Note: Descriptions are shown in the official language in which they were submitted.
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
RECOMBINANT HALOALIPHATIC DEHALOGENASES
Large quantities of short-chain halogenated aliphatic hydrocarbons (HAHs) are
produced for use as organic solvents , degreasing agents, pesticides,
intermediates for the
synthesis of various other organic compounds and as ingredients in the
manufacture of
plastics. The extensive use of these halogenated compounds in industrial
processes creates
a substantial opportunity for new technologies capable of upgrading and/or
recycling low-
value co-products.
Excess HAHs produced as co-products in chemical manufacturing process can be
burned to produce heat and, in some cases, be recycled to low value starting
materials, thus
yielding some recovery from a waste product or excess co-product. In complex
microbial
environments (nature, water treatment plants, etc.), HAH degradation occurs by
microbial
biodegradation. Biodegradation of HAHs results in the formation of carbon
dioxide, water,
and hydrochloric acid when the halogen is a chloride.
t 5 The biodegradation of HAHs to carbon dioxide, water, and hydrochloric acid
by select
microorganisms is disclosed in U.S. Patent Nos. 4.853.334 and 4,877,736. A
process for
the decomposition of chlorinated aliphatic hydrocarbons, without specifying
the
microorganism involved is described in U.S. Patent No. 4,749,491. In addition,
the aerobic
metabolism of trichforoethylene by Acinetobacter spp. has been reported by
Nelson et al.,
Appl. Environ. Microbiol., 52:383-384 (1986). An overview of the degradation
of halogenated
aliphatic compounds in the environment is given in Vogel et al., Environ. Sci.
Technol.,
21:722-736 (1987). U. S. Pat. No. 5,372,944 discloses a Rhodoccocus species
which
produces a dehalogenase which converts HAHs to haionydrins. However, these
references
largely rely on cellular systems and do not take advantage of the benefits
that may be
obtained from the use of an immobilized, activity-modified enzyme in a
continuous feed
process. Most relevantly, U.S. Patent No. 5,372,944 relies on Rhodococcus
cultures
comprising wild type or mutant cells. However, the mutation techniques taught
therein do
not take advantage of recombinant DNA methods and so fail to capitalize on the
benefits
these methods offer in terms of improvement in activity and expression of the
dehalogenase
enzyme.
Rather than depend on biodegradation of HAHs by cell cultures, it would be
advantageous to have an improved, recombinant enzyme that can be readily
adapted to
continuous-feed methods whereby the HAHs could be efficiently converted to
valuable
1
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
intermediates for use in production of other useful products, such as chemical
intermediates
in the preparation of polyethers to form polyurethanes or in the preparation
of glycols and
polyglycols to form lubricants, surfactants, emulsifiers, etc.
The present invention is directed to a recombinant enzyme, capable of
converting
HAHs to vicinal halohydrins, comprising an amino acid sequence substantially
homologous
with the amino acid sequence of residues 1-292 of Figure 2. Another object of
the invention
is to provide DNA sequences encoding a polypeptide comprising such an enzyme,
more
specifically to DNA sequences comprising a polynucleotide substantially
homologous with
the nucleotide sequence of bases 37-912 of Figure 2.
to Another object of the invention is to provide a vector containing the DNA
sequences)
and a method for producing the polypeptide comprising placing the vector into
a host cell
and growing the host cell under conditions allowing the transformant to
produce the
dehalogenase.
Further objects of the present invention are to provide an immobilized form of
the
5 enzyme on a solid support as well as a process for converting a HAH to an
alcohol or
halohydrin comprising contacting the HAH with the immobilized enzyme.
Brief Descrption of Drawincts
Figure 1 illustrates a plasmid map of the vector pEXPROK. Plasmid pEXPROK is
2o derived from the commercially available pPROK-1 plasmid (Clontech, Mountain
View, CA)
containing the Ptac promoter and the 5S, T1 T2 terminator sequences. In the
figure, the
T1T2 region is indicator by "Term." This plasmid was generated by replacing
the pPROK-1
multiple cloning site with a pair of oligonucleotides which introduced
restriction site Nco I,
Hind III, Xho I, Nhe I, and Not I into the linker. The "ATG" sequence of the
Nco I site
25 represents a functional in-frame start site. The Nhe I site is followed by
the EXFLAG linker
sequence. The sequence of the EXFLAG linker corresponds to nucleotides 919-975
in
Figure 2 and encodes amino acids 295-315 in the RDhI protein sequence shown in
Figure 2.
Figure 2 (i.e. Figures 2A and 2B) presents the nucleotide sequence encoding
the
putative Rhodococcus rhodochrous TDTM003 haloalkane dehalogenase enzyme and
the
3o amino acid sequence derived from this nucleotide sequence. Amino acid
residues 1-292
correspond to the Rhodococcus dehalogenase (RDhI) structural gene and are
encoded by
nucleotides 37-912. Amino acid residues -12 through -1 (nucleotides 1-36)
represent a
polyhistidine-containing amino-terminal tail, with residues -12 and -11
participating in the
2
r _.
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
formation of both the translational start site and the Nco I cloning site.
Amino residues 293-
294 (nucleotides 913-918) are encoded by the Nhe I cloning site and are
followed by amino
acids 295-305, which are referred to herein as the EXFLAG peptide. The EXFLAG
linker
(nucleotides 919-975) encodes the EXFLAG peptide and a dual-translational stop
site (each
indicated by an asterisk).
Figure 3 illustrates a plasmid map of the vector pEXPROK-RDhI.
Figure 4 (i.e. Figures 4A and 4B) presents an alignment comparison chart of
the
amino acid sequences of the putative Rhodococcus rhodochrous TDTM003
haloalkane
dehalogenase, the Xanthobacter autotrophicus GJ10 dehalogenase, the Renilla
reniformis
~c~ luciferin monooxygenase, and the Pseudomonas spp. LinB gene product (a
tetrachloro-
cyclohexadiene hydrolase).
Figure 5 presents a plasmid map of the vector pRDhl-K02.3-EXPROK comprising
the putative Rhodococcus rhodochrous TDTM003 haloalkane dehalogenase gene
under the
control of the IPTG-inducible Ptac transcription promoter.
Figure 6 illustrates a plasmid map of the high level expression vector pRSET-
RDhI
comprising the putative Rhodococcus rhodochrous TDTM003 haloalkane
dehalogenase
gene under the control of the T7 transcription promoter.
Figure 7 illustrates a plasmid map of the high level expression vector pTrcHis-
RDhI
comprising the putative Rhodococcus rhodochrous TDTM003 haloalkane
dehalogenase
2o gene under the control of the trc transcription promoter.
Figure 8 illustrates a plasmid map of the high level expression vector pTrxFus-
RDhI
comprising a modified version of the putative Rhodococcus rhodochrous TDTM003
haloalkane dehalogenase gene fused to the gene encoding E. coli thioredoxin,
the combined
fusion gene being under the control of the P~ transcription promoter.
Figure 9 presents an image of an SDS-PAGE gel of cell lysate samples from
cells
expressing the pEXPROK-RDhI clone, compared to the partially purified rRDhl
enzyme.
Figure 10 presents an image of an anti-FLAG antibody immunoblot of an SDS-PAGE
gel identical to that of Figure 9.
Figure 11 presents an image of an SDS-PAGE gel of cell-free extracts from
cells
3o expressing pRSET-RDhI.
Figure 12 presents an image of an anti-FLAG antibody immunoblot of an SDS-PAGE
gel identical to that of Figure 11.
3
CA 02281931 1999-08-12
WO 98/36080 PCT/IJS98/02776
Figure 13 presents an image of an SDS-PAGE gel of cell-free extracts from
cells
expressing pTrcHis-RDhI.
Figure 14 presents an image of an anti-FLAG antibody immunoblot of an SDS-PAGE
gel identical to that of Figure 13.
Figure 15 presents an image of an SDS-PAGE gel of cell-free extracts from
cells
expressing pTrxFus-RDhI.
Figure 16 presents a productivity profile for an immobilized enzyme bioreactor
acting
on the substrate, 1,2,3-Trichloropropane.
Figure 17 presents a bar chart of the activities of EPPCR-mutated Rhodococcus
o rhodochrous haloalkane dehalogenases.
Figure 18 presents a bar chart of the activities of EPPCR-mutated Rhodococcus
rhodochrous haloalkane dehalogenases.
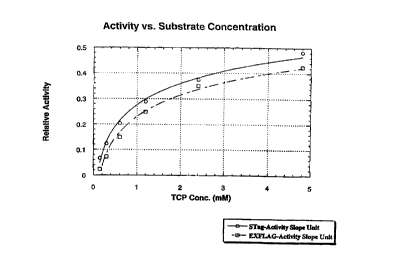
Figure 19 presents a graph of enzyme activity data for an RDhI enzyme bearing
a
carboxy-terminal S-Tag polypeptide tail and for an RDhI enzyme bearing a
carboxy-terminal
i ~ EXFLAG polypeptide tail.
The present invention results from intensive research into obtaining a DNA
sequence
encoding a polypeptide having haloaliphatic dehalogenase activity from a
microorganism
belonging to the genus Rhodococcus, making recombinant DNA sequences by
integrating
the DNA sequence per se - or as modified - into a vector, and transforming a
2o microorganism with the recombinant vector. Transformants were screened for
dehaiogenase activity levels and from those with heightened activity, the
dehalogenase
enzymes were isolated. Various solid support immobilization systems were then
evaluated
to identify enzyme-support combinations in which the enzyme could effectively
convert
halogenated aliphatic hydrocarbons to alcohols or halohydrins.
25 Halogenated aliphatic hydrocarbons (HAHs) subject to conversion using the
immobilized dehalogenase include C2-C,o aliphatic hydrocarbon molecules and
groups which
have two or more halogen atoms attached, wherein at least two of the halogens
are on
adjacent carbon atoms. Preferred HAHs are saturated hydrocarbons in which at
least one of
the halogens occupies a primary position on the molecule or group; more
preferred are
3o those in which no more than 1 halogen occupies the same carbon atom.
Especially
preferred HAHs are saturated hydrocarbons comprising 1,2-dihalo groups,
examples of
which are the 1,2-dihaloethane, 1,2-dihalopropane, 1,2-dihalobutane, and 1,2,3-
4
r
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
trihalopropane molecules and groups. These classes include, for example, 1,2-
dichloroethane, 1,2-dichloropropane, 1,2,-dichlorobutane, 1,2,3-
trichloropropane, and 1,2-
dibromo-3-chloropropane molecules and groups.
As used herein, the term "halogen" means chlorine, bromine, or iodine. The
preferred halogens are bromine and chlorine. The most preferred halogen is
chlorine and
among the most preferred HAHs are volatile chlorinated aliphatic hydrocarbon
(VCAH)
molecules and groups; especially preferred VCAHs include 1,2-dichloropropane
and 1,2,3-
trichioropropane molecules and groups.
As used herein, the term "halohydrin" means a vicinal halohydrin, i.e. any
aliphatic
io organic compound, other than a carboxylic acid, which contains both a
hydroxyl substituent
and a halogen substituent on adjacent carbon atoms of the molecule. a,~3-
halohydrins are
the most preferred vicinal halohydrins.
The terms "immunoblot" and "immunoblotting" are used herein to denote the
process
of: 1 ) transferring proteins) from an electrophoresis gel, e.g., a
polyacrylamide gel for use in
~ 5 PAGE, to a protein-binding membrane; and then 2) probing that membrane
with an antibody
specific to protein constituents that may be included among those transferred
to the
membrane; and then 3) determining the location of that antibody using any of
various
chromogenic methods well known in the art, e.g., developing color in a
colorable marker
which is directly or indirectly linked to the antibody. An example of an
immunoblotting
2o method is the Western blot.
The terms "permeablize," "permeablizing," and "permeablization" are used
herein to
denote the process of making something permeable, e.g., to make cell walls
permeable.
The term "sonicate" is used herein to denote the use of sonic waves to rapidly
vibrate the
contents of a test tube or other container, in order to thoroughly mix them.
The term "vortex"
25 is used herein to denote the action of mechanically gyrating a test tube
along its bottom
while manually holding the top of the test tube stationary, in order to mix
its contents.
The word "selectable" as used herein, means "able to be selected." For
example, the
phrase "selectable marker" or "dominant selectable marker" indicates a genetic
feature, such
as a gene encoding an antibiotic resistance enzyme, whose presence allows the
gene's host
3o cell to multiply in a corresponding selection medium, e.g., a growth medium
containing that
antibiotic. When such a genetic feature is incorporated into a plasmid
containing a gene
encoding a RDhI enzyme, and cells are then treated to receive the plasmid,
growing the cells
in a selection medium allows the cells actually receiving the plasmid to grow
selectively, in
5
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
contrast to those cells which did not receive or retain the plasmid. This
permits the ready
identification of cells which contain the RDhI gene.
As used herein, the phrase "expression construct" denotes a plasmid, virus,
virion,
viroid, transposable element, cos-construct, transfectable carrier-associated
DNA strand
(e.g., a DNA-coated "gene-gun" pellet or DNA-coated natural or synthetic
histone-like
particle), or other DNA-to-cell delivery system which is known in the art.
As used herein, in the context of describing amino acid sequences, the
following
single letter designations apply.
A, a Alanine (Ala) M, m Methionine (Met)
to C, c Cysteine (Cys) N, n Asparagine (Asn)
D, d Aspartic Acid (Asp)P, p Proline (Pro)
E, a Glutamic Acid (Glu)Q, q Glutamine (Gln)
F, f Phenylalanine (Phe)R, r Arginine (Arg)
G, g Glycine (Gly) S, s Serine (Ser)
is H, h Histidine (His) T, t Threonme (Thr)
I, i Isoleucine (Ile) V, v Vafine (Val)
K, k Lysine (Lys) W, w Tryptophan (Trp)
L, I Leucine (Leu) Y, y Tyrosine (Tyr)
2o As used herein, in the context of describing DNA sequences, the following
single
letter designations apply:
A Adenine G Guanine N A, C, G, or T
C Cytosine T Thymine R A or G
Y C or T
25 ,
The following
abbreviations
and definitions
are used herein:
@ At, e.g., @37C is "at 37C" and @60min. is "at
60 minutes"
A Angstroms (one angstrom is 1x10-' meters)
A Absorbance, e.g., A28 is "absorbance measured
at 280nm"
3o as Amino acid
Amp Ampicillin
2-AMP 2-Aminopropanol
AMPSO 3-[(1,1-dimethyl-2-hydroxyethyl)amino]-2-hydroxy-propanesulfonic
acid
35 ATCC American Type Culture Collection (Rockville,
MD, USA)
base A nucleotide which is part of a poiynucleotide
by Base pairs
CAPSO 3-(cyclohexylamino}-2-hydroxy-1-propanesulfonic
acid
CD Compact disc
4o CHES 2-(N-cyclohexylamino)ethanesulfonic acid
CM Carboxymethyl
CnBr Cyanogen bromide
O Change or difference, e.g., OA is "change in
absorbance"
dATP Deoxyadenosine triphosphate
45 DCB 1,4-Dichlorobutane
6
~ ~ .. _
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
DCH 2,3-Dichloro-1-propanol
dCTP Deoxycytidine triphosphate
DEAE Diethylaminoethyl
dGTP Deoxyguanosine triphosphate
dTTP Deoxythymidine triphosphate
EDTA Ethylenediamine tetraacetic acid or ethylenediamine
tetraacetate
EPPCR Error-prone polymerase chain reaction
GC Gas chromatography
GIA Glutaraldehyde
to gm Grams
hr Hours
Hz Hertz (a measure of frequency in units of cycles
per second)
ID Internal diameter
Ig Immunoglobulin, e.g., IgG is "immunoglobulin G"
is IPTG Isopropylthiogalactopyranoside
IUB International Union of Biochemistry
kbp Kiio-base pairs
kD Kilo-Daltons (one Dalton weighs '/12 of a ''O
atom)
K Inhibition constant
LB Luria broth
gg Micrograms
~L Microliters
~M Micromolar
mole Micromoles
~5 M Molar (moles of solute per liter of solution)
mg Milligrams
min. Minutes
mL Milliliters
mm Millimeters
3o mM Millimolar
MW Molecular weight
N Normal (moles of chemically active solute groups
per liter of solution
,
e.g., HZSO, has two acid hydrogens and so 1 M
H~S04 is a 2N solution)
n m Nanometers
35 ng Nanograms
NP-40 Nonoxynol; p-(n-C9H,9)-C6H,-(OCHZCH
)
OH; also called
z
~
nonylphenoxypolyethoxyethanol (a non-ionic detergent
surfactant)
OD Optical density, e.g., ODsoo "optical density
measured at 600nm"
oligo Oligonucleotide
4o p_ Plasmid, e.g., pRSET, pTrcHis, pTrxFus, or pUC
PAGE Polyacrylamide gel electrophoresis
PCR Poiymerase chain reaction
PEI Polyethyleneimine
pfu Plaque forming units
45 phage Bacteriophage
QAE Quaternized ethyl ammonium (an anion exchange
group)
RDhI Rhodococcus hafoalkane dehalogenase enzyme
residue An amino acid which is part of a poiypeptide
rpm Rotations per minute
5o rRDhl Recombinant Rhodococcus haloalkane dehalogenase
enzyme
SDS Sodium dodecyl sulfate
spp. Species
7
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
TCP 1,2,3-Trichloropropane
TM Trademark
Tris Tris(hydroxymethyl)aminomethane
tRNA Transfer RNA
U Units
Vmax Maximum enzymatic velocity
w/v Percent by weight per volume, i.e. number of grams of solute
per
100 mL of solution, also written as "% (w/v)"
to % w/w Percent by weight per weight, i.e. number of grams of a substance per
100 grams of a mixture containing that substance; also written as
"% (w/w)"
Approximately
~5 The following steps were carried out in the hope of obtaining an enzyme,
and an
immobilized enzyme, meeting the objectives of the present invention. These
steps were
performed using techniques known to those skilled in the art:
( 1 ) isolation and partial determination of the amino acid sequence of a
dehalogenase
enzyme;
20 ('' ) construction of oligonucleotide probes based on the partial sequence
determination;
(3> isolation of a dehalogenase-encoding DNA fragment by use of the
oligonucleotide
probes, followed by amplification the DNA;
(4) ligation of the fragment into a cloning vector having a suitable origin of
replication and a
gene encoding a dominant selectable marker;
2s (5) transformation and selection of a microorganism containing the
recombinant plasmid;
(6) transference of the DNA sequence to a suitable expression vector and using
this
recombinant vector to transform a host cell;
(7) production of the recombinant dehalogenase by the transformant; and
(8) purification of the dehalogenase; followed by
30 (9) immobilization of the dehalogenase onto a variety of splid supports;
(10) use of the immobilized dehalogenase in a process for conversion of HAHs
to alcohols or
halohydrins; and
( 11 ) selection of effective dehalogenase support systems.
Surprisingly, in the process of performing the above-outlined studies, novel
35 recombinant dehalogenase enzymes were obtained that have performance
characteristics
8
t ~
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
superior to those of the wild-type enzyme from which the recombinant enzymes
were
derived. fn addition, effective immobilized dehalogenase support systems were
identified.
The dehalogenase for use in the present invention is preferably derived from
Rhodococcus species ATCC 55388 and is capable of converting a HAH to a
halohydrin or
alcohol, preferably a halohydrin. The preferred recombinant enzyme comprises
an
enzymatically active polypeptide comprising the minimal functional portion of
the wild type
dehalogenase enzyme, i.e. the smallest possible segment thereof which, after
proper
folding, retains haloalkane dehalogenase activity. Preferably, this
polypeptide is
substantially homologous with the amino acid sequence of residues 1-292 of
Figure 2. More
o preferably, this polypeptide is at least about 90% homologous, even more
preferably at least
about 95% homologous, and yet more preferably at least about 99°o
homologous therewith.
Especially preferred are enzymatically active polypeptides having the amino
acid sequence
of residues 1-292 or of residues of Figure 2.
The preferred recombinant enzyme may also comprise one or more other units
such
~ 5 as labels, tags, tails, linkers, solid supports, chelants, other enzymes,
and so forth -
regardless of their size - which may either be produced with or linked to the
enzymatically
active polypeptide of the enzyme after it is formed. Such units may be excised
from the
enzyme after it has been properly folded and/or immobilized upon a solid
support. In a
preferred embodiment, the enzyme is produced with or linked to a substantially
hydrophilic
2o tail. This tail may be a hydrophilic oligopeptide expressed as part of the
enzyme or may be,
e.g., an oligosaccharide moiety attached by the host cell to the core enzyme
after expression
thereof. The tail must be of sufficient length and hydrophilicity as to allow
the core enzyme
to remain in suspension in an aqueous medium. A preferred tail is a
substantially hydrophilic
oligopeptide expressed as part of the enzyme. More preferably, the enzyme is
expressed
z5 with a highly hydrophilic oligopeptide tail. Most preferably, the
oligopeptide tail is expressed
at the carboxy terminus of the enzyme. A most preferred oligopeptide tail is a
hydrophilic,
carboxy-terminal tail which is rich in histidine and/or aspartic acid
residues, especially one
which is from about 5 to about 25 amino acids in length and contains at least
about 25%
histidine or aspartic acid residues, more preferably at least about 50% of
such residues.
3o The recombinant enzyme is preferably produced by a host cell containing at
least a section
of a polynucleotide having the nucleotide sequence of bases 37-912 of Figure
2.
The present invention is also directed to recombinant DNA sequences capable of
expressing the enzymes of the present invention. These DNA sequences include
those able
to express the novel haloalkane dehalogenase(s) by means of translation
systems not
9
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
following, or not fully following, the standard DNA code's codon-to-amino acid
correspondence pattern. Such systems include those in which certain codons are
"suppressed" relative to the standard DNA code. In one type of a "suppressed"
expression
system, at least one of the 20 or so amino-acid-specific classes of aminoacyl-
tRNA ("aa-
tRNA") molecules contains at least one tRNA molecule - having an anticodon
belonging to
that class - which is linked to the "wrong" amino acid, so as to predispose
the translation
system to produce a "violation" of the standard DNA code (i.e. by causing the
insertion, in at
least one position in the growing polypeptide chain, of an amino acid not
normally found in
correspondence with the mRNA codon governing that position). In another
variation on such
io a system, the pool of amino-acyl-tRNA molecules contains an aa-tRNA whose
anticodon is
complementary to an mRNA codon normally signaling initiation or termination of
translation,
thus suppressing the signal. These systems may exist, e.g., as a result of
mutations in one
or more tRNA molecules or aa-tRNA synthetases, a result of mistakes by non-
mutated aa-
tRNA synthetase(s), or a result of human intervention in forcing the non-
standard Imkage of
an amino acid to a tRNA.
In such a translation system, a DNA sequence of the present invention will
still
produce the novel haloalkane dehalogenase(s) either because the insertions) of
the "wrong"
amino acid do not cause the enzyme to lack activity or because the DNA
sequence contains
- at the positions) where an "incorrect" amino acid would otherwise be
inserted - a codon
2o that "anticipates" the change in the translation system so as to allow
either the insertion of
the "correct" amino acid or the "correct" signaling of the mRNA codon therein.
A preferred
DNA sequence comprises a polynucleotide substantially homologous with the
nucleotide
sequence of bases 37-912 of Figure 2. More preferably, this polynucleotide is
at least about
90% homologous, even more preferably at least about 95% homologous, and yet
more
25 preferably at least about 99% homologous therewith. Especially preferred
are
polynucleotides having the amino acid sequence of bases 37-912 of Figure 2.
As used herein, the phrase "substantially homologous" expresses the degree of
similarity of a subject sequence - i.e. a subject nucleotide sequence (of an
oligo- or poly-
nucleotide or DNA strand) or a subject amino acid sequence (of an oligo- or
poly-peptide or
3o protein) - to a related, reference nucleotide or amino acid sequence. This
phrase is defined
as at least about 75% "correspondence" (i.e. the state of identical elements -
nucleotides or
amino acids - being situated in parallel) between the subject and reference
sequences when
those sequences are in "alignment." In this context, "alignment" is said to
exist when a
minimal number of "null" elements have been inserted in the subject and/or
reference
..._._..___.~.___ .. .
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
sequences so as to maximize the number of existing elements in correspondence
between
the sequences. "Null" elements are not part of the subject and reference
sequences; also,
the minimal number of "null" elements inserted in the subject sequence may
differ from the
minimal number inserted in the reference sequence. Increased degrees of
homology of a
given sequence, which may be expressed as, e.g., "90% homologous," are
likewise defined
with reference to their degree of sequence identicality to a reference
sequence.
In this definition, a reference sequence is considered "related" to a subject
sequence
where either: 1 ) both nucleotide sequences encode proteins or portions of
proteins which
may be identified to the same IUB subclass or 2) both amino acid sequences
make up
~c~ proteins or portions of proteins which may be identified to the same IUB
subclass, regardless
of whether such identification is based on functional properties, sequence
homology, or
parental origin. "Parental origin" refers to the fact that a given enzyme may
initially be
grouped within an IUB subclass because of its recognized major or minor
function(s), but
after the DNA sequence encoding that enzyme accumulates one or more
mutation(s), the
i ~ encoded enzyme may exhibit functional capacities of a different IUB
subclass - whether or
not the enzyme also retains its original functionality; the "different IUB
subclass" may fall
within the same or a different IUB main class. The reference to "portions of
proteins"
signifies that bi- and multi-functional enzymes - including fusion proteins -
are also
contemplated as falling within a given IUB subclass based on the
identification to that
20 subclass of one of their functional domains.
In a preferred embodiment of the present invention, the haloalkane
dehalogenase at
least parentally belongs to IUB sub-subclass 3.8.1. The enzymes of the present
invention
have been found to possess unexpectedly superior properties to those of the
wild-type
haloalkane dehalogenase enzyme found in Rhodococcus as, e.g., was utilized in
U.S. Patent
25 No. 5,372,944. Generally, aside from its stability under reaction
conditions, two
characteristics of a given enzyme will determine its usefulness in commercial
processes: its
affinity for product, as well as its affinity for substrate. Where an enzyme's
affinity for
product molecules is relatively high, it will be extremely sensitive to
feedback inhibition by the
product. Such an enzyme inrill be less useful in commercial processes in which
enzymes are
30 often required to operate in the presence of significant product
concentrations. A convenient
indicator of an enzyme's relative affinity for product is its inhibition
constant measured at
90% inhibition ("K;(90}"), i.e. the product concentration at which the enzyme
retains only 10%
of its Vmax, the Vmax being measured when the concentration of product is 0.
In regard to
the present invention, whereas the wild type haloalkane dehalogenase has a
measured
11
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
K;(90) of 20 mM, the recombinant enzyme of the present invention (see Figure
2) has a
measured K;(90) of 50 mM. IM other words, the recombinant enzyme is much less
sensitive
to feedback inhibition by product and can therefore operate in the presence of
product
concentrations that would essentially shut off the wild type enzyme
altogether.
The enzyme of the present invention may be expressed alone, or covalently
attached, along its amino and/or carboxy terminus, to one or more polypeptide
tail(s). Such
tails may be encoded by exons separate from the enzyme-encoding exon or by DNA
sequences which are part of the enzyme-encoding exon. When the tail-encoding
DNA is to
be part of the enzyme-encoding exon, the tail-encoding DNA may be attached or
"fused" to
io the 3' and/or 5' end of the enzyme gene, e.g., either: 1 ) during enzyme
gene amplification
by including the tail-encoding nucleotide sequence in an oligonucleotide
primer or 2) during
plasmid construction by ligating the tail-encoding DNA directly into a plasmid
which contains
the enzyme gene (whether the enzyme gene is inserted into the plasmid before
or after
insertion of the tail-encoding DNA).
ns Under the influence of the appropriate genetic control elements - i.e.
enhancers,
promoters, transcription and translation start and stop sequences, and so
forth - expression
of such DNA (or mRNA) fusion genes results in production of dehalogenase
enzymes with
polypeptide tails on one or both ends. An example of a preferred tail-free
enzyme is that
having the amino acid sequence of residues 1-292 of Figure 2. Examples of some
preferred
?o polypeptide tails include poly-histidine sequences, polyacid (e.g., poly-
aspartic and/or -
glutamic'acid) sequences, cellulose binding domains, and the c-myc, S-Tag, and
FLAG
peptides. Antibodies and affinity columns that bind these exemplary tails are
commercially
available and may be readily used to purify or immobilize the expressed fusion
proteins.
However, many other tails may be used while retaining a functional
dehalogenase enzyme.
25 Whether or not a tail-encoding sequence is included in the expressed gene,
the gene must
include, in a position outside the enzyme gene or the enzyme-tail fusion gene,
a translation
start site, preferably ATG, and will also preferably include an endonuclease
restriction site.
In one preferred embodiment, the open reading frame of a single exon encodes a
functional dehalogenase enzyme having tails of up to about 30 amino acid
residues on the
3o amino and/or carboxy termini. In this embodiment, when both termini have
tails, the tails
may be of approximately equal length. In another preferred embodiment, the
enzyme is
expressed with both an amino and a carboxy terminal tail, but the carboxy
terminal tail is
significantly longer than the amino terminal one. In this embodiment,
preferably the amino-
terminal tail is up to about 25 amino acids in length and the carboxy-terminal
tail is about 2 to
12
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
about 150 amino acids in length. In any of these embodiments, preferably, the
amino-
and/or carboxy-terminal tail will contain a stretch of at least 5 adjacent
histidine residues. In
an alternate embodiment, the amino terminal tail is about 10-150 amino acids
in length and
preferably contains or is itself a poly-histidine sequence. In this
embodiment, the enzyme
s may be reversibly immobilized or reversibly inactivated by contact with a
surface coated with
chelated divalent metal ions, e.g., Mgz' or Ni7'. In this embodiment, the poly-
histidine-
containing amino-terminal tail may be so long as to partially or totally block
access to the
enzyme's active site. In an alternate version of this embodiment, the tail may
be designed to
contain one or more amino acid residues which change the configuration of the
tail from that
io found in a poly-histidine sequence to a bent, recurved, or flexible-joint
configuration allowing
increased access to the active site of the enzyme.
In a more preferred embodiment, the open reading frame encodes a functional
dehalogenase enzyme with an amino terminal tail of about 1 to about 25 amino
acids and a
carboxy-terminal extension having a polyhistidine sequence, a FLAG peptide
sequence
IS (available from KODAK Imaging Systems/VWR, Rochester, NY) andlor an S-Tag
peptide
sequence. In an especially preferred embodiment, the open reading frame
encodes a
functional dehalogenase enzyme having: 1 ) an amino-terminal tail of up to
about 10 amino
acids and a polyhistidine sequence and 2) a carboxy-terminal tail comprising
(i.e. containing)
the FLAG (see Figure 2) or S-Tag peptide sequence.
2o The enzymes and/or tails of the above-described dehalogenase enzymes may be
modified by use of the techniques of directed evolution, in order to improve
their productivity,
stability, and/or inhibition profiles. One directed evolution technique uses
the gene shuffling
method disclosed in U.S. Patent No. 5,605,793 to Stemmer et al., in which a
number of
similar DNA sequences are fragmented and reassembled in a random fashion to
generate
25 highly diverse libraries which can be screened for enzymes with the
attributes of interest.
Another version of this technology involves use of error-prone gene
amplification
technologies. A third version of directed evolution employs a combination of
these two
methodologies. A fourth version of directed evolution is the.so-called
"staggered extension"
process as disclosed in the publication by Zhao et al., in Nature
Biotechnology (1998)
30 (currently in press). In a preferred embodiment, error-prone gene
amplification is used to
introduce semi-random mutations into the dehalogenase gene (e.g., Figure 2,
residues 1-
292) at a rate of about 1-6 point mutations per gene copy per gene
amplification reaction,
following which the mutant library is introduced into bacteria, induced to
express protein, and
13
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
screened for activity, preferably in a spatially addressable grid format (such
as a 96 well or a
384 well plate}.
Effective use of directed evolution to improve an enzyme or enzyme family
requires
an optimized mutagenesis strategy as well as an expression system and a
screening
s strategy and screening conditions which effectively detect the desired
performance attributes
of the enzyme. For (non-random) primer-dependent mutagenesis methods (e.g.,
error-
prone gene amplification and defined primer-based recombination), specific
protein
subdomains can be easily targeted for mutagenesis by primer design and
positioning. In a
preferred embodiment, primers are used which allow mutagenesis of the entire
transcription
~c~ and translation domain as it occurs within the expression construct.
Preferably, primers are
directed exclusively to the protein coding region of the expression construct
or target DNA
(including tails). In a more preferred embodiment, primers are designed in
such a way as to
target mutagenesis to the dehalogenase enzyme gene while preserving the
sequence of the
tails. For example. in relation to Figure 2, the dehalogenase enzyme gene may
be the sole
mutagenesis target when an error-prone gene amplification technique employs
both a primer
complementary to nucleotides closely preceding nucleotide 36 and a primer
complementary
to nucleotides closely following nucleotide 912. Likewise, the entire Figure 2
coding region is
the mutagenesis target when the primers anneal outside of the region of
nucleotides 1-951;
the Figure 2 amino tail or carboxy tail, respectively, is targeted when the
primers anneal
20 outside of the region of nucleotides 1-36 or 913-951.
The DNA sequences) encoding the enzyme or fusion protein of the present
invention
will preferably be inserted into an expression vector, followed by
transfection of the vector
into a host cell, and growth of the host cell under conditions in which it
expresses the
enzyme. A wide variety of recombinant host-vector expression systems for
prokaryotic cells
2s are known and may be used in the invention. For example, commercially
available vectors
such as pKK233-2, pKK388-1, pSE380, pTrcHis (A, B, and C), pRSET (A, B, and
C),
pProEX-1, and bacteriophages Lambda (gtll), T3, and T7 are all capable of
directing
expression of heterologous proteins in Escherichia coli and other gram-
negative prokaryotes.
In these expression formats, a variety of strain-appropriate inducibfe
promoters can also be
3o used. In addition, other prokaryotes (such as those of the genus Bacillus,
Pseudomonas,
Actinomyces, Bacillus, or Rhodococcus), eukaryotic microorganisms (such as
yeast and
fungi, e.g., those of the genus Pichia, Saccharomyces, or Aspergillus, e.g.,
Pichia pastoris or
Saccharomyces cerevisiae), other eukaryotic cells and cell lines (such as Sf21
cells infected
with baculouvirus-derived vectors), and even algal cells are capable of
producing, in active
14
fi
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
form, heterologous proteins of prokaryotic origin; in the event these other
cells are utilized in
the present invention, appropriate expression vectors would be selected for
use therewith.
Whereas numerous prokaryotic expression vectors are available publicly and may
be used in
the present invention, expression of the novel enzymes is exemplified herein
with the use of
commercially available vectors from the pTrcHis, pRSET and pTrxFus series
(available from
Invitrogen of San Diego, CA, USA) in conjunction with E. coli host cells.
When a directed evolution technique, such as error-prone gene amplification
(e.g.,
error-prone PCR or "EPPCR), is employed, the DNA of the mutant gene pool
produced
thereby is digested with appropriate restriction enzymes (i.e. those
endonucleases having
m restriction sites located external to the mutagenesis target); next, the
mutant genes are
purified and ligated into prokaryotic expression vectors to form a plasmid
library. Competent
host cells, e.g., preferably E. coli cells, are then transformed with the
plasmid library and
grown in a suitable medium: in the case of E. coli, the cells are plated on
agar containing a
selective growth medium. The cells may then be diluted to form indwidual
clones, or m the
i s case of prokaryotes such as E. coli, they may undergo an initial growth
phase, after which
the cell colonies are picked individually and transferred to separate
containers, e.g., the wells
of a 96 well plate, such that each well contains an individual clone of
transformed cells.
From this library of clones, individual clones can be expanded, induced to
express the
protein of interest, and screened for the activity of interest.
2o Screening for the haloafiphatic dehalogenase activity of the novel enzymes
is
preferably accomplished by detecting the protons or the halide ions released
upon hydrolysis
of a carbon-halogen covalent bond in a substrate molecule. In a preferred
embodiment, the
pH change accompanying the proton release serves as a measure of enzyme
activity; this
pH change is preferably determined using a fluorescent or visible pH indicator
which
25 undergoes measurable color change over the functional pH range of the
target enzyme. In
an alternate method, multiple parallel pH probes may be utilized.
In the activity screening assay, the assayed mixture will contain: 1 ) whole
cells,
permeablized cells, cell lysate, or purified enzymes obtained from cells
expressing a mutant
dehalogenase, preferably from bacterial cells; 2) a substrate; and 3) a low
concentration of
3o buffer (typically < 10 mM). When use of permeablized cells is desired, a
chemical detergent
(e.g., sodium deoxycholate) or a physical freeze/thaw process may be used to
make
bacterial cells permeable. The substrate will preferably comprise one or more
halogenated
aliphatic hydrocarbons as discussed above. The buffer may be selected from any
known to
be effective or found to be effective over the pH range in which the enzyme
retains activity.
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
In some cases, the cell debris itself will be seen to provide sufficient
buffering capacity to
allow accurate quantitation of activity. Where an added buffer is used, it
will preferably have
a pKa in the range of about 6 to about 10, although other buffers may be used.
Examples of
preferred buffers include glycine, 2-AMP, CAPSO, ethanolamine, CHES, borate,
serine, and
AMPSO; especially preferred is CAPSO and even more preferred is a
concentration of about
5mM CAPSO.
The activity screening assay will also require the use of a detection method.
In a
preferred embodiment, a pH change is detected. Preferably, a pH indicator will
be included
in the assayed mixture. Any pH indicator having a color change in the pH range
in which the
t o enzyme is active may be used. Preferably, the pH indicator will undergo a
color change in
the range of about pH6 to about pHlO, more preferably in the range of about
pH7 to about
pH9. Examples of preferred visible pH indicators include m-cresol purple,
cresol red, phenol
red, bromthymol blue, and thymol blue; examples of preferred fluorescent pH
indicators
include cx-naphthol sulfonic acid, 1,4-naphthol sulfonic acid, coumaric acid,
3.6-dioxyphthalic
dinitrile, and orcinaurine. in an alternative embodiment, a pH probe may be
utilized to detect
the pH change. Especially preferred is the use of the visible pH indicator, m-
cresol purple,
and even more preferred is a concentration of about 50pM m-cresol purple.
In another preferred embodiment, detection is accomplished by measuring the
release of halide ions from the substrate by: 1 ) including in the assayed
mixture a halide-
2o sensitive fluorescent dye, such as lucigenin (available from Molecular
Probes of Eugene,
OR, USA) - lucigenin is quenched upon contact with halide ions and so a
decrease in
fluorescence in measured therefrom; or 2) utilizing a halide ion responsive
probe device,
such as a halide-selective electrode.
In a third preferred embodiment, detection of enzyme activity is accomplished
using a
25 coupled enzyme system. For example, a coupled enzyme system may be used to
detect the
production of product molecules: dehalogenation of haloalkanes results in
generation of
alcohols, and many alcohols are substrates for one or more commercially
available alcohol
dehydrogenase enzymes (whose activity is measured by disappearance of NADH).
Detection of alcohols via coupling to the NADH requirement of the
dehydrogenases is well
3o known in the art.
The enzymes of the present invention may be immobilized onto one or more solid
support(s). Enzyme immobilization technologies are most conveniently
classified into
covalent and non-covalent methods. Covalent methods utilize reactive groups
present on
16
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
certain amino acid side-chains to bond to a polymeric or inorganic support
either directly or
by using a bifunctional cross-linking agent. The primary advantage of this
approach is the
robustness of the linkage. Non-covalent immobilization methods are more
numerous and
range from direct and indirect (e.g., chelate- or chelant-mediated) ionic,
adsorptive, or
bioaffinity support associations (e.g., biotin-avidin) to gel-entrapment or
microencapsulation.
The choice of a particular immobilization technology for a commercial enzyme
process is based on a combination of factors. Of primary importance are the
cost of the
support matrix and its biocompatible linking or coupling chemistries. Next are
the recovery
of activity upon immobilization and the robustness of the immobilized support
under reaction
to conditions. Unfortunately, since each enzyme is unique, approaches to
finding the best
system are empirical. However, in conjunction with the enzymes of the present
invention, a
preferred method of immobilization involves covalently linking the enzyme to
the support by
means of reactive groups such as epoxides, activated nucleophiles, isourea,
and so forth.
These reactive groups may be present on the native surface of the support
material or the
15 support material may be modified to bear linkers containing such groups.
Preferred linkers
include those comprising at least one of: dialdehyde, diacid, diamino,
diisocyanate, cyanate,
and diimide groups: linkers comprising at least one carbodiimide group may
also be used,
provided that a diamino group is not used in conjunction with a carbodiimide.
Among the
preferred solid supports are alumina-based supports and silica-based supports;
more
2o preferred are polyethyleneimine-impregnated alumina- or silica-based
supports. A preferred
method of immobilization comprises pre-treating the solid support with
glutaraldehyde and
then contacting the support with the enzyme.
Once immobilized, the enzyme may be conveniently used to convert its
substrate/reactant into product. This conversion can be performed in any
suitable medium
25 which does not substantially affect the activity of the dehalogenase.
Preferably the
enzymatic conversion is done in a aqueous medium containing either a buffering
system or
one or more pH-control devices.
The halogenated hydrocarbon substrate is generally added to a reaction medium
to
the saturation point of the substrate, though in some cases, supersaturated
substrate
3o mixtures, substrate emulsions, or pure substrate preparations may also be
used. Given the
saturation point of most halogenated hydrocarbon substrates, the concentration
of
halogenated hydrocarbon used will generally range from about 0.005% to about
0.5% (w1v).
Preferably, the concentration of the halogenated hydrocarbon is from about
0.005% to about
0.25%. More preferred is a concentration of halogenated hydrocarbon from about
0.005% to
17
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
about 0.2% in medium. The substrate may be added to the reaction solution
initially, as in a
batch method, or be added into the liquid stream of a continuous feed process.
In such
continuous feed processes, the liquid stream may initially contain substrate
or the substrate
may be first added thereto as the stream is en route to the reactor. In either
case, more
substrate may be added directly to the liquid stream in the reactor in order
to ensure that a
high concentration of substrate is presented to the enzyme throughout the
reactor. The
liquid stream may be re-saturated with substrate at various intervals in the
process in order
to enable accumulation of product at concentrations higher than the solubility
limits of the
substrate. The batch method reaction is usually carried out with shaking or
stirring.
to Although the reaction time or reactor residence time may vary depending on
the reaction
conditions, such as the substrate concentration or the amount of enzyme, the
reaction
conditions are preferably selected so that the reaction is completed within a
maximum of 120
hours.
The invention will be further clarified by a consideration of the following
examples,
t 5 which are intended to be purely exemplary of the present invention. All
percents are percent
by weight unless otherwise indicated.
General Experimental
Materials and Media:
2o All oligonucleotides were synthesized and purified by Genosys
Biotechnologies Inc.
(Woodland, TX), Life Technologies, Inc. (Rockville, MD) or Integrated DNA
Technologies,
Inc. (Coralville, IA). Restriction enzymes and DNA modifying enzymes were
purchased from
Gibco-Bethesda Research Laboratories (Gaithersburg, MD), New England Biolab
Inc.
(Beverly, MA), or Stratagene Cloning Systems (La Jolla, CA) and were used
according to
25 manufacturer's protocols. Competent E. coli AG1 cells were purchased from
Stratagene
Cloning Systems, Competent E. coli JM109 cells and TOP 10F' cells were
purchased from
Invitrogen Corp. (San Diego, CA). Small scale plasmid DNA isolations were done
using the
Rapid Pure Miniprep (RPMT"') system (BIO 101, Inc., La Jolla, CA). DNA
ligations were
performed with pre-tested reagent kits purchased from Stratagene Cloning
Systems.
3o Purification of DNA fragments was with either QIAquick Gel Extraction Kits
and QIAquick
PCR Purification Kits both purchased from Qiagen Inc. (Chatsworth, CA). SDS-
polyacrylamide gels and associated buffers and stains, as well as electroblot
transfer
buffers, came from Integrated Separation System (ISS, Natick, MA). Antibodies,
anti-
18
r
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
FLAGT"~ monoclonal antibody M2, and goat anti-mouse IgG1 were obtained from
International Biotechnology Inc. (IBI, New Haven, CT) and Southern
Biotechnology
Associates (Birmingham, AL), respectively. Bacteria were cultured in Luria-
Broth ("LB")
using premixed reagents purchased from Gibco-Bethesda Research Laboratories (G-
BRL;
Gaithersburg, MD).
Reagents
1,4-Dichlorobutane, 60% perchloric acid, ferric nitrate, and mercuric
thiocyanate were
from Aldrich. Anhydrous ethanol was from Quantum/USI (Tuscola, IL, USA). 1,2,3-
o Trichloropropane was a gift from The Dow Chemical Company's Allylics Group
(Freeport,
TX, USA). Monobasic potassium phosphate, dibasic potassium phosphate,
imidazole,
guanidine hydrochloride, disodium EDTA, ammonium sulfate, and Tris free base
were Fisher
Biotech Grade. Sulfuric acid was from Fisher (ACS grade).
s Support Materials
The Tresyl-Toyopearl chromatography support was from TosoHaas (Lot #
65TRM72R). Sephadex G-25 prepackaged columns were from Pharmacia. Celite R-648
was from Manville. Polyethyleneimine, 50,000 MW and PEI-silica were from
Sigma.
Giutaraldehyde, Grade 1, as 25% aqueous solution, also from Sigma, was stored
at -20°C
2o until just prior to use. Other samples used in immobilization include:
Davison Low SA
Alumina, Norton SA 6176 Alumina, Calcicat Type C Alumina, Calcicat s-88-473
Type A
Silica, Shell 5980-F Silica, Davison 952-08-5X Silica, Borecker subunit
Carbon, and
AmCy 5701-Sn Carbon.
25 Methods:
PCR Reactions
DNA Amplification was performed using standard polymerase chain reaction
buffers
supplied by Perkin-Elmer-Cetus (Nutley, NJ). Typically, 50 pL reactions
include 1 x
concentration of manufacturer supplied buffer, 1.5 mM MgCl2, 125 pM dATP, 125
pM dCTP,
30 125 p.M dGTP, 125 ~M dTTP, 0.1-1.0 pM forward and reverse primers, 5U
AmpIiTaq DNA
Polymerase and <1 ng target DNA. Unless otherwise indicated, thermal profile
for
19
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
amplification of DNA is for 35 cycles of a thermal profile of 0.5 min.
Ca?94°C; 1 min. C~55°C; 1
min. C~72°C.
Protein Detection by Polyacylamide Gel Electroa~horesis
Soluble protein was mixed 1:1 with solubilization buffer (Tris/SDS/f3-
mercaptoethanol,
pH 6.8; ISS) and boiled for five minutes before being loaded on 10-20% gels
(Daiichi, Natick,
MA) and electrophoresed with Tris-glycine buffer (ISS). Gels were stained with
Pro-BIueT"~
(ISS).
Standard Chloride Detection Assay to Determine Units of Enzyme Activity
When using 1,4-dichlorobutane (Aldrich) as a substrate, 100 mM NaGlycinate pH
9
was added to each 9 mL capped vial to a final volume of 6 mL. When using 1,2,3-
trichloropropane as a substrate. 10 mM TrisSulfate/1 mM EDTA (pH 7.0) was
used. Six NL
substrate were then added and the contents were vortexed. Vials were incubated
at 30°C
for 1 hour with stirring. Sampling occurred at 5 time points by remomng 1 mL
of mixture and
placing it in an Eppendorf tube containing 100 pL 0.375 M Fe''(NO,), in 5.25 M
HCIO,.
i s Tubes were vortexed. When all samples had been collected, 100 NL
mercuric(II)
thiocyanate saturated in ethanol was added to each of the tubes. Once again,
samples were
vortexed, then centrifuged for 3 minutes. Optical densities were read at
460nm. Slopes
representing change in absorbance over time (DA/min) were determined and
divided by 1.52
(the extinction coefficient at 460nm using NaCI as standard in units of
DA/Nmole CI-) to give
2o Nmole CI-/min. One unit of enzyme activity is defined as the amount
required to
dehalogenate 1.0 Nmole of substrate/minute under the specified conditions.
Procedure for Error-Prone PCR Mutaaenesis
In this directed evolution procedure, an RDhI enzyme gene or RDhI fusion
protein
gene was provided as an EPPCR mutagenesis target, e.g., by using appropriate
restriction
25 enzymes to digest a plasmid containing the target DNA sequence. In most
cases, the target
DNA was purified by gel electrophoresis, followed by gel extraction of the
target DNA.
EPPCR involved performing a standard PCR gene amplification of the target
gene, using
appropriate oligonucleotide primers, except that the standard PCR buffer was
supplemented
with sufficient magnesium chloride and manganese chloride to bring the
reaction mixture to 7
3o mM magnesium chloride and 0.15 mM manganese chloride. This procedure may be
repeated upon one or more of the EPPCR products to introduce further mutations
therein.
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
The resulting EPPCR products were ligated into expression vectors (e.g.,
pTrcHis,
pTrxFus) and the vectors were then used to transform appropriate, competent
host cells,
e.g., E. coli AG1 or JM109 cells, for enzyme expression and enzyme activity
analysis.
Plasmid-containing clones were identified by selective growth on LB/Amp agar
plates.
Individual colonies were transferred by toothpick into the wells of a 96-well
plate containing a
selective growth medium and incubated at 37°C for -8-l2hr to allow for
growth. Following
the initial growth phase, replica plates were generated, expahded, and
individual clones
thereof were assayed for dehalogenase activity as described in the following
section.
Procedure for Measurina RDhI Enzyme Activity by Detection of pH Chanqe
o RDhI enzyme activity was measured by detecting the pH change resulting from
action of the enzyme in dehalogenating substrate. Prokaryotic host cells
expressing the
enzyme were grown in broth, quantitated, and permeablized prior to addition of
a pH
indicator, buffer, and substrate.
Each well of a 96-well microplate received 200pL of an SOB broth (obtained
from
Difco, Detroit, MI, USA) which had been supplemented with about 50-100ug/mL of
ampicillin
("SOB/Amp"). Cells from a single colony of enzyme-producing E. coli clone were
inoculated
into one well of the plate. When testing a library of rRDhl enzymes or rRDHL
fusion
proteins, each well was inoculated with cells from a different E. coli clone.
Six wells received
no cells, in order to serve as a negative control, and six additional wells
were inoculated with
2o an E. coli clone producing the wild-type RDhI enzyme, as a positive
control. The inoculates
were incubated overnight in a Psycrotherm oven at 37°C while being
shaken at 250 rpm.
After incubation, the cultures were induced by addition of IPTG to a final
concentration of 1 mM, followed by another 5 hours of incubation at
37°C in a Psycrotherm
oven with 150 rpm shaking. After the 5 hour incubation, the cell density of
each culture was
25 determined by use of a 1.573 Vmax/Kinetic Microplate Reader (Molecular
Devices,
Sunnyvale, CA, USA). 20pL aliquots of each of the induced cultures were then
transferred
the wells of a fresh 96-well plate and 2.2uL of pH8.0, 10x permeablization
buffer (10 mM
sodium deoxycholate, 1% NP-40, 50 mM Tris, and 50 mM EDTA) was added to each
aliquot, followed by shaking for 3-5 min. at moderate shaking speed. Each of
the cell culture
3o aliquots then received 200~.L of a DCB-saturated buffer (>1 ~,L DCB/mL
buffer system}, at pH
9.2-9.5, which contained 5mM CAPSO and 100pM cresol purple. The developing
color
change of the indicator was measured by use of a SpectraMaxPlus microplate
reader
21
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
(Molecular Devices, Sunnyvale, CA, USA) and the slope of the color change was
plotted to
extrapolate the initial enzyme activity.
Example 1
Isolation of dehaloaenase en~me from Rhodococcus.
Rhodococcus species ATCC 55388 was cultured as described in U.S. Patent No.
5,372,944. An enzyme extract was prepared from this culture as generally
described in U.S.
Patent No. 5,373,944 by taking a 25-75% ammonium sulfate cut, two ion exchange
chromatography steps (1. DEAE-Sephadex; 2. DEAE-Sephacryl) in which the salt
concentration was varied over the range of 0-400 millimolar sodium sulfate in
the form of a
tU gradient, gel filtration chromatography using Sephadex G-75, and then
concentration by
ultrafiltration to obtain an enzyme preparation containing greater than
65°o dehalogenase by
SDS-polyacrylamide analysis.
A portion (-25 mg) of the purified enzyme was subjected to cyanogen bromide
digestion. Peptide fragments were isolated using an RP-8 Macrosphere (Altech)
mixed mode
i s cation column with a 0-80% acetonitrile/water gradient containing 0.1 %
trifluoracetic acid.
Three purified protein and purified cyanogen bromide (CnBr) fragments were
subjected to sequencing by automated Edman degradation. The sequences of the N-
terminus and three CnBr fragments were determined. One of the CnBr fragments
was
identical to the N-terminus in sequence. The other two corresponded to unique
internal
2o dehalogenase sequences. Sequences of all the peptides are shown in Table 1.
Table 1: Sequences of N-terminal and Proteol~rtic Fragments Derived from
Purified
Rhodococcus Dehalogenase.
N Terminal Sequence:
SEIGT GFPFD PHYVE VLGER
Cvanog_en Bromide Fragment Seguences:
1. HYVDV GPRDG
2. DHYRE PFLKP VDRE
DNA Primer Desian
25 Primers RDhI 5.4 and RDhI 3.12 were designed to allow amplification and
cloning of
the open reading frame encoding the Rhodococcus dehalogenase (RDhI) gene in
expression
system pEXPROK. The sequence of RDhI 5.4 was derived from the N-terminal
sequence of
22
, _.
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
the protein whereas RDhI 3.12 was designed based on the actual DNA sequence.
Primers
RDhI 5.7 and RDhI 3.13 were designed to generate an RDhI gene in expression
system
pRSET and pTrcHis. Primers Trx2++ and Trx- were designed to generate an RDhI
gene in
expression system pTrxFus.
The sequences of these oligonucleotide primers are as follows:
Table 2: Seauences and Orientation of Oligonucleotide Primers Used in Cloning
of the
Rhodococcus Dehalogenase
Oltgo NameOrientationDesign Sequcnc:e'
hascd on
RDh 1 5 F=onsard Iv' ~ '~ ' GG'rT<.'C.'T,TG GGfJ': _' (
. 4 CT 1 CCf:T': 1 ~:-': i Gn t r"J
1 <'!';:: i. ( TC ) Tf.
termmaUhomology
RDh i 3 kevcrsc ?'.Scqucnce5 ' E~ m - ~A~~AC_~: A~~C ~n~ .
. i a lJat~ '~m;;:""" ",;':,~:~.
:iJt: ': Furw-ar.~ ~ ' cG: ACF.~AT';,:~,~..; Gw- .
5 . ~ . . ,.. ,.. , ...
Nc3c~ : Ncc : ~. ~ Y .. i
l.'AT CAT CT,T C;G'i A1'' .. ~ c~:.i..
F.... ,_'.' f,' .
FI H H
GGT T.. .:CC' TTC GA:' r'%'~ ..,.
TA
RJi:l 3.1~kcvCr.r ~ -cAT Gi,; AAr. TAi-. Tci, ~.;
~;~;.. i:,:c ..~ TT,c-
Na, C . Hl:,d : ; .
TrX,!++ 5'-CC GGG Gi;" CCC F.:,-; GCT :'C'1'
GAT. T,I'A i;C~T ACC GGT
BamH I Nco I
TTT CCC TTC GAC CC': CAT TA-3'
TrX- 5'-TCG ACT GC.'A GGC GGC CGC TCA
T'I'P. TTT GTC ATC-3'
Pst I Not I
acv=al~m~; w=r~., ', v, vi r; t ~=uezinea ease reaunaancy at a given
position. Underlined sequences correspond to 5' sequences intended to
introduce, into the amplified DNA products, restriction sites compatible
with the intended cloning vector (pEXPROK).
Cloning of the Partial Rhodococcus Dehalogenase Genes
Cloning of the Rhodococcus dehalogenase gene was accomplished by amplification
from a genomic DNA library as follows. Genomic DNA was isolated from the
Rhodococcus
ATCC strain 5538 using the methods of P.J. Asturias and K. Timmis (J.
Bacteriology
175:4631-4640 (1993)). Purified genomic DNA (100 fig) was sheared mechanically
to an
average size of <10 kbp. Fragments were ligated to BamH I linkers, followed by
BamH I
digestion and ligation into a BamH I digested preparation of bacteriophage
Lambda-ZAP
2o ExpressT"" DNA (obtained from Stratagene, Inc. of LaJolla, CA, USA). A
library containing
the genomic Rhodococcus DNA fragments was prepared commercially (Stratagene,
Inc.,
LaJolla, CA) and supplied at a titer of 1x10' pfu/pL (plaque forming units per
microliter). A
23
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
redundant DNA primer (RDhI 5.4) corresponding to the codons for amino acids 6-
13 of the
N-terminal sequence was synthesized using solid phase phosphoramidite
chemistry and
purified by HPLC (Table 2).
The RDhI 5.4 primer was used in combination with a commercially available
primer
which recognizes the T3 bacteriophage promoter sequence (and is contained
within the
Lambda ZAP ExpressT"~ vector) to amplify dehalogenase sequences from the
singly-
expanded genomic DNA bacteriophage library. Amplification was accomplished
using the
polymerase chain reaction (50 pL) containing 1 pM of RDhI 5.4 primer, 100nM
biotinylated
T3 Pro primer (New England Biolabs), 10x Amplitaq reaction buffer (Perkin-
Elmer-Cetus),
~0 1.5 mM MgClz, 5U of rAmpIiTaq DNA polymerase (Perkin-Elmer-Cetus), and 4 pL
of the
phage library (whole phage). Amplification was for 35 cycles of the following
thermal profile:
1 min. @94°C; 2 min. @ 55°C; 2 min. @72°C. PCR products
were separated by
electrophoresis through 1.0% agarose and a discrete band of 1.3 kbp was
identified, excised
from the gel, and isolated using a QiaQuick gel purification kit (Qiagen.
Inc.). After
~ 5 confirming that this DNA was also capable of being amplified by other
Rhodococcus
dehalogenase-specific primers, the fragment was digested with restriction
enzymes Nco I
and Pst I and ligated into Nco IlPst I digested pGEMSzf(+) (ProMega, Madison,
WI).
Sequencing of the 3'-untranslated region of the cloned segment allowed
identification of a
putative stop codon and subsequent amplification of the coding region with
primers RDhI 5.4
20 and RDhI 3.12.
Sequence and Restriction Enzyme Analyses. Double stranded sequencing of the
dehalogenase gene proceeded via successive rounds of the dideoxy method with
the
biotinylated primers (Table 3) designed for each successive round, based on
the sequencing
results in preceding rounds. Bands were separated on 5.5-6.0% polyacrylamide
urea
25 sequencing gels, the DNA transferred to nitrocellulose filters by capillary
transfer and
visualized using the well-known streptavidin-alkaline phosphatase development
protocols in
combination with chemiluminescent substrates.
24
r
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
Table 3' Sequences and Orientation of Oligonucleotide Primers Used in Sequel
ncing the
Rhodococcus Dehalc~genase Gene
Oligo Name OrientationSpecific Sequence*
for by
Dhl Seq Forward 697-714 5'Bio-CCTGTCCCGAAGTTGTTG
7
Dhl Seq Reverse 807-791 5'Bio-CGGGCCGATGTCCACTG
8
Dhl Seq Forward 186-202 5'Bio-TGCTCCAGACCTGATCG
11
Dhl Seq Reverse 496-480 5'Bio-TCTGATCGATGATCAAC
12
Dhl Seq Forward 404-422 5'Bio-TCCCGACGTGGACGAATG
13
Dhl Seq Reverse 663-646 5'Bio-GAGCGCGACGATGTTCGC
14
Dhl Seq Forward 725-742 5'Bio-CACCCGGCGTACTGATCC
15
Dhl Seq Reverse 951-934 5'Bio-GAGACCGGTCAGCATTCC
18
PROK-SE01 Forward PROMOTER 5'Bio-GAGCGGATAACAATTTCA
PROK-SE02 Reverse TERMINATOR 5'Bio-TCTCATCCGCCAAAACAG
rs~o=rs~oun: m=H, ~, u, or i ; ~ ~=aermea oase reaunaancy at a gmen posrt~on.
Does not include the
biotinylated primers already described in Table 2 which also were used to
determine the sequence of
s the gene. Commercially available (New England Biolabs) biotmylated primers
specific for the T3, T7,
and SP6 promoters were also used but are not listed here.
The vector pEXPROK (Figure 1 ) is a derivative of the commercially available
pPROK-1 vector (Clontech, Inc., Mountain View, CA). Whereas the pEXPROK
retains the
to functional elements of the pPROK vector (including the ampicillin
resistance marker, the
Ptac transcriptional promoter, and paired transcription termination signals
following the
polylinker), pEXPROK vector replaces the EcoR I-to- Hind III polylinker of
pPROK-1 with an
extended synthetic polylinker referred to as EXFLAG. The EXFLAG linker is
designed to
allow insertion of an open reading frame between an Nco I site and an Nhe I
site. In-frame
f 5 with the six-nucleotide Nhe I site is an 11 amino acid peptide, the final
octapeptide of which
corresponds to the well-known FLAG peptide (Kodak Imaging Systems, Rochester,
NY) to
which antibodies and affinity reagents are commercially available. Sequence
and features of
the EXFLAG linker are as follows:
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
EcoR I Nco I Hind III Xba I Xho I Nhe I I-----EXFLAG-
GAATTCAG CCATGGCATAAGCTT TCTAGA CTCGAGGGA GCTAGC GGC CTA GGT
Gly Leu Gly
peptide-_______________________-~ Not I
GAC TAC AAG GAC GAT GAT GAC AAA TAA TGA GCGGCCGC TAGCTT
Asp Tyr Lys Asp Asp Asp Asp Lys *** ***
PCR amplification of the RDhI 5.4/T3Pro gene from the pGEM5 construct with
to primers RDhI 5.4 and RDhI 3.12, followed by digestion with Nco I and Nhe I,
allows ligation
of the Rhodococcus dehalogenase gene into the appropriately digested
expression vector.
This procedure was used to insert the RDhI gene into pEXPROK. The plasmid maps
of
pEXPROK and pEXPROK-RDhI are shown in Figures 1 and 3. respectively. The DNA
sequence of the pEXPROK-RDhI construct was later confirmed by automated DNA
I S sequencing.
Sequence Analysis. The complete DNA and derived protein sequences for the
dehalogenase gene are shown in Figure 2. DNA Sequence data reveals an open
reading
frame of 876 bp, giving a deduced protein sequence of 292 amino acids and a
predicted
molecular weight of 33kD. This is similar to the molecular weight reported for
a number of
20 other hydrolytic dehalogenases.
To determine whether the isolated gene is likely to encode a dehalogenating
enzyme,
a MacVector v.4.5.2 (Kodak, Inc.) sequence analysis package was used to
compare the
derived protein sequence with those of all other known proteins contained in
the Entrez
Sequence Database (the Entrez Database is maintained by the National Center
for
25 Biotechnology Information). The RDhI polypeptide displays the greatest
similarity to
members of the so-called a1(3 hydrolase family of enzymes including several
haloalkane and
haloacid dehalogenases, epoxide hydroalses, arcd enzymes with a number of
diverse
catalytic functions. Alignment of the Dow Rhodococcus dehalogenase with two
other
dehalogenases and a non-dehalogenase (luciferin monooxygenase) enzyme is shown
in
3o Figure 4. The enzymes included in the figure and their publication
references are as follows:
Xanthobacter autotrophicus haloalkane dehalogenase - D.B. Janssen, et al., J.
Bacteriology
171:6791-6799 (1989); tetrachlorocyclohexadiene hydrolase (TCCH or LinB) - Y.
Nagata, et
al., J. Bacteriology 175(20):6403-6410 (1993); Renilla reniformis luciferin
monooxygenase
W.W. Lorenz, et al. Proc. National Acad. of Sciences, U.S.A. 88(10):4438-4442
(1992}.
35 More recent releases of the Entrez database also reveal significant
similarity between the
26
~ F
CA 02281931 1999-08-12
WO 98136080 PCT/US98/02776
Dow RDhI protein and two hypothetical mycobacterium tuberculosis proteins of
unknown
function (Entrez Database Accession numbers 1449324 and 1478233, submitted 7-
22-96
and 7-23-96, respectively, by K. Badcock and C.M. Churcher, et al.), as well
as with the
haloalkane dehalogenase isolated from Rhodococcus rhodochrous (Entrez Database
Accession number 1196824, submitted 2-15-96 by A.N. Kulakova, et al.).
Of the sequences aligned in Figure 4, only the Xanthobacter dehalogenase has
been
well characterized at a structural and mechanistic level. Notably, two of the
three residues
known to be involved directly in the Xanthobactercatalytic cycle (the two most
important
residues, Asp-124 and His-289) are conserved in the Rhodococcus sequence.
These
o similarities and those indicated in the Figure suggest a high degree of
structural and
mechanistic conservation among members of this family of proteins.
Dehalogenase Protein Expression.
To confirm the identity of the above, cloned enzyme as a dehalogenase, we
sought
to express the full-length protein in E. coli. To accomplish this, a 1300 by
Nco I/Spe I
5 restriction fragment, containing the RDhI gene was excised from the
pRDhIK02.1-pGEMS
construct and iigated with the Nco IlNhe I-digested pEXPROK vector. Because
Spe I and
Nhe 1 generate ligation-compatible restriction fragments, this resulted in the
generation of an
expression construct (Figure 5) containing the complete putative RDhI gene
under the
transcriptional control of the IPTG-inducible Ptac promoter and the
termination control of the
?o endogenous RDhI 3' untranslated region.
Colonies transformed with the resulting piasmid (pRDhIK02.3-EXPROK) were grown
overnight in 2 mL minicultures, following which 1 mL of each culture was
pelleted, washed,
and sonicated. Extracts were then assayed for their capacity to catalyze
release of chloride
following addition of the RDhI substrate, 1-chlorobutane. Chloride releasing
activity was
zs absent from cultures not containing the cloned gene; cultures with the
cloned gene exhibited
chloride releasing activity which increased when transcriptional activity of
the gene was
increased by the addition of IPTG. Thus, dehalogenase activity could be
induced in
overnight cultures of the recombinant E. coli containing the pRDhIK02.3-
pEXPROK
construct.
Example 2
The gene encoding this dehalogenating enzyme has been isolated and cloned into
the bacterium, E. coli. DNA sequence analysis revealed that this isolated gene
encodes a
27
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
protein with a high degree of sequence similarity to other known
dehalogenating enzymes.
In an effort to increase levels of biosynthesis to commercially meaningful
levels (i.e.
"expression"), a number of systems reported to enable high level production of
heterologous
proteins in E coli were examined.
To generate the expression vector pEXPROK-RDhI, plasmid pEXPROK was digested
with restriction enzymes Nco IlNhe I and then purified by a QIAquick Gel
Extraction Kit
(Qiagen, Inc., Chatsworth, CA). The RDhI open reading frame was amplified with
primer
RDHL 5.4 as the forward primer (containing an Nco I site to direct the start
of translation)
and primer RDHL 3.12 as the reverse primer (and containing an Nhe I site).
Following
to digestion of the amplified DNA with Nco I and Nhe I, the gene was ligated
into the pEXPROK
vector. The new construct was then transformed into E. coli AG1 competent
cells and
ampicillin resistant colonies were picked. Plasmids containing the RDhI gene
were identified
by analytical restriction enzyme digestion and referred to as pEXPROK-RDhI
construct. The
pEXPROK-RDhI plasmid map is shown in Figure 3.
Construction of pRSET-RDhI and pTrcHis-RDhI Expression Vectors
For construction of both pRSET-RDhI and pTrcHis-RDhI expression vectors, the
RDhI
gene was amplified from the pEXPROK-RDhI using oligonucleotide primers RDhI
5.4 and
RDhI 3.13 using standard PCR conditions. Amplification products were separated
on
agarose gets and purified using standard procedures.
2o Both pRSET and pTrcHis vectors are IPTG inducible expression vectors,
derived
from the pUC 18 and 19 series of cloning vectors. They both were purchased
from
Invitrogen Corp. (San Diego, CA) and contain the following features:
(a) Both are designed for high level protein expression and both carry an
ampicillin resistance gene.
(b) Both contain a sequence that encodes an N-terminal fusion peptide which
codes for six histidine residues. These residues function as a metal binding
domain and
may allow later purification of recombinant protein by affinity
chromatography.
(c) The vectors encode an enterokinase cleavage recognition sequence (the
FLAG and/or EXFLAG peptide) downstream of the dehalogenase coding region which
allows
3o detection by and immobilization upon anti-FLAG antibodies.
The high level expression property of the pRSET vector results from the
presence of
the T7 promoter upstream of the heterologous gene. Since E. coil does not
contain the T7
28
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
polymerase, however, an M13 phage containing the T7 RNA polymerase gene is
needed for
protein expression. In practice, bacteria containing a heterologous gene under
the control of
a T7 promoter are induced to produce the heterologous protein by infection of
recombinant
E. coli with T7 phage containing the T7 RNA polymerase. Alternatively,
commercially
available E. coli stably expressing the T7 RNA polymerase enzyme (i.e. BL21 )
can be
transformed with the pRSET construct.
The pRSET-RDhI expression vector was generated by digesting plasmid pRSET with
restriction enzymes Nco IlHind III and then incorporating an RDhI gene
fragment which
contains an Nco I site at the 5 ~ end and a Hind I II site at the 3' end. The
new construct was
io then transfected into E. coliJM109 competent cells and ampicillin resistant
colonies were
picked. Plasmids containing the RDhI gene were identified by analytical
restriction enzyme
digestion and referred to as the pRSET-RDhI construct. The pRSET-RDhI
expression
construct is shown in Figure 6. One such clone (Clone 16-4) was used to
characterize
protein expression using the pRSET system.
The pTrcHis vector contains another high level transcriptional promoter - the
trc
promoter, a fusion of the well-characterized trp promoter and the lac
promoter. The pTrcHis
vector also contains a mini-cistron upstream of the heteroiogous gene which
provides highly
efficient, repeat initiation of translation of the cloned protein in the
multiple cloning site.
Using a similar process, we cloned an RDhI gene fragment into the pTrcHis
vector to
2o generate the pTrcHis-RDhI expression vector. For expression studies, E.
coli TOP 10'
competent cells were transformed with the pTrcHis new construct. Both the
pRSET-RDhI
and pTrcHis-RDhI expression vectors contain an 11 amino acid EXFLAG peptide
downstream of the Nhe I site.
The EXFLAG peptide sequence is in-frame with the open reading frame of the
cloned
25 protein and is useful for analytical detection and affinity purification.
Figure 7 shows a map
of the completed pTrcHis-RDhI expression construct. One such clone (Clone 18-
3) was
identified as a high expressing clone and used for further characterization of
the TrcHis
expression system.
Construction of pTrxFus-RDhI Expression Vector
3o The ThioFusionT"" expression system (Invitrogen Corp., San Diego, CA)
provides a
means of expressing large amounts of heterologous protein by fusing the gene
encoding
such a protein to the gene encoding the E. coli protein, thioredoxin, in the
pTrxFus
expression vector. The thioredoxin moiety can confer both solubility and heat
stability to its
29
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
fusion partner, thereby opening up new options for purification by osmotic
shock or heat
treatment. The expression vector, pTrxFus, allows foreign genes to be inserted
into its
multiple cloning site. It uses the P~ promoter from bacteriophage lambda to
drive expression
and the cl repressor, also from bacteriophage lambda, to control the level of
transcription.
Expression of the cl repressor gene is under control of the trp promoter and
repressor.
Expression of a foreign gene is induced by adding tryptophan to the medium
which shuts
down cl repressor synthesis and allows transcription from the P~ promoter.
Primers Trx2++ and Trx- (see DNA Primer Design) were designed to modify the
RDhI
gene fragment with an enzyme restriction site unique to the TrxFus multiple
cloning site.
to Plasmid pEXPROK-RDhI was used as a template, and a gene fragment was
generated by
PCR, using primers Trx2++ and Trx-, which added a BamH I site to the 5~ end
and a Pst I
site to the 3' end. The fragment was purified using a OIAquick PCR
Purification Kit. Both the
pTrxFus vector and the gene fragment were enzyme-digested. agarose gel
purified, and
ligated. The new construct, pTrxFus-RDhI (Figure 8), was incorporated into
61174
is electrocompetent cells (Invitrogen Corp.) which had been prepared following
the
manufacturer's instructions.
Expression Analysis
Growth and Induction of Cell Cultures--For expression studies, clones
identified as
containing proper DNA constructs were cultured in 3 mL of Luria Broth (LB) or
SOB medium
20 (Difco, Detroit, MI, USA) containing 50 pg/mL ampicillin in 15 mL round-
bottom
polypropylene culture tubes. These culture tubes were incubated overnight at
37°C with
shaking (200 cycles/minute in a rotary shaker) or grown to an ODsoo of 0.6.
Afterward, 2 mL
of fresh medium with IPTG was added (to a final IPTG concentration of 1mM) and
the tubes
were incubated at 37°C with constant shaking for another 4-5 hours. For
recombinant
25 clones of pRSET, after 1 hour of IPTG induction the cell cultures were
infected with
previously titered M13/T7 phage and the incubation continued as described
previously.
For recombinant clones of pTrxFus, RDhI gene-containing clones were cultured
in 1
mL RM medium with 100 pg/mL ampicillin and incubated overnight at 30°C
with shaking
(200 cycles/minute in a rotary shaker). The next day, 9 mL fresh Induction
Medium were
3o added and growth continued at 30°C to an OD55o of 0.5. Then, cell
cultures were induced
with tryptophan (to a final concentration of 100 pg/mL) and transferred to a
37°C incubator
and shaken at 200 rpm for another 2 to 4 hours.
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
Cell Free Extract Preparation--For protein analysis, induced, overnight cell
cultures
were pelleted by centrifugation at 4°C (5000 rpm for 10 minutes in a
Sonrall SS-34 rotor).
Cell pellets were washed in cold 10 mM Tris sulfate buffer (pH 7.5) containing
1 mM disodium
EDTA and then centrifuged again. For clones of pEXPROK, pRSET, and pTrcHis,
final
suspensions were sonicated at 14 Hz on ice through 3 repetitions of a 20
seconds "on", 30
seconds "off" regimen, using a small-tip sonicating probe (Soniprep 150, MSE
Ltd., Crawley,
Sussex). Insoluble debris was removed by centrifugation at 10,000 rpm for 10
minutes.
Cell-free supernatants were then transferred to clean polypropylene tubes and
appropriate
assays performed. Final cell suspensions from clones of pTrxFus were sonicated
for three
to 10-second bursts and then flash-frozen in a dry ice/ethanol bath. Shortly
after freezing, the
cell lysates were quickly thawed at 37°C and two more, rapid sonication-
freeze-thaw cycles
were performed. After the last thaw, the procedures described above for
removing the
cellular and insoluble debris were continued.
expression and Purification of pEXPROK-RDhI
t5 Figure 9 shows a Pro-BIueT"" stained SDS-PAGE gel of cell lysate samples of
the
pEXPROK-RDhI clone 12-4 on the left side (lanes 2-5) and partially purified
rRDhl enzyme
on the right side (lanes 8-11 ). Lane 1 contains molecular weight standards
and lanes 6 and
7 contain single, 60 ng and 180 ng bands of the FLAG-peptide protein at a
molecular weight
of 55kD. Lanes 2-5 show all the soluble protein from cell-free extracts. Since
rRDhl enzyme
2o is not a major protein in the extracts, immunoblotting of an identical gel
was done to confirm
the presence of this recombinant enzyme. Figure 10 shows this recombinant
enzyme band
in each sample lane, as recognized by an anti-FLAG antibody at the predicted
molecular
weight of - 35kD. Lanes 6 and 7 in Figure 10 are 20 ng and 60 ng,
respectively, of the
FLAG-peptide protein. Affinity purified recombinant enzyme was analyzed on
both a Pro-
25 BIueT"~-stained SDS-PAGE gel and an immunoblotting membrane. Four
consecutive
fractions of affinity-purified rRDhl enzyme were run in lanes 8-11 of the Pro-
BIueT"~-stained
SDS-PAGE gel shown in Figure 10. In addition to a prominent band at -35kD
molecular
weight, other protein bands are visible on the gel. The immunoblot of the
partially purified
enzymes (Figure 10, lanes 8-11 ), however, confirms that the recombinant
enzyme at -35kD
3o is the only protein to stain with anti-FLAG antibodies and thus appears to
be the proper
translated rRDhl protein. This data suggests that rRDhl enzyme is stable both
in the E. coli
intracellular environment and throughout the purification process.
31
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
Expression of pRSET-RDhI and pTrcHis-RDhI
Cell free extracts obtained from clones of the pRSET recombinant enzyme
expression system and the pTrcHis recombinant enzyme expression system were
analyzed
for the presence of recombinant RDhI protein. Five clones containing the
correct size
Nco IlHind III DNA fragment were identified, cultured overnight, lysed, and
analyzed for
rRDhl expression by SDS-PAGE gel (Figure 11 ). Lane 1 shows molecular weight
standards
and lane 7 and 8 contain single 60 ng & 180 ng bands of the FLAG-peptide
protein at a
molecular weight of 55kD. Lanes 2-6 show samples of 1 pL of cell-free extracts
from the 5
clones and lanes 9-12 show samples of 0.1 pL of the cell-free extracts.
Immunoblots of
to these extracts reveal doublet bands (35kD and 38kD) in each sample lane,
when the anti-
FLAG antibody is used to stain the immunoblots (Figure 12). This may suggest
that there
are two start codons in the pRSET-RDhI system. The first start codon was
originally
designed in the pRSET vector system to be about 41 amino acids (123 bp) before
the actual
cloning site, which allows the initiation of translation from that Met ATG
codon followed by 6
is histidines. The second start codon may occur at the Nco I cloning site
itself, which was
designed into the original 5~ end primer of the RDhI gene fragment. However,
the presence
of an anti-FLAG antibody-reactive band confirms the presence of rRDhl enzyme.
Figure 13 shows the Pro-BIueT"~-stained SDS-PAGE gel with cell-free extracts
from
the pTrcHis system and Figure 14 shows the immunoblot of an identical SDS-PAGE
gel. All
2o clones of the pTrcHis system show an overloaded, anti-FLAG-reacted band at
a molecular
weight of -35 kD, which confirmed the presence of rRDhl enzyme in the
extracts. Since the
volumes of the initial culture and the cell-free extract preparations are the
same in all three
systems, these overloaded bands are an indication of higher enzyme production
in the
pTrcHis system.
25 Expression of pTrxFus-RDhI
The soluble protein, cell-free extracts from the pTrxFus system were examined
for
the presence of rRDhl enzyme, using reducing SDS-PAGE. Figure 15 shows a gel
stained
with Pro-BIueT"". Lane 12 shows molecular weight standards and lane 11 shows a
single
150ng band of the FLAG-peptide protein at a molecular weight of 55kD. The
thioredoxin
3o fusion bands are clearly visible as the major protein in lanes 1 to 9 at a
molecular weight of
47 kD. This size corresponds to the 12 kD of the thioredoxin protein and 35 kD
of the rRDhl
enzyme. In contrast, lane 10 has a sample of insoluble matter from cell lysis,
which shows
no presence of the high-level, expressed thioredoxin fusion protein. This
demonstrates that
32
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
all of the fusion protein is in a soluble state. Data from other experiments
indicate this fusion
protein band can be recognized by anti-FLAG antibody at the same molecular
weight in the
immunoblot membrane {data not shown).
Analysis of Hydrolytic Dehaloaenation Activity
To quantify the hydrolytic dechlorination activity of the recombinant enzyme,
a
colorimetric chloride-release assay at 460 rim was used.
Recombinant protein activity was measured by adding an appropriate amount of
cell-
free extract (prepared as described above) to 6.0 mL of reaction buffer in a
glass vial. 100
mM sodium glycinate buffer (pH 9.0) was used for measuring activity toward 1,4-
~o dichlorobutane (DCB) (Aldrich Chemical Co.), and 100 mM Tris-SO4 buffer (pH
7.0) was
used for measuring activity toward 1,2,3-trichloropropane (TCP). The
halogenated substrate
(6pL) and a micro stir bar were added and the vial was capped. Capped vials
were incubated
in a 30°C water bath with stirring.
Periodically, 1.0 mL samples were removed and assayed for free chloride.
Reagent
t 5 1, 0.375M Ferric Nitrate in 5.25 N Perchloric acid ( 10°.o v/v),
was added to stop the hydrolytic
reaction and reagent 2, saturated Mercuric Thiocyanate in ethanol (10% v/v),
was added to
develop color. Final samples were read in a Perkin-Elmer 552A UVNIS
Spectrophotometer
at 460 rim. Rates were determined after correcting for non-enzymatic
hydrolysis against a
blank.
2o Dehalogenating Activity of Recombinant Rhodococcus dehalogenase
While the preceding data suggest that the recombinant dehalogenase can be
synthesized at much higher levels in E. toll than in wild type Rhodococcus,
they do not
address the activity of the expressed protein. Indeed, enhancing production of
a
dechlorinating enzyme is the key objective of this work. For this reason, we
examined the
25 relative levels of dehaiogenase activity in representative clones from each
of the above
constructs. Activity was determined by the free chloride release assay and
compared with
protein expression as documented in Figures 13-15. Protein expression was
quantified on
SDS-PAGE gels by high resolution scanning densitometry and the measured amount
of
rRDhl was stated in terms of % of total soluble protein. The following table
shows the
3o relationship between dehalogenating activity and the percent of rRDhl
enzyme in the total
soluble protein among all four expression systems.
**
33
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
Expression% of Total DCB* Activity Clone Name
System Soluble Proteinper
mL of Culture
pEXPROK ~3 0.3 x 10-z EXPROK-RDhI
pRSET -10 0.8 x 102 RSET RDhI Clone
16-4
pTrcHis -15 2.4 x 10-2 TrcHis RDhI Clone
18-3
pTrxFus -30 4.8 x 102 TrxFus RDhI Clone
4
u~rs unns were measures as the aegree of indicator color change (oOD/min.) as
the
enzyme dechlorinated 1,4-dichlorobutane.
This data reveals a strong correlation between level of rRDhl protein
expression and
observed dehalogenase activity.
In this example, Rhodococcus haloalkane dehalogenase can be expressed at high
levels in E. Coli in 3 of the 4 systems examined. The recombinant Rhodococcus
dehalogenase is stably expressed in all four systems and recognized by anti-
FLAG
antibodies at the expected molecular weight. This recombinant enzyme exhibits
a
~o dehalogenase activity at a level similar to that of the wild type and
proportional to the level
of heterologous protein expression.
Example 3.
Preparation of Porous Alumina Sup,~~orts
Protein (385 mg) representing 22 TCP units of activity were immobilized on 2.0
g of
volatile-free alumina (lot #1587 of k-4 alumina from UOP of DesPlaines, IL,
USA) at 4°C with
mild agitation over the weekend. The procedure followed UOP's standard
practice of GIA-
activation of the polyethyleneimine coating, water washing, and enzyme
addition. The
bathing solution was decanted and the support was washed five times with 2 mM
Tris/1 mM
2o EDTA, pH 7.5.
Enzyme Purification and Preparation for Immobilization
Recombinant Rhodococcus dehalogenase was produced in E. coli using the pTrcHis
expression system. Enzyme preparations used for all immobilization studies
were first
partially purified using ammonium sulfate precipitation, using a cut of 45 to
70% saturation at
4 °C, followed by dialysis and clarification in 10 mM Tris sulfate, 1
mM EDTA, pH 7.5. This
basic buffer was used throughout all purification steps. These preparations
were routinely
4.5-fold purified from the lysate, as determined by absorbance at 280 nm, and
were
34
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
estimated to be 30-35% pure dehalogenase protein by SDS-PAGE. More highly
purified
enzyme preparations were achieved by an additional DEAE-Sepharose
chromatographic
step of eluting with a 0-400 mM ammonium sulfate gradient. This provided about
10-fold
purification from lysate, with 85-90% enzyme purity. This step was followed by
QAE-
Sepharose FF chromatography with a narrower 0-120 mM ammonium sulfate
gradient,
achieving about 12-fold purification from lysate, and SDS-PAGE which
demonstrated
enzyme homogeneity. Purified RDhI from the TrcHis RDhI expression system is
typically
referred to herein as "rRDhl."
Preparation of Supports
io All anion exchange supports were thoroughly hydrated according to
manufacturer's
instructions (if necessary), then exhaustively washed to remove ethanol and to
exchange
into the sulfate form by continuous rinsing withl0 mM Tris sulfate. 1 mM EDTA.
This same
starting buffer was used throughout to load enzyme preparations.
Inorganic supports were modified with polyethyleneimine and glutaraldehyde
i 5 according to well established protocols (U.S. Patent No. 4,268,410 and
Mosbach,
Immobilized Enzymes, in 44 Methods in Enzymoloav. (1976) (Academic Press,
NY)). Dry
samples of supports were weighed out and distributed into 12 mL capped vials.
An aqueous
solution of 2.5 % polyethyleneimine was added to a total of 10 mL per gram of
support.
Vials were capped, and then agitated gently on a rocking shaker for 1 hour at
room
2o temperature. Samples were transferred to a small Buchner funnel where
liquid was
removed by gentle vacuum. Supports were transferred to a watch glass and
allowed to air
dry at room temperature overnight (about 18 hr). Samples were transferred to a
new vial to
which was added a freshly thawed solution of 25% aqueous glutaraldehyde at a
ratio of 20
mL per gm of support. The mixture was capped and shaken intermittently for 1
hour in a
25 hood. The glutaraldehyde was removed by decantation and washed exhaustively
with water
until no aldehyde was detected by a fuchsin test. Prior to enzyme
immobilization, supports
were decanted, but not dried.
immobilization of Enzyme
All enzyme immobilization was performed in a cold room at 4°C using a
rocking
3o shaker to provide gentle agitation. Times used for binding of enzyme
preparations to
supports ranged from 1 hour for the ion-exchange supports to a maximum of 4
days for
experiments with the PEI-GIA modified inorganic supports. Buffer exchange was
used only
for the Celite R 648 binding capacity studies in which the Tris buffer was
exchanged for a 10
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/OZ776
mM potassium phosphate, 1 mM EDTA, pH 7.0 buffer on a pre-packed Sephadex G-25
column.
Enzyme Stripping from Ion-exchange Supports
Two sets of 250 NL aliquots of resin slurry were bound overnight with either
11.1 DCB
U (Toyopearl~) or 6.63 DCB U (PEI Cellulose) of enzyme. The binding
supernatants were
carefully removed and assayed. Resins were spun at 6,000 rpm for 8 minutes.
Additional
supernatant was removed and the resins were washed twice with 100 mM Na
Glycinate
buffer (pH 9). One tube from each set was treated with 0.5 M (NH4)zS04 for 1
hour to strip
the enzyme from the support. These resins were rinsed again with buffer.
Resins and
~o supernatants were assayed for activity using DCB as a substrate.
ion-Exchange Supports
Anion-exchange chromatography has been used extensively in the purification
and
characterization of both the wild type and the recombinant dehalogenase
enzymes. The
enzyme is anionic at neutral pH (where the dehalogenation reaction is
performed) and anion
a exchange supports often function well in immobilizing such proteins. This
approach was
also attractive because it allowed for simultaneous purification and
immobilization of the
enzyme. To confirm this potential utility, we examined binding and elution of
the rRDhl
protein to anion and cation exchange resins over a wide pH range. Figure 2
shows the
nearly quantitative retention of the dehalogenase on DEAE Sepharose anion
exchange resin
20 over a range of 5 pH units. For contrast, the CM-Sepharose rapidly loses
its binding
capacity above pH 5.
Immobilizations
A number of support materials were examined for their efficacy in immobilizing
rRDhl.
In these studies, a 40-70% ammonium sulfate cut of the dehalogenase enzyme was
used.
25 Duplicate sets of enzyme, immobilized on each of thirteen ion-exchange
supports were
prepared. One of each set was assayed immediately for dehalogenase activity
using the
chloride release assay. The second was treated with TCP-saturated 10 mM Tris
sulfate (pH
7.5) at 45°C for 1 hour. Following this treatment, supernatant was
removed and this set was
also assayed by the standard chloride method. Table 4 summarizes the results
of these
30 assays.
36
_.
CA 02281931 1999-08-12
WO 98136080 PCT/US98/02776
Table 4 Screen of TrcHis RDhI on 13 ion-exchange supports following incubation
at
45°C for 1 hour in the~~resence of substrate
Support Supplier Lot/Batch % Activity
#
after
TCP
Treatment
Silica Gel PE1-Silica Sigma 24H0810 0
DEAE Sephadex A-50 Sigma 24H0485 19
PEI Cellulose(med. mesh) Sigma 94H7200 54
Glass, Aminopropyl Sigma 34H8260 43
Toyopearl~ Super Q-650M TosoHaas 65QAM02RM 79
DEAETrisacryl Plus-M Sigma 92H0861 21
Spectra/Gel Ion Exchange 1 X8 Spectrum 16865 14
Dowex~ 1 X8-200 Ion Exchange Aldrich 12627-85-9 54'
Resin
DE52 Whatman 1152032 50
Quaternaryammonium Cellulose Whatman 9852032 2
DEAE Sepharose Sigma 53H0177 30
AG 3X4 100-200 Bio-Rad 52594A 18
AG 4X4 100-200 Bio-Rad 47426A g
*incubated at 37C
io These results indicate that there is marked heterogeneity in the efficacy
of these
matrices as supports for the dehalogenase enzymes. Similar heterogeneity will
be seen for
similar dehalogenases catalyzing similar reactions.
Four of the best candidates were screened for stability over time in the
presence of
TCP. These were: PEI cellulose, Toyopearl~ Super Q-650M, Glass Aminopropyl,
and
DEAE Sepharose. Duplicate sets were prepared, one to be used for an initial
chloride
detection assay, the second for assay after exposure to TCP. Table 5 shows
that 3 of the 4
lost significant activity in the first 24 hours but retained stable activity
out to at least 7 days
following the initial loss. Toyopearl~ underwent a similar but delayed loss at
48-120 hours
and then appeared to stabilize.
37
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
Table 5: Stability Study of TrcHis RDHL on 4 Ion-Exchange Supports in
Presence of TCP at Room Temperature
SUaaOrt % Activity Over Time
24 hr 48 hr 120 hr 192 hr
PEI Cellulose(med. mesh) Sigma 83 79 78 78
Glass, Aminopropyl Sigma 78 87 75 66
Toyopearl~ Super Q-650M TosoHaas 100 100 78 82
DEAE Sepharose Sigma 76 79 73 62
All four resins appear to be good candidates for immobilizing the dehalogenase
and
appear to provide a suitable surface for prolonged enzyme activity.
Covalent Coualina to Tresyl-Activated Po~acrylic Polymer
In order to determine the feasibility of covalently coupling rRDhl through
pendant
n> amino groups to any support, an activated resin, Tresyl-Toyopearl, was
evaluated. This
activated resin provides a stable secondary amino group linkage with the
enzyme:
o
II
-O-R-O-CHI-CH=OSOCH,CF; + H~N-Enzymc -> )-O-R-O-CH,-CH,-NH-Enzyme
The rRDhl preparation used for these studies had been affinity purified from
E. coli
lysate using the anti-FLAG antibody column, and was estimated to be about 20%
pure by
SDS-PAGE. 0.35 units of enzyme (1.96 mg total protein) were coupled to 40 mg
of the
Tresyl-Toyopearl under conditions described by the manufacturer. After 3
hours, 93% of
2o protein had been coupled as determined by the decrease observed at AzBo. An
additional 10
mg of resin were added and coupling continued for 1 hour to bind >98% of the
protein. Re-
assay of the washed gel for dehalogenase activity revealed recovery of 0.11
units of activity
(31 %). A second trial using 2.6 units of the same enzyme preparation and 1.0
gm of
activated resin demonstrated a recovery of 37% activity. According to
manufacturer's notes,
recoveries of activity from enzymes coupled to this support usually lie in the
40-60% range,
so 31-37% represents reasonable recovery and is sufficient to make
commercially practical
the coupling of the enzyme, via its amino groups, to an immobilization support
material for
use in a bench-size or industrial bioreactor.
38
a
CA 02281931 1999-08-12
WO 98136080 PCT/US98/02776
This covalent attachment to hydrophilic resins is also an effective means of
immobilizing the dehalogenase enzyme.
Polvethvleneimine Imoreanated Inoraanic Supports Cross Linked with
Glutaraldehyde
Inorganic supports have also found wide utility in the industrial enzyme arena
due to
availability, low cost, high loading capacity, ease of regeneration and reuse,
and the wide
range of pore sizes. Porous alumina, silica, and Celite have found widespread
use as
supports for immobilized enzymes, with titanium- and carbon-based supports
seeing more
limited application.
Enzymes can be immobilized to inorganic supports by three mechanisms. The
enzyme may associate with the inorganic support through ionic interactions or
may bind
through an ion-exchange mechanism to an ionic polymer which has been
impregnated into
the inorganic support, or be crosslinked to the ionic polymer using a
bifunctional chemical
linker. The first approach has not seen wide applicability because the weak
ionic
interactions frequently lead to enzyme leaching. Polyethyleneimine (PEI) is
the polymer of
choice for impregnation because of low cost. The amino groups allow a wide
range of
crosslinking chemistry to be applied. Glutaraldehyde is by far the most
studied and
inexpensive crosslinking agent used. Studies with rRDhl focused entirely on
this coupling
chemistry.
Established methods for the preparation of PEI-impregnated porous supports,
2o followed by glutaraldehyde crosslinking were used (U.S. Patent No.
4,268,410 and Mosbach,
Immobilized Enzymes, in 44 Methods in Enzymoloav, (1976) (Academic Press,
NY)). The
recovery of rRDhl activity was initially screened for two supports. Porous
silica already
impregnated with PEI was obtained from Sigma (nominal pore size of 250 A).
Celite R-648
was obtained from Manville (nominal pore size of about 150 ~) and impregnated
with PEI
25 (avg. 50,000 MW) from Sigma according to the methods of U.S. Patent No.
4,268,410. Both
supports were treated with glutaraldehyde (GIA) and then washed exhaustively
with water.
5.5 units (1.0 mL at 0.55 mg protein/mL) of a highly purified rRDhl enzyme
preparation
(>98% pure by SDS-PAGE) was used to couple to 500 mg each of the two
glutaraldehyde-
treated, PEI-impregnated supports. This loading level (0.11 % w/w) was assumed
to be at
30 least two orders of magnitude below the known loading capacity of the
supports. The
enzyme was incubated with the supports, with gentle shaking overnight (18 hr)
at 4 °C,
before washing exhaustively to remove unlinked protein. Re-assay of the two
supports with
DCB demonstrated recoveries of 40% for the PEI-Celite R-648 and 31 % for the
PEI-Silica.
39
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
These samples were stored at room temperature under reaction conditions
(saturating DCB)
for 1 week and re-assayed. The Celite immobilized enzyme preparation lost 49%
of activity
in a week while the Silica immobilized enzyme preparation lost 28% of its
activity.
In order to quickly determine which type of inorganic support would provide
the best
recovery of rRDhl activity, several commercially available porous supports
were screened.
As in the previous experiment, loading levels of the highly purified rRDhl
enzyme were set
greater than three orders of magnitude (0.0055 % w/w) below the expected
loading
capacities of the supports in order to compare the supports on the sole basis
of activity
recovered, independent of loading capacity, pore size and so forth.
io Three porous aluminas, three porous silicas, and two porous carbons were
screened.
Additionally, Sigma PEI-Silica and Celite R-648 (evaluated in the previous
screen) were
reevaluated under the same conditions. All supports were impregnated with PEI
and treated
with glutaraldehyde as before. 25 pL of enzyme preparation (13.8 pg) were
incubated with
each support for 72 hours at 4 °C with gentle agitation to ensure
maximum loading. Enzyme
loading in the bathing solution was monitored by measurement of AZe~, at 24
and 72 hours.
Activity against DCB was then determined in the bathing solution (unbound) and
on the
washed gels (bound) after the 72 hour incubation. Tables 6 and 7 show the
results of these
studies.
Table 6 rRDhl Enzyme Uptake into PEI-impregnated GIA treated Porous
Supports as Monitored by A280
Support %Loaded @ 24 hr % Loaded @ 72
hr
Alumina - Davison Low SA 83% 62%
?5 Alumina - Norton SA 6176 86% g4%
Alumina - Calcicat Type C 84% 67%
Silica - Calcicat S-88-473 69% 72%
TypeA
Silica - Shell 5980-F 81 % 93%
Silica - Davison 952-08-5X 91 % 92%
3o Carbon - Borecker Subunit 77% g1 %
Carbon - AmCy 5701-Sn 90% 95%
Celite - Manville 8648 82% g5%
PEI-Silica - Sigma 85% g3%
F
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
Table 7 Recovery of Enzyme Activityr on PEI-Impregnated GIA treated Supports
Support % Bound % Unbound %Lost*
Alumina - Davison Low SA 7% 38% 55%
Alumina - Norton SA 6176 3% 17% 80%
Alumina - Calcicat Type C 7% 34% 59%
Silica - Calcicat S-88-473 12% 28% 60%
Type A
Silica - Shell 5980-F 12% g% g0%
t0 Silica - Davison 952-08-5X 17% 11 % 72%
Carbon - Borecker Subunit 5% 9% 86%
Carbon - AmCy 5701-Sn 7% 5% 88%
Celite - Manville 8648 21 % 5% 74%
PEI-Silica - Sigma 6% 7~0 g7oo
5 '% Lost = 100% - (% Bound + % Unbound)
Each support exhibits a different uptake profile ranging from 62°o to
95°ro uptake after
72 hours. For most systems, 72 hours is adequate to achieve maximum loading of
protein
achievable at 4 °C. However, the three alumma systems actually showed
greater uptake at
~0 24 hours than at 72 hours. Recovery of bound enzyme actmit~es at 72 hours
ranged from
3% to 21 % as compared to an untreated soluble enzyme control. Considerable
activity was
unaccounted for or "lost" in all systems examined, ranging from 55°ro
to 87°,r. Also, the
previously run supports (Sigma PEI-Silica and Manville Celite 8648) showed
poorer bound
recoveries. This could be due to either the lower enzyme loading ratio or the
longer
25 incubation times used in this experiment. Given the efficiency of binding
and the recovery of
bound enzyme activity, Celite, silica, carbon, and alumina all function as
effective
immobilization support materials in the present invention, although Celite and
silicas
outperform alumina and carbon. In terms of stability, however, alumina
supports appear to
perform better (see below).
3o The bound samples were also submitted to a long term stability study.
Following
assay using DCB as a substrate, supports were rinsed and immersed in TCP-
saturated
buffer. At the given time-point, TCP buffer was removed, supports were rinsed
again and
assayed with DCB.
41
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
Table 8 Lona term stability study of PEI cross-linked sup~~orts at room
temperature
Support % ActivityMaintained
at 41 at 136
hr hr
Alumina 38 57
1
Afumina 66 67
2
Alumina 58 75
3
Silica 76 81
1
Silica 79 46
2
Silica 60 30
3
Carbon 75 0
1
Carbon 37 48
2
Celite 57 12
PEI-Silica43 60
Thus the two supports which exhibited intermediate levels of recovery in the
immobilization reaction, silicas and aluminas, proved to have the best
stabilities over time.
All of these supports were also screened for their ability to bind the enzyme
directly without
PEI or GIA modification. However, binding was very poor and irreproducible,
and allowed
easy removal of enzyme from the supports with washing.
Polyethyleneimine-Impregnated Inorganic Supports with Enz~rme Bound by Ion
Exchange
The molecular weight of PEI is also known to have an impact on the overall
yield and
stability of immobilized enzymes. In addition, PEI is capable of functioning
either as an ion
~5 exchange ligand on various supports or as a glutaraidehyde cross-link
acceptor. For these
reasons, PEIs of two different molecular weights were impregnated onto various
porous
inorganic supports following the method described in the previous section. In
these
experiments, however, enzyme (semi-purified preparations containing 1-2 U/mL)
was bound
by ion-exchange but the GIA crosslinking step was omitted. Samples were
submitted for a
2o stability screen to determine if the size of the PEI was an important
factor.
42
F
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
Table 9 Stability Screen of PEI-impregnated Porous Su~aports No Crosslinking
Suaaort Supoiier MW PEI % ActivityMaintained
at 24
hr
at
120
hr
1 Alumina Norton SA 6176 50,000 70 62
2 Alumina Calcicat Type C " 61 42
3 Silica Calcicat S-88-473 Type 101 64
A "
4 Silica She115980-F " 80 55
Carbon Borecker Subunit " 41 32
6 Carbon AmCy 5701-Sn " 39 17
7 Celite Manville 8648 " 83 50
8 Alumina Norton SA 6176 2000 82 55
9 Alumina Calcicat Type C " 91 61
10Silica Calcicat S-88-473 Type 94 57
A "
11Silica She115980-F " 76 55
12Carbon Borecker Subunit " 44 50
13Carbon AmCy 5701-Sn " 42 21
14Celite Manville 8648 " 58 2
S
With the exception of Celite, the different molecular weights of PEI did not
appear to
have a major impact on either immobilization efficiency or stability of the
enzyme.
Example 4
to Construction of pRSET-RDhLNde
The pRSET-RDhLNde expression vector was generated by digesting plasmid pRSET
RDhI clone 16-4 with the restriction enzymes, Nde I and Hind III, and then
incorporating into
the construct a RDhI gene fragment which contained a Nde I site at its 5' end
and a Hind III
site at its 3' end. The new construct was then transformed into E. coli JM109
competent
cells (from Invitrogen of Carlsbad, CA, USA) and ampicillin resistant colonies
were picked.
Plasmids containing the RDhI gene were identified by analytical restriction
enzyme digestion
and referred to as the pRSET-RDhLNde construct.
43
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
Production of Recombinant RDhI Protein
Both of the new constructs - pRSET-RDhl.Nde and pTrcHis-RDhI - were
transformed into E. coli B834(DE3) competent cells (from Novagen, Inc. of
Madison, WI,
USA). The production of active dehalogenase enzymes was confirmed by a
dehalogenation
activity assay and enzyme production levels were investigated with PAGE.
Dehalogenation
activity was measured by using a colorimetric chloride release assay at 460 nm
to assess
enzymatic dechlorination activity toward 1,4-dichlorobutane (DCB).
We observed the enhanced production of recombinant RDhI enzyme in this host-E.
coli B834(DE3) competent cell. The following table shows the relationship
between
to dehalogenating activity and the percent of rRDhl enzyme in the total
soluble protein among
different expression systems and host cells.
ExpressionCompetent rRDhl as % DCB' Activity per
S stem Host Cell of mL
Soluble Proteinof Culture ( x
10' )
EXPROK E. coli AG 1 -3 -0.3
'
RSET E. coli JM 109 -10 -0.8
TrcHis E. coli TOP -15 -2.4
10F"
TrxFus E. coli G! 174'-30 -4.8
TrcHis E. coli 8834 -42 -4.5 - 12.5
DE3
RSET E. coli 8834 -48 -14.8
DE3
* DCB unit is a measure of dechlorination activity toward 1,4-dichlorobutane
(DCB).
' E. coli AG 1 chemically competent cells were purchased from Stratagene (La
Jolla, CA, USA};
E. coli TOP 10F' chemically competent cells were purchased from Invitrogen
(Carlsbad, CA,
USA); E. coli GI 174 cells were purchased from Invitrogen (Carlsbad, CA) and
were made
electro-competent according to the supplier's instructions.
Example 5
Modified Rhodococcus Dehalogenase
Since the Rhodococcus dehalogenase being produced by the TrcHis RDhI construct
had been modified with additional amino acids at both the amino and carboxy
termini,
plasmid constructs were generated to test the effects each of these
modifications might have
on the activity of the enzyme. The amino terminal poly-histidine tail was
eliminated by
enzymatic digestion of the pTrcHis RDhI 18-3 plasmid with Ncol and Agel and
the ligation of
a 17 by oligo into the resulting gap.
44
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
The DNA sequences of the oligo pairs are as follows:
RDhI Delta His-6-F
5'-CATGGGTGAAATAGGTA-3'
RDhI Delta His-6-R
5'-CCGGTACCTATTTCACC-3'
Using standard molecular biology protocols, the His-6-F and His-6-R
oligonucleotides
were annealed, ligated into the digested 18-3 construct, and transformed into
competent E.
coli TOP10 F' cells. Transformed colonies were selected by growth on LB/Amp
agar plates.
The resulting amino terminal sequence was:
-12 -11 3
~ 5 ATG GGT GAA ATA GGT
Met Gly Ile
(shown with the amino acid numbering of the original, unmodified sequence).
Digestion and re-figation resulted in a construct in which the Ala-293 Ser-294
~o sequence (Figure 2) became an Ala-293 Arg-294 sequence. Following the Arg-
encoding
codon is a stop codon which corresponds to the TGA nucleotide tri-mer at bases
927-979 in
the original sequence.
The carboxy terminus EXFLAG was eliminated by digesting pTrcHis RDhI 18-3 with
Avr II and Nhel and re-ligating the plasmid.
25 Individual clones were screened by enzymatic digestion and gel
electrophoresis.
Candidate clones were grown at 37° C in 5 mL cultures, induced with
IPTG, and lysed by
sonication. The lysates were analyzed by PAGE, Western blot, and chloride
detection
assay. Those clones lacking the amino terminal poly-histidines or the carboxy
terminal
EXFLAG demonstrated catalytic activity equal to the original construct.
Example 6
Construction of pTrcHis RDhI-S-Tag and pRSET RDhI-S-Talc
Material and Methods:
CTERM S-Tag F (forward) and CTERM S-Tag R (reverse) are two primers that were
designed to change the FLAG polypeptide - an 11 amino acid sequence - to the S-
Tag
polypeptide, a 15 amino acid sequence. The sequences of these oligonucleotides
are as
follows (each strand of the S-Tag fragment is underlined):
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
---Avr II---
CTERM S-Tag F 5'-CTA GGT GAC AAA GAA ACC GCT GCT GCT AAA
___Nsp V--_
TTC GAA CGC CAG CAC ATG GAC AGC AAA TAA
GTT TAA ACA TCA TTCCAATTGC
1« ---Not I---
CTERM S-Tag R 5'-GGCCGCAATTGGAATGATGTTTA AAC TTA TTT GCT
---Nsp V---
GTC CAT GTG CTG GCG TTC GAA TTT AGC AGC AGC
GGT TTC TTT GTCAC
Construction of pTrcHis RDhI-S-Taa
To generate plasmid pTrcHis RDhI-S-Tag, the plasmid pTrcHis RDhI clone 18-3
was
2« digested with the restriction enzymes Avr II and Not I and ligated with the
S-Tag fragment
(the S-Tag fragment was prepared by annealing primer CTERM S-Tag F and primer
CTERM S-Tag R together at room temperature). The new construct, pTrcHis RDhI-S-
Tag,
was incorporated into E. coli AG 1 competent cells (from Stratagene of La
Jolla, CA, USA)
and ampicillin resistant colonies were picked. Plasmids containing the S-Tag
fragment were
identified by analytical restriction enzyme digestion.
Construction of pRSET RDhI-S-Taa
The same procedure that was used to construct pTrcHis RDhI-S-Tag was also used
to construct pRSET RDhI-S-Tag, but instead starting with the plasmid pRSET
RDhI clone
16-4 - digested with the restriction enzymes, Avr II and Not I - and ligating
that construct to
3o the S-Tag fragment described above.
Semi-purified rRDhl (the EXFLAG-tagged protein derived from TrcHis RDhI Clone
18-3) was compared kinetically with semi-purified RDhI-S-Tag protein produced
in this
example. Chloride-releasing activity was examined at TCP concentrations
ranging from
OmM to SmM. As shown in Figure 19, the S-Tag-modified protein exhibited a
consistent
increase of about 15% in Vmax over the EXFLAG-modified protein. The S-Tag
protein also
exhibits a ~25% lower Km for TCP than does the EXFLAG protein. These results
confirm
that changes at the C-terminal end of the TDhI enzyme can be used to modulate
and
improve activity of the enzyme.
46
._.. .....
~,.
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
Example 7
Reactor Desian and Performance
Reactor Set-Up:
A bench-scale reactor was assembled using all 316 stainless steel with ~/4
inch ID, in
a shell and tube design. Reactor inlet and outlet tubing were also stainless
steel. A Lauda
circulating water bath (with a thermostat) was used to maintain the reactor at
30°C. The
reactor was packed with immobilized rRDhl enzyme running in an up-flow
direction. The
immobilized rRDhl enzyme was first prepared by loading a partially purified
enzyme
preparation (approximately 70% purity by SDS-PAGE) onto PEI-impregnated
alumina (ISP
to 4000 grade from UOP) which had been pretreated with 25% (w/v)
glutaraldehyde for 2
hours: this was followed by extensive washing with distilled water. Sufficient
protein was
introduced to the alumina to provide 300 mg protein (by Lowry method) per gm
of support.
Binding was allowed to occur overnight at room temperature. Bound enzyme
activity was
estimated by measuring unbound enzyme activity in the bathing solution or by
final
i ~ absorbance at 280 nm.
The immobilized rRDhl enzyme was transferred to the reactor using 2 mm glass
beads as spacers at the inlet and outlet. Flow was initiated using an aqueous
feed of a pre-
warmed 10 mM sodium phosphate/10 uM EDTA buffer (pH 7.0). After several hours
of wash
to remove any unbound enzyme, the aqueous feed was saturated with 1,2,3-
20 trichloropropane (TCP) and delivered as a continuously stirred solution at
a flow rate of 0.15
mUmin. The reactor was allowed to equilibrate for - 2 residence times before
sampling the
inlet and outlet streams for analysis of reactant TCP and product 2,3-dichloro-
1-propanol
(DCH) concentrations by GC. In order to prepare the samples for GC, each
sample was first
saturated with sodium sulfate and then extracted with chloroform (2 volumes)
containing
25 lOmM each of two internal standards (1,1,1,2-tetrachloroethane and 3-chloro-
1-propanol).
TCP and DCH levels were then estimated from the GC data using the internal
standard
method and the productivity (the percent yield per volume per time) was
calculated
therefrom. This initial productivity was used as a measure of initial enzyme
activity.
Productivity of Bench Scale rRDhl Bioreactor
3o The bioreactor was run continuously for a period of three months, with
periodic
sampling of the inlet and outlet streams according to the above-described
method.
Volumetric productivity (product weight per fluid volume per minute) of the
enzyme was
determined at each time point and the percent conversion fell from about 60%
to about 40%
over this time period. Measurements are presented in Figure 16. Based on this
data, the
47
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
half life of the immobilized enzyme was estimated to be about 3500 hours. This
places the
immobilized dehalogenase among the most stable protein catalysts yet reported.
Example 8
Directed Evolution of Dehalogenases by EPPCR
Error-Prone PCR Mutagenesis was performed upon Agel- and Nhel-digests of
plasmid pTrc/His RDhI 18-3 which had been purified by agarose gel
electrophoresis and
extracted from the gel. The EPPCR products were ligated into expression
vectors having
Agel and Nhel cutting sites. The resulting plasmids were transformed into
competent AGI
cells which were grown into colonies on ampicillin-supplemented agar. The
resulting cell
clone "pTric/His RDhI" EPPCR library was tested using the procedure for
measuring RDhI
enzyme activity by detection of pH change, as follows: welts B1 to H12 of a 96-
well
microplate, each containing 200pL SOB/Amp broth, was inoculated with a single
colony from
the pTrc/His RDhI EPPCR library: wells A1-A6 contain only media as a negative
control, and
i s wells A7-A12 were inoculated with wild type pTrc/His RDhI 18-3 colonies as
a positive
control. Representative results are presented in Figures 17 and 18.
The results demonstrate that most of the clones produced by EPPCR mutagenesis
exhibit activities equal to or less than the activity range for the wild-type
RDhI produced by
TrcHis RDhI Clone 18-3. However, in each case in which 84 random clones from
an EPPCR
20 library were analyzed for dehafogenase activity, a few on each 96-well
plate exhibited activity
significantly higher than that of the wild-type enzymes.
On February 3, 1998, the three piasmids, pTrcHis RDhI clone 18-3, pRSET RDhI
clone 16-4, and pTrxFus RDhI clone 4, were deposited with the American Type
Culture
Collection (ATCC) in accordance with the Budapest Treaty and were respectively
given the
25 following designations: ATCC 209609, ATCC 209610, and ATCC 209611. On
February 3,
1998, the cell culture E. coli TrxFus RDhI clone 4, was deposited with the
American Type
Culture Collection (ATCC) in accordance with the Budapest Treaty and was given
the
following designation: ATCC 202087.
On January 30, 1998, the two cell cultures, E. coli TrcHis RDhI clone 18-3 and
E. coli
3o RSET RDhI clone 16-4 were deposited with the American Type Culture
Collection (ATCC) in
accordance with the Budapest Treaty and were respectively given the following
designations: ATCC 202086 and ATCC 202085.
48
CA 02281931 1999-08-12
WO 98/36080 PCT/US98/02776
Other embodiments of the invention will be apparent to those skilled in the
art from a
consideration of this specification or practice of the invention disclosed
herein. It is intended
that the specification and examples be considered as exemplary only, with the
true scope
and spirit of the invention being indicated by the following claims.
49
CA 02281931 1999-08-12
GENERATION INFORMATION:
APPLICANT:
NAME: The Dow Chemical Company
STREET: 1790 Bldg. Washington Street
CITY: Midland
STATE: MI
COUNTRY: U.S.A.
POSTAL CODE: 48674
TELEPHONE: 517-636-1687
TELEFAX: 517-638-9786
TITLE OF INVENTION: Recombinant Haloaliphatic Dehalogenases
NUMBER OF SEQUENCES: 26
COMPUTER READABLE FORM:
MEDIUM TYPE: 3-1/2" Diskette
COMPUTER: IBM PC compatible
OPERATING SYSTEM: MS-Windows 95, Ver. 4.00
SOFTWARE: MS-Word for Windows, Ver. 7.0
INFORMATION FOR SEQ ID N0:1:
SEQUENCE CHARACTERISTICS
LENGTH: 305 _
TYPE: amino acid
STRANDEDNESS: single
TOPOLOGY: linear
ORIGINAL SOURCE:
ORGANISM: Rhodococcus rhodocrous
INDIVIDUAL ISOLATE: TDTM003
FEATURE:
NAME/KEY: RDhl Enzyme
LOCATION: 1..292
FEATURE:
NAME/KEY: Carboxy-terminal EXFLAG tail
LOCATION: 295..305
FEATURE:
NAME/KEY: Amino-terminal poly-His tail
LOCATION: -10..-1
SEQUENCE DESCRIPTION: SEQ ID N0:1:
Met Gly Gly Ser His His His His His His Gly Met Ser Glu Ile Gly
-12 -10 -5 -1 1
Thr Gly Phe Pro Phe Asp Pro His Tyr Val Glu Val Leu Gly Glu Arg
10 15 20
Met His Tyr Val Asp Val Gly Pro Arg Asp Gly Thr Pro Val Leu Phe
25 30 35
Leu His Gly Asn Pro Thr Ser Ser Tyr Leu Trp Arg Asn Ile Ile Pro
40 45 50
His Val Ala Pro Ser His Arg Cys Ile Ala Pro Asp Leu Ile Gly Met
55 60 65
Gly Lys Ser Asp Lys Pro Asp Leu Asp Tyr Phe Phe Asp Asp His Val
70 75 80
CA 02281931 1999-08-12
Arg Tyr Leu Asp Ala Phe Ile Glu Ala Leu Gly Leu Glu Glu Val Val
85 90 95 100
Leu Val Ile His Asp Trp Gly Ser Ala Leu_Gly Phe His Trp A1a Lys
105 110 115
Arg Asn Pro Glu Arg Val Lys Gly I1e Ala Cys Met Glu Phe Ile Arg
120 125 130
Pro Ile Pro Thr Trp Asp Glu Trp Pro Glu Phe Ala Arg Glu Thr Phe
135 140 145
Gln Ala Phe Arg Thr Ala Asp Va1 Gly Arg Glu Leu Ile Ile Asp Gln
150 155 160
Asn Ala Phe Ile Glu Gly Val Leu Pro Lys Cys Val Val Arg Arg Leu
165 170 175 190
Thr Glu Val Glu Met Asp His Tyr Arg Glu Pro Phe Leu Lys Pro Val
185 190 195
Asp Arg Glu Pro Leu Trp Arg Phe Pro Asn Glu Ile Pro Ile Ala G1y
200 205 210
G1u Pro Ala Asn Ile Val Ala Leu Val Glu Ala Tyr Met Asn Trp Leu -
215 220 225
His Gln Ser Pro Val Pro Lys Leu Leu Phe Trp G1y Thr Pro Gly Va1
230 235 240
Leu Ile Pro Pro Ala Glu Ala Ala Arg Leu Ala Glu Ser Leu Pro Asn
245 250 255 200
Cys Lys Thr Val Asp Ile Gly Pro Gly Leu His Tyr Leu Gln Glu Asp
265 270 275
Asn Pro Asp Leu I1e Gly Ser Glu Ile Ala Arg Trp Leu Pro Gly Leu
280 285 290
Ala Ser Lys Leu Gly Asp Tyr Lys Asp Asp Asp Asp Lys
295 300 305
INFORMATION FOR SEQ ID N0:2:
SEQUENCE CHARACTERISTICS
LENGTH: 973
TYPE: DNA
STRANDEDNESS: double
TOPOLOGY: linear
ORIGINAL SOURCE:
ORGANISM: Rhodococcus rhodocrous
INDIVIDUAL ISOLATE: TDTM003
FEATURE:
NAME/KEY: RDhl DNA
LOCATION: 39..914
FEATURE:
NAME/KEY: Carbo:cy-terminal EXFLAG tail DNA
LOCATION: 921..953
FEATURE:
NAME/KEY: Amino-terminal poly-His tail DNA
LOCATION:. 9..38
51
CA 02281931 1999-08-12
SEQUENCE DESCRIPTION: SEQ ID N0:2:
CC ATGGGGGGT TCTCATCATCAT CATCAT CATGGTATG TCTGAAATA 47
GGT ACCGGTTTT CCCTTCGACCCT CATTAT GTGGAAGTC CTGGGCGAG 95
GGT ATGCACTAC GTCGATGTTGGA CCGCGG GATGGCACG CCTGTGCTG 143
TTC CTGCACGGT AACCCGACCTCG TCCTAC CTGTGGCGC AACATCATC 191
CCG CATGTAGCA CCGAGTCATCGG TGCATT GCTCCAGAC CTGATCGGG 239
ATG GGAAAATCG GACAAACCAGAC CTCGAT TATTTCTTC GACGACCAC 287
GTC CGCTACCTC GATGCCTTCATC GAAGCC TTGGGTTTG GAAGAGGTC 335
GTC CTGGTCATC CACGACTGGGGC TCAGCT CTCGGATTC CACTGGGCC 383
AAG CGCAATCCG GAACGGGTCAAA GGTATT GCATGTATG GAATTCATC 431
CGG CCTATCCCG ACGTGGGACGAA TGGCCG GAATTCGCC CGTGAGACC 479
TTC CAGGCCTTC CGGACCGCCGAC GTCGGC CGAGAGTTG ATCATCGAT 527
CAG AACGCTTTC ATCGAGGGTGTG CTCCCG AAATGCGTC GTCCGTCCG 575
CTT ACGGAGGTC GAGATGGACCAC TATCGC GAGCCCTTC CTCAAGCCT 623
GTT GACCGAGAG CCACTGTGGCGA TTCCCC AACGAGATC CCCATCGCC 671
GGT GAGCCCGCG AACATCGTCGCG CTCGTC GAGGCATAC ATGAACTGG 719
CTG CACCAGTCA CCTGTCCCGPAG TTGTTG TTC.TGGGGC ACACCCGuC 767
GTA CTGATCCCC CCGGCCGAAGCC GCGAGA CTTGCCGAA AGCCTCCCC 815
AAC TGCAAGACA GTGGACATCGGC CCGGGA TTGCACTAC CTCCAGGAA 863
GAC AACCCGGAC CTTATCGGCAGT GAGATC GCGCGCTGG CTCCCCGGA 911
CTC GCTAGCGGC CTAGGTGACTAC AAGGAC GATGATGAC AAATAATGA 959
973
GCG GCCGCAAGCTT
INFORMATION FOR SEQ ID N0:3:
SEQUENCE CHARACTERISTT_CS
LENGTH: 295
TYPE: amino acid
STRANDEDNESS: single
TOPOLOGY: linear
ORIGINAL SOURCE:
ORGANISM: Pseudomonas spp.
FEATURE:
NAME/KEY: tetrachloro-cyclohexadiene hydrolase
LOCATION: 1..295
SEQUENCE DESCRIPTION: SEQ ID N0:3:
Met Ser Leu Gly Ala Lys Pro Phe Gly Glu Lys Lys Phe I1e Glu I1e
1 5 lp 15
52
CA 1999-08-12 .
02281931
LysGlyArg ArgMetAla TyrIleAsp GluGlyThr GlyAspPro Ile
20 25 30
LeuPheGln HisGlyAsn ProThrSer SerTyrLeu TrpArgAsn Ile
35 40 45
MetProHis CysAlaGly LeuGlyArg LeuIleAla CysAspLeu I1e
50 55 60
GlyMetGly AspSerAsp LysLeuAsp ProSerGly ProGluArg Tyr
65 70 75 80
AlaTyrAla GluHisArg AspTyrLeu AspA1aLeu TrpGluAla Leu
85 90 95
AspLeuGly AspArgVal ValLeuVal ValHisAsp TrpGlySer Ala
100 105 110
LeuGlyPhe AspTrpAla ArgArgHis ArgGluArg ValGlnGly Ile
115 120 125
AlaTyrMet GluAlaIle AlaMetPro IleG1uTrp AlaAspPhe Pro
130 135 140
GluGlnAsp ArgAspLeu PheGlnAla PheArgSer GlnAlaG1y Glu -
145 150 155 160
GluLeuVal LeuGlnAsp AsnValPhe ValGluGln ValLeuPro Gly
165 170 175 -
LeuIleLeu ArgProLeu SerGluAla GluMetAla AlaTyrArg Glu
180 185 190
ProPheLeu AlaAlaGlu AlaArgArg ProThr.Leu SerTrpPro Arg
195 200 205
GlnIlePro IleAlaGly ThrProAla AspValVal AlaI1eAla Arg
210 215 220
AspTyrAla GlyTrpLeu SerGluSer ProIlePro LysLeuPhe I1e
225 230 235 240
AsnAlaGlu ProGlyAla LeuThrThr GlyArgMet ArgAspPhe Cys
245 250 255
ArgThrTrp ProAsnGln ThrGluIle ThrValAla GlyAlaHis Fhe
260 265 270
IleGlnGlu AspSerPro AspGluIle GlyAlaAla IleAlaAla Phe
275 280 285
ValArgArg LeuArgPro Ala
290 295
INFORMA TIONFORSEQID N0:4:
SEQUENCE CHARACTERISTICS
LENGTH: 311
TYPE: amino acid
STRANDEDNESS: single
TOPOLOGY: linear
ORIGINAL SOURCE:
ORGANISM: Renilla reniformis
53
CA 02281931 1999-08-12
FEATURE:
NAME/KEY: luciferin monooxygenase
LOCATION: 1..311
SEQUENCE DESCRIPTION: SEQ ID N0:4:
Met Thr Ser Lys Val Tyr Asp Pro Glu Gln Arg Lys Arg Met Ile Thr
,1 5 10 15
Gly Pro Gln Trp Trp Ala Arg Cys Lys Gln Met Asn Val Leu Asp Ser
20 25 30
Phe Ile Asn Tyr Tyr Asp Ser Glu Lys His Ala Glu Asn Ala Val I1e
35 40 45
Phe Leu His Gly Asn Ala Ala Ser Ser Tyr Leu Trp Arg His Val Val
50 55 60
Pro His Ile Glu Pro Val Ala Arg Cys Ile Ile Pro Asp Leu Ile Gly
65 70 75 80
Met Gly Lys Ser Gly Lys Ser Gly Asn Gly Ser Tyr Arg Leu Leu Asp
85 90 95
His Tyr Lys Tyr Leu Thr Ala Trp Phe Glu Leu Leu Asn Leu Pro Lys -
100 105 110
Lys Ile Ile Phe Val Gly His Asp Trp Gly Ala Cys Leu Ala Phe His
115 120 125
Tyr Ser Tyr Glu His Gln Asp Lys Ile Lys Ala Ile Val His Ala Glu
130 135 140
Ser Val Val Asp Val Ile Glu Ser Trp Asp Glu Trp Pro Asp Ile Glu
145 150 155 160
Glu Asp Ile Ala Leu Ile Lys Ser Glu Glu Gly Glu Lys Met Val Leu
165 170 175
Glu Asn Asn Phe Phe Val Glu Thr Met Leu Pro Ser Lys Ile Met Arg
180 185 190
Lys Leu Glu Pro Glu Glu Phe Ala Ala Tyr Leu Glu Pro Phe Lys Glu
195 200 205
Lys G1y Glu Val Arg Arg Pro Thr Leu Ser Trp Pro Arg Glu Ile Pro
210 215 220
Leu Val Lys Gly Gly Lys Pro Asp Val Val Gln Ile Val Arg Asn Tyr
225 230 235 240
Asn Ala Tyr Leu Arg Ala Ser Asp Asp Leu Pro Lys Met Phe Ile Glu
245 250 255
Ser Asp Pro Gly Phe Phe Ser Asn Ala Ile Val Glu Gly Ala Lys Lys
260 265 270
Phe Pro Asn Thr Glu Phe Val Lys Val Lys Gly Leu His Phe Ser Gln
275 280 285
Glu Asp Ala Pro Asp Glu Met Gly Lys Tyr Ile Lys Ser Phe Val Glu
290 295 300
Arg Val Leu Lys Asn Glu Gln
305 310
54
CA 02281931 1999-08-12
INFORMATION FOR SEQ ID N0:5:
SEQUENCE CHARACTERISTICS
LENGTH: 310
TYPE: amino acid
STRANDEDNESS: single
TOPOLOGY: linear
ORIGINAL SOURCE:
ORGANISM: Xanthobacter autotrophicus
INDIVIDUAL ISOLATE: GJ10
FEATURE:
NAME/KEY: dehalogenase
LOCATION: 1..310
SEQUENCE DESCRIPTION: SEQ ID N0:5:
Met Ile Asn Ala Ile Arg Thr Pro Asp Gln Arg Phe Ser Asn Leu Asp
1 5 10 15
Gln Tyr Pro Phe Ser Pro Asn Tyr Leu Asp Asp Leu Pro Gly Tyr Pro
20 25 30
Gly Leu Arg Ala His Tyr Leu Asp Glu Gly Asn Ser Asp Ala Glu Asp -
35 40 45
Val Phe Leu Cys Leu His Gly Glu Pro Thr Trp Ser Tyr Leu Tyr Arg -
50 55 60
Lys Met Ile Pro Val Phe Ala Glu Ser Gly Ala Arg Val I1e Ala Fro
65 70 75 80
Asp Phe Phe Gly Phe Gly Lys Ser Asp Lys Pro Val Asp Glu Glu Asp
85 90 95
Tyr Thr Phe Glu Phe His Arg Asn Phe Leu Leu Ala Leu Ile Glu Arg
100 105 110
Leu Asp Leu Arg Asn Ile Thr Leu Val Val Gln Asp Trp Gly Gly Fhe
115 120 125
Leu Gly Leu Thr Leu Pro Met Ala Asp Pro Ser Arg Phe Lys Arg Leu
135 140
130
Ile Ile Met Asn Ala Cys Leu Met Thr Asp Pro Val Thr Gln Pro Ala
150 155 160
145
Phe Ser Ala Phe Val Thr Gln Fro Ala Asp Gly Phe Thr Ala Trp Lys
165 170 175
Tyr Asp Leu Val Thr Pro Ser Asp Leu Arg Leu Asp Gln Phe Met Lys
180 185 190
Arg Trp Ala Pro Thr Leu Thr Glu Ala Glu Ala Ser Ala Tyr Ala Ala
195 200 205
Pro Phe Pro Asp Thr Ser Tyr Gln Ala Gly Val Arg Lys Phe Pro Lys
210 215 220
Met Val Ala Gln Arg Asp Gln Ala Cys Ile Asp Ile Ser Thr G1u Ala
230 235 240
225
Ile Ser Phe Trp Gln Asn Asp Trp Asn Gly Gln Thr Phe Met Ala Ile
245 250 255
CA 02281931 1999-08-12
Gly Met Lys Asp Lys Leu Leu Gly Pro Asp Val Met Tyr Pro Met Lys
260 265 270
Ala Leu Ile Asn Gly Cys Pro Glu Pro Leu Glu Ile Ala Asp Ala Gly
275 280 285
His Phe Val Gln Glu Phe Gly Glu Gln Val Ala Arg Glu Ala Leu Lys
290 295 300
His Phe Ala Glu Thr Glu .
305 310
INFORMATION FOR SEQ ID N0:6:
SEQUENCE CHARACTERISTICS
LENGTH: 32
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide RDhl 5.4
LOCATION: 1..32
SEQUENCE DESCRIPTION: SEQ ID N0:6:
GGTTCCATGG GNTTYCCNTT YGAYCCNCAY TA 32 -
INFORMATION FOR SEQ ID N0:7:
SEQUENCE CHARACTERISTICS .
LENGTH: 28
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide RDhl 3.12
LOCATION: 1..28
SEQUENCE DESCRIPTION: SEQ ID N0:7:
CAGAGCTAGC GAGTCCGGGG AGCCAGCG 28
INFORMATION FOR SEQ ID N0:8:
SEQUENCE CHARACTERISTICS
LENGTH: 87
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide RDh1 5.7
LOCATION: 1..87
SEQUENCE DESCRIPTION: SEQ ID N0:8:
CGTACATATG GCCATGGGGG GTTCTCATCA TCATCATCAT CATGGTATGT CTGAAATAGG 60
TACCGGTTTT CCCTTCGACC CTCATTA 87
56
CA 02281931 1999-08-12
INFORMATION FOR SEQ ID N0:9:
SEQUENCE CHARACTERISTICS
LENGTH: 33
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide RDhl 3.13
LOCATION: 1..33
SEQUENCE DESCRIPTION: SEQ ID N0:9:
GATGACAAAT AATGAGCGGC CGCAAGCTTG TAC 33
INFORMATION FOR SEQ ID N0:10:
SEQUENCE CHARACTERISTICS
LENGTH: 55
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide Trx2++
LOCATION: 1..55
SEQUENCE DESCRIPTION: SEQ ID N0:10:
CCGGGGATCC CATGGCTTCT GAAATACGTA CCGGTTTTCC CTTCGACCCT CATTA 55
INFORMATION FOR SEQ ID NO:11:
SEQUENCE CHARACTERISTICS
LENGTH: 33
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide Trx-
LOCATION: 1..33
SEQUENCE DESCRIPTION: SEQ ID N0:11:
TCGACTGCAG GCGGCCGCTC ATTATTTGTC ATC 33
INFORMATION FOR SEQ ID N0:12:
SEQUENCE CHARACTERISTICS
LENGTH: 18
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide Dhl Seq 7
LOCATION: 1..18
SEQUENCE DESCRIPTION: SEQ ID N0:12:
57
. CA 02281931 1999-08-12
CCTGTCCCGA AGTTGTTG 18
INFORMATION FOR SEQ ID N0:13:
SEQUENCE CHARACTERISTICS
LENGTH: 17
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide Dhl Seq 8
LOCATION: 1..17
SEQUENCE DESCRIPTION: SEQ ID N0:13:
CGGGCCGP.TC TCCACTG 17
INFORMATION FOR SEQ ID N0:14:
SEQUENCE CHARACTERISTICS
LENGTH: 17
TYPE: DNA _
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide Dhl Seq 11
LOCATION: 1..17
SEQUENCE DESCRIPTION: SEQ ID N0:14:
TGCTCCAGAC CTGATCG 17
INFORMATION FOR SEQ ID N0:15:
SEQUENCE CHARACTERISTICS
LENGTH: 17
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide Dhl Seq 12
LOCATION: 1..17
SEQUENCE DESCRIPTION: SEQ ID N0:15:
TCTGATCGAT GATCAAC 17
INFORMATION FOR SEQ ID N0:16:
SEQUENCE CHARACTERISTICS
LENGTH: 18
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide Dhl Seq 13
LOCATION: 1..18
58
CA 02281931 1999-08-12
SEQUENCE DESCRIPTION: SEQ ID N0:16:
TCCCGACGTG GACGAATG 18
INFORMATION FOR SEQ ID N0:17:
. SEQUENCE CHARACTERISTICS
LENGTH: 18
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide Dhl Seq 14
LOCATION: 1..18
SEQUENCE DESCRIPTION: SEQ ID N0:17:
GAGCGCGACG ATGTTCGC 18
INFORMATION FOR SEQ ID N0:18:
SEQUENCE CHARACTERISTICS _
LENGTH: 18
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear -
FEATURE:
NAME/KEY: Oligonucleotide Dhl Seq 15
LOCATION: 1..18
SEQUENCE DESCRIPTION: SEQ ID N0:18:
CACCCGGCGT ACTGATCC 18
INFORMATION FOR SEQ ID N0:19:
SEQUENCE CHARACTERISTICS
LENGTH: 18
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide Dhl Seq 18
LOCATION: 1..18
SEQUENCE DESCRIPTION: SEQ ID N0:19:
GAGACCGGTC AGCATTCC 18
INFORMATION FOR SEQ ID N0:20:
SEQUENCE CHARACTERISTICS
LENGTH: 18
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY:. Oligonucleotide PROK-Seql
59
CA 02281931 1999-08-12
LOCATION: 1..18
SEQUENCE DESCRIPTION: SEQ ID N0:20:
GAGCGGATAA CAATTTCA 18
INFORMATION FOR SEQ ID N0:21:
SEQUENCE CHARACTERISTICS
LENGTH: 18
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide PROK-Seq2
LOCATION: 1..18
SEQUENCE DESCRIPTION: SEQ ID N0:21:
TCTCATCCGC CAAAACAG 18
INFORMATION FOR SEQ ID N0:22:
SEQUENCE CHARACTERISTICS
LENGTH: 96
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide EXFLAG linker
LOCATION: 1..96
SEQUENCE DESCRIPTION: SEQ ID N0:22:
GAATTCAGCC ATGGCATAAG CTTTCTAGAC TCGAGGGAGC TAGCGGCCTA GGTGACTACA 60
GGACGATGAT GACAAATAAT GAGCGGCCGC TAGCTT 96
INFORMATION FOR SEQ ID N0:23:
SEQUENCE CHARACTERISTICS
LENGTH: 17
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide RDhl Delta His-6-F
LOCATION: 1..17
SEQUENCE DESCRIPTION: SEQ ID N0:23:
CATGGGTGAA ATAGGTA 17
INFORMATION FOR SEQ ID N0:24:
SEQUENCE CHARACTERISTICS
LENGTH: 17
TYPE: DNA
STRANDEDNESS: single
, , CA 02281931 1999-08-12
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide RDh1 Delta His-6-R
LOCATION: 1..17
SEQUENCE DESCRIPTION: SEQ ID N0:24:
CCGGTACCTA TTTCACC 17
INFORMATION FOR SEQ ID N0:25:
SEQUENCE CHARACTERISTICS
LENGTH: 82
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide CTERM S-Tag F
LOCATION: 1..82
SEQUENCE DESCRIPTION: SEQ ID N0:25:
CTAGGTGACA AAGAAACCGC TGCTGCTAAA TTCGAACGCC AGCACATGGA CAGCAP.ATP=. 60
GTTTAAACAT CATTCCAATT GC 82
INFORMATION FOR SEQ ID N0:26:
SEQUENCE CHARACTERISTICS
LENGTH: 82
TYPE: DNA
STRANDEDNESS: single
TOPOLOGY: linear
FEATURE:
NAME/KEY: Oligonucleotide CTERM S-Tag R
LOCATION: 1..82
SEQUENCE DESCRIPTION: SEQ ID N0:26:
GGCCGCAATT GGAATGATGT TTAAACTTAT TTGCTGTCCA TGTGCTGGCG TTCGAP_TTTP_ 60
GCAGCAGCGG TTTCTTTGTC AC 82
61