to prepare
succinate
ester 1, except substituting TBDMSO(CHZ)4I, for 4-bromo-1-butene.
Preparation of Succinate Ester 8
t-Bu.
t0
The desired compound was prepared in the same manner as succinate ester 5_,
except replacing acid
viii with 6-benzyloxy hexanoic acid.
Preparation of Succinate Ester 9
O
t-Bu~O OH
O
~OTBDMS
The desired compound was prepared in the same manner as succinate ester ~,
except replacing acid
viii with 4-pentenoic acid.
~OTBDMS
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
39
x 11
/ S02NH2
H I
HOHN N~~ N
O H
1 /
O
Exam lp a 1 A
O O
~~ O
H2~'~~ H ~
t-Bu0 OH + _ O ( \ ---.-~. t-Bu0 N~~O \
O ~ / O ~/
1 / TsOH
1 / 1~
HO HO
To a solution of succinate ester I_, benzyltyrosine tosylate salt (8.1 g, I
8.4 mmol, Aldrich
Chemical Co.), HOBT (2.5 g, 18.4 mmol) and NMM (4 mL, 36.8 mmol) in 80 mL DMF
at 0 °C
was added EDC (3.5 g, 18.4 mmol) in a single portion. The resulting solution
was allowed to
slowly warm to ambient temperature and was stirred for 3 days at which time it
was poured into a
sepratory funnel containing water and ethyl acetate. The separated aqueous
layer was extracted
with ethyl acetate (3x) and the combined organic layers were washed with
aqueous 1M NaHS04,
aqueous 1M NaHC03 and brine, dried over MgS04, filtered and concentrated in
vacuo. The
residue was flash chromatographed (CH2C12 to 2% methanol-CH2C12) to afford
8.26 g of
intermediate 1~ as a white foam.
Example I B
t-Bu0 N~ O ' \ t Bu0 N~ \
H ~ _ H
O ~ O
O
/ 1 / 1~ Ho~ 1 /
HO HO
To a solution of I~ (8.25 g, 15.4 mmol) dissolved in 77 mI. '1'~iF at 0
°C was added BH3
solution ( 1 M in THF, 51.3 mL, 51.3 mmol) dropwise by syringe over 10
minutes. The resulting
solution was stirred at 0 °C for 1.5 hours. Ethanol ( I 5.4 mL) was
added over 5 minutes followed
by pH 7 buffer (30 mL) and 30% H202 solution {30 mL). After 10 minutes, the
cooling bath was
CA 02277121 1999-07-06
WO 98/30551 PCT/US98I00144
removed and the cloudy mixture was stirred at ambient temperature for 2.5
hours at which time it
was concentrated to half the original volume and added to a mixture of brine
and and ethyl acetate.
The separated aqueous layer was extracted with ethyl acetate (3x) and the
combined organic layers
were washed with brine, dried with MgS04, filtered and concentrated in vacuo.
Flash
5 chromatography (2% methanol-CH2Cl2 then 5% methanol-CHzCl2) gave 7.62 g of
intermediate 1 c
as a white foam.
Example 1
O H ~ O H O
t-Bu0 N~ O I ~ t-Bu0 ~~~ O
O ~ O
HO~ ~ / L
HO O
To a solution of 1~ (4.54 g, 8.19 mmol) and Bu3P (4.1 mL, 16.4 mmol) in 800 mL
benzene at ambient temperature was added ADDP (4.13 g, 16.4 mmol) in a single
portion. The
solution was stirred at ambient temperature for 2 hours and then concentrated
in vacuo. The
residue was suspended in a minimal amount of CH2C12 and flash chromatographed
(30% ethyl
acetate-hexane) to give cyclic intermediate 1 d (3.1 g) as a white solid.
Exam ly a 1 D
O H ~O O H O
t-Bu0 N~~O ~ t-Bu0 ~~~OH
O I / O
1
/ ls! ~ / lg
v O
A mixture of ,],~ (3.1 g, 5.77 mmol) and 10% PdJC catalyst (620 mg) in 30 mL
methanol
was stirred under a positive hydrogen pressure for 3 hours. The reaction
mixture was then filtered
through Celite with methanol washings. The f:ltrate was concentrated in vacuo
to give cyclic
intermediate ~g (2.54 g) as a white solid which was used without further
purification.
CA 02277121 1999-07-06
WO 98/30551 PCTIUS98/00144
41
Example 1 E
S02NH2
O ~ O O O
~ H J~
t-Bu0 N~~ OH t-Bu0 NW N ~ I
O v ' = H
1L
O O
To a solution of 1 a (510 mg, 1.14 mmol) dissolved in 6 mL DMF at 0 °C
was added NMM
(150 pL, 1.37 mmol), HOBT (I85 mg, 1.37 mmol), EDC (263 mg, 1.37 mmol) and 4-
{2-
aminoethyl)benzenesulphonamide (274 mg, 1.37 mmol, Aldrich Chemical Co.). The
resulting
clear solution was stirred overnight at ambient temperature and then poured
into a mixture of water
and ethyl acetate. The separated aqueous layer was extracted twice v~zth ethyl
acetate and the
combined organic layers were washed with brine, dried with MgS04, filtered and
concentrated in
vacuo. Flash chromatography (3% methanol-CH~CI2) afforded ~f (758 mg) as a
white solid.
Example 1 F
/ I SOpNH2 O O / ( S02NH2
H ~ H
t-Bu0 O N~~ N HO ~~ N
H O H
1 / 1f t / .i~
0 0
A mixture of 1 f (717 mg, I .14 mmol), trifluoroacetic acid (5 mL) and CH2CI2
( I mL) was
stirred at ambient temperature for 1 hour and then concentrated under a stream
of nitrogen. The
residue was dissolved in 1:1 mix of CH2C12-methanol and concentrated in vacuo.
This was
repeated until a white solid formed to give 630 mg ~ which was used without
purification.
/ S02NH2 O / S02NH2
O \ H
HO NW N I HOHN N~~ N ~ I
O H O H
1 ',. la 1 / Ex1
0 0
To a 0 °C solution in DMF (8 mL) of 1~ (630 mg, 1.1 mmol) was added NMM
(242 pL,
2.2 mmol), HOBT (178 mg, 1.32 mmol) and EDC (253 mg, 1.32 mmol). After 15
minutes at 0
CA 02277121 1999-07-06
WO 98/30551 PCTlUS98/00144
42
°C, O-(tert-butyldimethylsilyl)hydroxyl amine ( 194 mg, 1.32 mmol) was
added in a single portion
and the mixture was allowed to warm to ambient temperature and stir overnight.
The solution was
then poured into a mixture of brine and CH2CI2. The aqueous layer was
extracted twice with
CH2CI2 and the combined organic layers were washed with brine, dried with
Na2SOa, filtered and
concentrated in vacuo. The crude solid was flash chromatogaphed (5% methanol-
CH2C12) to give
183 mg of the desired compound as a white solid. mp > 270 °C. 1H NMR
(DMSO) 8 -0.6-(-0.4)
(m, IH), 0.6-1.0 (m, 5H), 0.71 (d, 3H, J = 6.3 Hz), 0.80 (d, 3H, J = 6.3 Hz),
1.1-1.4 (m, 2H),
1.5-1.7 (m, 3H), 2.1 (dt, 1H) J = 10.8, 2.7 Hz), 2.5-2.6 (m, 1H), 2.75-2.85
(m, 2H), 3.02 (dd,
1 H, J = 12.9,4.8 Hz), 3.3-3.4 (m, 2H), 3.9-4.1 (m, 2H), 4.5-4.7 (m, I H),
6.90 (d, 2H, J = 8.7
Hz), 7.21 (t, 2H, J = 8.7 Hz), 7.29 (br s, 2H), 7.40 (d, 2H) J = 8.1 Hz), 7.75
(d, 2H, J = 8.I
Hz)) 7.80 (d, 1H, J = 9.6 Hz), 7.89 (t, 1H, J = 5.7 Hz), 8.66 (s, 1H)) 10.3
(s, 1H). 13C NMR
(DMSO) 8 21.9, 24.5, 25.0) 25.3) 28.4, 28.7, 35.I, 37.0, 39.8, 41.1, 46.3,
46.9, 53.8, 73.2,
121.3, 121.5, 125.9, 129.0, 129.4, 132.4, 132.7) 142.4, 143.8, 157.4, 170.4)
171.5, 173Ø
MS (CI) m/e 589 (M+1 ). Anal. Calcd for: C29H4pN40~S~0.4H20: C, 58.45; H,
6.90; N, 9.40.
Found: C, 58.49; H, 6.94; N, 9.20. [a) +55° (c 0.5, DMF).
Example 2
O H O / I
HOHN N~~ N
O H
O
The desired compound was prepared according to the method of Examples IE-G,
except
substituting phenethylamine for 4-(2-aminoethyl)benzenesulphonamide. mp > 270
°C. 1H NMR
(DMS O) b -0.6-(-0.4) (m, 1 H), 0.6-1.0 (m, 4H), 0.70 (d, 3H, J = 6.3 Hz),
0.80 (d, 3H, J = 6.3
Hz), l.l-1.4 (m, 2H), 1.5-1.7 (m, 3H), 2.0-2.1 (m, 1H), 2.5-2.6 (m, 1H), 2.7-
2.8 (m, 2H),
3.0-3.1 (m, 1 H), 3.2-3.4 (m, 2H ), 3.9-4.1 (m, 2H ), 4.55-4.65 (m) 1 H), 6.90
(d, 2H, J = 8.4
Hz), 7.15-7.35 (m, 7H), 7.8-7.9 (m, 2H), 8.68 {s, 1H), 10.3 (s, 1H). 13C NMR
(DMSO) 8
21.6, 24.2, 24.7, 24.9, 28.0, 28.4, 35.1, 36.7) 40.0, 40.8, 46.0, 46.6, 53.5,
72.9, 120.9,
121.1, 126.0, 128.2, 128.5, 128.7, 132.0, 132.4, 139.2, 157.0, 170.0, 171.1,
I72.7. MS (CIj
m/e 510 (M+1 ). Anal. Calcd for: C29H39N305: C, 68.34; H, 7.71; N, 8.24.
Found: C, 68.00;
H, 7.78; N, 8.05. [a) +18° (c 0.5, DMF).
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
43
Exam 1
H ~
HOHN O N~~ N
O H
O
The desired compound was prepared according to the method of Examples 1 E-G,
except
substituting cyclopropylamine for 4-(2-aminoethyl)benzenesulphonamide. mp >
270 °C. 1H
NMR (DMSO) 8 -0.6-(-0.4) (m, 1H), 0.3-0.4 (m, 2H), 0.6-0.7 (m, 2H), 0.71 (d,
3H, J = 6 Hz),
0.8 (d, 3H, J = 6 Hz), 0.7- I .0 (m, SH), 1.1-1.3 (m, 2H}, I .5-1.7 (m, 3H),
2.04 (apparent t, 1 H,
J = 12 Hz), 2.5-2.7 (m, 2H), 3.04 (dd, 1H, J = 12.8,4.1 Hz), 3.9-4.1 (m, 2H),
4.5-4.6 (m, 1H})
6.90 (d, 2H, J = 8.4 Hz), 7.15-7.25 (m, 2H), 7.7-7.85 (m) 2H), 8.69 (s, 1H),
I0.3 (s, 1H).
13C NMR (DMSO) S 5.60) 5.83, 21.6, 22.2, 24.2, 24.7, 24.9, 28.0, 28.4, 36.5,
40.8, 46.0,
46.5, 53.4, 72.9, 120.9) 121.2, 128.6, 132.0, 132.4, 157.0, 170.0, 172.2,
172.6. MS (CI) m/e
446 (M+1). Anal. Calcd for C~H35N305~0.3H20: C) 63.92; H, 7.96: N, 9.32.
Found: C,
63.86; H, 7.96; N, 9.39. [aJ +17° (c 0.5, DMF).
Ex~nle 4
H ~~ ~I
HOHN N~~ N
O H
O
The desired compound was prepared according to the method of Examples I E-G,
except
substituting aniline for 4-(2-aminoethyl)benzenesulphonamide. mp > 270
°C. tH NMR (DMSO)
8 -0.5-(-0.4) (m) 1H), 0.6-1.0 (m, 4H), 0.67 (d, 3H, J = 6.3), 0.81 (d, 3H, 3
= 6.3 Hz), 1.1-1.4
(m, 2H), 1.5-1.65 (m, 2H), 1.69 (dt, 1H, J = 10.8, 2.4 Hz), 2.12 (dt, 1H, J =
10.5, 2.7 Hz),
2.69 (br t, 1 H, J = 12.6 Hz), 3.9-4.0 (m, 1 H), 4.0-4.1 (m, 1 H), 4.75-4.90
(m, 1 H), 6.9-7.0 (m,
2H), 7.07 (t, I H, J = 7.2 Hz), 7.25-7.40 (m, 4H), 7.60 (d, 2H, J = 7.8 Hz))
7.98 (d, 1H, J =
9.6 Hz), 8.70 (s, 1 H), 10.0 (s, 1 H), 10.3 (s, 1 H). 13C NMR (DMSO) 8 21.6,
24.2, 24.7, 25.0,
28.1, 28.4, 36.7, 40.8, 46.0, 46.6, 54.4, 73.0, I 19.1, 121.0, 121.3, 123.3,
128.8, 132.1,
132.3, 138.8, 157.1, 170.0, 170.1, 172.9. MS (CI) m/e 482 (M+1). Anal. Calcd
for
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
44
C2~H35N305~H20: C, 64.91; H, 7.46; N, 8.41. Found: C. 64.85; H, 7.16; N, 8.29.
[a] +38° (c
0.4, MeOH).
Example 5
O H
HOHN N~ N
O = ~O
O
The desired compound was prepared according to the method of Examples I E-G,
except
substituting morpholine for 4-(2-aminoethyl)benzenesulphonamide.
mp > 270 °C. 1H NMR (DMSO) b -0.6-(-0.4) (m, IH), 0.6-1.0 (m, 4H), 0.71
(d) 3H, J = 6.3
Hz)) 0.78 (d, 3H, J = 6.3 Hz),1.14 (dt, 1 H, J = 10.8, 2.7 Hz), 1.2-1.4 (m, 1
H), 1.45- I .55 {m,
2H), 1.68 (dt, 1 H, J = 11.1, 3.0 Hz), 2.10 (dt, 1 H, J = 11.4, 3.3 Hz), 2.79
(t. 1 H, J = 12.6 Hz),
2.9-3.0 (m, 1 H), 3.4-3.8 (complex m, 8H), 3.9-4.1 (m, 2H), 5.0-5.1 (m, 1 H),
6.89 (d, 2H, J =
8.4 Hz), 7.27 (t, 2H, J = 7.3 Hz),8.00 (d, 1H, J = 9.6 Hz), 8.67 (s, 1H)) 10.3
(s, 1H). l3C
NMR (DMSO) 8 21.6, 24.0, 24.8, 25.0, 28.2, 28.5, 40.3) 40.9, 45.7, 46.5, 48.9,
66.1, 72.8,
120.7, 120.8, 129.1, I 3 I .9, 132.2, 157.1, 169.7, 170.0, 172.5. MS (CI) m/e
476 (M+1 }.
Anal. Calcd for C25H37N306~O.SH20: C, 61.96; H, 7.90; N, 8.67. Found: C,
61.86; H, 7.68;
N, 7.24. [a] +65° (c 0.4, MeOH).
E
O H ~O ~N~
HOHN N~~ N
O H
O
The desired compound was prepared according to the method of Examples 1 E-G,
except
substituting 2-aminopyridine for 4-(2-aminoethyl)benzenesulphonamide. mp > 270
°C. ~H NMR
(DMSO) S -0.6-(-0.4) (m,, IH), 0.6-1.0 (m, 4H), 0.68 (d, 3H, J = 6.3 Hz), 0.83
(d, 3H, J = 6.3
Hz), 1.1-1.4 (m, 2H)) 1.5-1.8 (m, 3H), 2.13 (dt, 1H, J = 10.8, 2.7 Hz), 2.66
(t, 1H, J = 12.0
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
Hz), 3.28 (dd) 1 H, J = 12.3, 6.0 Hz), 3.9-4.0 (m. 1 H), 4.0-4.1 (m, 1 H), 4.8-
5.0 (m, 1 H), 6.9-
7.0 (m, 2H), 7.1-7.2 (m, 1 H), 7.25-7.40 (m, 2H), 7.83 (dt, 1 H, J = 8.1, 1.8
Hz)) 7.99 (d, 1 H, J
= 9.6 Hz), 8.06 (d, 1 H, J = 8.4 Hz), 8.35 (dd, 1 H, J = 5.4, 2.1 Hz), 10.3
(br s, 1 H), 10.5 (s,
1 H). I3C NMR (DMSO) b 21.6, 24.2, 24.8, 25.0, 28.1 ) 28.4, 36.3, 40.8, 46.1,
46.7, 54.6,
5 73.1, 113.6, 119.6, 121.1, 121.6, 128.8, 132.2, I 32.4) 139.0, 147.4) 151.4,
157.3, 170.0,
171.1, 173.2. MS (C1) m/e 483 (M+1 ). Anal. Calcd for C26H34N405~H20: C,
62.38; H, 7.24;
N, 11.19. Found: C, 62.04; H, 7.44; N, 9.70. [a] +34° (c 0.6,
MeOH).
10 Ex 7
o N / \
HOHN N
O H
O
Example 7A
O H O O O / I
~ H ~
t-Bu0 N~~ OH t-8u0 N~~ N
O = O = H NH2
Z~.
O O
The desired compound was prepared using the procedure described for example 1
E, except
substituting 1,2-phenylenediamine for 4-(2-aminoethyl)benzenesulphonamide.
Example 7B
o ~ I
N O H lI \ I
~~ N
t-Bu0 _ t-Bu0 N~
0 H NH2 O
1 z~
0 0
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
46
A solution of ?b and camphorsulfonic acid (60 mg) in 15 mL toluene and 5 mL
THF was
stirred at reflux for 3 hours. The resulting brown solution was cooled,
concentrated and flash
chromatographed (3% methanol-CHZC12) to afford 7c ( 1.0 g) as a light tan
solid.
Example 7C
O H ~ ~ ~ O H
t-Bu0 N~ HOHN N~ N
p O H
/ ~ ~
0 0
The desired compound was prepared from 7c according to the method of Examples
1 F and
G. mp > 270 °C. 1H NMR (DMSO} 8 -0.5-(-0.3) (m, 1H), 0.6-1.0 (m, 4H),
0.59 (d, 3H, J =
6.3 H z)) 0.64 (d, 3H, J = 6.3 Hz), 1.1-1.4 (m, 2 H), 1.6-1.8 (m, 3H), 2.0-2.1
(m, 1 H ), 3.07 (t,
IH, J = 12.9 Hz), 4.0-4.2 (m, 2H), 5.4-5.5 (m, 1H), 6.96 (apparent d, 2H, J =
6.3 Hz), 7.1-7.2
(m, 2H), 7.32 (br t, 2H, J = 6.9 Hz), 7.4-7.5 (m, 2H), 8.18 (d, 1 H) J = 9.0
Hz), 8.69 (s, I H),
10.3 (s, 1 H), 12.2 (s, I H). 13C NMR (DMSO) b 21.6, 24.1, 24.7, 28.1, 28.5,
38.0, 40.9)
46.1, 46.6, 47.9, 72. 9, 111.3, 118.5, 121.1, 121. 2, 121.9, 128. 8, 132.2, 13
2.5, 154. 9, 157.2,
170.1, 172.6. MS (CI) m/e 479 (M+1 ). Anal. Calcd for CZ~H34N404~ 1.4H20: C,
64.37; H,
7.36; N, 11.12. Found: C, 64.47; H, 7.41: N, 10.35. [a] -39° (c 0.5,
DMF).
xam
O H_ ~
HOHN Nv _NHCHa
O
0
EXam l
O O
t-8u0~ N~ OH t-BuO~ N~ NHCH
O -"i I'O
w w
HO~ HO
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
47
To a 0 °C solution in DMF (40 mL) of BOC-L-tyrosine (2.81 g, 10 mmol)
and HOBT
(3.70 g, 27 mmol) was added methylamine hydrochloride (675 mg) 10 mmol) and
NMM (3.16
y mL, 2.9 mmol j and the mixture was stirred for 30 minutes. EDC (2.76 g, 14
mmol) was added
and stirring was continued for 2 hours in the ice bath and then at room
temperature for 3 days. The
i 5 reaction mixture was diluted with saturated aqueous ammonium chloride and
was extracted twice
with ethyl acetate. The organic extracts were combined, dried over sodium
sulfate, filtered and the
filtrate concentrated to a yellow oil. A minimum amount of ethyl acetate was
added and the
suspension heated until a solution resulted. Crystallization was allowed to
occur at room
temperature and $~ as a white solid was collected by filtration (1.97g, 67%
yield).
Exam In a 8B
O \ O O
----~ ----
t-Bu0 C02H t-Bu0 C02H t-Bu0 C02H
6:1 syn:and ~ 1:1.5 syn:anti
Step 1: To a -78 °C solution in THF (8 mL) of diisopropylamine (541 mg,
5.35 mmol) was added
butyllithium (2 ml, 5 mmol, 2.SM in hexanes) dropwise and the solution was
stirred for 15
minutes. A mixture of (R)-iso-butylsuccinic acid tent-butylester (O.Sg, 2.17
mmol) in DMPU ( 1
mL) and THF (3 mL) was added dropwise. The resulting yellow solution was
stirred for one hour
at -70 °C and a solution of 4-bromo-1-butene (354 mg, 2.62 mmol) in THF
(3 mL) was added
dropwise over 8 minutes. The reaction mixture was stirred for 1 hour at -70
°C and a solution of
LiI (35 mg, 2.62 mmol) in THF ( 1 mL) was added dropwise. The cold bath was
removed and
stirring was continued overnight at ambient temperature. The reaction mixture
was poured into
saturated aqueous ammonium chloride and ethyl acetate and the organic layer
set aside. The
aqueous layer was washed once with ethyl acetate and the organics combined,
dried over MgS04)
filtered and the filtrate concentrated to a yellow oil which was purified by
flash chromatography on
silica gel (15% ethyl acetate-hexanes) to yield 244 mg of 8~ as a yellow
gum(40% yield). Isomer
ratio approximately 6:1.
Ste~2: To a -78 °C solution in THF (7 mL) of diisopropylamine (523 mg,
5.17 mmol) was added
butyllithium (1.93 ml, 4.83 mmol, 2.5M in hexanes) dropwise and the solution
was stirred for 15
- 30 minutes. A solution of 8~ (611 mg, 2.15 mmoI) in THF (8 mL) was added
dropwise. The
reaction mixture was warmed to -20°C and stirred for 1 S minutes before
being cooled to -78C and
quenched with a solution of methanol(356 mg, 11 mmol) in 2 mI THF. The
reaction mixture was
warmed to room temperature and mixed with saturated aqueous ammonium chloride
and the pH
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
48
was adjusted to 4 with added dilute HCI. The solution was extracted twice with
ethyl acetate and
the organic extracts were combined, dried over MgS04) filtered and the
filtrate concentrated to a to
give 8b (594 mg) as a yellow gum having an isomer ratio determined by CMR to
be 1:1.5 syn
anti.
0
2H ~ NHCH3
To a solution of BOC-L-tyrosine-N-methyl amide (8a, 627 mg, 2.13 mmol) in THF
(2 ml)
was added 4M HCl-dioxane (6 mL) dropwise. The resulting yellow solution was
stirred for 4
hours at ambient temperature and then concentrated to dryness. The residue was
azeotroped twice
with toluene and once with ether leaving an off-white solid which was added to
an ice-bath-cooled
flask containing compound 8b (561 mg, 1.97 mmol)) HOBT (716 mg, 5.3 mmol), NMM
(562
mg, 5.6 mmol) and DMF (8 mL). The resulting orange solution was stirred for 30
and EDC (534
mg, 2.79 mmol) was added as a solid. Stirring in the ice bath was continued
for 2 hours and then
at room temperature overnight. The reaction mixture was diluted with saturated
aqueous
ammonium chloride and extracted 3 times with ethyl acetate. The organic
extracts were combined,
dried, filtered and the filtrate concentrated to a yellow oil which was
purified by flash
chromatography(4% methanol-methylene chloride) to give 8c (783 mg, 86% yield)
as a white
powder.
Exams a 8D
H O
N H
NHCH3-~ t-Bu0 N NHCH
O
$~ O $s~
H ~
HO
HO
The desired compound $~, was prepared from $~ by hydroboration/oxidation
according to
the method of Example 1 C.
Example 8C
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
49
EX ale 8E
O H O O O
N 11
t-Bu0 ~_ NHCH3 t-Bu0 Nv _ NHCH3
Ow) _ $d O
HO
HO
O
To a -5 °C solution of triphenylphosphine (419 mg, I.6 mmol) in
methylene chloride (20
mL) was added dropwise a solution of diethylazodicarboxylate (249 mg, 1.43
mmol) in methylene
chloride (20 mL) and the solution stirred for 30 minutes. A solution of $~
(465 mg, 0.97 mmol) in
methylene chloride (120 mL) was added dropwise and the solution was stirred
for 1 hour. The
reaction mixture was poured into brine and the organic layer set aside. The
aqueous phase was
washed with ethyl acetate and the organic layers were combined, dried and
concentrated to a
yellow gum which was purified by flash chromatography (50% ethyl acetate-
hexanes) to give the
desired compound ~ (75 mg).
Example 8F
O H O O O
N H
t-Bu0 ~ NHCH3 HO N NHCH
3
O
The desired compound $f was prepared by saponification of $~ using
trifluoroacetic acid in
dichloromethane according to the method of Example 1F.
Example 8 ,
O H O O O
N H
HO ~ NHCH3 HOHN N
NHCH3
O $f O
0
0
CA 02277121 1999-07-06
WO 98/30551 PCT/US98l00144
To a 0 °C solution of ~ (50 mg, 0.125 mmol) in DMF (0.8 mL) was added
NMM( I 9 mg)
0.19 mmol),a solution of HOBT ( 19 mg, 0.14 mmol) in DMF (0.2 mL) and EDC (27
mg, 0. I4
mmol). After stirring for 15 minutes, a solution of O-tert-
butyldimethylsilylhydroxylamine (21
mg, 0.14 mmol) in DMF (0.2 mL) was added and stirring was continued for 30
minutes in the ice
5 bath and then at ambient temperature for 3 days. The reaction mixture was
diluted with saturated
aqueous ammonium chloride and was extracted twice with ethyl acetate. The
combined organic
extracts were dried and concentrated to a clear gum. The gum was mixed with
diethyl ether and the
resulting suspension filtered to give 32 mg of a white solid (61 % yield). A
portion (9.8 mg) of
this material was dissolved in a mixture of 1.5 ml acetonitrile, 0.5 ml
methanol and 2.0 ml water
10 and loaded onto a C-18 reverse phase HPLC column and eluted with a gradient
from 10%
acetonitrile/90% water to 70% acetonitrile/30% water. The faster eluting peak
was collected and
concentrated to give the anti compound as a white powder (3.1 mg) and the
slower eluting peak
was collected and concentrated to give the syn compound as a white powder (3.4
mg).
Anti: 1H NMR (300 MHz, DMSO-d6) b 10.41 {s, 1H), 8.69 (s, 1H), 7.79-7.87 (c,
1H),
15 7.69-7.76 (c, 1 H), 7.51-7.67 (c, 1H), 7.16-7.28 (c, 1 H), 6.87-6.96 (c,
2H), 4.53-4.64 (c, 1H),
3.88-4. I 3 (c, 2H), 3.01-3.11 (c, 1 H), 2.54-2.69 (c, 4H), 2.00-2. I 3 (c, I
H), 1.5 I -1.73 (c, 3H),
I .12-1.36 (c, 2H), 0.84-0.95 (c) 1 H), 0.80 (d, 3H, J = 6Hz), 0.71 (d, 3H, J
= 6Hz), 0.50-0.67
(c, IH). 13C NMR (300 MHz, DMSO-d6) 8 172.68, 172.65, 171.56, 157.03, 132.05,
128.64,
121.26, 120.99, 97.75, 72.97, 53.49, 46.61, 45.97, 40. 83, 36.75, 28.3 8,
28.04, 25.45, 24.93,
20 24.75, 24.22, 21.60. IR, (KBr) 3300, 2950, 2940, I 640, 1530, 1510, 1210,
1190 cm-1. MS
(DCI/NH3) m/e 420(m+H)+.
Syn:lH NMR (300 MHz, DMSO-d6) S 9.90 (bs 1H), 7.94-8.01 (c, IH), 7.40 (d, 1H,
J =
6 Hz), 7.05 (dd, 1 H, J = 1.5, 4.5 Hz), 6.73-6.79 (c) 2H), 6.66-6.71 (c, 1 H),
5.66-5.74 (c, 1 H),
3.96-4.03 (c, 1H), 3.81-3.90 (c, 1H), 2.91 (dd, 1H, J = 3) 9 Hz), 2.45 (d, 3H,
J = 3 Hz), 2.23-
25 2.29 (c, 1 H), 1.94 (d, 1 H, J = 6 Hz), 1.60-1.70 (c, I H}, 1.21-1.31 (c,
2H), 1.00-1.10 (c, I H),
0.88-1.00 (c, 2H), 0.72-0.82 (c, 1H), 0.49 (d, 3H, J = 3 Hz), 0.43 (d, 3H, J =
3 Hz), -0.01-(-
0.09) (c, 1H). 13C NMR (300 MHz, DMSO-d6) d 171.8, 171.2, 170.5) 159.5, 130.8,
130.6,
130.0, 119.2, 119.1, 70.3, 52.0, 49, 45.7, 38.5) 33.4, 30.2, 25.9, 25.4, 23.4,
22.5, 21.5,
21.4. MS (DCI/NH3} m/e 420 (M+H)+.
x 19
O
H O N NHCH3
O
I\
O
CA 02277121 1999-07-06
WO 98130551 PCT/US98/00144
51
The desired compound was prepared according to the method of Examples 8C-F,
except
substituting allyl bromide for 4-bromo-1-butene in Example 8B.
Example 10
O H
HOHN N _ NHCH3
O
O
The desired compound was prepared from the compound of Example 9 according to
the
method of Example 8G. ~H NMR (300 MHz, DMSO-d6) 8 10.30 (s, 1H), 8.69 (bs,
IH), 7.81
(d, I H, J = 6 Hz), 7.62 (dd, 1 H, J = 3, 3 Hz), 7.05-7.17 (c, 3H), 6. 81 (dd,
1 H, J = 4.5, 1.5
Hz), 4.65-4.73 (c, 1 H), 4.08-4.15 (c, 1 H), 3.98-4.05 (c, 1 H), 3.15 (dd, 1
H, J = 3, 4.5 Hz),
2.62 (d, 3H) J = 3 Hz)) 2.54 (d, 1H, J = 7.5 Hz), 2.00 (dt, 1H, J = 6, 1.5
Hz), 1.59 (dt, IH, J =
7.5, 1.5 Hz), 1.10-1.27 (c, 4H), 0.73-0.79 (c, 7H), 0.70 (d, 2H, J = 3 Hz),
0.57-0.68 (c, 1H), -
0.61--0.72 (c, 1H). 13C NMR (300 MHz) DMSO-d6) d 172.7, 171.5) 169.6, 158.3,
I32.7,
132.5, 128.9, 122.0, 120.0, 72.9, 53.0, 46.7, 46.1, 39.9, 36.7) 29.9, 29.3,
25.4, 25.1, 24.0,
21.2. IR (KBr) 3300) 2960, 2920) 1660, 1640, 1530, 1510, 1220 cm-1. MS (FAB
(+)) m/e
428(M+Na)+, 406(M+H)+.
Example 11
~~ H
NHCH3
O
O
The desired compound was prepared according to the method of Example lA-C, F
and G,
except substituting succinate ester ~ for succinate ester ~, and substituting
Se for benzyltyrosine
tosylate salt. mp > 300C. 1H NMR (300 MHz, DMSO-d6) 8 -0.25-(-0.1 ) (m, 1 H),
0.6-0.71 (m,
1H), 0.72 (d, 3H, J = 6.9 Hz), 0.82 (d, 3H, J = 6.3 Hz), 0.85-1.39 (m, 8H),
1.4-1.59 (m) 1H),
1.6-1.75 (m, 1 H), 2.14-2.24 (m, 1 H), 2.58-2.60 (m, 1 H)) 2.61 (d, 3H, J =
4.8 Hz), 2.86-2.91
(m, 1 H), 4.08-4.22 (m, 2H), 4.4-4.55 (m, 1 H), 6. 8 I (d, 1 H, J = 8.1 Hz),
6.92 ( 1, 8.1 H ), 7.2-
7.24 (m, 2H), 7.71 (d, 1 H, J = 4.8 Hz)) 7.93 (d, 1 H, J = 9 Hz)) 8.69 (s, 1
H}, 10.3 (s, 1 H). MS
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
52
(DCI/NH3) m/e 434 (M+H)+. Anal. calcd for C23H35N30$~0.SH20: C, 62.42; H,
8.19: N, 9.49.
Found: C, 62.69; H, 8.12; N, 9.45. [ a ] + 31 ° (c 0.3, DMF).
Example 12
O H O
HOHN Nv 'NHCH3
O
S O
Example 12A
O ~ O
t-Bu0 C02H t-Bu0 ~ COpH
The desired compound 12a was prepared according to the method of Example 8B,
except
substituting 6-bromo-1-hexene for 4-bromo-1-butene.
Example 12B
O H O
O _ ~
HO Nv _NHMe
t-Bu0 CO~I-f
-.-.i O
O
The desired compound was prepared according to the method of Examples I A-F,
except
substituting 12a for succinate ester 1.
CA 02277121 1999-07-06
WO 98/30551 PCT/U598/00144
53
EX~Ie 12C
O H O O
N H
HO ~ NHMe HORN N
NHMe
O -~ O
O O
To a 0 °C solution of ~ (0.21, 0.49 mmol) in DMF (4 mL), was added NMM
( 200 pl,
1.8 mmol), HOBT (80mg, 0.59 mmol) and EDC ( 113mg, 0.59 mmol). After I 5
minutes at 0 C
O-benzylhydroxylamine hydrochloride (95mg, 0.59 mmoI) was added in a single
portion and the
mixture was allowed to warm to room temperature with stirring overnight. The
turbid solution
was added to water and 5% methanol/CH2C12. The aqueous layer was extracted
twice with
CH2Cl2 and the combined organic layers were concentrated. The crude material
was triturated in
5:1 ether/methanol and the solid collected by filtration to give 109mg of the
O-benzylhydroxamate.
This material was dissolved in 75m1 of 70:30 THF/MeOH and treated with lOmg of
10% Pd/C
under I atm of H2 for 2hours. The catalyst was filtered off and the solution
concentrated to give
the desired anti isomer (35mg) as a white solid. IH NMR (DMSO) b 0.1-0.2 (m,
1H), 0.73 (d,
3H, J = 6.6 Hz), 0.75-1.15 (m, 6H), 0.85 (d, 3H, J = 6.3 Hz), 1.2-1.44 (m,
3H), 1.5-1.8 (m,
4H), 2.2-2.32 (m, I H), 2.60 (d, 3H, .J = 4.8 Hz), 2.62-2.73 (m, I H), 2. 80-
2.90 (m, 1 H), 4. I 5-
4.23 (m, 2H), 4.33-4.50 (m) 1 H), 6.92 (d, 2H, J = 8.1 Hz), 7.23 (d, 2H, J =
8.4 Hz), 7.93 (d,
IH, J = 4.2 Hz), 8.10 (d, 1H, J = 8.4 Hz), 8.72 (s, IH), 10.38 (s, 1H). MS
(DCI/NH3) m/e 448
(M+H)+. Anal. calcd for C~H3~N3Og~0.25H20: C, 63.76; H, 8.36; N, 9.29. Found:
C, 63.74;
H, 8.32; N, 8.43. [a] +2° (c 0.2, DMF).
~~Inlhe 13
o ~ o
HOHN ~ NHCH3
O
CA 02277121 1999-07-06
WO 98/30551 PCT/L1S98/00144
54
Example 13A
O H O
~t-Bu0 N v _ NHCH3
a
O
/ I
The desired compound ~ was prepared by coupling of 12a and p-iodo-
phenylalanine-N-
methylamide hydrochloride using the described above for the preparation of
1_~.
Example 13B
H O
N
NHCH3 t-Bu0 NHCH3
O ~
I
To a solution of the compound of ~a (0.65g, 1.93 mmol) in acetonitrile (25 mL)
in a glass
bomb was added triethylamine ( 1.63 ml, 16 mmol). Argon was bubbled through
the solution for
five minutes foDowed by rapid addition of
tetralds(triphenylphosphine)palladium(0) ( 127mg, 10
mol%). The bomb was then sealed and heated at 80 C for 2hours. After cooling,
the solution was
concentrated and purified by flash chromatography (40% ethyl acetate-hexanes)
to give the desired
compound ~ (146mg) as a white solid.
Ex r~ple 13C
0
t-Bu0 ~ NHCH ~ N
3 HCH3
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
The desired compound was prepared from ~3b according to the method of Examples
1 F
and G. ~H NMR (DMSO) 8 -0.7-(-0.4) (m, 1H). 0.42-2.2 (series of m) 12H}) 0.6-
0.7 (narrow
m, 3H), 0.8-0.9 (narrow m. 3H), 2.6-2.7 (narrow m> 3H), 3.0-3.2 (m. 3H), .~.2-
5.0 (m, 3H))
7.0-8.0 (m, 6H)> 8.6-8.7 (m, 1H). MS (DCl/NH3} m/e 430 (M+H)+.
' 5
Examp a 14
O H O
H0. N
H O
\ /
v
10 Ex~rra ple 14A
O H
t-BuO~ N~ OH t-BuO
O --- O ~ i
/ \
OI-Bu Ot-Bu
To a 0 °C solution of nBuLi (2.SM/Hexanes, 14.25 mL) in diethyl ether
(SmL) was added
bromobenzene (S.SSg, 35.6mmol) over a few minutes. The resulting yellow
solution allowed to
15 stir cold for 45 minutes and then was cannulated into a -78 °C
solution of N-Boc-O-tBu-L-tyrosine
(3.Og, 8.9mmol) in diethyl ether (75mL). The reaction mixture was warmed to 0
°C over 1.5
hours and then was quenched with 2N citric acid. The aqueous layer was
extracted twice with
diethyl ether and the combined organic extracts were washed with saturated
aqueous NaHC03 and
brine, dried (MgS04) and concentrated in vacuo. Flash chromatography (hexane-
ethyl acetate 9:1 )
2U afforded the desired compound ~ ( 1.84 g) which was carried on without
further purification.
E~ l
H
' t-BuO
HCI~Hz
O Ii (/
_ ~ / \ ~ / \
Ot-Bu I OH
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
56
A solution of the 14a ( 1.8 g, 4.7mmo1 i in trifluoroacetic acid was stirred
at 0~ for 3()
minutes. The excess trifluoroacetic acid was evaporated in vacuo. The residue
was taken up in
1 N HCl in ether and stirred for 30 minutes. The mixture was diluted with
diethyl ether 170mL?
and the resulting solid filtered. The extremely hygroscopic solid was dried in
a vacuum oven for
several hours, transferred into a round bottom flask and dried under high
vacuum for 16 hours to
give the desired compound ~ (0.48 g) as a hygroscopic, white HCl salt.
Example 14C
O O H O
HCI~H2 ~ HO.
H O
\ /
OH
v
The desired compound was prepared from 14b and succinate ester 3_ according to
the
method of Examples lA-C. mp 210-220° (Dec). ~H NMR (300 MHz, DMSO-d6) 8
10.30 (s,
1 H), 8.63 (s, 1 H), 8.17-8.14 (d, 9.2H), 8.10-8.07 (d, 2H, J = 8.5 Hz), 7.66-
7.63 (t) 1 H, J =
7.3 Hz), 7.56-7.51 (t, 2H, J = 7.8 Hz)) 7.43-?.40 (d, 1H) J = 8.5 Hz), 7.25-
7.22 (d, 1H) J =
8.4 Hz), 6.96-6.94 (d, 1 H, J = 8.5 Hz), 6.85-6.83 (d, 1H, J = 8.5 Hz), 5.68-
5.61 (m. 2H),
3.09-3.03 (m, 1H), 2.77-2.68 (m, 1H), 2.17-2.13 (m, 1H)) 1.71-1.69 (m, 1H))
1.50-1.49 (m,
1H)) 1.34-1.31 (m, 2H)) 1.16-1.05 (m) 3H}, 0.78-0.62 (m, 5H)) 0.47-0.45 (d,
3H. J = 7.3 Hz).
0.00-(-)0.08 (m) 1 H). MS (DCI/NH3) m/e 4$1 (M+H)+. Anal calcd for
C2gH36N~05~O.SH20:
C, 68.68; H, 7.61; N. 5.72. Found: C, 68.88; H. 7.88; N) 5.10. [a]D:
+21.3°(c = 0.46, DMF).
Example 15
O H O
HONH N
O
H
O
Example 15A
H O
BOCN~ ~H BOCN /
3
HO I
Ho l.~a
CA 02277121 1999-07-06
WO 98130551 PCT/US98100144
57
To a solution of methylmagnesium bromide (35m1) 3.OM in Et20, 105.6 mmol) in
dry
toluene( I40m1) was added pyrrole ( 12m1, 17 I .9mmo1) dropwise at -40
°C under nitrogen. The
resulting solution was then stirred at -10 °C for 10 minutes and then
was cannulated into a solution
of BOC-L-tyrosine methyl ester (3.9g, 13.2mmol) in dry toluene (40m1) at -65
°C. The
- temperature was allowed to warm to -10 °C over 4 hours and the
reaction was quenched by
addition of 2N citric acid. The reaction mixture was extracted with CH2Cl2
(3x), dried over
Na2S 04, filtered and the solvent was evaporated. The crude product was
purified by flash
chromatography (30% ethyl acetate-hexanes) to give the desired compound _l~
(2.46 g, 56%) as a
light brown foam.
Exam In a 15B
H O O
BOCN / H /
I\ H I\ H
'TFA
HO ~ ~ HO ~
Compound 15a (720mg, 2. l8mmol) was dissolved in trifluoroacetic acid (Sml)
and stuTed
at room temperature for 5 minutes. The solvent was evaporated to give ~b
(900mg) as a brown
oil which was used without further purification.
Exam 1e~15r
O O H O
H2
/ ~ HONH - N -
N
I , H ~TFA O H
HO
p
The desired compound was prepared according to the method of Example 14C)
except
substituting 1~ for 14b. m.p. 242 °C (dec). 1H NMR (300 MHz, DMSO-d6) b
-0.02-(-0.15)
(m, 1 H), 0.53-0.88 (m) 4H), 0.61 (d) 3H, J = 3 Hz), 0.79 (d, 3H, J = 3 Hz),
0.90-1.04 (m,
1 H ), 1.07-1.40 (m, SH), 1.42-1.60 (broad ) 1 H), 1.65-1.76 (dt, 1 H, J = 3)
9 Hz), 2.19-2.30 (dt,
1H, J = 3, 12 Hz), 2.65-2.78 ( 1H), 3.0-3.1 (dd) 1H, J = 3, 15 Hz), 4.04-4.07
(m, 2H), 5.21-
5.32 (m, 1 H), 6.24-6.28 (m) 1 H ), 6.81-6.88 (dd, 1 H, J = 3, 9 Hz), 6.91-
6.99 (dd, 1 H, J = 3, 9
Hz), 7.14 ( 1 H), 7.25-7.34 (m) 2H), 7.39-7.46 (dd, 1 H, J = 3, 9 Hz), 8.12
(d, 1H, J = 9 Hz),
8.69 (s, 1H), 10.31 (s, 1H), 11.93 (s, 1H). MS (DCI/NH3) m/e 470 (M+H)+. Anal
calcd for
CA 02277121 1999-07-06
WO 98/30551 PGT/US98/00144
58
C26H35N305'H20: C, 64.04; H, 7.64; N, 8.61. Found: C, 64.00; H) 7.61; N, 8.44.
[a]D:
+97.3°(c = 0.26, EtOH).
Example 16
O H O
HOHN N~ NHCH3
O
N
O H
Example 16A
0 0
BocHN~ OH BocHN~ NHMe
t6a
N02 NOZ
To a solution of N-tert-butoxycarbonyl p-nitro-phenylalanirte (Sigma) (0.78g,
2.51 mmol)
in DMF ( 12.5 mL) was added EDC (0.53 g, 2.77 mmol)) HOBT (0.37 g, 2.77 mmol),
NMM
(0.30 g, 2.77 mmol) and methylamine hydrochloride (0.19 g, 2.77 mmol) and the
reaction mixture
was stirred at room temperature for 2 hours. The reaction mixture was
partitioned between ethyl
acetate and brine. The aqueous layer was separated and extracted twice with
ethyl acetate. The
combined ethyl acetate extracts were washed with brine, dried over MgS04,
filtered and
evaporated to dryness. The crude material was purified by flash chromatography
(60% ethyl
acetate-hexanes) to give lf~a (0.8 g, 98%).
Example 16B
O O H O
BocHN~ ~
NHCH3 ~_g~0 Nv ' NHCH3
a ,-a s O
NOz 1S
N02
OH
The desired compound was prepared by deprotection of _I ~ by treatment with
trifluoroacetic acid according to the procedure of Example 15B, followed by
coupling with
succinate ester ~ and hydroboration/oxidation using the method of Examples 1 A
and B.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
59
Example 16C
O H ~O O H O
t-Bu0 Nv ' NHCH3 t-Bu0 . ~ NHCH3
O ~ _ O
NOZ N02
OH OH
O
To a solution of the compound _1~~ (207 mg, 0.397 mmol) in CH2C12 was added
Jones'
reagent dropwise until the orange color of the reagent was preserved, then
ethyl alcohol was added
dropwise to quench the excess Jones' reagent (the color changed to green). The
mixture was
evaporated to a small volume and partitioned between CH2Cl2 and brine. The
aqueous phase pH
was adjusted to 2. The aqueous phase was extracted with CH2Cl2 (3x). The
combined CHZC12
extracts were dried (MgS04), filtered and evaporated to give the desired
compound 16c (209 mg)
which was used without further purification.
Example 16D
O H~ ~O _ O H O
t-Bu0 . Nv _ NHMe t-Bu0 Nv _ NHMe
3 O = i
----~ O
NOZ ~ NHz
OH OH
O O
A mixture of j,ø~ (205 mg) 0.383 mmol) and 10% Pd/C (40 mg) in EtOH was
stirred under
H2 ( 1 atm) for 2 hours. The reaction mixture was filtered through Celite and
the residue was
washed thoroughly with 10% methanol-CH2Cl2. The filtrate and washings were
collected and
evaporated to dryness to give ~ ( 193 mg) as a pale brown solid which was used
without further
purification.
CA 02277121 1999-07-06
WO 98/30551 PCT/ITS98/00144
Example 16E
O N
t-Bu0 NHCH3 t-Bu0 NHCH3
O ~ --~. O
1~
NHz
OH N
O O H
To a solution of j~ ( 190 mg, 0.376 mmol) in CHZC12 (4 mL) was added
triethylamine
5 ( 156.9 mL, 1.13 mmol) followed by bis(2-oxo-3-oxazolidinyl)phosphinic
chloride ( 143.7 mg,
0.564 mmol). After stirring at room temperature for 2 hours, the mixture was
poured into CHZCl2
and washed with saturated aqueous NaHC03 and brine. The aqueous layer was then
extracted
twice with CH2C12. The combined CH2Cl2 layers were dried (MgS04}, filtered and
evaporated to
dryness. Flash chromatography (2%-5% methanol-CH2CI2) gave ~ (87.7 mg).
Exam I
0
t-Bu0 N~ NHCHa HOHN N~ NHCH3
O --i O
O H O H
The desired compound was prepared from leg, according to the method of
Examples 1F
and G. mp: >250 °C. 1H NMR (300 MHz, DMSO-d6) 8 -0.09 (m) 1H), 0.78 (d,
3H, J = 6.2
Hz)) 0.85 (d, 3H, J = 6.2 Hz), 0.78-0.85 (m, 2H)) 1.10-1.40 (m, 6H), 1.60-1.75
(m, 2H}, 2.12
(m) 1 H), 2.22 (m, 1 H), 2.63 (d, 3 H, J = 4.5 Hz), 2.74 (t, 1 H, J = 13.2
Hz)) 2.99 (dd, 1 H, J =
13.2, 3 Hz), 4.5 (m, I H), 7.10 (m, 2H), 7.37 (m, 2H), 7.84 (bs, I H), 8.08
(bs, 2H), 9.09 (s,
1H), 10.15 (bs, 1H). MS (DCI-NH3) 447 (M+H)+, 429, 403, 283.
[a] _ +50.0 ° (c=0.11) CH30H).
CA 02277121 1999-07-06
WO 98130551 PCT/US98/00144
61
Exam~nle 17_
O H O
HO N ~ NHCH3
O
N
H
exam; lei 17A
O H O'' O H O
t-Bu0 N~ NHCH3 t-Bu0 ~ NHCH3
O --.-~ O
~ NO
2 N02
OH OMs
To a solution of 1 ~ (449 mg, 0.863 mmol) in CH2C12 was added triethylamine
(0.18 mL,
1.29 mmol) and methanesulfonyl chloride (0.080 mL) 1.04 mmol). After stirring
at room
temperature for 0.5 hours, the mixture was poured into CHZCl2 and washed with
NaHC03 and
brine. The CH2Cl2 was dried (MgS04), filtered and evaporated to dryness. Flash
chromatography (40-80% ethyl acetate-hexanes) gave ~ (428 mg, 83%) as white
crystals.
Example 17B
O H O' O H O'I
t-Bu0 N~ NHCH3 t-8u0 N~ NHCH3
o ---~ o
N02
OMs
A mixture of 17~ (420 mg, 0.701 mmol), 10%Pd/C (50 mg) and triethylamine
(0.098 mL)
0.701 mmol) in isopropanol (4 mL) was stirred under H2 ( 1 atm) for 10 hours.
The reaction
mixture was filtered through Celite and the residue was washed thoroughly with
10% methanol-
CHZC12. The filtrate and washings were combined and evaporated to dryness.
Flash
chromatography (2-5% methanol-CH2Cl2) provided 7b (280.4 mg, 84.4%) as white
crystals.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
62
Example 17C
O ~ Ou O ~ O
t-Bu0 N~ NHCH3 HO ~ NHCH3
O --" O
N N
H H
The desired compound was prepared from 17b according to the method of Example
1F.
mp 194-196 °C (dec). 1 H NMR (500 MHZ, DMS O-d6, 30 °C) 8 0.03
(t) 1 H, J = 11 Hz), 0.74
(d, 3H, J = 6.1 Hz), 0.67-0.84 (m, 3H), 0.85 (d, 3H, J = 6.1 Hz), 1.03-1.08
(m, 4H), 1.32-
1.38 (m, 3H)) 1.89 (dt, 1H, J = 11.2, 3.4 Hz), 3.0 (dt, 1H, J = 11.1, 3.4 Hz},
2.62 (d) 3H, J =
4.8 Hz), 2.66 (t, I H, J = 12.5 Hz), 2.98 (dd, 1H, J = 12.5, 4.2 Hz), 3.17 (m,
2H), 4.5 (m, 1 H),
7 (bs, 1 H), 7.09 (bs, 1 H), 7.18 (bs, 1 H), 7.31 (bs) 1 H), 7.76 (q, 1 H, J =
4.8 Hz), 8.05 (d, 1 H.
J = 9.1 Hz)) 12.07 (bs, IH). MS (DCI-NH3) 418 (M+H)+, 401, 229. Anal calcd for
C23H35N304~ I .8CF3COOH~ 1.8H20: C, 48.83; H, 6.22; N, 6.42. Found: C, 48.68;
H, 6.30;
N, 6.73. [a] _ -10.7 ° (c=0.14, CH30H)
Example 18
O H O
HOHN Nv _NHCH3
O
N
O~ O
Exam In a 18A
O H O O H O
t-Bu0 N~ NHCH3 t-Bu0 N~ NHCH
3
O _-.-~ O
N N
H O
CA 02277121 1999-07-06
WO 98/30551 PCT/I1S98/00144
63
To a solution of 17~ (230mg, 0.486 mmol) in THF (4 mL) was added saturated
NaHC03
(3 mL,) followed by benzyl chlvroformate (0.083 mL, 0.583 mmol). The mixture
was stzrrined at
ambient temperature for 2 hours and then was evaporated to a small volume. The
residue was
partitioned between CH2CI2 and brine. The aqueous layer was separated and
extracted with twice
with CHZCI2 and the combined CH2C12 layers were dried, filtered and evaporated
to dryness.
Flash chromatography (60%-80% ethyl acetate-hexanes) yielded _1$a (264 mg,
89.7%) as a white
solid.
Exam 1R a 18B
O H OI' O Og
t-Bu0 N~ NHCH3 HOHN N~ NHCH
O -~ O 3
N N
O~O O~O \\ d
I0
The desired compound was prepared from ~ according to the procedure of
Examples 1F
and G. mp: >250 °C. 1H NMR (DMSO) b 0.0 (m, 1H), 0.65-0.90 (m, 3H),
0.79 (d) 3H, J =
6.2 Hz), 0.89 (d, 3H, J = 6.2 Hz), I .02-1.40 (m, 7H), I .73 (dt, 1 H) J =
11.4, 3.0 Hz), 2.30 (dt,
1 H, J = 11.5, 3.0 Hz), 2.68 (d, 3 H, J = 4.8 Hz), 2.77 (d, 1 H, J = I 3.2
Hz), 3.02 (dd. 1 H, J =
I3.2, 3 Hz), 3.71 (m, 1H)) 3.93 (m, 1H), 4.57 (m, 1H), 5.14 (s, 2H), 7.16 (dd,
IH, J = 7.5,
0.6 Hz), 7.26 (m, IH)) 7.37-7.41 (m) 7H), 7.83 (q, 1H, J = 4.8 Hz), 8.13 (d,
1H, J = 9 Hz),
8.76 (s, I H), 10.36 (s, 1 H). MS (DCI-NH3) m/e 567 (M+H)+, 523, 356. Anal.
calcd for
C31 H42N406'0.4H20: C, 64.87; H, 7.51; N) 9.76. Found: C, 64.79; H, 7.32; N)
9.8. [a] _ -
42.4 ° (c~.13, CH30H)
E~cam 1» a 19
o ~H o
HOHN N V ' NHCH3
O
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/OOI44
64
A mixture of the compound of Example 18 (27.0 mg, 0.048 mmol) and 10% Pd/C (5
mg)
in THF-MeOH ( 10:1, 22 mL) was stirred under H2 ( 1 atm) for 4 hours. The
mixture was filtered
through Celite, and the residue was washed thoroughly with 10% MeOH/CH2Cl2.
The filtrate and
washings were combined and evaporated to dryness. Flash chromatography (5%-8%-
10%
methanol-CH2C12) gave the desired compound (I0.3 mg, 50%). mp 258-260
°C (dec}. 1H NMR
(S00 MHz, DMSO-d6) 8 -0.06 (t, 1H, J = 11 Hz)) 0.72 (d, 3H, J = 6.5 Hz), 0.82
(d, 3H, J = 6.5
Hz), 0.72-0.82 (m, 4H), 0.88-1.10 (m) 3H), 1.18-1.32 (m, 2H)) 1.52 (bs, 1 H),
1.69 (m, I H),
2. I 8 (m, 1 H), 2.53 (d, 1 H, J = 13 Hz), 2.60 (d, 3H, J = 4.5 Hz), 2.17 (dd,
1 H, J = I 3, 2.5 Hz),
3.08 (bs, 2H), 4.42 (m, 1 H), 5.11 (m, 1 H), 6.50 (d, 1 H) J = 7.5 Hz), 6.59
(d, 1 H, J = 7.5 Hz),
l 0 7.02 (t, 2H, J = 5.5 Hz), 7.60 (Q, 1 H, J = 4.5 Hz), 7.81 (d, 1 H, J = 9
Hz), 8.64 (s, 1 H), 10.27
(s) 1H). MS (DCI-NH3) m/e 433 (M+H)+, 415, 389. Anal calcd for C23H3
f,N4O4~0.8
CF3COOH~0.8 CH3COOH~0.8 THF: C) 56.60; H, 7.70; N) 9.23. Found: C, 56.37; H,
7.79; N,
9.09.
I S Example 20
o ~ o
HOHN v 'O \
O
O
Example 20A
O O H O
t-Bu O H t-Bu0 N v _O
O O
O
The desired compound was prepared according to the method of Examples 1 A-C, F
and G,
except substituting succinate ester ,~ for succinate ester 1_. mp > 270
°C. 1H NMR (DMSO) b -
0.3-(-0.1) (m, IH), 0.6-0.9 (m, 3H), 0.63 (d, 3H, J = 6.6 Hz), 0.72 (d, 3H, J
= 6.6 Hz)) 0.9-
1.0 (m, 1H), 1.0-1.6 (m) 6H), 0.6-0.7 (m, 1H)) 2.19 (dt, 1H, J = 11.1, 3.3
Hz), 2.67 (t, IH) J
= 13.2 Hz), 3.2 (dd, 1H) J = 13.2, 3.3 Hz), 4.I-4.3 (m, 2H), 4.7-4.8 (m, IH),
5.15 (apparent
AB, 2H, J = 12.6 Hz), 6.8-7.0 (m, 2H}) 7.22 (d) 2H, J = 8.4 Hz}, 7.3-7.4 (m,
SH), 8.12 (d,
IH, J = 9.0 Hz)) 8.69 (s, 1H), 10.3 (s, IH). MS (DCI/NH3) 511 (M+H)+. Anal
calcd for
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
C29H38N206~0.5H20: C, 67.03; H, 7.56; N, 5.39. Found: C, 67.18; H, 7.57; N,
5.32. [a] +9°
(c 0.4) MeOH).
xam 1 I
o p o
HOH v 'O H
O
O
The desired compound was prepared by hydrogenation of the compound of Example
20
using the procedure of Example 1 D except substituting THF for methanol. mp >
250 °C. 1H
NMR (DMSO) 8 -0.2-(-0.1 ) (m, 1 H), 0.6- l .4 (m, 9H), 0.72 (d, 3H, J = 6.6
Hz), 0.83 (d, 3H, J
10 = 6.6 Hz), 1.4-1.6 (m, 2H)) 1.6-1.75 (m) 1 H), 2.15-2.3 (m, 1 H), 2.62 (t,
1 H, J = 12.9 Hz),
3.1-3.2 (m, 1H), 4.1-4.25 (m, 2H), 4.5-4.6 (m, 1H), 6.8-7.0 (m, 2H), 7.20
(apparent t, 2H, J =
8.4 Hz), 7.99 (d, 1H, J = 9.3 Hz), 8.70 (s, 1H), 10.3 (s) 1H). 13C NMR (DMSO)
b 21.45,
21.53, 22.0, 22.1, 24.1, 24.3, 24.5, 25.4, 28.1, 35.7, 40.9, 46.1) 46.2, 53.3,
65.9, l I4.7,
117.7, 118.5, 128.3, 129.7, 130.4, I3I.1, 154.0, 170.0, 173.0, 173.1. MS
(DCI/NH3) m/e 421
15 (M+H)+. Anal calcd for C~~H32N206~ 1.4H20: C. 59.28; H, 7.87; N, 6.28.
Found: C, 59.36;
H, 7.51; N) 6.13. [a] +28° (c 0.3, MeOH).
Example 22
0 H 0
HOHN ~~~~N(CH3~
HO
20 0
The desired compound was prepared according to the method of Examples lE, F
and G,
except substituting N,N-dimethylethylenediamine for 4-(2-
aminoethyl)benzenesulphonamide. mp
121-I24°C. iH NMR (DMSO) 8 -0.6-(-0.4) (m, IH), 0.6-1.0 (m) 3H), 0.71
(d, 3H, J = 6.6
25 Hz), 0.79 (d, 3H, J = 6.6 Hz j) I .1-1.4 (m, 3H), 1.5-1.7 (m, 3H), 2.0-2.2
(m, 1 H), 2.62 (t, 1 H,
J = I3.5 Hz), 3.0-3.2 (m) 3H), 3.3-3.6 (m) 2H), 3.9-4.1 (m, 2H), 4.4 (br s)
1H), 4.6-4.7 {m,
1H}) 6.92 (d, 2H, J = 8.4 Hz), 7.15-7.30 (m, 2H), 7.84 (d) 1H) J = 9.3 Hz),
8.15 (t, 1H) J =
CA 02277121 1999-07-06
WO 98130551 PCT/(TS98/04144
66
5.7 Hz), 9.50 (br s, 1 H). MS (DCI/NH3) m/e 477 (M+H)+. Anal calcd for
C2gH40N405' 1.5
TFA~0.3H20: C, 51.50; H, 6.50; N, 8.58. Found: C, 51.53; H, 6.62; N, 8.39. [a]
+41 ° (c 0.3,
H20).
S Example 23
/ \
O H N
1i
HOHN NV 'N
O H
O
O N~ \ I O ~ \
tBuO N t8u0
H
O H O O
O O
A solution of 2 a ( 1.19g, 2.06 mmol) prepared according to the method of
Examples 1 A-
E, except substituting succinate ester 3_ for succinate ester _l, and
substituting commercially
available 2-aminoacetophenone hydrochloride for 4-(2-
aminoethyl)benzenesulphonamide) and
ammonium acetate (4.56g, 59.4 mmol) in 10 mL acetic acid at 115 °C was
stirred for 6 hours and
then cooled to ambient temperature. The acetic acid was removed under vacuum
and the orange
solid was redissolved in 100 mL ethyl acetate and 50 mL H20. The aqueous layer
was extracted
with ethyl acetate (2x) and the combined organic layers were washed with
saturated aqueous
NaHC03 and brine, dried with MgS04, filtered and concentrated. This material
was flash
chromatographed (CH2C12 then 2% MeOH/CH2C12) to give _2~b (856 mg) as a brick-
red foam.
CA 02277121 1999-07-06
WO 98/30551 PCTlUS98100144
67
Exam~l,~.23B
\ / \
O H~ ~ O H N
1I
tBuO N N HOHN N~N
O H O H
O O
The desired compound was prepared as a white solid from ~ using the procedure
of
Examples 1F and G. mp > 250°C. 1H NMR (DMSO) 8 -0.1-0 (m, 1H), 0.6-0.9
(m, 1H)) 0.68
(d, 3H, J = 6.3 Hz), 0.85 (d, 3H, J = 6.3 Hz), 0.9-1.4 (m, 7H), 1.4-1.8 (m)
3H), 2.2-2.3 (m,
1H), 2.88 (t, 1H, J = 12.9 Hz), 3.22 (dd, 1H, J = 13.5,4.2 Hz), 4.I-4.3 (m,
2H), 5.1-5.3 (m)
1H), 6.8-7.0 (m, 2H)) 7.1-7.2 (m, 1H), 7.25-7.35 (m, 4H)) 7.53 (d) 1H, J = 1.8
Hz), 7.76 (d,
2H, J = 6.9 Hz), 8.01 (d, 1H, J = 9.3 Hz), 8.66 (s, 1H), 10.3 (s, 1H), 11.8
(s, 1H). MS
(DCI/NH3) m/e 519 (M+H)+. Anal calcd for C3pH3gN404~ 1.3H20: C, 66.47; H)
7.55; N,
10.34. Found: C, 66.60; H, 7.60; N, 10.23. [a) -35° (c 0.8, MeOH).
~xam~e 24
O H
HOHN N
I
IS
The desired compound was prepared according to the method of Examples 1 A-C, F
and G,
except substituting commercially succinate ester 3_ for succinate ester 1, and
substituting
commercially-available O-benzyl-(L)-proline hydrochloride for 4-(2-
aminoethyl)benzenesulphonamide. mp > 250 °C. 1H NMR (DMSO) b -0.3-(-
0.1) (m, 1H)) 0.6-
0.9 (m, 3H), 0.70 (d, 3H, J = 6.6 Hz), 0.81 (d, 3H, J = 6.6 Hz), 0.9-1.0 (m,
1H), 1.0-1.4 (m,
SH)) 1.4-1.6 (m, 1 H)) 1.6- I .7 (m, 1 H), I .8-2.1 (m, 3H), 2. I -2.3 (m)
2H), 2.60 (t, 1 H, J = 13.2
Hz), 2.8-2.9 (m, 1 H), 3.75-3.85 (m) 2H), 4.1-4.3 (m) 2H), 4.37 (dd, 1 H, J =
8.7, 5.1 Hz}, 4.7-
4.8 (m, 1H), 5.13 (AB pattern, 2H), 6.8-7.0 (m) 2H), 7.26 (d, 2H, J = 9.0 Hz),
7.3-7.4 (m,
CA 02277121 1999-07-06
WO 98/30551 PCT/US98100144
68
SH), 8.07 (d, 1H, J = 9.0 Hz), 8.69 (s, 1H), 10.3 (s, 1H). MS (DCI/NH3) m/e
608 (M+H)+.
Anal calcd for: C34H45N30~~0.8H20: C. 65.64; H, 7.55; N. 6.75. Found: C,
65.69; H, 7.09; N)
6.68. [a] -9 ° (c 0.3> MeOH).
Example 25
o ~ o
HOHN
O
O"OH
O
The desired compound was prepared as an off white solid from the compound of
Example
24 using the hydrogenolysis procedure of Example 21. mp > 250 °C. 1H
NMR (DMSO) 8 -0.2-(-
0.1) (m) 1H), 0.6-0.8 (m, 3H), 0.70 (d, 3H, J = 6.6 Hz)) 0.81 (d, 3H, J = 6.6
Hz)) 0.9-1.0 (m,
1 H), 1.0-1.4 (m, SH), 1.4-1.6 (m, 1 H), 1.6-1.7 (m, 1 H), 1.8-2. I (m, 3H),
2.1-2.3 (m, 2H),
2.65 (t, 1H, J = I3.2 Hz), 2.9-3.0 (m, IH), 3.7-3.9 (m, 2H), 4.1-4.3 (m, 3H),
4.6-4.7 (m, 1H),
6.8-7.0 (m, 2H)> 7.2-7.3 (m, 2H), 8.06 (d, 1H, J = 9.3 Hz), 8.68 (s, IH), 10.3
(s, 1H), 12.1-
12.4 (br s, 1H). 13C NMR (DMSO) 8 2I.7, 22.2, 24.2, 24.7, 24.9, 25.5, 28.3,
28.7) 35.1,
40.8, 46.1, 46.3, 46.6, 52.4, 58.7, 66.1, 117.9, I 18.6, 128.7, 130.4, 131.4,
154.1, 169.8,
170.1, 173.0) 173.4. MS (DCI/NH3) m/e S 18 (M+H)+. Anal calcd for C27H39N30~7~
I.OH20:
C, 60.54; H) 7.71; N, 7.84. Found: C, 60.53; H) 7.73; N) 7.51. [a] +16°
(c 0.3, MeOH).
Exam 1,
o ~ o
HOHN v 'OCH3
O
O
The desired compound was prepared as an off-white solid according to the
method of
Examples 1 A-E, except substituting succinate ester ~ for succinate ester l
and onutting 4-(2-
aminoethyl)benzenesulphonamide and substituting methanol for DMF in Example
lE. mp >
250°C. 1 H NMR (DMSO) 8 -0.25-(-0.1 ) (m, 1 H), 0.6-1.0 (m, 4H), 0.75
(d) 3H, J = 6.6 Hz),
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
69
0. 84 (d, 3 H, J = 6.6 Hz)) I .0-1.2 (m, 2H), I .2-1.4 (m, 2H), 1.4-1.6 (m, 2H
)) 1.6-1.7 (m, 1 H),
2.22 (dt, 1 H, J = 1 I .4) 3.3 Hz}, 2.64 (t) 1 H, J = 13.2 Hz)) 3.1-3.2 (m) 1
H), 3.65 (s, 3H)) 4.1-
4.3 (m, 2H)) 4.6-4.7 (m, 1H), 6.8-7.0 (m, 2H), 7.2-7.25 (m, 2H), 8.10 (d, 1H,
J = 9.6 Hz),
8.71 (s, IH)) 10.33 (s) 1H). 13C NMR (DMSO) 8 21.4, 22.1, 24.0, 24.3, 24.6,
25.4, 28.1,
35.4, 40.9, 46.15) 46.24, 51.8, 52.9, 65.9, 117.8, 118.6, 128.3, 129.9, 131.2,
154.1, 169.9,
172.0) 173.2. MS (DCI/NH3) m/e 435 (M+H)+. Anal calcd for C23H34N206~0.4H20:
C,
62.54; H, 7.94; N) 6.34. Found: C, 62.59; H, 7.81; N, 6.22. [a] +24° (c
0.3, MeOH).
xam 27
0 H 0
HOHN NV'N
O
O
The desired compound was prepared as a white solid according to the method of
Example
I, except substituting succinate ester 3_ for succinate ester 1_ and
substituting piperidine for 4-(2-
aminoethyl)benzenesulphonamide. mp > 250°C. 1H NMR (DMSO) 8 -0.2-0 (m,
1H), 0.6-0.8
( m, 1 H), 0.71 (d, 3H, J = 6.6 Hz), 0.81 (d, 3H, J = 6.3 Hz), 0.9-1.05 (m, 1
H), 1.05-1.8 (m,
14H}, 2. I -2.2 (m, 1 H), 2.7-2.8 (m, 2H}. 3.2-3.3 (m, 1 H), 3.5-3.7 (m, 3H),
3.75-3.85 (m, 1 H),
4.0-4.3 (m, 2H), 4.9-5.0 (m, 1 H), 6.8-6.9 (m, 1 H), 6.9-7.0 (m, 1 H), 7.2-7.3
(m, 2H), 8.03 (d,
1H, J = 9.0 Hz), 8.67 (d, 1H, J = 1.5 Hz), 10.29 (d, 1H, J = 1.5 Hz}. 13C NMR
(DMSO) b
21.7) 22.1, 24.I, 24.2, 24.9, 25.3) 25.5, 26.3) 28.2, 35.9, 40.9, 42.5, 46.0,
46.3, 49.8, 66.0,
117.9, 118.3, 118.4, 128.8, 130.5, 131.4, 154.1, 169.5, 170.1, 172.7. MS
(DCI/1VH3) m/e 488
(M+H)+. Anal calcd for C~7H41N3Og~0.4H20: C, 65.54; H, 8.51; N, 8.49. Found:
C, 65.64;
H, 8.49; N, 8.39. [a] +78 ° (c 0.25) MeOH).
Example 28
O H O
HORN N~N~CH3
O C H3
O
CA 02277121 1999-07-06
WO 98J30551 PCT/US98J00144
The desired compound was prepared as a white solid according to the method of
Example
1, except substituting succinate ester 3_ for succinate ester 1 and
substituting dimethylamine
hydrochloride for 4-(2-aminoethyl)benzenesulphonamide. mp > 250 °C. 1H
NMR (DMSO) 8
5 0.2-0.0 (m, I H), 0.6-0.8 (m, 2H), 0.71 (d, 3H, J = 6.6 Hz), 0.81 (d, 3H) J
= 6.3 Hz), 0.9-1.0
(m, I H), 1.1-1.6 {m) 6H), 1.65- I .75 (m, 1 H)) 2.20 (dt, I H) J = 11.1, 3.0
Hz), 2.66 (t) I H, J =
12.9 Hz), 2.84 (s, 3H), 2.8-2.9 (m, 1H), 3.19 (s) 3H), 4.05-4.25 (m, 2H), 4.9-
5.0 (m, 1H),
6.8-7.0 (m, 2H), 7.2-7.3 (m, 2H), 8.03 (d, 1 H, J = 9.3 Hz), 8.67 (d) IH, J =
2.1 Hz), 10.30 (d,
1 H, J = 1.5 Hz). MS (DCI/NH3) m/e 448 (M+H)+. Anal calcd for
C~H3~N30g~0.7H20: C,
10 62.64; H, 8.41; N, 9.13. Found: C, 62.80; H, 8.30; N) 8.87. [a] +53°
(c 0.3, MeOH}.
Example 29
O H O _
HOH
0
N
H
U
Exam In a 29A
I -tent-butvldimeth~yl-3-bromoindole
To a cold (-78°) solution of indole (4.Og, 34mmo1) in THF (120mL) was
added nBuLi
(2.SM/Hexanes) over 5 minutes. The solution was warmed to -10°
(ice/salt bath) for 15 minutes,
re-cooled to -78° and TBDMS-Cl (5.8g, 38mmo1) was added in THF (30mL).
The solution was
held at 0° for 3 hours, cooled to -78° and N-Bromosuccinimide
(6.Og, 34mmol) was added in one
portion. The solution was stirred coldfor 2 hours and then was allowed to warm
to ambient
temperature at which time hexane/pyridine ( 100mL/1mL) was added to the
solution and the
resulting suspension was filtered over Celite. The organics were evaporated
and the residue
quickly purified by flash chromatography (hexanes/methylene chloride 2:1)
giving B.Sg of the
desired compound as a slightly purple solid.
CA 02277121 1999-07-06
WO 98/30551 PCT/L1S98/00144
71
Exam l
N O
t-BuO~ N
O N
S'r-
t-Bu0
To a 0 °C solution of nBuLi (2.SM/hexanes, 9.6 mL) in diethyl ether (10
mL) was added 1-
tert-butyldimethylsilyl-3-bromoindole (7.4g, 24mmol) in diethyl ether (5 mL).
The resulting pale
yellow solution was stirred cold for 25 minutes then was added to a -78
°C solution of N-tert-
butoxycarbony-O-tert-butyl-L-tyrosine (2.0g, 6mmo1) in diethyl ether ( 150
mL). The solution was
warmed to 0°, held for lhour, and quenched with saturated aqueous NH4Cl
(25mL). The aqueous
layer was extracted with diethyl ether (3x), and the combined organics washed
with saturated
aqueous NaHC03 and brine) dried (MgS04) and concentrated. Flash chromatography
(gradient
elution; 0.5-2% acetone/hexane) gave O.Sg of 29b as a reddish foam.
Example 29C
O N O
t-Bu0 O N 1 , i t-Bu0 O N
N H
Si-
\/ ~ \/
t-Bu0 ~ t-Buo
To a solution of 29~ ( 1.41 g, 2.56mmo1) in 30mL dry THF was added 2.6mL
tetrabutylammonium fluoride ( 1 M in THF) over 1 minute. The greenish solution
was stirred at
ambient temperature for 1 hour and diluted with diethyl ether. The organics
were washed with
water (2x) and brine, dried (MgS 04), filtered and concentrated in vacuo to
give 2~c ( 1.3 g) as a
reddish foam.
Example 29D
H O
t.BuO N
O N O
o H HORN O 1 ~ /
\ / H
t-8u0
c7
CA 02277121 1999-07-06
WO 98/30551 PCT/L1S98/00144
72
The desired compound was prepared according to the method of Examples 14B and
C,
except substituting 29c for 14a. mp 250° (dec). 1H NMR (DMSO-d6) 8
12.00 (s, IH), 10.28 (s,
1H), 8.65 (s, 1H), 8.59 (s, 1H), 8.21-8.15 (m, 2H), 7.48-7.44 (m, 2H), 7.29-
7.16 (m, 3H),
6.91-6.82 (m, 2H}) 5.41-5.38 (m) 1H), 4.31-4.05 (m, 2H), 3.06-3.01 (m, 1H),
2.88-2.80 (m,
1 H), 2.25-2.17 (m, I H), 1.72-1.71 (m, 1H), 1.81-1.67 (m, 1 H), 1.35-1.23 (m,
1 H), 1.16-106
(m, 4H)) 0.77-0.43 (m, 8H), 0.01-(-)0.06 (m, 1 H). MS (DCI/NH3) m/e 520
(M+H}+.
[a]D: +12.5°(c = 0.12, DMF).
Example 30
The desired compound was prepared according to the method of Examples I A, B,
C and
F, except substituting succinate ester 4_ for succinate ester 1, and
substituting L-tyrosine N-
methylamide hydrochloride for benzyltyrosine tosylate salt: mp >270°C.
IH NMR (300 MHz,
DMSO-d6) 8 0.02-(-0.11) (complex, 1H), 0.56-0.85 (complex, 2H), 0.91-1.56
(complex, lOH},
1.86-1.97 (m, 1H), 2.08-2.19 (m, 1H}, 2.24 (s, 3H), 2.30-2.64 (complex, 3H),
2.57 ~d, 3H, 3 =
5.1 Hz), 2.95 (dd, 1 H, J = 12.9, 3.0 Hz), 4.05-4.16 (m, 1 H), 4.16-4.26 (m, 1
H), 4.50 (m, 1 H},
6.81 (dd, 1H, J = 2.1) 8.4 Hz), 6.93 (dd, 1H, J = 2.1, 8.4 Hz}, 6.98-7.07
(complex, 4H), 7.15
(m, 1H)) 7.22 (m, 1H), 7.75 (m, 1H)) $.12 (d, 1H, J = 9.0 Hz), 12.08 (s, 1H).
MS (DCI/NH3)
495 {M+H)+, 391. Anal calcd for C29H3gN2O5: C, 70.41; H) 7.74; N, 5.66. Found:
C, 70.19; H,
7.66; N, 5.85.
Exam l
i
O H O
N
HORN ~ NHCH3
O
CA 02277121 1999-07-06
WO 98/30551 PCTI1JS98/OOI44
73
The desired compound was prepared from the compound of Example 30 according to
the
procedure of Example IG. mp >270°C. IH NMR (300 MHz, DMSO-d6) 8 -0.22-(-
0.12)
(complex, 1 H), 0.62-0.73 (complex, 2H)) 0.88- I .57 (complex, 9H), I.68-1.79
(complex, 1H),
2.14 (m, I H), 2.23-2.37 (m, 1 H), 2.25 (s, 3H), 2.41-2.64 (m, 2H), 2.54 (d,
3H) J = 4.2 Hz),
2.94 (m, 1H), 4.06-4.26 (m, 2H), 4.47 (m, IH), 6.81 (dd, 1H, J = 2.4, 7.8 Hz),
6.92 (dd) 1H,
J = 2.7, 8.1 Hz), 6.99-7.07 (complex, 4H), 7.16 (dd, 1 H, J = 2.4, 8.7 Hz),
7.22 (dd, 1 H, J =
2.4, 8.7 Hz), 7.66 (m, I H), 8.05 (d, 1 H, J = 9.0 Hz), 8.70 (s, I H), 10.33
(s, 1 H). MS (APCn
510 (M+H)+, 492, 477, 461. Anal. Calcd for C29H39N3O5: C, 68.34; H, 7.7I; N,
8.24. Found:
C, 68.07; H, 8.00; N, 8.16.
15 Exams lei 32A
~I ~I
o ~ o
t-8u0 O OH + HZN~OBn t-~O v 'OBn
O ~TsOH O
~OTBDINS
HO
~n~cn HO
The desired compound was prepared according to the method of Example 1A)
except
substituting succinate ester 5 for succinate ester _l.
Exam 1
CA 02277121 1999-07-06
WO 98/30551 PCT/LTS98/00144
74
Example 32B
~I
w _
0
0 0
t-Bu0 "v"OBn t-Bu0 O = OBn
O
OH ~ /
HO
TBDMSO HO
To a 0 °C solution of epimeric amides ~ ( 1.67g) 2.24 mmol) in THF ( I
S mL) was added
tetrabutylammonium fluoride ( 1.OM in THF, 6.5 mL, 6.5 mmol) dropwise via
syringe over 5
minutes. After 30 minutes the cooling bath was removed and the reaction
mixture was stirred at
room temperature for 1.5 hours at which time it was poured into a separatory
funnel containing
H20 and ethyl acetate. The aqueous phase was extracted with ethyl acetate and
the combined
organic layers were dried with Na2S04, filtered and concentrated. Flash
chromatography (50-80%
EtOAc-hexanes) afforded ~ ( 1.23 g} as an epimeric mixture of diols as a
colorless foam.
Ex~le 32D
i
H O O N O
N ~OB~ HOHN v _OH
O O
OH
O
HO
IS
The desired compound was prepared from ~ by ring closure, hydrolysis of the
tent-butyl
ester, conversion to the hydroxamic acid and debenzylation according to the
method of Examples
1 C, F, G and D. 1 H NMR (300 MHz, DMSO-d6) 8 -0.4-(-0.5 I ) (m, 1 H), 0.52-
0.93 (m, 3H),
1.12-1.20 (m, ZH), 1.30-1.42 (m, IH), 1.44-1.63 (m, 3H), 1.68-1.80 (m, 1H),
1.98-2.08 (m,
IH), 2.22 (s, 3H)) 2.31-2.39 (m, IH), 2.40-2.51 (m, IH}, 2.52-2.63 (m, 1H),
3.24 (dd, 2H, J
= 4.5) 4.8 Hz), 3.91-4.14 (m) 2H), 4.58-4.7 (m, 1H}, 6.89 (d, 2H, J=8.1 Hz),
7.01 (s, 4H),
7.18 (d, 2H, J = 8.4 Hz), 7.96 (d, 1 H, J = 9.9 Hz), 8.70 (bs) I H), 10.4 (s,
1 H). MS
(DCI/NH3) m/e 483 (m+H)+. [a ) (MeOH) _ +24° (MeOH).
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
Exam ly a 33
/ \
0 0
O N v _NHCH3
0
\/
0
5
y
0 0 0
OH
t-Bu0 t-Bu0 ~' _ NHCH3
O O \
OH
OTBDI~
OTBDMS
The desired compound was prepared according to the method of Example lA,
except
substituting succinate ester _6 for succinate ester _l, and substituting L-
tyrosine N-methylamide
10 hydrochloride for benzyltyrosine tosylate salt.
Examlle B
i
i ~ v
O H O O H O
t-Bu0 ~ NHCH3 Hp ~ NHCH3
O O
3~
OH
OTBDMS O
15 The desired compound was prepared from ~g by desilylation according to the
method of
Example 32D, followed by ring closure and saponification of the tert-butyl
ester according to the
method of Examples 1 C and F. mp 193-195°C. 1H NMR (DMSO-d6) b -0.30-(-
0.17) (complex)
1H)) 0.61-0.78 (complex, 1 H), 0.79-0.98 (complex, 2H), 0.96 (t, 3H) J = 7.4
Hz), 1.30-1.66
CA 02277121 1999-07-06
WO 98130551 PCTlUS98/00144
76
(complex) 6H), 1.96-2.17 (complex, 2H), 2.37 (m, 2H), 2.44-2.60 (complex, 3H),
2.64 (d, 3H,
J = 5.1 Hz), 3.10 (m, 1H), 4.05 (m, 2H), 4.66 (m) 1H), 6.89-6.96 (complex,
4H), 7.05 (d, 2H,
J = 8.4 Hz), 7.20 (m, 2H), 7.89 (m, 1H), 8.02 (d, 1H, J = 9.6 Hz), 12.07 (s,
1H). MS (ESI+)
495 (M+H), 464, 436. Anal calcd for C29H38N205~ 0.75 H20: C, 68.54; H, 7.83;
N, 5.51.
Found: C, 68.63; H, 7.73; N, 5.40.
Example 34
/ \
0 H 0
HOHN Nv 'NHCH3
O
\ /
O
The desired compound was prepared from the product of Example 33 according to
the
method of Example 1G. mp >260 °C. 1H NMR (DMSO-d6) b -0.30-(0.17)
(complex, 1H), 0.61-
0.95 (complex, 3H)) 0.86 (t, 3H, J = 7.5 Hz), 1.28-1.40 (complex, 2H), 1.47-
1.63 (complex,
4H), 1.73-1. 85 (m, 1 H), 2.06-2.16 (complex, 1 H), 2.26-2.40 (complex, 2H),
2.24-2.59
(complex, 3H), 2.63 (d) 3H) J = 4.7 Hz), 3.09 (m, 1H)) 3.91-4.18 (m, 2H), 4.62
(m, 1H), 6.89-
6.96 (m, 4H)) 7.05 (d) 2H) J = 8.1 Hz)) 7.18-7.24 (complex, 2H), 7.88 (m, 1H),
8.01 (d, 1H, J
= 9.1 Hz), 8.66 (s, 1 H)) 10.33 (s, 1 H). MS (ESI+) m/e 510 (M+H)+, 477. Anal
calcd for
C29H39N3O5: C, 68.34; H, 7.71; N, 8.24. Found: C, 68.06; H, 7.41; N, 8.14.
Example 35
O H O
N
HOHN ~ NHCH3
O
'
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
77
Example 35A
0
t-BuO~ N~ O H O
'I OH t-Bu0 ~ Ni
O
O H
NHCbz HO NHCbz
The desired compound was prepared according to the method of Examples 8A) C
and D
except substituting N-a-t-BOC-N-~-Cbz-L-lysine for BOC-L-tyrosine in Example
8A and
substituting succinate ester 2 for $~.
Example 35B
O ~ O H O
t-Bu0 NHCH ~' t-Bu0
s ~ NHCH3
O O
H ~!
HO NHCbz O NHCbz
To a -78 °C solution of oxalyl chloride ( 154 ul, 224 mg, 1.765 mmol)
in anhydrous
CH2Cl2 (3 mL) was added a solution of anhydrous DMSO (251 ul, 276 mg, 3.54
mmol) in
anhydrous CH2C12 (3 mL) dropwise. The suspension was stirred for lhour at -70
°C and a
solution of ~5 (500 mg, 0.887 mmol) in anhydrous CH2C12 (3 mL) was added
dropwise. The
reaction mixture was stirred for 6 hours at -70 °C and triethylamine
(390 ul, 283 mg, 2.80 mmol)
was added and the resulting yellow solution was stirred for 1 hour at ambient
temperature. The
reaction mixture was diluted with saturated aqueous NH4C1 and CHZC12. The
aqueous phase was
washed with 10% isopropanol-CHC13. the combined organic layers were dried over
MgS04 and
filtered and the filtrate was concentrated leaving a yellow gum which was
purled using flash
chromatography (3% methanol-CH2C12) to give ~ (411 mg) as a foamy white solid
(82% yield).
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
78
Example 35C
O . NV\ O . NV\
t-Bu0 " O NHCH3 ' t-Bu0 " O NHCH3
H ~ ~ N.
O NHCbz
The desired compound was prepared by catalytic hydrogenation of ,~5 (methanol,
palladium hydroxide on carbon, 1 atm. H2) for 3 days.
Exa-pm ie 35D
0 0 0 0
t ~ _ N~NHCH --~ t-Bu0
NHCH3
O - O
O
O
lU
The desired compound was prepared according to the method of Example 18,
except
substituting ~ for the compound of Example 17.
Exam 1R a 35E
0 0 0 0
N
t-Bu0 _ ~ NHCH ~N _ H~ NHCH
3 ~ O = 3
~ ''
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
79
The desired compound was prepared according to the method of Examples IF and
G,
except substituting ~5 for 8~. 1H NMR (300 MHz, DMSO-d6) b 10.55 (s, 1H), 8.80
(s, 1H),
8.31 (d, 1H, J = 12 Hz), 7.65 (d) 1H, J = 12 Hz), 7.24-7.39 (c, SH), 4.91-5.04
(c, 2H), 4.20-
4.30 (c, 1 H), 2.99-3.35 (c, 5 H), 2.76-2.85 (c) 1 H), 2.73 (d, 3H, J = 3 Hz),
1.94-2.04 (c, 1 H),
1.06-I.72 (c, 12H), 0.72-0.95 {c, 6H). 13C NMR (DMSO-d6): 173.1, 172.0, 169.7,
155.1,
136.2, 127.7, 127.0, 126.7, 65.4, 49.9, 49.4, 48.8, 48.7, 45.0, 28.8) 28.5,
27.I, 26.8, 26.6,
24.71, 24.70, 23.4, 22.1, 20.9. MS (ESI) m/e 527(M+Na)) 522(M+NH4), 505(M+H).
IR
(KBr) 3420, 2940, 1630, 1540 cm-1. HRMS (ESI) Theory: 505.3026. Found:
505.3023.
x~ m 1 ,~
O H O
HOHN N
~NHCH3
O
H
~-'N
The desired compound was prepared according to the method of Example 19,
except
substituting the compound of Example 35 for the compound of Example 18.
Example 37
O H O
HOHN N
NHCH3
~N O
i
OzS'
Example 37A
o) ~ ° o
H H
N N
t-Bu0
t-Bu0 NHCH
3 _ ~NHCH3
O O
HO
' 20 ~~~~ NHC8z Mg0 ~ NHCBz
CA 02277121 1999-07-06
WO 98/30551 PCTlUS98/00144
$0
To a 0 °C solution in dichloromethane ( 18 mL} of 5a ( 1.0 g, 1.77
mmol) was added
triethylamine (697 pl, 506 mg, 5.0 mmol) followed by the dropwise addition of
methanesulfonyl
chloride (310 ~1, 458 mg, 4.0 mmol). The reaction mixture was stirred at 0
°C for 30 minutes and
then at ambient temperature for 2 hours. The reaction mixture was diluted with
CH2C12 and
water. The aqueous phase was washed once with CH2C12. The combined organic
layers were
dried over MgS04, filtered and concentrated in vacuo to give 37a as a yellow
gum ( 1.13 g) which
was used without further purification.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
81
Example 37B
o ~ o o
H
t-Bu0 NHCH t-Bu0
O 3 ~ NHCH3
O
Ms0 ~ NHCBz Ms0 ~ NH2
The desired compound was prepared by hydrogenation of 37a {methanol, 10%
palladium
on carbon, 1 atm HZ) for 3 days.
Exam 1
O NV \ O H O
_ N
O 3 t-Bu0 ~ NHCH3
t ~ NHCH
O =
Ms0 ~ NHZ Ms0 STS N O
~S~
O
1 U o2N
To a solution of 37b (732 mg, 1.44 mmol) in dichloromethane (40 mL) was added
triethylamine (4I8 pL, 304 mg, 3.0 mmol) and a solution of 2-
nitrobenzenesulfonyl chloride (335
mg, 1.5 mmol) in dichloromethane (8 mL) was added dropwise and stirring at was
continued for 3
hours. The reaction solution was washed with water, saturated NaHC03 and
brine. The organic
layer was dried over Na2S04, filtered and the filtrate concentrated to give a
green-brown solid
which was purified using flash chromatography (3% MeOH-dichloromethane) to
give ~ as a
white solid (591mg, 59% yield).
CA 02277121 1999-07-06
WO 98/30551 PCTIUS98/00144
82
Examlle~ 37D
0
t-Bu0 NHCH3 t-Bu0 NHCH
3
H
Ms0 ~ ~ S p - ~.ZSI i ~O
tj' ~ ~ O' ~ \
02N O N /
2
S Compound ~(S90mg, 0.8Smmol) was dissolved in anhydrous DMF (4SmI) and to
that solution
was added K2C03 (23Smg, l.7mmo1) as a solid and the suspension stirred at RT
over 3 days.
The reaction mixture was diluted with 40m1 water and extracted 1 X 400m1 with
ether. The
organic layer was dried over MgS04) filtered and the filtrate concentrated to
a yellow solid which
was purified using flash chromatography eluting with 30% EtOAc/CH2Cl2 to give
a white solid
( 166mg, 33% yield}.
Exam In a 37E
o p
t-Bu0 - N " ' NHCH ~ Ha"~N N "
O 3 CH3
~~O i~O
~ O~ \ p'~ \
/
02N / 02N
1 S The desired compound was prepared according to the method of Examples 1 F
and G,
except substituting 37d for 1~ 1H NMR (300 MHz) DMSO-d6) 8 10.46(d, 1H, J =
I.S Hz), 8.78
(d, 1 H, J = 1.S Hz), 8.29 (bd) 1 H, J = 9 Hz), 7.77-7.97 (c) 4H), 7.45-7. S4
(c, 1 H), 4.20-4.33
(c, 1H), 2.93-3.16 (c, 2H), 2.61-2.85(c. 2H)) 2.56 (d. 3H, J = 4.S Hz), 1.97-
2.07 (c, 1H),
1.21-1.81 (c, 13H)) 0.81-0.94 (c, 4H), 0.78 (d, 3H, J = 6 Hz). MS (APCI) m/e
S73
(M+NH4)+, SS6 (M+H)+.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
83
Exam a 8
O CH3
i
HOI-IN~~~~~NwN'CH3
H
Example 38A
'I 't
O H O O H O
t-t3u0 ~ OBn t-Bu0 ~ OH
O '~ O
\ / \
S O O
To a methanol solution ( 150 mL) of (3.39 g, 5.52 mmol), prepared by ring
closure of
32c using the method of Example 1 C, was added dry 10%Pd/C (340 mg) and the
mixture was
stirred under 4 atm of hydrogen in a pressurized reaction vessel for 2 hours
at room temperature.
The mixture was filtered through a Celite pad with methanol washings. The
filtrate was
concentrated to provide 3~b (2.83 g) as a white foam.
Exam 31p a 38B
~I ~)
O H O O H O
t-Bu0 Nv _OH HOHN N~~.-~N'
O O H
\ / \ /
O O
The desired compound was prepared according to the method of Examples lE, F
and G,
except substituting ~ for 1 f and substituting N,N-dimethylethylenediamine for
4-(2-
aminoethyl)benzenesulphonamide. mp >250 °C. 1H NMR (DMSO) S-0.4-(-0.46)
(m, 1H),
0.52-0.97 (m, 3H)) 1.08-1.20 (m, 2H), 1.31-1.40 (m) 2H), 1.52-1.61 (m, 2H))
1.68-1.75 (m,
1 H), 2.0-2.08 (m, 1 H), 2.12 (s, 6H), 2.23 (s, 3H), 2.24-2.45 (m) 2H), 2.52-
2.61 (m, 1 H),
CA 02277121 1999-07-06
WO 98/30551 PCT/US98100144
84
3.03-3.20 (m, 3H), 3.3-3.39 (m, 2H), 3.90-4.10 (m, 2H), 4.52-4.64 (m, 1 H),
6.90 (d, 2H, J =
8.4 Hz)) 6.98-7.04 (m, 4H}, 7.17-7.22 (m, 2H), 7.63-7.66 (m, 1H), 7.95 (d, 1H,
J = 9.3 Hz))
8.69 (s, 1H)) 10.34 (s, 1H). MS (ESI) m/e 553 (M+H). Anal calcd for
C31H4qN405~0.75H20:
C, 65.75; H, 8.09; N, 9.89. Found: C, 65.72; H, 8.28; N, 9.80. [a] +28°
( c 1.04, MeOH).
Example 39
O H O
HOHN N~N~S~
O H
O
The desired compound was prepared according to the method of Examples 1 E-G,
except
substituting 2-(methylthio)ethylamine for 4-(2-aminoethyl)benzenesulphonamide.
mp > 270 °C.
1H NMR (DMSO) 8 -0.6-(-0.4) (m, 1H), 0.5-1.0 (m, 5H), 0.71 (d, 3H, J = 6.6
Hz), 0.80 (d)
3H, J = 6.6 Hz), 1.1-1.4 (m, 2H), 1.5-1.7 (m, 3H), 2.0-2.I (m, IH), 2.07 (s,
3H), 2.5-2.7 (m,
3H), 3.05-3.4 (m, 2H), 3.9-4.1 (m, 2H), 4.55-4.7 (m, 1H), 6.90 (d, 8.4H), 7.2-
7.3 (m, 2H),
7.8-7.9 (m) 2H), 8.68 (d, 1H, J = 1.5 Hz), 10.3 (d, 1H, J = 1.5 Hz). 13C NMR
(DMSO) 8
14.5, 21.6, 24.2. 24.7) 24.9, 28.0, 28.4, 32.8, 37.8, 40.3, 40.8, 46.0, 46.6,
53.5, 72.9, 121.0,
121.2, 128.7, 132.0, 132.4. 157.1. 170.0, 171.1, 172.7. MS (DCI/NH3) m/e 480
(M+H)+.
Anal calcd for C24H3~N305S~0.5H20: C, 58.99; H, 7.84; N, 8.60. Found: C,
59.05; H, 7.65;
N, 8.45.
[a] +41 ° (c 0.5, MeOH).
Exam lp a 40
o ~ o
HOHN N
O
~z~
O
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
Example A
0
HChH2
S~
4~ ~ \
OH
To a 0 °C solution of nBuLi (2.SM/hexanes, 14.2 mL) in diethyl ether
(50 mL) was added
5 4-bromothioanisole (7.2g, 35.6mmol) over a few minutes. The resulting
solution was allowed to
stir cold for 25 minutes and then was added to a -78 °C solution of N-
BOC-tBu(OH) tyrosine (3g,
8.9mmol) in diethyl ether (200 mL). The solution was stirred at -78 °C
for 25 minutes, warmed to
0° over 1 hour and quenched with an aqueous solution of NH4C1. The
aqueous layer was
extracted twice with diether ether and the combined organics were washed with
brine, dried
10 (Na2S04), filtered and concentrated in vacuo. Flash chromatography (8:1
hexane-ethyl acetate)
gave 2.Sg of product which was immediately taken up in lOmL 4N HCl-dioxane and
stirred for 30
minutes. The resulting slurry was diluted with diethyl ether) filtered and
dried for 16 hours under
high vacuum, to give 4~ ( 1.4 g) as a chalky-white solid.
15 Example 40B
O O H O
HCI'H
t-Bu0 'V
4Q~ \ / ~ O I ~ S(O)~Me
~ \
OH ' OH
HO
The desired compound was prepared as a mixture of the sulfide (n=0) and
sulfone (n=2)
using the procedure of Examples 1 A and B, except substituting ~ for
benzyltyrosine tosylate
20 salt.
Example 40C
H O
t-Bu0 N w
O
S(O)"Me
SOpMe
HO I OH
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
86
To a solution of Q (l.Og, l.7mmo1) in acetone (50 mL) was added OXONETM
(potassium peroxymonosulfate, Sg, 8mmo1). The resulting slurry was stirred for
3 days. The
reaction was diluted with ethyl acetate and water and the aqueous layer was
extracted twice with
ethyl acetate. The combined organics were washed with brine, dried (Na2S04),
filtered and
concentrated in vacuo. Flash chromatography (2-4% methanol-methylene chlorid)
gave 40c (0.9
g) as a white foam.
Exam 1R a 40D
O H O
t-Bu0 N _ ~ H0.
O ~i
S02Me SOzMe
4~
OH
HO
The desired compound was prepared according to the method of Examples 1 C, F
and G,
except substituting 4Qc for 1 c. mp 250-250 °C (dec. ). 1 H NMR (DMS O-
d6) 8 10.28 (s, 1 H),
8.65 (s, 1 H), 8.28-8.25 (d, 2H, J = 6.8 Hz), 8.19-8.16 (d, 1 H, J = 9.5 Hz))
8.07-8.04 (d, 2H, J
= 6. 8 Hz), 7.42-7.39 (d, 1 H, J = 8.5 Hz), 7.24-7.20 (d, 1 H, 7 = 8.5 Hz),
6.97-6.93 (d, 1 H, J =
8.1 Hz)) 6.87-6.83 (d, 1H, J = 8.2 Hz), 5.60-5.47 (m) 1H), 4.21-4.13 (m, 2H),
3.12 (s, 3H),
3.12-3.06 (m, 1 H), 2.81-2.73 (m, 1 H ), 2.15-2.12 (m, 1 H), 1.70-1.69 (m, 1
H), 1.47-1.40 (m,
1 H), 1.33-1.30 (m) 1 H), 1.15-0.99 (m, 4H)) 0.71-0.68 (m, 3H)) 0.50-0.38 (m,
7H), (-)0.02-(-
)0.09 (m, 1 H). Anal calcd for C29H3gN20~S~O.SH20: C, 61.35; H, 6.92; N, 4.93.
Found: C,
61.26; H, 6.83; N, 4.6I. [a]D: +11.4°(c = 0.21, DMF).
Exam eln a 41
O H O
HOHN Nv 'NHCH3
O j
O
Example 41 A
0
!+zN~ ~ t-Buo~ N
II OH
0
\ I 41a \
off off
CA 02277121 1999-07-06
wo 9sr3ossi rc rius98iooia4
87
To a solution of (L) p-hydroxyphenylglycine (5 g, 29.9 mmol) Sigma) in 50%
aqueous
dioxane ( 100 mL) and triethylamine (8.3 mL, 59.8 mmol) at ambient temperature
was added a
solution of Boc-anhydride (13.7 g, 59.$ mmol) in dioxane (10 mL) over 1
minute. The resulting
solution was stirred at ambient temperature for 2.5 days and then was poured
into a mixture of
aqueous 1M HCl (100 mL) and ether (75 mL). The aqueous layer was extracted
twice with ether
and the combined organic layers were washed with aqueous 1 M HCl and brine,
dried with
MgS04, filtered and concentrated to afford 4~1 ( 10.8 g) as a white foam which
was used without
further purification.
Exam In a 41B
H O
t-BuO~ t-BuO
OH ~ NHMe
O / O
/
OH OH
The desired compound was prepared according to the method of Example lE,
except
substituting methylamine hydrochloride for 4-(2-
aminoethyl)benzenesulphonamide.
Example 41 .
H O O
fBuO" _ ~
NHMe HZNv 'NHMe
O
/ TFA
w~
OH OH
A mixture of 41 b ( 1.13 g, 4.02 mmol), trifluoroacetic acid ( 10 mL) and
CH2C12 {2 mL)
was stirred at ambient temperature for 1 hour and then concentrated under a
stream of nitrogen.
The residue was dissolved in a 1:1 mix of CH2C12 and methanol (40 mL) and
concentrated in
vacuo. The dissolving-concentration sequence was repeated until a white foam
formed to give 41 c
( 1.2 g) which was used without purification.
Ex~le 41 D
O O O H O
t-Bu0 OH ~~ NHMe t-Bu0 ~ NHMe
O + i I ~1FA O /
$ ~ 41~ 31s! w ~
I off I off
CA 02277121 1999-07-06
WO 98130551 PCT/US98/00144
88
The desired compound was prepared according to the method of Example IA,
except
substituting succinate ester 3 for succintate ester 1_, and substituting 41c
for benzyltyrosine tosylate
salt.
Example 41 E
H~ O H O
hBuO N NHMe HOHN Nv _ NHMe
O / O
~ \ ~ \
OH
O
The desired compound was prepared according to the method of Examples IB, C, F
and
G, except substituting 41d for 1 b. mp > 270 °C. 1H NMR (DMSO) 8 -0.65-
(-0.5) (m, 1H), 0.4-
1. I (m, 6H), 0.77 (apparent t, 6H, J = 6.6 Hz), 1.2-1.5 (m, 4H), 1.6-1.7 (m,
1 H), 2.3-2.5 (m,
1H), 2.65 (d) 3H, J = 4.5 Hz), 4.1-4.2 (m, 2H), 5.30 (d; IH, J = 9.3 Hz}) 6.87
(dd, 1H, J =
8.1, 2.4 Hz), 6.96 (dd, 1 H, J = 8.1, 2.4 Hz), 7.24 (dd, 1 H, J = 8.1, 2.4
Hz), 7.38 (dd, 1 H, J =
8.1, 2.4 Hz), 8.0-8.1 (m, 1H), 8.19 (d, 1H, J = 9.3 Hz), 8.64 (d) 1H) J = I.2
Hz), 10.24 (d,
1H, J = 1.5 Hz). MS (DCI/NH3) m/e 420 {M+H)+. Anal calcd for
C22H33N305'O.1H20: C,
62.72; H) 7.94; N, 9.97. Found: C, 62.61; H, 7.73; N, 9.73. [a] +186°
(c 0.25, DMF).
Example 42
0
HOHN'
O
0
The desired compound was prepared according to the method of Examples 1 A-C, F
and G,
except substituting tyramine for benzyltyrosine tosylate salt. mp > 270
°C. 1H NMR (300 MHz,
DMS O-d6) 8 -0.5-(-0.4) (m, I H)) 0.5-1.0 (m, 4H), 0.73 (d, 3H, J = 6.3 Hz),
0.77 (d, 3H) J =
6.3 Hz), l . l -1.3 (m, 2H), 1.4-1.7 (m, 3H), 1.98 (dd, 1 H, J = 10.5 ) 3.3
Hz), 2.5-2.6 (m, I H),
2.8-3.0 (m, 2H)) 3.8-4.1 (m) 3H), 6.85-6.95 (m, 2H), 7.1-7.2 (m, 2H), 7.40 (d,
9.3H), 8.67 (s,
1 H), 10.3 (s, 1 H). 13C NMR (DMSO) 8 2 I.S, 24.1, 24.6) 25.2, 28.1, 28.5,
33.5, 38.3, 40.6,
46.6) 46.7, 72.7, 120.6, 120.9, 129.0, 131.6) 133.3, 156.9, 170.3, 172.6. MS
(CI NH3) m/e
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
89
363 (M+H)+. Anal calcd for C2pH3pN204~0.8H20: C, 63.74; H, 8.45; N, 7.43.
Found: C,
63.90; H) 8.53; N, 7.33. [a] +103° (c 0.3, MeOH).
Example 43
0
HOHN
Exalrn le 4 A
O
HCI~H2
i
4~ / _\
OH
The desired compound was prepared according to the method of Example 40A,
except
substituting 4-bromo-tert-butyl benzene for 4-bromothioanisole.
Exam 1R a 43B
_ o
O HC, Hp
OH v ~ H O
t-Bu0 O ~ / t-Bu0 N = W
\ O = ~ i
Z g~a ' H ~ \
TBDMSO
OH OH
1S
The desired compound is prepared by coupling of ~ and 7, and deprotection
using
tetrabutylammonium fluoride according to the method of Examples 32B and C.
Example 4 D
O H O O O
t-Bu0 N ~ HO,N
O E / O
2~ OH OH
O
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
The desired compound was prepared by ring closure according to the method of
Example
8E) followed by saponification of the tert-butyl ester and conversion to the
hydroxamate according
to the method of Examples 1 F and G, except substituting ~c for lc. mp 220-221
°C. 'H NMR
5 (CD30D) 8 -0.26 (m, 1H), 0.50 (d, 3H, J = 5.7 Hz), 0.64 (d) 3H, J = 5.8 Hz),
0.85 (m, 4H),
1.18 (m, 3H), 1.35 (s, 9H), 1.63 (m, 1H), 1.79 (m, 2H), 2.89 (t, 1H, J = 12.9
Hz), 3.21 (dd,
1H) J = 4.7, 12.9 Hz), 4.08 (m, 1H)) 4.18 (m, 1H), 5.93 (dd, 1H, J = 4.4, 12.2
Hz), 6.90 (dd,
1 H, J = 2.7, 8.1 Hz), 6.96 (dd, 1 H) J = 2.7, 8.4 Hz), 7.19 (dd> 1 H, J =
2.4, 8.1 Hz), 7.41 (dd,
1H, J = 1.7, 8.1 Hz), 7.55 (d, 2H) J = 8.5 Hz)> 8.05 (d, 2H, J = 8.4 Hz), 8.38
(d, 1H, J = 12.2
10 Hz). 13C NMR (CD30D) 8 21.35, 24.75, 25.99, 29.49, 30.24, 30.67, 31.48,
36.03, 36.83,
42.44, 47.90, 48.45, 55.04, 74.40, 122.14, 122.37, 126.77, 129.80, 130.55,
133.17, 133.23,
134.16, 158.65, 159.01, 173.25, 175.52, 199.27. MS (DCI/NH3) m/e 523 (M+H)+.
Anal calcd
for C31H42N205'H20~ C, 68.86; H, 8.20; N) 5.18. Found: C, 68.57; H, 8.05; N,
5.45.
Example 44
The desired compound was prepared as a white foam according to the method of
Examples
lE and F, except substituting piperidine for 4-(2-
aminoethyl)benzenesulphonamide, substituting
for ~g and substituting methanol for DMF in Example lE. 1H NMR (DMSO) 8 -0.40-
(-0.24)
(m, 1 H), 0.52-0.72 (m, 1 H), 0.73-1.0 (m) 2H)) 1.10-1.43 (m, 7H), 1.44-1.70
(m, 6H), 1.92-
2.08 (m, 2H), 2.22 (s, 3H)) 2.24-2.37 (m, 1H), 2.39-2.50 (m, 1H), 2.71-2.93
(m, 2H), 3.32-
3.40 (m, 2H)) 3.51-3.58 (m, 2H)) 4.0-4.08 (m) 2H), 5.0-5.12 (m, 1H), 6.88 (d)
2H, J = 8.4
Hz), 6.97 (d, 2H) J = 8.1 Hz)) 7.03 (d, 2H) J = 8.1 Hz), 7.15 (d, 1 H, J = 9.3
Hz), 7.24 (d) 1 H,
J = 9.2 Hz), 8.13 (d, 1H, J = 9.9 Hz). MS (DCI/NH3) 535 (M+H)+.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
91
Example 45
HOH
The desired compound was prepared as a white solid according to the method of
Example
1G, except substituting the compound of Example 44 for 1~. mp > 250 °C.
1H NMR (DMSO) 8
-0. SO-(-0.38 ) (m, 1 H)) 0.53- I .0 (m, 3 H), I .07-1.20 (m, 2H), 1.22-1.43
(m, SH), 1.45- I .64 (m,
SH), I .65-1.81 (m, 1 H), 1.98-2.10 (m) 1 H), 2.23 (s) 3H), 2.25-2.31 (m, 1
H), 2.38-2.45 (m)
1H), 2.71-2.90 (m, 2H), 3.30-3.42 (m, 2H), 3.50-3.62 (m, 2H)) 3.94-4.10 (m,
2H), 4.97-5.10
(m, 1 H), 6. 87 (d, 2H, J = 9.0 Hz), 6.93-7.07 (m, 4H), 7.15 (d, 1 H, J = 7.2
Hz), 7.24 (d, 1 H, J
= 7.1 Hz), 8.05 (d, 1 H, J = 9.3 Hz)) 8.69 (s, I H)) 10.36 (s, 1 H). MS (ES I-
) m/e 548 (M-1 ).
Anal calcd for C32H43N3Og: C, 69.91; H, 7.88; N, 7.64. Found: C) 69.85; H,
7.77; N) 7.57.
[a] + 56° ( c = 1.0, MeOH).
~x~
CH3
O H O N
NI 7S
HO ~ N
O H
O'
The desired compound was prepared according to the method of Examples 1 E and
F,
except subsituting ~ for fig, and substituting 2-aminothiazole for 4-(2-
aminoethyl)benzenesulphonamide. 1H NMR (300 MHz, DMSO-d6) 8 -0.34-(-0.20) (m)
1H),
0.60-0.74 (m, IH), 0.81-0.97 (m, 2H), 1.13-1.25 (m, 2H), 1.36-1.45 (m, 2H),
1.55-1.67 (m,
2H), 1.90-2.01 (m) 1 H), 2.05-2.16 (m, I H), 2.22 (s, 3H), 2.24-2.38 (m, 1 H),
2.40-2.45 (m)
1 H), 2.57-2.65 (m) 1 H), 3.24-3.34 (m) 1 H)) 3.94-4.05 (m) 1 H)) 4.07-4.16
(m, 1 H), 4.95-5.03
(m, 1 H)) 6.93-7.04 (m) 7H), 7.20-7.27 (m, 1 H), 7.31 (d, 1 H, J = 3.6 Hz),
7.35-7.38 (m, 1 H),
\ /
0
CA 02277121 1999-07-06
WO 98/30551 PCT/US98100144
92
7.53 (d, 1 H, J = 3.6 Hz), 8.22 (d, 1 H) J = 9.6 Hz), 12.40 (bs, 1 H). MS
(DCI/NH3) m/e 550
(M+H )+.
Example 47
CH3
O
HOH ~ N S
H
v 0
The desired compound was prepared according to the method of Example 1 G,
except
substituting the compound of Example 46 for 1 a. mp>250 °C. 1H NMR (300
MHz, DMSO-d6) 8
-0.48-(-0.33) (m, 1H), 0.60-0.93 (cm, 3H), 1.10-1.23 (m, 2H), 1.28-1.50 (m,
2H), 1.52-1.67
(m, 2H), 1.70-1.81 (m, 1H), 2.04-2.18 (m, IH), 2.21 (s, 3H), 2.23-2.31 (m,
1H), 2.40-2.48
(m, 1 H), 2. 58-2.67 (m, 1 H), 3.23-3.25 (m, 1 H), 3.90-4.00 (m, 1 H), 4.04-
4.16 (m, 1 H), 4.88-
4.98 (m, 1H), 6.90-7.01 (m, 6H), 7.22-7.28 (m, 1H), 7.29 (d, 1H, J = 3.9 Hz),
7.31-7.36 {m,
1 H), 7.50 (d) 1 H, J = 3.3 Hz)) 8.12 (d, I H, J = 9.3 Hz)) 8.68 (s, 1 H),
10.35 (s) 1 H), 12.30 (s,
1 H). MS (DCI/NH3) m/e 565 (M+H)+. Anal calcd for C3pH36N4OgS~0.5 H20: C,
62.80; H,
6.50; N, 9.76. Found: C, 62.95;, H, 6.33; N, 9.76. [a] + 26° ( c = 0.9,
MeOH).
Examrle 48
O H O
HOHN N ~ NHCH3
O
N
S02CH3
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
93
Example 48A
0 0
~O p ~~ NHMe ~p ~ ~ NHMe
O O
N,
S02Me
To a solution of I 7b ( 142mg, 0.300mmo1) in CHZCI2 (4 mL) was added
triethyiamirte
(0.059 tnL, 0.420 mmol) followed by methanesulfonyl chloride (0.028 mL, 0.360
mmol). The
mixture was left stirring at room temperature for 2 hours and then was poured
into brine. The
biphasic mixture was extracted with CH2CI2 (3x) and the combined CH2Cl2 layers
were dried
(MgS04), filtered and evaporated to dryness. Flash chromatography (2%-5%
MeOH/CHZCI2)
yielded 157.7 mg (95.2%) of 48a as pale yellow crystals.
0 0
t-Bu0 O ~~NHCH3 HORN ~~NHCH
O O a
N N
S02Me S02Me
The desired compound was prepared as a white solid according to the method of
Examples
1 S I F and G, except substituting 4~a, for 1 f. mp 242-244 °C (dec).
1H NMR (DMSO-D6) 8 -0. I 18
(m) 1H), 0.63 (m, 2H), 0.73 (d, 3H, J = 6.6 Hz)) 0.83 (d, 3H, J = 6.6 Hz),
0.79-1.40 (m) 7H),
1.66 (m, 1 H)) 2.24 (m, 1 H), 2.44-2.56 (m, 1 H), 2.62 (d, 3H, J = 4. S Hz),
2.50-2.75 (m, 2H),
2.93 (s, 3H), 3.01 (dd, 1H, J = 2.7) 12 Hz)) 3.60 (m, 2H), 4.52 (m, IH), 7.17
(dd, 1H, J = 8.1,
1.8 Hz), 7.32 (dd) IH) J = 8.1, 1.8 Hz), 7.41 (dd, 1H, J = 8.1, 1.8 Hz), 7.38
(dd, IH, J = 8.1)
1.8 Hz), 7.76 (q, 1H, J = 4.5 Hz), 7.06 (s, 1H, J = 9.3 Hz), 8.70 (s, 1H))
10.30 (s, 1H). MS
(ESI) 1043 (M+Na)+, 1021 (2M+H)+, 533 (M+Na)+) 51 I (M+H)+, 478, 215. Anal
calcd for
C24H38N406S'1.75 H20: C) 53.16; H, 7.71; N, 10.33. Found: C, 53.14; H, 7.32;
N) 9.90.
[a] _ +I4.6° (c=0.205) CH30H).
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
94
Exam In a 49
O H O
HOHN N V _ NHCH3
O
N
S '
~~ CH3
The desired compound was prepared according to the method of Example 48,
except
substituting p-toluenesulfonyl chloride for methanesulfonyl chloride. mp: 235-
237 °C (dec). 1H
NMR (DMSO-D6) 8 -O.I6 (m, 1H), 0.59 (m, 3H), 0.72 (d, 3H, J = 6.6 Hz), 0.82
(d, 3H, J =
6.6 Hz), 0.95-1.26 (m) 7H), 1.64 (m, 1 H), 2.22 (m, 1 H), 2.61 (d, 3H, J = 4.5
Hz), 2.69 (t, 1 H,
J = 13.2 Hz), 2.97 (dd, 1 H, J = 13.2, 2.4 Hz), 3.36 (m, 1H)) 3.51 (m, 1 H),
4.47 (m, 1 H), 6.82
(dd) 1H, J = 1.8, 8.1 Hz), 6.97 (dd, 1 H) J = 1.8, 8.1 Hz), 7.28 (m, 1 H, J =
1.2, 8.1 Hz), 7.33-
7.447.33-7.44 (m, SH), 7.72 (q, 1 H, J = 4.5 Hz), 8.05 (d) 1H, J = 8.7 Hz),
8.70 (s) 1 H), 10.30
(s, 1H). MS (DCI-NH3) m/e 587 (M+H)+, 543, 447) 391) 302, 258, 215. Anal calcd
for
C30H42N406S~0.75 MeOH: C, 60.46; H. 7.x.2; N, 9.17. Found: C. 60.33; H, 7.40;
N, 8.90.
[a] _ +41.5° (c=0.065, CH30H).
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
Example 50
O H O
HOHN N V ' NHCH3
O
N
N
Exams 1~ a SOA
H O H O
B~N~ BOCN
OH BOP-Cl, TEA NHCH3
+ MeNH2 HCI
~ CH2C12
Ts Ts
5
To a 0 °C solution of N-Boc-imidazolyl-tosyl-L-histidine (S.Og,
12.2mmol) in
dichloromethane ( 125m1) was added methylamine hydrochloride (990mg,
14.7mmol), BOP-CI
(3.7g, 14.7mmo1) and TEA (4m1, 29.4mmo1) under nitrogen, the ice-bath was
removed and the
10 mixture was stirred at room temperature for 23 hours. The reaction mixture
was diluted with brine
and extracted with three portions of dichloromethane) dried over sodium
sulfate) filtered and
concentrated in vacuo. The crude mixture was purified by flash chromatography(
6090 ethyl
acetate-hexanes, then 10% MeOH/CH2Cl2) to provide ~ (1.67g, 32%) as a white
solid.
15 Example SOB
H O
BOCN
NHCH3 .~A H2N~ NHCH3
Ts ~ Ts
A solution of SOa in trifluoroacetic acid was stirred for 10 minutes. The
trifluoroacetic acid
was evaporated and the mixture was partitioned between CH2C12 and saturated
aqueous NaHC03.
20 The aqueous phase was extracted with three portions of CH2C12 and the
combined extracts were
washed with brine, dried over sodium sulfate and concentrated in vacuo to give
~Qb ( 1.09g, 85 % )
as an off white solid.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
96
Exam l
0 0
H O ~ O
OH ~ NHCH3 N
t-Bu0 + But-O ~ NHCH3
O O
Ts
To a 0 °C solution of succinate ester 3_ (772mg, 2.59mmo1) and 5(~ (
1.Og, 3.1 mmol) in
dichloromethane (20m1) was added BOP-Cl (789mg, 3.lmmol) and triethylamine
(8621.1,1)
6.2mmo1) under nitrogen, the ice-bath was removed, and the mixture was stirred
at room
temperature for 23 hours. Additional 5~, BOP-Cl and triethylamine (0.8
equivalent) were then
added and stirring was continued for another 48 hours. The reaction mixture
was diluted with
brine and extracted with three portions of dichloromethane. The combined
organic extracts were
dried over sodium sulfate, filtered and concentrated in vacuo. The crude
mixture was purified by
flash chromatography( 5% MeOH-CH2C12) and (80% ethyl acetate-hexanes)to
provide ~r
(l.Olg, 65%) as a yellow foam.
Exam lp a SOD
O H O
t-Bu0 N~ NHCH3 t-Bu0 Nv _ NHCH
3
Ts '
~ OH Ts
The desired compound was prepared according to the method of Example 1 B,
except
substituting SOc for 1 b.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98100144
97
E
w
O H O O H
t-Bu0 ~ NHCH3 t-Bu0 N NHCH3
O O
~\ <\
OH Ts '
OMs Ts
To a 0 °C solution of 5~ ( 156mg, 0.25mmo1) in CH2C12(4ml) under
nitrogen was added
methanesulfonyl chloride(25~.1, 0.325mmo1), followed by NMM(41p,1, 0.375mmol),
the resulting
mixture was stirred at 0°c for 2hr and diluted with CH2C12/brine,
extracted with two portions of
CH2C12, dried over sodium sulfate, filtered, solvent was evaporated and the
crude mixture was
purified by flash chromatography(5% MeOHlCH2Cl2) to afford SOg (113 mg, 65%)
as a white
foam.
F~xample 50F
O H O
_ ~ O H O
t-Bu0 N~ NHCH3 N
O t-Bu0 ~ NHCH3
N O
N
N ~ \
OMs Ts
H
A mixture of ~ ( 166mg, 0.24mmol) and HOBT (64.8mg, 0.48mmo1) in THF (4mi) was
stirred at room temperature for 24 hours, the solvent was evaporated, and the
crude product was
purified by flash chromatography(5% -10% MeOH-CH2C12) to afford 50f (116 mg,
89%) as a
white solid.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
98
Example SOG
O H~ O H_ O
t-Bu0 N NHCH3 t-Bu0 Nv _ NHCH
O 3
O
,N
OMs H
A mixture of SOf ( 137mg, 0.25mmo1), LiI (SOmg, 0.375mmol) and Na2C03 (27 mg,
0.25
mmol) in acetone(Sml) was heated at 50 °C for 5 hours. The reaction
mixture was then cooled to
ambient temperature and stirring was continued for 17 hours. The solvent was
evaporated and the
crude product was purified by flash chromatography(10% MeOH-CH2C12) to afford
~Og (7lmg,
63%) as a white solid.
Example SOH
O H O
N O H O
t-Bu0 ~ NHCH3 N
O _ -~ HOHN ~ NHCH3
~N ~ N
The desired compound was prepared according to the method of Examples 1 F and
G,
except substituting 5~"g for 1 ~ 1 H NMR(300 MHz, DMSO-d6) 8 0.75 (d, 3H, J =
6.2 Hz), 0.81
(d, 3H, J = 6.2 Hz), 0.80 (m, overlaped, 4H), 1.14 (m, 1 H), 1.22-1.52 (m)
4H),1.56-1.80 (m,
2H), 2.24-2.47 (m. 2H), 2.58 (d, 3H, J = 5.1 Hz), 2.63-2.79 (m, 2H), 3.63-3.76
(m) 1H ),
3.87-3.96 (m, 1 H), 4.35-4.48 (m) 1 H), 6.92 (s, 1H)) 7.46 (bs, 2H), 8.12 (d,
1 H, J = 15 Hz),
8.735 (d, 1 H, J = 1.5 Hz), 10.36(d, 1 H) J = 1.5 Hz). MS(DCI/NH3), m/e 408
(M+H)+.
[a]D=+10.4°.(c=0.25, EtOH).
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
99
Examsle 51
O H O
0
1 ,
0
The compound was prepared using the method of Example 43, except substituting
succinate ester ~ for Z and substituting ketone 14~i for ,4~.
mp 211-212 °C. 1 H NMR (300 MHz, DMS O-d6) 8 -0.38 (m, 1 H), 0.64 (m, 1
H), 0. 81 (m, 1 H),
0.98 (m, 4H), 1.58 (m, 1 H ), 1.62 (m, 1 H), 1. 80 (dt, I H), 2.00 (m, 2H), 2.
I 8 (m, 1 H), 2.21 (s,
3H), 2.81 (t, 1 H), 3.07 (dd, 1 H), 4.04 (m, 2H), 5.73 (m, 1 H), 6.69 (d, 2H},
6.93 (m, 4H), 7.17
(dd, 1H), 7.38 (dd, 1H), 7.51 (t) 2H}, 7.64 (t, 1H), 8.11 (m) 3H), 8.68 (s,
1H), 10.38 (s, IH) .
MS (DCI/NH3) m/e 543 (M+H)+.
Anal. calcd for C33H3gN205~0.75 H20: C, 7 I.26; H, 7. ~16; N, 5.04. Found: C,
71.03; H, 7.09;
N, 5.16.
Exam 1
O H O
HO N
O
O
The desired compound was prepared by following the procedures described in
Examples 1 A-C and
1 F starting with succinate ester 7 and ketone 14~.
1H NMR (300MHz, DMSO-d6) 8 8.14-8.11 (d, 1H, J=9.8 Hz), 8.05-8.03 (d, 2H,
J=7.2 Hz),
7.66-7.64 (m, 1H), 7.54-7.51 (m) 2H), 7.41-7.38 (m, IH)) 7.21-7.18 (m, 1H),
6.98-6.89 (m)
2H), 5.82-5.75 (m, 1 H), 4.13-3.99 (m, 2H), 3.15-3.09 (m, 1 H), 2.87-2.79 (m,
1 H), 2.00-1.91
(m) 2H), 1.69- I .67 (m, 1 H), 1.59-1.58 (m, 1 H)) 1.3-.06 (mm, 7H), 0.55-0.37
(m, 7H), (-
)0.25-(-)0.33 (m, 1 H). MS (APCI) m/e 450 (M-H)-, 452 (M+H)+, 486 (M+Cl)-.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
100
Exam lp a 53
O H O
HOHN N
O
O
The desired compound was prepared by according to the method of Example 1G,
except
substituting acid 52 ,for l~. 1H NMR (300MHz, DMSO-d6) b 10.1 (s, 1H), 8.50
(s, 1H), 7.92-
7.85 (m, 3H), 7.52-7.47 (m, 1H), 7.39-7.34 (m, 2H), 7.27-7.24 (m, 1H), 7.09-
7.06 (m, 1H),
6.82-6.74 (m, 1H), 3.95-3.90 (m, 1H}, 3.00-2.92 (m, 1H), 2.72-2.64 (m, 1H),
1.88-1.80 (m,
1H), 1.59-1.52 (m, 3H), 0.94-0.47 (bm, 5H), 0.46-0.23 (mm) 6H), (-)0.55-(-
)0.57 (m, 1H).
MS (DCI/NH4) m/e 467 (M+H)+~. Anal. Calcd for: C2~H34N205~0.25H20: C, 68.84;
H, 7.38;
N, 5.94. Found: C) 68.53; H) 7.38; N, 5.66.
Example 54
HOHN~N I ~ O,
~ /\O
Example 54A
O
H2
O
HO
The desired compound was prepared according to the method of Example 40A
except
substituting 4-bromo-1,2-(methylenedioxy)benzene for 4-bromothioanisole.
CA 02277121 1999-07-06
WO 98/30551 PCT/LTS98/00144
I01
Example 54B
o H O
HOHN N I
O
O
O
The desired compound was prepared according to the method of Example 43,
except substituting
succinate _5 for 7 and ketone 54a for 4~a. 1H NMR (300MHz, DMSO-d6) b 10.36
(s> 1H), 8.70
(s, 1 H)) 8.16-8.13 (d, 1 H, J=9.5 Hz), 7.80-7.77 (d, 1 H, J=7.7), 7.55 (s, 1
H), 7.36-7.33 (d,
1 H, J=6.6 Hz), 7.11-7.13 (d, 1 H, J=8.1 Hz), 7.02-6.99 (d) 1 H, J=8.4 Hz),
6.96-6.87 (m, 4H),
6.62-6.60 (d> 2H, J=8.1 Hz)) 6.10-6.08 (d, 2H, J=4.1 Hz)) 5.64-5.60 (m, 1H))
4.06-4.03 (m,
2 H), 3.04-2.98 (m) 1 H), 2.87-2.78 (m, 1 H), 2.21 (s) 3H)) 2.07-1.96 (m, 3H),
1.84-1.80 (m,
2H), 1.70-1.60 (m, 1 H), 1.60-1.52 (m, 1 H), 1.05-0.96 (bm, 4H)) 0.87-0.80 (m,
1 H), 0.64-0.60
(m, 1H), (-)0.37-(-) 0.38 (m, 1H). MS (ESI) m/e 587 (M+H)+, 585 (M-H)-.
Anal. Calcd for: C34H3gN~0-~~0.25H20: C, 69.07; H, 6.59; N, 4.70. Found: C,
68.72; H,
6.41; N, 4.64.
Example 55
O H O
HOHN N
O ~ F
O
Example SSA
O
H2
F
HO
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
102
The desired compound was prepared according to the method of Example 40A,
except
substituting 4-bromofluorobenze for 4-bromothioanisole.
Exam ip a SSB
O H O
HOHN N
o I/
F
O
The desired compound was prepared according to the method of Example 43,
except substituting
succinate 5_ for 7 and ketone SSa for 43a. 1H NMR (300MHz, DMSO-d6) S 10.38
(s, 1H), 8.71
(s, 1H), 8.19-8.13 (m, 3H), 7.37-7.28 (m, 3H), 7.17-7.11 (d, 1H, J=7.3 Hz),
6.95-6.88 (m,
4H), 6.59-6.57 (d, 2H, J=7.1 Hz), 5.73-5.65 (m, 2H), 4.06-4.03 (m, 2H), 3.08-
3.03 {m, 1H),
2.89-2.73 (m, 2H), 2.2 i (s, 3H), 2.18-2.17 (m, 1H), 2.06-1.99 (m, 2H), 1.82-
1.76 (m, 1H),
1.64-1.55 (m, 2H), 1.09-0.59 (bm, 9H), (-)0.382-(-)0.385 (m, 1H). MS (ESI) m/e
561
(M+H)+, 559 (M-H)-. Anal. Calcd for: C33H3~FN205~0.25H20: C, 70.13; H, 6.68;
N, 4.95.
Found: C) 70.01: H, 6.59; N, 5.05.
16 Example 56
i I
O H O
HO N
O ~ O I
O
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
103
oacam le 6A
H2 \
OCH2Ph
HO
The desired compound was prepared according to the method of example 40A
except substituting
4-benzyloxybromobenzene for 4-bromothioanisole.
Exam lie 56B
> H O
N
I \
O '~O \
I/
O
The desired compound was prepared by following the procedures described in
Examples 1 A-C and
1 F starting with succinate ester ~ and ketone ~. 1 H NMR (300MHz, DMSO-d6) 8
8.21-8.18 (d,
1H, J=9.6 Hz), 8.10-8.07 (d, 2H, J=9.1 Hz), 7.44-7.31 (m, 6H)) 7.16-7.11 (m,
3H), 6.96-6.88
(m, 4H), 6.64-6.61 (d, 2H, J=7.8 Hz), 5.69-5.65 (m, 1H), 5.18 (s, 2H), 4.08-
4.07 (m, 2H),
3.07-3.01 (m, 1H), 2.87-2.78 (m, 1H), 2.19 (s, 3H), 2.20-1.98 (m, 3H), 1.71-
1.53 (m, 2H))
1.17-0.86 (mm, 7H), -0.20- (-) 0.41 (m, 1H). MS (DCI/1VH4) m/e 634 (M+H)~.
Exam a 57
o H O
HOHN N I \
O /
O
O
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
104
The desired compound was prepared by according to the method of Example 1 G,
except
substituting acid 5~ for la. iH NMR (300MHz, DMSO-d6) 8 10.38 (s, 1H)) 8.70
(s, IH), 8.16-
8.14 (m, 3H), 7.47-7.38 (m, 6H)) 7.21-7.18 (m, 3H), 6.96-6.91 (m, 3H), 6.65-
6.63 (d, 2H,
J=7.8 Hz), 5.75-5.65 (m, 1 H), 5.21 (s, 2H), 4.11-4.08 (m, 2H), 3.10-3.04 (m,
1 H), 2.90-2. 81
(m, IH), 2.23 (s, 3H), 2.13-2.OI (m, 2H), 1.88-1.87 (m, 1H), 1.77-1.65 (m,
2H), 1.07-0.96
(m, 6H)) 0.70-0.66 (m, 2H), (-)0.29-(-)0.32 (m, 1H).
Exam lie 58
O H O
HOHN N I
O
OH
O
The desired compound was prepared by removing the benzyl group of example 57
using to the
procedure described in Example 1D. 1H NMR (300MHz, DMSO-d6) 8 10.42 (s, 1H),
10.35 (s)
1H), 8.69 (s, 1H)) 8.11-8.08 {d) 1H) J=9.6 Hz), 8.03-8.00 (d, 2H, J=8.4 Hz),
7.36-7.33 (d,
1 H, J=8.4 Hz), 7. I 7-7.11 (d, 1 H, J=8 Hz}, 6.94-6.84 (m, 6H), 6.63-6.61 (d,
2H) J=8.1 Hz)>
5.66-5.59 (m, 1H), 4.06-4.00 {m, 2H), 3.03-2.97 (m, 1H), 2.84-2.76 (m, 1H),
2.12 (s, 3H),
2.08-1.96 (m, 2H), 1.82-1.78 (m, 1 H), 1.63- I .55 (m, 2H), 1.04-0.61 (mm,
6H), (-)0.21-(-
)0.41 (m, 1H).
MS (DCI/NH4) m/e 559 (M+H). Anal. Calcd for: C33H3gN206~0.25H20: C, 70.37; H,
6.89;
N, 4.97. Found: C, 70.20: H) 6.94; N, 4.86.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
105
x 1 ~9
I~
i
O H O
HOHN N
O
O
The desired compound was prepared according to the method of Example 43,
except substituting
succinate _8 for 7 and ketone 14~ for 4~,~. 1H NMR (300MHz, DMSO-d6) 8 10.31
(s, 1H), 8.65
(s, 1 H), 8.12-$.08 (d, 2 H, J=9. $ Hz}, 8.06-8.03 (d, 2H, J=7.1 Hz), 7.59-
7.54 (m, 1 H), 7.48-
7. 43 (m, 2H), 7.38-7.22 (m, 7H), 7.18-7.14 (m, 1H), 6.95-6.87 (m, 2H), 5.75-
5.69 (m, 1H),
4.29 (s, 2H), 4.07-4.03 (m, 2H), 3.13-3.07 (m, 1H}, 2.98-2.75 (m. 3H), 1.93-
1.88 (m, 1H),
1.82-0.56 (mm, 15H), (-) 0.036-(-) 0.030 (m, 1 H). MS (ESI} 573 (M+H)+, 571 (M-
H)-. Anal.
Calcd for: C34H4pN206: C) 71.30; H, 7.04; N) 4.89. Found: C, 71.16; H) 7.14;
N, 4.85.
CA 02277121 1999-07-06
WO 98/30551 PCTIUS98/00144
106
Example A
0
HG~Hz \
HO
The desired compound was prepared according to the method of Example 40A,
except substituting
4-benzyloxymethyl bromobenzene for 4-bromothioanisole.
0
~o \
= _
o
w
0
o /
The desired compound was prepared according to the methods of Examples 1 A,
coupling
succinate 5 with ketone 60A, followed by deprotection of the silyl ether as in
Example 32B and
subsequent cyclization as in Example 1C.
MS (DCI/NH3 j m/e 720 (M+H).
CA 02277121 1999-07-06
WO 98/30551 PGT/US98100144
107
Example 60C
A solution of ester øQø (0.050 g, 7.0 x 10-z mmol) in 4:1 CH3 CN/H20 (5 mL)
was treated
with ceric ammonium nitrate (0.19 g, 3.5 x 10-1 mmol) and stirred and 1.5 h.
The solution was
partitioned between ethyl acetate and water, the organic layer was dried
(MgS04) and concentrated
to a solid. The solid was purified on silica gel with 25% ethyl acetate/hexane
ramped to 60% to
provide 0.01 g(26%) of ~.
m
The desired compound was prepared according to the methods of Examples 1 F,
except substituting
Oc for lf.
1 H NMR (d6-DMSO) 8 8.21 (d, 1 H, J=10.3 Hz), 8.13 (d, 2H, J=8.5 Hz), 7.59 (d,
2H, J=8.1
Hz), 7.38 (dd, 1 H, J=1.4) 8.1 Hz), 7.15 (dd, 1 H, J=1.8, 8.1 Hz)) 7.02-6.87
(m, 4H), 6.61 (d,
2H, J=8.1 Hz), 5. 83-5.69 (m, 1 H), 5.48 (s, 2H), 4.07 (t, 1 H, J=1.5 Hz),
3.10 (dd, 2H) J=3.7,
13.6 Hz), 2.86-2.72 (m, 2H), 2.30-1.93 (m, 2H), 2.21 (s, 3 H), I .77-1.49 (m,
2H), 1.20-0.79
(m) 7H), 0.70-0.55 (m, 2H)) -0.16--0.32 (m) 1H).
MS (ESI) m/e 558 (M+H).
Example ~D
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
108
H
Na
S
O O
a
s
O
OH
° 61a
The desired compound was prepared according to the methods of Examples lA-C,
coupling
succinate 1 with ketone ~A_, followed by deprotection of the benzyl ether as
in Example 60C.
Example 61 B
H
)H
The desired compound was prepared according to the methods of Examples 1 F-G,
substituting
ester ~ for ,]~.
1 H NMR (300MHz, DMS O-d6) 8 10.29 (d, 1 H, J=1.7 Hz), 8.64 (d, 1 H, J=1.7
Hz), 8.02-7.98
(m, 3H), 7.46-7.38 (m, 3H), 7.22 (dd, 1H, J=8.1, 2 Hz), 6.96-6.88 (m, 2H),
5.80-5.68 (m,
Example 61
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
109
1 H), 5.34 (t, I H, J=5.8 Hz), 4.57 (d, 2H, J=5.4 Hz)) 4.05 (t, 2H, J=4.7 Hz),
3. I O (dd, 1 H,
J=13.5) 4.4 Hz), 2.79 (t, IH, J=12.9 Hz}, 2.05-1.94 (m) IH)) 1.77-1.46 (m,
3H), 1.13-0.54
(m, 6H), 0.48 (dd, 6H, J=45.1, 5 Hz), -0.34-0.50 (m, I H). MS (ESI) m/e 497 (M
+ H)+,
' Anal. Calcd for: C28H36N206~1.25H2U: C, 64.78; H, 7.47; N) 5.39. Found: C,
64.44; H,
7.23; N, 5.43.
Example 62
OMe
Me
Me
H ~ \
O
O
Exam»le 62A
O ~ O
O O
O \
The desired compound was prepared according to the methods of Examples 1 A,
coupling succinate
~ with ketone ,~46, followed by deprotection of the silyl ether as in Example
32B and subsequent
cyclization as in Example 1C.
CA 02277121 1999-07-06
WO 98/30551 PCTlUS98/00144
110
Example 62B
O H O
N
O O
O - / ~.
O O
62b
A solution of macrocycle olefin CL2a ( 1.544 g, 3.14 mmol) in THF ( 10 mL) at
0°C under argon
atmosphere was treated with 9-BBN ( 19.5 mL of 0.5 M solution in THF, 9.75
mmol) dropwise
and stirred at 0°C for 0.2 h then at ambient temperature for 5.0 h. The
resulting solution was
treated with DMF (80 mL), [ 1,1'-Bis(diphenylphosphino)-
ferroceneJdichloropalladium(II),
complex with dichloromethane (l:l) (0.30 g) 0.37 mmol), 3,4,5-trimethaxy-
bromobenzene (2.41
g, 9.75 mmol), and cesium carbonate (6.14 g, 18.84 mmol) and stirred at
60°C for 6 h then at
ambient temperature for 10 h. The solution was partitioned between ether and
water, the organic
layer was dried (MgS04), and concentrated to an oil. The oil was purified on
silica gel with ethyl
acetate/hexane) 1:2 to provide 0.86 g (42%) of ~2b. MS (ESI) m/e 660 (M+H)+.
OMe
OMe
O
HO
O -
O
The desired compound was prepared according to the methods of Example 1F,
except substituting
f Zb for 1 f. 1 H NMR (300 MHz DMSO-d f,) 8 12.08 (s,1 H), 8.25 (d, 1 H, J=9.3
Hz), 8.07 (d,
2H, J=7.4 Hz), 7.66-7.61 (m) 1 H)) 7.53-7.48 (m,2H), 7.42 (d, 1 H, J=8.1 Hz),
7.18 (d, 1 H,
Example 62C
CA 02277121 1999-07-06
WO 98/30551 PCT/ITS98/00144
111
J=8.1 Hz), 6.99-6.91 (m) 2H), 6.22 (s) 2H), 5.76 (m, 1H), 4.04-4.11 (m, 2H),
3.69 (s) 6H),
3.59 (s, 3H), 3.10-3.18 (m, I H), 2.75 (t, 1 H, J=12.3 Hz), 1.98-2.30 (m, 4H),
I .53-1.74 (m,
2H), 0.80-1.21 (m, 6H), 0.58-0.74 (m) 1 H), -0.16- -0.30 (m, 1 H). MS (ESI)
m/e 604
(M+H)+. Anal. Calcd for: C35H41 NOg~0.25H20: C, 69.11; H, 6.87; N, 2.30.
Found: C,
S 69.01; H, 6.90; N, 2.22.
xm
OMe
Me
OMe
o a o
H O
O /
/
O
The desired compound was prepared by according to the method of Example 1G,
except
substituting Example 62 62 for 1~. 1H NMR (300 MHz, DMSO-d6) 8 10.38 (s, 1H),
8.71 (s,
1H), 8.20 (d) 1H, J=9.6 Hz), 8.08 (d, 2H, J=7.3 Hz), 7.60-7.67 (m) 1H)) 7.47-
7.52 (m, 2H).
7.42 (d, 1 H, J=8.5 Hz)) 7.20 (d, 1 H, J=8.1 Hz), 6.90-6.97 (m) 2H), 6.21 (s,
2H), 5.66-5.77
(m, 1 H), 3.99-4.11 (m, 2H), 3.69 (s, 6H), 3.59 (s, 3H), 3.09-3.17 (m) 1 H),
2.73 (t, 1 H,
J=12.9 Hz)) I.98-2.29 (m) 3H), 1.74-1.84 (m) 1H), 1.50-1.70 (m) 2H), 0.59-1.I3
(m, 7H),
0.31- -0.44 (m, 1H).MS (ESI) m/e 619 (M+H)+. Anal. Calcd for:C35H42N2Og: C,
67.94; H.
6.84; N, 4.52. Found: C, 67.65; H, 6.75; N, 4.43.
Examlhe 64
OMe
OMe
O
H O~ H
/
O
The desired compound was prepared according to the method of Examples 62-63,
except
substituting 3,5-dimethoxy-bromobenzene for 3,4,5-trimethoxy-bromobenzene in
Example 62B.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
112
~ H NMR (300 MHz, DMS O-d6) 8 10.37 (s. 1 H), 8.70 (s, 1 H), 8. I7 (d, 1 H,
J=9.9 Hz), 8.07 (d,
2H, J=7.4 Hz), 7.58-7.66 (m, 1H), 7.44-7.51 (m, 2H), 7.40 (dd, 1H) J=8.4, 2
Hz), 7.18 (dd,
1 H, J=8.1, 2 Hz)) 6.88-6.97 (m, 2H), 6.23 (t, 1 H, J=2 Hz), 6.05 (d, 2H)
J=2.3 Hz), 5.66-5.77
(m, 1 H), 4.02-4.08 (m, 2H), 3.67 (s. 6H)) 3.07-3.16 (m, 1 H), 2.76 (t, 1 H,
J=12.6 Hz), 2.14-
2.28 (m, 1 H), 1.95-2.10 (m, 2H}, 1.75-1.85 (m, 1 H), 1.49-1.72 (m, 2H), 0.56-
1.11 (m, 7H), -
0.31- -0.44 (m, 1H). MS (ESI) m/e 589 (M + H)+. Anal. Calcd for: C34H4pN2O7:
C, 69.36;
H, 6.84; N, 4.75. Found: C, 69.27; H, 6.63; N) 4.61.
Exam In a 65,
Br
HO.N N
1f H _ O
H o _ I~
O'v
The desired compound was prepared according to the method of Examples 62-63,
except
substituting 1,3,5-tribromobenzene for 3,4,5-trimethoxy-bromobenzene in
Example 62B. 1 H
NMR (300 MHz, DMSO-d6} b 0.38 (s) 1H), 8.72 (s. 1H), 8.17 (d, 1H, J=9.6 Hz),
8.07 (d, 2H,
J=7.7 Hz), 7.55-7.61 (m. 2H)) 7.35-7.47 (m, 3H), 7.13-7.19 (m, 1H), 7.06 (d.
2H, J=1.8 Hz),
6.88-6.97 (m. 2H)) 5.68-5.79 (m, 1 H), 4.03-4.10 (m, 2H), 3.08-3.16 (m, 1 H),
2.85 (t, 1 H,
J=12.7 Hz)) 2.22-2.35 (m, 1 H), 1.91-2.10 (m, 2H), 1.75-1.85 (m, 1 H), 1.46-
1.75 (m, 2H),
0.57-1.16 (m, 7H}. -0.30- -0.43 (m, 1H). MS (ESI) m/e 687 (M + H)+. Anal.
Calcd for:
C32H34N2O5Br2: C, 55.99; H, 4.99; N, 4.08. Found: C) 56.14; H, 4.97; N, 4.01.
Example 66
OMe
~OMe
O
O
H O
H
O
CA 02277121 1999-07-06
WO 98/30551 PCT/LTS98100144
113
Exam lp a 66A
OMe OMe
/~ /
HO ~ OH HO ~ O/~OMe
A solution of 5-methoxyresorcinol (2.52 g, 18 mmol) in DMF (25 mL) at ambient
temperature was treated with K2C03 (9.94 g, 72 mmol) and 2-bromoethyi methyl
ether ( 1.72 mL,
18 mmol) and heated at 50°C for 16 h. The suspension was pardoned
between Et20 and water)
the organic layer was dried (MgS04)) and concentrated to the crude product.
Purification on silica
gel with ethyl acetate/hexane, 1:4 provided 1.26 g (35%) of the title
compound. MS (ESI) m/e
199 (M + H)+.
Example 66B
OMe OMe
/ ~ /
H O ~ O~/O M a Tf0 ~ ~ O~O M a
~~ 6b
The desired compound was synthesized following the procedure described in
J.Org.
Chem. 14I, 4102 (1976). MS (DCI/NH3) m/e 287 (M + H)+.
Example 66C
Me
~OMe
O
O H O
H O
O I/
/ I
O
The desired compound was prepared according to the method of Examples 62-63,
except
substituting fib for 3,4,5-trimethoxy-bromobenzene in Example 62B.1 H NMR (300
MHz,
DMSO-d6) b 10.35 (s, 1H), 8.66 (s, 1H), 8.13 (d, 1H, J=9.6 Hz)) 8.07 (d, 2H,
J=7.5 Hz))
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
114
7.57-7.66 (m, 1H)) 7.46-7.51 (m, 3H). 7.15-7.21 (m, IH), 6.87-6.97 (m, 2H),
6.22-6.25 (m,
1H), 6.02-6.07 (m, 2H), 5.65-5.75 (m, IH), 4.02-4.09 (m, 2H)) 3.96-4.02 (m,
2H), 3.67 (s)
3H)) 3.59-3.64 (m, 2H), 3.30 (s, 3H), 3.07-3.16 (m, 1H), 2.76 (t, IH, 12.9
Hz), 2.11-2.38 (m,
1H), 1.94-2.10 (m, 2H), 1.73-1.85 (m, 1H), 1.48-1.72 (m, 2H), 0.58-1.10 (m,
7H), -0.30- -
0.44 (m, 1H). MS )ESI) m/e 633 (M + H)+. Anal. Calcd for: C36H44N208~ H20: C)
66.44;
H, 7.12; N) 4.30. Found: C, 66.40; H, 6.97; N, 4.31.
Example 67
OMe
OMe
HO~N~~H
'''' ~~(( N
H O~ _
N
i
Ms
Example 67A
O O
BocHN~OH BocHN~,N.Me
OMe
I , I
02N 02N
67a
The described compound 7a was prepared according to the method of Example 16A
except substituted methylamine hydrochloride with N,O-dimetheylhydroxyamine
hydrochloride.
Exam 1R a 67B
BocHN~N.Me BocHN~ .Me
N
OMe -~ OMe
ON
2 H2N
67a 67b
A solution of 67a (0.52 g) 0.35 mole) and PdJC (52 mg) in EtOH ( 10 mL) was
stirred
under HZ for 2 hours, stirred for 30 minutes. The reaction mixture was
filtered through Celite and
CA 02277121 1999-07-06
WO 98!30551 PCT/ITS98/00144
115
the residue was washed thoroughly with 10% methanol-CH2C12. The filtrate and
washings were
collected and evaporated to dryness to give 0.47 g of 7~1 as a off white solid
{99%).
Exam le 7C
BocHN~,Me BocHN~ ,Me
N
OMe ,~ ~ OMe
H2N
CbzHN
7c
A solution of ~~ (0.47 g, 1.46 mmole) in Et20 ( 10 mL) at room temperature was
treated
with saturated aqeoud NaHC03 ( 10 mL), stirred for 10 minutes, treated with
benzyl
chloroformate (0.25 mL, 1.75 mmol), stirred for 2 hours. The mixture was
partitioned between
water and ether, the aqueous layer was separated and extracted twice with
ether. The etheral layer
(50 mL) were combined, dried (MgS04) and concentrated to provide 610.7 mg
(92%) of 67c.
Example 67D
O O
BocHN ~N . Me HCI~H2N
OMe
(/ ~ ~/
CbzHN
CbzHN
A solution of Example ø~ ( 1.67 g, 3.65 mmole) in. THF (40 mL) at -78
°C was treated
with phenyllithium ( 1.8 M in Et20 and cyclohexane, 7.0 mL, 36.6 g, 1.08
mole). The mixture
was warmed to -15 °C, stirred for 2 hours, and then quenched with
saturated ammonium chloride.
The mixture was partitioned between EtOAc and brine, the aqueous layer was
separated and
extracted three times with EtOAc. The combined organic extracts (100 mL) were
dried (MgS04),
and concentrated to an oil. The oil was purified on silica gel with 10-40%
EtOAc/hexane to
provide 1.17 g (67%) product which was taken up in HCl-dioxane (4N, 10 mL) and
stirred for 3
hours. The resulting slurry was diluted with Et20 (200 mL), filtered and dried
under vaccum to
give 1.0 g ~~ as a white solid {98%).
CA 02277121 1999-07-06
WO 98/30551 PCT/LTS98/00144
116
Exam lp a 67E
O H O
HCf~H2N N
/ i O O I/
/ ~ \
CbzHN
/ NHCbz
OTBS
67d
The desired compound 67e was prepared according to the methods of Examples lA,
coupling succinate 9_ with ketone ø~.
Exam In a 67F
O N O \ I \ OMe
O I ~ O H O
\ ----.. O N I \
O
OTBS NHCbz I \
/ NHCbz
OTBS
A mixture of Example ~7g (0.46 g, 0.608 mmol), P(o-tol)3 (37.0 mg, 0.122
mmol),
Pd(OAc)2 ( 14.7 mg, 0.061 mmol) and 3,4,5-trimethoxy bromobenzene (224.4 mg,
0.912 mmol)
in acetonitrile (8 mL) under Ar was heated at 75 °C for 14 hours. The
mixture was evaporated to
dryness and purified on silica gel with 10-40% EtOAc/hexane to provide 511 mg
(91%) of ~.
Example 67G
OMe OMe
OMe
OMe ~ ~ 'OMe
H . O ~ O H OH
O N I \ ~ O N I \
O / O /
OTBS NHCbz
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
117
The desired compound was prepared according to the previous methods.
Deprotecion of
the silyl ether as in Example 32B and subsequent cyclization as in Example 17
A-B.
Example 67H
OMe
'OMe ~ ~ OMe
O H OH O OH
O N -~~ ~O N
° ~ ° I/
CI
C.
N
i
Ms
~$
A solution of Example 67g (65.3 mg, 0.099 mole) in CH2C12 (4 mL) at 0
°C was treated
with pyridine (0.032 mL, 0.39 mmol) followed by (0.018 mL, 0.24 mol), warmed
to room
temperature for 7 hours. The mixture was poured into CH2C12 and washed with
brine and
saturated aqueous NaHC03. The organic layer was dried (MgS04) and concentrated
to give 73
mg of ~7h as an oil.
Example 67I
Me
OMe
OMe
~~ H . OH
° N
( ~ ~o ~ ( w
° / o I/
I
I
N
N
Ms Ms
A solution of ~,x(73 mg, 0.099 mmol) in CH2C12 ( 10 mL) room temperature was
treated
with Dess-Martin reagent (46.1 mg) stirred for 1 hour. The mixture was
partitioned between water
and CH2C12, the aqueous layer was separated and extracted twice with CH2C12.
The combined
organic extracts were washed with aqueous NaHC03 and brine, dried (MgS04), and
concentrated
to an oil. The oil was purified on silica gel with 10-409o EtOAc/hexane to
provide 39.1 mg
(53.7% for the last two steps) product.
CA 02277121 1999-07-06
WO 98/30551 PCT/US98/00144
118
Exam 1R a 67J
OMe OMe
~OMe ~ _OMe
OMe ~~OMe
_ I O H O O H O
O N \ ~. HO, N N \
O ~ H O I /
I ~ I \
N
N
Ms Ms
The desired compound was prepared by the methods of Examples 1F and G, except
substituting for 1F. 1H NMR (300 MHz, DMSO-d6) 8 -0.48 (m, 1H), 0.6-0.8 (m,
SH), 1.35 -
1.81 (m, 3H), 2.0-2.62 (m, 4H), 2.73 (m, 1H), 2.80 (t, 1 H, J = 12.5 Hz), 3.05
(s, 3H), 3.19
(dd, 1 H, J1= 12.5 Hz, J2 = 4.5 Hz)) 3.59 (s> 3H), 3.69 (s, 6H), 3.73-3.77 (m,
2H), 5.73 (m,
1 H)) 6.23 (s, 2H), 7.45 (d, 1 H, J = 8.6 Hz), 7.32-7.39 (m, 2H)) 7.51 (t, 2H,
J = 7.6 Hz), 7.56
(d, 1 H, J = 8.6 Hz), 7.64 ( t) 1 H, J = 7.6 H z ), 8.08 (d, 2H, J = 7.6 Hz),
8.29 (d, 1 H, J = 9.0
Hz), 8.71 (s, 1H), 10.37 (s, 1H); MS (ESI) m/e 718 (M+Na)+, 696 (M+H)+.