Note: Descriptions are shown in the official language in which they were submitted.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
REAGENTS AND METHODS USEFUL FOR DETECTING
DISEASES OF THE BREAST
Background of the Invention
This invention relates generally to detecting diseases of the breast.
Furthermore, the invention also relates to reagents and methods for detecting
diseases of the breast. More particularly, the present invention relates to
reagents
such as polynucleotide sequences and the polypeptide sequences encoded
thereby,
as well as methods which utilize these sequences. The polynucleotide and
polypeptide sequences are useful for detecting, diagnosing, staging,
monitoring,
prognosticating, preventing or treating, or determining predisposition to
diseases
or conditions of the breast, such as breast cancer.
Breast cancer is the most common form of cancer occurring in females
in the U.S. The incidence of breast cancers in the United States is projected
to
be 180,300 cases diagnosed and 43,900 breast cancer-related deaths to occur
during 1998 (American Cancer Society statistics). Worldwide, the incidence
of breast cancer increased from 700,000 in 1985 to about 900,000 in 1990.
G.N. Hortobagyi et al., CA Cancer J Clin 45: 199-226 (1995).
Procedures used for detecting, diagnosing, staging, monitoring,
prognosticating, preventing or treating, or determining predisposition to
diseases or conditions of the breast, such as breast cancer, are of critical
importance to the outcome of the patient. For example, patients diagnosed
with early breast cancer have greater than a 90% five-year relative survival
rate
as compared to a survival rate of about 20% for patients diagnosed with
distantly metastasized breast cancers. (American Cancer Society statistics).
Currently, the best initial indicators of early breast cancer are physical
examination of the breast and mammography. J.R. Harris et al. In: C ncer:
Principles and Practice of Oncology, Fourth Edition, pp. 1264-1332,
Philadelphia, PA: J/B. Lippincott Co. ( 1993). Mammography may detect a
breast tumor before it can be detected by physical examination, but it has
limitations. For example, mammography's predictive value depends on the
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-2-
observer's skill and the quality of the mammogram. In addition, 80 to 93% of
suspicious mammograms are false positives, and 10 to 15% of women with '
breast cancer have false negative mammograms. C.J. Wright et al., Lancet
346: 29-32 ( 1995). New diagnostic methods which are more sensitive and '
specific for detecting early breast cancer are clearly needed.
Breast cancer patients are closely monitored following initial therapy
and during adjuvant therapy to determine response to therapy, and to detect
persistent or recurrent disease, or early distant metastasis. Current
diagnostic
procedures for monitoring breast cancer include mammography, bone scan,
chest radiographs, liver function tests and tests for serum markers. The serum
tumor markers most commonly used for monitoring patients are
carcinoembryonic antigen (CEA) and CA 15-3. Limitations of CEA include
absence of elevated serum levels in about 40% of women with metastatic
disease. In addition, CEA elevation during adjuvant therapy may not be
related to recurrence but to other factors that are not clinically important.
CA
15-3 can also be negative in a significant number of patients with progressive
disease and, therefore, fail to predict metastasis. Both CEA and CA 15-3 can
be elevated in nonmalignant, benign conditions giving rise to false positive
results. Therefore, it would be clinically beneficial to find a breast
associated
marker which is more sensitive and specific in detecting cancer recurrence. J.
R. Harris et al., supra. M. K. Schwartz, In: Cancer: Principles and Practice
of Oncolo,~v. Vol. 1, Fourth Edition, pp. 531 - 542, Philadelphia, PA: JB.
Lippincott Co. 1993.
Another important step in managing breast cancer is to determine the
stage of the patient's disease because stage determination has potential
prognostic value and provides criteria for designing optimal therapy.
Currently, pathological staging of breast cancer is preferable over clinical
staging because the former gives a more accurate prognosis. J. R. Harris et
al., supra. On the other hand, clinical staging would be preferred were it at
least as accurate as pathological staging because it does not depend on an
invasive procedure to obtain tissue for pathological evaluation. Staging of
breast cancer could be improved by detecting new markers in serum or urine
which could differentiate between different stages of invasion. Such markers
could be mRNA or protein markers expressed by cells originating from the
primary tumor in the breast but residing in blood, bone marrow or lymph
nodes and could serve as sensitive indicators for metastasis to these distal
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-3-
organs. For example, specific protein antigens and mRNA, associated with
breast epithelial cells, have been detected by immunohistochemical techniques
and RT-PCR, respectively, in bone marrow, lymph nodes and blood of breast
cancer patients suggesting metastasis. K. Pantel et al., OnkoloQie 18: 394-401
( 1995).
Such diagnostic procedures also could include immunological assays
based upon the appearance of various disease markers in test samples such as
blood, plasma, serum or urine obtained by minimally invasive procedures which
are detectable by immunological methods. These diagnostic procedures would
provide information to aid the physician in managing the patient with disease
of
the breast, at low cost to the patient. Markers such as prostate specific
antigen
(PSA) and human chorionic gonadotropin (hCG) exist and are used clinically for
screening patients for prostate cancer and testicular cancer, respectively.
For
example, PSA normally is secreted by the prostate at high levels into the
seminal
fluid, but is present in very low levels in the blood of men with normal
prostates.
Elevated levels of PSA protein in serum are used in the early detection of
prostate
cancer or disease in asymptomatic men. See, for example, G.E. Hanks et al.,
In:
Cancer: Principles and Practice of Oncology, Vol. I, Fourth Edition, pp. 1073-
1 I 13, Philadelphia, PA: J.B. Lippincott Co. 1993. M. K. Schwartz et al., In:
Cancer: Principles and Practice of Oncology, Vol. 1, Fourth Edition, pp. 531-
542, Philadelphia, PA: J.B. Lippincott Co. 1993. Likewise, the management of
breast diseases could be improved by the use of new markers normally expressed
in the breast but found in elevated amounts in an inappropriate body
compartment
as a result of the disease of the breast.
Further, new markers which could predict the biologic behavior of
early breast cancers would also be of significant value. Early breast cancers
that threaten or will threaten the life of the patient are more clinically
important
than those that do not or will not be a threat. G.E. Hanks, supra. Such
markers are needed to predict which patients with histologically negative
lymph nodes will experience recurrence of cancer and also to predict which
cases of ductal carcinoma in situ will develop into invasive breast carcinoma.
More accurate prognostic markers would allow the clinician to accurately
identify early cancers localized to the breast which will progress and
metastasize if not treated aggressively. Additionally, the absence of a marker
for an aggressive cancer in the patient could spare the patient expensive and
non-beneficial treatment. J. R. Harris et al., supra. E. R. Frykberg et al.,
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-4-
Cancer 74: 350-361 ( 1994).
It therefore would be advantageous to provide specific methods and
reagents useful for detecting, diagnosing, staging, monitoring,
prognosticating, preventing or treating, or determining predisposition to
diseases or conditions of the breast. Such methods would include assaying a
test sample for products of a gene which are overexpressed in diseases and
conditions associated with the breast, including cancer. Such methods may
also include assaying a test sample for products of a gene which have been
altered by the disease or condition associated with the breast, including
cancer.
Such methods may further include assaying a test sample for products of a
gene whose distribution among the various tissues and compartments of the
body have been altered by a breast-associated disease or condition, including
cancer. Such methods would comprise making cDNA from mRNA in the test
sample, amplifying, when necessary, portions of the cDNA corresponding to
the gene or a fragment thereof, and detecting the cDNA product as an
indication of the presence of the disease or condition including cancer or
detecting translation products of the mRNAs comprising gene sequences as an
indication of the presence of the disease. Useful reagents include
polynucleotide(s), or fragments) thereof which may be used in diagnostic
methods such as reverse transcriptase-polymerise chain reaction (RT-PCR),
PCR, or hybridization assays of mRNA extracted from biopsied tissue, blood
or other test samples; or proteins which are the translation products of such
mRNAs; or antibodies directed against these proteins. Such assays would
include methods for assaying a sample for products) of the gene and detecting
the products) as an indication of disease of the breast. Drug treatment or
gene
therapy for diseases and conditions of the breast including cancer can be
based
on these identified gene sequences or their expressed proteins, and efficacy
of
any particular therapy can be monitored. Furthermore, it would be
advantageous to have available alternative, non-surgical diagnostic methods
capable of detecting early stage breast disease, such as cancer.
Summary of the Invention
The present invention provides a method of detecting a target BS202
polynucleotide in a test sample which comprises contacting the test sample
with at
least one BS202-specific polynucleotide and detecting the presence of the
target
BS202 polynucleotide in the test sample. The BS202-specific polynucleotide has
CA 02292843 1999-12-03
WO 99102559 PCT/US98114046
-5-
at least 50% identity with a polynucleotide selected from the group consisting
of
SEQUENCE ID NO 1, SEQUENCE ID NO 2, SEQUENCE ID NO 3,
SEQUENCE ID NO 4, SEQUENCE ID NO S, SEQUENCE ID NO 6,
SEQUENCE ID NO 7, and fragments or complements thereof. Also, the BS202-
specific polynucleotide may be attached to a solid phase prior to performing
the
method.
The present invention also provides a method for detecting BS202 mRNA
in a test sample, which comprises performing reverse transcription (RT) with
at
least one primer in order to produce cDNA, amplifying the cDNA so obtained
using BS202 oligonucleotides as sense and antisense primers to obtain BS202
amplicon, and detecting the presence of the BS202 amplicon as an indication of
the presence of BS202 mRNA in the test sample, wherein the BS202
oligonucleotides have at least 50% identity with a sequence selected from the
group consisting of SEQUENCE ID NO I, SEQUENCE ID NO 2, SEQUENCE
B7 NO 3, SEQUENCE ID NO 4, SEQUENCE ID NO S, SEQUENCE ID NO 6,
SEQUENCE ID NO 7, and fragments or complements thereof. Amplification can
be performed by the polymerase chain reaction. Also, the test sample can be
reacted with a solid phase prior to performing the method, prior to
amplification or
prior to detection. This reaction can be a direct or an indirect reaction.
Further,
the detection step can comprise utilizing a detectable label capable of
generating a
measurable signal. The detectable label can be attached to a solid phase.
The present invention further provides a method of detecting a target
BS202 polynucleotide in a test sample suspected of containing target BS202
polynucleotides, which comprises (a) contacting the test sample with at least
one
BS202 oligonucleotide as a sense primer and at least one BS202 oligonucleotide
as an anti-sense primer, and amplifying same to obtain a first stage reaction
product; (b) contacting the first stage reaction product with at least one
other
BS202 oligonucleotide to obtain a second stage reaction product, with the
proviso
that the other BS202 oIigonucleotide is located 3' to the BS202
oligonucleotides
utilized in step (a) and is complementary to the first stage reaction product;
and (c)
" detecting the second stage reaction product as an indication of the presence
of a
target BS202 polynucleotide in the test sample. The BS202 oligonucleotides
selected as reagents in the method have at least 50% identity with a sequence
selected from the group consisting of SEQUENCE ID NO 1, SEQUENCE ID NO
2, SEQUENCE ID NO 3, SEQUENCE ID NO 4, SEQUENCE ID NO 5,
SEQUENCE m NO 6, SEQUENCE ID NO 7, and fragments or complements
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-6-
thereof. Amplification may be performed by the polymerase chain reaction. The
test sample can be reacted either directly or indirectly with a solid phase
prior to
performing the method, or prior to amplification, or prior to detection. The
detection step also comprises utilizing a detectable label capable of
generating a
measurable signal; further, the detectable label can be attached to a solid
phase.
Test kits useful for detecting target BS202 polynucleotides in a test sample
are also
provided which comprise a container containing at least one BS202-specific
polynucleotide selected from the group consisting of SEQUENCE ID NO 1,
SEQUENCE ID NO 2, SEQUENCE ID NO 3, SEQUENCE ID NO 4,
SEQUENCE ID NO 5, SEQUENCE ID NO 6, SEQUENCE 117 NO 7, and
fragments or complements thereof. These test kits further comprise containers
with tools useful for collecting test samples (such as, for example, blood,
urine,
saliva and stool). Such tools include lancets and absorbent paper or cloth for
collecting and stabilizing blood; swabs for collecting and stabilizing saliva;
and
cups for collecting and stabilizing urine or stool samples. Collection
materials,
such as papers, cloths, swabs, cups, and the like, may optionally be treated
to
avoid denaturation or irreversible adsorption of the sample. The collection
materials also may be treated with or contain preservatives, stabilizers or
antimicrobial agents to help maintain the integrity of the specimens.
The present invention also provides a purified polynucleotide or fragment
thereof derived from a BS202 gene. The purified polynucleotide is capable of
selectively hybridizing to the nucleic acid of the BS202 gene, or a complement
thereof. The polynucleotide has at least 50% identity with a polynucleotide
selected from the group consisting of SEQUENCE ID NO l, SEQUENCE ID NO
2, SEQUENCE B7 NO 3, SEQUENCE ID NO 4, SEQUENCE B~ NO'S,
SEQUENCE ID NO 6, SEQUENCE ID NO 7, and fragments or complements
thereof. Further, the purified polynucleotide can be produced by recombinant
and/or synthetic techniques. The purified recombinant polynucleotide can be
contained within a recombinant vector. The invention further comprises a host
cell
transfected with the recombinant vector.
The present invention further provides a recombinant expression system
comprising a nucleic acid sequence that includes an open reading frame derived
from BS202. The nucleic acid sequence has at least 50% identity with a
sequence
selected from the group consisting of SEQUENCE ID NO 1, SEQUENCE 1D NO
2, SEQUENCE ID NO 3, SEQUENCE ID NO 4, SEQUENCE ID NO 5,
SEQUENCE ID NO 6, SEQUENCE ID NO 7, and fragments or complements
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
_7_
thereof. The nucleic acid sequence is operably linked to a control sequence
compatible with a desired host. Also provided is a cell transfected with this
recombinant expression system.
The present invention also provides a polypeptide encoded by BS202.
The polypeptide can be produced by recombinant technology, provided in
purified
form, or produced by synthetic techniques. The polypeptide comprises an amino
acid sequence which has at least 50% identity with an amino acid sequence
selected from the group consisting of SEQUENCE ID NO 14, SEQUENCE ID
NO 15, SEQUENCE ID NO 16, SEQUENCE ID NO 17, SEQUENCE ID NO
18, and fragments thereof.
Also provided is an antibody which specifically binds to at least one
BS202 epitope. The antibody can be a polyclonal or monoclonal antibody. The
epitope is derived from an amino acid sequence selected from the group
consisting
of SEQUENCE ID NO 14, SEQUENCE ID NO 15, SEQUENCE ID NO 16,
SEQUENCE ID NO 17, SEQUENCE ID NO 18, and fragments thereof. Assay
kits for determining the presence of BS202 antigen or anti-BS202 antibody in a
test sample are also included. In one embodiment, the assay kits comprise a
container containing at least one BS202 polypeptide having at least 50%
identity
with an amino acid sequence selected from the group consisting of SEQUENCE
ID NO 14, SEQUENCE ID NO 15, SEQUENCE ID NO 16, SEQUENCE ID NO
i 7, SEQUENCE ID NO 18, and fragments thereof. Further, the test kit can
comprise a container with tools useful for collecting test samples (such as
blood,
urine, saliva, and stool). Such tools include lancets and absorbent paper or
cloth
for collecting and stabilizing blood; swabs for collecting and stabilizing
saliva; and
cups for collecting and stabilizing urine or stool samples. Collection
materials
such as papers, cloths, swabs, cups, and the like, may optionally be treated
to
avoid denaturation or irreversible adsorption of the sample. These collection
materials also may be treated with or contain preservatives, stabilizers or
antimicrobial agents to help maintain the integrity of the specimens. Also,
the
polypeptide can be attached to a solid phase.
In another embodiment of the invention, antibodies or fragments thereof
against the BS202 antigen can be used to detect or image localization of the
antigen
in a patient for the purpose of detecting or diagnosing a disease or
condition.
Such antibodies can be polyclonal or monoclonal, or made by molecular biology
techniques, and can be labeled with a variety of detectable labels, including
but not
limited to radioisotopes and paramagnetic metals. Furthermore, antibodies or
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
_g_
fragments thereof, whether monoclonal, polyclonal, or made by molecular
biology
techniques, can be used as therapeutic agents for the treatment of diseases
characterized by expression of the BS202 antigen. In the case of therapeutic
applications, the antibody may be used without derivitization, or it may be
derivitized with a cytotoxic agent such as a radioisotope, enzyme, toxin,
drug,
prodrug, or the like.
Another assay kit for determining the presence of BS202 antigen or anti-
BS202 antibody in a test sample comprises a container containing an antibody
which specifically binds to a BS202 antigen, wherein the BS202 antigen
comprises at least one BS202-encoded epitope. The BS202 antigen has at least
about 60% sequence similarity to a sequence of a BS202-encoded antigen
selected
from the group consisting of SEQUENCE ID NO 14, SEQUENCE )D NO 1 S,
SEQUENCE ID NO 16, SEQUENCE ID NO 17, SEQUENCE ID NO 18, and
fragments thereof. These test kits can further comprise containers with tools
useful for collecting test samples (such as blood, urine, saliva, and stool).
Such
tools include lancets and absorbent paper or cloth for collecting and
stabilizing
blood; swabs for collecting and stabilizing saliva; cups for collecting and
stabilizing urine or stool samples. Collection materials, such as papers,
cloths,
swabs, cups and the like, may optionally be treated to avoid denaturation or
irreversible adsorption of the sample. These collection materials also may be
treated with, or contain, preservatives, stabilizers or antimicrobial agents
to help
maintain the integrity of the specimens. The antibody can be attached to a
solid
phase.
A method for producing a polypeptide which contains at least one epitope
of BS202 is provided, which method comprises incubating host cells transfected
with an expression vector. This vector comprises a poiynucleotide sequence
encoding a polypeptide, wherein the polypeptide comprises an amino acid
sequence having at least 50% identity with a BS202 amino acid sequence
selected
from the group consisting of SEQUENCE ID NO 14, SEQUENCE 117 NO 15,
SEQUENCE ID NO 16, SEQUENCE )D NO 17, SEQUENCE ID NO 18, and
fragments thereof.
A method for detecting BS202 antigen in a test sample suspected of
containing BS202 antigen also is provided. The method comprises contacting the
test sample with an antibody or fragment thereof which specifically binds to
at
least one epitope of BS202 antigen, for a time and under conditions sufficient
for
the formation of antibody/antigen complexes; and detecting the presence of
such
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-9-
complexes containing the antibody as an indication of the presence of BS202
antigen in the test sample. The antibody can be attached to a solid phase and
may
be either a monoclonal or polyclonal antibody. Furthermore, the antibody
specifically binds to at least one BS202 antigen selected from the group
consisting
of SEQUENCE )D NO 14, SEQUENCE 1D NO 15, SEQUENCE 1D NO 16,
SEQUENCE ID NO 17, SEQUENCE >D NO 18, and fragments thereof.
Another method is provided which detects antibodies which specifically
bind to BS202 antigen in a test sample suspected of containing these
antibodies.
The method comprises contacting the test sample with a polypeptide which
contains at least one BS202 epitope, wherein the BS202 epitope comprises an
amino acid sequence having at least 50% identity with an amino acid sequence
encoded by a BS202 polynucleotide, or a fragment thereof. Contacting is
carried
out for a time and under conditions sufficient to allow antigen/antibody
complexes
to form. The method further entails detecting complexes which contain the
polypeptide. The polypeptide can be attached to a solid phase. Further, the
polypeptide can be a recombinant protein or a synthetic peptide having at
least
50% identity with an amino acid sequence selected from the group consisting of
SEQUENCE )D NO 14, SEQUENCE )D NO 15, SEQUENCE 1D NO 16,
SEQUENCE ID NO 17, SEQUENCE )D NO 18, and fragments thereof.
The present invention provides a cell transfected with a BS202 nucleic acid
sequence that encodes at least one epitope of a BS202 antigen, or fragment
thereof. The nucleic acid sequence is selected from the group consisting of
SEQUENCE ID NO l, SEQUENCE ID NO 2, SEQUENCE ID NO 3,
SEQUENCE ID NO 4, SEQUENCE ID NO 5, SEQUENCE ID NO 6,
SEQUENCE 1D NO 7, and fragments or complements thereof.
A method for producing antibodies to BS202 antigen also is provided,
which method comprises administering to an individual an isolated immunogenic
polypeptide or fragment thereof, wherein the isolated immunogenic polypeptide
comprises at least one BS202 epitope. The immunogenic polypeptide is
administered in an amount sufficient to produce an immune response. The
isolated, immunogenic polypeptide comprises an amino acid sequence selected
from the group consisting of SEQUENCE m NO 14, SEQUENCE m NO 15,
SEQUENCE ID NO 16, SEQUENCE ID NO 17, SEQUENCE >D NO 18, and
fragments thereof.
Another method for producing antibodies which specifically bind to BS202
antigen is disclosed, which method comprises administering to an individual a
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-10-
piasmid comprising a nucleic acid sequence which encodes at least one BS202
epitope derived from an amino acid sequence selected from the group
consisting~f
SEQUENCE ID NO 14, SEQUENCE ID NO 15, SEQUENCE ID NO 16,
SEQUENCE DJ NO 17, SEQUENCE ID NO 18, and fragments thereof. The
plasmid is administered in an amount such that the plasmid is taken up by
cells in
the individual and expressed at levels sufficient to produce an immune
response.
Also provided is a composition of matter that comprises a BS202
polynucleotide of at least about 10-12 nucleotides having at least SO%
identity with
a polynucleotide selected from the group consisting of SEQUENCE ID NO 1,
SEQUENCE ID NO 2, SEQUENCE ID NO 3, SEQUENCE ID NO 4,
SEQUENCE ID NO 5, SEQUENCE ID NO 6, SEQUENCE ID NO 7, and
fragments or complements thereof. The BS202 polynucleotide encodes an amino
acid sequence having at least one BS202 epitope. Another composition of matter
provided by the present invention comprises a polypeptide with at least one
BS202
epitope of about 8-10 amino acids. The polypeptide comprises an amino acid
sequence having at least 50% identity with an amino acid sequence selected
from
the group consisting of SEQUENCE ID NO 14, SEQUENCE ID NO 15,
SEQUENCE 1D NO 16, SEQUENCE ID NO 17, SEQUENCE ID NO 18, and
fragments thereof. Also provided is a gene, or a fragment thereof, coding for
a
BS202 polypeptide which has at least 50% identity to SEQUENCE 1D NO 14; and
a gene, or a fragment thereof, comprising DNA having at least 50% identity
with
SEQUENCE ID NO 6 or SEQUENCE ID NO 7.
Brief Descr_ilption of the Drawines
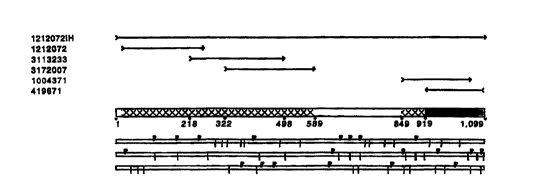
Figures lA-1B show the nucleotide alignment of clones 1212072
(SEQUENCE ID NO 1 ), 3113233 (SEQUENCE ID NO 2), 3172007
(SEQUENCE 1D NO 3), 1004371 {SEQUENCE ID NO 4), 419671
(SEQUENCE ID NO 5), the full-length sequence of clone 1212072 (designated as
clone 1212072IH (SEQUENCE ID NO 6)), and the consensus sequence
(SEQUENCE >D NO 7) derived therefrom.
Figure 2 shows the contig map depicting the formation of the consensus
nucleotide sequence (SEQUENCE ID NO 7) from the nucleotide alignment of
overlapping clones 1212072 (SEQUENCE ID NO 1 ), 3113233 (SEQUENCE 1D
NO 2), 3172007 (SEQUENCE m NO 3), 1004371 (SEQUENCE ID NO 4),
419671 (SEQUENCE ID NO 5), and 1212072IH {SEQUENCE ID NO 6).
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-11-
Detailed Description of the Invention
The present invention provides a gene, or a fragment thereof, which codes
for a BS202 polypeptide having at least about 50°lo identity to
SEQUENCE ID NO
i4. The present invention further encompasses a BS202 gene, or a fragment
thereof, comprising DNA which has at least about 50% identity with SEQUENCE
117 NO 6 or SEQUENCE m NO 7.
The present invention also provides methods for assaying a test sample for
products of a breast tissue gene designated as BS202, which comprises making
cDNA from nIRNA in the test sample, and detecting the cDNA as an indication of
the presence of breast tissue gene BS202. The method may include an
amplification step, wherein one or more portions of the mRNA from BS202
corresponding to the gene or fragments thereof, is amplified. Methods also are
provided for assaying for the translation products of BS202. Test samples
which
may be assayed by the methods provided herein include tissues, cells, body
fluids
and secretions. The present invention also provides reagents such as
oligonucleotide primers and polypeptides which are useful in performing these
methods.
Portions of the nucleic acid sequences disclosed herein are useful as
primers for the reverse transcription of RNA or for the amplification of cDNA;
or
as probes to determine the presence of certain mRNA sequences in test samples.
Also disclosed are nucleic acid sequences which permit the production of
encoded
polypeptide sequences which are useful as standards or reagents in diagnostic
immunoassays, as targets for pharmaceutical screening assays and/or as
components or as target sites for various therapies. Monoclonal and polyclonal
antibodies directed against at least one epitope contained within these
polypeptide
sequences are useful as delivery agents for therapeutic agents as well as for
diagnostic tests and for screening for diseases or conditions associated with
BS202, especially breast cancer. Isolation of sequences of other portions of
the
gene of interest can be accomplished utilizing probes or PCR primers derived
from
these nucleic acid sequences. This allows additional probes of the mRNA or
cDNA of interest to be established, as well as corresponding encoded
polypeptide
sequences. These additional molecules are useful in detecting, diagnosing,
staging, monitoring, prognosticating, preventing or treating, or determining
the
predisposition to diseases and conditions of the breast, such as breast
cancer,
characterized by BS202, as disclosed herein.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-12-
Techniques for determining amino acid sequence "similarity" are well-
known in the art. In general, "similarity" means the exact amino acid to amino
acid comparison of two or more polypeptides at the appropriate place, where
amino acids are identical or possess similar chemical and/or physical
properties
such as charge or hydrophobicity. A so-termed "percent similarity" then can be
determined between the compared polypeptide sequences. Techniques for
determining nucleic acid and amino acid sequence identity also are well known
in
the art and include determining the nucleotide sequence of the mRNA for that
gene
(usually via a cDNA intermediate) and determining the amino acid sequence
encoded thereby, and comparing this to a second amino acid sequence. In
general, "identity" refers to an exact nucleotide to nucleotide or amino acid
to
amino acid correspondence of two polynucleotides or poiypeptide sequences,
respectively. Two or more polynucleotide sequences can be compared by
determining their "percent identity." Two or more amino acid sequences
likewise
can be compared by determining their "percent identity." The percent identity
of
two sequences, whether nucleic acid or peptide sequences, is the number of
exact
matches between two aligned sequences divided by the length of the shorter
sequences and multiplied by 100. An approximate alignment for nucleic acid
sequences is provided by the local homology algorithm of Smith and Waterman,
Advances in Applied Mathemati~ 2:482-489 ( 1981 ). This algorithm can be
extended to use with peptide sequences using the scoring matrix developed by
Dayhoff, Atlas of Protein Sequences and Structure, M.O. Dayhoff ed., 5 suppl.
3:353-358, National Biomedical Research Foundation, Washington, D.C., USA,
and normalized by Gribskov, Nucl. Acids Res. 14(6):6745-6763 ( 1986). An
implementation of this algorithm for nucleic acid and peptide sequences is
provided by the Genetics Computer Group (Madison, WI) in their BestFit utility
application. The default parameters for this method are described in the
Wisconsin
Sequence Analysis Package Program Manual, Version 8 (1995) (available from
Genetics Computer Group, Madison, WI). Other equally suitable programs for
calculating the percent identity or similarity between sequences are generally
known in the art.
The compositions and methods described herein will enable the
identification of certain markers as indicative of a breast tissue disease or
condition; the information obtained therefrom will aid in the detecting,
diagnosing,
staging, monitoring, prognosticating, preventing or treating, or determining
diseases or conditions associated with BS202, especially breast cancer. Test
CA 02292843 1999-12-03
WO 99/02559 PCTNS98/14046
-3 3-
methods include, for example, probe assays which utilize the sequences)
provided herein and which also may utilize nucleic acid amplification methods
such as the polymerase chain reaction (PCR), the ligase chain reaction (LCR),
and
hybridization. In addition, the nucleotide sequences provided herein contain
open
reading frames from which an immunogenic epitope may be found. This epitope
is believed to be unique to the disease state or condition associated with
BS202. It
also is thought that the polynucleotides or polypeptides and protein encoded
by the
BS202 gene are useful as a marker. This marker is either elevated in disease
such
as breast cancer, altered in disease such as breast cancer, or present as a
normal
protein but appearing in an inappropriate body compartment. The uniqueness of
the epitope may be determined by (i) its immunological reactivity and
specificity
with antibodies directed against proteins and polypeptides encoded by the
BS202
gene, and (ii) its nonreactivity with any other tissue markers. Methods for
determining immunological reactivity are well-known and include, but are not
limited to, for example, radioimmunoassay (RIA), enzyme-linked
immunoabsorbent assay (ELISA), hemagglutination (HA), fluorescence
polarization immunoassay (FPIA), chemiluminescent immunoassay (CLIA) and
others. Several examples of suitable methods are described herein.
Unless otherwise stated, the following terms shall have the following
meanings:
A polynucleotide "derived from" or "specific for" a designated sequence
refers to a polynucleotide sequence which comprises a contiguous sequence of
approximately at least about 6 nucleotides, preferably at least about 8
nucleotides,
more preferably at least about 10-12 nucleotides, and even more preferably at
least
about IS-20 nucleotides corresponding, i.e., identical or complementary to, a
region of the designated nucleotide sequence. The sequence may be
complementary or identical to a sequence which is unique to a particular
polynucleotide sequence as determined by techniques known in the art.
Comparisons to sequences in databanks, for example, can be used as a method to
determine the uniqueness of a designated sequence. Regions from which
sequences may be derived, include but are not limited to, regions encoding
specific epitopes, as well as non-translated andlor non-transcribed regions.
The derived polynucleotide will not necessarily be derived physically from
the nucleotide sequence of interest under study, but may be generated in any
manner, including, but not limited to, chemical synthesis, replication,
reverse
transcription or transcription, which is based on the information provided by
the
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-14-
sequence of bases in the regions) from which the polynucleotide is derived. As
such, it may represent either a sense or an antisense orientation of the
original
polynucleotide. In addition, combinations of regions corresponding to that of
the
designated sequence may be modified in ways known in the art to be consistent
with the intended use.
A "fragment" of a specified polynucleotide refers to a polynucleotide
sequence which comprises a contiguous sequence of approximately at least about
6 nucleotides, preferably at least about 8 nucleotides, more preferably at
least
about 10-12 nucleotides, and even more preferably at least about 15-20
nucleotides corresponding, i.e., identical or complementary to, a region of
the
specified nucleotide sequence.
The term "primer" denotes a specific oligonucleotide sequence which is
complementary to a target nucleotide sequence and used to hybridize to the
target
nucleotide sequence. A primer serves as an initiation point for nucleotide
polymerization catalyzed by either DNA polymerase, RNA polymerase or reverse
transcriptase.
The term "probe" denotes a defined nucleic acid segment (or nucleotide
analog segment, e.g., PNA as defined hereinbelow) which can be used to
identify
a specific polynucleotide present in samples bearing the complementary
sequence.
"Encoded by" refers to a nucleic acid sequence which codes for a
polypeptide sequence, wherein the polypeptide sequence or a portion thereof
contains an amino acid sequence of at least 3 to 5 amino acids, more
preferably at
least 8 to 10 amino acids, and even more preferably at least 15 to 20 amino
acids
from a polypeptide encoded by the nucleic acid sequence. Also encompassed are
polypeptide sequences which are immunologically identifiable with a
polypeptide
encoded by the sequence. Thus, a "poiypeptide," "protein," or "amino acid"
sequence has at least about 50% identity, preferably about 60% identity, more
preferably about 75-85% identity, and most preferably about 90-95% or more
identity to a BS202 amino acid sequence. Further, the BS202 "polypeptide,"
"protein," or "amino acid" sequence may have at least about 60% similarity,
preferably at least about 75% similarity, more preferably about 85%
similarity,
and most preferably about 95% or more similarity to a polypeptide or amino
acid
sequence of BS202. This amino acid sequence can be selected from the group
consisting of SEQUENCE m NO 14, SEQUENCE 1D NO 15, SEQUENCE ID
NO 16, SEQUENCE l:D NO 17, SEQUENCE ID NO 18, and fragments thereof.
CA 02292843 1999-12-03
WO 99102559 PCTNS98/14046
-15-
A "recombinant polypeptide," "recombinant protein," or "a polypeptide
produced by recombinant techniques," which terms may be used interchangeably
herein, describes a polypeptide which by virtue of its origin or manipulation
is not
associated with all or a portion of the polypeptide with which it is
associated in
nature and/or is linked to a polypeptide other than that to which it is linked
in
nature. A recombinant or encoded polypeptide or protein is not necessarily
translated from a designated nucleic acid sequence. It also may be generated
in
any manner, including chemical synthesis or expression of a recombinant
expression system.
The term "synthetic peptide" as used herein means a polymeric form of
amino acids of any length, which may be chemically synthesized by methods well-
known to the routineer. These synthetic peptides are useful in various
applications.
The term "polynucleotide" as used herein means a polymeric form of
nucleotides of any length, either ribonucleotides or deoxyribonucleotides.
This
term refers only to the primary structure of the molecule. Thus, the term
includes
double- and single-stranded DNA, as well as double- and single-stranded RNA.
It also includes modifications, such as methylation or capping and unmodified
forms of the polynucleotide. The terms "polynucleotide," "oligomer,"
"oligonucleotide," and "oligo" are used interchangeably herein.
"A sequence corresponding to a cDNA" means that the sequence contains a
polynucleotide sequence that is identical or complementary to a sequence in
the
designated DNA. The degree (or "percent") of identity or complementarity to
the
cDNA will be approximately 50% or greater, preferably at least about 70% or
greater, and more preferably at least about 90% or greater. The sequence that
corresponds to the identified cDNA will be at least about 50 nucleotides in
length,
preferably at least about 60 nucleotides in length, and more preferably at
least
about 70 nucleotides in length. The correspondence between the gene or gene
fragment of interest and the cDNA can be determined by methods known in the
art
and include, for example, a direct comparison of the sequenced material with
the
cDNAs described, or hybridization and digestion with single strand nucleases,
followed by size determination of the digested fragments.
"Purified polynucleotide" refers to a polynucleotide of interest or fragment
thereof which is essentially free, e.g., contains less than about 50%,
preferably
less than about 70%, and more preferably less than about 90%, of the protein
with
which the polynucleotide is naturally associated. Techniques for purifying
CA 02292843 1999-12-03
WO 99102559 PCT/US98/14046
-16-
polynucleotides of interest are well-known in the art and include, for
example,
disruption of the cell containing the polynucleotide with a chaotropic agent
and
separation of the polynucleotide(s) and proteins by ion-exchange
chromatography,
affinity chromatography and sedimentation according to density.
"Purified polypeptide" or "purified protein" means a polypeptide of interest
or fragment thereof which is essentially free of, e.g., contains less than
about
50%, preferably less than about 70%, and more preferably less than about 90%,
cellular components with which the polypeptide of interest is naturally
associated.
Methods for purifying polypeptides of interest are known in the art.
The term "isolated" means that the material is removed from its original
environment (e.g., the natural environment if it is naturally occurring). For
example, a naturally-occurring polynucleotide or polypeptide present in a
living
animal is not isolated, but the same polynucleotide or DNA or polypeptide,
which
is separated from some or all of the coexisting materials in the natural
system, is
isolated. Such polynucleotide could be part of a vector and/or such
polynucleotide
or polypeptide could be part of a composition, and still be isolated in that
the
vector or composition is not part of its natural environment.
"Polypeptide" and "protein" are used interchangeably herein and indicate at
least one molecular chain of amino acids linked through covalent and/or non-
covalent bonds. The terms do not refer to a specific length of the product.
Thus
peptides, oligopeptides and proteins are included within the definition of
polypeptide. The terms include post-translational modifications of the
polypeptide, for example, glycosylations, acetylations, phosphorylations and
the
like. In addition, protein fragments, analogs, mutated or variant proteins,
fusion
proteins and the like are included within the meaning of polypeptide.
A "fragment" of a specified polypeptide refers to an amino acid sequence
which comprises at least about 3-S amino acids, more preferably at least about
8-
10 amino acids, and even more preferably at least about 15-20 amino acids
derived
from the specified polypeptide.
"Recombinant host cells," "host cells," "cells," "cell lines," "cell
cultures,"
and other such terms denoting microorganisms or higher eukaryotic cell lines
cultured as unicellular entities refer to cells which can be, or have been,
used as
recipients for recombinant vector or other transferred DNA, and include the
original progeny of the original cell which has been transfected.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98114046
-17-
As used herein "replicon" means any genetic element, such as a plasmid, a
chromosome or a virus, that behaves as an autonomous unit of polynucleotide
replication within a cell.
A "vector" is a replicon in which another polynucleotide segment is
attached, such as to bring about the replication and/or expression of the
attached
segment.
The term "control sequence" refers to a polynucleotide sequence which is
necessary to effect the expression of a coding sequence to which it is
ligated. The
nature of such control sequences differs depending upon the host organism. In
prokaryotes, such control sequences generally include a promoter, a ribosomal
binding site and terminators; in eukaryotes, such control sequences generally
include promoters, terminators and, in some instances, enhancers. The term
"control sequence" thus is intended to include at a minimum all components
whose
presence is necessary for expression, and also may include additional
components
whose presence is advantageous, for example, leader sequences.
"Operabiy linked" refers to a situation wherein the components described
are in a relationship permitting them to function in their intended manner.
Thus,
for example, a control sequence "operably linked" to a coding sequence is
ligated
in such a manner that expression of the coding sequence is achieved under
conditions compatible with the control sequence.
The term "open reading frame" or "ORF" refers to a region of a
polynucleotide sequence which encodes a polypeptide. This region may represent
a portion of a coding sequence or a total coding sequence.
A "coding sequence" is a polynucleotide sequence which is transcribed
into mRNA and translated into a polypeptide when placed under the control of
appropriate regulatory sequences. The boundaries of the coding sequence are
determined by a translation start codon at the 5' -terminus and a translation
stop
codon at the 3' -terminus. A coding sequence can include, but is not limited
to,
mRNA, cDNA and recombinant polynucleotide sequences.
The term "immunologically identifiable with/as" refers to the presence of
epitope(s) and polypeptide(s) which also are present in and are unique to the
designated polypeptide(s). Immunological identity may be determined by
antibody binding and/or competition in binding. These techniques are known to
the routineer and also are described herein. The uniqueness of an epitope also
can
be determined by computer searches of known data banks, such as GenBank, for
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-18-
the polynucleotide sequence which encodes the epitope and by amino acid
sequence comparisons with other known proteins.
As used herein, "epitope" means an antigenic determinant of a polypeptide
or protein. Conceivably, an epitope can comprise three amino acids in a
spatial
conformation which is unique to the epitope. Generally, an epitope consists of
at
least five such amino acids and more usually, it consists of at least eight to
ten
amino acids. Methods of examining spatial conformation are known in the art
and
include, for example, x-ray crystallography and two-dimensional nuclear
magnetic
resonance.
A "conformational epitope" is an epitope that is comprised of a specific
juxtaposition of amino acids in an immunologically recognizable structure,
such
amino acids being present on the same polypeptide in a contiguous or non-
contiguous order or present on different polypeptides.
A polypeptide is "immunologically reactive" with an antibody when it
binds to an antibody due to antibody recognition of a specific epitope
contained
within the polypeptide. Immunological reactivity may be determined by antibody
binding, more particularly, by the kinetics of antibody binding, and/or by
competition in binding using as competitors) a known polypeptide(s) containing
an epitope against which the antibody is directed. The methods for determining
whether a polypeptide is immunologically reactive with an antibody are known
in
the art.
As used herein, the term "immunogenic polypeptide containing an epitope
of interest" means naturally occurring polypeptides of interest or fragments
thereof, as well as poiypeptides prepared by other means, for example, by
chemical synthesis or the expression of the polypeptide in a recombinant
organism.
The term "transfection" refers to the introduction of an exogenous
polynucleotide into a prokaryotic or eucaryotic host cell, irrespective of the
method
used for the introduction. The term "transfection" refers to both stable and
transient introduction of the polynucleotide, and encompasses direct uptake of
polynucleotides, transformation, transduction, and f-mating. Once introduced
into
the host cell, the exogenous polynucleotide may be maintained as a non-
integrated
replicon, for example, a plasmid, or alternatively, may be integrated into the
host
genome.
"Treatment" refers to prophylaxis and/or therapy.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-19-
The term "individual" as used herein refers to vertebrates, particularly
members of the mammalian species and includes, but is not limited to, domestic-
animals, sports animals, primates and humans; more particularly, the term
refers
to humans.
S The term "sense strand" or "plus strand" (or "+") as used herein denotes a
nucleic acid that contains the sequence that encodes the polypeptide. The term
"antisense strand" or "minus strand" (or "-") denotes a nucleic acid that
contains a
sequence that is complementary to that of the "plus" strand.
The term "test sample" refers to a component of an individual's body
which is the source of the analyte (such as antibodies of interest or antigens
of
interest). These components are well known in the art. A test sample is
typically
anything suspected of containing a target sequence. Test samples can be
prepared
using methodologies well known in the art such as by obtaining a specimen from
an individual and, if necessary, disrupting any cells contained thereby to
release
target nucleic acids. These test samples include biological samples which can
be
tested by the methods of the present invention described herein and include
human
and animal body fluids such as whole blood, serum, plasma, cerebrospinal
fluid,
sputum, bronchial washing, bronchial aspirates, urine, lymph fluids, and
various
external secretions of the respiratory, intestinal and genitourinary tracts,
tears,
saliva, milk, white blood cells, myelomas and the like; biological fluids such
as
cell culture supernatants; tissue specimens which may be fixed; and cell
specimens
which may be fixed.
"Purified product" refers to a preparation of the product which has been
isolated from the cellular constituents with which the product is normally
associated and from other types of cells which may be present in the sample of
interest.
"PNA" denotes a "peptide nucleic acid analog" which may be utilized in a
procedure such as an assay described herein to determine the presence of a
target.
"MA" denotes a "morpholino analog" which may be utilized in a procedure such
as an assay described herein to determine the presence of a target. See, for
example, U.S. Patent No. 5,378,841. PNAs are neutrally charged moieties
which can be directed against RNA targets or DNA. PNA probes used in assays
in place of, for example, the DNA probes of the present invention, offer
advantages not achievable when DNA probes are used. These advantages include
manufacturability, large scale labeling, reproducibility, stability,
insensitivity to
changes in ionic strength and resistance to enzymatic degradation which is
present
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-20-
in methods utilizing DNA or RNA. These PNAs can be labeled with ("attached
to") such signal generating compounds as fluorescein, radionucleotides,
chemiluminescent compounds and the like. PNAs or other nucleic acid analogs
such as MAs thus can be used in assay methods in place of DNA or RNA.
Although assays are described herein utilizing DNA probes, it is within the
scope
of the routineer that PNAs or MAs can be substituted for RNA or DNA with
appropriate changes if and as needed in assay reagents.
"Analyte," as used herein, is the substance to be detected which may be
present in the test sample. The analyte can be any substance for which there
exists
a naturally occurring specific binding member (such as an antibody), or for
which
a specific binding member can be prepared. Thus, an analyte is a substance
that
can bind to one or more specific binding members in an assay. "Analyte" also
includes any antigenic substances, haptens, antibodies and combinations
thereof.
As a member of a specific binding pair, the analyte can be detected by means
of
naturally occurring specific binding partners (pairs) such as the use of
intrinsic
factor protein as a member of a specific binding pair for the determination of
Vitamin B 12, the use of folate-binding protein to determine folic acid, or
the use
of a lectin as a member of a specific binding pair for the determination of a
carbohydrate. The analyte can include a protein, a polypeptide, an amino acid,
a
nucleotide target and the like. The analyte can be soluble in a body fluid
such as
blood, blood plasma or serum, urine or the like. The analyte can be in a
tissue,
either on a cell surface or within a cell. The analyte can be on or in a cell
dispersed
in a body fluid such as blood, urine, breast aspirate, or obtained as a biopsy
sample.
The terms "diseases of the breast," "breast disease," and "condition of the
breast" are used interchangeably herein to refer to any disease or condition
of the
breast including, but not limited to, atypical hyperplasia, fibroadenoma,
cystic
breast disease, and cancer.
"Breast cancer," as used herein, refers to any malignant disease of the
breast including, but not limited to, ductal carcinoma in situ, lobular
carcinoma in
situ, infiltrating ductal carcinoma, medullary carcinoma, tubular carcinoma,
mucinous carcinoma, infiltrating lobular carcinoma, infiltrating
comedocarcinoma
and inflammatory carcinoma.
An "Expressed Sequence Tag" or "EST" refers to the partial sequence of a
cDNA insert which has been made by reverse transcription of mRNA extracted
from a tissue followed by insertion into a vector.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-21-
A "transcript image" refers to a table or list giving the quantitative
distribution of ESTs in a library and represents the genes active in the
tissue from
which the library was made.
The present invention provides assays which utilize specific binding
members. A "specific binding member," as used herein, is a member of a
specific
binding pair. That is, two different molecules where one of the molecules,
through chemical or physical means, specifically binds to the second molecule.
Therefore, in addition to antigen and antibody specific binding pairs of
common
immunoassays, other specific binding pairs can include biotin and avidin,
carbohydrates and lectins, complementary nucleotide sequences, effector and
receptor molecules, cofactors and enzymes, enzyme inhibitors, and enzymes and
the like. Furthermore, specific binding pairs can include members that are
analogs
of the original specific binding members, for example, an analyte-analog.
Immunoreactive specific binding members include antigens, antigen fragments,
I S antibodies and antibody fragments, both monoclonal and polyclonal and
complexes thereof, including those formed by recombinant DNA molecules.
The term "hapten," as used herein, refers to a partial antigen or non-protein
binding member which is capable of binding to an antibody, but which is not
capable of eliciting antibody formation unless coupled to a carrier protein.
A "capture reagent," as used herein, refers to an unlabeled specific binding
member which is specific either for the analyte as in a sandwich assay, for
the
indicator reagent or analyte as in a competitive assay, or for an ancillary
specific
binding member, which itself is specific for the analyte, as in an indirect
assay.
The capture reagent can be directly or indirectly bound to a solid phase
material
before the performance of the assay or during the performance of the assay,
thereby enabling the separation of immobilized complexes from the test sample.
The "indicator reagent" comprises a "signal-generating compound"
("label") which is capable of generating and generates a measurable signal
detectable by external means, conjugated ("attached") to a specific binding
member. In addition to being an antibody member of a specific binding pair,
the
indicator reagent also can be a member of any specific binding pair, including
either hapten-anti-hapten systems such as biotin or anti-biotin, avidin or
biotin, a
carbohydrate or a lectin, a complementary nucleotide sequence, an effector or
a
receptor molecule, an enzyme cofactor and an enzyme, an enzyme inhibitor or an
enzyme and the like. An immunoreactive specific binding member can be an
antibody, an antigen, or an antibody/antigen complex that is capable of
binding
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-22-
either to the polypeptide of interest as in a sandwich assay, to the capture
reagent
as in a competitive assay, or to the ancillary specific binding member as in
an
indirect assay. When describing probes and probe assays, the term "reporter
molecule" may be used. A reporter molecule comprises a signal generating
compound as described hereinabove conjugated to a specific binding member of a
specific binding pair, such as carbazole or adamantine.
The various "signal-generating compounds" (labels) contemplated include
chromagens, catalysts such as enzymes, luminescent compounds such as
fluorescein and rhodamine, chemiluminescent compounds such as dioxetanes,
acridiniums, phenanthridiniums and luminol, radioactive elements and direct
visual labels. Examples of enzymes include alkaline phosphatase, horseradish
peroxidase, beta-galactosidase and the like. The selection of a particular
label is
not critical, but it must be capable of producing a signal either by itself or
in
conjunction with one or more additional substances.
"Solid phases" ("solid supports") are known to those in the art and include
the walls of wells of a reaction tray, test tubes, polystyrene beads, magnetic
or
non-magnetic beads, nitrocellulose strips, membranes, microparticles such as
latex
particles, sheep (or other animal) red blood cells and Duracytes° (red
blood cells
"fixed" by pyruvic aldehyde and formaldehyde, available from Abbott
Laboratories, Abbott Park, IL) and others. The "solid phase" is not critical
and
can be selected by one skilled in the art. Thus, latex particles,
microparticles,
magnetic or non-magnetic beads, membranes, plastic tubes, walls of microtiter
wells, glass or silicon chips, sheep (or other suitable animal's) red blood
cells and
Duracytes° are ill suitable examples. Suitable methods for immobilizing
peptides
on solid phases include ionic, hydrophobic, covalent interactions and the
Iike. A
"solid phase," as used herein, refers to any material which is insoluble, or
can be
made insoluble by a subsequent reaction. The solid phase can be chosen for its
intrinsic ability to attract and immobilize the capture reagent.
Alternatively, the
solid phase can retain an additional receptor which has the ability to attract
and
immobilize the capture reagent. The additional receptor can include a charged
substance that is oppositely charged with respect to the capture reagent
itself or to
a charged substance conjugated to the capture reagent. As yet another
alternative,
the receptor molecule can be any specific binding member which is immobilized
upon (attached to) the solid phase and which has the ability to immobilize the
capture reagent through a specific binding reaction. The receptor molecule
enables
the indirect binding of the capture reagent to a solid phase material before
the
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-23-
performance of the assay or during the performance of the assay. The solid
phase
thus can be a plastic, derivatized plastic, magnetic or non-magnetic metal,
glass or
silicon surface of a test tube, microtiter well, sheet, bead, microparticle,
chip,
sheep (or other suitable animal's) red blood cells, Duracytes° and
other
configurations known to those of ordinary skill in the art.
It is contemplated and within the scope of the present invention that the
solid phase also can comprise any suitable porous material with sufficient
porosity
to allow access by detection antibodies and a suitable surface affinity to
bind
antigens. Microporous structures generally are preferred, but materials with a
gel
structure in the hydrated state may be used as well. Such useful solid
supports
include, but are not limited to, nitrocellulose and nylon. It is contemplated
that
such porous solid supports described herein preferably are in the form of
sheets of
thickness from about 0.01 to 0.5 mm, preferably about 0.1 mm. The pore size
may vary within wide limits and preferably is from about 0.025 to 15 microns,
especially from about 0.15 to 15 microns. The surface of such supports may be
activated by chemical processes which cause covalent linkage of the antigen or
antibody to the support. The irreversible binding of the antigen or antibody
is
obtained, however, in general, by adsorption on the porous material by poorly
understood hydrophobic forces. Other suitable solid supports are known in the
art.
R a en .
The present invention provides reagents such as polynucleotide sequences
derived from a breast tissue of interest and designated as BS202, polypeptides
encoded thereby and antibodies specific for these polypeptides. The present
invention also provides reagents such as oligonucleotide fragments derived
from
the disclosed polynucleotides and nucleic acid sequences complementary to
these
polynucleotides. The polynucleotides, polypeptides, or antibodies of the
present
invention may be used to provide information leading to the detecting,
diagnosing,
staging, monitoring, prognosticating, preventing or treating of, or
determining the
predisposition to, diseases and conditions of the breast, such as breast
cancer.
The sequences disclosed herein represent unique polynucleotides which can be
used in assays or for producing a specific profile of gene transcription
activity.
Such assays are disclosed in European Patent Number 0373203B 1 and
International Publication No. WO 95/11995.
Selected BS202-derived poiynucleotides can be used in the methods
described herein for the detection of normal or altered gene expression. Such
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-24-
methods may employ BS202 polynucleotides or oligonucleotides, fragments or
derivatives thereof, or nucleic acid sequences complementary thereto.
The polynucleotides disclosed herein, their complementary sequences, or
fragments of either, can be used in assays to detect, amplify or quantify
genes,
nucleic acids, cDNAs or mRNAs relating to breast tissue disease and conditions
associated therewith. They also can be used to identify an entire or partial
coding
region of a BS202 polypeptide. They further can be provided in individual
containers in the form of a kit for assays, or provided as individual
compositions.
If provided in a kit for assays, other suitable reagents such as buffers,
conjugates
and the like may be included.
The polynucleotide may be in the form of RNA or DNA. Polynucleotides
in the form of DNA, cDNA, genomic DNA, nucleic acid analogs and synthetic
DNA are within the scope of the present invention. The DNA may be double-
stranded or single-stranded, and if single stranded, may be the coding (sense)
strand or non-coding (anti-sense) strand. The coding sequence which encodes
the
polypeptide may be identical to the coding sequence provided herein or may be
a
different coding sequence which coding sequence, as a result of the redundancy
or
degeneracy of the genetic code, encodes the same polypeptide as the DNA
provided herein.
This polynucleotide may include only the coding sequence for the
polypeptide, or the coding sequence for the polypeptide and an additional
coding
sequence such as a leader or secretory sequence or a proprotein sequence, or
the
coding sequence for the polypeptide {and optionally an additional coding
sequence) and non-coding sequence, such as a non-coding sequence 5' and/or 3'
of the coding sequence for the polypeptide.
In addition, the invention includes variant polynucleotides containing
modifications such as polynucleotide deletions, substitutions or additions;
and any
polypeptide modification resulting from the variant polynucleotide sequence. A
polynucleotide of the present invention also may have a coding sequence which
is
a naturally occurring allelic variant of the coding sequence provided herein.
In addition, the coding sequence for the polypeptide may be fused in the
same reading frame to a polynucleotide sequence which aids in expression and
secretion of a polypeptide from a host cell, for example, a leader sequence
which
functions as a secretory sequence for controlling transport of a polypeptide
from
the cell. The polypeptide having a leader sequence is a preprotein and may
have
the leader sequence cleaved by the host cell to form the polypeptide. The
CA 02292843 1999-12-03
WO 99/02559 PCT/LJS98/14046
-25-
polynucleotides may also encode for a proprotein which is the protein plus
additional 5' amino acid residues. A protein having a prosequence is a
proprotein
and may, in some cases, be an inactive form of the protein. Once the
prosequence
is cleaved, an active protein remains. Thus, the polynucleotide of the present
invention may encode for a protein, or for a protein having a prosequence, or
for a
protein having both a presequence (leader sequence) and a prosequence.
The polynucleotides of the present invention may also have the coding
sequence fused in frame to a marker sequence which allows for purification of
the
polypeptide of the present invention. The marker sequence rnay be a hexa-
histidine tag supplied by a pQE-9 vector to provide for purification of the
polypeptide fused to the marker in the case of a bacterial host, or, for
example, the
marker sequence may be a hemagglutinin (HA) tag when a mammalian host, e.g. a
COS-7 cell line, is used. The HA tag corresponds to an epitope derived from
the
influenza hemagglutinin protein. See, for example, I. Wilson et al., Cell
37:767
( 1984).
It is contemplated that polynucleotides will be considered to hybridize to
the sequences provided herein if there is at least 50%, preferably at least
70%, and
more preferably at least 90% identity between the polynucleotide and the
sequence.
The present invention also provides an antibody produced by using a
purified BS202 polypeptide of which at least a portion of the polypeptide is
encoded by a BS202 polynucleotide selected from the polynucleotides provided
herein. These antibodies may be used in the methods provided herein for the
detection of BS202 antigen in test samples. The presence of BS202 antigen in
the
test samples is indicative of the presence of a breast disease or condition.
The
antibody also may be used for therapeutic purposes, for example, in
neutralizing
the activity of BS202 polypeptide in conditions associated with altered or
abnormal expression.
The present invention further relates to a BS202 polypeptide which has the
deduced amino acid sequence as provided herein, as well as fragments, analogs
and derivatives of such polypeptide. The polypeptide of the present invention
rnay
be a recombinant polypeptide, a natural purified polypeptide or a synthetic
polypeptide. The fragment, derivative or analog of the BS202 polypeptide may
be
one in which one or more of the amino acid residues is substituted with a
conserved or non-conserved amino acid residue (preferably a conserved amino
acid residue) and such substituted amino acid residue may or may not be one
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-26-
encoded by the genetic code; or it may be one in which one or more of the
amino
acid residues includes a substituent group; or it may be one in which the
polypeptide is fused with another compound, such as a compound to increase the
half-life of the polypeptide (for example, polyethylene glycol); or it may be
one in
which the additional amino acids are fused to the polypeptide, such as a
leader or
secretory sequence or a sequence which is employed for purification of the
polypeptide or a proprotein sequence. Such fragments, derivatives and analogs
are within the scope of the present invention. The polypeptides and
polynucleotides of the present invention are provided preferably in an
isolated
form and preferably purified.
Thus, a polypeptide of the present invention may have an amino acid
sequence that is identical to that of the naturally occurring polypeptide or
that is
different by minor variations due to one or more amino acid substitutions. The
variation may be a "conservative change" typically in the range of about 1 to
5
amino acids, wherein the substituted amino acid has similar structural or
chemical
properties, e.g., replacement of leucine with isoleucine or threonine with
serine.
In contrast, variations may include nonconservative changes, e.g., replacement
of
a glycine with a tryptophan. Similar minor variations may also include amino
acid
deletions or insertions, or both. Guidance in determining which and how many
amino acid residues may be substituted, inserted or deleted without changing
biological or immunological activity may be found using computer programs well
known in the art, for example, DNASTAR software (DNASTAR Inc., Madison
WI).
Probes constructed according to the polynucleotide sequences of the
present invention can be used in various assay methods to provide various
types
of analysis. For example, such probes can be used in fluorescent in sits
l~bridization (FISH) technology to perform chromosomal analysis, and used to
identify cancer-specific structural alterations in the chromosomes, such as
deletions or translocations that are visible from chromosome spreads or
detectable
using PCR-generated and/or allele specific oligonucleotides probes, allele
specific
amplification or by direct sequencing. Probes also can be labeled with
radioisotopes, directly- or indirectly- detectable haptens, or fluorescent
molecules,
and utilized for in situ hybridization studies to evaluate the mRNA expression
of
the gene comprising the polynucleotide in tissue specimens or cells.
This invention also provides teachings as to the production of the
polynucleotides and polypeptides provided herein.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-27-
Probe Assavs
The sequences provided herein may be used to produce probes which can
be used in assays for the detection of nucleic acids in test samples. The
probes
may be designed from conserved nucleotide regions of the polynucleotides of
interest or from non-conserved nucleotide regions of the polynucieotide of
interest. The design of such probes for optimization in assays is within the
skill of
the routineer. Generally, nucleic acid probes are developed from non-conserved
or unique regions when maximum specificity is desired, and nucleic acid probes
are developed from conserved regions when assaying for nucleotide regions that
are closely related to, for example, different members of a multi-gene family
or in
related species like mouse and man.
The polymerise chain reaction (PCR) is a technique for amplifying a
desired nucleic acid sequence (target) contained in a nucleic acid or mixture
thereof. In PCR, a pair of primers are employed in excess to hybridize to the
complementary strands of the target nucleic acid. The primers are each
extended
by a polymerise using the target nucleic acid as a template. The extension
products become target sequences themselves, following dissociation from the
original target strand. New primers then are hybridized and extended by a
polymerise, and the cycle is repeated to geometrically increase the number of
target sequence molecules. PCR is disclosed in U.S. Patents 4,683,195 and
4,683,202.
The Ligase Chain Reaction (LCR) is an alternate method for nucleic acid
amplification. In LCR, probe pairs are used which include two primary (first
and
second) and two secondary (third and fourth) probes, all of which are employed
in
molar excess to target. The first probe hybridizes to a first segment of the
target
strand, and the second probe hybridizes to a second segment of the target
strand,
the first and second segments being contiguous so that the primary probes abut
one another in 5' phosphate-3' hydroxyl relationship, and so that a ligase can
covalently fuse or ligate the two probes into a fused product. In addition, a
third
(secondary) probe can hybridize to a portion of the first probe and a fourth
(secondary) probe can hybridize to a portion of the second probe in a similar
abutting fashion. Of course, if the target is initially double stranded, the
secondary probes also will hybridize to the target complement in the first
instance.
Once the ligated strand of primary probes is separated from the target strand,
it
will hybridize with the third and fourth probes which can be ligated to form a
complementary, secondary ligated product. It is important to realize that the
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-28-
ligated products are functionally equivalent to either the target or its
complement.
By repeated cycles of hybridization and ligation, amplification of the target
sequence is achieved. This technique is described more completely in EP-A- 320
308 to K. Backman published June 16, 1989 and EP-A-439 182 to K. Backman
et al., published July 31, 1991.
For amplification of mRNAs, it is within the scope of the present invention
to reverse transcribe mRNA into cDNA followed by polymerase chain reaction
(RT-PCR); or, to use a single enzyme for both steps as described in U.S.
Patent
No. 5,322,770; or reverse transcribe mRNA into cDNA followed by asymmetric
gap ligase chain reaction (RT-AGLCR) as described by R.L. Marshall et al., PCR
Methods and Applications 4: 80-84 (1994).
Other known amplification methods which can be utilized herein include
but are not limited to the so-called "NASBA" or "3SR" technique described by
J.C. Guatelli et al., PNAS USA 87:1874-1878 (1990) and also described by J.
Compton, Nature 350 (No. 6313):91-92 ( 1991 ); Q-beta amplification as
described
in published European Patent Application (EPA) No. 4544610; strand
displacement amplification (as described in G.T. Walker et al., Clin. Chem.
42:9-
13 [1996]) and European Patent Application No. 684315; and target mediated
amplification, as described in International Publication No. WO 93/22461.
Detection of BS202 may be accomplished using any suitable detection
method, including those detection methods which are currently well known in
the
art, as well as detection strategies which may evolve later. See, for example,
Caskey et al., U.S. Patent No. 5,582,989, Gelfand et al., U.S. Patent No.
5,210,015. Examples of such detection methods include target amplification
methods as well as signal amplification technologies. An example of presently
known detection methods would include the nucleic acid amplification
technologies referred to as PCR, LCR, NASBA, SDA, RCR and TMA. See, for
example, Caskey et al., U.S. Patent No. 5,582,989, Gelfand et al., U.S. Patent
No. 5,210,015. Detection may also be accomplished using signal amplification
such as that disclosed in Snitman et al., U.S. Patent No. 5,273,882. While the
amplification of target or signal is preferred at present, it is contemplated
and
within the scope of the present invention that ultrasensitive detection
methods
which do not require amplification can be utilized herein.
Detection, both amplified and non-amplified, may be (combined) carried
out using a variety of heterogeneous and homogeneous detection formats.
Examples of heterogeneous detection formats are disclosed in Snitman et al.,
U.S.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-29-
Patent No. 5,273,882, Albarella et al., in EP-84114441.9, Urdea et al., U.S.
Patent No. 5,124,246, Ullman et al. U.S. Patent No. 5,185,243 and Kourilsky et
al., U.S. Patent No. 4,581,333. Examples of homogeneous detection formats are
disclosed in, Caskey et al., U.S. Patent No. 5,582,989, Gelfand et al., U.S.
Patent No. 5,210,015. Also contemplated and within the scope of the present
invention is the use of multiple probes in the hybridization assay, which use
improves sensitivity and amplification of the BS202 signal. See, for example,
Caskey et al., U.S. Patent No. 5,582,989, Gelfand et al., U.S. Patent No.
5,210,01 S.
In one embodiment, the present invention generally comprises the steps of
contacting a test sample suspected of containing a target polynucleotide
sequence
with amplification reaction reagents comprising an amplification primer, and a
detection probe that can hybridize with an internal region of the amplicon
sequences. Probes and primers employed according to the method provided
I S herein are labeled with capture and detection labels, wherein probes are
labeled
with one type of label and primers are labeled with another type of label.
Additionally, the primers and probes are selected such that the probe sequence
has
a lower melt temperature than the primer sequences. The amplification
reagents,
detection reagents and test sample are placed under amplification conditions
whereby, in the presence of target sequence, copies of the target sequence (an
amplicon) are produced. In the usual case, the amplicon is double stranded
because primers are provided to amplify a target sequence and its
complementary
strand. The double stranded amplicon then is thermally denatured to produce
single stranded amplicon members. Upon formation of the single stranded
amplicon members, the mixture is cooled to allow the formation of complexes
between the probes and single stranded amplicon members.
As the single stranded amplicon sequences and probe sequences are
cooled, the probe sequences preferentially bind the single stranded amplicon
members. This finding is counterintuitive given that the probe sequences
generally are selected to be shorter than the primer sequences and therefore
have a
lower melt temperature than the primers. Accordingly, the melt temperature of
the
amplicon produced by the primers should also have a higher melt temperature
than
the probes. Thus, as the mixture cools, the re-formation of the double
stranded
amplicon would be expected. As previously stated, however, this is not the
case.
The probes are found to preferentially bind the single stranded amplicon
members.
CA 02292843 1999-12-03
WO 99/02559 PCTNS98/14046
-30-
Moreover, this preference of probe/single stranded amplicon binding exists
even
when the primer sequences are added in excess of the probes.
After the probe/single stranded amplicon member hybrids are formed, they
are detected. Standard heterogeneous assay formats are suitable for detecting
the
hybrids using the detection labels and capture labels present on the primers
and
probes. The hybrids can be bound to a solid phase reagent by virtue of the
capture
label and detected by virtue of the detection label. In cases where the
detection
label is directly detectable, the presence of the hybrids on the solid phase
can be
detected by causing the label to produce a detectable signal, if necessary,
and
detecting the signal. In cases where the label is not directly detectable, the
captured hybrids can be contacted with a conjugate, which generally comprises
a
binding member attached to a directly detectable label. The conjugate becomes
bound to the complexes and the conjugate's presence on the complexes can be
detected with the directly detectable label. Thus, the presence of the hybrids
on
the solid phase reagent can be determined. Those skilled in the art will
recognize
that wash steps may be employed to wash away unhybridized amplicon or probe
as well as unbound conjugate.
Although the target sequence is described as single stranded, it also is
contemplated to include the case where the target sequence is actually double
stranded but is merely separated from its complement prior to hybridization
with
the amplification primer sequences. In the case where PCR is employed in this
method, the ends of the target sequences are usually known. In cases where LCR
or a modification thereof is employed in the preferred method, the entire
target
sequence is usually known. Typically, the target sequence is a nucleic acid
sequence such as, for example, RNA or DNA.
The method provided herein can be used in well-known amplification
reactions that include thermal cycle reaction mixtures, particularly in PCR
and gap
LCR (GLCR). Amplification reactions typically employ primers to repeatedly
generate copies of a target nucleic acid sequence, which target sequence is
usually
a small region of a much larger nucleic acid sequence. Primers are themselves
nucleic acid sequences that are complementary to regions of a target sequence.
Under amplification conditions, these primers hybridize or bind to the
complementary regions of the target sequence. Copies of the target sequence
typically are generated by the process of primer extension and/or ligation
which
utilizes enzymes with polymerase or ligase activity, separately or in
combination,
to add nucleotides to the hybridized primers and/or ligate adjacent probe
pairs.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-31-
The nucleotides that are added to the primers or probes, as monomers or
preformed oligomers, are also complementary to the target sequence. Once the -
primers or probes have been sufficiently extended and/or ligated, they are
separated from the target sequence, for example, by heating the reaction
mixture to
a "melt temperature" which is one in which complementary nucleic acid strands
dissociate. Thus, a sequence complementary to the target sequence is formed.
A new amplification cycle then can take place to further amplify the
number of target sequences by separating any double stranded sequences,
allowing primers or probes to hybridize to their respective targets, extending
and/or ligating the hybridized primers or probes and re-separating. The
complementary sequences that are generated by amplification cycles can serve
as
templates for primer extension or filling the gap of two probes to further
amplify
the number of target sequences. Typically, a reaction mixture is cycled
between
and 100 times, more typically, a reaction mixture is cycled between 25 and 50
15 times. The numbers of cycles can be determined by the routineer. In this
manner,
multiple copies of the target sequence and its complementary sequence are
produced. Thus, primers initiate amplification of the target sequence when it
is
present under amplification conditions.
Generally, two primers which are complementary to a portion of a target
20 strand and its complement are employed in PCR. For LCR, four probes, two of
which are complementary to a target sequence and two of which are similarly
complementary to the target's complement, are generally employed. In addition
to
the primer sets and enzymes previously mentioned, a nucleic acid amplification
reaction mixture may also comprise other reagents which are well known and
include but are not limited to: enzyme cofactors such as manganese; magnesium;
salts; nicotinamide adenine dinucleotide (NAD); and deoxynucleotide
triphosphates (dNTPs) such as, for example, deoxyadenine triphosphate,
deoxyguanine triphosphate, deoxycytosine triphosphate and deoxythymine
triphosphate.
While the amplification primers initiate amplification of the target
sequence, the detection (or hybridization) probe is not involved in
amplification.
Detection probes are generally nucleic acid sequences or uncharged nucleic
acid
analogs such as, for example, peptide nucleic acids which are disclosed in
International Publication No. WO 92/20702; morpholino analogs which are
described in U.S. Patents Nos 5,185,444, 5,034,506 and 5,142,047; and the
like. Depending upon the type of label carried by the probe, the probe is
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-32-
employed to capture or detect the amplicon generated by the amplification
reaction.
The probe is not involved in amplification of the target sequence and
therefore may
have to be rendered "non-extendible" in that additional dNTPs cannot be added
to
the probe. In and of themselves, analogs usually are non-extendible and
nucleic
acid probes can be rendered non-extendible by modifying the 3' end of the
probe
such that the hydroxyl group is no longer capable of participating in
elongation.
For example, the 3' end of the probe can be functionalized with the capture or
detection label to thereby consume or otherwise block the hydroxyl group.
Alternatively, the 3' hydroxyl group simply can be cleaved, replaced or
modified.
U.S. Patent Application Serial No. 07/049,061, filed April 19, describes
modifications which can be used to render a probe non-extendible.
The ratio of primers to probes is not important. Thus, either the probes or
primers can be added to the reaction mixture in excess whereby the
concentration
of one would be greater than the concentration of the other. Alternatively,
primers
and probes can be employed in equivalent concentrations. Preferably, however,
the primers are added to the reaction mixture in excess of the probes. Thus,
primer to probe ratios of, for example, 5:1 and 20:1, are preferred.
While the length of the primers and probes can vary, the probe sequences
are selected such that they have a lower melt temperature than the primer
sequences. Hence, the primer sequences are generally longer than the probe
sequences. Typically, the primer sequences are in the range of between 20 and
50
nucleotides long, more typically in the range of between 20 and 30 nucleotides
long. The typical probe is in the range of between 10 and 25 nucleotides long.
Various methods for synthesizing primers and probes are well known in
the art. Similarly, methods for attaching labels to primers or probes are also
well
known in the art. For example, it is a matter of routine to synthesize desired
nucleic acid primers or probes using conventional nucleotide phosphoramidite
chemistry and instruments available from Applied Biosystems, Inc., (Foster
City,
CA), DuPont (Wilmington, DE), or Milligen (Bedford MA). Many methods have
been described for labeling oligonucleotides such as the primers or probes of
the
present invention. Enzo Biochemical (New York, NY) and Clontech (Palo Alto,
CA) both have described and commercialized probe labeling techniques. For
example, a primary amine can be attached to a 3' oligo terminus using 3'-Amine-
ON CPGTM (Clontech, Palo Alto, CA). Similarly, a primary amine can be
attached to a 5' oligo terminus using Aminomodifier II~ (Clontech). The amines
can be reacted to various haptens using conventional activation and linking
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-33-
chemistries. In addition, copending applications U.S. Serial Nos. 625,566,
filed
December 1 I, 1990 and 630,908, filed December 20, 1990, teach methods for -
labeling probes at their 5' and 3' termini, respectively. International
Publication
Nos WO 92/10505, published 25 June 1992, and WO 92/11388, published 9 July
1992, teach methods for labeling probes at their S' and 3' ends, respectively.
According to one known method for labeling an oligonucleotide, a label-
phosphoramidite reagent is prepared and used to add the label to the
oligonucleotide during its synthesis. See, for example, N.T. Thuong et al.,
Tet.
tter 29(46):5905-5908 ( 1988); or J.S. Cohen et al., published U.S. Patent
Application 07/246,688 (NTIS ORDER No. PAT-APPL-7-246,688) ( 1989).
Preferably, probes are labeled at their 3' and 5' ends.
A capture label is attached to the primers or probes and can be a specific
binding member which forms a binding pair with the solid phase reagent's
specific
binding member. It will be understood that the primer or probe itself may
serve as
the capture label. For example, in the case where a solid phase reagent's
binding
member is a nucleic acid sequence, it may be selected such that it binds a
complementary portion of the primer or probe to thereby immobilize the primer
or
probe to the solid phase. In cases where the probe itself serves as the
binding
member, those skilled in the art will recognize that the probe will contain a
sequence or "tail" that is not complementary to the single stranded amplicon
members. In the case where the primer itself serves as the capture label, at
least a
portion of the primer will be free to hybridize with a nucleic acid on a solid
phase
because the probe is selected such that it is not fully complementary to the
primer
sequence.
Generally, probe/single stranded amplicon member complexes can be
detected using techniques commonly employed to perform heterogeneous
immunoassays. Preferably, in this embodiment, detection is performed according
to the protocols used by the commercially available Abbott LCx°
instrumentation
(Abbott Laboratories, Abbott Park, IL).
The primers and probes disclosed herein are useful in typical PCR assays,
wherein the test sample is contacted with a pair of primers, amplification is
performed, the hybridization probe is added, and detection is performed.
Another method provided by the present invention comprises contacting a
test sample with a plurality of polynucleotides, wherein at least one
polynucleotide
is a BS202 molecule as described herein, hybridizing the test sample with the
plurality of polynucleotides and detecting hybridization complexes.
Hybridization
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-34-
complexes are identified and quantitated to compile a profile which is
indicative of
breast tissue disease, such as breast cancer. Expressed RNA sequences may
further be detected by reverse transcription and amplification of the DNA
product
by procedures well-known in the art, including polymerase chain reaction
(PCR).
Dru creeni g and Gene Theranv.
The present invention also encompasses the use of gene therapy methods
for the introduction of anti-sense BS202 derived molecules, such as
polynucleotides or oligonucleotides of the present invention, into patients
with
conditions associated with abnormal expression of polynucleotides related to a
breast tissue disease or condition especially breast cancer. These molecules,
including antisense RNA and DNA fragments and ribozymes, are designed to
inhibit the translation of BS202 mRNA, and may be used therapeutically in the
treatment of conditions associated with altered or abnormal expression of
BS202
polynucleotide.
Alternatively, the oligonucleotides described above can be delivered to
cells by procedures known in the art such that the anti-sense RNA or DNA may
be
expressed i~ vivo to inhibit production of a BS202 polypeptide in the manner
described above. Antisense constructs to a BS202 polynucleotide, therefore,
reverse the action of BS202 transcripts and may be used for treating breast
tissue
disease conditions, such as breast cancer. These antisense constructs may also
be
used to treat tumor metastases.
The present invention also provides a method of screening a plurality of
compounds for specific binding to BS202 polypeptide(s), or any fragment
thereof, to identify at least one compound which specifically binds the BS202
polypeptide. Such a method comprises the steps of providing at least one
compound; combining the BS202 polypeptide with each compound under suitable
conditions for a time sufficient to allow binding; and detecting the BS202
polypeptide binding to each compound.
The polypeptide or peptide fragment employed in such a test may either be
free in solution, affixed to a solid support, borne on a cell surface or
located
intracellularly. One method of screening utilizes eukaryotic or prokaryotic
host
cells which are stably transfected with recombinant nucleic acids which can
express the polypeptide or peptide fragment. A drug, compound, or any other
agent may be screened against such transfected cells in competitive binding
assays. For example, the formation of complexes between a polypeptide and the
agent being tested can be measured in either viable or fixed cells.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-35-
The present invention thus provides methods of screening for drugs,
compounds, or any other agent which can be used to treat diseases associated
with
BS202. These methods comprise contacting the agent with a polypeptide or
fragment thereof and assaying for either the presence of a complex between the
agent and the polypeptide, or for the presence of a complex between the
polypeptide and the cell. In competitive binding assays, the polypeptide
typically
is labeled. After suitable incubation, free (or uncomplexed) polypeptide or
fragment thereof is separated from that present in bound form, and the amount
of
free or uncomplexed label is used as a measure of the ability of the
particular agent
to bind to the polypeptide or to interfere with the polypeptide/cell complex.
The present invention also encompasses the use of competitive screening
assays in which neutralizing antibodies capable of binding polypeptide
specifically
compete with a test agent for binding to the polypeptide or fragment thereof.
In
this manner, the antibodies can be used to detect the presence of any
polypeptide
in the test sample which shares one or more antigenic determinants with a
BS202
polypeptide as provided herein.
Another technique for screening provides high throughput screening for
compounds having suitable binding affinity to at least one polypeptide of
BS202
disclosed herein. Briefly, large numbers of different small peptide test
compounds are synthesized on a solid phase, such as plastic pins or some other
surface. The peptide test compounds are reacted with polypeptide and washed.
Polypeptide thus bound to the solid phase is detected by methods well-known in
the art. Purified polypeptide can also be coated directly onto plates for use
in the
screening techniques described herein. In addition, non-neutralizing
antibodies
can be used to capture the polypeptide and immobilize it on the solid support.
See, for example, EP 84/03564, published on September 13, 1984.
The goal of rational drug design is to produce structural analogs of
biologically active poiypeptides of interest or of the small molecules
including
agonists, antagonists, or inhibitors with which they interact. Such structural
analogs can be used to design drugs which are more active or stable forms of
the
polypeptide or which enhance or interfere with the function of a polypeptide
in
vivo. J. Hodgson, Bio/Technolo~v 9:19-21 ( 1991 ).
For example, in one approach, the three-dimensional structure of a
polypeptide, or of a polypeptide-inhibitor complex, is determined by x-ray
crystallography, by computer modeling or, most typically, by a combination of
the
two approaches. Both the shape and charges of the polypeptide must be
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-36-
ascertained to elucidate the structure and to determine active sites) of the
molecule. Less often, useful information regarding the structure of a
polypeptide
may be gained by modeling based on the structure of homologous proteins. In
both cases, relevant structural information is used to design analogous
polypeptide-like molecules or to identify efficient inhibitors
Useful examples of rationai drug design may include molecules which
have improved activity or stability as shown by S. Braxton et al.,
Biochemistry
31:7796-7801 ( 1992), or which act as inhibitors, agonists, or antagonists of
native peptides as shown by S.B.P. Athauda et al., J Biochem. fTok~cL 113
(6):742-746 (1993).
It also is possible to isolate a target-specific antibody selected by an assay
as described hereinabove, and then to determine its crystal structure. In
principle
this approach yields a pharmacophore upon which subsequent drug design can be
based. It further is possible to bypass protein crystallography altogether by
generating anti-idiotypic antibodies ("anti-ids") to a functional,
pharmacologically
active antibody. As a mirror image of a mirror image, the binding site of the
anti-
id is an analog of the original receptor. The anti-id then can be used to
identify
and isolate peptides from banks of chemically or biologically produced
peptides.
The isolated peptides then can act as the pharmacophore (that is, a prototype
pharmaceutical drug).
A sufficient amount of a recombinant polypeptide of the present invention
may be made available to perform analytical studies such as X-ray
crystallography. In addition, knowledge of the polypeptide amino acid sequence
which is derivable from the nucleic acid sequence provided herein will provide
guidance to those employing computer modeling techniques in place of, or in
addition to, x-ray crystallography.
Antibodies specific to a BS202 polypeptide (e.g., anti-BS202 antibodies)
further may be used to inhibit the biological action of the polypeptide by
binding to
the polypeptide. In this manner, the antibodies may be used in therapy, for
example, to treat breast tissue diseases including breast cancer and its
metastases.
Further, such antibodies can detect the presence or absence of a BS202
polypeptide in a test sample and, therefore, are useful as diagnostic markers
for
the diagnosis of a breast tissue disease or condition especially breast
cancer. Such
antibodies may also function as a diagnostic marker for breast tissue disease
conditions, such as breast cancer.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-37-
The present invention also is directed to antagonists and inhibitors of the
polypeptides of the present invention. The antagonists and inhibitors are
those
which inhibit or eliminate the function of the polypeptide. Thus, for example,
an
antagonist may bind to a polypeptide of the present invention and inhibit or
eliminate its function. The antagonist, for example, could be an antibody
against
the polypeptide which eliminates the activity of a BS202 polypeptide by
binding a
BS202 polypeptide, or in some cases the antagonist may be an oligonucleotide.
Examples of small molecule inhibitors include, but are not limited to, small
peptides or peptide-like molecules.
The antagonists and inhibitors may be employed as a composition with a
pharmaceutically acceptable carrier including, but not limited to, saline,
buffered
saline, dextrose, water, glycerol, ethanol and combinations thereof.
Administration of BS202 polypeptide inhibitors is preferably systemic. The
present invention also provides an antibody which inhibits the action of such
a
polypeptide.
Antisense technology can be used to reduce gene expression through
triple-helix formation or antisense DNA or RNA, both of which methods are
based on binding of a polynucleotide to DNA or RNA. For example, the 5'
coding portion of the polynucleotide sequence, which encodes for the
polypeptide
of the present invention, is used to design an antisense RNA oligonucleotide
of
from 10 to 40 base pairs in length. A DNA oligonucleotide is designed to be
complementary to a region of the gene involved in transcription, thereby
preventing transcription and the production of the BS202 polypeptide. For
triple
helix, see, for example, Lee et al., Nuc. Acids Res. 6:3073 ( 1979); Cooney et
al.,
Science 241:456 ( 1988); and Dervan et al., Sc'aence, 251:1360 ( 1991 ) The
antisense RNA oligonucleotide hybridizes to the mRNA in vivo and blocks
translation of a mRNA molecule into the BS202 polypeptide. For antisense, see,
for example, Okano, ~. Neurochem. 56:560 (1991); and "Oligodeoxynucleotides
as Antisense Inhibitors of Gene Expression," CRC Press, Boca Raton, Fla.
( 1988). Antisense oiigonucleotides act with greater efficacy when modified to
contain artificial internucleotide linkages which render the molecule
resistant to
nucleolytic cleavage. Such artificial internucleotide linkages include, but
are not
limited to, methylphosphonate, phosphorothiolate and phosphoroamydate
internucleotide linkages.
CA 02292843 1999-12-03
WO 99!02559 PCT/US98/14046
-38-
Recombinant Technolo~v.
The present invention provides host cells and expression vectors
comprising BS202 polynucleotides of the present invention and methods for the
production of the polypeptide(s) they encode. Such methods comprise culturing
the host cells under conditions suitable for the expression of the BS202
polynucleotide and recovering the BS202 polypeptide from the cell culture.
The present invention also provides vectors which include BS202
polynucleotides of the present invention, host cells which are genetically
engineered with vectors of the present invention and the production of
polypeptides of the present invention by recombinant techniques. ,.
Host cells are genetically engineered (transfected, transduced or
transformed) with the vectors of this invention which may be cloning vectors
or
expression vectors. The vector may be in the form of a plasmid, a viral
particle, a
phage, etc. The engineered host cells can be cultured in conventional nutrient
media modified as appropriate for activating promoters, selecting transfected
cells,
or amplifying BS202 gene(s). The culture conditions, such as temperature, pH
and the like, are those previously used with the host cell selected for
expression,
and will be apparent to the ordinarily skilled artisan.
The polynucleotides of the present invention may be employed for
producing a polypeptide by recombinant techniques. Thus, the polynucleotide
sequence may be included in any one of a variety of expression vehicles, in
particular, vectors or plasmids for expressing a polypeptide. Such vectors
include
chromosomal, nonchromosomal and synthetic DNA sequences, e.g., derivatives
of Simian Virus 40 (SV40); bacterial plasmids; phage DNA; yeast plasmids;
vectors derived from combinations of plasmids and phage DNA, viral DNA such
as vaccinia, adenovirus, fowl pox virus and pseudorabies. However, any other
plasmid or vector may be used so long as it is repiicable and viable in the
host.
The appropriate DNA sequence may be inserted into the vector by a variety
of procedures. In general, the DNA sequence is inserted into appropriate
restriction endonuclease sites by procedures known in the art. Such procedures
and others are deemed to be within the scope of those skilled in the art. The
DNA
sequence in the expression vector is operatively linked to an appropriate
expression control sequences) (promoter) to direct mRNA synthesis.
Representative examples of such promoters include, but are not limited to, the
LTR or the SV40 promoter, the E. cQli lac or trp, the phage lambda P sub L
promoter and other promoters known to control expression of genes in
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-39-
prokaryotic or eukaryotic cells or their viruses. The expression vector also
contains a ribosome binding site for translation initiation and a
transcription
terminator. The vector may also include appropriate sequences for amplifying
expression. In addition, the expression vectors preferably contain a gene to
S provide a phenotypic trait for selection of transfected host cells such as
dihydrofolate reductase or neomycin resistance for eukaryotic cell culture, or
such
as tetracycline or ampicillin resistance in E. coli.
The vector containing the appropriate DNA sequence as hereinabove
described, as well as an appropriate promoter or control sequence, may be
employed to transfect an appropriate host to permit the host to express the
protein.
As representative examples of appropriate hosts, there may be mentioned:
bacterial
cells, such as E. ~, Salmonella typhimurium; Streptom, ces sue.; fungal cells,
such as yeast; insect cells, such as Drosophila and Sf9; animal cells, such as
CHO, COS or Bowes melanoma; plant cells, etc. The selection of an appropriate
host is deemed to be within the scope of those skilled in the art from the
teachings
provided herein.
More particularly, the present invention also includes recombinant
constructs comprising one or more of the sequences as broadly described above.
The constructs comprise a vector, such as a plasmid or viral vector, into
which a
sequence of the invention has been inserted, in a forward or reverse
orientation.
In a preferred aspect of this embodiment, the construct further comprises
regulatory sequences including, for example, a promoter, operably linked to
the
sequence. Large numbers of suitable vectors and promoters are known to those
of
skill in the art and are conunercially available. The following vectors are
provided
by way of example. Bacterial: pINCY (Incyte Pharmaceuticals Inc., Palo Alto,
CA), pSPORT I (Life Technologies, Gaithersburg, MD), pQE70, pQE60, pQE-9
(Qiagen) pBs, phagescript, psiXl74, pBluescript SK, pBsKS, pNHBa, pNHl6a,
pNHl8a, pNH46a (Stratagene); pTrc99A, pKK223-3, pKK233-3, pDR540,
pRITS (Pharmacia); Eukaryotic: pWLneo, pSV2cat, pOG44, pXTI, pSG
(Stratagene) pSVK3, pBPV, pMSG, pSVL (Pharmacia). However, any other
plasmid or vector may be used as long as it is replicable and viable in the
host.
Plasmid pINCY is generally identical to the plasmid pSPORTI (available
from Life Technologies, Gaithersburg, MD) with the exception that it has two
modifications in the polylinker (multiple cloning site). These modifications
are ( 1 )
it lacks a HindIII restriction site and (2) its EcoRI restriction site lies at
a different
location. pINCY is created from pSPORTl by cleaving pSPORTI with both
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-40-
HindIll and EcoRI and replacing the excised fragment of the polylinker with
synthetic DNA fragments (SEQUENCE m NO 8 and SEQUENCE ID NO 9).
This replacement may be made in any manner known to those of ordinary skill in
the art. For example, the two nucleotide sequences, SEQUENCE ID NO 8 and
SEQUENCE ID NO 9, may be generated synthetically with S' terminal
phosphates, mixed together, and then ligated under standard conditions for
performing staggered end ligations into the pSPORTl plasmid cut with HindIII
and EcoRI. Suitable host cells (such as ~, coli DHSp. cells) then are
transfected
with the ligated DNA and recombinant clones are selected for ampicillin
resistance.
Plasmid DNA then is prepared from individual clones and subjected to
restriction
enzyme analysis or DNA sequencing in order to confirm the presence of insert
sequences in the proper orientation. Other cloning strategies known to the '
ordinary artisan also may be employed.
Promoter regions can be selected from any desired gene using CAT
1 S (chloramphenicol transferase) vectors or other vectors with selectable
markers.
Two appropriate vectors are pKK232-8 and pCM7. Particular named bacterial
promoters include lacI, lacZ, T3, SP6, T7, gpt, lambda P sub R, P sub L and
trp.
Eukaryotic promoters include cytomegalovirus (CMV) immediate early, herpes
simplex virus (HSV) thymidine kinase, early and late SV40, LTRs from
retroviruses and mouse metallothionein-I. Selection of the appropriate vector
and
promoter is well within the level of ordinary skill in the art.
In a further embodiment, the present invention provides host cells
containing the above-described construct. The host cell can be a higher
eukaryotic
cell, such as a mammalian cell, or a lower eukaryotic cell, such as a yeast
cell, or
the host cell can be a prokaryotic cell, such as a bacterial cell.
Introduction of the
construct into the host cell can be effected by calcium phosphate
transfection,
DEAE-Dextran mediated transfection, or electroporation [L. Davis et al.,
"Basic
Methods in Molecular Biology," 2nd edition, Appleton and Lang, Paramount
Publishing, East Norwalk, CT ( 1994)].
The constructs in host cells can be used in a conventional manner to
produce the gene product encoded by the recombinant sequence. Alternatively,
the polypeptides of the invention can be synthetically produced by
conventional
peptide synthesizers.
Recombinant proteins can be expressed in mammalian cells, yeast,
bacteria, or other cells, under the control of appropriate promoters. Cell-
free
translation systems can also be employed to produce such proteins using RNAs
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-4l -
derived from the DNA constructs of the present invention. Appropriate cloning
and expression vectors for use with prokaryotic and eukaryotic hosts are
described
by Sambrook et al., Molecular Cloning:: A Laborator~r Manual, Second Edition,
(Cold Spring Harbor, NY, 19$9}.
Transcription of a DNA encoding the polypeptide(s) of the present
invention by higher eukaryotes is increased by inserting an enhancer sequence
into
the vector. Enhancers are cis-acting elements of DNA, usually about from 10 to
300 bp, that act on a promoter to increase its transcription. Examples include
the
SV40 enhancer on the late side of the replication origin (bp 100 to 270), a
cytomegalovirus early promoter enhancer, a polyoma enhancer on the late side
of
the replication origin and adenovirus enhancers.
Generally, recombinant expression vectors will include origins of
replication and selectable markers permitting transfection of the host cell,
e.g., the
ampicillin resistance gene of E. coli and S. cerevisiae TRPI gene, and a
promoter
derived from a highly-expressed gene to direct transcription of a downstream
structural sequence. Such promoters can be derived from operons encoding
glycolytic enzymes such as 3-phosphoglycerate kinase (PGK), alpha factor, acid
phosphatase, or heat shock proteins, among others. The heterologous structural
sequence is assembled in appropriate phase with translation initiation and
termination sequences, and preferably, a leader sequence capable of directing
secretion of translated protein into the periplasmic space or extracellular
medium.
Optionally, the heterologous sequence can encode a fusion protein including an
N-
terminal identification peptide imparting desired characteristics, e.g.,
stabilization
or simplified purification of expressed recombinant product.
Useful expression vectors for bacterial use are constructed by inserting a
structural DNA sequence encoding a desired protein together with suitable
translation initiation and termination signals in operable reading phase with
a
functional promoter. The vector will comprise one or more phenotypic
selectable
markers and an origin of replication to ensure maintenance of the vector and
to, if
desirable, provide amplification within the host. Suitable prokaryotic hosts
for
transfection include E. coli, B ill s s ' 's, Salmonella typhimurium and
various
species within the genera Pseudomonas, Streptom,~ and Stab lococc~,
although others may also be employed as a routine matter of choice.
Useful expression vectors for bacterial use comprise a selectable marker
and bacterial origin of replication derived from plasmids comprising genetic
elements of the well-known cloning vector pBR322 (ATCC 37017). Other
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-42-
vectors include but are not limited to PKK223-3 (Pharmacia Fine Chemicals,
Uppsala, Sweden) and GEM 1 (Promega Biotec, Madison, WI). These pBR322-
"backbone" sections are combined with an appropriate promoter and the
structural
sequence to be expressed.
Following transfection of a suitable host and growth of the host to an
appropriate cell density, the selected promoter is derepressed by appropriate
means
(e.g., temperature shift or chemical induction), and cells are cultured for an
additional period. Cells are typically harvested by centrifugation, disrupted
by
physical or chemical means, and the resulting crude extract retained for
further
purification. Microbial cells employed in expression of proteins can be
disrupted
by any convenient method including freeze-thaw cycling, sonication, mechanical
disruption, or use of cell lysing agents. Such methods are well-known to the
ordinary artisan.
Various mammalian cell culture systems can also be employed to express
recombinant protein. Examples of mammalian expression systems include the
COS-7 lines of monkey kidney fibroblasts described by Gluzman, ell 23:175
( I 981 ), and other cell lines capable of expressing a compatible vector,
such as the
C 127, HEK-293, 3T3, CHO, HeLa and BHK cell lines. Mammalian expression
vectors will comprise an origin of replication, a suitable promoter and
enhancer
and also any necessary ribosome binding sites, polyadenylation sites, splice
donor
and acceptor sites, transcriptional termination sequences and 5' flanking
nontranscribed sequences. DNA sequences derived from the SV40 viral genome,
for example, SV40 origin, early promoter, enhancer, splice, and
polyadenylation
sites may be used to provide the required nontranscribed genetic elements.
Representative, useful vectors include pRc/CMV and pcDNA3 (available from
invitrogen, San Diego, CA).
BS202 polypeptides are recovered and purified from recombinant cell
cultures by known methods including affinity chromatography, ammonium sulfate
or ethanol precipitation, acid extraction, anion or canon exchange
chromatography, phosphocellulose chromatography, hydrophobic interaction
chromatography, hydroxyapatite chromatography or lectin chromatography. It is
preferred to have low concentrations (approximately 0.1-5 mM) of calcium ion
present during purification [Price, et al., J. Biol. Chem. 244:917 (1969)].
Protein
refolding steps can be used, as necessary, in completing configuration of the
polypeptide. Finally, high performance liquid chromatography (HPLC) can be
employed for final purification steps.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98114046
-43-
Thus, polypeptides of the present invention may be naturally purified
products expressed from a high expressing cell line, or a product of chemical
synthetic procedures, or produced by recombinant techniques from a prokaryotic
or eukaryotic host (for example, by bacterial, yeast, higher plant, insect and
S mammalian cells in culture). Depending upon the host employed in a
recombinant
production procedure, the polypeptides of the present invention may be
glycosylated with mammalian or other eukaryotic carbohydrates or may be non-
glycosyiated. The polypeptides of the invention may also include an initial
methionine amino acid residue.
The starting plasmids can be constructed from available plasmids in accord
with published, known procedures. In addition, equivalent plasmids to those
described are known in the art and will be apparent to one of ordinary skill
in the
art.
The following is the general procedure for the isolation and analysis of
1 S cDNA clones. In a particular embodiment disclosed herein, mRNA was
isolated
from breast tissue and used to generate the cDNA library. Breast tissue was
obtained from patients by surgical resection and was classified as tumor or
non-
tumor tissue by a pathologist.
The cDNA inserts from random isolates of the breast tissue libraries were
sequenced in part, analyzed in detail as set forth in the Examples, and are
disclosed in the Sequence Listing as SEQUENCE ID NO 1, SEQUENCE ID NO
2, SEQUENCE ID NO 3, SEQUENCE >I7 NO 4, SEQUENCE ID NO S. Also
analyzed in detail as set forth in the Examples, and disclosed in the Sequence
Listing, is the full-length sequence of clone 1212072 (designated as clone
2S 1212072IH (SEQUENCE ID NO 6)). The consensus sequence of these inserts is
presented as SEQUENCE ID NO 7. These polynucleotides may contain an entire
open reading frame with or without associated regulatory sequences for a
particular gene, or they may encode only a portion of the gene of interest.
This is
attributed to the fact that many genes are several hundred and sometimes
several
thousand bases in length and, with current technology, cannot be cloned in
their
entirety because of vector limitations, incomplete reverse transcription of
the first
strand, or incomplete replication of the second strand. Contiguous, secondary
clones containing additional nucleotide sequences may be obtained using a
variety
of methods known to those of skill in the art.
3S Methods for DNA sequencing are well known in the art. Conventional
enzymatic methods employ DNA polymerase, Klenow fragment, Sequenase (US
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-
Biochemical Corp, Cleveland, OH) or Tag polymerise to extend DNA chains
from an oligonucleotide primer annealed to the DNA template of interest.
Methods
have been developed for the use of both single-stranded and double-stranded
templates. The chain termination reaction products may be electrophoresed on
urea/polyacrylamide gels and detected either by autoradiography (for
radionucleotide labeled precursors) or by fluorescence (for fluorescent-
labeled
precursors). Recent improvements in mechanized reaction preparation,
sequencing and analysis using the fluorescent detection method have permitted
expansion in the number of sequences that can be determined per day using
machines such as the Applied Biosystems 377 DNA Sequencers (Applied
Biosystems, Foster City, CA).
The reading frame of the nucleotide sequence can be ascertained by several
types of analyses. First, reading frames contained within the coding sequence
can
be analyzed for the presence of start codon ATG and stop codons TGA, TAA or
TAG. Typically, one reading frame will continue throughout the major portion
of
a cDNA sequence while other reading frames tend to contain numerous stop
codons. In such cases, reading frame determination is straightforward. In
other
more difficult cases, further analysis is required.
Algorithms have been created to analyze the occurrence of individual
nucleotide bases at each putative codon triplet. See, for example J.W.
Fickett,
Nuc. Acids Res. 10:5303 ( 1982). Coding DNA for particular organisms
(bacteria, plants and animals) tends to contain certain nucleotides within
certain
triplet periodicities, such as a significant preference for pyrimidines in the
third
codon position. These preferences have been incorporated into widely available
software which can be used to determine coding potential (and frame) of a
given
stretch of DNA. The algorithm-derived information combined with start/stop
codon information can be used to determine proper frame with a high degree of
certainty. This, in turn, readily permits cloning of the sequence in the
correct
reading frame into appropriate expression vectors.
The nucleic acid sequences disclosed herein may be joined to a variety of
other polynucleotide sequences and vectors of interest by means of well-
established recombinant DNA techniques. See J. Sambrook et al., supra. Vectors
of interest include cloning vectors, such as plasmids, cosmids, phage
derivatives,
phagemids, as well as sequencing, replication and expression vectors, and the
like. In general, such vectors contain an origin of replication functional in
at least
one organism, convenient restriction endonuclease digestion sites and
selectable
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-45-
markers appropriate for particular host cells. The vectors can be transferred
by a
variety of means known to those of skill in the art into suitable host cells
which
then produce the desired DNA, RNA or polypeptides.
Occasionally, sequencing or random reverse transcription errors will mask
the presence of the appropriate open reading frame or regulatory element. In
such
cases, it is possible to determine the correct reading frame by attempting to
express
the polypeptide and determining the amino acid sequence by standard peptide
mapping and sequencing techniques. See, F.M. Ausubel et al., Current Protocols
in Molecular Biology, John Wiley & Sons, New York, NY ( 1989). Additionally,
the actual reading frame of a given nucleotide sequence may be determined by
transfection of host cells with vectors containing all three potential reading
frames.
Only those cells with the nucleotide sequence in the correct reading frame
will
produce a peptide of the predicted length.
The nucleotide sequences provided herein have been prepared by current,
state-of the-art, automated methods and, as such, may contain unidentified
nucleotides. These will not present a problem to those skilled in the art who
wish
to practice the invention. Several methods employing standard recombinant
techniques, described in J. Sambrook (s_upra) or periodic updates thereof, may
be
used to complete the missing sequence information. The same techniques used
for
obtaining a full length sequence, as described herein, may be used to obtain
nucleotide sequences.
Expression of a particular cDNA may be accomplished by subcloning the
cDNA into an appropriate expression vector and transfecting this vector into
an
appropriate expression host. The cloning vector used for the generation of the
breast tissue cDNA library can be used for transcribing mRNA of a particular
cDNA and contains a promoter for beta-galactosidase, an amino-terminal met and
the subsequent seven amino acid residues of beta-gaiactosidase. Immediately
following these eight residues is an engineered bacteriophage promoter useful
for
artificial priming and transcription, as well as a number of unique
restriction sites,
including EcoRI, for cloning. The vector can be transfected into an
appropriate
host strain of E. c i.
Induction of the isolated bacterial strain with isopropylthiogalactoside
(IPTG) using standard methods will produce a fusion protein which contains the
first seven residues of beta-galactosidase, about 15 residues of linker and
the
peptide encoded within the cDNA. Since cDNA clone inserts are generated by an
essentially random process, there is one chance in three that the included
cDNA
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-46-
will lie in the correct frame for proper translation. If the cDNA is not in
the proper
reading frame, the correct frame can be obtained by deletion or insertion of
an
appropriate number of bases by well known methods including i~ v'tro
mutagenesis, digestion with exonuclease III or mung bean nuclease, or
oligonucleotide linker inclusion.
The cDNA can be shuttled into other vectors known to be useful for
expression of protein in specific hosts. Oligonucleotide primers containing
cloning sites and segments of DNA sufficient to hybridize to stretches at both
ends
of the target cDNA can be synthesized chemically by standard methods. These
primers can then be used to amplify the desired gene segments by PCR. The
resulting new gene segments can be digested with appropriate restriction
enzymes
under standard conditions and isolated by gel electrophoresis. Alternately,
similar
gene segments can be produced by digestion of the cDNA with appropriate
restriction enzymes and filling in the missing gene segments with chemically
synthesized oligonucleotides. Segments of the coding sequence from more than
one gene can be ligated together and cloned in appropriate vectors to optimize
expression of recombinant sequence.
Suitable expression hosts for such chimeric molecules include, but are not
limited to, mammalian cells, such as Chinese Hamster Ovary (CHO) and human
embryonic kidney {HEK) 293 cells, insect cells, such as Sf9 cells, yeast
cells,
such as Saccharom, ces cerevisiae and bacteria, such as E. c2li. For each of
these
cell systems, a useful expression vector may also include an origin of
replication
to allow propagation in bacteria and a selectable marker such as the beta-
iactamase
antibiotic resistance gene to allow selection in bacteria. In addition, the
vectors
may include a second selectable marker, such as the neomycin
phosphotransferase
gene, to allow selection in transfected eukaryotic host cells. Vectors for use
in
eukaryotic expression hosts may require the addition of 3' poly A tail if the
sequence of interest lacks poly A.
Additionally, the vector may contain promoters or enhancers which
increase gene expression. Such promoters are host specific and include, but
are
not limited to, MMTV, SV40, or metallothionine promoters for CHO cells; trp,
lac, tac or T7 promoters for bacterial hosts; or alpha factor, alcohol oxidase
or
PGH promoters for yeast. Adenoviral vectors with or without transcription
enhancers, such as the Rous sarcoma virus (RSV) enhancer, may be used to drive
protein expression in mammalian cell lines. Once homogeneous cultures of
recombinant cells are obtained, large quantities of recombinantiy produced
protein
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-47-
can be recovered from the conditioned medium and analyzed using
chromatographic methods well known in the art. An alternative method for the
production of large amounts of secreted protein involves the transfection of
mammalian embryos and the recovery of the recombinant protein from milk
produced by transgenic cows, goats, sheep, etc. Polypeptides and closely
related
molecules may be expressed recombinantly in such a way as to facilitate
protein
purification. One approach involves expression of a chimeric protein which
includes one or more additional polypeptide domains not naturally present on
human polypeptides. Such purification-facilitating domains include, but are
not
limited to, metal-chelating peptides such as histidine-tryptophan domains that
allow purification on immobilized metals, protein A domains that allow
purification on immobilized immunoglobulin and the domain utilized in the
FLAGS extension/affinity purification system (Immunex Corp, Seattle, WA).
The inclusion of a cleavable linker sequence such as Factor XA or enterokinase
from Invitrogen (San Diego, CA) between the polypeptide sequence and the
purification domain may be useful for recovering the polypeptide.
Immunoassays.
BS202 polypeptides, including fragments, derivatives, and analogs
thereof, or cells expressing such polypeptides, can be utilized in a variety
of
assays, many of which are described herein, for the detection of antibodies to
breast tissue. They also can be used as immunogens to produce antibodies.
These antibodies can be, for example, polyclonal or monoclonal antibodies,
chimeric, single chain and humanized antibodies, as well as Fab fragments, or
the
product of an Fab expression library. Various procedures known in the art may
be used for the production of such antibodies and fragments.
For example, antibodies generated against a polypeptide comprising a
sequence of the present invention can be obtained by direct injection of the
polypeptide into an animal or by administering the polypeptide to an animal
such
as a mouse, rabbit, goat or human. A mouse, rabbit or goat is preferred. The
polypeptide is selected from the group consisting of SEQUENCE m NO 14,
SEQUENCE ID NO 15, SEQUENCE ID NO 16, SEQUENCE ID NO 17,
SEQUENCE ID NO 18, and fragments thereof. The antibody so obtained then
will bind the polypeptide itself. In this manner, even a sequence encoding
only a
fragment of the polypeptide can be used to generate antibodies that bind the
native
polypeptide. Such antibodies then can be used to isolate the polypeptide from
test
samples such as tissue suspected of containing that polypeptide. For
preparation
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-48-
of monoclonal antibodies, any technique which provides antibodies produced by
continuous cell line cultures can be used. Examples include the hybridoma
technique as described by Kohler and Milstein, Nature 256:495-497 ( 1975), the
trioma technique, the human B-cell hybridoma technique as described by Kozbor
et al., Immun. Todav 4:72 ( 1983) and the EBV-hybridoma technique to produce
human monoclonal antibodies as described by Cole et al., in Monoclonal
Antibodies and Cancer Therapy, Alan R. Liss, Inc, New York, NY, pp. 77-96
( 1985). Techniques described for the production of single chain antibodies
can be
adapted to produce single chain antibodies to immunogenic polypeptide products
of this invention. See, for example, U.S. Patent No. 4,946,778.
Various assay formats may utilize the antibodies of the present invention,
including "sandwich" immunoassays and probe assays. For example, the
antibodies of the present invention, or fragments thereof, can be employed in
various assay systems to determine the presence, if any, of BS202 antigen in a
test
sample. For example, in a first assay format, a polyclonal or monoclonal
antibody
or fragment thereof, or a combination of these antibodies, which has been
coated
on a solid phase, is contacted with a test sample, to form a first mixture.
This first
mixture is incubated for a time and under conditions sufficient to form
antigen/antibody complexes. Then, an indicator reagent comprising a monoclonal
or a polyclonal antibody or a fragment thereof, or a combination of these
antibodies, to which a signal generating compound has been attached, is
contacted
with the antigen/antibody complexes to form a second mixture. This second
mixture then is incubated for a time and under conditions sufficient to form
antibody/antigen/antibody complexes. The presence of BS202 antigen in the test
sample and captured on the solid phase, if any, is determined by detecting the
measurable signal generated by the signal generating compound. The amount of
BS202 antigen present in the test sample is proportional to the signal
generated.
In an alternative assay format, a mixture is formed by contacting: ( 1 ) a
polyclonal antibody, monoclonal antibody, or fragment thereof, which
specifically
binds to BS202 antigen, or a combination of such antibodies bound to a solid
support; (2) the test sample; and (3) an indicator reagent comprising a
monoclonal
antibody, polyclonal antibody, or fragment thereof, which specifically binds
to a
different BS202 antigen (or a combination of these antibodies) to which a
signal
generating compound is attached. This mixture is incubated for a time and
under
conditions sufficient to form antibody/antigen/antibody complexes. The
presence,
if any, of BS202 antigen present in the test sample and captured on the solid
phase
CA 02292843 1999-12-03
WO 99/01559 PCTIUS98/14046
-49-
is determined by detecting the measurable signal generated by the signal
generating
compound. The amount of BS202 antigen present in the test sample is
proportional to the signal generated.
In another assay format, one or a combination of at least two monoclonal
antibodies of the invention can be employed as a competitive probe for the
detection of antibodies to BS202 antigen. For example, BS202 polypeptides such
as the recombinant antigens disclosed herein, either alone or in combination,
are
coated on a solid phase. A test sample suspected of containing antibody to
BS202
antigen then is incubated with an indicator reagent comprising a signal
generating
compound and at least one monoclonal antibody of the invention for a time and
under conditions sufficient to form antigen/antibody complexes of either the
test
sample and indicator reagent bound to the solid phase or the indicator reagent
bound to the solid phase. The reduction in binding of the monoclonal antibody
to
the solid phase can be quantitatively measured.
In yet another detection method, each of the monoclonal or polyclonal
antibodies of the present invention can be employed in the detection of BS202
antigens in tissue sections, as well as in cells, by immunohistochemical
analysis.
The tissue sections can be cut from either frozen or chemically fixed samples
of
tissue. If the antigens are to be detected in cells, the cells can be isolated
from
blood, urine, breast aspirates, or other bodily fluids. The cells may be
obtained
by biopsy, either surgical or by needle. The cells can be isolated by
centrifugation
or magnetic attraction after labeling with magnetic particles or ferrofluids
so as to
enrich a particular fraction of cells for staining with the antibodies of the
present
invention. Cytochemical analysis wherein these antibodies are labeled directly
(with, for example, fluorescein, colloidal gold, horseradish peroxidase,
alkaline
phosphatase, etc.) or are labeled by using secondary labeled anti-species
antibodies (with various labels as exemplified herein) to track the
histopathology
of disease also are within the scope of the present invention.
In addition, these monoclonal antibodies can be bound to matrices similar
to CNBr-activated Sepharose and used for the affinity purification of specific
BS202 polypeptides from cell cultures or biological tissues such as to purify
recombinant and native BS202 proteins.
The monoclonal antibodies of the invention also can be used for the
generation of chimeric antibodies for therapeutic use, or other similar
applications.
The monoclonal antibodies or fragments thereof can be provided
individually to detect BS202 antigens. Combinations of the monoclonal
CA 02292843 1999-12-03
WO 99/02559 PCT/IJS98/I4046
-50-
antibodies (and fragments thereof) provided herein also may be used together
as
components in a mixture or "cocktail" of at least one BS202 antibody of the
invention, along with antibodies which specifically bind to other BS202
regions,
each antibody having different binding specificities. Thus, this cocktail can
include the monoclonal antibodies of the invention which are directed to BS202
polypeptides disclosed herein and other monoclonal antibodies specific to
other
antigenic determinants of BS202 antigens or other related proteins.
The polyclonal antibody or fragment thereof which can be used in the
assay formats should specifically bind to a BS202 polypeptide or other BS202
polypeptides additionally used in the assay. The polyclonal antibody used
preferably is of mammalian origin such as, human, goat, rabbit or sheep
polyclonal antibody which binds BS202 polypeptide. Most preferably, the
polyclonal antibody is of rabbit origin. The polyclonal antibodies used in the
assays can be used either alone or as a cocktail of polyclonal antibodies.
Since the
cocktails used in the assay formats are comprised of either monoclonal
antibodies
or polyclonal antibodies having different binding specificity to BS202
polypeptides, they are useful for the detecting, diagnosing, staging,
monitoring,
prognosticating, preventing or treating, or determining the predisposition to,
diseases and conditions of the breast, such as breast cancer.
It is contemplated and within the scope of the present invention that BS202
antigen may be detectable in assays by use of a recombinant antigen as well as
by
use of a synthetic peptide or purified peptide, which peptide comprises an
amino
acid sequence of BS202. The amino acid sequence of such a polypeptide is
selected from the group consisting of SEQUENCE ID NO 14, SEQUENCE ID
NO 15, SEQUENCE ID NO 16, SEQUENCE ID NO 17, SEQUENCE m NO
1$, and fragments thereof. It also is within the scope of the present
invention that
different synthetic, recombinant or purified peptides, identifying different
epitopes
of BS202, can be used in combination in an assay for the detecting,
diagnosing,
staging, monitoring, prognosticating, preventing or treating, or determining
the
predisposition to diseases and conditions of the breast, such as breast
cancer. In
this case, all of these peptides can be coated onto one solid phase; or each
separate
peptide may be coated onto separate solid phases, such as microparticles, and
then
combined to form a mixture of peptides which can be later used in assays.
Furthermore, it is contemplated that multiple peptides which define epitopes
from
different antigens may be used for the detection, diagnosis, staging,
monitoring,
prognosis, prevention or treatment of, or determining the predisposition to,
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-51-
diseases and conditions of the breast, such as breast cancer. Peptides coated
on
solid phases or labeled with detectable labels are then allowed to compete
with
those present in a patient sample (if any) for a limited amount of antibody. A
reduction in binding of the synthetic, recombinant, or purified peptides to
the
antibody (or antibodies) is an indication of the presence of BS202 antigen in
the
patient sample. The presence of BS202 antigen indicates the presence of breast
tissue disease, especially breast cancer, in the patient. Variations of assay
formats
are known to those of ordinary skill in the art and many are discussed herein
below.
In another assay format, the presence of anti-BS202 antibody and/or
BS202 antigen can be detected in a simultaneous assay, as follows. A test
sample
is simultaneously contacted with a capture reagent of a first analyte, wherein
said
capture reagent comprises a first binding member specific for a first analyte
attached to a solid phase and a capture reagent for a second analyte, wherein
said
capture reagent comprises a first binding member for a second analyte attached
to a
second solid phase, to thereby form a mixture. This mixture is incubated for a
time and under conditions sufficient to form capture reagent/first analyte and
capture reagent/second analyte complexes. These so-formed complexes then are
contacted with an indicator reagent comprising a member of a binding pair
specific
for the first analyte labeled with a signal generating compound and an
indicator
reagent comprising a member of a binding pair specific for the second analyte
labeled with a signal generating compound to form a second mixture. This
second
mixture is incubated for a time and under conditions sufficient to form
capture
reagendfirst analyte/indicator reagent complexes and capture reagent/second
analyte/indicator reagent complexes. The presence of one or more analytes is
determined by detecting a signal generated in connection with the complexes
formed on either or both solid phases as an indication of the presence of one
or
more analytes in the test sample. In this assay format, recombinant antigens
derived from the expression systems disclosed herein may be utilized, as well
as
monoclonal antibodies produced from the proteins derived from the expression
systems as disclosed herein. For example, in this assay system, BS202 antigen
can be the first analyte. Such assay systems are described in greater detail
in EP
Publication No. 0473065.
In yet other assay formats, the polypeptides disclosed herein may be
utilized to detect the presence of antibody against BS202 antigen in test
samples.
For example, a test sample is incubated with a solid phase to which at least
one
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-52-
polypeptide such as a recombinant protein or synthetic peptide has been
attached.
The polypeptide is selected from the group consisting of SEQUENCE ID NO 14,
SEQUENCE ID NO 15, SEQUENCE ID NO 16, SEQUENCE 117 NO 17,
SEQUENCE ID NO 18, and fragments thereof. These are reacted for a time and
under conditions sufficient to form antigen/antibody complexes. Following
incubation, the antigen/antibody complex is detected. Indicator reagents may
be
used to facilitate detection, depending upon the assay system chosen. In
another
assay format, a test sample is contacted with a solid phase to which a
recombinant
protein produced as described herein is attached, and also is contacted with a
monoclonal or polyclonal antibody specific for the protein, which preferably
has
been labeled with an indicator reagent. After incubation for a time and under
conditions sufficient for antibody/antigen complexes to form, the solid phase
is
separated from the free phase, and the label is detected in either the solid
or free
phase as an indication of the presence of antibody against BS202 antigen.
Other
assay formats utilizing the recombinant antigens disclosed herein are
contemplated. These include contacting a test sample with a solid phase to
which
at least one antigen from a first source has been attached, incubating the
solid
phase and test sample for a time and under conditions sufficient to form
antigen/antibody complexes, and then contacting the solid phase with a labeled
antigen, which antigen is derived from a second source different from the
first
source. For example, a recombinant protein derived from a first source such as
~
r~ is used as a capture antigen on a solid phase, a test sample is added to
the so-
prepared solid phase, and following standard incubation and washing steps as
deemed or required, a recombinant protein derived from a different source
(i.e.,
non-E co ') is utilized as a part of an indicator reagent which subsequently
is
detected. Likewise, combinations of a recombinant antigen on a solid phase and
synthetic peptide in the indicator phase also are possible. Any assay format
which
utilizes an antigen specific for BS202 produced or derived from a first source
as
the capture antigen and an antigen specific for BS202 from a different second
source is contemplated. Thus, various combinations of recombinant antigens, as
well as the use of synthetic peptides, purified proteins and the like, are
within the
scope of this invention. Assays such as this and others are described in U.S.
Patent No. 5,254,458, which enjoys common ownership herewith.
Other embodiments which utilize various other solid phases also are
contemplated and are within the scope of this invention. For example, ion
capture
procedures for immobilizing an immobilizable reaction complex with a
negatively
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-53-
charged polymer (described in EP publication 0326100 and EP publication No.
0406473), can be employed according to the present invention to effect a fast
solution-phase immunochemicai reaction. An immobilizable immune complex is
separated from the rest of the reaction mixture by ionic interactions between
the
negatively charged poly-anion/immune complex and the previously treated,
positively charged porous matrix and detected by using various signal
generating
systems previously described, including those described in chemiluminescent
signal measurements as described in EPO Publication No. 0 273,115.
Also, the methods of the present invention can be adapted for use in
systems which utilize microparticle technology including automated and semi-
automated systems wherein the solid phase comprises a microparticle (magnetic
or
non-magnetic). Such systems include those described in, for example, published
EPO applications Nos. EP 0 425 633 and EP 0 424 634, respectively.
The use of scanning probe microscopy (SPM) for immunoassays also is a
technology to which the monoclonal antibodies of the present invention are
easily
adaptable. In scanning probe microscopy, particularly in atomic force
microscopy, the capture phase, for example, at least one of the monoclonal
antibodies of the invention, is adhered to a solid phase and a scanning probe
microscope is utilized to detect antigen/antibody complexes which may be
present
on the surface of the solid phase. The use of scanning tunneling microscopy
eliminates the need for labels which normally must be utilized in many
immunoassay systems to detect antigen/antibody complexes. The use of SPM to
monitor specific binding reactions can occur in many ways. In one embodiment,
one member of a specific binding partner (analyte specific substance which is
the
monoclonal antibody of the invention) is attached to a surface suitable for
scanning. The attachment of the analyte specific substance may be by
adsorption
to a test piece which comprises a solid phase of a plastic or metal surface,
following methods known to those of ordinary skill in the art. Or, covalent
attachment of a specific binding partner (analyte specific substance) to a
test piece
which test piece comprises a solid phase of derivatized plastic, metal,
silicon, or
glass may be utilized. Covalent attachment methods are known to those skilled
in
the art and include a variety of means to irreversibly link specific binding
partners
to the test piece. If the test piece is silicon or glass, the surface must be
activated
prior to attaching the specific binding partner. Also, polyelectrolyte
interactions
may be used to immobilize a specific binding partner on a surface of a test
piece by
using techniques and chemistries. The preferred method of attachment is by
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-54-
covalent means. Following attachment of a specific binding member, the surface
may be further treated with materials such as serum, proteins, or other
blocking
agents to minimize non-specific binding. The surface also may be scanned
either
at the site of manufacture or point of use to verify its suitability for assay
purposes. The scanning process is not anticipated to alter the specific
binding
properties of the test piece.
While the present invention discloses the preference for the use of solid
phases, it is contemplated that the reagents such as antibodies, proteins and
peptides of the present invention can be utilized in non-solid phase assay
systems.
These assay systems are known to those skilled in the art, and are considered
to be
within the scope of the present invention.
It is contemplated that the reagent employed for the assay can be provided
in the form of a test kit with one or more containers such as vials or
bottles, with
each container containing a separate reagent such as a probe, primer,
monoclonal
antibody or a cocktail of monoclonal antibodies, or a polypeptide (e.g.
recombinantly, synthetically produced or purified) employed in the assay. The
polypeptide is selected from the group consisting of SEQUENCE ID NO 14,
SEQUENCE m NO I5, SEQUENCE ID NO 16, SEQUENCE m NO 17,
SEQUENCE m NO 18, and fragments thereof. Other components such as
buffers, controls and the like, known to those of ordinary skill in art, may
be
included in such test kits. It also is contemplated to provide test kits which
have
means for collecting test samples comprising accessible body fluids, e.g.,
blood,
urine, saliva and stool. Such tools useful for collection ("collection
materials")
include lancets and absorbent paper or cloth for collecting and stabilizing
blood;
swabs for collecting and stabilizing saliva; cups for collecting and
stabilizing urine
or stool samples. Collection materials, papers, cloths, swabs, cups and the
like,
may optionally be treated to avoid denaturation or irreversible adsorption of
the
sample. The collection materials also may be treated with or contain
preservatives,
stabilizers or antimicrobial agents to help maintain the integrity of the
specimens.
Test kits designed for the collection, stabilization and preservation of test
specimens obtained by surgery or needle biopsy are also useful. It is
contemplated that all kits may be configured in two components which can be
provided separately; one component for collection and transport of the
specimen
and the other component for the analysis of the specimen. The collection
component, for example, can be provided to the open market user while the
components for analysis can be provided to others such as laboratory personnel
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-55-
for determination of the presence, absence or amount of analyte. Further, kits
for
the collection, stabilization and preservation of test specimens may be
configured
for use by untrained personnel and may be available in the open market for use
at
home with subsequent transportation to a laboratory for analysis of the test
sample.
In Vivo Antibody Use.
Antibodies of the present invention can be used in vivo; that is, they can be
injected into patients suspected of having or having diseases of the breast
for
diagnostic or therapeutic uses. The use of antibodies for in vivo diagnosis is
well
known in the art. Sumerdon et al., Nucl. Med. Biol 17:247-254 (1990) have
described an optimized antibody-chelator for the radioimmunoscintographic
imaging of carcinoembryonic antigen (CEA) expressing tumors using Indium-111
as the label. Griffin et al., J. Clin. Onc. 9:631-640 (1991) have described
the use
of this agent in detecting tumors in patients suspected of having recurrent
colorectal cancer. The use of similar agents with paramagnetic ions as labels
for
magnetic resonance imaging is known in the art (R. B. Lauffer, Ma etic
Resonance in Medicine 22:339-342 ( 1991 ). Antibodies directed against BS202
antigen can be injected into patients suspected of having a disease of the
breast
such as breast cancer for the purpose of diagnosing or staging the disease
status of
the patient. The label used will depend on the imaging modality chosen.
Radioactive labels such as Indium-111, Technetium-99m, or Iodine-131 can be
used for planar scans or single photon emission computed tomography (SPELT).
Positron emitting labels such as Fluorine-19 can also be used for positron
emission tomography (PET). For MRI, paramagnetic ions such as Gadolinium
(III) or Manganese (II) can be used. Localization of the label within the
breast or
external to the breast may allow deterrrlination of spread of the disease. The
amount of label within the breast may allow determination of the presence or
absence of cancer of the breast.
For patients known to have a disease of the breast, injection of an antibody
directed against BS202 antigen may have therapeutic benefit. The antibody may
exert its effect without the use of attached agents by binding to BS202
antigen
expressed on or in the tissue or organ. Alternatively, the antibody may be
conjugated to cytotoxic agents such as drugs, toxins, or radionuclides to
enhance
its therapeutic effect. Garnett and Baldwin, Cancer Research 46:2407-2412
( 1986) have described the preparation of a drug-monoclonal antibody
conjugate.
Pastan et al., Cell 47:641-648 ( 1986) have reviewed the use of toxins
conjugated
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-56-
to monoclonal antibodies for the therapy of various cancers. Goodwin and
Meares, Cancer Supplement 80:2675-2680 ( 1997) have described the use of
Yttrium-90 labeled monoclonal antibodies in various strategies to maximize the
dose to tumor while limiting normal tissue toxicity. Other known cytotoxic
radionuclides include Copper-67, Iodine-131, and Rhenium-186 all of which can
be used to label monoclonal antibodies directed against BS202 antigen for the
treatment of cancer of the breast.
E. c i bacteria (clone 1212072) was deposited on November 4, 1997 with
the American Type Culture Collection (A.T.C.C.), 12301 Parkiawn Drive,
Rockville, Maryland 20852. The deposit was made under the terms of the
Budapest Treaty and will be maintained for a period of thirty (30) years from
the
date of deposit, or for five (S) years after the last request for the deposit,
or for the
enforceable period of the U.S. patent, whichever is longer. The deposit and
any
other deposited material described herein are provided for convenience only,
and
are not required to practice the present invention in view of the teachings
provided
herein. The cDNA sequence in all of the deposited material is incorporated
herein
by reference. Clone 1212072 was accorded A.T.C.C. Deposit No. 98571.
The present invention will now be described by way of examples, which
are meant to illustrate, but not to linut, the scope of the present invention.
EXAMPLES
Example 1 ~ Identification of Breast Tissue Library BS202 Gene Specific Clones
A. Library Comparison of Expressed Sequence Tags (EST'sl or
Transcript Images. Partial sequences of cDNA clone inserts, so-called
"expressed
sequence tags" (EST's), were derived from cDNA libraries made from breast
tumor tissues, breast non-tumor tissues and numerous other tissues, both tumor
and non-tumor and entered into a database (LIFESEQTM database, available from
Incyte Pharmaceuticals, Palo Alto, CA) as gene transcript images. She
International Publication No. WO 95/20681. (A transcript image is a listing of
the
number of EST's for each of the represented genes in a given tissue library.
EST's sharing regions of mutual sequence overlap are classified into clusters.
A
cluster is assigned a clone number from a representative 5' EST. Often, a
cluster
of interest can be extended by comparing its consensus sequence with sequences
of other EST's which did not meet the criteria for automated clustering. The
alignment of all available clusters and single EST's represent a contig from
which
a consensus sequence is derived.) The transcript images then were evaluated to
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-57-
identify EST sequences that were representative primarily of the breast tissue
libraries. These target clones then were ranked according to their abundance
(occurrence) in the target libraries and their absence from background
libraries.
Higher abundance clones with low background occurrence were given higher
study priority. EST's corresponding to the consensus sequence of BS202 were
found in 44.4% ( 16 of 36) of breast tissue libraries. EST's corresponding to
the
consensus sequence SEQUENCE ID NO 7 (or fragments thereof) were found in
only 0.14% ( 1 of 683) of the other, non-breast, libraries of the data base.
Therefore, the consensus sequence or fragment thereof was found more than 303
times more often in breast than non-breast tissues. Overlapping clones 1212072
{SEQUENCE ID NO 1), 3113233 (SEQUENCE )D NO 2), 3172007
(SEQUENCE ID NO 3), 1004371 (SEQUENCE ID NO 4), 419671
(SEQUENCE ID NO 5), respectively, were identified for further study. These
represented the minimum number of clones that (along with the full-length
sequence of clone 1212072 {hereinafter referred to as "clone 1212072IH
(SEQUENCE ID NO 6)") were needed to form the contig and from which the
consensus sequence provided herein (SEQUENCE ID NO 7) was derived.
B. Generation of a Consensus Sequence. The nucleotide sequences of
clones 1212072 (SEQUENCE ID NO 1 ), 3113233 (SEQUENCE ID NO 2),
3172007 (SEQUENCE ID NO 3), 1004371 (SEQUENCE ID NO 4), 419671
(SEQUENCE ID NO 5), and 1212072IH (SEQUENCE ID NO 6) were entered in
the SequencherTM Program (available from Gene Codes Corporation, Ann Arbor,
MI) in order to generate a nucleotide alignment (contig map) and then generate
their consensus sequence (SEQUENCE )D NO 7). Figures lA-1B show the
nucleotide sequence alignment of these clones and their resultant nucleotide
consensus sequence (SEQUENCE ID NO 7). Figure 2 presents the contig map
depicting the clones 1212072 (SEQUENCE ID NO 1 ), 3113233 (SEQUENCE
>D NO 2), 3172007 (SEQUENCE ID NO 3), 1004371 (SEQUENCE ID NO 4),
419671 (SEQUENCE m NO 5), and 1212072IH (SEQUENCE ID NO 6) which
form overlapping regions of the BS202 gene and the resultant consensus
nucleotide sequence (SEQUENCE ID NO 7) of these clones in a graphic display.
Following this, a three-frame translation was performed on the consensus
sequence (SEQUENCE m NO 7). The first forward frame was found to have an
open reading frame encoding a 97 residue amino acid sequence which is
presented
as SEQUENCE ID NO 14. The open reading frame corresponds to nucleotides 1-
291 of SEQUENCE ID NO 7. The 97 residue polypeptide sequence depicted in
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-58-
SEQUENCE ID NO 14 was compared with published sequences using software
and techniques known to those skilled in the art. A portion of the polypeptide
sequence of a bovine chromaffin granule APTase II homologue was found to be
partially homologous to the BS202 polypeptide of SEQUENCE ID NO 14. The
bovine chromaffin granule APTase II homologue is described by Tang et al.
Science 272:1495-1497 (1996), and the sequence has been deposited with
GenBank under Accession No U51100.
Example 2: Sequencing of BS202 EST-Specific Clones
The full-length DNA sequence of clone 1212072 of the BS202 gene contig
was determined (clone 1212072IH, SEQUENCE ID NO 6) using dideoxy
termination sequencing with dye terminators following known methods [F.
Sanger et al., PNAS U.S.A. 74:5463 ( 1977)).
Because vectors such as pSPORTI (Life Technologies, Gaithersburg,
MD) and pINCY (available from Incyte Pharmaceuticals, Inc., Palo Alto, CA)
contain universal priming sites just adjacent to the 3' and 5' ligation
junctions of
the inserts, the inserts were sequenced in both directions using two universal
primers (SEQUENCE ID NO 10 and SEQUENCE ID NO 11, available from New
England Biolabs, Beverly, MA, and Applied Biosystems Inc, Foster City, CA,
respectively). The sequencing reactions were run on a polyacrylamide
denaturing
gel, and the sequences were determined by an Applied Biosystems 377 Sequencer
(available from Applied Biosystems, Foster City, CA). Additional sequencing
primers (SEQUENCE ID NO 12 and SEQUENCE ff~ NO 13) were designed
from sequence information of the consensus sequence (SEQUENCE ID NO 7).
These primers then were used to determine the remaining DNA sequence of the
cloned insert from each DNA strand, as previously described.
Example 3: Nucleic Acid
A. RNA Extraction from Tissue. Total RNA is isolated from breast
tissues and from non-breast tissues. Various methods are utilized, including
but
not limited to the lithium chloride/urea technique, known in the art and
described
by Kato et al., 3. Virol. 61:2182-2191 (1987), and TRIzoITM (Gibco-BRL, Grand
Island, NY).
Briefly, tissue is placed in a sterile conical tube on ice and 10-15 volumes
of 3 M LiCI, 6 M urea, 5 mM EDTA, 0.1 M ~3-mercaptoethanol, 50 mM Tris-HCI
(pH 7.5) are added. The tissue is homogenized with a Polytron°
homogenizer
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-59-
(Brinkman Instruments, Inc., Westbury, NY) for 30-50 sec on ice. The solution
is transferred to a 15 mi plastic centrifuge tube and placed overnight at -
20°C. The
tube is centrifuged for 90 min at 9,000 x g at 0-4°C and the
supernatant is
immediately decanted. Ten ml of 3 M LiCI are added and the tube is vortexed
for
5 sec. The tube is centrifuged for 45 min at 11,000 x g at 0-4°C. The
decanting,
resuspension in LiCI, and centrifugation is repeated and the final pellet is
air dried
and suspended in 2 ml of 1 mM EDTA, 0.5% SDS, 10 mM Tris (pH 7.5).
Twenty microiiters (20 ~,l) of Proteinase K (20 mg/ml) are added, and the
solution
is incubated for 30 min at 37°C with occasional mixing. One-tenth
volume (0.22-
0.25 ml) of 3 M NaCI is added and the solution is vortexed before transfer
into
another tube containing 2 ml of phenol/chloroform/isoamyl alcohol (PCI). The
tube is vortexed for 1-3 sec and centrifuged for 20 min at 3,000 x g at
10°C. The
PCI extraction is repeated and followed by two similar extractions with
chloroform/isoamyl alcohol (CI). The final aqueous solution is transferred to
a
1 S prechilled 15 ml Corex glass tube containing 6 ml of absolute ethanol, the
tube is
covered with parafilm, and placed at -20°C overnight. The tube is
centrifuged for
30 min at 10,000 x g at 0-4°C and the ethanol supernatant is decanted
immediately.
The RNA pellet is washed four times with 10 ml of 75% ice-cold ethanol and the
final pellet is air dried for 15 min at room temperature. The RNA is suspended
in
0.5 ml of 10 mM TE (pH 7.6, 1 mM EDTA) and its concentration is determined
spectrophotometrically. RNA samples are aliquoted and stored at -70°C
as ethanol
precipitates.
The quality of the RNA is determined by agarose gel electrophoresis (see
Example 5, Northern Blot Analysis) and staining with 0.5 ~.g/ml ethidium
bromide for one hour. RNA samples that do not contain intact rRNAs are
excluded from the study.
Alternatively, for RT-PCR analysis, 1 ml of Ultraspec RNA reagent is
added to 120 mg of pulverized tissue in a 2.0 ml polypropylene microfuge tube,
homogenized with a Polytron~ homogenizer (Brinkman Instruments, Inc.,
Westbury, NY) for 50 sec and placed on ice for 5 min. Then, 0.2 ml of
chloroform is added to each sample, followed by vortexing for 15 sec. The
sample
is placed on ice for another S min, followed by centrifugation at 12,000 x g
for 15
min at 4°C. The upper layer is collected and transferred to another
RNase-free 2.0
ml microfuge tube. An equal volume of isopropanol is added to each sample, and
3S the solution is placed on ice for 10 min. The sample is centrifuged at
12,000 x g
for 10 min at 4°C, and the supernatant is discarded. The remaining
pellet is
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-6 I -
isolation of poly-adenylated RNA. Total RNA or mRNA can be dissolved in lysis
buffer (5 M guanidine thiocyanate, 0.1 M EDTA, pH 7.0) for analysis in the
ribonuclease protection assay.
C. RNA Extraction from pol, sue. Tissue is minced in saline at 4°C
and mixed with 2.5 volumes of 0.8 M sucrose in a TK,S~1VI ( 150 mM KCI, 5 mM
MgCl2, 50 mM Tris-HCI, pH 7.4) solution containing 6 mM 2-mercaptoethanol.
The tissue is homogenized in a Teflon-glass Potter homogenizes with five
strokes
at 100-200 rpm followed by six strokes in a Dounce homogenizes, as described
by
B. Mechler, Methods in Enz~mologx 152:241-248 (1987). The homogenate then
is centrifuged at 12,000 x g for 15 min at 4°C to sediment the nuclei.
The
polysomes are isolated by mixing 2 ml of the supernatant with 6 ml of 2.5 M
sucrose in TK,SOM and layering this mixture over 4 ml of 2.5 M sucrose in
TK,SOM in a 38 ml polyallomer tube. Two additional sucrose TK,SOM solutions
are successively layered onto the extract fraction; a first layer of 13 mi
2.05 M
I S sucrose followed by a second layer of 6 ml of 1.3 M sucrose. The polysomes
are
isolated by centrifuging the gradient at 90,000 x g for 5 hr at 4°C.
The fraction
then is taken from the 1.3 M sucrose/2.05 M sucrose interface with a
siliconized
pasteur pipette and diluted in an equal volume of TE {10 mM Tris-HCI, pH 7.4,
I
mM EDTA). An equal volume of 90°C SDS buffer ( 1 % SDS, 200 mM NaCI, 20
mM Tris-HCI, pH 7.4) is added and the solution is incubated in a boiling water
bath for 2 min. Proteins next are digested with a Proteinase K digestion (50
mg/ml) for 15 min at 37°C. The mRNA is purified with 3 equal volumes of
phenol-chloroform extractions followed by precipitation with 0. I volume of 2
M
sodium acetate (pH 5.2) and 2 volumes of 100% ethanol at -20°C
overnight. The
precipitated RNA is recovered by centrifugation at 12,000 x g for 10 min at
4°C.
The RNA is dried and resuspended in TE (pH 7.4) or distilled water. The
resuspended RNA then can be used in a slot blot or dot blot hybridization
assay to
check for the presence of BS202 mRNA (see Example 6).
The quality of nucleic acid and proteins is dependent on the method of
preparation used. Each sample may require a different preparation technique to
maximize isolation efficiency of the target molecule. These preparation
techniques
are within the skill of the ordinary artisan.
Example 4: Ribonuclease Protection Assav
A Synthesis of Labeled Complementary RNA IcRNAI Hybridization
Probe and Unlabeled Sense Strand. Labeled antisense and unlabeled sense
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-60-
washed twice with cold 75% ethanol, resuspended by vortexing, and the
resuspended material is then pelleted by centrifugation at 7500 x g for 5 min
at
4°C. Finally, the RNA pellet is dried in a Speedvac (Savant,
Farmingdale, NY)
for 5 min and reconstituted in RNase-free water.
B. RNA Extraction from Blood Mononuclear Cells. Mononuclear cells
are isolated from blood samples from patients by centrifugation using Ficoll-
Hypaque as follows. A 10 ml volume of whole blood is mixed with an equal
volume of RPMI Medium (Gibco-BRL, Grand Island, NY). This mixture is then
underlayed with 10 ml of Ficoll-Hypaque (Pharmacia, Piscataway, NJ) and
centrifuged for 30 minutes at 200 x g. The huffy coat containing the
mononuclear
cells is removed, diluted to 50 ml with Dulbecco's PBS (Gibco-BRL, Grand
Island, NY) and the mixture centrifuged for 10 minutes at 200 x g. After two
washes, the resulting pellet is resuspended in Dulbecco's PBS to a final
volume of
1 ml.
RNA is prepared from the isolated mononuclear cells as described by N.
Kato et al., J. Viroloev 61:2182-2191 ( 1987). Briefly, the pelleted
mononuclear
cells are brought to a final volume of 1 ml and then are resuspended in 250
p.L of
PBS and mixed with 2.5 ml of 3 M LiCI, 6 M urea, 5 mM EDTA, 0.1 M 2-
mercaptoethanol, 50 mM Tris-HCl (pH 7.5). The resulting mixture is
homogenized and incubated at -20°C overnight. The homogenate is
centrifuged at
8,000 RPM in a Beckman J2-21M rotor for 90 minutes at 0-4.°C. The
pellet is
resuspended in 10 ml of 3 M LiCI by vortexing and then centrifuged at 10,000
RPM in a Beckman J2-21M rotor centrifuge for 45 minutes at 0-4°C.
The
resuspending and pelleting steps then are repeated. The pellet is resuspended
in 2
ml of 1 mM EDTA, 0.5% SDS, 10 mM Tris (pH 7.5) and 400 ~.g Proteinase K
with vortexing and then it is incubated at 37°C for 30 minutes with
shaking. One
tenth volume of 3 M NaCI then is added and the mixture is vortexed. Proteins
are
removed by two cycles of extraction with phenol/ chloroform/ isoamyl alcohol
(PCI) followed by one extraction with chloroform/ isoamyl alcohol {CI). RNA is
precipitated by the addition of 6 ml of absolute ethanol followed by overnight
incubation at -20°C. After the precipitated RNA is collected by
centrifugation, the
pellet is washed 4 times in 75% ethanol. The pelleted RNA is then dissolved in
solution containing 1 mM EDTA, 10 mM Tris-HCl (pH 7.5). .
Non-breast tissues are used as negative controls. The mRNA can be
further purified from total RNA by using commercially available kits such as
oligo
dT cellulose spin columns (RediCoITM from Pharmacia, Uppsala, Sweden) for the
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-62-
riboprobes are transcribed from the BS202 gene cDNA sequence which contains a
5' RNA polymerase promoter such as SP6 or T7. The sequence may be from a
vector containing the appropriate BS202 cDNA insert, or from a PCR-generated
product of the insert using PCR primers which incorporate a 5' RNA polymerase
promoter sequence. For example, clone 1212072, or another comparable clone
containing the BS202 gene cDNA sequence flanked by opposed SP6 and T7 or
other RNA polymerase promoters, is purified using a Qiagen Plasmid
Purification
Kit (Qiagen, Chatsworth, CA). Then, 10 pg of the plasmid DNA are linearized
by cutting with an appropriate restriction enzyme such as DdeI for 1 hr at
37°C.
The linearized plasmid DNA is purified using the QIAprep Kit (Qiagen,
Chatsworth, CA) and used for the synthesis of antisense transcript from the
appropriate promoter using the Riboprobe~ in vitro Transcription System
(Promega Corporation, Madison, WI), as described by the supplier's
instructions,
incorporating either (alpha32P) CTP (Amersham Life Sciences, Inc. Arlington
Heights, IL) or biotinylated CTP as a label. To generate the sense strand, 10
p.g
of the purified plasmid DNA are cut with restriction enzymes, such as XbaI and
NotI, and transcribed as above from the appropriate promoter. Both sense and
antisense strands are isolated by spin column chromatography. Unlabeled sense
strand is quantitated by UV absorption at 260 nm.
B. Hybridization of Labeled Probe to Tar ret. Frozen tissue is pulverized
to powder under liquid nitrogen and 100-500 mg are dissolved in i ml of lysis
buffer, available as a component of the Direct ProtectT"'' Lysate RNase
Protection
Kit (Ambion, Inc., Austin, TX). Further dissolution can be achieved using a
tissue homogenizer. In addition, a dilution series of a known amount of sense
strand in mouse liver lysate is made for use as a positive control. Finally,
45 p.l of
solubilized tissue or diluted sense strand is mixed directly with either: ( 1
) 1 x 105
cpm of radioactively labeled probe; or (2) 250 pg of non-isotopically labeled
probe
in 5 p,l of lysis buffer. Hybridization is allowed to proceed overnight at
37°C.
See, T. Kaabache et al., Anal. Biochem. 232:225-230 ( 1995).
C. RNase Digestion. RNA that is not hybridized to probe is removed
from the reaction as per the Direct Protect~'~"'' protocol using a solution of
RNase A
and RNase T 1 for 30 min at 37°C, followed by removal of RNase by
Proteinase K
digestion in the presence of sodium sarcosyl. Hybridized fragments protected
from digestion are then precipitated by the addition of an equal volume of
isopropanol and placed at -70°C for 3 hr. The precipitates are
collected by
centrifugation at 12,000 x g for 20 min.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-63-
D. Fragment Analysis. The precipitates are dissolved in denaturing gel
loading dye (80% formamide, 10 mM EDTA (pH 8.0), 1 mg/ml xylene cyanol, 1
mg/ml bromophenol blue), heat denatured, and electrophoresed in 6%
polyacrylamide TBE, 8 M urea denaturing gels. The gels are imaged and analyzed
using the STORMS storage phosphor autoradiography system (Molecular
Dynamics, Sunnyvale, CA). Quantitation of protected fragment bands, expressed
in femtograms (fg), is achieved by comparing the peak areas obtained from the
test
samples to those from the known dilutions of the positive control sense strand
(see
Section B, su~~a). The results are expressed in molecules of BS202 RNA/cell
and
as a image rating score. In cases where non-isotopic labels are used, hybrids
are
transferred from the gels to membranes (nylon or nitrocellulose) by blotting
and
then analyzed using detection systems that employ streptavidin alkaline
phosphatase conjugates and chemiluminesence or chemifluoresence reagents.
Detection of a product comprising a sequence selected from the group
consisting of SEQUENCE ID NO 1, SEQUENCE m NO 2, SEQUENCE ID NO
3, SEQUENCE ID NO 4, SEQUENCE ID NO 5, SEQUENCE 117 NO 6,
SEQUENCE ID NO 7, and fragments or complements thereof, is indicative of the
presence of BS202 mRNAs, suggesting a diagnosis of a breast tissue disease or
condition, such as breast cancer.
Example 5: Northern Blotting
The Northern blot technique is used to identify a specific size RNA
fragment from a complex population of RNA using gel electrophoresis and
nucleic
acid hybridization. Northern blotting is well-known technique in the art.
Briefly,
5-10 p,g of total RNA (see Example 3) are incubated in 15 p,l of a solution
containing 40 mM morphilinopropanesulfonic acid (MOPS) (pH 7.0), 10 mM
sodium acetate, 1 mM EDTA, 2.2 M formaldehyde, 50% v/v formamide for 15
min at 65°C. The denatured RNA is mixed with 2 p,l of loading buffer
(50%
glycerol, 1 mM EDTA, 0.4% bromophenol blue, 0.4% xylene cyanol) and loaded
into a denaturing 1.0% agarose gel containing 40 mM MOPS (pH 7.0), 10 mM
sodium acetate, 1 mM EDTA and 2.2 M formaldehyde. The gel is electrophoresed
at 60 V for 1.5 hr and rinsed in RNAse free water. RNA is transferred from the
gel onto nylon membranes (Brightstar-Plus, Ambion, Inc., Austin, TX) for 1.5
hours using the downward alkaline capillary transfer method (Chomczynski,
Anal. Biochem. 201:134-139, 1992). The filter is rinsed with 1X SSC, and RNA
is crosslinked to the filter using a Stratalinker'~"'' (Stratagene, Inc., La
lolla, CA)
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
on the autocrosslinking mode and dried for 15 min. The membrane is then placed
into a hybridization tube containing 20 ml of preheated prehybridization
solution
(SX SSC, 50% formamide, SX Denhardt's solution, 100 p,g/ml denatured salmon
sperm DNA) and incubated in a 42°C hybridization oven for at least 3
hr. While
the blot is prehybridizing, a 3~P-labeled random-primed probe is generated
using
the BS202 insert fragment (obtained by digesting clone 1212072 or another
comparable clone with XbaI and NotI) using Random Primer DNA Labeling
System (Life Technologies, Inc., Gaithersburg, MD) according to the
manufacturer's instructions. Half of the probe is boiled for 10 min, quick
chilled
on ice and added to the hybridization tube. Hybridization is carried out at
42°C for
at least 12 hr. The hybridization solution is discarded and the filter is
washed in
30 ml of 3X SSC, 0.1 % SDS at 42°C for 15 min, followed by 30 ml of 3X
SSC,
0.1 % SDS at 42°C for 15 min. The filter is wrapped in Saran Wrap,
exposed to
Kodak XAR-Omat film for 8-96 hr, and the film is developed for analysis. High
level of expression of mRNA corresponding to a sequence selected from the
group
consisting of SEQUENCE m NO 1, SEQUENCE ID NO 2, SEQUENCE ID NO
3, SEQUENCE m NO 4, SEQUENCE ID NO 5, SEQUENCE ID NO 6,
SEQUENCE B7 NO 7, and fragments or complements thereof, is an indication of
the presence of BS202 mRNA, suggesting a diagnosis of a breast tissue disease
or
condition, such as breast cancer.
Example 6: Dot BIQt/Slot Blot
Dot and slot blot assays are quick methods to evaluate the presence of a
specific nucleic acid sequence in a complex mix of nucleic acid. To perform
such
assays, up to 50 pg of RNA are mixed in 50 p.l of 50% formamide, 7%
formaldehyde, 1X SSC, incubated 15 min at 68°C, and then cooled on ice.
Then,
100 p.l of 20X SSC are added to the RNA mixture and loaded under vacuum onto
a manifold apparatus that has a prepared nitrocellulose or nylon membrane. The
membrane is soaked in water, 20X SSC for 1 hour, placed on two sheets of 20X
SSC prewet Whatman #3 filter paper, and loaded into a slot blot or dot blot
vacuum manifold apparatus. The slot blot is analyzed with probes prepared and
labeled as described in Example 4, supra. Detection of mRNA corresponding to a
sequence selected from the group consisting of SEQUENCE ID NO 1,
SEQUENCE ID NO 2, SEQUENCE m NO 3, SEQUENCE ID NO 4,
SEQUENCE ID NO 5, SEQUENCE ID NO 6, SEQUENCE ID NO 7, and
fragments or complements thereof, is an indication of the presence of BS202,
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-65-
suggesting a diagnosis of a breast tissue disease or condition, such as breast
cancer.
Other methods and buffers which can be utilized in the methods described
in Examples 5 and 6, but not specifically detailed herein, are known in the
art and
are described in J. Sambrook et al., supra.
Example 7: In Situ Hybridization
This method is useful to directly detect specific target nucleic acid
sequences in cells using detectable nucleic acid hybridization probes.
Tissues are prepared with cross-linking fixative agents such as
paraformaldehyde or glutaraldehyde for maximum cellular RNA retention. See,
L. Angerer et al., Methods in Cell Biol. 35:37-71 ( 1991 ). Briefly, the
tissue is
placed in greater than 5 volumes of 1 % glutaraldehyde in 50 mM sodium
phosphate, pH 7.5 at 4°C for 30 min. The solution is changed with fresh
glutaraldehyde solution ( 1 % glutaraldehyde in 50 mM sodium phosphate, pH
7.5)
for a further 30 min fixing. The fixing solution should have an osmolality of
approximately 0.375% NaCI. The tissue is washed once in isotonic NaCI to
remove the phosphate.
The fixed tissues then are embedded in paraffin as follows. The tissue is
dehydrated though a series of increasing ethanol concentrations for 15 min
each:
50% (twice), 70% (twice), 85%, 90% and then 100% (twice). Next, the tissue is
soaked in two changes of xylene for 20 min each at room temperature. The
tissue
is then soaked in two changes of a 1:1 mixture of xylene and paraffin for 20
min
each at 60°C; and then in three final changes of paraffin for 15 min
each.
Next, the tissue is cut in 5 p.m sections using a standard microtome and
placed on a slide previously treated with a tissue adhesive such as 3-
aminopropyltriethoxysilane.
Paraffin is removed from the tissue by two 10 min xylene soaks and
rehydrated in a series of decreasing ethanol concentrations: 99% (twice), 95%,
85%, 70%, 50%, and 30%; and then in distilled water (twice). The sections are
pre-treated with 0.2 M HCl for 10 min and permeabilized with 2 ~,g/ml
Proteinase
K at 37°C for 15 min.
Labeled riboprobes transcribed from the BS202 gene plasmid (see
Example 4) are hybridized to the prepared tissue sections and incubated
overnight
at 56°C in 3X standard saline extract and 50% formamide. Excess probe
is
removed by washing in ZX standard saline citrate and 50% formamide followed
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-66-
by digestion with 100 ~.g/ml RNase A at 37°C for 30 min. Probe
fluorescence is
visualized by illumination with ultraviolet (UV) light under a microscope.
Fluorescence in the cytoplasm is indicative of BS202 mRNA. Alternatively, the
sections can be visualized by autoradiography. °
Example 8: Reverse Transcription PCR
A. One S~ RT-PCR Assay. Target-specific primers are designed to
detect the above-described target sequences by reverse transcription PCR using
methods known in the art. One step RT-PCR is a sequential procedure that
performs both RT and PCR in a single reaction mixture. The procedure is
performed in a 200 p,l reaction mixture containing 50 mM (N,N,-bis[2-
Hydroxyethyl]glycine), pH 8.15, 81.7 mM KOAc, 33.33 mM KOH, 0.01 mg/ml
bovine serum albumin, 0.1 mM ethylene diaminetetraacetic acid, 0.02 mg/ml
NaN;, 8% w/v glycerol, 150 p.M each of dNTP, 0.25 pM each primer, SU rTth
polymerase, 3.25 mM Mn(OAc)Z and S ~,1 of target RNA (see Example 3). Since
RNA and the rTth polymerase enzyme are unstable in the presence of Mn(OAc)2,
the Mn(OAc)2 should be added just before target addition. Optimal conditions
for
cDNA synthesis and thermal cycling readily can be determined by those skilled
in
the art. The reaction is incubated in a Perkin-Elmer Thermal Cycler 480.
Conditions which may be found useful include cDNA synthesis at 60°-
70°C for
15-45 min and 30-45 amplification cycles at 94°C, I min; 55°-
70°C, 1 min; 72°C,
2 min. One step RT-PCR also may be performed by using a dual enzyme
procedure with Taq polymerase and a reverse transcriptase enzyme, such as
MMLV (Moloney murine leukemia virus) or AMV (avian myeloblastosis virus)
RT (reverse transcriptase) enzymes.
B. Traditional RT-PCR. Alternatively, a traditional two-step RT-PCR
reaction may be performed, as described by K.Q. Hu et al., Virology 181:721-
726 ( 1991 }, as follows. The extracted mRNA is transcribed in a 25 ~.1
reaction
mixture containing 10 mM Tris-HCI, pH 8.3, 5 mM MgCl2, 500 p,M dNTP, 20
U RNasin, 1 ~tM antisense primer and 25 U AMV or MMLV reverse
transcriptase. Reverse transcription is performed at 37-45°C for 30-60
min,
followed by further incubation at 95°C for 5 min to inactivate the RT.
PCR is
performed using 10 ~,1 of the cDNA reaction in a final PCR reaction volume of
50
p.l containing 10 mM Tris-HCl (pH 8.3), 50 mM KCI, 2 mM MgCl2, 200 N.M
dNTP, 0.5 pM of each primer and 2.5 U of Taq polymerase. Optimal conditions
for cDNA synthesis and thermal cycling can be readily determined by those
skilled
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-67-
in the art. The reaction is incubated in a Perkin-Elmer Thermal Cycler 480 or
other comparable instrument. Conditions which may be found useful include 30-
45 cycles of amplification (94°C, 1 min; 55-70°C, 1 min;
72°C, 2 min), final
extension (72°C, 10 min) and soak at 4°C.
C. PCR Fragment Analysis. The correct products then can be verified by
size determination using gel electrophoresis with SYBR~ Green I nucleic acid
gel
stain (Molecular Probes, Eugene, OR) and imaged using a STORM imaging
system, or also verified by Southern, dot or slot blot analysis using a
labeled
probe against the internal sequences of the PCR product. The probes also may
be
polynucleotides analogs, such as morpholinos or peptide nucleic acids analogs
{PNAs). Detection of a product comprising a sequence selected from the group
consisting of SEQUENCE ID NO I, SEQUENCE ID NO 2, SEQUENCE ID NO
3, SEQUENCE ID NO 4, SEQUENCE ID NO 5, SEQUENCE ID NO 6,
SEQUENCE ID NO 7, and fragments or complements thereof, is indicative of the
presence of BS202 mRNA(s), suggesting a diagnosis of a breast tissue disease
or
condition, such as breast cancer.
~xamnle 9: OH-PCR
A. Probe selection and Labeling. Target-specific primers and probes are
designed to detect the above-described target sequences by oligonucleotide
hybridization PCR. International Publication Nos WO 92/10505, published June
25, 1992, and WO 92/11388, published July 9, 1992, teach methods for labeling
oligonucleotides at their 5' and 3' ends, respectively. According to one known
method for labeling an oligonucieotide, a label-phosphoramidite reagent is
prepared and used to add the label to the oligonucleotide during its
synthesis. For
example, set N. T. Thuong et al., Tet. Letters 29(46):5905-5908 ( 1988); or J.
S.
Cohen et al., published U.S. Patent Application 071246,688 (NTIS ORDER No.
PAT-APPL-7-246,688) ( 1989). Preferably, probes are labeled at their 3' end to
prevent participation in PCR and the formation of undesired extension
products.
For one step OH-PCR, the probe should have a TM at least 15°C below
the TM of
the primers. The primers and probes are utilized as specific binding members,
with or without detectable labels, using standard phosphoramidite chemistry
and/or post-synthetic labeling methods which are well-known to one skilled in
the
art.
B. One Step Oligo~,ybridization PCR. OH-PCR is performed on a 200
p.l reaction containing 50 mM (N,N,-bis[2-Hydroxyethyl]glycine), pH 8.15, 8I.7
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-68-
mM KOAc, 33.33 mM KOH, 0.01 mg/ml bovine serum albumin, 0. I mM
ethylene diaminetetraacetic acid, 0.02 mg/ml NaN~, 8% w/v glycerol, ISO p.IVI
each of dNTP, 0.25 p.M each primer, 3.75 nM probe, SU rTth polymerase, 3.25
mM Mn(OAc)z and 5 p,l blood equivalents of target (see Example 3). Since RNA
and the rTth polymerase enzyme are unstable in the presence of Mn(OAc)2, the
Mn(OAc)2 should be added just before target addition. The reaction is
incubated
in a Perkin-Elmer Thermal Cycler 480. Optimal conditions for cDNA synthesis
and thermal cycling can be readily determined by those skilled in the art.
Conditions which may be found useful include cDNA synthesis (60°C,
30 min),
30-45 amplification cycles (94°C, 40 sec; 55-70°C, 60 sec),
oligo-hybridization
(97°C, 5 min; 15°C, 5 min; 15°C soak). The correct
reaction product contains at
least one of the strands of the PCR product and an internally hybridized
probe.
C. OH-PCR Product Ana~sis. Amplified reaction products are detected
on an LCx~ Analyzer system (available from Abbott Laboratories, Abbott Park,
1 S IL). Briefly, the correct reaction product is captured by an antibody
labeled
microparticle at a capturable site on either the PCR product strand or the
hybridization probe, and the complex is detected by binding of a detectable
antibody conjugate to either a detectable site on the probe or the PCR strand.
Only
a complex containing a PCR strand hybridized with the internal probe is
detectable. The detection of this complex then is indicative of the presence
of
BS202 mRNA, suggesting a diagnosis of a breast disease or condition, such as
breast cancer.
Many other detection formats exist which can be used and/or modified by
those skilled in the art to detect the presence of amplified or non-amplified
BS202-
derived nucleic acid sequences including, but not limited to, ligase chain
reaction
(LCR, Abbott Laboratories, Abbott Park, IL); Q-beta replicase (Gene-TrakTM,
Naperville, Illinois), branched chain reaction (Chiron, Emeryville, CA) and
strand
displacement assays (Becton Dickinson, Research Triangle Park, NC).
Examt~le 10: Synthetic Peptide Production
Synthetic peptides were modeled and then prepared based upon the
predicted amino acid sequence of the BS202 polypeptide consensus sequence (see
Example 1 ). In particular, a number of BS202 peptides derived from
SEQUENCE ID NO 14 were prepared, including the peptides of SEQUENCE ID
NO 15, SEQUENCE 1D NO 16, SEQUENCE m NO 17, and SEQUENCE ID
NO 18. All peptides were synthesized on a Symphony Peptide Synthesizer
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-69-
(available from Rainin Instrument Co, Emeryville, CA) or similar instrument,
using FMOC chemistry, standard cycles and in-situ HBTU activation. Cleavage
and deprotection conditions were as follows: a volume of 2.5 ml of cleavage
reagent (77.5% v/v trifluoroacetic acid, IS% v/v ethanedithiol, 2.5% v/v
water,
5% v/v thioanisole, 1-2% w/v phenol) was added to the resin, and agitated at
room temperature for 2-4 hours. The filtrate was then removed and the peptide
was precipitated from the cleavage reagent with cold diethyl ether. Each
peptide
was filtered, purified via reverse-phase preparative HPLC using a
water/acetonitrile/0.1 % TFA gradient, and lyophilized. The product was
confirmed by mass spectrometry.
The purified peptides were used to immunize animals (see Example 14).
Example 1 I a: Expression of Protein in a Cell Line Using Plasmid 577
A. Construction of a BS202 Exnre ion Plasmid. Plasmid 577, described
IS in U.S. patent application Serial No. 08/478,073, filed June 7, 1995, has
been
constructed for the expression of secreted antigens in a permanent cell line.
This
plasmid contains the following DNA segments: (a) a 2.3 kb fragment of pBR322
containing bacterial beta-lactamase and origin of DNA replication; (b) a 1.8
kb
cassette directing expression of a neomycin resistance gene under control of
HSV-
1 thymidine kinase promoter and poly-A addition signals; (c) a 1.9 kb cassette
directing expression of a dihydrofolate reductase gene under the control of an
SV40 promoter and poly-A addition signals; (d) a 3.5 kb cassette directing
expression of a rabbit immunoglobulin heavy chain signal sequence fused to a
modified hepatitis C virus (HCV) E2 protein under the control of the SV40T-Ag
promoter and transcription enhancer, the hepatitis B virus surface antigen
(HBsAg) enhancer I followed by a fragment of Herpes Simplex Virus-1 (HSV-I )
genome providing poly-A addition signals; and (e) a residual 0.7 kb fragment
of
SV40 genome late region of no function in this plasmid. All of the segments of
the vector were assembled by standard methods known to those skilled in the
art
of molecular biology.
Plasmids for the expression of secretable BS202 proteins are constructed
by replacing the hepatitis C virus E2 protein coding sequence in plasmid 577
with
that of a BS202 polynucleotide sequence selected from the group consisting of
SEQUENCE ID NO 1, SEQUENCE ID NO 2, SEQUENCE ID NO 3,
SEQUENCE ID NO 4, SEQUENCE ID NO 5, SEQUENCE ID NO 6,
SEQUENCE ID NO 7, and fragments or complements thereof, as follows.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-70-
Digestion of plasmid 577 with XbaI releases the hepatitis C virus E2 gene
fragment. The resulting plasmid backbone allows insertion of the BS202 cDNA -
insert downstream of the rabbit immunoglobulin heavy chain signal sequence
which directs the expressed proteins into the secretory pathway of the cell.
The
BS202 cDNA fragment is generated by PCR using standard procedures. Encoded
in the sense PCR primer sequence is an XbaI site, immediately followed by a 12
nucleotide sequence that encodes the amino acid sequence Ser-Asn-Glu-Leu
("SNEL") to promote signal protease processing, efficient secretion and final
product stability in culture fluids. Immediately following this 12 nucleotide
sequence the primer contains nucleotides complementary to template sequences
encoding amino acids of the BS202 gene. The antisense primer incorporates a
sequence encoding the following eight amino acids just before the stop codons:
Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys (SEQUENCE D7 NO 19). Within this
sequence is incorporated a recognition site to aid in analysis and
purification of the
IS BS202 protein product. A recognition site (termed "FLAG") that is
recognized by
a commercially available monoclonal antibody designated anti-FLAG M2
(Eastman Kodak, Co., New Haven, CT) can be utilized, as well as other
comparable sequences and their corresponding antibodies. For example, PCR is
performed using GeneAmp° reagents obtained from Perkin-Elmer-Cetus, as
directed by the supplier's instructions. PCR primers are used at a final
concentration of 0.5 p,M. PCR is performed on the BS202 plasmid template in a
100 ~.1 reaction for 35 cycles (94°C, 30 seconds; 55°C, 30
seconds; 72°C, 90
seconds) followed by an extension cycle of 72°C for 10 min.
B. Transfection of Dihydrofolate Reductase Deficient Chinese Hamster
Ovarv Cells. The plasmid described supra is transfected into CHO/dhfr- cells
[DXB-111, Uriacio et al., PNAS 77:4451-4466 (1980)]. These cells are available
from the A.T.C.C., 12301 Parklawn Drive, Rockville, MD 20852, under
Accession No. CRL 9096. Transfection is carried out using the cationic
liposome-mediated procedure described by P. L. Felgner et al., PNAS 84:7413-
7417 (1987). Particularly, CHO/dhfr- cells are cultured in Ham's F-12 media
supplemented with 10% fetal calf serum, L-glutamine ( 1 mM) and freshly seeded
into a flask at a density of 5-8 x 105 cells per flask. The cells are grown to
a
confluency of between 60 and 80% for transfection. Twenty micrograms (20p,g)
of plasmid DNA are added to 1.5 ml of Opti-MEM I medium and 100 p.l of
Lipofectin Reagent (Gibco-BRL; Grand Island, NY) are added to a second I.5 mi
portion of Opti-MEM I media. The two solutions are mixed and incubated at room
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-71-
temperature for 20 min. After the culture medium is removed from the cells,
the
cells are rinsed 3 times with 5 ml of Opti-MEM I medium. The Opti-MEM I-
Lipofection-plasmid DNA solution then is overlaid onto the cells. The cells
are
incubated for 3 hr at 37°C, after which time the Opti-MEM I-Lipofectin-
DNA
solution is replaced with culture medium for an additional 24 hr prior to
selection.
C. Selection and Amplification. One day after transfection, cells are
passaged 1:3 and incubated with dhfr/G418 selection medium (hereafter, "F-12
minus medium G"). Selection medium is Ham's F-12 with L-glutamine and
without hypoxanthine, thymidine and glycine {JRH Biosciences, Lenexa, Kansas)
and 300 ~g per ml 6418 (Gibco-BRL; Grand Island, NY). Media volume-to-
surface area ratios of S ml per 25 cm2 are maintained. After approximately two
weeks, DHFR/G418 cells are expanded to allow passage and continuous
maintenance in F-12 minus medium G.
Amplification of each of the transfected BS202 cDNA sequences is
achieved by stepwise selection of DHFR+, 6418+ cells with methotrexate
(reviewed by R. Schimke, Cell 37:705-713 [ 1984]). Cells are incubated with F-
12 minus medium G containing 150 nM methotrexate (MTX) (Sigma, St. Louis,
MO) for approximately two weeks until resistant colonies appear. Further gene
amplification is achieved by selection of 150 nM adapted cells with 5 ~t.M
MTX.
D. Antigen Production. F-12 minus medium G supplemented with 5 N,M
MTX is overlaid onto just confluent monolayers for 12 to 24 hr at
37°C in 5%
CO2. The growth medium is removed and the cells are rinsed 3 times with
Dulbecco's phosphate buffered saline (PBS) (with calcium and magnesium)
(Gibco-BRL; Grand Island, NY) to remove the remaining media/serum which
may be present. Cells then are incubated with VAS custom medium (VAS custom
formulation with L-glutamine with HEPES without phenol red, available from
JRH Bioscience; Lenexa, KS, product number 52-08678P), for 1 hr at
37°C in
5% CO2. Cells then are overlaid with VAS for production at 5 ml per T flask.
Medium is removed after seven days of incubation, retained, and then frozen to
await purification with harvests 2, 3 and 4. The monolayers are overlaid with
V AS for 3 more seven day harvests.
E. Analysis of breast Tissue Gene BS202 Antigen Expression. Aliquots
of VAS supernatants from the cells expressing the BS202 protein construct are
analyzed, either by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) using
standard methods and reagents known in the art (Laemmli discontinuous gels),
or
by mass spectrometry.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-72-
F. Purification. Purification of the BS202 protein containing the FLAG
sequence is performed by immunoaffinity chromatography using an affinity
matrix
comprising anti-FLAG M2 monoclonal antibody covalently attached to agarose by
hydrazide linkage (Eastman Kodak Co., New Haven, CT). Prior to affinity
purification, protein in pooled VAS medium harvests from roller bottles is
exchanged into 50 mM Tris-HCl (pH 7.5), 150 mM NaCI buffer using a
Sephadex G-25 (Pharmacia Biotech Inc., Uppsala, Sweden) column. Protein in
this buffer is applied to the anti-FLAG M2 antibody affinity column. Non-
binding
protein is eluted by washing the column with 50 mM Tris-HCl (pH 7.5), 150 mM
NaCI buffer. Bound protein is eluted using an excess of FLAG peptide in 50 mM
Tris-HCl (pH 7.5), 150 mM NaCI. The excess FLAG peptide can be removed
from the purified BS202 protein by gel electrophoresis or HPLC.
Although plasmid 577 is utilized in this example, it is known to those
skilled in the art that other comparable expression systems, such as CMV, can
be
utilized herein with appropriate modifications in reagent and/or techniques
and are
within the skill of the ordinary artisan.
The largest cloned insert containing the coding region of the BS202 gene is
then sub-cloned into either (i) a eukaryotic expression vector which may
contain,
for example, a cytomegalovirus (CMV) promoter and/or protein fusible sequences
which aid in protein expression and detection, or (ii) a bacterial expression
vector
containing a superoxide-dismutase (SOD) and CMP-KDO synthetase (CKS) or
other protein fusion gene for expression of the protein sequence. Methods and
vectors which are useful for the production of polypeptides which contain
fusion
sequences of SOD are described in EPO 0196056, published October l, 1986,
and those containing fusion sequences of CKS are described in EPO Publication
No. 0331961, published September 13, 1989. This so-purified protein can be
used in a variety of techniques, including, but not limited to animal
immunization
studies, solid phase immunoassays, etc.
Example l lb' Expression of Protein in a Cell Line Using pcDNA3 1/M, c His
A. Construction of a BS202 Expression Plasmid. Plasmid
pcDNA3.1/Myc-His (Cat.# V855-20, Invitrogen, Carlsbad, CA) has been
constructed, in the past, for the expression of secreted antigens by most
mammalian cell lines. Expressed protein inserts are fused to a myc-his peptide
tag.
The myc-his tag is a 21 residue amino acid sequence having the following
sequence: Glu-Gln-Lys-Leu-Ile-Ser-Glu- Glu-Asp-Leu-Asn-Met-His-Thr-Glu-
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-73-
His-His-His-His-His-His (SEQUENCE ID NO 20) and comprises a c-myc
oncoprotein epitope and a polyhistidine sequence which are useful for the
purification of an expressed fusion protein by using either anti-myc or anti-
his
affinity columns, or metalioprotein binding columns.
Plasmids for the expression of secretable BS202 proteins are constructed
by inserting a BS202 polynucleotide sequence selected from the group
consisting
of SEQUENCE ID NO 1, SEQUENCE ID NO 2, SEQUENCE ID NO 3,
SEQUENCE m NO 4, SEQUENCE m NO 5, SEQUENCE m NO 6,
SEQUENCE lT7 NO 7, and fragments or complements thereof. Prior to
construction of a BS202 expression plasmid, the BS202 cDNA sequence is first
cloned into a pCR°-Blunt vector as follows:
The BS202 cDNA fragment is generated by PCR using standard
procedures. For example, PCR is performed procedures and reagents from
Stratagene°, Inc. (La Jolla, CA), as directed by the manufacturer. PCR
primers
are used at a final concentration of 0.5 p.M. PCR using 5 U of pfu polymerase
(Stratagene, La Jolla, CA) is performed on the BS202 plasmid template (see
Example 2) in a 50 p.l reaction for 30 cycles (94°C, 1 min;
65°C, 1.5 min; 72°C, 3
min) followed by an extension cycle of 72°C for 8 min. (The sense PCR
primer
sequence comprises nucleotides which are either complementary to the pINCY
vector directly upstream of the BS202 gene insert or which incorporate a 5'
EcoRI
restriction site, an adjacent downstream protein translation consensus
initiator, and
a 3' nucleic acid sequence which is the same sense as the 5'-most end of the
BS202 cDNA insert. The antisense PCR primer incorporates a 5' NotI restriction
sequence and a sequence complementary to the 3' end of the BS202 cDNA insert
just upstream of the 3'-most, in-frame stop codon.) Five microliters (5 ~,l)
of the
resulting blunted-ended PCR product are ligated into 25 ng of linearized
pCR°-
Blunt vector (Invitrogen, Carlsbad, CA) interrupting the lethal ccdB gene of
the
vector. The resulting Iigated vector is transformed into TOP10 E. cQli
(Invitrogen,
Carlsbad, CA) using a One ShotT"'' Transformation Kit (Invitrogen, Carlsbad,
CA)
following manufacturer's instructions. The transformed cells are grown on LB-
Kan (50 p.g/ml kanamycin) selection plates at 37°C. Only cells
containing a
plasmid with an interrupted ccdB gene will grow after transformation [Grant,
S.G.N., PNAS 87:4645-4649 ( 1990)]. Transformed colonies are picked and
grown up in 3 mI of LB-Kan broth at 37°C. Plasmid DNA is isolated by
using a
QIAprep° (Qiagen Inc., Santa Clarita, CA) procedure, as directed
by the
manufacturer. The DNA is cut with EcoRI or SnaBI, and NotI restriction
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-74-
enzymes to release the BS202 insert fragment. The fragment is run on 1
Seakem° LE agarose/0.5 p.g/m1 ethidium bromide/TE gel, visualized
by UV
irradiation, excised and purified using QIAquick~'~"' (Qiagen Inc., Santa
Clarita,
CA) procedures, as directed by the supplier's instructions.
The pcDNA3.1/Myc-His plasmid DNA is linearized by digestion with
EcoRI or SnaBI, and NotI in the polylinker region of the plasmid DNA. The
resulting plasmid DNA backbone allows insertion of the BS202 purified cDNA
fragment, supra, downstream of a CMV promoter which directs expression of the
proteins in mammalian cells. The ligated plasmid is transformed into DHS
alpha~'~''
cells (GibcoBRL Grand Island, NY), as directed by the manufacturer. Briefly,
10
ng of pcDNA3.IlMyc-His containing a BS202 insert are added to 50 p,l of
competent DHS alpha cells, and the contents are mixed gently. The mixture is
incubated on ice for 30 min, heat shocked for 20 sec at 37°C, and
placed on ice for
an additional 2 min. Upon addition of 0.95 ml of LB medium, the mixture is
incubated for 1 hr at 37°C while shaking at 225 rpm. The transformed
cells then
are plated onto 100 mm LB/Amp (SOp,g/ml ampicillin) plates and grown at
37°C.
Colonies are picked and grown in 3 ml of LB/Amp broth. Plasmid DNA is
purified using a QIAprep Kit. The presence of the insert is confirmed using
techniques known to those skilled in the art, including, but not limited to
restriction digestion and gel analysis. (J. Sambrook et al., supra. )
B. Transfection of Human Embryonic Kidn~ Cell 293 Ceps. The
BS202 expression plasmid described in section A, supra, is retransformed into
DHS alpha cells, plated onto LB/ampicillin agar, and grown up in 10 ml of
LB/ampicillin broth, as described hereinabove. The plasmid is purified using a
QIAfilter~""' Maxi Kit (Qiagen, Chatsworth, CA) and is transfected into HEK293
cells [F.L. Graham et al., J. Gen Vir 36'59-72 ( 1977)]. These cells are
available
from the A.T.C.C., 12301 Parklawn Drive, Rockville, MD 20852, under
Accession No. CRL 1573. Transfection is carried out using the cationic
lipofectamine-mediated procedure described by P. Hawley-Nelson et al., Focus
15.73 ( 1993). Particularly, HEK293 cells are cultured in 10 m1 DMEM media
supplemented with 10% fetal bovine serum (FBS), L-glutamine (2 n>IVI) and
freshly seeded into 100 mm culture plates at a density of 9 x 106 cells per
plate.
The cells are grown at 37 °C to a confluency of between 70% and
80% for
transfection. Eight micrograms (8 pg) of plasmid DNA are added to 800 p.l of
Opti-MEM h medium (Gibco-BRL, Grand Island, NY), and 48-96 p,l of
Lipofectamine'~"' Reagent (Gibco-BRL, Grand Island, NY) are added to a second
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-75-
800 pl portion of Opti-MEM I media. The two solutions are mixed and incubated
at room temperature for 15-30 min. After the culture medium is removed from
the
cells, the cells are washed once with 10 ml of serum-free DMEM. The Opti-MEM
I-Lipofectamine-plasmid DNA solution is diluted with 6.4 ml of serum-free
S DMEM and then overlaid onto the cells. The cells are incubated for 5 hr at
37°C,
after which time, an additional 8 ml of DMEM with 20% FBS are added. After
18-24 hr, the old medium is aspirated, and the cells are overlaid with 5 ml of
fresh
DMEM with 5% FBS. Supernatants and cell extracts are analyzed for BS202
gene activity 72 hr after transfection.
C Analysis of breast Tissue Gene BS202 Antigen Ex ression. The
culture supernatant, su,Rra, is transferred to cryotubes and stored on ice.
HEK293
cells are harvested by washing twice with 10 ml of cold Dulbecco's PBS and
lysing by addition of 1.5 ml of CAT lysis buffer (Boehringer Mannheim,
Indianapolis, IN), followed by incubation for 30 min at room temperature.
Lysate is transferred to 1.7 ml polypropylene microfuge tubes and centrifuged
at
1000 x g for 10 min. The supernatant is transferred to new cryotubes and
stored
on ice. Aliquots of supernatants from the cells and the lysate of the cells
expressing the BS202 protein construct are analyzed for the presence of BS202
recombinant protein. The aliquots can be run on SDS-polyacrylamide gel
electrophoresis (SDS-PAGE) using standard methods and reagents known in the
art. (J. Sambrook et al., supra) These gels can then be blotted onto a solid
medium such as nitrocellulose, nytran, etc., and the BS202 protein band can be
visualized using Western blotting techniques with anti-myc epitope or anti-
histidine monoclonal antibodies (Invitrogen, Carlsbad, CA) or anti-BS202
polyclonal serum (see Example 14). Alternatively, the expressed BS202
recombinant protein can be analyzed by mass spectrometry (see Example 12).
D. Purification. Purification of the BS202 recombinant protein containing
the myc-his sequence is performed using the Xpress° affinity
chromatography
system (Invitrogen, Carlsbad, CA) containing a nickel-charged agarose resin
which specifically binds polyhistidine residues. Supernatants from 10 x 100 mm
plates, prepared as described ra, are pooled and passed over the nickel-
charged
column. Non-binding protein is eluted by washing the column with SO mM Tris-
HCl (pH 7.5)/150 mM NaCI buffer, leaving only the myc-his fusion proteins.
Bound BS202 recombinant protein then is eluted from the column using either an
excess of imidazole or histidine, or a low pH buffer. Alternatively, the
recombinant protein can also be purified by binding at the myc-his sequence to
an
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-76-
affinity column consisting of either anti-myc or anti-histidine monoclonal
antibodies conjugated through a hydrazide or other linkage to an agarose resin
and
eluting with an excess of myc peptide or histidine, respectively.
The purified recombinant protein can then be covalently cross-linked to a
solid phase, such as N-hydroxysuccinimide-activated sepharose columns
(Pharmacia Biotech, Piscataway, NJ), as directed by supplier's instructions.
These columns containing covalently linked BS202 recombinant protein, can then
be used to purify anti-BS202 antibodies from rabbit or mouse sera (see
Examples
13 and 14).
E. Coating Microtiter Plates with BS202 Expressed Proteins Supernatant
from a 100 mm plate, as described supra, is diluted in an appropriate volume
of
PBS. Then, 100 ~,l of the resulting mixture is placed into each well of a
Reacti-
BindT"' metal chelate microtiter plate (Pierce, Rockford, IL), incubated at
room
temperature while shaking, and followed by three washes with 200 p,l each of
PBS with 0.05% Tween~ 20. The prepared microtiter plate can then be used to
screen polyclonal antisera for the presence of BS202 antibodies (see Example
17).
Although pcDNA3.1/Myc-His is utilized in this example, it is known to
those skilled in the art that other comparable expression systems can be
utilized
herein with appropriate modifications in reagent andlor techniques and are
within
the skill of one of ordinary skill in the art. The largest cloned insert
containing the
coding region of the BS202 gene is sub-cloned into either (i) a eukaryotic
expression vector which may contain, for example, a cytomegalovirus (CMV)
promoter and/or protein fusible sequences which aid in protein expression and
detection, or (ii) a bacterial expression vector containing a superoxide-
dismutase
(SOD) and CMP-KDO synthetase (CKS) or other protein fusion gene for
expression of the protein sequence. Methods and vectors which are useful for
the
production of polypeptides which contain fusion sequences of SOD are described
in published EPO application No. EP 0 196 056, published October 1, 1986, and
vectors containing fusion sequences of CKS are described in published EPO
application No. EP 0 331 961, published September 13, 1989. The purified
protein can be used in a variety of techniques, including, but not limited to
animal
immunization studies, solid phase immunoassays, etc.
Example 12: Chemical Analysis of breast Tissue Proteins
A. Analysis of Tryptic Peptide Fragments Using 1V~S. Sera from patients
with breast disease, such as breast cancer, sera from patients with no breast
CA 02292843 1999-12-03
WO 99/02559 PCTlUS98/14046
-77-
disease, extracts of breast tissues or cells from patients with breast
disease, such
as breast cancer, extracts of breast tissues or cells from patients with no
breast
disease, and extracts of tissues or cells from other non-diseased or diseased
organs of patients, are run on a polyacrylamide gel using standard procedures
and
stained with Coomassie Blue. Sections of the gel suspected of containing the
unknown polypeptide are excised and subjected to an in-gel reduction,
acetamidation and tryptic digestion. P. Jeno et al., al. Bio. 224:451-455
( 1995) and J. Rosenfeld et al., A . Bi . 203:173-179 ( 1992). The gel
sections
are washed with 100 mM NH4HC0~ and acetonitrile. The shrunken gel pieces are
swollen in digestion buffer (50 mM NH4HC0~, 5 mM CaClz and 12.5 p.g/ml
trypsin) at 4°C for 45 min. The supernatant is aspirated and replaced
with 5 to 10
~.1 of digestion buffer without trypsin and allowed to incubate overnight at
37°C.
Peptides are extracted with 3 changes of 5% fornuc acid and acetonitrile and
evaporated to dryness. The peptides are adsorbed to approximately 0.1 p.l of
POROS R2 sorbent (Perseptive Biosystems, Framingham, Massachusetts)
trapped in the tip of a drawn gas chromatography capillary tube by dissolving
them in 10 ~l of 5% formic acid and passing it through the capillary. The
adsorbed peptides are washed with water and eluted with 5% formic acid in 60%
methanol. The eluant is passed directly into the spraying capillary of an API
III
mass spectrometer (Perkin-Elmer Sciex, Thornhill, Ontario, Canada) for
analysis
by nano-electrospray mass spectrometry. M. Wilm et al., Int. J. Mass Spectrom.
Ion Process 136:167-180 (1994) and M. Wilm et al., Anal. Chem. 66:1-8 (1994).
The masses of the tryptic peptides are determined from the mass spectrum
obtained off the first quadrupole. Masses corresponding to predicted peptides
can
be further analyzed in MS/MS mode to give the amino acid sequence of the
peptide.
B. Peptide Fragment Analy is Using LC/MS. The presence of
polypeptides predicted from mRNA sequences found in hyperplastic disease
tissues also can be confirmed using liquid chromatography/tandem mass
spectrometry (LC/MS/MS). D. Hess et al., METHODS. A Companion to
Methods in Enz~rmolog3r 6:227-238 (1994). The serum specimen or tumor extract
from the patient is denatured with SDS and reduced with dithiothreitol ( 1.5
mg/ml)
for 30 min at 90°C followed by alkylation with iodoacetamide (4 mg/ml)
for 15
min at 25°C. Following acrylamide electrophoresis, the polypeptides are
electroblotted to a cationic membrane and stained with Coomassie Blue.
Following staining, the membranes are washed and sections thought to contain
the
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
_78_
unknown polypeptides are cut out and dissected into small pieces. The
membranes are placed in 500 ~.1 microcentrifuge tubes and immersed in 10 to 20
~,1 of proteolytic digestion buffer ( 100 mM Tris-HCI, pH 8.2, containing 0.1
M
NaCI, 10% acetonitrile, 2 mM CaCl2 and 5 p,g/ml trypsin) (Sigma, St. Louis,
MO). After 15 hr at 37°C, 3 ~l of saturated urea and 1 ~.I of 100 ~g/ml
trypsin are
added and incubated for an additional 5 hr at 37°C. The digestion
mixture is
acidified with 3 p,l of 10% trifluoroacetic acid and centrifuged to separate
supernatant from membrane. The supernatant is injected directly onto a
microbore, reverse phase HPLC column and eluted with a linear gradient of
acetonitrile in 0.05% trifluoroacetic acid. The eluate is fed directly into an
electrospray mass spectrometer, after passing though a stream splitter if
necessary
to adjust the volume of material. The data is analyzed following the
procedures set
forth in Example 12, Section A.
1 S Example 13: Gene Immunization Protocol
A. In Vivo Ant' etg n Expression. Gene immunization circumvents protein
purification steps by directly expressing an antigen in vivo after inoculation
of the
appropriate expression vector. Also, production of antigen by this method may
allow correct protein folding and glycosylation since the protein is produced
in
mammalian tissue. The method utilizes insertion of the gene sequence into a
plasmid which contains a CMV promoter, expansion and purification of the
plasmid and injection of the plasmid DNA into the muscle tissue of an animal.
Preferred animals include mice and rabbits. See, for example, H. Davis et al.,
Human Molecular Genetics 2:1847-1851 ( 1993). After one or two booster
immunizations, the animal can then be bled, ascites fluid collected, or the
animal's
spleen can be harvested for production of hybridomas.
B. Plasmid Preparation and Purification. BS202 cDNA sequences are
generated from the BS202 cDNA-containing vector using appropriate PCR
primers containing suitable 5' restriction sites following the procedures
described
in Example 11. The PCR product is cut with appropriate restriction enzymes and
inserted into a vector which contains the CMV promoter (for example, pRc/CMV
or pcDNA3 vectors from Invitrogen, San Diego, CA). This plasmid then is
expanded in the appropriate bacterial strain and purified from the cell lysate
using a
CsCI gradient or a Qiagen plasmid DNA purification column. All these
techniques
are familiar to one of ordinary skill in the art of molecular biology.
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-79-
C. Immunization Protocol. Anesthetized animals are immunized
intramuscularly with 0.1-100 p,g of the purified piasmid diluted in PBS or
other .
DNA uptake enhancers (Cardiotoxin, 25% sucrose). See, for example, H. Davis
et al., Human Gene Theraov 4:733-740 (1993); and P. W. Wolff et al.,
Biotechni ues 11:474-485 ( 1991 ). One to two booster injections are given at
monthly intervals.
D. Testing and Use of Antiserum. Animals are bled and the resultant sera
tested for antibody using peptides synthesized from the known gene sequence
(see
Example 16) using techniques known in the art, such as Western blotting or EIA
techniques. Antisera produced by this method can then be used to detect the
presence of the antigen in a patient's tissue or cell extract or in a
patient's serum by
ELISA or Western blotting techniques, such as those described in Examples 15
through 18.
Example 14: Production of Antibodies Against BS202
A. Production of Polvclonal Antisera Antiserum against BS202 was
prepared by injecting rabbits with peptides whose sequences were derived from
that of the predicted amino acid sequence of the BS202 consensus nucleotide
sequence (SEQUENCE ID NO 7). The synthesis of peptides SEQUENCE ID NO
15, SEQUENCE ff~ NO 16, SEQUENCE ID NO 17, and SEQUENCE ID NO 18
is described in Example 10. Peptides used as immunogens were not conjugated to
a carrier such as keyhole limpet hemocyanine, KLH, (i.e., they were
unconjugated.).
Animal Immunization. Female white New Zealand rabbits
weighing 2 kg or more were used for raising polyclonal antiserum. One animal
was immunized per unconjugated peptide, SEQUENCE ID NO 1 S, SEQUENCE
ID NO 16, SEQUENCE ID NO 17, and SEQUENCE ID NO 18. One week prior
to the first immunization, 5 to 10 mi of blood were obtained from the animal
to
serve as a non-immune prebleed sample.
Unconjugated BS202 peptides (SEQUENCE ID NO 15, SEQUENCE ID
NO 16, SEQUENCE ID NO 17, and SEQUENCE ID NO 18) were used to
prepare the primary immunogen by emulsifying 0.5 ml of the peptide at a
concentration of 2 mg/ml in PBS (pH 7.2) which contained 0.5 ml of complete
Freund's adjuvant (CFA) (Difco, Detroit, MI). The immunogen was injected into
several sites of the animal via subcutaneous, intraperitoneal, and
intramuscular
routes of administration. Four weeks following the primary immunization, a
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-80-
booster immunization was administered. The immunogen used for the booster
immunization dose was prepared by emulsifying 0.5 ml of the same unconjugated
peptide used for the primary immunogen, except that the peptide now was
diluted
to 1 mg/ml with 0.5 ml of incomplete Freund's adjuvant (IFA) (Difco, Detroit,
MI). Again, the booster dose was administered into several sites via
subcutaneous, intraperitoneal and intramuscular types of injections. The
animals
were bled (5 ml) two weeks after the booster immunizations and each serum was
tested for immunoreactivity to the peptide as described below. The booster and
bleed schedule was repeated at 4 week intervals until an adequate titer was
obtained. The titer or concentration of antiserum was determined by using
unconjugated peptides in a microtiter EIA as described in Example 17, below.
An
antibody titer of 1:500 or greater was considered an adequate titer for
further use
and study.
Table 1. Titer of rabbit anti-BS202 peptide antisera (11 week bleed)
Peutide Immuno~en Titer
SEQUENCE ID NO 15 >62,000
SEQUENCE m NO 17 11,300
SEQUENCE ID NO 18 10,500
B. Production of Monoclonal Antibody
1. Immunization Protocol. Mice are immunized using peptides
which can either be conjugated to a carrier such as KLH [prepared as described
hereinbelow, or unconjugated (i.e., not conjugated to a carrier such as KLH)]
except that the amount of the unconjugated or conjugated peptide for
monoclonal
antibody production in mice is one-tenth the amount used to produce polyclonal
antisera in rabbits. Thus, the primary immunogen consists of 100 pg of
unconjugated or conjugated peptide in 0.1 ml of CFA emulsion while the
immunogen used for booster immunizations consists of 50 p.g of unconjugated or
conjugated peptide in 0.1 ml of IFA. Hybridomas for the generation of
monoclonal antibodies are prepared and screened using standard techniques. The
methods used for monoclonal antibody development follow procedures known in
the art such as those detailed in Kohler and Milstein, Nature 256:494 { 1975)
and
reviewed in J.G.R. Hurrel, ed., Monoclonal Hybridoma Antibodies Techniques
and Applications, CRC Press, Inc., Boca Raton, FL ( i 982). Another method of
CA 02292843 1999-12-03
WO 99/02559 PGT/ITS98/14046
-81-
monoclonal antibody development which is based on the Kohler and Milstein
method is that of L.T. Mimms et al., Virology 176:604-619 ( 1990).
The immunization regimen (per mouse) consists of a primary
immunization with additional booster immunizations. The primary immunogen
used for the primary immunization consists of 100 p,g of unconjugated or
conjugated peptide in 50 p,l of PBS (pH 7.2) previously emulsified in 50 p,l
of
CFA. Booster immunizations performed at approximately two weeks and four
weeks post primary immunization consist of 50 pg of unconjugated or conjugated
peptide in 50 p,l of PBS (pH 7.2) emulsified with 50 ~,l IFA. A total of 100
~,1 of
this immunogen are inoculated intraperitoneally and subcutaneously into each
mouse. Individual mice are screened for immune response by microtiter plate
enzyme immunoassay (EIA) as described in Example 17 approximately four
weeks after the third immunization. Mice are inoculated either intravenously,
intrasplenically or intraperitoneally with 50 ~tg of unconjugated or
conjugated
peptide in PBS (pH 7.2) approximately fifteen weeks after the third
immunization..
Three days after this intravenous boost, splenocytes are fused
with, for example, Sp2/0-Ag 14 myeloma cells (Milstein Laboratories, England)
using the polyethylene glycol (PEG) method. The fusions are cultured in
Iscove's
Modified Dulbecco's Medium (IMDM) containing 10% fetal calf serum (FCS),
plus 1 % hypoxanthine, aminopterin and thymidine (HAT}. Bulk cultures were
screened by microtiter plate EIA following the protocol in Example 17. Clones
reactive with the peptide used an immunogen and non-reactive with other
peptides
(i.e., peptides of BS202 not used as the immunogen) are selected for final
expansion. Clones thus selected are expanded, aliquoted and frozen in IMDM
containing 10% FCS and 10% dimethyl sulfoxide, (DMSO).
2. Peptide Conjugation. Peptide is conjugated to maleimide
activated KLH (commercially available as Imject°, available from Pierce
Chemical
Company, Rockford, IL). Imject° contains about 250 moles of reactive
maleimide
groups per mole of hemocyanine. The activated KLH is dissolved in phosphate
buffered saline (PBS, pH 8.4) at a concentration of about 7.7 mg/ml. The
peptide
is conjugated through cysteines occurring in the peptide sequence, or to a
cysteine
previously added to the synthesized peptide in order to provide a point of
attachment. The peptide is dissolved in DMSO (Sigma Chemical Company, St.
Louis, MO) and reacted with the activated KLH at a mole ratio of about 1.5
moles
of peptide per mole of reactive maleimide attached to the KLH. A procedure for
CA 02292843 1999-12-03
WO 99/02559 PC'T/US98/14046
-82-
the conjugation of peptide is provided hereinbelow. It is known to the
ordinary
artisan that the amounts, times and conditions of such a procedure can be
varied to
optimize peptide conjugation.
The conjugation reaction described hereinbelow is based on
S obtaining 3 mg of KLH peptide conjugate ("conjugated peptide"), which
contains
about 0.77 p.moles of reactive maleimide groups. This quantity of peptide
conjugate usually is adequate for one primary injection and four booster
injections
for production of polyclonal antisera in a rabbit. Briefly, peptide is
dissolved in
DMSO at a concentration of 1.16 ~.moles/100 p.l of DMSO. One hundred
microliters ( 100 p,l) of the DMSO solution are added to 380 pl of the
activated
KLH solution prepared as described hereinabove, and 20 ~.1 of PBS (pH 8.4) are
added to bring the volume to S00 p.l. The reaction is incubated overnight at
room
temperature with stirring. The extent of reaction is determined by measuring
the
amount of unreacted thiol in the reaction mixture. The difference between the
I S starting concentration of thiol and the final concentration is assumed to
be the
concentration of peptide which has coupled to the activated KLH. The amount of
remaining thiol is measured using Ellman's reagent (S,S'-dithiobis(2-
nitrobenzoic
acid), Pierce Chemical Company, Rockford, IL.). Cysteine standards are made at
a concentration of 0, 0. I , O.S, 2, S and 20 mM by dissolving 3S mg of
cysteine
HCl (Pierce Chemical Company, Rockford, IL) in 10 ml of PBS (pH 7.2) and
diluting the stock solution to the desired concentration(s). The photometric
determination of the concentration of thiol is accomplished by placing 200 p.l
of
PBS (pH 8.4) in each well of an Immulon 2~ microwell plate (Dynex
Technologies, Chantilly, VA). Next, 10 p.l of standard or reaction mixture are
2S added to each well. Finally, 20 p,l of Ellman's reagent at a concentration
of 1
mg/ml in PBS {pH 8.4) are added to each well. The wells are incubated for 10
minutes at room temperature, and the absorbance of all wells is read at 41 S
nm
with a microplate reader (such as the BioRad Model 3SS0, BioRad, Richmond,
CA). The absorbance of the standards is used to construct a standard curve and
the thiol concentration of the reaction mixture is determined from the
standard
curve. A decrease in the concentration of free thiol is indicative of a
successful
conjugation reaction. Unreacted peptide is removed by dialysis against PBS (pH
7.2) at room temperature for 6 hours. The conjugate is stored at 2-8°C
if it is to be
used immediately; otherwise, it is stored at -20°C or colder.
3S 3. Production of Ascites Fluid Containing Monoclonal Antibodies.
Frozen hybridoma cells prepared as described hereinabove are thawed and placed
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14t146
-83-
into expansion culture. Viable hybridoma cells are inoculated
intraperitoneally into
Pristane treated mice. Ascitic fluid is removed from the mice, pooled,
filtered
through a 0.2 p, filter and subjected to an immunoglobulin class G (IgG)
analysis
to determine the volume of the Protein A column required for the purification.
4. Purification of Monoclonal Antibodies From Ascites Fluid.
Briefly, filtered and thawed ascites fluid is mixed with an equal volume of
Protein
A sepharose binding buffer ( 1.5 M glycine, 3.0 M NaCI, pH 8.9) and refiltered
through a 0.2 ~, filter. The volume of the Protein A column is determined by
the
quantity of IgG present in the ascites fluid. The eluate then is dialyzed
against
PBS (pH 7.2) overnight at 2-8°C. The dialyzed monoclonal antibody is
sterile
filtered and dispensed in aliquots. The immunoreactivity of the purified
monoclonal antibody is confirmed by determining its ability to specifically
bind to
the peptide used as the immunogen by use of the EIA microtiter plate assay
procedure of Example 17. The specificity of the purified monoclonal antibody
is
confirmed by determining its lack of binding to irrelevant peptides such as
peptides of BS202 not used as the immunogen. The purified anti-BS202
monoclonal thus prepared and characterized is placed at either 2-8°C
for short term
storage or at -80°C for long term storage.
5. Further Characterization of Monoclonal Antibody. The isotype
and subtype of the monoclonal antibody produced as described hereinabove can
be
determined using commercially avaiIabie kits (available from Amersham. Inc.,
Arlington Heights, IL}. Stability testing also can be performed on the
monoclonal
antibody by placing an aliquot of the monoclonal antibody in continuous
storage at
2-8°C and assaying optical density (OD) readings throughout the course
of a given
period of time.
C Use of Recombinant Proteins as Immuno$ens It is within the scope
of the present invention that recombinant proteins made as described herein
can be
utilized as immunogens in the production of polyclonal and monoclonal
antibodies, with corresponding changes in reagents and techniques known to
those skilled in the art.
Example 15~ Purification of Serum Antibodies Which Specifically
Bind to BS~.O_ 2 Peptides
Immune sera, obtained as described hereinabove in Examples 13 and/or
14, is affinity purified using immobilized synthetic peptides prepared as
described
in Example 10, or recombinant proteins prepared as described in Example 11. An
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-84-
IgG fraction of the antiserum is obtained by passing the diluted, crude
antiserum
over a Protein A column (Affi-Gel protein A, Bio-Rad, Hercules, CA). Elution
with a buffer (Binding Buffer, supplied by the manufacturer) removes
substantially all proteins that are not immunoglobulins. Elution with 0.1 M
buffered glycine (pH 3) gives an immunoglobulin preparation that is
substantially
free of albumin and other serum proteins.
Immunoaffmity chromatography is performed to obtain a preparation with
a higher fraction of specific antigen-binding antibody. The peptide used to
raise
the antiserum is immobilized on a chromatography resin, and the specific
antibodies directed against its epitopes are adsorbed to the resin. After
washing
away non-binding components, the specific antibodies are eluted with 0.1 M
glycine buffer, pH 2.3. Antibody fractions are immediately neutralized with I
.0
M Tris buffer (pH 8.0) to preserve immunoreactivity. The chromatography resin
chosen depends on the reactive groups present in the peptide. If the peptide
has an
amino group, a resin such as Affi-Gel 10 or Affi-Gel 15 is used (Bio-Rad,
Hercules, CA). If coupling through a carboxy group on the peptide is desired,
Affi-Gel 102 can be used (Bio-Rad, Hercules, CA). If the peptide has a free
sulfhydryl group, an organomercurial resin such as Affi-Gel 501 can be used
(Bio-Rad, Hercules, CA).
Alternatively, spleens can be harvested and used in the production of
hybridomas to produce monoclonal antibodies following routine methods known
in the art as described hereinabove.
Example 16: Western Blotting of Tissue Samples
Protein extracts are prepared by homogenizing tissue samples in 0.1 M
Tris-HCl (pH 7.5), 15% (w/v) glycerol, 0.2 mM EDTA, 1.0 mM 1,4-
dithiothreitol, 10 p.g/m! leupeptin and 1.0 mM phenylmethylsulfonylfluoride
[Kain
et al., Biotechniques, 17:982 (1994)]. Following homogenization, the
homogenates are centrifuged at 4°C for 5 minutes to separate
supernatant from
debris. Debris is reextracted by homogenization with a buffer that is similar
to
above also contains 0.1 M Tricine and 0.1 % SDS. The supernatant from the
second extraction is used for Western blotting. For protein quantitation, 2-5
p,l of
supernatant are added to 1.5 ml of Coomassie Protein Reagent (Pierce,
Rockford,
IL), and the resulting absorbance at 595 nm is measured.
For SDS-PAGE, samples are adjusted to desired protein concentration
with Tricine Buffer (Novex, San Diego, CA), mixed with an equal volume of 2X
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-85-
Tricine sample buffer (Novex, San Diego, CA), and heated for 5 minutes at
100°C
in a thermal cycler. Samples are then applied to a Novex 10-20% Precast
Tricine-
Gel for electrophoresis. Following electrophoresis, samples are transferred
from
the gels to nitrocellulose membranes in Novex Tris-Glycine Transfer buffer.
Membranes are then probed with specific anti-peptide antibodies using the
reagents and procedures provided in the Western Lights or Western Lights Plus
(Tropix, Bedford, MA) chemiluminescence detection kits. Chemiluminescent
bands are visualized by exposing the developed membranes to Hype~lm ECL
(Amersham, Arlington Heights, IL).
Competition experiments are carned out in an analogous manner as above,
with the following exception; the primary antibodies (anti-peptide polyclonal
antisera) are pre-incubated for 30 minutes at room temperature with varying
concentrations of peptide immunogen prior to exposure to the nitrocellulose
filter.
Development of the Western is performed as above.
After visualization of the bands on film, the bands can also be visualized
directly on the membranes by the addition and development of a chromogenic
substrate such as 5-bromo-4-chloro-3-indolyl phosphate (BLIP). This
chromogenic solution contains 0.016% BCIP in a solution containing 100 mM
NaCI, 5 mM MgCh and 100 mM Tris-HCl (pH 9.5). The filter is incubated in the
solution at room temperature until the bands develop to the desired intensity.
Molecular mass determination is made based upon the mobility of pre-stained
molecular weight standards (Novex, San Diego, CA) or biotinylated molecular
weight standards (Tropix, Bedford, MA).
Example 17: EIA Microtiter Plate AssaX
The immunoreactivity of antiserum preferably obtained from rabbits as
described in Example 14 was determined by means of a microtiter plate EIA, as
follows. Briefly, the synthetic BS202 peptides of SEQUENCE ID NO 15,
SEQUENCE ID NO 16, SEQUENCE ID NO 17, and SEQUENCE ID NO 18,
prepared as described in Example 10, were dissolved in carbonate buffer (50
mM,
pH 9.6) to a final concentration of 2 p.g/ml. Next, 100 ~.l of the peptide or
protein
solution were placed in each well of an Immulon 2° microtiter plate
(Dynex
Technologies, Chantilly, VA). The plate was incubated overnight at room
temperature and then washed four times with deionized water. The wells were
blocked by adding 125 pl of a suitable protein blocking agent, such as
Superblock° (Pierce Chemical Company, Rockford, IL), to each well
and then
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-86-
immediately discarding the solution. This blocking procedure was performed
three times. Antiserum obtained from immunized rabbits or mice, prepared as
previously described, was diluted in a protein blocking agent (e.g., a 3%
Superblock° solution) in PBS containing 0.05% Tween-20°
(monolaurate
polyoxyethylene ether) (Sigma Chemical Company, St. Louis, MO) and 0.05%
sodium azide at dilutions of 1:100, 1:500, 1:2500, 1:12,500, and 1:62,500 and
placed in each well of the coated microtiter plate. The wells then were
incubated
for three hours at room temperature. Each well was washed four times with
deionized water. One hundred microliters of alkaline phosphatase-conjugated
goat
anti-rabbit IgG or goat anti-mouse IgG antiserum (Southern Biotech,
Birmingham, AB) diluted 1:2000 in 3% Superblock° solution in
phosphate
buffered saline containing 0.05% Tween 20° and 0.05% sodium azide, were
added to each well. The wells were incubated for two hours at room
temperature.
Next, each well was washed four times with deionized water. One hundred
microliters of paranitrophenyl phosphate substrate (Kirkegaard and Perry
Laboratories, Gaithersburg, MD) then were added to each well. The wells were
incubated for thirty minutes at room temperature. The absorbance at 405 nm was
read in each well. Positive reactions were identified by an increase in
absorbance
at 405 nm in the test well above that absorbance given by a non-immune serum
(negative control). A positive reaction was indicative of the presence of
detectable
anti-BS202 antibodies. Titers of the anti-peptide antisera were calculated
from the
previously described dilutions of antisera and defined as the calculated
dilution,
where A4os~m= 0.5 OD.
Example 18: Coating of Solid Phase Particles
A. Coatine of Microparticles with Antibodies Which Specifically Bind to
BS202 Antigen. Affinity purified antibodies which specifically bind to BS202
protein (see Example 15) are coated onto microparticles of polystyrene,
carboxylated polystyrene, polymethylacrylate or similar particles having a
radius
in the range of about 0.1 to 20 ~.m. Microparticles may be either passively or
actively coated. One coating method comprises coating EDAC (1-(3-
dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (Aldrich Chemical Co.,
Milwaukee, WI) activated carboxylated latex microparticles with antibodies
which
specifically bind to BS202 protein, as follows. Briefly, a final 0.375% solid
suspension of resin washed carboxylated latex microparticles (available from
Bangs Laboratories, Carmel, IN or Serodyn, Indianapolis, IN) are mixed in a
CA 02292843 1999-12-03
WO 99/02559 PCTNS98/14046
_87_
solution containing 50 mM MES buffer, pH 4.0 and 150 mg/1 of affinity purified
anti-BS202 antibody (see Example 14) for 15 min in an appropriate container.
EDAC coupling agent is added to a final concentration of 5.5 ~.g/ml to the
mixture
and mixed for 2.5 hr at room temperature.
The nucroparticles then are washed with 8 volumes of a Tween
20°/sodium phosphate wash buffer (pH 7.2) by tangential flow filtration
using a
0.2 p,m Microgon Filtration module. Washed microparticles are stored in an
appropriate buffer which usually contains a dilute surfactant and irrelevant
protein
as a blocking agent, until needed.
B. Coating of 1/4 Inch Beads. Antibodies which specifically bind to
BS202-antigen also may be coated on the surface of 1/4 inch polystyrene beads
by
routine methods known in the art (Snitman et al., US Patent 5,273,882) and
used
in competitive binding or EIA sandwich assays.
Polystyrene beads first are cleaned by ultrasonicating them for about 15
seconds in 10 mM NaHCO~ buffer at pH 8Ø The beads then are washed in
deionized water until all fines are removed. Beads then are immersed in an
antibody solution in 10 mM carbonate buffer, pH 8 to 9.5. The antibody
solution
can be as dilute as 1 p.g/ml in the case of high affinity monoclonal
antibodies or as
concentrated as about 500 p.g/ml for polyclonal antibodies which have not been
affinity purified. Beads are coated for at least 12 hours at room temperature,
and
then they are washed with deionized water. Beads may be air dried or stored
wet
(in PBS, pH 7.4). They also may be overcoated with protein stabilizers (such
as
sucrose) or protein blocking agents used as non-specific binding blockers
(such as
irrelevant proteins, Carnation skim milk, 5uperblock°, or the like).
Example 19: Micro~article Enzyme Immunoassa~~MEIAI
BS202 antigens are detected in patient test samples by performing a
standard antigen competition EIA or antibody sandwich EIA and utilizing a
solid
phase such as microparticles (MEIA). The assay can be performed on an
automated analyzer such as the IMx° Analyzer (Abbott Laboratories,
Abbott Park,
IL).
A. Antibody Sandwich EIA. Briefly, samples suspected of containing
BS202 antigen are incubated in the presence of anti-BS202 antibody-coated
microparticles (prepared as described in Example 17) in order to form
antigen/antibody complexes. The microparticles then are washed and an
indicator
reagent comprising an antibody conjugated to a signal generating compound
(i.e.,
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
_88_
enzymes such as alkaline phosphatase or horseradish peroxide) is added to the
antigen/antibody complexes or the microparticles and incubated. The
microparticles are washed and the bound antibody/antigen/antibody complexes
are
detected by adding a substrate (e.g., 4-methyl umbelliferyl phosphate (MUP),
or
OPD/peroxide, respectively), that reacts with the signal generating compound
to
generate a measurable signal. An elevated signal in the test sample, compared
to
the signal generated by a negative control, detects the presence of BS202
antigen.
The presence of BS202 antigen in the test sample is indicative of a diagnosis
of a
breast disease or condition, such as breast cancer.
B. Competitive Binding Assay The competitive binding assay uses a
peptide or protein that generates a measurable signal when the labeled peptide
is
contacted with an anti-peptide antibody coated microparticle. This assay can
be
performed on the IMx° Analyzer (available from Abbott Laboratories,
Abbott
Park, IL). The labeled peptide is added to the BS202 antibody-coated
microparticles (prepared as described in Example 17) in the presence of a test
sample suspected of containing BS202 antigen, and incubated for a time and
under
conditions sufficient to form labeled BS202 peptide (or labeled protein) /
bound
antibody complexes andlor patient BS202 antigen / bound antibody complexes.
The BS202 antigen in the test sample competes with the labeled BS202 peptide
(or
BS202 protein) for binding sites on the microparticle. BS202 antigen in the
test
sample results in a lowered binding of labeled peptide and antibody coated
microparticles in the assay since antigen in the test sample and the BS202
peptide
or BS202 protein compete for antibody binding sites. A lowered signal
(compared to a control) indicates the presence of BS202 antigen in the test
sample.
The presence of BS202 antigen suggests the diagnosis of a breast disease or
condition, such as breast cancer.
The BS202 polynucleotides and the proteins encoded thereby which are
provided and discussed hereinabove are useful as markers of breast tissue
disease,
especially breast cancer. Tests based upon the appearance of this marker in a
test
sample such as blood, plasma or serum can provide low cost, non-invasive,
diagnostic information to aid the physician to make a diagnosis of cancer, to
help
select a therapy protocol, or to monitor the success of a chosen therapy. This
marker may appear in readily accessible body fluids such as blood, urine or
stool
as antigens derived from the diseased tissue which are detectable by
immunologicai methods. This marker may be elevated in a disease state, altered
in
CA 02292843 1999-12-03
WO 99/02559 PCT/US98/14046
-89-
a disease state, or be a normal protein of the breast which appears in an
inappropriate body compartment.
Example 20' Immunohistochemical Detection of BS202 Protein
Antiserum against a BS202 synthetic peptide derived from the consensus
peptide sequence ( SEQUENCE ID NO 14) described in Example 14, above, is
used to immunohistochemically stain a variety of normal and diseased tissues
using standard procedures. Briefly, frozen blocks of tissue are cut into 6
micron
sections, and placed on microscope slides. After fixation in cold acetone, the
sections are dried at room temperature, then washed with phosphate buffered
saline and blocked. The slides are incubated with the antiserum against a
synthetic
peptide derived from the consensus BS202 peptide sequence (SEQUENCE ID NO
14) at a dilution of 1:500, washed, incubated with biotinylated goat anti-
rabbit
antibody, washed again, and incubated with avidin labeled with horseradish
peroxidase. After a final wash, the slides are incubated with 3-amino-9-
ethylcarbazole substrate which gives a red stain. The slides are
counterstained
with hematoxylin, mounted, and examined under a microscope by a pathologist.