Note: Descriptions are shown in the official language in which they were submitted.
CA 02277545 1999-07-07
WO 98/30720
PCT/US98/00589
BIOCONJUGATION OF OLIGONUCLEOTIDES
FIELD OF THE INVENTION
This invention describes a novel method for conjugating oligonucleotides to
other molecular entities exclusively at the S'-position of the
oligonucleotide. The
method of this invention takes advantage of an enzymatic method of
synthesizing
S RNA via an RNA polymerase.
BACKGROUND OF THE INVENTION
A method for the in vitro evolution of nucleic acid molecules with highly
specific binding to target molecules has been developed. This method,
Systematic
Evolution of Ligands by Exponential Enrichment, termed SELEX, is described in
United States Patent Application Serial No. 07/536,428, filed June 11, 1990,
entitled
"Systematic Evolution of Ligands by Exponential Enrichment," now abandoned;
United States Patent Application Serial No. 07/714,131, filed June 10, 1991,
entitled
"Nucleic Acid Ligands," now United States Patent No. 5,475,096; United States
Patent Application Serial No. 07/931,473, filed August 17, 1992, entitled
"Methods
for Identifying Nucleic Acid Ligands," now United States Patent No. 5,270,163
(see
also, WO 91 /19813), each of which is specifically incorporated by reference
herein.
Each of these applications, collectively referred to herein as the SELEX
Patent
Applications, describes a fundamentally novel method for making a nucleic acid
ligand to any desired target molecule. The SELEX process provides a class of
products which are referred to as nucleic acid Iigands (also referred to in
the art as
"aptamers"), each Iigand having a unique sequence and property of binding
specifically to a desired target compound or molecule.
The SELEX method involves selection from a mixture of candidate
oligonucleotides and step-wise iterations of binding, partitioning and
amplification,
using the same general selection scheme, to achieve virtually any desired
criterion of
binding affinity and selectivity. Starting from a mixture of nucleic acids,
preferably
comprising a segment of randomized sequence, the SELEX method includes steps
of
contacting the mixture with the target under conditions favorable for binding,
partitioning unbound nucleic acids from those nucleic acids which have bound
specifically to target molecules, dissociating the nucleic acid-target
complexes,
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
2
amplifying the nucleic acids dissociated from the nucleic acid-target
complexes to
yield a ligand-enriched mixture of nucleic acids, then reiterating the steps
of binding,
partitioning, dissociating and amplifying through as many cycles as desired to
yield
highly specific high affinity nucleic acid ligands to the target molecule.
The basic SELEX method has been modified to achieve a number of specific
objectives. For example, United States Patent Application Serial No.
07/960,093,
filed October 14, I 992, entitled "Method for Selecting Nucleic Acids on the
Basis of
Structure," abandoned in favor of United States Patent Application Serial No.
08/198,670, describes the use of SELEX in conjunction with gel electrophoresis
to
select nucleic acid molecules with specific structural characteristics, such
as bent
DNA. United States Patent Application Serial No. 08/123,935, filed September
17,
1993, entitled "Photoselection of Nucleic Acid Ligands," describes a SELEX
based
method for selecting nucleic acid ligands containing photoreactive groups
capable of
binding and/or photocrosslinking and/or photoinactiviating a target molecule.
United
States Patent Application Serial No. 08/134,028, filed October 7, 1993,
entitled
"High-Affinity Nucleic Acid Ligands That Discriminate Between Theophylline and
Caffeine," abandoned in favor of United States Patent Application Serial No.
08/443,957, now United States Patent No. 5,580,737, describes a method for
identifying highly specific nucleic acid ligands able to discriminate between
closely
related molecules, which can be non-peptidic, termed Counter-SELEX. United
States Patent Application Serial No. 08/143,564, filed October 25, 1993,
entitled
"Systematic Evolution of Ligands by Exponential Enrichment: Solution SELEX,"
abandoned in favor of United States Patent Application Serial No. 08/461,069,
now
United States Patent No. 5,567,588, describes a SELEX-based method which
achieves highly efficient partitioning between oligonucleotides having high
and low
affinity for a target molecule. United States Patent Application Serial No.
08/434,425, filed May 3,1995, entitled "Systematic Evolution of Ligands by
Exponential Enrichment: Tissue SELEX" and United States Patent Application
Serial
No. 08/433,124, filed May 3, 1995, entitled "Nucleic Acid Ligands of Tissue
Target,"
describe the use of SELEX to identify and prepare Nucleic Acid Iigands to
Tissue
Targets.
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
3
The SELEX method encompasses the identification of high-affinity nucleic
acid ligands containing modified nucleotides conferring improved
characteristics on
the ligand, such as improved in vivo stability or improved delivery
characteristics.
Examples of such modifications include chemical substitutions at the ribose
and/or
phosphate and/or base positions. SELEX-identified nucleic acid ligands
containing
modified nucleotides are described in United States Patent Application Serial
No.
08/117,991, filed September 8, 1993, entitled "High Affinity Nucleic Acid
Ligands
containing Modified Nucleotides," abandoned in favor of United States Patent
Application Serial No. 08/430,709, now United States Patent No. 5,660,985,
that
describes oligonucleotides containing nucleotide derivatives chemically
modified at
the 5- and 2'-positions of pyrimidines. United States Patent Application
Serial No.
09/134,028, supra, describes highly specific nucleic acid ligands containing
one or
more nucleotides modified with 2'-amino (2'-NHZ), 2'-fluoro (2'-F), and/or 2'-
O-
methyl (2'-OMe). United States Patent Application Serial No. 08/264,029, filed
June
22, 1994, entitled "Novel Method of Preparation of Known and Novel 2'-Modified
Nucleosides by Intramolecular Nucleophilic Displacement," describes
oligonucleotides containing various 2'-modified pyrimidines.
The SELEX method encompasses combining selected oligonucleotides with
other selected oligonucleotides and non-oligonucleotide functional units as
described
in United States Patent Application Serial No. 08/284,063, filed August 2,
1994,
entitled "Systematic Evolution of Ligands by Exponential Enrichment: Chimeric
SELEX," now United States Patent No. 5,637,459 and United States Patent
Application Serial No. 08/234,997, filed April 28, 1994, entitled "Systematic
Evolution of Ligands by Exponential Enrichment: Blended SELEX," now United
States Patent No. 5,683,867, respectively. These applications allow the
combination
of the broad array of shapes and other properties, and the efficient
amplification and
replication properties, of oligonucleotides with the desirable properties of
other
molecules.
The SELEX method encompasses complexes of oligonucleotides. United
States Patent Application Serial No. 08/434,465, filed May 4, 1995 entitled
"Nucleic
Acid Ligand Complexes," describes a method for preparing a therapeutic or
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
4
diagnostic complex comprised of a nucleic acid ligand and a lipophilic
compound or
a non-immunogenic, high molecular weight compound.
Nucleic acid ligands derived by the SELEX process have been used in
diagnostic applications. (See e.g., United Stated Patent Application No.
08/487,425,
filed June 7, 1995, entitled "Enzyme Linked Oligonucleotide Assays (ELONAS),"
United States Patent Application No. 08/479,729, filed June 7, 1995, entitled
"Use of
Nucleic Acid Ligands in Flow Cytometry," and United States Patent Application
No.
08/628,356, filed April 5, 1996, entitled "Method for Detecting a Target
Compound
in a Substance Using a Nucleic Acid Ligand." The full text of the above
described
patent applications, including but not limited to, all definitions and
descriptions of the
SELEX process, are specifically incorporated by reference herein in their
entirety.
Considerable research is being directed to the application of oligonucleotides
and oligonucleotide analogs as diagnostic and research reagents and as
potential
therapeutic agents. There are currently at least three areas of exploration
regarding
the use of oligonucleotides as pharmaceutical compounds. In the most advanced
field, antisense oligonucleotides are used to bind to certain coding regions
in an
organism to prevent the expression of proteins or to block various cell
functions.
Additionally, the discovery of RNA species with catalytic functions --
ribozymes --
has led to the study of RNA species that serve to perform intracellular
reactions that
will achieve desired effects. And lastly, the discovery of the SELEX process
(~stematic Evolution of Ligands by Exponential Enrichment) (Tuerk and Gold
(1990) Science 249:505) has shown that oligonucleotides can be identified that
will
bind to almost any biologically interesting target.
The use of antisense oligonucleotides as a means for controlling gene
expression and the potential for using oligonucleotides as possible
pharmaceutical
agents has prompted investigations into the introduction of a number of
chemical
modifications into oligonucleotides to increase their therapeutic activity and
stability.
Such modifications are designed to increase cell penetration of the
oligonucleotides,
to stabilize them from nucleases and other enzymes that degrade or interfere
with the
structure or activity of the oligonucleotide analogs in the body, to enhance
their
binding to targeted RNA, to provide a mode of disruption (terminating event)
once
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
sequence-specifically bound to targeted RNA and/or to improve the
pharmacokinetic
properties of the oligonucleotides.
Recent research has shown that RNA secondary and tertiary structures can
have important biological functions (Tinoco et al. ( 1987) Cold Spring Harb.
Symp.
5 Quant. Biol. 52:135; Larson et al. (1987) Mol. Cell. Biochem. 74:5; Tuerk et
al.
(1988) Proc. Natl. Acad. Sci. USA 85:1364; Resnekov et al. (1989) J. Biol.
Chem.
264:9953). PCT Patent Application Publication WO 91/14436, entitled "Reagents
and Methods for Modulating Gene Expression Through RNA Mimicry," describes
oligonucleotides or oligonucleotide analogs which mimic a portion of RNA able
to
interact with one or more proteins. The oligonucleotides contain modified
internucleoside linkages rendering them nuclease-resistant, have enhanced
ability to
penetrate cells, and are capable of binding target oligonucleotide sequences.
The use of oligonucleotides as therapeutic and diagnostic agents is growing
rapidly with many compounds in preclinical and human clinical trials. In many
of
these applications the oligonucleotide is derivatized or conjugated with
another
molecular entity. These conjugations are typically performed for the purpose
of
attaching fluorescent dyes or other diagnostic reporter groups or for
attaching
compounds that modulate the activity or the pharmacokinetic behavior of the
oligonucleotide. For example, Smith et al. describe the synthesis of
fluorescent dye-
conjugated primers for use in fluorescence-based DNA sequence analysis (Smith
et
al. ( 1987) Methods Enzymol. I 55: 260-301 ). United States Patent No.
5,650,275 of
Pitner et al., describes the use of spectroscopically detectable labeled
nucleic acid
ligands to determine the presence or absence of a target compound in a sample
(see
also copending United Stated Patent Application No. 08/487,425, filed June 7,
1995,
entitled "Enzyme Linked Oligonucleotide Assays (ELONAS)," United States Patent
Application No. 08/479,729, filed June 7, 1995, entitled "Use of Nucleic Acid
Ligands in Flow Cytometry," and United States Patent Application No.
08/628.356,
filed April 5, 1996, entitled "Method for Detecting a Target Compound in a
Substance Using a Nucleic Acid Ligand"). United States Patent Application
Serial
No. 08/434,465, filed May 4, 1995, entitled "Nucleic Acid Ligand Complexes,"
describes the use of oligonucleotides conjugated to lipophilic compounds or
non-
CA 02277545 1999-07-07
WO 98/30720 PCT/US98t00589
6
immunogenic, high molecular weight compounds to modulate the activity or
pharmokinetic behavior of the oligonucleotides. A lipophilic compound
covalently
attached to an antisense oligonucleotide through a phosphoester bond has been
described in EP 462 145 B I of Bischofberger. Conjugation has also been used
to
S make oligonucleotide dimers and to attach oligonucleotides to multimeric
platforms.
(Jones et al. (1995) J. Med. Chem. 38:2138).
Several chemical methods exist to accomplish such conjugations. (For a
review, see Goodchild (1990) Bioconjugate Chemistry 1:165-187). The presence
of a
chemically reactive functional group, such as an amine or thiol, at the 5'-
terminus of
an oligonucleotide allows selective attachment of various conjugates,
including
reporter groups (Smith et al. (1987) Methods Enzymol. 155:260-301; Sproat et
al.
( 1987) Nucleic Acids Res. 15:6181-6196) and peptide epitopes (Tung et al. (
1991 )
Bioconjugate Chem. 2:464-465; Bruick et al. (1997) Chem. Biol. 3:39-56).
Oligodeoxynucleotides containing a terminal amino functionality have been
utilized
for the construction of bioconjugates with novel properties. In some of the
more
common methods of synthesizing these bioconjugates, a primary aliphatic amine
group is incorporated at the 5'-terminus of the oligonucleotide in the final
step of the
assembly of a synthetic oligonucleotide (Tung et al. (1991) Bioconjugate~Chem.
2:464-465; Smith et al. ( 1987) Methods Enzymol. 155:260-301 ). A commercial
reagent (actually a series of such linkers having various lengths of
polymethylene
connectors) for linking to the 5' terminus of an oligonucleotide is 5'-Amino-
Modifier
C6. These reagents are available from Glen Research Corp (Sterling, VA). These
compounds have been used by Krieg {Krieg et al. ( I 971 ) Antisense Res. and
Dev.1:161 ) to link fluorescein to the 5'-terminus of an oligonucleotide.
Since many
macromolecules of interest are hydrophilic, these reactions are generally done
in
water, requiring large excesses of reagent to overcome the competing
hydrolysis.
Usually the amine on the oligonucleotide is added to the terminus of the
molecule
and must compete with free amine and alcohol on the fully deprotected
oligonucleotide if this modification is done post-synthetically.
In another common method of conjugating oligonucleotides to other
molecular entities, particularly detector molecules, the molecular entity is
converted
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
7
into a phosphoramidite, which is then added to the free alcohol of the full
length
oligonucleotide which is attached to a solid support. This method is less than
ideal
due to the air and water sensitivity of the phosphoramidite, as well as the
fact that the
molecule can only be added to the terminus of the oligonucleotide.
Furthermore,
many detector molecules are not compatible with this method due to the harsh
conditions normally needed to fully deprotect and release the oligonucleotide
from
the support. A third method of conjugating oligonucleotides to other molecules
is the
coupling of an alkylthio derivatized oligonucleotide with a a-haloacetyl or
with a
maleimide containing compound. (Jones et al. (1995) J. Med. Chem. 38:2138).
An alternative method for the synthesis of oligodeoxynucleotides terminated
by 5'-amino-5'-deoxythymidine has been described (Bruick et al. (1997) Nucleic
Acids Res. 25:1309-1310). This method uses a DNA template to direct the
ligation
of a peptide to an oligonucleotide, in which the peptide is presented by a
second
oligonucleotide in the form of a reactive thioester-linked intermediate.
Oligodeoxynucleotides have been labeled for potential in vivo diagnostic
imaging by two methods. Hnatowich has synthesized oligodeoxynucleotides with a
primary amine on the 5'-terminus then coupled peptidyl Tc chelates via NHS
chemistry (Hnatowich (1995) J. Nucl. Med. 36:2306). Hayes et al. have
synthesized
5'-[fluorenylmethoxycarbonyl]-5-(E)-[2-tri-n-butylstannylvinyl]-2'-
deoxyuridine-3'-
cyanoethyl N,N-diisopropylphosphoramidite). (Hayes et al. ( I 997) Nucleic
Acid
Res. 25:2897-2901 ). This reagent is compatible with automated solid-phase
synthesis and has been incorporated into the thrombin aptamer. The modified
deoxynucleotide is readily iodinated with'23I as a potential thrombus imaging
agent.
Both of these techniques apply to synthetic deoxyoligonucleotides and are not
transferable to ribooligonucleotides produced synthetically.
Conjugates of oligonucleotides with peptides having specific functions can be
useful for various applications. Examples include the use of a nuclear
transport
signal peptide to direct intracellular trafficking (Eritja et al. ( 1991 )
Tetrahedron 47:
4113-4120); a hydrophobic peptide (Juby et al. ( 1991 ) Tetrahedron Lett.
32:879-822)
or polylysine (Leonetti et al. (1991) Bioconjugate Chem. 1:149-153) to
increase cell
penetrability, and polylysine to provide multiple attachment sites for
nonradioactive
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
8
reporting probes (Haralambidis et al. ( 1987) Tetrahedron Lett. 28:5199-5202;
Haralambidis et al. (1990) Nucleic Acids Res. 18:493-499).
Transcription from synthetic DNA templates using T7 RNA polymerise is a
convenient method for the synthesis of RNA oligonucleotides. The transcription
of
DNA by T7 RNA polymerise begins at a uniquely defined base relative to the
promoter DNA sequence (Chamberlin and Ring (1973) J. Biol. Chem. 248:2235-
2244). The first nucleotide transcribed is usually a purine. The transcription
of a
DNA template into an RNA is distinct in that it results in a new RNA having a
triphosphate at its 5' terminus. The effects of modifying the 5'-position in
the
initiating nucleotide in RNA synthesis was studied by Martin and Coleman.
(Martin
and Coleman( 1989) Biochemistry 28:2760-2762). Martin and Coleman discovered
that the first nucleotide incorporated into an RNA transcript is unique in
that the 5'-
triphosphate is not utilized in a bond-formation step. That is, while
Watson/Crick
base-pairing is involved, the 5' region of the initial nucleotide is not
involved in
binding to the protein and/or to the DNA template. Thus, it was observed that
initiation of DNA transcription by T7 RNA polymerise proceeds effectivelv
whether
initiated with guanosine triphosphate (GTP), guanosine monophosphate (GMP) or
guanosme.
The use of the modified guanosine, 5'-amino-5'-deoxyguanosine, as an
initiator in enzymatic RNA synthesis has been described by Lohse and Szostal<.
(Lohse and Szostak (1996) Nature 381:442-444). Synthesis of this molecule is
difficult.
To date, the use of a modified guanosine as an initiator in enzymatic RNA
synthesis, wherein the guanosine has a substituent at the 5'-position that is
larger than
a triphosphate, has not been demonstrated.
SUMMARY OF THE INVENTION
The present invention describes a novel and highly efficient method for
derivatizing or conjugating oligonucleotides with other molecular entities.
Specifically, the present invention describes a method for enzymatically
generating
oligonucleotides derivatized exclusively at the 5'-position of the
oligonucleotide,
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
using 5'-substituted guanosines as initiators in the enzymatic synthesis of
RNA. The
methods disclosed herein allow for the addition of a variety of molecular
entities --
including but not limited to reactive molecules, reporter molecules, reporter
enzymes,
lipophilic molecules, peptides and proteins -- to the 5'-terminus of nascent
RNA
S oligonucleotides.
In its most basic form the method of the instant can be described by the
following steps:
a) providing a DNA template; and b) combining the DNA template with
nucleotide triphosphates, a S'-substituted guanosine and an RNA polymerase
under
conditions suitable for transcription. In a preferred embodiment the
initiating base on
the RNA is a guanosine and the RNA polymerase is T7 RNA polymerase. The types
of nucleotide triphosphates used will depend on the composition of the
template and
the desired RNA product.
The method of this invention utilizes a 5'-modified guanosine monophosphate
(GAP) as the initiator in an RNA polymerase-catalyzed template-dependent
transcription. The guanosine initiator is modified at the 5'-position with a
molecular
entity whose chemical nature is compatible with RNA transcription. These
guanosines can be substituted at the 5'-position with molecular entities which
differ
greatly in size from the triphosphate group of a guanosine triphosphate.
Examples of
molecular entities that may be coupled to the oligonucleotide include, but are
not
limited to other macromolecules, such as oligonucleotides, lipophilic
compounds,
such as cholesterol, phospholipids, diacyl glycerols and dialkly glycerols,
proteins,
peptides or carbohydrates, polymers or resins, such as polystyrene, diagnostic
detector molecules, such as biotin or fluorescein, reporter enzymes,
photoaffinity
labels, steroids, pharmacokinetic modulators such as PEG, lipids or liposomes,
reactive moieties for post-transcriptional conjugation such as a hexylamine or
a dime
or dienophile, and chelates for binding metals.
The molecular entity can be designed to serve in a large variety of functions.
For example, a reporter group such as biotin or a fluorescent molecule may be
incorporated into the bioconjugate to provide reporter bioconjugates for use
as
diagnostic reagents. A macromolecule such as a polyethylene glycol may be
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
incorporated into the bioconjugate to provide a bioconjugate with improved
pharmacokinetics. Chelates for binding metals, particularly radioactive metals
such
as 99m Tc can be attached to the oligonucleotide for diagnostic imaging
purposes.
Other radioactive metals, such as rhenium-188, can be conjugated for directed
5 radiotherapy applications. Bioconjugates may also comprise peptides which
are
reactive to an active site on a protein. Other labeling haptens, such as the
Bolton-
Hunter reagent can be incorporated to facilitate radio-iodination. Structural
probes
such as fluorescent quenching agents or spin labels can be incorporated to
study
protein-nucleic acid interactions. To facilitate covalent SELEX a
photoaffinity label,
10 such as a psoralen, acridine, or a like molecule can be conjugated. A
chemical entity
such as a dime or Schiffs base could be incorporated for chemical covalent
SELEX.
In an embodiment of parallel SELEX the combinatorial small molecule library
can be
conjugated to the transcript. The molecular entity can also be designed to
serve as a
photoaffinity label. Histological probes may be incorporated into the
bioconjugate
1 S for visualization or anitbody staining.
This application further discloses a method for generating bioconjugates
comprising nucleic acid ligands derivatized with a molecular entity
exclusively at the
5'-position of the nucleic acid ligands. This particular embodiment takes
advantage
of the method for identifying nucleic acid ligands referred to as SELEX, an
acronym
for Systematic Evolution of Ligands by EXponential enrichment.
Briefly, bioconjugates to a target are identified by the SELEX method by the
steps comprising:
1 ) preparing a candidate mixture of bioconjugates by the steps comprising (a)
providing a DNA template having a sequence to be transcribed and (b) combining
the
DNA template with nucleotide triphosphates, a 5'-modified guanosine, and an
RNA
polymerase under conditions suitable for transcription;
2) contacting the bioconjugate candidate mixture with a target, wherein
bioconjugates having an increased affinity to the target relative to the
bioconjugate
candidate mixture may be partitioned from the remainder of the bioconjugate
candidate mixture;
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
11
3) partitioning the increased affinity bioconjugates from the remainder of the
bioconjugate candidate mixture; and
4) amplifying the increased affinity bioconjugates to yield a ligand-enriched
mixture of bioconjugates, whereby bioconjugates of the target are identified.
The 5'-substituted GAP can aid in (1) the SELEX partition step, e.g. BIA-SELEX
(see United States Application Serial No. 08/792,075, filed January 31, 1997,
entitled
"Flow Cell SELEX," which is incorporated herein by reference), plate SELEX
(Conrad et al. (1996) Methods of Enzymol. 267:336), and streptavidin column
partitions, (2) monitoring the progress of a SELEX using a reporter
substitution,
and/or (3) participating directly with the target protein, e.g., Blended SELEX
(United
States Patent No. 5,683,867, issued November 04, 1997, entitled "Systematic
Evolution of Ligands by Exponential Enrichment: Blended SELEX")
One embodiment of this invention is an extension of the Blended SELEX
methodology (United States Patent No. 5,683,867, issued November 04, 1997,
entitled "Systematic Evolution of Ligands by Exponential Enrichment: Blended
SELEX," which is incorporated herein by reference in its entirety), providing
a novel
means for identifying and generating oligonucleotides with specifically
selected
properties. This embodiment of the invention provides a method for identifying
and
synthesizing oligonucleotides derivatized with molecular entities, which are
selected
based upon the desired properties for the oligonucleotide, examples of which
are
described above.
In another embodiment of this invention the S'-derivatized guanosine contains
a reactive moiety which can be used for post-transcription conjugation of the
transcript. This embodiment of the invention can be described by the following
steps: a) providing a DNA template b) combining the DNA template with
nucleotide triphosphates, a 5'-substituted guanosine, wherein said 5'-
substituent
contains a reactive moiety and an RNA polymerase under conditions suitable for
transcription; and c) reacting the product from step b) with a molecular
entity
containing a moiety capable of reacting with the reactive moiety on said 5'-
substituent. Post transcription conjugation is necessary to obtain
oligonucleotides
derivatized with molecular entities which are not compatible with
transcription.
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
12
Examples of reactive moieties include but are not limited to amines, dimes,
dienophiles, thiols, vinylsulfones, photoaffmity labels and interchelators.
Also included in this invention are any novel conjugated oligonucleotides
which can be produced by the method of this invention.
BRIEF DESCRIPTION OF THE FIGURES
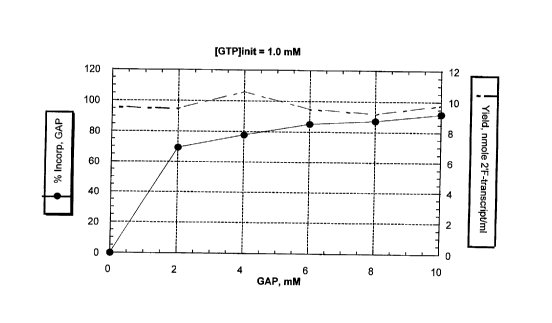
FIGURE 1 illustrates graphically the percent incorporation of GAP and the
yield of GAP transcript at concentrations of GAP in the range between 0 and 10
mM.
FIGURE 2 depicts the results of the GAP-TEG-biotin / 'y-3zP-GTP initiation
competition assay described in Example 2. The products of the transcription
reactions were analyzed by denaturing gel electrophoresis. All lanes are
labeled with
the ratio of GAP-biotin to GTP, with the exception of lane C which contains 6
mM
GTP as a control. As the concentration of GAP-biotin increased the y-32P-GTP
decreased.
FIGURE 3 shows the results of the Streptavidin shift assay described in
Example 2. The reaction products were combined with Streptavidin and analyzed
by
denaturing gel electrophoresis. All lanes are labeled with the ratio of GAP-
biotin to
GTP.
FIGURE 4A shows the results of the transcription reactions with GAP
analogs 11-16. Lanes 1 and 2 contain the transcript without GAP, lane 3 is the
GAP-
TEG transcript, lane 4 is the GAP-biotin transcript, lane 5 is the GAP-TEG-
biotin
transcript, lane 6 is the GAP-Tc chelate transcript, lane 7 is the GAP-TEG-Tc
chelate
transcipt, lane 8 is the GAP-fluorescein transcript and lane 9 is the GAP-TEG-
fluorescein transcript. The analysis was performed on an 8% polyacrylamide gel
containing 7 M urea.
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
13
FIGURE 4B depicts the transillumination of the fluorescein-GAP initiated
transcripts in lanes 8 and 9.
FIGURE 5 shows the results of the 99m Tc labeling of the GAP-Tc chelate
initiated transcript. The ~9m Tc labeled transcript was analyzed by 8%
polyacrylamide denaturing gel electrophoresis in the absence of EDTA. Lane 1
contains the 32P full length transcript control and lane 2 contains the ~9m Tc
labeled
GAP-Tc chelate.
DETAILED DESCRIPTION OF THE INVENTION
The present invention includes a novel method for enzymatically generating
oligonucleotide bioconjugates. Specifically, this invention describes a novel
method
for enzymatically generating bioconjugates comprising RNA oligonucleotides
derivatized specifically at the 5'-position with a molecular entity. This
method
1 S utilizes 5'-substituted guanosines as initiators in RNA polymerase
catalyzed template
directed synthesis of bioconjugates. The method of this invention can be used
to
conjugate an oligonucleotide prior to transcription or to incorporate a
reactive moiety
into the transcript which can then be used to bioconjugate the oligonucleotide
post-
transcription. This method may be applied to the synthesis of a variety of
conjugated
ribonucleotides including nucleic acid ligands, ribozymes and antisense RNA.
The molecular entity can be any molecule, including another macromolecule,
which is compatible with transcription. Examples of molecular entities that
may be
coupled to the oligonucleotide include, but are not limited to other
macromolecules,
such as oligonucleotides, lipophilic compounds, proteins, peptides or
carbohydrates,
polymers or resins, such as polystyrene, diagnostic detector molecules, such
as biotin
or fluorescein, reporter enzymes, photoaffmity labels, steroids,
pharmacokinetic
modulators such as PEG, lipids or liposomes, reactive moieties for post-
transcriptional conjugation such as a hexylamine or a dime or dienophile, and
chelates for binding metals.
The molecular entity can be designed to serve in a large variety of functions.
For example, a reporter group such as biotin or a fluorescent molecule may be
CA 02277545 1999-07-07
WO 98/30720 PCT/IJS98/00589
14
incorporated into the bioconjugate to provide reporter bioconjugates for use
as
diagnostic reagents. A macromolecule such as a polyethylene glycol may be
incorporated into the bioconjugate to provide a bioconjugate with improved
pharmacokinetics. Chelates for binding metals, particularly radioactive metals
such
as 99m Tc can be attached to the oligonucleotide for diagnostic imaging
purposes.
Other radioactive metals, such a rhenium-188, can be conjugated for directed
radiotherapy applications. Bioconjugates may also comprise peptides which are
reactive to an active site on a protein. Bioconjugates can also be used to
attach the
oligonucleotide to columns, solid support matrices, or surfaces such as
microtiter
plates.
Certain terms used to describe the invention herein are defined as follows:
"Nucleoside" means either a deoxyribonucleoside or a ribonucleoside or any
chemical modifications thereof. Modifications of the nucleosides include, but
are not
limited to, 2'-position sugar modifications, 5-position pyrimidine
modifications, 8-
position purine modifications, modifications at cytosine exocyclic amines,
substitution of 5-bromo-uracil, and the like.
"Nucleotide" as used herein is defined as a modified or naturally occurring
deoxyribonucleotide or ribonucleotide. Nucleotides typically include purines
and
pyrimidines, which include thymidine, cytidine, guanosine, adenine and
uridine.
"Oligonucleotide" refers to a polynucleotide formed from a plurality of
linked nucleotide units as defined above. The nucleotide units each include a
nucleoside unit linked together, typically via a phosphate linking group. The
term
oligonucleotide also refers to a plurality of nucleotides that are /inked
together via
linkages other than phosphate linkages. The oligonucleotide may be naturally
occurring or non-naturally occurring. In a preferred embodiment the
oligonucleotides
of this invention have between 1-1,000 nucleotides.
"Nucleic acid ligand" as used herein is a nucleic acid having a desirable
action on a target. A desirable action includes, but is not limited to,
binding of the
target, catalytically changing the target, reacting with the target in a way
which
modifies/alters the target or the functional activity of the target,
covalently attaching
to the target as in a suicide inhibitor, facilitating a reaction between the
target and
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
another molecule. In the preferred embodiment, the action is specific binding
affinity
for a target molecule, such target molecule being a three dimensional chemical
structure other than a polynucleotide that binds to the nucleic acid ligand
through a
mechanism which predominantly depends on Watson/Crick base pairing or triple
5 helix binding, wherein the nucleic acid ligand is not a nucleic acid having
the known
physiological function of being bound by the target molecule, In one
embodiment,
the nucleic acid ligand is a non-naturally occurring nucleic acid. In
preferred
embodiments of the invention, the nucleic acid ligands are identified by the
SELEX
methodology. Nucleic acid ligands includes nucleic acids that are identified
from a
10 candidate mixture of nucleic acids, said nucleic acid ligand being a ligand
of a given
target, by the method comprising a) contacting the candidate mixture with the
target,
wherein nucleic acids having an increased affinity to the target relative to
the
candidate mixture may be partitioned from the remainder of the candidate
mixture; b)
partitioning the increased affinity nucleic acids from the remainder of the
candidate
15 mixture; and c) amplifying the increased affinity nucleic acids to yield a
ligand-
enriched mixture of nucleic acids.
"Nucleic acid" means either DNA, RNA, single-stranded or double-stranded
and any chemical modifications thereof. Modifications include, but are not
limited
to, those which provide other chemical groups that incorporate additional
charge,
polarizability, hydrogen bonding, electrostatic interaction, and fluxionality
to the
nucleic acid ligand bases or the nucleic acid ligand as a whole. Such
modifications
include, but are not limited to, 2'-position sugar modifications, 5-position
pyrimidine
modifications, 8-position purine modifications, modifications at exocyclic
amines,
substitution of 4-thiouridine, substitution of S-bromo or 5-iodo-uracil,
backbone
modifications, methylations, unusual base-pairing combinations such as the
isobases
isocytidine and isoguanidine and the like. Modifications can also include 3'
and 5'
modifications such as capping.
"DNA template" refers to a deoxyribonucleotide which provides instructions
for an RNA polymerise to assemble a complementary ribonucleotide copy in a
process termed "transcription." The strand of DNA copied is called the "sense
strand." The DNA template strand also provides signals to initiate the copy
synthesis
CA 02277545 1999-07-07
WO 98/30720 PCT/US98J00589
16
by the enzyme at specific locations before the start of the sense strand and
to
terminate the copy synthesis at specific locations shortly after the end of
the sense
strand. The DNA template may be single-stranded or double-stranded. In a
preferred
embodiment, the DNA template is double-stranded.
"Non-immunogenic, high molecular weight compound" is a compound of
approximately 1000 Da or more that typically does not generate an immunogenic
response. An immunogenic response is one that induces the organism to produce
antibody proteins. Examples of non-immunogenic, high molecular weight
compounds include polyethylene glycol (PEG); polysaccharides, such as dextran;
polypeptides, such as albumin; and magnetic structures, such as magnetite.
As used herein a "macromolecule" refers to a large organic molecule.
Examples of macromolecules include, but are not limited to nucleic acids,
oligonucleotides, proteins, peptides, carbohydrates, polysaccharides,
glycoproteins,
lipophilic compounds, such as cholesterol, phospholipids, diacyl glycerols and
dialkyl
glycerols, hormones, drugs, non-immunogenic high molecular weight compounds,
fluorescent, chemiluminescent and bioluminescent marker compounds, antibodies
and biotin, etc without limitation.
"Bioconjugate" as defined herein refers to any oligonucleotide which has
been derivatized with another molecular entity. In a preferred embodiment the
oligonucleotide is derivatized with a macromolecule.
"Bioconjugation" or "Conjugation" refers to the derivatization of an
oligonucleotide with another molecular entity. The "molecular entity" can be
any
molecule and can include a small molecule or another macromolecule. Examples
of
molecular entities include but are not limited to other macromolecules,
polymers or
resins, such as polyethylene glycol (PEG) or polystyrene, diagnostic detector
molecules, such as biotin, fluorescein or coumarin, reporter enzymes,
photoaffinity
labels, steroids, pharmacokinetic modulators such as PEG, lipids or liposomes,
reactive moieties for post-transcriptional conjugation such as a hexylamine or
a dime
or dienophile, and chelates for binding metals or any other modifying group.
The
terms bioconjugation and conjugation are used interchangeably throughout the
Specification.
CA 02277545 1999-07-07
WO 98!30720 PCT/LTS98/00589
17
"Therapeutic Agent" means a compound which is used in the treatment or
prevention of diseases and disorders.
"Diagnostic Agent" means a bioconjugate which can be used for detecting
the presence or absence of and/or measuring the amount of a target in a
sample.
Detection of the target molecule is mediated by its binding to a nucleic acid
component of a bioconjugate specific for that target molecule. The
bioconjugate can
be labeled, for example radiolabeled, to allow qualitative or quantitative
detection.
"Improved pharmacokinetic properties" means that a bioconjugate shows a
longer circulation half life in vivo relative to a nucleic acid that is not
part of a
bioconjugate, or has other pharmacokinetic benefits such as improved target to
non-
target concentration ratio.
"Target" refers to any compound upon which a nucleic acid can act in a
predetermined desirable manner. A SELEX target molecule can be a protein,
peptide, nucleic acid, carbohydrate, lipid, polysaccharide, glycoprotein,
hormone,
receptor, antigen, antibody, virus, pathogen, toxic substance, substrate,
metabolite,
transition state analog, cofactor, inhibitor, drug, dye, nutrient, growth
factor, cell,
tissue, etc., without limitation. Virtually any chemical or biological
effector would
be a suitable SELEX target. Molecules of any size can serve as SELEX targets.
A
target can also be modified in certain ways to enhance the likelihood of an
interaction
between the target and the nucleic acid.
Transcription from synthetic DNA templates using T7 RNA polymerise and
the GAP molecules is a convenient and highly efficient method for synthesis of
RNA
derivatized exclusively at the 5'-position. In RNA polymerise catalyzed DNA
template-dependent transcription, the enzyme uses one strand of DNA as a
template
to assemble a complementary RNA copy. The transcription of DNA by T7 RNA
polymerise begins at a uniquely defined base relative to the promoter DNA
sequence.
No primer piece of RNA is required to start the copy synthesis. Successive
nucleotide triphosphates are condensed such that the growth of the RNA copy is
from
the 5'-end to the 3'-end. The enzyme positions the first nucleotide (usually
GTP or
ATP) and the 3'-hydroxyl group of this nucleotide then reacts with the 5'-
triphosphate
of the incoming nucleoside. The 3'-hydroxyl group of the dinucleotide then
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
18
condenses with the next nucleotide brought into position; and so on. The
synthesis is
driven forward by the hydrolysis of pyrophosphate. Finally, and central to
this
invention, it has been found that the 5'-triphosphate of the first nucleotide
in the
nascent RNA is not involved in the transcription. The inventors have exploited
this
finding to develop a novel method for rapidly and conveniently synthesizing
oligonucleotide bioconjugates.
The present invention provides a method for the enzymatic synthesis of
bioconjugates comprising RNA derivatized exclusively at the 5'-position with a
molecular entity. In its most basic form the method of the instant invention
can be
described by the following steps:
a) providing a DNA template and b) combining the DNA template with
nucleotide triphosphates, a 5'-substituted guanosine and an RNA polymerase
under
conditions suitable for transcription. The types of nucleotide triphosphates
used will
depend on the composition of the template and the desired RNA product.
Using the method of this invention the 5'-modified guanosine can only be
added at the initiating 5'-end of the transcript during the initiation phase
of
transcription. As stated above, transcript elongation is driven forward by the
hydrolysis of pyrophosphate, therefore it is necessary that the remaining
nucleotides
be nucleoside triphosphates. The 5'-substituted guanosine does not have a 5'-
triphosphate group and as such it can participate in initiation, but not
elongation.
Therefore, in contrast to other methods of enzymatically incorporating
substituted
nucleotide triphosphates during RNA synthesis, wherein substituted nucleotide
triphosphates are incorporated throughout the RNA transcript, the method of
the
present invention provides a unique method of synthesizing bioconjugates
comprising a molecular entity attached exclusively to the 5'-position of an
oligonucleotide.
Since the first nucleotide in the DNA template to be transcribed is a
cytosine,
a 5'-derivatized guanosine (referred to herein as GAP) will compete with a GTP
as
the first component of the nascent RNA transcript. Thus, a mixture of RNA
oligonucleotides containing bioconjugates comprising 5'-substituted RNA
oligonucleotides and 5'-unsubstituted RNA oligonucleotides will be obtained.
By
CA 02277545 1999-07-07
WO 98/30720 PCT/I1S98/00589
19
increasing the concentration of the 5'-derivatized guanosine in the
transcription
reaction relative to the GTP concentration, however, proportionally more
derivatized
guanosine will be incorporated into the transcript. As shown in Figure 1, a
ratio of
GAP:GTP of 10:1 results in 92 % of the transcript being initiated with GAP.
Theoretically, if GAP and GTP are used as initiating nucleotides with equal
efficiency, GAP should be present 90.91 % of the time. Depending on the
required
level of purity of the 5'-substituted transcript, the GAP-conjugate:GTP ratio
can be
varied.
The ability to use a S'-derivatized guanosine as a substrate in the enzymatic
synthesis of an oligonucleotide bioconjugate offers significant advantages
over
currently available methods or synthesizing these compounds. First, this
method
offers the ability to specifically incorporate a macromolecule at the 5'-
position of the
RNA oligonucleotide during enzymatic synthesis of the RNA oligonucleotide.
This
is in contrast to traditional and time-consuming methods of synthesizing
bioconjugates, in which the oligonucleotide must be first chemically
synthesized in a
manner which incorporates a modified nucleoside during the last step of the
synthesis, and then conjugating the modified oligonucleotide to a molecular
entity.
Second, the use of 5'-derviatized guanosines allows for the modification of
transcripts
which are too long to be chemically synthesized, greatly increasing the
possible
applications. Third, current methods known in the art for enzymatically
incorporating modified nucleotide triphosphates into an nascent
oligonucleotide
result in the synthesis of modified nucleotides having several substituted
nucleotides
within the oligonucleotide. In contrast, the method of the present invention
allows
for the controlled, enzymatic synthesis of a bioconjugate substituted
exclusively at
the 5'-end of the oligonucleotide.
One embodiment of the present invention includes a method for generating
high affinity bioconjugates to specific target molecules. In the preferred
method, the
nucleic acid ligand is identified by the SELEX method. The SELEX method is
described in United States Patent Application Serial No. 07/536,428, filed
June 11,
1990, entitled "Systematic Evolution of Ligands by EXponential Enrichment,"
now
abandoned; United States Patent Application Serial No. 07/714,131, filed June
10,
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
1991, entitled "Nucleic Acid Ligands," now United States Patent No. 5,475,096;
United States Patent Application Serial No. 07/931,473, filed August 17, 1992,
entitled "Methods of Identifying Nucleic Acid Ligands," now United States
Patent
No. 5,270,163 (See also PCT Application Publication No. WO 91 / 19813 ). These
5 applications, each specifically incorporated herein by reference, are
collectively
called the SELEX Patent Applications.
In its most basic form, the SELEX process may be defined by the following
series of steps:
1 ) A candidate mixture of nucleic acids of differing sequence is prepared.
10 The candidate mixture generally includes regions of fixed sequences (i.e.,
each of the
members of the candidate mixture contains the same sequences in the same
location)
and regions of randomized sequences. The fixed sequence regions are selected
either: (a) to assist in the amplification steps described below, (b) to mimic
a
sequence known to bind to the target, or (c) to enhance the concentration of a
given
I 5 structural arrangement of the nucleic acids in the candidate mixture. The
randomized
sequences can be totally randomized (i.e., the probability of finding a base
at any
position being one in four) or only partially randomized (e.g., the
probability of
finding a base at any location can be selected at any level between 0 and 100
percent).
2) The candidate mixture is contacted with the selected target under
20 conditions favorable for binding between the target and members of the
candidate
mixture. Under these circumstances, the interaction between the target and the
nucleic acids of the candidate mixture can be considered as forming nucleic
acid-
target pairs between the target and those nucleic acids having the strongest
affinity for
the target.
3) The nucleic acids with the highest affinity for the target are partitioned
from those nucleic acids with a lesser affinity to the target. Because only an
extremely small number of sequences (and possibly only one molecule of nucleic
acid) corresponding to the highest affinity nucleic acids exist in the
candidate
mixture, it is generally desirable to set the partitioning criteria so that a
significant
amount of the nucleic acids in the candidate mixture (approximately 5-50%) are
retained during partitioning.
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
21
4) Those nucleic acids selected during partitioning as having the relatively
higher affinity to the target are then amplified to create a new candidate
mixture that
is enriched in nucleic acids having a relatively higher affinity for the
target.
5) By repeating the partitioning and amplifying steps above, the newly
formed candidate mixture contains fewer and fewer unique sequences, and the
average degree of affinity of the nucleic acids to the target will generally
increase.
Taken to its extreme, the SELEX process will yield a candidate mixture
containing
one or a small number of unique nucleic acids representing those nucleic acids
from
the original candidate mixture having the highest affinity to the target
molecule.
The SELEX Patent Applications describe and elaborate on this process in
great detail. Included are targets that can be used in the process; methods
for
partitioning nucleic acids within a candidate mixture; and methods for
amplifying
partitioned nucleic acids to generate enriched candidate mixtures. The SELEX
Patent
Applications also describe ligands solutions obtained to a number of target
species,
1 S including both protein targets where the protein is and is not a nucleic
acid binding
protein. The SELEX Patent Applications describe a number of uses for nucleic
acid
ligands including numerous therapeutic and diagnostic uses.
In this embodiment the bioconjugate is prepared by the SELEX method as
described in the SELEX Patent Applications. Briefly, bioconjugates to a target
are
identified by the SELEX method by the steps comprising:
1 ) preparing a candidate mixture of bioconjugates by the steps comprising (a)
providing a DNA template having a sequence to be transcribed and (b) combining
the DNA template with nucleotide triphosphates, a modified guanosine, and an
RNA
polymerase under conditions suitable for transcription;
2) contacting the bioconjugate candidate mixture with a target, wherein
bioconjugates having an increased affinity to the target relative to the
bioconjugate
candidate mixture may be partitioned from the remainder of the bioconjugate
candidate mixture;
3) partitioning the increased affinity bioconjugates from the remainder of the
bioconjugate candidate mixture; and
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
22
4) amplifying the increased affinity bioconjugates to yield a ligand-enriched
mixture of bioconjugates, whereby bioconjugates of the target are identified.
The 5'-substituted GAP can aid in ( 1 ) the SELEX partition step, e.g. BIA-
SELEX
(see United States Application Serial No. 08/792,075, filed January 31, 1997,
entitled
"Flow Cell SELEX, which is incorporated herein by reference), plate SELEX
(Conrad et al. (1996) Methods of Enzymol. 267:336), and streptavidin column
partitions, (2) monitoring the progress of a SELEX using a reporter
substitution,
and/or (3) participating directly with the target protein, e.g., Blended SELEX
(United
States Patent No. 5,683,867, issued November 04, 1997, entitled "Systematic
I O Evolution of Ligands by Exponential Enrichment: Blended SELEX")
Using this method, nucleic acid ligands derivatized exclusively at the 5'-
position of the nucleic acid ligand with virtually any molecular entity which
is
compatible with transcription can be prepared and identified. Molecular
entities that
can be coupled to nucleic acid ligands include, but are not limited to
lipophilic
1 S molecules, proteins, peptides, reporter molecules, reporter enzymes and
steroids.
In another embodiment of this invention the 5'-derivatized guanosine contains
a reactive moiety which can be used for post-transcription conjugation of the
transcript. This embodiment of the invention can be described by the following
steps: a) providing a DNA b) combining the DNA template with nucleotide
20 triphosphates, a 5'-substituted guanosine, wherein said 5'-substituent
contains a
reactive moiety and an RNA polymerase under conditions suitable for
transcription;
and c) reacting the product from step b) with a molecular entity containing a
moiety
capable of reacting with the reactive moiety on said 5'-substituent. Post
transcription
conjugation is necessary to obtain oligonucleotides derivatized with molecular
25 entities which are not compatible with transcription. Examples of reactive
moieties
include but are not limited to amines, dimes, dienophiles, thiols,
vinylsulfones,
photoaffinity labels and interchelators.
In certain embodiments, the molecular entity may provide certain desirable
characteristics to the nucleic acid ligand, such as, increasing RNA
hydrophobicity and
30 enhancing binding, membrane partitioning and/or permeability. Additionally,
reporter molecules, such as biotin, fluorescein, or peptidyl metal chelates
for
CA 02277545 1999-07-07
WO 98130720 PCT/LTS98100589
23
incorporation of diagnostic radionuclides may be added, thus providing a
bioconjugate which may be used as a diagnostic agent.
Example 1 describes the synthesis of a variety of 5'-modified guanosine
monophosphates. For commonly used functional groups it is more efficient to
conjugate the moiety of interest to the GAP molecule prior to transcription.
This
allows for large scale synthesis of the initiator, pre-transcription
purification of the
initiator, and negates the need for post-transcriptional conjugations. The
modified
guanosines synthesized are set forth in Schemes 1, 2, 4 and S and include GAP
(5),
GAP-fluorescein (11), GAP-biotin (12), GAP-Tc chelate (13), GAP-TEG (10), GAP-
TEG-fluorescein (14), GAP-TEG-biotin (15) and GAP-TEG-Tc chelate (16). Biotin
and fluorescein are very common conjugates which provide very useful
properties for
RNAs. The GAP-TEG analogs were synthesized because they are less expensive,
less hydrophilic and potentially less immunogenic than the GAP analogs. When
conjugating GAP to more hydrophobic adducts, such as biotin, solubility in
aqueous
buffers becomes a serious consideration.
Example 2 illustrates the feasibility of using transcription with 5'-modified
guanosines to synthesize oligonucleotides modified exclusively at the 5'-
position.
This example demonstrates that both GAP and GAP conjugates can compete with
GTP for the initiation of RNA synthesis. As shown in Figure 1, a ratio of
GAP:GTP
of 10:1 results in 92 % of the transcript being initiated with GAP. Example 2
also
illustrates that the yield of full length product does not decrease as a
result of GAP-
Biotin inhibiting the transcription reaction.
Example 3 illustrates the post transcription conjugation of a GAP initiated
transcript. Post transcription conjugation is necessary to obtain
oligonucleotides
derivatized with molecular entities that are not compatible with
transcription.
Transcription with primary amine initiators allows for the post-
transcriptional
conjugation of RNA with a wide variety functional groups through easily
available
NHS chemistry. In this example a GAP initiated RNA was reacted with a biotin
NHS ester.
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
24
Example 4 demonstrates that GAP (5) and GAP-TEG (10) incorporate to the
same extent resulting in the same amount of full length 5'-modified
oligonucleotide
product.
Example 5 (Figure 4A) demonstrates that GAP conjugate compounds 11-16
incorporate to the same extent resulting in the same amount of full length
product as
GTP (see Table below). This example compares the transcription reactions run
with
GAP analogs l I-16 and a control run without GAP. The results are set forth in
the
table below. Figure 4B shows that the full length transcripts initiated with
GAP-
fluorescein (11) and GAP-TEG-fluorescein (14) result in a fluorescent signal
upon
irradiation with ultra violet light. This example clearly demonstrates that
virtually
any linker or conjugate attached to guanosine, which ultimately is compatible
with
the transcription enzymes, could be used to enzymatically derivatize the 5'-
terminal
end of an RNA molecule.
Lanes (Figure 4A) pmoles eluted
1,2 transcript without 1572
GAP
3 GAP-TEG 1948
4 GAP-Biotin 1810
5 GAP-TEG-Biotin 2369
6 GAP-Tc chelate 2032
7 GAP-TEG-Tc chelate 1984
8 GAP-Fluorescein 2426
9 GAP-TEG-Fluorescein 2614
Example 6 describes the labeling of a GAP-Tc chelate (13) initiated transcript
with 9~m Tc.
The following examples are presented for illustrative purposes only and are
not intended to limit the scope of the invention.
EXAMPLES
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
General methods. All reagents and solvents were used as received from the
manufacturer. 5'-dimethoxytrityl-'-N isobutyrylguanosine (1) was purchased
from
ChemGenes Corp. The 5' Amino-Modifier C6-TFA was purchased from Glen
Research. All reactions were carried out under anhydrous conditions with inert
5 atmosphere in oven-dried glassware. TLC was performed on Baker Si250F TLC
Plate-Silica Gel and the spots were rendered visible by UV light. Flash
chromatography was performed with a Biotage Flash 40 apparatus using a
Kiloprep
Column (KP-Sil, 60 A). RP-HPLC was performed on a Waters' Delta Pak 5 p C l 8
300 A, 3.9 x 150 mm column. Buffer A: 100 mM TEAA at pH 7.0; Buffer B: ACN.
10 The temperature was 30 ° C and the flow rate was 0.50 mL/min. NMR
spectra were
recorded on a Bruker ARX 300 spectrometer using CDC13 and (CD3)zS0 as solvents
with TMS as an internal standard. Electrospray mass spectrometry was performed
on
a Fissions Quattro II (Beverly, MA) using negative ion mode. The samples were
delivered in a 1:1 MeOH/Hz0 (v/v) containing 0.1 % TEA at 10 ~L/min to the
mass
15 spectrometer.
General in vitro transcription. T7 RNA polymerase was purchased from Enzyco,
Denver, Colorado. 2'-F-CTP and -UTP were purchased from USB Biochemicals.
The transcriptional template was a 104-by DNA amplified by PCR from a
linearized
20 pUC plasmid with the sequence:
TAA TAC GAC TCA CTA TAG GGA GAC AAG AAT AAA CGC TCA AGC
GGG ATT TTC CTG ATC ATC CCA CTG ATT CGG GGC CTT ACT TCG ACA
GGA GGC TCA CAA CAG GC (SEQ ID NO: l )
where the bold bases represent the T7 promoter sequence and the underlined
bases
25 represent the PCR primer regions. Ih vitro transcription was performed
under
standard conditions (Krupp and Soll ( 1987) FEB S 212:271-275; Milligan and
Uhlenbeck { 1989) Methods of Enzymology 180:51-62), which were modified for
the
incorporation of 2'-F pyrimidine triphosphates into the transcript (Lin et al.
( 1994)
Nucleic Acids Res. 22:5229-34). Briefly, transcription was performed in 40 mM
Tris-HCI, pH 8, 4% PEG 8000, 12 mM MgClz, 5 mM DTT, 1 mM spermadine, 0.002
Triton X-100 with a template concentration of 0.5 to 1.0 ~M, and a T7 RNA
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
26
polymerase concentration of 0.4 pM. 2'-F-CTP and 2'-F-UTP were added to 3mM
while ATP and GTP were added to a final concentration of 1 mM. Reactions were
incubated at 37 ° C for 16-20 hours.
Example 1. Synthesis 5' substituted ~uanosine monouhosphates
Small-scale solid phase synthesis of 5'-(O'-hexylamino)~uanosine mono~hosphate
IGAP). Initially, GAP (5) was synthesized with commercially available reagents
using a DNA/RNA synthesizer. Starting with acetate protected guanosine CPG
(Glen Research) a single coupling step was used to add 5'-Amino-modifier C6
phosophoramidite (Glen Research). The product was cleaved from the solid
support,
deprotected with NaOH and purified by reverse phase chromatography to yield
GAP
(5). The success of this experiment stimulated the larger scale production of
the
GAP molecule, discussed below.
Late scale solution phase synthesis of S'-(O'-hexylamino)~uanosine
monoph05Dhate
,~. Scheme 1 sets forth the large scale solution phase synthesis of GAP (5).
SCHEME 1
0 0
N N
~, ~NH O CHg AczO/Pyridinc ~ ~~ ~ CH3
DMTO O N N~H-~-CH-CH3 DCM DMTO O N N H-C-CH-CH3
1
2O HO OH Ac0 OAc
~ ~' r~,\'.
~ ,~l~~.p ~t~
O ~ o~~'O
5
n i-
N~ O F3C_O_N NC O
(i II NH O CH3 N~NH
I I i 1 ) TFA- C6 amine O ~~ ~ 0 CHg
HON N H-C-CH-CH3 tetrazole O ~-O N N~N-C-CH-CHg
' p ~ H
GS 2) TITRATE O.IM I,IfHF/Pyridinc
Ac0 'OAc Ac0 OAc
3
O
CIitNH~/NH~OH HZN N
-O ~, ~NH
O-~-O O N N~NH2
O
30 5 HO OH
4
CA 02277545 1999-07-07
WO 98/30720 PCT/I1S98/00589
27
2',3'-diacetvl-5'-dimethox trityl zN isobut~ryl~uanosine ,(2). The 5'-
dimethoxytrityl-
ZN-isobutyrylguanosine (1) (5 g, 7.63 mmol) was dissolved in 30 mL of pyridine
and
15 mL of dichloromethane (DCM ) at 23 °C and 3.6 mL (38 mmol) of acetic
anhydride was added. The reaction was complete in (5 hours) as determined by
TLC
(9.5:9.5:1 hexane/EtOAc/MeOH). The solution was then brought up in EtOAc (500
mL) and washed with saturated NaHC03 (3 x 300 mL). The NaHC03 fractions were
back extracted with EtOAc. The EtOAc fractions were combined, dried over MgS04
and concentrated to a dry precipitate in vacuo. This afforded 5.5 g (97.6%) of
pure
product 2 as determined by 'H NMR (300 MHz, (CD3)ZSO).
2'.3'-diacetyl-ZN isobutyrylsuano- sine (3). The 2',3'-diacetyl-5'-
dimethoxytrityl-'-N-
isobutyrylguanosine (2) (S.5 g, 7.44 mmol) was brought up in 10 mL of DCM and
loaded onto a Biotage Flash 40 silica gel column. The dimethoxytrityl was
removed
on the column using a of 3 % solution of trichloroacetic acid (TCA) and 0.5
MeOH in DCM.. After the dimethoxytrityl had eluted from the column, as
determined visually and by TLC ( 19:1 DCM/MeOH), the column was washed with
10 volumes of 0.5% MeOH in DCM. The product was then eluted with an increasing
gradient of 1 %-10 % MeOH in DCM. The appropriate fractions were combined and
concentrated to a solid in vacuo. This afforded 2.96 g (91 %) of pure product
3 as
determined by 'H NMR (300 MHz, {CD3)ZSO).
5'-(O'-hexylamino)auanosine monophosphate (GAP) (5). To a stirred solution of
3
(2.96 g, 6.76 mmol) in dry ACN (20 mL) was added 5' Amino-Modifier C6-TFA
(3.64 g, 1.3 equiv; Glen Research) and 0.47 M tetrazole in anhydrous ACN (3 74
mL,
26 equiv; Glen Research). The reaction was complete in four hours as
determined by
TLC. The solution was titrated with 0.1 M oxidizing solution (I,/THF/pyridine)
( 100
mL; Glen Research) until a brown color persisted. The solution was
concentrated in
vacuo to approximately one fifth of its volume and then brought up in EtOAc
(500
mL) and washed with 5 % NaHS03 (2 x 300 mL) and saturated NaHC03 (2 x 300
mL). The aqueous washes were back extracted with EtOAc (500 mL) and the
EtOAc fractions were combined, dried over MgSOa and then concentrated to
dtvness
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
28
in vacuo. This afforded a crude yield of 5.57 g ( 109.8%) of 4. This crude
material
was then treated with 1:1 NHQOH/CH3NH2 ( 125 equiv:125 equiv) for 30 minutes
at
65 ° C. The NH40H and CH3NH2 were removed in vacuo and the remaining
material
was purified via RP-HPLC to obtain 2.5 g (80%) of 5 as a solid. Product was
verified by 'H NMR (300 MHz, (CD3)ZSO), and MS calculated for Ct6H,6N608P
(M+1 ): 462.3.
Svnthesis of 5'-(O'-tetraethylene ~lycol)~uanosine mono hosphate~GAP TEG)
(10).
S'-(O'-tetraethylene glycol)guanosine monophosphate (GAP-TEG) (10) was
synthesized by reaction of compound 3 with TEG-Phthalimide phosphoramidite (8)
as outlined in Scheme 2. The synthesis of TEG-Phthalimide phosphoramidite is
outlined in Scheme 3 below.
SCHEME 2
O
N NH O NCO
HO O N I N H-O-CH 3CH3 + \ I N O ~O-P.N
O
t TEG-Phthalimide-Phosphoramidite
Ac0 OAc
3 tetrazo le/ACN 8
t~trate
0. I M Iz
~NH
/ N O -O <N I N ~ N-~- HH3
~O-O-O O H CH3
O
Aco oAc
NH~OI-I
CI-l3NHz O
~N~~
H2N~0~0~0~0-~-O O N N NH2
O
GAP-TEG HO OH
CA 02277545 1999-07-07
WO 98/30'720 PCT/US98/00589
29
5'-(O'-tetraethylene szlycol)euanosine monophosphate (GAP TEG) (10). To a
stirred
solution of 3 (0.941 g, 2.15 mmol) in dry ACN (20 mL) was added 0.47 M
tetrazole
in anhydrous ACN (120 mL, 56.4 mmol; Glen Research) and 1.89 g of crude 8. The
reaction was complete in four hours as determined by TLC. The solution was
titrated
with 0.1 M oxidizing solution (32 mL; Glen Research) until a brown color
persisted.
The solution was concentrated in vacuo to approximately one fifth of its
volume,
taken up in EtOAc (500 mL) and washed with 5 % NaHS03 (2 x 300 mL) and
saturated NaHC03 (2 x 300 mL). The aqueous washes were back extracted with
EtOAc (500 mL). The organic fractions were combined, dried over MgS04 and
concentrated to dryness in vacuo. This afforded a crude yield of 2.2 g (
118.8%) of
compound 9. The crude material was treated with 1:1 NH40H/CH3NH~ (125 equiv:
125 equiv) for 30 minutes at 65 °C. The NH40H and CH3NH2 were removed
in
vacuo and the remaining material was purified via RP-HPLC to obtain 0.900 g
(77.95%) of pure product compound 10 as determined by 'H NMR (300 MHz,
(CD3)zS0) and MS calculated for C,8Hz9N60"P (M+1): 537.1.
Tetraethylene glycol phthalimide phosphoramidite (8) was synthesized as
outlined in
Scheme 3.
SCHEME 3
HO O OH P-toluenesulfonylchloride ~ ~ $-O O OH
O
TEG pyridine TEG-Tosylate
G
phthalimide
DBU
DMF
O O NCO
N~O~OH Diisopropylamine \ I N~O~O,P.N~
O CN(CI-IZ),P(Cl)N(iPr)2 - /O
TEG-Phthalimide TFG-Phthalimide-Phosphoramidite
7
8
Tetraethylene~lycol monotos la~(6). Tetraethylene glycol (100 mL, 575 mmol)
was dissolved in 250 mL of pyridine and cooled to 0°C and treated with
11.0 g (0.1
CA 02277545 1999-07-07
WO 98!30720 PCT/US98/00589
eq., 57.5 mmol) p-toluenesulfonyl chloride. When solution was complete, the
reaction was stored in the refrigerator overnight. The reaction was complete
as
determined by TLC ( 19:1 EtOAc/MeOH). The reaction mixture was then
concentrated in vacuo. The residue was dissolved in 600 mL of EtOAc and
extracted
5 with Hz0 (3 x 200 mL). The H20 fractions were back-extracted with 400 mL of
EtOAc and the combined EtOAc fractions were extracted with saturated aqueous
Na,HP04. The organic phase was dried over MgS04 and concentrated in vacuo to
yield 18.0 g (90.2 % crude) of tetraethylene glycol monotosylate (6) as a
colorless oil.
'H NMR (300 MHz, CDC13) b 7.77 (d, J = 8.1 Hz, 2H), 4.13 (t, J = 4.8 Hz. 2H),
10 3.68-3.53 (m, 14 Hz), 2.58 (t, J = 5.6 Hz, 1H), 2.42 (s, 3H).
Tetraethylene glycol monophthalimide (7). To a stirred solution of 18.0 g
(51.7
mmol) of crude 6 in 225 mL of anhydrous DMF was added 8.0 g (1.05 eq., 54.3
mmol) of phthalimide and 8.12 mL (1.05 eq., 54.3 mmol) of 1,8-
15 diazabicyclo[5.5.0]undec-7-ene. The solution was heated at 70°C for
18 hours and
then concentrated in vacuo. The reaction was determined complete by TLC ( 19:1
EtOAc/MeOH). The crude yellow oil was purified by flash chromatography using a
Biotage Flash 40 silica gel column and eluting with 25% EtOAc/hexane, 50%
EtOAc/hexane, 75% EtOAc/hexane, EtOAc, and then 10% MeOH to afford 13.7 g
20 (82%) of compound 7 as an oil. Upon standing, 7 became a waxy white solid.
'H
NMR (300 MHz, CDC13) 8 7.84-7.78 (m, 2H), 7.70-7.66 (m, 2H), 3.86 (t, J = 5.6
Hz,
2H), 3.70 (t, J = 5.6 Hz, 2H), 3.64-3.51 (m, 12H), 2.67 (bs, 1 H).
1-Phthalimido-tetraethylene ~lycol(diiso ropy!amino)-(3-cyanoethoxy~hosphine
8JI.
25 A 1 g (3.1 mmol) aliquot of 7 was brought up in of THF (20 mL), dried in
vacaao to
an oil and then resuspended in THF (20 mL) under argon. To this stirred
solution
was added N,N diisopropylethylamine (702 q,L, 4.0 mmol) and 2-cyanoethyl
diisopropylchlorophosphoramidite (762 qL, 3.41 mmol; Aldrich). The reaction
was
complete in 30 minutes as determined by TLC. The solution was dried in vacuo
to
30 an oil, taken up in EtOAc (300 mL), washed with HBO (2 x 100 mL) and the
organic
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
31
phase was dried over MgS04. This afforded a crude yield of 1.89 g ( 116.5%) of
compound 8 which was used without further purification.
General method for con'u ation of NHS ester activated molecules to the 5'-
amino-
terminus of compounds 5 (Scheme 4) and 10 (Scheme 57. To a solution of 5 or 8
( 10
mmol) in DMSO (219.5 ~.L, 20 mg/mL) and TEA (11.5 p.L, 5%) was added the
desired NHS ester molecule (20 mmol). The reaction was complete in one hour as
determined by RP-HPLC. Purification of the 5' modified guanosine
monophosphates
was achieved by reverse phase chromatography. Separation was achieved by
loading
1 gram of crude reaction mixture on a Waters Delta Pak C-18 column (50 X 3 00
mm) column and running a 2% to 50% gradient (108 min) of acetonitrile in 100
mM
triethylamine carbonate pH 8 at 12 mL/min. Analytical separations were
determined
with a Waters Delta Pak C-18 (4.9 X 150 mm) column and running 2% to 50%
gradient (54 min) of acetonitrile in 100 mM triethylamine acetate pH 7 at 1
mL/min.
Purification by RP-HPLC afforded yields of ( 18 mmol, 90%) of compounds 1 I-16
(Schemes 3 & 4).
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
32
SCHEME 4
GAP
NHS-ester
DMSO/TEA
1 hour
O
N NH
~O-P-O ~N ~ N~NH2
p p p
HN~NH GAP-fluorescein HO OH
O O 11
S N~NH O
3 H 4
//N NH
O_P_O \N I
ii O N NH2
IS O
GAP-biotin
O 1y HO OH
/~'~O
~NH HN
O O
S HN
S \~ O \N I
O-P-O N N NH
:; ~ ,p. i
GAT-TEG-Tc Chelate HO
13
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
33
SCHEME 5
GAP-TEG
+ NHS-ester pMSO/TEA
1 hour
O ~ O ~ OH
w w ~ i
HO-C O
" ~ S
O ( / N~N~O~O~O~ o <N
H H O-P-01 ~ N N NH2
GAP-TEG-fluorescein
O HO OH
~ 14
1O HN- -NH O
O O N NH
S N~N~O~O~O~ _~
s H a H O P-O N N N HZ
O O
GAP-TEG-biotin HO OH
15
~O
O
~NH HN
0 0
S HN NIl NH
S ~O~O~O~ O Ci ~~
O-P-O N N NH2
O O
GAP-TEG-Tc chelate HO OH
16
Biotinamidocanroate 5'-(O'-hexylamino)-tsuanosine monophOSnhate~l4).
To a solution of S (1 mmol) in DMSO (21.85 mL, 20 mg/mL) and TEA (1.15 mL,
5%) was added biotinamidocaproate N hydroxysuccinimidyl ester (909 mg, 2
equiv).
The reaction was complete in one hour as determined by RP-HPLC. Purification
by
RP-HPLC afforded a yield of 719.8 mg {90%) of pure product compound 14.
Example 2. Initiation competition assa~rs
GAP / y 3zP-GTP initiation competition assay. A series of transcription
reactions
were performed using 'y-3zP-GTP to determine the extent to which GAP (5) would
compete with GTP for initiation of transcription reactions. The reactions were
run
under the standard conditions set forth above. The reactions were performed
with the
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
34
GAP molecule added to a final concentration of 0 to 10 mM while GTP ( 1 mM)
and
gamma labeled 3zP-GTP were held at fixed concentrations. Since the ''P is in
the
gamma position, only those GTP molecules which initiate transcription, will
result in
the incorporation of a radiolabel into the transcript. The reaction products
were
analyzed by denaturing gel electrophoresis. Full length transcript bands were
excised from the gel, the RNA was eluted from the gel slices and was
quantitated by
UV absorbance at 260 nm. Percent incorporation was calculated with the
following
equation:
(1-CPM~cA,,~/CPM~",a.~) X 100
where CPM~GA,,~ = the CPM of the full length band at a given GAP concentration
and
CPM~;~;,,~ = the CPM of the full length band at a GAP concentration of 0 mM.
The
results are depicted in Figure 1, which shows that an increase in
concentration of
GAP results in a corresponding decrease in y-32P GTP incorporation (Figure 1).
Each reaction resulted in the same yield of full length transcript.
Quantitation
indicates that GAP is used as an initiating nucleoside to the same extent as
GTP.
GAP-TEG-biotin / y-32P-GTP initiation competition assay. A second series of
reactions were performed using a fixed amount of GTP ( 1 mM) and y-3'P GTP and
an increasing concentration of GAP-TEG-biotin (15) to give ratios of: 0, 0.1,
0.5, 1,
2, 4, and 10 to 1 GTP. The reaction products were analyzed by denaturing gel
electrophoresis, which showed that the increase in GAP-TEG-biotin resulted in
a
corresponding decrease in y 'ZP GTP incorporation. (Figure 2). The competition
by
GAP-TEG-biotin is in agreement with the ratio of GAP-TEG-biotin to GTP.
GAP- Biotin inhibition assay. Transcription reactions were performed under the
standard conditions set forth above, except that a-3zP-ATP was added to
internally
label the transcription products. The same series of transcription reactions
with GTP
to GAP-biotin (12) ratios again between 0 and 10 fold excess was used. As the
amount of GAP in each reaction increased the amount of full length product
remained the same, demonstrating that GAP-biotin does not inhibit RNA
transcription.
Streptavidin Shift assay. Transcription reaction products for the 0 to 10
ratios of
GAP- biotin were combined with 10 pM streptavidin in 37.5 mM Tris, pH 7.5. The
CA 02277545 1999-07-07
WO 98/30720 PCT/US98/00589
products of the reaction were analyzed on a denaturing gel and quantified by
phosphoimager. The amount of Streptavidin shift was correlated to the
theoretical
amount of GAP-biotin that should have been incorporated (Figure 3).
GAP-biotin incorporation
5 The presence of the 5' primary amine was assessed by the ability to
conjugate
NHS-biotin to the GAP transcript. Transcripts were synthesized in the presence
of
increasing concentrations of GAP. The resulting transcripts were conjugated
with a
100-fold excess of NHS-biotin. After conjugation transcripts were admixed with
a 5-
fold excess of Streptavidin and applied to an 8% urea polyacrylamide gel.
These data
10 indicate that there is no biotin incorporation in the absence of GAP and
that the
percentage of gel-shifted transcript increases with increasing GAP added to
the
transcription reaction. The gel shift assay reaches a maximum at approximately
70
%, underestimating the GAP incorporation at the highest GAP:GTP ratios as
compared to the y-32P-ATP-GTP assay or HPLC analysis of the conjugated
15 transcript.
Repeating the same experiments with GAP-TEG instead of GAP will result
in an identical outcome.
Example 3. Post transcription biotinylation of GAP RNA transcripts
20 A GAP initiated RNA was transcribed at a GAP to GTP concentration of
15:1. The 5' amine on the terminus of the unpurified full length GAP RNA (TEA
salt) was then conjugated to a long chain biotin NHS ester in anhydrous DMSO
/10%
TEA. The biotin NHS ester was added at 5 eq. of biotin for one hour followed
by an
additional S eq. for one hour. The biotinylated transcript was purified by
ethanol
25 precipitation to remove free biotin. To verify post transcription
conjugation the
transcription reaction products were combined with a large excess of
streptavidin ( 10
~M streptavidin in 37.5 mM Tris, pH 7.5). The products were analyzed on a
denaturing gel to show a steptavidin shift. In the presence of streptavidin 50
percent
of the biotin conjugated RNA material shifted to the slower migrating
Streptavidin
30 complex.
CA 02277545 1999-07-07
WO 98/30720 PCT/US98l00589
36
Example 4. Transcription with GAP (5) and GAP-TEG (101
Transcription reactions were performed under standard conditions as set forth
above, with GAP (5) or GAP-TEG (10) added at a ratio of 10 to 1 over the
concentration of GTP. The reaction products were analyzed on a denaturing gel,
which showed that transcription with GAP and GAP-TEG resulted in the same
yield
of full length transcript.
Example 5. Transcription with GAP and GAP-TEG conju ates
This example describes the incorporation of GAP conjugates (11-13) and
GAP-TEG conjugates (14-16). Transcription reactions were performed in parallel
under standard conditions as set forth above using a 104-by transcriptional
template
with the addition of a-3zP-ATP. The GAP conjugates were added at a ratio of 10
to 1
over the concentration of GTP. A control reaction was run in which no modified
guanosine was added to the standard RNA transcription reaction. The reaction
products were analyzed on an 8% polyacrylamide gel containing 7 M urea. The
gel
was visualized by autoradiography. (Figure 4A). The bands corresponding to
full-
length transcript were cut out of the gel, eluted and quantitated by UV
spectroscopy.
Analysis showed that all of the modified guanosine compounds resulted in the
same
yield of full length transcript as the control. Lanes 8 and 9 were
transilluminated to
detect the presence of fluorescein in the full length GAP-fluorescein and GAP-
TEG-
fluorescein transcripts. In all cases the 10 fold excess of the modified
guanosine over
GTP concentration showed no diminution of transcription yield.
Example 6. Labeline of GAP-Tc chelate (13) transcript
To 1 nmole GAP-Tc chelate (13) initiated transcript was added 200 ~,L of 100
mM NaP04 buffer, pH 8.5, 23 mg/mL NaTartrate, and 50 ~L ~9m Tc pertechnetate
(5.0 mCi) eluted from a 99-Mo column within 12 hours of use. The labeling
reaction
was initiated by the addition of 10 pL 5 mg/mL SnCl2. The reaction mixture was
incubated for 15 minutes at 90 ° C. The reaction was separated from
unreacted 9~m
Tc by spin dialysis through a 30,000 MW cut-off membrane (Centrex, Schleicher
&
Scheull) with two 300 ~L washes. This labeling protocol results in 30-50% of
the
CA 02277545 1999-07-07
WO 98/30720
PCT/US98/00589
37
added 99m Tc being incorporated with a specific activity of 2-3 mCi/nmole RNA.
99m Tc labeled transcript was analyzed by 8% polyacrylamide denaturing gel
electrophoresis in the absence of EDTA. Quantitation indicates that over 97%
of the
99m Tc is associated with the full-length transcipt after spin dialysis
(Figure 5).