Note: Descriptions are shown in the official language in which they were submitted.
CA 02286824 1999-10-14
r
1
METHOD FOR PRODUCING VINYL PHOSPHONIC ACID COMPOUNDS
to
The present invention relates to a process for preparing vinylphosphonic
acid compounds using certain catalysts, and to the use of such catalysts
is for the preparation process.
Vinylphosphonic acid compounds, in particular dialkyl vinylphosphonates,
have importance as precursors for preparing vinylphosphonic acid and as
monomers for copolymerization for producing adhesives or flame-resistant
zo plastics.
Various processes for preparing them are known. In the process described
in DE-C-21 32 962, ethylene oxide is reacted with phosphorus trichloride
to give 2-chloroethanephosphonic dichloride, and this compound is con-
es verted into bis-2-chloroethyl 2-chloroethanephosphonate. The resulting
compound is then reacted with phosgene in the presence of a catalyst.
Amines, heterocyclic nitrogen compounds, as well as tertiary phosphines,
are used as catalyst.
so DE-A-30 O1 894 describes a process for preparing vinylphosphonic acid
derivatives, in which dialkyl 2-acetoxyethanephosphonates are cleaved in
the presence of acidic or basic catalysts. The basic catalysts proposed are
CA 02286824 1999-10-14
-2-
tertiary amines and phosphines, as well as ammonium salts or
phosphonium salts, besides heterocyclic compounds and amides. The
disadvantage of the process is the formation of a mixture of
vinylphosphonic acid derivatives. The maximum content of dialkyl
s vinylphosphonates is 23 % .
An improved variant of this process disclosed in DE-A-31 20 437 entails
a distillation followed by reaction of the bottom product mixture resulting
from the distillation with orthocarboxylates to give dialkyl vinylphos-
io phonates.
EP-A-0 722 948 discloses thermal cleavage of dimethyl 2-
acetoxyethanephosphonate in the gas phase to give acetic acid and
dimethyl vinylphosphonate. No catalyst is used in this case.
is
The disadvantages of the above processes are the formation of product
mixtures, elaborate, multistage synthetic processes, the need to use high
reaction temperatures, and the use of chlorinated starting compounds. The
large proportion of byproducts in particular considerably impairs the
2o economics of the process.
A simple addition reaction is advantageous for synthesizing
vinylphosphonic acid compounds and results in the required product in
high yields. One example of a reaction of this type is addition of dialkyl
is phosphites onto acetylene. US 3,673,285 describes the addition of alkynes
onto diethyl phosphite at from 130 to 200°C in the presence of nickel-
phosphine complexes. On addition of acetylene, the corresponding diethyl
vinylphosphonate is obtained in a yield of 30 % . The disadvantage of this
process is, besides the low yield, the tendency of the phosphorous esters
3o to decompose in a strongly exothermic reaction at temperatures as low as
CA 02286824 1999-10-14
-3-
130°C.
It is an object of the present invention to provide a process for preparing
vinylphosphonic acid compounds which avoids the disadvantages of known
s processes and makes the required products available with high selectivity
and yield under mild conditions from acetylene and phosphorous acid
compounds.
~Ve have found that this object is achieved by a process for preparing
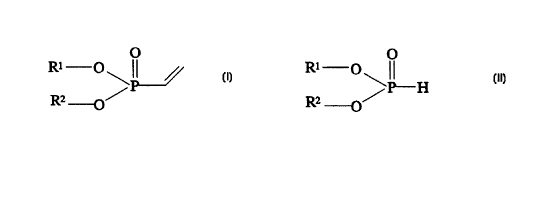
io vinylphosphonic acid compounds of the formula (I)
where R1 and R2 are, independently, H, C~_16-alkYl, C6-12-aryl, C~_12'
alkaryl or C~_12-aralkyl, it being possible for the organic radicals to be
zo substituted by one or more halogen atoms, hydroxyl, acyl or acetoxy
groups, by reacting phosphorous acid compounds of the formula (II)
RZ /
-O
where R1 and R2 have the above meaning, with acetylene in the presence
of a Pd(II) complex and/or of a Pd(0) complex or corresponding Pt
so complexes as catalyst.
CA 02286824 1999-10-14
-4-
We have found that the reaction of acetylene with phosphorous acid
compounds, in particular dialkyl phosphites, to give vinylphosphonic acid
compounds, in particular dialkyl vinylphosphonates, is possible with high
selectivity and yield by using a catalytic amount of a palladium or
s platinum complex, preferably palladium complex, especially in
homogeneous phase. Similar palladium complexes have been used for
hydrophosphorylation of terminal higher alkynes such as 1-octyne, see J.
Am. Chem. Soc., 118 (1996) 1571 - 1572. However, reaction with
io
acetylene is not mentioned or proposed.
~~e have found that acetylene can be reacted under very mild conditions
with very high selectivity using a palladium catalyst to give
vinylphosphonic acid compounds, in particular dialkyl vinylphosphonates,
directly without any dimerization, oligomerization or polymerization of
i5 acetylene or double reaction to give a tetraalkyl ethylenediphosphonate.
In the phosphorous acid compounds of the formula (II) employed for the
reaction, Rl and R'- are, independently, H, C1_16-alkyl, C6_1~-aryl, C~_1.,-
alkaryl or C~_12-aralkyl, it being possible for the organic radicals to be
2o substituted by one or more halogen atoms, hydroxyl, acyl or acetoxy
groups. Rl and R'- are preferably, independently, linear C1_t2-alkyl,
phenyl, (Ci_6-alkyl)phenyl or phenyl(C1_6-alkyl). Rl and R2 are particularly
preferably, independently, linear C1_6-alkyl radicals. These radicals are
zs
preferably unsubstituted.
If Rl and R' differ from hydrogen, then the compounds employed are
diesters of phosphorous acid. Reaction thereof results in diesters of
vinylphosphonic acid of formula (I). This reaction can be followed by
cleavage of the ester groups, resulting in vinylphosphonic acid, R10H and
3o RZOH.
CA 02286824 1999-10-14
- 5-
Conversion of the phosphorous acid compounds into the vinylphosphonic acid
compounds takes place in the presence of a Pd(I~ complex and/or of a Pd(0)
complex or corresponding Pt complex, but preferably Pd complex as catalyst.
The
catalyst is usually present in homogeneous phase for this purpose.
The Pd(0) complex employed preferably has phosphine ligands or phosphate
ligands.
A large number of ligands are suitable as phosphine ligands or phosphate
ligands.
For example, the ligands may have the formula PXYZ where X, Y and Z are,
independently, alkyl, aryl, allcoxy or aryloxy radicals having up to 18 carbon
atoms.
to Alkyl or aryl radicals are preferred in this connection, especially aryl
radicals.
Corresponding ligands are described, for example, in DE-A-1 593 277. They are
preferably triarylphosphines or triaryl phosphates in which the aryl groups
are
unsubstituted or substituted. Suitable substituents are C~.~-alkyl, acyl or
acetoxy
radicals. The triarylphosphine is preferably unsubstituted. Triphenylphosphine
is
t5 particularly employed as phosphine ligand. The catalyst used particularly
preferably
according to the invention is tetrakis(triphenylphosphine)palladium(0).
The complexes may be composed, for example, of monodentate or bidentate
ligands.
Examples of suitable complex structures are the following:
\ / R'
/P~ iX ~p~ ,P\ jp~ ~P
R\ P M\Y R\ M\ iR R\ M\
P P P X
R R'
R R ' R' R ' R R'
where the meanings are
2o M Pd, Pt, preferably Pd
R independently at each posirion organic radicals linked via O and/or C atoms
to the phosphorus atoms, in particular aryl radicals or aryloxy
CA 02286824 1999-10-14
a
-6-
radicals having 2 sites capable of linkage,
R,R" independently at each position monovalent organic radicals, in
particular aryl andlor aryloxy radicals
X, Y independently monovalent anionic ligands.
The charges therein are equalized, if necessary employing canons or
anions without coordinate links to equalize the charges.
io
It is preferred for the monovalent radicals to be derived from benzene or
phenol and for the divalent radicals to be derived from biphenyl, 1,1'-
binaphthyl, biphenyloxy and/or 1,1'-binaphthyloxy radicals. It is moreover
possible for all the aromatic radicals to be substituted, for example by
is one or more C1-6-alkyl radicals or corresponding alkoxy radicals. The
biphenyl and binaphthyl radicals, and radicals derived therefrom, are
linked to the phosphorus atom via 2 positions in the molecule. It is
possible for both positions to be linked to the same phosphorus atom. It
is also possible for them to be linked to different phosphorus atoms and
2o thus produce bridged structures which have, for example, 2 phosphorus
atoms and 3 of said radicals. Suitable corresponding bidentate phosphate
ligands are described in US 5',512,695. The phosphate ligands described
therein can also be employed in analogous form as phosphine ligands.
Further suitable monodentate and bidentate aromatic ligands are described
2s in WO 95/29153. The described ligands can in this case likewise be
employed as phosphine or phosphate ligands.
Pd(II) complexes which can be used according to the invention preferably
have the phosphine ligands, phosphate ligands, aryl cyanides or alkyl
3o cyanides described for Pd(0) complexes as ligands. The aryl cyanides can
CA 02286824 1999-10-14
moreover be substituted as above. Benzonitrile or acetonitrile are
preferably employed. Particularly preferred Pd(II) complexes are
Pd(C6HSCN)2C12, Pd(CH3CN)4(BF4)2 or Pd((C6H~)3P)2C12.
s The catalysts can moreover be formed in situ in the reaction.
The catalysts employed according to the invention are normally employed
in an amount of from 0.01 to 10% by weight, preferably in an amount
of from 0.5 to 3 % by weight, particularly preferably 1 to 2 % by weight,
io based on the amount of phosphorous acid compounds to be vinylated, in
particular dialkyl phosphites.
The temperature in the reaction is, as a rule, from 20 to 120°C,
preferably 20 to 80°C, particularly preferably 60 to 80°C.
is
The reaction can moreover be carried out without solvent or in the
presence of an inert solvent. Examples of inert solvents which can be
used are cyclic ethers such as THF, long-chain ethers such as triethylene
glycol dimethyl ether or tetraethylene glycol dimethyl ether.
The reaction is carried out under atmospheric pressure or elevated
pressure, preferably at from 1 to 20, particularly preferably 1.5 to 6, bar
(absolute). This preferably entails mixing the phosphorous acid compound
of the formula (II) and the catalyst, and passing in acetylene. Once the
2s reaction is complete, the product can be removed by distillation.
The novel process can be carried out continuously or batchwise.
The invention also relates to the use of the catalysts described above in
3o the preparation of vinylphosphonic acid compounds, in particular
CA 02286824 1999-10-14
_g_
vinylphosphonic esters, specifically dialkyl vinylphosphonates.
The invention is explained in detail by means of Examples shown
hereafter.
EXAMPLE 1
6 g of dimethyl phosphite were stirred with 20 ml of tetrahydrofuran in
a 4-neck flask which had a capacity of 500 ml and was equipped with an
io internal thermometer, dry-ice condenser and gas introduction tube, and
were degassed under argon. After addition of 2 mol % tetrakis- (tri-
phenylphosphine)palladium(0), 6 l/h acetylene were passed into the reaction
solution at 60°C for 24 h. Dimethyl vinylphosphonate was isolated in
90'~o yield after workup by distillation.
is
EXAMPLE 2
2~ g of diethyl phosphite were degassed under argon while stirring in a
4-neck flask which had a capacity of 500 ml and was equipped with an
zo internal thermometer, dry-ice condenser and gas introduction tube. After
addition of 1 mol % tetrakis(triphenylphosphine)palladium(0), 6 l/h
acetylene were passed into the reaction solution at 60°C for 24 h.
Diethyl vinylphosphonate was isolated in 95 % yield after workup by
distillation.
EXA,~iPLE 3
65 g of dimethyl phosphite were stirred with 110 ml of tetrahydrofuran
in an autoclave with a capacity of 300 ml. After addition of 0.7 mol %
so tetrakis(triphenylphosphine)palladium(0), initially 5 bar of nitrogen and
10
CA 02286824 1999-10-14
-9-
bar of acetylene were injected. After heating the autoclave to 65°C,
further acetylene was injected to 20 bar. The amount of acetylene taken
up at this temperature was replaced each hour for 20 h and then, after
cooling, the reaction discharge was flushed with nitrogen and distilled.
s Dimethyl vinylphosphonate was isolated in 95 % yield.