Note: Descriptions are shown in the official language in which they were submitted.
CA 02288640 1999-11-OS
WO 98/50530 PCT/US98/09249
1
DESCRIPTION
ENZYMATIC NUCLEIC ACID TREATMENT OF DISEASES OR CONDITIONS
RELATED TO LEVELS OF EXPRESSION OF C-RAF
Background Of The Invention
This invention relates to methods and reagents for the treatment of diseases
or
conditions relating to the levels of expression of raf genes.
The following is a discussion of relevant art, none of which is admitted to be
prior
art to the present invention.
The Raf family of serine/threonine kinases function as cytoplasmic signaling
proteins that transduce mitogenic signals in response to activation of various
growth
factor receptors (for reviews, see Daum, 1994 Trends in Biochem. Sci. 19, 474;
Katz,
1997, Cacrr. Opin. Genet. Devel. 7, 75; Marais, 1996, Cancer Surveys 27;
Naumann,
1997, Cancer Res. 143, 237). c-Raf is the cellular homolog of v-Raf, the
transforming
element of the murine sarcoma virus 3611. The Raf family consists of three
highly
conserved isozymes in vertebrates: c-Raf 1, which is constitutively expressed
in all
tissues, A-Raf, which is expressed in urogenital tissue and B-Raf which is
expressed in
and cerebrum and testes (Storm, 1990, Oncogene 5, 345). Inappropriate
expression of
these key genes involved in cell growth and differentiation can result in
uncontrolled cell
proliferation and/or propagation of damaged DNA, leading to hyperproliferative
disorders
such as cancer, restenosis, psoriasis and rheumatoid arthritis.
Raf is one of the major downstream effectors of Ras, a member of the class of
small GDP/GTP-binding proteins involved in cellular signal transduction
pathways
(figure 35; Marshall, 1995, Molec. Reprod. Devel., 42, 493). Appropriate
mitogenic
signals cause an increase in levels of the GTP-bound Ras. In its GTP-bound
active state,
Ras binds Raf and localizes it to the plasma membrane. This results in
activation of the
Raf kinase activity. Activated Raf in turn phosphorylates MEK, thereby
activating the
CA 02288640 1999-11-OS
WO 98/50530 2 PCT/US98/09249
MAP kinase signaling cascade leading to cell cycle progression. Amino terminal
truncation of Raf leads to constitutively active protein. Expression of either
constitutively
active Raf or constitutively active MEK is sufficient for oncogenic
transformation of
fibroblasts (Cowley, 1994, Cell 77, 81; Mansour, 1994, Science 265, 966;
Kolch, 1991,
Nature 349, 426). In normal cells, the expression level of Raf is limiting in
cellular
transformation (Cuadrado, 1993, Oncogene 8, 2443). The pivotal position that
the Ras
and Raf family of proteins occupy in cellular signal transduction pathways
emphasizes
their importance in the control of normal cellular growth.
Activation of Raf in mammalian cells is triggered by a variety of growth
factors
and cytokines. Raf activation has been observed in cardiac myocyte cultures
stimulated
by fibroblast growth factor (FGF), endothelin or phorbol ester (Bogoyevitch,
1995, J.
Biol. Chem. 270, 1 ). Activation has also been seen in Swiss 3T3 cells treated
with
bombesin and platelet derived growth factor (Mitchell, 1995, J. Biol. Chem.
270, 8623) or
with colony stimulating factor or lipopolysacchride (Reimann, 1994, J. Immun.
153, 398),
in L6 myoblasts stimulated with insulin-like growth factor (Cross, 1994,
Biochem J. 303,
21), as well as in B cells stimulated via the immunoglobulin receptor (Kumar,
1995,
Biochem J. 307, 215).
There is growing evidence from a number of laboratories that suggests that the
Ras/Raf pathway may also be involved in cell motility (Bar-Sagi and Feramisco,
1986
Science 233, 1061; Partin et al., 1988 Cancer Res. 48-6050; Fox et al., 1994
Oncogene 9,
3519). These studies show that cell lines transfected with activated Ras show
an increase
in ruffling, pseudopod extension and chemotactic response, all of which are
cell-motility-
related processes. Uncontrolled cell motility has been implicated in several
pathological
processes such as restenosis, angiogenesis and wound healing.
Raf activation leads to induction of several immediate early transcription
factors
including NF-kB and AP-1 (Bruder, 1992, Genes Devel. 6, 545; Finco, 1993, J.
Biol.
Chem. 268, 17676). AP-1 regulates expression of a variety of proteases (Sato,
1994
Oncogene 8, 395; Gaire, 1994, J Biol Cltem 269, 2032; Lauricell-Lefebvre,
1993,
CA 02288640 1999-11-OS
WO 98/50530 3 PCT/US98/09249
Invasion Metastasis 13, 289; Troen, 1991, Cell Growth Differ 2, 23). A cascade
of MMP
and serine proteinase expression is implicated in the acquisition of an
invasive phenotype
as well as in angiogenesis in tumors (MacDougall, 1995, Cancer and Metastasis
Reviews
14, 351 ). Thus, Raf signaling is expected to contribute to increased
invasiveness in tumor
cells, leading to metastasis.
Coexpression studies of Raf l and Bcl-2 have shown that these proteins bind
and
interact to synergistically suppress apoptosis (Wang, 1994, Oncogene 9, 2751).
Thus,
overexpression of Raf 1 in tumor cells is likely to contribute to malignant
transformation
and increased resistance to chemotherapeutic agents. Overexpression of c-Raf 1
is
observed in squamous cell carcinomas of the head and neck taken from patients
resistant
to radiation therapy (Riva, 1995, Oral Oncol., Eur. J. Cancer 31B, 384) and in
lung
carcinomas (Rapp, 1988, The Oncogene Handbook, 213). Activated (truncated) Raf
has
been detected in a number of human cancers including small-cell lung, stomach,
renal,
breast and laryngeal cancer (Rape, 1988, The Oncogene Handbook, 213).
1 S Therapeutic intervention in down-regulating Raf expression have focused on
antisense oligonucleotide approaches:
Antisense oligonucleotides targeting c-Raf 1 were used to demonstrate that IL-
2
stimulated growth of T cells requires c-raf (Riedel, 1993, Eur. J. Immunol.
23, 3146).
Antisense oligonucleotides targeting c-Raf 1 in SQ-20B cells showed reduced
Raf
expression and increased radiation sensitivity (Soldatenkov, 1997, The Cancer
J. from
Scientific American 3, 13). Rapp et al. have disclosed a method for inhibiting
c-Raf 1
gene expression using a vector expressing the gene in the antisense
orientation
(International PCT Publication No. WO 93/04170). Antisense oligonucleotides
targeting
c-Raf 1 in SQ-20B cells showed reduced DNA synthesis in response to insulin
stimulation in rat hepatoma cells (Tonxlcvist, 1994, J. Biol. Chem. 269,
13919). Monia et
al. have disclosed a method for inhibiting Raf expression using antisense
oligonucleotides
" (U.S. Patent No. 5,563,255) and shown that antisense oligonucleotides
targeting c-Raf 1
can inhibit Raf mRNA expression in cell culture, and inhibit growth of a
variety of tumor
CA 02288640 1999-11-OS
WO 98/50530 4 PCT/US98/09249
types in human tumor xenograft models (Monia et al., 1996, Proc. Natl. Acad.
Sci.93,
15481; Monia et al., 1996, Nature Med. 2, 668). No toxicity was observed in
these
studies following systemic administration of c-Raf antisense oligonucleotides,
suggesting
that at least partial down regulation of Raf in normal tissues is not overtly
toxic.
It has been proposed that synthetic ribozymes can be delivered to target cells
exogenously in the presence or absence of lipid delivery vehicles (Thompson et
al.,
International PCT Publication No. WO 93/23057; Sullivan et al., International
PCT
Publication No. WO 94/02595).
Recently Sandberg et al., 1996, Abstract, IBC USA Conferences on Angiogenesis
Inhibitors and other novel therapeutics for Ocular Diseases of
Neovascularization,
reported pharmacokinetics of a chemically modifies hammerhead ribozyme
targeted
against a vascular endothelial growth factor (VEGF) receptor RNA in normal and
tumor
bearing mice after daily bolus or continuous infusion.
Desjardins et al., 1996, J. Pharmacol. Exptl. Therapeutic, 27, 8, 1419,
reported
pharmacokinetics of a synthetic, chemically modified hammerhead ribozyme
against the
rat cytochrome P-450 3A2 mRNA after single intravenous injection.
The references cited above are distinct from the presently claimed invention
since
they do not disclose and/or contemplate the use of ribozymes to cleave Raf
RNA.
Furthermore, Applicant believes that the references do not disclose and/or
enable the use
of ribozymes to down regulate normal Raf gene expression in mammalian cells
and/or
whole animal.
Summary Of The Invention
This invention relates to identification, synthesis and use of nucleic acid
catalysts to
cleave RNA species that are required for cellular growth responses. In
particular,
applicant describes the selection and function of ribozymes capable of
cleaving RNA
CA 02288640 1999-11-OS
WO 98!50530 PCT/US98/09249
encoded by c-raf gene. Such ribozymes may be used to inhibit the hyper-
proliferation of
tumor cells in one or more cancers, restenosis, psoriasis, fibrosis and
rheumatoid arthritis.
In the present invention, ribozymes that cleave c-raf RNA are described.
Moreover,
applicant shows that these ribozymes are able to inhibit gene expression and
cell
5 proliferation in vitro and in vivo, and that the catalytic activity of the
ribozymes is
required for their inhibitory effect. From those of ordinary skill in the art,
it is clear from
the examples described herein, that other ribozymes that cleave target RNAs
required for
cell proliferation may be readily designed and are within the invention.
By "inhibit" is meant that the activity of c-raf or level of RNAs encoded by c-
raf is
reduced below that observed in the absence of the nucleic acid, particularly,
inhibition
with ribozymes is preferably below that level observed in the presence of an
inactive
RNA molecule able to bind to the same site on the mRNA, but unable to cleave
that
RNA.
By "nucleic acid catalyst" is meant a nucleic acid molecule capable of
catalyzing
(altering the velocity and/or rate of) a variety of reactions including the
ability to
repeatedly cleave other separate nucleic acid molecules (endonuclease
activity) in a
nucleotide base sequence-specific manner. Such a molecule with endonuclease
activity
may have complementarity in a substrate binding region to a specified gene
target, and
also has an enzymatic activity that specifically cleaves RNA or DNA in that
target. That
is, the nucleic acid molecule with endonuclease activity is able to
intramolecularly or
intermolecularly cleave RNA or DNA and thereby inactivate a target RNA or DNA
molecule. This complementarity functions to allow sufficient hybridization of
the
enzymatic RNA molecule to the target RNA or DNA to allow the cleavage to
occur.
100% complementarity is preferred, but complementarity as low as 50-75% may
also be
useful in this invention. The nucleic acids may be modified at the base,
sugar, and/or
phosphate groups. The term enzymatic nucleic acid is used interchangeably with
phrases
such as ribozymes, catalytic RNA, enzymatic RNA, catalytic DNA, catalytic
oligonucleotides, nucleozyme, DNAzyme, RNA enzyme, endoribonuclease,
CA 02288640 1999-11-OS
WO 98/50530 6 PCT/US98/09249
endonuclease, rninizyme, leadzyme, oligozyme or DNA enzyme. All of these
terminologies describe nucleic acid molecules with enzymatic activity. The
specific
nucleic acid catalysts described in the instant application are not limiting
in the invention
and those skilled in the art will recognize that all that is important in a
nucleic acid
catalyst of this invention is that it has a specific substrate binding site
which is
complementary to one or more of the target nucleic acid regions, and that it
have
nucleotide sequences within or surrounding that substrate binding site which
impart a
nucleic acid cleaving activity to the molecule.
By "substrate binding arm" or "substrate binding domain" is meant that
portion/region of a ribozyme which is complementary to {i.e., able to base-
pair with) a
portion of its substrate. Generally, such complementarity is 100%, but can be
less if
desired. For example, as few as 10 bases out of 14 may be base-paired. Such
arms are
shown generally in Figure 1 and 3. That is, these arms contain sequences
within a
ribozyme which are intended to bring ribozyme and target RNA together through
complementary base-pairing interactions. The ribozyme of the invention may
have
binding arms that are contiguous or non-contiguous and may be of varying
lengths. The
length of the binding arms) are preferably greater than or equal to four
nucleotides;
specifically 12-100 nucleotides; more specifically 14-24 nucleotides long. If
two binding
arms are chosen, the design is such that the length of the binding arms are
symmetrical
(i. e., each of the binding arms is of the same length; e.g., five and five
nucleotides, six and
six nucleotides or seven and seven nucleotides long) or asymmetrical (i. e.,
the binding
arms are of different length; e.g., six and three nucleotides; three and six
nucleotides long;
four and five nucleotides long; four and six nucleotides long; four and seven
nucleotides
long; and the like).
In one of the preferred embodiments of the inventions herein, the nucleic acid
catalyst is formed in a hammerhead or hairpin motif, but may also be formed in
the motif
of a hepatitis d virus, group I intron, group II intron or RNaseP RNA (in
association with
an RNA guide sequence) or Neurospora VS RNA. Examples of such hammerhead
motifs
CA 02288640 1999-11-05
WO 98/50530 7 PCT/US98/09249
are described by Dreyfus, supra, Rossl et al., 1992, AIDS Research and Human
Retroviruses 8, 183; of hairpin motifs by Hampel et al., EP0360257, Hampel and
Tritz,
1989 Biochemistry 28, 4929, Feldstein et al., 1989, Gene 82, 53, Haseloff and
Gerlach,
1989, Gene, 82, 43, and Hampel et al., 1990 Nucleic Acids Res. 18, 299; of the
hepatitis d
virus motif is described by Perrotta and Been, 1992 Biochemistry 31, 16; of
the RNaseP
motif by Guerrier-Takada et al., 1983 Cell 35, 849; Forster and Altman, 1990,
Science
249, 783; Li and Altman, 1996, Nucleic Acids Res. 24, 835; Neurospora VS RNA
ribozyme motif is described by Collins (Saville and Coliins, 1990 Cell 61, 685-
696;
Saville and Collins, 1991 Proc. Natl. Acad. Sci. USA 88, 8826-8830; Collins
and Olive,
1993 Biochemistry 32, 2795-2799; Guo and Collins, 1995, EMBO. J. 14, 363);
Group II
introns are described by Griffin et al., 1995, Chem. Biol. 2, 761; Michels and
Pyle, 1995,
Biochemistry 34, 2965; Pyle et al., International PCT Publication No. WO
96/22689; and
of the Group I intron by Cech et al., U.S. Patent 4,987,071. These specific
motifs are not
limiting in the invention and those skilled in the art will recognize that all
that is
important in a nucleic acid catalyst of this invention is that it has a
specific substrate
binding site which is complementary to one or more of the target gene RNA
regions, and
that it have nucleotide sequences within or surrounding that substrate binding
site which
impart an RNA cleaving activity to the molecule.
By "equivalent" RNA to c-raf is meant to include those naturally occurnng RNA
molecules associated with cancer in various animals, including human, rodent,
primate,
rabbit and pig. The equivalent RNA sequence also includes in addition to the
coding
region, regions such as 5'-untranslated region, 3'-untranslated region,
introns, intron-exon
junction and the like.
By "complementarity" is meant a nucleic acid that can form hydrogen bonds)
with
another RNA sequence by either traditional Watson-Crick or other non-
traditional types
" (for example, Hoogsteen type) of base-paired interactions.
In a preferred embodiment the invention provides a method for producing a
class of
enzymatic cleaving agents which exhibit a high degree of specificity for the
RNA of a
CA 02288640 1999-11-OS
WO 98/50530 PCT/US98/09249
8
desired target. The nucleic acid catalyst is preferably targeted to a highly
conserved
sequence region of a target mRNAs encoding c-raf proteins such that specific
treatment of
a disease or condition can be provided with either one or several enzymatic
nucleic acids.
Such nucleic acid catalysts can be delivered exogenously to specific cells as
required.
Alternatively, the ribozymes can be expressed from DNA/RNA vectors that are
delivered
to specific cells.
Such ribozymes are useful for the prevention of the diseases and conditions
discussed above, and any other diseases or conditions that are related to the
levels of c-
Raf activity in a cell or tissue.
By "related" is meant that the inhibition of c-raf RNAs and thus reduction in
the
level of respective protein activity will relieve to some extent the symptoms
of the disease
or condition.
In preferred embodiments, the ribozymes have binding arms which are
complementary to the target sequences in TablesXII-XIX. Examples of such
ribozymes
are also shown in Tables XII-XIX. Examples of such ribozymes consist
essentially of
sequences defined in these Tables.
By "consists essentially of is meant that the active ribozyme contains an
enzymatic center or core equivalent to those in the examples, and binding arms
able to
bind mRNA such that cleavage at the target site occurs. Other sequences may be
present
which do not interfere with such cleavage.
Thus, in a first aspect, the invention features ribozymes that inhibit gene
expression
and/or cell proliferation. These chemically or enzymatically synthesized RNA
molecules
contain substrate binding domains that bind to accessible regions of their
target mRNAs.
The RNA molecules also contain domains that catalyze the cleavage of RNA. The
RNA
molecules are preferably ribozymes of the hammerhead or hairpin motif. Upon
binding,
the ribozymes cleave the target mRNAs, preventing translation and protein
accumulation.
In the absence of the expression of the target gene, cell proliferation is
inhibited.
CA 02288640 1999-11-OS
WO 98/50530 9 PCT/US98/09249
In a preferred embodiment, ribozymes are added directly, or can be complexed
with
cationic lipids, packaged within liposomes, or otherwise delivered to target
cells. The
nucleic acid or nucleic acid complexes can be locally administered to relevant
tissues ex
vivo, or in vivo through injection, infusion pump or stmt, with or without
their
incorporation in biopolymers. In another preferred embodiment, the ribozyme is
administered to the site of c-raf expression (e.g., tumor cells) in an
appropriate liposomal
vehicle.
In another aspect of the invention, ribozymes that cleave target molecules and
inhibit c-raf activity are expressed from transcription units inserted into
DNA or RNA
vectors. The recombinant vectors are preferably DNA plasmids or viral vectors.
Ribozyme expressing viral vectors could be constructed based on, but not
limited to,
adeno-associated virus, retrovirus, adenovirus, or alphavirus. Preferably, the
recombinant
vectors capable of expressing the ribozymes are delivered as described above,
and persist
in target cells. Alternatively, viral vectors may be used that provide for
transient
1 S expression of ribozymes. Such vectors might be repeatedly administered as
necessary.
Once expressed, the ribozymes cleave the target mRNA. Delivery of ribozyme
expressing vectors could be systemic, such as by intravenous or intramuscular
administration, by administration to target cells ex-planted from the patient
followed by
reintroduction into the patient, or by any other means that would allow for
introduction
into the desired target cell (for a review see Couture and Stinchcomb, 1996,
TIG., 12,
510). In another aspect of the invention, ribozymes that cleave target
molecules and
inhibit cell proliferation are expressed from transcription units inserted
into DNA, RNA,
or viral vectors. Preferably, the recombinant vectors capable of expressing
the ribozymes
are locally delivered as described above, and transiently persist in smooth
muscle cells.
However, other mammalian cell vectors that direct the expression of RNA may be
used
for this purpose.
By "patient" is meant an organism which is a donor or recipient of explanted
cells
or the cells themselves. "Patient" also refers to an organism to which nucleic
acid
CA 02288640 1999-11-OS
WO 98/50530 1 ~ PCT/US98/09249
catalysts can be administered. Preferably, a patient is a mammal or mammalian
cells.
More preferably, a patient is a human or human cells.
By "vectors" is meant any nucleic acid- and/or viral-based technique used to
deliver
a desired nucleic acid.
These ribozymes, individually, or in combination or in conjunction with other
drugs, can be used to treat diseases or conditions discussed above. For
example, to treat a
disease or condition associated with c-raf levels, the patient may be treated,
or other
appropriate cells may be treated, as is evident to those skilled in the art.
In a further embodiment, the described ribozymes can be used in combination
with
other known treatments to treat conditions or diseases discussed above. For
example, the
described ribozymes could be used in combination with one or more known
therapeutic
agents to treat cancer.
In preferred embodiments, the ribozymes have binding arms which are
complementary to the sequences in the tables, shown as Seq. LD. Nos. 1-
501,1078-1152,
1461-1768, 1841-1912, 2354-2794 and 2846-2956. Examples of such ribozymes are
shown as Seq. LD. Nos. 502-1002, 1003-1077, 1153-1460, 1769-1840, 1913-2353
and
2795-2845. Other sequences may be present which do not interfere with such
cleavage.
Ribozymes that cleave the specified sites in Raf mRNAs represent a novel
therapeutic approach to treat tumor angiogenesis, ocular diseases, rhuematoid
arthritis,
psoriasis and others. Applicant indicates that ribozymes are able to inhibit
the activity of
Raf and that the catalytic activity of the ribozymes is required for their
inhibitory effect.
Those of ordinary skill in the art will find that it is clear from the
examples described that
other ribozymes that cleave Raf mRNAs may be readily designed and are within
the
invention.
Other features and advantages of the invention will be apparent from the
following
description of the preferred embodiments thereof, and from the claims.
CA 02288640 1999-11-05
WO 98/50530 11 PCT/US98/09249
Description Of The Preferred Embodiments
The drawings will first briefly be described.
Drawines:
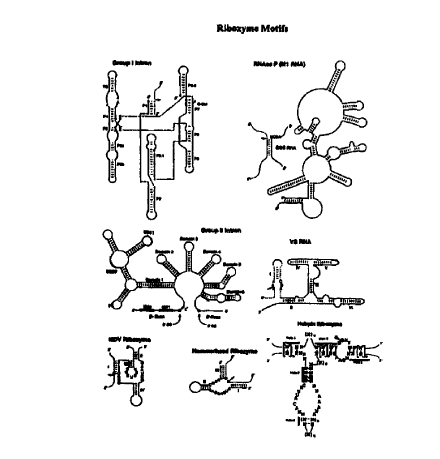
Figure 1 shows the secondary structure model for seven different classes of
nucleic acid catalysts. Arrow indicates the site of cleavage. ---------
indicate the target
sequence. Lines interspersed with dots are meant to indicate tertiary
interactions. - is
meant to indicate base-paired interaction. Group I Intron: P1-P9.0 represent
various
stem-loop structures (Cech et al., 1994, Nature Struc. Bio., 1, 273). RNase P
(M1 RNA):
EGS represents external guide sequence (Forster et al., 1990, Science, 249,
783; Pace et
al., 1990, J. Biol. Chem., 265, 3587). Group II Intron: 5'SS means 5' splice
site; 3'SS
means 3'-splice site; IBS means intron binding site; EBS means exon binding
site (Pyle et
al., 1994, Biochemistry, 33, 2716). VS RNA: i-VI are meant to indicate six
stem-loop
structures; shaded regions are meant to indicate tertiary interaction
(Collins, International
PCT Publication No. WO 96/19577). HDV °bozyme: : I-IV are meant to
indicate four
stem-loop structures (Been et al., US Patent No. 5,625,047). Hammerhead
Ribozyme:
I-III are meant to indicate three stem-loop structures; stems I-III can be of
any length and
may be symmetrical or asymmetrical (Usman et al., 1996, Curr. Op. Struct.
Bio., 1, 527).
Hairpin Ribozyme: Helix l, 4 and S can be of any length; Helix 2 is between 3
and 8
base-pairs long; Y is a pyrimidine; Helix 2 (H2) is provided with a least 4
base pairs (i.e.,
n is 1, 2, 3 or 4) and helix 5 can be optionally provided of length 2 or more
bases
(preferably 3 - 20 bases, i. e., m is from 1 - 20 or more). Helix 2 and helix
5 may be
covalently linked by one or more bases (i.e., r is 1 base). Helix 1, 4 or 5
may also be
extended by 2 or more base pairs (e.g., 4 - 20 base pairs) to stabilize the
ribozyme
structure, and preferably is a protein binding site. In each instance, each N
and N'
independently is any normal or modified base and each dash represents a
potential base
pairing interaction. These nucleotides may be modified at the sugar, base or
phosphate.
r Complete base-pairing is not required in the helices, but is preferred.
Helix 1 and 4 can
be of any size (i.e., o and p is each independently from 0 to any number,
e.g., 20) as long
CA 02288640 1999-11-OS
WO 98/50530 12 PCT/US98/09249
as some base-pairing is maintained. Essential bases are shown as specific
bases in the
structure, but those in the art will recognize that one or more may be
modified chemically
(abasic, base, sugar and/or phosphate modifications) or replaced with another
base
without significant effect. Helix 4 can be formed from two separate molecules,
i.e.,
without a connecting loop. The connecting loop when present may be a
ribonucleotide
with or without modifications to its base, sugar or phosphate. "q" is 2 bases.
The
connecting loop can also be replaced with a non-nucleotide linker molecule. H
refers to
bases A, U, or C. Y refers to pyrimidine bases. " " refers to a covalent bond.
(Burke et al., 199b, Nucleic Acids & Mol. Biol., 10, 129; Chowrira et al., US
Patent No.
S,G31,359).
Figure 2 shows a general approach to accessible site and target discovery
using
nucleic acid catalysts.
Figure 3 is a diagram of a hammerhead ribozyme. The consensus hammerhead
cleavage site in a target RNA is a "U" followed by "H" (anything but "G"). The
hammerhead ribozyme cleaves after the "H." This simple di-nucleotide sequence
occurs,
on average, every 5 nt in a target RNA. Thus, there are approximately 400
potential
hammerhead cleavage sites in a 2-Kb mRNA. Stems I and III are formed by
hybridization of the hammerhead binding arms with the complementary sequence
in
target RNA; it is these binding arms that confer specificity to the hammerhead
ribozyme
for its target. The binding arms of the hammerhead are interrupted by the
catalytic
domain that forms part of the structure responsible for cleavage.
Figure 4 shows a scheme for the design and synthesis of a Defined Library:
simultaneous screen of 400 different ICAM-targeted ribozymes is used as an
example.
DNA oligonucleotides encoding each ICAM-targeted ribozyme are synthesized
individually (A), pooled (B), then cloned and converted to retroviral vectors
as a pool.
The resulting retroviral vector particles are used to transduce a target cell
line that
expresses ICAM (B). Cells expressing ribozymes that inhibit ICAM expression
(ICAM-
low) are sorted from cells expressing ineffective ribozymes by FACS sorting
(C),
CA 02288640 1999-11-OS
WO 98/50530 13 PCTNS98/09249
effective ribozymes enriched in the ICAM-low population of cells are
identified by filter
hybridization (D).
Figure 5 A) shows randomization of the binding arms of a hammerhead ribozyme
to produce a Random Library. The binding arms can be of any length and any
symmetry,
i.e., symmetrical or assymmetrical. B) shows complexities of hammerhead Random
Ribozyme Libraries comprising a 6-nt or a 7-nt long binding arms.
Figure 6 is a schematic overview of Target Discovery strategy. An
oligonucleotide is prepared in a single reaction vessel in which all 4
standard nucleotides
are incorporated in a random fashion in the target binding arms) of the
ribozyme to
produce a pool of all possible ribozymes (A). This pool is cloned into an
appropriate
vector in a single tube to produce the Random Library expression vector (B)
and
retroviral vector particles are produced from this pool in a single tube (C).
The resulting
Random Ribozyme Library retroviral expression vector pool is then used to
transduce a
cell type of interest (D). Cells exhibiting the desired phenotype are then
separated from
the rest of the population using a number of possible selection strategies (E
and see text).
Genes that are critical for expression of the selected phenotype can then be
identified by
sequencing the target binding arms of ribozymes contained in the selected
population (F).
Figure 7 shows an example of application of Random Ribozyme Libraries to
identify genes critical for the induction of ICAM expression. Human Umbilical
Vein
Endothelial Cells (HUVECs) are transduced with a Random Ribozyme Library (A),
ICAM expression is induced using TNF-alpha (B), and cells expressing ribozymes
that
inhibit ICAM induction are selected from cells expressing ineffective
ribozymes by
sorting ICAM-low cells (C). Genes critical for ICAM induction are identified
by
sequencing the binding arms of the ribozymes that inhibit ICAM expression in
the ICAM
low cells.
Figure 8 is an example of an efficient cloning strategy for producing a
Defined or
Random Ribozyme Libraries. DNA oligos encoding ribozyme coding regions and
CA 02288640 1999-11-OS
WO 98/50530 14 PCT/US98/09249
restriction sites for cloning are designed to also contain a stem-loop
structure on the 3'
ends (1). This stem loop forms an intramolecular primer site for extension to
form a
double-stranded molecule by DNA polymerase (2). The double-stranded fragment
is
cleaved with appropriate restriction endonucleases to produce suitable ends
for
subsequent cloning (3).
Figure 9 shows molecular analysis of the PNP-targeted Defined Ribozyme
Library: sequence analysis. Plasmid DNA from the PNP-targeted Defined Ribozyme
Library was prepared and sequenced as a pool. The sequencing primer used reads
the
non-coding strand of the region encoding the ribozymes. Note that the sequence
diverges
at the binding arm, converges at the catalytic domain (5' -
TTTCGGCCTAACGGCCTCATCAG-3'), and then diverges at the other binding arm.
These results are consistent with those expected for a sequence of a
heterogeneous pool of
clones containing different sequences at the ribozyme binding arms.
Figure 10 shows molecular analysis of the PNP-targeted Defined Ribozyme
Library: sequence analysis after propagation in Sup T1 human T cells and
selection in 10
mmol 6-thioguanosine. Sup T1 cells were transduced with retroviral vector-
based
Defined Ribozyme Library comprised of 40 different PNP-targeted ribozyme
oligos
cloned into the U6+27 transcription unit (Figure 11D). The cells were
propagated for 2
weeks following transduction, then subjected to 16 days of selection in 10
mmol 6-
thioguanosine. Surviving cells were harvested, and ribozyme sequences present
in the
selected population of cells were amplified and sequenced. Note that, relative
to the
original Library where sequences of the binding arms were ambiguous due to the
presence
of 40 different ribozymes (Figure 9), the sequence of the binding arms in the
selected
population corresponded to only 1 of the 40 ribozymes included in the Library.
These
results suggest that this ribozyme was the most-potent ribozyme of 40
ribozymes tested.
Figure 11 is a schematic representation of transcription units suitable for
expression ribozyme library of the instant invention. A) is a diagrammatic
representation
of some RNA polymerase (Pol) II and III ribozyme (RZ) transcription units. CMV
CA 02288640 1999-11-OS
WO 98/50530 15 PCT/US98/09249
Promoter Driven is a Pol II transcript driven by a cytomegalovirus promoter;
the
transcript can designed such that the ribozyme is at the S'- region, 3'-region
or some
where in between and the transcript optionally comprises an intron. tRNA-DC is
a Pol III
transcript driven by a transfer RNA (tRNA) promoter, wherein the ribozyme is
at the 3'-
end of the transcript; the transcript optionally comprises a stem-loop
structure 3' of the
ribozyme. U6+27 is a Pol III transcript driven by a U6 small nuclear (snRNA)
promoter;
ribozyme is 3' of a sequence that is homologous to 27 nucleotides at the 5'-
end of a U6
snRNA; the transcript optionally comprise a stem-loop structure at the 3'-end
of the
ribozyme. VAI-90 is a Pol III transcript driven by an adenovirus VA promoter;
ribozyme
is 3' of a sequence homologous to 90 nucleotides at the S'-end of a VAI RNA;
the
transcript optionally comprises a stem-loop structure at the 3'-end of the
ribozyme. VAC
is a Pol III transcript driven by an adenovirus VAI promoter; the ribozyme is
inserted
towards the 3'- region of the VA RNA and into a S35 motif, which is a stable
greater than
or equal to 8 by long intramolecular stem formed by base-paired inteaction
between
sequences in the S'-region and the 3'-region flanking the ribozyme (see
Beigelman et al.,
International PCT Application No. WO 96/18736); the S35 domain positions the
ribozyme away from the main transcript as an independent domain. TRZ is a Pol
III
transcript diven by a tRNA promoter; ribozyme is inserted in the S35 domain
and is
positioned away from the main transcript (see Beigelman et al., International
PCT
Application No. WO 96/18736). B) shows various transcription units based on
the U1
small nuclear RNA (snRNA) system. C) is a schematic representation of a
retroviral
vectors encoding ribozyme genes. NGFR, nerve growth factor receptor is used as
a
selectable marker, LTR, long terminal repeat of a retrovirus, UTR,
untranslated region.
D) shows a U6+27 hammerhead ribozyme transcription unit based on the U6 snRNA.
The ribozyme transcript comprises the first 27 nt from the U6 snRNA which is
reported to
be necessary for the stability of the transcript. The transcript terminates
with a stretch of
uridine residues. The hammerhead ribozyme shown in the figure has random (I~
binding
arm sequence.
CA 02288640 1999-11-OS
WO 98/50530 16 PCT/US98/09249
Figure 12 is a schematic representation of a combinatorial approach to the
screening of r-ibozyme variants.
Figure 13 shows the sequence of a Starting Ribozyme to be used in the
screening
approach described in Figure 12. The Starting Ribozyme is a hammerhead (HH)
S ribozyme designed to cleave target RNA A (HH-A). Position 7 in HH-A is also
referred
to in this application as position 24 to indicate that U24 is . the 24th
nucleotide
incorporated into the HH-A ribozyme during chemical synthesis. Similarly,
positions 4
and 3 are also referred to as positions 27 and 28, respectively. s indicates
phosphorothioate substitution. Lower case alphabets in the HH-A sequence
indicate 2'-
O-methyl nucleotides; uppercase alphabets in the sequence of HH-A at positions
5, 6, 8,
12 and 1 S.1 indicate ribonucleotides. Positions 3, 4 and 7 are shown as
uppercase, large
alphabets to indicate the positions selected for screening using the method
shown in
Figure 12. ~ indicates base-paired interaction. iB represents abasic inverted
deoxy
ribose moiety.
Figure 14 shows a scheme for screening variants of HH-A ribozyme. Positions
24, 27 and 28 are selected for analysis in this scheme.
Figure 1 S shows non-limiting examples of some of the nucleotide analogs that
can
be used to construct ribozyme libraries. 2'-O-MTM-U represents 2'-O-
methylthiomethyl
uridine; 2'-O-MTM-C represents 2'-O-methylthiomethyl cytidine; 6-Me-U
represents 6-
methyl uridine (Beigelman et al., International PCT Publication No. WO
96/18736 which
is incorporated by reference herein).
Figure 16 shows activity of HH-A variant ribozymes as determined in a cell-
based
assay. * indicates the substitution that provided the most desirable attribute
in a
ribozyme.
Figure 17A shows the sequence and chemical composition of ribozymes that
showed the most desirable attribute in a cell.
CA 02288640 1999-11-OS
WO 98/50530 17 PCT/US98/09249
Figure 17B shows formulae for four different novel ribozyme motifs.
Figure 18 shows the formula foe a novel ribozyme motif.
- Figure 19 shows the sequence of a Starting Ribozyme to be used in the
screening
approach described in Figure 14. A HH ribozyme targeted against RNA B (HH-B)
was
chosen for analysis of the loop II sequence variants.
Figure 20 shows a scheme for screening loop-II sequence variants of HH-B
ribozyme.
Figure 21 shows the relative catalytic rates (k~,) for RNA cleavage reactions
catalyzed by HH-B loop-II variant ribozymes.
Figure 22 is a schematic representation of HH-B ribozyme-substrate complex and
the activity of HH-B ribozyme with either the 5'-GAAA-3' or the 5'-GUUA-3'
loop-II
sequence.
Figure 23 shows a scheme for using a combinatorial approach to identify
potential
ribozyme targets by varying the binding arms.
Figure 24 shows a scheme for using a combinatorial approach to identify novel
ribozymes by the varying putative catalytic domain sequence.
Figure 25 shows a table of accessible sites within a Bcl-2 transcript {(975
nucleotides) which were found using the combinatorial in vitro screening
process.
Figure 26 shows a table of accessible sites with a Kras transcript (796
nucleotides)
which were found using the combinatorial in vitro screening process as well as
a graphic
depiction of relative activity of ribozymes to those sites.
Figure 27 shows a table of accessible sites with a UPA transcript (400
nucleotides)
. which were found using the combinatorial in vitro screening process as well
as a graphic
depiction of relative activity of ribozymes to those sites.
CA 02288640 1999-11-OS
WO 98/50530 18 PCT/US98/09249
Figure 28 shows a graph displaying data from a ribonuclease protection assay
{RPA) after treatment of MCF-7 cells with ribozymes to targeted to site 549 of
the
transcript (Seq.ID #9). The Bcl-2 mRNA isolated from MCF-7 cells is normalized
to
GAPDH which was also probed in the RPA. The graph includes an untreated
control and
an irrelevant ribozyme (no complementarity with Bcl-2 mRNA).
Figure 29 displays a schematic representation of NTP synthesis using
nucleoside
substrates.
Figure 30 depicts a scheme for the synthesis of a xylo ribonucleoside
phosphoramidite.
. Figure 31 is a diagrammatic representation of hammerhead (HH) ribozyme
targeted against stromelysin RNA (site 617) with various modifications.
Figure 32 is a is a schematic representation of a one pot deprotection of RNA
synthesized using RNA phosphoramidite chemistry.
Figure 33 is a comparison of a one-pot and a two-pot process for deprotection
of
RNA.
Figure 34 shows the results of a one-pot deprotection with different polar
organic
reagents.
Figure 35 is a diagrammatic represention of ras signal transduction pathway.
Figure 36 is a diagrammatic representation of hammerhead ribozymes targeted
against c-raf RNA.
Figure 37 is a graphical representation of c-raf 2'-C-allyl 1120 hammerhead
(HH)
ribozyme-mediated inhibition of cell proliferation.
Figure 38 is a graphical representation of inhibition of cell proliferation
mediated by
c-raf 2'-C-allyl 1120 and 1251 hammerhead (HH) ribozymes.
CA 02288640 1999-11-OS
WO 98/50530 19 PCT/US98/09249
Figure 39 shows the effects of flt-1 ribozymes (active/inactive) on LLC-HM
primary tumor growth in mice.
Figure 40 shows the effects of flt-1 ribozymes on LLC-HM primary tumor volume
immediately following the cessation of treatment.
Figure 41 shows the effects of flt-1 ribozymes on lung metastatic indices
(number
of metastases and lung mass).
Figure 42 shows the effects of flk-1 ribozymes (active/inactive) on LLC-HM
primary tumor growth in mice.
Figure 43 shows the effects of flk-1 ribozymes on LLC-HM primary tumor volume
immediately following the cessation of treatment.
Figure 44 shows the effects of flk-1 ribozymes on lung metastatic indices
(number
of metastases and lung mass).
Nucleic Acid Catalysts:
Catalytic nucleic acid molecules (ribozymes) are nucleic acid molecules
capable
of catalyzing one or more of a variety of reactions, including the ability to
repeatedly
cleave other separate nucleic acid molecules in a nucleotide base sequence-
specific
manner. Such nucleic acid catalysts can be used, for example, to target
cleavage of
virtually any RNA transcript (Zaug et al., 324, Nature 429 1986 ; Cech, 260
JAMA 3030,
1988; and Jefferies et al., 17 Nucleic Acids Research 1371, 1989). Catalytic
nucleic acid
molecules mean any nucleotide base-comprising molecule having the ability to
repeatedly
act on one or more types of molecules, including but not limited to nucleic
acid catalysts.
By way of example but not limitation, such molecules include those that are
able to
repeatedly cleave nucleic acid molecules, peptides, or other polymers, and
those that are
able to cause the polymerization of such nucleic acids and other polymers.
Specifically,
such molecules include ribozymes, DNAzymes, external guide sequences and the
like. It
CA 02288640 1999-11-OS
WO 98/50530 2~ PCT/US98/09249
is expected that such molecules will also include modified nucleotides
compared to
standard nucleotides found in DNA and RNA.
Because of their sequence-specificity, traps-cleaving nucleic acid catalysts
show
promise as therapeutic agents for human disease (Usman & McSwiggen, 1995 Ann.
Rep.
Med. Chem. 30, 285-294; Christoffersen and Marr, 1995 J. Med. Chem. 38, 2023-
2037).
Nucleic acid catalysts can be designed to cleave specific RNA targets within
the
background of cellular RNA. Such a cleavage event renders the RNA non-
functional and
abrogates protein expression from that RNA. In this manner, synthesis of a
protein
associated with a disease state can be selectively inhibited. In addition,
nucleic acid
catalysts can be used to validate a therapeutic gene target and/or to
determine the function
of a gene in a biological system (Christoffersen, 1997, Nature Biotech. 15,
483).
There are at least seven basic varieties of enzymatic RNA molecules derived
from
naturally occurring self cleaving RNAs (see Table I). Each can catalyze the
hydrolysis of
RNA phosphodiester bonds in traps (and thus can cleave other RNA molecules)
under
physiological conditions. In general, enzymatic nucleic acids act by first
binding to a
substrate/target RNA. Such binding occurs through the substrate/target binding
portion of
an nucleic acid catalyst which is held in close proximity to an enzymatic
portion of the
molecule that acts to cleave the target RNA. Thus, the enzymatic nucleic acid
first
recognizes and then binds a target RNA through complementary base-pairing, and
once
bound to the correct site, acts enzymatically to cut the target RNA. Strategic
and
selective cleavage of such a target RNA will destroy its ability to direct
synthesis of an
encoded protein. After an enzymatic nucleic acid has bound and cleaved its RNA
target,
it is released from that RNA to search for another target and thus can
repeatedly bind and
cleave new targets.
In addition, several in vitro selection (evolution) strategies (Orgel, 1979,
Proc. R.
Soc. London, B 205, 435) have been used to evolve new nucleic acid catalysts
capable of
catalyzing a variety of reactions, such as cleavage and ligation of
phosphodiester linkages
and amide linkages, (Joyce, 1989, Gene, 82, 83-87; Beaudry et al., 1992,
Science 257,
CA 02288640 1999-11-OS
WO 98/50530 2I PCT/US98/09249
635-641; Joyce, 1992, Scientific American 267, 90-97; Breaker et al., 1994,
TIBTECH 12,
268; Bartel et al., 1993, Science 261:1411-1418; Szostak, 1993, TIBS 17, 89-
93; Kumar et
al., 1995, FASEB J., 9, 1183; Breaker, 1996, Curr. Op. Biotech., 7, 442;
Breaker, 1997,
Nature Biotech. I5, 427).
There are several reports that describe the use of a variety of in vitro and
in vivo
selection strategies to study structure and function of catalytic nucleic acid
molecules
(Campbell et al., 1995, RNA l, 598; Joyce 1989, Gene, 82,83; Lieber et al.,
1995, Mol
Cell Biol. 15, 540; Lieber et al., International PCT Publication No. WO
96/01314;
Szostak 1988, in Redesigning the Molecules of Life, Ed. S. A. Benner, pp 87,
Springer-
Verlag, Germany; Kramer et al., U.S. Patent No. 5,616,459; Draper et al., US
Patent No.
5,496,698; Joyce, U.S. Patent No. 5,595,873; Szostak et al., U.S. Patent No.
5,631,146).
The enzymatic nature of a ribozyme is advantageous over other technologies,
since the effective concentration of ribozyme sufficient to effect a
therapeutic treatment is
generally lower than that of an antisense oligonucleotide. This advantage
reflects the
ability of the ribozyme to act enzymatically. Thus, a single ribozyme
(enzymatic nucleic
acid) molecule is able to cleave many molecules of target RNA. In addition,
the
ribozyme is a highly specific inhibitor, with the specificity of inhibition
depending not
only on the base-pairing mechanism of binding, but also on the mechanism by
which the
molecule inhibits the expression of the RNA to which it binds. That is, the
inhibition is
caused by cleavage of the RNA target and so specificity is defined as the
ratio of the rate
of cleavage of the targeted RNA over the rate of cleavage of non-targeted RNA.
This
cleavage mechanism is dependent upon factors additional to those involved in
base-
pairing. Thus, it is thought that the specificity of action of a ribozyme is
greater than that
of antisense oligonucleotide binding the same RNA site.
The development of ribozymes that are optimal for catalytic activity would
contribute significantly to any strategy that employs RNA-cleaving ribozymes
for the
purpose of regulating gene expression. The hammerhead ribozyme, for example,
functions with a catalytic rate (k~at) of ~1 min-1 in the presence of
saturating (10 mM)
CA 02288640 1999-11-OS
WO 98!50530 22 PCT/US98/09249
concentrations of Mg2+ cofactor. However, the rate for this ribozyme in Mg2'~
concentrations that are closer to those found inside cells (0.5 - 2 mM) can be
I O- to 100-
fold slower. In contrast, the RNase P holoenzyme can catalyze pre-tRNA
cleavage with a
kcat of ~30 min-1 under optimal assay conditions. An artificial 'RNA ligase'
ribozyme
(Bartel et al., supra) has been shown to catalyze the corresponding self
modification
reaction with a rate of 100 min-1. In addition, it is known that certain
modified
hammerhead ribozymes that have substrate binding arms made of DNA catalyze RNA
cleavage with multiple turn-over rates that approach 100 min-1. Finally,
replacement of a
specific residue within the catalytic core of the hammerhead with certain
nucleotide
analogues gives modified ribozymes that show as much as a 10-fold improvement
in
catalytic rate. These findings demonstrate that ribozymes can promote chemical
transformations with catalytic rates that are significantly greater than those
displayed in
vitro by most natural self cleaving ribozymes. It is then possible that the
structures of
certain self cleaving ribozymes may not be optimized to give maximal catalytic
activity,
or that entirely new RNA motifs could be made that display significantly
faster rates for
RNA phosphoester cleavage.
By "nucleotide" as used herein is as recognized in the art to include natural
bases
(standard), and modified bases well known in the art. Such bases are generally
located at
the 1' position of a sugar moiety. Nucleotide generally comprise a base, sugar
and a
phosphate group. The nucleotides can be unmodified or modified at the sugar,
phosphate
and/or base moiety, (also referred to interchangeably as nucleotide analogs,
modified
nucleotides, non-natural nucleotides, non-standard nucleotides and other ; see
for
example, Usman and McSwiggen, supra; Eckstein et al., International PCT
Publication
No. WO 92/07065; Usman et al., International PCT Publication No. WO 93/15187;
all
hereby incorporated by reference herein). There are several examples of
modified nucleic
acid bases known in the art and has recently been summarized by Limbach et
al., 1994,
Nucleic Acids Res. 22, 2183. Some of the non-limiting examples of base
modifications
that can be introduced into enzymatic nucleic acids without significantly
effecting their
catalytic activity include, inosine, purine, pyridin-4-one, pyridin-2-one,
phenyl,
CA 02288640 1999-11-OS
WO 98/50530 PCT/US98/09249
23
pseudouracil, 2, 4, 6-trimethoxy benzene, 3-methyl uracil, dihydrouridine,
naphthyl,
aminophenyl, S-alkylcytidines (e.g., 5-methylcytidine), 5-alkyluridines (e.g.,
ribothymidine), S-halouridine (e.g., 5-bromouridine) or 6-azapyrimidines or 6-
alkylpyrimidines (e.g., 6-methyluridine) and others (Burgin et al., 1996,
Biochemistry,
S 35, 14090). By "modified bases" in this aspect is meant nucleotide bases
other than
adenine, guanine, cytosine and uracil at 1' position or their equivalents;
such bases may
be used within the catalytic core of the enzyme and/or in the substrate-
binding regions.
In another preferred embodiment, catalytic activity of the molecules described
in
the instant invention can be optimized as described by Draper et al., supra.
The details
will not be repeated here, but include altering the length of the ribozyme
binding arms, or
chemically synthesizing ribozymes with modifications (base, sugar and/or
phosphate) that
prevent their degradation by serum ribonucleases and/or enhance their
enzymatic activity
(see e.g., Eckstein et al., International Publication No. WO 92/07065;
Perrault et al., 1990
Nature 344, 565; Pieken et al., 1991 Science 253, 314; Usman and Cedergren,
1992
Trends in Biochem. Sci. 17, 334; Usman et al., International Publication No.
WO 93/15187; and Rossi et al., International Publication No. WO 91/03162;
Sproat, US
Patent No. 5,334,711; and Burgin et al., supra; all of these describe various
chemical
modifications that can be made to the base, phosphate and/or sugar moieties of
enzymatic
RNA molecules). Modifications which enhance their efficacy in cells, and
removal of
bases from stem loop structures to shorten RNA synthesis times and reduce
chemical
requirements are desired. (All of these publications are hereby incorporated
by reference
herein).
There are several examples in the art describing sugar and phosphate
modifications that can be introduced into nucleic acid catalysts without
significantly
effecting catalysis and with significant enhancement in their nuclease
stability and
efficacy. Ribozymes are modified to enhance stability and/or enhance catalytic
activity
by modification with nuclease resistant groups, for example, 2'-amino, 2'-C
allyl, 2'-
flouro, 2'-O-methyl, 2'-H, nucleotide base modifications (for a review see
Usman and
CA 02288640 1999-11-OS
WO 98/50530 PCT/US98/09249
24
Cedergren, 1992 TIBS 17, 34; Usman et al., 1994 Nucleic Acids Symp. Ser. 31,
163;
Burgin et al., 1996 Biochemistry 35, 14090). Sugar modification of nucleic
acid catalysts
has been extensively described in the art (see Eckstein et al., International
Publication
PCT No. WO 92/07065; Perrault et al. Nature 1990, 344, 565-568; Pieken et al.
Science
1991, 253, 314-317; Usman and Cedergren, Trends in Biochem. Sci. 1992, 17, 334-
339;
Usman et al. International Publication PCT No. WO 93/15187; Sproat, US Patent
No.
5,334,711 and Beigelman et al., 1995 J. Biol. Chem. 270, 25702; all of the
references are
hereby incorporated in their totality by reference herein).
Such publications describe general methods and strategies to determine the
location of incorporation of sugar, base and/or phosphate modifications and
the like into
ribozymes without inhibiting catalysis, and are incorporated by reference
herein. In view
of such teachings, similar modifications can be used as described herein to
modify the
nucleic acid catalysts of the instant invention.
In yet another preferred embodiment, nucleic acid catalysts having chemical
modifications which maintain or enhance enzymatic activity is provided. Such a
nucleic
acid is also, generally, more resistant to nucleases than the corresponding
unmodified
nucleic acid. Thus, in a cell and/or in vivo the activity may not be
significantly lowered.
As exemplified herein such ribozymes are useful in a cell and/or in vivo even
if activity
over all is reduced 10 fold (Burgin et al., 1996, Biochemistry, 35, 14090).
Such
modifications herein are said to "maintain" the enzymatic activity on all RNA
ribozyme.
In a preferred embodiment, the nucleic acid catalysts of the invention are
added
directly, or can be complexed with cationic lipids, packaged within liposomes,
or
otherwise delivered to smooth muscle cells. The RNA or RNA complexes can be
locally
administered to relevant tissues through the use of a catheter, infusion pump
or stmt, with
or without their incorporation in biopolymers. Using the methods described
herein, other
nucleic acid catalysts that cleave target nucleic acid may be derived and used
as described
above. Specific examples of nucleic acid catalysts of the instant invention
are provided
below in the Tables and figures.
CA 02288640 1999-11-OS
WO 98/50530 25 PCT/US98/09249
Sullivan, et al., WO 94/02595, describes the general methods for delivery of
nucleic acid catalysts. Ribozymes may be administered to cells by a variety of
methods
known to those familiar to the art, including, but not restricted to,
encapsulation in
liposomes, by iontophoresis, or by incorporation into other vehicles, such as
hydrogels,
cyclodextrins, biodegradable nanocapsules, and bioadhesive microspheres. For
some
indications, ribozymes may be directly delivered ex vivo to cells or tissues
with or without
the aforementioned vehicles. Alternatively, the RNA/vehicle combination is
locally
delivered by direct injection or by use of a catheter, infusion pump or stmt.
Other routes
of delivery include, but are not limited to, intravascular, intramuscular,
subcutaneous or
joint injection, aerosol inhalation, oral (tablet or pill form), topical,
systemic, ocular,
intraperitoneal and/or intrathecal delivery. More detailed descriptions of
ribozyme
delivery and administration are provided in Sullivan et al., supra and Draper
et al., WO
93/23569 which have been incorporated by reference herein.
Such nucleic acid catalysts can be delivered exogenously to specific cells as
required. In the preferred hammerhead motif the small size (less than 60
nucleotides,
preferably between 30-40 nucleotides in length) of the molecule allows the
cost of
treatment to be reduced.
Therapeutic ribozymes delivered exogenously must remain stable within cells
until translation of the target RNA has been inhibited long enough to reduce
the levels of
the undesirable protein. This period of time varies between hours to days
depending upon
the disease state. Clearly, ribozymes must be resistant to nucleases in order
to function as
effective intracellular therapeutic agents. Improvements in the chemical
synthesis of
RNA (Wincott et al., 1995 Nucleic Acids Res. 23, 2677; incorporated by
reference herein)
have expanded the ability to modify ribozymes by introducing nucleotide
modifications to
enhance their nuclease stability as described above.
CA 02288640 1999-11-OS
WO 98!50530 26 PCT/US98/09249
Synthesis, Deprotection, and Purification of Nucleic Acid Catal
Generally, RNA molecules are chemically synthesized and purified by
methodologies based on the use of tetrazole to activate the RNA
phosphoramidite,
ethanolic-NH4OH to remove the exocyclic amino protecting groups, tetra-n-
butylammonium fluoride (TBAF) to remove the 2'-OH alkylsilyl protecting
groups, and
gel purification and analysis of the deprotected RNA. Examples of chemical
synthesis,
deprotection, purification and analysis procedures for RNA are provided by
Usman et al.,
1987 J. Am. Chem. Soc., 109, 7845; Scaringe et al. Nucleic Acids Res. 1990,
18, 5433-
5341; Perreault et al. Biochemistry 1991, 30 4020-4025; Slim and Gait Nucleic
Acids
Res. 1991, 19, 1183-1188. All the above noted references are all hereby
incorporated by
reference herein.
Synthesis of nucleic acids greater than 100 nucleotides in length is difficult
using
automated methods, and the therapeutic cost of such molecules is prohibitive.
In this
invention, small nucleic acid motifs (e.g., antisense oligonucleotides,
hammerhead or the
hairpin ribozymes) are used for exogenous delivery. The simple structure of
these
molecules increases the ability of the nucleic acid to invade targeted regions
of the
mRNA structure. However, these nucleic acid molecules can also be expressed
within
cells from eukaryotic promoters (e.g., Izant and Weintraub, 1985 Science 229,
345;
McGarry and Lindquist, 1986 Proc. Natl. Acad. Sci.USA 83, 399;
SullengerScanlon et
al., 1991, Proc. Natl. Acad. Sci. USA, 88, 10591-5; Kashani-Sabet et al., 1992
Antisense
Res. Dev., 2, 3-15; Dropulic et al., 1992 J. Virol, 66, 1432-41; Weerasinghe
et al., 1991 J.
Virol, 65, 5531-4; Ojwang et al., 1992 Proc. Natl. Acad. Sci. USA 89, 10802-6;
Chen et
al., 1992 Nucleic Acids Res., 20, 4581-9; Sarver et al., 1990 Science 247,
1222-1225;
Thompson et al., 1995 Nucleic Acids Res. 23, 2259). Those skilled in the art
realize that
any nucleic acid can be expressed in eukaryotic cells from the appropriate
DNA/RNA
vector. The activity of such nucleic acids can be augmented by their release
from the
primary transcript by a ribozyme (Draper et al., PCT W093/23569, and Sullivan
et al.,
PCT W094/02595, both hereby incorporated in their totality by reference
herein; Ohkawa
et al., 1992 Nucleic Acids Symp. Ser., 27, 15-6; Taira et al., 1991, Nucleic
Acids Res., 19,
CA 02288640 1999-11-OS
WO 98/50530 27 PCT/US98/09249
5125-30; Ventura et al., 1993 Nucleic Acids Res., 21, 3249-55; Chowrira et
al., 1994 J.
Biol. Chem. 269, 25856).
The ribozymes were chemically synthesized. The method of synthesis used
follows the procedure for normal RNA synthesis as described in Usman et al.,
1987 J.
Am. Chem. Soc., 109, 7845; Scariilge et al., 1990 Nucleic Acids Res., 18,
5433; and
Wincott et al., 1995 Nucleic Acids Res. 23, 2677-2684 and makes use of common
nucleic
acid protecting and coupling groups, such as dimethoxytrityl at the 5'-end,
and
phosphoramidites at the 3'-end. Small scale synthesis were conducted on a 394
Applied
Biosystems, Inc. synthesizer using a modified 2.5 pmol scale protocol with a 5
min
coupling step for alkylsilyl protected nucleotides and 2.5 min coupling step
for 2'-O-
methylated nucleotides. Table II outlines the amounts, and the contact times,
of the
reagents used in the synthesis cycle. A 6.5-fold excess (163 ~.L of 0.1 M =
16.3 ~mol) of
phosphoramidite and a 24-fold excess of S-ethyl tetrazole (238 ~,L of 0.25 M =
59.5
~.mol) relative to polymer-bound 5'-hydroxyl was used in each coupling cycle.
Average
1 S coupling yields on the 394 Applied Biosystems, Inc. synthesizer,
determined by
colorimetric quantitation of the trityl fractions, were 97.5-99%. Other
oligonucleotide
synthesis reagents for the 394 Applied Biosystems, Inc.
synthesizer:detritylation solution
was 2% TCA in methylene chloride (ABI); capping was performed with 16% N
methyl
imidazole in THF {ABI) and 10% acetic anhydride/10% 2,6-lutidine in THF (ABI);
oxidation solution was 16.9 mM I2, 49 mM pyridine, 9% water in THF
(Millipore). B &
J Synthesis Grade acetonitrile was used directly from the reagent bottle. S-
Ethyl tetrazole
solution (0.25 M in acetonitrile) was made up from the solid obtained from
American
International Chemical, Inc.
Deprotection of the RNA was performed as follows. The polymer-bound
oligoribonucleotide, trityl-off, was transferred from the synthesis column to
a 4mL glass
screw top vial and suspended in a solution of methylamine (MA) at 65 °C
for 10 min.
After cooling to -20 °C, the supernatant was removed from the polymer
support. The
support was washed three times with 1.0 mL of EtOH:MeCN:H20/3:1:1, vortexed
and
CA 02288640 1999-11-OS
WO 98/50530 2g PCT/US98/09249
the supernatant was then added to the first supernatant. The combined
supernatants,
containing the oligoribonucleotide, were dried to a white powder.
The base-deprotected oligoribonucleotide was resuspended in anhydrous
TEA~HF/NMP solution (250 uL of a solution of I.SmL N methylpyrrolidinone, 750
~L
TEA and 1:0 mL TEA~3HF to provide a 1.4M HF concentration) and heated to
65°C for
1.5 h. The resulting, fully deprotected, oligomer was quenched with 50 mM TEAB
(9
mL) prior to anion exchange desalting.
For anion exchange desalting of the deprotected oligomer, the TEAB solution
was
loaded onto a Qiagen 500~ anion exchange cartridge (Qiagen Inc.) that was
prewashed
with 50 mM TEAB (10 mL). After washing the loaded cartridge with 50 mM TEAB
(10
mL), the RNA was eluted with 2 M TEAB (10 mL) and dried down to a white
powder.
Deprotection of RNA:
For high throughput chemical synthesis of oligoribonucleotides, it is
important
that the two main steps involved in the deprotection of oligoribonucleotides
(i.e., aqueous
basic treatment to remove exocyclic amino protecting groups and phosphate
protecting
groups and fluoride treatment to remove the 2'-OH alkylsilyl protecting groups
such as
the tButylDiMethylSilyl) are condensed.
Stinchcomb et al., supra describe a time-efficient (~ 2 hrs) one-pot
deprotection
protocol based on anhydrous methylamine and triethylamine trihydrogen
fluoride. Since
it has recently been reported that water contamination during fluoride
treatment may be
detrimental to the efficiency of the desilylation reaction (Hogrefe et al,
Nucleic Acids
Res. (1993), 21 4739-4741), it is necessary to use an anhydrous solution of
base such as a
33% methylamine in absolute ethanol followed by neat triethylamine
trihydrofluoride to
effectively deprotect oligoribonucleotides in a one-pot fashion. However it
may be
cumbersome to apply such a protocol to plate format deprotection where the
solid-
support is preferentially separated from the partially deprotected
oligoribonucleotides
prior to the 2'-hydroxyl deprotection. Indeed, because the methylamine
solution used is
CA 02288640 1999-11-OS
WO 98/50530 29 PCT/US98/09249
anhydrous, it may not be suitable to solubilize the negatively charged
oligoribonucleotides obtained after basic treatment. Therefore, applicant
investigated a
1:1 mixture of the ethanolic methylamine solution and different polar
additives such as
dimethylsulfoxide (DMSO), N,N-dimethylformamide (DMF), methanol,
hexamethylphosphoramide (HMPA), 1-methyl-2-pyrrolidinone (NMP) or 2-
methoxyethyl ether (glyme). Of all these additives, dimethylsufoxide is
capable of
efficiently solubilizing partially deprotected oligoribonucleotides (figure
34). A
comparison of the one pot and two pot deprotection methods are outlined and
demonstrated in figure 33.
The deprotection process commonly involves the deprotection of the exocyclic
amino protecting groups by NH40H, which is time consuming (6-24 h) and
inefficient.
This step is then followed by treatment with TBAF to facilitate the removal of
alkylsilyl
protecting groups, which again is time consuming and not very effective in
achieving
efficient deprotection.
A recent modification of this two-step strategy for oligoribonucleotide
deprotection has been reported by Wincott et al., (Nucleic Acids Res., 1995,
23, 2677-
2784) and by Vinayak et al., (Nucleic Acids Symposium series, 1995. 33, 123-
125). The
optimized conditions make use of aqueous methylamine at 65°C for 15
minutes in place
of the ammonium hydroxide cocktail to remove exocyclic amino protecting groups
while
the desilylation treatment needed to remove the 2'-OH alkylsilyl protecting
groups
utilizes a mixture of triethylamine trihydrogen fluoride (TEA.3HF), N-methyl-
pyrrolidinone and triethylamine at 65°C for 90 minutes, thereby
replacing tetrabutyl
ammonium fluoride.
Stinchcomb et al., International PCT Publication No. WO 95/23225 describe a
process for one pot deprotection of RNA. On page 73, it states that:
"In an attempt to minimize the time required for deprotection and to simplify
the
process of deprotection of RNA synthesized on a large scale, applicant
describes a one pot
CA 02288640 1999-11-OS
WO 98/50530 3o PCT/US98109249
deprotection protocol... According to this protocol, anhydrous methylamine is
used in
place of aqueous methyl amine. Base deprotection is carned out at 65 °C
for 15 minutes
and the reaction is allowed to cool for 10 min. Deprotection of 2'-hydroxyl
groups is then
carned out in the same container for 90 minutes in a TEA~3HF reagent. The
reaction is
quenched with 16 mM TEAB solution."
This invention concerns a one-pot process for the deprotection of RNA
molecules.
This invention features a novel method for the removal of protecting groups
from the
nucleic acid base and 2'-OH groups, which accelerates the process for
generating
synthetic RNA in a high throughput manner (e.g., in a 96 well format).
Chemical synthesis of RNA is generally accomplished using a traditional column
format on a RNA synthesizer where only one oligoribonucleotide is synthesized
at a time.
Simultaneous synthesis of more than one RNA molecule in a time efficient
manner
requires alternate methods to the traditional column format, such as synthesis
in a 96 well
plate format where up to 96 RNA molecules can be synthesized at the same time.
To
expedite this process of simultaneous synthesis of multiple RNA molecules, it
is
important to accelerate some of the time consuming processes such as the
deprotection of
RNA following synthesis (i.e., removal of base protecting group, such as the
exocyclic
amino protecting group and the phosphate protecting groups and the removal of
2'-OH
protecting groups, such as the tButylDiMethylSilyl). In a preferred
embodiment, the
invention features a one-pot process for rapid deprotection of RNA.
Stinchcomb et al., supra described a one-pot protocol for RNA deprotection
using
anhydrous methylamine and triethylamine trihydrogen fluoride. This procedure
involves
the use of an anhydrous solution of base such as a 33% methylamine in absolute
ethanol
followed by neat triethylamine trihydrofluoride to effectively deprotect
oligoribonucleotides in a one-pot fashion. However such a protocol rnay be
cumbersome
for deprotection of RNA synthesized on a plate format, such as a 96 well
plate, because it
may be necessary to separate the solid-support from the partially deprotected
RNA prior
to the 2'-hydroxyl deprotection. Also, since the methylamine solution used is
anhydrous,
CA 02288640 1999-11-OS
WO 98/50530 31 PCT/US98/09249
it may be difficult to solubilize the negatively charged oligoribonucieotides
obtained after
basic treatment. So, in a first aspect the invention features the use of a 1:1
mixture of the
ethanolic methylamine solution and a polar additive, such as dimethylsulfoxide
(DMSO),
N,N-dimethylformamide (DMF), methanol, hexamethylphosphoramide (HMPA), 1-
methyl-2-pyn olidinone (NMP), 2-methoxyethyl ether (glyme) or the like. More
specifically, dimethylsufoxide is used to partially deprotect
oligoribonucleotides (Figure
32). A comparison of the one pot and two pot deprotection methods are outlined
and
demonstrated in Figure 33.
This invention also concerns a rapid (high through-put) deprotection of RNA in
a
96-well plate format. More specifically rapid deprotection of enzymatic RNA
molecules
in greater than microgram quantities with high biological activity is
featured. It has been
determined that the recovery of enzymatically active RNA in high yield and
quantity is
dependent upon certain critical steps used during its deprotection.
In a preferred embodiment, the invention features a process for one-pot
deprotection of RNA molecules comprising protecting groups, comprising the
steps of a)
contacting the RNA with a mixture of anhydrous alkylamine (where alkyl can be
branched or unbranched, ethyl, propyl or butyl and is preferably methyl, e.g.,
methylamine), trialkylamine (where alkyl can be branched or unbranched,
methyl, propyl
or butyl and is preferably ethyl, e.g., ethylamine) and dimethylsulfoxide,
preferably in a
10:3:13, or 1:0.3:1 proportion at temperature 20-30 °C for about 30-100
minutes,
preferably 90 minutes, to remove the exocyclic amino (base) protecting groups
and the
phosphate protecting group (e.g., 2-cyanoethyl) (vs 4-20 h at 55-65 °C
using
NH40H/EtOH or NH3/EtOH, or 10-15 min at 65°C using 40% aqueous
methylamine)
under conditions suitable for partial deprotection of the RNA; b) contacting
the partially
deprotected RNA with anhydrous triethylamine~hydrogen fluoride (3HF~TEA) and
heating at about 50-70 °C, preferably at 65 °C, for about 5-30
min, preferably 1 S min to
remove the 2'-hydroxyl protecting group (vs 8 - 24 h using TBAF, or TEA~3HF
for 24 h
(Gasparutto et al. Nucleic Acids Res. 1992, 20, 5159-5166) (Other
alkylamine~HF
CA 02288640 1999-11-OS
WO 98150530 32 PCT/US98/09249
complexes may also be used, e.g., trimethylamine or diisopropylethylamine)
under
conditions suitable for the complete deprotection of the RNA. The reaction can
then be
quenched by using aqueous ammonium bicarbonate (1.4 M). Although some other
buffers can be used to quench the desilylation reaction (i.e.,
triethylammonium
S bicarbonate, ammonium acetate), the ammonium bicarbonate buffer is perfectly
suited to
retain the 5'-O-dimethoxytrityl group at the 5'-end of the oligoribonucleotide
thereby
facilitating a reverse phase-based solid-phase extraction purification
protocol.
By "one-pot" deprotection is meant that the process of deprotection RNA is
carried out in one container instead of multiple containers as in two-pot
deprotection.
In another preferred embodiment, the invention features a process for one pot
deprotection of RNA molecules comprising protecting groups, comprising the
steps of a)
contacting the RNA with a mixture of anhydrous alkylamine (where alkyl can be
branched or unbranched, ethyl, propyl or butyl and is preferably methyl, e.g.,
methylamine), and dimethylsulfoxide, preferably in a 1: i proportion at 20-30
°C
temperature for about 30-100 minutes, preferably 90 minutes, to remove the
exocyclic
amino (base) protecting groups and the phosphate protecting group (e.g., 2-
cyanoethyl)
(vs 4-20 h at 55-65 °C using NH40H/EtOH or NH3/EtOH, or 10-15 min at
65°C using
40% aqueous methylamine) under conditions suitable for partial deprotection of
the
RNA; b) contacting the partially deprotected RNA with anhydrous
triethylamine~hydrogen fluoride (3HF~TEA) and heating at about 50-70
°C, preferably at
65 °C, for about 5-30 min, preferably 15 min to remove the 2'-hydroxyl
protecting group
(Other alkylamine~HF complexes may also be used, e.g., trimethylamine or
diisopropylethylamine) under conditions suitable for the complete deprotection
of the
RNA. The reaction can then be quenched by using aqueous ammonium bicarbonate
(1.4
M). Although some other buffers can be used to quench the desilylation
reaction (i.e.,
triethylammonium bicarbonate, ammonium acetate), the ammonium bicarbonate
buffer is
perfectly suited to retain the 5'-O-dimethoxytrityl group at the S'-end of the
CA 02288640 1999-11-OS
WO 98/50530 33 PCTNS98/09249
oligoribonucleotide thereby facilitating a reverse phase-based solid-phase
extraction
purification protocol.
In another aspect the invention features a process for RNA deprotection where
the
exocyclic amino and phosphate deprotection reaction is performed with the
ethanolic
methylamine solution at room temperature for about 90 min or at 65°C
for 15 min ~ or at
45°C for 30 min or at 35°C for 60 min.
In a preferred embodiment, the process for deprotection of RNA of the present
invention is used to deprotect a ribozyme synthesized using a column format as
described
in (Scaringe et al., supra; Wicott et al., supra).
Inactive hammerhead ribozymes were synthesized by substituting a U for GS and
a U for A14 (numbering from Hertel, K. J., et al., 1992, Nucleic Acids Res.,
20, 3252).
The average stepwise coupling yields were >98% (Wincott et al., 1995 Nucleic
Acids Res. 23, 2677-2684).
Hairpin ribozymes are synthesized in two parts and annealed to reconstruct the
active ribozyme (Chowrira and Burke, 1992 Nucleic Acids Res., 20, 2835-2840).
Ribozymes are also synthesized from DNA templates using bacteriophage T7 RNA
polymerise (Milligan and Uhlenbeck, 1989, Methods Enzymol. 180, 51).
Ribozymes are modified to enhance stability and/or enhance catalytic activity
by
modification with nuclease resistant groups, for example, 2'-amino, 2'-C-
allyl, 2'-flouro,
2'-O-methyl, 2'-H, nucleotide base modifications (for a review see Usman and
Cedergren,
1992 TIBS 17, 34; Usman et al., 1994 Nucleic Acids Symp. Ser. 31, 163; Burgin
et al.,
1996 Biochemistry 6, 14090).
Ribozymes were purified by gel electrophoresis using general methods or are
purified by high pressure liquid chromatography (HPLC; See Stinchcomb et al.,
CA 02288640 1999-11-OS
WO 98/50530 34 PCT/US98/09249
International PCT Publication No. WO 95/23225, the totality of which is hereby
incorporated herein by reference) and are resuspended in water.
The sequences of the ribozyrnes that are chemically synthesized, useful in
this
study, are shown in Tables XII-XIX. Those in the art will recognize that these
sequences
are representative only of many more such sequences where the enzymatic
portion ~of the
ribozyme (all but the binding arms) is altered to affect activity. For
example, stem-loop II
sequence of hammerhead ribozymes can be altered (substitution, deletion,
and/or
insertion) to contain any sequences provided a minimum of two base-paired stem
structure can form. Similarly, stem-loop IV sequence of hairpin ribozymes, can
be
altered (substitution, deletion, and/or insertion) to contain any sequence,
provided a
minimum of two base-paired stem structure can form. Preferably, no more than
200
bases are inserted at these locations. The sequences listed in Tables XII-XIX
may be
formed of ribonucleotides or other nucleotides or non-nucleotides. Such
ribozymes
(which have enzymatic activity) are equivalent to the ribozymes described
specifically in
the Tables.
Nucleotide Triphosphates:
The use of modified nucleotide triphosphates would greatly assist in the
combinatorial chemistry. The synthesis of nucleoside triphosphates and their
incorporation into nucleic acids using polymerise enzymes has greatly assisted
in the
advancement of nucleic acid research. The polymerise enzyme utilizes
nucleoside
triphosphates as precursor molecules to assemble oligonucleotides. Each
nucleotide is
attached by a phosphodiester bond formed through nucleophilic attack by the 3'
hydroxyl
group of the oligonucleotide's last nucleotide onto the 5' triphosphate of the
next
nucleotide. Nucleotides are incorporated one at a time into the
oligonucleotide in a 5' to
3' direction. This process allows RNA to be produced and amplified from
virtually any
DNA or RNA templates.
Most natural polymerise enzymes incorporate standard nucleoside triphosphates
into nucleic acid. For example, a DNA polymerise incorporates dATP, dTTP,
dCTP, and
CA 02288640 1999-11-OS
WO 98/50530 35 PCTNS98/09249
dGTP into DNA and an RNA polymerise generally incorporates ATP, CTP, UTP, and
GTP into RNA. There are however, certain polymerises that are capable of
incorporating
non-standard nucleoside triphosphates into nucleic acids (Joyce, 1997, PNAS
94, 1619-
1622, Huang et al., Biochemistry 36, 8231-8242).
Before nucleosides can be incorporated into RNA transcripts using polyrnerase
enzymes they must first be converted into nucleoside triphosphates which can
be
recognized by these enzymes. Phosphorylation of unblocked nucleosides by
treatment
with POCl3 and trialkyl phosphates was shown to yield nucleoside 5'-
phosphorodichloridates (Yoshikawa et al., 1969, Bull. Chem. Soc. (Japan) 42,
3505).
Adenosine or 2'-deoxyadenosine 5'-triphosphate was synthesized by adding an
additional
' step consisting of treatment with excess tri-n-butylammonium pyrophosphate
in DMF
followed by hydrolysis (Ludwig, 1981, Acta Biochim. et Biophys. Acid Sci.
Hung. 16,
131-133).
Non-standard nucleoside triphosphates are not readily incorporated into RNA
transcripts by traditional RNA polymerises. Mutations have been introduced
into RNA
polymerise to facilitate incorporation of deoxyribonucleotides into RNA (Sousa
&
Padilla, 1995, EMBO J. 14,4609-4621 , Bonner et al., 1992, EMBO J. 11, 3767-
3775,
Bonner et al., 1994, J. Biol. Chem. 42, 25120-25128, Aurup et al., 1992,
Biochemistry 31,
9636-9641 ).
McGee et al., International PCT Publication No. WO 95/35102, describes the
incorporation of 2'-NHZ-NT'P's, 2'-F-NTP's, and 2'-deoxy-2'-benzyloxyamino UTP
into
RNA using bacteriophage T7 polymerise.
Wieczorek et al., 1994, Bioorganic & Medicinal Chemistry Letters 4, 987-994,
describes the incorporation of 7-deaza-adenosine triphosphate into an RNA
transcript
using bacteriophage T7 RNA polymerise.
- Lin et al., 1994, Nucleic Acids Research 22, 5229-5234, reports the
incorporation
of 2'-NHZ-CTP and 2'-NHz-UTP into RNA using bacteriophage T7 RNA polymerise
and
CA 02288640 1999-11-OS
WO 98/50530 36 PCT/US98/09249
polyethylene glycol containing buffer. The article describes the use of the
polymerase
synthesized RNA for in vitro selection of aptamers to human neutrophil
elastase (HNE).
The invention features NTP's having the formula triphosphate-OR, for example
the following formula I:
O O O
-O-P -O -P -O -P OR
O- O- O-
where R is any nucleoside; specifically the nucleosides 2'-O-methyl-2,6-
diaminopurine
riboside; 2'-deoxy-2'amino-2,6-diaminopurine riboside; 2'-(N-alanyl) amino-2'-
deoxy-
uridine; 2'-(N phenylalanyl)amino-2'-deoxy-uridine; 2'-deoxy -2'-(N (3-alanyl)
amino ;
2'-deoxy-2'-(lysiyl) amino uridine; 2'-C-allyl uridine; 2'-O-amino-uridine; 2'-
0-
methylthiomethyl adenosine; 2'-O-methylthiomethyl cytidine ; 2'-O-
methylthiomethyl
guanosine; 2'-O-methyIthiomethyl-uridine; 2'-Deoxy-2'-(N histidyl) amino
uridine; 2'-
deoxy-2'-amino-5-methyl cytidine; 2'-(N ~i-carboxamidine-(3-alanyl)amino-2'-
deoxy-
uridine; 2'-deoxy-2'-(N-(3-alanyl)-guanosine; and 2'-O-amino-adenosine.
In a second aspect, the invention features a process for the synthesis of
pyrimidine
nucleotide triphosphate (such as UTP, 2'-O-MTM-UTP, dUTP and the like)
including the
steps of monophosphorylation where the pyrimidine nucleoside is contacted with
a
mixture having a phosphorylating agent (such as phosphorus oxychloride,
phospho-tris-
triazolides, phospho-tris-triimidazolides and the like), trialkyl phosphate
(such as
triethylphosphate or trimethylphosphate or the like) and dimethylaminopyridine
(DMAP)
under conditions suitable for the formation of pyrimidine monophosphate; and
pyrophosphorylation where the pyrimidine monophosphate is contacted with a
pyrophosphorylating reagent (such as tributylammonium pyrophosphate) under
conditions suitable for the formation of pyrimidine triphosphates.
CA 02288640 1999-11-OS
WO 98/50530 3~ PCT/US98/09249
By "pyrimidines" is meant nucleotides comprising modified or unmodified
derivatives of a six membered pyrimidine ring. An example of a pyrimidine is
modified
or unmodified uridine.
By "nucleotide triphosphate" or "NTP" is meant a nucleoside bound to three
S inorganic phosphate groups at the 5' hydroxyl group of the modified or
unmodified ribose
or deoxyribose sugar where the 1' position of the sugar may comprise a nucleic
acid base
or hydrogen. The triphosphate portion may be modified to include chemical
moieties
which do not destroy the functionality of the group (i.e., allow incorporation
into an RNA
molecule).
In another preferred embodiment, nucleoside triphosphates (NTP's) of the
instant
invention are incorporated into an oligonucleotide using an RNA polymerase
enzyme.
RNA polymerases include but are not limited to mutated and wild type versions
of
bacteriophage T7, SP6, or T3 RNA polymerases.
In yet another preferred embodiment, the invention features a process for
1 S incorporating modified NTP's into an oligonucleotide including the step of
incubating a
mixture having a DNA template, RNA polymerase, NTP, and an enhancer of
modified
NTP incorporation under conditions suitable for the incorporation of the
modified NTP
into the oligonucleotide.
By "enhancer of modified NTP incorporation" is meant a reagent which
facilitates
the incorporation of modified nucleotides into a nucleic acid transcript by an
RNA
polymerase. Such reagents include but are not limited to methanol; LiCI;
polyethylene
glycol (PEG); diethyl ether; propanol; methyl amine; ethanol and the like.
In another preferred embodiment, the modified nucleoside triphosphates can be
incorporated by transcription into a nucleic acid molecules including
enzymatic nucleic
acid, antisense, 2-SA antisense chimera, oligonucleotides, triplex forming
oligonucleotide
(TFO), aptamers and the like (Stull et al., 1995 Pharmaceutical Res. 12, 465).
CA 02288640 1999-11-OS
WO 98/50530 38 PCT/US98/09249
By "antisense" it is meant a non-nucleic acid catalyst that binds to target
RNA by
means of RNA-RNA or RNA-DNA or RNA-PNA (protein nucleic acid; Egholm et al.,
1993 Nature 365, 566) interactions and alters the activity of the target RNA
(for a review
see Stein and Cheng, 1993 Science 261, 1004; Agrawal et al., U.S. Patent No.
5,591,721;
Agrawal, U.S. Patent No. 5,652,356).
By "2-SA antisense chimera" it is meant, an antisense oligonucleotide
containing a
S' phosphorylated 2'-5'-linked adenylate residues. These chimeras bind to
target RNA in a
sequence-specific manner and activate a cellular 2-SA-dependent ribonuclease
which, in
turn, cleaves the target RNA (Torrence et al., 1993 Proc. Natl. Acad. Sci. USA
90, 1300).
By "triplex forming oligonucleotides (TFO)" it is meant an oligonucleotide
that
can bind to a double-stranded DNA in a sequence-specific manner to form a
triple-strand
helix. Formation of such triple helix structure has been shown to inhibit
transcription of
the taxgeted gene (Duval-Valentin et al., 1992 Proc. Natl. Acad. Sci. USA 89,
504).
By "oligonucleotide" as used herein is meant a molecule having two or more
nucleotides. The polynucleotide can be single, double or multiple stranded and
may have
modified or unmodified nucleotides or non-nucleotides or various mixtures and
combinations thereof.
In yet another preferred embodiment, the modified nucleoside triphosphates of
the
instant invention can be used for combinatorial chemistry or in vitro
selection of nucleic
acid molecules with novel function. Modified oligonucleotides can be
enzymatically
synthesized to generate libraries for screening.
Nucleoside modifications of bases and sugars, have been discovered in a
variety of
naturally occurring RNA (e.g., tRNA, mRNA, rRNA; reviewed by Hall, 1971 The
Modified Nucleosides in Nucleic Acids, Columbia University Press, New York;
Limbach
et al., 1994 Nucleic Acids Res. 22, 2183). In an attempt to understand the
biological
significance, structural and thermodynamic properties, and nuclease resistance
of these
nucleoside modifications in nucleic acids, several investigators have
chemically
CA 02288640 1999-11-OS
WO 98/50530 39 PCT/US98/09249
synthesized nucleosides, nucleotides and phosphoramidites with various base
and sugar
modifications and incorporated them into oligonucleotides.
Uhlmann and Peyman, 1990, Chem. Reviews 90, 543, review the use of certain
nucleoside modifications to stabilize antisense oligonucleotides.
Usman et al., International PCT Publication Nos. WOl93/15187; and WO
95/13378; describe the use of sugar, base and backbone modifications to
enhance the
nuclease stability of nucleic acid catalysts.
Eckstein et al., International PCT Publication No. WO 92/07065 describe the
use
of sugar, base and backbone modifications to enhance the nuclease stability of
nucleic
acid catalysts.
Grasby et al., 1994, Proc. Indian Acad. Sci., 106, 1003, review the
"applications
of synthetic oligoribonucleotide analogues in studies of RNA structure and
function".
Eaton and Pieken, 1995, Annu. Rev. Biochem., 64, 837, review sugar, base and
backbone modifications that enhance the nuclease stability of RNA molecules.
Rosemeyer et al., 1991, Helvetica Chem. Acta, 74, 748, describe the synthesis
of
1-(2'-deoxy-~3-D-xylofuranosyl) thymine-containing oligodeoxynucleotides.
Seela et al., 1994, Helvetica Chem. Acta, 77, 883, describe the synthesis of 1-
(2'-
deoxy-j3-D-xylofuranosyl) cytosine-containing oligodeoxynucleotides.
Seela et al., 1996, Helvetica Chem. Acta, 79, 1451, describe the synthesis
xylose-
DNA containing the four natural bases.
In another preferred embodiment, catalytic activity of the molecules described
in
the instant invention can be optimized as described by Draper et al., supra.
The details
will not be repeated here, but include altering the length of the ribozyme
binding arms, or
chemically synthesizing ribozymes with modifications (base, sugar and/or
phosphate) that
CA 02288640 1999-11-OS
WO 98/50530 40 PCT/US98/09249
prevent their degradation by serum ribonucleases and/or enhance their
enzymatic activity
(see e.g., Eckstein et al., International Publication No. WO 92/07065;
Perrault et al., 1990
Nature 344, 565; Pieken et al., 1991 Science 253, 314; Usman and Cedergren,
1992
Trends in Biochem. Sci. 17, 334; Usman et al., International Publication No.
S WO 93/15187; and Rossi et al., International Publication No. WO 91/03162;
Sproat, US
Patent No. 5,334,711; and Burgin et al., supra; all of these describe various
chemical
modifications that can be made to the base, phosphate and/or sugar moieties of
enzymatic
RNA molecules). Modifications which enhance their efficacy in cells, and
removal of
bases from stem loop structures to shorten RNA synthesis times and reduce
chemical
requirements are desired. (All these publications are hereby incorporated by
reference
herein.).
By "enhanced enzymatic activity" is meant to include activity measured in
cells
andlor in vivo where the activity is a reflection of both catalytic activity
and ribozyme
stability. In this invention, the product of these properties is increased or
not significantly
1 S (less that 10 fold) decreased in vivo compared to an all RNA ribozyme.
In yet another preferred embodiment, nucleic acid catalysts having chemical
modifications which maintain or enhance enzymatic activity is provided. Such
nucleic
acid is also generally more resistant to nucleases than unmodified nucleic
acid. Thus, in a
cell and/or in vivo the activity may not be significantly lowered. As
exemplified herein
such ribozymes are useful in a cell and/or in vivo even if activity over all
is reduced 10
fold (Burgin et al., 1996, Biochemistry, 35, 14090). Such ribozymes herein are
said to
"maintain" the enzymatic activity on all RNA ribozyme.
In a most preferred embodiment the invention features a method of synthesizing
ribozyme libraries of various sizes. This invention describes methods to
chemically
synthesize ribozyme libraries of various sizes from suitable nucleoside
analogs.
Considerations for the selection of nucleotide building blocks and
determination
of coupling e~ciency: In addition to structural considerations (hydrogen bond
donors and
CA 02288640 1999-11-OS
WO 98/50530 41 PCT/US98/09249
acceptors, stacking properties, pucker orientation of sugars, hydrophobicity
or
hydrophilicity of some subgroups constitutive of the nucleotides) that may
lead to the
selection of a specific nucleotide to be included in the design of a ribozyme
library, one of
., the important features that needs to be considered when selecting
nucleotide building
blocks is the chemical compatibility of such building blocks with ribozyme
synthesis. A
"nucleotide building block" is a nucleoside or nucleoside analog that possess
a suitably
protected phosphorus atom at the oxidation state V reacting readily, upon
activation, to
give a P~-containing internucleoside linkage. A suitable nucleoside building
block may
also contain a phosphorus atom at the oxidation state III reacting readily,
upon activation,
to give a P"'-containing internucleoside linkage that can be oxidized to the
desired P''-
containing internucleoside linkage. Applicant has found that the
phosphoramidite
'chemistry (P"') is a preferred coupling method for ribozyme library
synthesis. There are
several other considerations while designing and synthesizing certain ribozyme
libraries,
such as: a) the coupling efficiencies of the nucleotide building blocks
considered for a
ribozyme library should not fall below 90% to provide a majority of full-
length ribozyrne;
b) the nucleotide building blocks should be chemically stable to the selected
synthesis and
deprotection conditions of the particular ribozyme library; c) the
deprotection schemes for
the nucleotide building blocks incorporated into a ribozyme library, should be
relatively
similar and be fully compatible with ribozyme deprotection protocols. In
particular,
nucleoside building blocks requiring extended deprotection or that cannot
sustain harsh
treatment should be avoided in the synthesis of a ribozyme library. Typically,
the
reactivity of the nucleotide building blocks should be optimum when diluted to
100 mM
to 200 mM in non-protic and relatively polar solvent. Also the deprotection
condition
using 3:1 mixture of ethanol and concentrated aqueous ammonia at 65 degrees C.
for 4
hours followed by a fluoride treatment as exemplified in Wincott et al. supra,
is
particularly useful for ribozyme synthesis and is a preferred deprotection
pathway for
such nucleotide building blocks.
In one preferred embodiment, a "nucleotide building block mixing" approach to
generate ribozyme libraries is described. This method involves mixing various
nucleotide
CA 02288640 1999-11-OS
WO 98/50530 42 PCT/I1S98/09249
building blocks together in proportions necessary to ensure equal
representation of each
of the nucleotide building blocks in the mixture. This mixture is incorporated
into the
ribozyme at positions) selected for randomization.
The nucleotide building blocks selected for incorporation into a ribozyme
library,
are typically mixed together in appropriate concentrations, in reagents, such
as anhydrous
acetonitrile, to form a mixture with a desired phosphoramidite concentration.
This
approach for combinatorial synthesis of a ribozyme library with one or more
random
positions within the ribozyme (X as described above) is particularly useful
since a
standard DNA synthesizer can handle a building block mixture similar to a
building block
solution containing a single building block. Such a nucleotide building block
mixture is
coupled to a solid support or to a growing ribozyme sequence attached to a
solid-support.
To ensure that the ribozyme library synthesized achieves the desired
complexity, the scale
of the synthesis is increased substantially above that of the total complexity
of the library.
For example, a 2.5 pmole ribozyme synthesis provides ~ 3x10" ribozyme
molecules
corresponding to sub-nanomolar amounts of each member of a billion compounds
ribozyme library.
Divinylbenzene highly cross-linked polystyrene solid-support constitutes the
preferred stationary phase for ribozyme library synthesis. However, other
solid-support
systems utilized in DNA or RNA synthesis can also be used for ribozyme library
synthesis. This includes silica-based solid-supports such as controlled-pore
glass (CPG)
or polymeric solid-supports such as all types of derivatized polystyrene
resins, grafted
polymers of chloromethylated polystyrene crosslinked with ethylene glycol,
oligoethylene
glycol.
Because of different coupling kinetics of the nucleotide building blocks
present in
a mixture, it is necessary to evaluate the relative incorporation of each of
the members of
the mixture and to adjust, if needed, the relative concentration of the
building blocks in
the mixture to get equimolar representation, compensating thereby the kinetic
parameter.
Typically a building block that presents a slow coupling kinetic will be over-
represented
CA 02288640 1999-11-OS
WO 98/50530 43 PCT/US98/09249
in the mixture and vice versa for a building block that presents a fast
coupling kinetic.
When equimolar incorporation is sought, acceptable limits for unequal
incorporation may
generally be +/-10%.
Synthesis of a random ribozyme library can be performed either with the
mixture
of desired nucleotide building blocks, or with a combination of certain random
positions
(obtained by using one or more building block mixtures) and one or more fixed
positions
that can be introduced through the incorporation of a single nucleotide
building block
reagent. For instance, in the oligonucleotide model S'-TT XXXX TTB-3' used in
example 2 infra, the positions from 3'-end 1 is fixed as 2'-deoxy-inverted
abasic ribose
(B), positions 2, 3, 8 and 9 have been fixed as 2'-deoxy-thymidine (T) while
the X
positions 4-7 correspond to an approximately equimolar distribution of all the
nucleotide
building blocks that make up the X mixture.
In another preferred embodiment, a "mix and split" approach to generate
ribozyme
libraries is described. This method is particularly useful when the number of
selected
1 S nucleotide building blocks to be included in the library is large and
diverse (greater than 5
nucleotide building blocks) and/or when the coupling kinetics of the selected
nucleotide
building blocks do not allow competitive coupling even after relative
concentration
adjustments and optimization. This method involves a mufti-step process
wherein the
solid support used for ribozyme library synthesis is "split" (divided) into
equal portions,
(the number of portions is equal to the number of different nucleotide
building blocks (n)
chosen for incorporation at one or more random positions within the ribozyme).
For
example, if there are 10 different nucleotide building blocks chosen for
incorporation at
one or more positions in the ribozyme library, then the solid support is
divided into 10
different portions. Each portion is independently coupled to one of the
selected
nucleotide building blocks followed by mixing of all the portions of solid
support. The
ribozyme synthesis is then resumed as before the division of the building
blocks. This
enables the synthesis of a ribozyme library wherein one or more positions
within the
ribozyme is random. The number of "splitting" and "mixing" steps is dependent
on the
CA 02288640 1999-11-OS
WO 98/50530 44 PCT/US98/09249
number of positions that are random within the ribozyme. For example if three
positions
are desired to be random then three different splitting and mixing steps are
necessary to
synthesize the ribozyme library.
Random ribozyme libraries are synthesized using a non-competitive coupling
procedure where each of the selected nucleotide analogs "n" separately couple
.to an
inverse "n" (1/n) number of aliquots of solid-support or of a growing ribozyme
chain on
the solid-support. A very convenient way to verify completeness of the
coupling reaction
is the use of a standard spectrophotometric DMT assay (Oligonucleotide
Synthesis, A
Practical Approach, ed. M. Gait, pp 48, IRC Press, Oxford, UK; incorporated by
reference
herein). These aliquots may be subsequently combined, mixed and split into one
new
aliquot. A similar approach to making oligonucleotide libraries has recently
been
described by Cook et al., (US Patent No. 5,587,471) and is incorporated by
reference
herein.
Nucleotide Svnthesis
Addition of dimethylaminopyridine (DMAP) to the phosphorylation protocols
known in the art can greatly increase the yield of nucleoside monophosphates
while
decreasing the reaction time (Fig. 29). Synthesis of the nucleosides of the
invention have
been described in several publications and Applicants previous applications
(Beigelman et
al., International PCT publication No. WO 96/18736; Dudzcy et al., Int. PCT
Pub. No.
WO 95/11910; Usman et al., Int. PCT Pub. No. WO 95/13378; Matulic-Adamic et
al.,
1997, Tetrahedron Lett. 38, 203; Matulic-Adamic et al., 1997, Tetrahedron
Lett. 38,
1669; all of which are incorporated herein by reference). These nucleosides
are dissolved
in triethyl phosphate and chilled in an ice bath. Phosphorus oxychloride
(POC13) is then
added followed by the introduction of DMAP. The reaction is then warmed to
room
temperature and allowed to proceed for 5 hours. This reaction allows the
formation of
nucleoside monophosphates which can then be used in the formation of
nucleoside
triphosphates. Tributylamine is added followed by the addition of anhydrous
acetonitrile
and tributylammonium pyrophosphate. The reaction is then quenched with TEAB
and
CA 02288640 1999-11-OS
WO 98/50530 45 PCT/US98/09249
stirred overnight at room temperature (about 20C). The triphosphate is
purified using
column purification and HPLC and the chemical structure is confirmed using NMR
analysis. Those skilled in the art will recognize that the reagents,
temperatures of the
reaction, and purif cation methods can easily be alternated with substitutes
and
S equivalents and still obtain the desired product.
The invention provides nucleoside triphosphates which can be used for a number
of different functions. The nucleoside triphosphates formed from nucleosides
found in
table III are unique and distinct from other nucleoside triphosphates known in
the art.
Incorporation of modified nucleotides into DNA or RNA oligonucleotides can
alter the
properties of the molecule. For example, modified nucleotides can hinder
binding of
nucleases, thus increasing the chemical half life of the molecule. This is
especially
important if the molecule is to be used for cell culture or in vivo. It is
known in the art
that the introduction of modified nucleotides into these molecules can greatly
increase the
stability and thereby the effectiveness of the molecules (Burgin et al., 1996,
Biochemistry
35, 14090-14097; Usman et al., 1996, Curr. Opin. Struct. Biol. 6, 527-533).
Modified nucleotides are incorporated using either wild type and mutant
polymerises. For example, mutant T7 polymerise is used in the presence of
modified
nucleotide triphosphate(s), DNA template and suitable buffers. Those skilled
in the art
will recognize that other polymerises and their respective mutant versions can
also be
utilized for the incorporation of NTP's of the invention. Nucleic acid
transcripts were
detected by incorporating radiolabelled nucleotides (a 'ZP NTP). The
radiolabeled NTP
contained the same base as the modified triphosphate being tested. The effects
of
methanol, PEG and LiCI were tested by adding these compounds independently or
in
combination. Detection and quantitation of the nucleic acid transcripts was
performed
using a Molecular Dynamics PhosphorImager. Efficiency of transcription was
assessed
by comparing modified nucleotide triphosphate incorporation with all-
ribonucleotide
incorporation control. Wild type polymerise was used to incorporate NTP's
using the
manufacturers buffers and instructions (Boehringer Mannheim).
CA 02288640 1999-11-OS
WO 98/50530 46 PCT/US98/09249
Transcription Conditions
Incorporation rates of modified nucleoside triphosphates into oligonucleotides
can
be increased by adding to traditional buffer conditions, several different
enhancers of
modified NTP incorporation. Applicant has utilized methanol and LiCI in an
attempt to
increase incorporation rates of dNTP using RNA polymerase. These enhancers of
modified NTP incorporation can be used in different combinations and ratios to
optimize
transcription. Optimal reaction conditions differ between nucleoside
triphosphates and
can readily be determined by standard experimentation. Overall however,
inclusion of
enhancers of modified NTP incorporation such as methanol or inorganic compound
such
as lithium chloride, have been shown by the applicant to increase the mean
transcription
rates.
Administration of Nucleoside mono, di or triphosphates
The nucleotide monophosphates, diphosphates, or triphosphates can be used as a
therapeutic agent either independently or in combination with other
pharmaceutical
1 S components. These molecules of the inventions can be administered to
patients using the
methods of Sullivan et al., PCT WO 94/02595. Molecules of the invention may be
administered to cells by a variety of methods known to those familiar to the
art, including,
but not restricted to, encapsulation in liposomes, by iontophoresis, or by
incorporation
into other vehicles, such as hydrogels, cyciodextrins, biodegradable
nanocapsules, and
bioadhesive microspheres. For some indications, ribozymes may be directly
delivered ex
vivo to cells or tissues with or without the aforementioned vehicles.
Alternatively, the
modified nucleotide triphosphate, diphosphate or monophosphate/vehicle
combination is
locally delivered by direct injection or by use of a catheter, infusion pump
or stmt. Other
routes of delivery include, but are not limited to, intravascular,
intramuscular,
subcutaneous or joint injection, aerosol inhalation, oral (tablet or pill
form), topical,
systemic, ocular, intraperitoneal and/or intrathecal delivery. More detailed
descriptions of
delivery and administration are provided in Sullivan et al., supra and Draper
et al., PCT
W093/23569 which have been incorporated by reference herein.
CA 02288640 1999-11-OS
WO 98/50530 4,~ PC'T/US98/09249
This invention further relates to a compound having the Formula II:
R20
O
X
R~
wherein, R, is OH, O-R3, where R3 is independently a moiety selected
from a group consisting of alkyl, alkenyl, alkynyl, aryl, alkylaryl,
carbocyclic aryl,
heterocyclic aryl, amide and ester; C-R3, where R3 is independently a moiety
selected
from a group consisting of alkyl, alkenyl, alkynyl, aryl, alkylaryl,
carbocyclic aryl,
heterocyclic aryl, amide and ester; halo, NHR4 (R4=alkyl (C1-22), acyl (C1-
22),
substituted or unsubstituted aryl), or OCH2SCH3 (methylthiomethyl), ONHRS
where RS
is independently H, aminoacyl group, peptidyl group, biotinyl group,
cholesteryl group,
lipoic acid residue, retinoic acid residue, folic acid residue, ascorbic acid
residue,
nicotinic acid residue, 6-aminopenicillanic acid residue, 7-
aminocephalosporanic acid
residue, alkyl, alkenyl, alkynyl, aryl, alkylaryl, carbocyclic aryl,
heterocyclic aryl, amide
or ester, ON=R6, where R6 is independently pyridoxal residue, pyridoxal-S-
phosphate
residue, 13-cis-retinal residue, 9-cis-retinal residue, alkyl, alkenyl ,
alkynyl, alkylaryl,
carbocyclic alkylaryl, or heterocyclic alkylaryl; B is independently a
nucleotide base or its
analog or hydrogen; X is independently a phosphorus-containing group; and RZ
is
independently blocking group or a phosphorus-containing group.
Specifically, an "alkyl" group refers to a saturated aliphatic hydrocarbon,
including straight-chain, branched-chain, and cyclic alkyl groups. Preferably,
the alkyl
group has I to 12 carbons. More preferably it is a lower alkyl of from 1 to 7
carbons,
more preferably 1 to 4 carbons. The alkyl group may be substituted or
unsubstituted.
When substituted the substituted groups) is preferably, hydroxy, cyano,
alkoxy, N02 or
N(CH3)2, amino, or SH.
The term "alkenyl" group refers to unsaturated hydrocarbon groups containing
at
least one carbon-carbon double bond, including straight-chain, branched-chain,
and cyclic
CA 02288640 1999-11-OS
WO 98/50530 48 PCT/US98/09249
groups. Preferably, the alkenyl group has 1 to 12 carbons. More preferably it
is a lower
alkenyl of from 1 to 7 carbons, more preferably 1 to 4 carbons. The alkenyl
group may be
substituted or unsubstituted. When substituted the substituted groups) is
preferably,
hydroxyl, cyano, alkoxy, N02, halogen, N(CH3)2, amino, or SH.
The term "alkynyl" refers to an unsaturated hydrocarbon group containing at
least
one carbon-carbon triple bond, including straight-chain, branched-chain, and
cyclic
groups. Preferably, the alkynyl group has 1 to 12 carbons. More preferably it
is a lower
alkynyl of from 1 to 7 carbons, more preferably 1 to 4 carbons. The alkynyl
group may
be substituted or unsubstituted. When substituted the substituted groups) is
preferably,
hydroxyl, cyano, alkoxy, =O, =S, N02 or N(CH3)2, amino or SH.
An "aryl" group refers to an aromatic group which has at least one ring having
a
conjugated p electron system and includes carbocyclic aryl, heterocyclic aryl
and biaryl
groups, all of which may be optionally substituted. The preferred
substituent(s) on aryl
groups are halogen, trihalomethyl, hydroxyl, SH, cyano, alkoxy, alkyl,
alkenyl, alkynyl,
and amino groups.
An "alkylaryl" group refers to an alkyl group (as described above) covalently
joined to an aryl group (as described above).
"Carbocyclic aryl" groups are groups wherein the ring atoms on the aromatic
ring
are all carbon atoms. The carbon atoms are optionally substituted.
"Heterocyclic aryl" groups are groups having from 1 to 3 heteroatoms as ring
atoms in the aromatic ring and the remainder of the ring atoms are carbon
atoms. Suitable
heteroatoms include oxygen, sulfur, and nitrogen, and include furanyl,
thienyl, pyridyl,
pyrrolyl, pyrrolo, pyrimidyl, pyrazinyl, imidazolyl and the like, all
optionally substituted.
An "amide" refers to an -C(O)-NH-R, where R is either alkyl, aryl, alkylaryl
or
hydrogen.
CA 02288640 1999-11-OS
WO 98/50530 49 PCTNS98/09249
An "ester" refers to an -C(O)-OR', where R is either alkyl, aryl, or
alkylaryl.
A "blocking group" is a group which is able to be removed after polynucleotide
synthesis and/or which is compatible with solid phase polynucleotide
synthesis.
A "phosphorus containing group" can include phosphorus in forms such as
dithioates, phosphoramidites and/or as part of an oligonucleotide.
In a preferred embodiment, the invention features a process for synthesis of
the
compounds of formula II.
In a preferred embodiment the invention features a process for the synthesis
of a
xylofuranosyl nucleoside phosphoramidite comprising the steps of a) oxidation
of a 2'
and 5'-protected ribonucleoside with a an oxidant such as chromium
oxide/pyridine/aceticanhydride, dimethylsulfoxide/aceticanhydride, or Dess-
Martin
reagent (periodinane) followed by reduction with a reducing agent such as,
triacetoxy
sodium borohydride, sodium borohydride, or lithium borohydride, under
conditions
suitable for the formation of 2' and 5'-protected xylofuranosyl nucleoside; b)
1 S phosphitylation under conditions suitable for the formation of
xylofuranosyl nucleoside
phosphoramidite.
In yet another preferred embodiment, the invention features the incorporation
of
the compounds of Formula II into polynucleotides. These compounds can be
incorporated into polynucleotides enzymatically. For example by using
bacteriophage T7
RNA polymerase, these novel nucleotide analogs can be incorporated into RNA at
one or
more positions (Milligan et al., 1989, Methods Enrymol., 180, 51 ).
Alternatively, novel
nucleoside analogs can be incorporated into polynucleotides using solid phase
synthesis
(Brown and Brown, 1991, in Oligonucleotides and Analogues: A Practical
Approach, p.
1, ed. F. Eckstein, Oxford University Press, New York; Wincott et al., 1995,
Nucleic
Acids Res., 23, 2677; Beaucage & Caruthers, 1996, in Bioorganic Chemistry:
Nucleic
- Acids, p 36, ed. S. M. Hecht, Oxford University Press, New York).
CA 02288640 1999-11-OS
WO 98/50530 50 PCT/US98/09249
The compounds of Formula II can be used for chemical synthesis of nucleotide-
tri-phosphates and/or phosphoramidites as building blocks for selective
incorporation into
oligonucleotides. These oligonucleotides can be used as an antisense molecule,
2-SA
antisense chimera, triplex forming oligonucleotides (TFO) or as an nucleic
acid catalyst.
The oligonucleotides can also be used as probes or primers for synthesis
and/or
sequencing of RNA or DNA.
The compounds of the instant invention can be readily converted into
nucleotide
diphosphate and nucleotide triphosphates using standard protocols (for a
review see
Hutchinson, 1991, in Chemistty of Nucleosides and Nucleotides, v.2, pp 81-160,
Ed. L. B.
Townsend, Plenum Press, New York, USA; incorporated by reference herein).
The compounds of Formula II can also be independently or in combination used
as an antiviral, anticancer or an antitumor agent. These compounds can also be
independently or in combination used with other antiviral, anticancer or an
antitumor
agents.
In one of the preferred embodiments of the inventions herein, the nucleic acid
catalyst is formed in a hammerhead or hairpin motif, but may also be formed in
the motif
of a hepatitis d virus, group I intron, group II intron or RNaseP RNA (in
association with
an RNA guide sequence) or Neurospora VS RNA. Examples of such hammerhead
motifs
are described by Dreyfus, supra, Rossi et al., 1992, AIDS Research and Human
Retroviruses 8, 183; of hairpin motifs by Hampel et al., EP0360257, Harnpel
and Tritz,
1989 Biochemistry 28, 4929, Feldstein et al., 1989, Gene 82, 53, Haseloff and
Gerlach,
1989, Gene, 82, 43, and Hampel et al., 1990 Nucleic Acids Res. 18, 299; of the
hepatitis d
virus motif is described by Perrotta and Been, 1992 Biochemistry 31, 16; of
the RNaseP
motif by Guerrier-Takada et al., 1983 Cell 35, 849; Forster and Altman, 1990,
Science
249, 783; Li and Altman, 1996, Nucleic Acids Res. 24, 835; Neurospora VS RNA
ribozyme motif is described by Collins (Saville and Collins, 1990 Cell 61, 685-
696;
Saville and Collins, 1991 Proc. Natl. Acad. Sci. USA 88, 8826-8830; Collins
and Olive,
1993 Biochemistry 32, 2795-2799; Guo and Collins, 1995, EMBO. J. 14, 363);
Group II
CA 02288640 1999-11-OS
WO 98/50530 51 PCT/US98/09249
introns are described by Griffin et al., 1995, Chem. Biol. 2, 761; Michels and
Pyle, 1995,
Biochemistry 34, 2965; Pyle et al., International PCT Publication No. WO
96/22689; and
of the Group I intron by Cech et al., U.S. Patent 4,987,071. These specific
motifs are not
limiting in the invention and those skilled in the art will recognize that all
that is
important in a nucleic acid catalyst of this invention is that it has a
specific substrate
binding site which is complementary to one or more of the target gene RNA
regions, and
that it have nucleotide sequences within or surrounding that substrate binding
site which
impart an RNA cleaving activity to the molecule.
In a preferred embodiment, a polynucleotide of the invention would bear one or
more 2'-hydroxylamino functionalities attached directly to the monomeric unit
or through
the use of an appropriate spacer. Since oligonucleotides have neither aldehyde
nor
hydroxylamino groups, the formation of an oxime would occur selectively using
oligo as
a polymeric template. This approach would facilitate the attachment of
practically any
molecule of interest (peptides, polyamines, coenzymes, oligosaccharides,
lipids, etc.)
directly to the oligonucleotide using either aldehyde or carboxylic function
in the
molecule of interest.
Scheme 1. Post synthetic Oxime Bond Formation
H
OLIGO ONH2 + H-C-R ~ OLIGO ~--p-N=C-R
CA 02288640 1999-11-OS
WO 98/50530 52 PCT/US98/09249
Scheme 2. Chemical Ligation of Oligonucleotides
Oligol ONH 2 + H-C-(CH 2)~ C~-1 + HZNO Olig02
ll
Oligo1 -N=C-(CH 2)~ C=N-O Olig02
Advantages of oxime bond formation:
~ The oximation reaction proceeds in water
~ Quantitative yields
~ Hydrolytic stability in a wide pH range (S - 8)
~ The amphoteric nature of oximes allows them to act either as
weak acids or weak bases.
~ Oximes exhibit a great tendency to complex with metal ions
In yet another preferred embodiment, the aminooxy "tether" in
oligonucleotides,
such as a ribozyme, is reacted with different compounds bearing carboxylic
groups (e.g.,
aminoacids, peptides, "cap" structures ,etc.) resulting in the formation of
oxyamides as
shown below.
CA 02288640 1999-11-OS
WO 98/50530 53 PCT/US98/09249
Scheme 3. Post synthetic oxyamide bond formation
OLIGO ONH 2 + HO-~c~-R ~ SLIGO p-N ~-R
Target Discover
Applicant has developed an efficient and rapid method for screening libraries
of
catalytic nucleic acid molecules capable of performing a desired function in a
cell. The
invention also features the use of a catalytic nucleic acid library to
modulate certain
attributes or processes in a biological system, such as a mammalian cell, and
to identify
and isolate a) nucleic acid catalysts from the library involved in modulating
the cellular
process/attribute of interest; and b) modulators of the desired cellular
process/attribute
using the sequence of the nucleic acid catalyst.
More specifically, the method of the instant invention involves designing and
constructing a catalytic nucleic acid library, where the catalytic nucleic
acid includes a
catalytic and a substrate binding domain, and the substrate binding domain
(arms) are
1 S randomized. This library of catalytic nucleic acid molecules with
randomized binding
arms) are used to modulate certain processes/attributes in a biological
system. The
method described in this application involves simultaneous screening of a
library or pool
of catalytic nucleic acid molecules with various substitutions at one or more
positions and
selecting for ribozymes with desired function or characteristics or
attributes. This
invention also features a method for constructing and selecting for catalytic
nucleic acid
molecules for their ability to cleave a given target nucleic acid molecule or
an unknown
target nucleic acid molecule (e.g., RNA), and to inhibit the biological
function of that
target molecule or any protein encoded by it.
It is not necessary to know either the sequence or the structure of the target
nucleic
acid molecule in order to select for catalytic nucleic acid molecules capable
of cleaving
the target in this cellular system. The cell-based screening protocol
described in the
CA 02288640 1999-11-OS
WO 98/50530 54 PCT/US98/09249
instant invention (i.e., one which takes place inside a cell) offers many
advantages over
extracellular systems, because the synthesis of large quantities of RNA by
enzymatic or
chemical methods prior to assessing the efficacy of the catalytic nucleic acid
molecules is
not necessary. The invention further describes a rapid method of using
catalytic nucleic
acid molecule libraries to identify the biological function of a gene sequence
inside a cell.
Applicant describes a method of using catalytic nucleic acid molecule
libraries to identify
a nucleic acid molecule, such as a gene, involved in a biological process;
this nucleic acid
molecule may be a known molecule with a known function, or a known molecule
with a
previously undefined function or an entirely novel molecule. This is a rapid
means for
identifying, for example, genes involved in a cellular pathway, such as cell
proliferation,
cell migration, cell death, and others. This method of gene discovery is not
only a novel
approach to studying a desired biological process but also a means to identify
active
reagents that can modulate this cellular process in a precise manner.
Applicant describes herein, a general approach for simultaneously assaying the
ability of one or more members of a catalytic nucleic acid molecule library to
modulate
certain attributes/process(es) in a biological system, such as plants, animals
or bacteria,
involving introduction of the library into a desired cell and assaying for
changes in a
specific "attribute," "characteristic" or "process." The specific attributes
may include cell
proliferation, cell survival, cell death, cell migration, angiogenesis, tumor
volume, tumor
metastasis, levels of a specific mRNA(s) in a cell, levels of a specific
proteins) in a cell,
levels of a specific protein secreted, cell surface markers, cell morphology,
cell
differentiation pattern, cartilage degradation, transplantation, restenosis,
viral replication,
viral load, and the like. By modulating a specific biological pathway using a
catalytic
nucleic acid molecule library, it is possible to identify the genes) involved
in that
pathway, which may lead to the discovery of novel genes, or genes with novel
function.
This method provides a powerful tool to study gene function inside a cell.
This approach
also offers the potential for designing novel catalytic oligonucleotides,
identifying
ribozyrne accessible sites within a target, and for identifying new nucleic
acid targets for
ribozyme-mediated modulation of gene expression.
CA 02288640 1999-11-OS
WO 98!50530 55 PCT/US98/09249
In another aspect the invention involves synthesizing a Random Binding Arm
Nucleic Acid Catalyst Library (Random Library) and simultaneously testing all
members
of the Random Library in cells. This library has ribozymes with random
substrate
binding arms) and a defined catalytic domain. Cells with an altered attribute
(such as
S inhibition of cell proliferation) as a result of interaction with the
members of the Random
Library are selected and the sequences of the ribozymes from these cells are
determined.
Sequence information from the binding arms) of these ribozymes can be used to
isolate
nucleic acid molecules that are likely to be involved in the pathway
responsible for the
desired cellular attribute using standard technology known in the art, e.g.,
nucleic acid
amplification using techniques such as polymerase chain reaction (PCR). This
method is
a powerful means to isolate new genes or genes with new function.
By "Random Library" as used herein is meant ribozyme libraries comprising all
possible variants in the binding arm (s) of a given ribozyme motif. Here the
complexity
and the content of the library is not defined. The Random Library is expected
to comprise
sequences complementary to every potential target sequence, for the ribozyme
motif
chosen, in the genome of an organism. This Random Library can be used to
screen for
ribozyme cleavage sites in a known target sequence or in a unknown target. In
the first
instance, the Random Library is introduced into the cell of choice and the
expression of
the known target gene is assayed. Cells with an altered expression of the
target will yield
the most effective ribozyme against the known target. In the second instance,
the
Random Library is introduced into the cell of choice and the cells are assayed
for a
specific attribute, for example, survival of cells. Cells that survive the
interaction with the
Random Library are isolated and the ribozyme sequence from these cells is
determined.
The sequence of the binding arm of the ribozyme can then be used as probes to
isolate the
genes) involved in cell death. Because, the ribozyme(s) from the Random
Library is able
to modulate (e.g., down regulate) the expression of the genes) involved in
cell death, the
cells are able to survive under conditions where they would have otherwise
died. This is
a novel method of gene discovery. This approach not only provides the
information about
mediators of certain cellular processes, but also provides a means to modulate
the
CA 02288640 1999-11-05
WO 98/50530 56 PCT/US98/09249
expression of these modulators. This method can be used to identify modulators
of any
cell process in any organism, including but not limited to mammals, plants and
bacteria.
The invention provides a method for producing a class of enzymatic cleaving
agents which exhibit a high degree of specificity for the nucleic acid
sequence of a desired
target. The nucleic acid catalyst is preferably targeted to a highly conserved
sequence
region of a target such that specific diagnosis and/or treatment of a disease
or condition
can be provided with a single enzymatic nucleic acid.
In a first aspect the invention features a method for identifying one or more
nucleic acid molecules, such as gene(s), involved in a process (such as, cell
growth,
proliferation, apoptosis, morphology, angiogenesis, differentiation,
migration, viral
multiplication, drug resistance, signal transduction, cell cycle regulation,
temperature
sensitivity, chemical sensitivity and others) in a biological system, such as
a cell. The
method involves the steps of a) providing a random library of nucleic acid
catalysts, with
a substrate binding domain and a catalytic domain, where the substrate binding
domain
has a random sequence, to the biological system under conditions suitable for
the process
to be altered; b) identifying any nucleic acid catalyst present in that
biological system
where the process has been altered by any nucleic acid catalyst; and c)
determining the
nucleotide sequence of at least a portion of the binding arm of such a nucleic
acid catalyst
to allow identification of the nucleic acid molecule involved in the process
in that
biological system.
In a related aspect the invention features a method for identification of a
nucleic
acid molecule capable of modulating a process in a biological system. The
method
includes: a) introducing a library of nucleic acid catalysts with a substrate
binding domain
and a catalytic domain, where the substrate binding domain has a random
sequence, into
the biological system under conditions suitable for modulating the process;
and b)
determining the nucleotide sequence of at least a portion of the substrate
binding domain
of any nucleic acid catalyst from a biological system where the process has
been
CA 02288640 1999-11-OS
WO 98/50530 5,~ PCT/US98109249
modulated to allow said identification of the nucleic acid molecule capable of
modulating
said process in that biological system.
In a second aspect, the invention the invention further concerns a method for
identification of a nucleic acid catalyst capable of modulating a process in a
biological
S system. This involves: a) introducing a library of nucleic acid catalysts
with a substrate
binding domain and a catalytic domain, where the substrate binding domain has
a random
sequence, into the biological system under conditions suitable for modulating
the process;
and b) identifying any nucleic acid catalyst from a biological system where
the process
has been modulated.
By "enzymatic portion" or "catalytic domain" is meant that portion/region of
the
ribozyme essential for cleavage of a nucleic acid substrate (for example see
Figure 3).
By "nucleic acid molecule" as used herein is meant a molecule having
nucleotides. The nucleic acid can be single, double or multiple stranded and
may
comprise modified or unmodified nucleotides or non-nucleotides or various
mixtures and
combinations thereof. An example of a nucleic acid molecule according to the
invention
is a gene which encodes for macromolecule such as a protein.
The "biological system" as used herein may be a eukaryotic system or a
prokaryotic system, may be a bacterial cell, plant cell or a mammalian cell,
or may be of
plant origin, mammalian origin, yeast origin, Drosophila origin, or
archebacterial origin.
This invention further relates to novel nucleic acid molecules with catalytic
activity, which are particularly useful for cleavage of RNA or DNA. The
nucleic acid
catalysts of the instant invention are distinct from other nucleic acid
catalysts known in
the art. This invention also relates to a method of screening variants of
nucleic acid
catalysts using standard nucleotides or modified nucleotides. Applicant has
determined
an efficient method for screening libraries of catalytic nucleic acid
molecules, particularly
- those with chemical modifications at one or more positions. The method
described in this
application involves systematic screening of a library or pool of ribozymes
with various
CA 02288640 1999-11-OS
WO 98/50530 5g PCT/US98/09249
substitutions at one or more positions and selecting for ribozymes with
desired function or
characteristic or attribute.
In one preferred embodiment, a method for identifying a nucleic acid molecule
involved in a process in a cell is described, including the steps of: a)
synthesizing a library
of nucleic acid catalysts, having a substrate binding domain and a catalytic
domain, where
the substrate binding domain has a random sequence; b) testing the library in
the cell
under conditions suitable to cause the process in the cell to be altered (such
as: inhibition
of cell proliferation, inhibition of angiogenesis, modulation of growth and
/or
differentiation, and others); c) isolating and enriching the cell with the
altered process; d)
identifying and isolating the nucleic acid catalyst in the altered cell; e)
using an
oligonucleotide, having the sequence homologous to the sequence of the
substrate binding
domain of the nucleic acid catalyst isolated from the altered cell, as a probe
to isolate the
nucleic acid molecule from the cell or the altered cell. Those nucleic acid
molecules
identified using the selection/screening method described above are likely to
be involved
I S in the process that was being assayed for alteration by the members) of
the ribozyme
library. These nucleic acid molecules may be new gene sequences, or known gene
sequences, with a novel function. One of the advantages of this method is that
nucleic
acid sequences, such as genes, involved in a biological process, such as
differentiation,
cell growth, disease processes including cancer, tumor angiogenesis,
arthritis,
cardiovascular disease, inflammation, restenosis, vascular disease and the
like, can be
readily identified using the Random Library approach. Thus theoretically, one
Random
Library for a given ribozyme motif can be used to assay any process in any
biological
system.
In another preferred embodiment the invention involves synthesizing a Defined
Arm Nucleic Acid Catalyst Library (Defined Library) and simultaneously testing
it
against known targets in a cell. The library includes ribozymes with binding
arms) of
known complexity (Defined) and a defined catalytic domain. Modulation of
expression
of the target gene by ribozymes in the library will cause the cells to have an
altered
CA 02288640 1999-11-OS
WO 98/50530 59 PCT/US98/09249
phenotype. Such cells are isolated and the ribozymes in these cells are the
ones most
suited for modulating the expression of the desired gene in the cell.
By "Defined Library" as used herein is meant a library of nucleic acid
catalysts,
wherein each member nucleic acid catalyst is designed and produced
independently, then
added to the library. Thus, the content, complexity (number of different
ribozyrnes
contained in the library) and ratios of library members are defined at the
outset. Defined
Library comprises > 2 ribozymes. The process involves screening the sequence
of the
known target RNA for all possible sites that can be cleaved by a given
ribozyme motif
and as described, for example in McSwiggen, US Patent No. 5,525,468,
incorporated by
reference herein. Synthesizing a representative number of different ribozymes
aginst the
target sequence. Combining the ribozyrnes and introducing the pooled ribozymes
into a
biological system comprising the target RNA under conditions suitable to
facilitate
modulation of the expression of the target RNA in said biological system.
Screening of Nucleic Acid Catalysts
Applicant describes herein, a general combinatorial approach for assaying
ribozyme variants based on ribozyme activity and/or a specific "attribute" of
a ribozyme,
such as the cleavage rate, cellular efficacy, stability, delivery,
localization and the like.
Variations of this approach also offer the potential for designing novel
catalytic
oligonucleotides, identifying ribozyme accessible sites within a target, and
for identifying
new nucleic acid targets for ribozyme-mediated modulation of gene expression.
In one preferred embodiment, the method relies upon testing mixtures
(libraries)
of ribozymes with various nucleotides, nucleotide analogs, or other analog
substitutions,
rather than individual ribozymes, to rapidly identify the nucleotide,
nucleotide analog, or
other analog that is variable at one or more positions within a ribozyme. In
the first step
{step 1, Figure 2), a desired number of positions (for example, 3 positions as
shown in
Figure 2) are chosen for variation in a first ribozyme motif (Starting
Ribozyme); there is
no requirement on the number of positions that can be varied and these
positions may or
CA 02288640 1999-11-OS
WO 98/50530 6~ PCT/US98/09249
may not be phylogenetically conserved for the ribozyme. In addition, these
position may
reside within the catalytic core, binding arms, or accessory domains. The
number of
positions that are chosen to be varied defines the number of "Classes" of
ribozyme
libraries that will be synthesized. In the example illustrated in Figure 2,
three positions
(designated positions 1, 2 and 3) are varied, so three different Classes of
ribozyme pool
are synthesized. In the next step (step 2), ribozyme pools are synthesized
containing a
random mixture of different nucleotides, nucleotide analogs, or other analogs
at all of the
desired positions (designated "X") to be varied except one, which is the
"fixed" position
(designated "F"). The fixed position contains a specific nucleotide,
nucleotide analog or
other analog. There is no requirement for the number of nucleotides, or
analogs be used.
The number of nucleotides or analogs defines the number of pools (designated
n) in each
Class. For example if ten different nucleotides or analogs are chosen, ten
different pools
(n=10) will be synthesized for each Class; each of the pools will contain a
specific
modification at one fixed position (designated F) but will contain an equal
mixture of all
ten modifications at the other positions (designated X). In a subsequent step
(step 3), the
different pools of ribozymes are tested for desired activity, phenotype,
characteristic or
attribute. For example, the testing may be determining in vitro rates of
target nucleic acid
cleavage for each pool, testing ribozyme-substrate binding affinities, testing
nuclease
resistance, determining pharmacodynamic properties, or determining which pool
is most
efficacious in a cellular or animal model system. Following testing, a
particular pool is
identified as a desired variant (designated " Desired Variant-1") and the
nucleotide or the
analog present at the fixed position within the Desired Variant-1 is made
constant
(designated "Z") for all subsequent experiments; a single position within a
ribozyme is
therefore varied, i.e., the variable nucleotide or analog at a single
position, when all other
X positions are random, is identified within a ribozyme motif. Subsequently,
new
ribozyme pools (Classes 2, 3 etc.) are synthesized containing an equal mixture
of all
nucleotides or analogs at the remaining positions to be optimized except one
fixed
position and one or more constant positions. Again, a specific nucleotide or
analog is
"fixed" at a single position that is not randomized and the pools are assayed
for a
CA 02288640 1999-11-OS
WO 98/50530 61 PCTNS98/09249
particular phenotype or attribute (step 4). This process is repeated until all
desired
positions have been varied and screened. For example if three positions are
chosen for
optimization, the synthesis and testing will need to be repeated three times
(3 Classes). In
the first two Classes, pools will be synthesized; in the final Class, specific
ribozymes will
be synthesized and tested. When the final position is analyzed (step 5), no
random
positions will remain and therefore only individual ribozymes are synthesized
and tested.
The resulting ribozyme or ribozymes (designated "second ribozyme motif') will
have a
defined chemical composition which will likely be distinct from the Starting
Ribozyme
motif (first ribozyme motif). This is a rapid method of screening for
variability of one or
more positions within a ribozyme motif.
In another preferred embodiment, the invention involves screening of chemical
modifications at one or more positions within a hammerhead ribozyme motif.
More
specifically, the invention involves variability in the catalytic core
sequence of a
hammerhead ribozyme. Particularly, the invention describes screening for
variability of
positions 3, 4 and 7 within a hammerhead ribozyme. The invention also features
screening for optimal loop II sequence in a hammerhead ribozyme.
In yet another preferred embodiment, the invention features a rapid method for
screening accessible ribozyme cleavage sites within a target sequence. This
method
involves screening of all possible sequences in the binding anm of a ribozyme.
The
sequence of the binding arms determines the site of action of certain
ribozymes. The
combinatorial approach can be used to identify desirable and/or accessible
sites within a
target sequence by essentially testing all possible arm sequences. The
difficulty with this
approach is that ribozyrnes require a certain number of base pairs (for
example, for
hammerhead ribozymes the binding arm length is approximately 12-16
nucleotides) in
order to bind functionally and sequence-specifically. This would require, for
example 12-
16 different groups of hammerhead ribozyme pools; 12-16 positions would have
to be
optimized which would require 12-16 different groups being synthesized and
tested.
Each pool would contain the four different nucleotides (A, C, U and G) or
nucleotide
CA 02288640 1999-11-OS
WO 98/50530 62 PCT/US98/09249
analogs (p = 4 for nucleotides). It would be very time consuming to test each
group,
identify the best pool, synthesize another group of ribozyme pools with one
additional
position constant, and then repeat the procedure until all 12-16 groups had
been tested.
However it is possible to decrease the number of Classes by testing multiple
positions
within a single Class. In this case, the number of pools within a Class equals
the number
of nucleotides or analogs in the random mixture (i.e., n) to the w power,
where w equals
the number of positions fixed in each Class. The number of Classes that need
to be
synthesized to optimize the final ribozyme equals the total number of
positions to be
optimized divided by the number of positions (w) tested within each Class. The
number
of pools in each Class = n'". The number of Class = total number of positions
/w.
In another preferred embodiment, the invention features a rapid method of
screening for new catalytic nucleic acid motifs by keeping the binding arms
constant and
varying one or more positions in a putative catalytic domain. Applicant
describes a
method to vary positions within the catalytic domain, without changing
positions within
1 S the binding arms, in order to identify new catalytic motifs. An example is
illustrated in
Figure 24. It is unclear how many positions are required to obtain a
functional catalytic
domain in a nucleic acid molecule, however it is reasonable to presume that if
a large
number of functionally diverse nucleotide analogs can be used to construct the
pools, a
relatively small number of positions could constitute a functional catalytic
domain. This
may especially be true if analogs are chosen that one would expect to
participate in
catalysis (e.g., acid/base catalysts, metal binding, etc.). In the example
illustrated, four
positions (designated 1, 2, 3 and 4) are chosen. In the first step, ribozyme
libraries (Class
1) are constructed: position 1 is fixed (F,) and positions 2, 3 and 4 are
random (X2, X3 and
X4, respectively). In step 2, the pools (the number of pools tested depends on
the number
of analogs used; n) are assayed for activity. This testing may be performed in
vitro or in a
cellular or animal model. Whatever assay that is used, the pool with the
desired
characteristic is identified and libraries (class 2) are again synthesized
with position 1
now constant (Z,) with the analog that was identified in class 1. In class 2,
position 2 is
fixed (FZ) and positions 3 and 4 are random (X, and X4). This process is
repeated until
CA 02288640 1999-11-OS
WO 98/SOS30 63 PCT/US98/09249
every position has been made constant and the chemical composition of the
catalytic
- domain is determined. If the number of positions in the catalytic domain to
be varied are
large, then it is possible to decrease the number of Classes by testing
multiple positions
within a single Class. The number of pools within a Class equals the number of
nucleotides or analogs in the random mixture (i.e., n) to the w power, where w
equals the
number of positions fixed in each Class. The number of Classes that need to be
synthesized to optimize the final ribozyme equals the total number of
positions to be
optimized divided by the number of positions (w) tested within each Class. The
number
of pools in each Class= n"'. The number of Classes= total number of positions
/w.
In a preferred embodiment a method for identifying variants of a nucleic acid
catalyst is described comprising the steps of: a) selecting at least three (3)
positions,
preferably 3-12, specifically 4-10, within said nucleic acid catalyst to be
varied with a
predetermined group of different nucleotides, these nucleotides are modified
or
unmodified (non-limiting examples of nucleotides that can used in this method
are shown
in Figure 15); b) synthesizing a first class of different pools of said
nucleic acid catalyst,
wherein the number of pools synthesized is equal to the number of nucleotides
in the
predetermined group of different nucleotides (for example if 10 different
nucleotides are
selected to be in the group of predetermined nucleotides then 10 different
pools of nucleic
acid catalysts have to be synthesized), wherein at least one of the positions
to be varied in
each pool comprises a defined nucleotide (fixed position; F) selected from the
predetermined group of different nucleotides and the remaining positions to be
varied
comprise a random mixture of nucleotides (X positions) selected from the
predetermined
group of different nucleotides; ~ c) testing the different pools of said
nucleic acid
catalyst under conditions suitable for said pools to show a desired attribute
(including but
not limited to improved cleavage rate, cellular and animal efficacy, nuclease
stability,
enhanced delivery, desirable localization) and identifying the pool with said
desired
attribute and wherein the position with the defined nucleotide (F) in the pool
with the
. desired attribute is made constant (Z position) in subsequent steps; d)
synthesizing a
second class of different pools of nucleic acid catalyst, wherein at least one
of the
CA 02288640 1999-11-OS
WO 98/50530 64 PCT/US98/09249
positions to be varied in each of the second class of different pools
comprises a defined
nucleotide (F) selected from the predetermined group of different nucleotides
and the
remaining positions to be varied comprise a random mixture (X) of nucleotides
selected
from the predetermined group of different nucleotides (this second class of
pools
therefore has F, X and Z positions); e) testing the second class of different
pools of said
nucleic acid catalyst under conditions suitable for showing desired attribute
and
identifying the pool with said desired attribute and wherein the position with
the defined
nucleotide in the pool with the desired attribute is made constant (Z) in
subsequent steps;
and fJ this process is repeated until every position selected in said nucleic
acid
catalyst to be varied is made constant.
In yet another preferred embodiment, a method for identifying novel nucleic
acid
molecules in a biological system is described, comprising the steps of a)
synthesizing a
pool of nucleic acid catalyst with a substrate binding domain and a catalytic
domain,
wherein said substrate binding domain comprises a random sequence; b) testing
the
pools of nucleic acid catalyst under conditions suitable for showing a desired
effect (such
as inhibition of cell proliferation, inhibition of angiogenesis, modulation of
growth and
/or differentiation, and others) and identifying the catalyst with said
desired attribute;
c) using an oligonucleotide, comprising the sequence of the substrate
binding domain of the nucleic acid catalyst showing said desired effect, as a
probe,
screening said biological system for nucleic acid molecules complementary to
said probe ;
and d) isolating and sequencing said complementary nucleic acid molecules.
These nucleic acid molecules identified using a nucleic acid screening method
described
above may be new gene sequences, or known gene sequences. The advantage of
this
method is that nucleic acid sequences, such as genes, involved in a biological
process,
such as differentiation, cell growth, disease processes including cancer,
tumor
angiogenesis, arthritis, cardiovascular disease, inflammation, restenosis,
vascular disease
and the like, can be readily identified.
CA 02288640 1999-11-OS
WO 98/50530 65 PCT/US98/09249
In a preferred embodiment, the invention features a nucleic acid molecule with
' catalytic activity having one of the formulae III-VII:
- Formula III
~(N) ~-- C-G-A-A -A M- 3°
L
~) n- G - A -G-N,rA - G -C4-U~Q- s,
Formula IV
M~ / Q s
z3
A
G
A A
G G z~
Z 3= 2' -O -MTM-U
C ~ G Z4= 2'-C-Allyl-U
(N)o (N)n Z~= 6-Methyl-U
L
CA 02288640 1999-11-OS
WO 98/50530 66 PCT/US98/09249
Formula V
M\
A z3
A
G
A A
G G z'
A
C ~ G Z3= 2'-O-MTM-U
(N)o (N)n Z4= 2'-O-MTM-C
L
Z 7= 6-M ethyl-U
Formula VI
My / Q s,
A z3
A z4
G
A A
G G z'
A Z 3= 2' -O -MTM-U
C ~ G Z 4= 2' -O -MTM-C
(N)o (N)n Z~= 2'-C-Allyl-U
L
CA 02288640 1999-11-OS
WO 98/50530 6~ PCTNS98/09249
Formula VII
- A
A
G
A A
G G z~
A
C ~ G Z 3= 2' -O -MTM-U
(N)o (N)n Z4= 2'-O-MTM-C
Z 7= P yridin-4-One
In each of the above formulae, N represents independently a nucleotide or a
non-
nucleotide linker, which may be same or different; M and Q are independently
oligonucleotides of length sufficient to stably interact (e.g., by forming
hydrogen bonds
with complementary nucleotides in the target) with a target nucleic acid
molecule (the
target can be an RNA, DNA or RNA/DNA mixed polymers); preferably the length of
Q is
greater than or equal to 3 nucleotides and the length of M is preferably
greater than or
equal to 5 nucleotides; o and n are integers greater than or equal to 1 and
preferably less
than about 100, wherein if (I~o and (N)n are nucleotides, (N)o and (l~n are
optionally
able to interact by hydrogen bond interaction; L is a linker which may be
present or
absent (i. e., the molecule is assembled from two separate molecules), but
when present, is
a nucleotide and/or a non-nucleotide linker, which may be a single-stranded
and/or
double-stranded region; and - represents a chemical linkage (e.g., a phosphate
ester
linkage, amide linkage or others known in the art). 2'-O-MTM-U and 2'-O-MTM-C
refers to 2'-O-methylthiomethyl uridine and 2'-O-methylthiomethyl-cytidine,
respectively. A, C, U and G represent adenosine, cytidine, uridine and
guanosine
nucleotides, respectively. The nucleotides in the formulae are unmodified or
modified at
the sugar, base, and/or phosphate portions as known in the art.
CA 02288640 1999-11-OS
WO 98/50530 68 PCT/US98/09249
In yet another embodiment, the nucleotide linker (L) is a nucleic acid
aptamer,
such as an ATP aptamer, HIV Rev aptamer (RRE), HIV Tat aptamer (TAR) and
others
(for a review see Gold et al., 1995, Annu. Rev. Biochem., 64, 763; and Szostak
&
Ellington, 1993, in The RNA World, ed. Gesteland and Atkins, pp 511, CSH
Laboratory
Press). A "nucleic acid aptamer" as used herein is meant to indicate nucleic
acid
sequence capable of interacting with a ligand. The ligand can be any natural
or a
synthetic molecule, including but not limited to a resin, metabolites,
nucleosides,
nucleotides, drugs, toxins, transition state analogs, peptides, lipids,
proteins, aminoacids,
nucleic acid molecules, hormones, carbohydrates, receptors, cells, viruses,
bacteria and
others.
In yet another embodiment, the non-nucleotide linker (L) is as defined herein.
In particular, the invention features modified ribozymes having a base
substitution
selected from pyridin-4-one, pyridin-2-one, phenyl, pseudouracil, 2, 4, 6-
trimethoxy
benzene, 3-methyluracil, dihydrouracil, naphthyl, 6-methyl-uracil and
aminophenyl.
In yet another embodiment, the non-nucleotide linker (L) is as defined herein.
The term "non-nucleotide" as used herein include either abasic nucleotide,
polyether,
polyamine, polyamide, peptide, carbohydrate, Lipid, or polyhydrocarbon
compounds.
Specific examples include those described by Seela and Kaiser, Nucleic Acids
Res. 1990,
18:6353 and Nucleic Acids Res. 1987, 15:3113; Cload and Schepartz, J. Am.
Chem. Soc.
1991, 113:6324; Richardson and Schepartz, J. Am. Chem. Soc. 1991,113:5109; Ma
et al.,
Nucleic Acids Res. 1993, 21:2585 and Biochemistry 1993, 32:1751; Durand et
al., Nucleic
Acids Res. 1990, 18:6353; McCurdy et al., Nucleosides & Nucleotides 1991,
10:287;
Jschke et al., Tetrahedron Lett. 1993, 34:301; Ono et al., Biochemistry 1991,
30:9914;
Arnold et al., International Publication No. WO 89/02439; Usman et al.,
International
Publication No. WO 95/06731; Dudycz et al., International Publication No. WO
95/11910 and Ferentz and Verdine, J. Am. Chem. Soc. 1991, 113:4000, all hereby
incorporated by reference herein. Thus, in a preferred embodiment, the
invention features
a nucleic acid catalyst having one or more non-nucleotide moieties, and having
enzymatic
CA 02288640 1999-11-OS
WO 98/50530 69 PCT/US98/09249
activity to cleave an RNA or DNA molecule. By the term "non-nucleotide" is
meant any
group or compound which can be incorporated into a nucleic acid chain in the
place of
one or more nucleotide units, including either sugar and/or phosphate
substitutions, and
allows the remaining bases to exhibit their enzymatic activity. The group or
compound is
abasic in that it does not contain a commonly recognized nucleotide base, such
as
adenosine, guanine, cytosine, uracil or thymine. The terms "abasic" or "abasic
nucleotide" as used herein encompass sugar moieties lacking a base or having
other
chemical groups in place of base at the 1' position.
In preferred embodiments, the enzymatic nucleic acid includes one or more
stretches of RNA, which provide the enzymatic activity of the molecule, linked
to the
non-nucleotide moiety. The necessary RNA components are known in the art, see,
e.g.,
Usman, supra. By RNA is meant a molecule comprising at least one
ribonucleotide
residue.
As the term is used in this application, non-nucleotide-containing enzymatic
nucleic acid means a nucleic acid molecule that contains at least one non-
nucleotide
component which replaces a portion of a ribozyme, e.g., but not limited to, a
double-
stranded stem, a single-stranded "catalytic core" sequence, a single-stranded
loop or a
single-stranded recognition sequence. These molecules are able to cleave
(preferably,
repeatedly cleave) separate RNA or DNA molecules in a nucleotide base sequence
specific manner. Such molecules can also act to cleave intramolecularly if
that is desired.
Such enzymatic molecules can be targeted to virtually any RNA transcript.
The specific nucleic acid catalysts described in the instant application are
not
limiting in the invention and those skilled in the art will recognize that all
that is
important in a nucleic acid catalyst of this invention is that it has a
specific substrate
binding site (e.g., M and/or Q of Formulae III-VII above) which is
complementary to one
or more of the target nucleic acid regions, and that it have nucleotide
sequences within or
surrounding that substrate binding site which impart a nucleic acid cleaving
activity to the
molecule.
CA 02288640 1999-11-OS
WO 98150530 70 PCT/US98/09249
Vector Expression of Enzymatic Nucleic Acid
The nucleic acid catalysts of the instant invention can be expressed within
cells
from eukaryotic promoters (e.g., Izant and Weintraub, 1985 Science 229, 345;
McGarry
and Lindquist, 1986 Proc. Natl. Acad. Sci.USA 83, 399; Scanlon et al., 1991,
Proc. Natl.
Acad. Sci. USA, 88, 10591-5; Kashani-Sabet et al., 1992 Antisense Res. Dev.,
2, 3-15;
Dropulic et al., 1992 J. Virol, 66, 1432-41; Weerasinghe et al., 1991 J.
Virol, 65, 5531-4;
Ojwang et al., 1992 Proc. Natl. Acad. Sci. USA 89, 10802-6; Chen et al., 1992
Nucleic
Acids Res., 20, 4581-9; Sarver et al., 1990 Science 247, 1222-1225; Thompson
et al.,
1995 Nucleic Acids Res. 23, 2259; Good et al., 1997, Gene Therapy, 4, 45; all
of the
references are hereby incorporated in their totality by reference herein).
Those skilled in
the art realize that any nucleic acid can be expressed in eukaryotic cells
from the
appropriate DNA/RNA vector. The activity of such nucleic acids can be
augmented by
their release from the primary transcript by a ribozyme (Draper et al., PCT WO
93/23569,
and Sullivan et al., PCT WO 94/02595; Ohkawa et al., 1992 Nucleic Acids Symp.
Ser.,
27, 15-6; Taira et al., 1991, Nucleic Acids Res., 19, 5125-30; Ventura et al.,
1993 Nucleic
Acids Res., 21, 3249-55; Chowrira et al., 1994 J. Biol. Chem. 269, 25856; all
of the
references are hereby incorporated in their totality by reference herein).
In another aspect of the invention, nucleic acid catalysts that cleave target
molecules
are expressed from transcription units (see for example Figure 11) inserted
into DNA or
RNA vectors. The recombinant vectors are preferably DNA plasmids or viral
vectors.
Ribozyme expressing viral vectors could be constructed based on, but not
limited to,
adeno-associated virus, retrovirus, adenovirus, or alphavirus. Preferably, the
recombinant
vectors capable of expressing the ribozymes are delivered as described above,
and persist
in target cells. Alternatively, viral vectors may be used that provide for
transient
expression of ribozymes. Such vectors might be repeatedly administered as
necessary.
Once expressed, the ribozymes cleave the target mRNA. The active ribozyme
contains an
enzymatic center or core equivalent to those in the examples, and binding arms
able to
bind target nucleic acid molecules such that cleavage at the target site
occurs. Other
CA 02288640 1999-11-OS
WO 98/50530 71 PCT/US98/09249
sequences may be present which do not interfere with such cleavage. Delivery
of
ribozyme expressing vectors could be systemic, such as by intravenous or
intramuscular
administration, by administration to target cells ex-planted from the patient
followed by
reintroduction into the patient, or by any other means that would allow for
introduction
into the desired target cell (for a review see Couture and Stinchcomb, 1996,
TIG., 12,
510).
In a preferred embodiment, an expression vector comprising nucleic acid
sequence
encoding at least one of the nucleic acid catalyst of the instant invention is
disclosed. The
nucleic acid sequence encoding the nucleic acid catalyst of the instant
invention is
operable linked in a manner which allows expression of that nucleic acid
molecule.
In one embodiment, the expression vector comprises: a transcription initiation
region {e.g., eukaryotic pol I, II or III initiation region); b) a
transcription termination
region (e.g., eukaryotic pol I, II or III termination region); c) a gene
encoding at least one
of the nucleic acid catalyst of the instant invention; and wherein said gene
is operably
linked to said initiation region and said termination region, in a manner
which allows
expression and/or delivery of said nucleic acid molecule. The vector may
optionally
include an open reading frame (ORF) for a protein operably linked on the 5'
side or the 3'-
side of the gene encoding the nucleic acid catalyst of the invention; and/or
an intron
{intervening sequences).
Transcription of the ribozyme sequences are driven from a promoter for
eukaryotic
RNA polymerise I (pol I), RNA polymerise II (pol II), or RNA polymerise III
(pol III).
Transcripts from pol II or pol III promoters will be expressed at high levels
in all cells;
the levels of a given pol II promoter in a given cell type will depend on the
nature of the
gene regulatory sequences (enhancers, silencers, etc.) present nearby.
Prokaryotic RNA
polymerise promoters are also used, providing that the prokaryotic RNA
polymerise
enzyme is expressed in the appropriate cells (Elroy-Stein and Moss, 1990 Proc.
Natl.
Acid. Sci.U S A, 87, 6743-7; Gao and Huang 1993 Nucleic Acids Res., 2i, 2867-
72;
Lieber et al., 1993 Methods Enzymol., 217, 47-66; Zhou et al., 1990 Mol. Cell.
Biol., 10,
CA 02288640 1999-11-OS
WO 98/50530 72 PCT/US98/09249
4529-37). Several investigators have demonstrated that ribozymes expressed
from such
promoters can function in mammalian cells (e.g., Kashani-Sabet et al., 1992
Antisense
Res. Dev., 2, 3-15; Ojwang et al., 1992 Proc. Natl. Acad. Sci. U S A, 89,
10802-6; Chen et
al., 1992 Nucleic Acids Res., 20, 4581-9; Yu et al., 1993 Proc. Natl. Acad.
Sci. U S A, 90,
6340-4; L'Huillier et al., 1992 EMBO J. 11, 4411-8; Lisziewicz et al., 1993
Proc. Natl.
Acad. Sci. U. S. A., 90, 8000-4; Thompson et al., 1995 Nucleic Acids Res. 23,
2259;
Sullenger & Cech, 1993, Science, 262, 1566). More specifically, transcription
units such
as the ones derived from genes encoding U6 small nuclear (snRNA), transfer RNA
(tRNA) and adenovirus VA RNA are useful in generating high concentrations of
desired
RNA molecules such as ribozymes in cells (Thompson et al., supra; Couture and
Stinchcomb, 1996, supra; Noonberg et al., 1994, Nucleic Acid Res., 22, 2830;
Noonberg
et al., US Patent No. 5,624,803; Good et al., 1997, Gene Ther. 4, 45;
Beigelman et al.,
International PCT Publication No. WO 96/18736; all of these publications are
incorporated by reference herein. Examples of transcription units suitable for
expression
of ribozymes of the instant invention are shown in Figure 11. The above
ribozyme
transcription units can be incorporated into a variety of vectors for
introduction into
mammalian cells, including but not restricted to, plasmid DNA vectors, viral
DNA
vectors (such as adenovirus or adeno-associated virus vectors), or viral RNA
vectors
(such as retroviral or alphavirus vectors) (for a review see Couture and
Stinchcomb, 1996,
supra).
In a preferred embodiment an expression vector comprising nucleic acid
sequence
encoding at least one of the catalytic nucleic acid molecule of the invention,
in a manner
which allows expression of that nucleic acid molecule. The expression vector
comprises
in one embodiment; a) a transcription initiation region; b) a transcription
termination
region; c) a gene encoding at least one said nucleic acid molecule; and
wherein said gene
is operably linked to said initiation region and said termination region, in a
manner which
allows expression and/or delivery of said nucleic acid molecule. In another
preferred
embodiment the expression vector comprises: a) a transcription initiation
region; b) a
transcription termination region; c) an open reading frame; d) a gene encoding
at least one
CA 02288640 1999-11-OS
WO 98/50530 ~3 PCT/US98/09249
said nucleic acid molecule, wherein said gene is operably linked to the 3'-end
of said open
reading frame; and wherein said gene is operably linked to said initiation
region, said
open reading frame and said termination region, in a manner which allows
expression
and/or delivery of said nucleic acid molecule. In yet another embodiment the
expression
vector comprises: a) a transcription initiation region; b) a transcription
termination region;
c) an intron; d) a gene encoding at least one said nucleic acid molecule; and
wherein said
gene is operably linked to said initiation region, said intron and said
termination region, in
a manner which allows expression and/or delivery of said nucleic acid
molecule. In
another embodiment, the expression vector comprises: a) a transcription
initiation region;
b) a transcription termination region; c) an intron; d) an open reading frame;
e) a gene
encoding at least one said nucleic acid molecule, wherein said gene is
operably linked to
the 3'-end of said open reading frame; and wherein said gene is operably
linked to said
initiation region, said intron, said open reading frame and said termination
region, in a
manner which allows expression and/or delivery of said nucleic acid molecule.
Delivery of Nucleic Acid Catalysts:
In a preferred embodiment, the nucleic acid catalysts are added directly, or
can be
complexed with cationic lipids, packaged within liposomes, or otherwise
delivered to
smooth muscle cells. The RNA or RNA complexes can be locally administered to
relevant tissues through the use of a catheter, infusion pump or stmt, with or
without their
incorporation in biopolyrners. Using the methods described herein, other
nucleic acid
catalysts that cleave target nucleic acid may be derived and used as described
above.
Specific examples of nucleic acid catalysts of the instant invention are
provided below in
the Tables and figures. ,
Sullivan, et al., supra, describes the general methods for delivery of
enzymatic
RNA molecules. Ribozymes may be administered to cells by a variety of methods
known
to those familiar to the art, including, but not restricted to, encapsulation
in Iiposomes, by
iontophoresis, or by incorporation into other vehicles, such as hydrogels,
cyclodextrins,
biodegradable nanocapsules, and bioadhesive microspheres. For some
indications,
CA 02288640 1999-11-OS
WO 98/50530 ~4 PCT/US98/09249
ribozymes may be directly delivered ex vivo to cells or tissues with or
without the
aforementioned vehicles. Alternatively, the RNA/vehicle combination is locally
delivered by direct injection or by use of a catheter, infusion pump or stmt.
Other routes
of delivery include, but are not limited to, intravascular, intramuscular,
subcutaneous or
joint injection, aerosol inhalation, oral (tablet or pill form), topical,
systemic, ocular,
intraperitoneal andlor intrathecal delivery. More detailed descriptions of
ribozyme
delivery and administration are provided in Sullivan et al., supra and Draper
et al., supra
which have been incorporated by reference herein.
The present invention also includes pharmaceutically acceptable formulations
of
the compounds described. These formulations include salts of the above
compounds, e.g.,
ammonium, sodium, calcium, magnesium, lithium, and potassium salts.
A pharmacological composition or formulation refers to a composition or
formulation in a form suitable for administration, e.g., systemic
administration, into a cell
or patient, preferably a human. Suitable forms, in part, depend upon the use
or the route
of entry, for example oral, transdermal, or by injection. Such forms should
not prevent
the composition or formulation to reach a target cell (i.e., a cell to which
the negatively
charged polymer is desired to be delivered to). For example, pharmacological
compositions injected into the blood stream should be soluble. Other factors
are known
in the art, and include considerations such as toxicity and forms which
prevent the
composition or formulation from exerting its effect.
By "systemic administration" is meant in vivo systemic absorption or
accumulation of drugs in the blood stream followed by distribution throughout
the entire
body. Administration routes which lead to systemic absorption include, without
limitations: intravenous, subcutaneous, intraperitoneal, inhalation, oral,
intrapulmonary
and intramuscular. Each of these administration routes expose the desired
negatively
charged polymers, e.g., NTP's, to an accessible diseased tissue. The rate of
entry of a
drug into the circulation has been shown to be a function of molecular weight
or size.
The use of a liposome or other drug carrier comprising the compounds of the
instant
CA 02288640 1999-11-OS
WO 98/50530 75 PCT/US98/09249
invention can potentially localize the drug, for example, in certain tissue
types, such as
the tissues of the reticular endothelial system (RES). A liposome formulation
which can
facilitate the association of drug with the surface of cells, such as,
lymphocytes and
macrophages is also useful. This approach may provide enhanced delivery of the
drug to
target cells by taking advantage of the specificity of macrophage and
lymphocyte immune
recognition of abnormal cells, such as the cancer cells.
The invention also features the use of the a composition comprising surface-
modified liposomes containing poly (ethylene glycol) lipids (PEG-modified, or
long-
circulating liposomes or stealth liposomes). These formulations offer an
method for
increasing the accumulation of drugs in target tissues. This class of drug
carriers resists
opsonization and elimination by the mononuclear phagocytic system (MPS or
RES),
thereby enabling longer blood circulation times and enhanced tissue exposure
for the
encapsulated drug (Lasic et al. Chem. Rev. 1995, 95, 2601-2627; Ishiwataet
al., Chem.
Pharm. Bull. 1995, 43, 1005-1011). Such liposomes have been shown to
accumulate
selectively in tumors, presumably by extravasation and capture in the
neovascularized
target tissues (Lasic et al., Science 1995, 267, 1275-1276; Oku et al., 1995,
Biochim.
Biophys. Acta, 1238, 86-90). The long-circulating liposomes enhance the
pharmacokinetics and pharmacodynamics of drugs, particularly compared to
conventional
cationic liposomes which are known to accumulate in tissues of the MPS (Liu et
al., J.
Biol. Chem. 1995, 42, 24864-24870; Choi et al., International PCT Publication
No. WO
96/10391; Ansell et al., International PCT Publication No. WO 96/10390;
Holland et al.,
International PCT Publication No. WO 96/10392; all of these are incorporated
by
reference herein). Long-circulating liposomes are also likely to protect drugs
from
nuclease degradation to a greater extent compared to cationic liposomes, based
on their
ability to avoid accumulation in metabolically aggressive MPS tissues such as
the liver
and spleen. All of these references are incorporated by reference herein.
The present invention also includes compositions prepaxed for storage or
administration which include a pharmaceutically effective amount of the
desired
CA 02288640 1999-11-OS
WO 98/50530 PCT/US98/09249
76
compounds in a pharmaceutically acceptable carrier or diluent. Acceptable
carriers or
diluents for therapeutic use are well known in the pharmaceutical art, and are
described,
for example, in Remington's Pharmaceutical Sciences, Mack Publishing Co. (A.R.
Gennaro edit. 1985) hereby incorporated by reference herein. For example,
preservatives,
stabilizers, dyes and flavoring agents may be provided. Id. at 1449. These
include
sodium benzoate, sorbic acid and esters of p-hydroxybenzoic acid. In addition,
antioxidants and suspending agents may be used.
A pharmaceutically effective dose is that dose required to prevent, inhibit
the
occurrence, or treat (alleviate a symptom to some extent, preferably all of
the symptoms)
of a disease state. The pharmaceutically effective dose depends on the type of
disease,
the composition used, the route of administration, the type of mammal being
treated, the
physical characteristics of the specific mammal under consideration,
concurrent
medication, and other factors which those skilled in the medical arts will
recognize.
Generally, an amount between 0.1 mg/kg and 100 mglkg body weight/day of active
1 S ingredients is administered dependent upon potency of the negatively
charged polymer.
In a one aspect, the invention provides nucleic acid catalysts that can be
delivered
exogenously to specific cells as required.
Local ribozyme administration offers the advantages of achieving high tissue
concentrations of ribozymes and limiting their exposure to catabolic and
excretory
mechanisms. Although local routes of administration provide access to
pathologies
involving a number of organ systems, systemic administration would make
ribozyme
treatment of several other major human diseases feasible.
It has been demonstrated that certain tissues accumulate oligonucleotides
and/or
oligonucleotide formulations following systemic administration. These tissues
include
sites of inflammation (Wu et al. 1993, Cancer Res. 53: 3765-3767), solid
tumors (Yuan et
al. 1994, Cancer Res. 54: 3352-3356), kidney (Cossum et al. 1993, J. Pharmaco.
and
Exp. Ther. 267: 1181-1190), brain (Wu et al. 1996, J. Pharmacol. Exp. Ther.
276: 206-
11) and those rich in reticulo-endothelial cells (liver, spleen, lymphatics;
Litzinger et al.
CA 02288640 1999-11-OS
WO 98/50530 PCT/US98/09249
77
1994, Biochim. Biophys. Acta 1190: 99-107; Agrawal et al. 1991, Proc. Natl.
Acad.
Sci. USA 88: 7595-7599; Agrawal et al. 1995, Clin. Pharmacology 28: 7-16;
Sands .et al.
1994, Molecular Pharmacol. 45: 932-943; Saijo et a1.1994, Oncology Research 6:
243-
249).
The kidney, as well as organs of the reticulo-endothelial system (RES), are
mainly
responsible for clearance of ribozymes following intravenous (i.v.)
administration.
Diseases involving these tissues are good candidates for systemic ribozyme
therapy by
virtue of their tendency to accumulate ribozymes.
In one preferred embodiment, the invention features method of treating
inflammation
using ribozymes. Inflammatory processes underlie the pathology of a large
number of
human diseases. Many of these processes can be blocked by inhibiting the
expression of
inflammatory mediators and/or their receptors (Cohen et al. 1995, Am. J. Med.
99: 45S-
52S). Systemic administration of monoclonal antibodies specific to these
mediators have
been shown to be efficacious in animal models of rheumatoid arthritis,
inflammatory
bowel disease, and acute respiratory distress syndrome {Arend et al. 1990,
Arthritis and
Rheumatism 33: 305-315). One potential way for systemic administration of
ribozymes
to impact systemic inflammatory disease is through inhibition of TNF-a
production by
macrophages. TNF-a has been shown participate in a variety of inflammatory
processes
and is produced mainly by macrophages which are known to accumulate cationic
lipid-
formulated ribozymes (Masahiro et al. 1990, J. Immunology. 144: 1425-1431).
Anti-
mouse TNF- a ribozymes were effective in cell culture, thus, it may be
possible that
systemic delivery of ribozymes by a liposome formulation could be an effective
therapeutic in the above mentioned inflammatory disease states.
In another preferred embodiment, the invention features methods of treating
diseases
involving RES using ribozymes. A number of studies have shown that
systemically
' administered oligonucleotides distribute to RES tissues (liver, spleen and
lymphatics).
Several studies with cationic lipid complexed oligonucleotides have also shown
specific
biodistribution to these. Pathology involving the RES includes a number of
infectious
CA 02288640 1999-11-OS
WO 98/50530 78 PCT/I1S98/09249
diseases of major importance, such as human immunodeficiency virus (HIV),
mycobacterium infections including tuberculosis (TB), avium, and leprae
(leprosy).
These diseases are all associated with, for example, overproduction of
interleukin-10 (IL-
10), a potent immunosuppressive cytokine (Barnes et al. 1993, Infect. Immun.
61: 3482-
9). Some of these infections can potentially be ameliorated by administration
of
neutralizing antibodies to IL-10.
In yet another preferred embodiment, the invention features method of treating
cancer using ribozymes. As evidence of the potential use of systemic
oligonucleotides as
anticancer agents, antisense phosphorothioates have been have been reported to
exhibit
antitumor efficacy in a murine model of Burkitt's lymphoma (Huang et al. 1995,
Mol.
Med. 1: 647-658). The molecular targets of systemic antineoplastic ribozymes
could
include oncogenes, protooncogenes, or angiogenic factors and receptors.
Although the
link between oncogenes and tumorigenesis is now well established, the specific
mutations
that lead to activation of a proto-oncogene can be widely diverse.
Upregulation of
1 S protooncogene products is also common in human cancer. Reducing the levels
of these
gene products may be beneficial in treatment of cancer. In addition, since
many tumors
are highly vascularized, angiogenic factors or receptors may provide good
alternate or
adjunct targets to oncogenes for the therapy of solid tumors and their
metastases.
Applicant, in a non-limiting example infra, show ribozymes targeting
angiogenic
mediators.
The potential number of molecular targets in cancer is quite large. Among
these
targets are oncogenes, protooncogenes, metalloproteinases, growth factors, and
angiogenic factors. However, a common denominator in many forms of metastatic
solid
tumors is extensive vascularization of the tumor. As tumors exceed about 1 mm
in
diameter, they require neovascularization for continued growth (Gimbrone et
al., 1972, J.
Exp. Med., 136, 261). In addition, the appearance of new blood vessels within
a tumor
correlates with the initiation of the process of metastasis (Martiny-Baron and
Marme,
1995). It is possible that by using a systemically administered ribozyme
targeting a key
CA 02288640 1999-11-OS
WO 98/50530 79 PCT/US98/09249
player in the process of angiogenesis would reduce both primary tumor growth,
tumor
progression and tumor metastasis.
"Angiogenesis" refers to formation of new blood vessels from existing blood
vessels which is an essential process in reproduction, development and wound
repair.
"Tumor angiogenesis" refers to the induction of the growth of blood vessels
from
surrounding tissue into a solid tumor. Tumor growth and tumor metastasis are
dependent
on angiogenesis (for a review see Folkman, 1985, Nature Med 1: 27-31; Folkman
1990
J. Natl. Cancerlnst., 82, 4; Foikman and Shing, 1992 J. Biol. Chem. 267,
10931).
"Tumor metastasis" refers to the transfer and/or migration of tumor cells,
, originating from a primary tumor, from one part of the body or organ to
another. Most
malignant tumors have the capacity to metastasize.
"Tumor" refers to a new growth of tissue wherein the cells multiply, divide
and
grow uncontrolled.
In a preferred embodiment, the invention features a method of treating non-
hepatic
ascites using ribozymes. Nonhepatic ascites or peritoneal fluid accumulation
resulting
from abdominal cancer and ovarian hyperstimulation syndrome (OHSS) can result
in
significant fluid loss from the intravascular space and hypovolemia. If
ascites volumes
are large, abdominal pain, hypovolemic hypotension, electrolyte abnormalities
and
respiratory difficulties can ensue. Thus, if ascites is left untreated, it can
be life
threatening. Evidence is now accumulating that nonhepatic ascites may be
induced, at
least in part, by vascular endothelial growth factor (VEGF). For this reason,
nonhepatic
ascites may be a potential therapeutic indication for ribozymes directed
against vascular
endothelial growth factor (VEGF) receptors delivered either systemically or
regionally to
the peritoneum.
Ovaries can be overstimulated by hormonal therapy during fertility treatment.
As a
result, women can experience ovarian hyperstimulation syndrome which is
associated
CA 02288640 1999-11-05
WO 98/50530 g~ PCT/US98/09249
with grossly enlarged ovaries and extreme ascites fluid accumulation. This
fluid
accumulation is thought to be induced by the release of a vascular
permeability agent
which may interact with vessels of the peritoneal cavity leading to plasma
extravasation.
Abramov and co-workers (1997, Fertil. Steril. 67: 261) have shown that plasma
VEGF
levels are elevated in DHSS and return to normal upon resolution of the
syndrome. An
earlier study has shown that VEGF is elevated in the serum and follicular
fluid of OHSS
patients and that the source of this VEGF may be the luteinizing granulosa
cells of the
ovary (Krasnow et al., 1996, Fertil. Steril. 65: 552). McClure et al. (1994,
Lancet 344,
235) concluded that VEGF is the key mediator of OHSS ascites production since
rhVEGF
increases DHSS ascites but not liver ascites and that this increase is
reversible by rhVEGF
antiserum. Thus, reducing the expression of VEGF receptors in the vasculature
of the
peritoneum may have a therapeutic benefit in OHSS by substantially reducing
OHSS-
stimulated ascites production. Since VEGF can interact with VEGF receptors on
vessels
throughout the peritoneum from ovarian release of VEGF into systemic
circulation,
systemic treatment may represent the best option for treating this syndrome.
Malignant ascites: Another form of ascites can be induced by malignancies of
the
peritoneum including breast, pancreatic, uterine and colorectal cancers. It is
thought that
certain cancers produce factors which influence peritoneal vascular
permeability leading
to plasma extravasation (Garrison et al., 1986; Ann. Surg. 203: 644; Garrison
et al., 1987,
J. Surg. Res. 42: 126; Nagy et al., 1993, Cancer Res. 53: 2631 ). Several
solid tumors
including some colorectal and breast carcinomas are known to secrete VEGF to
recruit
blood vessels for sustained growth and metastasis. This secreted VEGF may also
serve to
increase local vasculature permeability. In support of this hypothesis, Nagy
et al. (supra)
showed in mice that peritoneal fluid resulting from MOT and TTA3/St carcinomas
contained elevated levels of VEGF whose concentration correlated directly with
fluid
accumulation and development of hyperpermeable microvesseis. Therefore,
ribozymes
directed against VEGF receptors administered systemically may impact both the
tumor
growth and metastases of VEGF secreting tumors as well as ascites induced by
VEGF
interacting with the vasculature of the peritoneum.
CA 02288640 1999-11-OS
WO 98/50530 $1 PCT/US98/09249
Strateeies for Systemic DeliverX
Methods to enhance tissue accumulation
Tissue accumulation of ribozymes can be improved by formulation, conjugation,
or
further chemical stabilization of the ribozyme. Elimination due to glomeruiar
filtration
can be slowed by increasing the apparent molecular weight of the ribozyme,
e.g., by
liposome encapsulation or bioconjugation to PEG. Applicant has observed that
the rate of
catabolism can be slowed by a factor of 100 and lung accumulation increased
500 fold by
formulation with DMRIE/DOPE reagents. Liposomal encapsulation is likely to
have a
similar effect on the rate of catabolism. The rate of clearance into non-
target tissues could
also be reduced by encapsulation into liposomes, provided that the liposomes
were
surface modified with PEG such that RES clearance were avoided. Increasing the
rate of
uptake by target tissues can also be enhanced, for example, by conjugation of
cholesterol
to the ribozymes. Applicant has also observed that in tissues of the RES,
accumulation
has been increased several hundred fold by complexation with a cationic lipid
carrier.
Sustained release as a means to increase exposure
Sustained or continuous delivery devices, such as ALZET~ osmotic mini-pumps,
may also enhance accumulation in target tissues by increasing exposure
relative to bolus
i.v. administration. Sustained delivery from ALZET~ pumps has been shown to be
an
effective way of administering a phosphorothioate antisense molecule for
inhibition of
tumor growth in mice (Huang et al. 1995, supra). Applicant has observed that
the rate of
ribozyme catabolism in and rate of clearance from the circulation is
concentration
dependent and may relate to the equilibrium plasma protein binding of the
ribozyme.
Phosphorothioate DNA is rapidly cleared from circulation when its
concentration
exceeds the plasma protein binding constant, as is the case after i.v. bolus
administration.
Osmotic pumps administer oligonucleotides at a slower and constant rate, and
therefore
may maintain plasma levels near the equilibrium binding capacity. This would
result in
less of the administered dose being lost to glomerular filtration
(elimination) and hepatic
CA 02288640 1999-11-OS
WO 98/50530 82 PCT1US98/09249
extraction (catabolism); more of the administered dose may be available for
uptake into
target tissues.
Animal Models
Use of murine models
For a typical systemic study involving 10 mice (20 g each) per dose group, 5
doses
(1, 3, 10, 30 and 100 mg/kg daily over 14 days continuous administration),
approximately
400 mg of ribozyme, formulated in saline would be used. A similar study in
young adult
rats (200 g) would require over 4 g. Parallel pharmacokinetic studies may
involve the use
of similar quantities of ribozymes further justifying the use of murine
models.
Ribozymes and Lewis lung carcinoma and B-16 melanoma murine models
Identifying a common animal model for systemic efficacy testing of ribozymes
is an
efficient way of screening ribozymes for systemic efficacy.
The Lewis lung carcinoma and B-16 murine melanoma models are well accepted
models of primary and metastatic cancer and are used for initial screening of
anti-cancer.
These murine models are not dependent upon the use of immunodeficient mice,
are
relatively inexpensive, and minimize housing concerns. Both the Lewis lung and
B-16
melanoma models involve subcutaneous implantation of approximately 10~' tumor
cells
from metastatically aggressive tumor cell Iines (Lewis lung lines 3LL or D122,
LLc-LN7;
B-16-BL6 melanoma) in C57BL/6J mice. Alternatively, the Lewis lung model can
be
produced by the surgical implantation of tumor spheres (approximately 0.8 mm
in
diameter). Metastasis also may be modeled by injecting the tumor cells
directly i.v.. In
the Lewis lung model, microscopic metastases can be observed approximately 14
days
following implantation with quantifiable macroscopic metastatic tumors
developing
within 21-25 days. The B-16 melanoma exhibits a similar time course with tumor
neovascularization beginning 4 days following implantation. Since both primary
and
metastatic tumors exist in these models after 21-25 days in the same animal,
multiple
CA 02288640 1999-11-OS
WO 98/50530 83 PCT/US98/09249
measurements can be taken as indices of efficacy. Primary tumor volume and
growth
latency as well as the number of micro- and macroscopic metastatic lung foci
or number
of animals exhibiting metastases can be quantitated. The percent increase in
lifespan can
also be measured. Thus, these models would provide suitable primary efficacy
assays for
S screening systemically administered ribozymes/ribozyme formulations.
In the Lewis lung and B-16 melanoma models, systemic phanmacotherapy with a
wide
variety of agents usually begins 1-7 days following tumor
implantation/inoculation with
either continuous or multiple administration regimens. Concurrent
pharmacokinetic
studies can be performed to determine whether sufficient tissue levels of
ribozymes can
be achieved for pharmacodynamic effect to be expected. Furthermore, primary
tumors
and secondary lung metastases can be removed and subjected to a variety of in
vitro
studies (i. e., target mRNA reduction).
Anti-YEGF receptor ribozymes
Sustained tumor growth and metastasis depend upon angiogenesis. In fact, the
appearance of vessels in a growing tumor is correlated with the beginning of
metastatic
potential. Several studies have shown that antiangiogenic agents alone or in
combination
with cytotoxic agents reduce lung metastases and/or primary tumor volume in
the Lewis
lung and B-16 melanoma models (Borgstrom et al. 1995, Anticancer Res. 15: 719-
728;
Kato et al. 1994, Cancer Res. 54: S 143-5147; O'Reilly et al. 1994, Cell 79:
31 S-328;
Sato et al. 1995, Jpn. J. Cancer Res. 86: 374-382).
A major factor implicated in the induction of solid tumor angiogenesis is
vascular
endothelial growth factor (VEGF; Folkman, 1995, supra). Several human tumors
have
been shown to synthesize and secrete. With regard to treating lung metastasis,
VEGF and
VEGF receptors of both subtypes and their expression are upregulated in the
lung under
conditions of hypoxia (Tuder et al. 1994, J. Clin. Invest. 95: 1798-1807).
This may lead
to neovascularization which provides the means by which tumor cells gain
access to
CA 02288640 1999-11-OS
WO 98/50530 PCT/US98/09249
84
circulation (Mariny-Baron and Manne, 1995). Thus, VEGF and its receptors may
be
important targets in the treatment of metastatic disease.
Applicant has shown that a catalyticaliy active ribozyme targeting flt-1 RNA
inhibits VEGF-induced neovascularization in a dose-dependent manner in a rat
corneal
model of angiogenesis. Testing with cytotoxic agents in combination with
antiangiogenic
ribozymes may also prove useful.
Anti-K and H ras ribozymes
Mutations involving ras underlie a number of human cancers. Ras also plays a
role
in metastatic potential {Shekhar and Miller, 1994, Invasion Metastasis 14: 27-
37) and
may do so, in part, by influencing endothelial cell migration (Fox et al.
1994, Oncogene
9: 3519-26). With regard to lung cancer, ras has been shown to induce abnormal
mitoses
in lung f'ibroblasts (Lyubuski et al. 1994, Cytobios 80: 161-178) and is a
clinical marker
in non-small cell lung tumors (Niklinski and Forman, 1995, Eur. J. Cancer
Prev. 4: 129-
138). Studies in cells cultured from human small cell lung tumor xenografts
demonstrated overexpression of K-ras (Arvelo et al. 1994, Anticancer Res. 14:
1893-
1901}. This evidence provides ample support for the systemic testing of
ribozymes
directed against H- and K-ras in the murine cancer models (primary and
secondary
metastasis) discussed above.
Four of the current synthetic ribozymes directed against human K- ras will
cleave
homologous mouse K-ras targets at four sites and inhibit cultured rat aortic
smooth
muscle cell proliferation.
Anti-c fos ribozymes
The protein product of the proto-oncogene c fos is a nuclear transcription
factor
which is involved in tumorigenesis. In support of the possible use of
systemically
administered ribozymes directed against c fos, null mouse mutations of c fos
have been
shown to result in viable mice. Using this mouse model, it has been shown that
c fos is
CA 02288640 1999-11-OS
WO 98/50530 PCTNS98109249
8$
important in malignant conversion of papillomas. Additionally, c fos has been
shown to
up-regulate tumor metalloproteinases (Schonthal et al. 1988, Cell 54: 325-
334). . It is
possible that c:fos may play a role in tumor angiogenesis as evidenced by VEGF
mRNA
levels being significantly reduced in c fos deficient tumors. It has also been
shown that c-
fos is highly expressed in some B-16 cell and human melanoma cell lines
(Kroumpouzos
et al. 1994, Pigment Cell Res. 7: 348-353; Nakayama et al. 1995, J. Dermatol.
22: 549-
559; Pens et al. 1991, Arch. Dermatol. Res. 283: S00-505). The expression of c
fos may
be directly proportional to metastatic potential in B-16 melanoma cell lines.
With this
evidence, it is reasonable to conclude that c fos represents a suitable
systemic ribozyme
target in either the Lewis lung, B-16 melanoma, or human melanoma models.
Delivery of ribozymes and ribozvme formulations in the Lewis luns model
Several ribozyme formulations, including cationic lipid complexes which may be
useful for inflammatory diseases (e.g., DIMRIE/DOPE, etc.) and RES evading
liposomes
which may be used to enhance vascular exposure of the ribozymes, are of
interest in
cancer models due to their presumed biodistribution to the lung. Thus,
liposome
formulations can be used for delivering ribozymes to sites of pathology linked
to an
angiogenic response.
The sequences of the ribozymes that are chemically synthesized, useful in this
study, are non-limiting examples. Those in the art will recognize that these
sequences are
representative only of many more such sequences where the enzymatic portion of
the
ribozyme (all but the binding arms) is altered to affect activity. For
example, stem-loop II
sequence of hammerhead ribozymes can be altered (substitution, deletion,
and/or
insertion) to contain any sequences provided a minimum of two base-paired stem
structure can form. Similarly, stem-loop IV sequence of hairpin ribozymes
listed in
Tables IV (5'-CACGUUGUG-3') can be altered (substitution, deletion, and/or
insertion)
to contain any sequence, provided a minimum of two base-paired stem structure
can
form. Preferably, no more than 200 bases are inserted ax these locations. The
sequences
listed in Tables III and IV may be formed of ribonucleotides or other
nucleotides or non-
CA 02288640 1999-11-OS
WO 98/50530 g6 PCT/US98/09249
nucleotides. Such ribozymes (which have enzymatic activity) are equivalent to
the
ribozymes described specifically in the Tables.
Tar et sites
Targets for useful ribozymes can be determined as disclosed in Draper et al.,
WO
S 93/23569; Sullivan et al., WO 93/23057; Thompson et al., WO 94/02595; Draper
et al.,
WO 95/04818; McSwiggen et al., US Patent No. 5,525,468 and hereby incorporated
by
reference herein in totality. Rather than repeat the guidance provided in
those documents
here, below are provided specific examples of such methods, not limiting to
those in the
art. Ribozymes to such targets are designed as described in those applications
and
synthesized to be tested in vitro and in vivo, as also described. Such
ribozymes can also
be optimized and delivered as described therein.
The sequence of human c-raf mRNAs were screened for optimal ribozyme target
sites using a computer folding algorithm. Hammerhead or hairpin ribozyme
cleavage
sites were identified. These sites are shown in Tables XII-XIX (All sequences
are 5' to 3'
in the tables) The nucleotide base position is noted in the Tables as that
site to be cleaved
by the designated type of ribozyme. The nucleotide base position is noted in
the Tables
as that site to be cleaved by the designated type of ribozyme.
Because Raf RNAs are highly homologous in certain regions, some ribozyme
target
sites are also homologous (see Table XVIII and XIX). In this case, a single
ribozyme
will target different classes of Raf RNA. The advantage of one ribozyme that
targets
several classes of Raf RNA is clear, especially in cases where one or more of
these RNAs
may contribute to the disease state.
Hammerhead or hairpin ribozymes were designed that could bind and were
individually analyzed by computer folding (Jaeger et al., 1989 Proc. Natl.
Acad. Sci. USA,
86, 7706) to assess whether the ribozyme sequences fold into the appropriate
secondary
structure. Those ribozymes with unfavorable intramolecular interactions
between the
binding arms and the catalytic core are eliminated from consideration. Varying
binding
CA 02288640 1999-11-OS
WO 98/50530 g~ PCT/US98/09249
arm lengths can be chosen to optimize activity. Generally, at least 5 bases on
each arm
are able to bind to, or otherwise interact with, the target RNA. Ribozymes of
the
hammerhead or hairpin motif were designed to anneal to various sites in the
mRNA
message. The binding arms are complementary to the target site sequences
described
S above.
Examples
The following are non-limiting examples showing the selection, isolation,
synthesis and activity of enzymatic nucleic acids of the instant invention.
The following examples demonstrate the selection of ribozymes that cleave c-
raf
RNA. The methods described herein represent a scheme by which ribozymes may be
derived that cleave other RNA targets required for cell division. Also
provided is a
description of how such ribozymes may be delivered to cells. The examples
demonstrate
that upon delivery, the ribozymes inhibit cell proliferation in culture and
modulate gene
expression in vivo. Moreover, significantly reduced inhibition is observed if
mutated
ribozymes that are catalytically inactive are applied to the cells. Thus,
inhibition requires
the catalytic activity of the ribozymes.
Example 1: Identification of Potential Ribozyme Cleava,~e Sites in Human c-raf
RNA
The sequence of human c-raf RNA was screened for accessible sites using a
computer folding algorithm. Regions of the mRNA that did not form secondary
folding
structures and contained potential hammerhead and/or hairpin ribozyme cleavage
sites
were identified. The sequences of these cleavage sites are shown in tables XII-
XIX.
Example 2: Selection of Ribozyme Cleava~ ites in Human c-raf RNA
To test whether the sites predicted by the computer-based RNA folding
algorithm
corresponded to accessible sites in c-raf RNA, 20 hammerhead sites were
selected for
analysis. Ribozyme target sites were chosen by analyzing genomic sequences of
human
c-raf (GenBank Accession No. X03484; Bonner et al., 1986, Nucleic Acids
Research, 14,
CA 02288640 1999-11-OS
WO 98/50530 88 PCT/US98/09249
1009-1015) and prioritizing the sites on the basis of folding. Hammerhead
ribozymes
were designed that could bind each target (see Figure 1 ) and were
individually analyzed
by computer folding (Christoffersen et al., 1994 J. Mol. Struc. Theochem, 311,
273;
Jaeger et al., 1989, Proc. Natl. Acad. Sci. USA, 86, 7706) to assess whether
the ribozyme
sequences fold into the appropriate secondary structure. Those ribozymes with
unfavorable intramolecular interactions between the binding arms and the
catalytic core
were eliminated from consideration. As noted below, varying binding arm
lengths can be
chosen to optimize activity. Generally, at least 5 bases on each arm are able
to bind to, or
otherwise interact with, the target RNA. Ribozyme target sites within A-Raf
were chosen
by analyzing genomic sequences of human A-raf 1 (GenBank Accession No. X04790;
Beck et al., 1987, Nucleic Acids Research, 115, 595-609). Ribozyme target
sites within
B-Raf were chosen by analyzing genomic sequences of human B-raf 1 (GenBank
Accession No. M95712 M95720 X54072; Sitanandam et al., 1990, Oncogene, 5, 1775-
1780).
Example 3: Chemical Synthesis and Purification of Ribozymes for Efficient
Cleavage of
c-ra RNA
Ribozymes of the hammerhead or hairpin motif were designed to anneal to
various
sites in the RNA message. The binding arms are complementary to the target
site
sequences described above. The ribozymes were chemically synthesized. The
method of
synthesis used followed the procedure for normal RNA synthesis as described in
Usman
et al., (1987 J. Am. Chem. Soc., 109, 7845), Scaringe et al., (1990 Nucleic
Acids Res., 18,
5433) and Wincott et al., supra, and made use of common nucleic acid
protecting and
coupling groups, such as dimethoxytrityl at the S'-end, and phosphoramidites
at the 3'-
end. The average stepwise coupling yields were >98%.
Inactive ribozymes were synthesized by substituting a U for GS and a U for A14
(numbering from Hertel et al., 1992 Nucleic Acids Res., 20, 3252). Hairpin
ribozymes
were synthesized in two parts and annealed to reconstruct the active ribozyme
(Chowrira
and Burke, 1992 Nucleic Acids Res., 20, 2835-2840). Ribozymes were also
synthesized
CA 02288640 1999-11-OS
WO 98/50530 89 PCT/US98/09249
from DNA templates using bacteriophage T7 RNA polymerase (Milligan and
Uhlenbeck,
" 1989, Methods Enzymol. 180, 51 ). Ribozymes were modified to enhance
stability by
modification with nuclease resistant groups, for example, 2'-amino, 2'-C-
allyl, 2'-flouro,
2'-O-methyl, 2'-H (for a review see Usman and Cedergren, 1992 TIBS 17, 34).
Ribozymes were purified by gel electrophoresis using general methods or were
purified
by high pressure liquid chromatography (HPLC; See Wincott et al., supra; the
totality of
which is hereby incorporated herein by reference) and were resuspended in
water. The
sequences of the chemically synthesized ribozymes used in this study are shown
below in
Table XII-XIX.
Example 4: Ribozyme cleavage of c-raf RNA Target in vitro
Ribozymes targeted to the human c-raf RNA are designed and synthesized as
described above. These ribozymes can be tested for cleavage activity in vitro,
for
example using the following procedure. The target sequences and the nucleotide
location
within the c-raf mRNA are given in Table XII.
Cleavage Reactions: Full-length or partially full-length, internally-labeled
target
RNA for ribozyme cleavage assay is prepared by in vitro transcription in the
presence of
~a_32P~ CTP, passed over a G 50 Sephadex column by spin chromatography and
used as
substrate RNA without further purification. Alternately, substrates are 5'-32P-
end labeled
using T4 polynucleotide kinase enzyme. Assays are performed by pre-warming a
2X
concentration of purified ribozyme in ribozyme cleavage buffer (50 mM Tris-
HCI, pH 7.5
at 37°C, IO mM MgCl2) and the cleavage reaction was initiated by adding
the 2X
ribozyme mix to an equal volume of substrate RNA (maximum of 1-5 nM) that was
also
pre-warmed in cleavage buffer. As an initial screen, assays are carried out
for 1 hour at
37°C using a final concentration of either 40 nM or 1 mM ribozyme,
i.e., ribozyrne
excess. The reaction is quenched by the addition of an equal volume of 95%
formamide,
20 mM EDTA, 0.05% bromophenol blue and 0.05% xylene cyanol after which the
sample is heated to 95°C for 2 minutes, quick chilled and loaded onto a
denaturing
CA 02288640 1999-11-OS
WO 98/50530 90 PCT/US98/09249
polyacrylamide gel. Substrate RNA and the specific RNA cleavage products
generated
by ribozyme cleavage are visualized on an autoradiograph of the gel. The
percentage of
cleavage is determined by Phosphor Imagery quantitation of bands representing
the
intact substrate and the cleavage products.
Example 5: Ability of c-raf Ribozymes to Inhibit Smooth Muscle Cell
Proliferation:
Ribozymes targeting sites in c-Raf mRNA were synthesized using modifications
that confer nuclease resistance (Beigelman, 1995, J. Biol. Chem. 270, 25702).
The
ribozymes were screened for their ability to inhibit cell proliferation in
serum-starved
primary rat aortic smooth muscle cells as described by Jarvis et al. (199b,
RNA 2, 419;
incorporated by reference herein). The ribozyme targeting site represented by
Seq ID Nos
175 and 198 showed particularly high activity in inhibiting cell
proliferation. An inactive
control ribozyme was synthesized which had identical substrate binding arms
but
contained mutations in the catalytic core that eliminate cleavage activity.
Inhibition of
cell proliferation by active versus inactive c-Raf ribozymes is shown in
Figures 37 and
38. The data are presented as proliferation relative to the serum-stimulated
untreated
control cells. Clearly the active ribozyme is showing substantial inhibition
relative to
both the untreated control and its corresponding inactive control, thus
indicating that the
inhibition of proliferation is mediated by ribozyme-mediated cleavage of c-
Raf.
In several other systems, cationic lipids have been shown to enhance the
bioavailability of oligonucleotides to cells in culture (Bennet, C. F., et
al., 1992, Mol.
PharmacoloQV, 41, 1023-1033). In many of the following experiments, ribozymes
were
complexed with cationic lipids. The cationic lipid, Lipofectamine (a 3:1 (w/w)
formulation of DOSPA (2,3-dioleyloxy-N-[2(sperminecarboxamido)ethyl]-N,N-
dimethyl-1-propanaminium trifluoroacetate) and dioleoyl
phosphatidylethanolamine
(DOPE)), was purchased from Life Technologies, Inc. DMRIE (N [1-(2,3-
ditetradecyloxy)propyl]-N,N dimethyl-N hydroxyethylammonium bromide) was
obtained
from VICAL. DMRIE was resuspended in CHC13 and mixed at a 1:1 molar ratio with
dioleoyl phosphatidylethanolamine (DOPE). The CHCl3 was evaporated, the lipid
was
CA 02288640 1999-11-OS
WO 98/50530 91 PCT/US98/09249
resuspended in water, vortexed for 1 minute and bath sonicated for 5 minutes.
Ribozyme
and cationic lipid mixtures were prepared in serum-free DMEM immediately prior
to
addition to the cells. DMEM plus additives was warmed to room temperature
(about 20-
25°C), cationic lipid was added to the final desired concentration and
the solution was
S vortexed briefly. RNA oligonucleotides were added to the final desired
concentration and
the solution was again vortexed briefly and incubated for 10 minutes at room
temperature.
In dose response experiments, the RNA/lipid complex was serially diluted into
DMEM
following the 10 minute incubation.
Serum-starved smooth muscle cells were washed twice with PBS, and the
RNA/lipid complex was added. The plates were incubated for 4 hours at
37°C. The
medium was then removed and DMEM containing 10% FBS, additives and 10 pM
bromodeoxyuridine (BrdU) was added. In some wells, FBS was omitted to
determine the
baseline of unstimulated proliferation.
The plates were incubated at 37°C for 20-24 hours, fixed with 0.3%
H202 in
1 S 100% methanol, and stained for BrdU incorporation by standard methods. In
this
procedure, cells that have proliferated and incorporated BrdU stain brown; non-
proliferating cells are counter-stained a light purple. Both BrdU positive and
BrdU
negative cells were counted under the microscope. 300-600 total cells per well
were
counted. In the following experiments, the percentage of the total cells that
have
incorporated BrdU (% cell proliferation) is presented. Errors represent the
range of
duplicate wells. Percent inhibition then is calculated from the % cell
proliferation values
as follows: % inhibition = 100 - 100((Ribozyme - 0% serum)l(Control - 0%
serum)).
From this initial screen, hammerhead ribozyme targeted against c-raf site 1120
(Figure 36) was further tested. The active ribozyme was able to inhibit
proliferation of
smooth muscle cell, whereas, the control inactive ribozyme, that cannot cleave
c-raf RNA
due to alterations in their catalytic core sequence, fails to inhibit smooth
muscle cell
proliferation (Figure 37). Thus, inhibition of cell proliferation by these
hammerhead
CA 02288640 1999-11-OS
WO 98/50530 92 PCT/US98/09249
sequences is due to their ability to cleave c-raf RNA, and not because of any
non-
ribozyme activity.
Example 6: Oligonucleotide desi~reparation for cloning Defined and Random
Libraries
The DNA oligonucleotides used in this study to construct Defined and Random
Ribozyme Libraries were purchased from Life Technologies (BRL). A schematic of
the
oligonucleotide design used to construct said Defined or Comprehensive
Ribozyme
Libraries is shown in Figure 8. This example is meant to illustrate one
possible means to
construct such libraries. The methods described herein are not meant to be
inclusive of all
possible methods for constructing such libraries. The oligonucleotides used to
construct
the hammerhead ribozyme libraries were designed as follows:
5'-CGAAATCAATTG-(N 1 )X- { CatalyticCore } -(N2)x-CGTACGACACGAAAGTATCG-3'
Where NI = the Stem I target-specific binding arm of length x, Catalytic Core
=
the hammerhead catalytic domain 5'-CTGATGAGGCCGUUAGGCCGAAA-3', and N2
= the Stem III target specific binding arm of length x. The oligonucleotides
were
designed to self prime via formation of a stem-loop structure encoded at the
3' ends of the
oligos (Figure 8A). This intramolecuiar interaction favored an unbiased
extension of
complex pools of ribozyme-encoding oligonucleotides. In the case of Defined
Ribozyme
Library described below (Figures 9-10), N1 and N2 were 8 nt each and were
designed to
be complimentary to the RNA encoded by the purine nucleoside phosphorylase
(PNP)
gene. In the case of Random Ribozyme Libraries, N1 and N2 were randomized
during
synthesis to produce a single pool of all possible hammerhead ribozymes.
In the example shown (Figures 9-10), oligonucleotides encoding 40
different PNP-specific hammerhead ribozymes (greater than 40 ribozymes can be
used)
were pooled to a final concentration of 1 pM total oligonucleotides (2.5 nM
each
individual oligo). Oligos were heated to 68°C for 30 min and then
cooled to ambient
temperature to promote formation of the 3' stem-loop for self priming (Figure
8A). The
CA 02288640 1999-11-OS
WO 98/50530 93 PCT/US98/09249
3' stem loop was extended (Figure 8B) using Klenow DNA polymerase ( 1 p.M
total
oligonucleotides in 1 ml of 50 mM Tris pH 7.5, lOmM MgCl2, 100 pg/ml BSA. 25
~g M
dNTP mix, and 200 U Klenow) by incubating for 30 min at 37°C. The
reaction mixtures
were then heated to 65°C for 15 min to inactivate the polymerase. The
double-stranded
oligos (approximately 30 fig) were digested with the 100 U of the 5'
restriction
endonuclease Mfe I (NEB) as described by the manufacturer, then similarly
digested with
the 3' restriction endonuclease BsiWI (Figure 8C). To reduce the incidence of
multiple
ribozyme inserts during the cloning steps, the cleaved products were treated
with Calf
Intestinal Phosphatase (CIP, Boehringer Mannheim) as described by the
manufacturer to
remove the phosphate groups at the 5' ends. This step inhibits intra- and
intermolecular
ligation of the ribozyme-encoding fragments. Full-length product corresponding
to the
double-stranded, restriction digested and phosphatase-treated products was gel-
purified
following electrophoresis through 10% non-denaturing acrylamide gels prior to
cloning to
enrich for full-length material.
Example 7: Clonine of Defined and Random Libraries
The cloning vectors used contained the following cloning sites: 5'- MfeI - Cla
I -
BsiWI -3'. Vectors were digested with Mfe I and BsiWI prior to use. Thus,
vectors
cleaved with both enzymes should lack the Cla I site present between the
sites, while
vectors cleaved with only one of the enzymes should still retain the Cla I
site. Pooled
oligos were ligated to vector using a 2:1 or 5:1 molar ratio of double-
stranded oligo to
vector in SO-mL reactions containing 500 ng vector and S U ligase in lx ligase
buffer
(Boehringer Mannheim). Ligation reactions were incubated over night at
16°C, then
heated to 65°C 10 min to inactivate the ligase enzyme. The desired
products contain a
single ribozyme insert and lack the original Cla I site included between the
Mfe I and
BsiWI cloning sites. Any unwanted, background vector lacking ribozyme inserts
and thus
still containing the Cla I sites were inactivated by cleaving the product with
5 U of the
restriction endonuclease Cla I for 1 h at 37°C. Approximately 150 ng of
ligated vector
CA 02288640 1999-11-OS
WO 98!50530 94 PCTNS98/09249
was used to transform 100 ~1 XL-2 Blue competent bacteria as described by the
supplier
(Stratagene).
Example 8: Simultaneous screening of 40 different ribozymes tar etin~ PNP
usinu
Defined Ribozyme Libraries.
A Defined Ribozyme Library containing 40 different hammerhead ribozymes
targeting PNP was constructed as described above (Figures 8-10). PNP is an
enzyme that
plays a critical role in the purine metabolic/salvage pathways. PNP was chosen
as a target
because cells with reduced PNP activity can be readily selected from cells
with wild-type
activity levels using the drug 6-thioguanosine. This agent is not toxic to
cells until it is
converted to 6-thioguanine by PNP. Thus, cells with reduced PNP activity are
more
resistant to this drug and can be selectively grown in concentrations of 6-
thioguanosine
that are toxic to cells with wild-type activity levels.
The PNP-targeted Defined Ribozyme Library expression vectors were
converted into retroviral vector particles, and the resulting particles were
used to
transduce the Sup T1 human T cell line. A T-cell line was chosen for study
because T
lymphocytes are more dependent on the purine salvage pathway and thus are
highly
susceptible to 6-thioguanosine killing. Two weeks after transduction, the
cells were
challenged with 10 mmol 6-thioguanosine. Resistant cells began to emerge two
weeks
after initiation of selection. 6-Thioguanosine-resistant cells were harvested,
and the
ribozyme-encoding region of the expression vector was amplified using PCR and
sequenced. The sequence pattern of the ribozyme region in the selected cells
was
significantly different from that produced from the starting library shown in
Figure 9. In
the original library, sequences of the binding arms were ambiguous due to the
presence of
all 40 PNP-targeted ribozymes (Figure 9). However, the sequence of the
ribozyme-
encoding regions from the 6-thioguanosine selected cells was clearly weighted
towards
one of the ribozymes contained in the original pool - the ribozyme designed to
cleave at
nucleotide #32 of PNP mRNA. These data suggests that the ribozyme targeting
position
CA 02288640 1999-11-05
WO 98/50530 95 PCT/US98/09249
32 of the PNP mRNA appears to be more active than the other 39 PNP-targeted
ribozymes included in the pool.
Example 9: Optimizin~~oop II sequence of a Hammerhead Ribozvme lHH-Bl for
Enhanced Catalytic Rates
To test the feasibility of the combinatorial approach described in Figure 12
approach, Applicant chose to optimize the sequence of loop-II of a hammerhead
ribozyme
(HH-B) (see Figure 22). Previous studies had demonstrated that a variety of
chemical
modifications and different sequences within loop-II may have significant
effects on the
rate of cleavage in vitro, despite the fact that this sequence is not
phylogenetically
conserved and can in fact be deleted completely. According to the standard
numbering
system for the hammerhead ribozyme, the four positions within loop II are
numbered 12.1,
12.2, 12.3, and 12.4. The Starting Ribozyme (HH-B) contained the sequence
G,2., A,~.Z A
123 A 12.4' For simplicity, the four positions will be numbered 5' to 3':
G,Z,,= l; A,~.2 2; A
~z.3=3~ A ,z.a 4. The remainder of the hammerhead ribozyme "template" remained
constant and is based on a previously described hammerhead motif (Draper et
al.,
International PCT Publication No. WO 95/13380, incorporated by reference
herein).
A strategy for optimizing the four {number of Classes = 4) loop-II positions
is
illustrated in Figure 180. The four standard ribose nucleotides (A, C, U and
G) were
chosen to construct the ribozyme pools (n = 4). In the first step, four
different pools were
synthesized by the nucleotide building block mixing approach described herein.
Applicant first chose to "fix" (designated F} position 3 because preliminary
experiments
indicated that the identity of the base at this position had the most profound
effects on
activity; positions 1, 2 and 4 are random. The four pools were assayed under
stoichiometric conditions (1pM ribozyme; lp,M substrate), to help ensure that
the entire
population of ribozymes in each pool was assayed. Substrate and ribozyme were
pre-
annealed and the reactions were initiated with the addition of IOmM MgCl2. The
rate of
cleavage for each library was derived from plots of fraction of substrate
cleaved as a
function of time. Reactions were also performed simultaneously with the
starting
CA 02288640 1999-11-OS
WO 98!50530 96 PCT/US98/09249
ribozyme (i.e., homogenous, loop-II = GAAA). The relative rate of cleavage for
each
library (kre,) was calculated by dividing the observed rate of the library by
the rate of the
control/starting ribozyme and is plotted in Figure 21. The error bars indicate
the standard
error derived from the curve fits. The results show that all four pools had
similar rates
(k,~,); however, the library possessing "U" at position 3 was slightly faster.
Ribozyme pools were again synthesized (Class 2) with position 3 being made
constant (U3), position 4 was fixed (Fq) and positions 1 and 2 were random
(X). The four
pools were assayed as before; the pool containing "A" at position 4 was
identified as the
most desirable pool. Therefore, during the synthesis of the next pool (Class
3), positions
3 and 4 were constant with U3 and A4, position 2 was fixed (Fz) and position 1
was
random (X). The four pools were again assayed; all four pools showed very
similar, but
substantially elevated rates of cleavage. The pool containing U at position 2
was
identified as the fastest. Therefore, during the synthesis of the final four
ribozymes (Class
4), position 3, 4 and 2 were made constant with U,, A4 and Uz; position 1 was
fixed with
A, U, C or G. The final ribozyme containing G at position 4 was clearly
identified as the
fastest ribozyme, allowing the identification of G,z., U,z.z U,2.3 A,z.4 as
the optimized
ribozyme motif.
To confirm that the final ribozyme (G,z_, U,z,z U,z., A,z.4) was indeed faster
that the
starting ribozyme (G,z., A,z,z A,z.3 A,z.4), we compared the two ribozymes
(illustrated in
Figure 22) under single-turnover conditions at saturating ribozyme
concentrations. The
observed rates should therefore measure the rate of the chemical step, kz. The
fraction of
substrate remaining uncleaved as a function of time is shown in Figure 22
(lower panel),
and the derived rate contents are shown. The results show that the optimized
ribozyme
cleaves >10 times faster (3.7 miri' vs. 0.35 miri') than the starting
ribozyme.
Example 10: Optimizing Core Chemistry of a Hammerhead Ribozyme (HH-A)
To further test the feasibility of the approach described in Figure 12, we
chose to
optimize the three pyrimidine residues within the core of a hammerhead
ribozyme (HH-
CA 02288640 1999-11-OS
WO 98/50530 ~~ PCT/US98/09249
A). These three positions (shown in Figure 13 as U7, U4 and C3) were chosen
because
previous studies indicated that these positions are critical for both
stability (Beigelman et
al., 1995, supra) and activity {Ruffner et al." 1990, supra; Burgin et al.,
1996, supra) of
the ribozyme. According to the standard numbering system for the hammerhead
ribozyme, the three pyrimidine positions are 7, 4 and 3. For construction of
the libraries,
the ribozyme positions are numbered 3' to 5' : position 24 = 7, position 27 =
4, and
position 28 = 3 (see Figure 13). The remainder of the hammerhead ribozyme
"template"
remained constant and is based on a previously described hammerhead motif
(Thompson
et al., US Patent No. 5,610,052, incorporated by reference herein). The
starting ribozyme
template is targeted against nucleotide position 823 of k-ras mRNA (Site A).
Down
regulation of this message, as a result of ribozyme action, results in the
inability of the
cells to proliferate. Therefore in order to optimize a ribozyme, we chose to
identify
"variants" which were successful in inhibiting cell proliferation.
Cell Culture Assav:
Ribozyme: lipid complex formation
Ribozymes and LipofectAMINE were combined DMEM at final concentrations of
100 nM and 3.6 pM, respectively. Complexes were allowed to form for 15 min at
37 C
in the absence of serum and antibiotics.
Proliferation Assay
Primary rat aortic smooth muscle cells (RASMC) were seeded at a density of
2500
cells/well in 48 well plates. Cells were incubated overnight in DMEM,
supplemented
with 20% fetal bovine serum (FBS), Na-pyruvate, penicillin (50 U/ml), and
streptomycin
(SO pg/ml). Subsequently cells were rendered quiescent by a 48 h incubation in
DMEM
with 0.5% FBS.
Cells were incubated for 1.5 h with serum-free DMEM ribozyme:lipid complexes.
The medium was replaced and cells were incubated for 24 h in DMEM with 0.25%
FCS.
CA 02288640 1999-11-OS
WO 98/50530 98 PCT/US98/09249
Cells were then stimulated with 10% FBS for 24 h. 3H-thymidine (0.3 pCi//well)
was present for the last 12 h of serum stimulation.
At the end of the stimulation period the medium was aspirated and cells were
fixed in icecold TCA (10%) for 15 min. The TCA solution was removed and wells
were
washed once with water. DNA was extracted by incubation with 0.1 N NaOH at RT
for
min. Solubilized DNA was quantitatively transferred to minivials. Plates were
washed once with water. Finally, 'H-thymidine incorporation was determined by
liquid
scintillation counting.
A strategy for optimizing the three (number of Class = 3) pyrimidine residues
is
10 illustrated in Figure 20. Ten different nucleotide analogs (illustrated in
Figure 15) were
chosen to construct the ribozyme library (n = 10). In the first step, ten
different pools
(Class 1 ) were synthesized by the mix and split approach described herein.
Positions 24
and 27 were random and position 28 was fixed with each of the ten different
analogs. The
ten different pools were formulated with a cationic lipid (Jarvis et al.,
1996, RNA, 2,419;
15 incorporated by reference herein), delivered to cells in vitro, and cell
proliferation was
subsequently assayed (see Figure 16). A positive control (active ribozyme)
inhibited cell
proliferation by ~50% and an inactive control (inactive) resulted in a less
than 25%
reduction in cell proliferation. The ten ribozyme pools resulted in
intermediate levels of
reduction. However, the best pool could be identified as Xz4 XZ~ 2'-MTM-UZ$
(positions
24 and 27 random; 2'-O-MTM-U at position 28). Therefore, a second ribozyme
library
(Class 2) was synthesized with position 28 constant (2'-O-MTM-U); position 24
was
random (X24) and position 27 was fixed with each of the ten different analogs
(FZ,).
Again, the ten pools were assayed for their ability to inhibit cell
proliferation. Among
Class 2, two pools inhibited proliferation equally well: Xz4 2'-C-allyl-U2, 2'-
O-MTM-UZg
and X24 2'-O-MTM-C2, 2'-O-MTM-UzB. Because a single "winner" could not be
identified in Class 2, position 27 was made constant with either 2'-Gallyl-U
or with 2'-
O-MTM-C and the ten analogs were placed individually at position 24 (Class 3).
Therefore in Class 3, twenty different ribozymes were assayed for their
ability to inhibit
CA 02288640 1999-11-OS
WO 98/50530 99 PCT/US98/09249
cell proliferation. Because both positions 27 and 28 are constant, the final
twenty
ribozymes contain no random positions. Thus in the final group (Class 3), pure
ribozymes and not pools were assayed. Among the final groups four ribozymes
inhibited
cell proliferation to a greater extent than the control ribozyme (Figure 22).
These four
winners are illustrated in Figure 23A. Figure 23B shows general formula for
four
different motifs. A formula for a novel ribozyme motif is shown in Figure 18.
Example 11: Identifyina Accessible Sites for Ribozyme Action in a target
In the previous two examples {9 and 10), positions within the catalytic domain
of
the hammerhead ribozyme were optimized. The number of groups that needed to be
tested equals = the total number of positions within the ribozyme that were
chosen to be
tested. A similar procedure can be used on the binding arms of the ribozyme.
The
sequence of the binding arms determines the site of action of the ribozyme.
The
combinatorial approach can be used to identify those sites by essentially
testing all
possible arm sequences. The difficulty with this approach is that ribozymes
require a
certain number of base pairs (12-16) in order bind tightly and specifically.
According to
the procedure outlined above, this would require 12-16 different groups of
ribozyme
pools; 12-16 positions would have to be optimized which would require 12-16
different
groups being synthesized and tested. Each pool would contain the four
different
nucleotides (A, C, U and G) or nucleotide analogs (n = 4). It would be very
time
consuming to test each group, identify the best pool, synthesize another group
of
ribozyme pools with one additional position constant, and then repeat the
procedure until
all 12-16 groups had been tested. However it is possible to decrease the
number of
groups by testing multiple positions within a single group. In this case, the
number of
pools within a group equals the number of nucleotides or analogs in the random
mixture
(i.e., n) to the w power, where w equals the number of positions fixed in each
group. The
number of groups that need to be synthesized to optimize the final ribozyme
equals the
total number of positions to be optimized divided by the number of positions
(w) tested
CA 02288640 1999-11-OS
WO 98/50530 100 PCT/US98/09249
within each group. The number of pools in each group = nW. The number of
groups =
total number of positions / w.
For example, Figure 23 illustrates this concept on a hammerhead ribozyme
containing 12 base pair binding arms. Each of the two binding arms form 6 base
pairs
with it's corresponding RNA target. It is important to note that for the
hammerhead
ribozyme one residue (A15.1) must remain constant; A15.1 forms a base pair
with a
substrate nucleotide (U16.1) but is also absolutely required for ribozyme
activity. It is the
only residue within the hammerhead ribozyme that is part of both the catalytic
domain,
and the binding domain (arms). In the example this position is not optimized.
In the first
Group, three positions are fixed (designated F) with the four different 2'-O-
methyl
nucleotides (A, C, U and G). The 2'-O-methyl modification stabilizes the
ribozyme
against nuclease degradation and increases the binding affinity with it's
substrate. The
total number of pools in each group does not equal n, as in the previous
examples. The
number of pools in each group equals 4' _ (four analogs)~(number of positions
fixed; 3) _
64. In all 64 pools, all other positions in the arm are made random
(designated X) by the
nucleotide mixing building block approach. The catalytic domain is not
considered in
this example and therefore remains part of the ribozyme template (i.e.,
constant).
In the first step, all 64 ribozyme pools are tested. This test may be cleavage
in
vitro (see Example 9), or efficacy in a cellular (see Example 10) or animal
model, or any
other assayable end-point. This end-point however, should be specific to a
particular
RNA target. For example, if one wishes to identify accessible sites within the
mRNA of
GeneB, a suitable end-point would be to look for decreased levels of GeneB
mRNA after
ribozyme treatment. After a winning pool is identified, since each pool
specifies the
identity of three positions (w), three positions can be made constant for the
next group
(Class 2). Class 2 is synthesized containing 64 different pools; three
positions that were
fixed in Class 1 are now constant (designated Z), three more positions are
fixed (F), and
the remaining positions (X) are a random mix of the four nucleotides. The 64
pools are
assayed as before, a winning pool is identified, allowing three more positions
to be
CA 02288640 1999-11-OS
WO 98/50530 101 PCT/US98/09249
constant in the next Class of ribozyme pools (Class 3} and the process is
repeated again.
In the final Class of ribozymes (Class 4), only two positions are fixed, all
other positions
have been previously fixed. The total number of ribozymes is therefore n'" =
4'- = 16;
these ribozymes also contain no random positions. In the final step (step 4),
the 16
ribozymes are tested; the winning ribozyrne defines the sequence of the
binding arms for
a particular target.
Fixing multiple positions within a single group it is possible to decrease the
overall number of groups that need to be tested. As mentioned, this is
particularly useful
when a large number of different positions need to be optimized. A second
advantage to
this approach is that it decreases the complexity of molecules in each pool.
If one would
expect that many combinations within a given pool will be inactive, by
decreasing the
number of different ribozymes in each pool, it will be easier to identify the
"winning"
pool. In this approach, a larger number of pools have to be tested in each
group, however,
the number of groups is smaller and the complexity of each ribozyme pool is
smaller.
Finally, it should be emphasized there is not a restriction on the number of
positions or
analogs that can be tested. There is also no restriction on how many positions
are tested
in each group.
Example 12: Identifvin~ new RNA targets for Riboz~rmes
As described above for identifying ribozyme-accessible sites, the assayed used
to
identify the "winning" pool of ribozymes is not defined and may be cleavage in
vitro (see
Example 8), or efficacy in a cellular (see Example 9) or animal model, or any
other
assayable end-point. For identifying accessible sites, this end-point should
be specific to
a particular RNA target (e.g., mRNA levels). However, the . end-point could
also be
nonspecific. For example, one could choose a.disease model and simply identify
the
winning ribozyme pool based on the ability to provide a desired effect. In
this case, it is
not even necessary to know what the cellular target that is being acted upon
by the
ribozyme is. One can simply identify a ribozyme that has a desired effect. The
advantage
to this approach is that the sequence of the binding arms will be
complementary to the
CA 02288640 1999-11-OS
WO 98/50530 102 PCT/US98/09249
RNA target. It is therefore possible to identify gene products that are
involved in a
disease process or any other assayable phenotype. One does not have to know
what the
target is prior to starting the study. The process of identifying an optimized
ribozyme
(arm combinatorial) identifies both the drug {ribozyme) and the RNA target,
which may
S be a known RNA sequence or a novel sequence leading to the discovery of new
genes.
Example 13: Identifying New Ribozyme Catalytic Domains
In the previous two examples, positions within the binding domain of the
hammerhead ribozyme were varied and positions within the catalytic domain were
not
changed. Conversely, it is possible to vary positions within the catalytic
domain, without
changing positions within the binding arms, in order to identify new catalytic
motifs. An
example is illustrated in Figure 24. The hammerhead ribozyme, for example
comprises
about 23 residues within the catalytic domain. It is unclear how many of these
23
positions are required to obtain a functional catalytic domain, however it is
reasonable to
presume that if a large number of functionally diverse nucleotide analogs can
be used to
construct the pools, a relatively small number of positions could constitute a
functional
catalytic domain. This may especially be true if analogs are chosen that one
would expect
to participate in catalysis (e.g., acid/base catalysts, metal binding, etc.).
In the example
illustrated in Figure 24, four positions (designated 1, 2, 3 and 4) are
chosen. In the first
step, ribozyme libraries (Class 1) are constructed: position 1 is fixed (F,)
and positions 2,
3 and 4 are random (X2, X3 and X4, respectively). In step 2, the pools (the
number of
pools tested depends on the number of analogs used; n) are assayed for
activity. This
testing may be performed in vitro or in a cellular or animal model. Whatever
assay that is
used, the pool with the most activity is identified and libraries (class 2)
are again
synthesized with position 1 now constant (Z,) with the analog that was
identified in class
1. In class 2, position 2 is fixed (FZ) and positions 3 and 4 are random (X3
and X4}. This
process is repeated until every position has been made constant, thus
identifying the
catalytic domain or a new motif.
CA 02288640 1999-11-OS
WO 98/50530 103 PCT/US98/09249
EXAMPLE 14: Determination of CouulinE Efficiency of the nhosnhoramidite
derivatives
of 2'-C-allyl-uridine, l ; 4'-thio-cytidine. 2; 2'-methylthiomethyl-uridine,
3; 2'-
methylthiometh~ytidine, 4; 2'-amino-uridine, 5; N3-methy-uridine, 6: 1-b-D-
(ribofuranosyl)=pvridin-4-one, 7; 1-b-D-(ribofuranosvl)-pyridin-2-one, 8; 1-b-
D-
(ribofuranosyl)-phenyl, 9; 6-methyl-uridine. 10 to be used in a split and mix
approach.
The determination of the coupling efficiency of amidites 1 to 10 was assessed
using ten model sequences agacXGAuGa (where upper case represents
ribonucleotide
residues, lower case represents 2'-O-methyl ribonucleotide residues and X is
amidites 1 to
10, to be used in the construction of a hammerhead ribozyme library wherein
the
modified amidites 1 to 10 would be incorporated. Ten model sequences were
synthesized
using ten 0.112 g aliquots of 5'-O-DMT-2'-O-Me-Adenosine Polystyrene {PS)
solid-
support loaded at 22.3 pmol/g and equivalent to a 2.5 pmol scale synthesis.
Synthesis of
these ten decamers were performed on ABI 394 DNA synthesizer (Applied
Biosystems,
Foster City, Calif.) using standard nucleic acid synthesis reagents and
synthesis protocols,
with the exception of an extended (7.5 min) coupling time for the
ribonucleoside
phosphoramidites and phosphoramidites 1, 2, 3, 4, 6, 7, 8, 9, 10, 12.5 min
coupling time
for the 2'-amino-uridine phosphoramidite, amidite 5 and 2.5 min coupling time
for the 2'-
O-methyl nucleoside phosphoramidites.
Oligomers were cleaved from the solid support by treatment with a 3:1 mixture
of
ammonium hydroxide:absolute ethanol at 65 degree C for 4 hrs followed by a
desiiylation
treatment and butanol precipitation as described in Wincott et al. (Wincott et
al, Nucleic
Acids Res, 1995, 23, 2677-2684; incorporated by reference herein).
Oligonucleotides
were analyzed directly on an anion-exchange HPLC column (Dionex, Nucleopac, PA-
100, 4x250 mm) using a gradient of 50% to 80% of B over 12 minutes (A = 10 mM
sodium perchlorate, 1 mM Tris, pH 9.43; B = 300 mM sodium perchlorate, 1 mM
Tris,
- pH 9.36) and a Hewlett-Packard 1090 HPLC system.
The average stepwise yield (ASW~, indicating the coupling efficiency of
phosphoramidites, 1 to 10, were calculated from peak-area percentages
according to the
CA 02288640 1999-11-OS
WO 98/50530 104 PCT/US98/09249
equation ASWY = (FLP%)"° where FLP% is the percentage full-length
product in the
crude chromatogram and n the number of synthesis cycles. ASWY ranging from of
96.5% to 97.5% were obtained for phosphoramidites, 1 to 10. The experimental
coupling
efficiencies of the phosphoramidites 1 to 10, as determined using a standard
spectrophotometric dimethoxytrityl assay were in complete agreement with the
ASWY
and were judged satisfactory to proceed with the X24, X27, X28 ribozyme
library
synthesis.
EXAMPLE 15: Determination of optimal relative concentration of a mixture of 2'-
O-
methyl-~uanosine, cytidine uridine and adenosine uroviding-equal
representation of the
four nucleotides.
A mixture N, composed of an equimolar mixture of the four 2'-O-Me- nucleoside
phosphoramidites (mG=2'-O-methyl guanosine; mA=2'-O-methyl adenosine; mC=2'-O-
methyl cytidine; mU=2'-O-methyl uridine) was used in the synthesis of a model
sequence
TTXXXXTTB, where T is 2'-deoxy-thymidine and B is a 2'-deoxy-inverted abasic
1 S polystyrene solid-support as described in Example 14. After standard
deprotection
(Wincott et al., supra), the crude nonamer was analyzed on an anion-exchange
HPLC
column (see example 6). From the HPLC analysis, an averaged stepwise yield
(ASWY)
of 99.3% was calculated (see example 14) indicating that the overall coupling
efficiency
of the mixture N was comparable to that of 2'-deoxythymidine. To further
assess the
relative incorporation of each of the components within the mixture, N, the
full-length
product TTXXXXTTB (over 94.3% at the crude stage) was further purified and
subjected
to base composition analysis as described herein. Purification of the FLP from
the
failures is desired to get accurate base composition.
Base composition analysis summary:
A standard digestion/HPLC analysis was performed: To a dried sample containing
0.5 A260 units of TTXXXXTTB, 50 p,l mixture, containing 1 mg of nuclease
P1
(550 units/mg), 2.85 ml of 30 mM sodium acetate and 0.3 ml of 20 mM aqueous
zinc
chloride, was added. The reaction mixture was incubated at 50 degrees C
overnight.
CA 02288640 1999-11-OS
WO 98/50530 105 PCT/US98/09249
Next, 50 pl of a mixture comprising 500 ~.1 of alkaline phosphatase (1
units/p,l), 312 ~.l of
500 mM Tris pH 7.5 and 2316 p.l water was added to the reaction mixture and
incubated
at 37 degrees C for 4 hours. After incubation, the samples were centrifuged to
remove
sediments and the supernatant was analyzed by HPLC on a reversed-phase C18
column
equilibrated with 25 mM KH2P04. Samples were analyzed with a 5% acetonitrile
isocratic gradient for 8 min followed by a 5% to 70% acetonitrile gradient
over 8 min.
The HPLC percentage areas of the different nucleoside peaks, once corrected
for
the extinction coefficient of the individual nucleosides, are directly
proportional to their
molar ratios.
The results of these couplings are shown in Table IV.
Nucleoside dT 2'-OMe-C 2'-OMe-U 2'-OMe-G 2'-OMe-A
0.1 M 0.025M 0.025M 0.025M 0.025M
area 43.81 6.04 14.07 18.54 17.54
Epsilon 260 8800 7400 10100 11800 14900
nm
moles 0.00498 0.00082 0.00139 0.00157 0.00118
equivalent 4 0.656 1.119 1.262 0.946
As can be seen in Table IV, the use of an equimolar mixture of the four 2'-O-
methyl phosphoramidites does not provide an equal incorporation of all four
amidites, but
favors 2'-O-methyl-U and G and incorporates 2'-O-methyl-A and C to a lower
efficiency.
To alleviate this, the relative concentrations of 2'-O-methyl-A, G, U and C
amidite were
adjusted using the inverse of the relative incorporation as a guide line.
After several
iterations, the optimized mixture providing nearly identical incorporation of
all four
amidites was obtained as shown in Table V below. The relative representation
do not
exceed 12% difference between the most and least incorporated residue
corresponding to
a +/- 6% deviation from equimolar incorporation.
CA 02288640 1999-11-OS
WO 98/50530 106 PCT/US98/09249
NucleosidedT 2'-OMe-C 2'-OMe-U 2'-OMe-G 2'-OMe-A
O.1M 0.032M 0.022M 0.019M 0.027M
area 44.47 8.91 11.81 15.53 19.28
Epsilon 8800 7400 10100 11800 14900
260
nm
moles 0.00505 0.00120 0.00117 0.00132 0.00129
equivalent4 0.953 0.926 1.042 1.024
EXAMPLE 16: A Non-competitive coupling method for the preparation of the X24
X27
and N28 ribozyme library 5'- ascsasasa~ aFX GAX Gag ~c~ aaa Qcc Gaa Arc ccu cB
-3'
wherein 2'-C-allyl-uridine, I ; 4'-thio-cytidine, 2; 2'-methylthiomethyl-
uridine. 3: 2'-
S methylthiomethyl-cytidine, 4; 2'-amino-uridine. 5; N3-methyl-uridine, 6: 1-b-
D-
(ribofuranosyl)-pyrimidine-4-one, 7: 1-b-D-(ribofuranosyl)-pyrimidine-2-one.
8; 1-b-D-
~ofuranosyl)-phenyl, 9; and/or 6-methyl-uridine, 10 are incorporated at the
X24, X27
and F28 positions through the mix and split approach.
The synthesis of ten different batches of 2.5 ~mol scale Gag gcg aaa gcc Gaa
Agc
ccu cB sequence was performed on 2'-deoxy inverted abasic polystyrene solid
support B
on a 394 ABI DNA synthesizer (Applied Biosystems, Foster City, CA). These ten
aliquots were then separately reacted with phosphoramidite building blocks 1
to 10
according to the conditions described in example 11. After completion of the
individual
incorporation of amidites 1 to 10, their coupling efficiencies were determined
to be above
95 % as judged by trityl monitoring. The 10 different aliquots bearing the ten
different
sequences were mixed thoroughly and divided into ten equal subsets. Each of
these
aliquots were then successively reacted with ribo-A, ribo-G amidites and one
of the
amidites 1 to 10. The ten aliquots were combined, mixed and split again in 10
subsets.
At that point, the 10 different polystyrene aliquots, exhibiting the following
sequence: X
GAX Gag gcg aaa gcc Gaa Agc ccu cB, were reacted again with amidites 1 to 10
separately. The aliquots were not mixed, but kept separate to obtain a unique
residue at
CA 02288640 1999-11-OS
WO 98/50530 1~7 PCT/US98109249
the 28th position of each of the ten pools. The ribozyme synthesis was then
finished
' independently to yield ten random ribozymes pools. Each pool comprises a 3'-
terminal
inverted abasic residue B, followed by the sequence Gag gcg aaa gcc Gaa Agc
ccu c,
followed with one random position X in the 24th position corresponding to a
mixture of
S amidites 1 to 10, followed by the sequence GA, followed one random position
X in the
27th position corresponding to a mixture of amidites 1 to 10, followed by a
fixed
monomer F (one of the amidites 1 to 10) in the 28th position and finally the
5'-terminal
sequence aScsasasa g a. This is represented by the sequence notation 5'-
ascsasasag aFX
GAX Gag gcg aaa gcc Gaa Agc ccu cB-3', in which X are random positions and F
is a
unique fixed position. The total complexity of such a ribozyme library was 103
or 1,000
members separated in 10 pools of 100 different ribozyme sequences each.
EXAMPLE 17: Competitive coupling method (monomer mixing_approach~ for the
preparation of the x2_,~, and X30.3s "binding arms" ribozyme library
Synthesis of 5'-xsxsx xFF cuG Au G Agg ccg uua ggc cGA AAF xxx xB-3' is
described, with F being a defined 2'-O-methyl-ribonucleoside chosen among 2'-O-
methyl-ribo-adenosine (mA), -guanosine (mG), -cytidine (mC), -uridine (mL~ and
x
being an equal mixture of 2'-O-methyl-ribo-adenosine, -guanosine, -cytidine, -
uridine.
The syntheses of this ribozyme library was performed with an ABI 394 DNA
synthesizer (Applied Biosystems, Foster City, Calif.) using standard nucleic
acid
synthesis reagents and synthesis protocols, with the exception of an extended
(7.5 min)
coupling time for the ribonucleoside phosphoramidites (upper case) and 2'-
amino-uridine
phosphoramidite, u, (2.5 min) coupling time for the 2'-O-methyl-ribonucleoside
phosphoramidites (lower case) and the 2'-O-methyl-ribonucleoside
phosphoramidites
mixture, n.
Sixty four (64) batches of 0.086 g aliquots of 3'-O-DMT-2'-deoxy-inverted
abasic-Polystyrene (B) solid-support loaded at 29 p,mol/g and equivalent to a
2.5 ~mol
CA 02288640 1999-11-OS
WO 98150530 108 PCT1US98/09249
scale synthesis were individually reacted with a 27:32:19:22 / v:v:v:v
mixture, x, of
mA:mC:mG:mU diluted in dry acetonitrile to 0.1 M as described in example 7.
This
synthesis cycle was repeated for a total of four times. The 64 aliquots were
then grouped
into four subsets of sixteen aliquots (Class 1 ) that were reacted with either
mA, mG, mC,
S mU to synthesize the n6 position. This accomplished, the sequence: 5'- cuG
Au G Agg
ccg uua ggc cGA AA was added onto the 6 position of the 64 aliquots
constituting Class
1. Each subset of Class 1 was then divided into four subsets of four aliquots
(Class 2) that
were reacted with either mA, mG, mC, mU to synthesize the F30 position. Each
subset of
Class 2 was then divided into four subsets of one aliquot (Class 3) that were
reacted with
either mA, mG, mC, mU to synthesize the F31 position. Finally, the random
sequence 5'-
xsxsx x was added onto each of the 64 aliquots.
The ribozyme library yielded sixty four random ribozymes pools each having an
equal mixture of the four 2'-O-methyl-nucleoside at the position x2 to 6 and
x30 to 35,
and a defined 2'-O-methyl-nucleoside chosen among mA, mC, mG, mU at the
positions
F6, F30 and F31. The total complexity of such a "binding arms" ribozyme
library was
41 ~ or 4,194,304 members separated in 64 pools of 65,536 different ribozyme
sequences
each.
EXAMPLE 18: Competitive coupling method~monomer mixing approach) for the
preparation of the position 15 to 18 "loop II" ribozyme library
Synthesis of 5' UCU CCA UCU GAU GAG GCC XXF XGG CCG AAA AUC
CCU 3' is described, with F being a defined ribonucleoside chosen among
adenosine (A),
guanosine (G), cytidine (C), uridine (IJ) and X being an equal mixture of
adenosine (A),
guanosine (G), cytidine (C), uridine (IJ).
The syntheses of this ribozyme library was performed with an ABI 394 DNA
synthesizer (Applied Biosystems, Foster City, Calif.) using standard nucleic
acid
synthesis reagents and synthesis protocols, with the exception of an extended
(7.5 min)
CA 02288640 1999-11-OS
WO 98!50530 109 PCT/US98/09249
coupling time for the ribonucleoside phosphoramidites (A, G, C, U) and the
ribonucleoside phosphoramidite mixture, X.
Four batches (4) of 2.5 p,mol scale of GG CCG AAA AUC CCU sequence were
synthesized on 0.085 g samples of 5'-O-DMT-2'-O-TBDMS-3'-succinyl-uridine-
Polystyrene (U) solid-support loaded at 29.8 ~mol/g. To synthesize the
position X15, the
four aliquots of solid-supports were individually reacted with a 30:26:24:20 /
v:v:v:v
mixture, X, of A:C:G:U diluted in dry acetonitrile to 0.1 M according to the
optimized
conditions for the DNA phosphoramidites mixed-base coupling as described in
the DNA
Synthesis Course Manual published by Perkin-Elmer-Applied Biosystem Division.
(DNA Synthesis Course Manual : Evaluating and isolating synthetic
oligonucleotides, the
complete guide, p. 2-4, Alex Andrus, August 1995). The four aliquots of solid-
supports
were then individually reacted with either of the four ribonucleoside
phosphoramidites
(A, G, C, U) to create the F16 position. The position X17 and X18 were then
added onto
the F16 (either A, G, C or U) of the four aliquots of solid-supports by
repeating twice the
same procedure used for the position X15.
The synthesis of the ribozyme library was then ended by adding the sequence 5'-
UCU CCA UCU GAU GAG GCC on the position X18 of each of the four subsets of the
ribozyme library. The ribozyme library yielded four random ribozymes pools
that each
have an equal mixture of the four ribonucleoside (A, G, C and U) at the
position X15,
X17 and X18, and a discrete ribonucleoside chosen among A, C, G or U at the
positions
F16. The total complexity of such a loop II ribozyme library was 256 members
separated
in 4 pools of 64 different ribozyme sequences.
Example 19: Arm-Combinatorial Library Screening For Bcl-2, K-ras and Urokinase
plasmino~en Activator (UPA)
Substrate synthesis through in vitro transcription: Run-off transcripts for
Bcl-2
- and Kras were prepared using linearized plasmids (975 and 796 nucleotides
respectively).
Transcripts for UPA were produced from a PCR generated DNA fragment containing
a
T7 promoter (400 nucleotides). Transcription was performed using the T7
Megascript
CA 02288640 1999-11-OS
WO 98/50530 110 PCT/US98/09249
transcription kit (Ambion, Inc.) with the following conditions: a SOuI
reaction volume
containing 7.SmM each of ATP, CTP, UTP, and GTP, 2mM guanosine, Sul lOx T7
reaction buffer, Sul T7 enzyme mix, and O.Sug of linearized plasmid or PCR'd
DNA
template. The mixture was incubated at 37°C for 4 hours (6 hours for
transcripts < 500
bases). Guanosine was added to the transcription reactions so that the final
transcript
could be efficiently 5'-end labeled without prior phosphatase treatment.
Transcription
volume was then increased to 200u1 with buffer containing SOmM TRIS pH 7.5,
100mM
KCI, and 2mM MgClz and spin column purified over Bio-Gel P-60 (BioRad)
equilibrated
in the same buffer. 100u1 of transcript was then applied to 750u1 of packed
resin. Spin
column flow-through was used directly in a 5'-end labeling reaction as follows
(100u1
final volume): 82u1 of P-60 spin column purified transcript, 10u1 lOx
polynucleotide
kinase buffer, 4u1 l0U/ul Polynucleotide Kinase (Boehringer/Mannheim) and 4u1
150uCi/ul Gamma-32P-ATP (NEN) were incubated together at 37°C for one
hour. The
reaction volume was increased to 200u1 with buffer containing SOmM TRIS pH
7.5,
100mM KCl and 2mM MgCl2 and the sample was then purified over Bio-Gel P-60
packed spin column as described above. Approximate specific activities of the
5'-end
labeled transcripts were determined via BioScan and stored frozen at -
20°C.
Synthesis ofRibozyme pools:
In vitro ribozyme-transcript cleavage reactions: Cleavage reactions were
carried out as
follows: S'-end labeled transcript (~2-4 x 104 dpm/ul final) was incubated
with lOuM
ribozyme pool in SOmM TRIS pH 7.5, SOmM NaCI, 2mM MgCl2 and 0.01% SDS for 24-
48 hours at room temperature (~22°C). An equal volume of gel loading
dye (95%
formamide, O.O1M EDTA, 0.0375% bromophenol blue, and 0.0375% xylene cyanol)
was
added to stop the reaction and the samples are heated to 95°C.
Reactions (1-2 x 105 dpm
per lane) were run on a 5% denaturing polyacrylamide gel containing 7M urea
and lx
TBE. Gels are dried and imaged using the PhosphorImager system (Molecular
Dynamics). Ambion, Inc. RNA Century Marker Plus RNA standards body labeled in
a
T7 Megascript reaction as described above using 3u1 of lOmCi/ml Alpha-32P-ATP
(BioRad) and O.Sug Century RNA template and subsequently spin column purified
over
CA 02288640 1999-11-OS
WO 98/50530 111 PCT/US98/09249
Bio-Gel P-6 (Bio-Rad) were used as a size reference on the gel. Cleavage
product sizes
were determined using the RNA standards which provided an approximate site of
cleavage (est. Size in Figure). Because each of the ribozyme pools has three
positions
within the binding arms fixed, it is possible to identify all of the potential
ribozyme sites
that can potentially be cleaved by that pool. The estimated size of the
cleavage product is
therefore compared with the potential sites to identify the exact site of
cleavage.
This protocol has been completed on three different transcripts: Bcl-2 (figure
25), Kras
{figure 26), and UPA (figure 27). The data is summarized in the figures. All
potential
hammerhead ribozyme cleavage sites are indicated in the graph with a short
vertical line.
The actual sites identified are indicated in the graph. The size of the bar
reflects the
intensity of the cleavage product from the cleavage reaction. The
combinatorial pool used
to identify each site, the actual sequence of each site, the position of
cleavage within the
transcript (based on the known sequence), and the estimated size of the
cleavage product
(based on gel analysis) are listed.
Example 20: Reduction of Bcl-2 mRNA using Optimized Ribozymes
Two ribozymes targeted against the same site in the bcl-2 transcript
(Seq.ID#9,
figure 25) were synthesized, but the two ribozymes were stabilized using two
different
chemistries (U4/U7 amino and U4 c-allyl). Ribozymes (200 nM) were delivered
using
lipofectamine (7.2 mM) for 3 hours into MCF-7 cells (SO% confluency). Cellular
RNA
was harvested 24 hours after delivery, analyzed by RNase protectection
analysis {RPA)
and normalized to GAPDH mRNA in triplicate samples. Both ribozymes gave a
reduction in bcl-2 mRNA (see Figure 28). A ribozyme targeted against an
irrelevant
mRNA (c-myb) had no effect on the ratio of bcl-2 mRNA to GAPDH mRNA. All
reduction of bcl-2 RNA was statistically significant with respect to untreated
samples and
samples treated with the irrelevant ribozyme.
Example 21: Synthesis of purine nucleoside triphosphates: 2'-O-methyl-
uanosine-5'-
triphos_phate
2'-O-methyl guanosine nucleoside (0.25 grams, 0.84 mmol) was dissolved in
triethyl phosphate (5.0) ml by heating to 100 C for 5 minutes. The resulting
clear,
CA 02288640 1999-11-OS
WO 98/50530 112 PCT/US98/09249
colorless solution was cooled to 0 C using an ice bath under an argon
atmosphere.
Phosphorous oxychloride ( 1.8 eq., 0.141 ml) was then added to the reaction
mixture with
vigorous stirnng. The reaction was monitored by HPLC, using a sodium
perchlorate
gradient. After 5 hours at 0 C, tributylamine (0.65 ml) was added followed by
the
addition of anhydrous acetonitrile (10.0 ml), and after 5 minutes
(reequilibration to 0 C)
tributylammonium pyrophosphate (4.0 eq., 1.53 g) was added. The reaction
mixture was
quenched with 20 ml of 2M TEAB after 15 minutes at 0 C (HPLC analysis with
above
conditions showed consumption of monophosphate at 10 minutes) then stirred
overnight
at room temperature, the mixture was evaporated in vacuo with methanol co-
evaporation
(4x) then diluted in SO ml O.OSM TEAB. DEAE sephadex purification was used
with a
gradient of 0.05 to 0.6 M TEAB to obtain pure triphosphate (0.52 g, 66.0%
yield) (elutes
around 0.3M TEAB); the purity was confirmed by HPLC and NMR analysis.
Example 22: Svnthesis of Pvrimdine nucleoside trinhosnhates: 2'-O-
methvlthiomethvl-
uridine-5'-Mphosphate
2'-O-methylthiomethyl uridine nucleoside (0.27 grams, 1.0 mmol) was dissolved
in triethyl phosphate (S.0 ml). The resulting clear, colorless solution was
cooled to 0 C
with an ice bath under an argon atmosphere. Phosphorus oxychloride (2.0 eq.,
0.190 ml)
was then added to the reaction mixture with vigorous stirring.
Dimethylaminopyridine
(DMAP, 0.2eq., 25 mg) was added, the solution warmed to room temperature and
the
reaction was monitored by HPLC, using a sodium perchlorate gradient. After 5
hours at
20 C, tributylamine (1.0 ml) was added followed by anhydrous acetonitrile
(10.0 ml), and
after 5 minutes tributylammonium pyrophosphate (4.0 eq., 1.8 g) was added. The
reaction mixture was quenched with 20 ml of 2M TEAB after 15 minutes at 20 C
(HPLC
analysis with above conditions showed consumption of monophosphate at 10
minutes)
then stirred overnight at room temperature. The mixture was evaporated in
vacuo with
methanol co-evaporation (4x) then diluted in 50 ml O.OSM TEAB. DEAF fast flow
Sepharose purification with a gradient of 0.05 to 1.0 M TEAB was used to
obtain pure
triphosphate (0.40 g, 44% yield) (elutes around 0.3M TEAB) as determined by
HPLC and
NMR analysis.
CA 02288640 1999-11-OS
WO 98/50530 113 PCTNS98/09249
Example 23: Utilization of DMAP in Uridine 5'-Tn_phosphate Synthesis
The reactions were performed on 20 mg aliquots of nucleoside dissolved in
1 ml of triethyl phosphate and 19 ul of phosphorus oxychloride. The reactions
were
monitored at 40 minute intervals automatically by HPLC to generate yield-of
product
S curves at times up to 18 hours. A reverse phase column and ammonium acetate/
sodium
acetate buffer system {SOmM & 100mM respectively at pH 4.2) was used to
separate the
5', 3', 2' monophosphates (the monophosphates elute in that order) from the 5'-
triphosphate and the starting nucleoside. The data is shown in table VI. These
conditions
doubled the product yield and resulted in a 10-fold improvement in the
reaction time to
maximum yield (1200 minutes down to 120 minutes for a 90% yield). Selectivity
for S'-
monophosphorylation was observed for all reactions. Subsequent
triphosphorylation
occurred in nearly quantitative yield.
Materials Used in Bacteriophage T7 RNA Polymerase Reactions
BUFFER 1: Reagents are mixed together to form a lOX stock solution of buffer 1
(400
mM Tris-Cl (pH 8.1), 200 mM MgCIZ, 100 mM DTT, SO mM spermidine, and 0.1%
triton X-100. Prior to initiation of the polymerase reaction methanol, LiCI is
added and
the buffer is diluted such that the final reaction conditions for condition 1
consisted of
40mM tris pH (8.1 ), 20mM MgCl2, 10 mM DTT, 5 mM spermidine, 0.01 % triton X-
100,
10% methanol, and 1 mM LiCI.
BUFFER 2: Reagents are mixed together to form a lOX stock solution of buffer
2(400
mM Tris-Cl (pH 8.1), 200 mM MgClz, 100 mM DTT, 50 mM spermidine, and 0.1%
triton X-100. Prior to initiation of the polymerase reaction PEG, LiCI is
added and the
buffer is diluted such that the final reaction conditions for buffer 2
consisted of : 40mM
tris pH (8.1), 20mM MgClz, 10 mM DTT, 5 mM spermidine, 0.01% triton X-100, 4%
PEG, and 1 mM LiCI.
BUFFER 3: Reagents are mixed together to form a lOX stock solution of buffer 3
(400
mM Tris-Cl (pH 8.0), 120 mM MgCl2, 50 mM DTT, 10 mM spermidine and 0.02%
triton
X-100. Prior to initiation of the polymerase reaction PEG is added and the
buffer is
CA 02288640 1999-11-OS
WO 98/50530 114 PCT/US98/09249
diluted such that the final reaction conditions for buffer 3 consisted of :
40mM tris pH
(8.0), 12 mM MgClz, 5 mM DTT, 1 mM spermidine, 0.002% triton X-100, and 4%
PEG.
BUFFER 4: Reagents are mixed together to form a l OX stock solution of buffer
4 (400
mM Tris-Cl (pH 8.0), 120 mM MgCl2, 50 mM DTT, 10 mM spermidine and 0.02%
triton
X-100. Prior to initiation of the polymerise reaction PEG, methanol is added
and the
buffer is diluted such that the final reaction conditions for buffer 4
consisted of : 40mM
tris pH (8.0), 12 mM MgClz, 5 mM DTT, 1 mM spermidine, 0.002% triton X-100,
10%
methanol, and 4% PEG.
BUFFER 5: Reagents are mixed together to form a lOX stock solution of buffer 5
(400
mM Tris-Cl (pH 8.0), 120 mM MgCl2, 50 mM DTT, 10 mM spenmidine and 0.02%
triton
X-100. Prior to initiation of the polymerise reaction PEG, LiCI is added and
the buffer is
diluted such that the final reaction conditions for buffer 5 consisted of :
40mM tris pH
(8.0), 12 mM MgCl2, 5 mM DTT, 1 mM spermidine, 0.002% triton X-100, 1 mM LiCI
and 4% PEG.
BUFFER 6: Reagents are mixed together to form a lOX stock solution of buffer 6
(400
mM Tris-Cl (pH 8.0), 120 mM MgClz, 50 mM DTT, 10 mM spermidine and 0.02%
triton
X-100. Prior to initiation of the polymerise reaction PEG, methanol is added
and the
buffer is diluted such that the final reaction conditions for buffer 6
consisted of : 40mM
tris pH (8.0), 12 mM MgCl2, S mM DTT, 1 mM spermidine, 0.002% triton X-100,
10%
methanol, and 4% PEG.
Example 24: Screening of Modified Nucleoside triphosphates with Mutant T7 RNA
Polymerase
Each modified nucleotide triphosphate was individully tested in buffers 1
through
6 at two different temperatures (25 and 37°C). Buffers 1-6 tested at
25°C were designated
conditions 1-6 and buffers 1-6 test at 37°C were designated conditions
7-12 (table VII).
In each condition, Y639F mutant T7 polymerise (Sousa and Padilla, Supra) (0.3-
2 mg/20
ml reaction), NTP's (2 mM each), DNA template (10 pmol), inorganic
pyrophosphatase
(SU/ml) and a-'ZP NTP(0.8 mCi/pmol template) were combined and heated at the
designated temperatures for 1-2 hours. The radiolabeled NTP used was different
from the
CA 02288640 1999-11-OS
WO 98/50530 PCT/US98/09249
115
modified triphosphate being testing. The samples were resolved by
polyacrylamide gel
electrophoresis. Using a phosphorImager (Molecular Dynamics, Sunnyvale, CA),
the
amount of full-length transcript was quantified and compared with an all-RNA
control
reaction. The data is presented in Table VIII; results in each reaction is
expressed as a
percent compared to the all-ribonucleotide triphosphate (rNTP) control. The
control was
run with the mutant T7 polymerase using commercially available polymerase
buffer
(Boehringer Mannheim, Indianapolis, IN).
Example 25' Incorporation of Modified NTP's using Wild-type T7 RNA nolymerase
Bacteriophage T7 RNA polymerase was purchased from Boehringer Mannheim at
0.4 U/pL concentration. Applicant used the commercial buffer supplied with the
enzyme
and 0.2 p.Ci alpha-32P NTP in a 50 pL reaction with nucleotides triphosphates
at 2 mM
each. The template was double-stranded PCR fragment, which was used in
previous
screens. Reactions were carned out at 37°C for 1 hour. 10 pL of the
sample was run on a
7.5% analytical PAGE and bands were quantitated using a PhosphorImager.
Results are
1 S calculated as a comparison to an "all ribo" control (non-modified
nucleoside
triphosphates) and the results are in Table IX.
Example 26' Incorporation of Multiple Modified Nucleoside triphosphates Into
Oli ~onucleotides
Combinations of modified nucleoside triphosphates were tested with the
transcription protocol described in example 9, to determine the rates of
incorporation of
two or more of these triphosphates. Incorporation 2'-Deoxy-2'-{L-histidine)
amino
uridine {2'-his-NHZ UTP) was tested with unmodified cytidine nucleoside
triphosphates,
rATP and rGTP in reaction condition number 9. The data is presented as a
percentage of
incorporation of modified NTP's compared to the all rNTP control and is shown
in Table
Xa.
Two modified cytidines (2'-NH2-CTP or 2'dCTP) were incorporated along with
2'-his-NHZ-UTP with identical efficiencies. 2'-his-NHZ UTP and 2'-NHZ-CTP were
then
tested with various unmodified and modified adenosine triphosphates in the
same buffer
CA 02288640 1999-11-OS
WO 98/50530 116 PCT/US98/09249
(Table Xb). The best modified adenosine triphosphate for incorporation with
both 2'-his-
NHZ UTP and 2'-NHz CTP was 2'-NH2-DAPTP.
EXAMPLE 27: Optimization of Reaction conditions for Incorooration of Modified
Nucleotide Triphosphate
S The combination of 2'-his-NHZ UTP, 2'-NHZ-CTP, 2'-NHZ-DAP, and rGTP was
tested in several reaction conditions (Table XI) using the incorporation
protocol
described in example 14. The results demonstrate that of the buffer conditions
tested,
incorporation of these modified nucleoside triphosphates occur in the presence
of both
methanol and LiCI.
Example 28: Deprotection of Ribozvme in a 96 Well Plate
A ribozyme sequence {200nmole) was synthesized as described herein on a
polystyrene
solid support in a well of a 96 well plate. A 10:3:13 mixture (800 wL) of
anhydrous
methylamine (308p.L), triethylamine (92pL) and dimethylsulfoxide (DMSO) (400
p,L)
was prepared of which half (400 p.L) was added to the well and incubated at
room
temperature for 45 minutes. Following the reaction the solution was replaced
with the
remaining 400 p,L and incubated as before. At the end of the reaction, the
solid support
was filtered off, all 800 pL of MA/TEA/DMSO solution was collected together
and 100
p,L of TEA.3HF was added. The reaction was then heated at 65°C for 15
minutes and
then cooled to room temperature. The solution was then quenched with aqueous
NH_~+HCOz- (1mL) (see Figure 30). HPLC chromatography of the reaction mixture
afforded 32 O. D.u2~ "m of which 46% was full length ribozyme.
Example 29: Column Deprotection of Ribozvme
A ribozyme was synthesized using the column format as described herein. The
polystyrene solid-support with protected oligoribonucleotide or modified
oligoribonucleotide (200 nmole) was transferred into a glass vial equipped
with a screw
cap. A 10:3:13 mixture of anhydrous methylamine ( 308 p,L), triethylamine (92
~L) and
dimethylsulfoxide (DMSO) (400 pL) was added followed by vortexing of the glass
vial.
After allowing the reaction for 1.5 hours, the solid support was filtered off.
100 p,L of
TEA.3HF was added at room temperature to the vial and the reaction was mixed
causing
CA 02288640 1999-11-OS
WO 98/50530 PCT/US98/09249
117
the solution to gel. The reaction was then heated at 65 °C for 15
minutes and then cooled
to room temperature. The solution was then quenched with 1.5 M aqueous
NH4+H~CO~-
(1mL). HPLC chromatography of the reaction mixture afforded 32 O. D.uz~ nm of
which
46% was full length ribozyme.
Example 30: Column Deprotection of Ribozvme with anhvdrous ethanolic
methylamine
A ribozyme was synthesized using the column format as described herein. The
polystyrene solid-support with protected oligoribonucleotide , or modified
oligoribonucleotide (200 nmole) was transferred into a glass vial equipped
with a screw
cap. A 1:1 mixture of anhydrous ethanolic methylamine ( 400 pL) and
dimethylsulfoxide
(DMSO) (400 p.L) was added followed by vortexing of the glass vial. After
allowing the
reaction for 1.5 hours, the solid support was filtered off. 100 p.L of TEA.3HF
was added
at room temperature to the vial and the reaction was mixed causing the
solution to gel.
The reaction was then heated at 65 °C for 15 minutes and then cooled
to room
temperature. The solution was then quenched with 1.5 M aqueous NH4+HCO~-
(imL).
HPLC chromatography of the reaction mixture afforded 32 O. D.u2eo "", of which
46%
was full length ribozyme.
Example 31. Large-scale One-Pot Deprotection of Ribozvme
A ribozyme was synthesized at the 0.5 mmol scale using the column format as
described herein. The polystyrene solid-support (24 grs) with protected
oligoribonucleotide or modified oligoribonucleotide (500 p,mole) was
transferred into a
1L Schott bottle equipped with a screw cap. A 1:1.3 mixture of anhydrous
ethanolic
methylamine ( 150 mL) and dimethylsulfoxide (DMSO) (200 mL) was added followed
by
vortexing (200 rpm) of the glass bottle for 1.5 hours. The reaction mixture
was then
frozen at -70 °C for 30 minutes. 50 mL of neat TEA.3HF was then added
at room
temperature to the reaction mixture and the reaction was placed in a shaking
oven (200
rpm) where it was heated at 65 °C for 60 minutes and subsequently
frozen at -70 °C for
minutes. The solution was then quenched with 1.5 M aqueous NH~~iC03- (200 mL).
CA 02288640 1999-11-OS
WO 98/50530 11$ PCT/US98/09249
The reaction mixture was separated from the polystyrene solid-support by
filtration on a
sintered glass funnel (10-20 ~m porosity). U.V. spectrophotometric
quantification and
HPLC chromatography of the reaction mixture afforded 160,000 O.D.u2~ "", of
which
46.4% was full length ribozyme. After allowing the reaction for 1.5 hours, the
solid
support was filtered off
Example 32: Antitumor and antimetastatic efficacy of ribozymes directed
against the
mRNA encoding the two VEGF receptor subtyt~es, flt-1 and flk-1 in the mouse
Lewis
lung-HM carcinoma model of primary tumor growth and metastasis
The Lewis lung carcinoma (LLC) model is a syngeneic mouse model of metastatic
cancer commonly used for antitumor agent efficacy screening. According to
Folkman
(1995, supra), primary tumor growth and metastasis in this model is dependent
upon
VEGF-induced angiogenesis. Two variants of the LLC model exist. The low
metastatic
form involves the implantation of a tumor, usually subcutaneous, which sends
micrometastases to the lungs whose growth is suppressed by the presence of the
primary
tumor. The highly metastatic (HM) form differs from the low metastatic variant
in that
the growth of metastases is not suppressed by the presence of the primary
tumor. Thus,
the HM form is a model in which it is possible to measure pharmacologic
efficacy on both
primary tumor growth and metastasis in the same mouse without excision of the
primary
tumor.
Applicant selected the highly metastatic variant of the Lewis lung model for
antitumor/metastatic screening of ribozymes directed against VEGF receptor
(flt-1 and
Jlk-I ) mRNA. These ribozyrnes have been shown to reduce VEGF binding and VEGF-
stimulated proliferation in cultured MVEC's as well as VEGF-induced
neovascularization
of the rat cornea (Cushman et al., 1996, Angiogenesis Inhibitors and Other
Novel
Therapeutic Strategies for Ocular Diseases of Neovascularization, IBC
Conference
Abstract). Pharmacokinetically, Applicant has found that ribozymes distribute
systemically following continuous i.v. infusion (via Alzet osmotic minipumps)
at
significant concentrations within most tissues including subcutaneously
implanted
CA 02288640 1999-11-OS
WO 98/50530 119 PCT/US98/09249
tumors. This study examines the antitumor/antimetastatic efficacy of flt-1 and
flk-1
' ribozymes continuously infused i.v. in the LLC-HM mouse model.
Methods
Ribozymes
The ribozymes used in this study were hammerhead ribozymes comprising a 4 base
pair stem II, four phosphorothioate linkages at the 5'-end, a 2'-C-allyl
substitution at
position 4 (see Figure 1), and an inverted abasic nucleotide substitution at
the 3'-end. The
catalytically active and inactive ribozymes were RPL4610/4611
(activelinactive) and
RPL4733/4734 directed against Jlt-1 and flk-I messages, respectively.
Ribozymes
solutions were prepared in normal saline (USP).
Test solutions (ribozymes or saline control) were dispensed into Alzet~
osmotic
minipumps (Model # 1002--total volume capacity including excess = 200 ~.l)
which
dispense 0.5 ~.1/h at 37 °C when exposed to interstitial water. Pumps
were either filled
with normal saline (USP) or 167.0, 50.0, 16.7, 5.0, or 1.7 mg/ml ribozyme
solutions.
Prior to animal implantation, osmotic minipumps were placed in 37 °C
sterile water for at
least four hours to activate pumping.
Tumor inoculation
All animal procedures in this study were performed in accordance with the
National
Research Council's Guide for the Care and Use of Laboratory Animals ( 1996),
USDA
regulations, and the policies and procedures of the RPI Institutional Animal
Care and Use
Committee. A total of 210 female C57BL/6J mice weighing between 20-25 g were
used
in this study. All animals were housed under 12 h on/12 h off light cycles and
received
ad libitum food and water.
Highly metastatic variant Lewis lung carcinoma (LLC-HM) tumors were
propogated in vivo from an LLC-HM cell line. These tumors needed to be
propogated in
vivo because they can revert to the low metastatic phenotype in culture. LLC-
HM cells
CA 02288640 1999-11-OS
WO 98/50530 120 PCT/US98/09249
were initially cultured in DMEM + 10% FCS + 1 % GPS. For in vivo propogation,
S X
6 cells were injected subcutaneously in mice. Tumors were allowed to grow for
25
days at which time animals were euthanized by C02 inhalation and lung
macrometastases
were counted. Animals with the most macrometastases (approximately 15-20) were
5 selected for preparation of tumor breis and propogation. When tumors in
animals selected
for propogation reached a volume of approximately 1500 mm3, animals were
euthanized
by C02 inhalation and tumors were excised. Tumors were seived through a 100
~.m pore
size sterile nylon mesh. LLC-HM cells were resuspended in normal saline to a
final
concentration of S x 10G viable cells/ml (via hemocytometer). Three days prior
to
10 ribozyme dosing, all animals were subcutaneously inoculated on the right
flank with 5 x
105 cells (in a volume of 100 p.l).
Ribozyme or saline dosing
Each ribozyme solution was prepared to deliver 100, 30, 10, 3, or 1 mg/kg/day
in a
volume of 12 ml. A total of 10 animals per dose or saline control group were
surgically
implanted on the left flank with osmotic minipumps pre-filled with the
respective test
solution three days following tumor inoculation. Pumps were attached to
indwelling
jugular vein catheters. The specifications for the model #1002 Alzet osmotic
minipump
show that they accurately deliver aqueous solutions at 0.5 ~1/h for 14 days.
Table III
summarizes the experimental groups.
Tumor volume and metastatic index quantitation
Beginning four days and ending 24 days days following tumor inoculation, the
length and width of all primary tumors were measured every other day using
microcalipers. Tumor volumes were calculated using the standard formula for an
elipsoid
volume, (L~W2)/2. Tumor volumes were calculated in triplicate for each animal.
A mean
tumor volume was calculated for each animal. Group means and standard error of
the
group means were calculated from individual animal mean tumor volumes.
CA 02288640 1999-11-OS
WO 98/50530 121 PCT/US98I09249
Twenty-five days following tumor inoculation, all animals were euthanized by
C02
inhalation and lungs and primary tumors harvested. Lung macrometastases were
counted
under a dissecting microscope (2.5 X magnification). Lungs and primary tumors
were
also weighed on an analytical balance. Lung weights served as an index of
total lung
metastatic burden.
Statistical analysis
For all treatment groups, group tumor volume means on day 18 (end of
treatment)
as well as means of primary tumor and lung weight and numbers of lung
metastases were
evaluated for normality and subjected to analysis of variance. Statistical
differences
between group means were evaluated using the Tukey-Kramer post-hoc test (alpha
=
0.05). Comparisons with the control group (saline control) were made using the
Dunnett's test (alpha = 0.05).
Results
Flt-1
The effects of several doses of active and inactive Flt-1 ribozymes
(RPL4610/461 I,
respectively) on primary LLC-HM tumor growth are summarized in Figure 39 (A-
E). At
the lowest dose (Figure 39A), both active and inactive reduce primary tumor
growth
similarly throughout the entire time course compared to saline controls.
However, with
increasing dose, active ribozyme reduces primary tumor growth to a greater
extent than
the inactive ribozyme, with the largest difference observed at 30 mg/kg/day
(Figure 39D).
The magnitude of the maximal reduction compared to saline was approximately
four fold
with the active ribozyme RPL4610 at 30 mg/kg/day. It should be noted that this
observed
four fold reduction is still present at day 24 even though treatment ended 7
days earlier.
The growth curve data was subjected to exponential regression. The curve fits
show
that the tumor growth data fits an exponential curve with a high correlation
coefficient
(R>0.95). Thus, there appears to be no long lasting toxic effect on tumor
growth. Since
CA 02288640 1999-11-OS
WO 98!50530 122 PCT/US98/09249
the calculated slope of the exponential curve at any point indicates the rate
of tumor
growth, it should be possible to compare rates of growth between treatments.
Since the
curve fits do not assume that the tumor growth starts from the same point
(which is a
correct assumption since the all tumors start with the same tumor cell
inoculum
concentration), an accurate calculation of the slope of the exponential curve
is not
possible since the curve fitting algorithm extrapolates a t = 0 tumor size
which is then
used to calculate the slope. In the analysis, the saline tumor size at t = 0
is much greater
than the other treatment groups, thus comparisons with saline are not
necessarily accurate.
If the curve fit algorithm is restricted to the same tumor size, a dose-
dependent reduction
in the rate of tumor growth is observed with the active ribozyme. However, the
curve fits
show lower correlation coefficients in some cases.
In order to see whether a the ribozyme treatments statistically reduce primary
tumor
growth, primary tumor volume measurements at each dose immediately following
treatment (day 18) were compared (Figure 40). Active ribozyrne RPL4610
produced a
statistically significant (p < 0.05) and dose-dependent reduction in primary
tumor volume.
Although the inactive ribozyme (RPL4611) showed some reduction in primary
tumor
volume at the lowest and highest doses, there was no dose-dependent reduction
observed.
At doses between 3 and 30 mg/kg/day, the inactive ribozyme showed no
significant
reduction in primary tumor volume. There was a significant difference (p <
0.05)
between active and inactive ribozymes (Tukey-Kramer test) at doses of 10 and
30
mg/kg/day.
Applicant has also observed that the active ribozyme RPL4610 produced a
significant reduction in primary tumor mass at all doses tested (1-100
mg/kg/day) 25 days
following inoculation.
Figures 41 A and B illustrate that the active ribozyme reduced both the number
of
lung metastases and lung mass in a dose-dependent manner. The active alt-1
ribozyme
showed a significant reduction (p < 0.05 by Dunnetts) in the number of lung
metastases at
the 30 and 100 mg/kg/day doses compared to saline. There was also a
significant
CA 02288640 1999-11-05
WO 98/50530 123 PCT/US98/09249
difference between active and inactive ribozymes at these doses (p , 0.05 by
Student's t).
RPL4610 reduced the lung weight to almost normal levels at the highest dose (
100
mg/kg/day). There was no observable dose-related effect of the inactive
ribozyme on
either the number of lung metastases or lung weight. A significant reduction
(p < 0.05,
Student's t) in lung mass, an index of metastatic burden, was observed between
saline and
the active ribozyme. The lack of significance using more stringent statistical
tests
(Dunnet's or Tukey-Kramer), which take into account the variance within all
groups, was
due to high variability, especially in the inactive ribozyme group. However,
since five
doses were tested, it is possible to say that there is a dose-dependent trend
in the reduction
of lung metastases/lung weight.
Example 33: Effects of flk-1 ribozymes (active/inactive,) on LLC-HM primary
tumor
,growth in mice.
The dose-related effects of active and inactive flk-1 directed ribozymes
(RPL4733/4734, respectively) on primary LLCare shown in Figure 38 A-E.
The dose-related effects of active and inactive flk 1 directed ribozymes
(RPL4733/4734, respectively) on primary LLCare shown in Figure 42 A-E. At the
lowest
dose, there was no observable effect on primary tumor growth with the active
flk-I
ribozyme (Figure 42A). The inactive ribozyme showed a modest reduction in
primary
tumor growth. At higher doses (3-100 mg/kg/day, Figure 42B-E), the active flk-
1
ribozyme reduced primary tumor growth while the inactive ribozyme showed
little, if any,
antitumor efficacy over the dose range between 10 and 100 mg/kg/day (Figures
42C-E).
The antitumor efficacy of both active and inactive .flk-1 ribozymes are
similar at 3
mg/kg/day (Figure 42B).
As in the case of the flt-1 ribozymes, tumor growth followed exponential
growth
kinetics. Again, since the t = 0 tumor size could nvt accurately be estimated
by the curve
fit program, it is not possible to calculate the slope of the exponential
curve fits for the flk-
1 ribozymes.
CA 02288640 1999-11-OS
WO 98/50530 124 PCT/US98/09249
Immediately following the cessation of treatment (day 18), the active flk-1
ribozyme
showed a significant reduction in primary tumor volume from 3 to 100 mg/kg/day
(Figure
43). The magnitude of the reduction is approximately four fold and appeared to
be
maximal at 3 mg/kg/day. The lowest dose had no significant effect on primary
tumor
volume. The inactive flk-1 ribozyme had a significant antitumor effect at
doses of I and 3
mg/kg/day; however, this effect disappeared between 10 and 100 mg/kg/day.
The antimetastatic effects of the flk-1 ribozymes are illustrated in Figure 44
A and
B. Although neither ribozyme showed a statistically significant effect on the
number of
lung metastases at any dose, it appears that the active flk-I ribozyme showed
a significant
reduction in lung mass over the dose range between 3 and 100 mglkg/day.
Applicant has further observed that the lung mass was reduced to normal over
the
entire dose range. The inactive ribozyme reduced lung mass at I and 3
mg/kg/day (Figure
41 C); however, this trend was not observed at higher doses (3-100 mg/kg/day).
Example 34: Ribozyme-mediated decrease in vascularity of tumor
1 S Three tumors from each of three treatment groups (saline controls,
inactive
RPL4611 and active RPL4610, 30 mg/kg/day dose only) were analyzed for
vascularity
using an immunohistochemical assay which stains endothelial cells for CD31
(PECAM).
The vascularity was quantitated in a blinded fashion. From the raw data the
average
number of vessels per high magnification field (400X) were calculated. They
are as
follows: SALINE CONTROL = 24.1; RPL4611 (Inactive) = 27.6; RPL4610 (Active) _
16Ø
It is suggestive that ribozyme-specific antiangiogenic effect is exhibited by
the
active Flt-1 ribozyme in Lewis lung tumors. Thus, the mechanism of action for
the
observed reduction in the primary tumor volumes may be due to an
antiangiogenic effect.
Similar delivery strategies can be used to deliver c-raf ribozymes to treat a
variety of
diseases.
CA 02288640 1999-11-OS
WO 98/50530 125 PCT/US98/09249
Use of Ribozymes Tar eg tiny c-raf
Overexpression of the c-raf oncogene has been reported in a number of cancers
(see above). Thus, inhibition of c-raf expression (for example using
ribozymes) can
reduce cell proliferation of a number of cancers, in vitro and in vivo and can
reduce their
proliferative potential. A cascade of MMP and serine proteinase expression is
implicated
in the acquisition of an invasive phenotype as well as in angiogenesis in
tumors
(MacDougall & Matrisian, 1995, Cancer & Metastasis Reviews 14, 351;Ritchlin &
Winchester, 1989, Springer Semin Immunopathol., 11, 219).
A number of human diseases are characterized by the inappropriate
proliferation
of cells at sites of injury or damage to the normal tissue architecture. These
diseases
include restenosis, caused by the local proliferation of medial smooth muscle
cells at sites
of arterial wall disruption by surgery; psoriasis, caused by proliferation of
keratinocytes at
regions of endothelial cell damage in the skin, and various fibrosis, caused
by the
inappropriate replication of cells during wound healing processes. In certain
1 S inflammatory processes, cell proliferation may not be causative, yet it
exacerbates the
disease pathology. For example, in rheumatoid arthritis, synovial hyperplasia
leads to
accelerated cartilage damage due to secretion of proteases by the expanding
population of
synovial fibroblasts. Any number of these diseases and others which involve
cellular
proliferation or the loss of proliferative control, such as cancer, could be
treated using
ribozymes which inhibit the expression of the cellular Raf gene products.
Alternatively,
ribozyme inhibition of the cellular growth factor receptors could be used to
inhibit
downstream signalling pathways. The specific growth factors involved would
depend
upon the cell type indicated in the proliferative event.
Ribozymes, with their catalytic activity and increased site specificity (see
above),
are likely to represent a potent and safe therapeutic molecule for the
treatment of cancer.
In the present invention, ribozymes are shown to inhibit smooth muscle cell
proliferation.
From those practiced in the art, it is clear from the examples described, that
the same
CA 02288640 1999-11-05
WO 98/50530 126 PCT/US98/09249
ribozymes may be delivered in a similar fashion to cancer cells to block their
proliferation.
Diagnostic uses
Ribozymes of this invention may be used as diagnostic tools to examine genetic
drift and mutations within diseased cells or to detect the presence of c-raf
RNA in a cell.
The close relationship between ribozyme activity and the structure of the
target RNA
allows the detection of mutations in any region of the molecule which alters
the base-
pairing and three-dimensional structure of the target RNA. By using multiple
ribozymes
described in this invention, one may map nucleotide changes which are
important to RNA
structure and function in vitro, as well as in cells and tissues. Cleavage of
target RNAs
with ribozymes may be used to inhibit gene expression and define the role
(essentially) of
specified gene products in. the progression of disease. In this manner, other
genetic
targets may be defined as important mediators of the disease. These
experiments will
lead to better treatment of the disease progression by affording the
possibility of
combinational therapies (e.g., multiple ribozymes targeted to different genes,
ribozymes
coupled with known small molecule inhibitors, or intermittent treatment with
combinations of ribozymes and/or other chemical or biological molecules).
Other in vitro
uses of ribozymes of this invention are well known in the art, and include
detection of the
presence of mRNAs associated with c-raf related condition. Such RNA is
detected by
determining the presence of a cleavage product after treatment with a ribozyme
using
standard methodology.
In a specific example, ribozymes which can cleave only wild-type or mutant
forms
of the target RNA are used for the assay. The first ribozyme is used to
identify wild-type
RNA present in the sample and the second ribozyme will be used to identify
mutant RNA
in the sample. As reaction controls, synthetic substrates of both wild-type
and mutant
RNA will be cleaved by both ribozymes to demonstrate the relative ribozyme
efficiencies
in the reactions and the absence of cleavage of the "non-targeted" RNA
species. The
cleavage products from the synthetic substrates will also serve to generate
size markers
CA 02288640 1999-11-OS
WO 98/50530 127 PCT/US98/09249
for the analysis of wild-type and mutant RNAs in the sample population, Thus
each
analysis will require two ribozymes, two substrates and one unknown sample
which will
be combined into six reactions. The presence of cleavage products will be
determined
using an RNAse protection assay so that full-length and cleavage fragments of
each RNA
S can be analyzed in one lane of a polyacrylamide gel. It is not absolutely
required to
quantify the results to gain insight into the expression of mutant RNAs and
putative risk
of the desired phenotypic changes in target cells. The expression of mRNA
whose
protein product is implicated in the development of the phenotype (i.e., c-rah
is adequate
to establish risk. If probes of comparable specific activity are used for both
transcripts,
then a qualitative comparison of RNA levels will be adequate and will decrease
the cost
of the initial diagnosis. Higher mutant form to wild-type ratios will be
correlated with
higher risk whether RNA levels are compared qualitatively or quantitatively.
Additional Uses
Potential usefulness of sequence-specific nucleic acid catalysts of the
instant
1 S invention might have many of the same applications for the study of RNA
that DNA
restriction endonucleases have for the study of DNA (Nathans et al., 1975 Ann.
Rev.
Biochem. 44:273). For example, the pattern of restriction fragments could be
used to
establish sequence relationships between two related RNAs, and large RNAs
could be
specifically cleaved to fragments of a size more useful for study. The ability
to engineer
sequence specificity of the ribozyme is ideal for cleavage of RNAs of unknown
sequence.
The use of NTP's described in this invention have several research and
commercial applications. These modified nucleoside triphosphates can be used
for in
vitro selection (evolution) of oligonucleotides with novel functions. Examples
of in vitro
selection protocols are incorporated herein by reference (Joyce, 1989, Gene,
82, 83-87;
Beaudry et al., 1992, Science 257, 635-641; Joyce, 1992, Scientific American
267, 90-97;
Breaker et al., 1994, TIBTECH 12, 268; Bartel et al.,1993, Science 261:1411-
1418;
Szostak, 1993, TIBS 17, 89-93; Kumar et al., 1995, FASEB J., 9, 1183; Breaker,
1996,
Curr. Op. Biotech., 7, 442).
CA 02288640 1999-11-OS
WO 98/50530 12g PCT/US98/09249
Additionally, these modified nucleoside triphosphates can be employed to
generate modified oligonucleotide combinatorial chemistry libraries. Several
references
for this technology exist (Brenner et al., 1992, PNAS 89, 5381-5383, Eaton,
1997, Curr.
Opin. Chem. Biol. 1, 10-16) which are all incorporated herein by reference.
Nucleic acid molecules of the instant invention might have many of the same
applications for the study of RNA that DNA restriction endonucleases have for
the study
of DNA {Nathans et al., 1975 Ann. Rev. Biochem. 44:273). For example, the
pattern of
restriction fragments could be used to establish sequence relationships
between two
related RNAs, and large RNAs could be specifically cleaved to fragments of a
size more
useful for study. The ability to engineer sequence specificity of the ribozyme
is ideal for
cleavage of RNAs of unknown sequence. Nucleic acid molecules (e.g., ribozymes)
of the
invention can be used, for example, to target cleavage of virtually any RNA
transcript
(Zaug et al., 324, Nature 429 1986 ; Cech, 260 JAMA 3030, 1988; and Jefferies
et al., 17
Nucleic Acids Research 1371, 1989). Such nucleic acids can be used as a
therapeutic or
to validate a therapeutic gene target and/or to determine the function of a
gene in a
biological system (Christoffersen, 1997, Nature Biotech. 15, 483).
Various ligands can be attached to oligonucleotides using the componds
containing
zylo modification for the purposes of cellular delivery, nuclease resistance,
cellular
trafficking and localization, chemical ligation of oligonucleotide fragments.
Incorporation of one or more compounds of Formula II into a ribozyme may
increase its
effectiveness. Compounds of Formula II can be used as potential antiviral
agents.
Other embodiments are within the following claims.
CA 02288640 1999-11-OS
WO 98/50530 129 PCT/US98/09249
TABLE/
Characteristics of naturally occurring ribozymes
Group I Introns
~ Size: 150 to >1000 nucleotides.
~ Requires a U in the target sequence immediately 5' of the cleavage site.
~ Binds 4-6 nucleotides at the 5'-side of the cleavage site. .
~ Reaction mechanism: attack by the 3'-OH of guanosine to generate cleavage
products with
3'-OH and 5'-guanosine.
~ Additional protein cofactors required in some cases to help folding and
maintainance of
the active structure.
~ Over 300 known members of this class. Found as an intervening sequence in
Tetrahymena
thermophila rRNA, fungal mitochondria, chloroplasts, phage T4, blue-green
algae, and
others.
~ Major structural features largely established through phylogenetic
comparisons,
mutagenesis, and biochemical studies [',Z].
Complete kinetic framework established for one ribozyme [3,4~5~6].
~ Studies of ribozyme folding and substrate docking underway [',a,9].
~ Chemical modification investigation of important residues well established
['°,"].
' . Michel, Francois; Westhof, Eric. Slippery substrates. Nat. Struct. Biol.
(1994), 1 (1 ), 5-7.
2 . Lisacek, Frederique; Diaz, Yolande; Michel, Francois. Automatic
identification of group I
intron cores in genomic DNA sequences. J. Mol. Biol. (1994), 235{4}, 1206-17.
' . Herschlag, Daniel; Cech, Thomas R.. Catalysis of RNA cleavage by the
Tetrahymena
thermophila riboryme. 1. Kinetic description of the reaction of an RNA
substrate complementary
to the active site. Biochemistry (1990), 29(44), 10159-71.
4 . Herschlag, Daniel; Cech, Thomas R.. Catalysis of RNA cleavage by the
Tetrahymena
thermophila ribozyme. 2. Kinetic description of the reaction of an RNA
substrate that forms a
mismatch at the active site. Biochemistry {1990), 29(44), 10172-80.
5 . Knitt, Deborah S.; Herschlag, Daniel. pH Dependencies of the Tetrahymena
Ribozyme
Reveal an Unconventional Origin of an Apparent pKa. Biochemistry (1996),
35(5), 1560-70.
6 . Bevilacqua, Philip C.; Sugimoto, Naoki; Turner, Douglas H.. A mechanistic
framework for
the second step of splicing catalyzed by the Tetrahymena ribozyme.
Biochemistry (1996), 35(2),
648-58.
' . Li, Yi; Bevilacqua, Philip C.; Mathews, David; Turner, Douglas H..
Thermodynamic and
activation parameters for binding of a pyrene-labeled substrate by the
Tetrahymena ribozyme:
docking is not diffusion-controlled and is driven by a favorable entropy
change. Biochemistry
(1995), 34(44), 14394-9.
8 . Banerjee, Aloke Raj; Turner, Douglas H.. The time dependence of chemical
modfication
reveals slow steps in the folding of a group I ribozyme. Biochemistry (1995),
34(19), 6504-12.
9 . Zarrinkar, Patrick P.; Wifliamson, James R.. The P9.1-P9.2 peripheral
extension helps
guide folding of the Tetrahymena ribozyme. Nucleic Acids Res. (1996), 24(5),
854-8.
'° . Strobel, Scott A.; Cech, Thomas R.. Minor groove recognition of
the conserved G.cntdot.U
pair at the Tetrahymena ribozyme reaction site. Science (Washington, D. C.)
(1995), 267(5198),
CA 02288640 1999-11-OS
WO 98/50530 130 PCT/US98/09249
The small (4-6 nt) binding site may make this ribozyme too non-specific for
targeted RNA
cleavage, however, the Tetrahymena group I intron has been used to repair a
"defective" [3-
galactosidase message by the ligation of new (3-galactosidase sequences onto
the defective
message ['zJ.
RNAse P RNA (M1 RNA)
~ Size: 290 to 400 nucleotides.
~ RNA portion of a ubiquitous ribonucleoprotein enzyme.
~ Cleaves tRNA precursors to form mature tRNA ["].
~ Reaction mechanism: possible attack by M2+-OH to generate cleavage products
with 3'-
OH and S'-phosphate.
~ RNAse P is found throughout the prokaryotes and eukaryotes. The RNA subunit
has been
sequenced from bacteria, yeast, rodents, and primates.
~ Recruitment of endogenous RNAse P for therapeutic applications is possible
through
hybridization of an External Guide Sequence (EGS) to the target RNA
['°,'s]
~ Important phosphate and 2' OH contacts recently identified ['6,"]
Group II Introns
~ Size: >1000 nucleotides.
~ Trans cleavage of target RNAs recently demonstrated ['8'9].
~ Sequence requirements not fully determined.
~ Reaction mechanism: 2'-OH of an internal adenosine generates cleavage
products with 3'-
OH and a "lariat" RNA containing a 3'-5' and a 2'-5' branch point.
675-9.
" . Strobel, Scott A.; Cech, Thomas R.. Exocyclic Amine of the Conserved
G.cntdot.U Pair at
the Cleavage Site of the Tetrahymena Ribozyme Contributes to 5'-Splice Site
Selection and
Transition State Stabilization. Biochemistry (1996), 35(4), 1201-11.
'z. Sullenger, Bruce A.; Cech, Thomas R.. Ribozyme-mediated repair of
defective mRNA by
targeted traps-splicing. Nature (London) (1994), 371(6498), 619-22.
'3. Robertson, H.D.; Altman, S.; Smith, J.D. J. Biol. Chem., 247, 5243-5251
(1972).
'4. Forster, Anthony C.; Altman, Sidney. External guide sequences for an RNA
enzyme.
Science (Washington, D. C., 1883-) (1990), 249(4970), 783-6.
'S. Yuan, Y.; Hwang, E. S.; Altman, S. Targeted cleavage of mRNA by human
RNase P.
Proc. Natl. Acad. Sci. USA (1992) 89, 8006-10.
'6 . Harris, Michael E.; Pace, Norman R.. Identification of phosphates
involved in catalysis by
the ribozyme RNase P RNA. RNA (1995), 1 (2), 210-18.
" . Pan, Tao; L_oria, Andrew; Zhong, Kun. Probing of tertiary interactions in
RNA: 2'-hydroxyl-
base contacts between the RNase P RNA and pre-tRNA. Proc. Natl. Acad. Sci. U.
S. A. (1995),
92(26), 12510-14.
'e . Pyle, Anna Marie; Green, Justin B.. Building a Kinetic Framework for
Group If Intron
Ribozyme Activity: Quantitation of Interdomain Binding and Reaction Rate.
Biochemistry (1994),
33(9), 2716-25.
'9 . Michels, William J. Jr.; Pyle, Anna Marie. Conversion of a Group II
Intron into a New
Multiple-Turnover Ribozyme that Selectively Cleaves Oligonucleotides:
Elucidation of Reaction
Mechanism and StructurelFunction Relationships. Biochemistry (1995), 34(9),
2965-77.
CA 02288640 1999-11-OS
WO 98/50530 131 PCT/US98/09249
~ Only natural ribozyme with demonstrated participation in DNA cleavage
[zo,z'] in addition
to RNA cleavage and Iigation.
~ Major structural features largely established through phylogenetic
comparisons [ZZ].
~ Important 2' OH contacts beginning to be identified [zs]
S ~ Kinetic framework under development [za]
Neurospora VS RNA
~ Size: 144 nucleotides.
~ Trans cleavage of hairpin target RNAs recently demonstrated [zs].
~ Sequence requirements not fully determined.
~ Reaction mechanism: attack by 2'-OH 5' to the scissile bond to generate
cleavage products
with 2',3'-cyclic phosphate and 5'-OH ends.
~ Binding sites and structural requirements not fully determined.
~ Only 1 known member of this class. Found in Neurospora VS RNA.
Hammerhead Ribozyme
(see text for references)
~ Size: ~13 to 40 nucleotides.
~ Requires the target sequence UH immediately 5' of the cleavage site.
~ Binds a variable number nucleotides on both sides of the cleavage site.
~ Reaction mechanism: attack by 2'-OH 5' to the scissile bond to generate
cleavage products
with 2',3'-cyclic phosphate and 5'-OH ends.
~ 14 known members of this class. Found in a number of plant pathogens
(virusoids) that
use RNA as the infectious agent.
~ Essential structural features largely defined, including 2 crystal
structures [26,2']
~ Minimal ligation activity demonstrated (for engineering through in vitro
selection) [Z8]
z° . Zimmerly, Steven; Guo, Huatao; Eskes, Robert; Yang, Jian; Penman,
Philip S.;
Lambowitz, Alan M.. A group II intron RNA is a catalytic component of a DNA
endonuclease
involved in intron mobility. Cell (Cambridge, Mass.) (1995), 83(4), 529-38.
z' . Griffin, Edmund A., Jr.; Qin, Zhifeng; Michels, Williams J., Jr.; Pyle,
Anna Marie. Group II
intron ribozymes that cleave DNA and RNA linkages with similar efficiency, and
lack contacts with
substrate 2'-hydroxyl groups. Chem. Biol. (1995), 2(11 ), 761-70.
Michel, Francois; Ferat, Jean Luc. Structure and activities of group II
introns. Annu. Rev.
Biochem. (1995), 64, 435-61.
zs . Abramovitz, Dana L.; Friedman, Richard A.; Pyle, Anna Marie. Catalytic
role of 2'-hydroxyl
groups within a group Il intron active site. Science (Washington, D. C.)
(1996), 271(5254), 1410-
13.
za , Daniels, Danette L.; Michels, William J., Jr.; Pyle, Anna Marie. Two
competing pathways
for self-splicing by group II introns: a quantitative analysis of in vitro
reaction rates and products. J.
Mol. Biol. (1996), 256(1), 31-49.
zs Guo, Hans C. T.; Collins, Richard A.. Efficient trans-cleavage of a stem-
loop RNA
substrate by a ribozyme derived from Neurospora VS RNA. EMBO J. (1995), 14(2),
368-76.
ze . Scott, W.G., Finch, J.T., Aaron,K. The crystal structure of an all RNA
hammerhead
ribozyme:Aproposed mechanism for RNA catalytic cleavage. Cell, (1995), 81, 991-
1002.
z' McKay, Structure and function of the hammerhead ribozyme: an unfinished
story. RNA,
( 7 996), 2, 395-403.
ze . Long, D., Uhlenbeck, O., Hertel, K. Ligation with hammerhead ribozymes.
US Patent No.
CA 02288640 1999-11-OS
WO 98/50530 132 PCT/US98109249
Complete kinetic framework established for two or more ribozymes [z9].
Chemical modification investigation of important residues well established
('o]
S
Hairpin Ribozyme
~ Size: ~50 nucleotides.
~ Requires the target sequence GUC immediately 3' of the cleavage site.
~ Binds 4-G nucleotides at the 5'-side of the cleavage site and a variable
number to the 3'-side
of the cleavage site.
~ Reaction mechanism: attack by 2'-OH 5' to the scissile bond to generate
cleavage products
with 2',3'-cyclic phosphate and 5'-OH ends.
~ 3 known members of this class. Found in three plant pathogen (satellite RNAs
of the
tobacco ringspot virus, arabis mosaic virus and chicory yellow mottle virus)
which uses
RNA as the infectious agent.
~ Essential structural features largely defined ["~32~33~34~
~ Ligation activity (in addition to cleavage activity) makes ribozyme amenable
to
engineering through in vitro seiection (3s]
~ Complete kinetic framework established for one ribozyme (36].
~ Chemical modification investigation of important residues begun (3'~38~.
5,633,133.
zs . Hertel, K.J., Herschlag, D., Uhlenbeck, O. A kinetic and thermodynamic
framework for the
hammerhead ribozyme reaction. Biochemistry, (1994) 33, 3374-3385.Beigelman,
L., et al.,
Chemical modifications of hammerhead ribozymes. J. Biol. Chem., (1995) 270,
25702-25708.
'° . Beigelman, L., et aL, Chemical modifications of hammerhead
ribozymes. J. Biol. Chem.,
(1995) 270, 25702-25708.
" . Hampel, Arnold; Tritz, Richard; Hicks, Margaret; Cruz, Phillip. 'Hairpin'
catalytic RNA
model: evidence for helixes and sequence requirement for substrate RNA.
Nucleic Acids Res.
(1990), 18(2), 299-304.
32 . Chowrira, Bharat M.; Berzal-Herranz, Alfredo; Burke, John M.. Novel
guanosine
requirement for catalysis by the hairpin ribozyme. Nature (London) (1991 ),
354{6351 ), 320-2.
33 . gerzal-Herranz, Alfredo; Joseph, Simpson; Chowrira, Bharat M.; Butcher,
Samuel E.;
Burke, John M.. Essential nucleotide sequences and secondary structure
elements of the hairpin
ribozyme. EMBO J. (1993), 12(6), 2567-73.
Joseph, Simpson; Berzal-Herranz, Alfredo; Chowrira, Bharat M.; Butcher, Samuel
E..
Substrate selection rules for the hairpin ribozyme determined by in vitro
selection, mutation, and
analysis of mismatched substrates. Genes Dev. (1993), 7(1 ), 130-8.
ss . gerzal-Hen-anz, Alfredo; Joseph, Simpson; Burke, John M.. In vitro
selection of active
hairpin ribozymes by sequential RNA-catalyzed cleavage and ligation reactions.
Genes Dev.
(1992}, 6(1 ), 129-34.
Hegg, Lisa A.; Fedor, Martha J.. Kinetics and Thermodynamics of Intermolecular
Catalysis
by Hairpin Ribozymes. Biochemistry (1995), 34(48), 15813-28.
Grasby, Jane A.; Mersmann, Karin; Singh, Mohinder; Gait, Michael J.. Purine
Functional
Groups in Essential Residues of the Hairpin Ribozyme Required for Catalytic
Cleavage of RNA.
Biochemistry (1995), 34(12), 4068-76.
38 . Schmidt, Sabine; Beigelman, Leonid; Karpeisky, Alexander; Usman, Nassim;
Sorensen,
Ulrik S.; Gait, Michael J.. Base and sugar requirements for RNA cleavage of
essential nucleoside
residues in internal loop B of the hairpin ribozyme: implications for
secondary structure. Nucleic
Acids Res. (1996), 24(4), 573-81.
CA 02288640 1999-11-OS
WO 98/50530 133 PCT/US98/09249
Hepatitis Delta Virus (HDV) Ribozyme
~ Size: ~60 nucleotides.
~ Traps cleavage of target RNAs demonstrated ['9].
~ Binding sites and structural requirements not fully determined, although no
sequences 5' of
cleavage site are required. Folded ribozyme contains a pseudoknot structure
["°].
~ Reaction mechanism: attack by 2'-OH 5' to the scissile bond to generate
cleavage products
with 2',3'-cyclic phosphate and 5'-OH ends.
~ Only 2 known members of this class. Found in human HDV.
~ Circular form of HDV is active and shows increased nuclease stability ["]
3s . Perrotta, Anne T.; Been, Michael D.. Cleavage of oligoribonucleotides by
a ribozyme
derived from the hepatitis .delta. virus RNA sequence. Biochemistry (1992), 31
(1 ), 16-21.
ao _ perrotta, Anne T.; Been, Michael D.. A pseudoknot-like structure required
for efficient self-
cleavage of hepatitis delta virus RNA. Nature {London) (1991 ), 350(6317), 434-
6.
4' . Puttaraju, M.; Perrotta, Anne T.; Been, Michael D.. A circular traps-
acting hepatitis delta
virus ribozyme. Nucleic Acids Res. (1993), 21 (18), 4253-8.
CA 02288640 1999-11-OS
WO 98/50530 134 PCTNS98/09249
Table II: 2.5 ~mol RNA Synthesis Cycle
Reagent Equivalents Amount Wait
Time*
Phosphoramidites6.5 163 uL 2.5
S-Ethyl Tetrazole23.8 238 ~L 2.5
Acetic Anhydride100 233 ~L 5
sec
N Methyl Imidazole186 233 1zL 5
sec
TCA 83.2 1.73 mL 21
sec
Iodine 8.0 1.18 mL 45
sec
Acetonitrile NA 6.67 mL NA
* Wait time does not include contact time during delivery.
CA 02288640 1999-11-OS
WO 98/50530 13S PCT/US98/09249
TABLE III. NUCLEOSIDES USED FOR CHEMICAL SYNTHESIS
OF MODIFIED NUCLEOTIDE TRIPHOSPHATES
NUCLEOSIDES Abbreviation_ CHEMICAL STRUCTURE
I 2'-O-methyl-2,6- 2'-O-Me-DAP ~~ NH 2
diaminopurine liboside /N w N
HO \N I N ~ NH 2
O
HO OCH
2 2'-deoxy-2'amino-2,6- 2'-NHz DAP NH2
diaminopurine 1-iboside N ' N
i
HO ~N ~ N' _ NH2
/O
HO NH2
3 2'-(N alanyl)amino-2'- ala-2'- NHz U
deoxy-uridine ~ H
HO
O
HO HN~ ~C~ CH 3
C NH2
O
4 2'-(N phe-2'- NHZ-U
phenylalanyl)amino-2'- _ ,H
deoxy-uridine I N
HO N~O
O
HO HN~ ~C' CH2Ph
C NH2
O
CA 02288640 1999-11-OS
WO 98/50530 136 PCT/US98/09249
NUCLEOSIDES Abbreviation CHEMICAL STRUCTURE
2'-(N /3-alanyl) amino- 2'-J3-Ala-NHZ-U O
2'-deoxy uridine ~H
'N
HO N~O
O
O
HO HN.
NH2
6 2'-Deoxy-2'-(lysiyl) 2'-L-lys-NHz U
amino uridine NCH
Ho ~
NI ' O
O
O
HO HN
NH2
NH2
'7 2'-C-allyl uridine 2'-C-allyl-U O
~H
HO N O
HO /
8 2'-O-amino-uridine 2'-O-NHz-U O
N, H
HO
N O
O
t
HO O.
NH2
CA 02288640 1999-11-OS
WO 98/50530 13~ PCT/US98/09249
NUCLEOSIDES Abbreviation CHEMICAL STRUCTURE
9 2'-O-methylthiomethyl 2'-O-MTM-A NH2
adenosine N
HO
N N
HO O
S
2'-O-methylthiomethyl 2'-O-MTM-C NH2
cytidine ~ N
HO
O
O -
HO
S
11 2'-O-methylthiomethyl 2'-O-MTM-G O
guanosine N
HO' N N NH2
LO
HO
S~
CA 02288640 1999-11-OS
WO 98/50530 PCT/US98/09249
138
NUCLEOSIDES Abbreviation CHEMICAL STRUCTURE
12 2'-O-methylthiomethyl- 2'-O-MTM-U O
uridine ~H
HO
O
OH O
S
13 2'-(N histidyl) amino 2'-his-NH~-U O
uridine H
N~
HO ~
N" O
O
(~ O
HO HN H
N
NH2 ~,
N
14 2'-Deoxy-2'-amino-S- 5-Me-2'-NHZ-C NH2
methyl cytidine
~N
HO
N O
O
HO NH2
15 2'-(N (3-carboxamidine- (3-ala-CA-NH2-U O
(3-alanyl)amino-2'- ,H
deoxy-uridine . ~ _N
HO N" o
O
H2 N NH2
HO HN~C C'C' ~C~
H2 'NH
O
CA 02288640 1999-11-OS
WO 98/50530 139 PCT/US98/09249
NUCLEOSIDES Abbreviation CHEMICAL_STRUCTURE
16 2'-(N (3-alanyl) ~3-Ala-NHZ G
guanosine
NH
HO
'' ~N NH2
O
NH2
HO
O
17 2'-O-Amino-Adenosine 2'-O-NH2-A O
N, H
HO
N O
O
HO O~ NH
2
CA 02288640 1999-11-OS
WO 98/50530 140 PCT/US98/09249
Table VI. PHOSPHORYLATION OF URIDINE IN THE PRESENCE OF DMAP
0 equiv. 0.2 0.5 equiv. 1.0
DMAP equiv. DMAP equiv.
DMAP DMAP
Time ProductTime ProductTime Product Time Product
(min) % (min) % (min) % (min)
0 1 0 0 0 0 0 0
40 7 10 8 20 27 30 74
80 10 SO 24 60 46 70 77
120 12 90 33 100 57 110 84
160 14 130 39 140 63 150 83
200 17 170 43 180 63 190 84
240 19 210 47 220 64 230 77
320 20 250 48 260 68 270 79
1130 48 290 49 300 64 310 77
1200 46 1140 68 1150 76 1160 72
1210 69 1220 76 1230 74
CA 02288640 1999-11-OS
WO 98/50530 PCTNS98/09249
141
I /'~ Y..:
U
o
_
N
N
M M M
..
M
N
' -
N N
N
M
M
O
~x~~
~~
uJ 1 d' d' ~Y ~t d'
o 1 d' d' d' d' et.
I :
I a 4'!
...
~
r r 1 1 r r
.' t~ r 1 1 r r
J ~
r
J
Z
'
Q ~ O O
O
~ .I O O
O '
O
O
o ,
,
,
,
r r r r::...-:r r r r
0
O O O .s
O O O
+ ~O O O
.1 O O O
O
.
O O O O:w . . O O O O
oo
~X oooo;; oooo
b
0
U
.
O ~ r r r r ~:~ .
E . ~~ ~ ~ r r r T ()
~>c
a w
>> ':
y:::
0
,.,
O O 0
,
'.; O O ~ I,n tn tn
~ ~ ~ tf~ :
i.. KS:..; G3r
V ~
O O N N N N".~O O N N N
C7 N
~ N N r r r r :;-:,N N r
r
r
r
N:~
J /'~ /~ /~~ /1 /'~ /~'~.;t~~Gi
/'~ /~'~ /1 ~ /1 /~'~
O O r r T r:: ',O O T e-
r r
O O O O O O ~.:~0 O O O
O O
Z ~ Z Z Z Z ir~.:Z Z I ~r
Z Z Z
cn aaaaaa:::-.:~aaaaaa y
E
'I ~w./ ~.I ~/ ~.I Win/ ~"/,5
:.. ~ t ~.I ~.I ~./ ~.I
~/ 1"/
H O O O Oi O c~
O':i~s0
y
O
O
O O
O
~,
,
~
,~, ~
,
~.
Iyn...
:'
_ ~/.
w:i:. a H
W
'p r N M d' tf~ Cfl ~:fi.,;
Z Op O ~ r ~
O
~'~o-':.;
V
H
CA 02288640 1999-11-OS
WO 98/50530 142 PCT/US98/09249
O ~COO r enCflO N In1~ti7O ~CO00InODM d'O M ~t
'
O Nch~ f~ r ~ 00M N
N r d'
Z~
U
InOO a700Or O N d'r InO MO In1~I~M O O Inr
O N N MM tt) 00 rr ~ r ~ r
'
r - Wit N N M M
Z
~
U
r-~r OpO NN ctN ~M tnO OCOO OCI~N ~ O ~ M
r (pN r N rN r O N O O ~Y N
Z N 'cf Nr r r M r
p
U
O NO O ~f~f'00(DN ~fM ~ O ~CO~ r InM d'O ~ r
r Or N 00OM r O r CpI~I~ N
N N r N ~ M
U
N d'(fl00M I~cfl1~r ~r M O ODO M 00OM M O ~ r
~r Op NM r ~ s-r N N N
Z~
H
U
r CDM 00CDI~OD00r Nlf~d'r ~1'~1'O 00NM COr M O
N ~ N In r N 001~ r
N
Z
1~
U
OI~N d'f~N 00O '~i'00Inr ~Opr (p(pN ~ O r N
1~.-00N 'd'r r- ~ N r N
Z
~
H
N ~~ ~7O InO M O 3 ~fr ~ r M00r O f~M !fO r r
0 N 00 M r O ~t N O r hl
-
~
o ~ O
U u~
O M COl(700O ~ ~'00~ r NI~I~M OM InO r M
Cpr O N ~r r p M r O M
r r
Q O
U
M InCOd'O MO ~ O ~ d'O M r Ni~~ 00~N ~ O r r
~t r d' r M N O r
~
Z : W
M C
V ~ A
N 1~~ InM OO O O ~ MN CfO NN CON 'd'N 00r N rO
M ~t rr ' LO r el'N d'
ZN a
O O
U
d'O rCOO O V ~tntl'O N00LIBI~OM CGO N (~
r r ~f0" tp CON M O
H
U c
O
c
o U
a
v at ~ a a a a a
'-
aa a ~ ~ ~~ a a.?~ ~ a v ~
a v~ ~ a c~c~r o o =Z ~ H ~ ~ ~
-
?
Q a aa a d d dd d N z a a~ s f-f-t-
= NN N Q UC9~ ~ ~ ~~ ~ _ '~ Z Q C9ToZ ~ ~ ~::
ZZ Z N
Z ZZ Z .a.o.o-~Q O OO O Z ~= e~!tiliV O O O O
N N l C N N N N NE'~
N NN N N NN N N N N V 10Q V N N
CA 02288640 1999-11-OS
WO 98/50530 143 PCT/US98/09249
Table IX: INCORPORATION OF MODIFIED
NUCLEOTIDE TRIPHOSPHATES USING WILD TYPE
BACTERIOPHAGE T7 POLYMERASE
Modification label % ribo control
2'-NHZ-GTP ATP 4%
2'-dGTP ATP 3
2'-O-Me-GTP ATP 3%
2'-F-GTP ATP 4%
2'-_O-_M_TM-GTP ATP 3%
2'-NHZ-UTP _ 39%
~~~ ATP
2'-dTTP _ 5%
ATP
_ __ ATP 3
2'-O-Me-UTP
ala-2'-NHZ UTP ATP 2%
phe-2'-NHZ- UTP ATP 1
2'-~i-ala-NHZ UTP TP 3%
_ A
2'-C-allyl-UTP _ 2%
ATP
2'-O-NHZ-U'TP ATP 1
2'-O-MTM-UTP ATP 64%
2'-NHZ ATP _ _ GTP 1
2'-O-MTM-ATP GTP 1 % _
2'-NHZ-CTP GTP 59%
2'-dCTP GTP 40%
CA 02288640 1999-11-OS
WO 98/50530 144 PCT/US98/09249
Table Xa: Incorporation of 2'-his-UTP and Modified CTP's
modification 2'-his-UTP rUTP
CTP 16.1 100
2'-amino-CTP 9.5* 232.7
2'-deoxy-CTP 9.6* 130.1
2'-O M e-CTP 1.9 6.2
2'-MTM-CTP 5.9 5.1
control 1.2
Table Xb: Incorporation of 2'-his-UTP, 2-amino CTP, and Modified ATP's
2'-his-UTP and
modification 2'-amino-CTP rUTP and rCTP
ATP 15.7 100
2'-amino-ATP 2.4 28.9
2'-deoxy-ATP 2.3 146.3
2'-OMe-ATP 2.7 15
2'-F-ATP 4 222.6
2'-MTM-ATP 4.7 15.3
2'-OMe-DAP 1.9 5.7
~2'-amino-DAP 8.9* 9.6
Numbers shown are a percentage of incorporation compared to the all-RNA
control
* -Bold number indicates best observed rate of modified nucleotide
triphosphate
incorporation
CA 02288640 1999-11-OS
WO 98/50530 145 PCT/US98109249
Table XI. INCORPORATION OF 2'-his-UTP, 2'-NHZ CTP, 2'-NHZ DAP,
and rGTP USING VARIOUS REACTION CONDITIONS
Conditions compared to all
rNTP
7 8.7*
8 7*
9 2.3
2.7
11 1.6
12 _
2.5
5 Numbers shown are a percentage of incorporation compared to the all-RNA
control
* Two highest levels of incorporation contained both methanol and LiCl
CA 02288640 1999-11-OS
WO 98/50530 146 PCT/US98/09249
Table XII: Human C-raf Hammerhead Ribozyme and Target Sequences
nt Target SEQ Riboryme SEQ
positionSite ID. Sequence ID.
No. No.
17 GACCGCCUCCCGCUCCC1 GGGAGCGGCUGAUGAGX CGAA AGGCGGUC502
23 CUCCCGCUCCCUCACCC2 GGGUGAGGCUGAUGAGX CGAA AGCGGGAG503
27 CGCUCCCUCACCCGCCG3 CGGCGGGUCUGAUGAGX CGAA AGGGAGCG504
82 CAGGACGUUGGGGCGGC4 GCCGCCCCCUGAUGAGX CGAA ACGUCCUG505
97 GCCUGGCUCCCUCAGGU5 ACCUGAGGCUGAUGAGX CGAA AGCCAGGC506
101 GGCUCCCUCAGGUUUAA6 UUAAACCUCUGAUGAGX CGAA AGGGAGCC507
106 CCUCAGGUUUAAGAAUU7 AAUUCUUACUGAUGAGX CGAA ACCUGAGG508
107 CUCAGGUUU 8 CAAUUCUUCUGAUGAGX CGAA AACCUGAG509
AAGAAUUG
108 UCAGGUUUAAGAAUUGU9 ACAAUUCUCUGAUGAGX CGAA AAACCUGA510
119 UUAAGAAUUGUUUAAGC10 GCUUAAACCUGAUGAGX CGAA AUUCUUAA511
117 AGAAUUGUUUAAGCUGC11 GCAGCUUACUGAUGAGX CGAA ACAAUUCU512
118 GAAUUGUUU 12 UGCAGCUUCUGAUGAGX CGAA AACAAUUC513
AAGCUGCA
119 AAUUGUUUAAGCUGCAU13 AUGCAGCUCUGAUGAGX CGAA AAACAAUU514
128 AGCUGCAUC 19 GCUCCAUUCUGAUGAGX CGAA AUGCAGCU515
AAUGGAGC
141 GAGCACAUACAGGGAGC15 GCUCCCUGCUGAUGAGX CGAA AUGUGCUC516
151 AGGGAGCUUGGAAGACG16 CGUCUUCCCUGAUGAGX CGAA AGCUCCCU517
162 AAGACGAUCAGCAAUGG17 CCAUUGCUCUGAUGAGX CGAA AUCGUCUU518
172 GCAAUGGUUUUGGAUUC18 GAAUCCAA X CGAA ACCAUUGC519
CUGAUGAG
173 CAAUGGUUUUGGAUUCA19 UGAAUCCACUGAUGAGX CGAA AACCAUUG520
179 AAUGGUUUUGGAUUCAA20 UUGAAUCCCUGAUGAGX CGAA AAACCAUU521
179 UUUUGGAUUCAAAGAUG21 CAUCUUUGCUGAUGAGX CGAA AUCCAAAA522
180 UUUGGAUUC 22 GCAUCUUUCUGAUGAGX CGAA AAUCCAAA523
AAAGAUGC
194 UGCCGUGUUUGAUGGCU23 AGCCAUCACUGAUGAGX CGAA ACACGGCA524
195 GCCGUGUUUGAUGGCUC29 GAGCCAUCCUGAUGAGX CGAA AACACGGC525
203 UGAUGGCUCCAGCUGCA25 UGCAGCUGCUGAUGAGX CGAA AGCCAUCA526
213 AGCUGCAUCUCUCCUAC26 GUAGGAGACUGAUGAGX CGAA AUGCAGCU527
215 CUGCAUCUCUCCUACAA27 UUGUAGGACUGAUGAGX CGAA AGAUGCAG528
217 GCAUCUCUCCUACAAUA28 UAUUGUAGCUGAUGAGX CGAA AGAGAUGC529
220 UCUCUCCUACAAUAGUU29 AACUAUUGCUGAUGAGX CGAA AGGAGAGA530
225 CCUACAAUAGUUCAGCA30 UGCUGAACCUGAUGAGX CGAA AUUGUAGG531
228 ACAAUAGUUCAGCAGUU31 AACUGCUGCUGAUGAGX CGAA ACUAUUGU532
229 CAAUAGUUCAGCAGUUU32 AAACUGCUCUGAUGAGX CGAA AACUAUUG533
236 UCAGCAGUUUGGCUAUC33 GAUAGCCACUGAUGAGX CGAA ACUGCUGA534
237 CAGCAGUUUGGCUAUCA39 UGAUAGCCCUGAUGAGX CGAA AACUGCUG535
242 GUUUGGCUAUCAGCGCC35 GGCGCUGACUGAUGAGX CGAA AGCCAAAC536
244 UUGGCUAUCAGCGCCGG36 CCGGCGCUCUGAUGAGX CGAA AUAGCCAA537
257 CCGGGCAUCAGAUGAUG37 CAUCAUCUCUGAUGAGX CGAA AUGCCCGG538
273 GGCAAACUCACAGAUCC38 GGAUCUGUCUGAUGAGX CGAA AGUUUGCC539
280 UCACAGAUCCUUCUAAG39 CUUAGAAGCUGAUGAGX CGAA AUCUGUGA540
283 CAGAUCCUUCUAAGACA40 UGUCUUAGCUGAUGAGX CGAA AGGAUCUG591
284 AGAUCCUUCUAAGACAA41 UUGUCUUACUGAUGAGX CGAA AAGGAUCU592
286 AUCCUUCUAAGACAAGC92 GCUUGUCUCUGAUGAGX CGAA AGAAGGAU543
301 GCAACACUAUCCGUGUU93 AACACGGACUGAUGAGX CGAA AGUGUUGC549
303 AACACUAUCCGUGUUUU49 AAAACACGCUGAUGAGX CGAA AUAGUGUU545
309 AUCCGUGUUUUCUUGCC45 GGCAAGAA X CGAA ACACGGAU596
CUGAUGAG
310 UCCGUGUUUUCUUGCCG46 CGGCAAGACUGAUGAGX CGAA AACACGGA547
311 CCGUGUUUUCUUGCCGA97 UCGGCAAGCUGAUGAGX CGAA AAACACGG548
312 CGUGUUUUCUUGCCGAA98 UUCGGCAA X CGAA AAAACACG549
CUGAUGAG
314 UGUUUUCUUGCCGAACA99 UGUUCGGCCUGAUGAGX CGAA AGAAAACA550
339 ACAGUGGUC 50 CGCACAUUCUGAUGAGX CGAA ACCACUGU551
AAUGUGCG
362 AAUGAGCUUGCAUGACU51 AGUCAUGCCUGAUGAGX CGAA AGCUCAUU552
375 GACUGCCUUAUGAAAGC52 GCUUUCAUCUGAUGAGX CGAA AGGCAGUC553
376 ACUGCCUUAUGAAAGCA53 UGCUUUCACUGAUGAGX CGAA AAGGCAGU554
387 AAAGCACUC 54 CUCACCUUCUGAUGAGX CGAA AGUGCUUU555
AAGGUGAG
925 UGCAGUGUUCAGACUUC55 GAAGUCUGCUGAUGAGX CGAA ACACUGCA556
426 GCAGUGUUCAGACUUCU56 AGAAGUCUCUGAUGAGX CGAA AACACUGC557
432 UUCAGACUUCUCCACGA57 UCGUGGAGCUGAUGAGX CGAA AGUCUGAA558
CA 02288640 1999-11-OS
WO 98/50530 14~ PCT/US98/09249
nt Target SEQ Ribozyme SEQ
positionSite ID. Sequence ID.
No. No.
433 UCAGACUUCUCCACGAA5B UUCGUGGACU6AUGAGX CGAA AAGUCUGA559
435 AGACUUCUCCACGAACA59 UGUUCGUGCUGAUGAGX CGAA AGAAGUCU560
451 ACAAAGGUA 60 UGCUUUUUCUGAUGAGX CGAA ACCUUUGU561
AAAAAGCA
469 AGCACGCUUAGAUUGGA61 UCCAAUCUCUGAUGAGX CGAA AGCGUGCU562
465 GCACGCUUAGAUUGGAA62 UUCCAAUCCUGAUGAGX CGAA AAGCGUGC563
969 GCUUAGAUUGGAAUACU63 AGUAUUCCCUGAUGAGX CGAA AUCUAAGC569
975 AUUGGAAUACUGAUGCU64 AGCAUCAGCUGAUGAGX CGAA AUUCCAAU565
988 UGCUGCGUCUUUGAUUG65 CAAUCAAA X CGAA ACGCAGCA566
CUGAUGAG
990 CUGCGUCUUUGAUUGGA66 UCCAAUCACUGAUGAGX CGAA AGACGCAG567
991 UGCGUCUUUGAUUGGAG67 CUCCAAUCCUGAUGAGX CGAA AAGACGCA568
995 UCUUUGAUUGGAGAAGA68 UCUUCUCCCUGAUGAGX CGAA AUCAAAGA569
507 GAAGAACUUCAAGUAGA69 UCUACUUGCUGAUGAGX CGAA AGUUCUUC570
508 AAGAACUUC 70 AUCUACUUCUGAUGAGX CGAA AAGUUCUU571
AAGUAGAU
513 CUUCAAGUAGAUUUCCU71 AGGAAAUCCUGAUGAGX CGAA ACUUGAAG572
517 AAGUAGAUUUCCUGGAU72 AUCCAGGACUGAUGAGX CGAA AUCUACUU573
518 AGUAGAUUUCCUGGAUC73 GAUCCAGGCUGAUGAGX CGAA AAUCUACU574
519 GUAGAUUUCCUGGAUCA79 UGAUCCAGCUGAUGAGX CGAA AAAUCUAC575
526 UCCUGGAUCAUGUUCCC75 GGGAACAUCUGAUGAGX CGAA AUCCAGGA576
531 GAUCAUGUUCCCCUCAC76 GUGAGGGGCUGAUGAGX CGAA ACAUGAUC577
532 AUCAUGUUCCCCUCACA77 UGUGAGGGCUGAUGAGX CGAA AACAUGAU578
S37 GUUCCCCUCACAACACA78 UGUGUUGUCUGAUGAGX CGAA AGGGGAAC579
551 ACACAACUUUGCUCGGA79 UCCGAGCACUGAUGAGX CGAA AGUUGUGU580
552 CACAACUUUGCUCGGAA80 UUCCGAGCCUGAUGAGX CGAA AAGUUGUG581
556 ACUUUGCUCGGAAGACGB1 CGUCUUCCCUGAUGAGX CGAA AGCAAAGU582
566 GAAGACGUUCCUGAAGC82 GCUUCAGGCUGAUGAGX CGAA ACGUCUUC583
SS7 AAGACGUUCCUGAAGCU83 AGCUUCAGCUGAUGAGX CGAA AACGUCUU584
576 CUGAAGCUUGCCUUCUG89 CAGAAGGCCUGAUGAGX CGAA AGCUUCAG585
581 GCUUGCCUUCUGUGACABS UGUCACAGCUGAUGAGX CGAA AGGCAAGC586
582 CUUGCCUUCUGUGACAU86 AUGUCACACUGAUGAGX CGAA AAGGCAAG587
591 UGUGACAUCUGUCAGAA87 UUCUGACACUGAUGAGX CGAA AUGUCACAS88
595 ACAUCUGUCAGAAAUUC88 GAAUUUCUCUGAUGAGX CGAA ACAGAUGU589
602 UCAGAAAUUCCUGCUCA89 UGAGCAGGCUGAUGAGX CGAA AUUUCUGA590
603 CAGAAAUUCCUGCUCAA90 UUGAGCAGCUGAUGAGX CGAA AAUUUCUG591
609 UUCCUGCUC 91 AAUCCAUUCUGAUGAGX CGAA AGCAGGAA592
AAUGGAUU
617 CAAUGGAUUUCGAUGUC92 GACAUCGACUGAUGAGX CGAA AUCCAUUG593
618 AAUGGAUUUCGAUGUCA93 UGACAUCGCUGAUGAGX CGAA AAUCCAUU594
619 AUGGAUUUCGAUGUCAG94 CUGACAUCCUGAUGAGX CGAA AAAUCCAU595
625 UUCGAUGUCAGACUUGU95 ACAAGUCUCUGAUGAGX CGAA ACAUCGAA596
631 GUCAGACUUGUGGCUAC96 GUAGCCACCUGAUGAGX CGAA AGUCUGAC59?
638 UUGUGGCUACAAAUUUC97 GAAAUUUGCUGAUGAGX CGAA AGCCACAA598
649 CUACAAAUUUCAUGAGC98 GCUCAUGACUGAUGAGX CGAA AUUUGUAG599
645 UACAAAUUUCAUGAGCA99 UGCUCAUGCUGAUGAGX CGAA AAUUUGUA600
646 ACAAAUUUCAUGAGCAC100 GUGCUCAUCUGAUGAGX CGAA AAAUUUGU601
658 AGCACUGUAGCACCAAA101 UUUGGUGCCUGAUGAGX CGAA ACAGUGCU602
669 ACCAAAGUACCUACUAU102 AUAGUAGGCUGAUGAGX CGAA ACUUUGGU603
673 AAGUACCUACUAUGUGU103 ACACAUAGCUGAUGAGX CGAA AGGUACUU604
676 UACCUACUAUGUGUGUG104 CACACACACUGAUGAGX CGAA AGUAGGUA605
694 ACUGGAGUAACAUCAGA105 UCUGAUGUCUGAUGAGX CGAA ACUCCAGU606
699 AGUAACAUCAGACAACU106 AGUUGUCUCUGAUGAGX CGAA AUGUUACU607
708 AGACAACUCUUAUUGUU107 AACAAUAA X CGAA AGUUGUCU608
CUGAUGAG
710 ACAACUCUUAUUGUUUC108 GAAACAAUCUGAUGAGX CGAA AGAGUUGU609
711 CAACUCUUAUUGUUUCC109 GGAAACAA X CGAA AAGAGUUG610
CUGAUGAG
713 ACUCUUAUUGUUUCCAA110 UUGGAAACCUGAUGAGX CGAA AUAAGAGU611
716 CUUAUUGUUUCCAAAUU111 AAUUUGGACUGAUGAGX CGAA ACAAUAAG612
717 UUAUUGUUUCCAAAUUC112 GAAUUUGGCUGAUGAGX CGAA AACAAUAA613
718 UAUUGUUUCCAAAUUCC113 GGAAUUUGCUGAUGAGX CGAA AAACAAUA614
729 UUCCAAAUUCCACUAUU114 AAUAGUGGCUGAUGAGX CGAA AUUUGGAA615
725 UCCAAAUUCCACUAUUG115 CAAUAGUGCUGAUGAGX CGAA AAUUUGGA616
L ~ AUUCCACUAUUGGUGAU~ ~ AUCACCAA X CGAA AGUGGAAUT
730 116 CUGAUGAG 617
CA 02288640 1999-11-OS
WO 98/50530 14g PCT/US98/09249
nt Target SEQ Ribozyme SEQ
positionSite ID. Sequence 1D.
No. No.
732 UCCACUAUUGGUGAUAG117 CUAUCACCCUGAUGAGX CGAA AUAGUGGA618
739 UUGGUGAUAGUGGAGUC118 GACUCCACCUGAUGAGX CGAA AUCACCAA619
797 AGUGGAGUCCCAGCACU119 AGUGCUGGCUGAUGAGX CGAA ACUCCACU620
756 CCAGCACUACCUUCUUU120 AAAGAAGGCUGAUGAGX CGAA AGUGCUGG621
760 CACUACCUUCUUUGACU121 AGUCAAAGCUGAUGAGX CGAA AGGUAGUG622
761 ACUACCUUCUUUGACUA122 UAGUCAAA X CGAA AAGGUAGU623
CUGAUGAG
763 UACCUUCUUUGACUAUG123 CAUAGUCACUGAUGAGX CGAA AGAAGGUA629
764 ACCUUCUUUGACUAUGC124 GCAUAGUCCUGAUGAGX CGAA AAGAAGGU625
769 CUUUGACUAUGCGUCGU125 ACGACGCACUGAUGAGX CGAA AGUCAAAG626
?75 CUAUGCGUCGUAUGCGA126 UCGCAUACCUGAUGAGX CGAA ACGCAUAG627
778 UGCGUCGUAUGCGAGAG127 CUCUCGCACUGAUGAGX CGAA ACGACGCA628
7B8 GCGAGAGUCUGUUUCCA128 UGGAAACACUGAUGAGX CGAA ACUCUCGC629
792 GAGUCUGUUUCCAGGAU129 AUCCUGGACUGAUGAGX CGAA ACAGACUC630
793 AGUCUGUUUCCAGGAUG130 CAUCCUGGCUGAUGAGX CGAA AACAGACU631
799 GUCUGUUUCCAGGAUGC131 GCAUCCUGCUGAUGAGX CGAA AAACAGAC632
807 AUGCCUGUUAGUUCUCA132 UGAGAACUCUGAUGAGX CGAA ACAGGCAU633
808 UGCCUGUUAGUUCUCAG133 CUGAGAACCUGAUGAGX CGAA AACAGGCA634
811 CUGUUAGUUCUCAGCAC139 GUGCUGAGCUGAUGAGX CGAA ACUAACAG635
812 UGUUAGUUCUCAGCACA135 UGUGCUGACUGAUGAGX CGAA AACUAACA636
819 UUAGUUCUCAGCACAGA136 UCUGUGCUCUGAUGAGX CGAA AGAACUAA637
829 GCACAGAUAUUCUACAC137 GUGUAGAA X CGAA AUCUGUGC638
CUGAUGAG
826 ACAGAUAUUCUACACCU138 AGGUGUAGCUGAUGAGX CGAA AUAUCUGU639
827 CAGAUAUUCUACACCUC139 GAGGUGUACUGAUGAGX CGAA AAUAUCUG690
829 GAUAUUCUACACCUCAC140 GUGAGGUGCUGAUGAGX CGAA AGAAUAUC641
835 CUACACCUCACGCCUUC141 GAAGGCGUCUGAUGAGX CGAA AGGUGUAG642
892 UCACGCCUUCACCUUUA142 UAAAGGUGCUGAUGAGX CGAA AGGCGUGA643
843 CACGCCUUCACCUUUAA143 UUAAAGGUCUGAUGAGX CGAA AAGGCGUG644
848 CUUCACCUUUAACACCU149 AGGUGUUACUGAUGAGX CGAA AGGUGAAG645
899 UUCACCUUU 145 GAGGUGUUCUGAUGAGX CGAA AAGGUGAA646
AACACCUC
850 UCACCUUUAACACCUCC146 GGAGGUGUCUGAUGAGX CGAA AAAGGUGA647
857 UAACACCUCCAGUCCCU147 AGGGACUGCUGAUGAGX CGAA AGGUGUUA698
862 CCUCCAGUCCCUCAUCU148 AGAUGAGGCUGAUGAGX CGAA ACUGGAGG649
866 CAGUCCCUCAUCUGAAG199 CUUCAGAUCUGAUGAGX CGAA AGGGACUG650
869 UCCCUCAUCUGAAGGUU150 AACCUUCACUGAUGAGX CGAA AUGAGGGA651
877 CUGAAGGUUCCCUCUCC151 GGAGAGGGCUGAUGAGX CGAA ACCUUCAG652
878 UGAAGGUUCCCUCUCCC152 GGGAGAGGCUGAUGAGX CGAA AACCUUCA653
882 GGUUCCCUCUCCCAGAG153 CUCUGGGACUGAUGAGX CGAA AGGGAACC654
889 UUCCCUCUCCCAGAGGC159 GCCUCUGGCUGAUGAGX CGAA AGAGGGAA655
899 GCAGAGGUCGACAUCCA155 UGGAUGUCCUGAUGAGX CGAA ACCUCUGC656
905 GUCGACAUCCACACCUA156 UAGGUGUGCUGAUGAGX CGAA AUGUCGAC657
913 CCACACCUAAUGUCCAC157 GUGGACAUCUGAUGAGX CGAA AGGUGUGG658
918 CCUAAUGUCCACAUGGU158 ACCAUGUGCUGAUGAGX CGAA ACAUUAGG659
927 CACAUGGUCAGCACCAC159 GUGGUGCUCUGAUGAGX CGAA ACCAUGUG660
960 AGGAUGAUUGAGGAUGC160 GCAUCCUCCUGAUGAGX CGAA AUCAUCCU661
972 GAUGCAAUUCGAAGUCA161 UGACUUCGCUGAUGAGX CGAA AUUGCAUC662
973 AUGCAAUUCGAAGUCAC162 GUGACUUCCUGAUGAGX CGAA AAUUGCAU663
979 UUCGAAGUCACAGCGAA163 UUCGCUGUCUGAUGAGX CGAA ACUUCGAA664
989 CAGCGAAUCAGCCUCAC169 GUGAGGCUCUGAUGAGX CGAA AUUCGCUG665
995 AUCAGCCUCACCUUCAG165 CUGAAGGUCUGAUGAGX CGAA AGGCUGAU666
1000 CCUCACCUUCAGCCCUG166 CAGGGCUGCUGAUGAGX CGAA AGGUGAGG667
1001 CUCACCUUCAGCCCUGU167 ACAGGGCUCUGAUGAGX CGAA AAGGUGAG668
1010 AGCCCUGUCCAGUAGCC168 GGCUACUGCUGAUGAGX CGAA ACAGGGCU669
1015 UGUCCAGUAGCCCCAAC169 GUUGGGGCCUGAUGAGX CGAA ACUGGACA670
1027 CCAACAAUCUGAGCCCA170 UGGGCUCACUGAUGAGX CGAA AUUGUUGG671
1046 AGGCUGGUCACAGCCGA171 UCGGCUGUCUGAUGAGX CGAA ACCAGCCU672
1092 GCACCAGUAUCUGGGAC172 GUCCCAGACUGAUGAGX CGAA ACUGGUGC673
1099 ACCAGUAUCUGGGACCC173 GGGUCCCACUGAUGAGX CGAA AUACUGGU674
1119 AACAAAAUUAGGCCUCG179 CGAGGCCUCUGAUGAGX CGAA AUUUUGUU675
1120 ACAAAAUUAGGCCUCGU175 ACGAGGCCCUGAUGAGX CGAA AAUUUUGU676
CA 02288640 1999-11-OS
WO 98/50530 149 PCT/US98/09249
nt Target SEQ Ribozyme SEQ
positionSite ID. . Sequence ID.
No. No.
1126 UUAGGCCUCGUGGACAG176 _ CUGAUGAGX CGAA AGGCCUAA677
CUGUCCAC
1141 AGAGAGAUUCAAGCUAU177 AUAGCUUGCUGAUGAGX CGAA AUCUCUCU678
1192 GAGAGAUUC 178 AAUAGCUUCUGAUGAGX CGAA AAUCUCUC679
AAGCUAUU
1198 UUCAAGCUAUUAUUGGG179 CCCAAUAA X CGAA AGCUUGAA680
CUGAUGAG
1150 CAAGCUAUUAUUGGGAA180 UUCCCAAUCUGAUGAGX CGAA AUAGCUUG681
1151 AAGCUAUUAUUGGGAAA181 UUUCCCAA X CGAA AAUAGCUU682
CUGAUGAG
1153 GCUAUUAUUGGGAAAUA182 UAUUUCCCCUGAUGAGX CGAA AUAAUAGC683
1161 UGGGAAAUAGAAGCCAG183 CUGGCUUCCUGAUGAGX CGAA AUUUCCCA684
1189 GAUGCUGUCCACUCGGA189 UCCGAGUGCUGAUGAGX CGAA ACAGCAUC685
1189 UGUCCACUCGGAUUGGG185 CCCAAUCCCUGAUGAGX CGAA AGUGGACA686
1194 ACUCGGAUUGGGUCAGG186 CCUGACCCCUGAUGAGX CGAA AUCCGAGU687
1199 GAUUGGGUCAGGCUCUU187 AAGAGCCUCUGAUGAGX CGAA ACCCAAUC688
1205 GUCAGGCUCUUUUGGAA188 UUCCAAAA X CGAA AGCCUGAC689
CUGAUGAG
1207 CAGGCUCUUUUGGAACU189 AGUUCCAA X CGAA AGAGCCUG690
CUGAUGAG
1208 AGGCUCUUUUGGAACUG190 CAGUUCCACUGAUGAGX CGAA AAGAGCCU691
1209 GGCUCUUUUGGAACUGU191 ACAGUUCCCUGAUGAGX CGAA AAAGAGCC692
1218 GGAACUGUUUAUAAGGG192 CCCUUAUACUGAUGAGX CGAA ACAGUUCC693
1219 GAACUGUUUAUAAGGGU193 ACCCUUAUCUGAUGAGX CGAA AACAGUUC694
1220 AACUGUUUAUAAGGGUA199 UACCCUUACUGAUGAGX CGAA AAACAGUU695
1222 CUGUUUAUAAGGGUAAA195 UUUACCCUCUGAUGAGX CGAA AUAAACAG696
1228 AUAAGGGUA 196 GUGCCAUUCUGAUGAGX CGAA ACCCUUAU697
AAUGGCAC
1295 GGAGAUGUUGCAGUAAA197 UUUACUGCCUGAUGAGX CGAA ACAUCUCC698
1251 GUUGCAGUA 198 AGGAUCUUCUGAUGAGX CGAA ACUGCAAC699
AAGAUCCU
1257 GUAAAGAUCCUAAAGGU199 ACCUUUAGCUGAUGAGX CGAA AUCUUUAC700
1260 AAGAUCCUA 200 ACAACCUUCUGAUGAGX CGAA AGGAUCUU701
AAGGUUGU
1266 CUAAAGGUUGUCGACCC201 GGGUCGACCUGAUGAGX CGAA ACCUUUAG702
1269 AAGGUUGUCGACCCAAC202 GUUGGGUCCUGAUGAGX CGAA ACAACCUU703
1289 AGAGCAAUUCCAGGCCU203 AGGCCUGGCUGAUGAGX CGAA AUUGCUCU709
1290 GAGCAAUUCCAGGCCUU209 AAGGCCUGCUGAUGAGX CGAA AAUUGCUC705
1298 CCAGGCCUUCAGGAAUG205 CAUUCCUGCUGAUGAGX CGAA AGGCCUGG706
1299 CAGGCCUUCAGGAAUGA206 UCAUUCCUCUGAUGAGX CGAA AAGGCCUG707
1317 GUGGCUGUUCUGCGCAA207 UUGCGCAGCUGAUGAGX CGAA ACAGCCAC708
1318 UGGCUGUUCUGCGCAAA208 UUUGCGCACUGAUGAGX CGAA AACAGCCA709
1349 GUGAACAUUCUGCUUUU209 AAAAGCAGCUGAUGAGX CGAA AUGUUCAC710
1395 UGAACAUUCUGCUUUUC210 GAAAAGCACUGAUGAGX CGAA AAUGUUCA711
1350 AUUCUGCUUUUCAUGGG211 CCCAUGAA X CGAA AGCAGAAU712
CUGAUGAG
1351 UUCUGCUUUUCAUGGGG212 CCCCAUGACUGAUGAGX CGAA AAGCAGAA713
1352 UCUGCUUUUCAUGGGGU213 ACCCCAUGCUGAUGAGX CGAA AAAGCAGA714
1353 CUGCUUUUCAUGGGGUA219 UACCCCAUCUGAUGAGX CGAA AAAAGCAG715
1361 CAUGGGGUACAUGACAA215 UUGUCAUGCUGAUGAGX CGAA ACCCCAUG716
1386 CUGGCAAUUGUGACCCA216 UGGGUCACCUGAUGAGX CGAA AUUGCCAG717
1416 AGCAGCCUCUACAAACA217 UGUUUGUACUGAUGAGX CGAA AGGCUGCU718
1918 CAGCCUCUACAAACACC218 GGUGUUUGCUGAUGAGX CGAA AGAGGCUG719
1434 CUGCAUGUCCAGGAGAC219 GUCUCCUGCUGAUGAGX CGAA ACAUGCAG720
1948 GACCAAGUUUCAGAUGU220 ACAUCUGA X CGAA ACUUGGUC721
CUGAUGAG
1449 ACCAAGUUUCAGAUGUU221 AACAUCUGCUGAUGAGX CGAA AACUUGGU722
1450 CCAAGUUUCAGAUGUUC222 GAACAUCUCUGAUGAGX CGAA AAACUUGG723
1457 UCAGAUGUUCCAGCUAA223 UUAGCUGGCUGAUGAGX CGAA ACAUCUGA724
1958 CAGAUGUUCCAGCUAAU229 AUUAGCUGCUGAUGAGX CGAA AACAUCUG725
1969 UUCCAGCUAAUUGACAU225 AUGUCAAUCUGAUGAGX CGAA AGCUGGAA726
1467 CAGCUAAUU 226 GCAAUGUCCUGAUGAGX CGAA AUUAGCUG727
GACAUUGC
1473 AUUGACAUUGCCCGGCA227 UGCCGGGCCUGAUGAGX CGAA AUGUCAAU728
1489 AGACGGCUCAGGGAAUG228 CAUUCCCUCUGAUGAGX CGAA AGCCGUCU729
1502 AAUGGACUAUUUGCAUG229 CAUGCAAA X CGAA AGUCCAUU730
CUGAUGAG
1504 UGGACUAUUUGCAUGCA230 UGCAUGCACUGAUGAGX CGAA AUAGUCCA731
1505 GGACUAUUUGCAUGCAA231 UUGCAUGCCUGAUGAGX CGAA AAUAGUCC732
1521 AAGAACAUCAUCCAUAG232 CUAUGGAUCUGAUGAGX CGAA AUGUUCUU733
1529 AACAUCAUCCAUAGAGA233 UCUCUAUGCUGAUGAGX CGAA AUGAUGUU734
1528 UCAUCCAUAGAGACAUG234 CAUGUCUCCUGAUGAGX CGAA AUGGAUGA735
CA 02288640 1999-11-OS
WO 98/50530 150 PCT/US98/09249
nt Target SEQ Riboryme SEQ
positionSite ID. Sequence ID.
No. No.
1541 CAUGAAAUCCAACAAUA235 UAUUGUUGCUGAUGAGX~CGAA AUUUCAUG736
1599 CCAACAAUAUAUUUCUC236 GAGAAAUACUGAUGAGX CGAA AUUGUUGG737
1551 AACAAUAUAUUUCUCCA237 UGGAGAAA X CGAA AUAUUGUU738
CUGAUGAG
1553 CAAUAUAUUUCUCCAUG238 CAUGGAGACUGAUGAGX CGAA AUAUAUUG739
1554 AAUAUAUUUCUCCAUGA239 UCAUGGAGCUGAUGAGX CGAA AAUAUAUU740
1555 AUAUAUUUCUCCAUGAA240 UUCAUGGACUGAUGAGX CGAA AAAUAUAU741
1557 AUAUUUCUCCAUGAAGG241 CCUUCAUGCUGAUGAGX CGAA AGAAAUAU742
1568 UGAAGGCUU 242 UCACUGUUCUGAUGAGX CGAA AGCCUUCA793
AACAGUGA
1569 GAAGGCUUAACAGUGAA293 UUCACUGUCUGAUGAGX CGAA AAGCCUUC744
1581 GUGAAAAUUGGAGAUUU249 AAAUCUCCCUGAUGAGX CGAA AUUUUCAC795
1588 UUGGAGAUUUUGGUUUG295 CAAACCAA X CGAA AUCUCCAA746
CUGAUGAG
1589 UGGAGAUUUUGGUUUGG296 CCAAACCACUGAUGAGX CGAA AAUCUCCA747
1590 GGAGAUUUUGGUUUGGC247 GCCAAACCCUGAUGAGX CGAA AAAUCUCC748
1599 AUUUUGGUUUGGCAACA298 UGUUGCCACUGAUGAGX CGAA ACCAAAAU799
1595 UUUUGGUUUGGCAACAG249 CUGUUGCCCUGAUGAGX CGAA AACCAAAA750
1605 GCAACAGUA 250 CGUGACUUCUGAUGAGX CGAA ACUGUUGC751
AAGUCACG
1610 AGUAAAGUCACGCUGGA251 UCCAGCGUCUGAUGAGX CGAA ACUUUACU752
1624 GGAGUGGUUCUCAGCAG252 CUGCUGAGCUGAUGAGX CGAA ACCACUCC753
1625 GAGUGGUUCUCAGCAGG253 CCUGCUGACUGAUGAGX CGAA AACCACUC754
1627 GUGGUUCUCAGCAGGUU259 AACCUGCUCUGAUGAGX CGAA AGAACCAC755
1635 CAGCAGGUUGAACAACC255 GGUUGUUCCUGAUGAGX CGAA ACCUGCUG756
1645 AACAACCUACUGGCUCU256 AGAGCCAGCUGAUGAGX CGAA AGGUUGUU757
1652 UACUGGCUCUGUCCUCU257 AGAGGACACUGAUGAGX CGAA AGCCAGUA758
1656 GGCUCUGUCCUCUGGAU258 AUCCAGAGCUGAUGAGX CGAA ACAGAGCC759
1659 UCUGUCCUCUGGAUGGC259 GCCAUCCACUGAUGAGX CGAA AGGACAGA760
1680 GAGGUGAUCCGAAUGCA260 UGCAUUCGCUGAUGAGX CGAA AUCACCUC761
1693 UGCAGGAUAACAACCCA261 UGGGUUGUCUGAUGAGX CGAA AUCCUGCA762
1703 CAACCCAUUCAGUUUCC262 GGAAACUGCUGAUGAGX CGAA AUGGGUUG763
1704 AACCCAUUCAGUUUCCA263 UGGAAACUCUGAUGAGX CGAA AAUGGGUU764
1708 CAUUCAGUUUCCAGUCG264 CGACUGGACUGAUGAGX CGAA ACUGAAUG765
1709 AUUCAGUUUCCAGUCGG265 CCGACUGGCUGAUGAGX CGAA AACUGAAU766
1710 UUCAGUUUCCAGUCGGA266 UCCGACUGCUGAUGAGX CGAA AAACUGAA76?
1715 UUUCCAGUCGGAUGUCU267 AGACAUCCCUGAUGAGX CGAA ACUGGAAA768
1722 UCGGAUGUCUACUCCUA268 UAGGAGUACUGAUGAGX CGAA ACAUCCGA769
1724 GGAUGUCUACUCCUAUG269 CAUAGGAGCUGAUGAGX CGAA AGACAUCC770
1727 UGUCUACUCCUAUGGCA270 UGCCAUAGCUGAUGAGX CGAA AGUAGACA771
1730 CUACUCCUAUGGCAUCG271 CGAUGCCACUGAUGAGX CGAA AGGAGUAG772
1737 UAUGGCAUCGUAUUGUA272 UACAAUACCUGAUGAGX CGAA AUGCCAUA773
1740 GGCAUCGUAUUGUAUGA273 UCAUACAA X CGAA ACGAUGCC779
CUGAUGAG
1742 CAUCGUAUUGUAUGAAC279 GUUCAUACCUGAUGAGX CGAA AUACGAUG775
1795 CGUAUUGUAUGAACUGA275 UCAGUUCACUGAUGAGX CGAA ACAAUACG776
1767 GGGGAGCUUCCUUAUUC276 GAAUAAGGCUGAUGAGX CGAA AGCUCCCC777
1768 GGGAGCUUCCUUAUUCU277 AGAAUAAGCUGAUGAGX CGAA AAGCUCCC?78
1771 AGCUUCCUUAUUCUCAC278 GUGAGAAUCUGAUGAGX CGAA AGGAAGCU779
1772 GCUUCCUUAUUCUCACA279 UGUGAGAA X CGAA AAGGAAGC780
CUGAUGAG
1779 UUCCUUAUUCUCACAUC280 GAUGUGAGCUGAUGAGX CGAA AUAAGGAA781
1775 UCCUUAUUCUCACAUCA281 UGAUGUGACUGAUGAGX CGAA AAUAAGGA782
1777 CUUAUUCUCACAUCAAC282 GUUGAUGUCUGAUGAGX CGAA AGAAUAAG783
1782 UCUCACAUC 283 CGGUUGUUCUGAUGAGX CGAA AUGUGAGA784
AACAACCG
1795 ACCGAGAUCAGAUCAUC284 GAUGAUCUCUGAUGAGX CGAA AUCUCGGU785
1800 GAUCAGAUCAUCUUCAU285 AUGAAGAUCUGAUGAGX CGAA AUCUGAUC786
1803 CAGAUCAUCUUCAUGGU286 ACCAUGAA X CGAA AUGAUCUG787
CUGAUGAG
1805 GAUCAUCUUCAUGGUGG287 CCACCAUGCUGAUGAGX CGAA AGAUGAUC788
1806 AUCAUCUUCAUGGUGGG2B8 CCCACCAUCUGAUGAGX CGAA AAGAUGAU789
1823 CCGAGGAUAUGCCUCCC2B9 GGGAGGCACUGAUGAGX CGAA AUCCUCGG790
1829 AUAUGCCUCCCCAGAUC290 GAUCUGGGCUGAUGAGX CGAA AGGCAUAU791
1837 CCCCAGAUCUUAGUAAG291 CUUACUAA X CGAA AUCUGGGG792
CUGAUGAG
1839 CCAGAUCUUAGUAAGCU292 AGCUUACUCUGAUGAGX CGAA AGAUCUGG793
1890 CAGAUCUUAGUAAGCUA293 UAGCUUACCUGAUGAGX CGAA AAGAUCUG794
CA 02288640 1999-11-05
WO 98/50530 I51 PCT/US98/09249
nt Target SEQ Riboryme SEQ
positionSite ID. Sequence ID.
No. No.
1893 AUCUUAGUAAGCUAUAU294 AUAUAGCUCUGAUGAGXCGAA ACUAAGAU795
1848 AGUAAGCUAUAUAAGAA295 UUCUUAUACUGAUGAGXCGAA AGCUUACU796
1850 UAAGCUAUAUAAGAACU296 AGUUCUUACUGAUGAGXCGAA AUAGCUUA797
1852 AGCUAUAUAAGAACUGC297 GCAGUUCUCUGAUGAGXCGAA AUAUAGCU798
1889 AGGCUGGUAGCUGACUG298 CAGUCAGCCUGAUGAGXCGAA ACCAGCCU799
1905 AAGAAAGUA 299 UCUUCCUUCUGAUGAGXCGAA ACUUUCUU800
AAGGAAGA
1921 AGAGGCCUCUUUUUCCC300 GGGAAAAA XCGAA AGGCCUCU801
CUGAUGAG
1923 AGGCCUCUUUUUCCCCA301 UGGGGAAA XCGAA AGAGGCCU802
CUGAUGAG
1929 GGCCUCUUUUUCCCCAG302 CUGGGGAA XCGAA AAGAGGCC803
CUGAUGAG
1925 GCCUCUUUUUCCCCAGA303 UCUGGGGACUGAUGAGXCGAA AAAGAGGC809
1926 CCUCUUUUUCCCCAGAU304 AUCUGGGGCUGAUGAGXCGAA AAAAGAGG805
1927 CUCUUUUUCCCCAGAUC305 GAUCUGGGCUGAUGAGXCGAA AAAAAGAG806
1935 CCCCAGAUCCUGUCUUC306 GAAGACAGCUGAUGAGXCGAA AUCUGGGG807
1990 GAUCCUGUCUUCCAUUG307 CAAUGGAA XCGAA ACAGGAUC808
CUGAUGAG
1992 UCCUGUCUUCCAUUGAG308 CUCAAUGGCUGAUGAGXCGAA AGACAGGA809
1993 CCUGUCUUCCAUUGAGC309 GCUCAAUGCUGAUGAGXCGAA AAGACAGG810
1947 UCUUCCAUUGAGCUGCU310 AGCAGCUCCUGAUGAGXCGAA AUGGAAGA811
1956 GAGCUGCUCCAACACUC311 GAGUGUUGCUGAUGAGXCGAA AGCAGCUC812
1969 CCAACACUCUCUACCGA312 UCGGUAGACUGAUGAGXCGAA AGUGUUGG813
1966 AACACUCUCUACCGAAG313 CUUCGGUACUGAUGAGXCGAA AGAGUGUU819
1968 CACUCUCUACCGAAGAU319 AUCUUCGGCUGAUGAGXCGAA AGAGAGUG815
1977 CCGAAGAUC 315 CUCCGGUUCUGAUGAGXCGAA AUCUUCGG816
AACCGGAG
1990 GGAGCGCUUCCGAGCCA316 UGGCUCGGCUGAUGAGXCGAA AGCGCUCC817
1991 GAGCGCUUCCGAGCCAU317 AUGGCUCGCUGAUGAGXCGAA AAGCGCUC818
2000 CGAGCCAUCCUUGCAUC318 GAUGCAAGCUGAUGAGXCGAA AUGGCUCG819
2003 GCCAUCCUUGCAUCGGG319 CCCGAUGCCUGAUGAGXCGAA AGGAUGGC820
2008 CCUUGCAUCGGGCAGCC320 GGCUGCCCCUGAUGAGXCGAA AUGCAAGG821
2029 CUGAGGAUAUCAAUGCU321 AGCAUUGACUGAUGAGXCGAA AUCCUCAG822
2031 GAGGAUAUC 322 CAAGCAUUCUGAUGAGXCGAA AUAUCCUC823
AAUGCUUG
2038 UCAAUGCUUGCACGCUG323 CAGCGUGCCUGAUGAGXCGAA AGCAUUGA824
2054 GACCACGUCCCCGAGGC329 GCCUCGGGCUGAUGAGXCGAA ACGUGGUC825
2070 CUGCCUGUCUUCUAGUU325 AACUAGAA XCGAA ACAGGCAG826
CUGAUGAG
2072 GCCUGUCUUCUAGUUGA326 UCAACUAGCUGAUGAGXCGAA AGACAGGC827
2073 CCUGUCUUCUAGUUGAC327 GUCAACUACUGAUGAGXCGAA AAGACAGG828
2075 UGUCUUCUAGUUGACUU328 AAGUCAACCUGAUGAGXCGAA AGAAGACA829
2078 CUUCUAGUUGACUUUGC329 GCAAAGUCCUGAUGAGXCGAA ACUAGAAG830
2083 AGUUGACUUUGCACCUG330 CAGGUGCACUGAUGAGXCGAA AGUCAACU831
2089 GUUGACUUUGCACCUGU331 ACAGGUGCCUGAUGAGXCGAA AAGUCAAC832
2093 GCACCUGUCUUCAGGCU332 AGCCUGAA XCGAA ACAGGUGC833
CUGAUGAG
2095 ACCUGUCUUCAGGCUGC333 GCAGCCUGCUGAUGAGXCGAA AGACAGGU834
2096 CCUGUCUUCAGGCUGCC339 GGCAGCCUCUGAUGAGXCGAA AAGACAGG835
2136 GCACCACUUUUCUGCUC335 GAGCAGAA XCGAA AGUGGUGC836
CUGAUGAG
2137 CACCACUUUUCUGCUCC336 GGAGCAGACUGAUGAGXCGAA AAGUGGUG837
2138 ACCACUUUUCUGCUCCC337 GGGAGCAGCUGAUGAGXCGAA AAAGUGGU838
2139 CCACUUUUCUGCUCCCU338 AGGGAGCACUGAUGAGXCGAA AAAAGUGG839
2149 UUUCUGCUCCCUUUCUC339 GAGAAAGGCUGAUGAGXCGAA AGCAGAAA840
2148 UGCUCCCUUUCUCCAGA340 UCUGGAGACUGAUGAGXCGAA AGGGAGCA891
2149 GCUCCCUUUCUCCAGAG341 CUCUGGAGCUGAUGAGXCGAA AAGGGAGC842
2150 CUCCCUUUCUCCAGAGG342 CCUCUGGACUGAUGAGXCGAA AAAGGGAG843
2152 CCCUUUCUCCAGAGGCA393 UGCCUCUGCUGAUGAGXCGAA AGAAAGGG894
2171 ACACAUGUUUUCAGAGA349 UCUCUGAA XCGAA ACAUGUGU895
CUGAUGAG
2172 CACAUGUUUUCAGAGAA345 UUCUCUGACUGAUGAGXCGAA AACAUGUG896
2173 ACAUGUUUUCAGAGAAG346 CUUCUCUGCUGAUGAGXCGAA AAACAUGU897
2174 CAUGUUUUCAGAGAAGC347 GCUUCUCUCUGAUGAGXCGAA AAAACAUG898
2184 GAGAAGCUCUGCUAAGG348 CCUUAGCACUGAUGAGXCGAA AGCUUCUC899
2189 GCUCUGCUAAGGACCUU399 AAGGUCCUCUGAUGAGXCGAA AGCAGAGC850
2197 AAGGACCUUCUAGACUG350 CAGUCUAGCUGAUGAGXCGAA AGGUCCUU851
2198 AGGACCUUCUAGACUGC351 GCAGUCUACUGAUGAGXCGAA AAGGUCCU852
2200 ~ GACCUUCUAGACUGCUC~ ~ GAGCAGUCCUGAUGAGXCGAA AGAAGGUC_
352 j
853
CA 02288640 1999-11-05
WO 98/50530 152 PCT/LJS98/09249
nt Target SEQ Riboryme ~~
positionSite ID. Sequence SEQ
No. ID.
No.
2208 AGACUGCUCACAGGGCC353 GGCCCUGUCUGAUGAGX CGAA AGCAGUCU859
2218 CAGGGCCUU 359 AUGAAGUUCUGAUGAGX CGAA AGGCCCUG855
AACUUCAU
2219 AGGGCCUUAACUUCAUG355 CAUGAAGUCUGAUGAGX CGAA AAGGCCCU856
2223 CCUUAACUUCAUGUUGC356 GCAACAUGCUGAUGAGX CGAA AGUUAAGG857
2229 CUUAACUUCAUGUUGCC357 GGCAACAUCUGAUGAGX CGAA AAGUUAAG858
2229 CUUCAUGUUGCCUUCUU358 AAGAAGGCCUGAUGAGX CGAA ACAUGAAG859
2234 UGUUGCCUUCUUUUCUA359 UAGAAAAGCUGAUGAGX CGAA AGGCAACA860
2235 GUUGCCUUCUUUUCUAU360 AUAGAAAA X CGAA AAGGCAAC861
CUGAUGAG
2237 UGCCUUCUUUUCUAUCC361 GGAUAGAA X CGAA AGAAGGCA862
CUGAUGAG
2238 GCCUUCUUUUCUAUCCC362 GGGAUAGACUGAUGAGX CGAA AAGAAGGC863
2239 CCUUCUUUUCUAUCCCU363 AGGGAUAGCUGAUGAGX CGAA.AAAGAAGG864
2290 CUUCUUUUCUAUCCCUU369 AAGGGAUACUGAUGAGX CGAA AAAAGAAG865
2242 UCUUUUCUAUCCCUUUG365 CAAAGGGACUGAUGAGX CGAA AGAAAAGA866
2249 UUUUCUAUCCCUUUGGG366 CCCAAAGGCUGAUGAGX CGAA AUAGAAAA867
2248 CUAUCCCUUUGGGCCCU367 AGGGCCCACUGAUGAGX CGAA AGGGAUAG868
2249 UAUCCCUUUGGGCCCUG368 CAGGGCCCCUGAUGAGX CGAA AAGGGAUA869
2273 GAAGCCAUUUGCAGUGC369 GCACUGCACUGAUGAGX CGAA AUGGCUUC870
2279 AAGCCAUUUGCAGUGCU370 AGCACUGCCUGAUGAGX CGAA AAUGGCUU871
2290 UGGUGUGUCCUGCUCCC371 GGGAGCAGCUGAUGAGX CGAA ACACACCA872
2296 GUCCUGCUCCCUCCCCA372 UGGGGAGGCUGAUGAGX CGAA AGCAGGAC873
2300 UGCUCCCUCCCCACAUU373 AAUGUGGGCUGAUGAGX CGAA AGGGAGCA879
2308 CCCCACAUUCCCCAUGC374 GCAUGGGGCUGAUGAGX CGAA AUGUGGGG875
2309 CCCACAUUCCCCAUGCU375 AGCAUGGGCUGAUGAGX CGAA AAUGUGGG876
2318 CCCAUGCUC 376 UGGGCCUUCUGAUGAGX CGAA AGCAUGGG877
AAGGCCCA
2331 CCCAGCCUUCUGUAGAU377 AUCUACAGCUGAUGAGX CGAA AGGCUGGG878
2332 CCAGCCUUCUGUAGAUG378 CAUCUACACUGAUGAGX CGAA AAGGCUGG879
2336 CCUUCUGUAGAUGCGCA379 UGCGCAUCCUGAUGAGX CGAA ACAGAAGG880
2359 GUGGAUGUUGAUGGUAG380 CUACCAUCCUGAUGAGX CGAA ACAUCCAC881
2361 UUGAUGGUAGUACAAAA381 UUUUGUACCUGAUGAGX CGAA ACCAUCAA882
2364 AUGGUAGUACAAAAAGC382 GCUUUUUGCUGAUGAGX CGAA ACUACCAU883
2393 CCAGCUGUUGGCUACAU383 AUGUAGCCCUGAUGAGX CGAA ACAGCUGG884
2398 UGUUGGCUA 384 UACUCAUGCUGAUGAGX CGAA AGCCAACA885
CAUGAGUA
2906 ACAUGAGUAUUUAGAGG385 CCUCUAAA X CGAA ACUCAUGU886
CUGAUGAG
2908 AUGAGUAUUUAGAGGAA386 UUCCUCUACUGAUGAGX CGAA AUACUCAU887
2909 UGAGUAUUUAGAGGAAG387 CUUCCUCUCUGAUGAGX CGAA AAUACUCA888
2910 GAGUAUUUAGAGGAAGU388 ACUUCCUCCUGAUGAGX CGAA AAAUACUC889
2919 GAGGAAGUAAGGUAGCA389 UGCUACCUCUGAUGAGX CGAA ACUUCCUC890
2924 AGUAAGGUAGCAGGCAG390 CUGCCUGCCUGAUGAGX CGAA ACCUUACU891
2939 CAGGCAGUCCAGCCCUG391 CAGGGCUGCUGAUGAGX CGAA ACUGCCUG892
2462 CAUGGGAUUUUGGAAAU392 AUUUCCAA X CGAA AUCCCAUG893
CUGAUGAG
2463 AUGGGAUUUUGGAAAUC393 GAUUUCCACUGAUGAGX CGAA AAUCCCAU894
2469 UGGGAUUUUGGAAAUCA399 UGAUUUCCCUGAUGAGX CGAA AAAUCCCA895
2471 UUGGAAAUCAGCUUCUG395 CAGAAGCUCUGAUGAGX CGAA AUUUCCAA896
2476 AAUCAGCUUCUGGAGGA396 UCCUCCAGCUGAUGAGX CGAA AGCUGAUU897
2477 AUCAGCUUCUGGAGGAA397 UUCCUCCACUGAUGAGX CGAA AAGCUGAU898
2493 AUGCAUGUCACAGGCGG398 CCGCCUGUCUGAUGAGX CGAA ACAUGCAU899
2506 GCGGGACUUUCUUCAGA399 UCUGAAGACUGAUGAGX CGAA AGUCCCGC900
2507 CGGGACUUUCUUCAGAG400 CUCUGAAGCUGAUGAGX CGAA AAGUCCCG901
2508 GGGACUUUCUUCAGAGA401 UCUCUGAA X CGAA AAAGUCCC902
CUGAUGAG
2510 GACUUUCUUCAGAGAGU402 ACUCUCUGCUGAUGAGX CGAA AGAAAGUC903
2511 ACUUUCUUCAGAGAGUG403 CACUCUCUCUGAUGAGX CGAA AAGAAAGU904
2536 CCAGACAUUUUGCACAU409 AUGUGCAA X CGAA AUGUCUGG905
CUGAUGAG
2537 CAGACAUUUUGCACAUA905 UAUGUGCACUGAUGAGX CGAA AAUGUCUG906
2538 AGACAUUUUGCACAUAA406 UUAUGUGCCUGAUGAGX CGAA AAAUGUCU907
2595 UUGCACAUAAGGCACCA407 UGGUGCCUCUGAUGAGX CGAA AUGUGCAA908
2577 CCGAGACUCUGGCCGCC408 GGCGGCCACUGAUGAGX CGAA AGUCUCGG909
2600 AGCCUGCUUUGGUACUA409 UAGUACCACUGAUGAGX CGAA AGCAGGCU910
2601 GCCUGCUUUGGUACUAU410 AUAGUACCCUGAUGAGX CGAA AAGCAGGC911
2605 GCUUUGGUACUAUGGAA411 UUCCAUAGCUGAUGAGX CGAA ACCAAAGC912
CA 02288640 1999-11-OS
W0 98/50530 153 PCT/US98/09249
nt Target SEQ Ribozyme SEQ
positionSite ID. Sequeace ID.
No. No.
2608 UUGGUACUAUGGAACUU912 AAGUUCCACUGAUGAGX CGAA AGUACCAA913
2616 AUGGAACUUUUCUUAGG913 CCUAAGAA X CGAA AGUUCCAU914
CUGAUGAG
2617 UGGAACUUUUCUUAGGG414 CCCUAAGACUGAUGAGX CGAA AAGUUCCA915
2618 GGAACUUUUCUUAGGGG415 CCCCUAAGCUGAUGAGX CGAA AAAGUUCC916
2619 GAACUUUUCUUAGGGGA416 UCCCCUAA X CGAA AAAAGUUC917
CUGAUGAG
2621 ACUUUUCUUAGGGGACA417 UGUCCCCUCUGAUGAGX CGAA AGAAAAGU918
2622 CUUUUCUUAGGGGACAC918 GUGUCCCCCUGAUGAGX CGAA AAGAAAAG919
2633 GGACACGUCCUCCUUUC419 GAAAGGAGCUGAUGAGX CGAA ACGUGUCC920
2636 CACGUCCUCCUUUCACA920 UGUGAAAGCUGAUGAGX CGAA AGGACGUG921
2639 GUCCUCCUUUCACAGCU421 AGCUGUGACUGAUGAGX CGAA AGGAGGAC922
2640 UCCUCCUUUCACAGCUU422 AAGCUGUGCUGAUGAGX CGAA AAGGAGGA923
2691 CCUCCUUUCACAGCUUC423 GAAGCUGUCUGAUGAGX CGAA AAAGGAGG929
2698 UCACAGCUUCUAAGGUG929 CACCUUAGCUGAUGAGX CGAA AGCUGUGA925
2699 CACAGCUUCUAAGGUGU925 ACACCUUACUGAUGAGX CGAA AAGCUGUG926
2651 CAGCUUCUAAGGUGUCC926 GGACACCUCUGAUGAGX CGAA AGAAGCUG927
2658 UAAGGUGUCCAGUGCAU927 AUGCACUGCUGAUGAGX CGAA ACACCUUA928
2667 CAGUGCAUUGGGAUGGU428 ACCAUCCCCUGAUGAGX CGAA AUGCACUG929
2676 GGGAUGGUUUUCCAGGC429 GCCUGGAA X CGAA ACCAUCCC930
CUGAUGAG
2677 GGAUGGUUUUCCAGGCA430 UGCCUGGACUGAUGAGX CGAA AACCAUCC931
2678 GAUGGUUUUCCAGGCAA431 UUGCCUGGCUGAUGAGX CGAA AAACCAUC932
2679 AUGGUUUUCCAGGCAAG432 CUUGCCUGCUGAUGAGX CGAA AAAACCAU933
2693 AAGGCACUCGGCCAAUC433 GAUUGGCCCUGAUGAGX CGAA AGUGCCUU934
2701 CGGCCAAUCCGCAUCUC934 GAGAUGCGCUGAUGAGX CGAA AUUGGCCG935
2707 AUCCGCAUCUCAGCCCU435 AGGGCUGACUGAUGAGX CGAA AUGCGGAU936
2709 CCGCAUCUCAGCCCUCU936 AGAGGGCUCUGAUGAGX CGAA AGAUGCGG937
2716 UCAGCCCUCUCAGGAGC937 GCUCCUGACUGAUGAGX CGAA AGGGCUGA938
2718 AGCCCUCUCAGGAGCAG438 CUGCUCCUCUGAUGAGX CGAA AGAGGGCU939
2728 GGAGCAGUCUUCCAUCA939 UGAUGGAA X CGAA ACUGCUCC990
CUGAUGAG
2730 AGCAGUCUUCCAUCAUG490 CAUGAUGGCUGAUGAGX CGAA AGACUGCU941
2731 GCAGUCUUCCAUCAUGC991 GCAUGAUGCUGAUGAGX CGAA AAGACUGC942
2735 UCUUCCAUCAUGCUGAA992 UUCAGCAUCUGAUGAGX CGAA AUGGAAGA993
2795 UGCUGAAUUUUGUCUUC493 GAAGACAA X CGAA AUUCAGCA994
CUGAUGAG
2796 GCUGAAUUUUGUCUUCC494 GGAAGACACUGAUGAGX CGAA AAUUCAGC995
2797 CUGAAUUUUGUCUUCCA495 UGGAAGACCUGAUGAGX CGAA AAAUUCAG996
2750 AAUUUUGUCUUCCAGGA496 UCCUGGAA X CGAA ACAAAAUU947
CUGAUGAG
2752 UUUUGUCUUCCAGGAGC947 GCUCCUGGCUGAUGAGX CGAA AGACAAAA948
2753 UUUGUCUUCCAGGAGCU498 AGCUCCUGCUGAUGAGX CGAA AAGACAAA949
2768 CUGCCCCUAUGGGGCGG499 CCGCCCCACUGAUGAGX CGAA AGGGGCAG950
2795 CAGCCUGUUUCUCUAAC950 GUUAGAGACUGAUGAGX CGAA ACAGGCUG951
2796 AGCCUGUUUCUCUAACA951 UGUUAGAGCUGAUGAGX CGAA AACAGGCU952
2797 GCCUGUUUCUCUAACAA952 UUGUUAGACUGAUGAGX CGAA AAACAGGC953
2799 CUGUUUCUCUAACAAAC453 GUUUGUUACUGAUGAGX CGAA AGAAACAG959
2801 GUUUCUCUAACAAACAA959 UUGUUUGUCUGAUGAGX CGAA AGAGAAAC955
2825 AACAGCCUUGUUUCUCU955 AGAGAAACCUGAUGAGX CGAA AGGCUGUU956
2828 AGCCUUGUUUCUCUAGU456 ACUAGAGACUGAUGAGX CGAA ACAAGGCU957
2829 GCCUUGUUUCUCUAGUC457 GACUAGAGCUGAUGAGX CGAA AACAAGGC958
2830 CCUUGUUUCUCUAGUCA458 UGACUAGACUGAUGAGX CGAA AAACAAGG959
2832 UUGUUUCUCUAGUCACA459 UGUGACUACUGAUGAGX CGAA AGAAACAA960
2834 GUUUCUCUAGUCACAUC460 GAUGUGACCUGAUGAGX CGAA AGAGAAAC961
2837 UCUCUAGUCACAUCAUG961 CAUGAUGUCUGAUGAGX CGAA ACUAGAGA962
2842 AGUCACAUCAUGUGUAU462 AUACACAUCUGAUGAGX CGAA AUGUGACU963
2849 UCAUGUGUAUACAAGGA463 UCCUUGUACUGAUGAGX CGAA ACACAUGA964
2851 AUGUGUAUACAAGGAAG969 CUUCCUUGCUGAUGAGX CGAA AUACACAU965
2868 CCAGGAAUACAGGUUUU965 AAAACCUGCUGAUGAGX CGAA AUUCCUGG966
2879 AUACAGGUUUUCUUGAU966 AUCAAGAA X CGAA ACCUGUAU967
CUGAUGAG
2875 UACAGGUUUUCUUGAUG967 CAUCAAGACUGAUGAGX CGAA AACCUGUA968
2876 ACAGGUUUUCUUGAUGA968 UCAUCAAGCUGAUGAGX CGAA AAACCUGU969
2877 CAGGUUUUCUUGAUGAU469 AUCAUCAA X CGAA AAAACCUG970
CUGAUGAG
2879 GGUUUUCUUGAUGAUUU970 AAAUCAUCCUGAUGAGX CGAA AGAAAACC971
CA 02288640 1999-11-OS
WO 98/50530 154 PCT/US98/09249
nt Target SEQ ~ ~ Ribozyme SEQ
positionSite ID. Sequence _ ID.
No. No.
2886 UUGAUGAUU UGGGUUUU971 AAAACCCA CUGAUGAG X CGAA 972
AUCAUCAA
2887 UGAUGAUUU GGGUUUUA972 UAAAACCC CUGAUGAG X CGAA 973
AAUCAUCA
2892 AUUUGGGUU UUAAUUUU473 AAAAUUAA CUGAUGAG X CGAA 974
ACCCAAAU
2893 UUUGGGUUU UAAUUUUG479 CAAAAUUA CUGAUGAG X CGAA 975
AACCCAAA
2899 UUGGGUUUU AAUUUUGU975 ACAAAAUU CUGAUGAG X CGAA 976
AAACCCAA
2895 UGGGUUUUA AUUUUGUU976 AACAAAAU CUGAUGAG X CGAA 977
AAAACCCA
2898 GUUUUAAUU UUGUUUUU977 AAAAACAA CUGAUGAG X CGAA 978
AUUAAAAC
2899 UUUUAAUUU UGUUUUUA478 UAAAAACA CUGAUGAG X CGAA 979
AAUUAAAA
2900 UUUAAUUUU GUUUUUAU979 AUAAAAAC CUGAUGAG X CGAA 980
AAAUUAAA
2903 AAUUUUGUU UUUAUUGC480 GCAAUAAA CUGAUGAG X CGAA 981
ACAAAAUU
2909 AUUUUGUUU UUAUUGCA481 UGCAAUAA CUGAUGAG X CGAA 982
AACAAAAU
2905 UUUUGUUUU UAUUGCAC482 GUGCAAUA CUGAUGAG X CGAA 983
AAACAAAA
2906 UUUGUUUUU AUUGCACC483 GGUGCAAU CUGAUGAG X CGAA 984
AAAACAAA
2907 UUGUUUUUA UUGCACCU989 AGGUGCAA CUGAUGAG X CGAA 985
AAAAACAA
2909 GUUUUUAUU GCACCUGA485 UCAGGUGC CUGAUGAG X CGAA 986
AUAAAAAC
2929 GACAAAAUA CAGUUAUC986 GAUAACUG CUGAUGAG X CGAA 987
AUUUUGUC
2929 AAUACAGUU AUCUGAUG987 CAUCAGAU CUGAUGAG X CGAA 988
ACUGUAUU
2930 AUACAGUUA UCUGAUGG488 CCAUCAGA CUGAUGAG X CGAA 989
AACUGUAU
2932 ACAGUUAUC UGAUGGUC489 GACCAUCA CUGAUGAG X CGAA 990
AUAACUGU
2990 CUGAUGGUC CCUCAAUU990 AAUUGAGG CUGAUGAG X CGAA 991
ACCAUCAG
2949 UGGUCCCUC AAUUAUGU991 ACAUAAUU CUGAUGAG X CGAA 992
AGGGACCA
2948 CCCUCAAUU AUGUUAUU492 AAUAACAU CUGAUGAG X CGAA 993
AUUGAGGG
2999 CCUCAAUUA UGUUAUUU993 AAAUAACA CUGAUGAG X CGAA 994
AAUUGAGG
2953 AAUUAUGUU AUUUUAAU999 AUUAAAAU CUGAUGAG X CGAA 995
ACAUAAUU
2959 AUUAUGUUA UUUUAAUA495 UAUUAAAA CUGAUGAG X CGAA 996
AACAUAAU
2956 UAUGUUAUU UUAAUAAA996 UUUAUUAA CUGAUGAG X CGAA 997
AUAACAUA
2957 AUGUUAUUU UAAUAAAA497 UUUUAUUA CUGAUGAG X CGAA 998
AAUAACAU
2958 UGUUAUUUU AAUAAAAU998 AUUUUAUU CUGAUGAG X CGAA 999
AAAUAACA
2959 GUUAUUUUA AUAAAAUA499 UAUUUUAU CUGAUGAG X CGAA 1000
AAAAUAAC
2962 AUUUUAAUA AAAUAAAU500 AUUUAUUU CUGAUGAG X CGAA 1001
AUUAAAAU
2967 AAUAAAAUA AAUUAAAU501 AUUUAAUU CUGAUGAG X CGAA 1002
AUUUUAUU
CA 02288640 1999-11-05
WO 98/50530 PCT/US98/09249
155
A W01OriNM v~l7lOrOD01Or-INM v~tn0rDOO1O -iN MC N~Or OD01OriNM v~
C rh 00O ODODtODOD00ODODO141O101~l O~O~0101. OO OO OOO OO riri.1rlrt
00 O1 O
az oo oo o0 000 00 000 00 00 000 0.~.~.~ ~,~,~,~~.~.~,~,~ ,~.~ ,~
w
U~ C9~ FCU aFCU aaCUC7C7C9r.~C7a ~CaV U'~ UC7U'U U'Ua ECFC~C~CU'a a
U'U C7 U'o Uat7VU'UUC9U'U C7rCChUU FCV C9Ua U'FCUU UC7U'U'U'
' ' a aU C~aC9~VU rCU aU'a~Ca
Ua UC7UU aUC.7aa aC9a aU ~U UVC~t9U ~a UU'aU'a aa UC9UU rC
UU C7U ~U'r.Ct9D ar.~Ua~ U'FCU5 a~ ~U U ~CU'FCaa Ua U'a~aU C7
' '
y UU UU C7U UC7r.~Ua aUU aU UU UUa UC~(~a Ua aUa aC7U'U Ua U'
aU C7C7 a V'aU'Va oU' aC7aU DaU
", Ua UU UU aaa aU Uaa UaCaU UUU Ua~Ur.~Ua aaa UD Ua Ua U
~
~
o UU UU UU Uar.Cr,CV GC~CU r.Ca aa UUU aa Ua D~CaaU Ur.~FCU a5 D
y E C7C7C9C9UC~C7C7C~UC7C~U'C7C7C7C~C~C9C7C7C7C9C~C9C7C7C7C7C9C9C7C!U'UC~V
d
~ UU Ua UC~~CFCFC~Ca ~Caa rta a~ca~c~ca~cr~~ a~ aaa ~r~r~c~a~c~c
UU UU UU UUU UU UUU UU UU UUU UU UU UU UUU UU UU UU U
' '
r.~U UC7C7V'UC7a FC~Ca~CU a~ DU C7aa UU r.CU Ua ~C7a Va C7r.~aa V
C 5 a~~ ' ' a Ua Ur aU U'D ~a U't9Ua a
C r
C" C~a r.~ UC7Ur.r UC7aa~ UU Ua U~U C7U'a. FC~ C~aFC.a UFCC7rCa
aU U C9 UU aa U'a~a aC7~CU FC U U
MV'~l0rOD01O.-1NM V~U710rOD01O rlNM C~~ ~Or OD01OriN Mv~~lOrODC1
O OO OO OO Or-1.-1r-trie-Ir1riririrlN NNN NN NN NN MMM MM MM MM M
E-~ az oo oo o0 000 00 000 00 00 000 00 00 00 000 00 00 00 0
a ~'
E
a~c~~ aa a~~ ~~c~~~ ~~ aa a~~ ~a ~~ ~x~ ~~~ a~ ~a ~a a
o aa aa aa aaa aa aaa aa aa aaa aa aa aa aaa aa aa aa a
~c~c~c~~c~c~~c~c~c~~~c~c~c~t~t~c~c~c~~c~~~ c~c~c~c~c~~c~c~~ c~c~c~
,fl
aa aa aa aaa aa aaa aa aa aaa aa aa aa aaa aa aa aa a
UU UU UU UUU UU UUU UU UU UUV UU UU UU UUU UU UU UU U
UU VU UU UUU UU UUU UU UU UUV UU UU UU UUU UU UU UU U
.r GCd'.~Cd.KCECa'KCd
a'dØ'~C~C~CEC0.'FC~'FCFCFCA:~C~C~C~C~CFC~C~C~1',FCd.a'~Ca'
p, aa aa aa oaa aa aaa aa aa aaa aa aa aa aaa aa aa aa a
aa aa aa aaa aa aaa aa aa aaa aa aa aa aaa aa aa aa a
~~c~ca ~c~ aaa a~~ ~~d ~c~ ~~c~~c~ ~a ~~c~c~ ~~~c~ca ~r~~~ ~c
U U UU U U U UU UU UU U
xi UU UU UU UUU UUU U U UU U aa a a
~."~ aa aa aa aaa aa aaa aa aa aaa aa aa aa aaa aa c~~ c~a ~
c~c~~c~c~~ ~c~c~~c~c~c~c7~c~~c~~c~c~c~c~~c~c~c~c~~c~c~c~ c~
xx xx xx xxx xx xxx xx xx xxx xx xx xx xxx xx xx xx x
V UU UU UU UUU UU UUU UU UU UUU UU ~U UU UUU UU UU UU U
3 ~ C7U'C7C~C7C9UC7C7C9C9C9C9C9 C9C~C~C7U'C~ChC9C7UC~C9C7C9C9C~C7V'C~C9C~
~
x a~ ~~ ~~ ~~ ~~~ ~~ ~~~ ~~ ~~ ~~~ ~~ ~~ ~~ ~~~ ~~ ~~ ~~ ~
UU UU UU UUU UU UUU UU UU UUU UU UU UU UUU UU UU UU U
UU UU UU UUU UU UUU UU UU UUU UV VU UU UUU UU UU VU U
C7V'a U'U C7C~U'uC~ U~ rLU aU'aaU'UU ACU'aa Ur~a C~ U'a Ua
U~Caa r.CU ~aa U'U a~~ UU'Ur.~C7a U'~CC9a r.CC7aU~ a~ UU C9
aU'U'U UU C~Ur.~aa ~aU'r.Ca r.CC7Ur.~~ C9C7aC7Vr.~aU U Ua EC C7
UC~U'C9C7C9C7C7C9C9C9C9C~V'VC9C'JU'C7C7C7C7C7U'C9C7C9C7C9C~C9C9C9C7C7U C7
H
FCU'UV Ur.~Ua~U C~~ ~UC~~U cCC7r.CFCU'U'U Ur~U'r.Cr$C7 ~CU UC7C7ECU
C,~C7C7C7aU C~Ua U' U'a C9thU'UU'FCaU'aU'aU ar.C~ C7 Ua ~CC7U
~
U'C7VC7aU aUFC a V'r$a Ua t~~ aU UV'ar.CU'U ~CU FC V'U C9U'o
' ' ~ ' C' aU CV aU'C7aC7FCU ~Ca r
C
C7FCUU UCJFCUU FCFCU~C~U aC7 ~ FV aU F~ UU S~ ~D U .
~ ~ ~ a U
~S U~ Dt FC U'~a U'UU U~ U~ DFCU'U C ,7 U'~ ~C~
"J .'J C
G
o mMn~~oo rru o~navo~000N rN a,a,,-ao mwo ,~,~.-~~nMm N
N01.-1r1COD
w OMtnrr NrO N~ 00tnM01O OriaODOD01'-1~-1sr.1tn~OD~!7O v-1
ar
~ M~ ~O NNN NM V~C1fll0l0rOD0101O OO OO r-1'-1NMM VW C~C~~r r
T
O r-1r-Irle-W-ie-1ei'-Irl'-i'-W r-1rl'-i'-1e1
-i
CA 02288640 1999-11-OS
WO 98/50530 PCT/US98/09249
156
1 W O1~OD01O rlN Mv~~l0t~OD01O.-1N Mv~tnl0t~0~01OrlNM V'N tO~OD01O rlN
p -1-i~-i~ r1N NN NN NNN NN r'7MM MM MM MMM c~v~av~v'v'Iv'~a'c'N ~N
~ . v~
U C~aa C7C7Ua r.~a UaC7C7a U'UD C7a UU UC~C7r.~U aU'r.~C7r.~aC7Ur.~aU
r.~C7aC7 a ar.CUU a5rCC7a C7UC~UU C7rCUaFCC~FCUU'a~ Ur.~a aa aU
C aUC aU aU
a C~Ur.C~C7C7U ~FCC7Ua FCa r.~aa C~C7C7a Ur.CC~ChC7r~ C7a UUF a~ C7a'
U Uaa a aU U UaU VU UVU aFCaU C7C7C7aC7C~ C~ a
y a ~FC5 UC7Ua Ur.~r.~C7a aU r.~Ua r.CU aC7r.Caa Ur.CFCC7aU aaU C~U aC7
C~C7UU Ua Ua UU Uaa 5U UUa C7U C~C~UUC7aU C7U aa VaU aa aC7
a aaa UU aU 5U UUU Ua aaU rCU a~ UUa aU UU aa UUU Ua Ua
p r.~r.~~r.~Ur.~r.~a UU r.~Ua aU UVU aU Ua aUFCUU UU UU UDU Ua U~C
p O C7C7C7C7V C7V VC7C7C7V VC7C~C7V VV C~C7C~C~C7VC~C~V C9C7C7C9C9UV C7C~
E,
y
~ c~ar~~Caa Fca ar~aaa aa aar.Car.~~a r.Cr~o FCr~aU Or.~FCFCa ~ca ~Ca ,''
U UUU UU UU UU UUU UU UUU UU UU UUU UU UU UU UUU UU UU
'
a aU ~C7UU C~C7C7C7U Ua ~CUU aU UC7C9UU a r.CU Ur.~DC~C7UU a Q
~U ~ ~ ~ ~ U U U U CCa'~ ~ C7~ FCa aC~7~ C9~ a U
C C~ U CC7~C~C a ~CC~C C7U FJC
7 7 B
c~
N
A O ~-INM vwl7l0f~OD~ OriN MV~~l0f~0001Or-iNMvW17~D1~O~O~O .-iNM v~W DI'
p v~v~crC'V'c v~a~cvWf7~tnInN N~~ ~W Ot0l0tO\Ol0l0~O10v0t~t~I~!~I~c~f~f
az O OOO OO OO OO OOO OO OOO OO OO OOO OO OO OO OOO OO OO
~ ~~~ ~~ca~ ~a a~~ ~a ~~~ ~~c~a ~c~~ ~~ a~ ~a ~~~ a~ a~
a aaa aa aa aa aaa aa aaa aa aa aaa aa aa aa aaa aa aa
c~c~c~c~c~c~~c~~c~~c~c~~~ c~~c~~c~c~~ c~~t~c~c~~c~t~c~c~c~~ ~c~t~~ ~
c7c~c~c~~t~c~c~c~c~c~~~ c~~ t~~~ ~~ c~~ c~c~t~~c~c~c~c~c~t~c~c~c~c~t~~ o
a aaa aa aa aa aaa aa aaa aa aa aaa aa aa aa aaa aa aa
U UUU UU UU UV VUU UU UUU UU UU UUU UU UU UU UUU UU UU
U UUU UU UU UU VUU UU UUU UU UU UUU UU UU UU UUU UU UU
a ~aa ~a aa a~caa~ ~~ ~~~ ~~ a~ ~~~cr~~ ~~ ~~ ~~a aa aa do
a aaa aa aa aa aaa aa aaa aa aa aaa aa aa aa aaa aa aa
a aaa aa aa aa oaa aa aaa aa aa aaa aa aa aa aaa aa aa
~ aa~ ~~ ~~ ~~ a~~ ~~ ~~a ~a aa ~c~~ ~~ ~a ~ca ~~a aa ~~ -.
U UUU UU UV UU UUU UU UUU UU UU UUU UU UU UU UUU UV UU
a aaa aa aa aa aaa aa aaa aa aa aaa aa aa aa aaa aa aa
E--~
~ OC~c~~~ ~c~c~~ ~~~ c~c~~c~~ ~c~~~ c~~c~~~ c~c~c~~ ~c~~ ~c~~c~
x xxx xx xx xx xxx xx xxx xx xx xxx xx xx xx xxx xx xx
U UU UU UUU UU UUU UU UU UUU UU UU UU UUU UU UU
U UVU U
o C7C7C~C~U'C~U'V'U'U'C7C7C7(~CJC7C7C~C7U'UO
C~U'U'C7U'C9C~C7C.~(7C7C7C7C7C~U'
~ ~ ~~~c~a ~~ ~a ~~~ ~a ~~a ~a a~ a~~ ~~ ~~c~~ ~~~ aa ~~
c.
~ ~ c~c~~ c~~ c~~ c~~ c~c~~ c~c~c~c~~ c~~ ~~ ~~c~~~ ~~ ~c~c~~c~~~ c~~ ~
d
_
o~~~ ~~~ ~~ ~~ ~~ a~~ ~a a~~ a~ ~a ~a~ ~~ ~c~ ~~ ~~a a~ a~
UUU UU UU UU UU UUV UU UU UU UUU UV UU
U UUU UU UU UU UUU UU UVU UU UU VUU UU UU UU UUV UU UU .
U UUU UU UU UU ~~a a~ ~~U ~c~ a~ ~c~~ ~c~c~~ ~a ~c~~ ~~ ~ca
~ ~~a aa ~a ~~ a
aUU aa UFCa~ ~UU Ua a~
a UUC7Ur.GC~a UU aar.CC~~ aUU C9U C7C7U~CC7aa UU UC7C7Da VU aa cd
U aaC9aa C7r~aU C7UU a Ur.CU'~C7C7a C9a CUV C7C7ar
'' ~
a~a~CC~aU UU UU UU(9C7C7aC9C~C9CJC7U Ut~C~FCa aU C~C7r.C9C~C7C7C~.
C7UC~C~C7C7C7C7C9C~C~C9C~t~ C9C~C~ C~VC~UC9V VC7C7U C7 C~
p
V'U'C7C7C7C~U'U'C~V'U'C7U'U'C9C7UU'f.9U
C7C7C~C7C7C7C7UU'U'C~C~C7U'C7C7C~C7....
U UU'U U~CU' C9C~UFC C~C7O UU'UU C9C7U r.~C~UC7 r.CC7 C9r.CFC U N
r.~aa C~U U'~ t.7a aU U'aC7~ aC7 U ar.~FCU'a DV ~U'FC~C~UC7~U
~
C7C7r.C a~CFCC7aC7C7~CC7C7C7C~C7C7~Ca U'UUV FCU Ua Ur.CC7C7FC ~CUFC
r.~UC7a aU UU'aa Ut7r.Ca a~CFCUU Ur.~C~aU UU aa Ua r.CC7a C~ C7'"'
a U~U ar.Cr.Ca C7C~FC~a U~ UC7V C~C7Va C~~Ca Ua C7U r.~a C~ar.C r~ C7
7 UUU aU'FCU aU arU C7a C7
C
C~V FCUV C7FCa~CC7 U U UC~r.~U~ C7C . a~
O l0.-1t0M 00l~.-It~NM ~M1~OO v~N~DM.-I1~O rll0r-IN!~l~N lOV'Otnrl00N OM
y .-it(~01M ~r7ODMM tl1'1a'~O1DO~C'OO1N MODOD01MMC I~N 1OOD01V'v-1N~ONO~NM
ar I~hhCOODODO~0101O OOO Or-1NNM MM MM c~v~v'V'W f7W f1lDhfr 1~(~WO~
G
in
O n-~1rlri.-Irl~-1rl.-1rlN NNN NN NNN NN NN NNN NN NN NN NNN NN NN
~ N
Lw
it
N
CA 02288640 1999-11-OS
W0 98/50530 ~~.~ PCT/US98/09249
Table XIV. Hammerhead Ribozyme Sites for A-Raf
SEQ SEQ
Pos RZ ID. Substrate ID.
No. No.
UCCACCCUCUGAUGAGX CGAA AUUGGGUC1153 GACCCAAUAAGGGUGGA1461
28 CUCUGCGGCUGAUGAGX CGAA ACUCAGCC1154 GGCUGAGUCCCGCAGAG1462
92 ACUCUCGUCUGAUGAGX CGAA AUUGGCUC1155 GAGCCAAUAACGAGAGU1463
51 GCCUCUCGCUGAUGAGX CGAA ACUCUCGU1156 ACGAGAGUCCGAGAGGC1464
74 UCCUCACACUGAUGAGX CGAA AGUCCGCC1157 GGCGGACUCUGUGAGGA1965
123 CACGCCGCCUGAUGAGX CGAA ACAGCCGC1158 GCGGCUGUAGCGGCGUG1466
166 GAUGGGCUCUGAUGAGX CGAA AGGUGGGG1159 CCCCACCUCAGCCCAUC1467
179 UUUGUCAACUGAUGAGX CGAA AUGGGCUG1160 CAGCCCAUCUUGACAAA1468
176 AUUUUGUCCUGAUGAGX CGAA AGAUGGGC1161 GCCCAUCUUGACAAAAU1469
185 GAGCCUUACUGAUGAGX CGAA AUUUUGUC1162 GACAAAAUCUAAGGCUC1470
187 UGGAGCCUCUGAUGAGX CGAA AGAUUUUG1163 CAAAAUCUAAGGCUCCA1471
193 GCUCCAUGCUGAUGAGX CGAA AGCCUUAG1169 CUAAGGCUCCAUGGAGC1472
238 CUGCCCGGCUGAUGAGX CGAA AUGGCUCG1165 CGAGCCAUCCCGGGCAG1473
257 UAUACUUUCUGAUGAGX CGAA ACGGUGCC1166 GGCACCGUCAAAGUAUA1479
263 GGCAGGUACUGAUGAGX CGAA ACUUUGAC1167 GUCAAAGUAUACCUGCC1475
265 UGGGCAGGCUGAUGAGX CGAA AUACUUUG1168 CAAAGUAUACCUGCCCA1476
299 CCAUCCCGCUGAUGAGX CGAA ACAGUCAC1169.GUGACUGUCCGGGAUGG1477
317 GAGUCGUACUGAUGAGX CGAA ACACUCAU1170 AUGAGUGUCUACGACUC1978
319 GAGAGUCGCUGAUGAGX CGAA AGACACUC1171 GAGUGUCUACGACUCUC1479
325 UGUCUAGACUGAUGAGX CGAA AGUCGUAG1172 CUACGACUCUCUAGACA1480
327 CUUGUCUACUGAUGAGX CGAA AGAGUCGU1173 ACGACUCUCUAGACAAG1981
329 GCCUUGUCCUGAUGAGX CGAA AGAGAGUC1174 GACUCUCUAGACAAGGC1982
354 CUGAUUUACUGAUGAGX CGAA ACCCCGCA1175 UGCGGGGUCUAAAUCAG1483
356 UCCUGAUUCUGAUGAGX CGAA AGACCCCG1176 CGGGGUCUA 1484
AAUCAGGA
360 GCAGUCCUCUGAUGAGX CGAA AUUUAGAC1177 GUCUAAAUCAGGACUGC1485
377 AGUCGGUACUGAUGAGX CGAA ACCACACA1178 UGUGUGGUCUACCGACU1486
379 UGAGUCGGCUGAUGAGX CGAA AGACCACA1179 UGUGGUCUACCGACUCA1487
386 CCCUUGAUCUGAUGAGX CGAA AGUCGGUA1180 UACCGACUCAUCAAGGG1988
389 CGUCCCUUCUGAUGAGX CGAA AUGAGUCG1181 CGACUCAUCAAGGGACG1489
90? CAGGCAGUCUGAUGAGX CGAA ACCGUCUU1182 AAGACGGUCACUGCCUG1490
428 AGGGGAGCCUGAUGAGX CGAA AUGGCUGU1183 ACAGCCAUUGCUCCCCU1491
432 AUCCAGGGCUGAUGAGX CGAA AGCAAUGG1189 CCAUUGCUCCCCUGGAU1492
952 UCGACAAUCUGAUGAGX CGAA AGCUCCUC1185 GAGGAGCUCAUUGUCGA1493
455 ACCUCGACCUGAUGAGX CGAA AUGAGCUC1186 GAGCUCAUUGUCGAGGU1499
958 AGGACCUCCUGAUGAGX CGAA ACAAUGAG1187 CUCAUUGUCGAGGUCCU1495
964 UCUUCAAGCUGAUGAGX CGAA ACCUCGAC1188 GUCGAGGUCCUUGAAGA1496
967 ACAUCUUCCUGAUGAGX CGAA AGGACCUC1189 GAGGUCCUUGAAGAUGU1497
476 GUCAGCGGCUGAUGAGX CGAA ACAUCUUC1190 GAAGAUGUCCCGCUGAC1498
995 CCGUACAACUGAUGAGX CGAA AUUGUGCA1191 UGCACAAUUUUGUACGG1499
496 UCCGUACACUGAUGAGX CGAA AAUUGUGC1192 GCACAAUUUUGUACGGA1500
997 UUCCGUACCUGAUGAGX CGAA AAAUUGUG1193 CACAAUUUUGUACGGAA1501
500 GUCUUCCGCUGAUGAGX CGAA ACAAAAUU1199 AAUUUUGUACGGAAGAC1502
511 GGCUGAAGCUGAUGAGX CGAA AGGUCUUC1195 GAAGACCUUCUUCAGCC1503
512 AGGCUGAACUGAUGAGX CGAA AAGGUCUU1196 AAGACCUUCUUCAGCCU1504
519 CCAGGCUGCUGAUGAGX CGAA AGAAGGUC1197 GACCUUCUUCAGCCUGG1505
515 GCCAGGCUCUGAUGAGX CGAA AAGAAGGU1198 ACCUUCUUCAGCCUGGC1506
526 AGUCACAGCUGAUGAGX CGAA ACGCCAGG1199 CCUGGCGUUCUGUGACU1507
527 AAGUCACACUGAUGAGX CGAA AACGCCAG1200 CUGGCGUUCUGUGACUU1508
535 UAAGGCAGCUGAUGAGX CGAA AGUCACAG1201 CUGUGACUUCUGCCUUA1509
536 UUAAGGCACUGAUGAGX CGAA AAGUCACA1202 UGUGACUUCUGCCUUAA1510
542 AGAAACUUCUGAUGAGX CGAA AGGCAGAA1203 UUCUGCCUUAAGUUUCU1511
543 CAGAAACUCUGAUGAGX CGAA AAGGCAGA1204 UCUGCCUUAAGUUUCUG1512
CA 02288640 1999-11-05
WO 98/50530 158 PCT/US98/09249
SEQ SEQ
Pos RZ ID. ~ Substrate ID.
No. No.
597 GGAACAGACUGAUGAGX CGAA ACUUAAGG1205 CCUUAAGUUUCUGUUCC 1513
598 UGGAACAGCUGAUGAGX CGAA AACUUAAG1206 CUUAAGUUUCUGUUCCA 1514
549 AUGGAACACUGAUGAGX CGAA AAACUUAA1207 UUAAGUUUCUGUUCCAU 1515
553 AGCCAUGGCUGAUGAGX CGAA ACAGAAAC1208 GUUUCUGUUCCAUGGCU 1516
559 AAGCCAUGCUGAUGAGX CGAA AACAGAAA1209 UUUCUGUUCCAUGGCUU 1517
562 GGCAACGGCUGAUGAGX CGAA AGCCAUGG1210 CCAUGGCUUCCGUUGCC 1518
563 UGGCAACGCUGAUGAGX CGAA AAGCCAUG1211 CAUGGCUUCCGUUGCCA 1519
567 GGUUUGGCCUGAUGAGX CGAA ACGGAAGC1212 GCUUCCGUUGCCAAACC 1520
583 GGAACUUGCUGAUGAGX CGAA AGCCACAG1213 CUGUGGCUACAAGUUCC 1521
589 GCUGGUGGCUGAUGAGX CGAA ACUUGUAG1214 CUACAAGUUCCACCAGC 1522
590 UGCUGGUGCUGAUGAGX CGAA AACUUGUA1215 UACAAGUUCCACCAGCA 1523
600 GGAGGAACCUGAUGAGX CGAA AUGCUGGU1216 ACCAGCAUUGUUCCUCC 1524
603 CUUGGAGGCUGAUGAGX CGAA ACAAUGCU1217 AGCAUUGUUCCUCCAAG 1525
604 CCUUGGAGCUGAUGAGX CGAA AACAAUGC1218 GCAUUGUUCCUCCAAGG 1526
607 GGACCUUGCUGAUGAGX CGAA AGGAACAA1219 UUGUUCCUCCAAGGUCC 1527
614 ACUGUGGGCUGAUGAGX CGAA ACCUUGGA1220 UCCAAGGUCCCCACAGU 1528
623 UCAACACACUGAUGAGX CGAA ACUGUGGG1221 CCCACAGUCUGUGUUGA 1529
629 CUCAUGUCCUGAUGAGX CGAA ACACAGAC1222 GUCUGUGUUGACAUGAG 1530
639 GCGGUUGGCUGAUGAGX CGAA ACUCAUGU1223 ACAUGAGUACCAACCGC 1531
655 UGUGGUAGCUGAUGAGX CGAA ACUGUUGG1224 CCAACAGUUCUACCACA 1532
656 CUGUGGUACUGAUGAGX CGAA AACUGUUG1225 CAACAGUUCUACCACAG 1533
658 CACUGUGGCUGAUGAGX CGAA AGAACUGU1226 ACAGUUCUACCACAGUG 1534
668 AAAUCCUGCUGAUGAGX CGAA ACACUGUG1227 CACAGUGUCCAGGAUUU 1535
675 UCCGGACACUGAUGAGX CGAA AUCCUGGA1228 UCCAGGAUUUGUCCGGA 1536
676 CUCCGGACCUGAUGAGX CGAA AAUCCUGG1229 CCAGGAUUUGUCCGGAG 1537
679 AGCCUCCGCUGAUGAGX CGAA ACAAAUCC1230 GGAUUUGUCCGGAGGCU 1538
688 GCUGUCUGCUGAUGAGX CGAA AGCCUCCG1231 CGGAGGCUCCAGACAGC 1539
705 GUUCGAGGCUGAUGAGX CGAA AGCCUCAU1232 AUGAGGCUCCCUCGAAC 1590
709 GGCGGUUCCUGAUGAGX CGAA AGGGAGCC1233 GGCUCCCUCGAACCGCC 1591
730 GGGUUAGCCUGAUGAGX CGAA ACUCAUUC1239 GAAUGAGUUGCUAACCC 1592
739 UGGGGGGUCUGAUGAGX CGAA AGCAACUC1235 GAGUUGCUAACCCCCCA 1543
747 GGGGCUGGCUGAUGAGX CGAA ACCCUGGG1236 CCCAGGGUCCCAGCCCC 1594
789 GGAAGGGGCUGAUGAGX CGAA AGUGCUCC1237 GGAGCACUUCCCCUUCC 1545
785 GGGAAGGGCUGAUGAGX CGAA AAGUGCUC1238 GAGCACUUCCCCUUCCC 1546
790 GGGCAGGGCUGAUGAGX CGAA AGGGGAAG1239 CUUCCCCUUCCCUGCCC 1547
791 GGGGCAGGCUGAUGAGX CGAA AAGGGGAA1240 UUCCCCUUCCCUGCCCC 1548
815 AUGCGCUGCUGAUGAGX CGAA AGGGGGGC1241 GCCCCCCUACAGCGCAU 1549
829 GUGGAGCGCUGAUGAGX CGAA AUGCGCUG1242 CAGCGCAUCCGCUCCAC 1550
829 UGGACGUGCUGAUGAGX CGAA AGCGGAUG1293 CAUCCGCUCCACGUCCA 1551
835 UGGGAGUGCUGAUGAGX CGAA ACGUGGAG1249 CUCCACGUCCACUCCCA 1552
840 GACGUUGGCUGAUGAGX CGAA AGUGGACG1245 CGUCCACUCCCAACGUC 1553
848 ACCAUAUGCUGAUGAGX CGAA ACGUUGGG1246 CCCAACGUCCAUAUGGU 1554
852 GCUGACCACUGAUGAGX CGAA AUGGACGU1247 ACGUCCAUAUGGUCAGC 1555
857 GUGGUGCUCUGAUGAGX CGAA ACCAUAUG1248 CAUAUGGUCAGCACCAC 1556
880 UGAGGUUGCUGAUGAGX CGAA AGUCCAUG1249 CAUGGACUCCAACCUCA 1557
887 AGCUGGAUCUGAUGAGX CGAA AGGUUGGA1250 UCCAACCUCAUCCAGCU 1558
890 GUGAGCUGCUGAUGAGX CGAA AUGAGGUU1251 AACCUCAUCCAGCUCAC 1559
896 UGGCCAGUCUGAUGAGX CGAA AGCUGGAU1252 AUCCAGCUCACUGGCCA 1560
909 AGUGCUGACUGAUGAGX CGAA ACUCUGGC1253 GCCAGAGUUUCAGCACU 1561
910 CAGUGCUGCUGAUGAGX CGAA AACUCUGG1254 CCAGAGUUUCAGCACUG 1562
911 UCAGUGCUCUGAUGAGX CGAA AAACUCUG1255 CAGAGUUUCAGCACUGA 1563
930 UCCUCUACCUGAUGAGX CGAA ACCGGCAG1256 CUGCCGGUAGUAGAGGA 1564
933 ACCUCCUCCUGAUGAGX CGAA ACUACCGG1257 CCGGUAGUAGAGGAGGU 1565
~942~UCCAUCACCUGAUGAGX CGAA ACCUCCUC~ GAGGAGGUAGUGAUGGA 1566
1258
J
ji:.,..i'..
CA 02288640 1999-11-OS
WO 98150530 1~~ PCT/US98/09249
SEQ SEQ
Pos RZ ID. Substrate ID.
No. No.
985 UCCCCGAGCUGAUGAGX CGAA ACACGCUG1259CAGCGUGUC CUCGGGGA1567
988 UCCUCCCCCUGAUGAGX CGAA AGGACACG1260CGUGUCCUC GGGGAGGA1568
1000AAUGUGGGCUGAUGAGX CGAA ACUUCCUC1261GAGGAAGUC CCCACAUU1569
1008UGACUUGGCUGAUGAGX CGAA AUGUGGGG1262CCCCACAUU CCAAGUCA1570
1009GUGACUUGCUGAUGAGX CGAA AAUGUGGG1263CCCACAUUC CAAGUCAC1571
1015CUGCUGGUCUGAUGAGX CGAA ACUUGGAA1264UUCCAAGUC ACCAGCAG1572
1042CGGCCAAGCUGAUGAGX CGAA ACUUCCGC1265GCGGAAGUC CUUGGCCG1573
1045CAUCGGCCCUGAUGAGX CGAA AGGACUUC1266GAAGUCCUU GGCCGAUG'1574
1081ANUCCCGGCUGAUGAGX CGAA ACCCCAGG1267CCUGGGGUA CCGGGANU1575
1090AAUAGCCUCUGAUGAGX CGAA ANUCCCGG1268CCGGGANUC AGGCUAUU1576
1096CCCAGUAACUGAUGAGX CGAA AGCCUGAN1269NUCAGGCUA UUACUGGG1577
1098CUCCCAGUCUGAUGAGX CGAA AUAGCCUG1270CAGGCUAUU ACUGGGAG1578
1099CCUCCCAGCUGAUGAGX CGAA AAUAGCCU1271AGGCUAUUA CUGGGAGG1579
1109CUGGGUGGCUGAUGAGX CGAA ACCUCCCA1272UGGGAGGUA CCACCCAG1580
1192CCCGUCCCCUGAUGAGX CGAA AUCCUCUU1273AAGAGGAUC GGGACGGG1581
1153UGCCAAACCUGAUGAGX CGAA AGCCCGUC1274GACGGGCUC GUUUGGCA1582
1156CGGUGCCACUGAUGAGX CGAA ACGAGCCC1275GGGCUCGUU UGGCACCG1583
1157ACGGUGCCCUGAUGAGX CGAA AACGAGCC1276GGCUCGUUU GGCACCGU1584
1168GCCCUCGACUGAUGAGX CGAA ACACGGUG1277CACCGUGUU UCGAGGGC1585
1169CGCCCUCGCUGAUGAGX CGAA AACACGGU1278ACCGUGUUU CGAGGGCG1586
1170CCGCCCUCCUGAUGAGX CGAA AAACACGG1279CCGUGUUUC GAGGGCGG1587
1208GACACCUUCUGAUGAGX CGAA AGCACCUU1280AAGGUGCUC AAGGUGUC1588
1216UGGGCUGGCUGAUGAGX CGAA ACACCUUG1281CAAGGUGUC CCAGCCCA1589
1245AUUCUUGACUGAUGAGX CGAA AGCCUGGG1282CCCAGGCUU UCAAGAAU1590
1246CAUUCUUGCUGAUGAGX CGAA AAGCCUGG1283CCAGGCUUU CAAGAAUG1591
1297UCAUUCUUCUGAUGAGX CGAA AAAGCCUG1284CAGGCUUUC AAGAAUGA1592
1268GUCUUCCUCUGAUGAGX CGAA AGCACCUG1285CAGGUGCUC AGGAAGAC1593
1286AAGAUGUUCUGAUGAGX CGAA ACAUGUCG1286CGACAUGUC AACAUCUU1594
1292AACAGCAACUGAUGAGX CGAA AUGUUGAC1287GUCAACAUC UUGCUGUU1595
1299UAAACAGCCUGAUGAGX CGAA AGAUGUUG1288CAACAUCUU GCUGUUUA1596
1300AGCCCAUACUGAUGAGX CGAA ACAGCAAG1289CUUGCUGUU UAUGGGCU1597
1301AAGCCCAUCUGAUGAGX CGAA AACAGCAA1290UUGCUGUUU AUGGGCUU1598
1302GAAGCCCACUGAUGAGX CGAA AAACAGCA1291UGCUGUUUA UGGGCUUC1599
1309GGGUCAUGCUGAUGAGX CGAA AGCCCAUA1292UAUGGGCUU CAUGACCC1600
1310CGGGUCAUCUGAUGAGX CGAA AAGCCCAU1293AUGGGCUUC AUGACCCG1601
1327UGAUGGCACUGAUGAGX CGAA AUCCCGGC1299GCCGGGAUU UGCCAUCA1602
1328AUGAUGGCCUGAUGAGX CGAA AAUCCCGG1295CCGGGAUUU GCCAUCAU1603
1334UGUGUGAUCUGAUGAGX CGAA AUGGCAAA1296UUUGCCAUC AUCACACA1604
1337CACUGUGUCUGAUGAGX CGAA AUGAUGGC1297GCCAUCAUC ACACAGUG1605
1357AGAGGCUGCUGAUGAGX CGAA AGCCCUCA1298UGAGGGCUC CAGCCUCU1606
1369UGAUGGUACUGAUGAGX CGAA AGGCUGGA1299UCCAGCCUC UACCAUCA1607
1366GGUGAUGGCUGAUGAGX CGAA AGAGGCUG1300CAGCCUCUA CCAUCACC1608
1371AUGCAGGUCUGAUGAGX CGAA AUGGUAGA1301UCUACCAUC ACCUGCAU1609
1396CCAUGUCGCUGAUGAGX CGAA AGCGUGUG1302CACACGCUU CGACAUGG1610
1397ACCAUGUCCUGAUGAGX CGAA AAGCGUGU1303ACACGCUUC GACAUGGU1611
1406AUGAGCUGCUGAUGAGX CGAA ACCAUGUC1304GACAUGGUC CAGCUCAU1612
1412ACGUCGAUCUGAUGAGX CGAA AGCUGGAC1305GUCCAGCUC AUCGACGU1613
1415GCCACGUCCUGAUGAGX CGAA AUGAGCUG1306CAGCUCAUC GACGUG6C1619
1450CAUGGAGGCUGAUGAGX CGAA AGUCCAUG1307CAUGGACUA CCUCCAUG1615
1459UUGGCAUGCUGAUGAGX CGAA AGGUAGUC1308GACUACCUC CAUGCCAA1616
1969CGGUGGAUCUGAUGAGX CGAA AUGUUCUU1309AAGAACAUC AUCCACCG1617
1472UCUCGGUGCUGAUGAGX CGAA AUGAUGUU1310AACAUCAUC CACCGAGA1618
1982AGACUUGACUGAUGAGX CGAA AUCUCGGU1311ACCGAGAUC UCAAGUCU1619
[1484~UUAGACUUCUGAUGAGX CGAA AGAUCUCG~ ~ CGAGAUCUCAAGUCUAAr1620~
1312
CA 02288640 1999-11-OS
WO 98/50530 160 PCT/US98/09249
SEQ ~~ SEQ
Pos RZ ID. Substrate ID.
No. No.
1989UGUUGUUACUGAUGAGX CGAA ACUUGAGA1313 UCUCAAGUCUAACAACA 1621
1491GAUGUUGUCUGAUGAGX CGAA AGACUUGA1314 UCAAGUCUAACAACAUC 1622
1999UGUAGGAACUGAUGAGX CGAA AUGUUGUU1315 AACAACAUCUUCCUACA 1623
1501CAUGUAGGCUGAUGAGX CGAA AGAUGUUG1316 CAACAUCUUCCUACAUG 1624
1502UCAUGUAGCUGAUGAGX CGAA AAGAUGUU1317 AACAUCUUCCUACAUGA 1625
1505CCCUCAUGCUGAUGAGX CGAA AGGAAGAU1318 AUCUUCCUACAUGAGGG 1626
1517UUCACCGUCUGAUGAGX CGAA AGCCCCUC1319 GAGGGGCUCACGGUGAA 1627
1529AAGUCACCCUGAUGAGX CGAA AUCUUCAC1320 GUGAAGAUCGGUGACUU 1628
1537CCAAGCCACUGAUGAGX CGAA AGUCACCG1321 CGGUGACUUUGGCUUGG 1629
1538GCCAAGCCCUGAUGAGX CGAA AAGUCACC1322 GGUGACUUUGGCUUGGC 1630
1543CUGUGGCCCUGAUGAGX CGAA AGCCAAAG1323 CUUUGGCUUGGCCACAG 1631
1560GCUCCAUCCUGAUGAGX CGAA AGUCUUCA1329 UGAAGACUCGAUGGAGC 1632
1582GCUGCUCCCUGAUGAGX CGAA AGGGCUGG1325 CCAGCCCUUGGAGCAGC 1633
1594CAGAUCCUCUGAUGAGX CGAA AGGGCUGC1326 GCAGCCCUCAGGAUCUG 1634
1600ACAGCACACUGAUGAGX CGAA AUCCUGAG1327 CUCAGGAUCUGUGCUGU 1635
1628UGCAUACGCUGAUGAGX CGAA AUCACCUC1328 GAGGUGAUCCGUAUGCA 1636
1632GUCCUGCACUGAUGAGX CGAA ACGGAUCA1329 UGAUCCGUAUGCAGGAC 1637
1651GGAAGCUGCUGAUGAGX CGAA AGGGGUUC1330 GAACCCCUACAGCUUCC 1638
1657CUGACUGGCUGAUGAGX CGAA AGCUGUAG1331 CUACAGCUUCCRGUCAG 1639
1658UCUGACUGCUGAUGAGX CGAA AAGCUGUA1332 UACAGCUUCCAGUCAGA 1690
1663AGACGUCUCUGAUGAGX CGAA ACUGGAAG1333 CUUCCAGUCAGACGUCU 1641
1670UAGGCAUACUGAUGAGX CGAA ACGUCUGA1334 UCAGACGUCUAUGCCUA 1642
1672CGUAGGCACUGAUGAGX CGAA AGACGUCU1335 AGACGUCUAUGCCUACG 1643
1678CAACCCCGCUGAUGAGX CGAA AGGCAUAG1336 CUAUGCCUACGGGGUUG 1649
1685UAGAGCACCUGAUGAGX CGAA ACCCCGUA1337 UACGGGGUUGUGCUCUA 1645
1691AGCUCGUACUGAUGAGX CGAA AGCACAAC1338 GUUGUGCUCUACGAGCU 1646
1693UAAGCUCGCUGAUGAGX CGAA AGAGCACA1339 UGUGCUCUACGAGCUUA 1697
1700CCAGUCAUCUGAUGAGX CGAA AGCUCGUA1390 UACGAGCUUAUGACUGG 1698
1701GCCAGUCACUGAUGAGX CGAA AAGCUCGU1341 ACGAGCUUAUGACUGGC 1649
1711AAGGCAGUCUGAUGAGX CGAA AGCCAGUC1342 GACUGGCUCACUGCCUU 1650
1719GUGGCUGUCUGAUGAGX CGAA AGGCAGUG1343 CACUGCCUUACAGCCAC 1651
1720UGUGGCUGCUGAUGAGX CGAA AAGGCAGU1344 ACUGCCUUACAGCCACA 1652
1730CGGCAGCCCUGAUGAGX CGAA AUGUGGCU1345 AGCCACAUUGGCUGCCG 1653
1798AUAAAGAUCUGAUGAGX CGAA AUCUGGUC1346 GACCAGAUUAUCUUUAU 1654
1749CAUAAAGACUGAUGAGX CGAA AAUCUGGU1347 ACCAGAUUAUCUUUAUG 1655
1751ACCAUAAACUGAUGAGX CGAA AUAAUCUG1348 CAGAUUAUCUUUAUGGU 1656
1753CCACCAUACUGAUGAGX CGAA AGAUAAUC1399 GAUUAUCUUUAUGGUGG 1657
1754CCCACCAUCUGAUGAGX CGAA AAGAUAAU1350 AUUAUCUUUAUGGUGGG 1658
1755GCCCACCACUGAUGAGX CGAA AAAGAUAA1351 UUAUCUUUAUGGUGGGC 1659
1771GGGACAGACUGAUGAGX CGAA AGCCACGG1352 CCGUGGCUAUCUGUCCC 1660
1773CGGGGACACUGAUGAGX CGAA AUAGCCAC1353 GUGGCUAUCUGUCCCCG 1661
1777GGUCCGGGCUGAUGAGX CGAA ACAGAUAG1359 CUAUCUGUCCCCGGACC 1662
1787AUUUUGCUCUGAUGAGX CGAA AGGUCCGG1355 CCGGACCUCAGCAAAAU 1663
1796UUGCUGGACUGAUGAGX CGAA AUUUUGCU1356 AGCAAAAUCUCCAGCAA 1669
1798AGUUGCUGCUGAUGAGX CGAA AGAUUUUG1357 CAAAAUCUCCAGCAACU 1665
1834GGCAGUCACUGAUGAGX CGAA ACAGCAGG1358 CCUGCUGUCUGACUGCC 1666
1844UGGAACUUCUGAUGAGX CGAA AGGCAGUC1359 GACUGCCUCAAGUUCCA 1667
1849CCCGCUGGCUGAUGAGX CGAA ACUUGAGG1360 CCUCAAGUUCCAGCGGG 1668
1850UCCCGCUGCUGAUGAGX CGAA AACUUGAG1361 CUCAAGUUCCAGCGGGA 1669
1871UGGGGGAACUGAUGAGX CGAA AGGGGCCG1362 CGGCCCCUCUUCCCCCA 1670
1873UCUGGGGGCUGAUGAGX CGAA AGAGGGGC1363 GCCCCUCUUCCCCCAGA 1671
1874AUCUGGGGCUGAUGAGX CGAA AAGAGGGG1369 CCCCUCUUCCCCCAGAU 1672
1883GUGGCCAGCUGAUGAGX CGAA AUCUGGGG1365 CCCCAGAUCCUGGCCAC 16?3
1895AGCAGCUCCUGAUGAGX CGAA AUUGUGGC1366 GCCACAAUUGAGCUGCU 1674
CA 02288640 1999-11-OS
WO 98/50530 ~~1 , PCTNS98/09249
SEQ SEQ
Pos RZ ID. Substrate ID.
No. No.
1912UGGGGAGUCUGAUGAGX CGAA ACCGUUGC1367 GCAACGGUC ACUCCCCA1675
1916AUCUUGGGCUGAUGAGX CGAA AGUGACCG1368 CGGUCACUC CCCAAGAU1676
1925CUCCGCUCCUGAUGAGX CGAA AUCUUGGG1369 CCCAAGAUU GAGCGGAG1677
1939AGGGUUCCCUGAUGAGX CGAA AGGCACUC1370 GAGUGCCUC GGAACCCU1678
1948GGUGCAAGCUGAUGAGX CGAA AGGGUUCC1371 GGAACCCUC CUUGCACC1679
1951UGCGGUGCCUGAUGAGX CGAA AGGAGGGU1372 ACCCUCCUU GCACCGCA1680
1975AGGCAGGCCUGAUGAGX CGAA ACUCAUCG1373 CGAUGAGUU GCCUGCCU1681
1988GCGCUGAGCUGAUGAGX CGAA AGGCAGGC1374 GCCUGCCUA CUCAGCGC1682
1991GCUGCGCUCUGAUGAGX CGAA AGUAGGCA1375 UGCCUACUC AGCGCAGC1683
2006UAAGGCACCUGAUGAGX CGAA AGGCGGGC1376 GCCCGCCUU GUGCCUUA1684
2013CGGGGCCUCUGAUGAGX CGAA AGGCACAA1377 UUGUGCCUU AGGCCCCG1685
2014GCGGGGCCCUGAUGAGX CGAA AAGGCACA1378 UGUGCCUUA GGCCCCGC1686
2044AGGGCUGACUGAUGAGX CGAA AUUGGCUC1379 GAGCCAAUC UCAGCCCU1687
2096GGAGGGCUCUGAUGAGX CGAA AGAUUGGC1380 GCCAAUCUC AGCCCUCC1688
2053UUGGCGUGCUGAUGAGX CGAA AGGGCUGA1381 UCAGCCCUC CACGCCAA1689
2069UGGUGGGCCUGAUGAGX CGAA AGGCUCCU1382 AGGAGCCUU GCCCACCA1690
2089CGAACAUUCUGAUGAGX CGAA AUUGGCUG1383 CAGCCAAUC AAUGUUCG1691
2090CAGAGACGCUGAUGAGX CGAA ACAUUGAU1389 AUCAAUGUU CGUCUCUG1692
2091GCAGAGACCUGAUGAGX CGAA AACAUUGA1385 UCAAUGUUC GUCUCUGC1693
2099AGGGCAGACUGAUGAGX CGAA ACGAACAU1386 AUGUUCGUC UCUGCCCU1694
2096UCAGGGCACUGAUGAGX CGAA AGACGAAC1387 GUUCGUCUC UGCCCUGA1695
2113GGGAUCCUCUGAUGAGX CGAA AGGCAGCA1388 UGCUGCCUC AGGAUCCC1696
2119GAAUGGGGCUGAUGAGX CGAA AUCCUGAG1389 CUCAGGAUC CCCCAUUC1697
2126GGGUGGGGCUGAUGAGX CGAA AUGGGGGA1390 UCCCCCAUU CCCCACCC1698
2127AGGGUGGGCUGAUGAGX CGAA AAUGGGGG1391 CCCCCAUUC CCCACCCU1699
2151CACAUGGGCUGAUGAGX CGAA ACCCCCUC1392 GAGGGGGUC CCCAUGUG1700
2162AACUGGAACUGAUGAGX CGAA AGCACAUG1393 CAUGUGCUU UUCCAGUU1701
2163GAACUGGACUGAUGAGX CGAA AAGCACAU1394 AUGUGCUUU UCCAGUUC1702
2164AGAACUGGCUGAUGAGX CGAA AAAGCACA1395 UGUGCUUUU CCAGUUCU1703
2165AAGAACUGCUGAUGAGX CGAA AAAAGCAC1396 GUGCUUUUC CAGUUCUU1704
2170UCCAGAAGCUGAUGAGX CGAA ACUGGAAA1397 UUUCCAGUU CUUCUGGA1705
2171UUCCAGAACUGAUGAGX CGAA AACUGGAA1398 UUCCAGUUC UUCUGGAA1706
2173AAUUCCAGCUGAUGAGX CGAA AGAACUGG1399 CCAGUUCUU CUGGAAUU1T07
2174CAAUUCCACUGAUGAGX CGAA AAGAACUG1900 CAGUUCUUC UGGAAUUG1708
2181GGUCCCCCCUGAUGAGX CGAA AUUCCAGA1901 UCUGGAAUU GGGGGACC1709
2214AUGGAGGACUGAUGAGX CGAA ACAGGGGG1402 CCCCCUGUC UCCUCCAU1710
2216UGAUGGAGCUGAUGAGX CGAA AGACAGGG1403 CCCUGUCUC CUCCAUCA1711
2219AAAUGAUGCUGAUGAGX CGAA AGGAGACA1404 UGUCUCCUC CAUCAUUU1712
2223AACCAAAUCUGAUGAGX CGAA AUGGAGGA1905 UCCUCCAUC AUUUGGUU1713
2226GGAAACCACUGAUGAGX CGAA AUGAUGGA1406 UCCAUCAUU UGGUUUCC1714
2227AGGAAACCCUGAUGAGX CGAA AAUGAUGG1407 CCAUCAUUU GGUUUCCU1715
2231CAAGAGGACUGAUGAGX CGAA ACCAAAUG1408 CAUUUGGUU UCCUCUUG1716
2232CCAAGAGGCUGAUGAGX CGAA AACCAAAU1409 AUUUGGUUU CCUCUUGG1717
2233GCCAAGAGCUGAUGAGX CGAA AAACCAAA1410 UUUGGUUUC CUCUUGGC1718
2236AAAGCCAACUGAUGAGX CGAA AGGAAACC1911 GGUUUCCUC UUGGCUUU1719
2238CCAAAGCCCUGAUGAGX CGAA AGAGGAAA1912 UUUCCUCUU GGCUUUGG1720
2243UAUCCCCACUGAUGAGX CGAA AGCCAAGA1413 UCUUGGCUU UGGGGAUA1721
2294GUAUCCCCCUGAUGAGX CGAA AAGCCAAG1919 CUUGGCUUU GGGGAUAC1722
2251UUUAGAAGCUGAUGAGX CGAA AUCCCCAA1415 UUGGGGAUA CUUCUAAA1723
2254AAAUUUAGCUGAUGAGX CGAA AGUAUCCC1416 GGGAUACUU CUAAAUUU1724
2255AAAAUUUACUGAUGAGX CGAA AAGUAUCC1417 GGAUACUUC UAAAUUUU1725
2257CCAAAAUUCUGAUGAGX CGAA AGAAGUAU1418 AUACUUCUA 1726
AAUUUUGG
2261GCUCCCAACUGAUGAGX CGAA AUUUAGAA1419 UUCUAAAUU UUGGGAGC1727
(2262~AGCUCCCACUGAUGAGX CGAA AAUUUAGA~ ~ UCUAAAUUUUGGGAGCU1728j
1420 ~
CA 02288640 1999-11-OS
WO 98/50530 162 PCT/US98/09249
SEQ SEQ
Pos _ RZ ID. Substrate ID.
No. No.
2263GAGCUCCCCUGAUGAGX CGAA AAAUUUAG1421 CUAAAUUUUGGGAGCUC1729
2271AGAUGGAGCUGAUGAGX CGAA AGCUCCCA1422 UGGGAGCUCCUCCAUCU1730
2274UGGAGAUGCUGAUGAGX CGAA AGGAGCUC1423 GAGCUCCUCCAUCUCCA1731
2278CCAUUGGACUGAUGAGX CGAA AUGGAGGA1429 UCCUCCAUCUCCAAUGG1732
2280AGCCAUUGCUGAUGAGX CGAA AGAUGGAG1925 CUCCAUCUCCAAUGGCU1733
2294CUGCCACACUGAUGAGX CGAA AUCCCAGC1926 GCUGGGAUUUGUGGCAG1734
2295CCUGCCACCUGAUGAGX CGAA AAUCCCAG1427 CUGGGAUUUGUGGCAGG1735
2307CUGAGUGGCUGAUGAGX CGAA AUCCCUGC1428 GCAGGGAUUCCACUCAG1736
2308UCUGAGUGCUGAUGAGX CGAA AAUCCCUG1429 CAGGGAUUCCACUCAGA1737
2313GAGGUUCUCUGAUGAGX CGAA AGUGGAAU1430 AUUCCACUCAGAACCUC1738
2321AUUCCAGACUGAUGAGX CGAA AGGUUCUG1431 CAGAACCUCUCUGGAAU1739
2323AAAUUCCACUGAUGAGX CGAA AGAGGUUC1932 GAACCUCUCUGGAAUUU1790
2330CAGGCACACUGAUGAGX CGAA AUUCCAGA1433 UCUGGAAUUUGUGCCUG1741
2331UCAGGCACCUGAUGAGX CGAA AAUUCCAG1434 CUGGAAUUUGUGCCUGA1742
2347UCCAGUGGCUGAUGAGX CGAA AGGCACAU1435 AUGUGCCUUCCACUGGA1743
2348AUCCAGUGCUGAUGAGX CGAA AAGGCACA1436 UGUGCCUUCCACUGGAU1744
2357AACCCCAACUGAUGAGX CGAA AUCCAGUG1937 CACUGGAUUUUGGGGUU1745
2358GAACCCCACUGAUGAGX CGAA AAUCCAGU1438 ACUGGAUUUUGGGGUUC1746
2359GGAACCCCCUGAUGAGX CGAA AAAUCCAG1939 CUGGAUUUUGGGGUUCC1797
2365GUGCUGGGCUGAUGAGX CGAA ACCCCAAA1440 UUUGGGGUUCCCAGCAC1748
2366GGUGCUGGCUGAUGAGX CGAA AACCCCAA1941 UUGGGGUUCCCAGCACC1749
2385CCCCCCAACUGAUGAGX CGAA AUCCACAU1492 AUGUGGAUUUUGGGGGG1750
2386ACCCCCCACUGAUGAGX CGAA AAUCCACA1943 UGUGGAUUUUGGGGGGU1751
2387GACCCCCCCUGAUGAGX CGAA AAAUCCAC1444 GUGGAUUUUGGGGGGUC1752
2395ACAAAAGGCUGAUGAGX CGAA ACCCCCCA1445 UGGGGGGUCCCUUUUGU1753
2399AGACACAACUGAUGAGX CGAA AGGGACCC1496 GGGUCCCUUUUGUGUCU1754
2400GAGACACACUGAUGAGX CGAA AAGGGACC1497 GGUCCCUUUUGUGUCUC1755
2901GGAGACACCUGAUGAGX CGAA AAAGGGAC1498 GUCCCUUUUGUGUCUCC1756
2406GCGGGGGACUGAUGAGX CGAA ACACAAAA1499 UUUUGUGUCUCCCCCGC1757
2408UGGCGGGGCUGAUGAGX CGAA AGACACAA1450 UUGUGUCUCCCCCGCCA1758
2918AGUCCUUGCUGAUGAGX CGAA AUGGCGGG1451 CCCGCCAUUCAAGGACU1759
2419GAGUCCUUCUGAUGAGX CGAA AAUGGCGG1452 CCGCCAUUCAAGGACUC1760
2927AAAGAGAGCUGAUGAGX CGAA AGUCCUUG1453 CAAGGACUCCUCUCUUU1761
2930AAGAAAGACUGAUGAGX CGAA AGGAGUCC1454 GGACUCCUCUCUUUCUU1762
2432UGAAGAAACUGAUGAGX CGAA AGAGGAGU1455 ACUCCUCUCUUUCUUCA1763
2934GGUGAAGACUGAUGAGX CGAA AGAGAGGA1956 UCCUCUCUUUCUUCACC1764
2435UGGUGAAGCUGAUGAGX CGAA AAGAGAGG1457 CCUCUCUUUCUUCACCA1765
2936UUGGUGAACUGAUGAGX CGAA AAAGAGAG1458 CUCUCUUUCUUCACCAA1766
2438UCUUGGUGCUGAUGAGX CGAA AGAAAGAG1959 CUCUUUCUUCACCAAGA1767
2439UUCUUGGUCUGAUGAGX CGAA AAGAAAGA1460 UCUUUCUUCACCAAGAA1768
Where "X" represents stem II region of a HH ribozyme (Hertel et al., 1992
Nucleic Acids
Res. 20: 3252). The length of stem II may be > 2 base-pairs.
CA 02288640 1999-11-OS
WO 98/50530 163 PCT/US98/09249
0
z
r-iN MC~N~O1~m O1Oe-1NM spt!1l0I~m 01O r-1N MV~M ~O1'~mO~O r-fN MV~tnl0hm
p v~v~~rc av~cc c~numnu~~nww m nw wo tomo ~o~omoh rh rr hr rr
mm mm mm mm mmm mm mmm mm mm mm mmm mm mmm mm mm mm mm
w
c~c9c~U ao r~a ~dU ad aat9 a d~ dU ac9c9Ua ~dc~ ~ Uc9c~a Ud
DU UU Ua rd C7Ud dU ' ' ' ~ ' '
C
w C9C7C9U UU .C7aaC7Ut7C7C7U ~a Ut7UU UaU UC9r.~U~ C7d Ud Ua UU
U U a U'Ua C7f Uda U'd C7U C~dU U'd dU
~
ad t9C7da C~C7dC7Ua C9~d a dU U. dUU DD dU'U a Ut9aC7aa
y Ua Ud Ud ~U'aUC~dd C7aU UC7aU Ud UUd UC7add V dd dC9Ud
U
p aC7U'U UU UU C7aa U'U aDU Ua UV UU UUU C7C7(7UC7U'U'UU'aC9oU
Q'
UD dU UU UU aUU aU VUa aU aU UU aUa aU dUa aa Ua aU Ua
dU aU UU Ua UdU Ud UUa DD DU UU UUU dU aUd UU UU aU UU
y ChC7C7C~C9C~C9C7C7C7C9C9C7C~C9C~C~C7UV VC9C9C7C9C7C~C9C9U C7C9C~C7C~C9C9C9
~ c7~ aa dd aa aUa Ua daa Ud dU da Uc~d aa ~dU da dd ac~dd
V edUU UU UU UU UUU UU UUU UU UU UU UUU UU UUU UU UU UU UU
A ErU'C7C7U Ua Ud ddd UU'aoa ad ,~d UU adU r.~U'UUC7U'C7Ud C~U UU
CPU'U'd UU dC7U'aU aU aaa aU dd UU dUa Ua U'UC7ad UU aU aa
' '
dU UU C7U Da UUa ChU UUa UU UC7ao UUd C9d DUa UU aU Dd UC7
a z
,y 61o v-IN MV~~~Orm01or-INMV~U1101~m O~o ~NM ~~ l0rOD01o riN MV~WO
G ~r rr rr rr rrr mm mmm mm mm mo~o~o,o,o~o~o~a~a,o~0 00 00 00
c~r hr rr rr rc~r rr rhr.rt~r~t~hr t-rr t~r ht~r t~m mm mm mm
~b w
A
R
W
dd dd dd dd ddd dd ddd dd dd dd ddd dd ddd dd dd dd dd
o aa aa aa aa aaa aa aaa aa aa aa aaa aa aaa aa aa aa aa
c~~ c~~ c~c~cn~ c~c~~ c~c~c~c~~ c~c~~c~c~c~c~~c~c~~ ~~~ ~~ ~~ c~c~~c~
aa aa aa aa aaa aa aaa aa aa aa aaa aa oaa aa aa aa aa
UU UU UU UU UVU UU UUU UU UU UU UUU UU UUU VU UU UU UU
UU UU UU UU VUU UU UUU UV UU UU UUU UU UUU UU UU UU UU
dd dd dd dd ddd dd ddd dd dd dd ddd dd ddd dd dd dd dd
aa aa ao 5a oaa aa aaa aa aa aa aaa aa aaa aa aa aa aa
~ aa aa aa ao aaa aa aaa aa aa aa aaa aa aaa aa aa aa aa
."" dd dd dd dd ~dd dd d~d dd dd d~ ddd dd ddd dd dd dd dd
UU UU UU VU UU UU U U UU UU U UUU UU UUU UU UU UU UU
dd dd dd dd ddd dd ddd dd dd dd ddd dd ddd dd dd dd dd
aa aa aa aa aaa aa aaa aa aa aa aaa aa aaa aa aa aa aa
c~~ c~c~c~c~~c~~~c~c~t~~c~c~c~~ ~c~~c~c~ch~ ~~ c~c~c~c~c~c~c~~c~c~~
~ xx xx xx xx xxx xx xxx xx xx xx xxx xx xxx xx xx xx xx
c d dd dd dd ddd dd ddd dd dd dd ddd d ddd d dd dd dd
UU UU UU UU UUU UU UUU UU UU UU UUU ~U UUV ~U UU UU UU
C7C7C7C7C7C7C7C9C7C7C9C7C9C9C~C~C9C7 C9C~C9C9C9C9C9C9C9C9CJC9(7C7C9C9C9C9C~
~ dd dd dd dd ddd dd ddd dd dd dd ddd dd ddd dd dd dd dd
' ' '' ''
C7(.~C~C9C~C7C7C7C7C9C7UC7C9C7C9C9U C~C9C9t7C9C~U UC C7C9C9UV C9C7C9C9C9C~
~'dd dd dd dd ddd dd d d dd d dd ddd dJ dd dd d dd dd
UU UU VU UU UUU UU U d
o U UU U UU UUU UU UV VU U UU UU
a UU UU UU UU UUU UU UUU UU UV UU UUU UU UUU UU UU UU UU
' dd dr.Cr.Cd dd ddd dd ddd dd dC r.~d ddd dd ddd dd dd dd d~t
7 r
~y .
C a~ c~t~Uc~dd Uc~d U~ ~ c~caU dd c~c~a Ua dc~d Uc~dc~ a c~U
UU Ua U'C7aU UdU'dC9 ' ' 7 d ' d
~ UV UC7C9d C7a aaa (9U ~~ ~(.~aa C9U aUd C~d UCV Ua C,~C7UU dC7
D o5 U'U'da oU C7V'U U C7a C9C7
, C9C7C~C7C7C9t7C9C7U'C9C9U'U'C~C7C9C~C9C7UC7't9~ ~7 779 9~ U' 9~ UU
U CC CCC CC U CC
H
U'U'C'JC~C7U U'C~C7t7C7C9C7C9C7C7U'C9C9C'JC9C9C7C9C9C9C9 V(~U'C~C~C9C9 C9C~
dd dd dd dd ddd dd ddd dd dd dd ddd dd ddd dd dd dd dd
dU UC7C~V'U'U'Udd UU'd U'Ud U'U'C7U'UC9C9UU UU'U UU U'U FCU dC~
C9d U'a C7D aU dU'U aa U~ '' '
C7C7U dC7UU C9C9a UU daa C7a DD aU C9a
da UU ad DU U5U C7~ Uaa C7d aC7C7a aCJC9 d aUC9dD C7U dU dd
UU UC7U'U'C9U ddU C7 7 ~ ' '
C~d aU C9d UV C9ad D UUa UU aU Va aC7
dC7U'U'U'~ aa UtJa aU'UUU a U'V U'C7UdU C7U aC?a ao Ua U CPU
UV UC~d Dd Ua( da d ~ ' ~ '
~
. dU a aa DU dUU C9d UDU aU UU U Ua
C
0
N ~r-o.-~wr m~o~o.-io 00,moro co Na oc~~n~N r-M ro,' ~' '~ hm ~m
.-1N~ ~~D~O0110mv-1I'~m rlM~f1~ON ~.1U7O~NtOO~.-1N N~D~ NN ~-1N O1~-i~O
~~-1.-1rlri'-INN MMsrvcr~U'7N tn1D10h 1~1~mmm 01010101O r-1e-iNN NM Mv~
ri'-I'-1riv-1ririe-1ri
r
O
CA 02288640 1999-11-OS
WO 98/50530 PCT/US98/09249
164
0
z
~ O.-1NMa 1n101~OD01O.-INM c~1nto(~aD01O rlN Ma~N t0t~ODQ1O .-1N
A c~aom coaom ooaoaom 0~o~o
o0 vrno~ovo,o,rnovo 00 000 00 00~ ,...,.1
m aom mmo om mm ooao0 00o coo~aomaoa~m o~o~ovo~ovovo~avova,rno~
~r.-~~.-a,-r.--~~ ,-i.--i.-a.--i,-i,--i.--i~,-a~~ ,--i~~ .-a..-i.-i~ ~.-i~ ,--
~~ .-t.-~~ ~,--i
W
Ur.~C7C9C9aa C7U ar.~U C7GCC7U aaU C7C9rLr.~aUU UC7Ur.~C7Ua
U C~C7C7aC7C~a~~U aC7U'U'C7aa U'aC9Ua aU aC7U r.~a UC7a UU
U C7C7r.~U'rCr~a U~ UC7FC a UU UU' a Ua UaU'UrCU'C7U aU
U ' ~ ~ ~
C7a Ut9U UU ~U aUU U ~CU aa C9UU UC7 UC9arCa VC9
y C7FCa aFCC~Ua aa FCUa Ua C~C7UUU Ur.CC9rCC7a~ aa UUD Ua
D a UU UC7a aU'aC7aVU UU'aa aUU U'C7aa UaU UU C7aU ~C7
C
U UU UarCaU UU aUU Ua UU UUa aa UU UUU UU aUa Ua
r.CUU UUa Ur.~UU r.~a~CUU ar.CUUr.CUrLUU UUU UU rxUD a~
y C7C7U C7UC9C~C7VC7C7C9C7C7C~C9C~C7C7C9C7C9C~C7C9C7C9C9C~C7C~C7C7C7
~ ~Car.Cr.~FCU r.~r.Caa r.~aC7aa aa aC7rcaV aa ~CUU FCa aa~Caa
U UU UUU UU UU UUU UU UU UUU UU UU UUU UU UUU UU .~
E~C7r.~U C~C7a a~a FCU'UaU ~U U'a r.~C7U C~C7UU C7UU aa UUU UU cd
U ~ ~ ~ ~ ~ U
U rCU'7~C UFC~D U'~U UU US ~CU CFCb~ CFCCV'JD5 CU7FC~ Ua
C 'J .~ .7 .7
N
cQ
z 'V
h ODQ1On-1N Mc~tnlOI~OD01O~-INM cytllOl~OD01O e-1NM V~u~l0f~OO6~O
D O OO r-1.-irir-1.-1v-irlr-I'-I,--INN NN NNN NN NM MMM MM MMM MV~
"'~ODWODODODGDOD00f0ODOD0000OD07ODODODODODWN dDOD00ODaDODOD000000OD00"fl
w cv
~ ~ca ~a~ a~ aa ~a~ ~~ ~a ~~~ ~~ ~~ a~~ a~ a~a ~~
a aa aaa aa aa aaa aa aa aaa aa aa aaa aa aaa aa ~
c~~c~c~ ~c~ c~c~c~ csc~c~c~c~ c~ c~
~ c~c~c7 c~~ c~ c~c~c~c~ cs c~c~c~c~ c~c~c~..
a aa aaa aa aa aaa aa aa aaa aa aa aaa aa aoa aa p
U UU VUU UU UU UUU UU UU UUU UU UU UUU UU UUU UU
U UU UUU VU UU UUU UU UU UUU UU UU UUU UU UUV UU .~
a aa aaa aa aa aaa aa aa aaa aa aa aaa aa aaa aa
a aa aaa a
a aa aaa aa aa aaa aa aa aaa aa aaa aa
"""
U UU UUU UU UU UUU UU UU UVU UU UU UUU UU UUU UU
a aa aaa aa aa aaa aa aa aaa aa aa aaa aa aaa aa .~
~ c~ c~c~~ c~ c~ c~c~ c~c~c~c~H
~ x xx xxx xx xx xxx xx xx xxx xx xx xxx xx xxx xx
~ ~ a~ ~a~c~~ ~~ ~~~c~~ ~a aa~ ~~ a~ ~~~ ~~ ~a~ ~~
v W
a
d c~c~c~c~~c~~c~c~ c~ c~c~ c~c~ c~c~c~ ~c~ c~c~:
~ ~ ~~ ~c~~ ~~ ~a ~~~ ~~ ~~ a~~ ~~ ~a a~a ~ca a~~ a~
~ c~c~ cn c~ c~c~ c~c~~ c~c~c~ c~ c~c~c~c~ c~ c~
' ~ ~a ~~a ~~ ~~c~a~ ~a ~~ ~a~ ~~ ~ca ~~~ ~~ ~a~ a~ .
~
O U UU UUU UU UU UUU UU UU UUU UU UU UUU UU UUU UU
.OU UU UUU UU UU UUU UU UU UUU UU UU UUU UU UUU UU
'~r.CFC~ FCGCr~C~~CFCr~Cr.C~CFCr.CFCFCr~CFCr.C~CrL~ r.C~
FCr.CFCFCFCFC~C~C~C~C~c~
C7aU Ur.~U C~a ~C~CU~CC7C7t~U'r.~FCUC7Ua r.~r.~UaU FCFCUa C9r~x",
U UC7aUa r.~U C~U aaU'aU r.~U UDC9aU VU C7UU'U'U'U'r.~~ U'U
U aC7UUr.~a~CaU C7r.~C7aV'Ur.~aUC7UU C7C7UC7C7~Cr.CC7UC7C7C7
C~C7C7UVC7C7C9C~C9C7C7t7UC7C~C7C7C7C7C~C9C'JU'C7C7U t9C7C7C~C7C7C74a
C',
C~C7U'U'U'C9C7U'U'U'C7C9C9U'C7C7C7C7C7U'C7C7U'C7C7t'lC9C7C7U'C9C7C7U'O
_
U'C7C7Ur.CGCU U ~C7C7C9U FC~ ~CU'U'UV ~C~CU'~U'U'U'UFCC9sCU ~,O"
U a raU 7rL~~CD9$ C9C UU '' ' C '' '
~
~ . C Cr r UUC7Ua Ua U a r.FCUU UFC
C7U C7UC7C7C7aU rCC7U aC7aU'a~C~CaU C7U'U'UD C7U FCa U'U
C7UU aUa a~CC7a U'Ua ar.~C7C7aC7U a~ C~FCU~CU C7a UUC~aCC7~.,
U'UU UrU Ua a C ' '
~
. C9 UU UU r.FCV U C7 FCU VU ar.~C~V~CC~C9
a U'a UUU ~Cr.~UU'~aU'Ua UU'r.~~U'UU aa ~U'U'C7U U'aU U'r.~
N
H
N
r
O
0
o~M~cao~o~M~r~a~v~v~.a~aown a~c~o~or rnM r~o rr Nmr ,~r
tAN M1~OD~-iN WO riM V~hCOON MM Mv01~OlO1~0001ON v'C1OOt0v-1M iw
V~c~N t(7t0l0~O101~I~1'~1~f~OD00ODOD00ODCO6101O10101OO OO r-1rlr1NM Q,
" ri,-1'-i'-1r-1ri.-~rl.~.-1r-1rl.-I.-in-Iri.-1'-1~i~-i~-I.-1.-I.-IrlNN NN NNN
NN
N
a3
C
CA 02288640 1999-11-OS
WO 98/50530 165 PCT/US98/09249
Table XVI: Hammerhead Ribozyme Sites for B-raf
nt, SEQ SEQ
Position Ribozyme ID. Substrate ID.
No. No.
17 CGGGCGGGCUGAUGAGXCGAA AGGGGGCC1913 GGCCCCCUCCCCGCCCG2354
38 CGGGGCCCCUGAUGAGXCGAA AGCGGCCG1919 CGGCCGCUCGGGCCCCG2355
50 AUAACCGACUGAUGAGXCGAA AGCCGGGG1915 CCCCGGCUCUCGGUUAU2356
52 UUAUAACCCUGAUGAGXCGAA AGAGCCGG1916 CCGGCUCUCGGUUAUAA2357
56 CAUCUUAUCUGAUGAGXCGAA ACCGAGAG1917 CUCUCGGUUAUAAGAUG2358
57 CCAUCUUACUGAUGAGXCGAA AACCGAGA1918 UCUCGGUUAUAAGAUGG2359
59 CGCCAUCUCUGAUGAGXCGAA AUAACCGA1919 UCGGUUAUAAGAUGGCG2360
113 GUUGAACACUGAUGAGXCGAA AGCCUGGC1920 GCCAGGCUCUGUUCAAC2361
117 CCCCGUUGCUGAUGAGXCGAA ACAGAGCC1921 GGCUCUGUUCAACGGGG2362
118 UCCCCGUUCUGAUGAGXCGAA AACAGAGC1922 GCUCUGUUCAACGGGGA2363
165 CAGCCGAACUGAUGAGXCGAA AGGCCGCG1923 CGCGGCCUCUUCGGCUG2364
167 CGCAGCCGCUGAUGAGXCGAA AGAGGCCG1924 CGGCCUCUUCGGCUGCG2365
168 CCGCAGCCCUGAUGAGXCGAA AAGAGGCC1925 GGCCUCUUCGGCUGCGG2366
187 UCCUCCGGCUGAUGAGXCGAA AUGGCAGG1926 CCUGCCAUUCCGGAGGA2367
188 CUCCUCCGCUGAUGAGXCGAA AAUGGCAG1927 CUGCCAUUCCGGAGGAG2368
206 UUGUUUGACUGAUGAGXCGAA AUUCCACA1928 UGUGGAAUAUCAAACAA2369
208 AUUUGUUUCUGAUGAGXCGAA AUAUUCCA1929 UGGAAUAUCAAACAAAU2370
220 GUCAACUUCUGAUGAGXCGAA AUCAUUUG1930 CAAAUGAUUAAGUUGAC2371
221 UGUCAACUCUGAUGAGXCGAA AAUCAUUU1931 AAAUGAUUAAGUUGACA2372
225 CCUGUGUCCUGAUGAGXCGAA ACUUAAUC1932 GAUUAAGUUGACACAGG2373
239 GGCCUCUACUGAUGAGXCGAA AUGUUCCU1933 AGGAACAUAUAGAGGCC2374
241 AGGGCCUCCUGAUGAGXCGAA AUAUGUUC1939 GAACAUAUAGAGGCCCU2375
250 UUGUCCAACUGAUGAGXCGAA AGGGCCUC1935 GAGGCCCUAUUGGACAA2376
252 AUUUGUCCCUGAUGAGXCGAA AUAGGGCC1936 GGCCCUAUUGGACAAAU2377
261 CCCCACCACUGAUGAGXCGAA AUUUGUCC1937 GGACAAAUUUGGUGGGG2378
262 UCCCCACCCUGAUGAGXCGAA AAUUUGUC1938 GACAAAUUUGGUGGGGA2379
275 UGGUGGAUCUGAUGAGXCGAA AUGCUCCC1939 GGGAGCAUAAUCCACCA2380
278 UGAUGGUGCUGAUGAGXCGAA AUUAUGCU1940 AGCAUAAUCCACCAUCA2381
285 GAUAUAUUCUGAUGAGXCGAA AUGGUGGA1941 UCCACCAUCAAUAUAUC2382
289 UCCAGAUACUGAUGAGXCGAA AUUGAUGG1942 CCAUCAAUAUAUCUGGA2383
291 CCUCCAGACUGAUGAGXCGAA AUAUUGAU1943 AUCAAUAUAUCUGGAGG2389
293 GGCCUCCACUGAUGAGXCGAA AUAUAUUG1944 CAAUAUAUCUGGAGGCC2385
303 AUUCUUCACUGAUGAGXCGAA AGGCCUCC1945 GGAGGCCUAUGAAGAAU2386
312 UGCUGGUGCUGAUGAGXCGAA AUUCUUCA1996 UGAAGAAUACACCAGCA2387
325 AGUGCAUCCUGAUGAGXCGAA AGCUUGCU1947 AGCAAGCUAGAUGCACU2388
339 CUUUGUUGCUGAUGAGXCGAA AGUGCAUC1948 GAUGCACUCCAACAAAG2389
354 AUUCCAAUCUGAUGAGXCGAA ACUGUUGU1949 ACAACAGUUAUUGGAAU2390
355 GAUUCCAACUGAUGAGXCGAA AACUGUUG1950 CAACAGUUAUUGGAAUC2391
357 GAGAUUCCCUGAUGAGXCGAA AUAACUGU1951 ACAGUUAUUGGAAUCUC2392
363 UCCCCAGACUGAUGAGXCGAA AUUCCAAU1952 AUUGGAAUCUCUGGGGA2393
365 GUUCCCCACUGAUGAGXCGAA AGAUUCCA1953 UGGAAUCUCUGGGGAAC2394
383 AACAGAAACUGAUGAGXCGAA AUCAGUUC1954 GAACUGAUUUUUCUGUU2395
3B4 AAACAGAACUGAUGAGXCGAA AAUCAGUU1955 AACUGAUUUUUCUGUUU2396
385 GAAACAGACUGAUGAGXCGAA AAAUCAGU1956 ACUGAUUUUUCUGUUUC2397
386 AGAAACAGCUGAUGAGXCGAA AAAAUCAG195? CUGAUUUUUCUGUUUCU2398
387 UAGAAACACUGAUGAGXCGAA AAAAAUCA1958 UGAUUUUUCUGUUUCUA2399
391 GAGCUAGACUGAUGAGXCGAA ACAGAAAA1959 UUUUCUGUUUCUAGCUC2400
392 AGAGCUAGCUGAUGAGXCGAA AACAGAAA1960 UUUCUGUUUCUAGCUCU2401
393 CAGAGCUACUGAUGAGXCGAA AAACAGAA1961 UUCUGUUUCUAGCUCUG2402
395 UGCAGAGCCUGAUGAGXCGAA AGAAACAG1962 CUGUUUCUAGCUCUGCA2403
399 UUGAUGCACUGAUGAGXCGAA AGCUAGAA1963 UUCUAGCUCUGCAUCAA2409
405 UAUCCAUUCUGAUGAGXCGAA AUGCAGAG1964 CUCUGCAUCAAUGGAUA2905
413 UGUAACGGCUGAUGAGXCGAA AUCCAUUG1965 CAAUGGAUACCGUUACA2406
CA 02288640 1999-11-OS
WO 98/50530 166 PCT/IJS98/09249
nt. SEQ SEQ
Position Ribozyme ID. Substrate ID.
No. No.
418 GAAGAUGUCUGAUGAGX CGAA ACGGUAUC1966 _ ACAUCUUC2907
GAUACCGUU
419 AGAAGAUGCUGAUGAGX CGAA AACGGUAU1967 AUACCGUUACAUCUUCU2908
923 AGGAAGAACUGAUGAGX CGAA AUGUAACG1968 CGUUACAUCUUCUUCCU2409
425 AGAGGAAGCUGAUGAGX CGAA AGAUGUAA1969 UUACAUCUUCUUCCUCU2410
926 AAGAGGAACUGAUGAGX CGAA AAGAUGUA1970 UACAUCUUCUUCCUCUU2411
428 AGAAGAGGCUGAUGAGX CGAA AGAAGAUG1971 CAUCUUCUUCCUCUUCU2412
429 UAGAAGAGCUGAUGAGX CGAA AAGAAGAU1972 AUCUUCUUCCUCUUCUA2913
932 GGCUAGAACUGAUGAGX CGAA AGGAAGAA1973 UUCUUCCUCUUCUAGCC2414
439 AAGGCUAGCUGAUGAGX CGAA AGAGGAAG1974 CUUCCUCUUCUAGCCUU2415
935 AAAGGCUACUGAUGAGX CGAA AAGAGGAA1975 UUCCUCUUCUAGCCUUU2416
437 UGAAAGGCCUGAUGAGX CGAA AGAAGAGG1976 CCUCUUCUAGCCUUUCA2417
942 AGCACUGACUGAUGAGX CGAA AGGCUAGA1977 UCUAGCCUUUCAGUGCU2918
943 UAGCACUGCUGAUGAGX CGAA AAGGCUAG1978 CUAGCCUUUCAGUGCUA2919
944 GUAGCACUCUGAUGAGX CGAA AAAGGCUA1979 UAGCCUUUCAGUGCUAC2920
451 GAUGAAGGCUGAUGAGX CGAA AGCACUGA1980 UCAGUGCUACCUUCAUC2421
455 AAGAGAUGCUGAUGAGX CGAA AGGUAGCA1981 UGCUACCUUCAUCUCUU2422
456 AAAGAGAUCUGAUGAGX CGAA AAGGUAGC1982 GCUACCUUCAUCUCUUU2423
459 CUGAAAGACUGAUGAGX CGAA AUGAAGGU1983 ACCUUCAUCUCUUUCAG2424
461 AACUGAAACUGAUGAGX CGAA AGAUGAAG1984 CUUCAUCUCUUUCAGUU2425
463 AAAACUGACUGAUGAGX CGAA AGAGAUGA1985 UCAUCUCUUUCAGUUUU2426
969 AAAAACUGCUGAUGAGX CGAA AAGAGAUG1986 CAUCUCUUUCAGUUUUU2927
965 GAAAAACUCUGAUGAGX CGAA AAAGAGAU1987 AUCUCUUUCAGUUUUUC2928
969 UUUUGAAACUGAUGAGX CGAA ACUGAAAG1988 CUUUCAGUUUUUCAAAA2429
970 AUUUUGAACUGAUGAGX CGAA AACUGAAA1989 UUUCAGUUUUUCAAAAU2430
471 GAUUUUGACUGAUGAGX CGAA AAACUGAA1990 UUCAGUUUUUCAAAAUC2431
472 GGAUUUUGCUGAUGAGX CGAA AAAACUGA1991-UCAGUUUUUCAAAAUCC2432
473 GGGAUUUUCUGAUGAGX CGAA AAAAACUG1992 CAGUUUUUCAAAAUCCC2433
979 AUCUGUGGCUGAUGAGX CGAA AUUUUGAA1993 UUCAAAAUCCCACAGAU2439
510 UUUGUGGUCUGAUGAGX CGAA ACUUGGGG1994 CCCCAAGUCACCACAAA2435
524 UCUAACGACUGAUGAGX CGAA AGGUUUUU1995 AAAAACCUAUCGUUAGA2436
526 ACUCUAACCUGAUGAGX CGAA AUAGGUUU1996 AAACCUAUCGUUAGAGU2937
529 AAGACUCUCUGAUGAGX CGAA ACGAUAGG1997 CCUAUCGUUAGAGUCUU2438
530 GAAGACUCCUGAUGAGX CGAA AACGAUAG1998 CUAUCGUUAGAGUCUUC2439
535 GGCAGGAACUGAUGAGX CGAA ACUCUAAC1999 GUUAGAGUCUUCCUGCC2440
537 UGGGCAGGCUGAUGAGX CGAA AGACUCUA2000 UAGAGUCUUCCUGCCCA2441
538 UUGGGCAGCUGAUGAGX CGAA AAGACUCU2001 AGAGUCUUCCUGCCCAA2942
565 CUUGCAGGCUGAUGAGX CGAA ACCACUGU2002 ACAGUGGUACCUGCAAG2443
583 CGGACUGUCUGAUGAGX CGAA ACUCCACA2003 UGUGGAGUUACAGUCCG2444
584 UCGGACUGCUGAUGAGX CGAA AACUCCAC2004 GUGGAGUUACAGUCCGA2445
589 CUGUCUCGCUGAUGAGX CGAA ACUGUAAC2005 GUUACAGUCCGAGACAG2446
599 UUUCUUUACUGAUGAGX CGAA ACUGUCUC2006 GAGACAGUCUAAAGAAA2447
601 GCUUUCUUCUGAUGAGX CGAA AGACUGUC2007 GACAGUCUA 2448
AAGAAAGC
626 UGGGAUUACUGAUGAGX CGAA ACCUCUCA2008 UGAGAGGUCUAAUCCCA2449
628 UCUGGGAUCUGAUGAGX CGAA AGACCUCU2009 AGAGGUCUAAUCCCAGA2450
631 CACUCUGGCUGAUGAGX CGAA AUUAGACC2010 GGUCUAAUCCCAGAGUG2951
699 AUUCUGUACUGAUGAGX CGAA ACAGCACA2011 UGUGCUGUUUACAGAAU2452
650 AAUUCUGUCUGAUGAGX CGAA AACAGCAC2012 GUGCUGUUUACAGAAUU2453
651 GAAUUCUGCUGAUGAGX CGAA AAACAGCA2013 UGCUGUUUACAGAAUUC2954
658 CCAUCCUGCUGAUGAGX CGAA AUUCUGUA2014 UACAGAAUUCAGGAUGG2955
659 UCCAUCCUCUGAUGAGX CGAA AAUUCUGU2015 ACAGAAUUCAGGAUGGA2956
682 UCCCAACCCUGAUGAGX CGAA AUUGGUUU2016 AAACCAAUUGGUUGGGA2957
686 AGUGUCCCCUGAUGAGX CGAA ACCAAUUG2017 CAAUUGGUUGGGACACU2958
698 CCAGGAAACUGAUGAGX CGAA AUCAGUGU2018 ACACUGAUAUUUCCUGG2959
700 AGCCAGGACUGAUGAGX CGAA AUAUCAGU2019 ACUGAUAUUUCCUGGCU2960
L 701 AAGCCAGGCUGAUGAGX CGAA AAUAUCA~2020 CUGAUAUUUCCUGGCUU2461
~ j
CA 02288640 1999-11-OS
WO 98/50530 16~ PCT/US98/09249
nt. SEQ SEQ
Position Ribozyme ID. Subs ID.
No. tr No.
ate
702 UAAGCCAGCUGAUGAGX CGAA AAAUAUCA2021 _ _ 2462
UGAUAUUUCCUGGCUUA
709 UCUCCAGUCUGAUGAGX CGAA AGCCAGGA2022 UCCUGGCUUACUGGAGA2463
710 UUCUCCAGCUGAUGAGX CGAA AAGCCAGG2023 CCUGGCUUACUGGAGAA2464
723 CCACAUGCCUGAUGAGX CGAA AUUCUUCU2029 AGAAGAAUUGCAUGUGG2465
738 CAUUCUCCCUGAUGAGX CGAA ACACUUCC2025 GGAAGUGUUGGAGAAUG2466
798 GUAAGUGGCUGAUGAGX CGAA ACAUUCUC2026 GAGAAUGUUCCACUUAC2467
799 UGUAAGUGCUGAUGAGX CGAA AACAUUCU2027 AGAAUGUUCCACUUACA2468
759 UGUGUUGUCUGAUGAGX CGAA AGUGGAAC2028 GUUCCACUUACAACACA2469
755 GUGUGUUGCUGAUGAGX CGAA AAGUGGAA2029 UUCCACUUACAACACAC'2470
768 UUCGUACACUGAUGAGX CGAA AGUUGUGU2030 ACACAACUUUGUACGAA2971
769 UUUCGUACCUGAUGAGX CGAA AAGUUGUG2031 CACAACUUUGUACGAAA2472
772 GUUUUUCGCUGAUGAGX CGAA ACAAAGUU2032 AACUUUGUACGAAAAAC2473
783 AGGUGAAACUGAUGAGX CGAA ACGUUUUU2033 AAAAACGUUUUUCACCU2974
789 AAGGUGAACUGAUGAGX CGAA AACGUUUU2034 AAAACGUUUUUCACCUU2475
785 UAAGGUGACUGAUGAGX CGAA AAACGUUU2035 AAACGUUUUUCACCUUA2476
786 CUAAGGUGCUGAUGAGX CGAA AAAACGUU2036 AACGUUUUUCACCUUAG2477
787 GCUAAGGUCUGAUGAGX CGAA AAAAACGU2037 ACGUUUUUCACCUUAGC2478
792 AAAAUGCUCUGAUGAGX CGAA AGGUGAAA2038 UUUCACCUUAGCAUUUU2479
793 CAAAAUGCCUGAUGAGX CGAA AAGGUGAA2039 UUCACCUUAGCAUUUUG2980
798 AGUCACAACUGAUGAGX CGAA AUGCUAAG2040 CUUAGCAUUUUGUGACU2481
799 AAGUCACACUGAUGAGX CGAA AAUGCUAA2041 UUAGCAUUUUGUGACUU2482
800 AAAGUCACCUGAUGAGX CGAA AAAUGCUA2042 UAGCAUUUUGUGACUUU2483
807 UUCGACAACUGAUGAGX CGAA AGUCACAA2043 UUGUGACUUUUGUCGAA2484
808 UUUCGACACUGAUGAGX CGAA AAGUCACA2044 UGUGACUUUUGUCGAAA2485
809 CUUUCGACCUGAUGAGX CGAA AAAGUCAC2045 GUGACUUUUGUCGAAAG2486
812 CAGCUUUCCUGAUGAGX CGAA ACAAAAGU2046 ACUUUUGUCGAAAGCUG2987
823 CCCUGGAACUGAUGAGX CGAA AGCAGCUU2047 AAGCUGCUUUUCCAGGG2488
829 ACCCUGGACUGAUGAGX CGAA AAGCAGCU2048 AGCUGCUUUUCCAGGGU2489
825 AACCCUGGCUGAUGAGX CGAA AAAGCAGC2049 GCUGCUUUUCCAGGGUU2490
826 AAACCCUGCUGAUGAGX CGAA AAAAGCAG2050 CUGCUUUUCCAGGGUUU2491
833 ACAGCGGACUGAUGAGX CGAA ACCCUGGA2051 UCCAGGGUUUCCGCUGU2492
834 GACAGCGGCUGAUGAGX CGAA AACCCUGG2052 CCAGGGUUUCCGCUGUC2493
835 UGACAGCGCUGAUGAGX CGAA AAACCCUG2053 CAGGGUUUCCGCUGUCA2494
842 ACAUGUUUCUGAUGAGX CGAA ACAGCGGA2059 UCCGCUGUCAAACAUGU2495
859 AAAUUUAUCUGAUGAGX CGAA ACCACAUG2055 CAUGUGGUUAUAAAUUU2496
855 GAAAUUUACUGAUGAGX CGAA AACCACAU2056 AUGUGGUUAUAAAUUUC2497
857 GUGAAAUUCUGAUGAGX CGAA AUAACCAC2057 GUGGUUAUA 2498
AAUUUCAC
861 GCUGGUGACUGAUGAGX CGAA AUUUAUAA2058 UUAUAAAUUUCACCAGC2499
862 CGCUGGUGCUGAUGAGX CGAA AAUUUAUA2059 UAUAAAUUUCACCAGCG2500
863 ACGCUGGUCUGAUGAGX CGAA AAAUUUAU2060 AUAAAUUUCACCAGCGU2501
872 UGUACUACCUGAUGAGX CGAA ACGCUGGU2061 ACCAGCGUUGUAGUACA__
2502
875 UUCUGUACCUGAUGAGX CGAA ACAACGCU2062 AGCGUUGUAGUACAGAA2503
878 AACUUCUGCUGAUGAGX CGAA ACUACAAC2063 GUUGUAGUACAGAAGUU2504
886 AUCAGUGGCUGAUGAGX CGAA ACUUCUGU2069 ACAGAAGUUCCACUGAU2505
887 CAUCAGUGCUGAUGAGX CGAA AACUUCUG2065 CAGAAGUUCCACUGAUG2506
901 UCAUAAUUCUGAUGAGX CGAA ACACACAU2066 AUGUGUGUUAAUUAUGA2507
902 GUCAUAAUCUGAUGAGX CGAA AACACACA2067 UGUGUGUUAAUUAUGAC2508
905 UUGGUCAUCUGAUGAGX CGAA AUUAACAC2068 GUGUUAAUUAUGACCAA2509
906 GUUGGUCACUGAUGAGX CGAA AAUUAACA2069 UGUUAAUUAUGACCAAC2510
916 AGCAAAUCCUGAUGAGX CGAA AGUUGGUC2070 GACCAACUUGAUUUGCU2511
920 AAACAGCACUGAUGAGX CGAA AUCAAGUU2071 AACUUGAUUUGCUGUUU2512
921 CAAACAGCCUGAUGAGX CGAA AAUCAAGU2072 ACUUGAUUUGCUGUUUG2513
927 UGGAGACACUGAUGAGX CGAA ACAGCAAA2073 UUUGCUGUUUGUCUCCA2514
928 UUGGAGACCUGAUGAGX CGAA AACAGCAA2079 UUGCUGUUUGUCUCCAA2515
931 AACUUGGACUGAUGAGX CGAA ACAAACAG2075 CUGUUUGUCUCCAAGUU2516
CA 02288640 1999-11-05
WO 98/50530 16g PCT/US98/09249
nt. SEQ SEQ
Position Ribozyme ID. Substrate ID.
No. No.
933 AGAACUUGCUGAUGAGX CGAA AGACAAAC2076 GUUUGUCUCCAAGUUCU2517
939 GUUCAAAGCUGAUGAGX CGAA ACUUGGAG2077 CUCCAAGUUCUUUGAAC2518
940 UGUUCAAACUGAUGAGX CGAA AACUUGGA2078 UCCAAGUUCUUUGAACA2519
942 GGUGUUCACUGAUGAGX CGAA AGAACUUG2079 CAAGUUCUUUGAACACC2520
943 UGGUGUUCCUGAUGAGX CGAA AAGAACUU2080 AAGUUCUUUGAACACCA2521
958 UCCUGUGGCUGAUGAGX CGAA AUUGGGUG2081 CACCCAAUACCACAGGA2522
975 CUGCUAAGCUGAUGAGX CGAA ACGCCUCU2082 AGAGGCGUCCUUAGCAG2523
978 UCUCUGCUCUGAUGAGX CGAA AGGACGCC2083 GGCGUCCUUAGCAGAGA2524
979 GUCUCUGCCUGAUGAGX CGAA AAGGACGC2084 GCGUCCUUAGCAGAGAC'2525
999 CCAGAUGUCUGAUGAGX CGAA AGGGCAGU2085 ACUGCCCUAACAUCUGG2526
999 AUGAUCCACUGAUGAGX CGAA AUGUUAGG2086 CCUAACAUCUGGAUCAU2527
1005 AAGGGGAUCUGAUGAGX CGAA AUCCAGAU2087 AUCUGGAUCAUCCCCUU2528
1008 CGGAAGGGCUGAUGAGX CGAA AUGAUCCA2088 UGGAUCAUCCCCUUCCG2529
1013 GGGUGCGGCUGAUGAGX CGAA AGGGGAUG2089 CAUCCCCUUCCGCACCC2530
1014 CGGGUGCGCUGAUGAGX CGAA AAGGGGAU2090 AUCCCCUUCCGCACCCG2531
1026 UAGAGUCCCUGAUGAGX CGAA AGGCGGGU2091 ACCCGCCUCGGACUCUA2532
1032 GCCCAAUACUGAUGAGX CGAA AGUCCGAG2092 CUCGGACUCUAUUGGGC2533
1039 GGGCCCAACUGAUGAGX CGAA AGAGUCCG2093 CGGACUCUAUUGGGCCC2539
1036 UGGGGCCCCUGAUGAGX CGAA AUAGAGUC2094 GACUCUAUUGGGCCCCA2535
1048 CUGGUGAGCUGAUGAGX CGAA AUUUGGGG2095 CCCCAAAUUCUCACCAG2536
1049 ACUGGUGACUGAUGAGX CGAA AAUUUGGG2096 CCCAAAUUCUCACCAGU2537
1051 GGACUGGUCUGAUGAGX CGAA AGAAUUUG2097 CAAAUUCUCACCAGUCC2538
1058 AGGAGACGCUGAUGAGX CGAA ACUGGUGA2098 UCACCAGUCCGUCUCCU2539
1062 UUGAAGGACUGAUGAGX CGAA ACGGACUG2099 CAGUCCGUCUCCUUCAA2540
1064 UUUUGAAGCUGAUGAGX CGAA AGACGGAC2100 GUCCGUCUCCUUCAAAA2541
1067 GGAUUUUGCUGAUGAGX CGAA AGGAGACG2101 CGUCUCCUUCAAAAUCC2592
1068 UGGAUUUUCUGAUGAGX CGAA AAGGAGAC2102 GUCUCCUUCAAAAUCCA2543
1074 UUGGAAUGCUGAUGAGX CGAA AUUUUGAA2103 UUCAAAAUCCAUUCCAA2544
1078 GGAAUUGGCUGAUGAGX CGAA AUGGAUUU2104 AAAUCCAUUCCAAUUCC2545
1079 UGGAAUUGCUGAUGAGX CGAA AAUGGAUU2105 AAUCCAUUCCAAUUCCA2546
1084 GGCUGUGGCUGAUGAGX CGAA AUUGGAAU2106 AUUCCAAUUCCACAGCC2547
1085 GGGCUGUGCUGAUGAGX CGAA AAUUGGAA2107 UUCCAAUUCCACAGCCC2548
1095 CUGGUCGGCUGAUGAGX CGAA AGGGCUGU2108 ACAGCCCUUCCGACCAG2549
1096 GCUGGUCGCUGAUGAGX CGAA AAGGGCUG2109 CAGCCCUUCCGACCAGC2550
1115 AUUUCGAUCUGAUGAGX CGAA AUCUUCAU2110 AUGAAGAUCAUCGAAAU2551
1118 UUGAUUUCCUGAUGAGX CGAA AUGAUCUU2111 AAGAUCAUCGAAAUCAA2552
1129 CCCAAAUUCUGAUGAGX CGAA AUUUCGAU2112 AUCGAAAUCAAUUUGGG2553
1128 GUUGCCCACUGAUGAGX CGAA AUUGAUUU2113 AAAUCAAUUUGGGCAAC2559
1129 CGUUGCCCCUGAUGAGX CGAA AAUUGAUU2114 AAUCAAUUUGGGCAACG2555
1196 CUGAUGAGCUGAUGAGX CGAA AUCGGUCU2115 AGACCGAUCCUCAUCAG2556
1149 GAGCUGAUCUGAUGAGX CGAA AGGAUCGG2116 CCGAUCCUCAUCAGCUC2557
1152 UGGGAGCUCUGAUGAGX CGAA AUGAGGAU2117 AUCCUCAUCAGCUCCCA2558
1157 CACAUUGGCUGAUGAGX CGAA AGCUGAUG2118 CAUCAGCUCCCAAUGUG2559
1169 UGUGUUUACUGAUGAGX CGAA AUGCACAU2119 AUGUGCAUAUAAACACA2560
1171 AUUGUGUUCUGAUGAGX CGAA AUAUGCAC2120 GUGCAUAUA 2561
AACACAAU
1180 ACAGGUUCCUGAUGAGX CGAA AUUGUGUU2121 AACACAAUAGAACCUGU2562
1189 UCAAUAUUCUGAUGAGX CGAA ACAGGUUC2122 GAACCUGUCAAUAUUGA2563
1193 GUCAUCAACUGAUGAGX CGAA AUUGACAG2123 CUGUCAAUAUUGAUGAC2564
1195 AAGUCAUCCUGAUGAGX CGAA AUAUUGAC2124 GUCAAUAUUGAUGACUU2565
1203 CUCUAAUCCUGAUGAGX CGAA AGUCAUCA2125 UGAUGACUUGAUUAGAG2566
1207 UGGUCUCUCUGAUGAGX CGAA AUCAAGUC2126 GACUUGAUUAGAGACCA2567
1208 UUGGUCUCCUGAUGAGX CGAA AAUCAAGU2127 ACUUGAUUAGAGACCAA2568
1221 CACCACGACUGAUGAGX CGAA AUCCUUGG2128 CCAAGGAUUUCGUGGUG2569
1222 UCACCACGCUGAUGAGX CGAA AAUCCUUG2129 CAAGGAUUUCGUGGUGA2570
1223 AUCACCACCUGAUGAGX CGAA AAAUCCUU2130 AAGGAUUUCGUGGUGAU2571
CA 02288640 1999-11-OS
WO 98/50530 169 PCT/US98/09249
nt. SEQ .,_,._,-_ ~_gEQ
Position Ribo ID. Sub ID.
zyme No. str No.
ate
1239 CUGUGGUU_ XCGAA AUCCUCCA2131 _ _ 2572
CUGAUGAG UGGAGGAUC__ _
~AACCACAG
1250 AGCAGACACUGAUGAGXCGAA ACCUGUGG2132 CCACAGGUUUGUCUGCU2573
1251 UAGCAGACCUGAUGAGXCGAA AACCUGUG2133 CACAGGUUUGUCUGCUA2574
1254 GGGUAGCACUGAUGAGXCGAA ACAAACCU2134 AGGUUUGUCUGCUACCC2575
1259 AGGGGGGGCUGAUGAGXCGAA AGCAGACA2135 UGUCUGCUACCCCCCCU2576
1272 CAGGUAAUCUGAUGAGXCGAA AGGCAGGG2136 CCCUGCCUCAUUACCUG2577
1275 AGCCAGGUCUGAUGAGXCGAA AUGAGGCA2137 UGCCUCAUUACCUGGCU2578
1276 GAGCCAGGCUGAUGAGXCGAA AAUGAGGC2138 GCCUCAUUACCUGGCUC2579
1284 UAGUUAGUCUGAUGAGXCGAA AGCCAGGU2139 ACCUGGCUCACUAACUA'2580
1288 ACGUUAGUCUGAUGAGXCGAA AGUGAGCC2190 GGCUCACUAACUAACGU2581
1292 UUUCACGUCUGAUGAGXCGAA AGUUAGUG2191 CACUAACUAACGUGAAA2582
1305 AUUUCUGUCUGAUGAGXCGAA AGGCUUUC2192 GAAAGCCUUACAGAAAU2583
1306 GAUUUCUGCUGAUGAGXCGAA AAGGCUUU2143 AAAGCCUUACAGAAAUC2584
1314 GUCCUGGACUGAUGAGXCGAA AUUUCUGU2194 ACAGAAAUCUCCAGGAC2585
1316 AGGUCCUGCUGAUGAGXCGAA AGAUUUCU2145 AGAAAUCUCCAGGACCU2586
1325 UUCUCGCUCUGAUGAGXCGAA AGGUCCUG2146 CAGGACCUCAGCGAGAA2587
1341 AUGAAGAUCUGAUGAGXCGAA ACUUCCUU2147 AAGGAAGUCAUCUUCAU2588
1349 AGGAUGAACUGAUGAGXCGAA AUGACUUC2148 GAAGUCAUCUUCAUCCU2589
1346 UGAGGAUGCUGAUGAGXCGAA AGAUGACU2149 AGUCAUCUUCAUCCUCA2590
1347 CUGAGGAUCUGAUGAGXCGAA AAGAUGAC2150 GUCAUCUUCAUCCUCAG2591
1350 CUUCUGAGCUGAUGAGXCGAA AUGAAGAU2151 AUCUUCAUCCUCAGAAG2592
1353 UGUCUUCUCUGAUGAGXCGAA AGGAUGAA2152 UUCAUCCUCAGAAGACA2593
1367 UUUCAUUCCUGAUGAGXCGAA AUUCCUGU2153 ACAGGAAUCGAAUGAAA2594
1381 CGUCUACCCUGAUGAGXCGAA AGUGUUUU2154 AAAACACUUGGUAGACG2595
1385 GUCCCGUCCUGAUGAGXCGAA ACCAAGUG2155 CACUUGGUAGACGGGAC2596
1395 CAUCACUCCUGAUGAGXCGAA AGUCCCGU2156 ACGGGACUCGAGUGAUG2597
1406 AAUCUCCCCUGAUGAGXCGAA AUCAUCAC2157 GUGAUGAUUGGGAGAUU2598
1914 CCAUCAGGCUGAUGAGXCGAA AUCUCCCA2158 UGGGAGAUUCCUGAUGG2599
1915 CCCAUCAGCUGAUGAGXCGAA AAUCUCCC2159 GGGAGAUUCCUGAUGGG2600
1929 CCCACUGUCUGAUGAGXCGAA AUCUGCCC2160 GGGCAGAUUACAGUGGG2601
1930 UCCCACUGCUGAUGAGXCGAA AAUCUGCC2161 GGCAGAUUACAGUGGGA2602
1947 CCAGAUCCCUGAUGAGXCGAA AUUCUUUG2162 CAAAGAAUUGGAUCUGG2603
1452 AUGAUCCACUGAUGAGXCGAA AUCCAAUU2163 AAUUGGAUCUGGAUCAU2604
1458 UUCCAAAUCUGAUGAGXCGAA AUCCAGAU2164 AUCUGGAUCAUUUGGAA2605
1461 CUGUUCCACUGAUGAGXCGAA AUGAUCCA2165 UGGAUCAUUUGGAACAG2606
1462 ACUGUUCCCUGAUGAGXCGAA AAUGAUCC2166 GGAUCAUUUGGAACAGU2607
1471 CCCUUGUACUGAUGAGXCGAA ACUGUUCC2167 GGAACAGUCUACAAGGG2608
1473 UUCCCUUGCUGAUGAGXCGAA AGACUGUU2168 AACAGUCUACAAGGGAA2609
1512 UCACAUUCCUGAUGAGXCGAA ACAUUUUC2169 GAAAAUGUUGAAUGUGA2610
1529 CUGAGGUGCUGAUGAGXCGAA AGGUGCUG2170 CAGCACCUACACCUCAG2611
1535 UAACUGCUCUGAUGAGXCGAA AGGUGUAG2171 CUACACCUCAGCAGUUA2612
1542 AGGCUUGUCUGAUGAGXCGAA ACUGCUGA2172 UCAGCAGUUACAAGCCU2613
1543 AAGGCUUGCUGAUGAGXCGAA AACUGCUG2173 CAGCAGUUACAAGCCUU2614
1551 CAUUUUUGCUGAUGAGXCGAA AGGCUUGU2174 ACAAGCCUUCAAAAAUG2615
1552 UCAUUUUUCUGAUGAGXCGAA AAGGCUUG2175 CAAGCCUUCAAAAAUGA2616
1564 AGUACUCCCUGAUGAGXCGAA ACUUCAUU2176 AAUGAAGUAGGAGUACU2617
1570 UUCCUGAGCUGAUGAGXCGAA ACUCCUAC2177 GUAGGAGUACUCAGGAA2618
1573 GUUUUCCUCUGAUGAGXCGAA AGUACUCC2178 GGAGUACUCAGGAAAAC2619
1595 GAGUAGGACUGAUGAGXCGAA AUUCACAU2179 AUGUGAAUAUCCUACUC2620
1597 AAGAGUAGCUGAUGAGXCGAA AUAUUCAC2180 GUGAAUAUCCUACUCUU2621
1600 AUGAAGAGCUGAUGAGXCGAA AGGAUAUU2181 AAUAUCCUACUCUUCAU2622
1603 CCCAUGAACUGAUGAGXCGAA AGUAGGAU2182 AUCCUACUCUUCAUGGG2623
1605 AGCCCAUGCUGAUGAGXCGAA AGAGUAGG2183 CCUACUCUUCAUGGGCU2624
1606 UAGCCCAUCUGAUGAGXCGAA AAGAGUAG2189 CUACUCUUCAUGGGCUA2625
1614 UUGUGGAACUGAUGAGXCGAA AGCCCAUG2185 CAUGGGCUAUUCCACAA2626
CA 02288640 1999-11-OS
WO 98/50530 j ~~ PCT/US98/09249
nt. SEQ SEQ
Position Ribozyme ID. Substrate ID.
No. No.
1616 CUUUGUGGCUGAUGAGXCGAA AUAGCCCA2186 UGGGCUAUUCCACAAAG2627
1617 GCUUUGUGCUGAUGAGXCGAA AAUAGCCC2187 GGGCUAUUCCACAAAGC2628
1637 GGUAACAACUGAUGAGXCGAA AGCCAGUU2188 AACUGGCUAUUGUUACC2629
1639 UGGGUAACCUGAUGAGXCGAA AUAGCCAG2189 CUGGCUAUUGUUACCCA2630
1642 CACUGGGUCUGAUGAGXCGAA ACAAUAGC2190 GCUAUUGUUACCCAGUG2631
1693 CCACUGGGCUGAUGAGXCGAA AACAAUAG2191 CUAUUGUUACCCAGUGG2632
1662 ACAAGCUGCUGAUGAGXCGAA AGCCCUCA2192 UGAGGGCUCCAGCUUGU2633
1668 GGUGAUACCUGAUGAGXCGAA AGCUGGAG2193 CUCCAGCUUGUAUCACC2639
1671 GAUGGUGACUGAUGAGXCGAA ACAAGCUG2194 CAGCUUGUAUCACCAUC2635
1673 GAGAUGGUCUGAUGAGXCGAA AUACAAGC2195 GCUUGUAUCACCAUCUC2636
1679 GAUAUGGACUGAUGAGXCGAA AUGGUGAU2196 AUCACCAUCUCCAUAUC2637
1681 AUGAUAUGCUGAUGAGXCGAA AGAUGGUG2197 CACCAUCUCCAUAUCAU2638
1685 CUCAAUGACUGAUGAGXCGAA AUGGAGAU2198 AUCUCCAUAUCAUUGAG2639
1687 GUCUCAAUCUGAUGAGXCGAA AUAUGGAG2199 CUCCAUAUCAUVGAGAC2640
1690 UUGGUCUCCUGAUGAGXCGAA AUGAUAUG2200 CAUAUCAUUGAGACCAA2641
1701 UCAUCUCACUGAUGAGXCGAA AUUUGGUC2201 GACCAAAUUUGAGAUGA2642
1702 AUCAUCUCCUGAUGAGXCGAA AAUUUGGU2202 ACCAAAUUUGAGAUGAU2643
1711 AUAAGUUUCUGAUGAGXCGAA AUCAUCUC2203 GAGAUGAUCAAACUUAU2644
1717 AUAUCUAUCUGAUGAGXCGAA AGUUUGAU2204 AUCAAACUUAUAGAUAU2645
1718 AAUAUCUACUGAUGAGXCGAA AAGUUUGA2205 UCAAACUUAUAGAUAUU2646
1720 GCAAUAUCCUGAUGAGXCGAA AUAAGUUU2206 AAACUUAUAGAUAUUGC2647
1729 UCGUGCAACUGAUGAGXCGAA AUCUAUAA2207 UUAUAGAUAUUGCACGA2648
1726 UGUCGUGCCUGAUGAGXCGAA AUAUCUAU2208 AUAGAUAUUGCACGACA2649
1754 GUGUAAGUCUGAUGAGXCGAA AUCCAUGC2209 GCAUGGAUUACUUACAC2650
1755 CGUGUAAGCUGAUGAGXCGAA AAUCCAUG2210 CAUGGAUUACUUACACG2651
1758 UGGCGUGUCUGAUGAGXCGAA AGUAAUCC2211 GGAUUACUUACACGCCA2652
1759 UUGGCGUGCUGAUGAGXCGAA AAGUAAUC2212 GAUUACUUACACGCCAA2653
1770 GGAUGAUUCUGAUGAGXCGAA ACUUGGCG2213 CGCCAAGUCAAUCAUCC2659
1774 CUGUGGAUCUGAUGAGXCGAA AUUGACUU2214 AAGUCAAUCAUCCACAG2655
1777 UCUCUGUGCUGAUGAGXCGAA AUGAUUGA2215 UCAAUCAUCCACAGAGA2656
1789 UUACUCUUCUGAUGAGXCGAA AGGUCUCU2216 AGAGACCUCAAGAGUAA2657
1796 UAUAUUAUCUGAUGAGXCGAA ACUCUUGA2217 UCAAGAGUAAUAAUAUA2658
1799 AAAUAUAUCUGAUGAGXCGAA AUUACUCU2218 AGAGUAAUAAUAUAUUU2659
1802 AAGAAAUACUGAUGAGXCGAA AUUAUUAC2219 GUAAUAAUAUAUUUCUU2660
1804 UGAAGAAACUGAUGAGXCGAA AUAUUAUU2220 AAUAAUAUAUUUCUUCA2661
1806 CAUGAAGACUGAUGAGXCGAA AUAUAUUA2221 UAAUAUAUUUCUUCAUG2662
1807 UCAUGAAGCUGAUGAGXCGAA AAUAUAUU2222 AAUAUAUUUCUUCAUGA2663
1808 UUCAUGAACUGAUGAGXCGAA AAAUAUAU2223 AUAUAUUUCUUCAUGAA2664
1810 UCUUCAUGCUGAUGAGXCGAA AGAAAUAU2229 AUAUUUCUUCAUGAAGA2665
1811 GUCUUCAUCUGAUGAGXCGAA AAGAAAUA2225 UAUUUCUUCAUGAAGAC2666
1822 UUUACUGUCUGAUGAGXCGAA AGGUCUUC2226 GAAGACCUCACAGUAAA2667
1828 CCUAUUUUCUGAUGAGXCGAA ACUGUGAG2227 CUCACAGUA 2668
AAAAUAGG
1834 AAAUCACCCUGAUGAGXCGAA AUUUUUAC2228 GUAAAAAUAGGUGAUUU2669
1841 UAGACCAACUGAUGAGXCGAA AUCACCUA2229 UAGGUGAUUUUGGUCUA2670
1842 CUAGACCACUGAUGAGXCGAA AAUCACCU2230 AGGUGAUUUUGGUCUAG2671
1843 GCUAGACCCUGAUGAGXCGAA AAAUCACC2231 GGUGAUUUUGGUCUAGC2672
1847 UGUAGCUACUGAUGAGXCGAA ACCAAAAU2232 AUUUUGGUCUAGCUACA2673
1849 ACUGUAGCCUGAUGAGXCGAA AGACCAAA2233 UUUGGUCUAGCUACAGU2674
1853 UUUCACUGCUGAUGAGXCGAA AGCUAGAC2234 GUCUAGCUACAGUGAAA2675
1863 UCCAUCGACUGAUGAGXCGAA AUUUCACU2235 AGUGAAAUCUCGAUGGA2676
1865 ACUCCAUCCUGAUGAGXCGAA AGAUUUCA2236 UGAAAUCUCGAUGGAGU2677
1878 ACUGAUGGCUGAUGAGXCGAA ACCCACUC2237 GAGUGGGUCCCAUCAGU2678
1883 UUCAAACUCUGAUGAGXCGAA AUGGGACC2238 GGUCCCAUCAGUUUGAA2679
1887 ACUGUUCACUGAUGAGXCGAA ACUGAUGG2239 CCAUCAGUUUGAACAGU2680
1888 ~AACUGUUCCUGAUGAGXCGAA AACUGAUG~ ~CAUCAGUUUGAACAGUU2681
2240
CA 02288640 1999-11-OS
WO 98/50530 1 ~ 1 PCT/US98/09249
nt. SEQ SEQ
Position Ribozyme ID. Substrate ID.
No. No.
1896 AUCCAGACCUGAUGAGX CGAA ACUGUUCA2241 UGAACAGUUGUCUGGAU2682
1899 UGGAUCCACUGAUGAGX CGAA ACAACUGU2242 ACAGUUGUCUGGAUCCA2683
1905 ACAAAAUGCUGAUGAGX CGAA AUCCAGAC2243 GUCUGGAUCCAUUUUGU2684
1909 AUCCACAACUGAUGAGX CGAA AUGGAUCC2249 GGAUCCAUUUUGUGGAU2685
1910 CAUCCACACUGAUGAGX CGAA AAUGGAUC2245 GAUCCAUUUUGUGGAUG2686
1911 CCAUCCACCUGAUGAGX CGAA AAAUGGAU2296 AUCCAUUUUGUGGAUGG2687
1930 AUUCUGAUCUGAUGAGX CGAA ACUUCUGG2297 CCAGAAGUCAUCAGAAU2688
1933 UGCAUUCUCUGAUGAGX CGAA AUGACUUC2248 GAAGUCAUCAGAAUGCA2689
1946 UGGAUUUUCUGAUGAGX CGAA AUCUUGCA2249 UGCAAGAUA 2690
AAAAUCCA
1952 GCUGUAUGCUGAUGAGX CGAA AUUUUUAU2250 AUAAAAAUCCAUACAGC2691
1956 GAAAGCUGCUGAUGAGX CGAA AUGGAUUU2251 AAAUCCAUACAGCUUUC2692
1962 CUGACUGACUGAUGAGX CGAA AGCUGUAU2252 AUACAGCUUUCAGUCAG2693
1963 UCUGACUGCUGAUGAGX CGAA AAGCUGUA2253 UACAGCUUUCAGUCAGA2694
1969 AUCUGACUCUGAUGAGX CGAA AAAGCUGU2254 ACAGCUUUCAGUCAGAU2695
1968 AUACAUCUCUGAUGAGX CGAA ACUGAAAG2255 CUUUCAGUCAGAUGUAU2696
1975 AAUGCAUACUGAUGAGX CGAA ACAUCUGA2256 UCAGAUGUAUAUGCAUU2697
1977 CAAAUGCACUGAUGAGX CGAA AUACAUCU2257 AGAUGUAUAUGCAUUUG2698
1983 CAAUCCCACUGAUGAGX CGAA AUGCAUAU2258 AUAUGCAUUUGGGAUUG2699
1989 ACAAUCCCCUGAUGAGX CGAA AAUGCAUA2259 UAUGCAUUUGGGAUUGU2700
1990 UACAGAACCUGAUGAGX CGAA AUCCCAAA2260 UUUGGGAUUGUUCUGUA2701
1993 UCAUACAGCUGAUGAGX CGAA ACAAUCCC2261 GGGAUUGUUCUGUAUGA2702
1994 UUCAUACACUGAUGAGX CGAA AACAAUCC2262 GGAUUGUUCUGUAUGAA2703
1998 UCAAUUCACUGAUGAGX CGAA ACAGAACA2263 UGUUCUGUAUGAAUUGA2704
2004 CAGUCAUCCUGAUGAGX CGAA AUUCAUAC2264 GUAUGAAUUGAUGACUG2705
2019 AAUAAGGUCUGAUGAGX CGAA ACUGUCCA2265 UGGACAGUUACCUUAUU2706
2020 GAAUAAGGCUGAUGAGX CGAA AACUGUCC2266 GGACAGUUACCUUAUUC2707
2024 GUUUGAAUCUGAUGAGX CGAA AGGUAACU2267 AGUUACCUUAUUCAAAC2708
2025 UGUUUGAACUGAUGAGX CGAA AAGGUAAC2268 GUUACCUUAUUCAAACA2709
2027 GAUGUUUGCUGAUGAGX CGAA AUAAGGUA2269 UACCUUAUUCAAACAUC2710
2028 UGAUGUUUCUGAUGAGX CGAA AAUAAGGU2270 ACCUUAUUCAAACAUCA2711
2035 CUGUUGUUCUGAUGAGX CGAA AUGUUUGA2271 UCAAACAUCAACAACAG2712
2053 AUAAAAAUCUGAUGAGX CGAA AUCUGGUC2272 GACCAGAUAAUUUUUAU2713
2056 ACCAUAAACUGAUGAGX CGAA AUUAUCUG2273 CAGAUAAUUUUUAUGGU2714
2057 CACCAUAACUGAUGAGX CGAA AAUUAUCU2279 AGAUAAUUUUUAUGGUG2715
2058 CCACCAUACUGAUGAGX CGAA AAAUUAUC2275 GAUAAUUUUUAUGGUGG2716
2059 CCCACCAUCUGAUGAGX CGAA AAAAUUAU2276 AUAAUUUUUAUGGUGGG2717
2060 UCCCACCACUGAUGAGX CGAA AAAAAUUA2277 UAAUUUUUAUGGUGGGA2718
2076 GAGACAGGCUGAUGAGX CGAA AUCCUCGU2278 ACGAGGAUACCUGUCUC2719
2082 GAUCUGGACUGAUGAGX CGAA ACAGGUAU2279 AUACCUGUCUCCAGAUC2720
2084 GAGAUCUGCUGAUGAGX CGAA AGACAGGU2280 ACCUGUCUCCAGAUCUC2721
2090 CUUACUGACUGAUGAGX CGAA AUCUGGAG2281 CUCCAGAUCUCAGUAAG2722
2092 ACCUUACUCUGAUGAGX CGAA AGAUCUGG2282 CCAGAUCUCAGUAAGGU2723
2096 CCGUACCUCUGAUGAGX CGAA ACUGAGAU2283 AUCUCAGUAAGGUACGG2729
2101 UUACUCCGCUGAUGAGX CGAA ACCUUACU2289 AGUAAGGUACGGAGUAA2725
2108 UGGACAGUCUGAUGAGX CGAA ACUCCGUA2285 UACGGAGUAACUGUCCA2726
2119 GGCUUUUGCUGAUGAGX CGAA ACAGUUAC2286 GUAACUGUCCAAAAGCC2727
2133 CUGCCAUUCUGAUGAGX CGAA AUCUCUUC2287 GAAGAGAUUAAUGGCAG2728
2134 UCUGCCAUCUGAUGAGX CGAA AAUCUCUU2288 AAGAGAUUAAUGGCAGA2729
2149 UUCUUUUUCUGAUGAGX CGAA AGGCACUC2289 GAGUGCCUC 2730
AAAAAGAA
2176 UGGGGAAACUGAUGAGX CGAA AGUGGUCU2290 AGACCACUCUUUCCCCA2731
2178 UUUGGGGACUGAUGAGX CGAA AGAGUGGU2291 ACCACUCUUUCCCCAAA2732
2179 AUUUGGGGCUGAUGAGX CGAA AAGAGUGG2292 CCACUCUUUCCCCAAAU2733
2180 AAUUUGGGCUGAUGAGX CGAA AAAGAGUG2293 CACUCUUUCCCCAAAUU2734
2188 GAGGCGAGCUGAUGAGX CGAA AUUUGGGG-2294 CCCCAAAUUCUCGCCUC2735
2189 AGAGGCGACUGAUGAGX CGAA AAUUUGGG2295 CCCAAAUUCUCGCCUCU2736
CA 02288640 1999-11-05
W O 98/50530 1 ~2 PCT/US98/09249
nt. SEQ SEQ
Position Ribozyme ID. Substrate ID.
No. No.
2191 AUAGAGGCCUGAUGAGX CGAA AGAAUUUG2296 CAAAUUCUCGCCUCUAU2737
2196 GCUCAAUACUGAUGAGX CGAA AGGCGAGA2297 UCUCGCCUCUAUUGAGC2738
2198 CAGCUCAACUGAUGAGX CGAA AGAGGCGA2298 UCGCCUCUAUUGAGCUG2739
2200 AGCAGCUCCUGAUGAGX CGAA AUAGAGGC2299 GCCUCUAUUGAGCUGCU2790
2217 UUGGCAAUCUGAUGAGX CGAA AGCGGGCC2300 GGCCCGCUCAUUGCCAA2741
2220 UUUUUGGCCUGAUGAGX CGAA AUGAGCGG2301 CCGCUCAUUGCCAAAAA2742
2230 CUGCGGUGCUGAUGAGX CGAA AUUUUUGG2302 CCAAAAAUUCACCGCAG2743
2231 ACUGCGGUCUGAUGAGX CGAA AAUUUUUG2303 CAAAAAUUCACCGCAGU2744
2244 AGGGUUCUCUGAUGAGX CGAA AUGCACUG2304 CAGUGCAUCAGAACCCU2795
2253 GAUUCAAGCUGAUGAGX CGAA AGGGUUCU2305 AGAACCCUCCUUGAAUC2796
2256 CCCGAUUCCUGAUGAGX CGAA AGGAGGGU2306 ACCCUCCUUGAAUCGGG2797
2261 ACCAGCCCCUGAUGAGX CGAA AUUCAAGG2307 CCUUGAAUCGGGCUGGU2748
2270 UGUUUGGACUGAUGAGX CGAA ACCAGCCC2308 GGGCUGGUUUCCAAACA2749
2271 CUGUUUGGCUGAUGAGX CGAA AACCAGCC2309 GGCUGGUUUCCAAACAG2750
2272 UCUGUUUGCUGAUGAGX CGAA AAACCAGC2310 GCUGGUUUCCAAACAGA2751
2285 UAGACUAACUGAUGAGX CGAA AUCCUCUG2311 CAGAGGAUUUUAGUCUA2752
2286 AUAGACUACUGAUGAGX CGAA AAUCCUCU2312 AGAGGAUUUUAGUCUAU2753
2287 UAUAGACUCUGAUGAGX CGAA AAAUCCUC2313 GAGGAUUUUAGUCUAUA2754
2288 AUAUAGACCUGAUGAGX CGAA AAAAUCCU2319 AGGAUUUUAGUCUAUAU2755
2291 AGCAUAUACUGAUGAGX CGAA ACUAAAAU2315 AUUUUAGUCUAUAUGCU2756
2293 CAAGCAUACUGAUGAGX CGAA AGACUAAA2316 UUUAGUCUAUAUGCUUG2757
2295 CACAAGCACUGAUGAGX CGAA AUAGACUA2317 UAGUCUAUAUGCUUGUG2758
2300 AGAAGCACCUGAUGAGX CGAA AGCAUAUA2318 UAUAUGCUUGUGCUUCU2759
2306 UUUUGGAGCUGAUGAGX CGAA AGCACAAG2319 CUUGUGCUUCUCCAAAA2760
2307 UUUUUGGACUGAUGAGX CGAA AAGCACAA2320 UUGUGCUUCUCCAAAAA2761
2309 UGUUUUUGCUGAUGAGX CGAA AGAAGCAC2321 GUGCUUCUCCAAAAACA2762
2323 CCUGCCUGCUGAUGAGX CGAA AUGGGUGU2322 ACACCCAUCCAGGCAGG2763
2337 ACGCACCACUGAUGAGX CGAA AUCCCCCU2323 AGGGGGAUAUGGUGCGU2764
2396 GGACAGGACUGAUGAGX CGAA ACGCACCA2324 UGGUGCGUUUCCUGUCC2765
2347 UGGACAGGCUGAUGAGX CGAA AACGCACC2325 GGUGCGUUUCCUGUCCA2766
2348 GUGGACAGCUGAUGAGX CGAA AAACGCAC2326 GUGCGUUUCCUGUCCAC2767
2353 UUUCAGUGCUGAUGAGX CGAA ACAGGAAA2327 UUUCCUGUCCACUGAAA2768
2379 CUCUCCUGCUGAUGAGX CGAA ACUCUCUC2328 GAGAGAGUUCAGGAGAG2769
2380 ACUCUCCUCUGAUGAGX CGAA AACUCUCU2329 AGAGAGUUCAGGAGAGU2770
2389 UUUGUUGCCUGAUGAGX CGAA ACUCUCCU2330 AGGAGAGUAGCAACAAA2771
2406 UGUUCAUUCUGAUGAGX CGAA AUUUUCCU2331 AGGAAAAUA 2772
AAUGAACA
2416 AGCAAACACUGAUGAGX CGAA AUGUUCAU2332 AUGAACAUAUGUUUGCU2773
2920 UAUAAGCACUGAUGAGX CGAA ACAUAUGU2333 ACAUAUGUUUGCUUAUA2774
2421 AUAUAAGCCUGAUGAGX CGAA AACAUAUG2334 CAUAUGUUUGCUUAUAU2775
2425 UAACAUAUCUGAUGAGX CGAA AGCAAACA2335 UGUUUGCUUAUAUGUUA2776
2426 UUAACAUACUGAUGAGX CGAA AAGCAAAC2336 GUUUGCUUAUAUGUUAA2777
2428 AUUUAACACUGAUGAGX CGAA AUAAGCAA2337 UUGCUUAUAUGUUAAAU2778
2432 UUCAAUUUCUGAUGAGX CGAA ACAUAUAA2338 UUAUAUGUUAAAUUGAA2779
2433 AUUCAAUUCUGAUGAGX CGAA AACAUAUA2339 UAUAUGUUA 2780
AAUUGAAU
2437 UUUUAUUCCUGAUGAGX CGAA AUUUAACA2340 UGUUAAAUUGAAUAAAA2781
2442 GAGUAUUUGUGAUGAGX CGAA AUUCAAUU2391 AAUUGAAUA 2782
AAAUACUC
2947 AAAGAGAGCUGAUGAGX CGAA AUUUUAUU2342 AAUAAAAUACUCUCUUU2783
2950 AAAAAAGACUGAUGAGX CGAA AGUAUUUU2343 AAAAUACUCUCUUUUUU2789
2452 P~AAAAAAACUGAUGAGX CGAA AGAGUAUU2349 AAUACUCUCUUUUUUUU2785
2454 UAAAAAAACUGAUGAGX CGAA AGAGAGUA2345 UACUCUCUUUUUUUUUA2786
2455 UUAAAAAACUGAUGAGX CGAA AAGAGAGU2346 ACUCUCUUUUUUUUUAA2787
2456 CUUAAAAACUGAUGAGX CGAA AAAGAGAG2347 CUCUCUUUUUUUUUAAG2788
2457 CCUUAAAACUGAUGAGX CGAA AAAAGAGA2348 UCUCUUUUUUUUUAAGG2789
2458 ACCUUAAACUGAUGAGX CGAA AAAAAGAG2349 CUCUUUUUUUUUAAGGU2790
2459 CACCUUAACUGAUGAGX CGAA AAAAAAGA2350 UCUUUUUUUUUAAGGUG2791
CA 02288640 1999-11-OS
WO 98/50530 1 ~3 PCT/US98/09249
nt. SEQ SEQ
Position Ribozyme ID. Substrate ID.
No. No.
2960 CCACCUUACUGAUGAG X CGAA AAAAAAAG2351 CUUUUUUUU UAAGGUGG2792
2461 UCCACCUUCUGAUGAG X CGAA F~~AAAAAA2352 UUUUUUUUU AAGGUGGA2793
2462 UUCCACCUCUGAUGAG X CGAA AAAAAAAA2353 UUUUUUUUA AGGUGGAA2794
Where "X" represents stem II region of a HH ribozyme (Hertel et al., 1992
Nucleic Acids
Res. 20: 3252). The length of stem II may be z 2 base-pairs.
174
CA 02288640 1999-11-OS
WO 98/50530 1 ~S PCT/US98/09249
0
z
vm voraoo~o .-~N M~rin~
G1ocoaomm aoo~rnovo~ovavo~
~ aocoaomo0maomao0omcoao
~INN NNN NN NN NNN N
W
Ur~r~c~a aa ~ca c~c~U r~
~ DU ~~ U5
d FC a ~~ ~ D
G aU ~UC GC~ UU Ua U
U
" pc~caaa a~ Ua caa ..c
~ aa apa c~a aU UC~U U
~ aU aaa ar~Ua Uaa U
Ur.~aaU r.~a Dr.~aUU a
y C9C9C7C~U C~t7C7U C9C~C9C9
~ FCFCFC~Cr.~r.~D aFCaaU D
UU UUU UU UU UUU U tVd
UFCa~~CaD UU ~U'U U cd
oc~rt a c~a ra cc~a
~
. F ,s~
UU U(.~aC~CC9aU 5U'C7a
o N
z .s~
MV~Nl0r OD01Or-iNMC tn
A MfnMMM MM v~v~ccr~ C
OJ00dDODOD00~ WCOCDOD00OD
plNN NNN NN NN NNN N
H
~~r~
?
aa aaa aa aa aaa a :
c~~ c~pc~c~~ ~c~c~c~c~c~m
~c~~c~c~c~~ pc~c~~c~c~4.
aa aaa aa aa aaa a p
UU UUU UU UU UUU U
UU UUU UU UU UUU U Li
.
aa aaa aa aa aaa a
aa aaa aa aa aaa a
~Kcaa~ aa a~ ~~~ ~ a;
UU UUU UU UU UUU U
FC~CACFCFCFCFC~C~ FCr~~C~ N
aa aaa aa aa aaa a
~c~c~c~c~c~c~pc~pc~p c~
e xx xxx xx xx xxx x
UU UUU UU UU UUU U
G
FCr.Cr~Cr~C~C~C KCr~Cr.Cr.CFC~C
C7C7C~U'C7C9 C9C7C9C9C'!C9
~'FCFCFCr.~~CfC~CFCaCFCFC~CFC
O UU UUU UU UU UUU U
UU UUU UU UU UVU U
C~U'U'Ua ~U FCC7r.CUU
~U aar U a~ aaU ~
r C
.D FCa. r.C~ C~r.aUU'C~
C7 a C7
C7C'JC~U'C7C7C7C7U U'C7C'JC'l4r
C7C9C7C7C7C7CJU'C9U'CJC7C7O
FCFCr-CU~ UFC~CC7C7UU'C7
UU U~C ~U ClC aUD a
~Cc~ac~csr.a pr.ac~ c~
a c~
aa a~a r~a aa ac~ r~,..,,
~~ ~ ~
~~ ~ ~~ ~~~ a
a~
o _
v~v~crMdDOWI161\O.-INM O ~,~.a
W DM W41W O01r00.-1Ov-1tn
O l0r 00ODO~0101OO e1NN M
.-1i -1-I-il-1NN NNN N ~d
m e.~ r, (
~
b r
Y
N
4a
N
CA 02288640 1999-11-OS
WO 98/50530 1 ~6 PCT/US98/09249
Table XVIII. Hammerhead (HH) Ribozyme target with sequence homology between
c-raf and A-raf
nt. Position Target Seq LD. No.
452 AGGAGCU C AUUGUCG 2897
527 UGGCGUU C UGUGACU 2898
583 UGUGGCU A CAAGUUC 2899
658 ACAGUGU C CAGGAUU 2900
857 AUAUGGU C AGCACCA 2901
1096 UCAGGCU A UUACUGG 2902
1098 AGGCUAU U ACUGGGA 2903
1246 CAGGCUU U CAAGAAU 2904
1247 AGGCUUU C AAGAAUG 2905
1309 AUGGGCU U CAUGACC 2906
1327 CCGGGAU U UGCCAUC 2907
1357 GAGGGCU C CAGCCUC 2908
1412 UCCAGCU C AUCGACG 2909
1469 AGAACAU C AUCCACC 2910
1628 AGGUGAU C CGUAUGC 2911
1658 ACAGCUU C CAGUCAG 2912
1663 UUCCAGU C AGACGUC 2913
1748 ACCAGAU U AUCUUUA 2914
1749 CCAGAUU A UCUUUAU 2915
1751 AGAUUAU C UUUAUGG 2916
1753 AUUAUCU U UAUGGUG 2917
1754 UUAUCUU U AUGGUGG 2918
1871 GGCCCCU C UUCCCCC 2919
1874 CCCUCUU C CCCCAGA 2920
1951 CCCUCCU U GCACCGC 2921
2046 CCAAUCU C AGCCCUC 2922
2127 CCCCAUU C CCCACCC 2923
2174 AGUUCUU C UGGAAUU 2924
2251 UGGGGAU A CUUCUAA 2925
2400 GUCCCUU U UGUGUCU 2926
2432 CUCCUCU C UUUCUUC 2927
CA 02288640 1999-11-OS
WO 98/50530 1 ~~ PCT/US98/09249
Table XIX. Hammerhead Ribozyme Target with sequence homology between c-raf
and B-raf
nt. PositionTarget Sequence Seq. I. D. No.
17 _____GCCCCCUC CCCGCCC 2928
405 UCUGCAU C AAUGGAU 2929
426 ACAUCUU C UUCCUCU 2930
479 UCAAAAU C CCACAGA 2931
702 GAUAUUU C CUGGCUU 2932
759 UUCCACU U ACAACAC 2933
861 UAUAAAU U UCACCAG 2934
931 UGUUUGU C UCCAAGU 2935
1034 GGACUCU A UUGGGCC 2936
1259 GUCUGCU A CCCCCCC 2937
1349 AAGUCAU C UUCAUCC 2938
1603 UCCUACU C UUCAUGG 2939
1662 GAGGGCU C CAGCUUG 2940
1802 UAAUAAU A UAUUUCU 2941
1804 AUAAUAU A UUUCUUC 2992
1806 AAUAUAU U UCUUCAU 2943
1807 AUAUAUU U CUUCAUG 2944
1808 UAUAUUU C UUCAUGA 2945
1810 UAUUUCU U CAUGAAG 2946
1834 UAAAAAU A GGUGAUU 2947
1842 GGUGAUU U UGGUCUA 2948
1847 UUUUGGU C UAGCUAC 2949
1956 AAUCCAU A CAGCUUU 2950
2035 CAAACAU C AACAACA 2952
2059 UAAUUUU U AUGGUGG 2952
2090 UCCAGAU C UCAGUAA 2953
2092 CAGAUCU C AGUAAGG 2954
2200 CCUCUAU U GAGCUGC 2955
2256 CCCUCCU U GAAUCGG 2956
CA 02288640 1999-11-OS
WO 98150530 178 PCT/US98/09249
Table XX.
ExperimentalRibozyme Dose Sample Size
per
Group Activity/Target (mg/kg/day) dose
RPL4610 Active/flt-1 1, 3, 10, 30, 10
100
RPL4611 Inactive/flt-1 1, 3, 10, 30, 10
100
RPL4733 Active/Jlk-1 1, 3, 10, 30, 10
100
RPL4734 Inactive/flk-1 1, 3, 10, 30, 10
100
Saline NA 12 ~tl/day 10