Note: Descriptions are shown in the official language in which they were submitted.
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-1
IMPLANTABLE PROSTHETIC DEV1_C_.FS COATED WITH BIOACTIVE
MOLECULES
Background of the Invention
Implantable prosthetic devices have been used in the surgical repair or
replacement of
internal tissue for many years. The efficacy of many types of implants is
primarily dependent--
upon the surrounding tissue's adaptive reformation around and ability to bond
to the implant
surface. In orthopedic implants in particular, the geometry and the quality of
bone reformation
determines how much load the bone can resist. Orthopedic implants include a
wide variety of
to devices, each suited to fulfill particular medical needs. Examples of such
devices are hip joint
replacement devices, knee joint replacement devices, shoulder joint
replacement devices, and
pins, braces and plates used to set fractured bones. Some contemporary
orthopedic implants,
including hip and knee components, use high performance metals such as cobalt-
chrome and
titanium alloy to achieve high strength. These materials are readily
fabricated into the complex
shapes typical of these devices using mature metal working techniques
including casting and
machining.
At least two other methods are currently employed for bone and joint
replacement and
repair. Those methods include: (1) the use of grouting materials such as
poly(methyl
methacrylate) (PMMA) as bone cement between the bone and the prosthesis; and
(2) direct
2o opposition of bone tissue onto porous and non-porous implant surfaces. The
latter method is
known as the "cementless implant method."
In one example of the cementless implant method, a prosthesis is coated with
hydroxyapatite which is a major inorganic component of bone. The
hydroxyapatite-coated
prosthesis is then implanted in the bone cavity. The hydroxyapatite, which is
a calcium salt, is
believed to facilitate osteointegration with the bone tissues. After partial
integration of the
hydroxyapatite-coated prosthesis with the bone, layers of hydroxyapatite can
be detected
between the prosthesis and the bone tissues.
Despite the success of both metal and non-metal components in many patients,
long term
data has demonstrated an unacceptably high failure rate in more active
patients due to loosening
3o of the implant caused by bone resorption around the implant or failure to
achieve bone ingrowth.
Bone resorption results from stress shielding of the bone around the implant.
The failure to
achieve bone ingrowth into the surface of the implant to support implant
mechanical stability has
been a major problem with conventional implants. The metal orthopaedic
prostheses rely on
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-2-
poly(methyl methacrylate) for attachment and fixation to bone. Loosening of
such implants as a
result of cement failure has resulted in additional surgeries for securing the
implants. In order to
avoid-the problems associated with these prostheses, prostheses having porous
or centered
coatings have also been used. Although these materials encourage tissue
ingrowth, the process
of ingrowth occurs over a period of weeks to months, during which time the
implant may be
loosened and fail to function properly.
Summan~of~he Invention
It has been discovered according to the present invention that conventional
implants can
be improved by coating with a layer of gold and attaching to the gold a
bioactive molecule. The
bioactive molecule functions at the implant surface to promote a favorable,
local, environmental
response. Accordingly, the invention is an improved implantable prosthetic
device coated with a
bioactive molecule.
The prosthetic device provided according to the invention is convenient and
simple to
prepare. The bioactive molecules are directly coupled to the prosthetic device
surface through a
gold-sulfide bond using simple solution chemistry techniques. Prior art
methods for modifying
the surface of biomaterials were complex and cumbersome. For instance, in
order to conjugate a
molecule to a polymeric surface, the surface wauld first have to be modified
to add a functional
2o group to which the molecule could bind. In some cases the molecule would
require the addition
of a linking group which is capable of reacting with the functional group.
According to one aspect, the invention is a prosthetic device including a
shaped substrate
having a substrate surface, for implantation in a mammal, a layer of gold
attached to the substrate
surface and defining a tissue contacting surface, and a bioactive molecule
bound to the gold
layer. The shaped substrate can be, for example, a polymer, a metal, a
plastic, a fabric, a
ceramic, a biological material, or a composite of two or more materials. The
gold layer may be
any thickness but preferably the gold layer has a thickness of about 10 to
1000 Angstroms. The
bioactive molecule in turn can form a monolayer on the surface of the gold
which, depending on
the size of the bioactive molecule, is about 1 to 500 Angstroms in thickness.
The bioactive molecule can be virtually any molecule which can be attached to
the gold
layer and which can affect favorably the implant in its local environment once
implanted. The
bioactive molecule, therefore, can be natural or synthetic including a
protein, a peptide, a protein
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-3-
analog, a sugar, a lipid, a glycol protein, a glycolipid or a nucleic acid. In
one embodiment the
bioactive molecule is selected from the group consisting of a cell modulating
molecule, a
cherriotactic molecule, an anticoagulant moleucle, an antithrombotic molecule,
an anti-tumor
molecule, an anti-infectious molecule, a growth potentiating molecule, and an
anti-inflammatory
molecule. In one embodiment the cell modulating molecule is selected from the
group consisting
of an anti-integrin antibody, a bone morphogenic protein, an integrin binding
protein, and a
cadherin binding protein. In another embodiment the chemotactic molecule is an
extracellular
matrix molecule selected from the group consisting of collagen, fibronectin,
laminin, and
proetoglycan. In yet another embodiment the anti-tumor molecule is selected
from the group
1o consisting of methotrexate, adriamycin, cyclophosphamide, and taxol. The
anti-infectious
molecule is selected from the group consisting of antibiotics such as
penicillin according to
another embodiment. In another embodiment the growth potentiating molecule is
selected from
the group consisting of growth factors such as PDGF, EGF, FGF, TGF, NGF, CNTF,
and
GDNF. According to another embodiment the anti-inflammatory molecule is
selected from the
1 s group consisting of steroidal and non-steroidal compounds.
The layer of gold can be attached directly to the substrate surface. In
another
embodiment the layer of gold is attached to the substrate surface via
attachment to an
intermediate layer, such as a layer of titanium intermediate the gold layer
and the substrate
surface.
20 According to another embodiment the surface of the prosthetic device is
formed of a
porous material, wherein the layer of gold creates a gold surface that has
projections and
indentations and wherein the layer of gold has an approximately uniform
thickness across the
surface of the porous material.
According to another aspect, the invention is a prosthetic device including a
shaped
25 substrate having a substrate surface, for implantation in a mammal, a layer
of gold attached to the
substrate surface and defining a tissue contacting surface, and a bioactive
peptide bound to the
gold layer. The shaped substrate can be, for example, a polymer, a metal, a
plastic, a fabric, a
ceramic, a biological material, or a composite of two or more materials. The
gold layer may be
any thickness but preferably the gold layer has a thickness of about 10 to
1000 Angstroms. The
3o bioactive peptide forms a monolayer on the surface of the gold which,
depending on the size of
the peptide, is about 1 to 500 Angstroms in thickness.
The bioactive peptide can be any peptide which can be attached to the gold
layer and
CA 02301064 2000-02-14
WO 99/01089 PCT/tTS98/13792
-4-
which can affect favorably the implant in its local environment. It can be
natural or synthetic. In
one embodiment the bioactive peptide is selected from the group consisting of
a cell modulating
peptide, a chemotactic peptide, an anticoagulant peptide, an antithrombotic
peptide, an anti-
tumor peptide, an anti-infectious peptide, a growth potentiating peptide, and
an anti-
s inflammatory peptide. In one embodiment the cell modulating peptide is
selected from the group
consisting of an anti-integrin antibody fragment, a cadherin binding peptide,
a bone morphogenic
protein fragment, and an integrin binding peptide. Preferably the cell
modulating peptide is a
integrin binding peptide which is selected from the group consisting of RGDC,
RGEC, RGDT,
DGEA, DGEAGC, EPRGDNYR, RGDS, EILDV, REDV, YIGSR, SIKVAV, RGD, RGDV ,
to HRNRKGV, KKGHV, XPQPNPSPASPVVVGGGASLPEFXY, and ASPVVVGGGASLPEFX.
The peptides also may be any functionally active fragment of the proteins
disclosed herein as
being bioactive molecules useful according to the invention. In another
embodiment the
chemotactic peptide is selected from, the group consisting of functionally
active fragments of
collagen, fibronectin, laminin, and proteoglycan. In yet another embodiment
the anti-tumor
15 peptide is selected from the group consisting of functionally active
fragments of protein anti-
tumor agents. The anti-infectious peptide is selected from the group
consisting of functionally
active fragments of the protein anti-infectious agents according to another
embodiment. In
another embodiment the growth potentiating peptide is selected from the group
consisting of
functionally active fragments of PDGF, EGF, FGF, TGF, NGF, CNTF, GDNF, and
type I
2o collagen related peptides. According to another embodiment the anti-
inflammatory peptide is
selected from the group consisting of functionally active fragments of anti-
inflammatory agents.
The layer of gold can be attached directly to the substrate surface. In
another
embodiment the layer of gold is attached to the substrate surface via
attachment to an
intermediate layer, such as a layer of titanium intermediate the gold layer
and the substrate
25 surface.
According to another embodiment the surface of the prosthetic device is formed
of a
porous material, wherein the layer of gold creates a gold surface that has
projections and
indentations and wherein the layer of gold has an approximately uniform
thickness across the
surface of the porous material.
30 The invention in another aspect is a prosthetic device including a shaped
substrate formed
of a textured material having a substrate surface with first projections and
first indentations and a
layer of gold attached to the substrate surface of the textured material,
wherein the layer of gold
CA 02301064 2000-02-14
WO 99/01089 PCTNS98/13792
-5-
creates a gold surface that has second projections and second indentations
corresponding to the
first projections and first indentations. In one embodiment, the layer of gold
has an
approximately uniform thickness across the substrate surface of the textured
material. Preferably
the textured material is a porous material such as a porous titanium material,
a porous polymer,
or any other non-fabric porous material.
In one embodiment the textured material is a polymer. In another embodiment
the gold
layer has a thickness of about 10 to 1000 Angstroms.
According to yet another embodiment the prosthetic device also includes a
layer of
bioactive peptide attached to the gold surface through a gold-sulfide bond.
1 o in another aspect the invention is a prosthetic device including a shaped
substrate having
a substrate surface, a layer of gold attached to the substrate surface, and an
RGDC peptide
attached to the gold layer through a gold-sulfide bond. According to an
embodiment the shaped
substrate is a polymer, a metal, a plastic, a fabric, a ceramic, a biological
material, or a composite
of two or more materials. In one embodiment the gold layer has a thickness of
about 10 to 1000
Angstroms. In another embodiment the bioactive peptide forms a layer about 1
to 500
Angstroms in thickness.
The layer of gold is attached directly to the substrate surface in one
embodiment. In
another embodiment the layer of gold is attached to the substrate surface via
attachment to a
layer of titanium intermediate the gold layer and the substrate surface.
According to another embodiment the surface of the prosthetic device is formed
of a
porous material, wherein the layer of gold creates a gold surface that has
projections and
indentations. In one embodiment the layer of gold has an approximately uniform
thickness
across the surface of the porous material.
Brief Description of the Drawingg
Figure 1 is a graph depicting the observed reflectivity change upon incubation
of a clean
gold surface with a 0.2 mM solution of the RGDC peptide;
Figure 2 is a graph depicting the SPR spectra taken in an air ambient before
and after
adsorption of the RGDC peptide layer; and
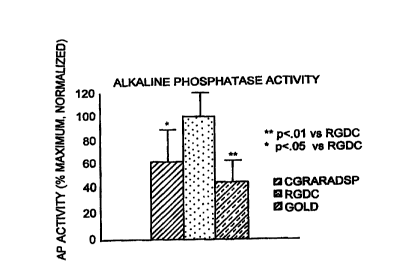
Figure 3 is a graph depicting alkaline phosphatase activity from osteoblasts
cultured on
RGDC-gold coated, CGRARADSP-gold coated, and plain gold surfaces.
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-6-
detailed Description of the Invention
According to the present invention, it was discovered that an implantable
device could be
coated with a bioactive molecule by first coating a substrate with a gold
layer and then attaching
the bioactive molecule through a simple reaction to the gold layer by forming
a gold-sulfide
bond. Prior art methods for attaching molecules to the surface of materials
are cumbersome. In
order to make a polymeric or other non-metal prosthetic device coated with
molecules using
these prior art methods the surface of the prosthetic device would have to be
modified and would
most likely require the addition of coupling reagents, making the preparation
of such devices
expensive, time consuming, and impractical. The preparation of metal implants
having
to molecules attached to surfaces of the implants has been a difficult
challenge in the prior art
because most metal surfaces have oxide layers which make binding of coupling
agents difficult.
The implantable prosthetic device coated with bioactive molecules disclosed
herein is prepared
by a simple technique for coupling bioactive molecules to biomaterial
surfaces.
The function performed by the surface is defined in part by the type of
bioactive molecule
bound to the surface. As used herein a "bioactive molecule" is any
biologically active molecule
which includes a sulfllydryl group or to which a sulfhydryl group can be
attached directly or
indirectly. Examples are a peptide, protein (e.g., apoprotein, glycoprotein,
antigen and antibody),
a protein analog containing at least one non-peptide linkage in place of a
peptide linkage, a
nucleic acid, etc. Nucleic acids include nucleotides; oligonucleotides; and
their art-recognized
2o and biologically functional analogs and derivatives including, for example,
oligonucleotide
analogs having phosphorothioate linkages.
Preferred bioactive molecules include a cell modulating molecule, a
chemotactic
molecule, anticoagulant moleucle, antithrombotic molecule, an anti-tumor
molecule, an anti-
infectious molecule, a growth potentiating molecule, and an anti-inflammatory
molecule.
A cell modulating molecule as used herein is a molecule that interacts with a
cell and
modifies the cell in any way e.g. alters gene expression, such as bone
morphogenic protein, anti-
integrin antibodies, integrin binding protein, and cadherin binding protein.
A chemotactic molecule as used herein is a molecule which attracts cells to a
surface or
aids in a cell's attachment to a surface and includes extracellular matrix
proteins such as
collagen, fibronectin, laminin, and proetoglycan.
An anti-tumor molecule as used herein is a molecule which decreases or
prevents a
further increase in growth of a tumor and includes anti-cancer agents such as
Acivicin;
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
7_
Aclarubicin; Acodazole Hydrochloride; Acronine; Adriamycin; Adozelesin;
Aldesleukin;
Altretamine; Ambomycin; Ametantrone Acetate; Aminoglutethimide; Amsacrine;
Anastrozole;
Anthramycin; Asparaginase; Asperlin; Azacitidine; Azetepa; Azotomycin;
Batimastat;
Benzodepa; Bicalutamide; Bisantrene Hydrochloride; Bisnafide Dimesylate;
Bizelesin;
Bleomycin Sulfate; Brequinar Sodium; Bropirimine; Busulfan; Cactinomycin;
Calusterone;
Caracemide; Carbetimer; Carboplatin; Carmustine; Carubicin Hydrochloride;
Carzelesin;
Cedefingol; Chlorambucil; Cirolemycin; Cisplatin; Cladribine; Crisnatol
Mesylate;
Cyclophosphamide; Cytarabine; Dacarbazine; Dactinomycin; Daunorubicin
Hydrochloride;
Decitabine; Dexormaplatin; Dezaguanine; Dezaguanine Mesylate; Diaziquone;
Docetaxel;
to Doxorubicin; Doxorubicin Hydrochloride; Droloxifene; Droloxifene Citrate;
Dromostanolone
Propionate; Duazomycin; Edatrexate; Eflornithine Hydrochloride; Elsamitrucin;
Enloplatin;
Enpromate; Epipropidine; Epirubicin Hydrochloride; Erbulozole; Esorubicin
Hydrochloride;
Estramustine; Estramustine Phosphate Sodium; Etanidazole; Etoposide; Etoposide
Phosphate;
Etoprine; Fadrozole Hydrochloride; Fazarabine; Fenretinide; Floxuridine;
Fludarabine
Phosphate; Fluorouracil; Flurocitabine; Fosquidone; Fostriecin Sodium;
Gemcitabine;
Gemcitabine Hydrochloride; Hydroxyurea; Idarubicin Hydrochloride; Ifosfamide;
Ilmofosine;
Interferon Alfa-2a; Interferon Alfa-2b; Interferon Alfa-nl; Interferon Alfa-
n3; Interferon Beta- I
a; Interferon Gamma- I b; Iproplatin; Irinotecan Hydrochloride; Lanreotide
Acetate; Letrozole;
Leuprolide Acetate; Liarozole Hydrochloride; Lometrexol Sodium; Lomustine;
Losoxantrone
Hydrochloride; Masoprocol; Maytansine; Mechlorethamine Hydrochloride;
Megestrol Acetate;
Melengestrol Acetate; Melphalan; Menogaril; Mercaptopurine; Methotrexate;
Methotrexate
Sodium; Metoprine; Meturedepa; Mitindomide; Mitocarcin; Mitocromin;
Mitogillin;
Mitomalcin; Mitomycin; Mitosper; Mitotane; Mitoxantrone Hydrochloride;
Mycophenolic Acid;
Nocodazole; Nogalamycin; Ormaplatin; Oxisuran; Paclitaxel; Pegaspargase;
Peliomycin;
Pentamustine; Peplomycin Sulfate; Perfosfamide; Pipobroman; Piposulfan;
Piroxantrone
Hydrochloride; Plicamycin; Plomestane; Porfimer Sodium; Porfiromycin;
Prednimustine;
Procarbazine Hydrochloride; Puromycin; Puromycin Hydrochloride; Pyrazofurin;
Riboprine;
Rogletimide; Safingol; Safingol Hydrochloride; Semustine; Simtrazene;
Sparfosate Sodium;
Sparsomycin; Spirogermanium Hydrochloride; Spiromustine; Spiroplatin;
Streptonigrin;
3o Streptozocin; Sulofenur; Talisomycin; Tecogalan Sodium; Tegafur;
Teloxantrone Hydrochloride;
Temoporfin; Teniposide; Teroxirone; Testolactone; Thiamiprine; Thioguanine;
Thiotepa;
Tiazofurin; Tirapazamine; Topotecan Hydrochloride; Toremifene Citrate;
Trestolone Acetate;
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
_g_
Triciribine Phosphate; Trimetrexate; Trimetrexate Glucuronate; Triptorelin;
Tubulozole
Hydrochloride; Uracil Mustard; Uredepa; Vapreotide; Verteporfin; Vinblastine
Sulfate;
Vincristine Sulfate; Vindesine; Vindesine Sulfate; Vinepidine Sulfate;
Vinglycinate Sulfate;
Vinleurosine Sulfate; Vinorelbine Tartrate; Vinrosidine Sulfate; Vinzolidine
Sulfate; Vorozole;
Zeniplatin; Zinostatin; Zorubicin Hydrochloride, and Taxol.
An anti-infectious molecule as used herein is a molecule which reduces the
activity of or
kills a microorganism and includes Aztreonam; Chlorhexidine Gluconate;
Imidurea; Lycetamine;
Nibroxane; Pirazmonam Sodium; Propionic Acid ; Pyrithione Sodium; Sanguinarium
Chloride ;
Tigemonam Dicholine; Acedapsone ; Acetosulfone Sodium; Alamecin; Alexidine;
Amdinocillin;
to Amdinocillin Pivoxil; Amicycline; Amifloxacin; Amifloxacin Mesylate;
Amikacin; Amikacin
Sulfate; Aminosalicylic acid; Aminosalicylate sodium; Amoxicillin; Amphomycin;
Ampicillin;
Ampicillin Sodium; Apalcillin Sodium; Apramycin; Aspartocin; Astromicin
Sulfate;
Avilamycin; Avoparcin; Azithromycin; Azlocillin; Azlocillin Sodium;
Bacampicillin
Hydrochloride; Bacitracin; Bacitracin Methylene Disalicylate; Bacitracin Zinc;
Bambermycins;
Benzoylpas Calcium; Berythromycin ; Betamicin Sulfate; Biapenem; Biniramycin;
Biphenamine
Hydrochloride ; Bispyrithione Magsulfex ; Butikacin; Butirosin Sulfate;
Capreomycin Sulfate;
Carbadox; Carbenicillin Disodium; Carbenicillin lndanyl Sodium; Carbenicillin
Phenyl Sodium;
Carbenicillin Potassium; Carumonam Sodium; Cefaclor; Cefadroxil; Cefamandole;
Cefamandole
Nafate; Cefamandole Sodium; Cefaparole; Cefatrizine; Cefazaflur Sodium;
Cefazolin; Cefazolin
Sodium; Cefbuperazone; Cefdinir; Cefepime; Cefepime Hydrochloride; Cefetecol;
Cefixime;
Cefmenoxime Hydrochloride; Cefmetazole; Cefmetazole Sodium; Cefonicid
Monosodium;
Cefonicid Sodium; Cefoperazone Sodium; Ceforanide; Cefotaxime Sodium;
Cefotetan;
Cefotetan Disodium; Cefotiam Hydrochloride; Cefoxitin; Cefoxitin Sodium;
Cefpimizole;
Cefpimizole Sodium; Cefpiramide; Cefpiramide Sodium; Cefpirome Sulfate;
Cefpodoxime
Proxetil; Cefprozil; Cefroxadine; Cefsulodin Sodium; Ceftazidime; Ceftibuten;
Ceftizoxime
Sodium; Ceftriaxone Sodium; Cefuroxime; Cefuroxime Axetil; Cefuroxime
Pivoxetil;
Cefuroxime Sodium; Cephacetrile Sodium; Cephalexin; Cephalexin Hydrochloride;
Cephaloglycin; Cephaloridine; Cephalothin Sodium; Cephapirin Sodium;
Cephradine;
Cetocycline Hydrochloride; Cetophenicol; Chloramphenicol ; Chloramphenicol
Palmitate ;
3o Chloramphenicol Pantothenate Complex ; Chloramphenicol Sodium Succinate;
Chlorhexidine
Phosphanilate; Chloroxylenol; Chlortetracycline Bisulfate ; Chlortetracycline
Hydrochloride ;
Cinoxacin; Ciprofloxacin; Ciprofloxacin Hydrochloride; Cirolemycin ;
Clarithromycin;
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-9-
Clinafloxacin Hydrochloride; Clindamycin; Clindamycin Hydrochloride;
Clindamycin Palmitate
Hydrochloride; Clindamycin Phosphate; Clofazimine ; Cloxacillin Benzathine;
Cloxacillin
Sodium; Cloxyquin; Colistimethate Sodium; Colistin Sulfate; Coumermycin;
Coumermycin
Sodium; Cyclacillin; Cycloserine; Dalfopristin; Dapsone ; Daptomycin;
Demeclocycline;
Demeclocycline Hydrochloride; Demecycline; Denofungin ; Diaveridine;
Dicloxacillin;
Dicloxacillin Sodium; Dihydrostreptomycin Sulfate; Dipyrithione;
Dirithromycin; Doxycycline;
Doxycycline Calcium ; Doxycycline Fosfatex; Doxycycline Hyclate; Droxacin
Sodium;
Enoxacin; Epicillin; Epitetracycline Hydrochloride; Erythromycin; Erythromycin
Acistrate;
Erythromycin Estolate; Erythromycin Ethylsuccinate; Erythromycin Gluceptate;
Erythromycin
l0 Lactobionate; Erythromycin Propionate; Erythromycin Stearate; Ethambutol
Hydrochloride;
Ethionamide; Fleroxacin; Floxacillin; Fludalanine; Flumequine; Fosfomycin;
Fosfomycin
Tromethamine; Fumoxicillin; Furazolium Chloride; Furazolium Tartrate; Fusidate
Sodium;
Fusidic Acid; Gentamicin Sulfate; Gloximonam; Gramicidin; Haloprogin;
Hetacillin; Hetacillin
Potassium; Hexedine; Ibafloxacin; Imipenem; Isoconazoie; Isepamicin;
Isoniazid; Josamycin;
Kanamycin Sulfate; Kitasamycin; Levofuraltadone; Levopropylcillin Potassium;
Lexithromycin;
Lincomycin; Lincomycin Hydrochloride; Lomefloxacin; Lomefloxacin
Hydrochloride;
Lomefloxacin Mesylate; Loracarbef; Mafenide; Meclocycline; Meclocycline
Sulfosalicylate;
Megalomicin Potassium Phosphate; Mequidox; Meropenem; Methacycline;
Methacycline
Hydrochloride; Methenamine; Methenamine Hippurate; Methenamine Mandelate;
Methicillin
Sodium; Metioprim; Metronidazole Hydrochloride; Metronidazole Phosphate;
Mezlocillin;
Mezlocillin Sodium; Minocycline; Minocycline Hydrochloride; Mirincamycin
Hydrochloride ;
Monensin ; Monensin Sodium ; Nafcillin Sodium; Nalidixate Sodium; Nalidixic
Acid;
Natamycin; Nebramycin; Neomycin Palmitate; Neomycin Sulfate; Neomycin
Undecylenate ;
Netilmicin Sulfate; Neutramycin; Nifuradene; Nifuraldezone; Nifuratel ;
Nifuratrone; Nifurdazil;
Nifurimide; Nifurpirinol; Nifurquinazol; Nifurthiazole; Nitrocycline;
Nitrofurantoin; Nitromide;
Norfloxacin; Novobiocin Sodium; Ofloxacin; Ormetoprim; Oxacillin Sodium;
Oximonam;
Oximonam Sodium; Oxolinic Acid; Oxytetracycline; Oxytetracycline Calcium;
Oxytetracycline
Hydrochloride; Paldimycin; Parachlorophenol; Paulomycin; Pefloxacin;
Pefloxacin Mesylate;
Penamecillin; Penicillin G Benzathine; Penicillin G Potassium; Penicillin G
Procaine; Penicillin
G Sodium; Penicillin V; Penicillin V Benzathine; Penicillin V Hydrabamine;
Penicillin V
Potassium; Pentizidone Sodium; Phenyl Aminosalicylate; Piperacillin Sodium;
Pirbenicillin
Sodium; Piridicillin Sodium; Pirlimycin Hydrochloride; Pivampicillin
Hydrochloride;
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
- 10-
Pivampicillin Pamoate; Pivampicillin Probenate; Polymyxin B Sulfate;
Porfiromycin ;
Propikacin; Pyrazinamide; Pyrithione Zinc; Quindecamine Acetate; Quinupristin;
Racephenicol;
Ramoplanin; Ranimycin; Relomycin; Repromicin; Rifabutin; Rifametane;
Rifamexil; Rifamide;
Rifampin; Rifapentine; Rifaximin; Rolitetracycline; Rolitetracycline Nitrate;
Rosaramicin;
Rosaramicin Butyrate; Rosaramicin Propionate; Rosaramicin Sodium Phosphate;
Rosaramicin
Stearate; Rosoxacin; Roxarsone; Roxithromycin; Sancycline; Sanfetrinem Sodium;
Sarmoxicillin; Sarpicillin; Scopafungin ; Sisomicin; Sisomicin Sulfate;
Sparfloxacin;
Spectinomycin Hydrochloride; Spiramycin; Stallimycin Hydrochloride;
Steffimycin;
Streptomycin Sulfate; Streptonicozid; Sulfabenz ; Sulfabenzamide;
Sulfacetamide;
1o Sulfacetamide Sodium; Sulfacytine; Sulfadiazine; Sulfadiazine Sodium;
Sulfadoxine; Sulfalene;
Sulfamerazine; Sulfameter; Sulfamethazine; Sulfamethizole; Sulfamethoxazole;
Sulfamonomethoxine; Sulfamoxole; Sulfanilate Zinc; Sulfanitran ;
Sulfasalazine; Sulfasomizole;
Sulfathiazole; Sulfazamet; Sulfisoxazole; Sulfisoxazole Acetyl; Sulfisoxazole
Diolamine;
Sulfomyxin; Sulopenem; Sultamicillin; Suncillin Sodium; Talampicillin
Hydrochloride;
Teicoplanin; Temafloxacin Hydrochloride; Temocillin; Tetracycline;
Tetracycline Hydrochloride
Tetracycline Phosphate Complex; Tetroxoprim; Thiamphenicol; Thiphencillin
Potassium;
Ticarcillin Cresyl Sodium; Ticarcillin Disodium; Ticarcillin Monosodium;
Ticlatone; Tiodonium
Chloride; Tobramycin; Tobramycin Sulfate; Tosufloxacin; Trimethoprim;
Trimethoprim Sulfate;
Trisulfapyrimidines; Troleandomycin; Trospectomycin Sulfate; Tyrothricin;
Vancomycin;
Vancomycin Hydrochloride; Virginiamycin ; Zorbamycin; Difloxacin Hydrochloride
; Lauryl
Isoquinolinium Bromide; Moxalactam Disodium; Ornidazole; Pentisomicin; and
Sarafloxacin
Hydrochloride.
A growth potentiating molecule as used herein is a molecule which stimulates
growth of
a cell and includes growth factors such as PDGF, EGF, FGF, TGF, NGF, CNTF, and
GDNF.
An anti-inflammatory molecule as used herein is a molecule which reduces an
inflammatory response and includes steroidal and non-steroidal
compounds;Alclofenac;
Alclometasone Dipropionate; Algestone Acetonide; Alpha Amylase; Amcinafal;
Amcinafide;
Amfenac Sodium; Amiprilose Hydrochloride; Anakinra; Anirolac ; Anitrazafen;
Apazone;
Balsalazide Disodium; Bendazac; Benoxaprofen ; Benzydamine Hydrochloride;
Bromelains;
3o Broperamole; Budesonide; Carprofen; Cicloprofen; Cintazone; Cliprofen;
Clobetasol Propionate;
Clobetasone Butyrate; Clopirac; Cloticasone Propionate; Cormethasone Acetate;
Cortodoxone;
Deflazacort; Desonide; Desoximetasone; Dexamethasone Dipropionate; Diclofenac
Potassium;
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-11-
Diclofenac Sodium; Diflorasone Diacetate; Diflumidone Sodium; Diflunisal ;
Difluprednate;
Diftalone; Dimethyl Sulfoxide; Drocinonide; Endrysone; Enlimomab ; Enolicam
Sodium ;
Epirizole ; Etodoiac; Etofenamate ; Felbinac; Fenamole; Fenbufen; Fenclofenac;
Fenclorac;
Fendosal; Fenpipalone; Fentiazac; Flazalone; Fluazacort; Flufenamic Acid;
Flumizole;
Flunisolide Acetate; Flunixin ; Flunixin Meglumine ; Fluocortin Butyl;
Fluorometholone
Acetate; Fluquazone; Flurbiprofen ; Fluretofen; Fluticasone Propionate;
Furaprofen; Furobufen;
Halcinonide; Halobetasol Propionate; Halopredone Acetate; Ibufenac ;
Ibuprofen; Ibuprofen
Aluminum; Ibuprofen Piconol; Ilonidap; Indomethacin; Indomethacin Sodium;
Indoprofen ;
Indoxole ; Intrazole; Isoflupredone Acetate; Isoxepac; Isoxicam; Ketoprofen;
Lofemizole
Hydrochloride ; Lornoxicam ; Loteprednol Etabonate; Meclofenamate Sodium;
Meclofenamic
Acid; Meclorisone Dibutyrate; Mefenamic Acid ; Mesalamine; MesecIazone;
Methylprednisolone Suleptanate; Morniflumate; Nabumetone; Naproxen ; Naproxen
Sodium ;
Naproxol ; Nimazone; Olsalazine Sodium; Orgotein ; Orpanoxin; Oxaprozin;
Oxyphenbutazone;
Paranyline Hydrochloride; Pentosan Polysulfate Sodium; Phenbutazone Sodium
Glycerate;
Pirfenidone ; Piroxicam; Piroxicam Cinnamate; Piroxicam Olamine; Pirprofen;
Prednazate;
Prifelone; Prodolic Acid; Proquazone; Proxazole; Proxazole Citrate ;
Rimexolone; Romazarit ;
Salcolex ; Salnacedin; Salsalate ; Sanguinarium Chloride ; Seclazone ;
Sermetacin; Sudoxicam;
Sulindac; Suprofen; Talmetacin; Talniflumate ; Talosalate ; Tebufelone ;
Tenidap; Tenidap
Sodium; Tenoxicam; Tesicam; Tesimide; Tetrydamine ; Tiopinac ; Tixocortol
Pivalate;
Tolmetin; Tolmetin Sodium; Triclonide; Triflumidate; Zidometacin; Zomepirac
Sodium.
An anticoagulant moleucle as used herien is a molceule that prevents clotting
of blood
and includes but is not limited to Ancrod; Anticoagulant Citrate Dextrose
Solution ;
Anticoagulant Citrate Phosphate Dextrose Adenine Solution; Anticoagulant
Citrate Phosphate
Dextrose Solution; Anticoagulant Heparin Solution; Anticoagulant Sodium
Citrate Solution;
Ardeparin Sodium; Bivalirudin ; Bromindione; Dalteparin Sodium ; Desirudin;
Dicumarol;
Heparin Calcium; Heparin Sodium; Lyapolate Sodium; Nafamostat Mesylate ;
Phenprocoumon;
Tinzaparin Sodium ; Warfarin Sodium.
An antithrombotic moleucle as used herien is a molceule that prevents
formation of a
thrombus and includes but is not limited to Anagrelide Hydrochloride;
Bivalirudin ; Dalteparin
Sodium ; Danaparoid Sodium; Dazoxiben Hydrochloride; Efegatran Sulfate;
Enoxaparin
Sodium; Ifetroban; Ifetroban Sodium; Tinzaparin Sodium ; Trifenagrel.
Preferably the bioactive molecule is a bioactive peptide. A "bioactive
peptide" as used
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
- 12-
herein refers to oligopeptides having a chain of less than or equal to fifty
amino acids and which
is capable of performing a desired biological function. In a preferred
embodiment the bioactive
molecule includes a cell modulating peptide, a chemotactic peptide, an
anticoagulant peptide, an
antithrombotic peptide, an anti-tumor peptide, an anti-infectious peptide, a
growth potentiating
peptide, and an anti-inflammatory peptide. A cell modulating peptide includes,
for example, an
antibody fragment or an integrin binding peptide. Bioactive peptides include
peptide fragments
of the proteins which are bioactive molecules disclosed herein and having the
functional
properties of those proteins.
A preferred use for the peptide-coated implantable device of the invention is
for
I o enhancing and/or accelerating bone growth in areas of damaged bone or in
bone replacement
surgery. Bone and joint replacement surgeries are commonly used, for instance,
to relieve pain,
improve function, and enhance the quality of life for patients with medical
conditions caused by
osteoarthritis, rheumatoid arthritis, post-traumatic degeneration, avascular
necrosis, and other
aging-related conditions. The prosthetic device of the invention which is
coated with bioactive
peptides that enhance or accelerate bone growth significantly improve the
ability of an implant to
remain attached to the bone surface. Preferred integrin binding peptides which
perform this
function are RGDC, RGEC, RGDT, DGEA, DGEAGC, EPRGDNYR, RGDS, EILDV, REDV,
YIGSR, SIKVAV, RGD, RGDV, and HRNRKGV.
Anti-infectious peptides include include antibiotic peptides such as those
disclosed in
2o U.S. Patent No. 5,602,097. Anti-tumor and anti-infectious peptides are also
disclosed in U.S.
Patent No. 5,516,755. U.S. Patent No. 5,484,885 discloses chemotactic,
antibiotic, and
lipopolysaccharide binding peptide fragments of CAP37 protein. These peptide
sequences are
approximately five consecutive amino acids long. US Pat No. 5,354,736
discloses several
collagen type I related peptides which are useful for promoting growth.
Growth potentiating peptides also include low molecular weight tibial growth
potentiating peptides such as those disclosed in U.S. Patent No. 5,576,301.
These peptides are
useful for potentiating tibial growth. These peptides have the following
sequences:
XPQPNPSPASPVVVGGGASLPEFXY and ASPVVVGGGASLPEFX.
Bioactive peptides such as those disclosed above are well known in the art.
Other
bioactive peptides useful according to the invention may be identified through
the use of
synthetic peptide combinatorial libraries such as those disclosed in Houghton
et al.,
Biotechniques, 13(3):412-421 (1992) and Houghton et al., Nature, 354:84-86
(1991) or using
CA 02301064 2000-02-14
WO 99/01089 PCT/IJS98/13792
-13-
phage display procedures such as those described in Hart, et al., J. Biol.
Chem. 269:12468
(1994). Hart et al. report a filamentous phage display library for identifying
novel peptide
ligands for mammalian cell receptors. In general, phage display libraries
using, e.g., M13 or fd
phage, are prepared using conventional procedures such as those described in
the foregoing
reference. The libraries display inserts containing from 4 to 80 amino acid
residues. The inserts
optionally represent a completely degenerate or a biased array of peptides.
Ligands that bind
selectively to a specific molecule such as a cell surface receptor are
obtained by selecting those
phages which express on their surface a ligand that binds to the specific
molecule. Ligands that
possess a desired biological activity can be screened in known biological
activity assays and
to selected on that basis. These phages then are subjected to several cycles
of reselection to identify
the peptide-expressing phages that have the most useful characteristics.
Typically, phages that
exhibit the binding characteristics (e.g., highest binding affinity or cell
stimulatory activity) are
further characterized by nucleic acid analysis to identify the particular
amino acid sequences of
the peptides expressed on the phage surface and the optimum length of the
expressed peptide to
achieve optimum biological activity. Alternatively, such peptides can be
selected from
combinatorial libraries of peptides containing one or more amino acids. Such
libraries can
further be synthesized which contain non-peptide synthetic moieties which are
less subject to
enzymatic degradation compared to their naturally-occurring counterparts. U.S.
Patent No.
5,591,646 discloses methods and apparatuses for biomolecular libraries which
are useful for
screening and identifying bioactive peptides. Methods for screening peptides
libraries are also
disclosed in U.S. Patent No. 5,565,325.
Peptides obtained from combinatorial libraries or other sources can be
screened for
functional activity by methods known in the art. For instance when the peptide
is a cell
modulating peptide, and in particular an integrin binding peptide, one of
ordinary skill in the art
can easily determine whether the peptide will modulate bone cell activity by
performing the in
vitro studies set forth in example 2 to measure osteoblast differentiation.
Likewise, similar
experiments can be conducted for other types of cells using cell specific
markers of
differentiation or growth. The type of assay of course, used for a particular
peptide depends on
the source of the peptide. For instance if a peptide is a fragment of an anti-
tumor molecule, the
3o peptide should be tested for functional activity in an anti-tumor assay.
Those of skill in the art
can easily choose an appropriate assay for testing functionality of a
particular peptide.
The bioactive molecules useful according to the invention are commercially
available
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
- 14-
from many sources and methods for making these molecules also are well known
in the art.
Bioactive peptides and proteins may easily be synthesized or produced by
recombinant means.
Such methods are well known to those of ordinary skill in the art. Peptides
and proteins can be
synthesized for example, using automated peptide synthesizers which are
commercially
available. Alternatively the peptides and proteins can be produced by
recombinant techniques by
incorporating the DNA expressing the peptide into an expression vector and
transforming cells
with the expression vector to produce the peptide.
The bioactive molecule is bound to a gold surface. Although many attempts have
been
made in the prior art to coat peptides, proteins and other biomaterials on
various surfaces, each of
1o these techniques has required the use of complex coupling techniques and
surface modification
including the use of coupling agents and linkers. It has been discovered
according to the present
invention that bioactive molecules can be attached to a prosthetic device via
a gold surface,
through a simple technique that results in the formation of a bond between a
gold and a
sulflrydryl group. The bond that forms between a sulfhydryl group and gold
only requires the
interaction between the sulfhydryl group and the gold in a solution. The
interaction does not
require coupling agents or linkers or surface activation or modification of
the gold.
The molecule is added to the gold surface using simple solution chemistry
techniques,
e.g., simply exposing the gold surface to a solution of molecule in a solvent
such as
ethanol:water. This approach is simple and is non-line of sight dependent. A
technique which is
line of sight dependent only coats an external surface and does not coat
internal pores or
interstices. Non-line of sight dependent methods are capable of coating the
internal surface area
such as pores. This technique produces an evenly coated layer of molecule on
any type of
device, even those having a porous, spongy, or textured surface.
Bioactive molecules can be attached to gold surfaces directly or via spacers.
If direct,
then bioactive molecules must have (or must be modified to have) a sulfhydryl
group. If
indirect, the bioactive molecule may or may not have sulfhydryl, but the
spacer will have a
sulfhydryl. In this instance the spacer is attached to the gold surface and
the bioactive molecule
is attached to the spacer, before or after attaching of the spacer to the gold
surface. Proteins or
peptides having endogenous cysteine groups already have a cysteine within the
molecule and do
3o not require the addition of another sulfhydryl group. If a protein or
peptide has more than one
cysteine and those cysteines have formed di-sulfide bridges the molecule can
be subjected to
reducing agents to ensure that the sulfllydryl group is free and available.
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-15-
Proteins or peptides without endogenous cysteine groups can easily be
manipulated to
incorporate a sulfhydryl group. For instance, peptides and proteins can be
subjected to site
directed mutagenesis to prepare a cysteine containing protein or peptide.
Additionally a cysteine
can be added to either the N-terminal or C-terminal of the peptide or protein
or incorporated
within the peptide or protein or within a branch of the peptide or protein. A
cysteine may be
added anywhere in the peptide or protein that does not affect the biological
activity of the peptide
or protein. This is demonstrated schematically as follows:
C-X-Y-X;
X-Y-Z-C;
to X-Y-C-Z; or
X-Y-Z
X
C
wherein X, Y, and Z are any amino acid and C is cysteine. Preferably a
cysteine group is added
to either the C-terminal or the N-terminal of the peptide. More preferably,
the cysteine group is
on the C terminal region of the peptide.
Proteins or peptides without endogenous cysteine and other non-sulfhydryl
containing
molecules can easily be manipulated to incorporate a non-cysteine sulfhydryl
group. For
example, sulfhydryl groups can be introduced into the molecules having a
primary amine (or
modified to have a primary amine) by reaction of the primary amine in the
molecule with 2-
iminothiolanc or Traut's reagent, or other commercially available reagents. A
variety of
commercially available reagents for coupling sulfliydryl groups to molecules
are available from
Pierce Chemical, Corp., such as Traut's reagent (Product No. 26101 ), SATA
(Product No.
26102) or SPDP (Products Nos. 21757, 21657, 21557). Traut's reagent is a water
soluble
reagent which reacts with primary amines at pH 7-10 to introduce sulfhydryl
groups, as disclosed
in Schram and Dulffer, Physiol.. Chem., 358, 137-139 (1977). Traut's reagent
has the following
structure:
"~' ~ ~w; cr
s
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
- 16
SATA is a reagent which adds protected sulfhydryls to molecules by reacting
with primary
amines. SATA has the following chemical structure:
o° 0 0
-0-C~~~II~-S (-~N,
fAfA
I~.w. I>t..l
SPDP, which includes LC-SPDP and Sulfo-LC-SPDP also is capable of adding a
sulfliydryl
group to primary amines. These mooecules have the following structures:
0
~.s_s_~1_~ _o_N~
a
o~
sror
~.w. n=..
tpm In 1.1 l
0 O ~ 0 0
~s-s-~-~ ~-~-IWIs-~-o ~ C~.s-.s_ov,-a, ~-r-Icx,~,-~-o ~
0
o so,w.
uaor s~u..u~s~o~
Preferably, the bioactive molecule is prepared with a sulfllydryl group at,
for example, the
carboxyl (C) or amino (N) terminus and then is coupled to the gold surface. In
an alternative
embodiment, a spacer is synthesized with a sulfhydryl group, preferably at or
near one end, and
then this spacer is attached at this end to the gold surface and via a
different functional group to
the bioactive molecule. The spacer molecule may be coupled for example to the
terminal amine
2o group or carboxyl group of the bioactive peptide or protein. Spacer
molecules can be selected,
for example, which contain (or which can be modified to contain) a functional
group that is
reactive with the peptide or protein N-terminal amine group and allowing the
functional group
and the peptide or protein N-terminal amine to form a linkage in accordance
with art-recognized
procedures. See, e.g., March, J., Advanced Organic Chemistry, 4th Ed., New
York, NY, Wiley
and Sons, 1985), pp.326-1120. In an analogous manner, the spacer molecule may
be coupled to
a reactive group in the C-terminus of the bioactive peptide or protein.
Additionally the spacer
molecule may be coupled to a branch of a molecule or an internally active
portion of a molecule
or any end group.
Thiol or amide groups may be added at any nucleotide of a nucleic acid. The
amine
group may be added so as to provide a point of attachment for a sulfhydryl
group by the above-
described reagents. Nucleic acids may also be synthesized with groups such as
amine groups.
The bioactive molecule is bound to a layer of gold which is attached to a
substrate surface
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-17-
of a shaped substrate. The layer of gold covers all or part of the prosthetic
device to define a
tissue contacting surface. The tissue contacting surface is the surface of the
gold to which the
molecules are bound. T'he layer of gold may be extremely thin or it may be
thick. The layer of
gold may actually be the entire prosthetic device. In this case the layer of
gold would encompass
the shaped substrate as well. Preferably the layer of gold is thin because of
the high cost of gold.
The layer of gold is attached to the shaped substrate surface by any means
known in the
art. For instance, the gold layer can be added to the implant using
evaporation, electroplating,
sputtering or electrodeposition. Using any of these techniques the gold can be
applied in a thin
layer to the surface of the implant. Preferably the gold is attached to the
substrate by
1 o electroplating or evaporation. Electroplating produces a gold layer which
is non-line of site
dependent. Using electroplating, therefore, a gold layer can be produced on an
uneven surface
such that the uneven nature of the surface is maintained.
A shaped substrate as used herein is a material which has the shape of an
implantable
prosthetic. The selection of the shape of the prosthetic is governed by the
physical requirements
of space, geometry and function at the region where the implant is to be
positioned in the body.
Implants can be made available in a range of sizes to fit the varying sizes in
the patient
population.
In some embodiments, the bioactive molecule coating is on and within the pores
of an
implantable prosthesis of the type where tissue ingrowth is contemplated,
wherein the bioactive
2o molecule encourages the ingrowth of the tissue into the pores or
facilitates attachment of tissue to
the prosthetic. In another embodiment, the coating is on a typical prosthesis
or on a 'temporary
implant', such as a long term but temporary catheter, and the coating is of an
antibacterial agent
to prevent colonization upon the prosthesis or catheter. Thus, the invention
is useful in
connection with prosthetic devices such as bone or joint replacement or repair
prosthetics,
vascular prostheses, including woven prostheses, catheters for implantation
and the like.
Virtually any implantable tissue contacting surface may be modified as
described herein.
The shaped substrate may be made from any material ordinarily used to prepare
implants.
For instance the shaped substrate may be made from any of a wide variety of
metals, such as,
pure titanium, titanium alloy, stainless steel, cobalt-chrome alloy, and gold.
The shaped
3o substrate may also be made from polymeric matrix composites, such as
continuous filament
carbon, graphite, glass and aramid fibers embedded within a polymer matrix,
such as
polysulfone, polyether-ether-ketone, polyether-ketone-ketone, polyimide, epoxy
or polycyanate,
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
- Ig -
polymers including polyethylene, polyetheretherketone {PEEK), polypropylene,
polymethylmethacrylate, polyamides, and polyester. Other polymeric matrix
composites include
but are not limited to polyethylene films, ultra-high molecular weight
polyethylene films and
fibers, polyvinylidene fluoride films, poly (methyl methacrylate) films,
polystyrene films, nylon
12 films and fibers, various polyesters and polyacrylates,
polyetherethereketones, aromatic
polyamides, polyethylene terephthalate fibers and films, poly (tetramethylene
terephthalate) -
fllms, and polyether-esters of poly (tetramethylene terephthalate).
The prosthetic device of the invention is useful for implantation in mammals.
Mammals
herein means humans, cats, dogs, mice, hamsters, pigs, goats, primates,
horses, cows, and sheep.
A preferred prosthetic device of the invention is a shaped substrate having a
substrate
surface, a layer of gold attached to the substrate surface, and an RGDC
peptide attached to the
gold layer through a gold-sulfide bond. The RGD peptide is a peptide found in
many
extracellular matrix proteins which is known to bind as(3, and a~(33 integrin
receptors. RGD
attached to surfaces has been demonstrated to increase osteoblast attachment
to the surface. It is
preferred that orthopedic prosthetic devices are coated with RGDC.
The prosthetic device with the bioactive molecule attached to the surface has
been found
to be extremely stable and as a result can be stored for extended periods of
time. The stability of
the device is important because it enables the device to be prepared in
advance and shipped to a
medical institution where it can be stored for future implantation. As a
result medical institutions
2o can store many prosthetic devices having various molecules already coated
on the surface for
various applications.
The prosthetic device of the invention may also be prepared and stored without
the
bioactive molecule attached to the device. The bioactive molecule can then be
added at a later
time point prior to use. The step of adding the bioactive molecule to the gold
surface is simple
and quick and may easily be performed immediately prior to a surgical process.
Accordingly,
the prosthetic device of the invention also includes a shaped substrate formed
of a textured
material and having a gold layer attached to the surface. More specifically
the shaped substrate
has a substrate surface with first projections and first indentations and a
layer of gold is attached
to the substrate surface of the textured material such that the layer of gold
creates a gold surface
3o that has second projections and second indentations corresponding to the
first projections and
indentations. The layer of gold optionally has an approximately uniform
thickness across the
substrate surface of the textured material.
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
- 19-
A "textured material" as used herein is a non-fabric material having small
(about 1-1000
microns in size) interstices throughout. The shaped substrate may be made
entirely of a textured
material or may optionally be made of a non-textured material but having a
surface which is
coated with a textured material to produce a textured surface. Preferably the
textured material is
a porous material such as a porous titanium material, a porous polymer, or any
other non-fabric
porous material. Porous metal surfaces have been created by plasma spraying
(U.S. Pat. No.
3,605,123) of fine metallic particles, or by sintering a loosely packed
coating of metallic particles
(U.S. Pat. No. 4,550,448, British Patent No. 1,316,809), or by diffusion
bonding kinked fiber
metal pads (U.S. Pat. No. 3,906,550). Plasma spraying employs super heated
gases to melt the
to metal particles to be sprayed. Sintering develops interparticle bonds in a
porous coating by
exposing the coating and implant metal to temperatures approaching their
melting point, while
diffusion bonding employs heat and pressure to promote atomic diffusion at the
coating implant
interface. Methods for preparing porous polymer materials are well known in
the art.
Additionally the shaped substrate may be made of a non-textured material but
having a surface
i5 which is at least partially coated with a textured material to produce a
partially textured surface.
Thus the invention also encompasses a prosthetic device having a shaped
substrate made from a
non-textured material but at least partially coated with a textured material
on which a layer of
gold is attached.
The substrate surface of the textured material has projections and
indentations.
20 "Projections and indentations" as used herein are microscopic cavities on
the surface of the
substrate defining a 'rough' surface microscopically. A substrate surface is
said to have
projections and indentations if it has a substantial region that is mostly
free of a flat smooth
surface, but instead is characterized by numerous indentations and projections
throughout the
region, numerous cavities having a diameter between 1 micron and 1 millimeter,
preferably
25 between 20 microns and 900 microns. In a preferred embodiment the gold
layer attached to the
textured material creates a gold surface that also has projections and
indentations and that has an
approximately uniform thickness across the substrate surface.
The following examples are provided to illustrate the methods and products of
the present
invention. As described above, many variations on these particular examples
are possible and,
3o therefore, the examples are merely illustrative and not limiting of the
present invention. As
demonstrated in the Examples below the implantable prosthetic device of the
invention has many
advantages over uncoated implants and even over peptide-coated implants that
do not have a
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-20-
gold surface.
Exam lies
The following examples describe experiments which were conducted on molecule-
coated
gold surfaces. Experiments were also carried out on molecule coated polymer
surfaces (FEP)
which serve as controls. By comparing the results obtained with the gold-
coated substrate with
those obtained with the FEP material it is possible to distinguish the effects
which occur as a
result of the immobilized peptide from those which occur as a result of the
surface context of the
immobilized peptide.
to Examule 1 ~ Immobilization o~ entides on Biomaterial Surfaces
Methods and Materials:
Peptides: The peptides used in the following studies are set forth in Table I.
All peptides
were synthesized commercially (QCB, Hopkinton, MA) to a purity of 98% or
greater by HPLC
and mass spectrometry. Peptides being coupled to FEP (the control substrate)
included G or
GGGG spacer sequence on their N- or C-terminus. Peptides being coupled to gold
coated
surfaces included a CG or CGGG spacer sequence on their N- or C-terminus.
Control peptides
were fabricated using scrambled sequences or, if known, amino acid
substitutions.
Table 1
Extracelluiar Matrix Peptide Ligand Integrin Receptors
Protein
2o Collagen I cRGD, RGDT, DGEA a, (3,, a2~3,,a3(3,
Bone Sialoprotein EPRGDNYR oc~(33
Osteopontin RGD oc~(33
Fibronectin RGDS, EILDV, REDV ar,3~3,, oc4(3,,oc5~3,,oc,,(3,,
a~a3~a~~s,a~~6~ a4~~
laminin YIGSR, SIKVAV, RGD a,~3,, ocz(3,,a,3(3,,
ab(3~,
Thrombospondin RGD a,,(33
Vitronectin RGDV, HRNRKGV a~(3,, a~(33,ac,,(3s
Osteonectin (SPARC) KKGHK ?
Human and rat osteoblasts/osteocytes express a range of integrins. These are
shown in
3o Table 2 below.
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-21 -
Table 2
Rat Calvarial Osteoblasts a,~3,, as(3,,a"(3,, a,,~33,a"~is
Human Osteoblasts a3(3,, a4~i,,a5(3,, a"(33
Preparation of Gold Coated Substrates: 12-mm diameter pre-cleaned circular
glass cover slips
were obtained from Fisher Scientific and placed into a custom mount consisting
of a 0.25 inch
thick aluminum plate. The samples were then suspended inside a four-source NRC
3177
electron beam evaporator with a Sloan 180° electron gun and Sloan
Six/Ten power supply. The
gold coating did not adhere well to plain glass, so titanium was used as an
intermediate. The
1o evaporator chamber was pumped down to achieve a vacuum in the low 10-6 to
high 10-' tort
range. Initial pumping was done with a mechanical pump and then a diffusion
pump was
brought on line to achieve and maintain the final pressure. A liquid nitrogen
trap was employed
to keep the system free of contaminating vapors from diffusion pump oil or
other contaminants.
The electron beam gun was activated and a 60 angstrom coating of Ti was put
onto the cover
is slips. The Ti source was then rotated away as the gold source was rotated
into place. A 500
angstrom layer of gold was applied. The samples were then removed from the
system and kept
under nitrogen or covered in Kimwipes and aluminum foil until ready for use.
Immobilization of Peptides on Gold Coated Substrates: Cysteine terminated
peptides
were solubilized in a 1:1 ethanol:distilled water solution at a concentration
of 0.22 mM. The
20 gold substrates were exposed to this solution for one hour. Plain gold
controls were made by
exposing samples to peptide-free ethanol:distilled water for one hour.
Reactions were carried out
in the dark to protect the light-sensitive cysteine.
Preparation of FEP Membranes with Immobilized Peptide: FEP membranes with
immobilized peptide are useful for comparison purposes. The FEP membranes were
prepared
25 using surface modification and coupling techniques. FEP films (Dupont) 25
micrometers thick
were cut into discs with a lathe ( 1.76cm diameters) and cleaned by sonication
in hexanes and
methanol for 20 seconds each. Surface hydroxyl (OH) groups were added to
cleaned FEP films
by a radio frequency glow discharge (RFGD) process. The films were placed in a
chamber and
brought to a pressure of 100 millitorr. The chamber was filled with hydrogen
and methanol
30 vapor at 500 millitorr for 10 minutes. The pressure was again reduced to
100 millitorr and the
radio frequency glow discharge was activated for 1 minute.
After rinsing the hydroxylated REP films in DMSO, the films were reacted with
CDI
CA 02301064 2000-02-14
WO 99/01089 PCTNS98/13792
-22-
(40mg/lml in DMSO) for 24 hours. To enhance. nucleophilic attack of the OH-
group to the CDI
substrate, the solution was supplemented with N-Hydroxy-succinimide (NHS,
Fluka) (1 mg/ml
in DMSO) (Frost, 1981) The excess CDI/NHS was rinsed off of the films with
DMSO before
applying the peptide solution. Films were placed in 0.22 M peptide in 1 M MES
buffer (pH 5)
for 48 hours (Hearn, 1987). Films were rinsed sequentially with 1 M MES
buffer, 1 M NaCI, and
distilled water. This stringent rinsing protocol was used to remove adsorbed
vs. linked peptide
from the surface.
The chemical reactions for the CDI Activation and the peptide coupling
reactions are as
follows:
CDI Activation:
OH + - ~ ~ Q""
FEP-OCDI complex
~.~lt' ~e.~~ i °~
aquoous solution
O-~ - + R NH,,
Immobilized peptide
The scheme for FEP and gold coated substrates are shown below.
P
8 E
Y p
I Z
D D
E E
I
LYS ~. FRC?BE
L Y5 .._ Ng ...... PF~OSE
1
CYS C'C' C
1
s ~ d
I I
~ilB~ . F E r
Methods for Surface Characterization
1. Contact Angle: Contact angles were measured with ethylene glycol, glycerol,
distilled
water, and ethanol on a goniometer. Each fluid was placed on the substrate
using a syringe with
a 30 gauge needle. At least three measurements per drop were taken. The
surface energy was
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
- 23 -
calculated using E. Sacher's method (Ratner, 1988; Kaelble, 1974; Kaelble,
1970). Contact
angle data provides information regarding the surface chemistry and surface
energetics of the top
Angstroms of a polymer substrate. A bead of pure liquid with a known surface
tension is
placed on the polymer surface. The resulting bead angle is measured using a
goniometer (an
5 alternate technique is to use a Cahn microbalance). A hydrophobic surface
causes liquid beading
and a high contact angle while a more hydrophilic surface is wettable and a
small contact angle is
observed. A range of fluids with polar (i.e. water) to non-polar (i.e. decane)
characteristics are
tested.
2. Surface-Plasmon Resonance: Surface plasmon resonance (SPR) was produced
when a
1o beam of p-polarized laser light impinges onto the surface of a thin metal
film. The light was
coupled to the metal film through a prism which was mounted on a rotating
turntable. At a
particular angle of incidence the E-field of the laser light interacts with
the surface bound free
electrons of the metal film in such a way that a charge density wave was
generated at the
interface of the metal and air. This excitation results in a sharp reduction
in the magnitude of the
~ 5 reflected light (measured with a photodiode). The angle at which this
occurs together with the
depth and half width of the minimum were determined by the thickness and
complex refractive
index of the metal film. The magnitude of the evanescent field which arises
from the charge
density wave decays exponentially in the direction normal to the surface.
Consequently, any
dielectric layer (such as a peptide overlayer or cell membrane) adhering to
the metal film will
2o cause a change in the condition for resonance. By fitting the Fresnel
equations, firstly to the date
for the uncoated metal film, and then to the metal plus thin film, the
thicknesses and complex
refractive indices of the metal and peptide overlayer were determined. Typical
thickness
resolution for the SPR were of the order of 0.01 nm making it an extremely
sensitive probe for
the surface chemistry of peptides and proteins. By observing changes in
reflectivity at a fixed
25 angle of incidence, it is possible to monitor the adsorption of peptides
from solution onto a
surface and thus obtain time resolved binding of molecules from the bulk to a
surface. Another
practical advantage of this method is that peptide chemistry can be determined
in aqueous
environments rather than the ultrahigh vacuums needed for other surface-
sensitive techniques
(e.g. ESCA).
30 3. Characterization of Peptide Stability:
a. Fluorescent tagging of immobilized peptides: S-(and-6)-
carboxythtramethylrhodamine succinimidyl ester, i.e. TAMRA SE (Molecular
Probes #C-1171),
CA 02301064 2000-02-14
WO 99/01089 PGT/US98/13792
-24-
reacts preferentially with amines. TAMRA SE has the advantage of maintaining
stability for
weeks and is stable in pH's ranging from 4 to 9. The excitation and emission
wavelengths of this
compound are 546, and 5'76,, respectively. TAMRA SE is made up as a 1mM
solution in
DMF. It is then mixed with a pH 8.5 sodium tetraborate buffer in a 1:1 ration
for a final solution
concentration of 0.5 mM. This is reacted with the samples on a stirrer plate
for four hours.
Rinsing is done overnight in 4 M urea + 0.6% Tween 60.
b. Analysis ofpeptide stability under physiologic conditions: Various
immobilized
peptides, tagged with fluorescent probes, are exposed to tissue culture media,
tissue culture
media with 10% serum, and osteoblasts. After 1, 7, 14 and 28 days of culture,
substrates are
to rinsed several times, and assessed for fluorescence using confocal
microscopy.
Results:
1. Contact Angle
Contact angles on pure gold are greatly effected by hydrocarbons adsorbed from
air, but
a 5 surface energy was performed on these contaminated surfaces anyway as any
implants are likely
to be maintained in air and are all going to be thus contaminated. Contact
angles were measured
with ethyleneglycol, glycerol, distilled water and ethanol on a goniometer.
Each fluid was
placed on the substrate using a syringe with a 26 gauge needle. The smallest
drop possible was
used to minimize gravitational effects. At least three measurements per liquid-
sample
2o combination were taken. The surface energy was calculated using Sacher's
method. The results
gave us the following surface energies:
Material Surface Energy
Plain Gold: 27.4 dyne/cm
Gold+RGD: 25.0 dyne/cm
25 Gold+CG: 81.9 dyne/cm
Gold+RGDC: 42.1 dyne/cm
On unmodified FEP, water generated a contact angle of approximately
105°indicating an
unwettable, low energy surface. RFGD treated FEP showed a contact angle of 60-
65° with water
confirming the presence of polar hydroxyl groups.
2. Surface Plasmon Resonance
The incubation of a pure gold surface with a 0.22 mM solution of the RGDC
peptide
CA 02301064 2000-02-14
WO 99/01089 PCTNS98/13792
-25-
results in rapid film formation. Presumably the rapid adsorption is driven by
the strong
interaction of the cysteine residue of the peptide with the gold surface. The
data is shown in
Figure 1.
Figure 1 is a graph of the observed reflectivity change upon incubation of a
clean gold
surface. The spectra were fitted using fresnel reflectivity theory. Fitting
the bare substrate
spectra yielded optical constants for the gold film of: Refractive index (n,k)
= 0.26708, -3.304,
Film thickness of 482, Fit error= 8.4 x 10-3. Using these constants for the
gold film, the SPR
spectra of the RGDC layer was analyzed to obtain the thickness and refractive
index of the
peptide layer. The refractive index and thickness of the RGDC layer were
allowed to vary
between sensible limits during the fitting procedure, the best fit to the data
yielded the following
parameters for the RGDC layer: Refractive index (n,k) = 1.4665, - 0.0992, Film
thickness of 23-
25~, Fit error= 4.36 x 10-3.
Figure 2 depicts a theoretical curve for the RDGC layer which was generated
using the
above parameters. The film thickness value of 23-25A indicates that the
peptide molecules are in
1 s an upright orientation.
Non-SH containing RGD failed to bind to the gold surface.
EXAMPLE 2' Evaluation of the E~'ects of Immobilized Pe~ntideS on Octeoblact
Differentiation In Vitro
Rat calvarial osteoblasts were used as a model system because they have been
used
extensively in in vitro for studies of bone cell differentiation. These cells
undergo a predictable,
temporal expression of biochemical and gene markers of the osteoblast
phenotype over a three to
four week period in culture (Aronow 1990, Harris 1994). Lian et al. have
described three phases
of osteoblast growth and differentiation in vitro (Breen 1994). The initial
phase (days 1-6)
involves active cell proliferation and increases in collagen type I gene
expression. Matrix
maturation occurs over the second week in culture and was accompanied by
increased alkaline
phosphatase mRNA expression and enzyme activity. The final phase involved cell
aggregation
into nodules with subsequent mineralization. This period included increased
osteocalcin and
osteopontin gene expression and protein synthesis. This standard sequence of
osteoblast
differentiation served as the reference by which experimental substrates were
evaluated.
In the proposed study, attachment, morphology, proliferation, biochemical
markers
CA 02301064 2000-02-14
WO 99/01089 PCTNS98/13792
-26-
(alkaline phosphatase and osteocalcin levels) and gene expression (see below
for details) for
calvarial osteoblasts were quantified after 7, 14, 21, and 28 days in culture.
Biochemical assays
and Northern analysis were performed using standard techniques such as those
set forth below.
Methods:
Rat Osteoblast Isolation: Primary rat calvarial osteoblasts (RCOB) were
isolated from
post-natal six day old rat pups. The crania were dissected using sterile
technique under the tissue
culture flow hood. Parietal and frontal bonds were dissected free from the
sutures and subjected
to collagenase digestion (4 x 20min; typeI: type II = 6:1 ) (Boden, 1996). The
specific activity of
to collagenase I and II (Worthington Enzymes, Freehold, NJ) was 42.5 IU/ml,
88.25 IU/ml in the
first digestion and 170 IU/ml, 353 IU/ml for the remaining two digestions
(Boden, supra 1996).
Cells from the second and third digestions were pooled to form an osteoblast
rich suspension.
These cells were rinsed, pelleted and plated in MEM (Gibco) with 10% FBS
(Hyclone) at a
density of 6,510 cells per cm z(Lian, 1990). After confluence, the media was
switched from
t5 MEM to a mineral rich BGJb media, to which 10% FBS, 50 mg/ml ascorbic acid,
and lOmM
Beta Glycerol Phosphate are added (Lian 1990). For sub-cultivated experiments,
primary cells
were expanded in T-75 flasks with MEM and 10% FBS. After reaching (about 80%)
confluence,
cells were sub-cultivated with 2.5% trypsin/EDTA and plated at 20,000 per cmz
in MEM +
10%FBS (Lian, supra 1990). At day 7, 50 mg/ml ascorbic acid was added to
induce collagen I
2o synthesis (Boden, supra 1996). At day 14, the media was switched to
BGJb+10% FBS, and
l OmM Beta Glycerol Phosphate.
Peptides: Peptides from Table I were synthesized as described above. Each
peptide was
coupled to a substrate at a concentration of 0.22M.
Competitive Binding Assays: Experimental and control peptides are added to a
25 suspension of 60,000 RCOB cells in serum free media, at a concentration of
0.05, 0.1 and 0.2
mM. Cells were incubated with soluble peptide for 45 minutes in a humidified
5% COz, 37°C
environment prior plating onto peptide-immobilized substrates or the
appropriate ECM protein.
All plated cells were maintained in F12 media with no serum. One or two hours
after the time of
initial plating, a cell count was completed for each of at least three wells.
Cell counting was
30 performed by rinsing several times with DMEM, and using the MTT assay (see
below) or by
fixing with formalin and performing and performing counts in ten different
high powered
microscopic fields on each individual substrate.
CA 02301064 2000-02-14
WO 99/01089 PCTNS98/13792
-27-
Cell Counting with MTT Assay: Standard curves were prepared by plating rat
calvarial
osteoblasts at densities of 100,000, 50,000, 25,000, 10,000 and 5,000
cells/well. Cells were
incubated in serum-free Dulbecco's Modified Eagie Media, i.e. DMEM {Gibco) for
two hours.
Then 3-[4.5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, i.e. MTT
(Sigma) in media
was added to a final concentration of 0.5 mg.ml. The plates were placed back
in the incubator
for three hours. Each well was then rinsed with Hanks' Balanced Salt and then
1 ml of 10%
Sodium Dodecyl Sulfate, i.e. SDS (Sigma) was added. Cells were covered in
aluminum foil to
protect it from light and left at room temperature for 12 hours. The SDS
solution was then
removed, placed into cuvettes, and examined in a Beckman DU-65
spectrophotometer at a
1o wavelength of 570nm.
For testing peptide-coated substrates, cells were plated at 50,000 cells per
well plates.
Attachment was evaluated at 20 minutes, 1 hour, 3 hours and 24 hours. At the
conclusion of
each time oint unattached cells were removed by rinsing three times with HBSS.
MTT in serum-
free media was added at a concentration of 0.5 mg/ml to perform cell counting
and incubator for
3 hours to allow the cells to process the MTT. The plates were then removed
and each well
given a single HBSS rinse followed by addition of 1 ml of 10% SDS for cell
lysis. After 12
hours the SDS solution was removed, placed into cuvettes, and examined in a
Beckman DU-65
spectrophotometer at a wavelength of 570nm.
Cell Morphology With Scanning Electron Microscopy: Substrates with cells were
treated
with 2% paraformaldehyde + 1% gluteralehyde for one hour. Then they were
rinsed in 1M PBS
and placed in 50%, 70%, 90% and 100% (twice) ethanol for ten minutes each to
dehydrate them.
The substrates were then immersed in 1:1 ethanol:hexamethyldisilazane (HMDS)
for 30 minutes,
and finally were treated with 100% HMDS for thirty minutes and allowed to dry.
Samples were
fixed onto SEM mounts using a conductive graphite adhesive and sputtered with
gold. SEM was
performed with a Hitachi 52700 scope.
Cell Proliferation Assay (~H Thymidine Incorporation): Primary rat calvarial
osteoblasts
were plated on substrates in 6-well plates at a density of 20,000 cells/cm2,
cultured in MEM
media (Gibco) + 10%FBS (Hyclone) for 2 days, rinsed with buffered saline, and
switched to
thymidine-free MEM (Gibco) + .2% BSA for 1 day (Kim, 1997). On day 4,
experimental
3o groups were exposed methyl 3H-thymidine (luCi/mmol; DuPont New England
Nuclear, Boston,
MA, USA) added 4 hours prior to harvest at 96 hours. Cells were harvested and
specific
radioactivity (cpm) measured using a scintillation counter. Briefly, cells
were washed 3 times
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-28-
with ice-cold PBS to remove excess label, trypsinized, spun down into a
pellet, and lysed with
120 ul phosphate buffered I % nonidet P-40 (Sigma). One hundred microliters of
each sample
were mixed with 800 ul 0.2% BSA, 100 ul 75% trichloroacetic acid (TCA), and
centrifuged.
Supernatant was removed and the pellet was centrifuged again with 1 ml 7.5%
TCA. The pellet
was then solubilized in 900 ml 0.1 N NaOH at 37°C overnight and
neutralized with 100 ul 1N
Hcl. Counts were normalized with DNA and expressed as cpm/ug DNA. All data was
normalized using total DNA. A fluorometric DNA assay (Arronow, 1990) was
performed on the
remaining 20 ul aliquots of cell lysate using a TKO 100 mini-fluorometer
(Hoefer, San
Francisco, CA, USA) to normalize cell counts. Samples were incubated with
benzimidazole
1o (Hoechst 33258; Pharmacia Biotech, Piscataway, NJ) and fluorescence
quantified. DNA content
was obtained using a calf thymus DNA (Pharmacia Biotech) standard curve.
Alkaline Phosphatase Activity: Alkaline phosphatase (AP) activity of cell
lysates was
determined by an established enzymatic conversion assay using p-nitrophenol
phosphate as a
substrate (Spiess). The enzyme activity was expressed as nanomoles of p-
nitrophenol produced
per minute per milligram of protein (nmol/min/mg protein). The protein content
was determined
using the Biorad protein assay kit (Biorad, Hercules, CA) using BSA as the
standard.
Alkaline Phosphatase Staining: Osteoblasts were fixed in buffered 2%
paraformaldehyde
for 24 hours. Before staining, cells were rinsed in distilled water. Alkaline
phosphatase staining
was visualized by incubating the cells for 30 min in 0.1 M Tris HCL pH 8.5
containing 0.4
mg/ml naphthol AS-MX phosphate + 1 mg/ml Fast Blue BB salt. Cells were then
rinsed in 1 M
PBS and preserved in PBS glycerol. The intensity of osteoblasts stained with
alkaline
phosphatase was qualitatively assessed by counting the number of osteoblasts
per l Ox field.
Osteocalcin RIA: Osteocalcin levels were assessed after removing aliquots of
conditioned media from cell cultures of experimental groups using a
radioimmunoassay (RIA)
for rat osteocalcin (rat osteocalcin kit, BTI, Stoughton, MA) according to a
previously described
method (Gundberg 1984). Purified rat osteocalcin, goat anti-rat osteocalcin
antibody, normal
goat nonimmune serum, donkey anti-goat 2nd antibody, RIA buffer and [I-125]
rat osteocalcin
were used as reagents for RIA as provided by BTI. The standards and the
samples were
incubated overnight with a known quantity of goat anti-rat osteocalcin
antibody followed by
another incubation with [I-125] rat osteocalcin (approx. 20,000 cpm). The
tubes were incubated
(2 hours) with donkey anti-goat 2nd antibody, centrifuged, and pellets counted
(cpm) using a
gamma counter. A standard curve was generated and sample concentration of
osteocalcin
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-29-
(ng/tube) obtained.
Extracellular Matrix Protein and Integrin Gene Expression by Northern
Analysis:
During the course of bone development and metabolism, a variety of osteoblast
growth and
differentiation factors are known to be expressed in vitro (Ibaraki, 1992) and
in vivo (Jingushi,
1991, Sandberg, 1993). Integrin gene expression is also modulated.
1. RNA extraction: RNAzoITM (Tel-Test, Friendswood, TX) reagent was added to
the cell cultures removed of media and then shaken gently until a viscous,
opaque liquid was
seen. The contents were transferred to ice cold tubes to which chloroform was
added and
vortexed. After centrifugation for 10 min. at 10,000 rpm, the top aqueous
phase was re-extracted
l0 with fresh RNAzoI and chloroform. After a series of re-extraction and
centrifugations, the cell
pellet was washed in 70% ethanol and resuspended in 50 ul TE buffer. The
concentrations and
purity of the RNA is measured with a spectrophotometer using the ratio of A'-
~° and A28°.
2. RNA gel: RNA ( 15-20 ug) from specific experimental groups was separated on
the basis of size with a denaturing 1.2% agarose (Formaldehyde) gel
electrophoresis. All RNA
gels were run for 3 hours at 100 V and photographed using an ultraviolet light
source. A
TurboBlot % kit was used to transfer the RNA from the gel to the nylon
(blotting) membrane.
The RNA was then cross linked and baked on permanently onto the membrane
(under a UV lamp
and baked at 120°C for 15 minutes).
3. Hybridization & Detection: cDNA probes for rat alkaline phosphatase and rat
osteocalcin were kindly provided by Dr. J. Lian (University of Massachusetts,
Worcester, MA).
The cDNA probes for a5 and X31 integrins were provided by Dr. E. Ruoslahti
(Cancer Institute,
La Jolla, CA). The cDNA probe for bone sialoprotein was provided by Dr. J.
Sodek (University
of Toronto, Toronto, Canada), while the cDNA probe for collagen was provided
by Dr. B. Kream
(University of Connecticut, New London, CT). The cDNA probes for GAPHDH, beta-
actin,
human osteopontin, and human osteonectin were obtained from the American Type
Culture
Collection (ATCC).
The membranes were treated with various buffers (SX SSPE, 50% formamide, 2%
SDS
and 10X Dengardt's solution). The hybridized probes were radioimmunodetected
through the
use of 32P. The membranes were then reacted with the cDNA probes and
hybridized with the
3o probes at 68° C overnight and washed through a series of (2X SCC &
0.1% SDS) solutions.
After the washing steps, the membrane were exposed with x-ray film. The mount
of RNA was
quantified through comparison with the known amount of RNA transcript that was
loaded in the
CA 02301064 2000-02-14
WO 99/01089 PCTNS98/13792
-30-
lane on the gel. The size of the RNA molecule was calculated by measuring the
distance
migrated and comparing it to the standard. All mRNA hybridization experiments
were
performed twice with each cDNA probe. All cDNA was normalized to GAPDH.
Results:
Cell Attachment
RCOBs in DMEM with 10% fetal bovine serum were plated at 25,000 per square
centimeter. Visual analysis revealed higher levels of attachment at 20 minutes
on the RGDC
treated substrates. This was quantitatively confirmed using an MTT assay which
showed that at
20 minutes there was 100% greater attachment to the RGDC surface compared with
gold and
RGD treated surfaces. Similar to the gold surface RCOBs showed much greater
attachment
when cultured on RGDC-FEP modified surfaces than on unmodified FEP.
Alkaline phosphatase Activity
Alkaline phosphatase activity revealed that RGDC coupled surfaces produced the
highest
levels of this enzyme. CGRARADSP (control peptide) and plain gold substrates
produced
values which were not significantly different from one another. The results
are shown in Figure
3
Osteocalcin mRNA Assay
At nine days Osteocalcin mRNA was heavily expressed on RGDC-gold coated
surfaces
but was not observed on Gold and CG-gold coated surfaces. After fourteen days
RGDC still
showed higher levels than the others, and at nineteen days all substrates were
virtually identical.
Additionally it was found that Osteocalcin mRNA expression was induced earlier
in cells which
were grown on the prosthetic implants of the invention having a peptide coated
gold surface than
on the polymeric prosthetic implants (FEP) coated with the identical peptide.
Osteocalcin is not
expressed at 9 days when cells are grown on the peptide coated polymer
surface. This finding
suggest that the gold surface is also important to the regulation of bone
morphogenesis.
Osteocalcin Protein Synthesis
It is also important to determine how immobilized proteins influence protein
synthesis.
CA 02301064 2000-02-14
WO 99/01089 PCTlUS98/13792
-31 -
Osteocalcin was evaluated since it is a marker of bone cell differentiation
and because
radioimmunoassays (RIA) are commercially available (Arono, 1990; Gundberg,
1984; Ibaraki.
1992).
After 14 days in culture, RGD-FEP coupled membranes induced significantly
higher
levels of osteocalcin synthesis compared with all other groups. The unique
ability of RGD-FEP
coated substrates to enhance osteocalcin synthesis is consistent with
increased RCOB mRNA 1
expression seen at day 14. The RGE is closest to RGD in stimulating
osteocalcin synthesis.
RAD peptide was similar to OH and TCP.
to ECM and Integrin Gene Expression for Sub-Cultivated RCOBs
Evaluation of matrix protein gene expression provides a quantitative method of
assessing
cell differentiation in a temporal fashion. The normal pattern of RCOB gene
expression has been
reported for primary and subcultured cells (Aronow, 1990; Breen, 1994; Lynch,
1995 and
others). It has been demonstrated that subcultured RCOB display a "right-
shifted" pattern of
gene expression compared to primary cells.
Alkaline Phosphatase
On day 9 Alkaline Phosphatase gene expression was observed on all substrates
at
minimal levels but was significantly higher on RGDC coated gold surfaces. No
change over the
time period studied in Alkaline Phosphatase levels was observed in the cells
cultured on FEP
surfaces.
Bone sialoprotein
Similar to Alkaline Phosphatase gene expression, bone sialoprotein gene
expression was
much higher on RGDC coated gold surfaces than on gold surfaces alone or gold
surfaces coated
with a control peptide. Bone sialoprotein gene expression was not observed in
cells cultured on
FEP surfaces.
(3, integrin
(3, integrin gene expression was observed on all substrates at minimal levels
but was
3o significantly higher on RGDC coated gold surfaces. By the 14th day of
culture, the mRNA
signal detected from cells cultured on RGDC coated gold surfaces had shifted
from one band to
two bands. This shift to two bands was not detected in RNA isolated from celis
cultured on any
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-32-
of the other surfaces.
as integrin
as integrin gene expression was observed in cells cultured on RGDC coated gold
surfaces
but was not detected in cells cultured on any other surfaces. Similar to (3,
integrin, the expression
pattern of a5 integrin was observed to shift on day 14 from a single band to a
double band.
Example 3: Pe tide odified Surfaces Sunnort Focal Adhesion Formation
The cytoplasmic domains of integrins are relatively short (approximately 50
amino
to acids), but are sufficiently long enough to interact with cytoskeletal
proteins in focal contacts (or
focal adhesions or adhesion plaques). Focal adhesions are connected to the
nucleus via
microspikes or bundles of actin filaments. Several experiments provide strong
evidence for these
connections between the exterior and interior of the cell. In fluorescence
photobleaching,
integrins were fluorescently labeled, then overexposed to form a bleached
spot. This bleached
area did not move, showing the restricted mobility of integrins in focal
contacts (Duband, 1986).
Solowska ( 1989) showed that expression of a mutant form of avian integrin
beta 1 subunit
lacking the cytoplasmic domain produces hybrid heterodimers which, while
efficiently exported
to the cell surface and still capable of binding fibronectin, do not localize
efficiently in focal
adhesions. This further implicates the cytoplasmic domain of the beta 1
subunit in interactions
2o required for cytoskeletal organization.
The cytoskeletal proteins present in focal adhesions are well-defined:
vinculin, talin, and
alpha actinin serve as links between integrins and the bundles of actin
filaments (stress fibers) of
the cytoskeleton. Evidence in the literature suggests that focal adhesions are
required for signal
transduction from the ECM to the nucleus of the cell. Upon integrin-mediated
adhesion to ECM
proteins, focal adhesion kinase {FAK), a tyrosine kinase, becomes
phosphorylated (Schneider,
1994). Activation of FAK is believed to initiate a signaling pathway to the
nucleus, resulting in
changes in gene expression.
Both fibronectin and type I collagen are present in the extracellular matrix.
We tested our
monolayer surface of active peptides to determine wather it can stimulate
focal adhesion
3o formation in the absence of serum in a similar manner to the interaction
between the cell and the
related parent protein of the peptide sequence. In order to evaluate the
influence of active
portions of these parent proteins on osteoblast cell response in short time
frames we modified
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-33-
gold coated coverslips with the fibronectin related peptide: RGDC or the
collagen related
peptide: DGEAGC, and evaluated the ability of these surfaces to support focal
adhesion
formation at two time points, 3 and 24 hours, under serum free conditions.
Methods:
FibronectinlRGDC Study: Experimental groups included RGDC, RADC, fibronectin
adsorbed to gold, plain gold, and plain glass surfaces. Gold substrates were
manufactured by
evaporating 80 angstroms of titanium to 12 mm glass coverslips (Fisher),
followed by a 500
angstrom layer of gold. To couple cysteine terminated peptides to the gold
substrates, a 0.22 mM
1o solution of the desired peptide was solubilized in a 1:1 mixture of
distilled water and ethanol.
These substrates were then incubated overnight. Plain gold controls were
exposed to ethanol and
distilled water as well. Fibronectin substrates were produced by incubating
gold coverslips with
pg/ml of fibronectin (Collaborative Biomedical Products, Bedford, MA) for 60
minutes,
followed by 10 mg/ml bovine serum albumin (BSA) (Sigma, St. Louis, MO) for 30
minutes to
cover any non-specific binding sites. All coverslips were then washed three
times in HBSS to
remove any non-adsorbed protein (Puleo, 1991 ). Primary rat calvarial
osteoblasts were isolated
according to protocol and seeded for periods of 3 or 24 hours in serum free or
serum conditions.
At each time point cells were rinsed in warm PBS, fixed in 3.7 %
paraformaldehyde for 30
minutes, and rinsed several times with HBSS. Vinculin and actin were labeled
via the following
2o protocol: nonspecific sites were blocked in S% BSA for 30 minutes, cells
were then
permeabilized with 0.2 % Triton X-100 (Fisher) for 10 minutes, incubated in a
1:50 mouse anti-
human vinculin antibody solution (Sigma St. Louis, MO}, blocked for 30
minutes, and incubated
with a anti-mouse rhodamine secondary antibody (1:50) and FITC conjugated
phalloidin
(Molecular Probes).
Type 1 CollagenlDGEAGC: Experimental groups included DGEAGC and rat tail type
I collagen adsorbed to gold, plain gold, and plain glass surfaces. Gold
substrates were
manufactured by evaporating 80 angstroms of titanium to 12 mm glass coverslips
(Fisher),
followed by a 500 angstrom layer of gold. To couple cysteine terminated
peptides to the gold
substrates, a 0.22 mM solution of the desired peptide was solubilized in a 1:1
mixture of distilled
water and ethanol. These substrates were then incubated overnight. Plain gold
controls were
exposed to ethanol and distilled water as well. Type I collagen substrates
were produced by
incubating gold coverslips with 10 ~,g/ml of collagen (Collaborative
Biomedical Products,
CA 02301064 2000-02-14
WO 99101089 PCT/US98/13792
-34-
Bedford, MA) for 60 minutes, followed by 10 mg/ml bovine serum albumin (BSA)
(Sigma, St.
Louis, MO) for 30 minutes to cover any non-specific binding sites. All
coverslips were then
washed three times in HBSS to remove any non-adsorbed protein (Puleo, 1991).
Primary rat
calvarial osteoblasts were isolated according to protocol and seeded for
periods of 3 or 24 hours
in serum free or serum conditions. At each time point cells were rinsed in
warm PBS, fixed in
3.7 % paraformaldehyde for 30 minutes, and rinsed several times with HBSS.
Vinculin and actin
were labeled via the following protocol: nonspecific sites were blocked in 5%
BSA for 30
minutes, cells were then permeabilized with 0.2 % Triton X-100 (Fisher) for 10
minutes,
incubated in a 1:50 mouse anti-human vinculin antibody solution (Sigma St.
Louis, MO),
1o blocked for 30 minutes, and incubated with an anti-mouse rhodamine
secondary antibody (1:50)
and FITC conjugated phalloidin (Molecular Probes).
Results:
At three hours, vinculin staining revealed the ability of RGDC peptide
modified surfaces
to support focal adhesion formation in the absence of serum. Fibronectin
coated surfaces also
supported focal adhesion formation. Cells on both surfaces tended to have
vinculin staining
located at the cell periphery in the form of distinct plaques at the cell
tips. No vinculin staining
was observed on cells plated on RADC, glass or plain gold.
At three hours, vinculin staining revealed the ability of DGEAGC peptide
modified
2o surfaces to support focal adhesion formation in the absence of serum.
Collagen coated surfaces
also supported focal adhesion formation. Cells on both surfaces tended to have
the brightest
vinculin staining located at the cell periphery in the form of either distinct
plaques or groups of
strands at the cell tips. No vinculin staining was observed on cells plated on
glass or plain gold.
Example 4: evaluation of the Effect of Immobilized Peptides on Osteobla.st
Differentiation
We have shown above that in vitro, peptide modified surfaces can influence
short and
long term cell responses like attachment, shape and function. We also
conducted a study to
evaluate the amount of bone formed in response to gold coated titanium rods
modified with the
peptide sequence Arg-Gly-Asp-Cys (RGDC). Titanium rods coated with gold, FEP
rods and
3o uncoated titanium rods were implanted bilaterally into the distal medial
femoral condyle of adult
rats and evaluated at 2, 4, 8, and 24 weeks post-implantation. The experiments
are discussed
below.
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-35-
In viva
Modification and Characterization of Peptide-grafted FEP Rods and Titanium:
Titanium
rods were generously donated by Osteonics Corporation (NJ, USA). Rods were
cleaned
according to ASTM standards before coating them with a 500 layer of gold using
an electron
beam evaporator. Rods were immersed in a 0.22 M solution ( 1:1 ethanol: water)
of RGDC
(American Peptide Company, Sunnyvale, CA) overnight at room temperature and
stored in
sterile PBS, using the techniques described above, until the time of surgery.
FEP rods were
coupled with peptides using the techniques described above. Un-coated titanium
rods are used as
a control. Briefly, the materials were initially cleaned in a radio frequency
glow discharge
1 o chamber using a flow-through system with an Argon atmosphere. The alloys
were immediately
transferred to a nitric acid bath for 30 min in order to passivate the surface
according to ASTM
standards (Puelo 1994). The samples were transferred to a gold evaporation
chamber and reacted
with peptides as described above. Characterization of gold coated titanium
materials, FEP and
titanium materials were performed as described above.
Peptide-coated Materials and uncoated Titanium Implanted in Rat Femur Sites:
Quantitative histornorphometric analysis and pull-out biomechanical testing
was
conducted at 2 and 4 weeks on implants inserted bilaterally into the femoral
canal of 20 adult
Sprague Dawley rats. Parameters evaluated included the area and thickness of
new bone formed
around the implants, the percent of the implant covered by new bone, and the
interfacial shear
strength at the bone/implant interface. The distal rat femur provides a well-
studied site for bone
material interactions and offers a sufficient bony area to implant small
specimens. Adult
Sprague Dawley rats weighed an average of 415 t 12 g at the time of surgery.
The rats were
anesthetized using a 0.5 ml intraperitoneal injection of Nembutal and 0.1 ml
of Cefazolin was
injected intramuscularly at the surgical site. Reaming of the distal end of
the femoral canal was
done first by inserting an 18 gauge needle down the femoral shaft, followed by
irrigation of the
femur with sterile saline, reaming with a 1.5 mm drill bit using a hand held
drill to prevent
thermal necrosis, irrigation, reaming with a 16 gauge needle, irrigation, and
final reaming of the
outer cortex with a 14 gauge needle. The rod was then press fit into place
with the outermost end
below the condylar surface, in each case. RGDC coated rods were placed at
random with one
3o control rod and one experimental rod being placed bilaterally in each
animal. Lateral and
anterior-posterior X-rays were taken postoperatively to assess rod position.
The fascia and skin
CA 02301064 2000-02-14
WO 99/01089 PCT/IJS98/13792
-36-
are closed in standard fashion using 5-1 vicryl bioresorbable sutures.
Histological evaluation: After mechanical testing, femurs were removed from
the dental
plaster and stored in phosphorous buffered saline for 24 hours until fixation
in 3.7%
paraformaldehyde for 48 hours at room temperature. Decalcification was
performed according to
a method described by Frank, et al, (1993). Briefly, bones were allowed to
demineralize over the
course of 2 weeks in 15% formic acid solution at 4 uC. Bones were rinsed and
permbealized in
6.8% sucrose/PBS solution overnight. Dehydration of bones was conducted as
follows: 20
minutes per ethanol concentration: 70, 80, 90, 95%. Bones were sectioned from
the growth plate
at 2, 5, 8, 12, and 15 mm and embedded in Historesin (Leica, Germany) for
histological analysis.
Briefly, bones were infiltrated for 48 hours at 4 uC and then embedded
overnight. 5 ~m sections
were made. Specimens were stained using Hematoxylin and Eosin and Gomori(~s
trichrome
stains. Quantitative histomorphometrical analysis was conducted on bone cross
sections
sectioned at 5mm using IP Lab Software. Images of bone cross sections were
captured by
microscope and imported into a computer via a CCD camera. Parameters measured
by two
independent observers included the perimeter of new bone formed, the area of
new bone, the
perimeter availabe for new bone formation and the diameter of the hole where
the implant was.
Some sections were not analyzed due to histological sectioning tears. Also, if
the amount of new
bone formed around the implant was not clear (e.g. implant abutting cortex)
that portion of the
cross section was not included for analysis.
Biomechanical Pull-out Testing: The biomechanical pull-out strength between
the
bone/RGDC and bone/Au was measured using the widely imployed pull-out test
(Chae et al in
1992 and Tisdel et al in 1994 , Branemark & Berzin~ All testing was performed
in a blinded
fashion.
Animals were sacrificed at 2 and 4 weeks postoperatively. Animals were first
anesthetized with a 0.5 ml intraperitoneal injection of Nembutal and then
sacrificed by a 0.5 ml
intracardial injection of Nembutal. Mechanical testing of all femurs was
conducted the same day
as sacrifice. Immediately after explantation, femurs were cleaned of all soft
tissue, x-rayed and
prepared for mechanical testing or histological evaluation. A total of 23
animals were evaluated.
Three animals were excluded, one because of death during surgery, and two
because of
3o pathologic fractures. For mechanical testing at 2 weeks, 7 animals were
assessed while 8 animals
were assessed at 4 weeks. Histological evaluations were carried out on 8
femurs used for
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-37-
mechanical testing at each time point. An alignment jig was designed in order
to insure a pure
tensile force was applied to the rod. Dental plaster was used to hold the
proximal portion of the
femur in place during testing. Modified needle-drivers gripped the end of the
implant as it was
pulled from the bone at a crosshead speed of 5 mm/min. The force required to
break the interface
was recorded and the portion of the implant estimated to be contact bone was
also recorded.
Results:
The in vivo studies involving implantation of FEP coated and uncoated
materials indicate
that implants coated with a bioactive molecule such as RGDC have accelerated
or enhanced bone
1o ingrowth. Briefly, the peptide coated implants demonstrated a significantly
greater percentage of
their surface perimeter covered with bone. Additionally the biomechanical pull-
out strength was
significantly greater for the peptide coated implants versus the uncoated
implants.
By 4 weeks the average pull-out force of peptide modified rods was 38% greater
than
gold control rods although this difference was not statistically significant
(Table 1). Furthermore,
at 4 weeks there was significantly (P < 0.01 ) more new bone area formed
around RGDC implants
and the thickness of this new bone formed around RGDC implants differed
significantly (P <
0.01 ) from Au controls at both 2 weeks (26.2 microns t 1.9 vs. 20.5 microns t
2.9) and 4 weeks
(32.7 microns t 4.6 vs. 22.6 microns t 4.0)
Biomechanical: No statistical differences were found between peptide modified
and gold
control rods for the interfacial shear strength at 2 and 4 weeks respectively.
It should be noted
however, that the mean of the peptide modified group at 4 weeks was 38% higher
than the
control group (Table 1 ).
Table 1
Interfacial hear Stren
th a
2 weeks Postimplantation4 weeks Postimplantation
Gold coated rods 0.17 t 0.09 0.13 t 0.06
RGDC modified rods 0.16 t 0.06 0.18 t 0.07
Histology: Although there were not a significant differences in the pull-out
forces
between groups, there were significant differences in the amount of bone
(thickness and area)
formed around the implants at two and four weeks. There were no differences in
the percent of
CA 02301064 2000-02-14
WO 99/01089 PCTNS98/13792
-38-
the implant cross section covered by bone (76 t 14%, 74 t 5%) for the RGDC and
Au groups
respectively. At four weeks more of the implant was covered by bone but the
percent of the
implant cross section covered by bone for the RGDC and Au groups did not
differ significantly
(92 t 4 % vs. 90 t 7 %). The area of new bone formed around RGDC implants was
not
significantly more compared to Au controls at 2 weeks (0.108 pmt t 0.026 vs.
0.082 ~m2 t
0.017), but by 4 weeks there was a significantly (P < 0.01 ) more area of new
bone formed around
RGDC implants (0.16 ~.mz t 0.016 vs. 0.108 urn2 t 0.023). The thickness of new
bone formed
around RGDC implants differed significantly (P < 0.01 ) from Au controls at
both 2 weeks (26.2
pm t 1.9 vs. 20.5 ~m t 2.9) and 4 weeks (32.7 ~,m t 4.6 vs. 22.6 pm t 4.0).
1o Exam,~jg~: Peptides act syner~istica ~v to increase bone cell
responsiveness.
The response of bone cells to peptide combinations showing synergy or high
individual
levels of activity is evaluated in vitro and in vivo using methods described
above with
combinations of peptides rather than a single peptide. Two non-adjacent
peptide sequences from
fibronectin, including RGD and PHSRN, a so-called synergy sequence, exhibit
synergistic
behavior (Aota 1994; Akiyama, 1995). These experiments can be used to
identify, track and
quantify different peptides on the same membrane.
References
1. Aota, S., et al. (1994), "The short amino acid sequence Pro-His-Ser-Arg-Asn
in human
fibronectin enhances cell-adhesive function," J. Biol. Chem. 269(40):24756-61.
2o 2. Akiyama, S.K., et al. (1995), "Function and Receptor Specificity of a
Minimal 20
Kiladalton Cell Adhesive Fragment of Fibronectin," Cell Adhesion and
Communication
3:13-25.
3. Akiyama, S.K., et al. (1995), "Fibronectin and integrins in invasion and
metastasis,"
Cancer Metastasis Rev. 14(3):173-189.
4. Aronow, M.A., et al. (1990), "Factors that Promote Aggressive Development
of the
Osteoblast Phenotype in Cultured Fetal Rat Calvaria Cells," Journal of
Cellular
Physiology 143:213-221.
5. Bartfeld, N.S., et al. (1993), "The v3 Integrin Associates with a 190-kDa
Protein That is
Phosphorylated on Tyrosine in Response to Platelet-Derived Growth Factor,"
Journal of
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-39-
Biological Chemistry 268(23): 17270-17276.
6. Bassuk, J.A., et al. (1993), "Molecular analysis of chicken embryo SPARC
(osteonectin)," Eur. J. Biochem 218(1):117-127.
7. Bautista, D.S., et al. (1994), "Inhibition of Arg-Gly-Asp (RGD)-mediated
Cell Adhesion
to Osteopontin by a Monoclonal Antibody against Osteopontin," Journal
ofBiological
Chemistry 269(37):23280-23285.
8. Beer, J.H., et al. (1992), "Immobilized Arg-Gly-Asp (RGD) peptides of
varying lengths
as structural probes of the platelet glycoprotein Iib/IIIa receptor," Blood
79:117-128.
9. Bengston, A., et al. "Anaphylatoxin release in association with
methylmethacryl fixation
of hip prostheses," JBJS 69; 46 ( 1987).
10. Boden, S.D. (1996), "Differential Effects and Glucocorticoid Potentiation
of Bone
Morphogenetic Protein Action During Rat Osteoblast Differentiation in vitro,"
Endocrinology 137(8):3401-3407.
11. Boudreau, N., et al. (1994), "From laminin to lamin: regulation of tissue-
specific gene
expression by the ECM," Forum.
12. Breen, E.C., et al. (1994), "TGFB Alters Growth and Differentiation
Related Gene
Expression in Proliferating Osteoblasts in vitro, Preventing Development of
the Mature
Bone Phenotype," .I. of Cellular Physiology 160:323-335.
13. Briggs, D., "Handbook of x-ray and photoelectron spectroscopy," London:
Heyden and
Sons (1977).
14. Brighton, C.T., et al. ( 1992), "Identification of Integrin Cell-
Substratum Adhesion
Receptors on Cultured Rat Bone Cells," J. Orthopaedic Research 10:766-773.
15. Callen, B.W., et al. (1993), "Behavior of primary bone cells on
characterized polystyrene
surfaces," Journal of Biomedical Materials Research 27:851-859.
16. Cameron, H.U. (1986), "Six year results with a microporous-coated metal
hip
prosthesis," Clin. Ortho. 208:81-83.
17. Cardarelli, P.M., et al. (1992), "The Collagen Receptor 21, from MG-63 and
HT1080
CA 02301064 2000-02-14
WO 99/01089 PCT/IJS98/13792
-40-
Cells, Interacts with a Cyclic RGD Peptide," Journal of Biological Chemistry
267(32):23159-23164.
18. Cheng, S.L., et al. (1995), "Expression and Regulation of Integrins During
the
Differentiation of Normal Human Osteoblasts and Human Bone Marrow Stromal
Cells,"
17th Annual Meeting of the American Society for Bone and Mineral Research
(T223).
19. Chen, S., et al. (1994), "Design and Synthesis of Novel Cyclic RGD
Containing Peptides
as Highly Potent and Selective Integrin aiibB3 Antagonists," J. of Medicinal
Chemistry
37(1).
20. Cheresh, D.A. (cd.), et al. (1994), Integrins: Molecular and Biological
Response~to the
Extracellular Matrix, Academic Press, Inc., San Diego, CA.
21. Chorev, M., et al. (1995), "Approach to discovering novel therapeutic
agents for
osteoporosis based on integrin receptor blockade," Biopolymers 37(6):367-375.
22. Clover, J., et al. (1992), "Integrin subunit expression by human
osteoblasts and
osteoclasts in situ and in culture," J. Cell Sci. 103:267-271.
23. Collins, D.N., et al., "Porous-coated anatomic total knee arthroplasty: a
prospect analysis
comparing cemented and cementless fixation," Clin. Orth., 267; 128 (1991).
24. Collier, J.P., et al. "Macroscopic and microscopic evidence of prosthetic
fixation with
porous-coated materials," In Instructional Course Lecture, The American
Academy of
Orthopaedic Surgeons, 40; 97-99 (1991).
25. Craig, W.S., et al. (1995), "Concept and Progress in the Development of
RGD-
Containing Peptide Pharmaceuticals," Biopolymers 37:157-175.
26. Danilov, Y.N., et al. (1989), "Arg-Gly-Asp)n-Albumin Conjugates as a Model
Substratum for Integrin-Mediated Cell Adhesion," Experimental Cell Research
182:186-
196.
27. Davies, J.E. (ed.) (1991), The Bone-Biomateria~Inter~f~e, University of
Toronto Press,
Toronto, Ont.
28. Dedhar, S. (1989a), "Regulation of expression of the cell adhesion
receptors, integrins, by
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-41 -
recombinant human interleukin-1 in human osteosarcoma cells. Inhibition of
cell
proliferation and stimulation of alkaline phosphatase activity," J. Cell.
Physiol. 138:291-
299.
29. Dedhar, S. (1989b), "Signal transduction via the beta-1 integrins is a
required
intermediate in interleukin-1 induction of alkaline phosphatase activity in
human
osteosarcoma cells," Exp. Cell Res. 183:207-214.
30. Dee, K.C., et al. (1996), "Conditions which promote mineralization at the
bone-implant
interface: a model in vitro study," Biomaterials 17:209-215.
31. Dieckgraefe, B.K. ( 1996), "Immunolocalization of alpha-integrin subunits
and extra
cellular matrix components during human colonic organogenesis,"
Gastroenterolo~
110( 1 ):58-71.
32. Dresner-Pollak, et al. ( 1994), "Blockade of Osteoclast-Mediated Bone
Resorption
Through Occupancy of the Integrin Receptor: A Potential Approach to the
Therapy of
Osteoporosis," Journal of Cellular Biochemistry 56:323-330.
33. Ducheyne, P., et al. "Influence of a functional dynamic loading on bone
ingrowth into
surface pores of orthopedic implants," J. Biomed. Mater. Res. 11; 811-8,,
1974.
34. Duschl, C., et al. ( 1994), "Biological Addressable Monolayer Systems
Formed by
Templates of Sulfur-Bearing Molecules," Biophysical Journal 67:1229-1237.
35. Ekblom, P., et al. (1991), "Laminin isoforms and their receptors in the
developing
kidney," Am-J Kidney-Dis. 17(6):603-5.
36. Frenette, P.S., et al. (1996), "Molecular Medicine: Adhesion Molecules -
Part I," New
England Journal ofMedicine 334(23):1526-1529.
37. Freshney, R.I. (1987), Culture of Animal Cells: A Manual of Basic
Techni~~, Wiley-
Liss, New York, NY.
38. Friedman, R.J., et al. (1993), "Current concepts in orthopaedic
biomaterials and implant
fixation," J. Bone Joint Surg. 75A:1086-1109.
39. Frost, R.G. (1981), "Covalent immobilization of proteins to N-
hydroxysuccinimide ester
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-42-
derivatives of agarose. Effect of protein charge on immobilization," Biochem-
Biophys-
Acta 670(2):163-169.
40. Galante, J.O., et al. "The biologic effects of implant materials," J.
Orth. Res. 9; 760,
1991.
41. Gehsen, K., et al. (1988), "Inhibition of in vitro tumor cell invasions by
Arg-Gly-Asp- -
containing synthetic peptides," J. Cell. Biol. 106:925-930.
42. Genovese, C., et al. ( 1994), "Construction of DNA sequences complementary
to rat a,
and a~ collagen mRNA and their use in studying the regulation of type I
collagen
synthesis by 1,25-di-hydroxyvitamin D.", Biochemistry 23:6216-6221.
to 43. Giancotti, F.G., et al. (1990), "Elevated levels of the as~i,
fibronectin receptor suppress
the transformed phenotype of Chinese hamster ovary cells," Cell 60:849-859.
44. Ginsberg, M., et al. (1988), "Cytokines, integrins, and platelets,"
Thromb. Haemost. 59:1-
6.
45. Ginsberg, M., et al. (1985), "Inhibition of fibronectin binding to
platelets by proteolytic
fragments and synthetic peptides which support fibroblast adhesion," Journal
of
Biological Chemistry 260(7):3931-3936.
46. Glass, J.R., et al. (1996), "Characterization of a hyaluronic acid-Arg-Gly-
Asp peptide cell
attachment matrix," Biomaterials 17:1101-1108.
47. Gohel, A., et al. (1993), "Involvement of integrins in osteocyte
formation: Modulation by
glucocorticoids and insulin-like growth factor I," Molec. Biol. Cell 4:292a.
48. Good, R.J., "Contact angles and surface free energy of solid," New York:
Plenum Press,
1979.
49. Graf, J., et al. (1987), "Identification of an Amino Acid Sequence in
Laminin Mediating
Cell Attachment, Chemotaxis, and Receptor Binding," Cell 48:989-996.
50. Gronowicz, G., (1995a), "Integrin Regulation of c-fos Expression in
Osteoblasts," 17th
Annual Meeting of the American Society for Bone and Mineral Research (T234).
51. Gronowicz, G., et al. ( 1995b), "Glucocorticoids Inhibit the Attachment of
Osteoblasts to
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
- 43 -
Bone Extracellular Matrix Proteins and Decrease B 1 Integrin Levels,"
Endocrinology
136(2):598-608.
52. Gronowicz, G.A., et al. (1994), "Synthetic peptide containing Arg-Gly-Asp
inhibits bone
formation and resorption in a mineralizing organ culture system feral rat
parietal bones,"
J. Bone Mineral Res. 9(2):193-201. ,
53. Grzesik, W., et al. (1994), "Bone matrix RGD glycoproteins:
immunolocalization and
interaction with human primary osteoblastic bone cells in vitro," J. Bone
Mineral
Research 9{4):487-496.
54. Grzesik, W., et al. (1995), "Interaction of Human Bone Cells with
Synthetic Peptides
to Derived from Bone Salioprotein Differing in their Spatial Conformation,"
17th Annual
Meeting of the American Society for Bone and Mineral Research (M268).
55. Gundberg, C.M., et al. (1984), "Osteocalcin isolation, characterization
and detection,"
Methods Enrymol. 107:517-544.
56. Haddad, R.J., et al. "Current concepts review: Biological fixation of
porous-coated
implants," J. Bone and Joint Surg., 69-A; 1459-1466, Dec. 1987.
57. Han, LL, et al. (1994), "A Study of the reproducibility of the MTT test,"
Journal of
Materials Science: Materials in Medicine 5:154-159.
58. Hart, L, et al. (1995), Cell Adhesion and Ca_n_cer, Cold Spring Harbor
Laboratory Press:
Plainview.
59. Healy, K.E., et al. ( 1996), "Kinetics of bone cell organization and
mineralization on
materials with patterned surface chemistry," Biomaterials 17(2):195-208.
60. Hearn, M.T.W. (1987), "1,1-Carbonyldiimidazole-Mediated Immobilization of
Enzymes
and Affinity Ligands," Methods in Enzymology 135:102-117.
61. Hirano, Y., et al. (1991), "Synthesis and cell attachment activity of
bioactive
oligopeptides: RGD, RGDS, RGDV, RGDT," J. Biomedical Materials Research
25:1523-1534.
62. Hoffman, A.S. (1992), "Molecular engineering of biomaterials in the 1990's
and beyond:
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-44-
a growing liaison of polymers with molecular biology," Artificial Organs
16(1):43-49.
63. _ Horowitz, S.M., et al., "Study of the mechanism by which mechanic
failure of
methylmethacrylate leads to bone resorption," JBJS 75A; 802, June 1993.
64. Howlett, R., et al. ( 1994), "Mechanism of initial attachment of cells
derived from human
bone to commonly used prosthetic materials during cell culture," Biomaterials
15(3):213-
222.
65. Hughes, D.E., et al. (1993), "Integrin expression in human bone," J. Bone
Mineral
Research 8(5):527-533.
66. Humphries, M.J., et al. (1986), "A synthetic peptide from fibronectin
inhibits
experimental metastasis of murine melanoma cells," Science 233:467-470.
67. Hynes, R.O. (1992), "Integrins: Versatility, Modulation, and Signalling in
Cell
Adhesion," Cell 69:11-25.
68. Hynes, R.O. ( 1987), "Integrins: a family of cell surface receptors," Cell
48:549-54.
69. Ibaraki, K., et al. ( 1992), "Bone matrix mRNA expression in
differentiating fetal bovine
osteoblasts," Journal of Bone and Mineral Research 7(7):743-754.
70. Ito, Y., et al. ( 1991 ), "Materials for enhancing cell adhesion by
immobilization of cell-
adhesive peptide," Journal of Biomedical Materials Research 25:1325-1337.
71. Iwamoto, Y., et al. (1988), "YIGSR, a synthetic laminin pentapeptide,
inhibits
experimental metastasis formation," Science 238:1132-34.
72. Jingushi, S., et al. (1991), "Biological cascades of fracture healing as
modules for bone-
biomaterial interfacial reactions," The Bone-Biomaterial Interface, Ed.
Davies, J.E.,
University of Toronto Press, Toronto, 250-262.
73. Jones, H.C., et al., "Cement Disease," Clin. Orth. 225:192, 1987.
74. Juliano, R.L., et al. (1993), "Signal Transduction from the Extracellular
Matrix," Journal
of Cell Biology 120{3):577-585.
75. Kaelble, D.H. (1979), "Dispersion-Polar Surface Tension Properties of
Organic Solids,"
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
- 45 -
Journal of Adhesion 2:66-81.
76. _ Kaelble, D.H., et al. (1974), "Interfacial Bonding and Environmental
Stability of Polymer
Matrix Composites," Journal of Adhesion 6:23-48.
77. Kallos, T., et al. "Intramedullary pressure and pulmonary embolization of
femoral
medullary contents in dogs during insertion of bone cement and a prosthesis,"
JBJS 56
1363, 1974.
78. Kim, H.D., et al. ( 1997), "Human Osteoblast Response in vitro to PDGF and
TGF-~i
Delivered from Controlled Release Polymer Rods," Biomaterials (in press).
79. Lane, T.F., et al. ( 1992), "Regulation of Gene Expression by SPARC during
Angiogenesis in vitro: Changes in Fibronectin, Thrombospondin-1, and
Plasminogen
Activator-Inhibitor-1," Journal of Biol. Chem. 267(23):16736-16745.
80. Lane, T.F., et al. (1994), "SPARC is a source of copper-binding peptides
that stimulate
angiogenesis," J. Cell Biol. 125(4):929-43.
81. Langer, R., et al. (1990), "Future directions in biomaterials,"
Biomaterials 11:738-744.
82. Langer, R., et al. (1993), "Tissue Engineering," Science 260:920-926.
83. Lian, J., et al. (1989), "Structure of the rat osteocalcin gene and
regulation of vitamin D-
dependent expression," PNAS 86:1143-1147.
84. Lin, H., et al. (1994), "Synthesis, surface, and cell-adhesion properties
of polyurethanes
containing covalently grafter RGD-peptides," Journal of Biomedical Materials
Research
28:329-342.
85. Lynch, M.P., et al. (1995), "The Influence of Type I Collagen on the
Development and
Maintenance of the Osteoblast Phenotype in Primary and Passaged Rat Calvarial
Osteoblasts: Modification of Expression of Genes Supporting Cell Growth,
Adhesion,
and Extracellular Matrix Mineralization," Experimental Cell Research 216:35-
45.
86. Makohliso, S.A, et la. (1993), "The magnitude and polarity of a
fluoroethylene propylene
electrode substrate charge influence neurite outgrowth in vitro," J. Biomed.
Mater. Res.
27(8):1075-85.
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-46-
87. Maquart, F.X., et al. (1993), "In vivo stimulation of connective tissue
accumulation by the
tripeptide-copper complex glycyl-L-histidyl-L-lysine-Cu2+ in rat experimental
wounds,"
J. Clin. Invest. 92(5):2368-76.
88. Massia, S.P., et al. (1990a), "Covalent surface immobilization of Arg-Gly-
Asp and Tyr-
Ile-Gly-Ser-Arg-containing peptides to obtain well-defined cell-adhesive
substrates,"
Anal. Biochem. 187:292-301.
89. Massia, S.P., et al. ( 1990b), "Covalently attached GRGD on polymer
surfaces promotes
biospecific adhesion of mammalian cells," Annals of the New York Academy of
Sciences
589:261.
l0 90. Matsuda, T., et al. (1987), "The in vitro response of osteoblasts to
bioactive glass,"
Biomaterials 8:275-284.
91. Meredith, J.E., et al. {I996), "The Regulation of Growth and Intracellular
Signalling by
Integrins," Endocrine Reviews 17(3):207-213.
92. Morgan, H., et al. ( 1992), "A surface plasmon resonance immunosensor
based on the
streptavidin-biotin complex," Biosensors and Bioelectronics 7:405-410.
93. Newens, A.F., et al., "Severe hypotension during prosthetic hip surgery
with acrylic bone
cement Anesthes.," 36; 298, 1972.
94. Noda, M., et al. (1987), "cDNA cloning of alkaline phosphatase from rat-
osteosarcoma
(ROS 17/2.8) cells," J. Bone Min. Res: 2:161-164.
95. Oldberg, A., et al. {1988), "The primary structure of cell-binding
salioprotein," J. Biol.
Chem 263:19430-19432.
96. Oldberg, A., et al. (1986), "Cloning and sequence analysis of rat bone
salioprotein
(osteopontin) cDNA reveals an Arg-Gly-Asp cell-binding sequence," Proc. Natl.
Acad.
Sci., USA 83(23):8819-8823.
97. Patterson, R., et al. (1995), "Effects of radio frequency glow discharge
and oligopeptides
on the attachment of human endothelial cells to polyurethane," ASAIO J.,
41:234-252.
98. Peel, S.A.F., et al. (1992), "Polymer surface modification causes changes
in phenotypic
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-47-
expression of primary bone cells," Mat. Res. Soc. Symp. Proc. 252:71-77.
99. _ Pesakova, V., et al. (1995), "Effect of the tripeptide glycyl-L-histidyl-
L-lysine on the
proliferation and synthetic activity of chick embryo chondrocytes,"
Biomaterials
16(12):911-91 S.
100. Pettit, L.D., et al. ( 1992), "The coordination of copper(II) to 1-
hydroxy-4-(glycyl-
histidyl-lysine)-anthraquinone; a synthetic model of anthraquinone anti-cancer
drugs," J.
Inorg. Biochem. 45(3):203-210.
1 O 1. Pfaff, M., et al. ( 1994), "Selective recognition of cyclic RGD
peptides of NMR defined
conformation by aiibB3, aVB3, and a5B 1 Integrins," J. Biol. Chem.
269(32):20233-
20238.
102. Pierschbacher, M.D., et al. ( 1984), "Cell attachment activity of
fibronectin can be
duplicated by small synthetic fragments of the molecule," Nature 309:30-33.
103. Pierschbacher, M.D., et al. (1987), "Influence of Stereochemistry of the
Sequence of Arg-
Gly-Asp-Xaa on Binding Specificity in Cell Adhesion," Journal of Biological
Chemistry
262(36):17294-17298.
104. Pilliar, R.M. (1987), "Porous-surface metallic implants for orthopaedic
applications," J.
Biomed. Meter. Res. 21(A1 Suppl):1-33.
105. Pistone, M., et al. (1996), "Integrin synthesis and utilization in
cultured human
osteoblasts," Cell Biology International 20(7):471-479.
106. Pohunkova, H., et al. (1995), "Reactivity and the fate of some composite
bioimplants
based on collagen in connective tissue," Biomaterials 16( 1 ):67-71.
107. Prime, K.L., et al. {1991), "Self Assembled Organic Monolayers: Model
Systems for
Studying Adsorption of Proteins at Surfaces," Science 252:1164-1167.
108. Puleo, D.A. (1994), "Activity of covalently immobilized enzyme on
silanized Co-Cr-
Mo," In: 19th Annual Meeting of the Society for Biomaterials.
109. Puelo, D.A. (1996), "Biochemical surface modification of Co-Cr-Mo,"
Biomaterials
17:217-222.
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-48-
110. Puelo, D.A., et al. ( 1992), "Formation of focal contacts by osteoblasts
on orthopedic
biomaterials," J. of Biomedical Research 26:291-301.
111. Puelo, D.A., et al. (1991), "RGDS Tetrapeptide Binds to Osteoblasts and
Inhibits
Fibronectin-Mediated Adhesion," Bone 12:271-276.
112. Pytela, R., et al. ( 1987), "Arginine-glycine-aspartic acid adhesion
receptors," Methods in
Enzymology 144:475-489.
113. Ranieri, J.P., et al. (1995), "Neuronal cell attachment to fluorinated
ethylene propylene
films with covalently immobilized laminin oligopeptides YIGSR and IKVAV II,"
Journal of Biomedical Research 29: 779-785.
114. Ratner, B.D. (ed.) (1988), Surface Characterization of Biomaterials,
Elsevier Science
Publishing Company; New York, New York.
115. Robey, P.G., et al. (1993), "Structure and molecular regulation of bone
matrix protein," J.
Bone Min. Res. 2:S483-7.
116. Roskelley, C.D., et al. (1994), "Extracellular matrix-dependent tissue-
specific gene
expression in mammary epithelial cells requires both physical and biochemical
signal
transduction," Proc. Natl. Acad. Sci., USA 91:12378-12382.
117. Ruoslahti, E., et al. (1987), "New Perspectives in cell adhesion: RGD and
integrins,"
Science 238:491-97.
i 18. Saito, T., et al., (1994), "Identification of integrin receptors on
cultured human bone
2o cells," J. Orthopaedic Research 12(3):384-394.
119. Sandberg, M.M., et al. (1993), "Gene expression during bone repair,"
Clinical
Orthopaedics and Related Research 289:292-312.
120. Schaller, M.D. (1996), "The focal adhesion kinase," Journal
ofEndocrinology 150:1-7.
121. Schneider, G., et al. (1994), "Formation of Focal Adhesions by
Osteoblasts Adhering to
Different Substrata," Experimental Cell Research 214:264-269.
122. Stephel, G.C., et al. (1989), "Laminin. A chain synthetic peptide which
supports neurite
outgrowth," Biochemical and Biophysical Research Communications 162(2):821-
829.
CA 02301064 2000-02-14
WO 99/01089 PCTNS98/13792
-49-
123. Sinnghvi, R., et al. (1994), "Engineering Cell Shape and Function,"
Science 264:696-698.
124. _ Soballe, K., et al., "Tissue ingrowth into titan and hydroxyapatite-
coated implants during
stable and unstable mechanical conditions," J. Orthop. Res., 10; 2-299, 1992.
125. Spector, M. (1987), "Historical review of porous-coated implants," J.
Arthroplasty 2:163-
77.
126. Spector, M., et al., "Advances in our understand of the implant bone
interface: factors
affecting formation and degeneration. In Instructional Course Lectures, The
American
Academy of Orthopaedics, 40; 101-113, 1991.
127. Staatz, W.D., et al. ( 1991 ), "Identification of a tetrapeptide
recognition sequence for the
l0 alpha-2, beta-1 integrin in collagen," Journal of Biological Chemistry
266:7363-7367.
128. Takatsuka, M. (1992), "Preparation of an RGD ALB conjugate. In vitro
analysis of
cellular responses," ASAIO-J 38(3):M275-8.
129. Takeuchi, Y., et al. (1996), "Differentiation and cell surface expression
of transforming
growth factor-beta receptors are regulated by interaction with matrix collagen
in marine
osteoblastic cells," J. Biol. Chem. 271(7):3938-44.
130. Tenenbaum, H.C., et al. (1982), "Differentiation of Osteoblasts and
Formation of
Mineralized Bone in vitro," Calcif. Tissue Int. 34:76-29.
131. Tisdel, C.L., et al., "The Influence of a hydroxyapatite and tricalcium
phosphate coating
on bone growth into titanium fiber-metal implants," J. Bone Joint Sur.
76A:159, 1994.
132. Valentine, R.F., et al. (1997), "Increased Bone Formation and Pull-Out
Strength with
RGD-Coated vs. Uncoated Implants," Soc. Biomaterials Abstracts in press.
133. Valentine, R.F., et al. (1992), "Electrically charged polymeric
substrates enhance nerve
fiber outgrowth in vitro," Biomaterials 13:183-90.
134. Valentine, R.F., et al. (1993), "Patterned neuronal attachment and
outgrowth on surface
modified, electrically charged fluoropolymer substrates," J. Biomater. Sci.
Polymer Edn.
5:13-36.
135. Valentine, R.F., et al. (1994), "Enhanced osteoblast attachment to amine-
and RGD-
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/13792
-50-
immobilized fluoropolymer substrates," Ortho. Res. Soc. Abstracts 40.
136. _ Valentini, R.F., et al. ( 1997), "Increased and Differential Gene
Expression of Cultured
Osteoblasts to RGD- and RGE-Containing Peptides," Ortho. Res. Soc. Abstracts
in press.
137. Valentini, R.F., et al. (1995), "Increased osteocalcin synthesis by rat
calvarial osteoblasts
on RGI grafted substrates," Soc. Biomaterials Abstracts 21:65. '
138. van Dijk, et al. (1993), "Evidence that a non-RGD domain in rat
osteopontin is involved
in cell attachment," Langmuir 8:130-138.
139. Journal of Cell Biology 121: 461-468.
140. Vukicevic, S., et al. (1990), "Differentiation of canalicular cell
processes in bone cells by
1o basement membrane matrix components: regulation by discrete domains of
laminin," Cell
63:437-45.
141. Vuori, K., et al. (1994), "Association of Insulin Receptor Substrate-1
with Integrins,"
Science 266:1576-1578.
142. Walsh, W.R., et al. (1995), "Controlled release of platelet-derived
growth factor using
ethylene vinyl acetate copolymer (EVAc) coated on stainless steel wires,"
Biomaterials
16:1319-1325.
143. Wang, N., et al. (1993), Mechanotransduction across the cell surface and
through the
cytoskeleton," Science 260:1124-27.
144. Whitesides, G.M., et al., "Molecular Self Assembly and Nanochemistry: A
Chemical
Strategy for the Synthesis of Nanostructures," Science 254:1312-1319.
145. Wu, J.E., et al. ( 1994), "Complex patterns of expression suggest
extensive roles for the
alpha 2 beta 1 integrin in marine development," Dev. Dyn. 199(4):292-314.
146. Yasuda, T., et al. ( 1996), "Possible Involvement of RGD (Arg-Gly-Asp)-
Containing
Extracellular Matrix Proteins in Rat Growth Plate Chondrocyte Differentiation
in
Culture," Journal of Bone and Mineral Research 11(10):1430-1437.
147. Zhang, Z., et al. (1995), "The 51 integrin supports survival of cells on
fibronectin and up-
regulates Bcl2 expression," Proc. Natl. Acad. Sci. USA 92:6161-6165.
CA 02301064 2000-02-14
WO 99/01089 PCT/US98/1379Z
-51 -
Each of the foregoing patents, patent applications and references is herein
incorporated by
reference in its entirety. Having described the presently preferred
embodiments in accordance
with the present invention, it is believed that other modifications,
variations and changes will be
suggested to those skilled in the art in view of the teachings set forth
herein. It is, therefore, to be
understood that all such variations, modifications, and changes are believed
to fall within the
scope of the present invention as defined by the appended claims.
What I claim is: