Note: Descriptions are shown in the official language in which they were submitted.
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
METHODS OF MODULATING P33ING1 MEDIATED APOPTOSIS
FIELD OF THE INVENTION
This invention relates to a method for modulating apoptosis in cells. The
- 5 method of modulation involves using a protein, ING1, and related proteins
and
peptides, nucleic acids encoding this and related proteins and peptides, and
nucleic
acids complementary to nucleic acids encoding p33'NC' .
This invention also relates to a method for determining the apoptotic
characteristic of cells using oligonucleotides from the ING1 gene, p33ING1
peptides and
antibodies directed against p33INC' peptides.
BACKGROUND OF THE INVENTION
Apoptosis is a form of programmed cell death which occurs through the
activation of cell-intrinsic suicide machinery. The biochemical machinery
responsible
for apoptosis is expressed in most, if not all, cells. Apoptosis is primarily
a
physiologic process necessary to remove individual cells that are no longer
needed or
that function abnormally. Apoptosis is a regulated event dependent upon active
metabolism and protein synthesis by the dying cell.
Apoptosis plays a major role during development and homeostasis. Apoptosis
can be triggered in a variety of cell types by the deprivation of growth
factors, which
appear to repress an active suicide response. Apoptosis is particularly
important for the
physiology of the immune system. Apoptosis is the mode of death of
centroblasts with
low affinity for antigen within germinal centers, cells killed by specific
cytotoxic T
lymphocytes or natural killer cells, as well as thymocytes bearing high-
affinity T-cell
receptors for self antigens that are clonally deleted during thymus
development
(negative selection).
The morphological and biochemical characteristics of cells dying by apoptosis
differ markedly from those of cells dying by necrosis. During apoptosis, cells
decrease
in size and round up. The nuclear chromatin undergoes condensation and
fragmentation. Cell death is preceded by DNA fragmentation. The DNA of
apoptotic
-1-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98I00277
cells is nonrandomly degraded by endogenous calcium and magnesium-dependent
endonuclease(s) inhibited by zinc ions. This enzymes) gives fragments of
approx. 200
base pairs (bp) or multiples of 200 by by cutting the linker DNA running
between
nucleosomes. Thus DNA appears to be one of the most important targets of the
process
that leads to cell suicide. The apoptotic cell then breaks apart into many
plasma
membrane-bound vesicles called "apoptotic bodies," which contain fragments of
condensed chromatin and morphologically intact organelles such as
mitochondria.
Apoptotic cells and bodies are rapidly phagocytosed, thereby protecting
surrounding
tissues from injury. The rapid and efficient clearance of apoptotic cells
makes
apoptosis extremely difficult to detect in tissue sections.
In contrast, necrosis is associated with rapid metabolic collapse that leads
to cell
swelling, early loss of plasma membrane integrity, and ultimate cell rupture.
Cytosolic
contents leach from the necrotic cell causing injury and inflammation to
surrounding
tissue.
Recent studies show that multiple cytotoxic stimuli well known to cause
necrosis
can lead to apoptosis instead when cells are exposed to the same noxious
agents at
lower concentrations. This insight has led to an interest in the role of
apoptosis in the
pathogenesis of renal diseases that result primarily from injury to renal
tubular
epithelial cells. These diseases include acute and chronic renal failure from
exposure of
the kidney to ischemia or to cytotoxic agents. There is also an interest in
the role of
apoptosis in Alzheimers and other neurological diseases.
Several genes associated with apoptosis have been identified. Examples include
the p53 gene, which is involved in osteosarcoma and adrenocortical, breast and
brain
cancers;
The gene cloned and sequenced as described herein, ING1 (formerly called
p33I°'), represents a new gene which is expressed in normal mammary
epithelial cells,
but expressed only at lower levels in several cancerous mammary epithelial
cell lines
and is not expressed in many primary brain tumors.
-2-
CA 02284730 1999-09-23
WO 98!44102 PCTICA98/00277
SUMMARY OF THE INVENTION
The present invention is directed to a method to potentiate apoptosis in a
eukaryotic cell, which method comprises selecting said eukaryotic cell; and
increasing
the p33INC1 biological activity in said eukaryotic cell. It is contemplated
that the p33INC'
biological activity in said eukaryotic cell may be increased by introducing
into said
eukaryotic cell an effective amount of a peptide comprising p33'NC' biological
activity
or by introducing into said eukaryotic cell an effective amount of an
oligonucleotide
which codes for a peptide comprising p33'NC' biological activity under
conditions such
that a peptide comprising p33INC' biological activity is expressed by the
cell.
Another aspect of the invention provides a method to inhibit apoptosis of a
eukaryotic cell, which method comprises selecting said eukaryotic cell; and
reducing
the effective quantity of p33'NCl biological activity in said eukaryotic cell.
It is
contemplated that the effective quantity of p33INC' biological activity may be
reduced by
administering to the cell a single-stranded oligonucleotide of at least 20
nucleotides
which comprises a sequence substantially identical to the complement of the
cDNA
sequence of Figure 3 or by administering to the cell a double-stranded
oligonucleotide
which comprises a sequence substantially identical to the cDNA sequence of
Figure 3
under conditions such that a nucleic acid of at least 20 nucleotides having a
sequence
which is substantially identical to the complement of the cDNA sequence of
Figure 3 is
expressed. It is also contemplated that the effective quantity of p33'NCl
biological
activity may be reduced by inactivating the ING1 gene in the cell.
Another aspect of the invention provides a method for determining the
apoptotic
characteristic of a eukaryotic cell which method comprises selecting said
cell; and
determining the level of expression of native p33'NC' in said cell and
comparing it to the
level of expression of native p33INC' in a non-apoptotic cell wherein an
increased level
of p33'NC' expression in the cell denotes a cell having the potential to be
apoptotic. It is
contemplated that the level of expression may be determined by exposing the
cell to
detestably labelled anti-ING1 antibodies or by exposing the cell to a
detestably labelled
single-stranded oligonucleotide of at least 100 nucleotides which comprises a
sequence
substantially identical to the complement of the cDNA sequence of Figure 3
under
-3-
CA 02284730 1999-09-23
WO 98I44I02 PCT/CA98/00277
conditions such that the oligonucleotide will bind to any ING1 mRNA present in
the
cell to determine the level of expression of mRNA ING1 in the cells. It is
also
contemplated that the level of expression may be determined by exposing the
cell to two
single-stranded oligonucleotides of from 15 to 25 nucleotides in length,
wherein the
first oligonucleotide binds to one region of the INGI mRNA and the second
oligonucleotide binds to a sequence complementary to another region of the
ING1
cDNA under conditions wherein polymerase chain reaction can occur and any ING1
mRNA in the cell will be amplified and labelled.
A still further aspect of the invention provides an assay for determining the
level
of p33'NC' activity in a eukaryotic cell, which assay comprises selecting the
eukaryotic
cell; determining the p33Inra' biological activity in the cell; and
correlating the
biological activity against a curve of the p33INC~ biological activity in
certain cell types.
A still further aspect of the invention is an isolated eukaryotic cell
substantially
free of p33'N~' biological activity.
BRIEF DESCRIPTION OF THE DRAWINGS
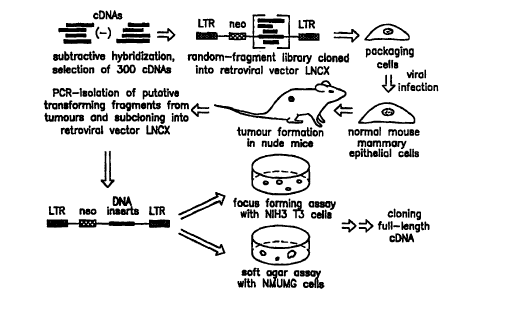
Figures lA to 1C illustrate the strategy and biological assays used for
cloning
ING1.
Figure 2 sets forth the partial cDNA sequence of ING1 (SEQ ID NO: 1) and the
predicted amino acid sequence (SEQ ID NO: 2) of p33'Nm
Figure 3 sets forth the complete cDNA sequence of INGl (SEQ ID NO: 9) and
the predicted amino acid sequence (SEQ ID NO: 10) of p33'NC' .
Figures 4A to 4C illustrate the effects of P33'NC' overexpression.
Figure 5 illustrates the changes in p33'NCl protein levels in breast cancer
cell
lines. Figure 5A is a Western blot. Figure 5B is a picture of the coomassie-
blue
stained gel of Figure 5A.
Figure 6A illustrates Western blotting of neuroblastoma cell lines with anti-
p33INC~ antibody. Figure 6B illustrates a Southern blot of neuroblastoma cell
line
compared to a normal diploid cell strain for ING1 DNA. Figure 6C illustrates
the RT-
PCR reaction on a neuroblastoma cell line compared to a control diploid
fibroblast.
-4-
_ -.~ -...._-__..... r , , .- _..
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
Figure 7 illustrates the level of ING1 mRNA in control (c) tissue,
glioblastoma
(GB), astrocytoma {AS) and meningioma (MN) tumors as determined by RT-PCR.
Figures 8A and 8B illustrate the expression of ING1 mRNA and p33INC1 in
proliferation competent (y) and in senescent human fibroblasts (o).
Figure 9 illustrates the level of p33'NCl protein through the cell cycle.
Panel A
has anti-cdk2 antibody as a positive control. Panel B shows the results with
anti-p33
antibodies. Panel C shows cell cycle profile at each point as determined by
FACS:
Figure 10 illustrates the number of cells per colony of cells blocked for ING1
expression.
Figure 1 I illustrates that ING 1 protein levels increase during serum
starvation-
induced apoptosis of P19 teratocarcinoma cells. Figure 11A shows a Western
Blot of
protein homogenates probed with a polyclonal antibody specific for ING1.
Figure 11B
shows a trypan blue dye exclusion assay of serum starved P19 cells infected
with
retroviral constructs expressing antisense ING1 (aS), sense ING1 (s) ,bcl-2
sense (S-
bcl-2), bcl-2 antisense (aS-bcl-2) and vector only (V) constructs.
Figure 12A illustrates a Western blot of human c-myc and ING1 expression in
NIH 3T3 tet-myc cells retrovirally infected with constructs containing vector
(v),
antisense (aS) and sense (S) ING1 constructs grown in the presence (+) or
absence (-)
or tetracycline. Figures 12B - 12D are pictures of cells having vector only
(Fig. 12B},
ING1 antisense construct (Fig. 12C) or ING1 sense construct (Fig. 12D) fixed
and
stained with labelled anti-ING1 antibody.
Figure 13 illustrates that expression of ING1 enhances c-myc-induced
apoptosis.
Figure 13A illustrates cell viability of NIH-3T3 tet-myc cells containing
antisense (aS),
sense (S) INGl or vector only (v) constructs. Figure 13B illustrates the
amount of
DNA laddering in tet-myc cells expressing INGI sense (S} , antisense (aS),
vector (v),
bcl-2 and control (c) without vector. Figure 13C illustrates the effect on
cell viability
of microinjection of GST-ING1 protein.
-5-
CA 02284730 1999-09-23
w0 98/44102 PCT/CA98/00277
DETAILED DESCRIPTION OF THE INVENTION
The invention described herein relates to the discovery of a method for the
modulation of apoptosis in eukaryotic cells. The invention also relates to a
method for
detecting cells having and/or lacking the potential to undergo apoptosis.
Using a strategy based upon subtractive hybridization of normal and cancerous
mammary epithelial cell mRNAs and the selection of genetic suppressor elements
[3], a
novel gene was isolated encoding a 33 kDa protein that is a potent inhibitor
of cell
growth. Acute expression of transfected constructs encoding p33~NG1 induced
apoptosis
in cells. Inhibition of translation of the mRNA for this gene prevented
apoptosis in
cells.
A. Definitions
As used herein the following terms have the following meanings:
"Antibody" means a molecule that binds to a known antigen. An "anti-p33'NC'
antibody" means an antibody molecule that binds to one or more epitopes of the
p33'Nm
protein.
"Antisense" and "Antisense nucleotides" means DNA or RNA constructs which
block the expression of the naturally-occurring gene product. For example, in
the
present invention, use of a DNA construct that produces ING1 antisense RNA
blocks
the expression of p33INC' by destroying or inactivating ING1 mRNA.
"Apoptosis" means a form of programmed cell death.
The "apoptotic characteristic" of a cell is the ability of a cell to undergo
apoptosis when exposed an apoptotic triggering event. Such events include, for
example, exposure to certain toxins and/or expression of a tumorigenic gene or
activation of cell surface proteins.
"Biological sample" means a sample of eukaryotic cells. These cells may be
part of a tissue or organ sample obtained, for example, by biopsy, or they
rnay be
individual cells, for example, blood cells or cells grown in tissue culture.
-6-
~ i
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
"Cancerous cell" means a cell in or from a neoplasm. Preferably the cancerous
cells is breast cancer, brain cancer, gastric cancer, hematologic neoplasms
and head and
neck squamous cell carcinomas.
"Breast cancer" means any of various malignant neoplasms of the breast or
mammary tissue.
"Brain cancer" means any of various malignant neoplasrns of the brain,
neuroglial cells or meninges .
"Cell cycle" means the cyclic biochemical and structural events occurring
during
growth of cells. The cycle is divided into periods called : quiescence (G6),
Gap, (G,),
DNA synthesis (S), Gape (GZ), and mitosis (M).
"Cell division" means mitosis, i.e., the usual process of cell reproduction.
"Code" or "encode", when used with reference to a nucleotide's relation to a
protein, mean the system whereby particular combinations of adjacent
nucleotides
control the insertion of particular amino acids in equivalent places in a
protein
molecule.
"Expression" means the production of a protein or polynucleotide in the cell.
A "eukaryotic cell" is a cell having a membrane-bound nucleus containing DNA
in the form of chromosomes, either in a tissue or organ or in tissue culture.
More
preferably, it is a mammalian cell.
"Growth" means progression through the cell cycle with the result that two
daughter cells are formed from each mother cell. "Actively growing" means that
state
wherein cells exhibit growth and cell division.
"Hyperplasticity" means an increase in cell number, excluding tumor formation.
"Immunohistochemistry" means a method to localize a specific protein within a
cell using cell sections or thin sections of a specific tissue and an antibody
directed
against the target protein. It is contemplated that the antibody may be
directly labelled,
radioactively, biotinylated or labelled with a fluorochrome. It is also
contemplated that
the antibody may be indirectly labelled.
"Inactivating" a gene means rendering the gene incapable of producing an
active
protein normally encoded by that gene. Methods of inactivating a gene include,
for
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
example, deleting the gene in the cell or introducing a deletion or mutation
into the
gene such that the gene no longer expresses an active protein. Preferably
cells having
such inactivated genes are substantially free of the active protein.
"In situ hybridization" means a method to identify and localize certain
specific
transcripts of specific genes in cells and tissue sections. The cells are
fixed to a solid
support and hybridized to a labelled nucleic acid probe complementary to the
sequence
to be identified. The probe may be directly labelled or indirectly labelled.
"Label" means to incorporate into a compound a substance that is readily
detected. Such substances include radioactive substances and fluorescent dyes,
for
example.
"Mammalian cell" means a cell in or from a mammal, either in a tissue or organ
or in tissue culture.
"Neoplasia" means the process resulting in the formation and growth of an
abnormal tissue that grows by cellular proliferation more rapidly than normal,
and
continues to grow after the stimuli that initiated the new growth cease.
"Neoplastic" describes the abnormal tissue that grows by cellular
proliferation
more rapidly than normal, and continues to grow after the stimuli that
initiated the new
growth cease.
"Normal cell" means a non-cancerous cell.
"Proliferation" means growth and reproduction, i.e., division of cells.
"Actively
proliferating" means cells that are actively growing and reproducing.
"Native" means the nucleic acid of a non-mutated gene or peptide sequence
encoded by such a gene as found in a phenotypically normal cell.
"Substantially identical" means that the polynucleotide or nucleic acid of
interest
is able to hybridize to the complement of the known sequence under stringent
conditions. Such stringent conditions preferably require at least 85 %
homology, more
preferably the conditions require at least 90 % homology and most preferably
the
conditions require at least 95 % homology. When used in relation to peptides
and
proteins, "substantially identical" means that the amino acid sequence of the
peptides
_g_
_. . . _. _.. ,
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
share at least 85 % homology, more preferably at least 90 % homology and most
preferably at least 95 % homology.
"Senescent cells" means cells that are no longer actively dividing and
reproducing. Such cells are typically characterized by an inability to respond
to growth
factors and by altered morphology including increased size and decreased
saturation
density in tissue culture.
B. Synthesis and Methodolo~v
A novel positive selection procedure that combines subtractive hybridization
with an in vivo selection assay was used to identify putative growth-
suppressor
elements. An overview of the strategy used is shown in Figure lA.
Following a modified subtractive hybridization protocol [4,5], total cDNA from
a normal mammary epithelial cell line [6] was hybridized independently with
cDNAs
from the breast cancer cell lines MCF-7, BT-483, BT-474, Hs-578T, ZR-75, MD-MB-
468, MD-MB-435 and BT-20 which were obtained from the American Type Culture
Collection. Subtracted cDNA, theoretically containing sequences more highly
expressed in the phenotypically normal epithelial cells, was then used as a
probe to
screen a normal human fibroblast cDNA library.
Following screening, 300 cDNA clones were isolated, and their inserts were
digested into fragments of 200-800 base pairs. The fragments were then
recloned into
the retroviral plasmid vector pLNCX [7]. After passage through the packaging
line
BOSC 23 [3], retroviruses containing the isolated fragments were used to
infect normal
mouse mammary epithelial cells {NMuMG). The infected cells were subsequently
injected into nude mice.
Within 45 days, several mice developed tumors from which the cloned inserts
were recovered by amplification using primers specific for pLNCX in polymerase
chain
reactions {PCR). Two different sequences were isolated from tumors, one of
which
was subsequently shown to be expressed in the antisense orientation.
The antisense sequence isolated, when introduced into normal fibroblast cells,
consistently showed the biological effects of increased cell proliferation in
soft agar and
-9-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
in focus forming assays (Fig. 1B and Table 1). This 182 by fragment
represented
nucleotides 781 to 963 of the cDNA shown in Figure 2 and nucleotides 942 to
1124 of
Figure 3. This cDNA encodes a 33 kDa protein called p33I"cl for INhibitor of
Growth.
This was formerly designated p33'c' (see U.S. Serial No. 08/569,721 which is
incorporated herein by reference in its entirety).
After plating NMuMG cells infected with either control virus or with virus
containing an insert of the antisense orientation of ING1 in soft agar, cells
receiving the
insert formed, on average, at least 50 times the number of colonies as cells
infected
with virus alone. Similar results were obtained following transfection of the
retroviral
construct into NIH3T3 cells, where pLNCX containing the insert of the
antisense
orientation of INGl resulted in the formation of 2.3 times the number of
generally
larger foci than vector alone.
These results corroborated the observations made in the nude mouse assay that
the ING1 sequence corresponds to a gene whose product plays a significant role
in
regulating cell growth.
In order to isolate the gene corresponding to the fragment showing biological
effects, normal human fibroblast and HeLa cell libraries were screened with
the
fragment, resulting in the isolation of 11 positive clones. Two clones
contained cDNA
whose sequence is shown in Figure 2. The complete cDNA sequence (Figure 3) was
obtained using rapid amplification of cDNA ends (RACE) by methods known in the
art.
Comparison of the sequence of p33INC' shown in Figure 3 to the available
protein and nucleotide data bases showed no significant homology to any
sequence
encoding a known protein and very limited similarity to retinoblastoma binding
protein
2 (RbBP2) [9] and to several zinc finger transcription factors. Regions of the
p33INC'
protein that show homology to retinoblastoma binding protein 2 were identified
using
the Blast program available from the National Centre for Biological
Information
(address: www.ncbi.nim.nih.gov).
Use of a polyclonal antibody raised against a glutathione-S-transferase (GST)
fusion with p33ING~ revealed a protein of 33 kDa by Western blot analysis of
human and
mouse cell extracts (Figure 1C).
-10-
~ ,
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
To determine whether the level of p33INC1 was decreased in cells infected with
viral constructs containing the antisense orientation, lysates were prepared
from control
NMuMG cells and from NMuMG cells infected with antisense INGl that had grown
and formed colonies in semi-solid medium. Results of Western blot analysis
showed
that chronic expression of antisense construct reduced the expression of the
endogenous
p33'NC' protein by approximately 90% in the cells (Figure 1C, lane 6) compared
to
control parental cells (Figure 1C, lane 5).
The ING1 cDNA contains several AU-rich elements (AREs) in the 3'
untranslated region of the clone (Figure 2) which are believed to be involved
in the
destabilization of specific mRNAs [10].
Since the ING1 gene was originally isolated by subtractive hybridization
between normal and transformed epithelial cDNAs, the levels of ING1 mRNA
expression in different normal, breast cancer, and brain cancer cell lines
were
examined. Results from Northern blot analysis show that ING1 is expressed at
considerably lower levels (approximately 2-8 fold as estimated by scanning
densitometry) in BT-20, ZR-75, MDA-MB-435 and T-47D breast cancer cells
compared to MDA-MB-468 and SK-BR-3 breast cancer cells and to normal Hs68
fibroblasts. Results from reverse transcription polymerase chain reaction (RT-
PCR)
showed that ING1 is not expressed or is expressed at very low levels in
glioblastomas,
astrocytomas and meningiomas.
Isolation of a DNA fragment that was capable of inducing tumors, foci and
growth in soft agar when expressed in the antisense orientation, suggested
that the
cellular role of p33'NC' is to negatively regulate growth. To test this idea,
part of the
ING1 cDNA was cloned into the mammalian expression vector pBK in the sense
orientation (pINGI-S). This construct and the plasmid vector, both of which
contain
neomycin resistance genes and a cytomegalovirus (CMV) promoter, were
transfected
into human breast cancer (Hs578T) and normal fibroblast (Hs68) cells.
Following
growth of the cells in antibiotic for 3 weeks, a large number of stable
transformants
were recovered from cells transfected with vector (1 +3), whereas very few
colonies
-11-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
were visible in plates of cells transfected with the sense orientation of INGl
cDNA
(2+4)(Figure 4A).
To corroborate the results of these chronic assays, the effect of
microinjecting
these constructs, together with non-specific antibodies into fibroblasts was
examined.
Arrows in panels 1 and 3 of Figure 4B identify cells injected with sense (S)
and
antisense (aS) constructs, respectively, which were visualized by staining for
the
presence of coinjected non-specific antibodies using indirect
immunofluorescence.
Arrows in panels 2 and 4 of Figure 4B show that cells injected with pINGI-S
failed to incorporate bromodeoxyuridine (BrdU) (panel 2) over a 36 hour time
course
after injection. In contrast, those injected with pINGl-aS entered S phase
(panel 4) as
estimated by staining with anti-BrdU antibodies.
Figure 4C shows the combined results of 5 separate experiments, which
indicated that injection of the pBK vector or of pINGl-aS constructs had no
appreciable
effect upon the ability of injected cells to incorporate BrdU, whereas
injection of
pINGl-S blocked the ability of cells to enter into and proceed through S
phase.
Similar results were obtained in larger populations of cells that were
electroporated with vector, sense and antisense construct DNAs together with a
construct encoding the CD20 surface marker. Such co-transfections allowed the
analysis of DNA content in cells that had taken up DNA by staining for CD20
and
subsequent analysis by fluorescence activated cell sorting (FACS). As shown in
Table
2, the CD20-expressing population co-transfected with pINGI-S had, on average,
63.1 % of cells in GOIG1 whereas those co-transfected with vector had 33.6% of
cells in
GOIG1 when cells were fixed and stained 48 hours after electroporation.
These results, using several independent methods, indicate that the
overexpression of p33'NGl inhibits cell growth and DNA synthesis in both
transient and
chronic assays, most likely by arresting cells in the G1 phase of the cell
cycle.
Since the activity of the tumor suppressor genes increases in senescent cells
[20], p33IN~' activity in low and high passage cells was checked. As shown in
Figures
8A and 8B, ING1 expression (and the level of the p33'NGl protein) increased
several-
fold when cells approached the end of their in vitro replicative lifespan.
-12-
CA 02284730 1999-09-23
WO 98/44102 PCTICA98100277
These data demonstrate that p33INC' is a novel inhibitor of cell growth and a
candidate tumor suppressor. Additional experiments also indicate that p33'NG'
is
localized in the nucleus of cells, which is consistent with p33'"c''s
functioning as a
tumor suppressor. Further data showed that ING1 gene is localized to the 13q33-
34
S chromosome region. A number of human cancers have been mapped to this region
including primary gastric cancer; hematologic neoplasms; head and neck
squamous cell
carcinomas. Alternatively, p33'NC' might play a role in the regulation of
cyclin-
dependent kinases (CDKs), as reported recently for the family of CDK
inhibitors
including p18[11], p21[12,13] and the candidate tumor suppressor pl6MTS'[8] to
which
a portion of the p33'"c' sequence shows some homology, and which has been
reported
to be the MTS 1 multiple tumor suppressor locus of human chromosome 9p21 that
is
inactivated in many types of human tumors [ 14,15] .
Indications that the ING1 gene may be involved in the modulation of apoptosis
first came from the conspicuous presence of p33'"c' in regressing tail and its
absence
from growing hindlimbs of metamorphosing Xenopus tadpoles. Accordingly, P19
cells
were infected with sense (S), antisense (aS) constructs and cell viability was
examined
under serum starvation. As shown in Figure 12B, the antisense construct
conferred a
moderate level of protection against death (70% viability). Conversely, P19
cells
expressing the sense ING1 construct exhibited greater cell death (44%
viability)
suggesting that the ING1 gene confers cellular susceptibility to death induced
by serum
starvation.
When c-myc was induced in the NIH 3T3 cells by tetracycline withdrawal and
the cells were serum starved for 72 hours, they showed a viability of 65 %
compared
with controls where myc was not induced (Figure 13A). Apoptosis was minimized
in
cells coexpressing the antisense INGl construct with only 5% of cells dying
upon
serum withdrawal. Cells expressing the ING1 protein alone showed 70% viability
after
serum starvation and apoptosis was magnified when c-myc was also expressed
(40%
viable). This showed that with both P19 and NIH 3T3 tet-myc cells expression
of the
ING1 gene is involved in regulation of apoptosis in a manner that is
synergistic with the
action of the myc gene.
-13-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
Without being restricted to a theory, it appears that a loss of p3fNC' or its
function appears to have similar consequences to those observed for p53 (33,
34). Since
ING1 appears to be important in the control of the G1 to S phase transition,
it is
possible that 1NG1 could modulate or be modulated by p53. Conversely, ING1
could
act independently, perhaps providing an activity where p53-independent
mechanisms
are at work. Alterations in the proper functioning of p331NC1 may contribute
to
tumorigenesis by rendering cells refractory to normal apoptotic pathways.
It is expected that several p33'NC'-related peptides will be useful in the
present
invention. In particular, p33'NC', its analogs and related proteins and
peptides which
are effective in potentiating apoptosis in eukaryotic cells are preferred.
Included within the scope of the p33"~cl, as that term is used herein, are
p33'NC's
having the amino acid sequence set forth in Figures 2 and 3, homologous amino
acid
sequence variants of the sequence of Figures 2 and 3, and homologous in vitro-
generated variants and derivatives of p33'NC', which are capable of exhibiting
a
IS biological activity in common with the p33'"cl of Figure 3. Also included
are p33'NC'
variants having post-translational modifications, such as acetylation,
glycosylation or
phosphorylation.
p33INC' biological activity is defined as the possession of at least one cell
proliferation, cell regulatory or tumor suppressive function qualitatively in
common
with native p33'NCIs. One example of the qualitative biological activity of
p33'NC' is its
ability to inhibit cell growth as estimated by decreasing the S-phase fraction
in a
population of cells.
The effective quantity of p33'NC' biological activity is that level of p331NC'
biological activity necessary to potentiate apoptosis. The methods of this
invention may
reduce the effective quantity to less than 50% of the p3fNC' biological
activity of
normal cells more preferably less than 30 % of the p3 f Ncl biological
activity, most
preferably there will be no p33'NCl biological activity.
p33'NCl immunological activity is defined herein as the possession of
immunological cross-reactivity with at least one epitope of native p3fNC'
Immunologically cross-reactive, as used herein, means that the candidate
polypeptide is
-14-
, , ,
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
capable of competitively inhibiting the qualitative biological activity of the
native
p33'"c' having this activity, with polyclonal antisera raised against the
known active
analog. Such antisera are prepared in conventional fashion by injecting goats
or
rabbits, for example, subcutaneously with the known active analog in complete
Freund's adjuvant, followed by booster intraperitoneal or subcutaneous
injection in
incomplete Freunds.
This invention is concerned with amino acid sequence variants of native
p33'"cj,
Amino acid sequence variants of the p33'"c' are prepared with various
objectives in
mind, including increasing the affinity of the p33'"c' for its binding
partner, facilitating
the stability, purification and preparation of the p33'"c', modifying its
biological half-
life, improving therapeutic efficacy, and lessening the severity or occurrence
of side
effects during therapeutic use of the p33'"c'.
Amino acid sequence variants of p33'"°' fall into one or more of three
classes:
insertional, substitutional, or deletional variants including those variations
that arise
from variable splicing of transcripts from the chromosomal gene. Additional
variants
may also be prepared by site specific mutagenesis of nucleotides in the DNA
encoding
the p33'"c', by which DNA encoding the variant is obtained, and thereafter
expressing
the DNA in recombinant cell culture. However, variant p33'"c' fragments having
up to
about 100 to 150 amino acid residues are prepared conveniently by in vitro
synthesis.
The amino acid sequence variants of the p33'"c' may be predetermined variants
not found in nature or naturally occurring alleles. The p33'"c' variants
typically exhibit
the same qualitative biological activity as naturally occurring p33'"c'.
However, the
p33'"c' variants and derivatives that are not capable of exhibiting
qualitative biological
activity similar to native p33'"c', may nonetheless be useful as reagents in
diagnostic
assays for p33'"c' or antibodies to p33'"cl. Further, when insolubilized in
accordance
with known methods, they may be used as agents for purifying anti-p33'"c'
antibodies
from antisera or hybridoma culture supernatants. Further, they may be used as
immunogens for raising antibodies to p33'"c' or as a component in an
immunoassay kit
(labeled so as to be a competitive reagent for native p33'"cl or unlabeled so
as to be
-15-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
used as a standard for the p33INC' assay) so long as at least one p33'NCl
epitope remains
active in these analogs.
While the site for introducing an amino acid variation may be predetermined,
the mutation, per se, need not be predetermined. For example, in order to
optimize the
performance of a mutation at a given site, random or saturation mutagenesis
(where all
20 possible residues are inserted) is conducted at the target codon and the
expressed
p33'"c' variant is screened for the optimal combination of desired activities.
Such
screening is within the ordinary skill of the art.
Amino acid insertions will usually be on the order of from about one to about
ten amino acid residues; substitutions are typically introduced for single
residues and
deletions will range from about one to about thirty residues. Deletions or
insertions
preferably are made in adjacent pairs. That is, a deletion of two residues or
insertion of
two residues. Substitutions, deletions, insertions or any combination thereof
may be
introduced or combined to arrive at a final construct.
Insertional amino acid sequence variants of the native p33'NC' are those in
which
one or more amino acid residues extraneous to native p33INC' are introduced
into a
predetermined site in the target p33i~'GI and which displace the pre-existing
residues.
Commonly, insertional variants are fusions of heterologous proteins or
polypeptides to
the amino or carboxyl terminus of the p33ING1. Such variants are referred to
as fusions
of the p33'NC' and a polypeptide containing a sequence which is other than
that which is
normally found in the p33INC' at the inserted position. Several groups of
fusions are
contemplated for carrying out the invention described herein.
Immunologically active p33'"c1 derivatives and fusions comprise the p33'NCl
and
a polypeptide containing a non-p33INC' epitope. Such immunologically active
derivatives and fusions of p33'NC' are within the scope of this invention. The
non-
p33'NCl epitope rnay be any immunologically competent polypeptide, i.e., any
polypeptide which is capable of eliciting an immune response in the animal in
which the
fusion is to be administered, or which is capable of being bound by an
antibody raised
against the non-p33'NC' polypeptide.
-lb-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
Substitutional variants are those in which at least one residue in the Figure
3
sequence has been removed and a different residue inserted in its place. Novel
amino
acid sequences as well as isosteric analogs (amino acid or otherwise) are
included
within the scope of this invention.
Some deletions, insertions and substitutions will not produce radical changes
in
the characteristics in the p33'NC' molecule. However, while it is difficult to
predict the
exact effect of the substitution, deletion or insertion in advance of doing
so, for
example, when modifying an immune epitope on the p33'"~' protein, one skilled
in the
art will appreciate that the effect will be evaluated by routine screening
assays. For
example, a change in the immunological character of the p33'N°'
protein, such as
affinity for a given antibody, is measured by a competitive-type
intrnunoassay.
Modifications of protein properties such as redox or thermal stability,
hydrophobicity,
susceptibility to proteolytic degradation, or the tendency to aggregate with
carriers or
into multimers may be assayed by methods well known to one of skill in the
art.
Deletions of cysteine or other labile amino acid residues may also be
desirable.
For example, they may increase the oxidative stability of the p3fNG' protein.
Deletion
or substitution of potential proteolysis sites, e.g., Arg Arg, is accomplished
by deleting
one of the basic residues or substituting one with glutaminyl or histidyl
residues.
Covalent modifications of the p33'N~' protein are included within the scope of
the present invention. Such modifications are introduced by reacting targeted
amino
acid residues with an organic derivatizing agent that is capable of reacting
with selected
side chains or terminal amino acid residues. The resulting covalent
derivatives of
p33'N~' are useful to identify residues important for p33'NG' ~ s biological
activity, for
immunoassays of the p33'NC' or for preparation of anti-p33'NC' antibodies for
immuno-
affinity purification of recombinant p33'NC'. Such modifications are within
the ordinary
skill of the art and are performed without undue experimentation.
In general, prokaryotes are used for cloning of DNA sequences and in
constructing the vectors useful in the present invention. For example, E. coli
HB 101,
DHSa and XL1-blue are particularly useful. These examples are meant to be
-17-
CA 02284730 1999-09-23
WO 98/44102 PCT1CA98/00277
illustrative and do not limit the present invention. Alternatively, in vitro
methods of
cloning such as the polymerase chain reaction may be used.
Expression hosts typically are transformed with DNA encoding the p33'NC'
protein which has been ligated into an expression vector. Such vectors
ordinarily carry
a replication site, although this is not necessary where chromosomal
integration will
occur. Expression vectors may also include marker sequences which are capable
of
providing phenotypic selection in transformed cells. Expression vectors also
optimally
will contain sequences which are useful for the control of transcription and
translation.
Expression vectors used in eukaryotic host cells will also contain sequences
necessary for the termination of transcription which may affect mRNA
expression.
Expression vectors may contain a selection gene as a selectable marker.
Examples of
suitable selectable markers for eukaryotic cells are dihydrofolate reductase,
thymidine
kinase, neomycin or hygromycin.
Antibodies to p33'NC' may be prepared in conventional fashion [18] by
injecting
goats or rabbits, for example, subcutaneously with the complete p33'NC'
protein or a
peptide consisting of at least 10 amino acids similar to the p33'NC' protein
in complete
Freund's adjuvant, followed by booster intraperitoneal or subcutaneous
injection in
incomplete Freund's adjuvant, The anti-p33'NC' antibodies may be directed
against one
or more epitopes on p33'NC'. Monoclonal antibodies against p33'NC' can be
prepared by
methods known in the art [18]. The antibodies are preferably labelled with a
marker,
for example, with a radioactive or fluorescent marker. It is contemplated that
the
antibodies would be labelled indirectly by binding them to an anti-goat or
anti-rabbit
antibody covalently bound to a marker compound.
C. Pharmaceutical Compositions
The present invention may be used to potentiate apoptosis in eukaryotic cells
by
increasing expression of p33'"c'. For example, the potential for apoptosis is
increased
by introducing into cells a drug or other agent which can increase, directly
or
indirectly, expression of p33'"c'. Inducing apoptosis in cancer cells is of
obvious
importance. Inducing apoptosis in tissue culture cells provides a model system
for
-18-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
studying the effects of certain drugs for triggering, reversing or halting the
apoptotic
pathway. Accordingly, increasing a cell's potential to enter the apoptotic
pathway is
useful.
In one embodiment, a protein or a peptide having p33'"c' biological activity
is
introduced directly. In a preferred embodiment the peptide possesses at least
apoptosis
modulating function qualitatively in common with native p33'"c'.
In another embodiment nucleotides coding for p33'NC' are introduced by
retroviral or other means. In one embodiment the nucleotide coding for p33'NC'
comprises a nucleotide sequence which codes for a peptide having p33'NC'
biological
activity. In one embodiment the oligonucleotide comprises an oligonucleotide
which
codes for the amino acid sequence set forth in Figure 3. In another embodiment
the
oligonucleotide sequence comprises a nucleotide sequence which codes for the
amino
acid sequence set forth in Figure 2. Preferably the nucleotide sequence is
substantially
identical to the cDNA sequence of Figure 3, more preferably the sequence is
substantially identical to the cDNA sequence of Figure 2 and most preferably
the
sequence is substantially identical to nucleotides 161 to 1143 of the cDNA
sequence of
Figure 3.
Apoptosis is inhibited or substantially decreased by preventing transcription
of
INGl DNA and/or translation of RNA. This can be carried out by introducing
antisense oligonucleotides of the ING1 sequence into cells, in which they
hybridize to
the p33'NC'-encoding mRNA sequences, preventing their further processing. It
is
contemplated that the antisense oligonucleotide can be introduced into the
cells by
introducing antisense single-stranded nucleic acid which is substantially
identical to the
complement of the cDNA sequence in Figures 2 or 3. It is also contemplated
that an
antisense oligonucleotide can be expressed in the cells by introducing a
single- or
double-stranded polynucleotide into the cell under conditions wherein a single-
stranded
nucleic acid sequence which is substantially identical to the complement of
the cDNA
sequence in Figures 2 or 3 is expressed in the cell, for example, by placing
the
polynucleotide in the antisense direction under the control of a strong
promoter. It is
contemplated that the antisense oligonucleotide introduced to the cell or
expressed in
-19-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
the cell is at least 20 nucleotides, more preferably it is at least 50
nucleotides and most
preferably it is at least 100 nucleotides in length. Preferably the antisense
oligonucleotide sequence is substantially identical to the complement of
nucleotides 942
to 1124 of the cDNA sequence set forth in Figure 3.
It is also possible to inhibit expression of p33'~'c' by the addition of
agents which
degrade p33'NC'. Such agents include a protease or other substance which
enhances
p33'NC' breakdown in cells. In either case the effect is indirect, in that
less p33'N°' is
available than would otherwise be the case.
It is also possible to inhibit expression of p33IN~1 by inactivating the gene
coding for ING1 in the cell. Such inactivation can occur by deleting the gene
in the cell
or by introducing a deletion or mutation into the gene thereby inactivating
the gene.
The gene may also be inactivated by inserting into the gene another DNA
fragment
such that expression of the native p33'NC' protein does not occur. Methods for
introducing mutations, deletions and insertions into genes in eukaryotic cells
are known
in the art. See, for example, U.S. Patent No. 5,464,764 which is incorporated
herein in
its entirety.
It is contemplated that the ability to inhibit apoptosis in a eukaryotic cell
in
tissue culture provides a model system for testing certain proteins and
factors for their
role in the apoptotic pathway. It also provides a model system for testing
compounds
suspected of being tumorigenic.
Viral or plasmid vectors may be used to deliver various constructs to target
cells
in vitro or in vivo. Such viral vectors may include retroviruses, adenovirus
or
adenovirus-associated viruses. Such methods are known in the art [19].
In vitro such peptides or vectors may be administered by infection,
microinjection, electroporation and by other methods known in the art.
In vivo parenteral administration of the nucleic acids is preferred with
subdermal
or intramuscular administration most preferred. Intravenous administration or
use of
implanted milliosmol pumps (available from Alza) may also be used.
When used for parenteral administration, which is preferred, the nucleic acids
of
the present invention may be formulated in a variety of ways. Aqueous
solutions of the
-20-
._. r
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
nucleic acids of the present invention may be encapsulated in polymeric beads,
liposomes, nanoparticles or other injectable depot formulations known tv those
of skill
in the art. (Examples thereof may be found, for example, in Remington's
Pharmaceutical Sciences, 18th Edition, 1990.) The nucleic acids may also be
encapsulated in a viral coat. Doses are selected to provide effective
inhibition of cancer
cell growth and/or proliferation.
The methods of this invention may also be achieved by using a pharmaceutical
composition comprising one or more of the following apoptosis inducing
compounds:
p33'N~', its analogs and related proteins and peptides. Doses are selected to
provide
effective induction of apoptosis.
Parenteral administration of the proteins or peptides is preferred, with
subdermal or intramuscular administration most preferred. Intravenous
administration
or use of implanted milliosmol pumps (available from Alza) may also be used.
When used for parenteral administration, which is preferred, the proteins and
peptides of the present invention may be formulated in a variety of ways.
Aqueous
solutions of the proteins or peptides of the present invention may be
encapsulated in
polymeric beads, Iiposomes, nanoparticles or other injectable depot
formulations known
to those of skill in the art. (Examples thereof may be found, for example, in
Remington's Pharmaceutical Sciences, 18th Edition, 1990.)
Compositions including a liquid pharmaceutically inert carrier such as water
may also be considered for both parenteral and oral administration. Other
pharmaceutically compatible liquids may also be used. The use of such liquids
is well
known to those of skill in the art. (Examples thereof may be found, for
example, in
Remington's Pharmaceutical Sciences, 18th Edition, 1990.)
The dose level and schedule of administration may vary depending on the
particular p33'"c'_related compounds) andlor compositions used, the method of
administration, and such factors as the age and condition of the subject.
As discussed previously, parenteral administration is preferred, but
formulations
may also be considered for other means of administration such as orally, per
rectum,
-2 I -
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
and transdermally. The usefulness of these formulations may depend on the
particular
compound used and the particular subject receiving the p33'N°'-related
compound.
Oral formulations of p33INC'_related compounds may optionally and
conveniently be used in compositions containing a pharmaceutically inert
carrier,
including conventional solid carriers, which are conveniently presented in
tablet or
capsule form. Formulations for rectal or transdermal use may contain a liquid
carrier
that may be oily, aqueous, emulsified or contain certain solvents suitable to
the mode of
administration. Suitable formulations are known to those of skill in the art.
(Examples
thereof may be found, for example, in Remington's Pharmaceutical Sciences,
18th
Edition, 1990. )
D. Use of ING1 DNA and RNA and p33'N~' and Related Proteins and Peptides for
Diagnosis
The present invention also has diagnostic use, since simple immunochemical
staining of cells or sections of cells should give an accurate estimate of the
portion of
cells expressing p33'N°'. Such a test based on the use of anti-p33'Nm
antibodies or
ING1 polynucleotides and other standard secondary techniques of visualization
will be
useful in determining the apoptotic characteristic of eukaryotic cells. Such a
test might
also be useful to the scientific research community.
Antibodies specifically reactive with p33'NCi can be produced, using known
methods [18]. For example, anti-p33'NC' antisera can be produced by injecting
an
appropriate host (e.g., rabbits, mice, rats, pigs) with p33'N~' and
withdrawing blood
from the host animal after sufficient time for antibodies to have been formed.
Monoclonal antibodies can also be produced using known techniques [18]. Such
antibodies to p33'NCi will generally be detectably labelled (e.g., with a
radioactive label,
a fluorescent material, biotin or another member of a binding pair or an
enzyme) by
methods known in the art. It is also contemplated that the anti-p3fN~'
antibodies may
be indirectly labelled as follows. The unlabelled primary antibody binds to
the target
protein. Then a secondary antibody (anti-antibody) directed against the
primary
-22-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
antibody and tagged with a detectable label binds to the antigen-primary
antibody
complex.
In a diagnostic method of the present invention, cells obtained from an
individual or a culture are processed in order to determine the extent to
which p33'NC' is
present in cells, in a specific cell type or in a body fluid. This can be
determined using
known techniques and an antibody specific for p33'NG'. Comparison of results
obtained
from cells or a body fluid being analyzed with results obtained from an
appropriate
control (e.g., cells of the same type known to have normal p33'NC' levels or
the same
body fluid obtained from an individual known to have normal p33'NG' levels) is
carried
out. Decreased p33'NG' levels are indicative of an increased probability of
abnormal
cell proliferation or oncogenesis or of the actual occurrence of abnormal
proliferation
or oncogenesis. It is contemplated that the levels of p33'"°' in
cancerous cells will be at
least 50 % less than the level of p33'N°' in non-cancerous cells, more
preferably the
levels will be less than 30 % of normal levels, most preferably p33'NC' will
not be
expressed. Increased p33'NC' levels are indicative of an increased probability
that the
cell is capable of entering the apoptotic pathway. It is contemplated that the
levels of
p33'NC' in apoptotic cells will be at least 50% greater than the level of
p33'NC' in normal
cells, more preferably at least 70%o greater.
It is contemplated that a diagnostic kit could include a solid support for
attaching
the cell or tissue to be tested and a delectably labelled anti-p33'N~'
antibody. It is further
contemplated that the anti-p33'NC' antibody may not be labelled but the kit
would
additionally contain another delectably labelled antibody capable of binding
to the anti-
p33'NC' antibody.
A hybridization probe comprising RNA, ING1 cDNA or ING1 genomic DNA
having a sequence substantially identical to Figure 3 ("ING1 polynucleotide")
may be
employed as a means for determining the sequence of the ING1 gene present in
the
genomic DNA of a given sample, or the level of ING1 mRNA expressed in cells of
such sample. Such hybridization probes will generally be delectably labelled
(eg. with
a radioactive label, a fluorescent label, biotin, etc). It is also
contemplated that the
ING1 polynucleotide may be indirectly labelled by methods known in the art. It
is
-23-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
contemplated that in situ hybridization of labelled nucleic acid to the cell
will be
employed to detect the presence of ING 1 mRNA in the cell.
A tissue sample or cell sample can be prepared by conventional means and
probed with the labelled ING1 ~ polynucleotide probe to determine the level of
expression of ING1 mRNA in the cells. The ING1 polynucleotide probe may also
be
used to determine whether the genomic DNA from the cell sample has a mutation
or
rearrangement of its ING1 gene by methods known in the art (i.e. PCR
sequencing or
restriction fragment length polymorphism analysis). The polynucleotide probe
may
also be used to determine whether the genomic DNA from the cell sample has a
mutation/deletion rearrangement of the chromosome region of 13q33-34.
The oligonucleotide probe useful in these methods may comprise at least about
nucleotides which sequence is substantially identical to the sequence of
Figure 3,
more preferably it will comprise at least about 100 nucleotides, and most
preferably it
will comprise at least 400 nucleotides. In the case of PCR sequencing it is
15 contemplated that one of the two INGI oligonucleotide primers will be
substantially
identical to one region of the sequence of Figure 3 and that the second
oligonucleotide
primer will be substantially identical to the complement of a second region of
the
sequence of Figure 3. The size of these primers is preferably from 15-25
nucleotides,
more preferably from 15-20 nucleotides. Most preferably the oligonucleotide
probes
20 and primers will be substantially identical to the coding region of the
cDNA sequence
of Figure 3. Such nucleotides can be generated synthetically by conventional
means.
Comparison of the results obtained from cells or a body fluid being analyzed
with results obtained from an appropriate control (eg. cells of the same type
known to
have abnormal or native p33'NCl or fluid from an individual known to have
normal
p33'NG') is carried out. Decreased INGl mRNA levels are indicative of an
increased
probability of abnormal cell proliferation or oncogenesis or of the actual
occurrence of
abnormal proliferation or oncogenesis. It is contemplated that the levels of
ING1
mRNA in cancerous cells will be at least 50% less than the level of INGI mRNA
in
non-cancerous cells, more preferably the levels will be less than 30% of
normal levels,
most preferably ING1 mRNA will not be expressed. Increased INGl mRNA levels
are
-24-
,,
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98I00277
indicative of an increased probability of cell apoptosis. It is contemplated
that the
levels of ING1 mRNA in cells having apoptotic potential will be at least 50%
more than
the level of ING1 mRNA in native non-senescent cells, more preferably at least
70%
more than native non-senescent cells.
The presence of a mutation/deletion in one copy of the ING1 gene in a diploid
cell is also indicative of an increased probability that abnormal cell
proliferation will
occur. The presence of mutations/deletions in both copies of the ING1 gene is
indicative of possible or actual oncogenesis.
The following examples are offered to illustrate this invention and are not
meant
to be construed in any way as limiting the scope of this invention.
EXAMPLES
The methods described as follows were used to perform the studies described
herein. In addition, the generally known methods set forth in laboratory
manuals for
molecular cloning and antibody techniques [e.g., 17,18] may advantageously be
used
by one of skill in the art to produce additional embodiments of the invention.
m le 1
Strategy for Cloning and Biological Assay
Subtractive hybridization of breast cancer cell line cDNAs with cDNA from
normal mammary epithelial cells, subcloning of subtracted cDNAs into the pLNCX
retroviral vector [7] and injection into nude mice was done essentially as
described [3]
with the modifications noted below. The cloning of full length cDNA was done
using
standard methods [17]. The strategy is shown in Figure lA.
cDNA was prepared from an non-transformed mammary epithelial cell line
(184A1) [6] and digested with the restriction enzyme ai.3A. cDNAs from the
breast
cancer cell lines MCF-7, BT-483, BT-474, Hs-578T, ZR-75, MD-MB-468, MD-MB-
435 and BT-20 (obtained from the American Type Culture Collection, Bethesda
MD)
were also digested with ~3A. Fragments of tester DNA (cDNA from normal
-25-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
epithelial cells) were Iigated to "a" adaptors. Fragments of driver DNA (cDNA
from
breast tumor cells) were ligated to "b" adaptors. Adaptors were prepared by
annealing
the synthetic oligonucleotides:
5'-GACCTGGCTCTAGAATTCACGACA-3' (SEQ ID NO: 3) with 5'-
GATCTGTCGTGAATTCTAGAGCCAGG-3' {SEQ ID NO: 4) (adaptor "a"); and
5'-GACTCGACGTTGTAACACGGCAGT-3' (SEQ ID NO: 5) with 5'-
GATCACTGCCGTGTTACAACGTCGAG-3' (SEQ ID NO: 6) (adaptor "b").
The mixture of driver DNA and tester DNA was denatured, then hybridized at
66 ° C for 18 hours. After hybridization, mixtures were treated with
Mung bean
nuclease to eliminate single-stranded adaptor-derived ends from "heterozygous"
hybrids
(hybrids containing both a and b adaptors). Resultant double-stranded
molecules were
then selectively amplified by PCR using primer "a".
The "amplicons" were then subjected to five successive rounds of
hybridization,
selective degradation and PCR amplification using 40 ~cg of driver cDNA
containing
adaptors and 200 ng, 5 ng and 5 pg of tester amplicons in respective rounds.
This
procedure allowed for a significant enrichment of sequences that were more
highl j~
expressed in the phenotypically normal epithelial cells as determined by slot
blot
hybridization.
All subtracted fractions were combined and used as a probe to screen a near-
senescent human diploid fibroblast cDNA library. Following screening, 300 cDNA
clones were isolated and their inserts were randomly fragmented {200-800 bps).
These
were then ligated with adaptors prepared by annealing two oligonucleotides
5'-AATCATCGATGGATGGATGG-3' (sense) (SEQ ID NO: 22)
5'-CCATCCATCCATCGATGATTAAA-3' (SEQ ID NO: 23) and
were amplified by PCR using the "sense" strand of the adaptor as the PCR
primer.
PCR amplified DNA was recloned into the ~I site of the retroviral plasmid
vector
pLNCX [7] with synthetic adaptors carrying initiation codons in all reading
frames.
This library of about 105 clones, enriched in tumor suppressor sequences was
then used
for the isolation of transforming genetic suppressor elements (GSEs).
-26-
CA 02284730 1999-09-23
WO 98!44102 PCTICA98I00277
After transfection of the recombinant retroviral plasmids into the packaging
line
BOSC 23 [25), retroviruses containing the isolated cDNA fragments were used to
infect
non-tumorigenic immortalized mouse mammary epithelial cells (NMuMG) which were
subsequently injected subcutaneously into nude mice. Subcloning into the
retroviral
S vector, packaging into the BOSC 23 virus-packaging cell line and assays
using nude
mice were performed as described [3].
After 45 days, two mice developed tumors from which two cDNA inserts were
recovered by PCR, one of which is subsequently shown to be expressed in the
antisense
orientation. The primers used in the PCR amplification were:
5'-CCAAGCTTTGTTTACATCGATGGATG-3' (SEQ ID NO: 7) (sense); and
5'-ATGGCGTTAACTTAAGCTAGCTTGCCAAACCTAC-3' (SEQ ID NO: 8)
(antisense). The recovered cDNA insert which was in the antisense orientation
was
digested with ~1 I and ~'ndIII and recloned back into the retroviral vector,
pLNCX, in
the same position and orientation and then tested individually in vitro.
NMuMG cells were infected with retrovirus produced from pLNCX vector
containing or not containing the ING1 insert (nucleotides 942 to 1,124 of the
ING
cDNA set forth in Figure 3). The soft agar culture was comprised of two
layers: an
underlay (DMEM, 10 % FCS, 0.6 % agar) and an overlay {DMEM, 10 % FCS, 0.3
agar). 5 X 104 cells were plated in soft agar in 10 cm plates and were left at
37 °C for 6-
7 weeks before being counted. 5 x 105 transfected NIH 3T3 cells were plated
per 10
cm dish. Transfected NIH 3T3 cells were grown in 5 % serum for 4 weeks prior
to
fixing and visualizing foci. pLNCX-S and pLNCX-aS represent sense and
antisense
orientations of the ING1 cDNA insert, respectively.
-27-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
TABLE 1
Results of the soft agar and focus forming assa~,y~
Soft Focus
agar forming
assay assay
Trial Number 1 2 3 4 mean 1 2 mean
pLNCX 0 0 0 0 0 9 13 11
(vector)
pLNCX-Ras 224 248 208 (-) 226.7 (-) (-) (-)
pLNCX-aS 42 46 41 82 52.8 18 34 26
pLNCX-S (-) (-) 0 0 0 (-) (-) (-)
(-) = not determined
These results showed that the antisense ING1 cDNA insert caused increased cell
proliferation.
Panel 1 of Figure 1B shows NMuMG cells infected with the retroviral vector
pLNCX and panel 2 of Figure 1B shows cells infected with the retroviral vector
pLNCX containing the antisense ING1 insert. The bar equals 1 mm. Panel 3 of
Figure
1B shows NIH 3T3 cells transfected with vector alone and panel 4 of Figure 1B
shows
cells transfected in parallel with pLNCX containing the antisense INGl insert.
Example 2
cDNA of ING1 and Predicted Amino Acid Seauence of p33~NC'
In order to isolate the gene corresponding to the fragment showing biological
effects, normal human fibroblast and HeLa cell cDNA libraries were screened
with the
ING1 cDNA fragment from Example 1, resulting in the isolation of 11 positive
clones.
Two clones containing the largest cDNA inserts were sequenced on both strands
using
an Applied Biosystems automated sequencer, yielding the sequence shown in
Figure 2.
In order to obtain the 5' end of the iNGl gene, 5' RACE (rapid amplification
of
cDNA ends) was used. Total cDNAs isolated following reverse transcription had
the
synthetic adaptor
-28-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98I00277
5'-GTACATATTGTCGTTAGAACGCGTAATACGCCTCACTATAGGGA-3' (SEQ
ID NO: 11) ligated to them and PCR reactions using nested primers from both
the
adaptor and the ING1 gene were used to amplify the 5' ends of all RT-generated
cDNAs. The primers used in the amplification were:
5'-CTGGATCTTCTCGTCGCC-3' (SEQ ID NO: 12) and
5'-AGTGCAGCATCGGCCGCTTC-3' (SEQ ID N0: I3) from the ING1 sequence and:
5'-GTACATATTGTCGTTAGAACGCG-3 ' (SEQ ID NO: 14) and
5'-TAATACGCCTCACTATAGGGA-3' (SEQ ID NO: 15) from the adaptor sequence.
The largest PCR products were recovered from agarose gels following
electrophoresis
and were subcloned and sequenced to generate the full-length sequence shown in
Figure
3.
The predicted coding region of INGI begins at nucleotide 16 and ends at
nucleotide 898, as shown in Figure 3, predicting a translation product of
33,350
daltons. Comparison of the sequence of p33'NC' to the available protein and
nucleotide
data bases showed no significant homology to any sequence encoding a known
protein
and very limited similarity to retinoblastoma binding protein 2 {RbBP2) [9]
and to
several zinc finger transcription factors. Regions of the p33'"c' protein that
show
homology to different members of the p16"'TS' family of cyclin-dependent
kinase
inhibitors and to retinoblastoma binding protein 2 were identified using the
Blast
program available from the National Centre for Biological Information
(address:
www.ncbi.nim.nih.gov).
F~pression of a GST-p33'N~l fusion~rotein and creation of anti-p_33 ~lvclonal
antibody
In order to generate polyclonal antibodies, a fragment of ING1 containing
nucleotides 161-1146 of Figure 3 was subcloned into the SRI-.~~ol sites of the
bacterial expression vector pGEX-4T1 (Pharmacia Biotech, Inc., Quebec, Canada)
containing the glutathione-binding portion of glutathione-S-transferase (GST).
Plasmids
were sequenced to verify that the correct reading frame was obtained and the
constructs
were electroplated into E. coli XLl-Blue. Following selection, bacterial
cultures were
-29-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
induced to express the fusion protein by the addition of O.1mM isopropyl thio-
galactopyranoside (IPTG) and fusion protein was purified by standard
glutathione-
Agarose column affinity chromatography. Eluted GST-p33'NC' fusion protein was
dialyzed and used in immunogen in female New Zealand white rabbits. After four
boosters, rabbits were bled and their serum tested for reactivity against the
fusion
protein. All animals showed reactivity and the bleeds showing the highest
titer were
chosen for subsequent use in Western blot, immunoprecipitation and
immunofluorescence protocols.
Example 4
Effect of the antisense ING1 fragment on expression of p33ING~ in tissue
culture cells
Analysis of p33"~~' protein levels in cell samples was performed by Western
blotting using anti-p33'NG' antibodies raised against the GST-p33'N~' fusion
protein.
Proteins were separated by electrophoresis in 12.5 % polyacrylamide/SDS gels,
and
electrophoretically transferred to membranes for 1 hour. The membranes were
blocked
in TBS (100 mM Tris, 150 rnM NaCI) containing 10% non-fat dried milk and 0.1%
Tween-20, for 2 hours. Incubation of the membranes with p33'NC' antiserum was
performed in TBS containing 5 % nonfat milk and 0.1 % Tween 20 (TBST) for 30
minutes. Horseradish peroxidase-conjugated goat anti-rabbit antibody was then
applied
to the filters for 1 hour in TBST. Peroxidase activity was detected using a
chemiluminescence system (Amersham Canada, Oakville Ontario Canada)
As shown in Figure 1C, NMuMG (lane 1} and ZR-75 (lane 2) cell lines were
tested. The Western blot analysis of human and mouse cell lysates revealed a
protein of
33 kD. Preincubation of antibodies with GST-p33'N~' fusion protein blocked
recognition of p33"~cl in a parallel blot using lysates from the same cells
(lanes 3 and
4).
To determine whether the level of p33ING~ was decreased in cells infected with
viral constructs containing the antisense orientation, Iysates were prepared
from control
NMuMG cells and from NMuMG cells infected with antisense ING1 (pLCNX-aS) that
had grown and formed colonies in semi-solid medium. A Western blot of lysates
from
-30-
_. ~ ,.
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
NMuMG cells infected with pLNCX vector (lane 5) or pLNCX vector containing
antisense ING1 insert (lane 6) by the method set out above is shown in Figure
1C.
Results of the Western blot analysis showed that chronic expression of
antisense
construct reduced the expression of the endogenous ING 1 gene by approximately
90 %
compared to control parental cells.
Example 5
Effects of p33'NG' Overexpression
Isolation of a DNA fragment that was capable of inducing foci and growth in
soft agar when expressed in the antisense orientation, suggested that the
cellular role of
INGI might be to negatively regulate growth. To test this idea, part of the
ING1
cDNA (basepairs 161 to 1143 of Figure 3) was cloned into the mammalian
expression
vector pBK (Stratagene, Aurora, Ontario Canada) in the sense orientation
(pINGI-S).
This construct and the plasmid vector, both of which contain neomycin
resistance genes
and a cytomegalovirus (CMV) promoter, were transfected into human breast
cancer
(Hs578T) and normal flbroblast (Hs68) cells. Following growth for 3 weeks in
medium containing 6418, plates were fixed and stained with Coomassie Brilliant
Blue
to identify surviving colonies. A large number of stable transformants were
recovered
from cells transfected with vector whereas very few colonies were visible in
plates of
cells transfected with the sense orientation of the cDNA of ING1. Figure 4A
shows
the results when human Hs578T breast cancer cells (panels 1 and 2) and normal
fibroblasts (panels 3 and 4) were transfected with the plasmids pBK (panels 1
and 3) or
pINGl-S containing the ING1 cDNA in the sense orientation (panels 2 and 4).
In order to corroborate the results of these chronic assays, we next examined
the
effect of microinjecting these constructs on the ability of normal diploid
fibroblasts to
initiate DNA synthesis.
Hs68 cells were plated on glass coverslips, deprived of serum for 12 hours,
rnicroinjected with the indicated mixture of plasmid DNA (0.1 ~,g/ml) plus
nonspecific
IgG (2 ,uglml) and were then incubated for 36 hours in complete medium
containing
BrdU. Fixed cells were identified by staining for injected IgG and for the
incorporation
-31-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
of BrdU. Microinjection, fixation and staining were done as described
previously [16].
Figure 4C shows the combined results of 5 separate experiments. Each group
represents 110-200 injected cells. As shown in Figure 4B, normal Hs68 HDFs
were
injected with solutions containing pINGl-S plus non-specific rabbit IgG
(panels 1 and
2) or with pINGl-aS containing the ING1 cDNA in the antisense orientation plus
non-
specific rabbit IgG {panels 3 and 4). Injected cells were grown in the
presence of BrdU
and were fixed and stained for the presence of co-injected IgG (panels 1 and
3) or
incorporated BrdU (panels 2 and 4). Arrows identify injected cells. Arrows in
panels
2 and 4 show that cells injected with pINGl-S failed to incorporate
bromodeoxyuridine
(BrdU, panel 2) over a 36 hour time course after injection, whereas those
injected with
pINGl-aS entered S phase (panel 4) as estimated by staining with anti-BrdU
antibodies.
Figure 4C shows the results of 5 separate experiments which indicate that
injection of
the pBK vector or of pINGl-aS constructs had no appreciable effect upon the
ability of
cells to proceed through S phase.
Similar results were obtained in larger populations of cells that were
electroporated with vector, sense and antisense construct DNAs together with a
construct encoding the CD20 surface marker. Such co-transfections allowed the
analysis by flow cytometry of DNA content in transfected cells that were
positive for
CD20 staining. Hs68 cells were co-transfected with pCMV-CD20 together with pBK-
p33'"~'-S or with pBK vector as a negative control. Cells were fixed and
stained for
CD20 expression using commercially available antibodies and with propidium
iodide 48
hours after electroporation. Cell cycle distribution was determined by flow
cytometry
using fluorescence-activated cell sorting. The percentage of the CD20+ cells
in
different phases of the cell cycle is shown for two independent experiments.
-32-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
TABLE 2
Overexnression of n33INC1 arrested cells in GO/Gl
pBK (vector) pBK-INGl-S
G 1160 S G21M G 1/G0 S G2IM
Triall 32.7 38.5 28.8 53.3 19.9 26.8
Trial2 34.5 35.9 29.6 72.8 19.7 7.5
mean 33.6 37.2 29.2 63.1 19.8 17.2
As shown in Table 2, the CD20-expressing population that was cotransfected
with pINGl-S had, on average, 63.1 % of cells in GO/G1 whereas those co-
transfected
with vector had 33.6% of cells in GO/G1 when cells were fixed and stained 48
hours
after electroporation. These results, using several independent methods,
indicate that
the overexpression of ING1 inhibits cell growth in both transient and chronic
assays,
most likely be arresting cells in the G1 phase of the cell cycle.
Examm~le 6
Alterations of INGl in cancer cell lines
Since ING1 was originally isolated by subtractive hybridization between normal
and transformed epithelial cDNAs, the ING1 gene and its expression in breast
cancer
cell lines was also examined. In Figure SA, lane 1 is MCFIOA phenotypically
normal
epithelial cell line from mammary gland; lane 2 is MDA-MB-468; lane 3 is ZR-
75; lane
4 is BT-20; lane 5 is SK-BR-3; lane 6 is MCFT; lane 7 is Hs578T and lane 8 is
BT-474
(breast cancer cell lines). Figure SB shows the coomassie-blue stained gel
corresponding to Figure SA. The expression of p33~NC1 in the cell lines was
tested by
preparing lysates of cell lines and Western blotting using anti-p33INC'
antibodies by the
method in Example 4. Although analysis of genomic fragments containing the
ING1
gene did not reveal any structural changes in breast cell lines, results from
Western blot
analyses shown in Figure 5 suggest that the p33INC1 pxotein was expressed at
considerably lower levels in some breast cancer cells compared to a
phenotypically
-33-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
normal epithelial cell line. This observation of reduced expression in the
absence of
mutation is similar to the expression of BRCA-1 reported to occur in non-
hereditary
forms of breast cancer [25] .
Normal diploid control cell strains and neuroblastoma cell lines were analyzed
by Western Blot analysis in a manner similar to that set forth for the breast
cancer cell
lines. Figure 6A illustrates the Western blotting results of IMR-5 (lane 1) SK-
L-C6
(lane 2); SK-N-SH (lane 3) all neuroblastoma cell lines, and W138 (lane 4) a
normal
diploid lung fibroblast cell line. Normal diploid fibroblast cells expressed
low levels of
p33'NCl while immortalized neuroblastoma cells expressed considerably higher
levels
and in the case of the SK-N-SH neuroblastoma line a truncated protein was
observed.
To investigate the nature of the changes} responsible for truncating p33'NC'
in
this neuroblastoma cell line, two complementary approaches were taken.
Southern blot
analysis of DNA, from neuroblastomas and from normal fibroblasts that was
digested
with different restriction endonucleases and probed with a ING1 nucleic acid
probe,
clearly indicated that p33'NC' was rearranged in the neuroblastoma cell line.
Human
genomic DNAs were digested with HindIII, I~r I or P~~l, electrophoresed
through a
0.7 % agarose gel, transferred to a nitrocellulose membrane and hybridized
with [32P]-
labelled p33'NCl cDNA. Hybridization was performed using standard procedures
[17].
Lanes 1-6 show the results for neuroblastoma SK-N-SH (2, 4 and 6) and fox
normal
diploid W138 cells (1, 3 and 5}. Patterns such as those shown by W138 cells
were also
seen in other normal diploid cell strains.
To confirm that changes in the p33'NCl gene had occurred in the neuroblastoma
cell line and to determine their nature by an independent method, reverse
transcription
polymerase chain reaction (RT-PCR) with RNA from SK-N-SH neuroblastoma cell
line
and from a phenotypically normal epithelial cell line (MCF-10) was performed
as
described [20] . Neuroblastoma cDNA was amplified with PCR primers specific
for the
p33 gene (direct(d) and reverse (r) primers). These are numbered and shown
underlined in Figure 3 and the PCR products were compared with PCR fragments
generated in parallel from control cell cDNA. Figure 6C, lanes 1 (primers ld-
4r), 3
(ld-2r), 5 (2d-4r) and 7 (2d-3r) show the results for W138, and lanes 2 (ld-
4r), 4 (ld-
-34-
,.
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
2r), 6 (2d-4r) and 8 (2d-3r) show the results for the neuroblastoma cell line.
Primers
were
ld: GTAGCGCAGTCTGACAAGCC (nucleotides 474-494 of SEQ ID NO: 9)
2d: TGGTTCCACTTCTCGTGCGT (763-782 of SEQ ID NO: 9)
2r: ACGCACGAGAAGTGGAACCA (SEQ ID NO: 16)
3r: TTTGGATTTCTCCAGGGCTT (SEQ ID NO: 17) and
4r: TACCTGTTGTAAGCCCTCTC (SEQ ID NO: 18). M shows a 1 kb ladder
molecule weight marker. All primer pairs gave similar results in both cell
lines except
for those using primers beyond nucleotide 858. For example, using primers 3r
and 4r
give no PCR product using neuroblastoma cDNA which is consistent with data
indicating that a deletion or a rearrangement had occurred within the p3fNC'
gene.
These experiments corroborate the idea that the 3' region of the p3fNC' gene
was
mutated in this neuroblastoma.
Normal diploid control cell strains and brain cancer cell lines were analyzed
by
RT-PCR analysis. Reverse transcription with total RNA from each of the cell
lines was
performed by the method set out in Example 9. The same primer pairs set forth
in
Example 9 were used. Figure 7 illustrates the RT-PCR results of glioblastoma
(lanes
GB1-GB4) astrocytoma (lanes AS1-AS3) and meningioma (MN1-MN3) as compared to
a control cell line (Cl-C2). The INGl mRNA was expressed at considerably lower
levels, or not expressed at all, in the glioblastomas, astrocytomas and
meningiomas as
compared to the normal cell line.
m le 7
Nuclear Localization of 33p 'NC'
The experiments described below were performed with a rabbit polyclonal
antibody (ap33) which was raised against a bacterially expressed
glutathione-S-transferase (GST)-p33'NC' fusion protein and which reacted with
a 33 kDa
protein in human and mouse cell lysates as prepared by the method in Example
3.
In the first series of experiments, we determined the location of p3fNC' in
fibrobiasts by examining the staining pattern of anti-p33 antibody in
fibroblasts by
-35-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
indirect immunofluorescence. For indirect immunofluorescence normal human
diploid
fibroblasts (Hs68 cells) were grown on glass coverslips for 48 hours at
37°C to 60%
confluence. The cells were fixed in 3.7% formaldehyde, washed in 0.5% Triton X-
100
and in 0.05 % Tween 20 for 10 minutes each at room temperature. Formaldehyde
and
detergents were diluted in phosphate buffered saline (PBS) pH 7.5. The cells
were
incubated with a 1:100 dilution of rabbit p33'NC' antiserum for 30 min, washed
in PBS
with 0.05 % Tween, incubated with goat anti-rabbit IgG-biotin antibody and
then with
streptavidin conjugated Texas Red [16]. Samples were examined with a Zeiss
Axiophot
fluorescence microscope and images were photographed on Kodak TMAX 100 film.
Staining with polyclonal rabbit antibody alone was observed both in nuclear
and
cytoplasmic compartments. Similar results were obtained with anti-p33
antibodies
which were preincubated with 5 fcg of GST protein indicating that the signal
was
specific for p33'NG'. When the anti-p33 serum was preincubated with 5 ~.g of
GSTp33
fusion protein, nuclear staining was lost completely but cytoplasmic staining
remained,
indicating that the vast majority of p33'NG' staining was nuclear.
To confirm the nuclear localization of the 33 kDa protein. The pINGl-s
construct of Example 5 was microinjected into normal Hs68 fibroblast cells
which were
fixed and stained with anti-p33-antibody 24 hours after injection. Strong
staining was
clearly localized to the nucleus. These results corroborate staining patterns
in uninfected
cells and show that p33'NG' is localized primarily, and possibly exclusively,
throughout
the nucleoplasm.
Exa
Chromosomal Localization of the INGI Vie, ne
To identify the chromosomal localization of the INGI gene, a genomic 18-kb
DNA insert containing the gene was labelled with digoxygenin-dUTP and
hybridized to
synchronized human lymphocyte metaphase spreads.
A genomic clone of the INGl gene was isolated from a lambda FIX II placental
human genomic library (Stratagene, Aurora, Ontario, Canada) with nucleotides
161 to
-36-
CA 02284730 1999-09-23
WO 98144102 PCT/CA98/00277
1143 of the ING1 sequence of Figure 3 using high stringency (65 °C 0.
1X SSC, 0.1 %
SDS) screening. The identity of the clone was confirmed by partial sequence
analysis.
FISH was performed using established methods on methotrexate/thymidine
synchronized, phytohemagglutinin stimulated, normal peripheral blood
lymphocytes
[21]. Approximately 50 metaphase spreads were examined for probe localization.
Suppression for 30 minutes with a mixture of sonicated human DNA {Sigma
Diagnostics, Mississauga, Ontario, Canada) and cot! DNA (Gibco/BRL,
Burlington,
Ontario, Canada) was required to reduce the background. The stained slides
were
counterstained with DAPI and actinomycin D (for a DA-DAPI banding pattern) and
were mounted in antifade medium and visualized utilizing a Zeiss Axioplan 2
microscope. Images of representative mitoses were captured using a cooled CCD
camera (Photometrics PXL 1400). Digital alignment of the images from each
fluor was
done after registration calibration through a triple bandpass filter
(F1TCITexas
RedIDAPI) to minimize registration error, utilizing commercial software
(Electronic
Photography v1.3, Biological Detection Inc., Pittsburgh PA).
The results clearly showed localization of the probe to chromosomal region
13q33-34. At least one specific probe signal was present in more than 90% of
the
mitoses examined. Approximately 80% of the cells had two chromatids of a
single
chromosome. Approximately, 40% showed labelling of both chromatids of both
chromosomes. More than 90% of the signals were localized to a single band. In
addition, cohybridization of p33INC' with a commercial 13121 alpha-satellite
probe
(Oncor, Gaithersberg MD) showed hybridization to the same chromosome.
The INGI gene has been localized to an area near known sites of genomic
alteration
in several human cancers: primary gastric cancer [22], hematologic neoplasms
[23] and
head and neck squamous cell carcinomas [24].
Expression levels of ING1 in ~g and senescent fibroblasts.
The normal human diploid fibroblast cell strain Hs68 (ATCC CRL#1635) and a
phenotypically normal mouse epithelial cell line from mammary gland {NMuNG)
were
-37-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine
serum. Hs68 cells were used at 30 ("young"), 70 ("pre-aged") and 80 ("old")
mean
population doublings (MPDs) for expression and life span experiments. After
retroviral
infection, the human diploid fibroblast cells (HDFs) were repeatedly passaged
in 10 cm
plates, splitting at a ratio of 1:2 when confluent.
For infection of fibroblasts, the retroviral vector (pLNCX) was used. The
highly
efficient ecotropic (BOSC23) and amphotropic (CAKB) packaging cell lines were
used
[2b]. pLNCX-aS or pLNCX alone, were transfected into the BOSC23 virus-
packaging
cell line. Ecotropic and amphotropic packaging lines, and the retroviral
vector were
kindly provided by Dr. A. Gudkov (University of Illinois at Chicago). The
amphotropic
cells were infected by viruses from the BOSC23 supernatant. Fibroblasts were
plated
at 105 cells per 10 cm plate and infected with undiluted viral supernatant
from
amphotropic producer cells. Infection efficiencies ranged from 85 to 95 % in
individual
trials .
Since the activity or expression levels of several tumor suppressors increase
in
senescent cells, the levels of ING1 expression in low and high passage cells
were
checked. All experiments were performed on the Hs68 strain of primary normal
human
diploid fibroblasts. Senescent cells were obtained by passaging early-passage
("young")
fibroblasts continuously to a point at which one population doubling required
from 2 - 3
weeks to complete compared to 24 hours, on average, for young HDFs. Hs68s at
80
MPDs exhibited characteristics typical of senescent cells, including an
inability to
respond to growth factors and altered morphology including increased size and
decreased saturation density.
To study the level of expression of ING1 mRNA, RT-PCR using total RNA
isolated from young and old cells was performed (Figure 8A). The relative
levels of
ING1 transcript were compared to the internal control gene glyceraldehyde-3-
phosphate
dehydrogenase (GAPDH) using PCR primers specific for the p33 and GAPDH genes.
INGI and GAPDH were amplified in the same reaction tube using the "primer
dropping" approach [27] which internally controls for efficiency of reverse
transcription and amplification by PCR.
-38-
CA 02284730 1999-09-23
WO 98144102 PCT/CA98/00277
Reverse transcription (RT) with 1 ~,g of total RNA from young and old Hs68
cells was performed using SO U of RNasin (Pharmacia Biotech, Inc., Quebec
Canada)
and 200 U of MMLV reverse transcriptase for 50 min. at 42°C in 20 ~,1
reaction
volumes. Two ~,1 of each RT reaction was amplified using 2 U of Taq
polymerase. The
two sets of primer pairs for the INGl gene and for the GAPDH gene that were
used,
were:
5'-GAAGCGGCGGATGCTGCACT-3' (SEQ ID NO: 19); and
S'-ACGCACGAGAAGTGGAACCA-3'(SEQ ID NO: 16) for the ING1 gene and
5' CGGAGTCAACGGATTTGGTCGTAT -3'(SEQ ID NO: 20); and
5' - AGCCTTCTCCATGGTGGTGAAGAC 3' (SEQ ID NO: 21 ) for the GAPDH gene.
Thirty two PCR cycles for ING1 and twenty two PCR cycles for GAPDH were
performed using standard conditions [17]. Primers for GAPDH were added to PCR
tubes at the end of the 10th cycle [27) .
The levels of ING1 mRNA were estimated by scanning densitometry to be
approximately ten fold higher in senescent fibroblasts compared to young
fibroblasts. In
order to see if increased mRNA levels resulted in increased protein levels,
Western
blotting experiments were performed with a rabbit polyclonal antibody that was
raised
against a bacterially expressed glutathione-S-transferase (GST)-p33'NCl fusion
protein
and that reacted with a 33 kDa protein in human and mouse cell lysates.
Hs68 and NMuMG cells were harvested and 20 ~,g of total protein was used in
each lane. Proteins were separated by electrophoresis in 12.5 %
polyacrylamide/SDS
gels, and transferred to membranes for 1 hour using an electroblotter. The
membranes
were blocked in TBS(100 n~IVI Tris, 150 mM NaCI} containing 10% nonfat dried
milk
and 0.1 % Tween-20 for 2 hours. Incubation of the membranes with p33INC~
antiserum
was performed in TBS containing 5 % nonfat milk and 0.1 % Tween-20 for 1 hour
and
then membranes were washed with TBST solution for 30 minutes. Horseradish
peroxidase-conjugated goat anti-rabbit antibody was then applied to the
filters for 1
hour in TBST. Peroxidase activity was detected using ECL (Amersham Canada,
Oakville, Ontario, Canada) and relative band intensities were determined by
scanning
densitometry.
-39-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
As shown in Figure 8B, the level of p33'NC' protein increases approximately 8
fold when cells approach the end of their in vitro replicative lifespan,
consistent with
results obtained using RT-PCR.
Since ING1 appears to arrest cells in G1 when overexpressed and senescent
cells
are primarily arrested in the G1 phase of the cell cycle [28], the level of
p33'NC~ protein
was tested during the cell cycle..Quiescent, proliferation-competent NMuMG
cells were
serum stimulated, lysates were prepared at different times after serum
addition, and
samples were analyzed by Western blotting with anti-p33 antibodies by the
method set
forth above. The level of p33INC~ was found to decrease as cells exited from
G0, to
increase during late G1 and to reach a maximum in S phase. This was followed
by a
decrease in G2 of the cell cycle (Figure 9B). CDK2 expression was used as a
control
for cell cycle progression and changed as reported previously (Figure 9A)
[29]. Figure
9C shows the results of DNA content analysis by fluorescence-activated cell
sorting
(FACS) in parallel cultures indicating that cells enter S phase at 16 hours
under these
experimental conditions. These results indicate that ING1 is regulated
following
mitogen addition to quiescent cells, with expression reaching a peak during
DNA
synthesis.
To determine the effects of reducing the levels of ING1 mRNA on the
replicative lifespan of HDFs, cells were infected with a pLNCX-aS (the
construct
carrying a 182 by fragment in the antisense orientation and representing
nucleotides 942
to 1,124 of the INGI cDNA (Figure 3)). This antisense fragment effectively
inhibits
translation of ING1 mRNA as shown previously where chronic expression of the
antisense construct resulted in 90 % inhibition of the expression of the
endogenous
p33I'~G' protein in cells.
Amphotropic and ecotropic packaging cells that were used for infection are
capable
of producing retroviruses with titers higher than 10~ per ml upon transient
transfection
which allows delivery of the retroviral construct to HDFs with efficiencies of
approximately 90 % as monitored by expression from a retroviral -p-
galactosidase
construct.
-40-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
"Young" HDFs at 30 MPDs were "pre-aged" by continuous subculturing until
reaching 70 MPDs. Hs68 cells at 70 mean population doublings (MPDs) were
infected
with the retroviral vector pLNCX as a control or with pLNCX-aS and were
subcultured
in parallel using subculturing ratios of 1:2. Infected cells were propagated
an additional
10 MPDs after which 105 control pLCNX and 105 pLCNX-aS cells at 80 MPD were
split into twelve 10 cm plates and cultivated for two months, with weekly
refeeding
using complete medium. Some of the cells infected with retrovirus alone were
observed
to divide once during this time, while cells containing the ING1-aS fragment
continued
to grow and created visible colonies.
To confirm the effect of the antisense fragment of ING1 in cells, indirect
immunofluorescence with a rabbit polycional antibody that was raised against
p33'NC'
was performed. Senescent vector-infected fibroblasts and fibroblasts from
colonies
resulting from ING1-aS retrovirus infection were grown on glass coverslips for
48
hours at 37 ° C to 60 % confluence. Then the cells were fixed in 3 .7 %
formaldehyde,
washed in 0.5 % Triton X100 and in 0.05 % Tween 20 in PBS solution for 10
minutes
each at room temperature. The cells were incubated with a 1:100 dilution of
rabbit
p331'~c' antiserum for 30 min, washed in PBS with 0.05 % Tween, incubated with
goat
anti-rabbit IgG-biotin antibody and then with streptavidin conjugated Texas
Red.
Samples were examined with a Zeiss Axiophot fluorescence microscope and images
were photographed on Kodak TMAX 400 film.
Staining with anti-p33 antibody was observed in the nuclear compartment of
senescent cells containing control virus but not in cells obtained from
colonies that had
received antisense p33 retrovirus. These results corroborate the previous
observations
that p33'NG' is a nuclear protein and confirmed that the levels of p33iNC1
protein
decrease in cells from colonies resulting from INGI-aS retrovirus infection.
Similar
results were seen in cells from 3 individual colonies and from 20 independent
senescent
cells containing control retrovirus.
To estimate the efficiency with which down regulation of the ING1 gene by
infection with PLCNX-aS was able to extend the proliferative lifespan of
normal
fibroblasts, the number of cells in each colony was counted. Results of these
-41-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
calculations are shown in Figure 10 in which colonies were divided into 4
groups
depending upon the number of cells in the colony. Most colonies contained 100-
159
cells, therefore if cells divided in an arithmetic progression (2,4,8...n}
this class
corresponds to approximately 7 additional MPDs (2'=128}. Colonies in the
largest
category (220-280) correspond to 8 cell doublings (28=25b). Similar results
were
obtained in two separate trials and strongly indicate that down regulation of
p33'NC'
protein is sufficient to extend the proliferative lifespan of normal
fibroblasts by
approximately 10 % , as previously reported for the p53 tumor suppressor gene
[30] .
EXAMPLE 10
Indications that ING1 may be involved in the modulation of apoptosis first
came
from the conspicuous presence of ING1 in regressing tail and absence from
growing
hindlimbs of metamorphosing Xenopus tadpoles and second, the serum deprivation-
induced elevation of ING1 levels in P19 teratocarcinoma cells that correlated
with the
induction of apoptosis within 48-72 hours (Figure 11A; 35).
In order to establish the effect of ING1 expression on apoptosis, we infected
P19 cells with sense (S), antisense (aS) and retroviral vector alone (V) and
examined
cell viability 72 hours after serum withdrawal.
P19 mouse teratocarcinoma cells were grown in alpha MEM medium with 10%
fetal calf serum (FCS: GIBCO/BRL, Burlington, Ontario, CANADA). For serum
starvation experiments, the cells were washed twice in PBS {phosphate buffered
saline;
137 mM NaCI, 2.7 mM KCI, 10.1 mM NazHP04, 1.8 mM KH2P04, pH 7.4} and
serum-free alpha MEM (GIBCO/BRL) was added.
Retroviral infection of target cells was used to introduce ING1 expression
constructs. Because the ING1 protein appears to block entry into S-phase of
the cell
cycle the use of standard drug resistance selection methods is precluded.
Retroviral
infection also has a higher efficiency than standard calcium phosphate
transfection
procedures. The retroviral vector, pLNCX (7), containing sense {S), antisense
(aS;
nucleotides 942 to 1124 of the ING1 cDNA) or vector alone were transfected
into a
-42-
CA 02284730 1999-09-23
WO 98/44102 PCTlCA98/00277
highly-efficient BOSC23 ecotropic virus-packaging cell line. The BOSC23
supernatant
was then used to infect ecotropic producer cell lines. The target P19 cells
were plated
at 104 cells per 10 cm plate and infected with undiluted viral supernatant
from ecotropic
producer cells. Infection efficiency was determined to be about 60% with the
P19 cells
because these cells were prone to clumping.
Cell viability was assessed using a trypan blue dye exclusion assay. Cell
suspensions were mixed 4:1 with 0.5% trypan blue:saline solution (GIBCO/BRL).
The
cells were incubated at room temperature for 5 minutes and counted with a
hemacytometer .
As shown in Figure 11B, the antisense ING1 construct conferred a moderate
level of protection against death (70%o viable) compared to that conferred by
a retroviral
construct constitutively expressing the bcl-2 gene (84% viable). Conversely,
P19 cells
expressing the sense ING1 construct exhibited greater death (44% viable) than
the
vector only (54 % viable) or antisense bcl-2 controls {56 % viable),
suggesting that ING1
confers cellular susceptibility to death induced by serum starvation. These
numbers
represent a minimal estimate of the effects of ING1 and bcl-2 due to the
modest
transfection efficiency of P19 cells as outlined above.
Since the infection efficiency was low for P19 cells, we turned to another
model
system of apoptosis developed in our laboratory, that of rodent fibroblasts
(Rat 1 and
NIH 3T3) containing a tetracycline-controlled human c-myc gene (tet-myc
cells).
Because these cells form a monolayer, transfection efficiencies of greater
than 90 % are
obtainable.
The human c-myc gene was inserted into a plasmid containing a minimal
cytomegalovirus promoter preceded by several binding sites for the
tetracycline
transcriptional protein (TA) (36). This construct (hTIC-myc) was stably
transfected
into NIH3T3 or Ratl cells previously selected for the stable expression of the
TA
protein that is capable of binding to, and activating transcription of the myc
gene on the
hTIC-myc construct, in the absence of tetracycline. In the presence of
tetracycline, the
TA protein is blocked from binding and very little, if any transcription is
seen from the
c-myc expression construct. If tetracycline is washed from the cells,
expression of the
-43-
CA 02284730 1999-09-23
WO 98!44102 PCT/CA98100277
c-myc gene increases and expression can be regulated by altering the level of
tetracycline to give up to 100 fold higher expression levels. Transformed cell
lines that
showed low background expression of c-myc and a high degree of inducibility
were
chosen for future studies.
The transformed rodent fibroblasts, either NIH 3T3- or rat 1-derived cells
containing the tetracycline-controlled human c-myc gene (tet-myc cells) were
maintained in high glucose Dulbecco's MEM (GIBCO/BRL) supplemented with 10 %
FCS and 2 ~,g/ml tetracycline {Sigma, St. Louis, MO) to repress premature
human
c-myc gene repression. Removal of tetracycline from the medium results in the
rapid
accumulation of human c-myc protein (Figure 12A) and subsequent apoptosis. The
advantage of such a system is that control and experimental cells possess an
identical
genetic background and the potential for cellular adaptation to constitutive c-
myc
overexpression is minimized.
The tet-myc NIH3T3 or Ratl cells were plated at 104 cells per 10 cm plate and
infected with undiluted viral supernatant from ecotropic BOSC23 cells
described above.
Infection efficiency was determined to be greater than 90% for NIH 3T3 tet-myc
cells
using a retroviral ~3-galactosidase construct.
Tet-myc cells containing the retroviral constructs were seeded at a density of
approximately 104 cells/cm2 on glass coverslips and grown at 37°C for
48 hours before
processing as described (lb).
The cells were subsequently treated to induce c-myc expression and apoptosis.
Briefly, this involved washing out the tetracycline inhibitor, elevating c-myc
expression
by up to 100 fold, and then transferring cells to medium without serum where
apoptosis
rapidly ensued. Nuclear proteins were isolated at certain timepoints and
immunoblotted.
DNA laddering was assessed using the method of Smith et al {32). Tet-myc cells
were plated at equal densities as described above. At 72 hours after exposure
to 0.1 %
FCS, DNA was isolated from the floating cells on a per plate basis. Equal
volumes of
lysate were run on a 2 % agarose gel and stained with ethidium bromide.
-44-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
When c-myc was induced in the NIH 3T3 tet-myc cells by tetracycline
withdrawal and the cells were then serum starved for 72 hours, they showed a
viability
of 65 % compared with controls where c-myc was not induced (Figure 13A).
Apoptosis
was minimized in cells coexpressing the antisense INGl construct with only 5 %
of cells
S dying upon serum withdrawal. Cells expressing INGI protein alone showed 70%
viability after serum starvation and apoptosis. This was magnified when both
the ING1
and the c-myc gene were coexpressed (40% viability). These cells exhibited
several
hallmarks of apoptosis including shrinkage, loss of substrate adhesion and
chromatin
condensation. In addition, internucleosomal DNA fragmentation was greatly
enhanced
in tet-myc cells expressing p33'"~t compared to vector only, antisense ING1 or
bcl-2-expressing tetmyc cells (Figure 13B).
To confirm that ING1 enhances apoptosis during serum starvation, we
microinjected p33'NC' or a CMV-ING1 expression constructs into rat tet-myc
fibroblasts
and counted the number of remaining injected cells at various times after
serum
deprivation. Rat tet-myc cells were seeded on coverslips as described above
and
injected with about 25 p,g/ml of GST-ING1 protein or GST protein, or with 25
~,g/ml of
CMV-driven ING1 expression construct or vector alone (31}. After allowing the
cells
to recover for 4-6 hours, the coverslips were washed twice with PBS and the
cells were
serum starved. Injected cells were identified through coinjection and
subsequent
immunostaining of a coinjected nonspecific antibody. Because the history of
each
injected cell could be followed, a more dramatic effect of ING1 expression
compared to
the previous experimental approaches was apparent (45 % viable at 24h), with a
further
decrease in surviving cells when c-myc and ING1 were coexpressed (9% viable at
24h).
The results of the microinjection of the GST-INGI protein are shown in Figure
13A.
Similar results were obtained with microinjection of the CMV-ING1 construct.
These
results support the data obtained with both the P19 and transformed NIH3T3 tet-
myc
cells and show that p33'NCl is involved in regulation of apoptosis, in a
manner that is
synergistic with the action of c-myc.
It therefore appears that in at least three independent cell lines, ING1
expression
confers an increased susceptibility to death upon serum starvation.
Conversely,
-45-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
decreasing endogenous INGl levels afforded some protection against cell death.
INGI
markedly influences the outcome of c-myc-induced apoptosis and may, therefore,
participate in guiding the response pathway whereby c-myc overexpression
activates
either apoptosis or tumorigenesis.
Modification of the above-described modes of carrying out various embodiments
of this invention will be apparent to those skilled in the art following the
teachings of
this invention as set forth herein. The examples described above are not
limiting, but
are merely exemplary of this invention, the scope of which is defined by the
following
claims .
REFERENCES
The following references are cited in the application as numbers in brackets
([])
at the relevant portion of the application.
1. Levine, A.J., "The Tumor Suppressor Genes", Annu. Rev. Biochem.
62:623-651 (1993).
2. Hunter, T. et al., "Cyclins and Cancer II: Cyclin D and CDK Inhibitors
Come of Age", J. Cell 79:573-582 (1994).
3. Gudkov, A.V. et al., "Isolation of genetic suppressor elements, inducing
resistance to topoisomerase II-interactive cytotoxic drugs, from human
topoisomerase II
cDNA", Natl. Acad. Sc. USA 90:3231-3235 (1993).
4. Straus, D. et al. , "Genomic subtraction for cloning DNA corresponding to
deletion mutations", Proc. Natl. Acad. Sc. USA 87:1889-1893 (1990}.
5. Lisitsyn, N. et al., "Cloning the Differences Between Two Complex
Genomes", Science 259:946-951 (1993).
6. Yaswen, P. et al. , "Down-regulation of a calmodulin-related gene during
transformation of human mammary epithelial cells", Proc. Natl. Acad. Sc. USA
87:7360-7364 (1990).
7. Miller, A.D. et al., "Improved Retroviral Vectors for Gene Transfer and
Expression", Biotechniques 7:980-986 (1989).
-46-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
8. Serrano, M. et al., "A new regulatory motif in cell-cycle control causing
specific inhibition of cyclin D/CDK4", Nature 366:704-707 (1993).
9. Defeo-Jones, D. , "Cloning of cDNAs for cellular proteins that bind to the
retinoblastoma gene product", Nature 352:251-254 (1991).
10. Aharon, T. et al. , "Selective Destabilization of Short-Lived mRNAs with
the Granulocyte-Macrophage Colony-Stimulating Factor AU-Rich 3' Noncoding
Region
is Mediated by a Cotranslational Mechanism", Mol. Cell. Biol. 13:1971-1980
(1993).
11. Guan, K. et al., "Growth suppression by p18, a p16'NK4/MTS1 and
pl4I"K4s~MTS2_related CDK6 inhibitor, correlates with wild-type pRb function",
Genes &
Dev. 8:2939-2952 (1994).
12. Harper, J.W. et al., "The p21 Cdk-Interacting Protein Cipl is a Potent
Inhibitor of Gl Cyclin-Dependent Kinases", Cell 75:805-816 (1993}.
13. El-Deiry, W.S. et al., "WAF1, a Potential Mediator of p53 Tumor
Suppression", Cel175:817-825 (1993).
14. Kamb, A. et al., "A Cell Cycle Regulator Potentially Involved in Genesis
of Many Tumor Types", Science 264:436-440 (1994).
I5. Nobori, T. et al., "Deletions of the cyclin-dependent kinase-4 inhibitor
gene in multiple human cancers", Nature 368:753-756 (1994).
16. Riabowol, K. et al., "The cdc2 Kinase Is a Nuclear Protein That Is
Essential for Mitosis in Mammalian Cells", Cell 57:393-401 (1989).
17. Sambrook, J. et al., "Molecular Cloning" (2nd.Ed.), A Laboratory Manual,
Cold Spring Harbor Laboratory Press (1989).
18. Harlow, E. et al., "Antibodies", A Laboratory Manual, Cold Spring
Harbor Laboratory (1988).
19. Yang, Y. et al., "An approach for treating the hepatobiliary disease of
cystic fibrosis by somatic gene transfer" Proc. Nat'l. Acad. Sci. USA 90:4601-
4605
(1993). .
20. Atadja, P. et al., "Increased activity of p53 in senescing fibroblasts"
Proc.
Nat'l. Acad. Sci. USA 92:8348-8352 (1995).
-47-
CA 02284730 1999-09-23
WO 98/44102 PCT/CA98/00277
21. . Demetrick, D.J. "Fluorescence in situ hybridization and human cell cycle
genes" In the Cell Cycle - Materials and Methods M. Pagano (ed.) Springer
Verlag
Press, 29-45 {1995).
22. Motomura et al., "Loss of alleles at loci on chromosome 13 in human
primary gastric cancers" Genomics 2, 180-184 (1988).
23. Mitelman et al. , "Report of the committee on chromosome changes in
neoplasia" Cytogenet Cell Genet 55:358-386 (1990).
24. Maestro et al., "Chromosome 13q deletion mapping in head and neck
squamous cell carcinomas: identification of two distinct regions of
preferential loss"
Cancer Research 56:1146-1150 (1996).
25. Thompson, M.E. et al., "Decreased expression of BRCA-1 accelerates
growth and is often present during sporadic breast cancer progression" Nature
Genetics
9:444-450 (1995).
26. Pear, W . S . et al . , "Production of high titer helper-free retroviruses
by
transient transfection" Proc. Natl. Acad. Sci. 90:8392-8396 {1993).
27. along, H. et al., "Monitoring mRNA expression by polymerase chain
reaction: the "primer-dropping" method" Anal. Biochem. 223:251-258 (1994).
28. Schneider E.L and Fowlkes, B.J., "Measurement of a DNA content and
cell volume in senescent human fibroblasts utilizing flow miltiparameter
single cell
analysis" Exp. Cell. Res. 98:298-302 (1976).
29. Tsai, L.H. et al., "The cdk2 kinase is required for the G1- to -S
transition in mammalian cells" Oncogene 8:1593-1602 (1993).
30. Bond, et al., "Escape from senescence in human diploid fibroblasts
induced directly by mutant p53" Oncogene 9:1885-1889 (1994).
31. Graessmann and Graessmann, "Microinjection of tissue culture cells",
Methods in Enzymology 101:482-492 (1983)
32. Smith et aI. , "antibodies to CD3/T-cell receptor complex induce death by
apoptosis in immature T cells in thymic cultures", Nature 337:181-184 (1989)
33. Meikrantz and Schlegel "Apoptosis and the cell cycle" J. Cell. Biochem
58:160-174 (1995)
-48-
CA 02284730 1999-09-23
WO 98144102 PCTICA98/00277
34. Lee and Bernstein "Apoptosis, cancer and the p53 tumor suppressor
gene" Cancer Met Rev. 14:149-161 (1995)
35. Galli & Fratelli, "activation of apoptosis by serum deprivation in a
teratocarcinoma cell line: inhibition by Lacetycarnitine", Exp. Cell Res.
204:54-60
( 1993)
36. Gossen and Bujard, "Tight control of gene expression in mammalian
cells by tetracycline-responsive promoters", Proc. of Natl Acad. Sci.
89(12):5547-5551
(1992)
37. Garkavtsev et aI. "Suppression of the novel growth inhibitor INGl
promotes neoplastic transformation" Nature Gen. 14:415-420 (1996)
The disclosure of the above publications, patents and patent applications are
herein incorporated by reference in their entirety to the same extent as if
the language
of each individual publication, patent and patent application were
specifically and
individually included herein.
-49-