Note: Descriptions are shown in the official language in which they were submitted.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 1 -
Prevention of Breast Cancer
with Selective Estrogen
Receptor Modulators
Technical Field
The present application relates to medical methods of
treatment. More particularly, the present invention
concerns the use of a class of substituted benzo[b]thiophene
compounds for the prophylaxis or prevention of breast
carcinoma in a patient in need of such treatment.
Background of the Invention
Breast carcinoma or breast cancer is the most common
form of cancer and the second most common cause of death in
women in the United States. In the years 1994 and 1995, an
estimated 182,000 new cases of breast cancer in women
occurred with mortality estimated at 46,000.
Currently, it is estimated that, statistically, most
women have a one in ten chance of developing this disease in
their lifetime. Breast carcinoma is a major cause of
mortality in women, as well as a cause of disability,
psychological trauma, and economic loss. A large percentage
of women contracting this disease eventually die from its
effects either directly or indirectly from complications,
e.g., metastasis, loss of general health, or collateral
effects from therapeutic interventions, such as surgery,
radiation, or chemotherapy.
The epidemiology of this disease, although the subject
of intense investigation, is still poorly understood. There
appears to be a substantial genetic component which
. predisposes some patients to contract the disease. Yet it
is not clear whether this genetic component is causative or
permissive to the disease or only predictive of the disease
process. It has been known for a long time that breast
carcinoma tends to occur more frequently in some families,
IF
CA 02286207 1999-10-08
WO 98/45286 PCTlUS98/06989
- 2 -
although such analysis is not accurately predictive of
disease occurrence in other family members.
A great deal of clinical and pharmacological
40 investigation has been carried out attempting to elucidate
the relationship between the hormone estrogen, and the
cause, maintenance, and treatment of breast carcinoma.
Although a great deal is known about the relationship of
estrogen in the maintenance and treatment of the disease,
45 there is a great deal of controversy associated with the
effect of estrogen on the epidemiology of this disease,
i.e., whether estrogen is a causative agent (carcinogen) or
an obligatory co-factor (permissive) in the initiation of
the disease.
50 The estrogens, which include 17b-estradiol, estrone,
and their active metabolites, are major sex-related hormones
in women, but additionally, appear to be important
homeostatic hormones in both men and women throughout their
adult life. Normally, everyone has some level of estrogen.
55 Cancer of the male breast is a rare disease, accounting
for less than to of all cancers in males. The American
Cancer Society reported that in 1994 an estimated 1,000 men
in the United States were diagnosed as having breast cancer,
with mortality estimated at 300.
60 Ductal carcinoma in situ (DCIS) of the breast is an
early form of breast cancer in which malignant epithelial
cells proliferate in the ductile system without microscopic
evidence of invasion through the basement membrane into the
surrounding breast tissue. The median age of patients with
65 DCIS at the time of diagnosis is about 52 years of age. The
increasingly widespread use of mammography has led to the
earlier detection of DCIS since most cases are detected in
otherwise asymptomatic women undergoing screening
mammography .
Hormone Replacement Therapy (HRT), recommended for
postmenopausal and peri-menopausal women to alleviate
cardiovascular disease, osteoporosis, and other menopausal
sequelae, has generated a great deal of debate as to the
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 3 -
potential of this therapy to increase the risk of
75 contracting breast carcinoma. Currently, the conclusions
from HRT studies, most of which were postspective studies,
appear to indicate a small increase in risk.
In contrast to the problematic role of estrogen in the
initiation of this disease, a great deal (although
80 incomplete) of understanding has been achieved relating
estrogen with established breast carcinoma. Estrogen is a
growth factor required by most breast carcinoma cells in the
early stages of this disease. Also it has been established,
but yet not fully understood why, that during the course of
85 this disease the cancer cells lose their sensitivity to the
effects of estrogen. Eventually, a majority of carcinoma
cells become no longer dependent on estrogen for growth and
are no longer responsive to any hormonally based therapy,
which included "anti-estrogens", GNRH agonists, progestins,
90 and androgens.
A great deal of benefit has been achieved with the use
of hormonally based therapeutic interventions. The most
widely used therapy is the use of tamoxifen. The five-year
survival rate for women with breast carcinoma has been
95 dramatically improved with this therapy; however, the
longer-term survival (ten-year+) rate has not improved to
the same extent. This lack of improvement in the long-term
rate has been attributed to the gradual evolution of the
carcinoma cells from estrogen dependence to independence.
100 Thus, even with the best combinations of treatment
modalities, (surgery, radiation, and/or chemotherapy), the
long-term prognosis for patients is poor, especially if
metastatic disease is present. Clearly, there is a great
need for improved therapies and, perhaps, more importantly,
105 a need for the prevention of the disease in the first
instance (de novo).
For the last decade it has been argued that "anti-
estrogen" therapy, especially the use of tamoxifen, should
be examined for its potential to prevent de novo breast
110 carcinoma. However, partially because of the lack of
CA 02286207 1999-10-08
WO 98145286 PCT1US98106989
_ q _
evidence of a benefit, and known and potential toxicity of
tamoxifen, no prospective prevention trials have been
conducted in healthy women.
Clearly, a great need exists for a breast cancer
115 prevention therapy useful for the entire population,
including individuals at high or no particular risk, and
including both men and women.
Brief Summary of the Invention
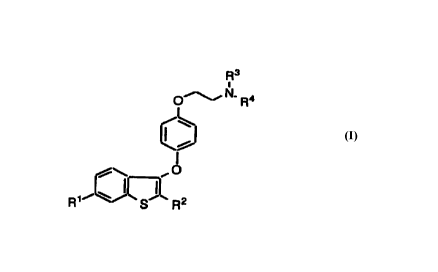
120 In accordance with the present invention, there is
provided a method for the prophylaxis or prevention of
breast carcinoma in a patient in need of such treatment
comprising administering a therapeutically effective amount
of a compound having the structure
125
R3
O~N,R4
I
O
R~ \ S ( \
R2
I
or a pharmaceutically acceptable salt or pro-drug thereof.
130 The invention further relates to a method for
preventing breast cancer by administrating to human for a
sufficient term an effective dose of a compound of formula I
or pharmaceutically acceptable salt or pro-drug thereof,
where the human has not been diagnosed with, but is
135 determined to be at risk for, developing breast cancer.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 5 -
In the structure shown above, R1 and R2 are
independently selected from the group consisting of hydroxy
and alkoxy of one to four carbon atoms.
R3 and R4 are independently selected from methyl or
140 ethyl, or R3 and R4, taken together with the nitrogen atom
to which they are attached, form a pyrrolidino,
methylpyrrolidino, dimethylpyrrolidino, piperidino,
morpholino, or hexamethyleneimino ring.
The compounds of the present invention are selective
145 estrogen receptor modulators (SERM's), that is, compounds
which produce estrogen agonism in one or more desired target
tissues while producing estrogen antagonism and/or minimal
(i.e. clinically insignificant) agonism in reproductive
tissue such as the breast or uterus.
150
Detailed Description
The current invention concerns the discovery that
compounds of formula I above are useful for preventing
breast cancer. The methods provided by this invention are
155 practiced by administering to a patient in need thereof a
dose of a compound of the present invention or a
pharmaceutically acceptable salt or solvate thereof, that is
effective to prevent breast cancer.
Throughout this specification and the appended claims,
160 general terms bear their usual meanings.
The terms "prevention of", "prophylaxis" and "prevent"
includes reducing the likelihood of a patient incurring or
developing breast cancer.
The term "de novo", as used in the current invention,
165 means the lack of transformation or metamorphosis of normal
breast cells to cancerous or malignant cells in the first
instance. Such a transformation may occur in stages in the
same or daughter cells via an evolutionary process or may
occur in a single, pivotal event. This de novo process is
170 in contrast to the metastasis, colonization, or spreading of
already transformed or malignant cells from the primary
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 6 -
tumor site to new locations. This invention also relates to
the administration of a compound of formula I to a patient
who is at risk of developing de novo breast cancer.
175 A person who is at no particular risk of developing
breast cancer is one who may develop de novo breast cancer,
has no evidence or suspicion of the potential of the disease
above normal risk, and who has never had a diagnosis of
having the disease. The greatest risk factor contributing
180 to the development of breast carcinoma is a personal history
of suffering from the disease, or an earlier occurrence of
the disease, even if it is in remission with no evidence of
its presence. Another risk factor is family history of the
disease.
185 The term "alkyl" denotes a monovalent radical derived
by removal of one hydrogen atom from methane, ethane, or a
straight or branched hydrocarbon and includes such groups as
methyl, ethyl, propyl, iso-propyl, n-butyl, sec-butyl, iso-
butyl, tert-butyl and the like.
190 "Alkoxy" means an alkyl group, as defined above.
attached to the parent molecular moiety through an oxygen
atom and includes such groups as methoxy, ethoxy, propoxy,
iso-propoxy, n-butoxy, sec-butoxy, iso-butoxy, tert-butoxy
and the like. In the present invention, methoxy is the
195 preferred alkoxy group.
The term "pro-drug," as used herein means a compound of
the present invention bearing a group which is metabolically
cleaved in a human to produce a therapeutically active
compound of the present invention. In particular, such pro-
200 drug compounds include those in which either or both of the
substituent groups R1 and R2 of the structure shown above
are hydroxy groups which have been protected by a
pharmaceutically acceptable hydroxy protecting group which
is metabolically cleaved in the body to yield a
205 corresponding monohydroxy or dihydroxy compound of the
present invention. Hydroxy protecting groups are described
in Chapter 2 of T. W. Greene, et al., "Protective Groups in
Organic Synthesis," Second Edition, John Wiley & Sons, Inc.,
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
-
New York, 1991. Simple ether and ester groups are preferred
210 as pro-drug hydroxy protecting groups.
Preferred compounds of the present invention include
6-hydroxy-2-(4-hydroxyphenyl)-3-[4-(2-piperidino-
ethoxy)phenoxy]benzo[b]thiophene or a pharmaceutically
acceptable salt or pro-drug thereof; and
215 6-hydroxy-2-(4-methoxyphenyl)-3-[4-(2-piperidino-
ethoxy)phenoxy]benzo[b]thiophene or a pharmaceutically
acceptable salt or pro-drug thereof.
Preparation of Compounds of the Invention
220 The starting material for one route for preparing
compounds of the present invention is prepared essentially
as described by C. D. Jones in U.S. Patents. No's.
4,418,068, and 9,133,814. The starting materials have the
formula l:
225
R$O \ S
OR 6
1
wherein R5 and R6 are independently -H or a hydroxy
230 protecting group.
The R5 and R6 hydroxy protecting groups are moieties
which are intentionally introduced during a portion of the
synthetic process to protect a group which otherwise might
react in the course of chemical manipulations, and is then
235 removed at a later stage of the synthesis. Since compounds
bearing such protecting groups are of importance primarily
as chemical intermediates (although some derivatives also
exhibit biological activity), their precise structure is not
critical. Numerous reactions for the formation, removal,
240 and reformation of such protecting groups are described in a
number of standard works including, for example, Protective
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
_ g _
Groups in Organic Chemistry, Plenum Press (London and New
York, 1973); Greene, T.W., Protective Groups in Organic
Synthesis, Wiley (New York, 1981) ; and The Peptides, Vol. I,
245 Schrooder and Lubke, Academic Press, (London and New York,
1965).
Representative hydroxy protecting groups include, for
example, -C1-C4 alkyl, -C1-Cq alkoxy, -CO-(Cl-C6 alkyl), -
S02-(Cq-C6 alkyl), and -CO-Ar in which Ar is benzyl or
250 optionally substituted phenyl. The term "substituted
phenyl" refers to a phenyl group having one or more
substituents selected from the group consisting of C1-Cq
alkyl, C1-Cg alkoxy, hydroxy, nitro, halo, and tri(chloro or
fluoro) methyl. The term "halo" refers to bromo, chloro,
255 fluoro, and iodo.
For compounds of formula 1, preferred R5 and R6
substituents are methyl, isopropyl, benzyl, and
methoxymethyl. Compounds in which R5 and R6 each are methyl
are prepared via the procedure described in the above-
260 referenced Jones patent.
Compounds of formula _1 are also prepared in which the
RS hydroxy protecting group is selectively removed, leaving
R6 as a hydroxy protecting group as part of the final
product. The same is true in the case in which the R6
265 hydroxy protecting group is selectively removed, leaving the
R5 hydroxy protecting group in place. For example, R5 can
be isopropyl or benzyl and R6 methyl. The isopropyl or
benzyl moiety is selectively removed via standard
procedures, and the R6 methyl protecting group is left as
270 part of the final product.
As shown in Reaction Scheme I, the first steps of the
present process for preparing certain compounds of the
present invention include selectively placing a leaving
group, R~ at the 3 position of a compound of formula _1, to
275 form a compound of formula 2, coupling the product of that
reaction with a 4-(protected-hydroxy)phenol, _3, to form a
compound of formula 4, and selectively removing the R8
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
_ g _
hydroxy protecting group to form a compound of formula 5.
In the sequence of steps shown in Reaction Scheme I, the
280 hydroxy protecting groups R5, R6 and R8 are chosen in such a
manner that, in the final step, the hydroxy protecting group
R8 can be selectively removed in the presence of hydroxy
protecting groups R5 and R6.
285 Reaction Scheme I
R~
I I ,. I I
R50 S I R50 S '
1 ~ OR 6 I
OR6
ORB
I
I
OH
OH ORB
i
I I
I I O ~ O
Rs0 ~ S ~ R50 ~ I S i
I
~ OR6 4 ~ OR6
In the first step of Reaction Scheme I, an appropriate
290 leaving group is selectively placed at the 3-position of the
formula 1 starting material via standard procedures.
Appropriate R~ leaving groups include the sulfonates such as
methanesulfonate, 4-bromobenzenesulfonate, toluenesulfonate,
ethanesulfonate, isopropanesulfonate, 4-methoxybenzene-
295 sulfonate, 4-nitrobenzenesulfonate, 2-
chlorobenzenesulfonate, triflate, and the like, halogens
such as bromo, chloro, and iodo, and other related leaving
groups. However, to insure proper placement of the leaving
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 10 -
group, the named halogens are preferred, and bromo is
300 especially preferred.
The present reaction is carried out using standard
procedures. For example, when the preferred halogenating
agents are used, an equivalent of such a halogenating agent,
preferably bromine, is reacted with an equivalent of the
305 formula 1 substrate, in a suitable solvent such as, for
example, chloroform or acetic acid. The reaction is
typically run at a temperature from about 40°C to about
80°C.
The reaction product from the above process step, a
310 compound of formula 2, is then reacted with a 4-(protected-
hydroxy)phenol, 3, to form compounds of formula _4 in which
Re is a selectively removable hydroxy protecting group.
Generally, the 4-hydroxy protecting moiety of the phenol may
be any known protecting group which can be selectively
315 removed without removing, in this instance, the RS and, when
present, R6 moieties of a formula 3 compound. Preferred R8
protecting groups include methoxymethyl, when R5 and/or R6
are not methoxymethyl, and benzyl. Of these, benzyl is
especially preferred. The 4-(protected-hydroxy)phenol
320 reactants are commercially available or can be prepared via
standard procedures.
The coupling reaction between compounds of formula _2
and those of formula 3 is known in the art as an Ullman
reaction and is generally run according to standard
325 procedures [see, e.g., "Advanced Organic Chemistry;
Reactions, Mechanisms, and Structure," Fourth Edition, 3-16,
(J. March, ed., John Wiley & Sons, Inc. 1992); Jones, C.D.,
J. Chem. Soc. Perk. Trans. I, 4:407 (1992)].
In general, equivalent amounts of the two aryl
330 substrates, in the presence of up to an equimolar amount of
a copper(I) oxide catalyst and an appropriate solvent, are
heated to reflux under an inert atmosphere. Preferably, an
equivalent of a formula 2 compound in which R~ is bromo is
reacted with an equivalent amount of,4-benzyloxyphenol in
335 the presence of an equivalent of cuprous oxide.
CA 02286207 1999-10-08
WO 98!45286 PCT/US98/06989
- 11 -
Appropriate solvents for this reaction are those
solvents or mixture of solvents which remain inert
' throughout the reaction. Typically, organic bases,
particularly a hindered base such as, for example, 2,4,6-
340 collidine, are preferred solvents.
The temperature employed in this step is generally
sufficient to effect completion of this coupling reaction,
and will influence the amount of time required therefore.
When the reaction mixture is heated to reflux under an inert
345 atmosphere such as nitrogen, the time-to-completion is
usually from about 20 to about 60 hours.
Following coupling of a compound of formula _2 with one
of formula 3, to form a formula 4 compound, formula _5
compounds are prepared by selectively removing the R8
350 hydroxy protecting group of a formula _4 compound via well
known reduction procedures. It is imperative that the
selected procedure will not affect the R5 and, when present,
R6 hydroxy protecting groups.
When R8 is the preferred benzyl moiety, and R5 and,
355 when present, R6 each are methyl, the present process step
is carried out via standard hydrogenolysis procedures.
Typically, the formula 4 substrate is added to a suitable
solvent or mixture of solvents, followed by the addition of
a proton donor to accelerate the reaction and an appropriate
360 hydrogenation catalyst.
Appropriate catalysts include noble metals and oxides
such as palladium, platinum, and rhodium oxide on a support
such as carbon or calcium carbonate. Of these, pailadium-
on-carbon, particularly 10~ palladium-on-carbon, is
365 preferred. Solvents for this reaction are those solvents or
mixture of solvents which remain inert throughout the
reaction. Typically, ethylacetate and C1-CQ aliphatic
alcohols, particularly ethanol, is preferred. For the
present reaction, hydrochloric acid serves as an adequate
370 and preferred proton donor.
When run at ambient temperature and a pressure ranging
form about 30 psi (206.8 kilopascals) to about 50 psi 344.7
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 12 -
kilopascals), the present reaction runs quite rapidly.
Progress of this reaction may be monitored by standard
375 chromatographic techniques such as thin layer
chromatography.
As shown in Reaction Scheme II, upon preparation of a
formula 5 compound, it is reacted with a compound of formula
380 R4R5N-(CH2)2-Q
6
wherein R4 and R5 are as defined above, and Q is a bromo or,
preferably, chloro, to form a compound of formula 7. The
385 formula 7 compound is then deprotected to form a compound of
formula I.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 13 -
Reaction Scheme II
OH R~ R4, N~\O
N
~ ~ Ra
I
p I
O
R50 ~' gI \ 5 ~ I I
R O S
OR6 I ~ OR6
R3
R4.N~\O
I
O
I
Rs0 .' S ~ \
OR6
z
Ta, R' - R° - H
I b , RS - H
Ic, R6 - H
390 In the first step of the process shown in Reaction
Scheme II, the reaction is carried out via standard
procedures. Compounds of formula 6 are commercially
available or are prepared by means well known to one of
ordinary skill in the art. Preferably, the hydrochloride
395 salt of a formula 6 compound is used. In a particularly
preferred case of the compounds of the present invention, 2-
- chloroethylpiperidine hydrochloride, is used.
Generally, at least about 1 equivalent of a formula _5
substrate is reacted with 2 equivalents of a formula _6
400 compound in the presence of at least about 4 equivalents of
CA 02286207 1999-10-08
WO 98145286 PCT/US98/06989
- 14 -
an alkali metal carbonate, preferably cesium carbonate, and
an appropriate solvent.
Suitable solvents for this reaction are those solvents
or mixture of solvents which remain inert throughout the
405 reaction. N,N-dimethylformamide, especially the anhydrous
form thereof, is preferred. The temperature employed in
this step should be sufficient to effect completion of this
alkyiation reaction. Typically, ambient temperature is
sufficient and preferred. The present reaction preferably
410 is run under an inert atmosphere, particularly nitrogen.
Under the preferred reaction conditions, this reaction
will run to completion in about 16 to about 20 hours. The
progress of the reaction can be monitored via standard
chromatographic techniques.
415 In an alternative process for preparing compounds of
the present invention, shown in Reaction Scheme III below, a
formula 5 compound is reacted in an alkali solution with an
excess of an alkylating agent of formula 8:
420 Q-(CH2)n-Q'
8
in which Q and Q' are the same or different leaving groups.
Appropriate leaving groups are those mentioned above.
CA 02286207 1999-10-08
WO 98/45286 PCTNS98/06989
- 15 -
425 Reaction Scheme III
Ohi QUO
' Q
I ~ ~Q, I ~
O
/ I I ~~' ~I I O
RIO S I ~ R~O~S
ORs ~ s
OR
R3R4NH
3
R R3 r
R4~Nw/'O R4~N1/'O
I'
i
O
i ~ O
I I
w S I RIO ~ I S I
_ORs ~ s
OR
I a , R5 - R6 - H
Ib, R5 - H
Ic, R6 - H
A preferred alkali solution for this alkylation
430 reaction contains potassium carbonate in an inert solvent
such as, for example, methyethyl ketone (MEK) or DMF. In
this solution, the unprotected hydroxy group of the formula
compound is converted to a phenoxide ion which displaces
one of the leaving groups of the alkylating agent.
435 This reaction proceeds best when the alkali solution
containing the reactants and reagents is brought to reflux
and allowed to run to completion. When using MEK as the
preferred solvent, reaction times range from about 6 hours
to about 20 hours.
CA 02286207 1999-10-08
WO 98145286 PCT/US98/06989
- 16 -
440 The reaction product from this step, a compound of
formula 9 is then reacted with a compound of formula _10
selected from 1-piperidine, 1-pyrrolidine, methyl-1-
pyrrolidine, dimethyl-1-pyrrolidine, 4-morpholine,
dimethylamine, diethylamine, diisopropylamine, or 1-
445 hexamethyleneimine, via standard techniques, to form
compounds of formula 7. Preferably, the hydrochloride salt
of a compound of formula 10 is employed, with piperidine
hydrochloride being particularly preferred. The reaction is
typically carried out with the alkylated compound of formula
450 9 in an inert solvent, such as anhydrous DMF, and heated to
a temperature in the range from about 60°C to about 110°C.
When the mixture is heated to a preferred temperature of
about 90°C, the reaction only takes about 30 minutes to
about 1 hour. However, changes in the reaction conditions
455 will influence the amount of time this reaction needs to be
run for completion. The progress of this reaction step can
be monitored via standard chromatographic techniques.
Certain preferred compounds of formula I are obtained
by cleaving the R5 and, when present, R6 hydroxy protecting
460 groups of formula I compounds via well known procedures.
Numerous reactions for the formation and removal of such
protecting groups are described in a number of standard
works including, for example, Protective Groups in Organic
Chemistry, Plenum Press (London and New York, 1973); Greene,
465 T.W., Protective Groups .in Organic Synthesis, Wiley, (New
York, 1981 ) ; and The Peptides, Vol . I, Schrooder and Lubke,
Academic Press (London and New York, 1965). Methods for
removing preferred R~ and/or R8 hydroxy protecting groups,
particularly methyl and methoxymethyl, are essentially as
470 described in the Examples, infra.
An alternative, and preferred, method for the
preparation of compounds of the present invention is shown
in Reaction Scheme IV. In the process shown there, the
sulfur atom of a formula 2 compound is oxidized to form a
475 sulfoxide, 11, which is then reacted with a nucleophilic
group to introduce the oxygen atom linker of formula I
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 17 -
compounds. The sulfoxide moiety of formula 12 compounds is
then reduced to provide certain compounds of the present
invention.
480
Reaction Scheme IV
r R~ i R~
I -> ~ ~ I
R50 S I ~ R50 S
ORs O ~ORs
1~
R3
I ~ O~ N~ Ra
HO
Ra Rs
t
Ra~N~O R4~N~0
I
i .r-- i
i O i O
I ~ I
R50 S ~~ R50 ~' S
I s O ~~ s
OR OR
14
I a , R5 - R6 - H
Ib, R5 _ H
Ic, R6 - H
485 In the first step of this process, a compound of
formula 2 is selectively oxidized to the sulfoxide, _12. A
number of known methods are available for the process step
[see, e.g., Madesclaire, M., Tetrahedron, _42 (20); 5459-5495
- (1986); Trost, B.M., et _al., Tetrahedron Letters, 22 (14);
490 1287-1290 (1981); Drabowicz, J., et al., Synthetic'
Communications, 11 (12); 1025-1030 (1981); Kramer, J.B., et
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 18 -
al., 34th National Orcfanic Sym osium, Williamsburg, VA.,
June 11-15, 1995]. However, many oxidants provide only poor
conversion to the desired product as well as significant
495 over-oxidation to the sulfone. The preferred process,
however, converts a formula 2 compound to a sulfoxide of
formula 12 in high yield with little or no formation of
sulfones. This process involves the reaction of a formula _2
compound with about 1 to about 1.5 equivalents of hydrogen
500 peroxide in a mixture of about 20o to about 500
trifluoroacetic acid in methylene chloride. The reaction is
run at a temperature from about 20° C to about 50° C, and
usually required from about 1 to about 2 hours to run to
completion.
505 Next, the 3-position leaving group, R~, is displaced by
the desired nucleophilic derivative of formula 13. Such
nucleophilic derivatives are prepared via standard methods.
In this step of the process, the acidic proton of the
nucleophilic group is removed by treatment with a base,
510 preferably a slight excess of sodium hydride or potassium
tertbutoxide, in a polar aprotic solvent, preferably DMF or
tetrahydrofuran. Other bases that can be employed include
potassium carbonate and cesium carbonate. Additionally,
other solvents such as dioxane or dimethylsulfoxide can be
515 employed. The deprotonation is usually run at a temperature
between about 0° C and about 30° C, and usually requires
about 30 minutes for completion. A compound of formula XIV
is then added to the solution of the nucleophile. The
displacement reaction is run at a temperature between 0° C
520 and about 50° C, and is usually run in about 1 to about 2
hours. The product is isolated by standard procedures.
In the next step of the present process, the sulfoxide
of formula 14 is reduced to a benzothiophene compound of
formula I.
525 When desired, the hydroxy protecting group or groups of
the products of the process shown in Reaction Scheme IV can
be removed, and a salt of the product of any step of the
process.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 19 -
Pro-drug ester compounds of formula I are prepared by
530 replacing the 6- and/or 4'-position hydroxy moieties, when
present, with a moiety of the formula -OCO(C1-C6 alkyl), or
-OS02(C2-C6 alkyl) via well known procedures. See, e.g.,
U.S. Pat. No. 4,358,593.
For example, when an -OCO(C1-C6 alkyl) group is
535 desired, a mono- or dihydroxy compound of formula I is
reacted with an agent such as acyl chloride, bromide,
cyanide, or azide, or with an appropriate anhydride or mixed
anhydride. The reactions are conveniently carried out in a
basic solvent such as pyridine, lutidine, quinoline or
540 isoquinoline, or in a tertiary amine solvent such as
triethylamine, tributylamine, methylpiperidine, and the
like. The reaction also may be carried out in an inert
solvent such as ethyl acetate, dimethylformamide,
dimethylsulfoxide, dioxane, dimethoxyethane, acetonitrile,
545 acetone, methyl ethyl ketone, and the like, to which at
least one equivalent of an acid scavenger (except as noted
below), such as a tertiary amine, has been added. If
desired, acylation catalysts such as 4-dimethylaminopyridine
or 4-pyrrolidinopyridine may be used. See, e.g., Haslam, _et
550 al., Tetrahedron, 36:2409-2433 (1980).
These reactions are carried out at moderate
temperatures, in the range from about -25° C to about 100°
C, frequently under an inert atmosphere such as nitrogen
gas. However, ambient temperature is usually adequate for
555 the reaction to run.
Acylation of a 6-position and/or 4'-position hydroxy
group also may be performed by acid-catalyzed reactions of
the appropriate carboxylic acids in inert organic solvents.
Acid catalysts such as sulfuric acid, polyphosphoric acid,
560 methanesulfonic acid, and the like are used.
The aforementioned ester pro-drug compounds also may be
provided by forming an active ester of the appropriate acid,
such as the esters formed by such known reagents such as
dicyclohexylcarbodiimide, acylimidazoles, nitrophenols,
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 20 -
565 pentachlorophenol, N-hydroxysuccinimide, and 1-
hydroxybenzotriazole. See, e.g., Bull. Chem. Soc. Ja an,
38:1979 (1965), and Chem. Ber., 788 and 2024 (1970).
Each of the above techniques which provide -OCO(C2-C6
alkyl) moieties are carried out in solvents as discussed
570 above. Those techniques which do not produce an acid
product in the course of the reaction, of course, do not
call for the use of an acid scavenger in the reaction
mixture.
When a formula I compound is desired in which the 6-
575 and/or 4'-position hydroxy group of a formula I compound is
converted to a group of the formula -OS02(CZ-C6 alkyl), the
mono- or dihydroxy compound is reacted with, for example, a
sulfonic anhydride or a derivative of the appropriate
sulfonic acid such as a sulfonyl chloride, bromide, or
580 sulfonyl ammonium salt, as taught by King and Monoir, J. Am.
Chem. Soc., 97:2566-2567 (1975). The dihydroxy compound
also can be reacted with the appropriate sulfonic anhydride
or mixed sulfonic anhydrides. Such reactions are carried
out under conditions such as were explained above in the
585 discussion of reaction with acid halides and the like.
Preparation of Pharmaceutically Acceptable Salts of
Compounds of the Present Invention
Although the free-base form of formula I compounds can
590 be used in the medical methods of treatment of the present
invention, it is preferred to prepare and use a pharma-
ceutically acceptable salt form. The compounds used in the
methods of this invention primarily form pharmaceutically
acceptable acid addition salts with a wide variety of
595 organic and inorganic acids. Such salts are also
contemplated as falling within the scope of the present
invention.
The term "pharmaceutically acceptable salts" as used
throughout this specification and the appended claims
600 denotes salts of the types disclosed in the article by
Berge, et al., J. Pharmaceutical Sciences, 66(1): 1-19
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 21 -
(1977). Suitable pharmaceutically acceptable salts include
salts formed by typical inorganic acids such as
hydrochloric, hydrobromic, hydroiodic, nitric, sulfuric,
605 phosphoric, hypophosphoric, and the like as well as salts
derived from organic acids, such as aliphatic mono and
dicarboxylic acids, phenyl substituted alkanoic acids,
hydroxyalkanoic and hydroxyalkandioic acids, aromatic acids,
aliphatic and aromatic sulfonic acids. 'Such
610 pharmaceutically acceptable organic acid addition salts
include acetate, phenylacetate, trifluoroacetate, acrylate,
ascorbate, benzoate, chlorobenzoate, dinitrobenzoate,
hydroxybenzoate, methoxybenzoate, methylbenzoate, o-
acetoxybenzoate, naphthalene-2-benzoate, bromide,
615 isobutyrate, phenylbutyrate, b-hydroxybutyrate, butyne-1,4-
dioate, hexyne-1,4-dioate, caprate, caprylate, chloride,
cinnamate, citrate, formate, fumarate, glycollate,
heptanoate, hippurate, lactate, malate, maleate,
hydroxymaleate, malonate, mandelate, mesylate, nicotinate,
620 isonicotinate, nitrate, oxalate, phthalate, terephthalate,
phosphate, monohydrogenphosphate, dihydrogenphosphate,
metaphosphate, pyrophosphate, propiolate, propionate,
phenylpropionate, salicylate, sebacate, succinate, suberate,
sulfate, bisulfate, pyrosulfate, sulfite, bisulfate,
625 sulfonate, benzenesulfonate, p-bromophenylsulfonate,
chlorobenzenesulfonate, ethanesulfonate, 2-
hydroxyethanesulfonate, methanesulfonate, naphthalene-1-
sulfonate, naphthalene-2-sulfonate, p-toluene-sulfonate,
xylenesulfonate, tartarate, and the like. Preferred salts
630 are the hydrochloride and oxalate salts.
The pharmaceutically acceptable acid addition salts are
typically formed by reacting a compound of formula I with an
equimolar or slight molar excess of acid. The reactants are
generally combined in a mutual solvent such as diethyl ether
635 or ethyl acetate. The salt normally precipitates out of
solution within about one hour to 10 days and can be
isolated by filtration or the solvent can be stripped off by
conventional means.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 22 -
640
The pharmaceutically acceptable salts generally have
enhanced solubility characteristics compared to the compound
from which they are derived, and thus are often more
amenable to formulation as liquids or emulsions.
645
Pharmaceutical Formulations
The compounds of this invention are administered by a
variety of routes including oral, rectal, transdermal,
subucutaneus, intravenous, intramuscular, and intranasal.
650 These compounds preferably are formulated prior to
administration, the selection of which will be decided by
the attending physician. Thus, another aspect of the
present invention is a pharmaceutical composition comprising
an effective amount of a compound of Formula I, or a
655 pharmaceutically acceptable salt thereof, optionally
containing an effective amount of estrogen or progestin, and
a pharmaceutically acceptable carrier, diluent, or
excipient.
The total active ingredients in such formulations
660 comprises from O.lo to 99.90 by weight of the formulation.
By "pharmaceutically acceptable" it is meant the carrier,
diluent, excipients and salt must be compatible with the
other ingredients of the formulation, and not deleterious to
the recipient thereof.
665 Pharmaceutical formulations of the present invention
are prepared by procedures known in the art using well known
and readily available ingredients. For example, the
compounds of Formula I, either alone, or in combination with
an estrogen or progestin compound, are formulated with
670 common excipients, diluents, or carriers, and formed into
tablets, capsules, suspensions, solutions, injectables,
aerosols, powders, and the like.
The total active ingredients in such formulations
comprises from O.lo to 99.9°s by weight of the formulation.
675 By "pharmaceutically acceptable" it is meant the carrier,
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 23 -
diluent, excipients and salt must be compatible with the
other ingredients of the formulation, and not deleterious to
the recipient thereof.
The formulations may be specially formulated for oral
680 administration, in solid or liquid form, for parenteral
injection, topical or aerosol administration, or for rectal
or vaginal administration by means of a suppository.
The pharmaceutical compositions of this invention can
be administered to humans and other mammals orally,
685 rectally, intravaginally, parenterally, topically (by means
of powders, ointments, creams, or drops), bucally or
sublingually, or as an oral or nasal spray. The term
"parenteral administration" refers herein to modes of
administration which include intravenous, intramuscular,
690 intraperitoneal, instrasternal, subcutaneous, or
intraarticular injection or infusion.
Pharmaceutical compositions of this invention for
parenteral administration comprise sterile aqueous or non-
aqueous solutions, dispersions, suspensions, or emulsions,
695 as well as sterile powders which are reconstituted
immediately prior to use into sterile solutions or
suspensions. Examples of suitable sterile aqueous and non-
aqueous carriers, diluents, solvents or vehicles include
water, physiological saline solution, ethanol, polyols (such
700 as glycerol, propylene glycol, polyethylene glycol), and
the like), and suitable mixtures thereof, vegetable oils
(such as olive oil), and injectable organic esters such as
ethyl oleate. Proper fluidity is maintained, for example,
by the use of coating materials such as lecithin, by the
705 maintenance of proper particle size in the case of
dispersions and suspensions, and by the use of surfactants.
Parenteral compositions may also contain adjuvants such
as preservatives, wetting agents, emulsifying agents, and
dispersing agents. Prevention of the action of
710 microorganisms is ensured by the inclusion of antibacterial
and antifungal agents, for example, paraben, chlorobutanol,
phenol sorbic acid, and the like. It may also be desirable
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 24 -
to include isotonic agents such as sugars, sodium chloride,
and the like. Prolonged absorption of injectable
715 formulations may be brought about by the inclusion of agents
which delay absorption such as aluminum monostearate and
gelatin.
In some cases, in order to prolong the effect of the
drug, it, is desirable to slow the absorption of the drug
720 following subcutaneous or intramuscular injection. This may
be accomplished by the use of a liquid suspension or
crystalline or amorphous material of low water solubility or
by dissolving or suspending the drug in an oil vehicle. In
the case of the subcutaneous or intramuscular injection of a
725 suspension containing a form of the drug with low water
solubility, the rate of absorption of the drug depends upon
its rate of dissolution.
Injectable "depot" formulations of the compounds of
this invention are made by forming microencapsulated
730 matrices of the drug in biodegradable polymers such as
poly(lactic acid), poly(glycolic acid), copolymers of lactic
and glycolic acid, poly (orthoesters), and poly
(anhydrides) these materials which are described in the art.
Depending upon the ratio of drug to polymer and the
735 characteristics of the particular polymer employed, the rate
of drug release can be controlled.
Injectable formulations are sterilized, for example, by
filtration through bacterial-retaining filters, or by
presterilization of the components of the mixture prior to
740 their admixture, either at the time of manufacture or just
prior to administration (as in the example of a dual chamber
syringe package).
Solid dosage forms for oral administration include
capsules, tablets, pills, powders, and granules. In such
745 solid dosage forms, the active component is mixed with at
least one inert, pharmaceutically acceptable carrier such as
sodium citrate, or dicalcium phosphate, and/or (a) fillers
or extenders such as starches, lactose, glucose, mannitol,
and silicic acid, (b) binding agents such as carboxymethyl-
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 25 -
750 cellulose, alginates, gelatin, poly(vinylpyrrolidine),
sucrose and acacia, (c) humectants such as glycerol, (d)
disintegrating agents such as agar-agar, calcium carbonate,
potato or tapioca starch, alginic acid, silicates and sodium
carbonate, (e) solution retarding agents such as paraffin,
755 (f) absorption accelerating agents such as quaternary
ammonium compounds, (g) wetting agents such as cetyl alcohol
and glycerin monostearate, (h) absorbents such as kaolin and
bentonite clay, and (i) lubricants such as talc, calcium
stearate, magnesium stearate, solid polyethylene glycols),
760 sodium lauryl sulfate, and mixtures thereof. In the case of
capsules, tablets and pills, the dosage form may also
contain buffering agents.
Solid compositions of a similar type may also comprise
the fill in soft or hard gelatin capsules using excipients
765 such as lactose as well as high molecular weight
polyethylene glycols) and the like.
Solid dosage forms such as tablets, dragees, capsules,
pills and granules can also be prepared with coatings or
shells such as enteric coatings or other coatings well known
770 in the pharmaceutical formulating art. The coatings may
contain opacifying agents or agents which release the active
ingredients) in a particular part of the digestive tract,
as for example, acid soluble coatings for release of the
active ingredients) in the stomach, or base soluble
775 coatings for release of the active ingredients) in the
intestinal tract.
The active ingredients) may also be microencapsulated
in a sustained-release coating, with the microcapsules being
made part of a pill of capsule formulation.
780 Liquid dosage forms for oral administration of the
compounds of this invention include solution, emulsions,
suspensions, syrups and elixirs. In addition to the active
components, liquid formulations may include inert diluents
commonly used in the art such as water or other
785 pharmaceutically acceptable solvents, solubilizing agents
and emulsifiers such as ethanol, isopropanol, ethyl
CA 02286207 1999-10-08
WO 98/45286 PCT/U598/06989
- 26 -
carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate,
propylene glycol, 1,3-butylene glycol, dimethyl formamide,
oils (in particular, cottonseed, ground nut, corn, germ,
790 olive, castor, and sesame oils), glycerol,
tetrahydrofurfuryl alcohol, polyethylene glycols), fatty
acid esters of sorbitol, and mixtures thereof.
Besides inert diluents, the liquid oral formulations
may also include adjuvants such as wetting agents,
795 emulsifying and suspending agents, and sweetening,
flavoring, and perfuming agents.
Liquid suspension, in addition to the active
ingredients) may contain suspending agents such as
ethoxylated isostearyl alcohols, polyoxyethylene sorbitol
800 and sorbitan esters, microcrystalline cellulose, aluminum
metahydroxide, bentonite clay, agar-agar, and tragacanth,
and mixtures thereof.
Compositions for rectal or intravaginal administration
are prepared by mixing one or more compounds of the present
805 invention with suitable non-irritating excipients such as
cocoa butter, polyethylene glycol or any suppository wax
which is a solid at room temperature, but liquid at body
temperature and therefore melt in the rectum or vaginal
cavity to release the active component(s). The compounds
810 are dissolved in the melted wax, formed into the desired
shape, and allowed to harden into the finished suppository
formulation.
Compounds of the present invention may also be
administered in the form of liposomes. As is know in the
815 art, liposomes are generally derived from phospholipids or
other lipid substances. Lipososome formulations are formed
by mono- or multilamellar hydrated liquid crystals which are
dispersed in an aqueous medium. Any non-toxic,
pharmaceutically acceptable, and metabolizable lipid capable
820 of forming liposomes can be used. The present compositions
in liposome form can contain, in addition to one or more
active compounds of the present invention, stabilizers,
excipients, preservatives, and the like. The preferred
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 27 -
lipids are phospholipids and the phosphatidyl cholines
825 (lecithins), both natural and synthetic.
Methods for forming liposomes are know in the art as
described, for example, in Prescott, Ed., Methods in Cell
Biology, Volume XIV, Academic Press, New York, N. Y. (1976),
p. 33 et seq.
830
Method of the Present Invention
Induction of mammary tumors in rats by administration
of the carcinogen N-nitroso-N-methylurea is a well-accepted
animal model for the study of breast cancer and has been
835 found suitable for analyzing the effect of chemopreventive
agents.
In two separate studies, 55-day old female Sprague-
Dawley rats were given an intravenous (Study 1) or
intraperitoneal (Study 2) dose of 50 mg of N-nitroso-N-
840 methylurea per kilogram of body weight one week prior to
feeding ad libitum a diet into which varying amounts of a)
6-hydroxy-2-(4-methoxyphenyl)-3-[4-(2-piperidinylethoxy)-
phenoxy]benzo[b]thiophene hydrochloride, b) (Z)-2-[4-(1,2-
diphenyl-1-butenyl)phenoxy]-N,N-dimethylethanamine base
845 (tamoxifen base), or c) control were blended. The control
comprised the vehicle employed in combination with the
active compounds.
In Study l, the dietary doses of 60 mg/kg of diet and
20 mg/kg of diet translated into roughly comparable doses of
850 3 and 1 mg/kg of body weight for the test animals.
In Study 2, the dietary doses of 20, 6, 2, and 0.6
mg/kg of diet translated roughly into comparable doses of 1,
0.3, 0.1 and 0.03 mg/kg of body weight for the test animals.
Rats were observed for evidence of toxicity and were
855 weighed and palpated for tumor formation once a week. The
animals were sacrificed after thirteen weeks (Study 1) or
eighteen weeks (Study 2) and tumors were confirmed and
weighed at autopsy. The results of these studies are shown
in Table 1 (Study 1) and Table 2 (Study 2) below.
860
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 28 -
Table 1
Prevention of Mammary Cancer in
Female Sprague-Dawley Rats by Administration of
6-Hydroxy-2-(4-methoxyphenyl)-3-[4-(2-piperidinylethoxy)-
865 phenoxy]benzo[b]thiophene Hydrochloride (Example 15)
Treatment Tumor-Free Avg. No. of Avg. Tumor
Rats Tumors Per Burden Per
Rat Rat
9m)
Control 3/24 (12%) 3.0 11.0
Cmpd. of Ex. 15 11/12 (92%) 0.08 0.05
60 mg/kg of
diet
Compd. of Ex. 11/12 (92%) 0.08 0.03
15
20 mg/kg of
diet
Table 2
Prevention of Mammary Cancer in
870 Female Sprague-Dawley Rats by Administration of
6-hydroxy-2-(4-Methoxyphenyl)-3-[4-(2-piperidinylethoxy)-
phenoxy]benzo[b]thiophene Hydrochloride (Example 15)
or Tamoxifen Base
Treatment Tumor-Free Avg. No. of Avg. Tumor
Rats Tumors Per Burden Per
Rat Rat
gm?
Control 6/23 (26%) 1.8 9.0
Cmpd. of Ex. 15 11/12 (92%) 0.08 0.43
20 mg/kg of
diet
Compd. of Ex. 7/12 (58%) 0.50 1.50
15
6 mg/kg of diet
Cmpd. of Ex. 15 8/12 (67%) 0.50 3.40
2 mg/kg of diet
Cmpd. of Ex. 15 6/I2 (50%) 0.75 I.60
0.6 mg/kg of
diet
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 29 -
Tamoxifen 7/11 (64%.) 0 0
33 52
base . .
2 mg/kg of diet
Tamoxifen 7/12 (5B%) 09 2
83 10
base . .
0.6 mg/kg of
diet
875
Examination of the data in Table 1 shows that
administration of the compound of Example 15 of the present
invention resulted in a significant decrease in tumor
incidence (87%), average numbers of tumors per rat (97%) and
880 average tumor burden per rat (99%) compared to control.
Examination of the data appearing in Table 2 shows that
administration of doses as low as 0.6 mg/kg of diet of the
compound of Example 15 of the present invention were
sufficient to significantly reduce the incidence of tumor
885 formation, number of tumors per rat, and average tumor
burden per rat when compared with control. The observed
effects were dose dependent and comparable to those observed
with tamoxifen.
The observation of an effect at all doses tested
890 prevented a definitive comparison between the two compounds
since a plateau-like effect was observed at the lower doses
for both compounds. However, the data from Tables 1 and 2
indicate that the compound of Example 15 is at least as
effective, or more effective than tamoxifen as an agent for
895 the inhibition or prevention of breast cancer.
Thus, administration of an effective amount of a
compound of the present invention, in particular 6-hydroxy-
2-(4-methoxyphenyl)-3-[9-(2-piperidinoethoxy)phenoxy]-
benzo[b)thiophene, is a useful method for the prophylaxis,
900 prevention or inhibition of breast tumor formation.
As used herein, the term "effective amount" means an
amount of compound of the present invention which is capable
of inhibiting or preventing breast tumor formation. The
specific dose of a compound administered according to this
905 invention is determined by the particular circumstances
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 30 -
surrounding each situation including, for example, the
potency of the compound administered, the route of
administration, the prior medical history of the patient,
and the pathological condition being treated. A typical
910 daily dose will contain a nontoxic dosage level of from
about 5 mg to about 600 mg/day of a compound of the present
invention. Preferred daily doses generally will be from
about 15 mg to about 80 mg/day.
The exact dose is determined, in accordance with the
915 standard practice in the medical arts of dose titrating the
patient; that is, initially administering a low dose of the
compound, and gradually increasing the does until the
desired therapeutic effect is observed.
920 The following examples are presented to further
illustrate the preparation of compounds of the present
invention. The Examples are not to be read as limiting the
scope of the invention as it is defined by the appended
claims.
925 NMR data for the following Examples were generated on a
GE 300 MHz NMR instrument, and anhydrous hexadeutero-
dimethylsulfoxide was used as the solvent unless otherwise
indicated.
930 Example 1
Preparation of [6-methoxy-3-[4-[2-(1-piperidinyl)ethoxy]-
phenoxy]-2-(4-methoxyphenyl)]benzo[b]thiophene oxalate salt
~ (COOH) 2
NCO
i
O
I
H3C0 ~ S
I
OCH3
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 31 -
935
Step a: Preparation of [6-methoxy-2-(4-methoxy-phenyl)-3-
bromo]benzo[b]thiophene
Br
I
H3C0 S
OCH3
940
To a solution of [6-methoxy-2-(4-methoxyphenyl)]benzo-
[b]thiophene (27.0 g, 100 mmol)in 1.10 L of chloroform at
60° C was added bromine (15.98 g, 100 mmol) dropwise as a
solution in 200 mL of chloroform. After the addition was
945 complete, the reaction was cooled to room temperature, and
the solvent removed in vacuo to provide 34.2 g (1000) of [6-
methoxy-2-(4-methoxyphenyl)-3-bromo]benzo[b]thiophene as a
white solid. mp 83-85° C. 1H NMR (DMSO-d6) d 7.70-7.62 (m,
4H) , 7. 17 (dd, J = 8. 6, 2.0 Hz, 1H) , 7.09 (d, J = 8. 4 Hz,
950 2H). FD mass spec: 349, 350. Anal. Calcd. for C16H1302SBr:
C, 55.03; H, 3.75. Found: C, 54.79; H, 3.76.
Step b): Preparation of [6-methoxy-2-(4-methoxyphenyl)-3
(4-benzyloxy)phenoxy]benzo[b]thiophene
955
O
I/
/ O
I
H3C0 S
I
OCH3
To a solution of [6-methoxy-2-(4-methoxyphenyl)-3-
bromo] benzo[b]thiophene (34.00 g, 97.4 mmol) in 60 mL of
960 collidine under N2 was added 4-benzyloxyphenol (38.96 g,
194.8 mmol) and cuprous oxide (14.5 g, 97.4 mmol). The
resultant mixture was heated to reflux for 48 hours. Upon
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 32 -
cooling to room temperature, the mixture was dissolved in
acetone (200 mL), and the inorganic solids were removed by
965 filtration. The filtrate was concentrated in vacuo, and the
residue dissolved in methylene chloride (500 mL). The
methylene chloride solution was washed with 3N hydrochloric
acid (3 X 300 mL), followed by 1N sodium hydroxide (3 x 300
mL). The organic layer was dried (sodium sulfate), and
970 concentrated in vacuo. The residue was taken up in 100 mL
of ethyl acetate whereupon a white solid formed that was
collected by filtration [recovered [6-methoxy-2-(4-
methoxyphenyl)]benzo-[b]thiophene (4.62 g, 17.11 mmol]. The
filtrate was concentrated in vacuo, and then passed through
975 a short pad of silica gel (methylene chloride as eluant) to
remove baseline material. The filtrate was concentrated in
vacuo, and the residue crystallized from hexanes/ethyl
acetate to provide initially 7.19 g of [6-methoxy-2-(4-
methoxyphenyl)-3-(4-benzyloxy)phenoxy]benzo[b]-thiophene as
980 an off-white crystalline solid. The mother liquor was
concentrated and chromatographed on silica gel
(hexanes/ethyl acetate 80:20) to provide an additional 1.81
g of product. Total yield of [6-methoxy-2-(4-methoxyphenyl)-
3-(4-benzyloxy)phenoxy]-benzo[b]thiophene was 9.00 g (240
985 based on recovered starting material). The basic extract was
acidified to pH = 4 with 5N hydrochloric acid, and the
resultant precipitate collected by filtration and dried to
give 13.3 g of recovered 4-benzyloxyphenol. mp 100-103° C.
1H NMR (CDC13): d 7.60 (d, J = 8.8 Hz, 2H), 7.39-7.24 (m,
990 7H), 6.90-6.85 (m, 7H), 4.98 (s, 2H), 3.86 (s, 3H) 3.81 (s,
3H). FD mass spec: 468. Anal. Calcd. for C2gH2qOq5: C,
74.34; H, 5.16. Found: C, 74.64; H, 5.29.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 33 -
Step c): Preparation of [6-methoxy-2-(4-methoxyphenyl)-3-
995 (4-hydroxy)phenoxy]benzo[b]thiophene
OH
~J
O
H3C0 S
OCH3
To a solution of [6-methoxy-2-(4-methoxyphenyl)-3-(4-
1000 benzyloxy)phenoxy]benzo[b]thiophene (1.50 g, 3.20 mmol) in
50 mL of ethyl acetate and 10 mL of to concentrated
hydrochloric acid in ethanol was added loo palladium-on-
carbon (300 mg). The mixture was hydrogenated at 40 psi for
20 minutes, after which time the reaction was judged
1005 complete by thin layer chromatography. The mixture was
passed through Celite to remove catalyst, and the filtrate
concentrated in vacuo to a white solid. The crude product
was passed through a pad of silica gel (chloroform as
eluant). Concentration provided 1.10 g (910) of [6-methoxy-
1010 2-(4-methoxyphenyl)-3-(4-hydroxy)phenoxy]benzo[b]-thiophene
as a white solid. mp 123-126° C. 1H NMR (DMSO-d6) d 9.10
(s, 1H), 7.59 (d, J = 8.8 Hz, 2H), 7.52 (d, J = 2.1 Hz, 1H),
7 . 14 (d, J = 8 . 8 Hz, 1H) , 6.95 (d, J = 8. 8 Hz, 2H) , 6. 89
(dd, J = 8 . 8, 2. 1 Hz, 1H) , 6.72 (d, J = 9. 0 Hz, 2H) , 6. 63
1015 (d, J = 9.0 Hz, 2H) , 3.78 (s, 3H) , 3.72 (s, 3H) . FD mass
spec: 378. Anal. Calcd. for C22H1e09S: C, 69.82; H, 4.79.
Found: C, 70.06; H, 4.98.
Step d): Preparation of [6-methoxy-3-[4-[2-(1-piperidinyl)-
1020 ethoxy]phenoxy]-2-(4-methoxyphenyl)]benzo[b]-
thiophene oxalate salt
To a solution of [6-methoxy-2-(4-methoxyphenyl)-3-(4-
hydroxy)phenoxy]benzo[b]thiophene (1.12 g, 2.97 mmol) in 7
mL of anhydrous N,N-dimethylformamide under N2 was added
CA 02286207 1999-10-08
WO 98/45286 PCT/US98l06989
- 34 -
1025 cesium carbonate (3.86 g, 11.88 mmol). After stirring for
minutes, 2-chioroethylpiperidine hydrochloride (1.10 g,
1.48 mmol) was added. The resultant mixture was stirred for
18 hours at ambient temperature. The reaction was the
distributed between chloroform/water (100 mL each). The
1030 layers were separated and the aqueous extracted with
chloroform (3 x 50 mL). The organic was combined and washed
with water (2 x 100 mL). Drying of the organic (sodium
sulfate) and concentration provided an oil that was
chromatographed on silica gel (2o methanol/chloroform). The
1035 desired fractions were concentrated to an oil that was
dissolved in 10 mL of ethyl acetate and treated with oxalic
acid (311 mg, 3.4 mmol). After stirring for 10 minutes, a
white precipitate formed and was collected by filtration and
dried to provide 1.17 g (70%) overall of [6-methoxy-3-[4-[2-
1040 (1-piperidinyl)ethoxy]-phenoxy]-2-(4-methoxyphenyl)]benzo[b]
thiophene as the oxalate salt. mp 197-200° C (dec). 1H NMR
(DMSO-d6~ d 7.60 (d, J = 8.7 Hz, 2H), 7.55 (d, J = 1.1 Hz,
1H), 7.14 (d, J = 8.8 Hz, 1H), 7.06 (d, J = 8.8 Hz, 2H),
6.91 (dd, J = 8.8, 1.1 Hz, 1H), 6.87 (s, 4H), 4.19 (broad t,
1045 2H) , 3.78 (s, 3H) , 3.72 (s, 3H) , 3.32 (broad t, 2H) , 3.12-
3.06 (m, 4H), 1.69-1.47 (m, 4H), 1.44-1.38 (m, 2H). FD mass
spec: 489 . Anal. Calcd. for C29H31NOqS~0.88 H02CC02H: C,
64.95; H, 5.80; N, 2.46. Found: C, 64.92; H, 5.77; N, 2.54.
1050
_ _ _ _ __.___~_ __
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 35 -
Example 2
Preparation of[6-methoxy-3-[4-[2-(1-piperidinyl)ethox
phenoxy]-2-(4-methoxyphenyl)]benzo[b]thiophene hydrochloride
1055 salt
~ HCI
~N~
O
I
i
O
I
H3C0 S
OCH3
Treatment of the oxalate salt from Example 1 with
aqueous base to produce the free base, followed by reaction
1060 with diethyl ether saturated with HC1 yielded the title
salt, mp 216-220° C. 1H NMR (DMSO-d6~ d 10.20 (bs, 1H),
7.64 (d, J = 8.7 Hz, 2H), 7.59 (d, J = 1.5 Hz, 1H), 7.18 (d,
J = 9. 0 Hz, 1H) , 7. 00 (d, J = 8. 7 Hz, 1H) , 6. 96 (dd, J =
9. 0, 1 . 5 Hz, 1H) , 6. 92 (q, JAB = 9. 0 Hz, 4H) , 4 . 31 (m, 2H) ,
1065 3.83 (s, 3H) , 3.77 (s, 3H) , 3.43 (m, 4H) , 2.97 (m, 2H) , 1.77
(m, 5H), 1.37 (m, 1H). FD mass spec: 489 . Anal. Calcd.
for C2gH31NOqS~1.0 HC1: C, 66.21; H, 6.13; N, 2.66. Found:
C, 66., 46; H, 6.16; N, 2.74.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 36 -
1070 Example 3
Preparation of [6-Methoxy-3-[4-[2-(1-pyrolodinyl)ethoxy]-
phenoxy]-2-(4-methoxyphenyl)]benzo[b]thio hene
~N~
O
i
O
I
H3C0 S
1075 OCH 3
The title compound was prepared in the same manner as
the compound of Example l, mp 95-98° C. 1H NMR (DMSO-d6) c1
7.64 (d, J = 9.0 Hz, 2H), 7.58 (d, J = 2.0 Hz, 1H), 7.18 (d,
1080 J = 9. 0 Hz, 1H) , 7. 00 (d, J = 9. 0 Hz, 2H) , 6.94 (dd, J =
9. 0, 2. 0 Hz, 1H) , 6. 86 (s, 4H) , 3.97 (t, J = 6. 0 Hz, 2H) ,
3.83 ( s, 3H), 3.76 (s, 3H), 2.73 (t, J = 6.0 Hz, 2H), 2.51
(m, 4H), 1.66 (m, 4H). FD mass spec: 477. Anal. Calcd. for
C28H2gNOqS: C, 70.71; H, 6.15; N, 2.99. Found: C, 70.59; H,
1085 6.15; N, 3.01.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98106989
- 37 -
Example 4
Preparation of [6-Methoxy-3-[4-[2-(1-hexamethyleneimino)-
1090 ethoxy]phenoxy]-2-(4-methoxyphenyl)lbenzo[b]thiophene
hydrochloride
~HCI
N'.~ O
i
O
I
H3C0 S
OCH3
1095 The title compound was prepared in the same manner as
the compound of Example l, mp 189-192° C. 1H NMR (DMSO-d6)
d 10. 55 (bs, 1H) , 7 . 64 (d, J = 9. 0 Hz, 2H) , 7. 58 (d, J = 2 . 0
Hz, 1H), 7.19 (d, J = 9.0 Hz, 1H), 7.00 (d, J = 9.0 Hz, 2H),
6.95 (dd, J = 9.0, 2. 0 Hz, H) , 6.86 (s, 4H) , 3.94 (t, J =
1100 6.0 Hz, 2H), 3.83 (s, 3H), 3.76 (s, 3H), 2.80 (t, J = 6.0
Hz, 2H), 2.66 (m, 4H), 1.53 (m, 8H). Anal. Calcd. for
C3oH33NOqS~1.0 HC1: C, 66.71; H, 6.35; N, 2.59. Found: C,
66.43; H, 6.46; N, 2.84.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 38 -
1105 Example 5
Preparation of [6-Methoxy-3-[4-[2-(1-N,N-diethylamino)-
ethoxy]phenoxy)-2-(4-methoxyphenyl))benzo[b)thiophene
hydrochloride
1110
1 .HCi
~N~O
i
O
H3C0 S ~
OCH3
The title compound was prepared in the same manner as
the compound of Example 1, mp 196-198° C. 1H NMR (DMSO-d6)
1115 d 10.48 (bs, 1H), 7.69 (d, J = 9.0 Hz, 2H), 7.59 (d, J = 2.0
Hz, 1H) , 7 . 19 (d, J = 9. 0 Hz, 1H) , 7. 00 (d, J = 9. 0 Hz, 2H) ,
6.97 (dd, J = 9. 0, 2.0 Hz, 1H) , 6. 87 (q, JAB = 9. 0 Hz, 4H) ,
4.25 (m, 2H) , 3. 83 (s, 3H) , 3.77 (s, 3H) , 3.54 (m, 2H) , 3. 0~
(m, 4H), 2.00 (m, 3H), 1.88 (m, 3H). Anal. Calcd. for
1120 C28H31N04S~1.5 HC1: C, 63.18; H, 6.15; N, 2.63. Found: C,
63.46; H, 5.79; N, 2.85.
__._ _ . _ . ___.___--.____T _.,.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 39 -
Example 6
1125 Preparation of {6-Methoxy-3-[4-[2-(morpholino)ethoxy]-
phenoxyJ-2-(4-methoxyphenyl)Jbenzo[bJthiophene hydrochloride
0'1 ~HCi
I
0
~i I
H3C0 S
OCH3
1130 The title compound was prepared in the same manner as
the compound of Example 1, mp 208-211° C. 1H NMR (DMSO-d6)
d 10.6 (bs, 1H), 7.63 (d, J = 9.0 Hz, 2H), 7.60 (d, J = 2.0
Hz, 1H) , 7.20 (J = 9.0 Hz, 1H) , 7. 00 (d, J = 9. 0 Hz, 2H) ,
6.97 (dd, J = 9.0, 2. 0 Hz, 1H) , 6. 91 (q, JpB = 9. 0 Hz, 4H) ,
1135 4.29 (m, 2H), 4.08-3.91 (m, 4H), 3.82 (s, 3H), 3.77 (s, 3H),
3.59-3.42 (m, 4H), 3.21-3.10 (m, 2H). Anal. Calcd. for
C28HZgN05S~1.0 HC1: C, 63.09; H, 5.73; N, 2.65. Found: C,
63.39; H, 5.80; N, 2.40.
CA 02286207 1999-10-08
WO 98/45286 PCTIUS98/06989
- 40 -
1140 Example 7
Preparation of [6-Hydroxy-3-[4-[2-(1-piperidinyl)ethoxy]-
phenoxy]-2-(4-hydroxyphenyl)]benzo[b]thiophene
O
O
HO S
1145 ~ OH
[6-methoxy-3-[4-[2-(1-piperidinyl)ethoxy]phenoxy]-2-(4-
methoxyphenyl)]benzo(b]thiophene hydrochloride (10.00 g,
19.05 mmol) was dissolved in 500 mL of anhydrous methylene
1150 chloride and cooled to 8° C. To this solution was added
boron tribromide (7.20 mL, 76.20 mmol). The resultant
mixture was stirred at 8° C for 2.5 hours. The reaction was
quenched by pouring into a stirring solution of saturated
sodium bicarbonate (1 L), cooled to 0° C. The methylene
1155 chloride layer was separated, and the remaining solids were
dissolved in methanol/ethyl acetate. The aqueous layer was
then extracted with 5o methanol/ethyl acetate (3 x 500 mL).
All of the organic extracts (ethyl acetate and methylene
chloride) were combined and dried (sodium sulfate).
1160 Concentration in vacuo provided a tan solid that was
chromatographed (silicon dioxide, 1-7% methanol/chloroform)
to provide 7.13 g (81 0) of [6-hydroxy-3-[4-[2-(1-
piperidinyl) ethoxy]phenoxy]-2-(4-hydroxyphenyl)]benzo[b]-
thiophene as a white solid. mp 93° C. 1H NMR (DMSO-d6) a
1165 9.73 (bs, 1H), 9.68 (bs, 1H), 7.45 (d, J = 8.6 Hz, 2H), 7.21
(d, J = 1 . 8 Hz, 1H) , 7 . 04 (d, J= 8 . 6 Hz, 1H) , 6. 84 (dd, J =
8 . 6, 1 . 8 Hz, 1H (masked) ) , 6. 81 (s, 4H) , 6.75 (d, J = 8. 6
Hz, 2H), 3.92 (t, J = 5.8 Hz, 2H), 2.56 (t, J = 5.8 Hz, 2H),
2.36 (m. 4H), 1.43 (m, 4H), 1.32 (m, 2H). FD mass spec:
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 41 -
1170 462. Anal. Calcd. for C2~H2~NOqS: C, 70.20; H, 5.90; N,
3.03. Found: C, 69.96; H, 5.90; N, 3.14.
Example 8
1175 Preparation of [6-Hydroxy-3-[4-[2-(1- iperidinyl)ethoxy]-
phenoxy]-2-(4-hydroxyphenyl)]benzo[b]thio hene oxalate salt
~N ~~(C\OOH) 2
O
I
I
O
I
HO S
OH
1180 The title compound was prepared in 80% yield from the
free base, mp 246-249° C (dec) . 1H NMR (DMSO-d6) ct ?.45 (d,
J = 8.6 Hz, 2H), 7.22 (d, J = 1.8 Hz, 1H), 7.05 (d, J = 8.6
Hz, 1H) , 6.87 (dd, J = 8. 6, 1 .8 Hz, 1H (masked) ) , 6.84 (s,
4H) , 6.75 (d, J = 8 . 6 Hz, 2H) , 4. 08 (bt, 2H) , 3. 01 (bt, 2H} ,
1185 2.79 (m, 4H), 1.56 (m, 4H), 1.40 (m, 2H). FD mass spec 462.
Anal. Calcd. for C2~H2~NOqS~0.75 H02CCOzH: C, 64.63; H, 5.42;
N, 2.64. Found: C, 64.61; H, 5.55; N, 2.62.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 42 -
1190
Example 9
Preparation of [6-Hydroxy-3-[4-[2-(1-piperidinyl)ethoxy]-
phenoxy]-2-(4-hydroxyphenyl)]benzo[b]thiophene hydrochloride
~ HCI
O
I
O
I
HO S
OH
1195
The title compound was prepared in 91o yield by
treatment of the corresponding free base with HCl saturated
diethyl ether, mp 158-165° C. 1H NMR (DMSO-d6) d 9.79 (s,
1H) , 9.74 (s, 1H) , 7 .40 (d, J = 8 . 6 Hz, 2H) , 7.23 (d, J =
1200 2.0 Hz, 1H) , 7.04 (d, J = 8. 6 Hz, 1H) , 6.86 (q, JAB = 9.3
Hz, 4H) , 6.76 (dd, J = 8.6, 2.0 Hz, 1) , 6.74 (d, J = 8.6 Hz,
2H) , 4 .26 (bt, 2H) , 3.37 (m, 4H) , 2.91 (m, 2H) , 1 .72 (m, 5
H), 1.25 (m, 1H). FD mass spec 461. Anal. Calcd. for
C27H27NOqS~1.0 HCl: C, 65.11; H, 5.67; N, 2.81. Found: C,
1205 64.84: H, 5.64; N, 2.91.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 43 -
Example 10
Preparation of [6-Hydroxy-3-[4-[2-(1-pyrolidinyl)ethoxy]-
1210 phenoxy]-2-(4-hydroxyphenyl)]benzo[b]thiophene
~N..~
O
'J
O
W
HO S
OH
The title compound was prepared from the product of
1215 Example 3 in a manner similar to that employed in Example 7
above; mp 99-113° C. 1H NMR (DMSO-d6) r1 9.75 (s, 1H) , 9.71
(s, 1H) , 7. 50 (d, J = 9. 0 Hz, 2H) , 7 .25 (d, J = 2 . 0 Hz, 1H) ,
7.09 (d, J = 9.0 Hz, 1H) , 6.85 (s, 1H) , 6.80 (dd, J = 9.0,
2.0 Hz, 1H), 6.79 (d, J = 9.0 Hz, 2H), 3.93 (m, 2H), 2.73
1220 (m, 2H) , 2. 53 (m, 4H) , 0. 96 (t, J = 7. 0 Hz, 4H) . Anal.
Calcd. for C26H25NOqS~0.5 H20: C, 68.40; H, 5.74; N, 3.07.
Found: C, 68.52; H, 6.00; N, 3.34.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 44 -
Example 11
1225
Preparation of [6-Hydroxy-3-[4-[2-(1-hexamethyleneimino)-
ethoxy]phenoxy]-2-(4-hydroxyphenyl)]benzo[b]thiophene
N~ O
~J
O
W
Ho s ~
OH
1230
The title compound was prepared from the product of
Example 4 in a manner similar to that employed in Example 7
above; mp 125-130° C. 1H NMR (DMSO-d6) d 9.75 (s, 1H), 9.71
(s, 1H) , 7. 50 (d, J = 9.0 Hz, 2H) , 7.26 (d, J = 2.0 Hz, 1H) ,
1235 7.09 (d, J = 9.0 Hz, 1H), 6.85 (s, 3H), 6.80 (dd, J = 9.0,
2.0 Hz, 1H) , 6. 79 (d, J = 9.0 Hz) , 3.94 (t, J = 6.0 Hz, 2H) ,
2.80 (t, J = 6.0 Hz, 2H), 2.66 (m, 4H), 1.53 (m, 8H). Anal.
Calcd. for CZ8H2gNOqS: C, 70.71; H, 6.15; N, 2.94. Found:
C, 70.67; H, 6.31; N, 2.93.
1240
_._-_. T ___-.__. _.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/Ob989
- 45 -
Example 12
Preparation of [6-Hydroxy-3-[4-[2-(1-N,N-
diethylamino ) ethoxy] phenoxy] -2- ( 4-
1245 hydroxyphenyl)]benzo[b]thin hene
w.N~O
I
I
O
I
HO S
OH
The title compound was prepared from the product of
1250 Example 5 in a manner similar to that employed in Example 7
above; mp 137-14I° C. 1H NMR (DMSO-d6) d 9.75 (s, 1H) , 9.71
(s, 1H), 7.49 (d, J = 9.0 Hz, 1H), 7.25 (d, j - 2.0 Hz, 1H),
7.09 (d, J = 9. 0 Hz, 1H) , 6. 85 (s, 4H) , 6. 80 (dd, J = 9. 0,
2.0 Hz, 1H), 6.79 (d, J = 9.0 Hz, 2H), 3.95 (t, J = 6.0 Hz,
1255 2H) , 2. 74 ( t, J = 6. 0 Hz, 2H) , 2 .51 (m, 9H) , 1 . 66 (m, 6H) .
Anal. Calcd. for C26H2-7NOqS: C, 69.46; H, 6.05; N, 3.12.
Found: C, 69.76; H, 5.85; N, 3.40.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 46 -
1260
Example 13
Preparation of [6-Hydroxy-3-[4-[2-(morpholino)ethoxy]-
phenoxy]-2-(4-hydroxyphenyl)]benzo[b]thiophene hydrochloride
o'~ ~HCi
~'N'~'o
~J
0
W
Ho s
OH
1265
The title compound was prepared from the product of
Example 6 in a manner similar to that employed in Example 7
above; mp 157-162° C. 1H NMR (DMSO-d6) a 10.60 (bs, 1H),
9.80 (s, 1H), 9.75 (s, 1H), 7.50 (d, J = 9.0 Hz, 2H), 7.28
1270 (d, J = 2. 0 Hz, 1H) , 7.10 (d, J = 9.0 Hz, 1H) , 6.92 (q, JRB
- 9. 0 Hz, 4H) , 6. 81 (dd, J = 9. 0, 2 . 0 Hz, 1H) , 6. 80 (d, J =
9.0 Hz, 2H), 4.30 (m, 2H), 3.95 (m, 2H), 3.75 (m, 2H), 3.51
(m, 4H), 3.18 (m, 2H). Anal. Calcd. for C26H25N05S~HC1: C,
62.46; H, 5.24; N, 2.80. Found: C, 69.69; H, 5.43; N, 2.92.
1275
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 47 -
Example 14
Preparation of [6-Hydroxy-3-[4-[2-(1-pi eridinyl)ethoxy]-
phenoxy]-2-(4-methoxyphenyl)]benzo[b]thiophene
1280
~N~~
__ O
i
O
HO S
OCH3
Step a): Preparation of 6-Methoxybenzo[b]thiophene-2-
boronic acid
1285
i
H3C0 S B-OH
OH
To a solution of 6-methoxybenzo[b]thiophene (18.13 g,
0.111 mol) in I50 mL of anhydrous tetrahydrofuran (THF) at -
1290 60° C was added n-butyllithium (76.2 mL, .122 mol, 1.6 M
solution in hexanes), dropwise via syringe. After stirring
for 30 minutes, triisopropyl borate (28.2 mL, .122 mol) was
introduced via syringe. The resulting mixture was allowed
to gradually warm to 0° C and then distributed between 1N
1295 hydrochloric acid and ethyl acetate (300 mL each). The
layers were separated, and the organic layer was dried over
sodium sulfate. Concentration in vacuo produced a white
solid that was triturated from ethyl ether hexanes.
Filtration provided 16.4 g (710) of 6-methoxybenzo[b]
1300 thiophene-2-boronic acid as a white solid. mp 200° C (dec).
1H NMR (DMSO-d6) d 7.83 (s, 1H) , 7.78 (d, J = 8. 6 Hz, 1H) ,
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 48 -
7.51 (d, J = 2. 0 Hz, 1H) , 6. 97 (dd, J = 8 . 6, 2. 0 Hz, 1H) ,
3.82 (s, 3H). FD mass spec: 208.
1305 Step b): Preparation of [6-Methoxy-2-(4-methanesulfonyl-
oxyphenyl)]benzo[b] thiophene
i
H3C0 S
OSO 2CH 3
1310 To a solution of 6-methoxybenzo[b]thiophene-2-boronic
acid (3.00 g, 14.4 mmol) in 100 mL of toluene was added 4-
(methanesulfonyloxy)phenylbromide (3.98 g, 15.8 mmol)
followed by 16 mL of 2.0 N sodium carbonate solution. lifter
stirring for 10 minutes, tetrakistriphenylphosphinepalladium
1315 (0.60 g, 0.52 mmol) was added, and the resulting mixture was
heated to reflux for 5 hours. The reaction mixture was then
allowed to cool to ambient temperature whereupon the product
precipitated from the organic phase. The aqueous phase was
removed and the organic layer was concentrated in vacuo to a
1320 solid. Trituration from ethyl ether yielded a solid that
was filtered and dried in vacuo to provide 3.70 g (77%) of
[6-methoxy-2-(4-methanesulfonyloxy-phenyl)]benzo[b]thiophene
as a tan solid. mp 197-201° C. ~H NMR (DMSO-d6) d 7.82-
7.77 (m, 3H), 7.71 (d, J = 8.8 Hz, 1H), 7.54 (d, J = 2.3 Hz,
1325 1H) , 7 . 40 (d, J = 8 . 7 Hz, 2H) , 6. 98 (dd, J = 8. 7, 1 . 5 Hz,
1H), 3.80 (s, 3H), 3.39 (s, 3H). FD mass spec 334. Anal.
Calcd. for C16H14~4S2: C, 57.46; H, 4.21. Found: C, 57.76; H,
4.21.
1330 Step c): Preparation of [6-Hydroxy-2-(4-methanesulfonyl-
oxyphenyl)]benzo[b] thiophene
__._____- T
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 49 -
HO S
OSO 2CH 3
1335 To a solution of [6-methoxy-2-(4-methanesulfonyloxy-
phenyl)]benzo[b]thiophene (9.50 g, 28.40 mmol) in anhydrous
methylene chloride (200 mL) at room under nitrogen gas was
added boron tribromide (14.20 g, 5.36 mL, 56.8 mmol). The
resulting mixture was stirred at ambient temperature for 3
1340 hours. The reaction was quenched by slowly pouring into
excess ice water. After vigorously stirring for 30 minutes,
the white precipitate was collected by filtration, washed
several times with water, and then dried in vacuo to provide
8.92 g (980) of [6-hydroxy-2-(4-methanesulfonyloxyphenyl)]
1345 benzo[b]thiophene as a white solid. mp 239-243° C. 1H NMR
(DMSO-d6) a 9.70 (s, 1H) , 7.76 (d, J = 8.7 Hz, 2H) , 7.72 (s,
1H), 7.62 (d, J = 8.7 Hz, 1H), 7.38 (d, J = 8.7 Hz, 2H),
7.24 (d, J = 1.7 Hz, 1H), 6.86 (dd, J = 8.7, 1.7 Hz, 1H),
3.38 (s, 3H). FD mass spec 320. Anal. Calcd. for C15H1204S2:
1350 C, 56.23; H, 3.77. Found: C, 56.49; H, 3.68.
Step d): Preparation of [6-Benzyloxy-2-(4-methanesulfonyl-
oxyphenyl)]benzo[b]thiophene
i
s ~.
OSO 2CH 3
1355
To a solution of [6-hydroxy-2-(4-methanesulfonyloxy-
phenyl)] benzo[b]thiophene (3.20 g, 10.0 mmol) in 75 mL of
anhydrous DMF was added Cs2C03 (5.75 g, 17.7 mmol) followed
1360 by benzylchloride (1.72 mL, 11.0 mmol). The resulting
mixture was stirred vigorously for 24 hours. The solvent
was removed in vacuo, and the solid residue was suspended in
CA 02286207 1999-10-08
WO 98/45286 PCT/US98106989
- 50 -
200 mL of water. The white precipitate was collected by
filtration and washed several times with water. Upon drying
1365 in vacuo, the crude product was suspended in 1:1
hexanes:ethyl ether. The solid was collected to provide
3.72 g (910) of [6-benzyloxy-2-(4-methanesulfonyloxy-
phenyl)]benzo[b]thiophene as a white solid. mp 198-202° C.
1H NMR (pMSO-d6) a 7.81-7.78 (m, 3H), 7.72 (d, J = 8.7 Hz,
1370 1H) , 7. 64 (d, J = 2.2 Hz, 1H) , 7.47-7.30 (m, 7H) , 5.15 (s,
2H), 3.39 (s, 3H). FD mass spec 410.
Step e): Preparation of [6-Benzyloxy-2-(4-hydroxyphenyl)]-
1375 benzo[b]thiophene
O S
OH
i
To a solution of [6-benzyloxy-2-(4-methanesulfonyloxy-
1380 phenyl)]benzo[b]thiophene (12.50 g, 30.50 mmol) in 300 mL of
anhydrous THF under nitrogen gas at ambient temperature was
added lithium aluminum hydride (2.32 g, 61.0 mmol) in small
portions. The mixture was then stirred at ambient
temperature for 3 hours and then quenched by carefully
1385 pouring the mixture into an excess of cold 1.0 N
hydrochloric acid. The aqueous phase was extracted with
ethyl acetate. The organic was then washed several times
with water and then dried (sodium sulfate) and concentrated
in vacuo to a solid. Chromatography (silicon dioxide,
1390 chloroform) provided 8.75 g (870) of [6-benzyloxy-2-(4-
hydroxyphenyl)]benzo[b] thiophene as a white solid. mp 212-
216° C. 1H NMR (DMSO-d6) d 9.70 (s, 1H) , 7.63 (d, J = 8.7
Hz, 1H) , 7 . 56 (d, J = 2. 2 Hz, 1H) , 7. 51-7 . 30 (m, 8H) , 7 . 00
(dd, J = 8.7, 2.2 Hz, 1H) , 6. 80 (d, J = 8 . 6 Hz, 2H) , 5. 13
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 51 -
1395 (s, 2H). FD mass spec 331. Anal. Calcd. for C21H1602S: C,
75.88; H, 4.85. Found: C, 75.64; H, 4.85.
Step f): Preparation of [6-Benzyloxy-2-(4-methoxyphenyl)]-
- benzo[b]thiophene
1400
i
E I
s I
OCH3
i
To a solution of [6-benzyloxy-2-(4-hydroxyphenyl)]
benzo[b]thiophene (8.50 g, 26.40 mmol) in 200 mL of
1405 anhydrous DMF under nitrogen gas at ambient temperature was
added sodium hydride (1.66 g, 41.5 mmol) in small portions.
Once gas evolution had ceased, iodomethane (3.25 mL, 52.18
mmol) was added dropwise. The reaction was stirred for 3
hours at ambient temperature. The solvent was then removed
1410 in vacuo, and the residue distributed between water/ethyl
acetate. The layers were separated, and the organic phase
was washed several times with water. The organic layer was
then dried (sodium sulfate) and concentrated in vacuo to
provide 9.00 g (980) of [6-benzyloxy-2-(4-methoxyphenyl)]
1415 benzo[b]thiophene as a white solid. mp 180-185° C. 1H NMR
(DMSO-d6) d 7. 67-7.58 (m, 5H) , 7.46-7.29 (m, 5H) , 7. 02 (dd,
J = 8. 8, 2.2 Hz, 1H) , 6. 98 (d, J = 8.7 Hz, 2H) , 5. 13 (s,
2H), 3.76 (s, 3H). FD mass spec 346. Anal. Calcd. for
C22H1802S : C, 76.27; H, 5.24 . Found: C, 76.54; H, 5. 43.
1420
Step g): Preparation of [6-Benzyloxy-2-(4-methoxyphenyl)-3-
bromo]benzo[b]thiophene
Br
I
OCH3
i
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 52 -
1425
[6-Benzyloxy-2-(4-methoxyphenyl)]benzo[b]thiophene
(10.0 g, 28.9 mmol) was placed in 200 mL of chloroform along
with 10.0 g of solid sodium bicarbonate at ambient
temperature. Ta this suspension was added bromine (1.50 mL,
1430 29.1 mmol) dropwise over 30 minutes as a solution in 100 mL
of chloroform. Upon completion of the addition, water (200
mL) was added and the layers were separated. The organic
phase was dried (sodium sulfate) and concentrated in vacuo
to a white solid. Crystallization from methylene chloride/
1435 methanol provided 10.50 g (850) of [6-benzyloxy-2-(4-
methoxyphenyl)-3-bromo]benzo-[b]thiophene as a white solid.
mp 146-150° C. 1H NMR (DMSO-d6) d 7.70 (d, J = 2.2 Hz, 1H),
7.65-7.60 (m, 3H), 7.47-7.30 (m, 5H), 7.19 (dd, J = 8.8, 2.2
Hz, 1H) , 7. 06 (d, J = 8.7 Hz, 2H) , 5.17 (s, 2H) , 3.78 (s,
1440 3H). FD mass spec 346. Anal. Calcd. for C22H1~02SBr: C,
62.13; H, 4.03. Found: C, 61.87; H, 4.00.
Step h): Preparation of [6-Benzyloxy-2-(4-methoxyphenyl)-3-
bromo]benzo[b]thiophene-(S-oxide)
1445
Br
I
S
OCH3
i
The title compound was prepared by oxidation of the
product from step g) with 1.5 equivalents of hydrogen
1450 peroxide in a mixture of trifluoroacetic acid in methylene
chloride. The product was isolated as a yellow solid by
crystallization from ethyl acetate. mp 202-205° C. 1H NMR
(DMSO-d6) d 7. 80 (d, J = 2.2 Hz, 1H) , 7. 68 (d, J = 8.7 Hz,
2H), 7.55(d, J = 8.4 Hz, 1H) 7.47-7.32 (m, 6H), 7.10 (d, J =
1455 8.7 Hz, 2H) , 5.23 (s, 2H) , 3.80 (s, 3H) . FD mass spec 441.
Anal. Calcd. for C22H1~03SBr: C, 59.87; H, 3.88. Found: C,
59.59; H, 3.78.
__.-_ _... T. .. __..-._. ~ ......__.~__.-,.,.--_ ___._._....... .. _. .. _...
._.._._....--._.__---.____ .T__._.._.....
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 53 -
Step i): Preparation of [6-Benzyloxy-3-[4-[2-(1-
1460 piperidinyl)ethoxy]phenoxy]-2-(4-methoxyphenyl)]-
benzo [b] thiophene- (S-oxide)
CND
O
lw
r
O
I
w O ~~.
OCH3
r
1465 Reaction of the product of step i) above with 4-(2-
piperidinoethoxy)phenol in base yielded the title compound
as a yellow oil. 1H NMR (DMSO-d6) d 7.76 (d, J = 2.2 Hz,
1H) , 7. 62 (d, J = 8. 8 Hz, 2H) , 7.44-7. 30 (m, 5H) , 7. 12 (dd,
J = 8.6, 2.2 Hz, 1H) , 7. 03-6.93 (m, 5H) , 6. 85 (d, J = 8.8
1470 Hz, 2H) , 5. 18 (s, 2H) , 3. 94 (bt, J = 5.8 Hz, 2H) , 3.73 (s,
3H), 2.56 (bt, J = 5.8 Hz, 2H), 2.37-2.34 (m, 4H), 1.45-1.32
(m, 6H). FD mass spec 592. Anal. Calcd. for C35H35N05S: C,
72.26; H, 6.06; N, 2.41. Found: C, 72.19; H, 5.99; N, 2.11.
1475
CA 02286207 1999-10-08
WO 98/45286 PCT/iJS98/06989
- 54 -
Step j): Preparation of [6-Benzyloxy-3-[4-[2-(1-
piperidinyl)ethoxy]phenoxy]-2-(4-methoxyphenyl)]-
benzo[b]thiophene
~N~
O
I
i
O
I
J \ s
OCH3
i
1480
Reduction of the product of step i) above yielded the
title compound, isolated in 950 overall yield. Purification
by chromatography (Si02, 1-5o methanol/chloroform) provided
1485 an off-white solid, mp 105-108°C. 1H NMR (DMSO-dg) d 7.62
(d, J = 2.2 Hz, 1H), 7.59 (d, J = 8.8 Hz, 2H), 7.45-7.30 (m,
5H) , 7. 15 (dd, J = 8 . 6 Hz, 1H) , 7. 00-6. 94 (m, 3H) , 6. 82 (s,
4H) , 5.13 (s, 2H) , 3.92 (bt, J = 5. 8 Hz, 2H) , 3.72 (s, 3H) ,
2.55 (bt, J = 5.8 Hz, 2H), 2.37-2.34 (m, 4H), 1.44-1.31 (m,
1490 4H). FD mass spec 565. Anal. Calcd. for C35Hg5NOqS: C,
74.31; H, 6.24; N, 2.48. Found: C, 74.35; H, 6.07; N, 2.76.
_.._ ~_~_.__ _.__. _ __ __T __.
CA 02286207 1999-10-08
WO 98/45286 PCT/ITS98/06989
- 55 -
Step k): Preparation of [6-Hydroxy-3-[4-[2-(1-piperidinyl)-
ethoxy]phenoxy]-2-(4-methoxyphenyl)]benzo[b]-
1495 thiophene
~N~.
O
i
O
I
Ho ~ s I
~OCH3
To a solution of [6-benzyloxy-3-{4-[2-(1-piperidinyl)
1500 ethoxy]phenoxy]-2-(4-methoxyphenyl)]benzo[b]thiophene (8.50
g, 15.0 mmol) in 300 mL of 5:1 ethanol/ethyl acetate was
added palladium black (1.50 g), ammonium formate (3.50 g,
55.6 mmol), and 30 mL of water. The resulting mixture was
heated to reflux and monitored by TLC. After approximately
1505 3 hours, the reaction was judged complete and the solution
was cooled to ambient temperature. The reaction was
filtered through a pad of Celite to remove catalyst, and the
filtrate was concentrated in vacuo to a solid. The
concentrate was distributed between saturated sodium
1510 bicarbonate solution and 5s ethanol/ethyl acetate. The
layers were separated, and the organic phase was dried
(sodium sulfate) and concentrated in vacuo. The crude
product was chromatographed (silicon dioxide, 1-50
methanol/chloroform) to provide 6.50 g (910) of [6-hydroxy-
1515 3-[4-[2-(1-piperidinyl) ethoxy]phenoxy]-2-(4-
methoxyphenyl)]benzo{b]thiophene as foam that converted to
solid upon trituration with hexanes. mp 174-176° C. 1H NMR
(DMSO-d6) d 9.77 (s, 1H) , 7.56 (d, J = 8. 8 Hz, 2H) , 7.23 (d,
J = 2.0 Hz, 1H), 7.07 (d, J = 8.6 Hz, 1H), 6.93 (d, J = 8.8
1520 Hz, 2H) , 6. 81 (s, 4H) , 6.76 (dd, J = 8. 6, 2.0 Hz, 1H) , 3.91
(bt, J = 5.9 Hz, 2H) , 3.71 (s, 3H) , 2.55 (bt, J = 5.9 Hz,
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 56 -
2H), 2.38-2.33 (m, 4H), 1.46-1.28 (m, 6H). FD mass spec
475. Anal. Calcd. for C2gH2gNOqS: C, 70.71; H, 6.15; N,
2.99. Found: C, 70.46; H, 5.93; N, 2.71.
1525
Example 15
Preparation of [6-Hydroxy-2-(4-methoxyphenyl)3-[4-(2-
piperidin-1-ylethoxy)phenoxy]benzo[b]thiophene
1530 hydrochloride salt
~ HCI
~N~.
O
i
O
I
HO S
OCH3
The product of Example 14 was converted to the corres-
1535 ponding hydrochloride salt in 85o yield by treatment with a
mixture of HCl saturated diethyl ether in ethyl acetate
followed by crystallization from ethanol/ethyl acetate; mp
156-160° C. 1H NMR (DMSO-dg) d 10.28 (bs, 1H), 9.85 (s, 1H),
7.56 (d, J = 8.8 Hz, 2H), 7.25 (d, J = 2.0 Hz, 1H), 7.06 (d,
1540 J = 8 . 7 Hz, 1H) , 6. 93 (d, J = 8 . 8 Hz, 2H) , 6. 87 (q, J,~ _
9. 3 Hz, 4H) , 4 .27 (bt, J = 5. 9 Hz, 2H) , 3 . 71 ( s, 3H) , 3. 44-
3.31 (m, 4H), 2.98-2.88 (m, 2H), 1.74-1.60 (m, 5H), 1.36-
1.29 (m, 1H) FD mass spec 475. Anal. Calcd. for
C2gH2gNOqS~1.0 HC1: C, 65.68; H, 5.90; N, 2.73. Found: ~C,
1545 65.98; H, 6.11; N, 2.64.
_ _____ _ T _ _.__ __ _ _ ._.___ _._ __. ___ __-T _
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 57 _
Example 16
1550 Preparation of [6-methoxy-3-[4-[2-(1-piperidinyl)-
ethoxy]phenoxy]-2-(4-hydroxyphenyl)]benzo[b]thio hene
O
I
O
I
H3C0 S
OH
1555 Step a): Preparation of [6-methoxy-2-(4-benzyloxyphenyl)]-
benzo[b]thiophene
i
I
H3C0 S I ~
O
I
1560 Following the general procedures of steps a) through g)
of Example 14, the title compound was obtained in 73o yield,
mp 217-221°C. 1H NMR (DMSO-d6) c1 7.63-7.60 (m, 3H), 7.59-
7 .26 (m, 7H) , 7. 02 (d, J = 8.7 Hz, 2H) , 6. 96 (dd, J = 8 . 8,
2.2 Hz, 1H), 5.11 (s, 2H), 3.88 (s, 3H). FD mass spec 346.
1565 Anal. Calcd. for C22H1g02S: C, 76.27; H, 5.24. Found: C,
76.00; H, 5.25.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 58 -
1570
Step b): [6-methoxy-2-(4-benzyloxyphenyl)-3-bromo]benzo-
[b]thiophene
Br
I I
H3C0 S
O
I
The title compound was obtained in 91o yield, mp 125-
127°C. 1H NMR (DMSO-dg) d 7.64-7.61 (m, 4H), 7.46-7.31 (m,
1575 5H), 7.15-7.09 (m, 3H), 5.15 (s, 2H), 3.82 (s, 3H). FD mass
spec 346. Anal. Calcd. for C2zH1~02SBr: C, 62.13; H, 4.03.
Found: C, 62.33; H, 3.93.
Step c): [6-Methoxy-2-(9-benzyloxyphenyl)-3-bromo]benzo[b]-
1580 thiophene-(S-oxide)
Br
I I
H3C0 S
O ~ O
The title compound was isolated as a yellow solid by
1585 chromatography (Si02, CHC13). mp 119-123° C. 1H NMR (DMSO-
d6) d 7.73 (d, J = 2.2 Hz, 1H) , 7 . 68 (d, J = 8. 8 Hz, 2H) ,
7.55 (d, J = 8.5 Hz, 1H) 7. 46-7.31 (m, 5} , 7 .26 (dd, J =
8.5, 2.2 Hz, 1H), 7.18 (d, J = 8.8 Hz, 2H), 5.16 (s, 2H),
3.86 (s, 3H). FD mass spec 441. Anal. Calcd. for
1590 C22H1~03SBr: C, 59.87; H, 3.88. Found: C, 60.13; H, 4.10.
___-~__. __T _ _..__________..~. __._.__~,._ .T__ ._.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 59 -
Step d): [6-Methoxy-3-[4-[2-(1-piperidinyl)ethoxy]phenoxy]
2-(4-benzyloxyphenyl)]benzo[b]thiophene-(S-oxide)
~N~
O
i
O
H3C0 ~ S
O O
i
1595
The title compound was obtained as a yellow solid, mp
89-93° C. 1H NMR (DMSO-d6) d 7. 68 (d, J = 2.2 Hz, 1H) , 7. 62
(d, J = 8.8 Hz, 2H), 7.42-7.28 (m, 5H), 7.08-6.92 (m, 6H),
1600 6.86 (d, J = 8.8 Hz, 2H) , 5.09 (s, 2H) , 3.94 (bt, J = 5.8
Hz, 2H), 3.81 (s, 3H), 2.56 (bt, J = 5.8 Hz, 2H), 2.37-2.34
(m, 4H), 1.45-1.31 (m, 6H). FD mass spec 592. Anal. Calcd.
for C35H35N05S~0.25 EtOAc: C, 71.52; H, 6.18; N, 2.32.
Found: C, 71.32; H, 5.96; N, 2.71.
1605
CA 02286207 1999-10-08
WO 98145286 PCT/US98/06989
- 60 -
Step e): [6-Methoxy-3-[4-[2-(1-piperidinyl)ethoxy]phenoxy]
2-(4-benzyloxyphenyl)]benzo[b]thiophene
~N~.
O
i
O
I
H3C0 ~ S
I
O
1610
The title compound was obtained in 91% yield, mp 106-
110°C. 1H NMR (DMSO-d6) a 7.59 (d, J = 8.8 Hz, 2H), 7.54
(d, J = 2.2 Hz, 1H), 7.42-7.28 (m, 5H), 7.13 (d, J = 8.8 Hz,
1H) , 7 . 03 (d, J = 8. 8 Hz, 2H) , 6. 82 (s, 4H) , 5. 08 (s, 2H) ,
1615 3.92 (bt, J = 5.8 Hz, 2H) , 3.78 (s, 3H) , 2.55 (bt, J = 5.8
Hz, 2H), 2.37-2.33 (m, 4H), 1.44-1.31 (m, 4H). FD mass spec
565. Anal. Calcd. for C35H35N04S: C, 74.31; H, 6.24; N,
2.48. Found: C, 74.26; H, 6.17; N, 2.73.
1620 Step f): Preparation of [6-methoxy-3-[4-[2-(1-piperidinyl)
ethoxy]phenoxy]-2-(4-hydroxyphenyl)]benzo[b]thiophene
H3C0
OH
CND.
O
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 61 -
1625 The title compound was obtained in 88$ yield, mp 147-
150° C. 1H NMR (DMSO-d6) d 9.72 (s, 1H), 7.51 (d, J = 2.0
Hz, 1H), 7.48 (d, J = 8.6 Hz, 2H), 7.11 (d, J = 8.8 Hz, 1H),
6. 88 (dd, J = 8. 8, 2.2 Hz, 1H) , 6. 81 (s, 4H) , 6.76 (d, J =
8 . 6, 2H) , 3 . 91 (bt, J = 5. 9 Hz, 2H) , 3. 77 (s, 3H) , 2.55 (bt,
1630 J = 5.9 Hz, 2H), 2.38-2.33 (m, 4H), 1.46-1.28 (m, 6H). FD
mass spec 475. Anal. Calcd. for C28H2gNOqS: C, 70.71; H,
6.15; N, 2.94. Found: C, 71.00; H, 6.17; N, 2.94.
Example 17
1635
Preparation of [6-methoxy-3-[4-[2-(1- i eridinyl)ethoxy]-
phenoxy]-2-(4-hydroxyphenyl)]benzo[b]thiophene hydrochloride
~O
I
0
I I
H3C0 S
OH
1640
The title compound was prepared in a manner analogous
to that employed in Example 15 to yield the title compound,
mp 215-217° C. ~H NMR (DMSO-d6) d 10.28 (bs, 1H), 9.80 (s,
1H) , 7.52 (d, J = 2.2 Hz, 1H) , 7. 47 (d, J = 8 . 6 Hz, 2H) ,
1645 7.12 (d, J = 8.4 Hz, 1H) , 6. 91-6. 80 (m, 5H) , 6.78 (d, J =
8.6 Hz, 2H), 4.27 (bt, J = 5.8 Hz, 2H), 3.78 (s, 3H), 3.43-
3.34 (m, 4H), 2.97-2.91 (m, 2H), 1.78-1.61 (m, 5H), 1.36-
1.29 (m, 1H). FD mass spec 475. Anal. Calcd. for
C2gH2gNOqS~1.0 HC1: C, 65.68; H, 5.90; N, 2.73. Found: C,
1650 65.87; H, 5.79; N, 2.99.
~ HCI
N
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 62 -
Formulation Examples
In the formulations which follow, "active ingredient"
1655 means a compound of formula I, or a salt or solvate thereof.
Formulation Example 1
Gelatin Capsules
Ingredient Quantity (mg/capsule)
Active ingredient 0.1 - 1000
Starch, NF 0 - 650
Starch flowable powder 0 - 650
Silicone fluid 350 centistokes 0 - 15
1660
Formulation Example 2
Tablets
Ingredient Quantity (mg/tablet)
Active ingredient 2.5 - 1000
Cellulose, microcrystalline 200 - 650
Silicon dioxide, fumed 10 - 650
Stearate acid 5 - 15
1665 Formulation Example 3
Tablets
Ingredient Quantity (mg/tablet)
Active ingredient 25 - 1000
Starch 45
Cellulose, microcrystalline 35
Polyvinylpyrrolidone 4
(as loo solution in water)
Sodium carboxymethyl cellulose 4.5
Magnesium stearate 0.5
Talc 1
_- _.. __- .. .....__ T.._ _ ..-__ _ ._..___. ___.. _ __ .T __ _.
CA 02286207 1999-10-08
WO 98/45286 PCT/IJS98/06989
- 63 -
The active ingredient, starch, and cellulose are passed
1670 through a No. 45 mesh U.S. sieve and mixed thoroughly. The
solution of polyvinylpyrrolidone is mixed with the resultant
powders which are then passed through a No. 14 mesh U.S.
sieve. The granules so produced are dried at 50°-60° C and
passed through a No. 18 mesh U.S. sieve. The sodium
1675 carboxymethyl starch, magnesium stearate, and talc,
previously passed through a No. 60 U.S.~sieve, are then
added to the granules which, after mixing, are compressed on
a tablet machine to yield tablets.
1680 Formulation Example 4
Suspensions
Ingredient Quantity (mg/5 ml)
Active ingredient 0.1 - 1000 mg
Sodium carboxymethyl cellulose 50 mg
S yrup
1.25 mg
Benzoic acid solution 0.10 mL
Flavor q.v,
Color q
.v.
Purified water to 5 mL
The medicament is passed through a No.45 mesh U.S.
1685 sieve and mixed with the sodium carboxymethyl cellulose and
syrup to form a smooth paste. The benzoic acid solution,
flavor, and color are diluted with some of the water and
added, with stirring. Sufficient water is then added to
produce the required volume.
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 64 -
1690 Formulation Example 5
Aerosol
Ingredient Quantity (o by
weight)
Active ingredient 0.25
Ethanol 25.75
Propellant 22 (Chlorodifluoromethane) 70.00
The active ingredient is mixed with ethanol and the
1695 mixture added to a portion of the propellant 22, cooled to
30° C, and transferred to a filling device. The required
amount is then fed to a stainless steel container and
diluted with the remaining propellant. The valve units are
then fitted to the container.
1700
Formulation Example 6
Suppositories
Ingredient Quantity (mg/suppository)
Active ingredient 2S0
Saturated fatty acid 2,000
glycerides
1705 The active ingredient is passed through a No. 60 mesh
U.S. sieve and suspended in the saturated fatty acid
glycerides previously melted using the minimal necessary
heat. The mixture is then poured into a suppository mold of
nominal 2 g capacity and allowed to cool.
1710
Formulation Example 7
Injectable Formulations
Ingredient Quantity
Active ingredient 50 mg
Isotonic saline 1,000 mL
CA 02286207 1999-10-08
WO 98/45286 PCT/US98/06989
- 65 -
1715 The solution of the above ingredients is intravenously
administered to a patient at a rate of about 1 mL per
minute.