Note: Descriptions are shown in the official language in which they were submitted.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-1-
A PORIN GENE FROM Campylobacter Zeiuni
RELATED PRODUCTS AND USES THEREOF
TECHNICAL FIELD
This invention relates to a porin gene from
Campylobacter jejuni, to related products and to the uses
BACKGROUND ART
In the following discussion, the numbers shown in
brackets refer to the articles identified in the
"REFERENCES" section provided later in this specification.
Campylobacter jejuni is recognized as a cause of
bacterial-induced diarrhea in both developing and
underdeveloped countries (39, 41). Active surveys
conducted in the United States have estimated the number
of cases of campylobacteriosis to be 2.5 million per year,
making it a multi-million dollar disease (39). Symptoms
caused by C. jejuni can range from watery to bloody
diarrhea (28, 39). In most cases campylobacteriosis is a
self-limiting disease but in the more severe cases,
antibiotic intervention with macrolids or fluoroquinolones
or rehydration therepy is necessary to eradicate the
infection (28).
The organism has been reported to possess several
virulence factors that may be responsible for disease (11,
26, 40) but little is known regarding the genetic
processes that surround their production. One virulence
factor, a toxin, has been cloned and sequenced
successfully and this is the cytolethal distending toxin
(CLDT) of C. jejuni (34). The CLDT operon was found to
contain three open reading frames (ORF) designated cdtA,
cdtB and cdtC and these correspond with 30.1 kDa, 28.9 kDa
and 21.1 kDa proteins respectively (34}. Escherichia coli
minicell experiments have shown that all three genes are
necessary for the production of the active toxin.
Screening of multiple strains of Campylobacter sp. by
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-2-
polymerase chain reaction (PCR) for the presence of the
cdtB gene and HeLa cell assays for the expression of CLDT
revealed that all the strains tested carried the gene and
tested positive in the cell culture assay (34). Johnson
and Lior (19) originally reported that 410 of 718
isolates of Campylobacter sp. screened for the production
of CLDT were positive; however, isolates screened for the
cdtB gene suggested that this percentage may be higher
than was previously reported (19, 34). Genetic studies
involving the production of an enterotoxin by C. j ejuni
revealed DNA similarities between a postulated GM1 binding
site on the toxB gene from Vibrio cholerae and the eltB
gene from E. coli. Despit this an enterotoxin gene has
not been successfully cloned and sequenced from C. jejuni
(5) .
C. jejuni has a genome estimated to be 1.7 Mb in size
as determined by pulsed field gel electrophoresis (PFGE)
(43) while the a percentage of guanidine+cytosine ranged
between 29-36 mol o (42, 43). The organism has the
capability to transform free DNA as well as to be
transduced by bacteriophages and to transfer DNA between
strains by conjugation (42, 44). These genetic exchange
mechanisms could facilitate the spread of antibiotic
resistant determinants between strains (44) and may result
in the aquistion of toxin production by one strain from
anther (32). A number of genes have been sequenced from
C. jejuni (42); however, the majority of these take the
form of highly conserved or "housekeeping" genes such as
serine hydroxylmethyl-transferase (glyA) (7) and
(-glutamyl phosphate reductase gene (proA) (22) . In
addition, genes such as flaA and flag encoding flagella
proteins (15) and peb4A, an antigenic surface protein (4)
have been cloned and sequenced. Difficulties such as gene
instability and failure to express functional products
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-3-
have been encountered and this has made genetic analysis
of C. jejuni problematic (34, 42) .
' Several porin genes from various bacterial species
have been purified, cloned and sequenced (6, 14, 16,17,
27). These porin usually exist as a single monomeric
protein (16, 29) or homotrimers (3, 6) and all show a
variation in their relative pores sizes (3, 17). Porins
are functional components of the outer membrane of
bacteria and they allow for the exchange of solutes as
well as permit the excretion of waste products to occur.
One characterized porin from E. coli has been found to
occur at a frequency of 105 on each bacterial cell (27, 46)
making it the most abundant molecule present on the cell
surface (36, 46). Porins have also been found to induce
morphologic changes in HEp-2 cells as a result of
alterations in the cytoskeleton following incubation with
increasing concentration of the purified protein (8).
The major outer membrane protein (MOMP) of C. jejuni
was first isolated and reconstituted into lipid bilayer
membranes and found to form small channels consistent with
that of a porin (18). The MOMP has an apparent molecular
weight of 45 kDa under native conditions and since 3-
folder monomers are needed to form the functional porin it
was confirmed to be part of the trimeric porin family (3).
The N-terminal sequence has been elucidated and been found
to contain little homology with other bacterial porin
proteins (3) but it did share homology with two outer
membrane proteins from W. recta (20). In this thesis it
was reported that the porin-LPS complex from C. j ejuni
possessed a heat-labile cytotoxic activity and was capable
of inducing apoptosis in HEp-2 cells but not in Vero
cells.
Thus, while the characterization of the protein with
respect to its pore capabilities has already been reported
(18), the corresponding gene and its sequence have not
previously identified.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-4 -
DISCLOSURE OF THE INVENTION
An object of the present invention is to identify and
sequence the a porin gene of C. j ejuni responsible for the
production of a cytotoxic protein-LPS complex so that the
gene can be cloned and expressed, and so that useful
products and methods can be developed.
Another object of the invention is to identify a gene
responsible for cytotoxic activity of C. jejuni to
facilitate the identification and treatment of infections
of mammals by the organism, and to enable prophylaxis
against such infection.
According to one aspect of the invention, there is
provided an isolated and purified porA gene from
Campylobacter jejuni, characterized in that said gene
expresses a 424 amino acid cytotoxic protein having a
calculated molecular weight of 45.6 kDa and a pI of 4.44.
Another aspect of the invention comprises
a DNA probe, characterized in that said probe has a
nucleotide sequence corresponding to a part of a target
sequence SEQ ID NO:1, wherein the nucleotide sequence of
the probe encompasses nucleotide substitutions, additions
and deletions that do not affect the ability of the probe
to bind specifically to said target.
The invention also relates to a method of detecting
the presence of Campylobacter jejuni infection,
characterized by the steps of: a) contacting a sample
obtained from a patient suspected of infection, with a
detectable amount of a purified cytotoxic rotein encoded
by at least a portion of the nucleic acid of the
invention, for a time sufficient to allow formation of a
complex between said protein and any anti-Campylobacter
jejuni antibodies present in said sample; and b) detecting
the presence of, and optionally the quantity of, said
complex formed during step (a).
In another form, the invention comprises an isolated
expression vector, characterized by a region encoding a
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-5-
porA protein of Campylobacter jejuni, or an antigenic
fragment thereof.
. Included within the invention is a method of inducing
an immune response in a human or animal host by
administering to the host a foreign protein, characterized
in that said protein has an amino acid sequence SEQ ID
N0:2, wherein the amino acid sequence encompasses amino
acid substitutions, additions and deletions that do not
alter the ability of the protein to raise antibodies when
introduced into said human or animal body.
Yet another aspect of the invention is a method of
producing antibodies for testing for infection by
Campylobacter jejuni, characterized in that a protein
having an amino acid sequence of SEQ ID N0:2 is introduced
into a human or animal body to raise antibodies, and said
antibodies are subsequently isolated from said body,
wherein said amino acid sequence encompasses amino acid
substitutions, additions and deletions that do not alter
the ability of the protein to raise antibodies when
introduced into said human or animal body.
BRIEF DESCRIPTION OF THE DRAWINGS
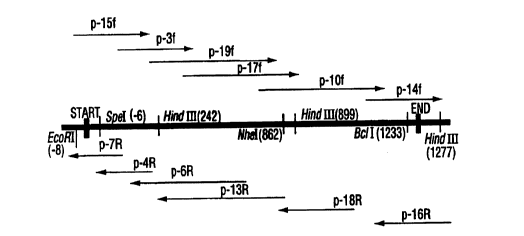
Figure 1 is a schematic diagram of the sequencing
reactions and restriction map of porA from C. jejuni
strain 2483 showing the restriction sites for the enzymes
used in the generation of a vectorette library (the arrows
designate the direction and primer used in the sequencing
of the intact gene).
Figure 2 shows a southern blot analysis of genomic
digests using a digoxigenin-labeled 650 by probe. Lanes 1
and 10: Hind III digested lambda DNA; Lane 2: Hind III
digested C. jejuni genomic DNA; Lane 3: BamHI digested C.
' jejuni genomic DNA; Lane 4: Bg1 I digested C. jejuni
genomic DNA; Lane 5: Nhe I digested C. jejuni genomic DNA;
Lane 6: EcoRI digested C. jejuni genomic DNA; Lane 7: Bc1
I digested C. jejuni genomic DNA; Lane 8: Spe I digested
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-6-
C. jejur2i genomic DNA; Lane 9: E. coli Xba I digested
genomic DNA.
Figure 3 shows the complete open reading frame and
translated protein of the porA gene. The single
underlined sequence represents the putative Shine-Dalgarno
ribosome binding site (RBS), -10 and -35 sequences, and
double lines represents a stem loop structure which may
indicate a rho-independent transcription termination site
with a 5 by loop followed by a poly-T region of DNA. Bold
face letters represent the initiation codon and "*"
represents the termination codon and the numbering is for
the nucleotide and amino acid count.
Figure 4 shows alignment of C. jejuni PorA with H.
influenzae P2, E. cloacae PhoE, K. pneumoniae PhoE, S.
typhi OmpC, and E. coli PhoE using GCG (Genetics Computer
Group). Capital letters represent identical or conserved
changes, small letters represent mismatches in the
sequences and spaces (...) were inserted in order to
achieve the best alignment. "*" represents termination
codon.
Figure 5 shows a stem loop structure of the
termination sequence of the porA gene. The numbering
represents the position of the loop in the 1450 by
fragment of Fig. 3.
Figure 6A shows morphological changes induced in HEp-
2 cells after 48 h treatment with C. jejuni cytotoxic
porin-LPS complex for control cells;
Figure 6B shows the morphological changes for cells
intoxicated with 1 ug of isolated C, jejuni cytotoxic
complex; note cytoplasmic vacuoles (arrowed);
Figure 6C shows the morphological changes for cells
intoxicated with 10 ~.g of isolated C. jejuni cytotoxic
porin-LPS complex. (magnification X 150);
Figure 7A shows an elution profile and silver stain
of the cytotoxic complex for a fraction from a G75 gel
filtration column;
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
_7_
Figure 7B shows an elution profile and silver stain
of the cytotoxic complex for a fractionation of peak A on
a TSK DEAF-5PW column. +++, >70°s of Hep-2 cells rounded
by 48 h; ++50-70o cell rounded by 48 h; +, <50% cell
~ 5 rounded by 48 h;
Figure 8 shows western blot analysis of the isolated
cytotoxic porin-LPS complex from Campylobacter sp using 40
~.g of crude, concentrated filtrate and homologous rabbit
antiserum. Lanes 1 and 9: Prestained standards (kDa)
(Gibco BRL); Lane 2: uninoculated broth; Lane 3:
Aeromonas veronii LCDC A2297 (used as a negative control);
Lane 4: C. coli strain 8682; Lane 5: C. jejuni LCDC
16336; Lane 6: C. j ejuni LCDC 3969; Lane 7: C. jejuni
strain 2483; Lane 8: E. coli (VT1) LCDC 3787.
Figure 9 shows double staining of native-PAGE (lanes
1 and 2) and SDS-PAGE (lanes 3 and 4) gels with periodic
acid Schiff (PAS) and Coomassie blue. Lane 1: native low
molecular weight standards (Pharmacia); Lane 2: 10 ~.g of
native carbohydrate co-purified with C. jejuni isolated
cytotoxic porin-LPS complex; Lane 3: 10 /,cg of heat
denatured carbohydrate which co-purified with C. jejuni
isolated cytotoxic porin-LPS complex; Lane 4: kaleidoscope
prestained standards (kDa) (BioRad).
Figure 10 shows western blot analysis of the isolated
cytotoxic complex with the lectin GNA. Lanes 1 and 4:
kaleidoscope prestained standards (kDa) (BioRad); Lane 2:
10 /cg of carbohydrate from the isolated C. jejuni
cytotoxic porin-LPS complex; Lane 3: 15 ,ug
carboxypeptidase Y;
~ 30 Figure 11A shows hydrophobic profiles and beta sheet
propensities as determined by the method of Novotny using
~ PC/Gene software package for C. jejuni strain 2483 PorA;
Figure 11B shows the hydrophobic profiles and beta
sheet propensities for H. influenzae P2; and
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
_g_
Figure 11C shows the hydrophobic profiles and beta
sheet propensities for C. jejuni FlaA.
BEST MODES FOR CARRYING OUT THE INVENTION
The present invention is based on the identification
ofla porin-lipopolysaccharide tLPS) complex from
Campylobacter jejuni that is an endotoxin and that is
fairly well conserved amongst strains of the organism, but
not widely found in other Campylobacter species. The
complex has been isolated and a corresponding porin gene,
designated "porA," has been identified, isolated,
sequenced and cloned by the inventors of the present
invention.
Specifically, the complex was obtained from strain
2483 of C. jejuni. While this strain is common in nature
and can be identified by designing a suitable probe from
the sequence disclosed herein, the inventors and assignee
of this application have deposited a sample of the strain
with the American Type Culture Collection of 12301
Parklawn Drive, Rockville, MD 20852 USA. The deposit was
made on March 19, 1998 under the terms of the
Budapest Treaty, and has been awarded the accession
no. ATCC 202,101.
The sequencing and cloning of the gene makes possible
various medical and industrial uses. For example,
knowledge of the DNA code makes it possible to design DNA
probes for identification of the gene in samples for
testing. A positive result indicates the presence of the
gene in the sample and is a strong indicator of the
presence of C. j ejuni. Such probes can also be used to
isolate the corresponding cDNA, that may then be amplified
by polymerase chain reaction. The development of DNA
probes based on a known sequence is a known procedure that
is familiar to persons skilled in the art and it will be
possible for such persons to develop suitable probes
without undue experimentation. Normally, such probes
R~CT~F'IED SHEET (RULE ~1 ~
ISA/EP
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
_g_
would consist of at least 15 consecutive nucleotides from
the cDNA sequence.
Furthermore, expression of the gene, or a significant
part thereof, in a suitable transformed host (e. g.
transformed E. coli or the like) makes it possible to
produce usable quantities of an expressed protein that
induces the immune system to raise antibodies. This can
be used to vaccinate the host against the effects of
intoxication with C. jejuni without causing harmful
effects. For this purpose, the protein may be used in
conjunction with suitable pharmacuetically-acceptable
carriers and may be used in concentrations of the protein
in the composition to achieved the desired protective
effect. Suitable modes of administration may be employed,
e.g. oral or parenteral administration.
The protein can also be used to produce antibodies
(e.g. in rabbit) useful in testing blood samples for
patients infected with C. jejuni.
It will be appreciated by persons skilled in the art
that the sequence of the porA gene identified herein may
undergo modification by substitution, addition or deletion
of a certain number of nucleotides without affecting the
uses of the present invention indicated above. The
present invention therefore also extends to isolated and
purified nucleic acid exhibiting such substitutions,
additions or deletions, and expression products thereof,
and probes designed for the identification thereof.
The teachings of International (PCT) Patent
Publication No. WO 95/05850 (published on March 2, 1995;
inventor: Martin J. Blaser; Applicant: Enteric Research
Laboratories, Inc) are also relevant to the isolation and
uses of the porA gene and the products derived therefrom.
The following information and procedures are specifically
mentioned.
The "isolated" nucleic acid is separated from other
nucleic acids found in the naturally occurring organism.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-10-
This specific nucleic acid can be used to detect C. jejuni
possessing the porA antigen in methods such as polymerase
chain reaction, ligase chain reaction and hybridization.
The isolated sequence or appropriate fragments
thereof can be utilized to produce a porA protein, by
splicing the sequence into an appropriate vector and
transfecting an appropriate host. In addition, the
nucleic acid can be homologous with nucleotide sequences
present in other bacteria. Such an amino acid sequence
shared with other bacteria can be used for example to
simultaneously detect related strains or as a basis for a
multiprotective vaccine.
An isolated nucleic acid capable of selectively
hybridizing with or selectively amplifying a nucleic acid
encoding the porA antigen or fragments thereof is also
contemplated. An isolated nucleic acid complementary to
the above nucleic acid is also provided. The sequences
can be selected based on the nucleotide sequence and the
utility of the particular sequence.
Modifications to the nucleic acids of the invention
are also contemplated as long as the essential structure
and function of the polypeptide encoded by the nucleic
acids is maintained. Likewise, fragments used as primers
or probes can have substitutions so long as enough
complementary bases exist for selective hybridization.
Purified antigenic polypeptide fragments encoded by
the nucleic acids of the present invention are also
contemplated. The "purified" antigen is sufficiently free
of contaminants or cell components with which the antigen
normally occurs to distinguish the antigen from the
contaminants or components.
An antigenic fragment of the antigen can be isolated
from the whole antigen by chemical or mechanical
disruption. The purified fragments thus obtained can be
tested to determine their antigenicity and specificity by
the methods taught herein. Antigenic fragments of the
antigen can also be synthesized directly. An
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-11-
immunoreactive fragment is an amino acid sequence of at
least abut 5 consecutive amino acids derived from the PorA
antigen.
The polypeptide fragments of the present invention
' 5 can also be recombinant proteins obtained by cloning
nucleic acids encoding the polypeptide in an expression
system capable of producing the antigenic polypeptide or
fragments thereof.
Once the amino acid sequence of the antigen is
provided, it is also possible to synthesize, using
standard peptide synthesis techniques, peptide fragments
chosen to be homologous to immunoreactive regions of the
antigen and to modify these fragments by inclusion,
deletion or modification of particular amino acids
residues in the derived sequences. Thus, synthesis or
purification of an extremely large number of peptides
derived from the antigen is possible.
The amino acid sequences of the present polypeptides
can contain an immunoreactive portion of PorA antigen
attached to sequences designed to provide for some
additional property, such as solubility. The amino acid
sequences of an PorA antigen can include sequences in
which one or more amino acids have been substituted with
another amino acid to provide for some additional
property, such as to remove/add amino acids capable of
disulfide bonding, to increase its biolongevity, alter
enzymatic activity, or alter interactions with gastric
acidity. In any case, the peptide must posses a bioactive
property, such as immunoreactivity, immunogenicity, etc.
CA 02284844 1999-09-24
WO 98/42842 PC't/CA98/00272
-12-
Determining Immunogenicity
The purified polypeptide fragments thus obtained can
be tested to determine their immunogenicity and
specificity by techniques known in the art. Various
concentrations of a putative immunogenically specific
fragment are prepared and administered to an animal and
the immunological response (e.g., the production of
antibodies or cell mediated immunity) of an animal to each
concentration is determined. The amounts of antigen
administered depend on the subject e.g. a human or a
guinea pig, the condition of the subject, the size of the
subject, etc. Thereafter an animal so inoculated with the
antigen can be exposed to the bacterium to test the
potential vaccine effect of the specific immunogenic
fragment. The specificity of a putative immunogenic
fragment can be ascertained by testing sera, other fluids
or lymphocytes from the inoculated animal for cross
reactivity with other closely related bacteria.
Vectors and Hosts
A vector comprising the nucleic acids of the present
invention is also provided. The vectors of the invention
can be in a host capable of expressing the antigen.
There are numerous E. coli expression vectors known to one
of ordinary skill in the art useful for the expression of
the antigen. Other microbial hosts suitable for use
include bacilli, such as Bacillus subtilus, and other
enterobacteriaceae, such as Salmonella, Serratia, and
various Pseudomonas species.
In these prokaryotic hosts one can also make
expression vectors, which will typically contain
expression control sequences compatible with the host cell
(e. g., an origin of replication). In addition, any number
of a variety of well-known promoters will be present, such
as the lactose promoter system, a tryptophan (Trp)
promoter system, a beta-lactamase promoter system, or a
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-13-
promoter system from phage lambda. The promoters will
typically control expression, optionally with an operator
sequence, and have ribosome binding site sequences for
example, for initiating and completing transcription and
translation. If necessary an amino terminal methionine can
be provided by insertion of a Met codon 5' and in-frame
with the antigen. Also, the carboxyl terminal extension of
the antigen can be removed using standard oligonucleotide
mutagenesis procedures.
Additionally, yeast expression can be used. There are
several advantages to yeast expression systems. First,
evidence exists that proteins produced in a yeast
secretion systems exhibit correct disulfide pairing.
Second, post-translational glycosylation is efficiently
carried out by yeast secretory systems. The Saccharomyces
cerevisiae pre-pro-alpha-factor leader region (encoded by
the MFa-1 gene) is routinely used to direct protein
secretion from yeast (Brake et al., 1984). The leader
region of pre-pro-alpha-factor contains a signal peptide
and a pro-segment which includes a recognition sequence
for a yeast protease encoded by the KEX2 gene: this enzyme
cleaves the precursor protein on the carboxyl side of a
Lys-Arg dipeptide cleavage-signal sequence. The antigen
coding sequence can be fused inframe to the pre-pro-alpha-
factor leader region. This construct is then put under the
control of a strong transcription promoter, such as the
alcohol dehydrogenase I promoter or a glycolytic promoter.
The antigen coding sequence is followed by a translation
termination codon
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-14-
which is followed by transcription termination signals.
Alternatively, the antigen coding sequences can be
fused to a second protein coding sequence, such as Sj26 or
(3 galactosidase, used to facilitate purification of the
fusion protein by affinity chromatography. The insertion
of protease cleavage sites to separate the components of
the fusion protein is applicable to constructs used for
expression in yeast.
Mammalian cells permit the expression of proteins in
an environment that favors important post-translational
modifications such as folding and cysteine pairing,
addition of complex carbohydrate structures, and secretion
of active protein. Vectors useful for the expression of
antigen in mammalian cells are characterized by insertion
of the antigen coding sequence between a strong viral
promoter and a polyadenylation signal. The vectors can
contain genes conferring either gentamicin or methotrexate
resistance for use as selectable markers. The antigen and
immunoreactive fragment coding sequence can be introduced
into a Chinese hamster ovary cell line using a
methotrexate resistance-encoding vector. Presence of the
vector DNA in transformed cells can be confirmed by
Southern analysis and production of an RNA corresponding
to the antigen coding sequence can be confirmed by
Northern analysis. A number of other suitable host cell
lines capable of secreting intact human proteins have been
developed in the art, and include the CHO cell lines, HeLa
cells, myeloma cell lines, Jurkat cells, etc. Expression
vectors for these cells can include expression control
sequences, such as an origin of replication, a promoter,
an enhancer, and necessary information processing sites,
such as ribosome binding sites, RNA splice sites,
polyadenylation sites, and transcriptional terminator
sequences. Preferred expression control sequences are
promoters derived from immunoflogulin genes, SV40,
Adenovirus, and Bovine Papilloma Virus, etc. The vectors
containing the DNA segments of interest can be transferred
CA 02284844 1999-09-24
WO 98/42842 PCTlCA98/00272
-15-
into the host cell by well-known methods, which vary
depending on the type of cellular host. For example,
calcium chloride transfection is commonly utilized for
prokaryotic cells, whereas calcium phosphate treatment or
electroporation may be used for other cellular hosts.
Materials and methods for baculovirus/insect cell
expression systems are commercially available in kit form
from, inter alia, Invitrogen, San Diego, CA ("MaxBac"T"~
kit). These techniques are generally known to those
skilled in the art and fully described in Summers and
Smith, Texas Agricultural Experiment Station Bulletin No.
1555 (1987) (hereinafter "Summers and Smith").
Recombinant baculovirus expression vectors have been
developed for infection into several insect cells. For
example, recombinant baculoviruses have been developed
for, inter alia, Aedes aegypti, Autographa Californica,
Bombyx mori, Drosophila melanogaster, Spodoptera
frugiperda, and Trichoplusia ni (PCT Pub. No. WO
89/046699; Carbonell et al., J. Virol., 56:153 (1985);
Wright, Nature, 321:718 (1986); Smith et al., Mol. Cell.
Biol., 3:2156 {1983), and see generally, Fraser, et al.,
In vitro Cell. Dev. Biol., 25:225 {1989).
Alternative vectors for the expression of antigen in
mammalian cells can also be employed, e.g those similar to
those developed for the expression of human gamma-
interferon, tissue plasminogen activator, clotting Factor
VIII, hepatitis B virus surface antigen, protease Nexinl,
and eosinophil major basic protein. Further, the vector
can include CMV promoter sequences and a polydenylation
signal available for expression of inserted DNAs in
mammalian cells {such as COS7).
The DNA sequences can be expressed in hosts after the
sequences have been operably linked to, i.e., positioned
to ensure the functioning of, an expression control
sequence. These expression vectors are typically
replicable in the host organisms either as episomes or as
an integral part of the host chromosomal DNA. Commonly, a
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-16-
selectable marker such as genes for tetracycline
resistance or hygromycin resistance are utilized to permit
detection and/or selection of those cells transformed with
the desired DNA sequences (see, e.g., U.S. Patent
4,704,362).
Polynucleotides encoding a variant polypeptide may
include sequences that facilitate transcription
(expression sequences) and translation of the coding
sequences such that the encoded polypeptide product is
produced. Construction of such polynucleotides is well
known in the art. For example, such polynucleotides can
include a promoter, a transcription termination site
(polyadenylation site in eukaryotic expression hosts), a
ribosome binding site, and, optionally, an enhancer for
use in eukaryotic expression hosts, and, optionally,
sequences necessary for replication of a vector.
Purified Antibodies
A purified monoclonal antibody specifically reactive
with PorA is also provided. The antibodies can be
specifically reactive with a unique epitope of PorA or
they can also react with epitopes of other organisms. The
term "reactive" means capable of binding or otherwise
associating nonrandomly with an antigen. "Specifically
reactive" as used herein describes an antibody or other
ligand that does not cross react substantially with any
antigen other than the one specified, in this case,
usually PorA antigen, or antigenic fragments thereof.
Antibodies can be made as described in the Examples (see
also, Marlow and Lane, Antibodies; A Laboratory Manual,
Cold Spring Harbor Laboratory, Cold Spring Harbor, New
York, 1988). Briefly, purified antigen can be injected
into an animal in an amount and in intervals sufficient to
elicit an immune response. Antibodies can either be
purified directly, or spleen cells can be obtained from
the animal. The cells are then fused with an immortal cell
line and screened for antibody secretion.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98100272
-17-
The antibody can be bound to a substrate or labeled
with a detectable moiety, or both bound and labeled. The
. detectable moieties contemplated with the composition of
the present invention are those listed below in the
. 5 description of the diagnostic methods, including
fluorescent, enzymatic and radioactive markers.
Antigen Bound to Substrate
A purified PorA antigen bound to a substrate and a
ligand specifically reactive with the antigen are also
contemplated. Such a purified ligand specifically reactive
with the antigen can be an antibody. The antibody can be a
monoclonal antibody obtained by standard methods and as
described herein. The monoclonal antibody can be secreted
by a hybridoma cell line specifically produced for that
purpose (Harrow and Lane, 1988). Likewise, nonhuman
polyclonal antibodies specifically reactive with the
antigen are within the scope of the present invention. The
polyclonal antibody
can also be obtained by the standard immunization and
purification protocols (Harrow and Lane, 1988).
Serological Detection (Diaynosis) Methods Detecting
Antibody with the Antigen
The present invention provides a method of detecting
the presence of C. jejuni strain possessing the PorA
antigen in a subject, comprising the steps of contacting
an antibody-containing sample from the subject with a
detectable amount of the PorA antigenic fragment of the
present invention and detecting the reaction of the
fragment and the antibody, the reaction indicating the
presence of the C. jejuni strain or previous infection
with the C. jejuni strain.
Detecting Antigen with Antibody/Ligand
One example of the method of detecting C. jejuni
possessing the PorA antigen is performed by contacting a
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-18-
fluid or tissue sample from the subject with an amount of
a purified antibody specifically reactive with the
antigen, and detecting the reaction of the ligand with the
antigen. It is contemplated that the antigen will be on
intact cells containing the antigen, or will be fragments
of the antigen. As contemplated herein, the antibody
includes any ligand which binds the antigen, for example,
an intact antibody, a fragment of an antibody or another
reagent that has reactivity with the antigen. The fluid
sample of this method can comprise any body fluid which
would contain the antigen or a cell containing the
antigen, such as blood, plasma, serum, saliva and urine.
Other possible examples of body fluids include sputum,
mucus, gastric juice and the like.
ELISA
Immunofluorescence assays (IFA) and enzyme
immunoassays such as enzyme linked immunosorbent assays
(ELISA) and immunoblotting can be readily adapted to
accomplish the detection of the antigen. An ELISA method
effective for the detection of the antigen can, for
example, be as follows: (1) bind the antibody to a
substrate; (2) contact the bound antibody with a fluid or
tissue sample containing the antigen; (3) contact the
above with a secondary antibody bound to a detectable
moiety (e. g., horseradish peroxidase enzyme or alkaline
phosphatase enzyme); (4) contact the above with the
substrate for the enzyme; (5) contact the above with a
color reagent; (6) observe color change. The above method
can be readily modified to detect antibody as well as
antigen.
Competitive Inhibition Assay
Another immunologic technique that can be useful in
the detection of C. jejuni expression PorA or previous C.
jejuni infection utilizes monoclonal antibodies (MAbs) for
detection of antibodies specifically reactive with PorA
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-19-
antigen. Briefly, sera or other body fluids from the
subject is reacted with the antigen bound to a substrate
(e. g. an ELISA 96-well plate). Excess sera is thoroughly
washed away. A labeled (enzyme-linked, fluorescent,
radioactive, etc.) monoclonal antibody is then reacted
with the previously reacted antigen-serum antibody
complex. The amount of inhibition of monoclonal antibody
binding is measured relative to a control (no patient
serum antibody). The degree of monoclonal antibody
inhibition is a very specific test for a particular
variety or strain since it is based on monoclonal antibody
binding specificity. MAbs can also be used for detection
directly in cells by IFA.
Micro-Agglutination Assay
A micro-aggulatination test can also be used to
detect the presence of the C. jejuni strain in a subject.
Briefly, latex beads (or red blood cells) are coated with
the PorA and mixed with a sample from the subject, such
that antibodies in the tissue or body fluids that are
specifically reactive with the antigen crosslink with the
antigen, causing agglutination. The agglutinated antigen-
antibody complexes form a precipitate, visible with the
naked eye or by spectrophotometer. In modification of the
above test, antibodies specifically reactive with the
antigen can be bound to the beads and antigen in the
tissue or body fluid thereby detected.
Sandwich Assay/Flow Cytometry/Immunoprecipitation
In addition, as in a typical sandwich assay, the
antibody can be bound to a substrate and reacted with the
antigen. Thereafter, a secondary labeled antibody is
bound to epitopes not recognized by the first antibody and
the secondary antibody is detected. Since the present
invention provides PorA antigen for the detection of C.
jejuni or previous C. jejuni infection, other serological
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-20-
methods such as flow cytometry and immunoprecipitation can
also be used as detection methods.
In the diagnostic methods taught herein, the antigen
can be bound to a substrate and contacted by a fluid
sample such as serum, urine, saliva or gastric juice. This
sample can be taken directly from the patient or in a
partially purified form. In this manner, antibodies
specific for the antigen (the primary antibody) will be
specifically react with the bound antigen. Thereafter, a
secondary antibody bound to, or labeled with, a detectable
moiety can be added to enhance the detection of the
primary antibody. Generally, the secondary antibody or
other liyand which is reactive, either specifically with a
different epitope of the antigen or nonspecifically with
the ligand or reacted antibody, will be selected for its
ability to react with multiple sites on the primary
antibody. Thus, for example, several molecules of the
secondary antibody can react with each primary antibody,
making the primary antibody more detectable.
Detectable Moieties
The detectable moiety will allow visual detection of
a precipitate or a color change, visual detection by
microscopy, or automated detection by spectrometry,
radiometric measurement or the like. Examples of
detectable moieties include fluorescein and rhodamine (for
fluorescence microscopy), horseradish peroxidase (for
either light or electron microscopy and biochemical
detection), biotin-streptavidin (for light or electron
microscopy) and alkaline phosphatase (for biochemical
detection by color change). The detection methods and
moieties used can be selected, for example, from a list
above or other suitable examples by the standard criteria
applied to such selections (Harrow and Lane, 1988).
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-21-
Treatment Methods
Methods of treating C. jejuni enteritis in a subject
' using the compositions of the present invention are
provided. For example, in one such method an amount of
ligand specifically reactive with the PorA antigen of C.
jejuni sufficient to bind the antigen in the subject and
improve the subject's clinical condition is administered
to the subject. Such improvement results from the ligand
interfering with the antigen's normal function in inducing
cell adherence inflammation and cellular damage. The
ligand can be purified monoclonal antibody specifically
reactive with the antigen, a purified polyclonal antibody
derived from a nonhuman animal, or other reagent having
specific reactivity with the antigen. Additionally,
cytotoxic moieties can be conjugated to the
ligand/antibody by standard methods. Examples of
cytotoxic moieties include ricin A chain, diphtheria toxin
and radioactive isotopes.
Another method of treating C. j ejuni enteritis subject
comprises administering to the subject an amount of a
ligand/antagonist for a receptor for the PorA antigen of
C. jejuni sufficient to react with the receptor and
prevent the binding of the PorA antigen to the receptor.
The result is an improvement in the subject's clinical
condition. Alternatively, the treatment method can include
administering to the subject an amount of an analogue of a
PorA receptor to result in competitive binding of the PorA
antigen, thus inhibiting binding of the PorA antigen to
its wild type receptor. The receptor is localized on cells
present in the intestinal mucosa, such as epithelial
cells, inflammatory cells, or endothelial cells.
Vaccines
The PorA antigen of this invention can be used in the
construction of a vaccine comprising an immunogenic amount
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-22-
of the antigen and a pharmaceutically acceptable carrier.
The vaccine can be the entire antigen, the antigen on an
intact C. jejuni, E. cold or other strain. The vaccine
can then be used in a method of preventing C. jejuni
infection. As mentioned, supra, mutant forms of C. j ejuni
may also be used.
Immunogenic amounts of the antigen can be determined using
standard procedures. Briefly, various concentrations of a
putative specific immunoreactive epitope are prepared,
administered to an animal and the immunological response
(e. g., the production of antibodies) of an animal to each
concentration is determined.
The pharmaceutically acceptable carrier in the
vaccine of the instant invention can comprise saline or
other suitable carriers (Arnon, R. (Ed.) Synthetic
Vaccines I:L 83-92, CRC Press, Inc., Boca Raton, Florida,
1987). An adjuvant can also be a part of the carrier of
the vaccine, in which case it can be selected by standard
criteria based on the antigen used, the mode of
administration and the subject (Arnon R. (Ed.), 1987).
Methods of administration can be by oral or sublingual
means, or by injection, depending on the particular
vaccine used and the subject to whom it is administered.
It can be appreciated from the above that the vaccine can
be used as a prophylactic (to prevent infection) or a
therapeutic (to treat disease after infection) modality.
Thus, the invention provides methods of preventing or
treating C. jejuni infection and the associated diseases
by administering the vaccine to a subject.
Such vaccines comprise antigen or antigens, usually
in combination with "pharmaceutically acceptable
carriers," which include any carrier that does not itself
induce the production of antibodies harmful to the
individual receiving the composition. Suitable carriers
are typically large, slowly metabolized macromolecules
such as proteins, polysaccharides, polylactic acids,
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-23-
polyglycolic acids, polymeric amino acids, amino acid
copolymers, lipid aggregates (such as oil droplets or
liposomes), and inactive virus particles. Such carriers
are well known to those of ordinary skill in the art.
. 5 Additionally, these carriers may function as
immunostimulating agents ("adjuvants"). Furthermore, the
antigen may be conjugated to a bacterial toxoid, such as a
toxoid from diphtheria, tetanus, cholera, H. pylori, etc.
pathogens.
Preferred adjuvants to enhance effectiveness of the
composition include, but are not limited to: (1) aluminum
salts (alum), such as aluminum hydroxide, aluminum
phosphate, aluminum sulfate, etc.; (2) oil-inwater
emulsion formulations (with or without other specific
immunostimulating agents such as muramyl peptides, or
bacterial cell wall components), such as for example (a)
MF59 (PCT Publ. No. WO 90/14837), containing 5% Squalene,
0.5% TweenT"~ 80, and 0.5% SpanT"~ 85 (optionally containing
various amounts of MTP-PE, although not required)
formulated into submicron particles using a microfluidizer
such as Model 110Y microfluidizer Microfluidics, Newton,
MA), t b) SAF, containing 10% Squalane, 0.4% Tween 80, 50
pluronic-blocked polymer L121, and thr-MDP either
microfluidized into a submicron emulsion or vortexed to
generate a larger particle size emulsion, and (c) Ribi
adjuvant system (RAS), (Ribi Immunochem, Hamilton, MT)
containing 2% Squalene, 0.2~ Tween 80, and one or more
bacterial cell wall components from the group consisting
of monophosphorylipid A (MPL), trehalose dimycolate (TDM),
and cell wall skeleton (CWS), preferably MPL ~ CWS
(Detox); (3) saponin adjuvants, such as Stimulon
(Cambridge Bioscience, Worcester, MA) may be used or
particles generated therefrom such as ISCOMs
(immunostimulating complexes); (4) Complete Freunds
Adjuvant and Incomplete Freunds Adjuvant (IFA); (5)
Cytokines, such as interleukins (IL-1, IL-2, etc.),
macrophage colony stimulating factor (M-CSF), tumor
CA 02284844 1999-09-24
WO 98142842 PCT/CA98/OOZ72
-24-
necrosis factor (TNF), etc ; and (6) other substances that
act as immunostimulating agents to enhance the
effectiveness of the composition. Alum and MF59 are
preferred.
Muramyl peptides include, but are not limited to, N-
acetyl-muramyl-L-threonyl-D-isoglutamine (thr-MDP), N-
acetyl-normuramyl-L-alanyl-D-isoglutamine (nor-MDP), N-
acetylmuramyl-L-alanyl-D-isoglutaminyl-L-alanine-2-(1'2'-
dipalmitoyl-sn-glycero-3-huydroxyphosphoryloxy)ethylamine
(MTP-PE) , etc .
The immunogenic compositions (e. g., the antigen,
pharmaceutically acceptable carrier, and adjuvant)
typically will contain diluents, such as water, saline,
glycerol, ethanol, etc. Additionally, auxiliary
substances, such as wetting or emulsifying agents, pH
buffering substances, and the like, may be present in such
vehicles.
Typically, the immunogenic compositions are prepared
as injectables, either as liquid solutions or suspensions;
solid forms suitable for solution in, or suspension in,
liquid vehicles prior to injection may also be prepared.
The preparation also may be emulsified or encapsulated in
liposomes for enhanced adjuvant effect.
Typical immunogenic compositions used as vaccines
comprise-an immunologically effective amount of antigenic
polypeptides, as well as any other of the above-mentioned
components, as needed. By "immunologically effective
amount," it is meant that the administration of that
amount to an individual, either in a single does or as
part of a series, is effective for treatment or
prevention. This amount varies depending upon the health
and physical condition of the individual to be treated,
the taxonomic group of individual to be treated (e. g.,
nonhuman primate, primate, etc.), the capacity of the
individual's immune system to synthesize antibodies, the
degree of protection desired, the formulation of the
vaccine, the treating doctor's assessment of the medical
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-25-
situation, and other relevant factors. It is expected that
the amount will fall in a relatively broad range that can
- be determined through routine trials.
The immunogenic compositions are conventionally
- 5 administered parenterally, e.g., by injection, either
subcutaneously or intramuscularly. Additional formulations
suitable for other modes of administration include oral
and pulmonary formulations, suppositories, and transdermal
applications. Dosage treatment may be a single dose
schedule or a multiple dose schedule. The vaccine may be
administered in conjunction with other immunoregulatory
agents.
Nucleic Acid Detection (Diagnosis) Methods
The presence of the PorA antigen and C. jejuni
possessing the PorA antigen can also be determined by
detecting the presence of a nucleic acid specific for the
antigen. The specificity of these sequences for the
antigen can be determined by conducting a computerized
comparison with known sequences, catalogued in GenBank, a
computerized database, using the computer programs Word
Search or FASTA of the Genetics Computer Group (Madison,
WI?, which search the catalogued nucleotide sequences for
similarities to the gene in question.
The nucleic acid specific for the antigen can be
detected utilizing a nucleic acid amplification technique,
such as polymerase chain reaction or ligase chain
reaction. Alternatively, the nucleic acid is detected
utilizing the direct hybridization or by utilizing a
restriction fragment length polymorphism. For example, the
present invention provides a method of detecting the
presence of C. j ejuni, possessing the PorA antigen,
comprising ascertaining the presence of a nucleotide
sequence associated with a restriction endonuclease
cleavage site. In addition, PCR primers which hybridize
only with nucleic acids specific for the antigen can be
utilized. The presence of amplification indicates the
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-26-
presence of the antigen. In another embodiment, a
restriction fragment of a DNA sample can be sequenced
directly using for example, Sanger ddNTp sequencing or 7-
deaza-2'-deoxyguanosine 5'-triphosphate and Taq
polymerase, and compared to the known unique sequence to
detect C. jejuni . In a further embodiment, the present
invention provides a method of detecting the presence of
C. jejuni by selective amplification by the methods
described above. In yet another embodiment, C. jejuni can
be detected by directly hybridizing the unique sequence
with a PorA selective nucleic acid probe.
Furthermore, the nucleotide sequence could be
amplified prior to hybridization by the methods described
above.
Once specific sequences are shown to be associated
with C. jejuni, the methods to detect specific sequences
are standard in the art. Detection of specific sequences
using direct probing involves the use of oligonucleotide
probes which may be prepared, for example, synthetically
or by nick translation. The probes may be suitably labeled
using, for example, a radio label, enzyme label,
fluorescent label, biotin-avidin label and the like for
subsequent visualization in the example of Southern blot
hybridization procedure. The labeled probe is reacted with
a bound sample DNA, e.g., to a nitrocellulose sheet under
conditions such that only fully complementary sequences
hybridize. The areas that carry DNA sequences
complementary to the labeled DNA probe become labeled
themselves as a consequence of the reannealing reaction.
The areas of the filter that exhibit such labeling
may then be visualized, for example, by autoradiography.
The label probe is reacted with a DNA sample bound to, for
example, nitrocellulose under conditions such that only
fully complementary sequences will hybridize. The
stringency of hybridization is usually ioc below the Ti
(the irreversible melting temperature of the hybrid formed
CA 02284844 1999-09-24
WO 98/42842 PCTlCA98/00272
-27-
between the probe and its target sequence) for the given
chain length. For 20mers, the recommended hybridization
temperature is about 58°C. The washing temperatures are
unique to the sequence under investigation and need to be
- 5 optimized for each variant.
Alternative probing techniques, such as a ligase
chain reaction (LCR), involve the use of mismatch probes,
i.e., probes which are fully complementary with
the target except at the point of the mutation. The target
sequence is then allowed to hybridize both with
oligonucleotides which are fully complementary and have
oligonucleotides containing a mismatch, under conditions
which will distinguish between the two. By manipulating
the reaction conditions, it is possible to obtain
hybridization only where there is full complementarily. If
a mismatch is present, there is significantly reduced
hybridization.
The polymerase chain reaction (PCR) is a technique
that amplifies specific DNA sequences with remarkable
efficiency. Repeated cycles of denaturation, primer
annealing and extension carried out with polymerase, e.g.,
a heat stable enzyme Taq polymerase, leads to exponential
increases in the concentration of desired DNA sequences.
Given a knowledge of the nucleotide sequence of a
mutation, synthetic oligonucleotides can be prepared which
are complementary to sequences which flank the DNA of
interest. Each oligonucleotide is complementary to one of
the two strands. The DNA can be denatured at high
temperatures (e.g., 95°C) and then reannealed in the
presence of a large molar excess of oligonucleotides. The
oligonucleotides, oriented with their 3' ends pointing
towards each other, hybridize to opposite strands of the
target sequence and prime enzymatic extension along the
nucleic acid template in the presence of the four
deoxyribonucleotide triphosphates. The end product is then
denatured again for another cycle. After this three-step
cycle has been repeated several times, amplification of a
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/002'72
-28-
DNA segment by more than one millionfold can be achieved.
The resulting DNA may then be directly sequenced in order
to locate any genetic alteration. Alternatively, it may
be possible to prepare oligonucleotides that will only
bind to altered DNA, so that PCR will only result in
multiplication of the DNA if a mutation is present.
Following PCR, direct visualization of allele-specific
oligonucleotide hybridization may be used for typing
C. jejuni strain associated with an outbreak.
Alternatively, an adaptation of PCR called amplification
of specific alleles (PASA) can be employed; this uses
differential amplification for rapid and reliable
distinction between alleles that differ at a single base
pair. Other techniques, such as 3SR, which utilize RNA
polymerase to achieve high copy number, can also be used
where appropriate.
In yet another method, PCR may be followed by
restriction endonuclease digestion with subsequent
analysis of the resultant products. Nucleotide
substitutions can result in the gain or loss of specific
restriction endonuclease site. The gain or loss of a
restriction endonuclease recognition site facilitates the
typing of the C. jejuni strains associated outbreak using
restriction fragment length polymorphism (RFLP) analysis
or by detection of the presence or absence of a
polymorphic restriction endonuclease site in a PCR product
that spans the sequence of interest.
For RFLP analysis, DNA is obtained, for example from
the stool of the subject suspected of containing C.
jejuni, or C. jejuni isolated from subject, is digested
with a restriction endonuclease, and subsequently
separated on the basis of size by agarose gel
electrophoresis. The Southern blot technique can then be
used to detect, by hybridization with labeled probes, the
products of endonuclease digestion. The patterns obtained
from the Southern blot can then be compared. Using such an
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-29-
approach, PorA DNA is detected by determining the number
of bands detected and comparing this number to the DNA
from C. jejuni strains that are not associated with the C.
jejuni outbreak. Restriction endonucleases can also be
utilized effectively to detect mutations in the PorA gene.
Similar creation of additional restriction sites by
nucleotide substitutions at the disclosed mutation sites
can be readily calculated by reference to the genetic code
and a list of nucleotide sequences recognized by
restriction endonucleases.
In general, primers for PCR and LCR are usually about
by in length and the preferable range is from 15-25 bp.
Better amplification is obtained when both primers are the
same length and with roughly the same nucleotide
15 composition. Denaturation of strands usually takes place
at 94°C and extension from the primers is usually at 72°C.
The annealing temperature varies according to the sequence
under investigation. Examples of reaction times are: 20
mins denaturing; 35 cycles of 2 min, 1 min, 1 min for
20 annealing, extension and denaturation; and finally a 5 min
extension step. PCR amplification of specific alleles
(PASA) is a rapid method of detecting single-base
mutations or polymorphisms. PASA (also known as allele
specific amplification) involves amplification with two
oligonucleotide primers such that one is allele-specific.
The desired allele is efficiently amplified, while
the other alleles) is poorly amplified because it
mismatches with a base at or near the 3' end of the
allele-specific primer. Thus, PASA or the related method
of PAMSA may be used to specifically amplify the mutation
sequences of the invention. Where such amplification is
done on C. j ejuni isolates or samples obtained from an
individual during outbreak, it can serve as a method of
detecting the presence of the mutations in the strain
responsible for the cause of the outbreak.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-30-
As mentioned above, a method known as ligase chain
reaction tLCR) can be used to successfully detect a
single-base substitution. LCR probes may be combined or
multiplexed for simultaneously screening for multiple
different mutations. Thus, LCR can be particularly useful
where, as here, multiple mutations are predictive of the
C. jejuni strain that is specifically associated with an
outbreak.
Antigen-Detecting Kit
The present invention provides a kit for the
diagnosis of infection by strains of C. jejuni.
Particularly, the kit can detect the presence of PorA
antigen specifically reactive with an antibody or an
immunoreactive fragment thereof. The kit can include an
antibody bound to a substrate, a secondary antibody
reactive with the antigen and a reagent for detecting a
reaction of the secondary antibody with the antigen. Such
a kit can be an ELISA kit and can comprise the substrate,
primary and secondary antibodies when appropriate, and any
other necessary reagents such as detectable moieties,
enzyme substrates and color reagents as described above.
The diagnostic kit can, alternatively, be an
immunoblot kit generally comprising the components and
reagents described herein.
Antibody-Detecting Kit
The diagnostic kit of the present invention can be
used to detect the presence of a primary antibody
specifically reactive with PorA or an antigenic fragment
thereof. The kit can include the antigen bound to a
substrate, a secondary antibody reactive with the antibody
specifically reactive with the PorA antigen and a reagent
for detecting a reaction of the secondary antibody with
the primary antibody. Such a kit can be an ELISA kit and
can comprise the substrate, antigen, primary and secondary
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-31-
antibodies when appropriate, and any other necessary
reagents such as detectable moieties, enzyme substrates
and color reagents as described above. The diagnostic kit
can, alternatively, be an immunoblot kit generally
comprising the components and reagents described herein.
Nucleic Acid Detection Diagnostic) Kits
Once the nucleotide sequence of the PorA antigen is
determined, the diagnostic kit of the present invention
can alternatively be constructed to detect nucleotide
sequences specific for the antigen comprising the standard
kit components such as the substrate and reagents for the
detection of nucleic acids. Because C. jejuni infection
can be diagnosed by detecting nucleic acids specific for
the antigen in intestinal tissue and stool, it will be
apparent to an artisan that a kit can be constructed that
utilizes the nucleic acid detection methods, such as
specific nucleic acid probes, primers or restriction
fragment length polymorphisms in analyses. It is
contemplated that the diagnostic kits will further
comprise a positive and negative control test.
The particular reagents and other components included in
the diagnostic kits of the present invention can be
selected from those available in the art in accord with
the specific diagnostic method practiced in the kit. Such
kits can be used to detect the antigen in tissue and fluid
samples from a subject.
The following examples are intended to illustrate,
but not limit, the invention. While they are typical of
those that might be used, other procedures known to those
skilled in the art may be alternatively employed.
The following Experimental Section discloses the
testing and experimentation on which the present invention
is based.
CA 02284844 1999-09-24
WO 98/42842 PCTlCA98/00272
-32-
EXPERIMENTAL SECTION
EXAMPLE 1
MATERIALS AND METHODS
Bacterial Strains and Media.
C. jejuni, strain 2483, was isolated from a patient
with gastroenteritis (23, 24) and was used for the
localization and sequencing of the porin gene (porA). The
organism was passed twice on tryptic soy agar containing
5o sheep blood (TSA) following isolation from the patient
and was subsequently stored at -70°C in tryptic soy broth
containing 5% sheep blood. Strains of Campylobacter sp.
and related organisms were maintained at -80°C in
glycerol-peptone water as part of the reference collection
at the Laboratory Centre for Disease Control, Ottawa,
Canada.
Vectorette Polymerase Chain Reaction (PCR).
Genomic DNA from C. jejuni strain 2483 was purified
as described previously (25). A total of 15 ng of genomic
DNA was digested for 2 h at 37°C with 120 U of BamHI,
2 0 EcoRI , NheI , SpeI , XbaI , HindI I I , BglI I and BclI
restriction enzymes (Boehringer Mannheim, Laval, Quebec,
Canada). The vectorette oligonucleotides, vectorette
universal primer and degenerate primers were synthesized
on an OligoT~" 1000M DNA synthesizer (Beckman, Fullerton,
Ca.) and are listed in Table 1.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-33-
Table 1
Primers used for vectorette PCR and for sequencing porA gene in
C. jejuni strain 2483 porin gene.
R and F are for reverse and forward direction of the primers.
Primer Sequence SEQ
designation ID
NO:
3'-VP 5'-CTCTCCCTTCTCGAATCGTAACCGTTCGTACGAGAATC-GCTGTCCTCTCCTTC-4
3'
5'-Bam $'-GATCGAAGGAGAGGACGCTGTCTGTCGAAGGTAAGGAAC- 5
HI GGAGGAGAGAAGGGAGAG-3'
5'-ECO $'-ppTTGAAGGAGAGGACGCTGTCTGTCGAAGGTAAGGAACG-6
RI GAGGAGAGAAGGGAGAG-3'
5'-Hind 5'-AGCTGAAGGAGAGGACGCTGTCTGT'CGAAGGTAAGGAAC-7
III GGAGGAGAGAAGGGAGAG-3'
5'-Nhe 5'-CTAGGAAGGAGAGGACGCTGTCTGTCGAAG(iTAAGGAAC-8
I GGAGGAGAGAAGGGAGAG-3'
UVP 5'-CGAATCGTAACCGTTCGTACGAGAATCGCT-3' 9
p-1 F 5'-GGTAATTTTGATAAAAATTT-3' 1 O
p-2F 5'-GATACAGGTAAATTTGATAA-3' i 1
p-3F 5'-GAAGAAGCTATCAAAGATGT-3' ] 7
p-4R 5'-TGCCACCATCAACAGCGTTG-3' 13
p-6R 5'-TAAGTAAGCACCTTCAAGTG-3' 14
p-7R 5'-ACTTGTGCTCTATATTTGTG-3' I $
p-lOF S'-TGATAGCGAACTTGATGATA -3' 16
p-I3R 5'-AGCATCCCAACCATTTACTT-3' 17
p-14F 5'-TGACTTCGTATATGGTGGAA-3' 1 g
p-ISF 5'-CTCCAAATTTATGTGCTACA-3' 19
p-16R 5'-CTATCAAATTTCCAACTTCT-3' 20
p-17F 5'-TGAAGATGTTGCTACAAGTG3' 2I
p-18R 5'-CTACTCTTGCAACAGCTTCA-3' 22
p-19F 5'-CTTCAAAGCTTTCATTCAGT-3' 23
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-34-
Common linkers were allowed to anneal as outlined
previously (37) by adding 10 mM concentration of each
dephosphorylated 57-mer top strand with the 53-mer bottom
strand (3'-VP) at 65°C for 2 min followed by cooling to
37°C over the next 20 min. A 30 ~cl ligation mixture was
made with each digest and each contained 2.5 /,cg of
digested genomic DNA, 1 /.cl of annealed common linkers with
the corresponding compatible ends (i.e. BamHI digested
genomic DNA with 5'-BamHI-), 1 mM ATP, 10 U T4 DNA ligase
(Boehringer Mannheim) and 10 mM DTT and incubated
overnight at 15°C.
Polymerase chain reaction of vectorette library and
inverse PCR.
Primers p-1F, p-2F and p-3F were generated from the
sequenced amino-residues (3, section 3, page XX) number 23
to 29, 21 to 27 and 4 to 10 respectively. Design of the
primers was aided by a codon usage chart available through
Genebank enabling a degeneracy of 2, 4 and 6 to be
obtained for each, respectively. Three separate PCR
reaction mixtures were prepared by adding 1 ,uM of each
degenerative primer (Table 1) with 1 /.cM universal
vectorette primer (UVP) , 100 ~.1 lOX PCR buffer (500 mM
KC1, 100 mM Tris-HC1 (pH 8.3), to TritonT"" X-100, 30 mM
MgCl2), 200 ~cl of a stock solution containing 200 mM of
each dNTP, 20 U Taq DNA polymerase (Promega, Madison, WI).
The final volume was raised to 1 ml with sterile ddH20.
Five microliters of each ligation mixture were added to
50 /,cl of each of the PCR reaction mixtures followed by
amplification in a PE9600 thermocycler (Perkin-Elmer,
Foster City, Ca.) with initial melting temperature set at
95°C for 2 min followed by 35 cycles at 95°C for 30s,
55°C
for 30s and 72°C for 2 min with a final extension at 72°C
for 2 min. The PCR reactions and a 100 by ladder (Gibco
BRL, Grand Island, NY) were electrophoresed on 1% low
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-35-
melting point agarose (LMP) (Gibco BRL) in 1X TAE buffer
and stained with ethidium bromide for 30 min. PCR
products were excised from the agarose, extracted using
the PromegaT~~ PCR Preps DNA purification system (Promega).
Inverse PCR was performed by first adding 2.5 ,ug of
Hind III digested genomic DNA with 6 ,ul of 100 mM DTT, 6
(1 of 10 mM ATP, 5 U of ligase (Boehringer Mannheim), 6 ~cl
of ligase buffer (Boehringer Mannheim) and sterile ddH,O to
give a final volume of 60 ~1. The mixture was allowed to
ligate overnight at 15°C. Following ligation, PCR was
performed as stated above using p-3F and p-7R primers
(Table 1) for 35 cycles. The PCR reaction was run on a 1%
LMP gel and stained with ethidium bromide. Amplicons were
extracted with PromegaT~~ PCR Preps DNA purification system
(Promega) and DNA sequenced. The amplicons were DNA
sequenced on an ABI 377 automated DNA sequences (Applied
Biosystems, Foster City, Ca) using the PrismT"' dye
terminator cycle sequencing kit (Applied Biosystems).
Analysis of DNA sequences were performed using SequencherT"~
3.0 (Gene Codes Corporation, Ann Arbor, MI) and PC/Gene
(Intelligenetics, Mountain View, Ca.).
Southern blot analysis of genomic DNA.
C. jejuni genomic digests using the restriction
enzymes outlined above, were set up and allowed to digest
overnight at 37°C. Prior to electrophoresis, Hind III
digested lambda DNA was labeled with digoxigenin-11-
uridine-5'-triphosphate using a random labeling kit
(Boehringer Mannheim) for 1 h at 37°C. Digested DNA was
electrophoresed on a 1 % agarose gel (Gibco) in 1X TAE
buffer together with the Hind III digested lambda DNA
ladder (Boehringer Mannheim). Following gel
electrophoresis, the gel was Southern blotted using
established procedures (38) by first placing the gel in
denaturing solution (0.5 M NaOH, 1.5 M NaCl) for 1 h at
room temperature on an orbital shaker followed by 1 h in
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-36-
neutralizing solution (0.5 Tris-HC1, pH 7.5, 1.5 M NaCl)
under the same conditions. The genomic DNA was
transferred to HybondT"~-N+ nylon membranes (Amersham,
Arlington, Heights, IL.) using a PosiblotT"" system at 75 mm
Hg for 90 min (Stratagene, Aurora, Ontario, Canada).
After transferring, the membrane was washed once with
2X SSC before being UV cross-linked in a UV StratalinkerT~~
2400 (Stratagene). Crossed linked membranes were
prehybridized at 55°C for 1 h in 10 ml prehybidization
solution (Gibco BRL). The PCR amplicon generated from the
p-3F and p-6R primers (Table 1) was extracted from a 1%
LMP and 10-25 ng was digoxigenin-labeled using a PCR
digoxigenin-labeling kit (Boehringer Mannheim) with the
same PCR conditions as for the vectorette PCR with
exception that 15 cycles were used instead of 35.
Approximately 50 ng of lambda ladder probe and the
digoxigenin-labeled cytotoxic protein probe were heat
denatured at 100°C for 10 min, placed on ice for 5 min and
added to the hybridization solution at 55°C overnight.
Following hybridization, the membrane was washed
twice for 15 min each in 2X SSC in 0.1% SDS at room temp
followed by two 15 min washes first at 55°C in 1X SSC in
0.1% SDS and then O.1X SSC in0.1% SDS. The washed
membrane was blocked in 5% blocking reagent (Boehringer
Mannheim) for 1 h on an orbital shaker prior to the
addition of anti-digoxigenin antibody conjugated to
alkaline phosphatase (Boehringer Mannheim) used at a
dilution of 1:5000 for 1 h. The membrane was washed 3
times for 5 min each in low salt Tris-buffered saline
(TBS) and placed in a 1:20 dilution of CPD-Star lumigen
substrate (Tropix, Bedford, Ma) in washing buffer
containing O.1M Tris-HCL at pH. 9.5, 0.1 M NaCl and 50 mM
MgCl2 for 5 min. The membrane was exposure to high
performance autoradiography film (Hyperfilm-MPT'")
(Amersham) until a suitable band intensity was achieved.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-37-
Cloning and DNA sequencing of porin gene.
The region of the genomic DNA digested with Spe I
' which reacted with the cytotoxic probe, was excised from a
subsequent LMP gel, and extracted using GenecleanT"" (Biol
101, Lajolla, Ca). To determine the optimal concentration
of insert to vector, various concentrations of inserts
were ligated to 50 ng of Xba I digested and alkaline
phosphatase (Promega) treated pUC 19 (Pharmacia Biotech,
Uppsala, Sweden) with 2.5 U of T4 DNA ligase and 1mM ATP
(Promega) at 15°C overnight. A total of 50 ng of vector
was used to transform Epicurian coli XL1-blue competent
cells as outlined by the manufacturer (Stratagene) and
100 ,ul was plated to Luria broth (LB) agar plates
containing 200 ,ug/ml ampicillin. For color development,
plates were covered with 50 ~1 halogenated indolyl-/3-D-
galactoside (Bluo-gal) at 20 mg/ml (Gibco BRL) and 15 ul
isopropylthio-(3-galactoside (IPTG) used at 0.5 M (Gibco
BRL). These were allowed to dry prior to the addition of
transformants.
Transformants were picked from the plates and grown
overnight in 3 ml of LB with 200 ,ug/ml ampicillin.
Plasmid preps were performed on 1.5 ml of culture using
the PromegaT"" miniprep DNA purification system (Promega).
A total of 50 ng of purified plasmid from the Spe I
ligation was added to 50 ,ul PCR mixture containing p-3F
and p-6R and amplified for 20 cycles using the same method
outlined above. Reactions were electrophoresed on a 1%
agarose gel in TAE buffer and stained with ethidium
bromide. Plasmids from positive clones were sequenced as
described above using the primers given in Table 1. PCR
was performed on genomic DNA using primers p-15F and p-16R
using the same method as for the porin probe.
CA 02284844 1999-09-24
WO 98/42842 PCTlCA98/00272
-38-
Screening of C. jejuni isolates for porin gene and
cytotoxin production.
A total of 30 strains of C. jejuni and related
organisms, including strain 2483 (Table 2), were grown on
trypic-soy agar containing loo sheep blood for 48 h in a
micro-aerobic environment.
CA 02284844 1999-09-24
WO 98!42842 PCT/CA98/00272
-39-
Table 2
Screening of 20 strains of C. jejuni for phenotypic expression of a cytotoxin
and presence of porA using primers specific for the porin gene
sequenced from C. jejuni strain 2483
(In the Table NT = not tested; ND= not determined)
Organism LCDC Source Lior BiotypeToxin PCR
number serot ~ positiveI positive
a
'. jejuni 3454 human 4 Ia + +
3969 ND untypableI + +
4951 human 7 I + +
4966 human 7 I + +
6847 human 1 Ia + +
7099 chicken 61 + +
7288 water 9 II + +
8916 human 94 IIa + +
9214 human 2 Ia + +
9541 water 82 II + +
9543 water 82 II + +
9555 human 23 I + +
10403 human 36 Ia + _
10673 human 82 II + +
14040 human 82 II + +
14906 human 82 I + +
_ 15151 human 82 I + +
16323 beef 82 I + +
16334 human 82 II + +
16336 human 82 II + +
16388 human 82 II + +
(2483)
1 ND 4 I + +
2074 ND 36 II +
Lari 729 ND 31 I + _
. coli 348 ND 14 I + _
'. sputorum 5754 ND NT NT + _
ubsp. fecalis
. fetus subsp.7055 ND NT NT + _
etus
. hyointestinalis8494 human NT NT +
'. jejuni 9365 ND NT NT + _
subsp.
oylei
. butzleri 13220 human 7 IIIA +
coli VTl+ ND NT NT + _
. col i VT2+H 19 human NT NT + _
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-40-
Using a bacteriologic sample loop of each strain were
removed and the DNA extracted as previously described
(25). PCR conditions were as stated above except 50 ng of
genomic DNA was used in the reaction with p-3F and p-6R
for 35 cycles at an annealing temperature of 55°C. The
PCR reactions were then mixed with 6X sample buffer and
20 ,ul of each were electrophoresed on a 1% agarose gel in
TAE buffer and stained with ethidium bromide. Each strain
was also screened for phenotypic expression of a cytotoxin
in a biphasic system using a 12-well cell culture plate
(Costar). A loop of each strain was inoculated into 2 ml
of minimal essentail media (MEM) without fetal bovine
serum (FBS) and used to overlay 1 ml of Meuller-Hinton
agar present in the bottom of the wells. The organisms
were grown for 48 h at which time the liquid media was
removed and centrifuged to remove the bacteria. HEp-2
cells were subcultured into 96 well plates at a density of
Z X 10q cells/well with 200 ,ul MEM supplemented with 10 %
FBS 24 h prior to the addition of the toxic filtrate. The
supernatant was assayed for cytotoxic activity by
replacing the 200 /.cl of growth media used in subculturing
the Hep-2 cells with 200 /,cl of the organisms free
filtrate. The cells were monitored over a 48 h period for
cytological changes. E. coli 0157: H7 strain 3787 (H19),
positive for verotoxin type 1 (VT1), and strain 90-2380,
positive for verotoxin type 2 (VT2), were used as postive
controls for the cell culture assay while uninoculated
media was used as the negative control.
Genebank accession number.
Results
Vectorette PCR.
Vectorette PCR was performed using the genomic DNA
digested with Nhel ligated to its corresponding common
oligonucleotide and these generated amplicons suitable for
CA 02284844 1999-09-24
WO 98/42$42 PCT/CA98/00272
-41-
DNA sequencing. The universal primer (UVP) and p-1F, p-2F
and p-3F yielded amplicons of similar size of
approximately 800 by in length which was consistent with
the position of the Nhe I restriction site (Fig 1) and the
' S position of the primers (Table 1). DNA sequencing of the
three amplicons revealed the same sequence which, when
translated, contained an ORF corresponding to the protein
sequence obtained from the N-terminus of the cytotoxin.
No other amplicons were seen with the remaining genomic
digests when the PCR conditions were maintained. From the
DNA sequence, primer p-6R was designed from nucleic acid
positions 768 to 749 of the sequenced amplicon. The new
primer, along with p-3F were subsequently used to amplify
the 650 by probe used for Southern blot analysis and
localization of the cytotoxic porin gene.
Partial cloning and sequencing of cytotoxic porin protein.
Southern blot analysis of the digested genomic DNA
using the digoxigenin-labeled cytotoxic porin probe
yielded several discrete bands when probed with the 650 BP
probe (Fig 2). The Spe I fragment was chosen, purified and
ligated to pUC 19 and used to transform Epicurian coli
XL1-blue competent cells. One colony from the Spe I
digested and extracted genomic DNA, which was positive by
PCR for the 650 by product was designated Cj08 and this
was sequenced.
Inverse PCR was only performed on Hind III digested
DNA because of the results obtained from the Southern blot
analysis using the 650 by probe and the restriction map
(Fig 2) of the amplicon initially generated from the Nhe I
vectorette library. The Southern blot showed a weak
reaction between the digoxigenin-labeled probe and an
approximate 800 by fragment making it a potential
candidate for amplification by inverse PCR. A new primer,
p-7R, which was generated from positions 5'-209-->190-3~ of
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-42-
the sequenced amplicon from the vectorette PCR, together
with p-3F produced an 800 by product with the ligated Hind
III digested genomic DNA. When sequenced the amplicon was
found to contain the N-terminus of the porin protein along
with the entire leader sequence with the start codon and
the ribosome binding site. The sequence data and
translated protein obtained from the clone and the inverse
PCR are shown in Figure 3. The restriction map revealed a
Spe I restriction site at position 6 of the function gene;
therefore the Spe I clone only contained part of the
functional gene, but the remainder of the gene was
elucidated from the sequence the amplicon generated from
the inverse PCR reaction.
The entire gene was found to be 1275 by in length
[SEQ ID N0:3] and was designated porA. The protein
encoded was 424 amino acids in length (Fig 3) [SEQ ID
N0:2] and had a calculated molecular weight of 45.6 kDa
and a pI of 4.44. The leader sequence was found to be 22
amino acid residues in length and was cleaved from the
active protein conforming to the -3,-1 rule (2) between
the Ala--'22 (A) and Thr--X23 (T) , which was the first amino
acid residue in the sequenced protein. The mature
protein, minus the leader sequence has a calculated
molecular weight of 43.5 kDa and had a pI of 4.35. These
finding were consistent with previous reports (3,18, 21)
regarding the size and pI of the porin protein from C.
jejuni. Primers, p-15F and p-16R were subsequently
designed to PCR amplify the entire porin gene (Fig. 4) as
well as for use in the sequencing reactions.
Sequence analysis.
Sequence homology searches were performed on the
entire ORF and translated protein using GCG (Genetics
Computer Group, Madison, WI). The translated porin
protein from C. jejuni strain 2483 had no significant
homology with any characterized protein except with the
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-43-
previously described C. jejuni porin protein (3) and the
45 kDa and 51 kDa outer membrane proteins form Wolinella
recta (20). However, the DNA sequence had the greatest
similarity with H. influenzae outer membrane protein P2
over short stretches following a BLAST data base search
(BLAST; Beckman Center for Molecular and Genetic
Medicine, Stanford University of Medicine). A comparative
analysis of the translated porin from C. jejuni strain
2483 and several bacterial outer membrane proteins
revealed that C. j ejuni porin protein had a 50% sequence
similarity but only 23% sequence identity with the H.
influenzae major outer membrane protein P2. In addition,
in those regions of the DNA where the homology was
greatest, the protein sequence identity was as much as
72%. The porin from C. jejuni also had a 46% similarity
and 21% identity with the Enterobacter cloacae pore
forming outer membrane protein PhoE, 44% similarity and
21% identity with Klebsiella pneumoniae PhoE, 43%
similarity and 17% identity with Salmonella typhi ompC,
and 42% similarity and 19% identity with E. coli PhoE.
When the porin from C. jejuni was compared to the
consensus sequence obtained from an alignment of ompF,
ompC, PhoE and Lc of E. coli, PhoE of K. pneumoniae and
ompC of S. typhi (29), a 45% similarity and 20% identity
was found.
Screening Campylobacter sp. for porA and cytotoxin
production.
Results of screening C. jejuni for phenotypic and
genotypic expression of the cytotoxin gene are summarized
in Table 2. It was found that all 32 strains of
Campylobacter sp. and related organisms produced a
cytotoxic component when the filtrate from the biphasic
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-44-
growth system was assayed in tissue culture but only 22 of
32 (69%) were PCR positive using the primers (p-3F and p-
6R) specific for porA. However, 19 of 20 (95%) of the C.
jejuni strains screened for porA were PCR positive for the
650 by product showing that the porin gene was highly
conserved among strains of C. jejuni, especially Lior
serotype 82, but was not conserved between related species
of Campylobacter.
DISCUSSION
The 1275 by ORF had a %guanosine+cytosine content of
36.8 mol o (Fig. 3) which is slightly higher than the
range previously described for C. jejuni chromosomal DNA
(42, 43). A putative Shine-Dalgarno (SD) sequence which
has previously been described (33) with a sequence of 5'-
AGGAG-3', lies centered 10 by upstream for the initiation
codon ATG. A putative -35 region, which has been
previously described (33) is centered 87 by upstream from
the initiation codon with a sequence of 5'-TTTACT-3' while
a putative -10 region, 5'-TTAAGA-3' is centered 57 by
upstream (Fig 3). Both the -35 and -10 sequences were
predicted as putative sites using PC/Gene software package
(Intelligenetics, Inc.) and comparative analysis with
putative sites from published sequences from C. jejuni. A
potential termination sequence does existed 25 by down
stream from the stop codon 5'-TAA-3' (Fig 5). It
consisted of a 9 by dyad stem loop with a predicted
stability of -19.2 kcal/mol separated by 5 unpaired bases
which could comprise the loop structure. This is followed
by a poly-T region and could signify a rho-independent
termination sequence (1).
With very few exceptions, codon usage in the coding
region of porA gene are consistent with the compilation
available through Genebank (Table 3 below).
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-45-
Tahle 3
Codon usage chart for the 1275 by open reading frame porA
from C. jejuni strain 2483.
UAA - Ile AUC 9 Arg CGU ~
1
UAG - AUA 2 AGA 7
UGA - AUU 4 CGC 1
Ala GCU 35 Lys AAA 22 CGA 0
GCC 1 AAG 0 AGG 0
GCG 1 Leu CUU 17 CGG 0
GCA 11 CUA 4 Ser UCA 6
Cys UGU 0 CUG U UCC 0
UGC 0 UUG 0 UCG 0
Asp GAU 30 UUA 17 UCU 6
GAC 2 CUC 0 AGC 7
Glu GAG 2 Met AUG 2 AGU 6
GAA 15 Asn AAC 19 Thr ACG 1
Phe UUU 10 AAU 15 ACC 0
Gly GGC 3 Pro CCC 0 ACU i
4
GGA 9 CCU I ACA 14
GGG 0 CCA 3 Val GUC 1
GGU 32 CCG 0 GUG 2
His CAC 2 Gln CAG 2 GUU 12
CAU 2 CAA 13 GUA 20
Trp UGG 5
Tyr UAC 11
UAU 12
For instance, Tyr was equally encoded by UAU and UAC
while GUA was used more frequently to encode Val rather
than GUU and AUC encoded Ile instead of AUU. The most
striking difference was the number of Phe encoded by UUC
which had previously been shown to be encoded more
frequently by UUU while AAC rather than AAU encoded Asn.
These frequencies were most likely due to the quantity of
G+C residues in the coding region and these may confer an
increase in gene stability at increased temperatures
especially as the organism is considered to be
thermophilic.
The ORF [SEQ ID NO: 3] was found to produce a 45.6
kDa protein with a pI of 4.44 both of which are consistent
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-46-
with previous reports on the C. jejuni porin protein (3,
18, 21). The translated protein was also found to contain
several hydrophobic regions as determined by the method of
Novotny and Auffray (31) (PC/Gene). Structural prediction
using the method of Garnier (12) (PC/Gene) and Novotny
(31) indicated there was also considerable secondary
structure associated with the porin with 50% of the amino
acids forming extended or (3-pleated sheets conformations.
This was consistent with previous circular dichroism (CD)
findings (3) as well as with other bacterial porin
proteins (29). The number of residues necessary to span
the membrane has been estimated from Rhodobacter
capsulatus porin to be between 6 to 17 residues in length
(35). Based on this assumption, together with the
~i-pleated sheets diagram and hydrophobic chart, there may
be as many as 12 membrane spanning domains while the
enterobacterial consensus sequence (29) and R. capsulatus
both contain 8(3-strands (29) .
The relative amount of sequence identity was low
compared to the sequence similarity. This indicated that
although the primary structure was quite distinct, the
properties associated with the 424 amino acid protein were
similar to those of other well characterized porins. For
instance, the relative amounts of basic, polar and acidic
residues are similar to that of H. influenzae P2 as well
as the enterobacterial consensus sequence; however, there
was a greater frequency of hydrophobic residues in the
porin from C. jejuni (Table 4) .
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-47-
Table 4
Comparison of the amino acid composition of porA from G jejuni strain 2483,
H. influenzae P2 and the consensus sequence from enterobacterial porin.
Numbers in parenthesis represent residues in the leader sequence.
No. Residues/mol
in:
Amino Acid C. jejuni H. influenza Consensus
por P2a
Group A Enterobacterial
porinb
Basic
Lys 22 (2) 30 (2) 23
Arg 9 16 11
His 4 7 1
Acidic
Asp 32 17 34
Glu 17 24 17
Polar
Asn 34 (1) 25 (1) 24
Cys 0 0 0
Gln 15 14 17
Gly 44 ( 1 ) 40 ( 1 ) 40
Ser 25 (2) 17 (1) 18
Thy 29 24 ( 1 ) 22
Tyr 23 23 23
Hvdro~hobic
Ala 48 (8) 24 (8) 26
Ile 15 15 (1) 9
Leu 38 (4) 24 (2) 23
Met 2 (1) 1 (1) 5
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-48-
Phe 23 (1) 13 (1) 21
Pro 4 3 4
Trp 5 0 3
Val 35 (2) 24 (1) 15
a Data derived from Hansen et al. ( 17)
b Data derived from H. Nikaido (29)
This could coincide with more membrane spanning
regions leading to a more extensive secondary structure.
When the C. jejuni porin sequence was compared to the
hypothetical folding pattern of the enterobacterial
consensus sequence, there was no more significant
similarities in the membrane spanning regions of the
consensus sequence than in the remaining sections.
Comparison of the N-terminal sequences of H. pylori porin
proteins (9, 10) showed very little sequence identity with
the porin from C. jejuni. However, when the sequences were
compared for similarities, the porin from C. jejuni had
the greatest similarity with HopC (57%) followed by HopE
(550) then HopD and HopB (50%) and the least similarity
with HopA (470). The conductance levels of the channels
formed by the H. pylori porin range from 0.25 to 0.36 nS
(10) which is considerably lower than the conductance of
8.82 nS reported for the C. jejuni porin (18). The
molecular weights of each of the H. pylori porins are
greater than the porin from C. jejuni and also appear to
present as monomers in lipid bilayers (10) instead of the
trimeric form similar to C. jejuni porin (3).
The MOMP has been found to elicit an immune response
both in humans (30) and rabbits (11) making it a suitable
candidate for vaccine development. PCR studies to
determine the frequency of the porin gene in other strains
of C. jejuni showed that 95% of these contained at least
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-49-
part, if not all of the intact gene while the other
Campylobacter sp. and related organism were PCR negative.
Previous reports indicated that only 60% of pathogenic
strains possessed a protein of similar size as determined
by SDS-PAGE and Western blot analysis using antiserum
against the MOMP from C. jejuni strain 85H (21) (a sample
of which was deposited at the American Type Culture
Collection of 12301 Parklawn Drive, Rockville, MD 20852
USA on March 19, 1998 under the terms of the Budapest
Treaty, accession no. ATCC 202,102). The PCR results
outlined above are valuable, and provide a new and
efficient method to identify C. jejuni from other
Campylobacter sp. The potential for the development of a
recombinant vaccine using the porin protein is also
noteworthy. Previous studies by Gonzales et al.(13) have
shown that T-cell activation occurred through the
induction of lymhokines by S. typhi porin and, as a
consequence, this may play a role in protective immunity.
Protection in guinea pigs was seen by using the
enterobacterial outer membrane protein PhoE as a vector to
express B-cell epitopes on the surface of E. coli
providing a vehicle for live vaccine development (45).
EXAMPLE 2
MATERIALS AND METHODS
Bacterial strains and culture media
C. j ejuni strain 2483 was isolated from a patient
with gastroenteritis and was characterized as Lior
serotype 82, biotype 1 and Penner serotype 0:11. The
organism was passed twice on tryptic soy agar containing
5% sheep blood (TSA) following isolation from the patient
and was subsequently stored at -70°C in tryptic soy broth
containing 5% sheep blood. Thawed aliquots were cultured
RECT~F~ED SHEET (RULE 91)
ISA/EP
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-49a-
on TSA with 5°s sheep blood prior to inoculation into
Brucella broth (BBL, Cockeysville, MD, USA) pre-
equilibrated in an atmosphere containing 5% Oz, lo% COZ and
85% N2. For batch preparation, a suspension of the
RECTIFIES SHEET (RULE 91)
ISAIEP
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-50-
organism was made equivalent in density to a McFarland
number 8 standard and inoculated into 4 L of Brucella
broth at a density of 2 ml/L. Inoculated broths were
incubated under stationary conditions in the gas mixture
for 48 h at 37°C.
Isolation of cytotoxic complex
Bacteria were harvested by centrifugation at 12,000 x
g for 20 min at 4°C and the 4 L of organism-free filtrate
were concentrated by ultrafiltration at 4°C with a stirred
cell apparatus using a YM30 membrane (30,000 NMWL)
(Amicon, Beverly, MA, USA). The filtrate was initially
concentrated approximately 40-fold by ultrafiltration and
further by the addition of ammonium sulfate to 80%
saturation at 4°C. The ammonium sulfate precipitated
proteins were collected by centrifugation at 12,000 x g
for 30 min and resuspended in 50 mM Tris-HCL buffer, pH
7Ø Purification of the cytotoxic protein was performed
using a Hewlett Packard 1050 series high performance
liquid chromatograph (HPLC) equipped with a diode-array
detector. Purification was initiated by adding
concentrated filtrate at 1% of the total bed volume to a
HiLoadT"' 16/60 SuperdexTM 75 sizing column (Pharmacia
Biotech, Uppsala, Sweden) and eluting with phosphate
buffered saline, pH 7.0 (PBS) at a flow rate of 1 ml/min.
Fractions were collected on a Gilson fraction collector
and 50 ,ul of each was evaluated for cytotoxic activity
using HEp-2, HeLa and CHO cells. The molecular mass of
the native cytotoxic complex was determined by calibrating
the column using low molecular weight standards (Pharmacia
Biotech) dissolved in PBS. Cytotoxic-containing fractions
were pooled, concentrated using Centraprep-30 units
(Amicon), and applied to a 7.5 X 75 mm TSK DEAE-5PW column
(Pharmacia Biotech). Proteins were eluted using a linear
gradient of 0.2-0.25 M NaCl in 50 mM Tris-HC1, pH 7.0 at a
flow rate of 1 ml/min. Fractions were collected, desalted
by spin dialysis using Centricon-30 units (Amicon) and 50
CA 02284844 1999-09-24
WO 98/42842 PCT'/CA98/00272
-51-
/,cl of each sample was applied to monolayers of HEp-2, HeLa
and CHO cells for assessment of cytotoxic activity.
The bacterial pellet removed from the filtrate was
placed on ice and sonicated using a Bronson Sonifier 450T'"
sonicator (Branson Ultrasonic Corporation, Danbury, CT),
centrifuged at 12,000 X g for 10 min and the supernatant
assayed in the same manner as the filtrate.
Polymerase chain reaction
Genomic DNA was isolated from C. jejuni strain 2483
by standard procedures (70). Polymerase chain reaction
(PCR) was conducted as outlined previously (71) using
50 ng of genomic DNA with E. coli verotoxin VTla primers
(GAAGAGTCCGTGGGATTACG) [SEQ ID N0:24] and VTlb
(AGCGATGCAGCTATTAATAA) [SEQ ID N0:25] and VT2a
(TTAACCACACCCACGGCAGT) [SEQ ID N0:26] and VT2b
(GCTCTGGATGCATCTCTGGT) [SEQ ID N0:27) at 42°C, 45°C, and
50°C annealing temperatures. PCR was also conducted using
primers DZ3 (AGTAAGGAGAAACAATGA) [SEQ ID N0:28] and 8009
(AATAAGCCTTAGAGTCTTTTTGGAATCC) [SEQ ID N0:29] specific for
Helicobacter pylori cagA and primers F6
(GCTTCTCTTACCACCAATGC) [SEQ ID N0:30] and R20
(TGTCAGGGTTGTTCACCATG) [SEQ ID N0:31] specific for
H: pylori vacA gene as outlined previously (72). The
H. pylori Penner reference serotypes 05 and 06 were used
in PCR reactions as positive controls for the cagA and
vacA respectively. The PCR methodology used was as
described for cagA (72) except that 100 ng/reaction of
chromosomal DNA was used for amplification of the vacA
gene using 35 cycles of 95°C for 1 min, 58°C for 1 min and
72°C for 2 min with a final extension of 10 min. PCR
reactions were electrophoresed~ on a 1% agarose gel in
Tris-acetate, EDTA-containing buffer (pH 8.3), stained
with ethidium bromide and visualized on a transilluminator
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-52-
(Ultra-violet Products, Inc, San Gabriel, CA, USA).
Cytotoxic activity and protein determination
The amount of protein at each stage of the isolation
procedure was quantified using the BCA protein assay
(Pierce, Rockford, IL, USA). HEp-2, HeLa and CHO cells
were grown in T-75 cell culture flasks (Costar, Cambridge,
MA) using Eagle's minimal essential media (MEM)
supplemented with 10% fetal bovine serum (FBS) (Sigma, St.
Louis, MO, USA). Cells were subcultured into 96-well
plates 24 h prior to determination of cytotoxic activity.
Cytotoxic activity was quantified in the three cell lines
as described previously (54) and activities expressed
after 48 h incubation as tissue culture dose 50 (TCDso). A
TCDSO was defined as the amount of toxin required to cause
cytotoxic changes in 500 of the cells. Cell cultures were
fixed for 10 min in absolute methanol and stained for 30
min with GiemsaT"~ (Gibco BRL, Grand Island, NY, USA). The
specific activities were determined at each step of the
isolation and expressed as TCDso/ug of protein. E. coli
0157:H7 strain LCDC 3787 (H19), positive for VT1, and
strain LCDC 90-2380, positive for VT2, were used as
controls for TCDSO determination in HEp-2 and HeLa cells.
V. cholerae 01, strain 755, an enterotoxin-producing
isolate, was used as a control in the CHO cell assay.
Molecular weight and physical characterization of the
cytotoxic complex
One ,ug of the isolated cytotoxic material was mixed
with equal volumes of 2X sample buffer containing i3-
mercaptoethanol and sodium dodecyl-sulphate. The sample
was boiled for 5 min and separated by sodium dodecyl
sulphate polyacrylamide gel electrophoresis (SDS-PAGE) on
a 12% homogeneous gel along with low molecular weight
standards and silver stained using a commercial kit
(BioRad, Hercules, CA, USA). One /.cg aliquots of the
isolated cytotoxic material were either heated at 70°C for
30 min or treated with trypsin in PBS at concentrations
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-53-
ranging from 0.03 to 1.25% for 2 h at 37°C. Residual
trypsin activity was inactivated by addition of FBS to
give a final concentration of 20% for 1 h at 37°C. Heated
and trypsin-treated samples were serially diluted 2-fold
in PBS prior to cell culture assay to determine the degree
of activity remaining after the treatments. Heat
inactivated trypsin and FBS alone were used as negative
controls.
N-terminal sequencing of cytotoxin
The cytotoxic component was isolated and denatured
with SDS and i3-mercaptoethanol and electrophoresed along
with kaleidoscope prestained molecular weight standards
(BioRad) on a 12o gel SDS-PAGE. Following
electrophoresis, the protein and standards were
electrophoretically transferred to polyvinylidene
diflouride (PVDF) (BioRad) for 18 h at 100 mA in 10 mM 3-
[cyclohexylamino]- 1-propanesulfonic acid (CAPS) (Sigma)
buffer, pH 11.0 containing 10% methanol. Following
transfer, the blot was stained with 0.1% Coomassie blue R-
250 (BioRad) in 50% methanol for 5 min and destained with
50% methanol and 10% acetic acid. The immobilized
cytotoxic protein was excised from the PVDF and sequenced
by Edman degradation on an Applied Biosystems model 473A
protein sequencer (CHUL Research Center, Saint-Foy,
Quebec, Canada). Protein analysis was performed using
Lasergene (DNAStar, Madison, WI, USA).
Neutralization and Western blot analysis
Neutralization studies were performed on 1 ,ug
aliquots of the isolated complex using polyclonal antisera
raised against the cytotoxic complex from C. jejuni, as
well as against E. coli VT1, E. coli VT2, CDT from
C. jejuni (54) and CDT from E. coli (54) , enterotoxin from
V. cholerae (Sigma) and the cytotoxin from C. difficile
(Techlab, Blacksburg, VA, USA). Normal rabbit serum was
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-54-
used as a negative control. Homologous antiserum was
raised by intramuscular inoculation of New Zealand white
rabbits with 0.5 ml of a 5 ,ug/ml preparation of isolated
cytotoxic material emulsified in 0.5 ml Freund's
incomplete adjuvant (FIA). This was followed at weekly
intervals for 4 weeks by subcutaneous injection of the
same antigen preparation in FIA. A 1:10 dilution of each
antiserum was added to serial two fold dilutions of 1 ,ug
of the isolated protein. After a 1 h incubation at 37°C,
aliquots of each were added to HEp-2 cells and incubated
for 48 h at 37°C. Each antiserum was also assayed using
Western blot analysis. C. jejuni cytotoxic complex, E.
coli VT1 and VT2, C. jejuni CDT, E. coli CDT, C. difficile
cytotoxin and V. cholera enterotoxin were each separated
on SDS-PAGE gels along with kaleidoscope prestained
molecular weight standards (BioRad) and transferred to 0.2
~cm pore size PVDF membranes for 18 h at 100 mA. Membranes
were washed with 5% skim milk for 1 h to prevent
nonspecific binding of the antibodies and then washed 3
times for 5 min each with PBS. A 1:500 dilution of each
of the antisera in a 1% skim milk solution containing
0.05% TweenT~~-20 was prepared and added to the membranes
for 2 h at room temperature. This was followed by a 1 h
treatment with 200 mU/ml of goat anti-rabbit alkaline
phosphatase conjugated antibody (Boehringer Mannheim,
Laval, Quebec, Canada) at room temperature. Membranes
were developed using 5-Bromo-4-chloro-3-indolyl-phosphate
(BCIP) and nitro blue tetrazolium (NBT) (Boehringer
Mannheim) .
Western blot analysis was also performed using crude
concentrated filtrates from C. jejuni strain 2483, C.
jejuni LCDC 3969, C. jejun.i LCDC 16336, C. coli strain
8682, Aeromonas veronii LCDC A2297 (negative control) and
E. coli 3787 (positive control for VT1) . A total of 40 ,ug
of each crude filtrate was electrophoresed and transferred
to PVDF as stated previously and probed with anti-
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98100272
-55-
cytotoxic complex from C. jejuni. In addition, each
filtrate was tested for cytotoxic activity in HEp-2 cells
after 48 h of incubation.
Carbohydrate characterization
A total of 2 lcg of the isolated cytotoxic material
was assayed for the presence of lipopolysaccharide by the
Limulus amebocyte lysate (LAL) test according to the
package insert (Pyrotell, Associates of Cape Cod, Inc.,
MA, USA). The toxic material along with E.coli LPS were
each diluted 10-fold with pyrogen free water in duplicate
and 100 /.cl of each dilution were incubated with 100 ,ul of
PyortellT"~ in a 37°C water bath for 1 h. Tubes were
inverted and those containing a solid clot were considered
positive. To determine whether the lipopolysaccharide
contributed the activity of the toxin, 1 ~g of the
isolated cytotoxic material was incubated for 1 h at 37°C
with 5 U neuraminidase (Sigma) at pH 5.0, 3 U of N-
glycosidase F (Boehringer Mannheim) at pH 7.2 and 10 mM
sodium metaperiodate (Sigma) for 90 min at room
temperature. The residual cytotoxic activity was then
assayed in HEp-2 and HeLa cells using serial twofold
dilutions.
Identification of the carbohydrate moiety was made
using a glycan differentiation kit (Boehringer Mannheim)
containing five unique digoxigenin-labeled lectins
(Table 5). Approximately 1 ~g of the isolated cytotoxic
protein and 5 /,cg of each carbohydrate standard were
spotted on PVDF membranes and allowed to dry overnight at
37°C. Membranes were probed for the co-purifying LPS
according to the manufacturer's insert instructions.
Those lectins that gave positive results were further
examined by Western blot analysis using 15 ,ug of each
carbohydrate standard and 8 lcg of the test carbohydrate
from the purified preparation. The isolated cytotoxic
material from three separate batches was assayed in ,ug for
CA 02284844 1999-09-24
WO 9$/42842 PCT/CA98/00272
-56-
the carbohydrate concentration using a phenol-sulphuric
acid assay measured at 490 nm (73). This was expressed as
a ratio to the number of ,ug of purified carbohydrate per
tcg of purified protein. SDS-PAGE and native PAGE were
performed using 10 ,ug of the carbohydrate and gels were
double stained, first with periodic acid-Schiff (PAS) (74)
then with Coomassie blue.
Table 5
Specificities and the reactions of the lectins
used in the carbohydrate determination
Lectins Specificity (linkage) Reactivity*
Galanthus nivalis Mana(1-3), a(1-6) or a(1-2)-Man+++
agglutinin (GNA) (terminally linked mannose)
Maackia amurensis NeuSAca(2-3)-Gal (sialic acid+
agglutinin (MAA) terminally linked a(2-3) to
galactose)
Daturastramonium Gal~3(1-4)GicNac(galactosc-(3(1-4)-N-+
agglutinin (DSA) acetylglucosamine)
Arachis hypogaea Gal(3(1-3)GalNac (galactose-X3(1-3)-N--
(peanut)
agglutinin (PNA) acetylgalactosamine)
Sambucus nigra agglutininNeuSAca(2-6)-Gal or GalNac -
(sialic
(SNA) acid terminally linked a(2-6)
to
galactose or N-acetylgalactosamine)
*+++ strong positive result; + weak positive result; - negative result
Results
Identification and molecular characterization of cytotoxic
complex
Cytological signs of intoxication caused by the
5 cytotoxic complex included the formation of vacuoles in
the cytoplasm of HEp-2 cells as compared with normal
unaffected cells (Fig. 6a and 6b). Similar results were
found with HeLa cells. The number of vacuoles in each
cell ranged from 1 to 5 with 50% or more of the cytoplasm
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-57-
of some cells being affected. After 24 h, the vacuoles
diminished in size and the cells developed a rounded,
highly refractile appearance. By 48 h, cytoplasmic
blebbing and nuclear condensation became more evident
along with cell loss from the monolayer (Fig, lc and
inset). Toxicity was dose-dependent and was detected
using 2-fold serial dilutions of the isolated material.
At the lower cytotoxin concentration of 1 ,ug of
protein/well, the vacuoles persisted up to 48 h while
rounding occurred up to 72 h. When higher cytotoxin
concentrations of 10 ~g protein/well were used; vacuoles
formed and dissipated within the first 12 h following
intoxication and greater than 50% of the cells were
rounded and refractile by 24 h. By 48 h, 80-100°s of the
cells had become rounded (Fig. 6c). Similar cytological
changes were observed in all of the cell lines when the
whole bacterial cell sonicate was assayed for toxicity.
Strain 2483 produced low levels of CDT in the crude
concentrate; however, this was neutralizable with
polyclonal antisera raised against either C. jejuni or E.
coli CDT (data not shown).
The organisms were grown for 48 h at 37°C in Brucella
broth at which time the bacteria were in the stationary
phase of growth. Concentrated proteins from the culture
supernatant possessing high levels of cytotoxic activity
were found to elute from the G75 column in the void volume
with a calculated native molecular mass of greater than
100 kDa (Fig 7a). This peak (peak A) was collected and
applied to the TSK DEAE-5PW column. Cytotoxic activities
of the TSK DEAE-5PW fractions showed the toxin eluted at
approximately 0.21-0.22 M NaCl (Fig 7b). The two-column
purification procedure produced a single silver-stained
protein with a molecular size of 45 kDa calculated by Rf
under denaturing conditions. The toxic activities at each
stage of the purification procedure are shown in Table 6.
The isolated cytotoxic complex demonstrated highest toxic
activity for HEp-2 cells and the lowest for CHO cells.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-58-
The cytotoxin was inactivated by heat treatment at 70°C
for 30 min but was resistant to trypsin at the
concentrations tested.
Table 6
Specific activity expressed as TCDso/,ug of
protein at each step of the purification
in Hep-2, HeLa and CHO cells.
Cell
Culture
Specific
Activity*
Toxin Hep-2 HeLa CHO
Preparation
C. jejuni Strain 2483
crude concentrate 1.56 0.51 0.51
Superdex 75 16/60 1.61 3.88 0.97
TSK DEAE-5PW 20.1 7.49 1.87
E. coli VT1+ Strain LCDC 3787 (H19) 0.35 0.17 NA
E. coli VT2+ Strain LCDC 90-2380 1.48 2.89 NA
V. cholerae O1, Strain 755 NA NA 0.48
*TCDso/,ug of protein; NA=no activity
Polymerase chain reaction
Oligonucleotide primers specific for E. coli VT1 and
VT2 failed to produce amplicons corresponding to A- and B-
subunits of mature verotoxin types 1 and 2. Also, primers
specific for the cagA and vacA genes of H. pylori failed
to generate amplicons.
Protein Alignment
The cytotoxic protein consisted of a single protein
with a calculated molecular mass of 45 kDa. The excised
band was subjected to N-terminal sequencing and a total of
31 amino acid resides were elucidated (Table 7). The
protein was found to contain several hydrophobic and
charged residues and had a predicted isoelectric point of
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-59-
4.35. The protein had 97% homology with the major outer
membrane protein (MOMP) from C. jejuni which has been
characterized as a porin (68) and the single amino acid
difference at residue 30 was conserved. The cytotoxic
porin also shared 56% and 63% sequence homology with 45
kDa and 51 Kda outer membrane proteins respectively from
Wolinella recta (75).
Table 7
Sequence homologies of the C. jejami 2483 cytotoxic porin and
related sequences as ascertained by BLAST' searches
Protein designation Sequence'-
C. jejuni cytotoxic porin protein TPLEEAIKDVDVSGVLRYRYDTGNFDKNFVN
C. jejuni MOMP; porin protein TPLEEAIKDVDVSGVLRYRYDTGNFDKNF*N
W. recta 45 kDa outer membrane
protein TPLEEAIKDVD-SG---XY-*---X-N--
W. recta 51 kDa outer membrane
protein TPLEEAIK*VD*SG--XYXY*----KN--
' Beckman Center for Molecular and Genetic Medicine, Stanford University
School of
Medicine.
Z Capital letters represent identical residues; "*" represent conserved
changes; "-" represents
mismatch in sequences; "X" represents unknown residue.
Neutralization and Western blot analysis
Polyclonal antisera raised against E. coli VT 1 and
VT 2, C. jejuni and E. coli CDT, V. cholerae enterotoxin
and C. difficile cytotoxin failed to neutralize the
5 cytotoxic effects elicited by the C. jejuni toxic complex
in cell culture. However, when this cytotoxic complex was
serially diluted, incubated with rabbit polyclonal
antiserum raised against the cytotoxic protein and added
to HEp-2 cells, the TCDSO occurred at a dilution of 1:2
whereas that of the normal rabbit serum was at a dilution
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-60-
of 1:32. Neutralization was defined as a decrease in the
TCDSOat 24 h post-intoxication. Antiserum raised against
the cytotoxic protein showed immunological reactivity in
Western blots with the purified 45 kDa cytotoxic protein.
Antisera raised to the other toxins showed no cross
reactivity with either the cytotoxin or carbohydrate on
immuno-blot analysis. Western blots of crude
concentrated filtrates from various cytotoxic strains of
Campylobacter species showed the presence of a protein
l0 with a molecular mass similar to that of the porin
(Fig. 8) while no bands were detected in the blots from
the uninoculated broth, and filtrates from A. veronii and
E. coli VT1 strains.
Lipopolysaccharide identification and carbohydrate
analysis
The isolated cytotoxic material and E.coli LPS were
assayed for the presence of endotoxin by incubating serial
dilutions with Limulus amebocyte lysate for 1 h at 37°C.
The cytotoxic material produced a strong positive result
at a dilution of 1:128,000 signifying that the isolated
cytotoxic material contained LPS. The E.coli LPS also
gave a positive result. To determine whether or not the
cytotoxic activity associated with the complex reside in
the protein component, the complex was incubated with 10
mM sodium metaperiodate, to oxidize the free hydroxyl
groups present on visceral hexoses, with 5 U
neuraminidase, to cleave sialic acid residues and with 3 U
N-glycosidase F to cleave asparagine bound N-glycans. The
complex was then assayed for cytotoxic activity in HEp-2
and FieLa cells and expressed as TCDSOendpoints. Titers of
32 were observed in the HEp-2 cells while a titer of 8 was
found in the HeLa cells as well as for the control cells
inoculated with untreated toxin.
The carbohydrate component of the LPS was
characterized using digoxigenin-labeled lectins (Table 5)
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-51-
and the data revealed a complex of different subunits.
Lectin Galanthus nivalis agglutinin (GNA) reacted strongly
with the purified material and suggested a high proportion
of terminally-linked mannose. The lectins Maackia
amurensis agglutinin (MAA) and Datura stramonium
agglutinin (DSA) also gave positive but weaker results,
indicating the presence of sialic acid terminally linked
a(2-3) galactose and galactose-f~(1-4)-N- acetylglucosamine
in the complex as well as hybrid N-glycan structures. The
remaining lectins showed no reactivity for the
carbohydrate complex. The proportion of carbohydrate to
protein in the purified material was calculated at a ratio
of 4:1. PAS staining revealed a high molecular weight
carbohydrate which did not appear as a discrete band as
did the protein component of the complex but, instead,
occupied a broad range of sizes (Fig. 9). Double staining
of purified cytotoxin in native PAGE gels showed no
protein component in contrast to samples boiled in
denaturing buffer prior to gel electrophoresis (Fig. 9).
Western blots performed with the lectins (Fig. 10) showed
that the high molecular mass smear seen following PAS
staining (Fig. 9) was carbohydrate in nature with high
reactivity for GNA (Fig. 10).
Discussion
A cytotoxin from strains of C. jejuni that was heat-
labile, trypsin-sensitive and induced characteristic
rounding of FiEp-2, HeLa and MRC-5 cells was first
documented by Yeen et aI. (76). Guerrant et aI. (58) also
described a cytotoxic component which was heat labile at
60°C, was partially sensitive to 0.25% trypsin and had a
molecular weight greater than 14 kDa. The cytotoxic
component identified by these workers could not be
neutralized using antisera raised against E. coli
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-62-
verotoxins or C. difficile toxin (58). A subsequent report
indicated the presence of a Shiga-like cytotoxin from
C. jejuni which could be neutralized with monoclonal
antibodies directed against the B subunit of the mature
Shiga-toxin; however, these workers also detected a
cytotoxin which could not be neutralized by the same
monoclonal antibody (77). In addition, Guerrant and
colleagues (58), unlike Yeen et a1. (76), found cytotoxic
activity in sonicated whole bacterial cell preparations.
In the present study cytotoxic activity was detected both
in culture and in sonicated filtrates of whole C. j ejuni
strain 2483 bacterial cells.
A cytotoxic complex comprising a porin and LPS was
isolated and characterized. Previous studies showed a
cytotoxic factor present in LPS-rich fractions from
C. jejuni (60); however, it was not known what role the
LPS played in toxicity. Misawa et a1. (78) found that the
expression of their cytotoxin was elevated when the
C. jejuni was grown in Brucella broth. However, contrary
to the findings of these workers, it was determined in the
present work that HEp-2 cells showed the highest
sensitivity to the cytotoxic complex and that these
activities were consistently higher whether or not the
cell cultures were grown in media supplemented with FBS
(78). The increase in activities observed in the
different cell lines may be due to the relative amounts of
the receptor required for binding of the porin-LPS
complex. Previous reports have implicated LPS in the
adhesion of C. jejuni to epithelial cells as well as to
intestinal mucus and also showed that this process could
be inhibited by periodate oxidation (49). Since the
cytotoxic activity of the porin-LPS was maintained
following treatment with periodate in both HEp-2 and HeLa
cells, it would appear that adhesion of the toxic complex
is facilitated by components other than LPS. It is
possible that expression of the porin protein may be
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/002'72
-63-
involved in binding the organism to host cells; however,
Fauchere et a1. (79) indicated that the MOMP was not
involved in adherence to HeLa cells. From these studies
it would appear that, although the LPS mediates attachment
of organisms to host cells (49), the porin component binds
the cytotoxic complex.
Although the mode of action of the cytotoxic porin
remains unclear, the morphological changes induced by it
are similar in nature to other well characterized
bacterial cytotoxins. During early stages of intoxication
the cytotoxic porin induced vacuole formation in HEp-2 and
HeLa cells and this was similar in appearance to those
produced in response to H. pylori vacuolating toxin (80).
Even though vacuolation following intoxication with
C. jejuni cytotoxin was shown here, no PCR products were
generated using primers specific for cagA and vacA genes
(72) suggesting that the genes encoding vacuole induction
by the C. jejuni porin are unique from the genes carried
and expressed in H. pylori .
When intoxication of host cells with the C. jejuni
cytotoxic porin was extended beyond 24 h, vacuoles
dissipated while the cytoplasmic blebbing and nuclear
condensation typical of verotoxin and diphtheria toxin
became more evident. Verotoxin and diphtheria toxin are
both known to interfere with protein synthesis leading to
programmed cell death or apoptosis (81,82). PCR-based
screening of C. jejuni using verotoxin-specific primers
was negative and confirmed the low stringency
hybridization experiments of Moore et aI. (77) and this
suggested that the C. jejuni cytotoxic complex was
distinct from verotoxin. It is possible that the porin
from C. j ejuni induces holes in the cell membrane in a
manner similar to that resulting from Staphylococcus
aureus a-toxin (82). Recently, the cytotoxic effects
elicited by Salmonella Typhimurium porin suggested that
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-64-
porins directly affect the cytoskeleton and the membrane
ultrastructure of HEp-2 cells (83). In addition, porins
from Neisseria sp. have also been shown to inhibit
polymerization of actin in human neutrophils (84) while
porins from S. Typhimurium have been found to induce both
an inflammatory response (85) and the release of cytokines
from human monocytes and lymphocytes (86).
Attempts to determine the isoelectric point of the
cytotoxic protein by Coomassie blue staining and probing
by Western blots of isoelectric focusing (IEF) gels with
antisera raised against the complex were unsuccessful.
Isolation of the cytotoxic porin protein using a
chromatofocusing column and polybuffer 7-4 (Pharmacia
Biotech) was difficult to reproduce due to probable
interference from LPS. The isolation protocol was also
applied to a cell-free filtrate from C. jejuni strain 3969
which had previously been reported to produce a cytotoxin
(59,78,87). Although the strain produced a lower
cytotoxic activity, similar morphological changes were
observed in HEp-2 cells and a protein of similar size was
observed following SDS-PAGE. A protein of comparable
molecular weight was also present in crude concentrated
filtrates from other cytotoxic strains of Campylobacter
sp., indicating that the release of the porin-LPS complex
was not unique to C. jejuni strain 2483. Carbohydrates
were also present in the cytotoxic product isolated from
C. jejuni strain 3969. Although this strain was
untypeable with available Lior antisera, it proved to be a
biotype l, Penner serotype 0:50. The differential in
cytotoxic activity between strain 3969 (low toxin
activity) and 2483 (high toxin activity) could be a
growth-rate dependent phenomenon since the release of the
porin-LPS complex may occur most avidly during cell death
or may be lost during active replication of the organism.
Strains with a higher growth rate could therefore produce
quantitatively more complex (88). Recently a vacuolating
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-65-
cytotoxin similar to that produced by H. pylori was
detected in the stools of children with diarrhea, even
though no etiologic diarrheal agent was identified (89).
Although many organisms have been attributed with the
ability to induce vacuole formation in host cells, this
process may be porin-mediated following release from dead
or dying organisms (88).
Lectin studies showed that the carbohydrate portion
of the LPS which co-purified with the porin possessed
terminally-linked mannose as well as sialic acid
terminally-linked to a(2-3} galactose and galactose-f~(1-
4)-N-acetylglucosamine complexed together with hybrid N-
glycan structures. Based on the thermostable somatic (O)
antigen, the strain of C. jejuni used in this study was
type 0:11. The positive result with the lectin MAA
suggests that the strain may be related to serotype 0:19
(90}. The LPS from the 0:19 serostrain of C. jejuni has
core structures that mimic those present on GM1 and Gola
gangliosides and other strains of 0:19 have been linked to
post infectious neuropathies (91). The presence of
terminally-linked mannose in the LPS of the 0:11 serotype
in this study may have significance. Treatment of the
isolated complex with sodium meta-periodate, neuraminidase
and N-glycosidase F had no effect on the toxicity elicited
by the complex, suggesting that the LPS is not an integral
component of the cytotoxic activity but that it may play a
protective role. Indeed, it is possible that it may have
interfered with the enzymatic degradation by trypsin and
may offer an explanation for the disparity in trypsin
inactivation data of previous reports. Under native
conditions, the LPS likely forms complexes with the porin
and protects it from discrete staining with Commassie
blue. The cytotoxic protein is only revealed by Coomassie
blue or silver staining after boiling in sample buffer
containing SDS and !3-mercaptoethanol prior to SDS-PAGE.
Since the porin from C. jejuni has been classified as part
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-66-
of the trimeric porin family, it was not unexpected that
it must be heat denatured in order to resolve the protein
component of the cytotoxic complex (63).
When characterization of the cytotoxic porin is more
complete and the encoding gene has been cloned and
sequenced, a fuller understanding of the role of the porin
in clinical campylobacteriosis will be forthcoming. Such
evaluations may suggest potential roles for the porin-LPS
complex as a diagnostic tool for the detection of either
the organism or its cytotoxin or additionally as a
recombinant vaccine for prevention and control of
Campylobacter disease.
EXAMPLE 3
Screenings were conducted of 23 strains of C. jejuni
and 9 strains of related organisms for phenotypic
expression of a cytotoxin and presence of porA using
primers specific for the porin gene sequenced from C.
jejuni strain 2483. The results are shown in Table 8
below.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-67-
Table 8
Organism LCDC SourceLior BiotypeToxin PCR
number Serotype positivepositive
C. jejuni 3454 human 4 Ia + +
3969 NA untypableI + +
4951 human 7 I + +
4966 human 7 1 + +
6847 human 1 Ia + +
7099 chicken61 + +
7288 water 9 11 + +
8916 human 94 Ila + +
9214 human 2 la + +
9541 water 82 II + +
9543 water 82 lI + +
9555 human 23 1 + +
10403 human 36 la + -
10673 human 82 II + +
14040 human 82 II + +
14906 human 82 I + +
15151 human 82 1 + +
16323 beef 82 1 + +
16334 human 82 II + +
16336 human 82 II + +
16388 human 82 11 + +
(2483)
1 NA 4 I + +
2074 NA 36 II + -
C.lari 729 NA 31 1 + -
S C. coli 348 NA 14 I + -
C. sputorum 5754 NA NT NT + -
subsp.
fecalis
C. fetus subsp.7055 NA NT NT +
fetus
C. hyointestinalis8494 human NT NT +
1 C. jejuni subsp.9365 NA NT NT + -
~ doylei
A. burileri 13220 human 7 IILA +
E. coli VTI+ 3787 (19)NA NT NT + -
E. coli VT2+ H19 human NT NT + -
NT not tested; NA information not available
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-68-
The above results show that the porin of this invention is
conserved in C. jejuni and C. coli both by phenotypic
expression (toxin positive) and genotypic presence (ICR
positive). This is a significant advantage over the
Bleser gene which is not highly conserved.
EXAMPLE 4
PorA from C. jejuni strain 2483 according to this
invention was compared against H. influenzae P2 and C.
jejuni FlaA. This was done by obtaining hydrophobic
profiles and beta sheet propensities as determined by the
method of Novotny (31) using the PC/Gene
CA 02284844 1999-09-24
WO 98142842 PCT/CA98/00272
-69-
SEQUENCE LISTING
(1) GENERAL INFORMATION:
(i) APPLICANT:
(A) NAME: Her Majesty the Queen in Right of Canada as
represented by the Minister of Health and Welfare, Canada
(B) STREET: Brook Klaxton Buildings
(C) CITY: Ottawa
(D) STATE: Ontario
(E) COUNTRY: Canada
(F) POSTAL CODE (ZIP): K1A OK9
(A) NAME: JOHNSON, Wendy M.
(B) STREET: 1 - 590 MacLaren St.
(C) CITY: Ottawa
(D) STATE: Ontario
(E) COUNTRY: Canada
(F) POSTAL CODE (ZIP): K1R 5K9
(A) NAME: BACON, David J.
(B) STREET: 3201 Whispering Pines Drive, Apt. #12
(C) CITY: Silver Springs
(D) STATE: MD
(E) COUNTRY: U.S.A.
(F) POSTAL CODE (ZIP): 20906
(A) NAME: RODGERS, Frank G.
(B) STREET: 9 Browning Drive
(C) CITY: Dover
(D) STATE: New Hampshire
(E) COUNTRY: U.S.A.
(F) POSTAL CODE (ZIP): 03820
(A) NAME: BOLLA, Jean-Michel
(B) STREET: 30 rue Rabutin Chantal
(C) CITY: Marseille
(D) STATE:
(E) COUNTRY: France
(F) POSTAL CODE (ZIP): 13009
(ii) TITLE OF INVENTION: A PORIN GENE FROM CAMPYLOBACTER JEJUNI,
RELATED PRODUCTS AND USES THEREOF
(iii) NUMBER OF SEQUENCES: 31
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-70-
(iv) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
(C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: PatentIn Release #1.0, Version #1.30 (EPO)
(v) CURRENT APPLICATION DATA:
APPLICATION NUMBER: WO PCT/CA98/
(vi) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: US 60/041,200
(B) FILING DATE: 25-MAR-1997
(2) INFORMATION FOR SEQ ID NO: 1:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 1950 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: double
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(iii) HYPOTHETICAL: NO
(iv) ANTI-SENSE: NO
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Campylobacter jejuni
(B) STRAIN: 2483
(xi) SEQUENCE DESCRIPTION: 5EQ ID N0: 1:
CCTTCGGATT TAAAATTTTT ACTATTTTAA GTGCTTCTTA AGAAAAAACT CCAAATTTAT 60
GTGCTACAAT TACAATGTTT TATTAATTTT TGACAAGGAG AATTCTCATG AAACTAGTTA 120
AACTTAGTTTAGTTGCAGCTCTTGCTGCAGGTGCTTTTTC AGCAGCTAAC GCTACTCCAC180
TTGAAGAAGCTATCAAAGATGTTGATGTATCAGGTGTATT AAGATACAGA TACGATACAG240
GTAATTTTGATAAAAATTTCGTTAACAACTCAAATTTAAA CAACAACAAA CAAGATCACA300
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-71-
AATATAGAGCACAAGTTAAC TTCAGTGCTGCTATAGCTGATAACTTCAAA GCTTTCATTC360
AGTTTGACTACAACGCTGTT GATGGTGGCACTGGTGTTGATAACGTAACA AATGCCGAAA420
AAGGACTTTTTGTTCGTCAA TTATACTTAACTTATACAAATGAAGATGTT GCTACAAGTG980
TAATCGCTGGTAAACAACAA TTAAACCTTATCTGGACGGATAACGCTATT GATGGTTTAG540
TAGGAACAGGTATCAAAGTA GTAAACAACAGCATCGATGGTTTAACTCTA GCTGCTTTTG600
CTGTAGATAG CTTTATGGCG GAAGAGCAAG GTGCAGATTT ATTAGGACAA AGTACTATAT 660
CTACAACACAGAAAGCAGCTCCTTTTAAAGTGGATTCAGT AGGAAATCTTTATGGTGCTG720
CTGCTGTAGGTTCTTATGATCTTGCTGGCGGACAATTTAA TCCACAATTATGGTTAGCTT780
ACTGGGATCAAGTAGCATTCTTCTATGCTGTAGATGCAGC TTATAGTACAACTATCTTTG840
ATGGAATCAACTGGACACTTGAAGGTGCTTACTTAGGAAA TAGCCTTGATAGCGAACTTG900
ATGATAAAACACACGCTAATGGCAATTTATTTGCTTTAAA AGGTAGCATTGAAGTAAATG960
GTTGGGATGC TAGCCTTGGTGGTTTATACTACGGTGATAA TCTACAGTCG1020
AGAAAAAGCT
TAATCGAAGA TCAAGGTAATCTTGGTTCTTTACTTGCAGGTGAGGAAATTTTCTATACTA1080
CTGGTTCAAG ACTAAATGGTGATACTGGTAGAAATATCTTCGGTTATGTAACTGGTGGAT1140
ATACTTTCAA CGAAACAGTTCGCGTTGGTGCTGACTTCGTATATGGTGGAACAAAAACAG1200
AAGATACTGC TCATGTAGGTGGTGGTAAAAAACTTGAAGCTGTTGCAAGAGTAGATTACA1260
AATACTCTCC AAAACTTAACTTCTCAGCATTCTATTCTTATGTGAACCTAGATCAAGGTG1320
TAAACACTAA TGAAAGTGCT GATCATAGCA CTGTAAGACT TCAAGCTCTT TACAAATTCT 1380
AAGAAGCTTT CAAGTCTAAC TTCAAGGCGG AGTTTTGCTC CGCCTTTTTT TATGCCTGAT 1490
TTTTAAAACT 1450
(2) INFORMATION FOR SEQ ID NO: 2:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 424 amino acids
(B) TYPE: amino acid
(C) STRANDEDNESS: unknown
CA 02284844 1999-09-24
WO 98142842 PCT/CA98/00272
-72-
(D) TOPOLOGY: unknown
(ii) MOLECULE TYPE: protein
(v) FRAGMENT TYPE: N-terminal
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Campylobacter jejuni
(B) STRAIN: 2983
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 2:
Met Lys Leu Val Lys Leu Ser Leu Val Ala Ala Leu Ala Ala Gly Ala
1 5 10 15
Phe Ser Ala Ala Asn Ala Thr Pro Leu Glu Glu Ala Ile Lys Asp Val
20 25 30
Asp Val Ser Gly Val Leu Arg Tyr Arg Tyr Asp Thr Gly Asn Phe Asp
35 40 95
Lys Asn Phe Val Asn Asn Ser Asn Leu Asn Asn Asn Lys Gln Asp His
50 55 60
Lys Tyr Arg Ala Gln Val Asn Phe Ser Ala Ala Ile Ala Asp Asn Phe
65 70 75 80
Lys Ala Phe Ile Gln Phe Asp Tyr Asn Ala Val Asp Gly Gly Thr Gly
85 90 95
Val Asp Asn Val Thr Asn Ala Glu Lys Gly Leu Phe Val Arg Gln Leu
100 105 110
Tyr Leu Thr Tyr Thr Asn Glu Asp Val Ala Thr Ser Val Ile Ala Gly
115 120 125
Lys Gln Gln Leu Asn Leu Ile Trp Thr Asp Asn Ala Ile Asp Gly Leu
130 135 140
Val Gly Thr Gly Ile Lys Val Val Asn Asn Ser Ile Asp Gly Leu Thr
145 150 155 160
Leu Ala Ala Phe Ala Val Asp Ser Phe Met Ala Glu Glu Gln Gly Ala
165 170 175
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-73-
Asp Leu Leu Gly Gln Ser Thr Ile Ser Thr Thr Gln Lys Ala Ala Pro
180 185 190
Phe Lys Val Asp Ser Val Gly Asn Leu Tyr Gly Ala Ala Ala Val Gly
195 200 205
Ser Tyr Asp Leu Ala Gly Gly Gln Phe Asn Pro Gln Leu Trp Leu Ala
210 215 220
Tyr Trp Asp Gln Val Ala Phe Phe Tyr Ala Val Asp Ala Ala Tyr Ser
225 230 235 240
Thr Thr Ile Phe Asp Gly Ile Asn Trp Thr Leu Glu Gly Ala Tyr Leu
245 250 255
Gly Asn Ser Leu Asp Ser Glu Leu Asp Asp Lys Thr His Ala Asn Gly
260 265 270
Asn Leu Phe Ala Leu Lys Gly Ser Ile Glu Val Asn Gly Trp Asp Ala
275 280 285
Ser Leu Gly Gly Leu Tyr Tyr Gly Asp Lys Glu Lys Ala Ser Thr Val
290 295 300
Val Ile Glu Asp Gln Gly Asn Leu Gly Ser Leu Leu Ala Gly Glu Glu
305 310 315 320
Ile Phe Tyr Thr Thr Gly Ser Arg Leu Asn Gly Asp Thr Gly Arg Asn
325 330 335
Ile Phe Gly Tyr Val Thr Gly Gly Tyr Thr Phe Asn Glu Thr Val Arg
390 345 350
Val Gly Ala Asp Phe Val Tyr Gly Gly Thr Lys Thr Glu Asp Thr Ala
355 360 365
His Val Gly Gly Gly Lys Lys Leu Glu Ala Val Ala Arg Val Asp Tyr
370 375 380
Lys Tyr Ser Pro Lys Leu Asn Phe Ser Ala Phe Tyr Ser Tyr Val Asn
385 390 395 400
Leu Asp Gln Gly Val Asn Thr Asn Glu Ser Ala Asp His 5er Thr VaI
405 410 915
Arg Leu Gln Ala Leu Tyr Lys Phe
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-74-
920
(2) INFORMATION FOR SEQ ID NO: 3:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 1275 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: double
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Campylobacter jejuni
(B) STRAIN: 2983
(xi)
SEQUENCE
DESCRIPTION:
SEQ
ID N0:
3:
ATGAAACTAGTTAAACTTAGTTTAGTTGCAGCTCTTGCTGCAGGTGCTTTTTCAGCAGCT60
AACGCTACTCCACTTGAAGAAGCTATCAAAGATGTTGATGTATCAGGTGTATTAAGATAC120
AGATACGATACAGGTAATTTTGATAAAAATTTCGTTAACAACTCAAATTTAAACAACAAC180
AAACAAGATCACAAATATAGAGCACAAGTTAACTTCAGTGCTGCTATAGCTGATAACTTC240
AAAGCTTTCATTCAGTTTGACTACAACGCTGTTGATGGTGGCACTGGTGTTGATAACGTA300
ACAAATGCCGAAAAAGGACTTTTTGTTCGTCAATTATACTTAACTTATACAAATGAAGAT360
GTTGCTACAAGTGTAATCGCTGGTAAACAACAATTAAACCTTATCTGGACGGATAACGCT920
ATTGATGGTTTAGTAGGAACAGGTATCAAAGTAGTAAACAACAGCATCGATGGTTTAACT480
CTAGCTGCTTTTGCTGTAGATAGCTTTATGGCGGAAGAGCAAGGTGCAGATTTATTAGGA590
CAAAGTACTA TATCTACAAC ACAGAAAGCA GCTCCTTTTA AAGTGGATTC AGTAGGAAAT 600
CTTTATGGTG CTGCTGCTGT AGGTTCTTAT GATCTTGCTG GCGGACAATT TAATCCACAA 660
TTATGGTTAG CTTACTGGGA TCAAGTAGCA TTCTTCTATG CTGTAGATGC AGCTTATAGT 720
ACAACTATCT TTGATGGAAT CAACTGGACA CTTGAAGGTG CTTACTTAGG AAATAGCCTT 780
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-75-
GATAGCGAAC TTGATGATAA AACACACGCT TATTTGCTTT AAAAGGTAGC890
AATGGCAATT
ATTGAAGTAA ATGGTTGGGA TGCTAGCCTT ACTACGGTGA TAAAGAAAAA900
GGTGGTTTAT
GCTTCTACAG TCGTAATCGA AGATCAAGGT CTTTACTTGC AGGTGAGGAA960
AATCTTGGTT
ATTTTCTATA CTACTGGTTC AAGACTAAAT GTAGAAATAT CTTCGGTTAT1020
GGTGATACTG
GTAACTGGTG GATATACTTT CAACGAAACA GTGCTGACTT CGTATATGGT1080
GTTCGCGTTG
GGAACAAAAA CAGAAGATAC TGCTCATGTA AAAAACTTGA AGCTGTTGCA1140
GGTGGTGGTA
AGAGTAGATT ACAAATACTC TCCAAAACTT CATTCTATTC TTATGTGAAC1200
AACTTCTCAG
CTAGATCAAG GTGTAAACAC TAATGAAAGT GCACTGTAAG ACTTCAAGCT1260
GCTGATCATA
CTTTACAAAT TCTAA 1275
(2) INFORMATION FOR SEQ ID N0:
4:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 53 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID N0: 4:
CTCTCCCTTC TCGAATCGTA ACCGTTCGTA CGAGAATCGC TGTCCTCTCC TTC 53
(2) INFORMATION FOR SEQ ID NO: 5:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 57 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-76-
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 5:
GATCGAAGGA GAGGACGCTG TCTGTCGAAG GTAAGGAACG GAGGAGAGAA GGGAGAG 57
(2) INFORMATION FOR SEQ ID NO: 6:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 57 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 6:
AATTGAAGGA GAGGACGCTG TCTGTCGAAG GTAAGGAACG GAGGAGAGAA GGGAGAG 57
(2) INFORMATION FOR SEQ ID NO: 7:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 57 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 7:
AGCTGAAGGA GAGGACGCTG TCTGTCGAAG GTAAGGAACG GAGGAGAGAA GGGAGAG 57
(2) INFORMATION FOR SEQ ID N0: 8:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 57 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
_77_
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 8:
CTAGGAAGGA GAGGACGCTG TCTGTCGAAG GTAAGGAACG GAGGAGAGAA GGGAGAG 57
(2) INFORMATION FOR SEQ ID NO: 9:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 30 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 9:
CGAATCGTAA CCGTTCGTAC GAGAATCGCT 30
(2) INFORMATION FOR SEQ ID NO: 10:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 10:
GGTAATTTTG ATAAAAATTT 20
(2) INFORMATION FOR SEQ ID NO: 11:
(i) SEQUENCE CHARACTERISTICS:
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
_78_
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 11:
GATACAGGTA AATTTGATAA 20
(2) INFORMATION FOR SEQ ID N0: 12:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomicl
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 12:
GAAGAAGCTA TCAAAGATGT 20
(2) INFORMATION FOR SEQ ID N0: 13:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(H) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID N0: 13:
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-79-
TGCCACCATC AACAGCGTTG 20
(2) INFORMATION FOR SEQ ID NO: 14:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 14:
TAAGTAAGCA CCTTCAAGTG 20
(2) INFORMATION FOR SEQ ID NO: 15:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 15:
ACTTGTGCTC TATATTTGTG 20
(2) INFORMATION FOR SEQ ID NO: 16:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-80-
y (xi) SEQUENCE DESCRIPTION: SEQ ID NO: 16:
TGATAGCGAA CTTGATGATA 20
(2) INFORMATION FOR SEQ ID N0: 17:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 17:
AGCATCCCAA CCATTTACTT 20
(2) INFORMATION FOR SEQ ID NO: 18:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 18:
TGACTTCGTA TATGGTGGAA 2p
(2) INFORMATION FOR SEQ ID NO: 19:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-81-
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 19:
CTCCAAATTT ATGTGCTACA 20
(2) INFORMATION FOR SEQ ID NO: 20:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 20:
CTATCAAATT TCCAACTTCT 20
(2) INFORMATION FOR SEQ ID NO: 21:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic ecid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 21:
TGAAGATGTT GCTACAAGTG 20
(2) INFORMATION FOR SEQ ID NO: 22:
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-82-
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 22:
CTACTCTTGC AACAGCTTCA 20
(2) INFORMATION FOR SEQ ID N0: 23:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID N0: 23:
CTTCAAAGCT TTCATTCAGT 20
(2) INFORMATION FOR SEQ ID N0: 24:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID N0: 24:
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-83-
GAAGAGTCCG TGGGATTACG 20
(2) INFORMATION FOR SEQ ID NO: 25:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 25:
AGCGATGCAG CTATTAATAA 20
(2) INFORMATION FOR SEQ ID NO: 26:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 26:
TTAACCACAC CCACGGCAGT 20
(2) INFORMATION FOR SEQ ID NO: 27:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-84-
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 27:
GCTCTGGATG CATCTCTGGT 20
(2) INFORMATION FOR SEQ ID NO: 28:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 18 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 28:
AGTAAGGAGA AACAATGA 18
(2) INFORMATION FOR SEQ ID NO: 29:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 28 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 29:
AATAAGCCTT AGAGTCTTTT TGGAATCC 28
(2) INFORMATION FOR SEQ ID N0: 30:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
CA 02284844 1999-09-24
WO 98J4Z842 PCT/CA98/00272
-85-
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 30:
GCTTCTCTTA CCACCAATGC 20
(2) INFORMATION FOR SEQ ID N0: 31:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 20 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 31:
TGTCAGGGTT GTTCACCATG 20
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-86-
REFERENCES
1. Adhya, S., and m. Gotteman. 1978. Control of
transcription termination. Ann. Rev. Biochem.
47:967-996.
2. Austen, B., and O. Westwood. 1991. Bacterial
protein translocation. In Protein targeting and
secretion. D. Rickwood, editor. IRL Press, Oxford,
UK. p. 13-23.
3. Bolla, J., E. Loret, M. Zalewski, and J. Pages. 1995.
Conformational analysis of the Campylobacter jejuni
porin. J.Bacteriol. 177:4266-4271.
4. Burucoa, C., C. Fremaux, Z. Pei, M. Tummuru, M.
Blaser, Y. Cenatiempo, and J. Fauchere. 1995.
Nucleotide sequence and characterization of peb4A
encoding an antigenic protein in Campylobacter
jejuni. Res. Microbiol. 146:467-476.
5. Calva, E., J. Torres, M. Vazquez, V. Angeles, H. de
la Vega, and G. Ruiz-Palacious. 1989. Campylobacter
jejuni chromosomal sequences that hybridize to Vibrio
cholerae and Escherichia coli LT enterotoxin genes.
Gene. 75:243-251.
6. Chakrabarti, S., K. Chaudhuri, K. Sen, and J. Das.
1996. Porins of Vibrio cholerae: Purification and
characterization of OmpU. J. Bacteriol. 178:524-
530.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
_87_
7. Chan, V.L., H. Bingham, A. Kibue, P.R.V. Nayudu and
J.L. Penner. 1988. Cloning and expression of the
Campylobacter jejuni glyA gene in Escherichia coli.
Gene. 73:185=191.
8. DE Martino, L., C. Nazzro, S. Concilio, and M.
Galdiero. 1995. Morphological changes induced in
HEp-2 cells by Salmonella typhimurium porins. J.
Submicrosc. Cytol. Pathol. 27:445-449.
9. Doig, P, M. Exner, R. Hancock, and T. Trust. 1995.
Isolation and characterization of a conserved porin
protein from Helicobacter pylori. J. Bacteriol.
177:5447-5452.
10. Exner, M., P. Diog, T. Trust, and R. Hancock. 1995.
Isolation and characterization of a family of porin
proteins from Helicobacter pylori. Infect. Immun.
63:1567-1572.
11. Fauchere, J-L, M. Kervella, A. Rosenau, J. pages,
and C. Fendri. 1992. In vitro study of virulence
factors of enteric Campylobacter spp. In:
Campylobacter jejuni: Current Status and Future
Trends. I. Nachamkin, M. Blaser and L. Tompkins,
eds. ASM, Washington, DC. pg. 168.
12. Gamier, J., D. Osguthorpe, and B. Robson. 1978.
Analysis of the accuracy and implications of simpe
method for predicting the secondary structure of
globular proteins. J. Mol. Biol. 120:97-120.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
_88_
13. Gonzales, C., A. Isibasi, V. Ortiz-Navarrete, J.
Paniagua, J. Garcia, F. Blanco, and J. Kumate.
Lymphocyte proliferative response to outer-membrane
proteins isolated from Salmonella. Microbiol.
Immuno. 37:793-799.
14. Gotschlich, E., M. Seiff, M. Blake, and M. Koomey.
1987. Porin proein of Neisseria gonorrhoeae:
Cloning and gene structure. Pro. Natl. Acad. Sci.
84:8135-8139.
15. Guerry, P., R. Alm. M. Power, and T. Trust.
Molecular and structural analysis of campylobacter
flagellin. p. 267-281. In I. Nachamikin, M. Blaser,
and L. Tompkins (ed.), Campylobacter jejuni: Current
Status and Future Trends, ASM, Washington DC.
16. Hansen, E., F. Gonzales, N. Chamberlain, M. Norgard,
E. Miller, L. Cope, S. Pelzel, B. Caddy, A. Clausell.
1988. Cloning and the gene encoding the major outer
membrnae protein of Haemophilus influnzae type b.
Infect. Immun. 56:2709-2716.
17. Hansen, E., C. Hasemann, A. Clausell, J. Capra, K.
Orth, C. Moomaw, C. Slaughter, J. Latimer, and E.
Miller. 1989. Primary structure of the porin
protein of Haemophilus influnzae type b determined by
nucleotide sequence analysis. Infect. Immun.
57:1100-1107.
18. Huyer, M., T. Parr, R. Hancock, and W. Page. 1986.
Outer membrane protein of Campylobacter jejuni. FEMS
Microbiol.Lett. 37:247-250.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
_89_
19. Johnson, W., and H. Lior. 1988. A new heat-labile
cytolethal.distending toxin (CLDT) produced by
Campylobacter sp. Microb. Pathog. 4:115-126.
20. Kennell, W., and S. Holt. 1991. Extraction,
purification and characterization of major outer
membrane proteins from Wolinella recta ATCC 33238.
59:3740-3749.
21. Kervella, M., J.-L. Fauch(re, D. Fourel, and J.-M.
Pag(s. 1992. Immunological cross-reactivity between
outer membrane pore proteins of Campylobacter j ejuni
and Escherichia coli. FEMS microbiology Letters.
99:281-286.
22. Louie, H., and V. Chan. 1993. Cloning and
characterization of the gamma-glutamyl phosphate
reductase gene of Campylobacter jejuni. Mol. Gen.
Genet. 240:29-35.
23. Mahajan, S., and F. G. Rodgers. 1989. Virulence of
Campylobacter jejuni for chicken embryos. J. Clin.
Microbiol. 27:1377-1379.
24. Mahajan, S., and F. G. Rodgers. 1990. Isolation,
characterization, and host cell-binding properties of
a cytotoxin from Campylobacter j ejuni. J. Clin.
Microbiol. 28:1314-1320.
25. Marmur, J. 1961. A procedure for the isolation of
deoxyribonucleic acid from microorganisms. J. Mol.
Biol. 3:208-218.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-90-
26. Mizuno, K., K. Takama, and S. Suzuki. 1994.
Characteristics of cytotoxin produced by
Campylobacter jejuni strains. Microbios. 78: 215-
228.
27. Movva, N., K. Nakamura, and M. Inouye. 1980. Gene
structure of the OmpA protein, major surface protein
of Eschericha coi required for cell-cell interaction.
J. Miol. Biol. 143:317-328.
28. Nachamkin, I. 1995. Campylobacter and Arcobacter.
In Manual of Clinical Microbiology. P. Murray, E.
Baron, M. Pfaller, F. Tenover, and R. Yolken, eds.
ASM Press, Washington, DC. Pg. 483-491.
29. Nakaido, H. 1992. Porins and specific channels of
bacterial outer membranes. Mol. Microbiol. 6:435-
442.
30. Newell, D., and I. Nachamkin. 1992. Immune response
directed aginst Campylobacter jejuni. In
Campylobacter jejuni: Current status and Future
Trends. I. Nachamkin, M. Blaser, and L. Tompkins,
editors. ASM, Washington, DC. 201-206
31. Novotny, J., and C. Auffray. 1984. Nucl. Acids.
Res. 12:243-255.
32. 0'Brien, A., J. Newland, S. Miller, R. Holmes, H.
Smith, and S. Formal. 1984. Shiga-like toxin-
converting phage from Escherichia coli strains that
cause hemorrhagic colitis or infantile diarrhea.
Science. 226:694-696.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-91-
33. Pesci, E., D. Cottle, and C. Pickett. 1994.
Genetic, enzymati, and pathogenic studies of the iron
superoxide dismutase of Campylobacter jejuni.
Infect. Immun. 62:2687-2694.
34. Pickett, C., E. Pesci, D. Cottle, G. Russell, A.
Erdem, and H. Zeytin. 1996. Prevalence of
cytolethal distending toxin production in
Campylobacter jejuni and relatedness of Campylobacter
sp. cdtB genes. Infect. Immun. 64:2070-2078.
35. Popot, J., C. De Vtry, and A. Atteia. 1994. Folding
and assembly of intergral membrane proteins: an
introduction. In Membrane protein and structure:
Experimental approaches. S. White, ed. Oxford
University Press, New York. pg. 41-96.
36. Qi, H., J. Tai, and M. Blake. 1994. Expression of
large amounts of Neisserial porin proteins in
Escherichia coli and refolding of the proteins into
native trimers. Infect. Immun. 62:2432-2439.
37. Riley, J., R. Butler, D. Ogilvie, R. Finniear, D.
Jenner, S. Powell, R. Anand, J. Smith, and A.
Markham. 1990. A novel, rapid method for the
isolation of terminal sequences from yeast artificial
chromosomal (YAC) clones. Nucl. Acids Res. 18:2887-
2890.
38. Sambrook, J., E.F. Fritsch and T. Maniatis. 1989.
Molecular cloning: a laboratory manual, 2nd ed.
Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, NY.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-92-
39. Skirrow, M. and M. Blaser. 1992. Clinical and
epidemiologic considerations, p.3-8. In I.
Nachamikin, M. Blaser, and L. Tompkins (ed.),
Campylobacter jejuni: Current Status and Future
Trends, ASM, Washington DC.
40. Suzuki, S., M. Kawaguchi, K. Mizuno, K. Takama, and
N. Yuki. 1994. Immunological properties and
ganglioside recognitions by Campylobacter jejuni-
enterotoxin and cholera toxin. FEMS Immunol. Med.
Microbiol. 8:207-211.
41. Tauxe, R. 1992. Epidemiology of Campylobacter
infections in the United States and other
industrialized nations. In: Campylobacter jejuni:
Current Status and Future Trends. I. Nachamkin,
M. Blaser and L.Tompkins, eds. ASM, Washington, DC.
Pg~ 9.
42. Taylor, D. 1992. Genetics of Campylobacter and
Helicobacter. Annu. Rev. microbiol. 46:35-64.
43. Taylor, D., M. Eaton, W. Yan, and N. Chang. 1992.
Genome maps of Campylobacter jejuni and Campylobacter
coli. J. Bacteriol. 174:2332-2337.
44. Tenover " F., C. Fennell, L. Lee, and D. Leblanc.
1992. Characterization of two plasmids from
Campylobacter jejuni isolates thatcarry the aphA-7
kanamycin resistence determinant. Antimicrob.
Agents. Chemother. 36:712-716.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-93-
45. Tommassen, J., M. Agterberg, R. Jansen, and G.
Spierings. 1993. Use of the enterobacterial outer
membrane protein PhoE in the development of new
vaccines and DNA probes. Int. J. Med. Microbiol.
Virol. Parasitol. Infect. Dis. 278:396-406.
46. Welte, W., U. Nestel, T. Wacker, and K. Diederichs.
1995. Structure and functon of the porin channel.
Kid. Intern. 48:930-940.
47. Skirrow, M. and Blaser, M. Clinical and epidemiologic
considerations. In: Campylobacter jejuni: Current
Status and Future Trends, edited by Nachamikin, I.,
Blaser, M. and Tompkins, L. Washington DC: ASM,
1992, p. 3-8.
48. Tauxe, R. Epidemiology of Campylobacter jejuni
infections in the United States and other
industrialized nations. In: Campylobacter jejuni:
Current Status and Future Trends, edited by
Nachamkin, I., Blaser, M. and Tompkins, L.
Washington DC: ASM, 1992, p. 9-19.
49. McSweegan, E. and Walker, R. Identification and
characterization of two Campylobacter jejuni
adhesions for cellular and mucus substrates.
Am.J.Epidemiol. 1986; 53:141-148.
50. Sears, C. And Kaper, J. Enteric bacterial toxins:
mechanisms of action and linkage to intestinal
secretion. Microbiol. Rev. 1996; 60: 167-215.
51. Wassenaar, T. Toxin production by Campylobacter spp.
Clin.Microbiol.Rev. 1997; 10:466-476.
CA 02284844 1999-09-24
WO 98/42842 PCTlCA98/00272
-94-
52. Kawaguchi, M., Takama, K, and Suzuki, S.
Distribution and solubilization of Campylobacter
jejuni toxins. Microbios Letters 1989; 42:113-
118.
53. Suzuki, S., Kawaguchi, M., Mizuno, K., Takama, K. and
Yuki, N. Immunological properties and ganglioside
recognitions by Campylobacter jejuni-enterotoxin and
cholera toxin. FEMS Immunol.Med.Microbiol. 1994;
6:207-212.
54. Johnson, W. and Lior, H. A new heat-labile
cytolethal distending toxin (CLDT) produced by
Camplyobacter spp. Micro.Pathog. 1988; 4:115-126.
55. Pickett, C., Pesci, E., Cottle, D., Russell, G.,
Erdem, A. and Zeytin, H. Prevalence of cytolethal
distending toxin production in Campylobacter jejuni
and relatedness of Campylobacter sp. cdtB genes.
Infect.Immun. 1996; 64:2070-2078.
56. Mahajan, S. and Rodgers, F.G. Virulence of
Campylobacter jejuni for chicken embryos.
J.Clin.Microbiol. 1989; 27:1377-1379.
57. Mahajan, S. and Rodgers, F.G. Isolation,
Characterization, and Host-cell-binding properties of
a cytotoxin from Campylobacter jejuni.
J.Clin.Microbiol. 1990; 28:1314-1320.
58. Guerrant, R., Wanke, C., Pennie, R., Barrett, L.,
Lima, A. and O'Brien, A. Production of a unique
cytotoxin by Campylobacter jejuni. Infect.Immun.
1987; 55:2526-2530.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-95_
59. Johnson, W. and Lior, H. Cytotoxic and cytotonic
factors produced by Campylobacter jejuni,
Campylobacter coli, and Campylobacter Iaridis.
J.Clin.Microbiol. 1986; 24:275-281.
60. Mizuno, K., Takama, K. and Suzuki, S.
Characteristics of cytotoxin produced by
Campylobacter jejuni strains. Microbios 1994;
78:215-228.
61. Welte, W., Nestel, U., blacker, T. and Diederichs, K.
Structure and function of the porin channel.
Kid. Inter. 1995; 48:930-940.
62. Mowa, R., Nakamura, K. and Inouye, M. Gene
structure of the ompA protein,a major surface protein
of Escherichia coli required for cell-cell
interaction. J.MoI.Biol. 1980; 143:317-328.
63. Nikaido, H. Porins and specific channels of
bacterial outer membranes. Mol.Microbiol. 1992;
6:435-442.
64. Sommese, L., Donnarumma, G., Cipollaro de L'ero, G.,
Marcatili, A., Vitiello, M. and Galdiero, M. Growth
hormone modulates IL-a and INF-Y release by murine
splenocytes activated by LPS or porins of Salmonella
typhimurium. J.Med.Microbiol. 1996; 45:40-47.
65. Galdiero, F., Cipollaro de L' ero, G., Donnarumma, G.
and Marcatili, A. Interleukin-1 and interleukin-6
gene expression in human monocytes stimulated with
Salmonella typhimurium porins. Immunology. 1995;
86:612-619.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-96-
66. Gonzales, C., Isibasi, A., Ortiz-Navarrete, V.,
Paniagua, J., Garcia, J., Blanco, F., and Kumate, J.
' Lymphocytic proliferative response to outer-membrane
proteins isolated from Salmonella.
Microbiol.Immunol. 1993; 37:793-799.
67. Kervella, M., Fauchere, J-L., Fourel, D. and Pages,
J.-M. Immunological cross-reactivity between outer
membrane pore proteins of Campylobacter jejuni and
Escherichia coli. FEMS Microbiol.Lett. 1992;
99:281-286.
68. Bolla, J-M., Loret, E., Zalewski, M. and Pages, J-M.
Conformational analysis of the Campylobacter jejuni
porin. J.Bacteriol. 1995; 177:4266-4271.
69. Huyer, M., Parr, T., Hancock, R. and Page, W. Outer
membrane protein of Campylobacter jejuni. FEMS
Microbiol.Lett. 1986; 37:247-250.
70. Sambrook, J., Fritsch, E.F. and Maniatis, T.
Molecular cloning: A laboratory manual, Cold Spring
Harbor:Cold Spring Harbor Laboratory, 1992. Ed. 2
71. Pollard, D., Johnson, W., Lior, H., Tyler, S. and
Rozee, K. Rapid and specific detection of verotoxin
genes in Escherichia coli by the polymerase chain
reaction. J.Clin.Microbiol. 1990; 28:540-545.
72. Xiang, Z., Censini, S., Bayell, P.,Telford, J.,
Figura, N., Rappuoli, R., and Covacci, A. Analysis
of expression of CagA and VacA virulence factors in
43 strains of Helicobacter pylori reveal that
clinical isolates can be divided into two major
types and that CagA is not necessary for expression
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-97-
of the vacuolating cytotoxon. Infect.Immun. 1995;
63:94-98.
73. Dubois, M., Gilles, K., Hamilton, J., Rebers, P. and
Smith, F. Colormetric method for detection of sugars
and related substances. Anal.Chem 1956; 28:350-
356.
74. Segrest, J. and Jackson, R. Molecular weight
determination of glycoproteins by polyacrylamide gel
electrophoresis in sodium dodecyl sulfate. In:
Methods of Enzymology, edited by Ginsburg, V. New
York: Academic Press, 1972, p. 54-63.
75. Kennell, W. and Holt, S. Extraction, purification
and characterization of major outer membrane proteins
from Wolinella recta ATCC 33238. Infect.Immun.
1991; 59:3740-3749.
76. Yeen, W., Puthucheary, S. and Pang, T. Demonstration
of a cytotoxin from Campylobacter jejuni.
J.CIin.Pathol. 1983; 36:1237-1240.
77. Moore, M., Blaser, M., Perez-Perez, G. and O'Brien,
A. Production of a Shiga-like cytotoxin by
Campylobacter . Microb.Pathog. 1988; 4:455-462.
78. Misawa, N., Ohnishi, T., Itoh, K. and Takahashi, E.
Development of a tissue culture assay system for
Campylobacter jejuni cytotoxin and the influence of
culture conditions on cytotoxin production.
J.Med.Microbiol. 1994; 41:224-230.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-98-
79. Fauchere, J., Kervella, M., Rosenau, A., Pages, J-M.
and Fendi, C. In vitro study of virulence factors of
enteric Campylobacter spp. In: Camplylobacter
jejuni: Current Status and Future Trends, edited by
Nachamkin, I., Blaser, M. and Tompkins, L.
Washington DC: ASM, 1992, p. 168-175.
80. Weel, J., van der Hulst, R., Gerritis, Y., Roorda P.,
Feller M., Dankert J., Tytgat G., and van der Ende,
A. The interrelationship between cytotoxin-
associated gene A, vacuolating cytotoxin, and
Helicobacter pylori-related diseases.
J.Infect.Dis. 1996; 173:1171-1175.
81. Inward, C., Williams, J., Chant, I., Crocker, J.,
Milford, D., Rose, P., and Taylor, C. Verocytotoxin-
1 induces apoptosis in Vero cells. J.Infect.
1995; 30:213-218.
82. Chen , Y. and Zychlinsky, A. Apoptosis induced by
bacterial pathogens. Microb.Pathog. 1994; 17:203-
212.
83. De Martino, L., Nazzaro, C. and Galdiero, M.
Morphological changes induced in HEp-2 cells by
Salmonella typhimuriurn porins.
J.Submicrosc.Cytol.Pathol. 1995; 27:445-449.
84. Bjerknes, R., Guttormsen, H., Solberg, C. and
Wetzler, L. Neisserial porins inhibit human
neutrophil actin polymerization, degranulation,
opsonin receptor expression, and phagocytosis but
prime the neitrophils to increase their oxidative
burst. Infect.Immun. 1995; 63:160-167.
k
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
_99_
85. Galdiero, F., Tufano, M., Galdiero, M., Masiello, S.
and Di Rosa, M. Inflammatory effects of Salmonella
typhimuriurn porins. Infect.Immun. 1990; 58:3183-
3186.
86. Galdiero, F., Cipollaro de L'ero, G., Benedetto, N.,
Galdiero, M. and Tufano, M. Release of cytotokines
induced by Salmonella typhimurium porins.
Infect.Immun. 1993; 61:155-161.
87. Misawa, N., Ohnishi, T., Itoh, K. and Takahashi, E.
Cytotoxin detection in Campylobacter jejuni strains
of human and animal origin with three tissue
culture assay systems. J. Med. Microbiol. 1995; 43:
354-359.
88. Menzl, K., Maier, E., Chakraborty, T. and Benz, R.
HlyA hemolysin of Vibrio cholerae O1 biotype El Tor
identification of the hemolytic complex and evidence
for the formation of anion-selective ion-permeable
channels. Eur.J.Biochem. 1996; 240:646-654.
89. Luzzi, I., Covacci, A., Censini, S., Pezzella, C.,
Crotti, D., Facchini, M., Giammanco, A.,
Guglielmetti, P., Piersimoni, C., Bonamico, M.,
Mariani, P., Rappuoli, R., and Caprioli, A..
Detection of a vacuolating cytotoxin in stools from
children with diarrhea. Clin.Infect.Dis. 1996;
23:101-106.
90. Penner, J. and Aspinall, G. Diversity of
lipopolysaccharide structures in Campylobacter
jejuni. J. Infect. Dis. 1997; 176 (Suppl 2): 5135-
138.
CA 02284844 1999-09-24
WO 98/42842 PCT/CA98/00272
-100-
91. Allos, B.M. Association between Campylobacter
infection and Guillain-Barre syndrome. ~T. Infect.
Dis. 1997; 176 (Suppl 2): 5125-128.