Note: Descriptions are shown in the official language in which they were submitted.
CA 02283704 1999-09-15
WO 98/42342 PCT/US98105486
TITLE OF THE INVENTION
THROMBIN INHIBITORS
BACKGROUND OF THE INVENTION
Thrombin is a serine protease present in blood plasma in
the form of a precursor, prothrombin. Thrombin plays a central role in
the mechanism of blood coagulation by converting the solution plasma
protein, fibrinogen, into insoluble fibrin.
Edwards et al., J. Amer. Chem. Soc. (1992) vol. 114, pp.
1854-63, describes peptidyl a-ketobenzoxazoles which are reversible
inhibitors of the serine proteases human leukocyte elastase and porcine
pancreatic elastase.
European Publication 363 284 describes analogs of peptidase
substrates in which the nitrogen atam of the scissile amide group of the
substrate peptide has been replaced by hydrogen or a substituted
carbonyl moiety.
Australian Publication 86245677 also describes peptidase
inhibitors having an activated electrophilic ketone moiety such as
fluoromethylene ketone or a-keto carboxyl derivatives.
Thrombin inhibitors described in prior publications contain
sidechains of arginine and lysine. These structures show low selectivity
for thrombin over other trypsin-like enzymes. Some of them show
toxicity of hypotension and liver toxicity.
European Publication 601 459 describes sulfonamido
heterocyclic thrombin inhibitors, such as N-[4-[(aminoimino-
methyl)amino]butyl]-1-[N-(2-naphthalenylsulfonyl)-L-phenylalanyl]-L-
prolinamide.
WO 94/29336 describes compounds which are useful as
thrombin inhibitors.
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
SUMMARY OF THE INVENTION
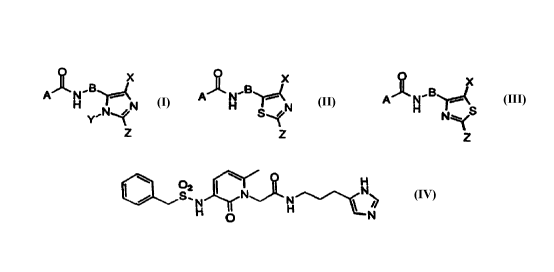
The invention relates to compounds of the formula:
O X O X O X
A~N~B ~ A~N~B ~~C A~N~B
H ~ I-I ' N H
Y~N~N S~ N~S
or Z or
and pharmaceutically acceptable salts thereof, wherein
A is selected from the group consisting of
N
W W~ N
f,,,. , ~ ,
O .....,.
X3
R2~X2 \ R3 N \ R3
Wv N Nw/~ , or Wv N I Nw/
H O H O
wherein
W1 is
H
X~
b
-2-
~ y. ., ......
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
wherein
Ra and Rb are independently selected from
hydrogen,
a heterocyclic group which is a stable 5- to 7-membered mono-
or bicyclic or stable 7- to 10-membered bicyclic heterocyclic
ring system any ring of which may be saturated or
unsaturated, and which consists of carbon atoms and from
one to three heteroatoms selected from the group consisting
of N, O and S, and wherein the nitrogen and sulfur
heteroatoms may optionally be oxidized, and the nitrogen
heteroatom may optionally be quaternized, and including
any bicyclic group in which any of the above-defined
heterocyclic rings is fused to a benzene ring,
C1_4 alkyl unsubstituted or substituted with CH3 or C3_7
cycloalkyl,
aryl,
substituted aryl with one or two substituents selected from
C1_4 alkyl,
C1_4 alkoxy,
methylenedioxy,
halogen or
hydroxy,
C3_7 cycloalkyl,
Cg_10 bicycloalkyl, or
Ra and Rb, along with the carbon to which they are attached,
form a Cg-7 cycloalkyl ring or
Rio
R11
where R10 is H or -OH, and
R11 is H or -OCHg, and
X1 is selected from the group consisting of
-3-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98105486
-OH
-~2
-NHCHg,
-NH(CH2)1-3CH3,
-NH(CH2)2-40H,
-NH(CH2)1-3COOH,
-NH(CH2)1-3COOR6, where R6 is C1_4alkyl,
-NH(CH2)1-3CONR7R8,
where R7 and R8 are independently hydrogen or
C 1_4alkyl,
n
- NH(CH2)j.~CO NOD
,
where D is 1, 2, 3, or 4 carbon atoms unsubstituted or
any 1, 2, 3, or 4 of which are substituted with OH,
-NHS02(CH2)1-3aryl,
-NH(CH2)1-3NH2,
-NHC3-7 cycloalkyl ring unsubstituted or substituted
with -OH, -C(O)OH, or -C(O)ORc, where Rc is
C 1_4 alkyl,
2
-NH(CH2)1-s Y Rs
where
Y2 is O or NH,
W4 is C or N,
Z1 is C or N, and
R6 is -CH20H or -N(CHg)2 provided that W4 and Z1
2~ are not the same,
O Ra
-NH(CH2)~-3C-N
~NR~
where
R7 is H or CH3, and
R8 is H or
-4-
._._____..-.~........_~_..._._ ... t ,. . .
CA 02283704 1999-09-15
WO 98/42342 PCT/US98105486
O
-CNH(tBu)
- NH S02- (C H2)1-2 N
-NHS02-(CH2)1-2
Rg
-NHS02-{CH2)~-2 -N
CH3
CH
S-N-
02 H ,
CH3 CH3
O S-N-
02 H
or
-NHS02-(CH2)1-2-NH-(CH2)2NH2
where RO is H, NH2, or OH;
or
W1 is
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
OH
B~
wherein B 1 is a bond, O, -CH2-O-, or -O-CH2-;
W2 is
hydrogen,
R1_~
R10C(O)-,
R1C(O)-,
R1S02-,
(R1)2CH(CH2)0-4NHC(O)-,
(R1)m(CH2)nNHqC(O)-,
where n is 0-4, m is 1 or 2, wherein R1 is same or different, and
q is 0 or 1, with the proviso that where n is 1-4, q is 1 and m is 1,
and where n is 0, m is 1 or 2, and q is 0 or 1, and where n is 0,
mis2andqis0;
R1 is
R17(CH2)t-, where t is 0-4,
(R17)(OR17)CH(CH2)p-, where p is 1-4,
(R17)2CH(CH2)r-, where r is 0-4 and each R17 can be the
same or different, and wherein (R17)2 can also form a
ring with CH represented by C3-7 cycloalkyl, C7-I2
bicylic alkyl, C10-16 t~cylic alkyl, or a 5- to 7- membered
mono- or bicyclic heterocyclic ring which can be
saturated or unsaturated, and which contains from one
to three heteroatoms selected from the group consisting
of N, O and S,
R170(CH2)p-, wherein p is 1-4;
-6-
CA 02283704 1999-09-15
WO 98142342 PCT/US98I05486
R2 and R17 are independently selected from
-phenyl, unsubstituted or substituted with one or more of
C 1-4 alkyl,
C1_4 alkoxy,
halogen,
hydroxy,
COOH, or
CONH2~
naphthyl,
biphenyl,
a 5- to 7- membered mono- or a 9- to 10-membered bicyclic
heterocyclic ring which can be saturated or unsaturated, and
which contains from one to four heteroatoms selected from the
group consisting of N, O and S,
-C1-7 alkyl, unsubstituted or substituted with one or more of
hydroxy,
COOH,
amino,
aryl,
C3_7 cycloalkyl,
heteroaryl, or
heterocycloalkyl,
-CFg
Cg-7 cycloalkyl,
C7-12 bicyclic alkyl, or
C10-16 tHCyclic alkyl;
X2 is
CF2,
CR15~R16
wherein R1~ and R16 are independently
hydrogen,
Cg-7 cycloalkyl,
_7_
CA 02283704 1999-09-15
WO 98/42342 PC'T/fJS98/05486
C1_4 alkyl unsubstituted or substituted with one or more of
hydroxy,
COOH,
ammo,
aryl,
heteroaryl, or
heterocycloalkyl,
aryl,
heteroaryl,
heterocycloalkyl, or
R1~ and R16 are joined to form a four to seven
membered cycloalkyl ring unsubstituted or
substituted with hydroxy, amino or aryl, or
S(O)r, where r is 0-2;
X3 is hydrogen or halogen;
R3 and R18 are independently selected from the group consisting of
hydrogen,
C 1-4 alkyl,
C3_7 cycloalkyl, or
trifluoromethyl;
B is selected from the group consisting of
C 1-4 alkyl,
C3_4 alkenyl, and
C3_4 alkynyi;
X is selected from the group consisting of
hydrogen,
halogen,
-CF3>
-CH2CFg,
-Cg-b cycloiakyl,
-CH2C3-5 cycloalkyl, and
-C1_4 alkyl;
_g_
CA 02283704 1999-09-15
WO 98142342 PCTIUS98/05486
Z is selected from the group consisting of
hydrogen,
-NH2>
-C1_4 alkylamino,
-C1_4 alkanol,
-C1_4 alkyl; and
Y is selected from the group consisting of
hydrogen, and
-C 1_7 alkyl,
-CH2CH2CH2CH2C3_6 cycloalkyl,
-CH2CH=CHCH2C3_g cycloalkyl,
-CHIC=CCH2C3_6 cycloalkyl,
-C H2C = CC H2C H2C3_6 cycioalkyl
-CH2CH2CH2CH2CH2Cg_g cycloalkyl,
-CH2CH=CHCH2CH2C3_6 cycloaikyl, and
-CH2COY1, wherein Y1 is selected from the group consisting of
-OC1_7 alkyl,
-OH,
-C 1-6 alkyl,
-Cg_g cyclolalkyl,
-CH2C3_6 cycloalkyl,
-CH2CH2C3-6 cycloalkyl,
-benzyl
-CH2benzyl
-NH2~
-NHCl_5 alkyl,
-NHC1_4 alkylCFg,
-NHC2_4 alkanol,
-NHC2_4 alkylamino,
-N NH
H
-N
-NHCH2 ~ ~ ,
3~5
_g_
I
CA 02283704 1999-09-15
WO 98142342 PCT/US98105486
H
N
-N
H
w
-NHCH2 ~ /N ,
~ /N ,
N-
-H \ /
-N
H \ / ,
N
-NHCH2 ~ / ,
-10-
CA 02283704 1999-09-15
WO 98142342 PCT/US98/05486
NH
-N
H
CH3
-N~N
H
-H N-CH3
CH3
N
-N
H
-11-
CA 02283704 1999-09-15
WO 98!42342 PCT/US98/05486
-N O
V
-N ~--Y2
wherein Y2 is H, NH2 or OH,
-N
3 '
Y
wherein Y3 is H, NH2 or OH, and
-N Y4
Y~
wherein Y4 is hydrogen, and
Y5 is NH2 or OH, or H
Y~ is hydrogen, and
Y4 is NH2 or OH.
The invention includes a composition for inhibiting loss of
blood platelets, inhibiting formation of blood platelet aggregates,
inhibiting formation of fibrin, inhibiting thrombus formation, and
inhibiting embolus formation in a mammal, comprising a compound of
the invention in a pharmaceutically acceptable carrier. These
compositions may optionally include anticoagulants, antiplatelet agents,
and thrombolytic agents. The compositions can be added to blood, blood
products, or mammalian organs in order to effect the desired
inhibitions.
The invention also includes a composition for preventing or
treating unstable angina, refractory angina, myocardial infarction,
-12-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
transient ischemic attacks, atrial fibrillation, thrombotic stroke, embolic
stroke, deep vein thrombosis, disseminated intravascular coagulation,
ocular build up of fibrin, and reocclusion or restenosis of recanalized
vessels, in a mammal, comprising a compound of the invention in a
pharmaceutically acceptable carrier. These compositions may
optionally include anticoagulants, antiplatelet agents, and thrombolytic
agents.
The invention also includes a method for reducing the
thrombogenicity of a surface in a mammal by attaching to the surface,
either covalently or noncovalently, a compound of the invention.
1~
DETAILED DESCRIPTION OF THE INVENTION
In one class of the invention, the compounds have the
formula
O X O X O X
A~N~B ~ A~N~g -- A~N~B
H ~ H
Y, N 1 N S ~N H N ~S
or Z or Z
and pharmaceutically acceptable salts thereof, wherein
A is selected from the group consisting of
-13-
ICA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
C H3 O
CHs~O~ N''~ N
C H3 H O ,,,,.~ ,
I ~ I
N
H2N~',.
O ,~''v' ,
N
S - N ~'~
O
/ ~
N
~S-N''
H3COOC O2 f"~ O ,,,r.r ,
-14-
W.
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
' N
S-N''
02 H p
O
O N
S-N'
O ,..
O
HgCO
H2N',,. N
O
-15-
CA 02283704 1999-09-15
WO 98142342 PCT/US98105486
N
H N
O ,r
N
3 \'
CH N O
1 / N
HO
O
-16-
r i
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
CH3
O
2
\ I s_ I N
N
H O ,
CI / ~ CH3
i
\ S~N N~
0 ,
C
02
\ I S, I N
N
H O
H3
CH3
02 I I
~S, N~~,
O
O
CH3
O
2
O S ~ N I N~/
i
O
-17-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
CH3
02 I I
S,N N~
H
CH3
02 ~ N~
H3C
O
~2
\ I S, I N '~.,
N
H O ,
CH3
/I
\ N N~
H O
/I
N~CH3
N N~~' , and
O
C H3
~Il -
wN~ N N
In one group of this class of the invention, the compounds
have the formula
-18-
,.
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
O X O X O X
A~ N~B ~ A~ N~B ~ A' _ N~B --
H ~ H
Y,N~N S~N H N~S
Z or Z or Z
and pharmaceutically acceptable salts thereof, wherein
B is selected from the group consisting of:
-CH2CH =CH- ,
-CH2CH2CH2- ,
-CH2C =C- , and
_CH2_
X is
H,
-CH3
-Cl
Z is
H,
-NH2, and
Y is selected from the group consisting of
-19-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
hydrogen,
-CH2COOC(CH3)s ,
-CH2COOH,
-CH2CONHC(CH3)s ,
-CH2CONHCH2CH3,
-CH2CONH -
,
-CH2CONHCH2-
,
CH2CONH J ,
N
H
-CH2CONH NH
-N
-CH2CONHCH2
-20-
,,
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
-CH2CONHC(CH3)2CH2CH3,
-CH2CONHCH2C(CH3)3,
-CH2CONHCH2CF3,
-CH2C0 ~ ,
-CH2CON~~OH
-CH2CON~ NH2
-CH2CONHCH2CH20H,
-CH2CONHCH2CH2NH2,
-CH2CONHC(CH3)2CH2NH2,
-CH2CONHCH2C(CH3)2NH2
-CH2COOC(CH3)3 .
Specific embodiments of the class include
\ /
/ \
'N ".
O N
H
/ /
O H N
H
-21-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
/ /
N
H
/ \
H2N",,.... O
O /
H N
H
H
-22-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
N
H
Me00C
~~_ N,u..~ ~ N
H O
O
/ /
O
H N
H
Me00C
O
N
H
-23-
CA 02283704 1999-09-15
WO 98/42342 PCTNS98/05486
H
N~,,~-~N
~o H
0
O
H
H
Me
N
H N
H
,
-24-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
M
H
\ /
/ \
H N"N~~~~ N
2
N
H
-25-
Image
Image
Image
-26-
CA 02283704 1999-09-15
WO 98142342 PCTIUS98/05486
O
I \ 02 I \~ H
/ S,N N~N N
H O H
N
I \ 02 \~ O H
/ S,N N~N N
H O H
N
CI I ~ O
O
/ S,N N~N H
N
H O H
N
I \ 02 I ~~ O ~-COO~Bu
S,N N~N N
H O H I /~
N
3.0
-27-
CA 02283704 1999-09-15
WO 98/42342 PCTIUS98105486
\ 02 I ~~ O ~-COOH
/ S.N N~N N
H O H
N
02 I \Y O r CONHtBu
/ S,N N~N N
H O H I /~
N
I ~ 02 \~ O rCONHEt
/ S,N N~N N
H O H
N
H
--~N
\ 02 I \~ O
/ S, N~ N O
H O H
'N
H
N
\ O2 \~ O
I / S, I N' ~ o
N v _N
H O H
N
-28-
CA 02283704 1999-09-15
WO 98/42342 PCT/pS98/05486
H
N
\ 02 ~ O
I / s. I N' ~ o
N v _N N
H O H
N
H
N
\ 02 \~ O
I / S. I N_ ~ p
N v 'N N
H O H I i
N
H ~ Fs
N
\ Oz \~ O
I / S. I N' ~ O
N v _N N
H O H
N
CN
\ O ~
N O
/ S2N N~N
H O H
N
-29-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
OH
\ O \ N
O
2
I ~ s_ I N' ~ N o
N v _N
H O H I /
N
H~OH
~/N
\ 02 \~ O
I / s_ I N' ~ o
N v _N
H O H I y
N
N~NH2
\ 02 \~ O
I / S, ( N' ~
N v N N
H O H
N
CI
I \ 02 I \~ O r-CONHtBu
/ S_N N~N N
H O H
N
-30-
CA 02283704 1999-09-15
WO 98/42342 PCTIUS98/05486
02 I ~~ O ~-CONH'Bu
/ S,N N~N N
H O H
N
02 I ~1' o r coo'Bu
O S,N N~N N
H O H
N
02 ( ~~ O ~CONHtBu
O S, N N~ N N
H H I
O N
02 I ~~ O ~-CONHtBu
O S, N N~ N N
H O H
N
O ~~ O rCONHtBu
2
S,N ~ N~ N
H O H I
'N
-31-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
O T-COOtBu
S,N N~N N
H O H
N
I ~ N~ O ~- COOtBu
N N~ N N
H O H
N
I ~ N~ O r CONHtBu
/ N N~ N N
H O H
N
I ~ i~ ~-COOtBu
N~N N~N N
H O H I y
N
I ~ N~ O r CON HtBu
IV N Nv _N N
H O H
N
-32-
CA 02283704 1999-09-15
WO 98/42342 PCT/IJS98/05486
H
N~ O
/ N N N N O
H O H
N
H ~ F3
N~ O
/ N N N N O
H O H I /
N
H
N
~ ~ N~_ ~o r
NI v N N~N N O
H O . H I />
N
H
N
O ~ NH
/ S,N N N N O
H O H I
N
-33-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
N
i
H
O ~N
N O
/ S~N N v -N
H O H I
N
H
\ ~ N
/ _O
S2 O
H O H
~N
NH2
H
~ N
I~ O I
/ N N N N O
H O H
N
H
I \ 02 ~ \ O / \vN
/ S,N N~N N O H
H O H
N
-34-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
NH
HN
w
N O
/HO
O H
N
N
N
I ~ IV O ~--.~ H
N N N N O
H O H
N
N NH2
O ~ O
2
/ S , I N~L O
H O H I ~>
~N
H J
N
N
02 ~ ~~ O ~ H
S,N N~N N O
H O H I /~
N
-35-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
H
CI \ \ N
O
I / S, I N~ O
N
O H I ~> H
'N
NH2
N-'
\ O \~ O
2
I ~ S, ( N~ O
0
'N
N,~NH2
\ 02 \~ O
s_ I N' ~ o
N v N
H O H
N
N ~~~ N H2
\ N~ O
N N N N O
H O H
N
-36-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
H
\ 02 ~ \ O ~N
/ S.N N~N N O H
H O H
N
/ ~ O
i
O ~S. N N ~ N
O H ~ H S ,.N
NH2
NH2
S --
\ N
N
H
NH2
\ ~ \ ~ s~
\ N
H N~~~~ N
2
O
O N
H
-37-
CA 02283704 1999-09-15
WO 98142342 PCT/US98/05486
W / I
/
hi2N~''~ N S
O I ~--NH2
O N N
H
O H
O
N / ~I
O N~ S
N I ~NH2
N
O
O N
S_ H
O
O NI \ S
I ~NH2
N
O
O_H NH2
n / I g--
O
O N' \ ~ N
H
N
O
-38-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
O N
S_H
O
I / O N' \ S
N ~ i/ NH2
N
O
S
,SO,-N~,,.. N I ~~--NH2
O H O N
I ~ H
or
S
H2N,,,,. N I ~ NH2
O O N
H
and pharmaceutically acceptable salts thereof.
Compounds of the present invention, which are thrombin
inhibitors, are useful in anticoagulant therapy. Anticoagulant therapy
is indicated for the treatment and prevention of a variety of thrombotic
conditions, particularly coronary artery and cerebrovascular disease.
Those experienced in this field are readily aware of the circumstances
requiring anticoagulant therapy. The term "patient" used herein is
-39-
CA 02283704 1999-09-15
WO 98142342 PCTlUS98I05486
taken to mean mammals such as primates, including humans, sheep,
horses, cattle, pigs, dogs, cats, rats, and mice.
Thrombin inhibition is useful not only in the anticoagulant
therapy of individuals having thrombotic conditions, but is useful
whenever inhibition of blood coagulation is required such as to prevent
coagulation of stored whole blood and to prevent coagulation in other
biological samples for testing or storage. Thus, thrombin inhibitors can
be added to or contacted with any medium containing or suspected of
containing thrombin and in which it is desired that blood coagulation be
inhibited, e.g. when contacting the mammal's blood with material
selected from the group consisting of vascular grafts, stems, orthopedic
prothesis, cardiac prosthesis, and extracorporeal circulation systems
The compounds of the invention can be administered in
such oral forms as tablets, capsules (each of which includes sustained
release or timed release formulations), pills, powders, granules, elixers,
tinctures, suspensions, syrups, and emulsions. Likewise, they may be
administered in intravenous (bolus or infusion), intraperitoneal,
subcutaneous, or intramuscular form, all using forms well known to
those of ordinary skill in the pharmaceutical arts. An effective but non-
~0 toxic amount of the compound desired can be employed as an anti-
aggregation agent. For treating ocular build up of fibrin, the compounds
may be administered intraocularly or topically as well as orally or
parenterally.
The compounds can be administered in the form of a depot
injection or implant preparation which may be formulated in such a
manner as to permit a sustained release of the active ingredient. The
active ingredient can be compressed into pellets or small cylinders and
implanted subcutaneously or intramuscularly as depot injections or
implants. Implants may employ inert materials such as biodegradable
polymers or synthetic silicones, for example, Silastic, silicone rubber or
other polymers manufactured by the Dow-Corning Corporation.
The compounds can also be administered in the form of
liposome delivery systems, such as small unilamellar vesicles, large
unilamellar vesicles and multilamellar vesicles. Liposomes can be
-40-
CA 02283704 1999-09-15
WO 98142342 PCT/US98/05486
formed from a variety of phospholipids, such as cholesterol,
stearylamine or phosphatidylcholines.
The compounds may also be delivered by the use of
monoclonal antibodies as individual carriers to which the compound
molecules are coupled. The compounds may also be coupled with
soluble polymers as targetable drug carriers. Such polymers can
include polyvinlypyrrolidone, pyran copolymer, polyhydroxy-propyl-
methacrylamide-phenol, polyhydroxyethyl-aspartamide-phenol, or
polyethyleneoxide-polylysine substituted with palmitoyl residues.
Furthermore, the compounds may be coupled to a class of biodegradable
polymers useful in achieving controlled release of a drug, for example,
polylactic acid, polyglycolic acid, copolymers of polylactic and
polyglycolic acid, polyepsilon caprolactone, polyhydroxy butyric acid,
polyorthoesters, polyacetals, polydihydropyrans, polycyanoacrylates and
cross linked or amphipathic block copolymers of hydrogels.
The dosage regimen utilizing the compounds is selected in
accordance with a variety of factors including type, species, age, weight,
sex and medical condition of the patient; the severity of the condition to
be treated; the route of administration; the renal and hepatic function of
the patient; and the particular compound or salt thereof employed. An
ordinarily skilled physician or veterinarian can readily determine and
prescribe the effective amount of the drug required to prevent, counter,
or arrest the progress of the condition.
Oral dosages of the compounds, when used for the indicated
effects, will range between about 0.1 mg per kg of body weight per day
(mglkg/day) to about 100 mg/kg/day and preferably 1.0-100 mg/kg/day
and most preferably 1-20 mg/kg/day. Intravenously, the most preferred
doses will range from about 0.01 to about 10 mg/kg/minute during a
constant rate infusion. Advantageously, the thrombin inhibitors may be
administered in divided doses of two, three, or four times daily.
Furthermore, they can be administered in intranasal form via topical
use of suitable intranasal vehicles, or via transdermal routes, using
those forms of transdermal skin patches well known to those of ordinary
skill in that art. To be administered in the form of a transdermal
-41-
CA 02283704 1999-09-15
WO 98142342 PCT/US98/05486
delivery system, the dosage administration will, or course, be
continuous rather than intermittent throughout the dosage regime.
For example, oral tablets can be prepared which contain an
amount of active compound of between 100 and 500 mg, e.g. 100, 200, 300,
400 or 500 mg. Typically, a patient in need of thrombin inhibitor
compound, depending on weight and metabolism of the patient, would be
administered between about 100 and 1000 mg active compound per day.
For a patient requiring 1000 mg per day, two tablets containing 250 mg of
active compound can be administered in the morning and two tablets
containing 250 mg of active compound can again be administered in the
evening. For a patient requiring 500 mg per day, one tablet containing
250 mg of active compound can be administered in the morning and one
tablet containing 250 mg of active compound can again be administered
in the evening.
The compounds are typically administered as active
ingredients in admixture with suitable pharmaceutical diluents,
excipients or carriers (collectively referred to herein as "carrier"
materials) suitably selected with respect to the intended form of
administration, that is, oral tablets, capsules, elixers, syrups and the
like, and consistent with convention pharmaceutical practices.
For instance, for oral administration in the form of a tablet
or capsule, the active drug component can be combined with an oral,
non-toxic, pharmaceutically acceptable, inert carrier such as lactose,
starch, sucrose, glucose, methyl cellulose, magnesium stearate,
dicalcium phosphate, calcium sulfate, mannitol, sorbitol and the like;
for oral administration in liquid form, the oral drug components can be
combined with any oral, non-toxic, pharmaceutically acceptable inert
carrier such as ethanol, glycerol, water and the Iike. Moreover, when
desired or necessary, suitable binders, lubricants, disintegrating agents
and coloring agents can also be incorporated into the mixture. Suitable
binders include starch, gelatin, natural sugars such as glucose or beta-
lactose, corn-sweeteners, natural and synthetic gums such as acacia,
tragacanth or sodium alginate, carboxymethylcellulose, polyethylene
glycol, waxes and the like. Lubricants used in these dosage forms
include sodium oleate, sodium stearate, magnesium stearate, sodium
-42-
r .. ......,.. ...............
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
benzoate, sodium acetate, sodium chloride and the like. Disintegrators
include, without limitation, starch methyl cellulose, agar, bentonite,
xanthan gum and the like.
The compounds can also be co-administered with suitable
anti-coagulation agents or thrombolytic agents such as plasminogen
activators or streptokinase to achieve synergistic effects in the treatment
of various ascular pathologies. For example, the compounds enhance
the efficiency of tissue plasminogen activator-mediated thrombolytic
reperfusion. The compounds may be administered first following
thrombus formation, and tissue plasminogen activator or other
plasminogen activator is administered thereafter. They may also be
combined with heparin, aspirin, or warfarin.
Specific embodiments of compounds of the invention inhibit
thrombin with a Ki range of less than 1.0 nM according to in vitro
measurements.
In vitro assay for determin~proteinase inhibition
Assays of human a-thrombin and human trypsin were
performed at 25°C in 0.05 M TRIS buffer pH 7.4, 0.15 M NaCI, 0.1% PEG.
Trypsin assays also contained 1 mM CaCl2.
In assays wherein rates of hydrolysis of a p-nitroanilide
(pna) substrate were determined, a Thermomax 96-well plate reader
was used to measure (at 405 nm) the time dependent appearance ofp-
nitroaniline. sar-PR-pna (sarcosine-Pro-Arg-p-nitroanilide) was used to
assay human a-thrombin (Km=125 N.M) and human trypsin (Km=59
~,M). p-Nitroanilide substrate concentration was determined from
measurements of absorbance at 342 nm using an extinction coefficient of
8270 cm-1M-1.
In certain studies with potent inhibitors (Ki < 10 nM) where
the degree of inhibition of thrombin was high, a more sensitive activity
assay was employed. In this assay the rate of thrombin catalyzed
hydrolysis of the fluorogenic substrate Z-GPR-afc (Cbz-Gly-Pro-Arg-7-
amino-4-trifluorornethyl coumarin) (Km=27 ~,M) was determined from
the increase in fluorescence at 500 nm (excitation at 400 nm) associated
with production of 7-amino-4-trifluoromethyl coumarin. Concentrations
-43-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
of stock solutions of Z-GPR-afc were determined from measurements of
absorbance at 380 nm of the 7-amino-4-trifluoromethyl coumarin
produced upon complete hydrolysis of an aliquot of the stock solution by
thrombin.
Activity assays were performed by diluting a stock solution
of substrate at least tenfold to a final concentration <_ 0.5 Km into a
solution containing enzyme or enzyme equilibrated with inhibitor.
Times required to achieve equilibration between enzyme and inhibitor
were determined in control experiments. Initial velocities of product
formation in the absence (Vo) or presence of inhibitor (Vi) were
measured. Assuming competitive inhibition, and that unity is
negligible compared Km/[S], [I]/e, and [I]/e (where [S], [I], and a
respectively represent the total concentrations, of substrate, inhibitor
and enzyme), the equilibrium constant (Ki) for dissociation of the
inhibitor from the enzyme can be obtained from the dependence of Vo/Vi
on [I] shown in equation 1.
Vo/Vi = 1 + [I]/Ki (1)
The activities shown by this assay indicate that the
compounds of the invention are therapeutically useful for treating
various conditions in patients suffering from unstable angina,
refractory angina, myocardial infarction, transient ischemic attacks,
atrial fibrillation, thrombotic stroke, embolic stroke, deep vein
thrombosis, disseminated intravascular coagulation, and reocclusion or
restenosis of recanalized vessels.
Some abbreviations that may appear in this application are
as follows.
~ i ..
CA 02283704 1999-09-15
WO 98142342 PCTIUS98/05486
Designation
BOC (Boc) t-butyloxycarbonyl
HBT(HOBT or HOBt) 1-hydroxybenzotriazole hydrate
BBC reagent benzotriazolyloxy-bis(pyrrolidino)-
carbonium hexafluorophosphate
PyCIU 1,1,3,3-bis(tetramethylene)-
chlorouronium hexafluorophosphate
EDC 1-ethyl-3-(3-dimethylaminopropyl)
carbodiimide hydrochloride
(BOC)20 di-t-butyl dicarbonate
DMF dimethylformamide
Et3N or TEA triethylamine
EtOAc ethyl acetate
TFA trifluoroacetic acid
DMAP dimethylaminopyridine
DME dimethoxyethane
BH3-THF Borane-tetrahydrofuran complex
D-Phe(3,4-C12) D-3,4-Dichlorophenylalanine
D-3,3-dicha D-3,3-Dicyclohexylalanine
Pro Proline
Ar g Arginine
Gly Glycine
D-3,3,-diphe D-3,3-Diphenylalanine
LAH lithium aluminum hydroxide
Cy cyclohexyl
POC13 phosphorous oxychloride
MeCN acetonitrile
BnEt3N+Cl- benzyl triethyl ammonium chloride
NaH sodium hydride
DMF dimethylformamide
BrCH2COOtBu tert butyl bromoacetate
EtOH ethyl alcohol
Pd(C) palladium on activated carbon catalyst
CF3COOH trifluoroacetic acid
DCM dichloromethane
-45-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98J05486
DIPEA diisopropylethylamine
The compounds of the present invention may have chiral
centers and occur as racemates, racemic mixtures and as individual
diastereomers, or enantiomers with all isomeric forms being included
in the present invention.
The term "alkyl" means straight or branched alkane
containing 1 to about 10 carbon atoms, e.g., methyl, ethyl, n-propyl,
isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl, iso-amyl, hexy,
octyl radicals and the Like. The term "alkenyl" means straight or
branched alkene containing 2 to about 10 carbon atoms, e.g., propylenyl,
buten-1-yl, isobutenyl, pentenylen-1-yl, 2,2-methylbuten-1-yl, 3-
methylbuten-1-yl, hexen-1-yl, hepten-1-yl, and octen-1-yl radicals and the
like. The term "alkynyl" means straight or branched alkyne containing
2 to about 10 carbon atoms, e.g., ethynyl, propynyl, butyn-1-yl, butyn-2-yl,
pentyn-1-yl, pentyn-2-yl, 3-methylbutyn-1-yl, hexyn-1-yl, hexyn-2-yl,
hexyn-3-yl, 3,3-dimethylbutyn-1-yl radicals and the like. Cycloalkyl
means a cyclic, saturated ring containing 3 to 8 carbon atoms, e.g.,
cyclopropyl, cyclohexyl, etc. Halogen means chloro, bromo, fluoro or
iodo. The term "aryl" means a 5- or 6-membered aromatic ring
containing 0, 1, or 2 heteroatoms selected from O, N, and S, e.g. phenyl,
pyridine, pyrimidine, imidazole, thiophene, oxazole, isoxazole, thiazole,
and amino- and halogen- substituted derivatives thereof.
The pharmaceutically-acceptable salts of the compounds of
the invention (in the form of water- or oil-soluble or dispersible products)
include the conventional non-toxic salts or the quaternary ammonium
salts which are formed, e.g., from inorganic or organic acids or bases.
Examples of such acid addition salts include acetate, adipate, alginate,
aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate,
camphorate, camphorsulfonate, cyclopentanepropionate, digluconate,
dodecylsulfate, ethanesulfonate, fumarate, glucoheptanoate,
glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride,
hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maieate,
methanesulfonate, 2-naphthalenesulfonate, nicotinate, oxalate,
pamoate, pectinate, persulfate, 3-phenylpropionate, picrate, pivalate,
-46-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
propionate, succinate, tartrate, thiocyanate, tosylate, and undecanoate.
Base salts include ammonium salts, alkali metal salts such as sodium
and potassium salts, alkaline earth metal salts such as calcium and
magnesium salts, salts with organic bases such as dicyclohexylamine
salts, N-methyl-D-glucamine, and salts with amino acids such as
arginine, lysine, and so forth. Also, the basic nitrogen-containing
groups may be quaternized with such agents as lower alkyl halides,
such as methyl, ethyl, propyl, and butyl chloride, bromides and iodides;
dialkyl sulfates like dimethyl, diethyl, dibutyl and diamyl sulfates, long
chain halides such as decyl, lauryl, myristyl and stearyi chlorides,
bromides and iodides, aralkyl halides like benzyl and phenethyl
bromides and others.
-47-
CA 02283704 1999-09-15
WO 98/42342 PCTIUS98/05486
SCHEME 1
H i) TrCI, Et3N Tr
'N~ ii) LiAIH4
I I
N m) DPPA, DBU N
Me00C iv) Ph3P, H20 H2N
i) EDC coupling
ii) deprotection
~I wI
N
N
H2N
O
O N
H
hydrogenation
~I ~I
N
H N ~~~~ N
2
O
O N
H
Scheme 1 shows a general procedure for preparing
compounds of the invention where B is a three carbon alkane or alkene
moiety. In Scheme I, imidazole-4-acrylic acid methyl ester is protected
-48-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98105486
as its trityl derivative by treatment with trityl chloride in the presence of
triethylamine. Reduction of the ester with lithium aluminum hydride
affords the corresponding alcohol which is then converted to an azide
using DBU and diphenylphosphoryl azide. The allyl azide is then
reduced to the corresponding allyl amine using the standard Staudinger
reduction/hydrolysis protocol. The resulting amine is then coupled to A-
COOH via routine EDC coupling techniques. Removal of the protecting
groups under acidic conditions followed by hydrogenation of the double
bond as necessary afforded compounds of the invention.
SCHEME 2
N-1 12, Nal
~N Na2C03 ~ N
i
Boc20, Et3N
Boc
N---1
N
I
o If
N Cul, (Ph3P)2PdCl2
O
-49-
CA 02283704 1999-09-15
WO 98/42342 PCT/US9$/05486
BOC~
N-
\ N
O
N
N2H4
O
H
N~ H
\ N N
i) Boc-D-Diphenylala-Pro \ N
N ~ ~ EDC, HOST
ii) TFA, CH2C12
O N
H H2N
Scheme 2 shows a procedure for preparing compounds of
the invention where B includes an alkyne. 4-Methylimidazole was
iodinated under basic conditions and then protected as its Boc derivative.
Following bis triphenylphosphinepalladium dichloride and copper
iodide mediated coupling to N-propargylphthalimide the protecting
groups were removed via treatment with hydrazine. The resulting
imidazole propargylamine is coupled to A-COOH via a standard EDC
coupling to form a compound of the invention.
-50-
CA 02283704 1999-09-15
WO 98/42342 PCTlUS98/05486
SCHEME 3
\~OH K2C03' BrCH2C02~Bu ~~OH
HN~N DMF N~~(N
C02tBu
2:1 mixture
i) Chromatography
ii) DPPA, DBU, DMF
-51-
_......._..__...~. ..~..,.__ .. r _......~~. ~. ...r .,
CA 02283704 1999-09-15
WO 98/42342 PCTlUS98/05486
~Ns
NON
i) H2, C02tBu -
10% PdIC major isomer
i) HCI,
ii) EDC, EtOAc
HOBt,
P3P2COOH ii ) EDC,
HOBt,
RNH2
iii) H2,
10% PdIC
\ \
;O I ~ Ou ~C02tBu
/ S_ N N~ N N
H O H I y
N
~~ NH2
i) HCI,
EtOAc NON
ii) EDC, CIONHR
HOBt,
RNH2
EDC,
HOBt,
P3P2COOH
\ \
O. ,O ~ O ~CONHR
/ S; N N~ N N
H O H
N
-52-
CA 02283704 1999-09-15
WO 98/42342 PCTIUS98105486
To form compounds of the invention with substituents X
and Y at the 2- and 4- positions of the imidazole ring system, 4-methyl-5-
imidazolemethanol is alkylated under basic conditions with an alkyl
haloformate such as t-butylbromoacetate. Following chromatographic
separation of the regioisomers, the major alcohol isomer is converted to
the corresponding azide by treatment with diphenylphosphoryl azide
and DBU.
Two different routes may be used to form a finished
compound of the invention. In one, the azido functionality is first
reduced in the presence of hydrogen and palladium on carbon and the
resulting amine coupled to the A-COOH via standard EDC methodology.
The t-butyl group is then removed under acidic conditions and the
compound of the invention secured via EDC coupling to the requisite
amine. Alternatively, the azidomethylimidazole t-butyl ester is first
deprotected under acidic conditions and the resulting acid coupled to an
amine using EDC. Reduction of the azide and EDC coupling to the A-
COOH affords the compound of the invention.
Unless otherwise stated, all NMR determinations were
made using 400 MHz field strength.
-53-
CA 02283704 1999-09-15
WO 98142342 PCT/US98/05486
EXAMPLE I
Preparation of Boc-D-3,3-diphenylalanine-L-proline-N-(trc~ns-4-
imidazvleallvl) amide
H
Step A: Boc-D-3.3-diphenvlalanine-L-proline benzvl ester
To a solution of Boc-D-3,3-diphenylalanine (5.0 g, 14.6
mmol), L-proline benzyl ester (3.30 g 16.0 mmol) and HOBT (2.56 g 19.0
mmol) in DMF (150 mI) was added EDC (3.62 g, 19.0 mmol) and
triethylamine (8.16 ml, 58 mmol). After stirring at room temperature
overnight, the solvent was removed in vacuo and the resulting residue
partitioned between chloroform and 1M citric acid. The aqueous layer
was extracted with chloroform and the combined organics were washed
with water, 10% Na2C03 solution and dried over MgSO4. The solution
was then filtered and the solvent removed in uacuo to give the title
compound: 1H NMR (CDCl3) d 1.20-1.90 (m, 4 H), 1.30 (s, 9 H), 2.80 (q, J =
6 Hz, 1 H), 3.70-3.80 (m, 1 H), 4.10-4.18 (m, 1 H), 4.35 (d, J = 11.5 Hz, 1 H)
5.03-5.28 (m, 4 H), 7.10-7.40 (m, 15 H).
Step B: Boc-D-3,3-diphenylalanine-L-proline
A solution of Boc-D-3,3-diphenylalanine-L-proline benzyl
ester (8.0 g, 15.0 mmol) in ethanol (200 ml) was hydrogenated in the
presence of 10% Pd/C (2.0 g) at atmospheric pressure for 24 h. The
reaction mixture was then filtered through Celite and the filtrate
concentrated in vacuo to give the title compound as a white solid: 1H
NMR (CDCl3) d 1.20-1.40 (m, 1 H), 1.35 (s, 9 H), 1.40-1.60 (m, 1 H), 1.70-
-54-
CA 02283704 1999-09-15
WO 98142342 PCT/US98/05486
1.90 (m, 1H),2.21-2.30 (m, 1H),2.70(q,J=9Hz, 1H),3.75(brt,J=9Hz,
1H),4.16(d,J=9Hz, 1H),4.35(d,J=11.5 Hz, 1H),4.97-5.18(m,2H),
7.22-7.39 (m, 10 H).
Step C: Trans-4-imidazoleacrvlic acid methyl ester hydrochloride
A solution of trans-4-imidazoleacrylic acid (20.0 g 145 mmol)
in methanol (300 ml) was saturated with anhydrous HCl and the
resulting solution was then refluxed for 90 min. After cooling to room
temperature the solvent was removed in uacuo to give the title
compound as a white solid: 1H NMR (CD30D) d 3.80 (s, 3 H), 6.61 (d, J =
16 Hz, 1 H), 7.60 (d, J = 16 Hz, 1 H), 7.92, {s, 1 H), 9.05, {s, 1 H).
Step D: Trans-1-tritvl-4-imidazoleacrylic acid methyl ester
To a solution of trans-4-imidazoleacrylic acid methyl ester
hydrochloride (20.0 g, 106 mmol) and triethylamine {44 ml, 318 mmol) in
chloroform (500 ml) was added a solution of trityl chloride (29.5 g, 318
mmol) in chloroform (100 ml). The resulting suspension was stirred at
room temperature for 24 h. The reaction mixture was then washed with
water and dried aver MgS04. Filtration and removal of the solvent in
uacuo to gave the title compound as a tan solid: 1H NMR (CDC13) d 3.75
(s,3H),6.54(d,J=l6Hz,lH)7.03(s,lH),7.I3-7.34(m,l5H)7.46(s,l
H),7.51(d,J=16Hz1H).
Step E: Trans-1-tritvl-4-imidazoleallvl alcohol
A 1M solution of LAH in ether (30 ml, 30 mmol) was added
dropwise to a cooled (-45°C) solution of trans-1-trityl-4-
imidazoleacrylic
acid methyl ester (23 g, 58.3 mmol) in THF (300 ml). After stirring at
-45°C for 3 h, additional 1M LAH solution (30 ml) was added. Stirring
was continued at -45°C for 1 h, then the solution was warmed to and
stirred at 0°C for 30 min. The reaction was quenched with ethyl
acetate,
then saturated NH4Cl and allowed to stir at raom temperature
overnight. The layers were separated and the aqueous layer was
extracted twice with ethyl acetate. The combined organics were dried
over MgS04, filtered and the solvents removed in uacuo. The crude
product was purified by flash chromatography on silica gel (ethyl acetate
-55-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
elution) to give the title compound: 1H NMR (DMSO) d 4.03 (m, 2 H), 4.68
(t, J = 5 Hz, 1 H) 6.20-6.40 (m, 2 H), 6.89 (s, 1 H} 7.00-7.50 (m, 16 H).
Steu F:F: Traps-1-tritvl-4-imidazoleall 1 azide
DBU (5.4 ml 35.7 mmol) was added dropwise to a cooled
{0°C) solution of traps-1-trityl-4-imidazoleallyl alcohol (8.76 g, 23.8
mmol)
and diphenylphosphoryl azide (7.7 ml, 35.7 mmol) in THF {300 ml). The
reaction mixture was allowed to warm gradually to and then stirred at
room temperature overnight. The solvent was then removed in uacuo
and the resulting residue purified by flash chromatography on silica gel
(2:1 hexane / ethyl acetate) to give the title compound: 1H NMR (CDCl3) d
3.88 (d, J = 6 Hz, 2 H), 6.25-6.50 (m, 2 H), 6.79 (s, 1 H), 7.14-7.34 (m, 15
H),
7.40 (s, 1 H).
Step G: Traps-1-tritvl-4-imidazoleallylamine
A solution of traps-1-trityl-4-imidazoleallyl azide (6.4 g, 16.8
mmol) and triphenylphosphine (11.0 g, 42.0 mmol) in THF (100 ml) was
heated to reflux for 2 h, after which water (1 ml) Was added. The
resulting solution was then heated at reflux for 24 h. After cooling to
room temperature the solvent was removed in uacuo and the residue
purified by flash chormatography on silica gel ( 19:1 chloroform / 10%
NH40H in MeOH) to give the title compound as white solid: 1H NMR
(CDCl3) d 1.50 (br s, 2 H), 3.41 (d, J = 5 Hz, 2 H), 6.30-6.48 (m, 2 H), 6.73
(s,
1H), 7.13-7.38 (m, 15 H), 7.40 {s, 1 H).
Step H: Traps-4-imidazoleallvlamine dihvdrochloride
A solution of traps-1-trityl-4-imidazoleallylamine {1.0 g, 2.7
mmol) in 1N HCl (50 ml) was heated to reflux for 30 min, cooled to room
temperature and filtered. The filter cake was washed with water.
Concentration of the filtrate in uacuo gave the title compound: 1H NMR
(DMSO) d 3.80 (br s, 2 H), 6.50-6.72 (m, 2 H), 7.81 (s, 1 H)~ g_50 (br s, 3
H),
9.15 (s, 1 H).
-56-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
t I~ Boc-D-3,3-diphenylalanine-L-praline-N-(trans-4-
imidazoleallvl) amide
Boc-D-3,3-diphenylalanine-L-praline and trans-4-
imidazoleallylamine dihydrochloride were coupled using essentially the
same procedure described in EXAMPLE I, Step A except that no citric
acid wash was preformed. The final compound was purified by
preparative HPLC. 1H NMR (CH30D) d 1.23 (s, 9 H), 1.40-1.65 (m, 2 H),
1.70-1.90 (m, 2 H), 2.90 (q, J = 6 Hz, 1 H), 3.75-3.85 (br m, 1 H), 3.90 (d, J
=
6Hz,2H),4.10(d,J=8Hz,lH)4.38(d,J=llHz,lH)5.10(d, J=llHz,
1 H), 6.00-6.10 (br m, 1 H), 6.41 (d, J = 16 Hz, 1 H), 7.01 (s, 1 H) 7.20-7.45
(m, 10 H), 7.60 (s, 1 H); MS (FAB) 544 (M+1)+.
EXAMPLE II
Preparation of D-3,3-Diphenylalanine-L-praline-N-(trans-4-imidazole
allvl) amide
~N H
N
A solution of Boc-D-3,3-diphenylalanine-L-praline-N-(trans-
4-imidazoleallyl) amide (55 mg, 0.1 mmol) in 2:1 methylene chloride /
TFA (30 ml) was stirred at room temperature for 4 h. The solvents were
removed in uacuo and the residue was purified by preparative HPLC.
1H NMR (CH30D) d 1.10-1.37 (m, 2 H), 1.70-1.90 (br s, 2 H), 2.70-2.90 (br
m, 1 H), 3.50-3.65 (br m, 1 H) 3.95 (br s, 2 H), 4.00-4.10 (br m, 1 H), 4.45
(d,
J=11.5Hz,lH),4.95(d,J=11.5Hz,lH)6.30-6.40 (m,lH),6.58(d,J=16
Hz, 1 H), 7.25-7.70 (m, 11 H), 7.85 (s, 1 H); MS (FAB) 444 (M+1)+.
-57-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE III
Preparation of D-3,3-Diphenylalanine-L-proline-N-(4-imidazolepropyl)
amide
H
Step A: Boc-D-3,3-diphenylalanine-L-proline-N-(4-imidazole
nronvl) amide
A solution of Boc-D-3,3-diphenylalanine-L-proline-N-(trc~ns-
4-imidazoleallyl) amide (200 mg, 0.36 mmol) in ethanol ( 100 ml)
containing 10% Pd/C (100 mg) was hydrogenated at 45 psi for 24 h. The
solution was subsequently filtered through Celite and the solvent
removed in uacuo to give the title compound as a solid: 1H NMR
(CD30D) d 1.28-1.65 (m, 3 H), 1.30 (s, 9 H), 1.70-1.87 (m, 3 H), 2.55 (t, J =
8
Hz, 2 H), 2.88 (q, J = 8 Hz, 1 H), 3.05-3.30 (m, 2 H), 3.70-3.80 (m, 1 H),
3.95-
4.05 (m, 1 H), 4.35 (d, J = 11.5 Hz, 1 H), 5.I0 (d, J = 11.5 Hz, 1H}, 6.80 (s,
1
H), 7.19-7.42 (m, 10 H), 7.60 (s, 1 H).
Step B: D-3,3-Diphenylalanine-L-proline-N-(4-imidazolepropyl)
amide
The title compound was prepared from Boc-D-3,3-
diphenylalanine-L-proline-N-(4-imidazolepropyl) amide using the
procedure of EXAMPLE II: 1H NMR {CD30D) d 1.22-1.35 (m, 2 H), 1.70-
1.95 (m, 4 H), 2.75-2.88 (m, 1 H), 2.78 (t, J = 8 Hz, 2 H), 3.12-3.33 (m, 2
H),
3.50-3.60 (m, 1 H), 4.00 (t, J = 7 Hz, 1 H), 4.42 (d, J = 12 Hz, I H), 4.99
(d, J
= 12 Hz, 1 H), 7.25-7.62 {m, 11 H), 8.80 (s, 1 H); MS (FAB) 446 {M+1)+.
-58-
CA 02283704 1999-09-15
WO 98/42342 PCTIUS98/05486
EXAMPLE IV
Preparation of N-Benzylsulfonyl-D-3,3-diphenylalanine-L-proline-N-
~trans-4-imidazoleallyl) amide
H
to A: D-3.3-Diphenvlalanine-L=proline benzvl ester
The title compound was prepared from Boc-D-3,3-
diphenylalanine-L-proline benzyl ester using the procedure described for
EXAMPLE II: 1H NMR (CDC13) d 1.35-1.48 (m, 1 H), 1.70-1.83 (m, 3 H),
2.70-2.82 (m, 1 H), 3.50-3.60 (m, 1 H), 4.15-4.25 (m, 3 H), 5.15 (ab q, J = 32
and 12 Hz, 2 H), 7.12-7.40 (m, 15 H).
Step B: N-Benzylsulfonyl-D-3,3-diphenylalanine-L-proline benzyl
ester
A solution of benzylsulfonyl chloride (177 mg, 0.934 mmol)
in methylene chloride (10 ml) was added dropwise to a solution of D-3,3-
diphenylalanine-L-proline benzyl ester (200 mg, 0.47 mmol) in
methylene chloride at -78°C. After stirring at -78°C for 45 min,
triethylamine (0.13 ml, 0.93 mmol) was added and the reaction mixture
was allowed to stir at -78°C for an additional 30 min. It was then
warmed to room temperature for 24 h. The reaction mixture was
washed with water, dried over MgS04, filtered and the solvent removed
in Uacuo. The residue was purified by flash chromatography on silica
gel (7:3 hexane / ethyl acetate). 1H NMR (CDCl3) d 1.40-1.55 (m, 1 H),
1.55-1.77 (m, 1 H), 1.77-1.90 (m, 2 H), 2.70-2.80 (m, 1 H), 3.72-3.83 (m, 1
H),
4.10 (d, J = 11 Hz, 1 H), 4.15-4.25 (m, 1 H), 4.33 (d, J = 11 Hz, 1 H), 4.65
(d,
-59-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98I05486
J = 6 Hz I H), 5.10 (ab q, J = 56 and 14 Hz, 2 H), 5.10-5.19 (m, 1 H), 7.20-
7.41 (m, 20 H).
Step C: N-Benzvlsulfonvl-D-3.3-diphen~lalanine-L-proline
The title compound was prepared from N-benzylsulfonyl-D-
3,3-diphenylalanine-L-proline benzyl ester using the procedure
described in EXAMPLE I, Step B: 1H NMR (CDCl3) d 1.30-1.53 (m, 2 H),
1.70-1.87 (m, 1 H), 2.07-2.17 (m, 1 H), 2.68 (q, J = 9 Hz, 1 H), 3.52-3.65 (m,
1
H), 3.95-4.15 (m, 3 H), 4.30 (d, J = 12 Hz, 1 H), 4.80-4.95 (m, 1 H), 5.00-
5.12
(m, 1 H), 7.10-7.45 (m, 15 H).
Sten D_:, N-Benzylsulfonyl-D-3,3-diphenylalanine-L-proline-N-
(traps-1-trityl-4-imidazoleallyl) amide
The title compound was prepared from N-benzylsulfonyl-D-
3,3-diphenylalanine-L-proline and traps-1-trityl-4-imidazoleallylamine
using the procedure described in EXAMPLE 1, Step A: 1H NMR (CDCl3)
d 1.30-1.85 (m, 4 H), 2.02-2.15 (m, 1 H), 2.55-2.70 (m, 1 H), 3.60-4.27 (m, 5
H), 4.33 (d, J = 12 Hz, 1 H), 4.80-4.90 (m, 1 H), 4.95 (d, J = 12 Hz, 1 H),
6.15-
6.37 (m, 2 H), 6.70 (s, 1 H), 6.85 (t, J = 8 Hz, 1 H), 7.05-7.40 (m, 31 H).
Ste : N-Benzylsulfonyl-D-3,3-diphenylalanine-L-proline-N-
(trans-4-imidazoleallvl) amide
N-Benzylsulfonyl-D-3,3-diphenylalanine-L-proline-N-(trart,s-
1-trityl-4-imidazoleallyl) amide (180 mg, 0.2 mmol) was dissolved in 2:1
methylene chloride / TFA (75 ml). Triethylsilane was then added
dropwise until the bright yellow color had disappeared. The reaction
was allowed to stir at room temperature overnight. The solvents were
removed in vacuo and the residue was purified by preparative HPLC. 1H
NMR (CD30D) d 1.40-1.50 (m, 1 H), 1.50-1.70 (m, 1 H), 1.70-1.90 (m, 2 H),
2.90-3.02 (m, 1 H), 3.72-3.83 (m, 2 H), 3.95-4.08 (m, 2 H), 4.21 (ab q, J = 16
and2.7Hz,2H),4.34(d,J=11.5 Hz,lH),5.06(d,J=11.5 Hz, 1H),6.22(d
of t, J = 16 and 6 Hz, 1 H), 6.50 (d, J = 16 Hz, 1 H), 7.10-7.60 (m, 16 H),
8.77
(s, 1 H); MS (FAB) 598 (M+1)+.
-60-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE V
Preparation of N-Benzylsulfonyl-D-3,3-diphenylalanine-L-proline-N-
(4-imidazolepropvl) amide
H
A solution of N-Benzylsulfonyl-D-3,3-diphenylalanine-L-
proline-N-(tr~ns-4-imidazoleallyl) amide (55 mg, 0.092 mmol) in ethanol
(50 ml) was hydrogenated at 40 psi in the presence of 10°lo Pd/C (50
mg)
for 4 h. The solution was filtered through Celite and the solvent removed
in vacuo. The product was purified by preparative HPLC. 1H NMR
(CD30D) d 1.40-1.90 (m, 6 H), 2.70 (t, J = 6 Hz, 2 H), 2.92-3.18 (m, 2 H),
3.20-3.30 (m, 1 H), 3.72-3.82 (br m, 1 H), 3.90-4.00 (m, 1 H), 4.20-4.38 (m, 3
H), 5.05 (d, J = 11.5 Hz, 1H), 7.10-7.60 (m, 16 H), 8.60 (s, 1 H); MS (FAB)
600 (M+1)+.
-61-
CA 02283704 1999-09-15
WO 98/42342 PCT/IJS98/05486
EXAMPLE VI
Preparation of N-Methoxycarbonylmethanesulfonyl-D-3,3-diphenyl
alanine-L-proline-N-ftrans-4-imidazoleallvl) amide
M
H
Step A: N-Methoxycarbonylmethanesulfonyl-D-3,3-diphenyl
alanine-L-proline benzyl ester
To a solution of D-3,3-diphenylalanine-L-proline benzyl ester
(200 mg, 0.47 mmol) in methylene chloride (20 ml) at -78°C was added
dropwise a solution of chlorosulfonylacetic acid methyl ester (100 mg,
0.56 mmol) in methylene chloride {5 ml). The reaction was stirred at
-78°C for 10 min and then triethylamine (0.13 ml, 0.93 mmol) was added.
Stirring at -78°C was continued for an additional 5 min followed
by
warming to room temperature for 16 min. The reaction was quenched
with water, dried over MgS04, filtered and the solvent removed in vacuo.
The residue was purified by flash chromatography on silica gel (3:2
hexane / ethyl acetate). 1H NMR (CDC13) d 1.30-1.50 (m, 1 H), 1.60-1.82
{m, 3 H), 2.60-2.70 (m, 1 H), 3.60-3.70 (m, 1 H), 3.73 (s, 3 H), 4.00 (ab q, J
=
108 and 16 Hz) 4.15-4.20 (m, 1 H), 4.35 (d, J = 12 Hz, 1 H), 5.00-5.20 (m, 3
H), 5.66 {d, J = 12 Hz, 1 H), 7.10-7.42 {m, 15 H).
-62-
f... ~. .
CA 02283704 1999-09-15
WO 98142342 PCT/US98/05486
Step B: N-Methoxycarbonylmethanesulfonyl-D-3,3-diphenyl
alanine-L-nroline
The title compound was prepared from N-methoxycarbonyl-
methanesulfonyl-D-3,3-diphenylalanine-L-proline benzyl ester using the
procedure described in EXAMPLE I, Step B. 1H NMR (CDCl3) d 1.45-
1.55 (m, 2 H), 1.72-1.82 (m, 1 H), 2.02-2.10 (m, 1 H), 2.62-2.75 (m, 1 H),
3.68-3.72 (m, 1 H), 3.75 (s, 3 H), 3.92 (ab q, J = 75 and 14 Hz, 2 H), 4.10-
4.18
(m, 1 H), 4.35 (d, J=12 Hz, 1 H), 5.07 (t, J = 12 Hz, 1 H), 5.80 (d, J = 12
Hz, 1
H), 7.20-7.42 (m, 10 H).
Step C: N-Methoxycarbonylmethanesulfonyl-D-3,3-diphenyl
alanine-L-proline-N-(trc~ns-1-tr~"vl-4-imidazoleallvl) amide
The title compound was prepared from N-methoxycarbonyl-
methanesulfonyl-D-3,3-diphenylalanine-L-proline and traps-1-trityl-4-
imidazoleallylamine essentially using the procedure described in
EXAMPLE I, Step A. 1H NMR (CDC13) d 1.40-1.90 (m, 3 H), 1.95-2.10 (m,
1 H), 2.58-2.70 (q, J = 8 Hz, 1 H), 3.57-3.62 (m, 1 H), 3.65 (s, 3 H), 3.75-
4.05
(m, 2 H), 3.98 (ab q, J = 65 and 15 Hz, 2 H), 4.10-4.18 (m, 1 H), 4.35 (d, J =
12 Hz, 1 H), 5.02 (d, J = 12 Hz, 1 H), 6.15-6.40 (m, 2 H}, 6.65 (t, J = 6 Hz,
1
H}, 6.75 (s, 1 H), 7.10-7.45 (m, 26 H).
Step D: N-Methoxycarbonylmethanesulfonyl-D-3,3-diphenyl-
alanine-L-nroline-N-(traps-4-imidazoleallvl) amide
The title compound was prepared from N-methoxycarbonyl-
methanesulfonyl-D-3,3-diphenylalanine-L-proline-N-(trccns-1-trityl-4-
imidazoleallyl) amide using the procedure described in EXAMPLE IV,
Step E. 1H NMR (CD30D) d 1.30-1.50 (m, 1 H), 1.50-1.70 (m, 1 H), 1.70-
1.90 (m, 2 H), 2.82-2.95 (m, 1 H), 3.64 (s, 3 H), 3.62-3.78 (m, 1 H), 3.80-
3.95
(m, 1 H), 3.95-4.19 (m, 4 H), 4.35 (d, 11.5 Hz, 1 H), 5.13 (d, J = 11.5 Hz, 1
H), 6.30 (d of t, J = 16 and 6 Hz, 1 H), 6.54 (d, J = 16 Hz, 1 H), 7.20-7.58
(m,
11 H), 7.81 (t, J = 6 Hz, 1 H), 8.83 (s, 1 H); MS (FAB) 580 (M+1)+.
-63-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98105486
EXAMPLE VII
Preparation of N-Methoxycarbonylmethanesulfonyl-D-3,3-diphenyl
alanine-L-proline-N-(4-imidazoleprowl) amide
Me00C
-N
O H
H
The title compound was prepared from N-methoxycarbonyl-
methanesulfonyl-D-3,3-diphenylalanine-L-proline-N-(tracn.s-4-imidazole-
allyl) amide using the procedure of EXAMPLE III, Step A. The final
product was purified by preparative HPLC. 1H NMR (CD30D) d 1.36-1.45
(m, 1 H), 1.55-1.70 (m, 1 H), 1.70-1.90 (m, 4 H), 2.70-2.80 (m, 2 H), 2.85-
2.98
(m, 1 H), 3.10-3.20 (m, 1 H), 3.21-3.30 (m, 1 H), 3.68 (s, 3 H), 3.70-3.80 (m,
1
H), 3.92-3.99 (m, 1 H), 4.16 (s, 2 H), 4.34 (d, J = 11.5 Hz, 1 H), 5.13 (d, J
=
I1.5 Hz, 1 H), 7.22-7.55 (m, 11 H), 8.76 (s, 1 H); MS (FAB) 582 (M+1)+.
CA 02283704 1999-09-15
WO 98142342 PCT/US98/05486
EXAMPLE VIII
Preparation of N-[(1R)-10-Camphorsulfonyl]-D-3,3-diphenylalanine-L-
nroline-N-(trans-4-imidazoleallvl) amide
L~'~ U U " p/~ H ~ ~ N H
N-=-
Step A: N-[(1R)-10-Camphorsulfonyl]-D-3,3-diphenylalanine-L-
nroline benzvl ester
The title compound was prepared from D-3,3-
diphenylalanine-L-proline benzyl ester and (1R)-(-)-10-camphorsulfonyl
chloride using the procedure described in EXAMPLE IV, Step B: 1H
NMR (CDC13) d 0.90 (s, 3 H), 0.95 (s, 3 H), 1.32-1.42 (m, 2 H), 1.70-1.78 (m,
2 H), 1.85 (d, J = 16 Hz, 1 H), 1.90-2.08 (m, 4 H), 2.35-2.48 (m, 1 H), 2.70
(q,
J=6Hz, 1H),2.97(d,J=l6 Hz, 1H),3.41(d,J=l6 Hz, 1H),3.72-3.82 (m,
1H),4.10-4.17(m,lH),4.35(d,J=l2 Hz,lH),5.00-5.18(m,3H),6.57(d,
J = 12 Hz, 1 H), 7.10-7.45 (m, 15 H).
Step B: N-[(1R)-10-Camphorsulfonyl]-D-3,3-diphenylalanine-L-
proline
The title compound was prepared from N-[( 1R)-10-
camphorsulfonyl]-D-3,3-diphenylalanine-L-proline benzyl ester using
the procedure described in EXAMPLE I, Step B. 1H NMR (CDC13) d 0.87
(s, 3 H), 0.93 (s, 3 H), 1.22-2.20 (m, 9 H), 2.35-2.45 (m, 1 H), 2.70 (q, J =
6
Hz, 1 H), 2.87 (d, J = 16 Hz, 1 H), 3.25 (d, J = 16 Hz, 1 H), 3.70-3.80 (m, 1
H),4.05(d,J=lOHz,lH),4.37(d,J=l2 Hz,lH),5.08(t,J=l2 Hz,lH),
6.50(d,J=l2 Hz, 1H)7.17-7.42 (m, lOH).
-65-
CA 02283704 1999-09-15
WO 98/42342 PCT/L1S98105486
Ste ~ N-[(1R)-10-Camphorsulfonyl]-D-3,3-diphenylalanine-L-
proline-N-(trans-1-trit~l-4-imidazoleallvl) amide
The title compound was prepared from N-[(1R)-10-
camphorsulfonyl]-D-3,3-diphenylalanine-L-proline and traps-1-trityl-4-
imidazoleallylamine using the procedure described in EXAMPLE I, Step
A. 1H NMR (CDCl3) d 0.80 (s, 3 H), 0.88 (s, 3 H), 1.22-2.09 (m, 11 H), 2.28-
2.40(m, 1H),2.65(q,J=9Hz, IH),2.75(d,J=l6 Hz, 1H),3.50(d,J=16
Hz, 1 H), 3.62-3.78 (m, 1 H), 3.85-3.95 (m, 2 H), 4.22 (d, J = 9 Hz, 1 H),
4.35
(d, J = 12 Hz, 1 H), 4.98 (t, J = 12 Hz, 1 H), 6.15-6.40 (m, 2 H), 6.75 (s, 1
H),
6.85 (t, J = 6 Hz, 1 H), 7.10-7.47 (m, 26 H).
Step D: N-[(IR)-10-Camphorsulfonyl]-D-3,3-diphenylalanine-L-
proline-N-(traps-4-imidazoleal~l) amide
The title compound was prepared from N-[(1R)-10-
camphorsulfonyl]-D-3,3-diphenylalanine-L-proline-N-(traps-1-trityl-4-
imidazoleallyl) amide using the procedure described in EXAMPLE IV,
Step E. 1H NMR (CD30D) d 0.80 (s, 3 H), 0.95 (s, 3 H), 1.30-2.10 (m, 11 H),
2.25-2.37 (m, 1H),2.85(d,J=l5 Hz, IH),2.98(q,J=7Hz, 1H),3.35(d,J
= 15 Hz, 1 H), 3.75-3.90 (m, 2 H), 3.97-4.10 (m, 2 H), 4.32 (d, J = 11.5 Hz, 1
H), 5.09 (d, J = 11.5 Hz, 1 H), 6.28 (d of t, J = 16 and 6 Hz, 1 H), 6.53 (d,
J =
16 Hz, 1 H), 7.20-7.60 (m, 11 H), 8.83 (s, 1 H); MS (FAB) 658 (M+1)+.
-66-
,. , ,
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/0548b
EXAMPLE IX
Preparation of N-[(1S)-10-Camphorsulfonyl]-D-3,3-diphenylalanine-L-
proline-N-(trans-4-imidazoleallyl) amide
O
,.
O~H ~ ~ NH
N-=J
The title compound was prepared from D-3,3-
diphenylalanine-L-proline benzyl ester and ( 1S)-(+)-10-camphorsuifonyl
chloride using the procedure described for EXAMPLE VIII: 1H NMR
(CD30D) d 0.83 {s, 3 H), 0.96 (s, 3 H), 1.25-2.40 (m, 11 H), 2.91 (d, J = 15
Hz,
1H),3.00(q,J=7Hz,lH),3.35(d,J=lSHz, 1H)3.75-3.90(m,2H),4.00-
4.10(m,2H),4.33(d,J=11.5 Hz, 1H),5.05(d,J=11.5 Hz, 1H),6.30(dof
t, J = 16 and 6 Hz, 1 H), 6.51 (d, J = 16 Hz, 1 H) 7.20-7.60 (m, 11 H) 7.60-
7.70
(m, 1 H), 8.83 (s, 1 H); MS (FA$) 658 (M+1)+
-67-
CA 02283704 1999-09-15
WO 98/42342 PCTIUS98/05486
EXAMPLE X
Preparation of D-3,3-Diphenylalanine-L-homoproline-N-(traps-4-
imidazoleallvl) amide
~N H
N
Step A: Boc-L-homoproline benzvl ester
DBU (2.2 ml, 15 mmol) was added to a solution of Boc-L-
homoproline {3 g, 13 mmol) in acetonitrile (30 ml). After stirring for 10
min, benzyl bromide (1.7 ml, 14 mol) was added and stirring was
continued for 3 h. The solvent was then rotovapped off and the residue
partitioned between ethyl acetate and water. The organic phase was
washed with 1 M citric acid, water then 10% aqueous Na2C03 and dried
(Na2S04). Concentration gave the title compound as a yellow oil (4 g).
1H NMR (CDC13) d 1.19-1.68 (m, 15 H), 2.24 (br m, 1 H), 2.90-2.97 (br m, 1
H), 3.91-4.02 (br m, 1 H), 5.20 (br m, 2 H), 7.35 (m, 5 H).
Step B: L-Homoproline benzvl ester hydrochloride
Anhydrous HCl was bubbled through a cooled (0°C) solution
of Boc-L-homoproline benzyl ester {3 g) in ethyl acetate (100 ml) for 10
min. The resulting mixture was allowed to warm gradually to room
temperature over 4 h. The solution was then purged with argon and
rotavapped down. Trituration of the residue with ether gave the title
compound as a white solid (2.4 g). 1H NMR (CD30D) d 1.55-1.92 (m, 6 H),
2.30 (br m, 1 H), 3.20 (br m, 1 H), 3.42 (br m, 1 H), 4.08 (br m, 1 H), 5.29
(br
m, 2 H), 7.36-7.40 (m, 5 H).
-68-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
Step C: Boc-D-3,3-dinhenvlalanine-L-homonroline benzyl ester
The title compound was prepared from Boc-D-3,3-diphenyl-
alanine and L-homoproline benzyl ester hydrochloride essentially
according to the procedure described for EXAMPLE I, Step A. 1H NMR
(CDC13) d 1.09-1.58 (m, 15 H), 2.05 (m, 1 H), 3.13 (br m, 1 H), 3.88 (br m, 1
H), 4.42 (br m, 1 H), 5.06 (br m, 1 H), 5.16 (m, 2 H), 5.55 (m, 1 H), 7.11-
7.40
(m, 15 H).
Step D: Boc-D-3.3-dinhenvlalanine-L-homoproline
A solution of Boc-D-3,3-diphenylalanine-L-homoproline
benzyl ester (4.2 g, 7.8 mmol) in ethyl acetate (50 ml) was hydrogenated
at 1 atm in the presence of 10°lo Pd/C ( 1 g} for 4 h. Removal of the
catalyst
by filtration through Celite and concentration of the filtrate an uacuo gave
the title compound as a white solid (3.5 g). 1H NMR (CDCI3} d 1.17-1.38
(m, 15 H), 2.15 (m, 1 H), 3.09 (m, 1 H), 3.84 (m, 1 H), 4.41 (m, 1 H), 5.15
(m, 1 H), 5.37 (m, 1 H), 7.17-7.40 (m, 10 H).
Step E: Boc-D-3,3-diphenylalanine-L-homoproline-N-(trccns-1-
tritvl-4-imidazoleallvl) amide
Boc-D-3,3-diphenylalanine-L-homoproline was coupled to
tr~ns-1-trityl-4-imidazoleallylamine essentially according to the
procedure described for EXAMPLE ~ I, Step A. 1H NMR (CDC13) d 1.20-
1.34 (m, 15 H), 2.30 (m, 1 H), 3.02 (m, 1 H), 3.73 (m, 1 H), 3.85 (m, 1 H),
4.10 (m, 1 H), 4.38 (m, 1 H), 4.92 (m, 1 H), 5.19 (m, 2 H), 6.23-6.34 (m, 2
H),
6.70 (s, 1 H), 7.13-7.37 (m, 26 H).
Step F: D-3,3-diphenylalanine-L-homoproline-N-(trans-4-
imidazole-allvl) amide
Simultaneous removal of the protecting groups from Boc-D-
3,3-diphenylalanine-L-homoproline-N-(traps-1-trityl-4-imidazoleallyl)
amide was accomplished essentially according to the procedure
described in EXAMPLE IV, Step E affording the title compound which
was purified by preparative HPLC. 1H NMR (CD30D) d 0.24 (m, 1 H),
1.12-1.36 (m, 4 H), 2.03 (br d, J = 13.6 Hz, 1 H), 3.22 (m, 1 H), 3.74 (br d,
J =
11.7 Hz, 1 H), 4.01 (m, 2 H), 4.44 (d, J = 11.3 Hz, 1 H), 4.95 (br s, 1 H),
5.34
-69-
CA 02283704 1999-09-15
WO 98142342 PCT/US98/05486
(d,J=11.3 Hz, 1H),6.35(dt,J=5.2, 16.2 Hz, 1H),6.47(d,J=16.2 Hz, 1
H), 7.25-7.64 (m, lI H), 8.85 (d, J = 1.3 Hz, 1 H); MS (FAB) 458 (M+1)+.
EXAMPLE XI
Preparation of D-3,3-Bis-(4'-methoxyphenyl)alanine-L-proline-N- trans-
4-imidazoleallvl) amide
a
H2NH",~..
p H / ~N H
N
Sten A: Bis-(4-methoxyphenvl)methanol
To a stirred solution of 4,4'-dimethoxybenzophenone (5.0 g,
20.6 mmol) in 1:1 THF / ethanol (100 ml) was added sodium borohydride
(946 mg, 25 mmol) and the mixture stirred overnight at ambient
temperature. Additional borohydride (200 mg) was added, and the
mixture heated to 40°C for 1 h, cooled and quenched with acetone. The
mixture was rotavapped to dryness, partitioned between EtOAc and cold
1M citric acid, the organic layer washed with 10°lo Na2C03, brine,
treated with activated carbon and concentrated to give the title compound
as a colorless solid.
Step B: 3,3-Bis-(4'-methoxyphenyl)-2-nitropropionic acid ethyl ester
To a stirred (0°C) solution of bis-(4-methoxyphenyl)methanol
(2.5 g, 10.2 mmol) and ethyl nitroacetate (2.26 ml, 20.4 mmol) in CH2C12
(50 mI) under argon was added AlCl3 (1.36 g, 10.2 mmol) in one portion.
After 1 h, the reaction mixture was allowed to warm to approximately
10°C and poured into a mixture of ice and 2M HCl. The resulting
mixture was stirred for 45 min, and the aqueous layer extracted with
_70_
CA 02283704 1999-09-15
WO 98142342 PCTIUS98/05486
CH2C12. The combined organic layers were washed with 10% Na2C03,
dried over MgS04, treated with activated carbon, and concentrated to
give a reddish oil that was chromatographed (CHC13 elution) to afford
the title compound as a colorless oil.
Step C: Boc-DL-3,3-bis-(4'-methoxyphen,vl)alanine ethyl ester
Amalgamated zinc was prepared by treating zinc dust ( 11.4
g) with 2M HCl (83 ml) for 5 min and then decanting the supernatant.
To this was then added a solution of 3,3-bis-(4'-methoxyphenyl)-2-
nitropropionic acid ethyl ester (3.0 g, 8.3 mmol) in 1:1 THF / CH30H ( 166
ml), followed by the addtion of 2M HCl (43 ml). This mixture was heated
at reflux under argon for 2 h, cooled to approximately 40°C, filtered
through a glass-fiber filter and concentrated by rotavap. The resulting
residue was diluted with brine, basified with 2M NaOH and extracted
with 3 portions of CH2C12. The combined organic layers were dried over
Na2S04 and concentrated to give a colorless oil (2.62 g) that was
dissolved in dioxane (50 ml) containing di-t-butyldicarbonate (2 g, 9.1
mmol). To this stirred solution under argon was added 1M NaOH (9 ml)
and stirred overnight at room temperature. The reaction mixture was
concentrated and the residue partitioned between EtOAc and 1M citric
acid. The organic layer was washed with 10% Na2C03, brine, dried over
MgS04 and concentrated to give a yellow oil that was chromatographed
(1:4 to 2:3 ethyl acetate / hexanes) to provide the title compound as a
colorless foam.
Step D: Boc-DL-3,3-bis-(4'-methoxvphenyl)alanine
To a stirred solution of Boc-DL-3,3-bis-(4'-methoxy-
phenyl)alanine ethyl ester (2.32 g, 5.4 mmol) in dimethoxyethane (50 ml)
was added 1M LiOH (8.1 ml) and the mixture stirred overnight under
argon. Additonal 1M LiOH (4 ml) was added, and stirring continued for
48 h. The reaction mixture was concentrated by rotavap, the residue
partitioned between 1M citric acid and EtOAc, and the aqueous layer
extracted with EtOAc. The combined organic layers were washed with
water, brine, dried over Na2S04 and concentrated by rotavap to give the
title compound as a colorless foam.
-71-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
Step E: Boc-DL-3,3-bis-(4'-methoxyphenyl)alanine-L-praline
benzvl ester
The title compound was prepared from Boc-DL-3,3-bis-(4'-
methoxyphenyl)alanine and L-praline benzyl ester essentially according
to the procedure described for EXAMPLE I, Step A.
Step F: Boc-DL-3.3-bis-(4'-methoxvnhenvl)alanine-L-praline
The title compound was prepared from Boc-DL-3,3-bis-(4'-
methoxyphenyl)alanine-L-praline benzyl ester essentially according to
the procedure described for EXAMPLE I, Step B.
Step G: Boc-DL-3,3-bis-(4'-methoxyphenyl)alanine-L-praline-N
(trans-1-tritvl-4-imidazoleallyl) amide
The title compound was prepared from Boc-DL-3,3-bis-(4'-
methoxyphenyl)alanine-L-praline and trans-1-trityl-4-
imidazoleallylamine using the procedure described in EXAMPLE I, Step
A.
Step H: D-3,3-Bis-(4'-methoxyphenyl)alanine-L-praline-N-(trans-
4-imidazoleallvl) amide
The title compound was prepared from Boc-DL-3,3-bis-(4'-
methoxyphenyl)alanine-L-praline-N-(traps-1-trityl-4-imidazoleallyl)
amide using the procedure described in EXAMPLE IV, Step E. The
diastereomers were separated by preparative HPLC. The title compound
is the less polar diastereomer. 1H NMR (CD30D) d 1.30-1.42 (m, 1 H),
1.82 (br s, 3 H), 2.72-2.88 (m, 1 H), 3.50-3.63 (m, 1 H), 3.75 (s, 3 H), 3.81
(s, 3
H), 4.00 (br s, 2 H), 4.05-4.17 (m, 1 H), 4.34 (d, J = 11.2 Hz, 1 H), 6.35 (d
of t,
J=l6and6Hz,lH),6.58(dJ=l6 Hz,lH),6.85(d,J=8.5Hz,2H),7.03
3(l (d,J=8Hz,2H),7.20(dJ=8.5Hz,2H),7.48(d,J=8.3Hz,2H),7.53(s,l
H), 8.86 (s, 1 H); MS (FAB) 504 M+1)+.
-72-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE XII
Preparation of D-3,3-Bis-(4'-methoxyphenyl)alanine-L-proline-N-(4-
imidazolenrowll amide
~H
~ N
The title compound was prepared from D-3,3-bis-(4'-
methoxyphenyl)alanine-L-proline-N-(traps-4-imidazoleallyl) amide
using the procedure described in EXAMPLE I, Step B. 1H NMR
(CD30D) d 1.10-1.40 (br m, 2 H), 1.70-1.95 (br m, 4 H), 2.70-2.85 (br m, 3
H}, 2.90-3.10 (br m, 2 H), 3.45-3.60 (br m, 1 H), 3.70 (br s, 3 H), 3.80 (br
s, 3
H), 3.95-4.08 (br m, 1 H), 4.25-4.40 (br m, 1 H), 6.75-6.85 (br m, 2 H}, 6.90-
7.05 (br m, 2 H), 7.10-7.22 (br m, 2 H), 7.30-7.37 (br m, 1 H), 7.42-7.53 (br
m, 2 H), 8.75 (br s, 1 H); MS (FAB) 506 (M+1)+
-73-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE XIII
Preparation of D-3,3-Diphenylalanine-L-proline-N-(traps-5-methyl-4-
imidazoleallvl~ amide
H
Ste~A: 1- and 3-Tritvl-4-methyl-5-imidazolecarboxaldehvde
Trityl chloride (28 g, 100 mmol) was added to a cooled (0°C)
solution of 4-methyl-5-imidazolecarboxaldehyde (10 g, 91 mmol) and
t;riethylamine (16 ml, 115 mmol) in methylene chloride (300 ml). After
stirring for 30 min, the reaction mixture was warmed to room
temperature and stirred there for 2 h. The reaction mixture was then
washed well with water and saturated NaHC03. Drying over Na2S04
and removal of the solvent in uaccuo gave a l:l mixture of the title
compounds as a white powder. 1-trityl isomer 1H NMR (CDC13) d 2.55
(s, 3 H), 7.12-7.38 (m, 15 H), 9.11 (s, 1 H); 3-trityl isomer 1H NMR (CDCl3)
d 1.83 (s, 3 H), 7.10-7.42 {m, 15 H), 10.02 (s, 1 H).
Step B: Traps-1-trityl-4-methyl-5-imidazoleacrylic acid methyl
ester
A mixture of 1- and 3-trityl-4-methyl-5-imidazolecarbox-
aldehyde (7 g, 20 mmol) and methyl
(triphenylphosphoranylidene)acetate (6.7 g, 20 mmol) in toluene (40 ml)
was heated at reflux for 24 h. After cooling to room temperature, silica
gel was added and the solvent removed in vacuo. The residue was
transferred to a flash column and eluted with 3:2 hexane / ethyl acetate
to give the title compound. 1H NMR (CDC13) d 1.58 (s, 3 H), 3.77 (s, 3 H),
-74-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
6.59 (d, J = 15.3 Hz, 1 H), 7.13-7.16 (m, 6 H), 7.33-7.36 (m, 10 H), 7.57 (dd,
J
= 0.4, 15.3 Hz, 1 H).
Step C: Trans-1-trityl-4-methyl-5-imidazoleallvl alcohol
A 1M solution of DIBAL in methylene chloride (20 ml, 20
mmol) was added to a cooled (-78°C) solution of the ester (2.5 g, 6.1
mmol)
in THF (30 ml). After stirring at -78°C for 30 min, the reaction
mixture
was stored at -25°C for 19 h. The reaction mixture was then warmed to
0°C and carefully quenched sequentially with methanol (1.4 ml) and 1M
sodium hydroxide (2.8 ml). After stirring for 10 min, 30% sodium
potassium tartrate (7 ml) was added. The reaction mixture was then
stirred until precipitation was complete. The precipitate was filtered off
and washed well with ether and ethyl acetate. The filtrate was washed
with brine and dried over Na2S04. Filtration and concentration afforded
the title compound as a crystalline foam. 1H NMR (CDCl3) d 1.49 (s, 3
H), 4.30 (d, J = 5.1 Hz, 2 H), 6.46-6.57 (m, 2 H), 7.13-7.17 (m, 6 H), 7.29
(s, 1
H), 7.31-7.37 (m, 9 H).
Step D:, Trans-1-trill-4-methyl-5-imidazoleallvl azide
Trans-1-trityl-4-methyl-5-imidazoleallyl alcohol was
converted to the corresponding azide essentially according to the
procedure described for EXAMPLE I, Step F. 1H NMR (CDCl3) d 1.50 (s,
3 H), 3.93 (d, J = 6.2 Hz, 2 H), 6.40-6.51 (m, 2 H), 7.13-7.18 (m, 6 H), 7.29
(s,
1 H), 7.32-7.35 (m, 9 H).
Step E: Trans-1-tritvl-4-methyl-5-imidazoleallvlamine
Trans-1-trityl-4-methyl-5-imidazolealiyl azide was converted
to the corresponding amine essentially according to the procedure
described for EXAMPLE I, Step G. 1H NMR (CDCl3) d 1.48 (s, 3 H), 1.82
(br s, 2 H), 3.46 (d, J = 6.0 Hz, 2 H), 6.36 (d, J = 15.4 Hz, 1 H), 6.47 (m, 1
H),
7.12-7.18 (m, 6 H), 7.27 (s, 1 H), 7.31-7.37 (m, 9 H).
Step F: D-3,3-Diphenylalanine-L-proline-N-(trans-5-methyl-4-
imidazoleallvl) amide
Boc-D-3,3-diphenylalanine-L-proline was coupled to trans-1-
trityl-4-methyl-5-imidazoleallylamine essntially according to the
-75-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98105486
procedure described for EXAMPLE I, Step A then the protecting groups
were simultaneously removed essentially according to the procedure of
EXAMPLE IV, Step E. 1H NMR (CD30D) d 1.18 (m, 1 H), 1.79 (m, 3 H),
2.42(s,3H),2.84(m,lH),3.58(m,lH),3.96-4.11(m,3H),4.45(d,J=
11.4 Hz, 1 H), 4.99 (d, J = 11.4 Hz, 1 H), 6.25 (dt, J = 5.1, 16.3 Hz, 1 H),
6.55
(d, J = 16.3 Hz, 1 H), 7.27-7.60 (m, 10 H), 8.76 (s, 1 H); MS (FAB) 458
(M+1)+
EXAMPLE XIV
Preparation of D-3,3-Diphenylalanine-L-proline-N-(5-methyl-4-
imidazolenropvl> amide
H
A solution of D-3,3-diphenylalanine-L-proline-N-(trans-5-
methyi-4-imidazoleallyl) amide (280 mg) in ethanol (10 ml) containing
5°lo Pd/C was stirred at room temperature under an atmosphere of
hydrogen overnight. The catalyst was removed by filtration through
Celite. The filtrate was concentrated and the residue purified by
preparative HPLC to give the title compound. 1H NMR (CD30D) d 1.30
(m, 1 H), 1.73-1.87 (m, 5 H), 2.30 (s, 3 H), 2.71 (m, 2 H), 2.82 (m, 1 H),
3.22
(m,2H), 3.56 (m, 1H),4.01(m, 1H),4.44(d,J=11.5 Hz, 1H),4.98(d,J=
11.5 Hz, 1 H), 7.27-7.62 (m, 10 H), 8.67 (s, 1 H); MS (FAB) 460 (M+1)+.
-76-
,.
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE XV
Preparation of D-3,3-Diphenylalanine-L-proline-N-(4-imidazole-
nronarwl) amide
H
V
\ ''N
H
Ste,~ N-Propargvlphthalimide
A mixture of propargylamine (7 ml, 102 mmol) and phthalic
anhydride (15 g, 101 mmol) in chloroform (200 ml) was heated together at
70°C overnight. Filtration gave a 1:1 mixture of the phthalimide and
the
acid-amide (20 g). This was suspended in THF (100 ml) and heated at
70°C until complete dissolution had been effected. Powdered 4A sieves
(15 g) was added and the heating continued for 24 h. The reaction
mixture was cooled and filtered through Celite washing the residue well
with methanol. The filtrate was concentrated, redissolved in chloroform
and then adsorbed onto silica gel and chromatographed (8:1:1 to 7:1.5:1.5
hexane / chloroform / ethyl acetate) to give the title compound as a white
solid. 1H NMR (CDC13) d 2.23 (m, 1 H), 4.47 (m, 2 H), 7.75 {m, 2 H), 7.88
(m, 2 H).
Step B: 4.5-Diodoimidazole
A solution of iodine {49.5 g, 195 mmol) and sodium iodide
(53.5 g, 360 mmol) in water (350 ml) was added dropwise over 3 h to a
solution of imidazole {6.8 g, 100 mmol) and sodium carbonate (23 g, 220
mmol) in water (650 ml). After an additional 3 h, the diiodide was
filtered off and washed well with water. Drying yielded a cream colored
_77_
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
solid which was recrystallized from aqueous acetone with the aid of
decolorizing carbon. 1H NMR (CD30D) d 7.75 (s, 1 H).
Step C: 4-Iodoimidazole
A hot saturated solution of the diodide (12 g) in ethanol (60
ml) was mixed together with a solution of sodium thiosulfate (29 g) in
water (10 ml). A white precipitate separated. The resulting mixture
was heated at 100°C for 24 h. then cooled and filtered. The filtrate
was
evaporated and the residue boiled three times with chloroform (400 ml
portions) each time followed by a hot filtration. Concentration of the
filtrate gave 4-iodoimidazole as a white solid. 1H NMR (CD30D) d 7.20
(s, 1 H), 7.63 (s, 1 H).
Step D: 1-Tritvl-4-iodoimidazole
Trityl chloride (3.8 g, 13.6 mmol) was added to a cooled (0°C)
solution of 4-iodoimidazole (2.25 g, 11.6 mmol) and triethylamine (2 ml,
14.3 mmol) in methylene chloride (25 ml). After stirring for 30 min the
reaction mixture was warmed to room temperature and stirred ther for
2 h. The reaction mixture was washed well with water and saturated
NaHC03 then dried (Na2S04). Concentration gave the product as a
white solid. 1H NMR (CDC13) d 6.92 (s, 1 H), 7.08-7.20 (m, 6 H), 7.28-7.40
(m, 10 H).
t E: 1-Tritvl-4-imidazoleproparevl nhthalimide
A suspension of N-propargylphthalimide (450.9 mg, 2.4
mmol) and 1-trityl-4-iodoimidazole (875.5 mg, 2 mmol) in diethylamine
(20 ml) was heated to 55°C. Dissolution was incomplete. Bis triphenyl-
phosphinepalladium dichloride (15.6 mg) and copper (I) iodide (a
smidgen) were added and heating continued overnight. The reaction
mixture was cooled and the solvent rotavapped off. The residue was
redissolved in methylene chloride and ether (twice the volume). It was
washed with saturated NaHC03 and water then dried (1:1 Na2S04 /
K2C03). Concentration, adsorption onto silica gel and chromatography
(5:4:1 ethyl acetate / hexane / chloroform) gave the title compound as a
_78_
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
cream powder. 1H NMR (CDC13) d 4.65 (s, 2 H), 7.01 (s, 1 H), 7.10 (m, 6
H), 7.33 (m, 10 H), 7.72 (m, 2 H), 7.86 (m, 2 H).
Step r: 1-Tritvl-4-imidazolepropar~vlami~e
Hydrazine monohydrate (3 ml) was added to a suspension of
the phthalimido compound (734.2 mg, 1.5 mmol) in ethanol (15 ml). The
bulk of the starting material dissolved. The reaction mixture was heated
at 80°C for 2 h. It was then cooled and rotavapped down. After
azeotroping with toluene the residue was adsorbed onto silica gel and
purified by flash chromatography (19:1 to 9:1 chlorofrom / 10°lo NH40H
in methanol) to give the amine as a white solid. 1H NMR (CDC13) d 3.60
(s, 2 H), 6.98 (s, 1 H), 7.13 (m, 6 H), 7.35 (m, 9 H), 7.39 (s, 1 H).
Step G: D-3,3-Diphenylalanine-L-proline-N-(4-imidazolepro
parevl) amide
D-3,3-Diphenylalanine-L-proline and 1-trityl-4-imidazole-
propargylamine were coupled essentially according to the procedure for
Example I, Step A then the protecting groups were removed essentially
according to the procedure of Example IV, Step E. 1H NMR (CD30D) d
1.30-1.39 (m, 1 H), 1.77 (m, 3 H), 2.79 (m, 1 H), 3.56 (m, 1 H), 4.06 (m, 1
H),
4.25(dd,J=17.9Hz,2H),4.44(d,J=11.4 Hz, 1H),4.98(d,J=11.4 Hz, 1
H), 7.27-7.70 (m, 11 H), 8.78 (s, 1 H); MS (FAB) 442 (M+1)+.
-79-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE XVI
Preparation of N-Benzylsulfonyl-D-3,3-diphenylalanine-L-proline-N-(4-
imidazolepropargvl) amide
H
N
~_ N w"~~ N
o H O
1 / ~ H
Step A: N-Benzylsulfonyl-D-3,3-diphenylalanine-L-proline-N-(1
trit~l-4-imidazoleproparg3rl) amide
The title compound was prepared from N-benzylsulfonyl-D-
3,3-diphenylalanine-L-proline and 1-trityl-4-imidazolepropargylamine
using the procedure described in EXAMPLE I, Step A.
Step B: N-Benzylsulfonyl-D-3,3-diphenylalanine-L-proline-N-(4-
imidazole~ro~ar~l) amide
The title compound was prepared from N-benzylsulfonyl-D-
3,3-diphenylalanine-L-proline-N-(1-trityl-4-imidazolepropargyl) amide
using the procedure described in EXAMPLE IV, Step E. 1H NMR
(CD30D) d 1.40-1.50 (m, 1 H), 1.50-1.68 (m, 1 H), 1.72-1.90 (m, 2 H), 2.95-
3.05 (m, 1 H), 3.72-3.82 (m, 1 H), 4.00-4.08 (m, 1 H), 4.10-4.18 (m, 2 H),
4.24
(d,J=2.7Hz,2H),4.33(d,J=11.7 Hz,lH),5.07(d,J=11.2 Hz,lH},
7.15-7.58 (m, 16 H), 8.07 (m, 1 H), 8.72 (s, 1 H); MS (FAB) 596 (M+1}+
_g0_
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE XVII
Preparation of D-3,3-diphenylalanine-L-homopraline-N-(4-imidazole-
pranare-vl) amide
H
N--1
\ N
J
H
Sten AA: Boc-D-3,3-diphenylalanine-L-homoproline-N-(1-trityl-4-
imidazolepro~ar~vl) amide
The title compound was prepared from Boc-D-3,3-
diphenylalanine-L-homoproline and 1-trityl-4-imidazolepropargylarnine
using the procedure described in EXAMPLE I, Step A.
Ste,~B: D-3,3-Diphenylalanine-L-homoproline-N-(4-imidazole
~r_opareyl) amide
The title compound was prepared from Boc-D-3,3-
diphenylalanine-L-homoproline-N-( 1-trityl-4-imidazoleprapargyl) amide
using the procedure described in EXAMPLE IV, Step E. 1H NMR
(CD30D) d 0.20 (m, 1 H), 0.80-1.40 (m, 4 H), 1.95-2.05 (m, 1 H), 3.18-3.30
(m, 1H),3.62-3.80 (m, 1H),4.25(s,2H),4.41(d,J=11.5 Hz, 1H),5.34(d,
J = 11.5 Hz, 1 H), 7.20-7.65 (m, 11 H), 8.46 (br s, 1 H); MS (FAB) 456
(M+1)+
-81-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE XVIII
Preparation of D-3,3-Diphenylalanine-L-proline-N-(5-methyl-4-
imidazole-pronar~vl) amide
~NH
/ \ /
H2N'~~'..
O
~ H
Step A: 4-Iodo-5-methvlimidazole
To a solution of 4-methylimidazole (8.20 g, 100 mmol) and
sodium carbonate (21.2 g, 200 mmol) in water (650 ml) was added a
solution of sodium iodide (26.5 g, 180 mmol) and iodine (25.4 g, 100 mmol)
in water (350 ml) over 90 min at room temperature. The reaction was
stirred a furthur 30 min and filtered. The resulting white solid was
washed with water and dried in uacuo at 50°C. 1H NMR (CD30D) d 2.20
(s, 3 H), 4.86 (br s, 1 H), 7.57 (s, 1 H).
Step B: 1-Boc-4-iodo-5-methvlimidazole
A suspension of 4-iodo-5-methylimidazole (4.16 g, 20 mmol)
and di-t-butyldicarbonate (5.24 g, 24 mmol) in methylene chloride (100
ml) containing triethylamine (4.0 ml, 28.7 mmol) was stirred at room
temperature until homogeneity was achieved (2 h). The reaction
mixture was then washed well with water, dried (Na2S04) and
concentrated. The residue was chromatographed (5:1 hexane / ethyl
acetate) to afford the title compound as a crystalline white solid. 1H
NMR (CDCl3) d 1.62 (s, 9 H), 2.43 (s, 3 H), 8.00 (s, 1 H).
-82-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/0548b
Sten C:C: 1-Boc-5-methvl-4-imidazoleproua~gvl nhthalimide
A miture of 1-Boc-4-iodo-5-methylimidazole (936.8 mg, 3
mmol), copper (I) iodide (30 mg) and bis triphenylphosphinepalladium-
dichloride (217.5 mg, 0.3 mmol) in triethylamine {30 ml) was heated at
60°C for 10 min. There was incomplete dissolution of the catalyst. N-
Propargylphthalirnide (651.4 mg, 3.5 mrnol) was added and the heating
continued. Most of the solids went in then triethylamine hydroiodide
was gradually deposited. After 4 h more alkyne (384.5 mg) was added.
After 1 h the reaction mixture became diff'lcult to stir and was then
evaporated to dryness. The residue was redissolved in chloroform and
adsorbed onto silica gel. Chromatography (5:4:1 ethyl acetate / hexane /
chloroform) gave the title compound. 1H NMR (CDCl3) d 1.61 (s, 9 H),
2.46 (s, 3 H), 4.70 (s, 2 H), 7.74 (m, 2 H), 7.89 (m, 2 H), 7.91 {s, 1 H).
Step D: 5-Methvl-4-imidazole"~roparg-vlamine
The phthaloyl group was removed from 1-Boc-5-methyl-4-
imidazolepropargyl phthalimide as for EXAMPLE XV, Step F with the
exception that the reaction was run at room temperature. The Boc
group was labile under these conditions. 1H NMR (CD30D) d 2.25 (s, 3
H), 3.62 {s, 2 H), 4.95 (br s, 2 H), 7.47 (s, 1 H).
Sten E: D-3,3-Diphenylalanine-L-proline-N-(5-methyl-4-
imidazole-proparQVl) amide
Boc-D-3,3-diphenylalanine-L-proline and 5-methyl-4-
imidazolepropargylamine were coupled essentially according to the
procedure for EXAMPLE I, Step A then the Boc group was removed
essentially according to the procedure of EXAMPLE IV, Step E. 1H
NMR (CD30D) d 1.32 (m, 1 H}, 1.77 (m, 3 H), 2.39 (s, 3 H), 2.79 (m, 1 H),
3.57 (m, 1 H), 4.06 (m, 1 H), 4.27 (dd, J = 17.9 Hz, 2 H), 4.45 (d, J = 11.4
Hz,
1 H), 4.98 (d, J = 11.4 Hz, 1 H), 7.26-7.60 (m, 10 H), 8.75 Es, 1 H); MS (FAB)
456 (M+1)+.
-83-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE XIX
Preparation of 3-Benzylsulfonylamino-6-methyl-1-(4-methylene-
carboxamidopropviimidazolvl)-2wridinone
O
Oz ~ \~ [~ H
S_N N~N N
H H
O N
Step A: 3-Benzyloxvcarbonvlamino-6-methyl-2-nvridinone
DPPA (70 ml, 320 mmol) was added to a stirred solution of 2-
hydroxy-6-methylpyridine-3-carboxylic acid (49 g, 320 mmol ) and
triethylamine (45 ml, 320 mmol) in dry dioxane (500 ml) and the
resulting solution was heated to reflux. After 16 h more triethylamine
(45 ml, 320 mmol) and benzyl alcohol (32 ml, 310 mmol) were added and
the solution was refluxed for a further 24 h. The reaction was
concentrated in uacuo to remove most of the volatiles. The residue was
partitioned between 1:I methylene chloride / chloroform and cold 1:1
brine / 1M citric acid. There was partial formation of an emulsion. The
aqueous phase was washed once with ether and the combined organics
filtered through Celite to break up the colloid. The filtrate was washed
with 1:1 saturated NaHC03 / 10% aqueous Na2C03 solution and dried
2U (Na2S04). Concentration in vacuo gave a tan solid. Methanol was added
to the crude product to give a slurry which was then filtered. The
residue was washed with methanol until the filtrate was clear. Drying
gave the title compound as an off white powder. A further crop was
obtained by evaporating the washings and chromatographing the
residue (7:1.5:1.5 ethyl acetate / hexanes / chloroform): 1H NMR (CDC13)
d 2.29 (s, 3H, CH3), 5.20 (s, 2 H, PhCH2), 6.06 (d, J = 7.6 Hz, pyridinone-5-
H), 7.32-7.43 (m, 5 H, Ph), 7.67 (br s, 1 H, CbzNH), 8.03 (br d, pyridinone-
4-H).
-84-
,,
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
Step B: 3-Benzyloxycarbonylamino-6-methyl-1-(t-butylmethylene-
carboxv)-2-pvridinone
Sodium hydride (5.3 g, 220 mmol) was added proportionwise
to a well stirred slurry of 3-benzyloxycarbonylamino-6-methyl-2-
pyridinone (53 g, 200 mmol) in THF (300 ml) at 0°C. By the end of the
addition a brown solution had resulted. t-Butyl bromoacetate (45 ml, 270
mmol) was then added. Within minutes NaBr started separating out.
After 1 h a thick white precipitate had formed. The reaction was stirred
for an additional hour (bath temperature then 15°C) then the THF was
rotovapped off. The residue was partitioned between THF / methylene
chloride (600 ml / 100 ml) and half saturated brine (200 ml). The organic
phase was dried (Na2S04), filtered and concentrated. The resulting
cream colored solid was triturated with hexane to give of N-alkylated
material as a white microcrystalline solid: 1H NMR (CDC13) d 1.47 {s, 9
H), 2.25 (s, 3 H), 4.75 (s, 2 H), 5.19 (s, 2 H), 6.09 (d, J = 7.8 Hz), 7.30-
7.40
(m, 5 H), 7.75 (br s, 1 H), 7.94 (br d, 1 H).
Ste : 3-Amino-6-methyl-1-(t-butylmethylenecarboxy)-2-
uvridinone
A mixture of 3-benzyloxycarbonylamino-S-methyl-1-(t-butyl-
methylenecarboxy)-2-pyridinone (10 g, 27 mmol) and Pearlman's
catalyst (2 g) in 4:1 ethanol/water (250 ml) was shaken in a Parr
apparatus under H2 (50 psi) for 3 h. The reaction mixture was filtered
through Celite and evaporated an vacuo. The solid residue was
triturated with ether to give the title compound as a pale yellow
crystalline solid: 1H NMR (CDCl3) d 1.46 (s, 9 H, t-Bu), 2.18 (s, 3 H, Me),
4.02 (br s, 2 H, NH2), 4.74 (s, 2 H, CH2), 5.90 (d, J = 7.3 Hz, 1 H,
pyridinone H-5), 6.47 (d, J = 7.3 Hz, 1 H, pyridinone H-4).
Step D: 3-Benzylsulfonylamino-6-methyl-1-(t-butylmethylene-
carboxv)-2-pvridinone
Benzylsulfonyl chloride (5.2 g, 27 mmol) was added to a
solution of 3-amino-6-methyl-1-(t-butylmethylenecarboxy)-2-pyridinone (6
g, 25 mmol) in pyridine (50 ml) at 0°C and as the resulting solution
was
stirred a thick precipitate formed. After 1 h the reaction mixture was
-85-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
evaporated in uacuo to a thick paste. This was partitioned between
methylene chloride and 10% potassium hydrogen sulfate solution. The
organic layer was dried (Na2S04) and evaporated in uacuo to give a
yellow solid which triturated first with hexane then ether. This gave the
title compound as an off white solid: 1H NMR (CDC13) d 1.51 (s, 9 H, t-
Bu), 2.26 {s, 3 H, Me), 4.31 (s, 2 H, PhCH2), 4.75 (s, 2 H, NCH2), 6.01 (d, J
= 7.7 Hz, 1 H, pyridinone H-5), 7.22-7.34 (m, 7 H, remaining H).
Step E: 3-Benzylsulfonylamino-6-methyl-1-methylenecarboxy-2-
pyridinone
HCl gas was bubbled through a stirred suspension of 3-
benzylsulfonylamino-6-methyl-1-(t-butylmethylenecarboxy)-2-pyridinone
(7.5 g, 19 mmol) in ethyl acetate (250 ml) at 0°C until a solution had
formed which was saturated with HCl. After 1 h at room temperature a
thick suspension had formed. The mixture was degassed with argon
and filtered to give the title compound as a pink solid: 1H NMR (CD30D)
d 2.32 (s, 3 H, Me), 4.43 (s, 2 H, PhCH2), 4.89 (s, 2 H, NCH2), 6.14 (d, J =
7.7 Hz, 1 H, pyridinone H-5), 7.28-7.33 (m, 6 H, remaining H).
a F: 3-Benzylsulfonylamino-6-methyl-1-(trans-1-trityl-4-
methvlenecarboxamidoallylimidazolvl)-2-nvridinone
The title compound was prepared from 3-benzylsulfonyl-
amino-6-methyl-1-methylenecarboxy-2-pyridinone and trans-1-trityl-4-
imidazoleallylamine using the procedure described in EXAMPLE I, Step
A. 1H NMR (CDCl3) d 2.40 (s, 3 H), 4.00 (t, J = 6 Hz, 2 H), 4.30 (s, 2 H),
4.68(s,2H),6.02(d,J=8Hz, 1H),6.20-6.40(m,2H),6.60(t,J=6Hz, 1
H), 6.73 (s, 1 H), 7.08-7.40 (m, 21 H), 8.02 (s, 1 H).
Step G: 3-Benzylsulfonylamino-6-methyl-1-(trans-4-methylene-
carboxamidoallvlimidazolvl)-2-pvridinone
The title compound was prepared from 3-benzylsulfonyl-
amino-6-methyl-1-(trans-1-trityl-4-
methylenecarboxamidoallylimidazolyl)-2-pyridinone using the
procedure described in EXAMPLE IV, Step E. 1H NMR {CD30D) d 2.35
(s,3H),4.05(brm,2H),4.45(s,2H),4.85(s,2H),6.19(d,J=8Hz,lH),
-86-
, i .
CA 02283704 1999-09-15
WO 98/42342 PCTIUS98/05486
6.38 (d oft,J=l6and6Hz, 1H),6.58(brd,J=l6 Hz, 1H),7.20-7.37 (m,6
H),7.52(s,iH),8.60(t,J=6Hz,lH),8.81(s,lH).
Step H: 3-Benzylsulfonylamino-6-methyl-1-(4-methylenecarbox
amido-progvlimidazolyl)-2-nvridinone
The title compound was prepared from 3-benzylsulfonyl-
amino-6-methyl-1-(trans-4-methylenecarboxamidoallylimidazolyl )-2-
pyridinone using the procedure described in EXAMPLE I, Step B. 1H
NMR (CD30D) d 1.15-1.40 (m, 2 H), 1.89 (t, J = 6.6 Hz, 2 H), 2.33 (s, 3 H),
2.77(t,J=7Hz,2H),4.43(s,2H),4.79(s,2H),6.16(d,J=7.3 Hz,lH),
7.15-7.40 (m, 7 H), 8.71 (s, 1 H); MS (FAB) 444 (M+1)+
EXAMPLE XX
Preparation of 3-Benzylsulfonylamino-6-methyl-1-(4-methyl-5-
methvlene-carboxamidomethvlimidazolvl)-2-pvridinone
\ 02 \~ O
/ S,N I N~N N
H O H
N
Step A: 1-Trityl-4-methyl-5-imidazolemethanol
A solution of 4-methyl-5-imidazolemethanol hydrochloride
(6.0 g, 40 mmol), trityl chloride (12.3 g, 44 mmol), and triethylamine (16.4
ml, 120 mmol) in chloroform (200 ml) was stirred at room temperature
overnight. The reaction mixture was washed with water, dried over
MgS04, filtered and the solvents removed in uacuo. The crude material
was purified by flash chromatography (24:1 CHC13 / MeOH). iH NMR
(CDC13) d 1.42 (s, 3 H), 3.80 (br s, 1 H), 4.55 (s, 2 H), 7.10-7.40 (m, 16 H).
Step B: 5-Azidomethvl-4-methyl-1-tritvlimidazole
To a solution of 1-trityl-4-methyl-5-imidazolemethanol (3.7 g,
10 mmol) and DPPA (3.0 ml, 13 mmol) in THF (100 ml) was added DBU
_87-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
{2.0 ml, 13 mmol). The resulting solution was heated to 60°C for 2 h.
The solvents were removed in uacuo and the residue purified by flash
chromatography (2:1 hexane / ethyl acetate) to give of the title compound.
1H NMR (CDC13) d 1.42 (s, 3 H), 4.22 (s, 2 H), 7.10-7.35 (m, 16 H).
Step C: 1-Tritvl-4-methyl-5-imidazolemethvlamine
To a solution of 5-azidomethyl-4-methyl-1-tritylimidazole
(3.0 g, 7.9 mmol) in THF (70 ml) was added triphenylphosphine (5.18 g,
19.8 mmol). The resulting solution was refluxed for 1.5 h then water (5
ml) was added. Refluxing was continued for an additional 24 hours.
After cooling to room temperature, the solvents were removed in uacuo
and the crude reaction mixture was purified by chromatography on
silica gel (19:1 chloroform / 10% NH40H in methanol) to give of the title
compound. 1H NMR (CDC13) d 1.40 (s, 3 H), 1.70 (br s, 2 H), 3.?0 (s, 2 H),
7.10-7.35 (m, 16 H).
Step D: 3-Benzylsulfonylamino-6-methyl-1-(1-trityl-4-methyl-5-
methylenecarboxamidomethvlimidazo~l )-2-gvridinone
The title compound was prepared from 3-benzylsulfonyl-
amino-6-methyl-1-methylenecarboxy-2-pyridinone and 1-trityl-4-methyl-
5-imidazolemethylamine using the procedure described in EXAMPLE I,
Step A. 1H NMR (CDCl3) d 1.40 (s, 3 H), 2.39 (s, 3 H), 4.25-4.35 (m, 4 H),
4.78 (s, 2 H), 6.01 (d, J = 8 Hz, 1 H), 6.85-7.05 (br s, 1 H), 7.10-7.35 (m,
22
H).
Step E: 3-Benzylsulfonylamino-6-methyl-I-(4-methyl-5-methyl-ene-
carboxamidomethvlimidazolvl)-2-pvridinone
The title compound was prepared from 3-benzylsulfonyl-
amino-6-methyl-1-( 1-trityl-4-methyl-5-methylenecarboxamidomethyl-
imidazolyl)-2-pyridinone using the procedure described in EXAMPLE
IV, Step E. IH NMR (CD30D) d 2.31 (s, 3 H), 2.34 (s, 3 H), 4.40 (s, 2 H),
4.42(s,2H),4.80(s,2H),6.16(d,J=7Hz, 1H),7.20-7.35(m,6H),8.65(s,
1 H); MS (FAB) 430 (M+1)+.
_88_
CA 02283704 1999-09-15
WO 98/42342 PCT/US98105486
EXAMPLE XXI
Preparation of 3-(4-Chlorobenzylsulfonylamino)-6-methyl-1-(4-
methvl-5-methvlenecarboxamidomethvlimidazohrl)-2-nvridinone
CI ~ O ~ O
S~N I N
N
H O H
'N
Step A: Sodium 4-chlorobenzvlthiosulfate
To a solution of 4-chlorobenzyl chloride (10.0 g, 62 mmol) in
1:1 methanol / water (100 mlj was added sodium thiosulfate (9.81 g, 62
mmol). The resulting solution was then refluxed for 24 h. After cooling
to room temperature, the solvents were removed i.n vacuo and the
residue triturated with ether to give the title compound as a white solid.
IH NMR (CD30D) d 4.25 (s, 2 H), 7.32 (q, J = 33, 9 Hz, 4 H).
Steg B: 4-Chlorobenzylsulfonvl chloride
Chlorine gas was slowly bubbled through a cooled (0°C)
solution of sodium 4-chlorobenzylthiosulfate (11.2 g, 56 mmol) in acetic
acid (100 ml) to which ice (10 g) had been added. Additional ice was
added as necessary to maintain the temperature <10°C. After 0.5 h the
addition of chlorine was stopped and the resulting yellow solution was
allowed to stir at 0°C for I h. The solution was then extracted with
ether
and the organics washed twice with cold 5% sodium bisulfite solution.
Drying over MgS04, filtration and removal of the solvents in uczcuo gave
the title compound as a white solid. 1H NMR (CDC13) d 4.82 (s, 2 H), 7.45
(m, 4 H).
Ste : 3-(4-Chlorobenzylsulfonylamino)-6-methyl-1-(t-butyl-
methvlenecarboxv)-2-Rvridinone
To a solution of 3-amino-6-methyl-1-(t-butylmethylene-
carboxy)-2-pyridinone (600 mg, 2.5 mmol) in pyridine (50 ml) was added
_89-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
4-chlorobenzylsulfonyl chloride (830 mg, 3.75 mmol). The resulting red
solution was stirred at room temperature overnight. The solvents were
removed in uacuo and the crude material purified by preparative HPLC.
1H NMR (CDC13) d 1.51 (s, 9 H), 2.27 (s, 3 H), 4.27 (s, 2 H), 4.75 (s, 2 H),
6.03 (d, J = 8 Hz, 1 H), 7.17-7.30 (m, 4 H), 7.35 (d, J = 8 Hz, 1 H).
Step D: 3-(4-Chlorobenzylsulfonylamino)-6-methyl-1-methylene-
car oxv-2-nvridinone
Hydrogen chloride gas was bubbled through a suspension of
3-(4-chlorobenzylsulfonylamino)-6-methyl-1-(t-butyl-methylenecarboxy)-
2-pyridinone (850 mg, 2.0 mmol) in ethyl acetate (100 ml} that had been
cooled to 0°C. After I5 min the addition of HCl gas was stopped and the
solution warmed to room temperature for 1 h. The mixture was then
purged with nitrogen and the solvent removed in uacuo to give the title
I5 compound as a solid. 1H NMR (CH30D) d 2.33 (s, 3 H), 4.43 (s, 2 H), 4.88
(s,2H),6.12(d,J=7.6Hz,lH),7.29(s,4H),7.34(d,J=7.8 Hz,lH).
Step E: 3-(4-Chlorobenzylsulfonylamino)-6-methyl-1-(1-trityl-4-
methyl-5-methylenecarboxamidomethylimidazolyl)-2-
pvridinone
The title compound was prepared from 3-(4-chlorobenzyl-
sulfonylamino)-6-methyl-1-methylenecarboxy-2-pyridinone and 1-trityl-4-
methyl-5-imidazolemethylamine using the procedure described in
EXAMPLE I, Step A. 1H NMR (CDCl3) d 1.40 (s, 3 H), 2.39 (s, 3 H), 4.25-
4.35 (m, 4 H), 4.78 (s, 2 H), 6.01 (d, J = 8 Hz, 1 H), 6.85-7.05 (br s, 1 H),
7.10-
7.35 (m, 22 H).
Step F: 3-(4-Chlorobenzylsulfonylamino-6-methyl-1-(4-methyl-5-
methvlenecarboxamidomethvlimidazolvl)-2-pvridinone
The title compound was prepared from 3-(4-chlorobenzyl-
sulfonylamino-6-methyl-1-( 1-trityl-4-methyl-5-methylenecarboxamido-
methyl-imidazolyl)-2-pyridinone using the procedure described in
EXAMPLE IV, Step E. 1H NMR (CD30D) d 2.33 (s, 3 H), 2.35 (s, 3 H),
4.43 (s, 2 H), 4.46 (s, 2 H), 4.81 (s, 2 H), 6.17 (d, J = 8 Hz, 1 H), 7.20-
7.30 (m,
4 H), 7.35 (d, J = 7.5 Hz, 1 H), 8.68 (s, 1 H); MS (FAB) 464 (M+1)+.
-90-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE XXII
Preparation of 3-Benzylsulfonylamino-6-methyl-1-(1'-t-butoxycarbonyl-
methyl-4-methyl-5-methylenecarboxamidomethyl-imidazolyl)-2-
pvridinone
02 ~ ~~ o r-cootB~
/ S, N N~ N N
H O H
N
Step A: 1-t-Butoxycarbonylmethyl-4-methyl-5-hydroxymethyl-
imidazole
t-Butyl bromoacetate (35 ml, 240 mmol} was added to a
mixture of 4-methyl-5-imidazolemethanol hydrochloride (30 g, 200
mmol) and potassium carbonate (80 g, 580 mmol) in N,N-
dimethylformamide (500 ml) and the resulting heterogenous mixture
stirred at room temperature for 24 h. The reaction mixture was filtered
through Celite and the DMF was then removed in uacuo from the
filtrate. The residue was dissolved in a minimum quantity of methylene
chloride and the resulting solution diluted several fold with ether and
ethyl acetate. This solution was washed well with cold water and dried
over sodium sulfate. Filtration and concentration gave a cream colored
solid ( 16 g) which was determined by NMR analysis to be a 2:1 mixture of
1-t-butoxycarbonyl-methyl-4-methyl-5-hydroxymethylimidazole and 3-t-
butoxycarbonyl-methyl-4-methyl-5-hydroxymethylimidazole respectively.
The two isomers displayed very similar mobility on TLC but were
separable by careful gradient elution chromatography on silica gel (99:1
to 19:1 chloroform / methanol): N-1 isomer 1H NMR (CDCl3) d 1.46 (s, 9
H), 2.23 (s, 3 H), 4.55 (s, 2 H), 4.63 (s, 2 H}, 7.35 (s, 1 H); N-3 isomer 1H
NMR(CDC13}d1.48(s,9H),2.17(s,3H),4.47(s,2H),4.56(s,2H),7.39
(s, 1 H).
-91-
CA 02283704 1999-09-15
WO 98/42342 PCT/i1S98/05486
Step B: 1-t-Butoxvcarbonvlmethvl-4-methyl-5-azido_methvlimidazole
A solution of 1-t-butoxycarbonylmethyl-4-methyl-5-hydroxy-
methylimidazole {4.61 g, 20 mmol) in DMF (100 ml) was cooled to 0°C and
treated sequentially with diphenylphosphoryl azide (5.4 ml, 25 mmol)
and DBU (3.7 ml, 25 ml). The resulting solution was allowed to warm
15
gradually to room temperature and was then stirred there overnight.
The DMF was rotavapped off, the residue dissolved in a minimum
quantity of methylene chloride and the resulting solution diluted several
fold with ether and ethyl acetate. This solution was washed sequentially
with 1M citric acid, water, 10°lo sodium carbonate, brine and then
dried
over magnesium sulfate. Filtration and concentration gave an oil which
was purified by flash chromatography (19:1 chloroform / methanol) to
give the title compound as an oil: 1H NMR (CDCl3) d 1.48 (s, 9 H), 2.27 (s,
3H),4.28(s,2H),4.56{s,2H),7.43(s,lH).
Stgn C: 1-t-Butoxycarbonylmethyl-4-methyl-5-
aminomethvlimidazole
A solution of 1-t-butoxycarbonylmethyl-4-methyl-5-azido-
methylimidazole (2.9 g) in ethyl acetate (100 ml) containing 10°~0
palladium on carbon ( 1.5 g) was stirred at room temperature under an
atmosphere of hydrogen for 3 h. After removal of the catalyst by
filtration through Celite, the filtrate was concentrated to give the amine
as a colorless oil: 1H NMR (CDgOD) d 1.48 (s, 9 H), 2.19 {s, 3 H), 3.69 (s, 2
H), 4.81 (s, 2 H), 7.49 (s, 1 H).
-92-
,,
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
Step D: 3-Benzylsulfonamino-6-methyl-1-(1-t-butoxycarbonylmethyl-
4-methyl-5-methvlenecarboxamidomethvlimidazolvl )-2-
pvridinone
The title compound was prepared from 3-benzylsulfonyl-
amino-6-methyl-1-methylenecarboxy-2-pyridinone and 1-t-butoxy-
carbonylmethyl-4-methyl-5-aminomethylimidazole essentially according
to the procedure of EXAMPLE I, Step A: 1H NMR (CD30D) d 1.41 (s, 9 H,
t-Bu), 2.36 (s, 3 H, pyridinone Me), 2.37 (s, 3 H, imidazole Me), 4.27 (s, 2
H, PhCH2), 4.36 (d, J = 5.7 Hz, 2 H, imidazoleCH2N), 4.51 (s, 2 H,
pyridinoneNCH2), 4.86 (s, 2 H, imidazoleNCH2), 6.06 (d, J = 7.9 Hz, 1 H,
pyridinone H5}, 7.15-7.26 (m, 6 H, Ph and pyridinone H6}, 8.08 (br s, 1 H,
S02NH), 8.28 (br s, 1 H, CONH), 8.48 (s, 1 H, imidazole H2); MS (FAB)
544 (M+1)+
EXAMPLE XXIII
Preparation of 3-Benzylsulfonylamino-6-methyl-1-(1-carboxymethyl-4-
methvl-5-methvlenecarboxam,'_domethvlimidazolvl)-2 pvridinone
02 I ~~ O ~-COOH
/ S,N N~N N
H O H
N
The title compound was prepared from 3-
benzylsulfonamino-6-methyl-1-(1-t-butoxycarbonylmethyl-4-methyl-5-
methylenecarboxamido-methylimidazolyl)-2-pyridinone essentially
according to the procedure of EXAMPLE XXI, Step D: 1H NMR (CD30D)
d2.31(s,3H),2.41(s,3H),4.44(s,2H),4.46(m,2H),4.73(s,2H),5.17 (s,
2H),6.13(d,J=7Hz,lH),7.30(m,6H),8.74(brs,lH),8.81 (s,lH);MS
(FAB) 488 (M+1)+.
-93-
CA 02283704 1999-09-15
WO 98/42342 PCT/~JS98/05486
EXAMPLE XXIV
Preparation of 3-Benzylsulfonylamino-6-methyl-1-(I-t-butylamino-
carbonylmethyl-4-methyl-5-methylenecarboxamidomethylimidazolyl )-
2-nvridinone
I ~ 02 I \~ O[I ~-CONHtBu
/ S,N N~N N
H O H
N
The title compound was prepared from 3
benzylsulfonamino-6-methyl-1-( 1-carboxymethyl-4-methyl-5-
methylenecarboxamidomethyl-imidazolyl)-2-pyridinone and t-
butylamine essentially according to the procedure of EXAMPLE I, Step
A: 1H NMR (CD30D) d 1.33 (s, 9 H), 2.32 (s, 3 H), 2.39 (s, 3 H), 4.41 (m, 2
H),4.45(s,2H),4.75(s,2H),4.99(s,2H},6.15(d,J=7.5 Hz, 1H),7.26-
7.34 (m, 6 H), 7.89 (br s, 1 H), 8.67 (br t, 1 H), 8.74 (s, 1 H); MS (FAB) 544
{M+1}+
EXAMPLE XXV
Preparation of 3-Benzylsulfonylamino-6-methyl-1-(1-
ethylaminocarbonyl-methyl-4-methyl-5-
methvlenecarboxamidomethvlimidazolyl)-2-t~vridinone
02 I ~~ O r CONHEt
/ S,N N~N N
H O H
N
The title compound was prepared from 3-
benzylsulfonamino-6-methyl-I-( 1-carboxymethyl-4-methyl-5-
methylenecarboxamidomethyl-imidazolyl)-2-pyridinone and ethylamine
hydrochloride essentially according to the procedure of EXAMPLE I,
-94-
r r .
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
Step A: 1H NMR (CD30D) d 1.13 (t, J = 7.3 Hz, 3 H), 2.32 (s, 3 H), 2.39 (s, 3
H),3.26(m,2H),4.43(m,2H),4.45(s,2H),4.75(s,2H},5.05(s,2H),6.15
(d, J = 7.7 Hz, 1 H}, 7.26-7.34 {m, 6 H), 8.25 (br s, 1 H), 8.64 (br t, 1 H),
8.77
(s, 1 H); MS (FAB) 515 (M+1)+.
EXAMPLE XXVI
Preparation of 3-Benzylsulfonylamino-6-methyl-1-(1-cyclopropylamino-
carbonylmethyl-4-methyl-5-methylenecarboxamidomethylimidazolyl )-2-
pvridinone
H
-~JN
O
I ~ S, I N~ O
H O H
~N
The title compound was prepared from 3-
benzylsulfonamino-6-methyl-1-( 1-carboxymethyl-4-methyl-5-
methylenecarboxamidomethyl-imidazolyl)-2-pyridinone and
cyclopropylamine essentially according to the procedure of EXAMPLE I,
Step A: 1H NMR (CD30D) d 0.72 (m, 4 H), 2.32 (s, 3 H), 2.40 {s, 3 H), 2.70
(m,lH),4.43(m,2H),4.45(s,2H),4.74{s,2H),5.02(s,2H),6.15(d,J=
7.7 Hz, 1 H), 7.24-7.36 (m, 6 H), 8.39 (br s, 1 H), 8.67 (br t, 1 H), 8.76 (s,
1
H); MS (FAB} 527 (M+1)+.
_95_
CA 02283704 1999-09-15
WO 98/42342 PCT/CIS98/05486
EXAMPLE XXVII
Preparation of 3-Benzylsulfonylamino-6-methyl-1-(1-cyclopropylmethyl-
aminocarbonylmethyl-4-methyl-5-methylenecarboxamidomethyl-
imidazolvl)-2-uvridinone
H
\ 02 ~ \ O
S,N N N N O
H O H
N
The title compound was prepared from 3-
benzylsulfonamino-6-methyl-1-( 1-carboxymethyl-4-methyl-5-
methylenecarboxamidomethyl-imidazolyl)-2-pyridinone and
cyclopropylmethylamine essentially according to the procedure of
EXAMPLE I, Step A: 1H NMR (CD30D) d 0.20 (d, J = 4.7 Hz, 2 H), 0.49
(d, J = 6.9 Hz, 2 H), 0.96 (br m, 1 H), 2.31 (s, 3 H), 2.39 (s, 3 H), 3.07 (d,
J =
7Hz,2H),4.43(s,2H),4.44(s,2H),4.74(s,2H),5.07(s,2H),6.15(d,J=
7.9 Hz, 1 H), 7.28 (m, 6 H), 8.77 (s, 1 H); MS (FAB) 541 (M+1)+
-96-
CA 02283704 1999-09-15
WO 98/42342 PCTNS98/05486
EXAMPLE XXVIII
Preparation of 3-Benzylsulfonylamino-6-methyl-1-[1-(1,1-dimethylpropyl-
aminocarbonylmethyl)-4-methyl-5-methylenecarboxamidomethyl-
imidazolvll-2-nyridinone
H
N
\ 02 \~ O
S~ I N' ~ O
N V _N
H O H I
N
The title compound was prepared from 3-
benzylsulfonamino-6-methyl-1-( 1-carboxymethyl-4-methyl-5-
methylenecarboxamidomethyl-imidazolyl)-2-pyridinone and t-
amylamine essentially according to the procedure of EXAMPLE I, Step
A: 1H 1VMR, (CD30D) d 0.85 (t, J = 7.5 Hz, 3 H), 1.28 (s, 6 H), 1.70 (q, J =
7.5Hz,2H),2.32(s,3H),2.38(s,3H),4.40(s,2H),4.44(s,2H},4.75(s,2
H),5.01(s,2H),6.15(d,J=7.7Hz,lH),7.28(m,6H),7.78(s,lH),8.74
I5 (s, 1 H); MS (FAB) 557 (M+1)+.
-97-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE XXIX
Preparation of 3-Benzylsulfonylamino-6-methyl-1-(1-t-butylmethylamino-
carbonylmethyl)-4-methyl-5-methylenecarboxamidomethyl-imidazolyl]-
2-DVridinone
H
N
O
2
N ~N N O
H O H
N
Step A: 1-Carbox~vl-4-methvl-5-azidomethvlimidazole
The title compound was prepared from 1-t-butoxycarbonyl
methyl-4-methyl-5-azidomethylimidazole using the procedure described
in EXAMPLE XXI, Step D. 1H NMR (CD30D) d 2.43 (s, 3 H), 4.61 (s, 2 H),
5.13 (s, 2 H), 8.94 (s, 1 H).
Step B: 1-t-Butylmethylaminocarbonylmethyl-4-methyl-5-
azidomethylimidazole
The title compound was prepared from 1-carboxymethyl-4-
methyl-5-azidomethylimidazole and t-butylmethylamine using the
procedure described in EXAMPLE I, Step A. 1H NMR (CDC13) d 0.84 (s,
9H),2.30(s,3H),3.05(d,J=6.4Hz,2H),4.35(s,2H),4.62(s,2H),5.45
(br s, 1 H), 7.50 (s, 1 H).
Ste : 1-t-Butylmethylaminocarbonylmethyl-4-methyl-5-
aminomethvlimidazole
A solution of 1-t-butylmethylaminocarbonylmethyl-4-
methyl-5-azidomethylimidazole (400 mg) and 10% Pd/C ( 400 mg) in ethyl
acetate (60 ml) was hydrogenated at atmospheric pressure for 4 h. The
solution was filtered through Celite and the solvents removed in Uacuo to
give the title compound. 1H NMR (CDC13) d 0.80 (s, 9 H), 1.61 (br s, 2 H),
_98_
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
2.23 (s, 3 H), 2.99 (d, J = 6.1 Hz, 2 H), 3.86 (s, 2 H), 4.63 (s, 2 H), 7.50
(s, 1
H).
to D: 3-Benzylsulfonylamino-6-methyl-1-(1-t-butylmethylamino-
carbonylmethyl-4-methyl-5-methylenecarboxamidomethyl-
imidazolvl)-2-pvridinone
The title compound was prepared from 3-
benzylsulfonamino-6-methyl-1-methylenecarboxy-2-pyridinone and 1-t-
butylmethylamino-carbonylmethyl-4-methyl-5-aminomethylimidazole
essentially according to the procedure of EXAMPLE I, Step A: 1H NMR
(CD30D)d0.90(s,9H),2.31(s,3H),2.39(s,3H),3.04(d,J=6.1Hz,2H),
4.43(m,4H),4.88(s,2H),5.12(s,2H),6.15(d,J=7.5 Hz, 1H),7.26-7.34
{m, 6 H), 8.25 (br s, 1 H), 8.71 (br t, 1 H), 8.77 (s, 1 H); MS (FAB) 557
{M+1)+.
EXAMPLE XXX
Preparation of 3-Benzylsulfonylamino-6-methyl-1-[1-(2,2,2-trifluoroethyl-
aminocarbonylmethyl)-4-methyl-5-methylenecarboxamidomethyl-
imidazolvll-2-l7vridinone
N ~ Fs
\ 02 \~ O
S,N N N N O
H O H
N
The title compound was prepared from 3-
benzylsulfonamino-6-methyl-1-( 1-carboxymethyl-4-methyl-5-
methylenecarboxamidomethyl-imidazolyl)-2-pyridinone and 2,2,2-
trifluoroethylamine hydrochloride essentially according to the procedure
of EXAMPLE I, Step A: 1H NMR (CD30D) d 2.30 (s, 3 H), 2.39 (s, 3 H),
3.95(q,J=9.1Hz,2H),4.41(s,2H),4.44(s,2H),4.74(s,2H),5.16(s,2
-99-
CA 02283704 1999-09-15
WO 98/42342 PCTNS98/05486
H), 6.14 (d, J = 7.5 Hz, 1 H), 7.26-7.31 (m, 6 H), 8.77 (s, 1 H); MS (FAB) 569
(M+1}+.
EXAMPLE XXXI
Preparation of 3-Benzylsulfonylamino-6-methyl-1-[1-(N-morpholino-
carbonylmethyl)-4-methyl-5-methylenecarboxamidomethylimidazolyl]-2-
pvridinone
NJ
O ~~ O
2
S, I N~ O
H O H
~N
The title compound was prepared from 3-
benzylsulfonamino-6-methyl-1-( 1-carboxymethyl-4-methyl-5-
methylenecarboxamidomethyl-imidazolyl)-2-pyridinone and morpholine
essentially according to the procedure of EXAMPLE I, Step A: 1H NMR
(CD30D)d2.32(s,3H),2.40(s,3H),3.40(d,J=4.4Hz,2H),3.57(d,J=
16.3 Hz, 2 H), 3.63 (dd, J = 5.4 Hz, 4 H), 4.45 (s, 2 H), 4.46 (s, 2 H}, 4.70
(s, 2
H),5.25(s,2H),6.14(d,J=7.5 Hz, 1H),7.24(d,J=7.7 Hz, 1H),7.32(m,
5 H}, 8.64 (br t, 1 H), 8.71 (s, 1 H); MS (FAB) 557 (M+1}+.
-100-
CA 02283704 1999-09-15
WO 98/42342 PCT/LTS98/05486
EXAMPLE XXXII
Preparation of 3-Benzylsulfonylamino-6-methyl-1-[1-(3-hydroxyazetidine
1-carbonylmethyl)-4-methyl-5-methylenecarboxamidomethylimidazolyl]
2-uvridinone
OH
N-'
\ 02 \~ O
/ S_ ' N' ~ p
N v _N
O
N
The title compound was prepared from 3-
benzylsulfonamino-6-methyl-1-( 1-carboxymethyl-4-methyl-5-
methylenecarboxamidomethyl-imidazolyl)-2-pyridinone and 3-
hydroxyazetidine essentially according to the procedure of EXAMPLE X,
Step Y: 1H IVMR (CD30D) d 2.31 (s, 3 H), 2.39 (s, 3 H), 3.82 (m, 1 H), 4.03
(m, 1 H), 4.24 (m, 1 H), 4.42 (m, 1 H), 4.44 (s, 6 H), 4.50 (m, 1 H), 4.73 (s,
2
H),5.04(s,2H),6.15(d,J=7.7 Hz, 1H),7.30(m,6H),8.72(s, 1H);MS
(FAB) 543 (M+1)+.
-101-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE XXXIII
Preparation of 3-Benzylsulfonylamino-6-methyl-1-[1-(2-hydroxyethyl-
aminocarbonylmethyl)-4-methyl-5-methylenecarboxamidomethyl-
imidazolvll-2-nvridinone
H~OH
N
0
~ s_ I N~ o
H O H
'N
The title compound was prepared from 3-
benzylsulfonamino-6-methyl-1-( 1-carboxymethyl-4-methyl-5-
methylenecarboxamidomethyl-imidazolyl)-2-pyridinone and
hydroxyethylamine essentially according to the procedure of EXAMPLE
I, Step A: 1H NMR (CD30D) d 2.30 (s, 3 H), 2.38 (s, 3 H), 3.34 (t, J = 5 Hz,
2H),3.61(t,J=5.5Hz,2H),4.44(s,4H),4.74(s,2H),5.06(s,2H),6.15
(d, J = 7.5 Hz, 1 H), 7.28 (m, 6 H), 8.76 (s, 1 H); MS (FAB) 531 (M+1)+.
-102-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
SAMPLE XXXIV
Preparation of 3-Benzylsulfonylamino-6-methyl-1-[1-(2-aminoethyl-
aminocarbonylmethyl)-4-methyl-5-methylenecarboxamidomethyi-
irnidazolvll-2-nvridinone
H~NH2
~/N
\ 02 \~ O
/ S, I N' ~ p
N ~N
H O H
N
Step A: Bocaminoeth~Iamine
A solution of di-t-butyl dicarbonate (6.98 g, 32 mmol) in
dioxane (50 ml) was added in a slow dropwise manner to a cooled (0°C)
solution of ethylenediamine (2.2 ml, 32 mmol) in dioxane (30 ml). Upon
completion of the addition, the reaction mixture was stirred overnight at
room temperature. After filtration to remove a white precipitate, the
filtrate was concentrated. The residue was partitioned between water
and methylene chloride. The organic phase was dried (Na2S04) and
concentrated to give the title compound as an oil which partially
crystallized. 1H NMR (CDC13) d 1.45 (s, 9 H), 2.80 (m, 2 H), 3.16 (m, 2 H).
Step B: 3-Benzylsulfonylamino-6-methyl-1-[1-(2-aminoethyl-
aminocarbonylmethyl)-4-methyl-5-methylenecarbox-
amidomethylimidazolyll-2-pyridinone
3-Benzylsulfonamino-6-methyl-1-( 1-carboxymethyl-4-
methyl-5-methylenecarboxamidomethylimidazolyl)-2-pyridinone and
Bocamino-ethylamine were coupled essentially according to the
procedure of EXAMPLE I, Step A. Removal of the Boc protecting group
according to the procedure of EXAMPLE XXI, Step D gave the title
compound: 1H NMR (CD30D) d 2.32 (s, 3 H), 2.39 (s, 3 H), 3.04 (t, J = 5.7
Hz,2H),4.49(s,2H),4.51(s,2H),4.76(s,2H),5.12(s,2H),5.31(t,J=
-103-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
5.7Hz,2H),6.15(d,J=7.5Hz,lH),7.20{d,J=7.7 Hz,lH),7.34(m,5
H), 8.77 (s, 1 H); MS (FAB) 531 (M+1)+.
EXAMPLE XX~~V
Preparation of 3-(4-Chlorobenzylsulfonylamino)-6-methyl-1-(1-t-butyl-
aminocarbonylmethyl-4-methyl-5-methylenecarboxamidomethyl-
imidazolyl)-2-pyridinone
CI
I ~ 02 ~ ~~ O~ r-CONHtBu
/ S,N N~N N
H O H
N
The title compound was prepared from 3-(4-chlorobenzyl-
sulfonylamino)-6-methyl-1-methylenecarboxy-2-pyridinone and 1-t-
butylaminocarbonylmethyl-4-methyl-5-aminomethylimidazole using the
procedure described in EXAMPLE I, Step A. 1H NMR (CD30D) d 1.33 (s,
9 H), 2.32 (s, 3 H), 2.39 (s, 3 H), 4.40 (br s, 2 H), 4.44 (s, 2 H), 4.75 (s,
2 H),
5.00 (s, 2 H), 6.I5 (d, J = 7.8 Hz, 1 H), 7.20-7.35 (m, 5 H), 7.93 (br s, 1
H),
8.70 (br m, 1 H), 8.77 (s, 1 H); MS (FAB) 577 (M+1)+.
-104-
,,
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE X~~XVI
Preparation of 3-Benzylsulfonylamino-6-propyl-1-(1-t-butylamino-
carbonylmethyl-4-methyl-5-rnethylenecarboxamidomethylimidazolyl)-2-
pyridinone
I ~ 02 ~~ OI, ~-CONHtBu
S,N N~N N
H O H
N
Step A: b-N.N-Dimethvlaminoethenvlcvclopropvl ketone
A mixture of cyclopropyl methyl ketone (5.88 ml, 59 mmol)
and N,N-dimethylformamide dimethyl acetal (7.83 ml, 59 mmol) was
heated in the presence of a catalytic quantity of p-toluenesulfonic acid for
48 h. The resulting crude sample of the title compound (a pale yellow oil)
was used in subsequent reactions without further purification: 1H NMR
(CDClg) d 0.74 (m, 2 H), 1.00 (m, 2 H), 1.75 (m, 1 H), 3.48 (s, 3 H), 3.50 (s,
3
H), 5.20 (d, 1 H), 7.55 (d, 1 H).
Step B: 6-Cvclopropyl-3-nitro-2-pyridinone
A mixture of crude b-N,N-
dimethylaminoethenylcyclopropyl ketone (12 g, < 86 mmol),
nitroacetamide (9 g, 86 mmol) and aqueous piperidinium acetate (10 ml)
[prepared from glacial acetic acid (42 ml), water (100 ml) and piperidine
(72 ml)] was stirred at room temperature overnight. Following dilution
with water (20 ml), the yellow precipitate was isolated via filtration and
drying in Uacuo to yield the title compound: 1H NMR (CDCl3) d 1.15 (m,
2H),1.36(m,2H),2.10(m,lH),6.02(brd,J=8.O Hz,lH),8.41(d,J=8.0
Hz, 1 H).
-105-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
St, e~ C: 3-Amino-6-progvl-2-pvridi~one
A mixture of 6-cyclopropyl-3-vitro-2-pyridinone (2 g, 13.3
mmol) and 10% palladium on carbon (600 mg) in ethyl acetate (100 ml)
was stirred at room temperature under an atmosphere of hydrogen
overnight. The catalyst was removed by filtration through a bed of Celite
and the filtrate concentrated to yield product as a white microcrystalline
solid. 1H NMR (CDC13) d 0.94 (t, J = 7.3 Hz, 3 H), 1.67 (m, 2 H), 2.49 (t, J =
7.5Hz,2H),4.00(brs),5.88(d,J=7.1 Hz,lH),6.59(d,J=7.1 Hz,lH).
Step D: 3-Benzvloxycarbonylamino-6-pro~yl-2-pvridinone
Benzyl chloroformate (1.8 ml, 12.6 mmol) was added to a
solution of 3-amino-6-propyl-2-pyridinone (1.63 g, 10.8 mmol) in a
mixture of dioxane (25 ml) and 1N NaOH at 0°C. Within minutes a
white precipitate formed. The reaction mixture was stirred at the same
temperature for 1 h then at room temperature for 1 h. The reaction
mixture was diluted with water and extracted with ethyl acetate then
methylene chloride. Each extract was washed with brine then combined
and dried over magnesium sulfate. Removal of the solvents in Uacuo
gave a yellow semi-solid which was a mixture of starting material and
product. This was redissolved in a mixture of dioxane {24 ml) and 10%
aqueous sodium carbonate (12 ml) and cooled to 0°C. Benzyl
chloroformate (1.5 ml, 10.5 mmol) was once again added and after
stirring for 0.5 h, the reaction mixture was allowed to stir at room
temperature for 3 h. After dilution with water, the precipitate was
filtered off and washed thoroughly, first with water and then with ether.
Drying gave the title compound as a white powder: 1H NMR (CDC13) d
0.96(t,J=?.3Hz,3H), 1.68(m,2H),2.52(t,J=7.5Hz,2H),5.22(s,2H),
6.07 (d, J = 7.5 Hz, 1 H), 7.34-7.43 (m, 4 H), 7.68 (s, 1 H), 8.05 (br d, J =
5.3
Hz, 1 H).
Step E 3-Benzyloxycarbonylamino-6-propyl-1-(t-butylmethylene-
carboxv)-2-pyridinone
Solid 3-benzyloxycarbonylamino-6-propyl-2-pyridinone (2.64
g, 9.3 mmol) was added in small portions to a suspension of sodium
hydride (269 mg, 11.2 mmol) in THF (30 ml) at 0°C. The reaction
-106-
CA 02283704 1999-09-15
WO 98/42342 PCTIUS98/05486
mixture was the stirred at room temperature for 20 min by which time
an almost completely homogeneous solution had been obtained. Tert-
butyl-bromoacetate (2.2 ml, 14.9 mmol) was then added. Within minutes
a white precipitate started forming. Stirring was continued overnight,
then the THF was evaporated in Uacuo. Ice was carefully added to the
residue to destroy any unreacted sodium hydride. Brine was added and
the resulting miture was extracted with 2:1:I ethyl acetate / ether /
chloroform and the combined extracts were dried over magnesium
sulfate. Filtration and evaporation of the filtrate gave a cream solid
which was purified by flash column chromatography eluting with 3:1:1
hexane / chloroform / ethyl acetate. This gave the title compound as a
white crystalline solid: 1H NMR (CDC13) d 1.00 (t, J = 7.3 Hz, 3 H), 1.48
(s,9H), 1.64(m,2H),2.46(t,J=7.6Hz,2H),4.72(s,2H),5.20(s,2H),
6.08 (d, J = 7.7 Hz, 1 H), 7.31-7.40 (m, 4 H), 7.77 (s, 1 H), 7.97 (br d, J =
7.0
Hz, 1 H).
Ste ~ 3-Amino-6-propyl-1-(t-butylmethylenecarboxy)-2-pvridinone
3-Benzyloxycarbonylamino-6-propyl-1-(t-butylmethylene-
carboxy)-2-pyridinone (2.38 g, mmol) was dissolved in a 1:1 mixture of
ethyl acetate and ethanol (100 ml) and then stirred in the presence of 20%
palladium hydroxide on carbon (800 mg) under an atmosphere of
hydrogen for 2 h. The catalyst was removed by filtration through Celite
and the filtrate concentrated to give the title compound as an orange oil:
1H NMR (CDC13) d 0.98 (t, J = 7.3 Hz, 3 H), 1.47 (s, 9 H), 1.57 (m, 2 H), 2.40
(t,J=7.7Hz,2H),4.73{s,2H),5.91{d,J=7.3 Hz,lH),6.55(d,J=7.3
Hz, 1 H).
to G' 3-Benzylsulfonylamino-f-propyl-1-(t-butylmethylene-
carboxv)-2-nvridinone
Benzylsulfonyl chloride (880 mg, 4.6 mmol) was added to a
solution of 3-amino-6-propyl-1-(t-butylmethylenecarboxy)-2-pyridinone
(1.1 g, 4.1 mmol) in pyridine (20 ml) at 0°C and as the resulting
solution
was stirred a thick precipitate formed. After 1 h the reaction mixture
was evaporated in Uacuo to a thick paste. This was partitioned between
-107-
CA 02283704 1999-09-15
WO 98/42342 PCTlUS98105486
methylene chloride and 10% potassium hydrogen sulfate solution. The
organic layer was dried (Na2S04) and evaporated in Uacuo to give a red
solid which was purified by flash chromatography (2:1:1 hexane /
chloroform / ethyl acetate) to give the desired product as a mustard
crystalline solid: 1H NMR (CDC13) d 1.03 (t, J = 7.3 Hz, 3 H), 1.51 (s, 9 H),
1.63(m,2H),2.46{t,J=7.7Hz,2H),4.32(s,2H),4.73(s,2H),5.99(d,J=
7.7 Hz, 1 H), 7.22-7.32 (m, 5 H}, 7.35 (d, J = 7.7 Hz, 1 H).
Step H: 3-Benzylsulfonylamino-6-propyl-1-methylenecarboxy-2-
pvridinone
HCl gas was bubbled through a stirred suspension of 3-
benzylsulfonylamino-6-propyl-1-(t-butylmethylenecarboxy)-2-pyridinone
(1.1 g, 2.8 mmol) in ethyl acetate (20 ml) at 0°C until a solution had
formed which was saturated with HCl. After 4 h at room temperature
the mixture was degassed with nitrogen and filtered to give the title
compound as a pale pink solid: 1H NMR (CDC13) d 1.02 (t, J = 7.3 Hz, 3
H),1.63(m,2H),2.57(t,J=7.7Hz,2H),4.44(s,2H),4.85(s,2H),6.11 (d,
J = 7.7 Hz, 1 H), 7.26-7.33 (m, 5 H}, 7.34 (d, J = 7.7 Hz, 1 H).
Step I: 1-Carbaxvmethvl-4-methyl-5-azidomethvlimidazole
The title compound was prepared from I-t-butoxycarbonyl-
methyl-4-methyl-5-azidomethylimidazole using the procedure described
in EXAMPLE XXI, Step D. 1H NMR (CD30D) d 2.43 (s, 3 H), 4.61 (s, 2 H),
5.14 (s, 2 H), 8.97 (s, 1 H).
Step J: 1-t-Butylaminocarbonylmethyl-4-methyl-5-azidamethyl-
imidazole
The title compound was prepared from 1-carboxymethyl-4-
methyl-5-azidomethylimidazole and tent -butylamine using the
procedure described in EXAMPLE I, Step A. 1H NMR {CDCl3) d 1.32 (s,
9 H), 2.29 (s, 3 H), 4.31 (s, 2 H), 4.48 (s, 2 H), 5.25 (br s, 1 H), 7.47 (s,
1 H).
-108-
~ i
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
Step K: 1-t-Butylaminocarbonylmethyl-4-methyl-5-aminomethyl-
imidazole
A solution of 1-t-butylaminocarbonylmethyl-4-methyl-5-
azidomethylimidazole (1.27 g, 5.4 mmol) and 10°~o Pd/C (700 mg) in
ethyl
acetate (100 ml) was hydrogenated at atmospheric pressure for 5 h. The
catalyst was removed by filtration through Celite and the solvents
removed in vacuo to give the title compound. 1H NMR (CDC13) d 1.27 (s,
9 H), 2.23 (s, 3 H), 3.83 {s, 2 H), 4.50 (s, 2 H), 7.08 (br s, 1 H), 7.46 (s,
1 H).
Step L: 3-Benzylsulfonamino-6-propyl-1-(1-t-butylaminocarbonyl-
methyl-4-methyl-5-methylenecarboxamidomethylimida-
zolyl )-2-pvridinone
The title compound was prepared from 3-benzylsulfonyl-
amino-6-propyl-1-methylenecarboxy-2-pyridinone and 1-t-butylamino-
carbonylmethyl-4-methyl-5-aminomethylimidazole essentially according
to the procedure of EXAMPLE I, Step A: 1H NMR (CD3~D) d 1.00 (t, J =
7.4Hz,3H), 1.32(s,9H),1.61(m,2H),2.38(s,3H),2.56(t,J=7.7Hz,2
H),4.41(d,J=3.9Hz,2H),4.44(s,2H),4.72(s,2H),4.99{s,2H),6.13{d,
J = 7.7 Hz, 1 H), 7.26-7.34 (m, 6 H), 8.74 (s, 1 H); MS (FAB) 571 (M+1)+.
EXAMPLE XXXVII
Preparation of 3-(2-Tetrahydropyranylmethanesulfonylamino)-6-methyl-
1-( 1'-t-butoxycarbonylmethyl-4-methyl-5-methylenecarboxamidomethyl-
imidazolvl)-2-pvridinone
02 I ~~ O ~-COO'Bu
O S, N N~ N N
H O H
N
to A: 2-Tetrahvdropvranvlmethanethioacetate
To a solution of 2-bromomethyltetrahydropyran (10.5 g, 59
mmol) in THF (100 ml) was added potassium thioacetate (7.35 g, 65
-109-
CA 02283704 1999-09-15
WO 98/42342 PCTNS98l05486
mmol). The resulting suspension was refluxed for 48 h. After cooling to
room temperature, the solvents were removed in vacuo and the residue
dissolved in chloroform and washed with water. The organics were
dried over MgS04, filtered and the solvents removed in uacuo. The crude
product was purified by chromatography (9:1 hexane / ethyl acetate). 1H
NMR (CDC13) d 1.21-1.40 (m, 1 H), 1.45-1.60 (m, 3 H), 1.73 (br d, J = 14 Hz,
1 H), 1.80-1.90 (m, 1 H), 2.35 (s, 3 H), 2.81-2.95 (m, 1 H), 3.01-3.15 (m, 1
H),
3.35-3.45 (m, 2 H), 3.95-4.07 (m, 1 H).
Steg B: 2-Tetrahvdrouvranvlmethanesulfonvl chloride
The title compound was prepared from 2-tetrahydropyranyl-
methanethioacetate using the procedure described in EXAMPLE XXI,
Step B. 1H NMR (CDC13) d 1.35-1.50 (m, 1 H), 1.50-1.70 (m, 3 H), 1.75 (br
d, J = 14 Hz, 1 H), 1.83-1.95 (m, 1 H) 3.42-3.57 (m, 1 H), 3.70-3.80 (m, 1 H),
3.90-4.12 (m, 3 H).
to C: 3-(2-Tetrahydropyranylmethanesulfonylamino)-6-methyl-1-
t-butvlmethvlenecarboxv)-2-pvridinone
The title compound was prepared from 2-tetrahydropyranyl-
methanesulfonyl chloride and 3-amino-6-methyl-1-(t-butylmethylene-
carboxy)-2-pyridinone using the procedure described in EXAMPLE XXI,
Step C. 1H NMR (CDC13) d 1.25-1.48 (m, 4 H), 1.45 (s, 9 H), 1.62 (br d, J =
14 Hz, 1 H), 1.78-1.85 (m, 1 H), 2.25 (s, 3 H), 3.00-3.10 (m, 1 H), 3.25-3.35
(m, 1 H), 3.35-3.45 (m, 1 H), 3.80-3.87 (m, 1 H), 3.87-3.95 (m, 1 H), 4.75 (s,
2
H), 6.05 (d, J = 8 Hz, 1 H), 7.45 (d, J = 8 Hz, 1 H) 7.49 (s, 1 H).
Step D: 3-(2-Tetrahydropyranylmethanesulfonylamino)-6-methyl-I-
methvlenecarboxv-2-pvridinone
The title compound was prepared from 3-{2-tetrahydro-
pyranylmethanesulfonylamino)-6-methyl-1-{t-butylmethylenecarboxy)-2-
pyridinone using the procedure described in EXAMPLE XXI, Step D. 1H
NMR, (CD30D) d 1.25-1.58 (m, 4 H), 1.63 (br d, J = 14 Hz, 1 H), 1.75-1.85
(m, 1 H), 2.32 {s, 3 H), 3.17-3.40 (m, 3 H), 3.75-3.90 (m, 2 H), 4.90 (s, 2
H),
6.22(d,J=8Hz, 1H),7.50(d,J=7.5 Hz, 1H).
3,5
-110-
CA 02283704 1999-09-15
WO 98/42342 PCTIUS98/05486
Std E: 3-(2-Tetrahydropyranylmethanesulfonylamino)-6-methyl-1-
( 1'-t-butoxycarbonylmethyl-4-methyl-5-methylene-carbox-
ammidomethvlimidazolvl)-2-pyridinone
The title compound was prepared from 3-(2-tetrahydro-
pyranylmethanesulfonylamino)-6-methyl-1-methylenecarboxy-2-
pyridinone and 1-t-butoxycarbonylmethyl-4-methyl-5-aminomethyl-
imidazole using the procedure described in EXAMPLE I, Step A. 1H
NMR (CD30D) d 1.20-1.58 (m, 4 H), 1.50 (s, 9 H), 1.65 (br d, J = 14 Hz, 1
H), 1.72-1.87 (m, 1 H), 2.31 (s, 3 H), 2.40 (s, 3 H), 3.16-3.40 (m, 3 H), 3.75-
3.87(m,2H),4.41(s,2H),4.75(s,2H),5.12(s,2H),6.22(d,J=7.3Hz,I
H), 7.45 (d, J = 7.5 Hz, 1 H), 8.80 (s, 1 H); MS (FAB) 552 (M+1)+
EXAMPLE XXXVIII
Preparation of 3-(2-Tetrahydropyranylmethanesulfonylamino)-6-methyl-
1-( 1'-t-butylaminocarbonylmethyl-4-methyl-5-
methvlenecarboxamidomethyl-imidazolvl)-2-pyridinone
02 I ~~ O ~-CONHtBu
O S,N N~N N
H O H
N
The title compound was prepared from 3-(2-tetrahydro-
pyranylmethanesulfonylamino)-6-methyl-1-methylenecarboxy-2-
pyridinone and 1-t-butylaminocarbonylmethyl-4-methyl-5-aminomethyl-
imidazole using the procedure described in EXAMPLE I, Step A. 1H
NMR (CD30D) d 1.20-1.40 (m, 2 H), 1.30 (s, 9 H), 1.40-1.60 (m, 2 H), 1.65
(br d, J = 14 Hz, 1 H), 1.75-1.90 (m, 1 H), 2.33 (s, 3 H), 2.39 (s, 3 H), 3.15-
3.40(m,3H),3.75-3.87(m,2H),4.40(s,2H),4.76(s,2H),5.00(s,2H),
6.24 (d, J = 7.6 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1 H), ?.95 (br s, 1 H), 8.70
(m, 1
H), 8.78 (s, 1 H) MS (FAB) 551 (M+1)+.
-111-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE _X_X_X_IX
Preparation of 3-(2-Tetrahydropyranmethanesulfonylamino)-6-propyl-1-
( 1'-t-butylaminocarbonylmethyl )-4-methyl-5-methylenecarboxamido-
methvlimidazolvl)-2-Qvridinone
O ~-CONHtBu
O S.N N~N N
H O H
N
The title compound was prepared using the procedure
described for Example XXXVI by substituting 2-
tetrahydropyranmethane-sulfonyl chloride for benzylsulfonyl chloride in
Step G. 1H NMR, (CD30D) d 1.00 (t, J = 7.2 Hz, 3 H), 1.34 (s, 9 H), 2.39 (s, 3
H), 2.58 (t, J = 7.6 Hz, 2 H), 3.83 (d, J = 10.5 Hz, 2 H), 4.39 (s, 2 H), 4.74
(s, 2
H), 5.01 (s, 2 H), 6.24 (d, J = 7.6 Hz, 1H), 7.50 (d, J = 7.6 Hz, 1 H), 8.78
(s, 1
H); MS (FAB) 579 (M+1)+.
-112-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/0~486
EXAMPLE XL
Preparation of 3-Cyclohexylmethanesulfonylamino-6-methyl-1-(1'-t-
butyl-aminocarbonylmethyl-4-methyl-5-methylenecarboxamidomethyl-
imidazolvl)-2-nvridinon
02 I ~~ O r CON HtBu
S_N N~N N
H O H I i
N
Step A: Sodium cvclohexvlmethanethiosulfate
The title compound was prepared from bromomethyl-
cyclohexane using the procedure described in EXAMPLE XXI, Step A.
1H NMR (CDC13) d 0.90-1.05 (m, 2 H), 1.15-1.39 (m, 3 H), 1.60-1.80 (m, 4
H),1.85(brd,J=l2Hz,2H)2.95(d,J=7Hz,2H).
Step B: Cvclohexvlmethanesulfonyl chloride
The title compound was prepared from sodium cyclohexyl-
methanethiosulfate using the procedure described in EXAMPLE XXI,
Step B. 1H NMR (CDCl3) d 1.10-1.42 (m, 5 H), 1.62-1.80 (m, 3 H), 1.97 (br
d,J=l2Hz,2H),2.15-2.30 (m, 1H),3.75(d,J=6Hz,2H).
to : 3-Cyclohexanemethylsulfonylamino-6-methyl-1-(t-butyl-
methvlenecarboxv)-2-pvridinone _
The title compound was prepared from cyclohexylmethane-
sulfonyl chloride and 3-amino-6-methyl-1-(t-butylmethylenecarboxy)-2-
pyridinone using the procedure described in EXAMPLE XXI, Step C. 1H
NMR (CDCl3) d 0.95-1.37 {m, 5 H), 1.45 (s, 9 H), 1.58-1.95 (m, 6 H), 2.27 (s,
3 H), 2.95 (d, J = 7 Hz, 2 H), 4.77 (s, 2 H), 6.10 (d, J = 8 Hz, 1 H), 7.20
(s, 1
H),7.45(d,J=BHz, 1H).
-113-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
Step D: 3-Cyclohexanemethylsulfonylamino-6-methyl-1-methylene-
carboxv-2-pvridinone
The title compound was prepared from 3-
cyclohexanemethyl-sulfonylamino-6-methyl-1-(t-
butylmethylenecarboxy)-2-pyridinone using the procedure described in
EXAMPLE XXI, Step D. 1H NMR (CD30D) d 0.95-1.40 (m, 5 H), 1.60-1.75
(m, 3 H), 1.85-1.95 (m, 3 H), 2.35 (s, 3 H), 2.97 (d, J = 6 Hz, 2 H), 4.90 {s,
2
H),6.22(d,J=8Hz,lH),7.43(d,J=8Hz,lH).
St, ep E: 3-Cyclohexylmethanesulfonylamino-6-methyl-1-( 1'-t-butyl-
aminocarbonylmethyl-4-methyl-5-methylenecarboxamido-
methvlimidazolvl )-2-pvridinone
The title compound was prepared from 3
cyclohexanemethyl-sulfonylamino-6-methyl-1-methylenecarboxy-2
pyridinone and 1-t-butyl-aminocarbonylmethyl-4-methyl-5-
aminomethylimidazole using the procedure described in EXAMPLE I,
Step A. 1H NMR (CD30D) d 0.95-1.42 (m, 5 H), 1.34 (s, 9 H), 1.60-1.75 {m,
3H),1.81-1.98(m,3H),2.33(s,3H),2.39{s,3H),3.00(d,J=5.6Hz,2H},
4.40(s,2H),4.74(s,2H),5.00(s,2H)6.24(d,J=7.4 Hz,lH),7.44(d,J=
7.6 Hz, 1 H), 7.95 (br s, 1 H), 8.75 (br m, 1 H), 8.78 (s, 1 H); MS (FAB) 549
(M+1 )+
-I14-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98I05486
EXAMPLE XLI
Preparation of 3-Pentanesulfonylamino-6-methyl-1-(1'-t-butoxycarbonyl-
methyl-4-methyl-5-methylenecarboxamidomethyl-imidazolyl)-2-
pvridinone
02 I ~~ O ~-COOtBu
S,N N~N N
H O H
N
Step A: Pentanesulfonvl chloride
The title compound was prepared from 1-pentanethiol using
the procedure described in EXAMPLE XXI, Step B. Ice water was used
as the solvent instead of ice water / acetic acid. 1H NMR (CDC13) d 0.95
(t, J = 7 Hz, 3 H), 1.32-1.45 (m, 4 H), 1.95-2.10 (m, 2 H), 3.60-3.70 (m, 2
H).
Step B: 3-Pentanesulfonylamino-6-methyl-1-(t-butylmethylene-
carboxv)-2-pyridinone
The title compound was prepared from pentanesulfonyl
chloride and 3-amino-6-methyl-1-(t-butylmethylenecarboxy)-2-pyridinone
using the procedure described in EXAMPLE XXI, Step C. It was
purified by chromatography (2:1 hexane / ethyl acetate). 1H NMR
(CDClg) d 0.85 (t, J = 7 Hz, 3 H), 1.20-1.40 (m, 4 H), 1.45 (s, 9 H), 1.74-
1.86
(m,2H),2.25(s,3H),2.98-3.05(m,2H),4.75(s,2H),6.08(d,J=8Hz, 1
H), 7.20 (s, 1 H), 7.45 (d, J = 8 Hz, 1 H).
to ~ 3-Pentanesulfonylamino-6-methyl-1-methylenecarboxy-2-
pyridinone
The title compound was prepared from 3-pentanesulfonyl-
amino-6-methyl-1-(t-butylmethylenecarboxy)-2-pyridinone using the
procedure described in EXAMPLE XXI, Step D. 1H NMR (CD3UD) d 0.85
(t, J = 7 Hz, 3,H), 1.22-1.40 (m, 4 H), 1.70-1.82 (m, 2 H), 2.35 (s, 3 H),
3.02-
3.12(m,2H),4.87(s,2H),6.25(d,J=8Hz,lH),7.47(d,J=8Hz,lH).
-115-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
St, e~ D: 3-Pentanesulfonylamino-6-methyl-1-(1'-t-butoxycarbonyl-
methyl-4-methyl-5-methylenecarboxamidomethylimida-
zolvl)-2-pvridinone
The title compound was prepared from 3-pentanesulfonyl-
amino-6-methyl-1-methylenecarboxy-2-pyridinone and 1-t-butoxy-
carbonylmethyl-4-methyl-5-aminomethylimidazole using the procedure
described in EXAMPLE I, Step A. 1H NMR (CD30D) d 0.89 (t, J = 6.8 Hz,
3 H), 1.25-1.40 (m, 4 H), 1.49 (s, 9 H), 1.70-1.81 (m, 2 H), 2.31 (s, 3 H),
2.40
(s,3H),3.08-3.18(m,2H),4.41(s,2H),4.76(s,2H),5.11(s,2H),6.23 (d,
J = 7.6 Hz, 1 H), 7.45 (d, J = 7.6 Hz, 1 H), 8.81 (s, 1 H); MS (FAB) 524
(M+1)+.
EXAMPLE XLII
Preparation of 3-(2-Phenethylamino)-6-methyl-1-(1'-t-butoxycarbonyl-
methyl-4-methyl-5-methylenecarboxamidomethylimidazolyl )pyraz-
inone
O r COOtBu
/ N N~ N N
H O H
N
Step A: N-(1-Cvanoeth~glvcine benzvl ester hydrochloride
TMSCN (4.27 ml, 32 mmol) was added cautiously (reaction
is exothermic) to a stirred solution of glycine benzyl ester (5.3 g, 32
mmol, prepared from the HCl salt by partitioning between EtOAc and
NaHC03 solution) and acetaldehyde (1.8 mI, 32 mmol) in methylene
chloride (11 ml). After 4 h the volatiles were removed in uacuo and the
residue was taken up in EtOAc and was washed with brine, dried
(Na2S04) and evaporated in ur~cuo to an oil. The oil was redissolved in
EtOAc and 9.9 M HCl in EtOH (38.4 mmol) was added to give a
crystalline precipitate which was isolated by filtration and washing with
-116-
CA 02283704 1999-09-15
WO 98/42342 PCTlUS98/05486
EtOAc, to give the title compound: 1H NMR (CDC13) d 1.49 (d, J = 7.1 Hz,
3H,CH3),3.54(d,J=17.3 Hz, 1H,CHAHB),3.64(d,J=17.3 Hz, 1H,
CHAHB), 3.74 (q, J = 7.0 Hz, 1 H, a-CH), 5.18 (s, 2 H, CH20), 7.36 (s, 5 H,
Ph).
Step B: 1-Benzyloxycarbonylmethyl-3,5-dichloro-6-
methvlnvrazinone
A stirred mixture of oxalyl chloride (9.3 ml, 107 mmol) and
N-(1-cyanoethyl)glycine benzyl ester hydrochloride (6.8 g, 26.7 mmol) in
1,2-dichlorobenzene (25 ml) was heated to 100°C for 15 h. The excess
reagent was evaporated in vacuo and the residue was purified by flash
chromatography (eluting first with hexanes to remove the
dichlorobenzene, then with 3:2 hexanes / ethyl acetate) to give a solid
which was triturated with 1:1 hexanes / ethyl acetate to give the title
compound as a pale green crystalline solid: 1H NMR (CDC13) d 2.35 (s, 3
H, CH3), 4.88 (s, 2 H, CH2), 5.24 (s, 2 H, CH2), 7.38 (m, 5 H, Ph).
Ste : 3-(2-Phenethylamino)-5-chloro-6-methyl-1-(benzyloxy-
carbonvlmeth~Ryrazinone
2-Phenethylamine (0.38 ml, 3.0 mmol) was added to a
stirred mixture of 1-benzyloxycarbonylmethyl-3,5-dichloro-6-
methylpyrazinone (327 mg, 1.00 mmol) in EtOAc (2 ml) and the resulting
mixture was heated to reflux under argon. After 2 h the reaction was
cooled, diluted with EtOAc (the product is sparingly soluble), washed
with 10~1~ citric acid solution and brine, dried (Na2S04) and evaporated
in vacuo to give the title compound as a crystalline solid. 1H NMR
(CDC13) d 2.21 (s, 3 H, CH3), 2.93 (t, J = 7.1 Hz, 2 H, PhCH2), 3.67 (q, J =
7.1 Hz, 2 H, CH2NH), 4.79 (s, 2 H, CH2), 5.21 (s, 2 H, CH2), 6.10 (br t, 1 H),
7.20-7.39 (m, 10 H, 2 Ph).
Step D: 3-(2-Phenethylamino)-5-chloro-6-methyl-1-(methylene-
carboxv)nvrazinone
Water (1 ml) was added to a stirred solution of 3-(2-
phenethylamino)-5-chloro-6-methyl-1-(benzyloxycarbonylmethyl)-
pyrazinone (436 mg) in l:l THF / MeOH (6 ml) and LiOH.H20 was added
-117-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/0548G
to the resulting mixture. After 2 h, the reaction mixture was diluted
with water and washed with EtOAc. The aqueous layer was acidified
with 10% KHS04 solution to give a cloudy mixture which was extracted
with methylene chloride. The organic layer was dried (Na2S04) and
evaporated in uacuo to give the title compound as a crystalline solid: 1H
NMR (DMSO-dg) d 2.19 (s, 3 H, Me), 2.84 (t, J = 7.0 Hz, 2 H, PhCH2), 3.45
(q, J = 7.0 Hz, 2 H, CH2NH), 4.70 (s, 2 H, CH2C02), 7.18-7.31 (m, 5 H, Ph),
7.46 (br s, 1 H, NH).
Step E: 3-(2-Phenethylamino)-6-methyl-1-methylenecarboxy-
pvrazinone
3-(2-Phenethylamino)-5-chloro-6-methyl-1-(methylene-
carboxy)pyrazinone (13.4 g, 41.6 mmol) was added to a stirred solution of
potassium hydroxide (7.28 g, 1I0 mmol, assuming 15% water in the
pellets) in water (600 ml). After degassing the resulting solution with
argon, 10% Pd/C (6.3 g) was added and the mixture then stirred under a
balloon of hydrogen. After 16 h, HPLC analysis showed that 1% of the
starting material remained. The mixture was filtered through Celite
and the filtrate was adjusted to pH 2 with 3N KHS04 solution. The
resulting precipitate was collected by filtration and washed with water.
Drying for 16 h at 0.5 mm Hg gave the title compound as a crystalline
solid: 1H NMR (DMSO-dg) d 2.11 (s, 3 H, Me), 2.87 (t, J = 7.6 Hz, 2 H,
PhCH2), 3.53 (br s, 2 H, CH2NH), 4.68 {s, 2 H, CH2C02), 6.68 (s, 1 H,
pyrazinone H-5), 7.20-7.31 (m, 5 H, Ph), 8.16 (br s, I H, NH).
Sten F_ 3-(2-Phenethylamino)-6-methyl-1-(1'-t-butoxycarbonyl-
methyl-4-methyl-5-
methvlenecarboxamidomethylimidazol~pvrazinone
The title compound was prepared from 3-(2-
phenethylamino)-6-methyl-1-methylenecarboxypyrazinone and 1-t-
butoxycarbonylmethyl-4-methyl-5-aminomethylimidazole essentially
according to the procedure of EXAMPLE I, Step A: 1H NMR (CD30D) d
1.44{s,9H),2.18(s,3H),2.19(s,3H),2.92(t,J=7.1Hz,2H),3.60(m,2
H),4.33(d,J=5.5Hz,2H),4.55(d,J=10.8Hz,4H),5.93(t,J=5.7Hz,1
H), 6.73 (s, 1 H), 7.2 (m, 1 H); MS (FAB) 495 (M+1)+.
-118-
CA 02283704 1999-09-15
WO 98142342 PCT/LTS98/05486
EXAMPLE XLIII
Preparation of 3-(2-Phenethylamino)-6-methyl-1-( 1'-t-butylamino-
carbonylmethyl-4-methyl-5-methylenecarboxamidomethylimidazolyl)-
pvrazinone
I ~ N~ O r-CONHtBu
/ N N~ N N
H O H
N
Step A: 3-(2-Phenethylamino)-6-methyl-1-( 1-carboxymethyl-4-
methyl-5-
methvlenecarboxamidomethylimidazolvl )pvrazinone
The title compound was prepared from 3-(2-
phenethylamino)-6-methyl-1-( 1'-t-butoxycarbonyl-methyl-4-methyl-5-
methylenecarbox-amidomethylimidazolyl)pyrazinone essentially
according to the procedure of EXAMPLE XXI, Step D: 1H NMR (CD30D)
d2.16(s,3H),2.41(s,3H),2.99(t,J=7.1Hz,2H),3.6?(t,J=7.1Hz,2H),
4.4?(m,2H),4.68(s,2H),5.15(s,2H),6.56(s,lH),7.21-7.33(m,SH),
8.84 (s, 1 H).
Step B: 3-(2-Phenethylamino)-6-methyl-1-(1'-t-butylaminocarbonyl-
methyl-4-methyl-5-
methvlenecarboxamidomethvlimidazolvl)-pvrazinone
The title compound was prepared from 3-(2-
phenethylamino)-6-methyl-1-( 1-carboxymethyl-4-methyl-5-
methylenecarboxamidomethyl-imidazolyl)pyrazinone and t-butylamine
essentially according to the procedure of EXAMPLE I, Step A: 1H NMR
(CD30D)d1.36(s,9H),2.17(s,3H),2.39(s,3H),2.98(t,J=7.4Hz,2H),
3.60(t,J=7.4Hz,2H),4.42(m,2H),4.70(s,2H),4.99(s,2H),6.59 (s, 1
H), 7.21-7.33 (m, 4 H), 7.97 (br s, 1 H), 8.78 (s, 1 H); MS (FAB) 495 (M+1)+.
-119-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/~5486
EXAMPLE XLIV
Preparation of 3-(2-Pyridylethylamino)-6-methyl-1-(1'-t-butoxycarbonyl-
methvl-4-methvl-5-methvlenecarboxamidomethylimidazolvl)gvrazinone
N O ~- COOtBu
N N N~N N
O
N
The title compound was prepared according to the
procedure described for EXAMPLE XLII by substituting 2-(2-
aminoethyl)pyridine for 2-phenethylamine in Step C: 1H NMR (CD30D)
d1.45(s,9H},2.12{s,3H),2.20(s,3H),3.08(t,J=7.OHz,2H),3.69(t,J=
7.0 Hz, 2 H), 4.32 (s, 2 H), 4.62 {s, 2 H), 4.79 (s, 2 H), 6.66 (s, 1 H), 7.25
(m, 1
H),7.35(d,J=4.O Hz, 1H),7.52(s, 1H), 7.74{t,J=7.5 Hz, 1H),8.44(d,J
= 4.0 Hz, 1 H); MS (FAB) 495 (M+1)+.
EXAMPLE XLV
Preparation of 3-(2-Pyridylethylamino)-6-methyl-1-(1'-t-butylamino-
carbonylmethyl-4-methyl-5-methylenecarboxamidomethylimidazolyl )-
l7vrazlnone
I ~ N~ O ~ CON HtBu
iV N N~N N
H O H
N
The title compound was prepared according to the
procedure described for EXAMPLE XLIV by substituting 1-t-
butylmethylamino-carbonylmethyl-4-methyl-~-aminomethylimidazole
for 1-t-butoxycarbonyl-methyl-4-methyl-5-aminomethylimidazole in Step
F: 1H NMR, (CD30D) d 1.35 (s, 9 H), 2.15 (s, 3 H), 2.38 (s, 3 H), 3.82 (t, J =
6.8Hz,2H),3.82(t,J=6.8Hz,2H),4.41(d,J=3.7Hz,2H),4.69(s,2H),
4.87 (s, 2 H), 4.99 (s, 2 H), 6.66 (s, 1 H), 7.77 (t, J = 6.50 Hz, 1 H), 7.85
(d, J =
-120-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
8.1 Hz, 1 H), 7.98 (s, 1 H), 8.68 (d, J = 5.7 Hz, 1 H), 8.78 (s, 1 H); MS
(FAB)
495 (M+1)+.
EXAMPLE XLVI
Preparation of 3-(2-Phenethylamino)-6-methyl-1-(1'-t-butylmethylamino-
carbonylmethyl-4-methyl-5-methylenecarboxamidomethylimidazolyl )-
nvrazinone
H
\ N~ O
N N N N O
H O H
N
The title compound was prepared according to the
procedure described for EXAMPLE XLIII by substituting 1-t-
butylmethylamino-carbonylmethyl-4-methyl-5-aminomethylimidazole
for 1-t-butoxycarbonyl-methyl-4-methyl-5-aminomethylimidazole in Step
F: 1H NMR (CD30D) d 0.93 (s, 9 H), 2.17 (s, 3 H), 2.39 (s, 3 H), 2.99 (t, J =
7.5Hz,2H),3.07,(d,J=6.1Hz,2H),3.68(t,J=7.5Hz,2H),4.43(s,2H),
4.71 (s, 2 H), 5.12 (s, 2 H), 6.56 (s, 1 H), 7.22-7.34 (m, 6 H), 8.34 (br t, 2
H),
8.82 (s, 1 H); MS (FAB) 508 (M+1)+.
-121-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98105486
EXAMPLE XLyII
Preparation of 3-(2-Phenethylamino)-6-methyl-I-[1'-(2,2,2-trifluoroethyl-
aminocarbonylmethyl)-4-methyl-5-methylenecarboxamidomethyl-
imidazolvllnvrazinone
H ~ F3
N
w N~ o
N N N N O
H O H
N
The title compound was prepared according to the
procedure described for EXAMPLE XLIII by substituting 1-(2,2,2-
trifluoroethyl-aminocarbonylmethyl)-4-methyl-5-aminomethylimidazole
(prepared according to the procedure for EXAMPLE XXIX, Steps A to C
by substituting 2,2,2-trifluoroethylamine hydrochloride for t-butylmethyl-
amine in Step B) for 1-t-butoxycarbonylmethyl-4-methyl-5-aminomethyl-
imidazole in Step F: 1H NMR (CD30D) d 2.16 (s, 3 H), 2.41 (s, 3 H), 2.98
(t,J=7.4Hz,2H),3.65(t,J=7.4Hz,2H),4.43(s,2H),4.68(s,2H),5.17
(s, 2 H), 6.59 (s, 1 H), 7.23-7.33 (m, 4 H), 8.83 (s, 1 H); MS (FAB} 520
(M+1)+
EXAMPLE XLVIII
Preparation of 3-(2-Phenethylamino)-6-methyl-1-[1'-(3-piperidineamino)-
carbonylmethyl]-4-methyl-5-methylenecarboxamido-methylimidazolyl]-
pvrazinone
H
N
I ~ N O
/ N N N N O
H O H
N
-I22-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
Step A: 3-Hvdroxv-N-t-butoxvcarbonvlgi~peridine
Di-t-butyldicarbonate (21 g, 96 mmol) was added to a
mixture of 3-hyroxypiperidine hydrochloride (12 g, 87 mmol) and
triethylamine (24.5 ml, 176 mmol) in methylene chloride (500 ml) at
0°C.
After stirring for 2 h, the reaction mixture was washed well with water
and dried (MgS04). Concentration gave the title compound. 1H NMR
(CDCl3) d 1.46 (s, 9 H), 1.60-1.89 (m, 4 H), 3.04-3.16 (m, 2 H), 3.52 (br s, 1
H), 3.73 (m, 2 H).
Ste~B_: 3-Methanesulfonyloxv-N-t-butoxvcarbonvlpiDPridine
Methanesulfonic anhydride (996 mg, 5.72 mmol) was added
to a mixture of 3-hydroxy-N-t-butoxycarbonylpiperidine (959 mg, 4.76
mmol) and triethylamine (0.86 ml, 6.19 mmol) in methylene chloride (30
ml) at 0°C. After stirring for 1 h, the reaction mixture was washed
with
staurated NaHC03 and dried (Na2S04). Concentration gave the title
compound. 1H NMR (CDC13) d 1.46 (s, 9 H), 1.77-2.17 (m, 4 H), 3.05 (s, 3
H), 3.14-3.49 (m, 2 H), 3.62 (m, 2 H), 4.72 (br s, 1 H).
Step C: 3-Azido-N-t-buto~vcarbonvlniberidine
Lithium azide (1.35 g, 27.6 mmol) was added to a solution of
3-methanesulfonyloxy-N-t-butoxycarbonylpiperidine (1.54 g, 5.51 mmol)
in DMF (20 ml) and the resulting mixture heated at 60°C for 48 h.
Removal of the solvent in uacuo and chromatographic purification (3:1
hexane / ethyl acetate) of the residue afforded the title compound. 1H
NMR (CDC13) d 1.46 {s, 9 H), 1.75 (m, 2 H), 1.96 (m, 2 H), 3.13 (br s, 1 H),
3.46(m,2H),3.57(m,2H).
Step D: 3-Amino-N-t-butoxycarbon~pi~eridine
A solution of 3-azido-N-t-butoxycarbonylpiperidine (850 mg)
in ethyl acetate (50 ml) was hydrogenated in the presence of 10% Pd/C
(550 mg) at atmospheric pressure for 2.5 h. The reaction mixture was
then filtered through Celite and the filtrate concentrated in vacuo to give
the title compound. 1H NMR (CDCl3) d 1.46 (s, 9 H), 1.66-2.05 (m, 4 H),
2.57 (br s, 1 H), 2.78 (m, 4 H), 3.80 (br d, 2 H).
-123-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
Step E: 1-[(N-t-butoxycarbonyl-3-piperidineamino)carbonyl-meth 1 -
4-methyl-5-azidomethylimidazole
The title compound was prepared from 1-carboxymethyl-4-
methyl-5-azidomethylimidazole and 3-amino-N-t-
butoxycarbonylpiperidine using the procedure described in EXAMPLE I,
Step A. 1H NMR (CDC13) d 1.45 (s, 9 H), 1.53 (br m, 2 H), 1.63 (m, 1 H),
1.80 (br m, 1 H), 2.28 (s, 3 H), 3.23-3.49 (br m, 4 H), 3.95 (br s, I H), 4.32
(dd, J = 14.6, 21.4 Hz, 2 H), 4.57 (s, 2 H), 7.47 (s, 1 H).
StegF: 1-[(N-t-butoxycarbonyl-3-piperidineamino)carbonyl-meth 1 -
4-methyl-5-aminomethviimidazole
A solution of 1-[(N-t-butoxycarbonyl-3-piperidine-
amino)carbonylmethyl]-4-methyl-5-azidomethylimidazole (3.77 g) in
ethanol (100 ml) was hydrogenated in the presence of 20% Pd(OH)2/C
(850 mg) at atmospheric pressure for 5 h. The reaction mixture was
then filtered through Celite and the filtrate concentrated in uacuo to give
the title compound.
Ste : 3-(2-Phenethylamino)-6-methyl-1-[1'-(N-t-butoxycarbonyl-3-
piperidineamino)carbonylmethyl]-4-methyl-5-methylene-
carboxamidomethvlimidazolvllnvrazinone
The title compound was prepared from 3-(2-
phenethylamino)-6-methyl-1-methylenecarboxypyrazinone and 1-[(N-t-
butoxycarbonyl-3-piperidineamino)carbonylmethyl]-4-methyl-5-
aminomethylimidazole using the procedure described in EXAMPLE I,
Step A.
Stgp H: 3-(2-Phenethylamino)-6-methyl-1-[1'-(3-piperidineamino)-
carbonylmethyl]-4-methyl-5-methylenecarboxamidomethyl-
imidazolyl_lpvrazinone
The title compound was prepared from 3-(2-
Phenethylamino)-6-methyl-1-[1'-(N-t-butoxycarbonyl-3-
piperidineamino)carbonylmethyl]-4-methyl-5-
methylenecarboxamidomethylimidazolyl]pyrazinone using the
procedure described in EXAMPLE XXI, Step D. 1H NMR (CD30D) d
-124-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98I05486
1.63-2.04(m,4H),2.16(s,3H),2.40(s,3H),2.99(m,4H),3.30(m,2H),
3.66 (m, 2 H), 4.05 (br s, 1 H), 4.44 (m, 2 H}, 4.68 (s, 2 H), 5.12 (m, 2 H>,
6.59
(s, 1 H), 7.20-7.32 (m, 5 H), 8.81 (s, 1 H); MS (FAB) 521 (M+1)+.
EXAMPLE XLIX
Tablet Preparation
Tablets containing 100.0, 200.0, and 300.0 mg, respectively,
of 3-Benzylsulfonylamino-6-methyl-1-[1-(2-hydroxyethyl-
aminocarbonylmethyl)-4-methyl-5-
methylenecarboxamidomethylimidazolyl]-2-pyridinone (example
XXXIII) active compound are prepared as illustrated below:
Ingredient Amount-m~
Active compound 100.0 200.0 300.0
Microcrystalline cellulose 160.0 150.0 200.0
Modified food corn starch 20.0 15.0 10.0
Magnesium stearate 1.5 1.0 1.5
All of the active compound, cellulose, and a portion of the
corn starch are mixed and granulated to 20°lo corn starch paste. The
resulting granulation is sieved, dried and blended with the remainder of
the corn starch and the magnesium stearate. The resulting granulation
is then compressed into tablets containing 100.0, 200.0, and 300.0 mg,
respectively, of active ingredient per tablet.
-125-
CA 02283704 1999-09-15
WO 98/42342 PCT/US98/05486
EXAMPLE L
An intravenous dosage form of the above-indicated active
compound is prepared as follows:
Active compound 0.5-lO.Omg
Sodium Citrate 5-50mg
Citric Acid 1-l5mg
Sodium Chloride 1-8mg
Water for Injection (USP) q.s. to 1 L
Utilizing the above quantities, the active compound is
dissolved at room temperature in a previously prepared solution of
sodium chloride, citric acid, and sodium citrate in Water for Injection
(USP, see page 1636 of United States Pharmacopeia/National Formulary
for 1995, published by United States Pharmacopeial Convention, Inc.,
Rockville, Maryland, copyright 1994.
-126-