Note: Descriptions are shown in the official language in which they were submitted.
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
HUMAI'I COMPLEMENT C3-DEGRADING PROTEINASE
s FROM .STREPTOCOCCUS PNEUMONIAE
Field of the Invention
1o This invention relates to Streptococcus pneumnniae and in particular this
invention relates to the identification of an S. pneumoniae protein that is
capable
of degrading human complement protein, C3.
l3ack~round of the Invention
This application claims the benefit of a provisional application (Serial
~ 5 No. 60/044.316) filing on April 24, 1997 entitled ''Human complement C 3-
degrading proteinase frorr.~ Streptococcus pncumoniae."
Respiratory infection with the bacterium Streptococcu.r pneur?TOrzlaL' (S.
pneumoniae) leads to an estimated X00,000 cases of pneumonia and 47,000
deaths annually. Those persons at highest risk of bacteremic pneumococcal
2o infection are infants under two years of age and the elderly. In these
populations, S. pneumonicre is the leading cause of bacterial pneumonia and
meningitis. Morf:over. S. pneumoniae is the major bacterial cause of ear
infections in children of al.l ages. Both children and the elderly share
defects in
the synthesis of protective antibodies to pneumococcal capsular polysaccharide
?s after either bacterial colonization, local or systemic infection, or
vaccination with
purified polysaccharides. S. pneumonicre is the leading cause of invasive
bacterial respiratory disease in both adults and children with H1V infection
and
produces hematogenous infection in these patients (Connor et al. C.'Irrrenl
Topics
in AIDS 1987;1:185-209 and Janoff et al. Ann. Intern. Med. 1992;117(4):314-
30 324).
Individuals who demonstrate the greatest risk for severe infection are not
able to make antibodies to. the current capsular polysaccharide vaccines. As a
result, there are now four ~~onjugate vaccines in clinical trial. Conjugate
vaccines
consist of pneumococcal capsular polysaccharides coupled to protein carriers
or
CA 02283755 1999-09-21
WO 98148022 PCTIUS98108281
adjuvants in an attempt to boost the antibody response. However, there are
other
potential problems with conjugate vaccines currently in clinical trials. For
example, pneumococcal serotypes that are most prevalent in the United States
are different from the serotypes that are most common in places such as
Israel,
- 5 Western Europe, or Scandinavia. Therefore, vaccines that may be useful in
one
geographic locale may not be useful in another. The potential need to modify ,
currently available capsular polysaccharide vaccines or to develop protein
conjugates for capsular vaccines to suit geographic serotype variability
entails
prohibitive financial and technical complications. Thus, the search for
immunogenic, surface-exposed proteins that are conserved worldwide among a
variety of virulent serotypes is of prime importance to the prevention of
pneumococcal infection and to the formulation of broadly protective
pneumococcal vaccines. Moreover, the emergence of penicillin and
cephalosporin-resistant pneumococci on a worldwide basis makes the need for
~5 effective vaccines even more exigent (Baquero et al. J. Antimicrob.
Chemnther.
1991;28S;31-8).
Several pneumococcal proteins have been proposed for conjugation to
pneumococcal capsular polysaccharide or as single immunogens to stimulate
immunity against S. pneumoniae. Surface proteins that are reported to be
2o involved in adhesion of S. pneumoniae to epithelial cells of the
respiratory tract
include PsaA, PspC/CBP112, and IgAI proteinase (Sampson et al. If~fect.
Immun. 1994;62:319-324, Sheffield et al. Microb. Pathogen. 1992; 13: 261-9,
and Wani, et al. Infect. Immun. 1996; 64:3967-3974). Antibodies to these
adhesins could inhibit binding of pneumococci to respiratory epithelial cells
and
25 thereby reduce colonization. Other cytosolic pneumococcal proteins such as
pneumolysin, autolysin, neuraminidase, or hyaluronidase are proposed as
vaccine antigens because antibodies could potentially block the toxic effects
of
these proteins in patients infected with S. pneumoniae. However. these
proteins
are typically not located on the surface of S. pnea~moniae, rather they are
secreted
30 or released from the bacterium as the cells lyse and die (Lee et al.
Vaccine 1994;
12:875-8 and Berry et al. Infect. Immun. 1994; 62:1101-1108). While use of
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
these cytosolic proteins as immunogens might ameliorate late consequences of
S. pneumoniae infection. antibodies to these proteins would neither promote
pneumococcal death nor prevent initial or subsequent pneumococcal
colonization.
A prototypic surface protein that is being tested as a pneumococcal
vaccine is the p:neumococcal surface protein A (PspA). PspA is a heterogeneous
protein of about: 70-140 lkDa. The PspA structure includes an alpha helix at
the
amino terminus, followed by a proline-rich sequence, and terminates in a
series
of 11 choline-binding repeats at the carboxy-terminus. Although much
information regarding it s structure is available, PspA is not structurally
conserved among a varieay of pneumococcal serotypes, and its function is
entirely unknovm (Yothe:r et al. J. Bacterial. 1992;174:601-9 and Yother J.
Bacteriol. 1994;176:2976-2985). Studies have confirmed the immunogenicity of~
PspA in animals (McDaniel et al. Microb. Pathogen. 1994; 17;323-337).
~ 5 Despite the immunogenicity of PspA, the heterogeneity of PspA, its
existence in
four structural groups (or ciades), and its uncharacterized function
complicate its
ability to be used as a vaccine antigen.
In patients who cannot make protective antibodies to the type-specific
polysaccharide capsule, the third component of complement, C3, and the
20 associated proteins of the alternative complement pathway constitute the
first
line of host defi~nse against S. pneumoniae infection. Because complement
proteins cannot penetrate the rigid cell wall of S. pneumoniae, deposition of
opsonic C3b on the pneumococcal surface is the principal mediator of
pneumococcal ~~learance:. Interactions of pneumococci with plasma C3 are
25 known to occur during pneumococcal bacteremia, when the covalent binding of
C3b, the opsonically active fragment of C3, initiates phagocytic recognition
and
ingestion (John.ston et all. J. Exp. Med 1969:129:1275-1290, Hasin HE, J.
Immunol. 1972; 109:26-31 and Hostetter et al. .I. Infect. Dis. 1984; 150:653-
61 ).
C3b deposits on the pneumococcal capsule, as well as on the cell wall. This
3o method for controlling li. pneumoniae infection is fairly inefficient.
Methods for
augmenting S. pneumor~iae opsonization could improve the disease course
_3_
CA 02283755 1999-09-21
WO 98148022 PCT/US98/08281
induced by this organism. There currently exists a strong need for methods and
therapies to limit S. pneumoniae infection.
Summary of the Invention
- 5
This invention relates to the identification and use of a family of human
complement C3-degrading proteinases expressed by S. pneumoniae. The protein
has a molecular weight of about 24 kD to about 34 kD as determined on a 10%
SDS polyacrylamide gel. The invention includes a number of proteins isolatable
from different C3-degrading strains of S. pnea~moniae.
In one aspect of the invention, the invention relates to an isolated protein
comprising at least an 80% sequence identity of SEQ ID N0:2 and capable of
degrading human complement protein C3. In a preferred embodiment, the
protein is isolated from S. pneumoniae or alternatively the protein is a
15 recombinant protein. Preferably the protein binds human complement protein
C3. In a preferred embodiment, the protein has a molecular weight as
determined on a 10% polyacrylamide gel of between about 24 kDa to about 34
kDa. A preferred protein of this invention is an isolated protein including
SEQ
ID N0:2.
2o The invention also relates to peptides from the C3-degarding proteinase
of this invention and preferably peptides of at least 15 sequential amino
acids
from an isolated protein comprising at least an 80% sequence identity of SEQ
ID
N0:2 and capable of degrading human complement protein C3 and more
preferably peptides of at least 15 sequential amino acids from SEQ ID N0:2.
25 The protein of claim 9, wherein the protein is a recombinant protein. In
another aspect of this invention, the invention relates to a peptide of at
least 1 ~
sequential amino acids from SEQ ID N0:2.
The protein of this invention can comprise SEQ ID N0:2, and preferably
has a molecular weight as determined on a 10% polyacrylamide gel of between
3o about 24 kDa to about 34 kDa. Also preferably the protein degrades human
complement protein C3. Preferred protein or polypeptides of this invention
-4-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
include a protein, comprising amino acids I-50 of SEQ ID N0:2 and a nucleic
acid fragment co~mprisin~; nucleic acids 1246 to 1863 of FIG. 1 A.
In another aspect of the invention the invention relates to a protein that
degrades human complennent protein C3 and wherein nucleic acid encoding the
S protein hybridizes to SEQ ID NO:1 under hybridization conditions of 6XSSC,
~X Denhardt, O.:p% SDS, and 100 pg/ml fragmented and denatured salmon
sperm DNA hybridized overnight at 65°C and washed in 2X SSC, 0.1% SDS
one time at room temperature for about i 0 minutes followed by one time at,
65°C for about 15 minutes followed by at least one wash in 0.2XSSC, 0.1
% SDS
at room temperature for ait least 3-5 minutes.
The invention also relates to an immune-system stimulating composition
comprising an ei:fective amount of an immune system-stimulating peptide or
polypeptide corr~prising apt least 15 amino acids from a protein comprising at
least an 80% sequence identity with SEQ ID N0:2 and capable of degrading
~ 5 human complement protein C3.
Preferabliy the protein is isolatable from S. pnearmoniae. In one
embodiment, thf: immune: system stimulating composition further comprises at
least one other immune stimulating peptide, polypeptide or protein from S.
pneumoniae.
2o The invention further relates to an antibody capable of specifically
binding to a protein comprising at least a 80% sequence identity with SEQ ID
N0:2 and capable of degrading human complement protein C3. In one
embodiment, the antibody is a monoclonal antibody an din an other embodiment,
the antibody is a polyclonal antibody. In another embodiment the antibody is
an
25 antibody fragmf;nt. The antibody or antibody fragments can be obtained from
a
mouse, a rat, human or a rabbit.
The invention also relates to a nucleic acid fragment capable of
hybridizing to S~EQ ID I'l0: l under hybridization conditions of 6XSSC, SX
Denhardt, 0.5% SDS, and 100 ~g/ml fragmented and denatured salmon sperm
30 DNA hybridizes overni~;ht at 65°C and washed in 2X SSC, 0.1% SDS one
time
at room temperature for about 10 minutes followed by one time at, 65°C
for
-5-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
about 15 minutes followed by at least one wash in 0.2XSSC, 0.1 % SDS at room
temperature for at least 3-5 minutes. In one embodiment the nucleic acid
fragment is isolated from an S. pneumoniue genome and in another embodiment.
the nucleic acid fragment encodes at least a portion of a protein. In one
embodiment, the protein degrades human complement C3 and in another
embodiment, the nucleic acid fragment encodes a protein that does not degrade
human complement C3.
In another embodiment, the nucleic acid fragment is in a nucleic acid
vector and the vector can be an expression vector capable of producing at
least a
I o portion of a protein. Cells containing the nucleic acid fragment are also
contemplated in this invention. In one embodiment. the cell is a bacterium or
a
eulcarvotic cell.
The invention further relates to an isolated nucleic acid fragment
comprising the nucleic acid sequence
15 gctcccagtatgcgtactcgtaaggtagagggaagaaaaaaactagctag.
In another aspect of this invention, the invention relates to a method for
producing an immune response to S. pneumoniue in an animal including the
steps of: administering a composition comprising a therapeutically
effective amount of at least a portion of a protein to an animal, wherein
nucleic
2o acid encoding the protein hybridizes to SEQ ID NO:1 under hybridization
conditions of 6XSSC, SX Denhardt, 0.5% SDS, and 100 pg/ml fragmented and
denatured salmon sperm DNA, hybridized overnight at 65°C and washed in
2x
SSC, 0.1% SDS one time at room temperature for about 10 minutes followed by
one time at 65°C for about 15 minutes followed by at least one wash in
0.2xSSC,
25 0.1 % SDS at room temperature for at least 3-5 minutes; and obtaining an
immune response to the protein. In one embodiment the immune response is a B
cell response and in another embodiment, the immune response is a T cell
response. In a preferred embodiment, the composition is a vaccine composition.
Preferably the at least a portion of the protein is at least 15 amino acids in
length
3o and also preferably the composition further comprises at least one other
protein
_6_
CA 02283755 1999-09-21
WO 98/48022 PCT/LJS98/08281
from S. pneumoniue. In one embodiment, the protein comprises at least 15
amino acids of SEQ ID I\f0:2.
In a further embodiment, the invention relates to a bacteria comprising an
insertional mutaaion, wherein the insertion mutation is in a gene encoding a
protein capable of degrading human complement C3. In one embodiment, the
bacteria comprises an insertional duplication mutation.
The invention further relates to an isolated protein of about 24 kDa to
about 34 kDa from Streptococcus pneumoniae that is capable of binding to and
degrading human complement C3 and to a method for inhibiting Streptococcus
pneumoniae-me~3iated C=s degradation comprising the step of : contacting a
Streptococcus pneumonia bacterium with antibody capable of binding to a
protein with at lf~ast 80% amino acid sequence identity to SEQ ID N0:2. The
invention further relates t:o an isolated nucleic acid fragment comprising the
nucleic acid sequence of SEQ ID NO:1 and to an RNA fragment transcribed by a
t 5 double-stranded DNA sequence comprising SEQ ID NO:1.
Etrief Description of the Figures
Figure l.A provides a gene sequence and Figure I B provides an amino
acid sequence o f a C3 degrading proteinase of this invention.
2o Figure 2 is a diagram of an insertion duplication mutant according to this
invention.
Figure 3 is a diagram of the restriction analysis of an insert from an
insertion duplic,~tion mutant of this invention.
25 Detailed Description of the Preferred ICmbodiments
The present invention relates to the identification and isolation of a C3
degrading proteinase wil.h a molecular weight of about 29 kDa (~ 5 kDa) on a
10% SDS-PAGE gel (wiith a predicted size of about 27.5 kDa based on SEQ ID
3o NO:1 ) and nucleic acid encoding the C3 degrading proteinase. The protein
was
originally identified by electrophoresis of pneumococcal lysates on SDS-PAGE
-7-
CA 02283755 1999-09-21
WO 98/48022 PCTIUS98/08281
gels impregnated with C3. It has been observed that exponentially growing
cultures of pneumococci from several serotypes were able to first degrade the
(3-
chain then degrade the a chain of C3 without producing defined C3 cleavage
fragments (Angel, et al. J. Infect. Dis. 170:600-608, 1994). This pattern of
degradation without cleavage differs substantially from other microbial
products
such as the elastase moiety of P.seudomona.s aerugino.sa and the cysteine
proteinase of Entamoeba hi.stolytica. The gene sequence (SEQ ID NO:1 )
encoding a C3 degrading protein according to this invention is provided in
Figure 1 A and the amino acid sequence (SEQ ID N0:2) of the protein is
provided in Figure 1 B.
The term "degrade" is used herein to refer to enzymes that are capable of
cleaving proteins into amino acids, peptides and/or polypeptide fragments. The
proteins of this invention degrade C3 without producing specific cleavage
fragments as observed on a polyacrylamide gel.
A C3-degrading proteinase of about 29 kDa was isolated from a library
of insertionally interrupted pneumococcal genes by identifying those clones
that
had increased C3 degrading activity as compared to wild type S. pnei~moniae.
There is at least some preference of the C3-degrading proteinases of this
invention for C3 in that, for example, the C3-degrading proteinase does not
2o degrade other proteins, such as albumin, to a large extent. Exemplary
methods
for performing insertion duplication mutagenesis and for the identification of
clones with elevated C3 degrading activity is provided in Example 1.
A gene encoding a C3-degrading proteinase is contained within a region
that includes four open reading frames and interruption of the third open
reading
frame by homologous recombination severely impaired C3 degradation. ORF3
includes about 726 nucleotides and the sequence of the translated protein
shares
no substantial homology with proteins registered in either the GenBank or
SwissProt databases.
The full length gene encoding a C3-degrading proteinase of this
invention was inserted into a gene expression vector for expression in E.
toll.
Recombinant C3-degrading proteinase was isolated as described in the examples.
_g_
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
Those of ordinary skill in the art recognize that, given a particular gene
sequence
such as that provided in Figure 1, there are a variety of expression vectors
that
could be used to express t:he gene. Further, there are a variety of methods
known
in the art that could be used to produce and isolate the recombinant protein
of
' 5 this invention and those of ordinary skill in the art also recognize that
the C3
degrading assay of this invention will determine whether or not a particular
expression system, in addition to those expression systems provided in the
examples, is fun~~tioning, without requiring undue experimentation. A variety
of
molecular and immunolo;;ical techniques can be found in basic technique texts
such as those of Sambrook et al. (~Llolecular Cloning, A Laboratory ~Llanuul.
1989 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY) and
Harlow et al. (Antibodie.s: .A Laboratory ~l~lanual. Cold Spring Harbor. NY;
Cold
Spring harbor Laboratory Press, 1988).
The gene' encoding the C3 degrading protein of this invention was
15 identified using a plasmicl library made with pneumococcal genomic DNA
fragments from strain CP1200. Although there are a variety of methods known
for obtaining a F~lasmid library; in a preferred strategy, a plasmid library
was
constructed with. Sau 3A digested pneumococcal genomic DNA fragments (0.5
-4.0 kb) from pneumococ:cal strain CP 1200 (obtained from D.A. Morrison,
2o University of Illinois, Champagne-Ilrbana, Illinois and described in
Havarstein
LF, et al. Proe. ~Vcrtl. Acad. Sci. (U,SA) 1995;92:11140-1 1144) and inserted
into
the Bam HI site of the integrative shuttle vector pVA 891 (erm', cm'; has
origin
of replication for E. coli). This library was transformed into an E. cnli DHLa
MCR strain by e;lectropo:ration. A total of 14000 E. coli transformants were
25 obtained by elec.troporation. Plasmid extractions of some randomly selected
E.
coli transformarns revealed that all of them contained recombinant plasmids.
Plasmid library DNA was extracted from the E. coli transformants and
was used to transform the CP 1200 parent pneumococcal strain using insertional
mutatgenesis homologocus recombination.
3o The pne;umococc:al strain CP 1200 cells were made competent using a
pH shift with HC1 procedure in CTM medium. 'the competent cells were frozen
_g_
CA 02283755 1999-09-21
WO 98148022 PCT/US98108281
at -70° C in small aliquots until needed. Eight thousand pneumococcal
transformants were produced using these methods.
Individual pneumococcal transformants were screened by ELISA for
their altered phenotypic character based on their ability to degrade C3.
Bacterial
- 5 cultures were incubated with C3 (0.83 ~g of C3/ml of culture) for about 2
hrs to
about 4 hrs and the amount of undegraded C3 left in the samples was detected
by
enzyme linked immunoadsorbent assays (ELISA) using HRP-conjugated goat
polyclonal antibody specific to human complement C3. The assay was
standardized so that wells containing undegraded C3 had an O.D. 490 = ~ 1Ø
to Wells containing degraded C3 had a reduced optical density resulting from
their
reduced ability to bind anti-C3 antibodies. The optical densities of the
mutant
and parent strains were compared to that of negative controls. The negative
controls were culture medium containing different concentrations of C3. The
percent of C3 degrading activity was determined as a ratio of optical density
of
t 5 sample to control. Four mutants (SN3, SN4, SN5 and SN6) were identified
with
elevated C3 degrading activity (about 2-2.2 fold higher activity) as compared
with the activity of the about 29 kDa C3-degrading protein from parent strain
CP1200. This finding was confirmed by Western Blot analysis.
Total DNA from mutants SN3, SN4, SNS and SN6 was isolated and used
2o for electroporation into E. coli DHSa MCR. Low excision rates of plasmid
DNA
from integrated plasmids within the pneumonocci genome can produce small
amounts of free plasmid DNA and this DNA can be recovered when the DNA is
transformed into E. coli. This allows further characterization of the plasmid.
Retransformation of the plasmid back into pneumococcus verifies the phenotype
25 of the original mutant.
Protein samples fiom the native C3-degrading protein and from mutants
SN3, SN4, SNS, SN6 were incubated with C3 and separated on a 7.5%
SDS-PAGE gel under reducing conditions. C3 degrading activity was assessed
using western blot analysis employing HRP-conjugated antibody to C3. Mutant
3o SN4 and mutant SN4-4G were used in further experiments. Mutant SN4-4G was
identified after CP 1200 was retransformed with the recombinant plasmid
CA 02283755 1999-09-21
WO 98!48022 PCTlUS98/08281
pLSN4a rescued from SN4. Both mutant SN4 and mutant SN4-4G almost
completely degraded C3 after a 4 hr incubation. While the native C3 degrading
protein degraded C3, after a 4 hr incubation, C3 degradation was incomplete as
compared with a comparable incubation using mutants SN4 and mutant SN4-4G.
The plasmid encoding the protein from mutant SN4 was chosen for
further investigation. Plasmid pLSN4a (encoding mutant SN4) was used to
retransform the 'wild type CP 1200 strain. This resulted in 48 pneumococcal
mutants with elevated C.4 degrading activity. Digestion with restriction
endonuclease >-Lind III demonstrated that plasmid pLSN4a was about ~ 7.8 kb
to and included an insert that was about ~ 2.3 kb.
Plasmid pLSN4a was used as a hybridization probe in southern
hybridization experimenia to verify the presence of the insert in chromosomal
DNA samples from the pneumococcal mutants. The results confirmed that the
vector with insert (pLSN4a) and also the origin of the inserts in the mutants
SN3
and SN4 were integrated in the chromosomal DNA. Both mutants SN3 and SN4
consisted of two hybridizing junction fragments of sizes about ~2.2 kb and
about
5.8 kb. These fragments were also present in their parent strain CP 1200.
There
were two other hybridizing fragments at about ~ 4.2 kb and about ~ 3. 5 kb and
these two fragments together gave a total of about ~7.8 kb (pLSN4a is ~ 7.8
kb).
2o These two bands were also present in the vector with insert sample. Both
insert
and vector included EcoR I sites and represent the recombinant plasmid.
Analysis indicated that a gene duplication had occurred in the SN4 mutant
strain:
therefore, the improved C3-degradation activity could be attributed to
increased
C3-degrading protein in the SN4 mutants.
The sequence of about 1 kb of the 2338 by insert was determined using
whole pLSN4a plasmid .as a template. The remaining sequence (about 1338 bp)
with just insert (PCR product) as a template, was sequenced by ICBR ,
University of F'aorida. Both complementary strands were sequenced. The results
indicated that there were four open reading frames with the relative locations
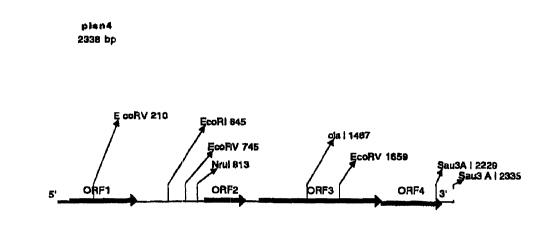
3o provided in the schematic below:
CA 02283755 1999-09-21
WO 98/48022 PCTlUS98/08281
ORF 1 ORF2 ORF3 -----
(462bp) (144 bp) (726 bp) ORF4 (358bp)
5'
3"
3'
5"
No significant homology was found between the derived amino acid
t o sequence of the above ORFs and protein sequences from the protein
databases
tested. The ORF3 nucleic acid sequence encoding a C3 degrading proteinase of
this invention is provided in Figure 1 A and is designated SEQ ID NO:1. The
amino acid sequence of this C3 degrading proteinase is provided in Figure 1 B
and is designated SEQ ID N0:2.
t 5 Out of four opening reading frames (three full and one partial) in the
insert, the ORF3 was chosen for ftirther examination because it contained the
largest insert. A 620 by internal portion (from nucleic acid 1246 to nucleic
acid
1863 of Figure 1 A) of ORF3 (PCR product) region was ligated into the Hind III
site of plasmid pVA 891 and the construct was transformed into CP 1200
2o competent cells to knock out the proteinase activity. The transformants
were
tested for their ability to degrade C3 after separation on SDS-page gels using
western blot analysis. The ORF3 disruption mutant had poor activity in
comparison with its parent strain CP 1200.
The entire ORF3 gene (PCR product) was cloned into Nde I and Bam H I
25 sites of pet-28b(+). The vector positions a His-Tag at the N-terminus of
the
protein. The plasmid construct was transformed into an E. coli (DHLa MCR)
strain for stabilization before it was transformed into an E. toll (BL 21 DE3)
protease deficient strain for protein expression.
The BL 21 DE3 strain that included the construct (pet 28b(+) with
3o ORF3) was induced for ORF3 protein expression. Total cell protein extracts
of
the induced and uninduced cultures were tested for C3 degrading activity. The
expressed His-tagged ORF3 protein was about ~ 29 kDa (~ 5 kDa) on 10%
SDS-PAGE gels in the induced samples from the insoluble protein fraction.
-12-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
Solubiliz anon of the ORF 3 protein from induced BL21 DE3 cultures
was performed by treating the sample with: a) TES {SOmM, 1 mM, 1 M}; b)
6mM G-HCI + 1 mM DTT; c) 6mM G-HC1 -~ 1 mM DTT + 1 % Tween 20; and
d) 6mM G-HC1 + 1mM DTT + 1% Triton X -100. Both treatments "c" and "d"
resulted in soluble protein. Treatment "c" was used to produce solubilized
recombinant C3 degrading protein that was used for further protein studies.
Guanidine-HCl and DTT were removed from the expressed His-'fagged
ORF3 protein samples b;y dialysis. The protein was subjected to Nickel column
purification and the eluted His-Tagged protein was visualized on a 10%
I o SDS-PAGE gel.
The isolated protein encoded by ORF 3 was incubated with human
complement C3 for 4 hrs at 37°C in the presence of PBS. Control samples
without the protein same>les were used as negative controls for comparative
purposes. The samples were run on SDS-PAGE gel under reducing conditions
and analyzed for the structure of C3 by Western Blot assay using polyclonal
antibodies to human complement C3. The results indicated that the samples
contained a protein encoded by the ORF 3 region and that the protein degraded
human C3 protf;in. Both a and (3 chains of C3 molecules were susceptible to
degradation. In these experiments while the a chain was almost completely
?o degraded, the (3 chain was also degraded, but to a somewhat lesser extent.
The C3 degrading proteins of this invention were designated CppA
proteinases and the genea of this invention are designated cppA. The proteins
of
this invention have an apparent molecular weight on a 10% 5DS-polyacrylamide
gel of about 29 kDa {~5 kDa) and preferably has a molecular weight of about 24
kDa to about 3~l kDa. A,s described above, Example 5 indicates that the
proteinase is conserved throughout S. pneumaniae strains. However. those of
ordinary skill in the art will recognize that some variability in amino acid
sequence is expected and that this variability should not detract from the
scope
of this invention. For e:Kample, conserved mutations do not detract from this
3o invention nor d.o variations in amino acid sequence identity of less than
about 80
amino acid :sequence identity and preferably less than about 90% amino acid
-13-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
sequence identity where the protein is capable of degrading human complement
protein C3, and particularly where the protein is isolated or originally
obtained
from an S. pneumoniae bacterium.
Some nucleic acid sequence variability is expected among the strains as
is some amino acid variability. Conserved amino acid substitutions are known
in
the art and include, for example, amino acid substitutions using other members
from the same class to which the amino acid belongs. For example, the nonpolar
(hydrophobic) amino acids include alanine, leucine, isoleucine, valine,
proline,
phenylalanine, tryptophan, and tyrosine. The polar neutral amino acids include
glycine, serine, threonine, cysteine, tyrosine, asparagine and glutamine. The
positively charged (basic) amino acids include arginine, lysine and histidine.
The negatively charged (acidic) amino acids include aspartic acid and glutamic
acid. Such alterations are not expected to affect apparent molecular weight as
determined by polyacrylamide gel electrophoresis or isoelectric point.
t 5 Particularly preferred conservative substitutions include, but are not
limited to,
Lys for Arg and vice verse to maintain a positive charge: Glu for Asp and vice
versa to maintain a negative charge; Ser for Thr so that a tree -OH is
maintained;
and Gln for Asn to maintain a free NH,. A preferred protein of this invention
includes a protein with the amino acid sequence of SEQ ID N0:2. Other
2o proteins include those degrading human complement protein C3 and having
nucleic acid encoding the protein that hybridizes to SEQ ID NO:1 under
hybridization conditions of 6XSSC, SX Denhardt, 0.5% SDS, and 100 pg/ml
fragmented and denatured salmon sperm DNA hybridized overnight at 65°C
and
washed in 2X SSC, 0.1% SDS one time at room temperature for about 10
25 minutes followed by one time at, 65°C for about 15 minutes followed
by at least
one wash in 0.2XSSC, 0.1% SDS at room temperature for at least 3-5 minutes
are also contemplated in this invention. Polypeptides or peptide fragments of
the
protein can also be used and a preferred protein of this invention comprises
amino acids 1-50 of SEQ ID N0:2.
3o The proteins of this invention can be isolated or prepared as recombinant
proteins. That is, nucleic acid encoding the protein, or a pouion of the
protein.
- 14-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
can be incorporated into aul expression vector or incorporated into a
chromosome
of a cell to express the protein in the cell. The protein can be purified from
a
bacterium or another cell, preferably a eukaryotic cell and more preferably an
animal cell. Alternatively, the protein can be isolated from a cell expressing
the
protein, such as a S. pneu,moniae cell. Peptides of the CppA proteinase are
also
considered in this invention. The peptides are preferably at least 15 amino
acids
in length and preferred peptides are peptides with at least 15 sequential
amino
acids from SEQ ID N0:2. Another preferred protein fragment includes amino
acids I-50 of SEQ ID NO:2.
Nucleic acid encoding CppA proteinase is also part of this invention.
SEQ ID NO: I is a preferred nucleic acid fragment encoding a CppA proteinase.
Those of ordinary skill in the art will recognize that some substitution will
not
alter the CppA proteinase sequence to an extent that the character or nature
of the
CppA proteinase~ is substantially altered. For example, nucleic acid with an
~ 5 identity of at least 80% to SEQ ID NO: I is contemplated in this
invention. A
method for determining whether a particular nucleic acid sequence falls within
the scope of this invention is to consider whether or not a particular nucleic
acid
sequence encoders a C3-degrading proteinase and has a nucleic acid identity of
at
least 80% as compared with SEQ ID NO: l . Other nucleic acid sequences
2o encoding the CppA proteinase includes nucleic acid encoding CppA where the
CppA has the same sequence or at least a 90% sequence identity with SEQ ID
N0:2 but which includes degeneracy with respect to the nucleic acid sequence.
A degenerate codon means that a different three letter codon is used to
specify
the same amino acid. For example, it is well known in the art that the
following
25 RNA codons (and therefore, the corresponding DNA codons, with a T
substituted for a U) can be used interchangeably to code for each specific
amino
acid:
Phenylalanine (Plhe or F) UUU or UUC
Leucine (Leu or L) UUA, UUG, CUU, CUC, CUA or CUG
3o Isoleucine (Ile or I) AUU, AUC or AUA
Methionine (Met or M) AUG
-IS-
CA 02283755 1999-09-21
WO 98/48022 PCTliJS98/08281
Valine (Val or V) GUU, GUC, GUA, GUG
Serine (Ser or S) UCU, UCC, UCA, UCG, AGU, AGC
Proline (Pro or P) CCU. CCC, CCA, CCG
Threonine (Thr or T) ACU, ACC, ACA, ACG
_ 5 Alanine (Ala or A) GCU, GCG, GCA, GCC
Tyrosine (Tyr or Y) UAU or UAC
Histidine (His or H) CAU or CAC
Glutamine (Gln or Q) CAA or CAG
Asparagine (Asn or N) AAU or AAC
to Lysine (Lys or K) AAA or AAG
Aspartic Acid (Asp or D) GAU or GAC
Glutamic Acid (Glu or E} GAA or GAG
Cysteine (Cys or C) UGU or UGC
Arginine (Arg or R) CGU, CGC, CGA, CGG, AGA, AGC
t 5 Glycine (Gly or G) GGU or GGC or GGA or GGG
Termination codon UAA, UAG or UGA
Further, a particular DNA sequence can be modified to employ the
codons preferred for a particular cell type. For example, the preferred codon
usage for E. coli is known, as are preferred codons for animals and humans.
2o These changes are known to those of ordinary skill in the art and therefore
these
gene sequences are considered part of this invention. Other nucleic acid
sequences include nucleic acid fragments of at least 30 nucleic acids in
length
from SEQ ID NO: I or other nucleic acid fragments of at least 30 nucleic acids
in
length where these fragments hybridize to SEQ ID NO:1 under hybridization
25 conditions of 6XSSC, SX Denhardt, 0.5% SDS, and 100 g.g/ml fragmented and
denatured salmon sperm DNA hybridized overnight at 65°C and washed in
2X
SSC, 0.1% SDS one time at room temperature for about 10 minutes followed by
one time at, 65°C for about 15 minutes followed by at least one wash in
0.2XSSC, 0.1% SDS at room temperature for at least 3-5 minutes.
3o The nucleic acid fragments of this invention can encode all, none (i.e.,
fragments that cannot be transcribed, fragments that include regulatory
portions
- 16-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
of the gene, or the like) or a portion of SEQ ID NO:2 and preferably
containing a
contiguous nucleic acid fragment that encodes at least nine amino acids from
SEQ ID N0:2. Because nucleic acid fragments encoding a portion of the CppA
proteinase are contemplated in this invention, it will be understood that not
all of
the nucleic acid j:ragments will encode a protein, polypeptide or peptide with
C3
degrading activity. Further, the nucleic acid of this invention can be mutated
to
remove or otherwise inactivate the C3 degrading activity of this protein.
Therefore, fragments without C3 degrading activity that meet the hybridization
requirements de~~cribed above are also contemplated. Methods for mutating or
otherwise altering nucleic acid sequences are well described in the art and
the
production of an immunogenic, but enzymatically inactive protein can be tested
for therapeutic utility. Preferred nucleic acid fragments include get ccc agt
atg
(Claim 34).
The nucleic acid fragments of this invention can be incorporated into
nucleic acid vectors or stably incorporated into host genomes to produce
recombinant protein inclu~,ding recombinant chimeric protein. A variety of
nucleic acid vectors are known in the art and include a number of commercially
available expression plasmids or viral vectors. The use of these vectors is
well
within the scope of what :is ordinary skill in the art. Exemplary vectors are
2o employed in the examples, but should not be construed as limiting on the
scope
of this invention.
This invention also relates to antibody capable of binding specifically to
a protein of about 29 kDa., and preferably a protein of about 24 kDa to about
34
kDa, from S. pneumoniae and preferably where the protein is capable of
degrading huma~a complement C3. Polyclonal antibody can be prepared to a
portion of the protein or to all of the protein. Similarly, monoclonal
antibodies
can be prepared to all or t:o a peptide fragment of the about 29 kDa C3
degrading
protein of this invention. Methods for preparing antibodies to protein are
well
known and well described, for example, by Harlow, et al. (supra). In a
preferred
3o example, the antibodies c.an be human derived, rat derived, mouse derived
or
rabbit derived. 1?rotein-binding antibody fragments and chimeric fragments are
- 17-
CA 02283755 1999-09-21
WO 98/48022 PCT/LTS98/08281
also known and are within the scope of this invention.
The invention also relates to the use of immune stimulating
compositions. The term "immune stimulating" or "immune system stimulating'
refers to protein or peptide compositions according to this invention that
activates at least one cell type of the immune system. Preferred activated
cells of
the immune system include phagocytic cells such as macrophages, as well as T
cells and B cells. Immune stimulating compositions comprising the peptides.
polypeptides or proteins of this invention can be used to produce antibody in
an
animal such as a rat, mouse, rabbit, a human or an animal model for studying
S.
pnea~moniae infection. Preferred immune stimulating compositions include an
immune stimulating amount of at least a peptide including at least 15 amino
acids from the CppA proteinase. The immune stimulating composition can
further include other proteins in a pharmaceutically acceptable buffer, such
as
PBS or another buffer recognized in the art as suitable and safe for
introduction
t 5 of proteins into a host to stimulate the immune system. The immune
stimulating
compositions can also include other immune system stimulating proteins such as
adjuvants or immune stimulating proteins or peptide fragments from S.
pneumoniae or other organisms. For example, a cocktail of peptide fragments
may be most useful for controlling S. pneumoniae infection. Preferably one or
2o more fragments of the proteins of this invention are used in a vaccine
preparation
to protect against or limit S. pneumonicre colonization or the pathogenic
consequences of S. pneumoniae colonization.
This invention also relates to a method for inhibiting Streptococct.~.s
pneumoniae-mediated C3 degradation comprising contacting a Streptococcus
25 pneumonia bacterium with a protein, such as an antibody or another protein
that
is capable of binding to an isolated protein of about 24 kDa to about 34 kDa
from Streptococcus pneumoniae. The protein capable of binding to an isolated
protein of about 24 kDa to about 34 kDa can be an antibody or a fragment
thereof or the protein can be a chimeric protein that includes the antibody
3o binding domain, such as a variable domain, from antibody that is capable of
specifically recognizing an isolated protein of about 24 kDa to about 34 kDa
_ lg _
CA 02283755 1999-09-21
WO 98148022 PCT/US98/08281
from Streptococcus pneu,moniae having C3 degrading activity.
The isol~~ted S. pnearmoniae protein of this invention can be isolated and
purified and the isolated protein or immunogenic fragments thereof can be used
to produce antibody. Peptide fragments or polypeptide fragments of the protein
without C3 degrading ability can be tested for their ability to limit the
effects of
S. pneumoniae infection. Similarly, the protein of this invention can be
modified, such a.s through mutation to interrupt or inactivate the C3
degrading
capacity of the protein. Isolated protein can be used in assays to detect
antibody
to ,S. pneumoniae or as part of a vaccine or a multi-valent or multiple
protein or
peptide-containing vaccine for S. pneumoniae therapy.
It is further contemplated that the proteins of this invention can be
surface expressed on vertebrate cells and used to degrade C3, for example,
where
complement deposition (or activation) becomes a problem, such as in
xenotransplantation or in complement-mediated glomerulonephritis. For
example, the recombinant protein, or a portion thereof, can be incorporated
into
xenotransplant c.elis and expressed as a surface protein or as a secreted
protein to
prevent or minimize complement deposition (and/or complement-mediated
inflammation).
All references and publications cited herein are expressly incorporated by
reference into this disclosure. There are a variety of alternative techniques
and
procedures available to those of skill in the art which would similarly permit
one
to successfully perform the intended invention in view of the present
disclosure.
It will be appreciated by those skilled in the art that while the invention
has been
described above in connection with particular embodiments and examples, the
invention is not necessarily so limited and that numerous other embodiments,
examples, uses, modifications and departures from the embodiments. examples
and uses may bf; made without departing from the inventive scope of this
application.
- 19-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
Example 1
Generation of Insertional Duplication Mutants and Recovery of
Recombinant Plasmids from Selected Mutants
In a preferred example, insertion-duplication mutagenesis was used to
- isolate a gene encoding the C3 degrading proteinase from Streptococcus
pneumoniae of this invention. A plasmid library was created with 0.5 - 4.0 kb
chromosomal fragments of pneumococcal strain CP 1200 (derivative of RX1;
Morrison, D. A., et al. J. Bacteriol., 156:281-290,1983) originally obtained
from
1 o Dr. Morrsion's lab, University of Illinois at Chicago and inserted into
the Bam
HI shuttle vector pVA 891 (erm', cm' Marcina, F.L. et. al. Gene 25:115-150,
1983, obtained from Dr. Marcina (Virginia Commonwealth University,
Richmond, VA). pVA891 has resistance markers for erythromycin (erm) and
chloramphenicol (cm). The vector has an origin of replication for E. coli, but
the
origin is non-replicative in Streptococci. Recombinant plasmid can survive
when it integrates into the pneumococcal chromosomal DNA by homologous
recombination .
E. coli DHS oe MCR competent cells were made according to the
procedure given in the Bio-Rad Laboratories manual (Richmond, CA) and the
library was transformed into the competent cells with Bio-Rad Gene Pulser
apparatus (Bio-Rad Laboratories, Richmond, CA) by electroporation.
E. coli cells were maintained as freezer stocks in small aliquots at -
80°C,
in LB broth in the presence of 10% glycerol. The cells were grown either in LB
or TB broth or on LB agar plates containing appropriate antibiotics
(erythromycin 200 ~g/ml or chloramphenicol 15 or 30 pg /ml or kanamycin 30
~g/ml).
Electroporation was conducted in 0.1 cm cuvette at 1- 2 kV/cm voltages
and a capacitance of 200 S2. Transformants were selected on LB medium
containing either chloramphenicoi (cm, 30 p,g /ml) or erythromycin (enn, 300
~g /ml) or combination of erm and cm (200ug/mI + I Sug/ml).
-20-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
A total of 14000 E. toll transformants were obtained from the library.
Plasmid extractions and :restriction analysis of randomly selected E. toll
transformants revealed the presence of recombinant plasmids.
Plasmids or recombinant plasmids were extracted from E. toll strains by
polyethylene gl~~col precipitation procedure (Kreig. P. and Melton. D., in
Promega Protocols and .Application.s p. 106, 1985-86) or a modified alkaline .
lysis miniprep protocol (Xiang. C., et al., Biotechnigues. 17:30-32, 1994) a
modified alkaline extraction procedure (Birnboim H C and J Doly.,1\rucl. Acids
Res. 7:1513-15~ 3, 1979), or CsCI-ethidium bromide gradient method or Qiagen
1o kit (Plasmid midi kit., Chartsworth, CA). Solutions containing DNA were
cleaned directly from agarose gels by GeneClean II kit (BIO 101, La Jolla. CA)
or Qiagen kit (DNA extraction from gels., Chartsorth, CA). DNA was cleaned by
Wizard DNA clean up ki.t (Promega Corp., Madison, WI). Amplified gene
products were also cleaned by Wizard PCR clean up kit (Promega Corp.,
~ 5 Madison, WI).
The plasmids were transformed into Pneucnococcal cells. The
pneumococcal strains were always maintained as freezer stocks in small
aliquots
at -80°C, in THl3 in the presence of 10% glycerol. Pneumococcal cells
were
grown without shaking in CAT (Morrison, D. A., et al., 1983, szrprcr) or THB
2o medium (broth or agar). For transformation experiments, either complete
transformation (CTM) broth (Morrison, D. A., et al.. 1983) or THB+ 0.~% Yeast
broth (Mother Janet., et al. J. Bacteriol. 168:1463-1465, 1986) and for ELISA
experiments, SI'rIP, a synthetic medium (see Table 1 ) were used. Erythromycin
(0.05 pg /ml) was employed as a selective antibiotic marker for pneumococcal
25 mutants.
-21 -
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
Table 1. SMP - a synthetic medium
SMP solution # I (final volume 2 liters):
NaCI 10.08; NH4CI 4.Og; KCl 0.88: Na2HP04 0.248; MgS04 0.048e; CaCl2 0.0208;
PeS04.7H20
0.0001 lg; Tribase 9.688 : add d.H2() up to I liter pH to 7.55 and then add
the following amino acids:
l.-Arginine 400 mg; L-Asparagine 20 mg (monohydrate 22.8 mg): Glycine 240 mg:
I,-1-listidinc 300 mg;
L-Isoieucine 13.10 mg; L-Leucine 13. (0 mg: L-Lysine 840 mg; 1.-methionine 360
mg: (low-methi.
2.6mg); L-valine ( 1.70 mg; llracil 2 mg. Make it up to a final volume of 2
liters.
SMP solution 2 (vitamins):
Biotin 0.07 mg; Choline 25 mg; Nictinamidc 3.0 mg; ca pantothenatc 12.0 mg;
Pyridoxal HCI 3.0 me:
Ribotlavin l.~ mg; Thiamine 3.0 mg: L-Cysteine I-ICI 0.5 e; L-Glutamine 0.1 g;
Na Pyruvatc 4.Og: add'
water and then make up to ~Oml.
Reconstituting SMP:
Start with: ml
Solution # 1 100
Add:
Solution #2 I
Solution #3(2~% Glucose) 1.6
Solution # 4f4% BSA) 2
Pneumococcal strain CP 1200 cells were made competent by
''competence induction by pH shift" (procedure obtained from Dr. Morrison~s
lab, Univ. of Illinois at Chicago, I11.) in CTM medium and the competent cells
were frozen at -70 ° C in small aliquots until required. In this
procedure, to 125
ml of CTM added 1.20 ml of 1 M HCl (final concentration 9mM) and 4 ml of 0.2
O.D. (550 nm) of frozen pneumococcal stock cells. This culture was incubated
at 37°C and O.D. readings of the culture were taken at 20 minute
intervals
beginning after 3 hrs of incubation. When the culture reached an O.D. of 0.156
(550 nm), 1.2 ml of 1N NaOH was added at 37"C. After mixing the culture
gently, 1 ml of culture was removed as a '0' time point sample, mixed with 100
Ltl of glycerol and kept on a prechilled metal block. Similarly. ten ml
samples
were drawn at each time point of 13, 17, 21 and 25 min and each sample was
added directly to prechilled 1 ml of glycerol. Each time point sample was
frozen in small aliquots at - 70°C. Competence was tested for each time
point
sample by adding 1 yl of DNA ( about 250 ng) to 100 ~tl of cells and
incubating
at 37°C for 25 min for transformation. The transformation culture was
diluted
and plated on selective medium (erythromycin 0.05~tg/ml). The time point
sample that showed the highest transformation efficiency was used for future
transformation experiments. Transformation of the extracted recombinant
plasmid library from E. coli transformants into pneumococcal strain CP1200
-22-
CA 02283755 1999-09-21
WO 98/48022 PCT/LTS98/08281
yielded about 8,000 pneumococcal transformants indicating that the plasmid was
inserted into the CP1200 chromosome via homologous recombination.
Extraction of pnc:umococcal chromosomal DNA was performed by a
slight modificavtion of the method used in the laboratory of Dr. Donald A.
- 5 Morrison, University of Illinois at Chicago. Pneumococcal cells were grown
in
THB to an O.D. at 550 run from 0.3-0.4, then rapidly chilled on ice and O.SM.
EDTA was added to a final concentration of 10 mM, the cells were spun at
10,000g for 10 minutes .at 4°C, and the pellets were resuspended in
1:10 volume
of cold STE (50mM Tris-HCI (pH 8.0), IOmM EDTA (pH 8.0), and O.1M
Io NaCI). After a second cf:ntrifugation, cells were resuspended in 1/100
volume of
cold STE, lysecl with 1°io Triton X-100, and incubated at 37°C
for 5-10 minutes
for autolysis. After the addition of 1 % SDS, the cells were swirled in water
bath
at 50-60°C for '.i min. RNase ( 100 pg/ml) and proteinase K (50 yg/ml)
were
added sequenti;~lly with incubations of 2 hours and 1 hours. respectively. The
~ 5 cells were extracted twice with one volume of phenol/ehloroform and once
with
one volume of chloroform and the supernatant was collected for ethanol
precipitation. T'he precipitate was washed twice with 70% ethanol, and the
pellet
was collected and resuspended in TE ( l OmM Tris-HC1 pH 8.0, 1 mM EDTA) or
water as required.
2o The plasmid library DNA was extracted by polyethylene glycol
precipitation procedure (Kreig. P. and Melton. D. 1985 szrpra), from pooled E.
coli transformants and used to transform CP 1200, the parent pneumococcal
strain following the method that was obtained from Dr. Morrison, University of
Illinois at Chicago. For pneumococcal transformation, frozen pneumococcal
25 competent cells were thawed on ice and to 100 pl of these competent cells.
200
ng to 1000 ng of plasmid library was added in a separate eppendorf tube. This
tube was incubated at 3'7°C in a water bath for about 25 min to 35 min
and the
mixture was diluted 1/10 in CAT medium and incubated further for about 1-1.7
hrs. Following the final incubation, the mixture was plated by overlay
procedure
30 (method was obtained from Dr. Morrison University of Illinois at Chicago).
The
overlay procedure involved pouring four different layers of agar (THB or CAT)
-23-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
in a small petri dish as follows: a) first or base layer: 3 ml of agar; b)
second or
cells' layer: mixture of 1.5 ml of agar and 1.5 ml of broth containing
required
concentration of bacterial cells; c) third layer: 3 ml of agar; 4) fourth
layer or top
layer: 3 ml of agar containing 4X required concentration of antibiotic
(erythromycin, at 0.05pg/ml x 4 = 6 pg /ml ). The plates were incubated at
37°C.
Individual transformants were transferred by stab inoculation to individual
wells
of microtitre plates containing 100y1 of THB and erythromycin (O.OSyg/ml). The
recovered transformants in microtitre plates were diluted 1:10 in SMP medium
and grown until early log phase, and screened for their ability to degrade C3
by
~ o ELISA.
Spontaneous excision of recombinant plasmids occur in these kind of
pneumococcal mutants with low frequency and therefore, chromosomal DNA
preparations of these mutants often include low levels of plasmid DNA (Pearce
B J., et al., Mol. Microb. 9(5):1037-1050, 1993). Electroporation of E. coli
is a
t5 highly efficient way of isolating the plasmid constructs in E. coli for
further
study. Chromosomal DNA ( 100 ng-200 ng in a final volume of 2 ~ls) fcom the
individual pneumococcal mutants of interest was electroporated into E. coli
DHS
a MCR competent cells to obtain E. coli transformants with recombinant
plasmids. One of the recovered recombinant plasmids (pLSN4a) (see Table 2}
2o was introduced back into wild type CP 1200 pneumococcal strain by
transformation. The transformant SN4-4G was again evaluated for its C3
degrading activity by ELISA.
DNA fragments were analyzed by horizontal electrophoresis in agarose
gels (0.5 % to I .0%) with Tris-borate EDTA (TBE) buffer or Tris-acetic acid
25 EDTA (TAE) buffer (Sambrook, J. E. Fritsch and T.Maniatis.1989). One kb
ladder from Gibco BRL or Hindi III or Hind III lEcoRl digested lamda DNA
from Boehringer Mannheim, was employed as a molecular weight standard.
Restriction endonucleases, calf intestinal phosphatase, T4 DNA ligase,
from Gibco BRL Life Technology, Grand Island, NY., Boehringer Mannheim ,
3o Indianapolis, IN., Promega Corp., Madison, WI., Bethesda Research
-24-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
Laboratories, G;zithersburg, MD., or New England Biolabs., Inc., Beverly, MA.,
were used as described by the manufacturers' instructions.
DNA fragments were analyzed by Southern hybridization. DNA was
transferred from. gels to PvISI Magnagraph nylon membranes (Micron
Separations, Inc., Westboro, MA) for hybridization and detection using Genius
nonradioactive DNA labeling and detection kit (Boehringer Mannheim
Biochemicals, hadianapolis, IN) following the instructions provided with the
kit.
Chromosomal or plasmid DNA either from the pneumococcal or E. coli culture
was isolated as described in earlier sections. About 100 ng - 400 ng of each
sample was digested with required restriction enzymes and run on 0.7% agarose
gel, transblotted onto Magnagraph-nylon membrane overnight. The rest of the
procedure was performed as instructed by the manufacturer.
Bacterial strains <~nd plasmids used in this example and the examples that
follow are summarized in Table 2 below.
- 25 -
CA 02283755 1999-09-21
WO 98148022 PCT/US98/08281
0
-n
L
N
L
.7
~
C ~n
C
v = 0 ~
'L ca
_ : E a o
E
a ~
C a
fU L ~ U
v
ca * 9 >
C M W U d
O fi
O U
. . N '~ O
C~ ~ ._
~ M ".f ~1 \O ~ ~ ~ z z z z
~ 00 ~T ~ z
~
Z '
Z
~
Z
' Q n f- ~ vpn cn ~n >
n Q f- d c ~ on
c
n
c
v > .~ ~t .-..1 .~ v = ,~ .~ _l
> w > .1 w _1
G ~..fl. a. C1 n. a. L O. G t1, f1 U
'O O. f1 O. Ll O. LL
s
E
E
0
f~
li
U
...
O
C
_
_
O
U
O
U
_N
U
'fl
C
C
E
-
~
..
C w
~
O
CC
CC
_
y
C * CO O
~ N
,"" ca C '_-
= M bD O
m M '~
M
C C ~ N
~ i. w
~ ~
' ~"'~ W
j ~ ~ O ' n
~ ~ a ~
., ~ a cn ~' ..". o
O
G G ~ -7 (11 ~ ~ Cn N
n p
p a N
pp v N
M V r1 ~' ~W c0
. O ~ ~'t
Q a. L2
x Cz/7 (~/7 N ~ ~ p.
x ~ .~ ~' .~ Z
Z
~
Z
v~o D ww..y.~D~mmm 6 nc U
nc
nr
n~
~c
C
L
D
~
.
c
o
on
L
o
~
~
L
C
C C ~
~
a
L .c aF E
, ~ ~n ~ .~
~ v
0
~
~
~
vc !f~~N CL ~~o
' '~
'~
'
L' ~ L .- .C .~ O
'~ ~ ,~ .~ O ~
~ ~ ~n
C C .C C
~ 9 v ~ .. . . :: y
~
.v
.-,~.,~_. _ ~TTO~OvrtO~U~.~
~~~~ CC
>e ~ ~~~~z~ E~~=
aaaa ~ aQaa~,a t"o
>
v
~>
L a n, a n. ' n. a. o. ~...
G a. n. n. ~
~
~r
'
.
"'
n.
,~
w_..
L ~
D
~
O
r~''n * .w. x~ O
C .~. M . C
r~1 '~
E
O
4~
N
~ O .
p
fi l)
O N ~ C
V
~
'
L ~ ., ~
~ . ~ ~
cLC
rn a. O ~. a. ~- c",
~
4-.
cC '~ ._ y. - .~ O
N .~
p
L
ca
9 0 ~ N~
L ~ C
0 O (
0 ~ C
~NO _N
r
C ~ N
.
_
~
~-
-
O ~ a > w > J L1.~ _
-
~
H c. . n, a n. n. a 3
u n.
~
~
'n
'R ~ 0 0 0 0 "~
0 o a~
a.
y
s
~,
'
L O O O O
O O
N N N ~ ~
~ N N '
~ L
P~..CG C~.. 2 ~ ..
LL~ OG 0. G ~"
M M M 0. N j
0. 0 ~O
Q a-
.. w
~ . .
. O
.
y ~ ~ ~ ~ ~ ~ ~ ~ ~
~ D N
a i LS LS Zi Li Zi U 0! m N "'
~ o~ U
Ct tS
d
y ~7 N N ~ is CS ~
m v, ~n ~n vn v, ~n v Q 0..
. x x x z x x x -~ ~ r = ~' ~' ai
-~ .-~ r a~
o 0 0 0 U
.o o .~
c
3
~ p Ga O L C~ ~ L7 O _ '
~ 07 m ; ~ ~ z
fi ~
N : ~,
N ~ .n
U
0 ~00~~~~~0 'J' ~~~~~~ _,V7U
.G ~ U U U U U U U U C (y ~ p~ " ~"
U U iy p" Q
x ti7~t~ lV ~7 ~t~ Or ti7 t~ t~ a "
~C7 ~t7 W ~7 t~ V~ c~ ~L..
t-L~ 'D
x
-26-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
Example 2
Identification of Mutants with altered C3-degrading activity
Individu;~l pneumococcal transformants were screened by ELISA for
their altered C3 degradin;7 activity. The pneumococcal transformants were
grown
individually in THB in the presence of erythromycin (0.05 yg/ml) in microtitre
plates up to log ~~hase and diluted 1/0 in SNIP medium (O.OS~g of
erythromycin/ml). The S1VIP bacterial cultures were grown up to log phase and
incubated with C:3 (0.83 ~_~g of C3/ml of culture) for 2-4 hrs. After
incubation
1 o with C3, 100 p.I s of each individual transformant was transferred to an
ELISA
binding plate and incubated overnight at 4°C. The plates were washed
with PBS
(10 pM phospha.te buffer saline + 0.05% Tween-20) three times. 100 yl of HRP-
conjugated goat polyclonal antibody specific to human complement Cs (1:10000
dilution of 48m~;/ml) was added to each well and the plates were incubated for
I -
~5 2 hrs at 37°C. Each microtitre plate was washed with PBS as
described above.
100 pl of 30% t~PD (12 mg of O-Phenylenediamine (Zymed, South San
Francisco, CA) i.n 30m1 of Citrate buffer (200mM Na,HPOa and I OOmM citric
acid-pH 5.0), and 12 pl of 30% H,O,) was added to each well and the plates
were
incubated for 30 min in dark. The reaction was stopped by the addition of 50
GIs
20 of 2.SM H,SOa t.o each well. The amount of undegraded C3 left in the
samples
was detected by HRP-conjugated goat polyclonal antibody specific to human
complement C3. The assay was standardized so that wells containing
undegraded C3 had an O.D. 490 = ~ 1Ø Wells with degraded C3 had reduced
optical density readings resulting from decreased binding of anti-C3
antibodies.
25 The optical den:~ities of t)!le mutant and parent strains were compared to
that of
negative controls (medium with different concentrations of C3) to calculate
the
percent of C3 degrading activities. There were four mutants, SN3. SN4, SNS
and SN6, with elevated C.'3 degrading activity (2.2 fold- Table 3) compared to
the activity of their parent strain CP1200. This finding was confirmed later
by
3o Western immunoblotting, for the pneumococcal mutant SN4. SN4-S 10
(disrupted
cppA gene) were also mutants of CP1200 with reduced C3 degrading activity.
-27-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
Table 3. )JLISA results for C3 degradation by parent and hyper active mutants
of
pneumococcal strains -
strains ELISA reading at 490nm *Percentage of C3 degraded in
each sample
or controls
**negative control 0.608 0%
CP 1200 (parent) 0.30 5 f
SN3 (mutant) 0.20 67.2%
SN4 (mutant) 0.162 73.4%
SN5 (mutant) 0.23 60.0%
SNS (mutant) 0.23 60.0%
*, averaee of minimum of 4 individual expts. conducted at different times
**, negative sample is medium only (THB or SMP) with C3 and without bacterial
cells
Immunoblotting was performed using ECL Western blotting protocols
(Amersham Life Sciences, Arlington Heights, IL). The pneumococcal mutants or
E. coli cultures with or without plasmids were grown from freezer stock
cultures.
in THB or LB up to log phase and incubated with C3 (0.83 pg of C3 / ml) for 2-
4 hrs, the cultures were spun down (2,500 rpm for 1 min RT or 4°C) and
the
supernatants were collected. The optical densities of the cultures were
carefully
monitored and samples were equalized before being subjected to incubation with
C3. Equal amounts of all collected supernatants containing undegraded C3 were
applied to 7.5% or 10% SDS-PAGE gels under reducing conditions. The gel was
transblotted to nitrocellulose membrane (75 volts; 4°C) for 1 hr.
Proteins were
transferred in this example and in subsequent examples from gels to
nitrocellulose membranes using a Hoeffer transfer apparatus in Towbin buffer
(3.03g Tris, 14.4g glycine and 200m1 Methanol in 1 litre volume pH.8.3; Towbin
et al. (1979) PNAS:4350-4354) for lhr at 70 volts or gels were stained with
0.125% Coomassie Brilliant Blue R-250 (Pierce, Rockford, IL) made in 50%
Methanol and 10% Acetic acid.
The blot was incubated in 10% skim milk (skim milk powder) for 1 hr
(room temperature) or overnight (4°C) with gentle shaking. The blot was
washed
in TTBS (0.1% Tween, 20 mM Tris, 137mM Saline Buffer) several times and
incubated with a 1:1000 dilution of HRP- conjugated goat antihuman C3,
polyclonal antibody, IgG fraction (ICN Pharmaceuticals/Cappel, Costa Mesa,
CA) made in 3X TTBS+3% BSA for 1 hr with gentle shaking. The incubated
-28-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
blot was washed again several times in TTBS and incubated for one minute in
chemiluminescent reagents ( 1:1 ratio of 2X luminol/Enhancer and 2X stable
peroxide solutions, Pierce, Rockford, IL). This blot was exposed to films for
~
sec to several seconds in the dark and the films were developed. The SDS-
- 5 PAGE gels alw;~ys contained pre-stained high molecular weight markers
(Bethesda Rese;~rch laboratories, life Sciences, Grand Island, NY) ranging
from
200 kd to 19 kd. The washes and incubations were performed at room
temperature with a gentle shaking unless stated otherwise.
Electroporation of chromosomal DNA from the hyper-active
to pneumococcal mutants, SN3, SN4, SNS and SN6 into E. coli DHS a. MCR
competent cells gave risf° to E. coli transformants with rescued
recombinant
plasmids. E. coli DHS cx MCR transformants. LSNp, LSN4. LSNS, LSN6,
LSN4G contained plasmids (Table 2 from pneumococcal mutants, SN3, SN4
SNS, SN6 and SN4-4G mutants respectively). Details of E. coli strains
~ 5 containing different constructs are listed in Table 2 (supra). Restriction
analysis
(Hind III) revealed that the inserts were indeed recombinant plasmids.
Different
sizes of recombinant plasmids were obtained from each hyperactive
pneumococcal mutant. Recombinant plasmids, pLSN3 and pLSN4 recovered
from mutants SN3 and S'~N4 were the same size (~7.8kb) and their insert size
was
20 ~2.4kb. The size of the insert of an ~1 lkb recombinant plasmid, pLSNS,
obtained from the pneumcoccal mutant SNS was about 5.6 kb. The fourth
pneumococcal mutant, SN6, gave two different, ~6.Skb and ~10.5kb
recombinant plasmids, pLSNci;, and pLSN6~, which had inserts of 1.1 kb and
S.1 kb respectively. These pneumococcal mutants were also examined by
25 southern hybridization. 'rhe hyperactive pneumococcal mutant SN4 was chosen
for further studies of C3 degradation and therefore, the recombinant plasmid
pLSN4 which was rescued from the mutant SN4 was subjected to a full
investigation.
Plasmid pLSN4 was used as a probe against EcoRl digested
30 chromosomal L>NA samples of the pneumococcal mutants and this confirmed the
integration of the vector + insert (pLSN4) in the mutants SN3 and SN4. Both
-29-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
SN3 and SN4 hyperactive mutants included two hybridizing fragments of sizes
2.2 kb and ~ 5.8 kb which were also present in parent strain CP1200. There
were
two other hybridizing vector/insert junction fragments at ~ 4.2 and ~ 3.5 and
these two together gave a total of ~ 7.8 kb (pLSN4 is ~ 7.8 kb). These two
bands
were also present in the EcoRl digested pLSN4 DNA sample. Both insert and
vector had EcoRl sites and represented recombinant plasmid. The pattern of the
other hyperactive mutants, SNS and SN6, suggested that these mutants may have
had different inserts in their integrated recombinant plasmids.
The same plasmid pLSN4 was used to retransform the parent
pneumococcal strain CP1200 to confirm its involvement in hyperactivity. As
expected, the obtained mutant SN4-4G (Table 2) reproduced the phenotype of
enhanced C3 degradation.
example 3
Isolation and identification of C3-degrading gene
Double stranded DNA sequence analysis was performed on the insert
part of the recombinant plasmid pLSN4. Since this insert was associated with
C3
degrading hyper-activity, we expected t:~ see insertion either in regulatory
region
of the corresponding gene or duplication of the gene; however, there was no
2o indication of insertion in a regulatory region on the basis of the protein
data base
search. This suggested the possibility of gene duplication. There were three
full
open reading frames (ORFs) and one partial open reading frame with no
significant homology between the derived amino acid sequences of the above
ORFs and the proteins as provided in searches of GenBank, Blast and SwissProt
databases. Preliminary data (Cathryn A S., et al., J. Inf. Dis. 170:600-608,
1994)
suggested that the C3 degrading proteinase might be cell wall associated
(exported protein) and therefore, we looked for the presence of a signal
sequence, a proline rich domain or a LPXTG motif. None of the four ORFs had
these sequence patterns and we chose ORF3, the largest ORF for further
3o analysis.
-30-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
Double-stranded DNA of plasmid pLSN4a was prepared using CsCI
gradient /ethidium bromide isolation and used as a template. Oligonucleotide
primers were synthesized using an applied Biosysterns 391 automated
synthesizer, by ~3ibco BRL, or by Oligo I DOOM DNA synthesizer (Beckman
Instruments Inc. La Brea, CA) Using the dideoxy chain terminator method
(Sanger F., et al., Proc. 1'Jcrtl. Acad. Sci. (USA) 82:1074-1078, 1977) and
employing Sequenase 2.0 (U.S. Biochem) and [a-;'S] dATP (Amersham life
Sciences, Arlington Heights, IL) sequencing was done with an apparatus: 20110
Macrophor Electrophoresis unit (LKB Bromma) as indicated by Sequenase
version 2.0 (Amersham life sciences).
The insert (see Fig 3) in the recombinant plasmid pLSN4, recovered from
the hyper active pneumococcal homologous-recombinant mutant SN4 seemed to
have restriction sites for Hinc II, Nru I, EcoR I. Cla I, EcoR V and Hpa I out
of
about 20 enzymes tested and this data correlated with the sequence data.
After reviewing the sequencing data, an internal fragment, 620bp, of the
cppA gene (ORh3 of the insert) was generated by gene amplification (see Table
4 for primers) with overhangs containing Hind III restriction sites. This
fragment
was subcloned into Hind. III sites in the vector pVA891, electroporated into
E.
coli and tested for the pr~°sence of the insert. Finally, this subclone
was
2o transformed into wt CP1200 pneumococcal competent cells to inactivate the
original cppA gene in thc: wild type CP 1200.
DNA amplifications were carried out using a Hybaid Omnigene machine
with primers (sc:e Table 4 for primers' sequences and amplification cycle
conditions) complimentary to the 5' and 3' ends of the required DNA fragments.
All the primers were constructed to include a restriction site on both ends.
The
amplification reaction (final volume 0.1 ml volume) utilized 10 pl of l OX
vent
buffer (final concentration, 1X contains: IOmM KC1, lOmM (NH~)SO~. 20mM
Tris-HCI (pH 8.8 at 25°(:), 2mM MgS04, 0.1 % triton X-100), 4p.ls
of 100mM
MgS04(final cancentrati.on 4 mM), 3 yl of l OmM dNTP s {final concentration
300pM), SOng 'template. 1 ~M primers and 1 ~1 of 2000units/ml of vent
polymerase (final concentration 2 units; enzyme was supplied in l OmM KCI,
-31 -
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
0.1M EDTA, lOqM Tris-HCl (pH 7.4), 1mM DTT. 0.1% Triton X-100) in a
final volume of 100 ~l with water. Vent buffer, Vent polymerase enzyme and
MgSO~ were purchased from New England Biolabs, MA, and dNTPs were
bought from Gibco BKL.
' 5 Additional sequence was generated via fluorescent sequencing usW g
Applied Biosystems Model 373a DNA sequencer (DNA Sequencing Core
Facility, Interdisciplinary Center for Biotechnology Research (ICBR),
University
of Florida, Gainesville, FL). A Robotlo Workstation (ABI Catalyst 800) and a
Perkin Elmer-Cetus PEC 9600 thermocycler were used in cycle sequencing
reactions. The template, an amplified gene product that represented the whole
insert from plasmid pLSN4a, was cleaned directly from 0.7% agarose gel by
Qiagen kit before it was used for automated sequencing. The sequencing
analysis
was conducted with programs (fasta, blast and other programs) available in the
GCG software package.
-32-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
N
0.
v
.n
3
a~
c
b)
~)
tn
fr7
b
- r,
O
Q.
U
O _ U _N U 'O
~
.G V U U ~' U
S ~ U r
O
~ U U U 4-
f%~ ~p t~ o c ,~ ,n ~ )
0
O N C ~ .- ~ p O'
oa z O ~ -~ -~
~ zz ~' ~ ~
C
NV ~V
IN IN
O 0 o O
V ~
' as ~ '
o ~ ~ o ~ '-
~o
O . ~~i' ;i) ~ '.
~
V V V o .
O' U o
V V ~ ~
~ ~ U ..
d E
-
U U d d
U ~ d
U
V U V ~
d
O
d d d d
i- E- N
d d V Q ; ~ N '
U U v
'
d d , ~ N
m
c
U V .G v r, v ~, .
E"' d
~ H d
d U U
,n
~-d ~.~ ~d ~ ~ ~''
'>
[- O C aJ C C)
r v
d
dE- E' '.'~ NNN V ~ ~.
" l~t
d l7
oo~n oov, E 'o
d d d f~1 f1 M M
E~ ~ M ~
~Y
V ~ d M )
V
~ ~. U U U U U U "
E~ ~ U U c~
U ~ ~ ~, ~r c~ i
E- O o~ c~i c~ m ~
U ~ ~ ~
~
a d a n ~ a, r U
U d o n c
H
f U d c
- V ~."' -o
d U y
d v
d
E.. ~a
U a
a.
~
V
f- U o
U j v
d V ~ ~
d ~
N
VV V F ~ c
- n
o d U .Y .-1
U d d d d ~ ~a
a
v
dV dU J ~p ~' a~ 0.
dU fn U -' -
-
~
c a U V d V M ~ T >, >
U -
U
U N ~ .
V
V t, C
U 0 c
'
U C7 ,-~~ ~-
'
zz NN -~ ~ = v
~ ~
~ C4 r~ .S E N N y G
J V V N s
c
a
0 0 ~
U a rr f, n ~.,
a, U f, f,
<r
v , ~ a z
a E ~ c~ ,~ V) U U U U
oG p o U
)
~= 'C U a~ _ ~ oo ~ n N o
U ' a) ri co
' =
C. n. ...a .~ G a. o~ ~n c~
0. 0.. C. r
y
v
' N
c
=
>
c '
D
o
y y
~
s
fr7
'fl v..
4-
y
C 'O
O
O
C E
v
..,
V
V ~
a
y ~ w
c d ~ 4, o c
on
y
y 8 f, w V G
V7 =
t
~i .:c ~ ~ E
c ~
fl.
L ... N a WD , O
V ~
v
V V W p N U
~ N ~ C
N_
. drn Ob _ p
' N O cc1
U O
O
N- ~ ~' ~ bn bn bn "
~ n o
c
v cc ' u '~ o --. on ~.
, Z ~ ~ i~ 'C .~ W ~
~ .fi , 'o
~ ~ ~
. U
,_ f~ M _' = a LV a.=J '
. v~ fG 4.
'9 ~ v
7
~
c. ~ rY ~ a c c c c c N ..
~ ~ 4 ~ == ~ ~
~
. ,
~ O Q. a~ N C C~ C ~_ G
O O ;~ ~' U
~ ~ ~ d cct
w i
f"'~ a3 s. nS d Li~ (1. ~
~ O
-33-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
EXAMPLE 4
C3-degrading Protein Isolation and Studies
Log phase cultures of hyperactive mutants and their parent strains were
incubated with C3 for 2-4 hrs and the culture supernatants were run on 7. S%
SDS-Page gels under reducing conditions and were checked for their increased
C3 degrading activity by immunoblotting with HRP-conjugated polyclonal
antibody to C3. This experiment demonstrated that mutants SN4 and SN4-4G
(obtained by retransformation of CP 1200 with the recombinant piasmid pLSN4
i o rescued from SN4) were more active than their parent strain CP 1200 in C3
degradation. Both oc and (3 chains C3 were almost completely degraded by th~~
mutants after 4 hours incubation whereas the degradation was incomplete for
the
parent strain. The CppA protein appeared to preferentially degrade the C3 a
chain.
1S A 620 by internal portion of the cppA gene was Iigated into Hind III site
of pVA 891 and the construct was transformed into CP 1200 competent cells.
The obtained transformant was tested for its ability to degrade C3. The ORF3
mutant was found to have a poor activity. The oc chain of the C3 molecule was
degraded and the [3-chain was less degraded, by SDS-PAGE and western blotting
2o analysis in comparison with its parent strain CP 1200. The reduced activity
rather than a complete absence of activity in the mutant indicated that the
potential for the presence of another fully functional gene encoding another
C3
degrading proteinase in the mutant.
The entire cppA gene was amplified and cloned into Nde I and Bam H I
2S sites of pet-28b(+) (Novagen, INC. Madison, WI) and the gene was
incorporated
with a His-Tag in its N-terminus region. The entire gene was positioned in the
vector in frame as confirmed by sequence analysis. The plasmid construct was
transformed into E. coli DHS ~ MCR strain for stabilization and the presence
of
the insert was verified before the vector and insert were transformed into E.
coli
3o BL 21 D3 (Novagen) protease deficient strain for expression. The colonies
containing the plasmid constructs were selected on LB medium containing
kanamycin (30qg/ml).
-34-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
Protein was isolated according to the Pet System manual (Madison, WI)
for small scale o~r large scale preparations. The BL 21 DE3 strain containing
the
construct (pet 28b(+)::OItF3 (cppA gene) was induced by IPTG and the
expressed protein, CppA., was solubilized. For solubilization, the induced
bacterial cultures were centrifuged and the pellet was resuspended in TES (50
mM Tris; 1 mM EDTA; 100 mM NaCI). The resuspension was sonicated (6x. i 5
sec pulses at a high output setting: about 50 watts) on ice and spun down to
collect the peller.. The pel',let was washed in TES (50 mM Tris; 1 mM EDTA;
100
mM NaCI) twice and finally the pellet was treated with 6mM G-HC1 + 1mM
DTT + I % Twe~en-20 for 3 hrs at 4°C.
The solubilized protein was diluted 1:10 in TTS (1% Tween, 50 mM
Tris, 0.7M NaCI) and dialyzed against TTS (1% Tween. 50 mM Tris Ø7M
NaCI) to remove Guanidine-HC1, DTT and EDTA. The dialysed CppA protein
was purified by Nickel column chromatography using the Pet system manual
instructions (Novagen, INC. Madison, WI). Nickel column (2.5 ml) was poured
and after removal of Guanidine-HCI, DTT and EDTA, the expressed His-Tagged
CppA protein was applied to the Nickel column for purification. The eluted
fractions were tested for His-Tagged-CppA protein by I O% SDS-PAGE gel and
Coomassie Brilliant Blue: R- 250 staining. The protein was kept on ice at
4°C or
2o frozen in small ~~liquots at -80°C until required.
The CpfrA protein (about 600 ng per ml of the reaction mixture] was
incubated with l.mman complement C3 (0.83 pg of C3 per ml of the reaction
mixture) for 4 hrs at 37°C in the presence of PBS and a negative
control without
protein was simultaneously set up. The samples were analyzed by 7.5% or 10%
SDS-PAGE gel under reducing conditions and western-blotting (ECL Western
blotting protocols -Amersham Life Sciences, Arlington Heights, IL).
As descn.°ibed above, the PCR product of the whole ORF3 gene was
subcloned into het vector pET28b(+) (Novagen, Madison, WI) with a His-tag in
the amino terminus position and the construct was introduced into protease
3o deficient strain E. toll BL 21 DE3 (Table 2) after it was stabilized in E.
cola
DHSa MCR. The E. toll BL 2I DE3 with the construct was subjected to
~-35-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
induction by IPTG. Total cell protein extracts of the induced and uninduced
cultures were tested. The expressed His-tagged ORF3 protein ( ~ 29 kd) was
identified in the insoluble fraction of the induced protein sample on 10% SDS-
Page gel.
The following reagents were used for solubilization for 3 hrs at
4°C or I
hr at room temperature: TES (50mM Tris, 1 mM EDTA, 1 M NaCI; (b) 6mM G-
HC1 + 1 mM DTT ; (c) 6mM G-HC1 + I mM DTT + 1 % Tween 20; (d) 6mM G-
HCl + 1 mM DTT + 1 % Triton X -100. Both "c" and "d" treatments made the
expressed protein soluble as it was observed on 10 % SDS-PAGE gel. The
treatment with "c" reagent was chosen for subsequent large scale preparations.
The solubilized protein was dialysed followed by purification through the
nickel
column and examined for its function against C3.
For SDS-PAGE gels used in this example and above, total cell proteins
or soluble or insoluble protein fractions were extracted according to Pet-
system
manual (Madison, WI). The proteins were separ-ated by SDS-PAGE gels (7.5%
or 10% or I S% resolving gel and 4.5% stacking gel) in the discontinuous
system
of Laemmli (Laemmli, U. K., Nature 227:680-685,1970). Briefly, samples were
combined with loading buffer (final concentration in samples was 7.57 mg/ml of
Tris, 2% SDS, 10% Glycerol and 1.25mg/ml of Bromphenol blue, ~ 5% (3-
2o mercaptoethanol) and either boiled 5 min or loaded directly on the
resolving
gels. Pre-stained high molecular weight standards (Protein markers (kd)
lysozyme, 14,300; (3-lactoglobulin, 18,400; carbonic anhydrase, 29,000;
ovalbumin, 43,000; bovine serum albumin, 68,000; phophorylase B, 97,400
myosin, 200,00 (Bethesda research laboratories, life sciences, Grand Land, NY)
were included on the gels. The large SDS-PAGE gels were electrophoresed at
I SmA for 14 hrs or l OmA for 20I1r. Mini gels were electrophoresed around 2-3
hrs at a constant voltage (100 -150 volts).
The expressed protein was incubated with C3 and the amount of C3
present was assessed by Western immunoblotting.
3o Immunoblotting analysis suggested that the samples that contained the
expressed protein degraded C3 molecules. The undegraded C3 was detected by
-36-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
polyclonal antibodies specific to human complement C3 and this was clearly
seen on the developed film in the case of the negative sample. Both oc and (3
chains of C3 molecules vrere seemed to be susceptible to the activity of the
ORF3 protein in comparison with the negative control which did not contain any
- 5 ORF3 protein; however, 'the oc chain was almost completely degraded while
the
(3 chain was partially degraded in the ORF3 samples.
Example 5
Conservation of the C3-degrading gene in Clinical Isolates
to
To examine the conservation of the gene cppA, an internal fragment of
cppA was used a.s a probe: to determine the presence of gene cppA in EcoRl
digested genomic DNA of different clinical (serotypes) of pneumococcal
isolates by southern hybridization. In the same experiment, the pneumococcal
15 parent CP 1200 and the hyperactive mutants SN4 and SN4-4G (both mutants
containing the same plasmids-see Table 2) were also included to confirm the
duplication of th.e cppA gene in the mutants. Southern hybridization was
performed using non-radiioactive DIG labeled internal fragment of the gene as
a
probe. The clinical isolates, typel, type3, type 14F and virulent type 23F
2o showed a hybridized band of about 2.3kb which was also present in the
control
pneumococcal strain CP1200 and in the SN4 mutants. This common band
indicates that they cppA gf.ne was present in all isolates tested. The SN4
mutants
also contained a second band with a size of about 3.Skb indicating the
presence
of a gene duplication. The 3.5 kb size is consistent with the observation that
25 plasmid pLSN4 has two restriction endonuclease recognition sites for EcoRl.
one
in the insert region and a second in the vector. Hence the restriction
digestion
with EcoRl produces two fragments of about 4.175 kb ( 3.531 kb of vector +
0.649 kb of insert) and 3.539kb (-~-1.67kb form insert + about 1.869 kb from
vector) from the recombinant plasmid. The cppA gene was located on the 1.67kb
30 portion of the insert and hence the 3.539 kb restricted fragment of the
recombinant pla.smid contained the cppA gene and only this band would
hybridize to the probe wl.iich was an internal fragment of the cppA gene;
-37-
CA 02283755 1999-09-21
WO 98/48022 PCT/US98/08281
therefore, in the case of the mutants with duplicated cppA gene, the second
hybridized band at ~ 3.Skb represented the duplicated cppA gene.
-38-