Note: Descriptions are shown in the official language in which they were submitted.
CA 02301875 2000-02-25
WO 98/12355 1 PCTIITS97/17101
- DESCRIPTION
METHODS OF PREPARING NUCLEIC ACIDS FOR
MASS SPECTROMETRIC ANALYSIS
ACKNOWLEDGMENTS
This invention was supported in part by a Financial Assistance Award from the
United
States Department of Commerce, Advanced Technology Program, Cooperative
Agreement
#70NANBSH1029. The U.S. Government may have rights in this invention.
CROSS-REFERENCE TO RELATED APPLICATIONS
This application is a continuation of U.S. Application Serial No. 60/032,369
filed
December ?, 1996 and U.S. Application Serial No. 08/759,993 filed December 2,
1996 which is
a continuation-in-part of U.S. Application Serial No. 08/715,582 filed
September 19, 1996.
INTRODUCTION
Approximately 4,000 human disorders are attributed to genetic causes. Hundreds
of
genes responsible for various disorders have been mapped, and sequence
information is being
accumulated rapidly. A principal goal of the Human Genome Project is to find
all genes
associated with each disorder. The definitive diagnostic test for any specific
genetic disease (or
predisposition to disease) will be the identification of polymorphic
variations in the DNA
sequence of affected cells that result in alterations of gene function.
Furthermore, response to
specific medications may depend on the presence of polymorphisms. Developing
DNA (or
RNA) screening as a practical tool for medical diagnostics requires a method
that is inexpensive,
accurate. expeditious, and robust.
Genetic polymorphisms and mutations can manifest themselves in several forms,
such as
point poiymorphisms or point mutations where a single base is changed to one
of the three other
bases, deletions where one or more bases are removed from a nucleic acid
sequence and the
bases flanking the deleted sequence are directly linked to each other.
insertions where new bases
are inserted at a particular point in a nucleic acid sequence adding
additional length to the overall
sequence. and expansions and reductions of repeating sequence motifs. Large
insertions and
deletions, often the result of chromosomal recombination and rearrangement
events, can lead to
CA 02301875 2000-02-25
WO 98/12355 PCTNS97l17101
2
partial or complete loss of a gene. Of these forms of polymorphism, in general
the most difficult
type of change to screen for and detect is the point polymorphism because it
represents the
smallest degree of molecular change.
Although a number of genetic defects can be linked to a specific single point
mutation
within a gene, e.g. sickle cell anemia, many are caused by a wide spectrum of
different mutations
throughout the gene. A typical gene that might be screened could be anywhere
from 1,000 to
100,000 bases in length, though smaller and larger genes do exist. Of that
amount of DNA, only
a fraction of the base pairs actually encode the protein. These discontinuous
protein coding
regions are called exons and the remainder of the gene is referred to as
introns. Of these two
types of regions, exons often contain the most important sequences to be
screened. Several
complex procedures have been developed for scanning genes in order to detect
polymorphisms.
These procedures are applicable to both exons and introns.
In terms of current use, most of the methods to scan or screen genes employ
slab or
capillary gel electrophoresis for the separation and detection step in the
assays. Gel
I S electrophoresis of nucleic acids primarily provides relative size
information based on mobility
through the gel matrix. If calibration standards are employed, gel
electrophoresis can be used to
measure absolute and relative molecular weights of large biomolecules with
some moderate
degree of accuracy; even then typically the accuracy is only 5% to 10%. Also
the molecular
weight resolution is limited. In cases where two DNA fragments with the
identical number of
base pairs can be separated. using high concentration polyacrylamide gels, it
is still not possible
to identify which band on a gel corresponds to which DNA fragment without
performing
secondary labeling experiments. Thus, gel electrophoresis techniques can only
determine size
and cannot provide any information about changes in base composition or
sequence without
performing more complex sequencing reactions. Gel-based techniques, for the
most part, are
dependent on labeling or staining methods to visualize and discriminate
between different
nucleic acid fragments.
All of the methods in use today capable of screening broadly for genetic
polymorphisms
suffer from technical complication and are labor and time intensive. Single
strand
conformational polymorphism (SSCP) (Orita et al., 1989), denaturing gradient
gel
electrophoresis (DGGE) (Abrams et al., 1990), chemical cleavage at mismatch
(CCM) (Saleeba
and Cotton, 1993), enzymatic mismatch cleavage (EMC) (Youil et al., 1995), and
"cleavase"
fragment length polymorphism (CFLP) procedures are currently gel-based, making
them
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
3
cumbersome to automate and perform efficiently. There is a need for new
methods that can
provide cost effective and expeditious means for screening genetic material in
an effort to reduce
medical expenses.
The late 1980's saw the rise of two new mass spectrometric techniques for
successfully
- 5 measuring the masses of intact very large biomolecules, namely, matrix-
assisted laser
desorption/ionization (MALDI) time-of flight mass spectrometry (TOF MS)
(Tanaka et al.,
1988; Spengler et al., 1989) and electrospray ionization (ESI) combined with a
variety of mass
analyzers (Fenn et al., 1989). Both of these methods are suitable for genetic
screening tests. The
MALDI mass spectrometric technique can also be used with methods other than
time-of flight,
IO for example, magnetic sector, Fourier-transform ion cyclotron resonance,
quadrupole, and
quadrupole trap. One of the advances in MALDI analysis of polynucleotides was
the discovery
of 3-hydroxypicolinic acid ("3-HPA") as a matrix for mixed-base
oligonucleotides (Wu, et al.,
1993).
MALDI-TOF MS involves laser pulses focused on a small sample plate comprising
15 analyte molecules (i.e. nucleic acids) embedded in either a solid or liquid
matrix which is
typically a small, highly absorbing material. The laser pulses transfer energy
to the matrix
causing a microscopic ablation and concomitant ionization of the analyte
molecules, producing a
gaseous plume of intact, charged nucleic acids in single-stranded form. If
double-stranded
nucleic acids are analyzed, the MALDI-TOF MS typically results in mostly
denatured single-
20 strand detection. The ions generated by the laser pulses are accelerated to
a fixed kinetic energy
by a strong electric field and then pass through an electric field-free region
in vacuum, traveling
with a velocity corresponding to their respective mass-to-charge ratios (m/z).
Thus, the smaller
m/z ions will travel through the vacuum region faster than the larger m/z ions
thereby causing a
separation. At the end of the electric field-free region, the ions collide
with a detector that
25 generates a signal as each set of ions of a particular mass-to-charge ratio
strikes the detector.
Usually for a given assay, 10 to 100 mass spectra resulting from individual
laser pulses are
summed together to make a single composite mass spectrum with an improved
signal-to-noise
ratio.
The mass of an ion (such as a charged nucleic acid) is measured by using its
velocity to
30 determine the mass-to-charge ratio by time-of flight analysis. In other
words, the mass of the
molecule directly correlates with the time it takes to travel from the sample
plate to the detector.
The entire process takes only microseconds. In an automated apparatus, tens to
hundreds of
CA 02301875 2000-02-25
WO 98/12355 4 PCT/US97/17101
samples can be analyzed per minute. In addition to speed, MALDI-TOF MS has one
of the
largest mass ranges for mass spectrometric devices. The current mass range for
MALDI-TOF
MS is from 1 to 1,000,000 Daltons (Da) (measured recently for a protein)
(Nelson et al., I995).
The performance of a mass spectrometer is measured by its sensitivity, mass
resolution
and mass accuracy. Sensitivity is measured by the amount of material needed;
it is generally
desirable and possible with mass spectrometry to work with sample amounts in
the femtomole
and low picomole range. Mass resolution, m/~m, is the measure of an
instrument's ability to
produce separate signals from ions of similar mass. Mass resolution is defined
as the mass, m, of
an ion signal divided by the full width of the signal, 0m, usually measured
between points of
IO half maximum intensity. Mass accuracy is the measure of error in
designating a mass to an ion
signal. The mass accuracy is defined as the ratio of the mass assignment error
divided by the
mass of the ion and can be represented as a percentage.
To be able to detect any point polymorphism directly by MALDI-TOF mass
spectrometry, one would need to resolve and accurately measure the masses of
nucleic acids in
which a single base change has occurred (in comparison to the wild type
nucleic acid). A single
base change can be a mass difference of as little as 9 Da. This value
represents the difference
between the two bases with the closest mass values, A and T (A = 2'-
deoxyadenosine-5'-
phosphate - 313.19 Da; T = f-deoxythymidine-5'-phosphate - 304.20 Da; G =2'-
deoxyguanosine-5'-phosphate = 329.21 Da; and C = 2'-deoxycytidine-5'-phosphate
= 289.19 Da).
If during the mutation process, a single A changes to T or a single T to A,
the mutant nucleic
acid containing the base transversion will either decrease or increase by 9 Da
in total mass as
compared to the wild type nucleic acid. For mass spectrometry to directly
detect these
transversions, it must therefore be able to detect a minimum mass change, Om,
of approximately
9 Da.
For example, in order to fully resolve (which may not be necessary) a point-
mutated
(A to T or T to A) .heterozygote 50-base single-stranded DNA fragment having a
mass, m, of
-- 15,000 Da from its corresponding wild type nucleic acid, the required mass
resolution is ml0m
= 15,000/9 ~ 1,700. However, the mass accuracy needs to be significantly
better than 9 Da to
increase quality assurance and to prevent ambiguities where the measured mass
value is near the
half way point between the two theoretical masses. For an analyte of 15,000
Da, in practice the
mass accuracy needs to be Dm ~ ~3 Da = 6 Da. In this case, the absolute mass
accuracy required
is (6I15.000)* 100 = 0.04%. Often a distinguishing level of mass accuracy
relative to another
CA 02301875 2000-02-25
WO 98/12355 5 PCTIUS97/17101
known peak in the spectrum is sufficient to resolve ambiguities. For example,
if there is a known
mass peak 1000 Da from the mass peak in question, the relative position of the
unknown to the
known peak may be known with greater accuracy than that provided by an
absolute, previous
calibration of the mass spectrometer.
In order for mass spectrometry to be a useful tool for screening for
polymorphisms in
nucleic acids, several basic requirements should be met. First, any nucleic
acids to be analyzed
should be purified to minimize the presence of salt ions and other molecular
contaminants.
These impurities may reduce the intensity and quality of the mass
spectrometric signal to a point
where either (i) the signal is undetectable or unreliable, or (ii) the mass
accuracy and/or
resolution is below the value necessary to detect the type of polymorphism
expected. Second,
the size of the nucleic acids to be analyzed should be within the range where
there is sufficient
mass resolution and accuracy. Mass accuracy and resolution significantly
degrade as the mass of
the analyte increases. Currently, the detection of single nucleotide
polymorphisms (SNPs) above
said mass value is difficult above a mass of approximately 30,000 Da for
oligonucleotides (~ 100
bases). Third, because all molecules within a sample are visualized during
mass spectrometric
analysis (i.e. it is not possible to selectively label and visualize certain
molecules and not others
as one can with gel electrophoresis methods), nucleic acid samples should be
partitioned prior to
analysis to remove unwanted nucleic acid products from the spectrum. Fourth,
the mass
spectrometric methods for generalized nucleic acid screening must be efficient
and cost effective
in order to screen a large number of nucleic acid bases in as few steps as
possible.
The methods for detecting nucleic acid polymorphisms known in the art do not
satisfy
these four requirements. For example, current methods for mass spectrometric
analysis of DNA
fragments have focused on double-stranded DNA fragments which result in
complicated mass
spectra, making it difficult to resolve mass differences between two
complementary strands (see,
e.g., Tang et al., 1994). Thus, there is a need for cost and time effective
methods of detecting
genetic polymorphisms using mass spectrometry, preferably MALDI or ESI, and
with mass
accuracy of a few parts in 10,000 or better.
SUMMARY OF THE INVENTION
This invention provides novel methods and kits for the screening of target
nucleic acids
and the identification of changes in base composition that might result from a
genetic
polymorphism. The present invention discloses novel processes focusing on the
use of mass
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
6
spectrometry as a genetic analysis tool and employing the unique properties of
mass
spectrometry and MALDI-TOF MS, in particular, to separate different amplified
single-stranded
target nucleic acids and identify their mass exactly. Significantly, mass
spectrometry requires
only minute samples, provides extremely detailed information about the
molecules being
analyzed including high mass accuracy, and is easily automated.
The present invention encompasses several embodiments, such as { 1 )
procedures for
reducing the length of target nucleic acids by removing one or more flanking
polynucleotide
regions that "flank," or are adjacent to or near, the regions of interest; (2)
procedures for isolating
either single-stranded or double-stranded target nucleic acids for mass
spectrometric analysis; {3)
procedures combining these two aspects; and (4} kits for the methods described
herein.
The present invention encompasses several embodiments, such as ( 1 )
procedures for
preparing a double-stranded target nucleic acid for mass spectrometric
analysis; (2) procedures
for determining the mass of target nucleic acids, where the target nucleic
acid may be either
single-stranded or double-stranded; and (3) kits for preparing a double-
stranded target nucleic
acid for mass spectrometric analysis. It will be understood by those of skill
in the art that where
the nucleic acid is double-stranded, the two strands are complementary to each
other and are
connected via hydrogen bonds along the strands.
An embodiment of the present invention encompasses a method of determining the
mass
of a target nucleic acid by mass spectrometric analysis. This method generally
includes:
identifying a target nucleic acid; reducing the length of the target nucleic
acid by cleaving at least
a portion of one or more of the flanking regions to produce a reduced-length
target nucleic acid;
obtaining a single-stranded reduced-length target nucleic acid; and
determining the mass of the
single-stranded reduced-length target nucleic acid using a mass spectrometer.
Typically, the
target nucleic acid will contain a region of interest and one or more flanking
regions.
A preferred embodiment encompasses amplifying the target nucleic acid prior to
reducing
the length of the target nucleic acid to produce an amplified target nucleic
acid. The amplified
target nucleic acid may be subsequently reduced in length and obtained in
single-stranded form,
free of its complement, for mass spectral analysis. The target nucleic acid
may be amplified by
any method known by one of skill in the art. for example, polymerise chain
reaction ("PCRT"'",
with PCRT~' being a preferred amplification method. These methods are well
known by those of
skill in the art.
CA 02301875 2000-02-25
WO 98!12355
PCT/US97/17101
It is contemplated that one of skill in the art may use the methods of this
invention to
analyze more than one target nucleic acid simultaneously. As used herein "a"
will be
understood to mean one or more. Thus, "a target nucleic acid" may refer, for
example, to one,
two, three, four, five or more target nucleic acids. Aspects of this
invention, therefore, include
- 5 determining .the mass of one single-stranded reduced-length target nucleic
acid as well as
determining the masses of multiple single-stranded reduced-length target
nucleic acids
simultaneously or in seriatim. Where the masses of multiple single-stranded
reduced-length
target nucleic acids are being determined, each of the target nucleic acids
may be reduced in
length by the same or a different method. Similarly, the single-stranded
reduced-length target
nucleic acids may be obtained from the reduced-length target nucleic acids by
the same or
different methods. For example, if two target nucleic acids are identified, or
selected, for
analysis, then these two target nucleic acids may both be reduced in length by
an endonuclease,
or one may be reduced in length by an endonuclease and the other by cleaving
at a chemically
cleavable site, and so on.
The target nucleic acids encompassed by this invention will generally contain
a region of
interest and one or more flanking regions. A "region of interest" refers to
the region for which
one is interested in determining the mass. For example, when the methods
disclosed in this
invention are employed to detect or screen for polymorphisms, the region of
interest would be
the region containing, or that is suspected of containing, a polymorphism. The
flanking regions
are the portions of DNA sequence on either side of the region of interest.
For embodiments employing PCRTM primers and polymerases to amplify a target
nucleic
acid, the primer is often complementary to a portion of one or more flanking
regions of the target
nucleic acid to allow the primer to effectively anneal to the target nucleic
acid and provide a site
to extend a complement to the target nucleic acid via PCRTM. Therefore, for
the methods
comprising amplification, it is preferred that at least one of the primers is
complementary to a
portion of a flanking region that is preferably adjacent to or close to the
polynucleotide region of
interest, generally within 40 nucleotides.
When the methods of this invention are used to detect a polymorphism, the
target nucleic
acids employed in this invention may include any polynucleotide sequence that
contains or is
suspected of containing a polymorphism. including but not limited to short
tandem repeats
(STRs), simple sequence length polymorphisms (SSLP), single nucleotide
polymorphisms
(SNPs), and any of a multitude of disease markers, for example, markers for
sickle cell anemia,
CA 02301875 2000-02-25
WO 98/12355 PCT/US97J17101
8
fragile X disorder, cystic fibrosis, Tay Sachs disease, Gaucher disease,
thalassemias, and cancer-
related genes. While the target nucleic acids for use in conjunction with the
present invention
may be double- or single-stranded, it is preferable that the nucleic acids be
obtained in single-
stranded form, free of its complementary strand prior to MS analysis. These
single-stranded
target nucleic acids may be any size that can be adequately resolved by mass
spectrometric
analysis. Preferably, in cases where a SNP is to be detected, the final
product single-stranded
amplified target nucleic acids are less than about 100 bases in length. More
preferably, the final
product, single-stranded amplified target nucleic acids are from about 10 to
90 bases in length.
As used in this context, "about" means anywhere from ~ 1 to 10 base pairs, and
all the integers in
between, for example, ~ 1, t2, ~3, t4, ~5, ~6, f7, t8, ~9, or t 10 base pairs.
However, one of ordinary skill in the art will appreciate that as mass
spectrometric
techniques for analysis of nucleic acids improve, the sizes of single-stranded
amplified target
nucleic acids useful in this invention can be increased. The nature of the
mutation to be detected
is also a factor in the size limitations for optimum mass resolution. For
example, as described
above for SNPs, the maximum size limit may be approximately 100 nucleotides in
length.
However, for microsatellite repeats and other two nucleotide repeats, the
maximum size limit
may be approximately 200 nucleotides in length, and the maximum size limit for
four-nucleotide
repeats may be approximately 300 nucleotides.
The target nucleic acids of this invention may be either double-stranded or
single-
stranded. As used herein, the phrase "obtaining a single-stranded reduced-
length target nucleic
acid" refers to isolating a single-stranded nucleic acid free from its
complement for purposes of
mass spectral analysis. Where the target nucleic acid is single-stranded, it
will be understood by
those of skill in the art that no further steps are required to obtain the
single-stranded reduced-
length target nucleic acid from the reduced-length target nucleic acid.
However, where the target
nucleic acid is double-stranded, one of the two complementary strands must be
separated or
isolated from the other such that only one of the two strands is subjected to
mass spectrometry,
e.g., by binding one of the strands to a solid support, denaturing the double-
stranded nucleic acid
and isolating either the bound or unbound strand free from its complement.
This allows for
greater mass resolution, simplifies the spectrum. and eliminates the
collection of cumulative
information.
CA 02301875 2000-02-25
WO 98/12355 9 PCT/US97/17101
- - The term complementary refers to the formation of sufficient hydrogen
bonding between
two nucleic acids to stabilize a double-stranded nucleotide sequence formed by
hybridization of
the two nucleic acids.
The methods for reducing the length of target nucleic acids eliminate
unnecessary
sequences and reduce the mass of the resulting single-stranded or double-
stranded target nucleic
acids, resulting in increased mass resolution and accuracy.
Exemplary methods of reducing length include: cleaving at endogenous
restriction
endonuclease cleavable sites present in one or more flanking regions but
absent in the region of
interest; cleaving at restriction endonuclease cleavable sites which are at or
adjacent to restriction
I ~ endonuclease recognition sites incorporated into one or more of the
flanking regions where the
cleavabIe sites are introduced into the flanking regions using of one or more
cleavable primers
containing restriction endonuclease recognition sites within their sequences;
cleaving at a
combination of restriction endonuclease cleavable sites where the sites are
endogenous and/or
introduced using mismatch or overhanging primers; selective digestion of one
or more flanking
regions using exonuclease and an exonuclease blocking moiety to protect the
regions of interest
from digestion; and chemically cleaving at a chemically cleavable site. For
embodiments where
cleavable sites are employed, the cleavable sites are often located in or near
a flanking region.
However, the target nucleic acids may be reduced in length by any of the
methods known by
those of skill in the art for cleaving within one or more flanking regions
preferably without
cleaving within the region of interest.
Another aspect of the invention involves the use of cleavable primers to
reduce the length
of an amplified target nucleic acid. An amplified target nucleic acid may be
reduced in length by
cleaving at least a portion of one or more of the flanking regions having a
cleavable site. In this
context, the cleavable site may be introduced via a cleavable primer and may
be located outside
of the region of interest. Cleavable primers of the invention may include
those having an
exonuclease blocking moiety, a Type IIS restriction endonuclease recognition
site, a Type II
restriction endonuclease recognition site, and sites capable of being
chemically cleaved.
" The restriction endonucleases employed with the present invention may
include type II
- and type IIS restriction endonucleases. The restriction endonuclease
recognition sites may be
either within a primer region, or outside the primer region, so long as the
restriction
endonuclease cleavable sites are within or near one or more of the flanking
regions. The
restriction endonuciease recognition sites are preferably not within a region
of interest. For type
CA 02301875 2000-02-25
WO 98/12355 1 ~ PCT/US97/17101
II -restriction endonucleases, the restriction endonuclease recognition site
is the same as the
restriction endonuclease cleavable site. For Type IIS restriction
endonucleases, the cleavable site
is at a defined distance away from one side of the recognition site, usually
from about 14 to about
20 base pairs away. Thus, if the Type IIS recognition site is contained within
a flanking region,
S the endonuclease cleaving site must be within about 20 bases of that
flanking region and is
preferably within 14 about bases of that flanking region. Thus, the term
"near" as employed in
this aspect of the invention means "within about 20 bases."
Another embodiment of the invention involves reducing the length of an
amplified target
nucleic acid and isolating a single-stranded amplified target nucleic acid at
the same time by
using a cleavable primer having an exonuclease blocking moiety. After
amplification of the
target nucleic acid, the amplified target nucleic acid will include an
exonuclease blocking
moiety. The amplified target nucleic acid is then treated with a 5' to 3'
exonuclease, which
degrades the strand containing the exonuclease blocking moiety in a 5' to 3'
direction only up to
the blocking moiety. The 5' to 3' exonuclease may optionally degrade the other
complementary
strand of the amplified target nucleic acid, in cases where the other strand
does not have an
exonuclease blocking moiety. The treatment with the 5' to 3' exonuclease
leaves a reduced-
length, single-stranded amplified target nucleic acid for mass spectrometric
analysis.
Cleavable sites within cleavable primers may include chemically cleavable
groups
incorporated within the phosphate backbone linkage (e.g. replacement of
phosphate with a
phosphoramidate) or as a substituent on or replacement of one of the bases or
sugars of the
oligonucleotide primer (e.g. a modified base or sugar. for example, a more
labile glycosidic
linkage). Such chemically cleavable groups would be apparent to one of skill
in the art in light
of the present disclosure and include, for example, dialkoxysilane, 3'-(S)-
phosphorothioate, 5'-
(S)-phosphorothioate, 3'-(N)-phosphoroamidate, S'-(N)-phosphoroamidate, and
ribose. FIGS.
16A and 16B depict a 3'-(S)-phosphorothioate and 5'-(S)-phosphorothioate,
respectively as
defined in this invention. Note that these linkages are often referred to as
thiophosphates as well.
A similar nomenclature is employed for 3'-(N)-phosphoroamidate, 5'-(N)-
phosphoroamidate.
The chemically cleavable site should generally be stable under the
amplification, hybridization
and washing conditions to be employed and is preferably within one or more of
the flanking
regions.
In a preferred embodiment, the cleavable site is located near the 3' end of
the primer used
to bind the amplified target nucleic acid to the solid support. By locating
the cleavable site near
CA 02301875 2000-02-25
WO 98/12355 I 1 PCT/US97/17101
the -3' end, it is possible to fiu-ther reduce the length of the amplified
target nucleic acid,
eliminating a flanking region from the polynucleotide region of interest.
Cleavable primers are
. described in PCT/LJS96/06116, filed April 26, 1996 (incorporated herein by
reference).
Accordingly, cleavable primers may contain one or more restriction recognition
sites of
one or more different restriction endonucleases; one or more cleavable sites
of one or more
different restriction endonucleases; one or more exonuclease blocking
moieties; one or more sites
capable of chemical cleavage; or a combination thereof.
The present invention also provides methods for obtaining single-stranded or
double-
stranded amplified target nucleic acids. The isolation methods include direct
attachment of one
of the two strands of a double-stranded amplified target nucleic acid or a set
of such molecules,
to a solid support. The isolation methods further include indirect attachment
of a single-stranded
or double-stranded amplified target nucleic acid, or a set thereof, to a solid
support via an
attachment capable of attaching to a solid support via covalent or noncovalent
attachment.
Methods of direct attachment include for example, biotin/avidin interactions,
as well as other
methods known by those of skill in the art.
For example, in one embodiment, a strand of an amplified target nucleic acid
may be
bound or attached to a solid support to permit rigorous washing and
concomitant removal of salt
adducts, unwanted oligonucleotides and enzymes. Either a double-stranded
amplified target
nucleic acid or a single-stranded amplified target nucleic acid may be
isolated for mass
spectrometric analysis. The single-stranded amplified target nucleic acid
analyzed by MS may
be either the strand bound or not bound to the solid support.
When the unbound strand is used for MS analysis, it is typically purified by
first washing
the bound strand and its attached complement under conditions not sufficiently
rigorous to
disrupt the strand's attachment to its bound complement. After unwanted
biomolecules and salts
are removed, the complement may then be released under more rigorous
conditions (see FIG.
11 ).
In contrast. when the bound strand is to be analyzed, it is typically washed
under more
vigorous conditions such that the interactions between the bound strand and
its unbound
- complement is disrupted. This allows the unbound strand to be washed away
with the other salts
and unwanted biomolecules. Cleavable linkers or cleavabte primers may be used
to release the
bound strand from the solid support prior to MS analysis.
CA 02301875 2000-02-25
WO 98/12355 PCT/US97l17101
12
The isolation methods described herein provide significantly improved mass
resolution
and accuracy in large mass ranges. Such isolation of either single-stranded or
double-stranded
amplified target nucleic acids generally occurs prior to the application of
the nucleic acids to the
matrix solution, resulting in well-defined mass spectral peaks and enhanced
mass accuracy. The
matrix solution can be any of the known matrix solutions used for mass
spectrometric analysis,
including 3-hydroxypicolinic acid ("3-HPA"), nicotinic acid, picolinic acid,
2,5-
dihydroxybenzoic acid, and nitrophenol.
The reducing and obtaining steps may occur consecutively in any order or
simultaneously. Thus, this invention encompasses (i) reducing the length of a
target nucleic acid
prior to isolating the single-stranded reduced-length target nucleic acid from
its complement acid
to obtain the single-stranded reduced-length target nucleic acid; (ii)
isolating a single-strand of
the full-length target nucleic acid free from its complement and then reducing
the length of the
single-stranded target nucleic acid to obtain the single-stranded reduced-
length target nucleic
acid; {iii) simultaneously reducing the length of the target nucleic acid and
isolating it free from
I 5 its complementary strand acid to obtain the single-stranded reduced-length
target nucleic acid; or
(iv) any combination of the above steps so long as acid a single-stranded
reduced-length target
nucleic acid is obtained free of its complementary strand prior to mass
spectral analysis.
Another aspect of this invention encompasses a method of determining the mass
of a
target nucleic acid, where the target nucleic acid generally contains a first
strand and a second
complementary strand. The method of the invention includes: identifying a
target nucleic acid;
amplifying the target nucleic acid prior to reducing the length of the target
nucleic acid to
produce an amplified target nucleic acid; reducing the length of the target
nucleic acid by
cleaving at least a portion of one or more of the flanking regions to produce
a reduced-length
target nucleic acid; obtaining a single-stranded reduced-length target nucleic
acid; and
determining the mass of the single-stranded reduced-length target nucleic acid
using a mass
spectrometer where the target nucleic acid further comprises a region of
interest and one or more
flanking regions and where the obtaining step comprises attaching the first
strand of the
amplified target nucleic acid to a solid support and separating the first
strand from the second
strand to produce a bound first strand and an unbound second strand. In this
embodiment, the
mass of the unbound second strand is determined.
The present invention additionally encompasses primers and methods for using
primers
that are capable of being "attached" or bound to a solid support. Generally,
this is accomplished
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
13
by -attaching a binding group or moiety to the primer or to a modified
nucleotide during
amplification, where the binding group or moiety is capable of attaching or
binding the
oligonucleotide to the solid support. This binding moiety may be attached to
the oligonucleotide
primer or amplification product either directly, through an intervening
linking group or by
specific hybridization to an intermediary oligonucleotide which is itself
bound to a solid support.
Binding moieties include functional groups for covalent bonding to a solid
support, ligands that
attach to the solid support via a high-affinity, noncovalent interaction (such
as biotin with
streptavidin), a series of bases complementary to an intermediary
oligonucleotide which is itself
attached to the solid support, as well as other means that are well-known to
those of skill in the
art, such as those described in PCT WO 96/37630, incorporated herein by
reference.
The first strand is typically separated from the second strand by washing
under conditions
rigorous enough to disrupt the double-stranded base pairing structure, but not
rigorous enough to
disrupt the attachment of the bound first strand to the solid support. The
solution-phase (or
washings) containing the unbound strand can then be prepared for mass spectral
analysis.
Cleavable primers and sites as discussed above are also employed in this
embodiment.
However, the cieavable site should preferably not be between the binding
moiety, i.e. the group
attaching the first bound strand to the solid support, and the region of
interest. Alternatively, the
cleavable site should be incorporated into the second strand only, and not
into the first strand that
is to be attached to the solid support.
A preferred embodiment encompasses the use of a cieavable primer having a
chemically
cleavable group of 3'-(S)-phosphorothioate or 5'-(S)-phosphorothioate, where
the frst strand is
biotinylated and bound to a solid support via a biotin:avidin interaction (i.
e. where streptavidin
beads are used for a solid support). It is also preferable to employ mass-
modified nucleotides
with this aspect of the invention.
Alternatively, the obtaining step may include (a) attaching the first strand
of the amplified
target nucleic acid to a solid support, (b) separating the first strand from
the second strand to
produce a bound first strand and an unbound second strand, (c) removing the
unbound second
strand, and (d) releasing the bound first strand from the solid support to
produce a single-
stranded reduced-length amplified target nucleic acid for mass spectral
analysis. In this
embodiment, the mass of the bound first strand is determined using a mass
spectrometer.
Several methods may be employed to release the reduced-length single-stranded
amplified target nucleic acid from the solid support. Generally, the methods
used must either
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
14
employ reversible chemical interactions between the binding group and the
solid support, that is,
a "cleavable linker," or a separate chemically or enzymatically cleavable site
somewhere within
the bound product. Thus, these methods for releasing the bound strand include
all of the
methods that may be used for reducing the length of the bound strand as well.
For example, an
exonuclease blocking group, endonuclease recognition site, or a chemically
cleavable site may be
incorporated into the bound strand between the binding moiety and the region
of interest,
cleaving at one of these sites through use of an exonuclease, endonuclease, or
a chemical agent
accomplishes both the releasing and the reduction in length simultaneously.
When more than
one target nucleic acid is identified for analysis, the target nucleic acids
may be released and
analyzed at the same time or consecutively.
This invention also encompasses methods for release that do not include
reducing the
length of the amplified or unamplified target nucleic acids depending on the
method used to bind
the amplified target nucleic acid to the solid support. For example, both the
hybridization and
biotin/streptavidin methods employ means such as denaturation to disrupt the
noncovalent
interactions and cause the release of the bound single-stranded target nucleic
acids. It may be
preferred to use a chemically cleavable site with the biotin/streptavidin
method so that release of
the target nucleic acids can be performed under relatively mild conditions.
Another embodiment of this invention encompasses a method of preparing a
double-
stranded target nucleic acid for mass spectrometric analysis. This method
generally includes
comprising: amplifying a target nucleic acid to produce an amplified target
nucleic acid;
attaching the first strand of the amplified target nucleic acid to a solid
support to produce a bound
first strand and an unbound second strand; removing, or detaching, the unbound
second strand
from the bound first strand; releasing the bound first strand from the solid
support to form a
single-stranded amplified target nucleic acid; and determining the mass of the
single-stranded
amplified target nucleic acid using a mass spectrometer where the amplified
target nucleic acid
comprises a first strand and a second complementary strand. In this
embodiment, the unbound
second strand is typically removed from the bound first strand by denaturing
and washing.
A preferred embodiment encompasses employing a cleavable linker during the
releasing
step, wherein the determining step preferably does not involve sequencing the
amplified target
nucleic acid.
The present invention also provides methods of detecting polymorphisms in one
or more
target nucleic acids. This embodiment generally includes: amplifying at least
one target nucleic
CA 02301875 2000-02-25
WO 98/!2355 PCTlUS97/l7101
acid; reducing the length of at least one of the amplified target nucleic
acids comprising cleaving
off a portion of one or more flanking regions, and determining the masses of
each of the reduced-
length amplified target nucleic acids using a mass spectrometer wherein said
amplified target
nucleic acid comprises a region of interest and one or more flanking regions.
This method may
. 5 be used to detect polymorphisms in a single target nucleic acid compared
to a wild type target
nucleic acid by detecting variability in mass. Other "alleles" of the target
nucleic acid may also
be detected using the methods of the invention.
In the present disclosure, "wild type" is the standard or reference nucleotide
sequence to
which variations are compared. Thus, by definition, any variation from wild
type is considered a
10 polymorphism, including naturally occurring sequence variations and
pathogenic mutations.
In another embodiment, methods are provided for detecting polymorphisms in at
least
one target nucleic acid. These methods may include: amplifying at least one
target nucleic acid;
isolating either a positive or negative strand of the amplified target nucleic
acid to form a single-
stranded amplified target nucleic acid; and determining the masses of each
single-stranded
i 5 amplified target nucleic acid using a mass spectrometer where the
amplified target nucleic acid
comprises a region of interest and one or more flanking regions.
In yet another embodiment, methods are provided for detecting polymorphisms in
at least
one target nucleic acid by amplifying at least one target nucleic acid;
reducing the length of at
least one of the amplified target nucleic acids comprising cleaving off a
portion of one or more
flanking regions; isolating either a positive or negative strand of said
amplified target nucleic
acid to form an amplified target nucleic acid; and determining the mass of
each single-stranded
amplified target nucleic acid using a mass spectrometer where the amplified
target nucleic acid
comprises a region of interest and optionally one or more flanking regions
The methods described in the present invention may also be used to detect
polymorphisms in a set of different target nucleic acids. In this context, the
methods should
generally include: amplifying each of the target nucleic acids; reducing the
length and/or
isolating a single-strand of each of said amplified target nucleic acids; and
determining the mass
of each of the single-strands of said amplified target nucleic acids using
mass spectrometry.
Thus, these methods can he used to detect polymorphisms in a plurality of
different target
nucleic acids simultaneously.
Using the methods described herein, one can uniquely identify a genomic sample
by
amplifying the target nucleic acids; isolating single-stranded amplified
target nucleic acids: and
CA 02301875 2000-02-25
WO 98/12355 PCT/ITS97/17101
16
defermining the masses of the single-stranded amplified target nucleic acids
using mass
spectrometry. The resulting mass determination or mass spectrum may provide
information
which may be used to indicate a disease state, or propensity to disease,
uniquely identify the
source of the sample, or map locations in a genome.
S In yet another embodiment. methods are provided for detecting polymorphisms
in at least
one amplified target nucleic acid further comprising removing at least one
flanking
polynucleotide region, if present, from at least one of the amplified target
nucleic acids before
the isolating step.
In a further embodiment, methods for detecting polymorphisms are described
wherein the
isolating step comprises binding the amplified target nucleic acid to a solid
support and the
removing step comprises using one or more restriction endonucleases to cleave
off one or more
flanking polynucleotide regions.
The mass of a preferably single-stranded amplified target nucleic acid may be
compared
with the known or predicted mass of the corresponding wild type single-
stranded amplified target
nucleic acid, that is, the wild type version of the target nucleic acid that
is being screened for
polymorphism. Alternatively, the masses of more than one amplified target
nucleic acid can be
compared with the known or predicted masses of the corresponding wild type
amplified target
nucleic acids.
The amplified target nucleic acid or set thereof, can optionally have one or
more
nucleotides replaced with mass-modified nucleotides, including mass-modified
nucleotide
analogs. For example, FIG. 2A and FIG. 2B illustrate the increase in
resolution for a A to T
mutation where the mass-modified nucleotide heptynyideoxyuridine has been used
in place of T
during PCR amplification. The use of this mass-modified nucleotide results in
a separation of
mass spectral peaks of 65 mass units instead of only 9 mass units. As this
example illustrates,
mass-modified nucleotides of the present invention may effect substantial
increases in spectral
resolution with only relatively small modifications in mass. Other examples of
mass-modified
nucleotides useful in the present invention include 5-(3-aminoallyl)-2'-dUTP,
5-bromo-dCTP, 5-
iodo-dCTP, 7-methyl-dGTP, 7-deaza-dGTP, dITP, 5-bromo-dUTP, 1,N6-etheno-dATP,
mercuri-dCTP, aminomethylcoumarin-6-dUTP, biotin-16-dUTP, 5-methyl-dCTP, 7-
deaza-
dATP, alphathio-dNTPs, n6-aminohexyl-dATP, 5-iodo-dUTP.
CA 02301875 2000-02-25
WO 9$I1Z35S PCT/U597/17101
17
Another optional aspect of the invention is the inclusion of internal
calibrants or internal
self calibrants in the amplified target nucleic acid or set thereof to be
analyzed by mass
spectrometry to provide improved mass accuracy.
A preferred aspect of the invention includes the methods of detecting
polymorphisms
where the determining step further includes utilizing internal self calibrants
to provide improved
mass accuracy. The isolation methods separately or together may also be
combined with the use
of internal self calibrants.
The above methods, separately or in combination, may also be combined with the
use of
mass-modified nucleotides and mass-modified nucleotide analogs incorporated in
the single
stranded or double-stranded amplified target nucleic acid or set of single-
stranded or double
stranded amplified target nucleic acids to improve mass resolution between
mass peaks. The
methods of detecting polymorphisms may also include at least one single-
stranded amplified
target nucleic acid optionally having one or more nucleotides replaced with
mass-modified
nucleotides.
In another embodiment, kits for preparing amplified target nucleic acids for
mass
spectrometric analysis are provided. The kits of the invention may include a
first primer capable
of binding a first strand of one of the target nucleic acids at a region 5' to
a region of interest of
said target nucleic acid; a second primer capable of binding a second strand
complementary to
the first strand at a region 5' to the region of interest of the target
nucleic acid; a DNA
polymerase capable of extending the primers to form primer extension products
of the first and
second primers; and a restriction endonuclease capable of reducing the length
of amplified target
nucleic acids where the first and second primers and said DNA polymerase are
provided in a
concentration and buffer suitable for increasing the number of target nucleic
acids to form
amplified target nucleic acids
Another embodiment encompasses a kit for preparing a double-stranded target
nucleic
acid having a first strand and a second complementary strand for mass
spectrometric analysis
including: a first primer capable of binding the first strand of the target
nucleic acid 5' to a region
of interest of the target nucleic acid; a second primer capable of binding the
second strand of the
target nucleic acid 5' to the region of interest of the target nucleic acid; a
DNA polymerase
capable of extending the primers to form an amplified target nucleic acid; and
a restriction
endonuclease capable of reducing the length of the amplified target nucleic
acid.
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/1~101
18
The first and second primers and DNA polymerase may be provided in a
concentration
and buffer suitable for increasing the number of target nucleic acids to form
amplified target
nucleic acids. The restriction endonucleases may be Type II or Type IIS
restriction
endonucleases. Preferably, the first primer is biotinylated, preferably at or
near the 5' end and the
kit further comprises a solid support capable of selectively binding the first
strand of the
amplified target nucleic acid. Thus, where the first primer is biotinylated,
the solid support could
be a streptavidin bead. Kits included in this invention may preferably also
comprise a matrix,
such as 3-hydroxypicolinic acid.
An aspect of the present invention also includes a kit for preparing a double-
stranded
target nucleic acid having a first strand and a second complementary strand
for mass
spectrometric analysis comprising: a first primer capable of binding the first
strand of the target
nucleic acid 5' to a region of interest of the target nucleic acid; a second
primer capable of
binding the second strand 5' to the region of interest of the target nucleic
acid; and a DNA
polymerase capable of extending the primers to form an amplified target
nucleic acid, where the
first primer comprises a cleavable primer cleavable by chemical or enzymatic
treatment.
Preferred cleavable primers include those having an exonuclease blocking
moiety, a Type II or
Type II restriction endonuclease recognition site, or a chemically cieavable
site, such as a
modif ed base, a modified sugar, or a chemically cleavable group incorporated
into the phosphate
backbone. Preferred chemically cleavable groups are dialkoxysilane, 3'-(S)-
phosphorothioate,
5'-(S)-phosphorothioate, 3'-(N)-phosphoroamidate, or 5'-(N)-phosphoroamidate.
Preferably, the kit may also contain a solid support capable of selectively
binding the first
strand of the amplified target nucleic acid. For example, if the first strand
preferably comprises a
biotin, the solid support could comprise a streptavidin bead. These kits may
also preferably
further comprise a matrix, such as 3-hydroxypicolinic acid.
Another embodiment is a kit containing: a first primer capable of binding a
first strand of
one of the target nucleic acids at a region 5' to a region of interest of the
target nucleic acid; a
second primer capable of binding a second strand complementary to the first
strand at a region 5'
to the region of interest of the target nucleic acid; a DNA polymerase capable
of extending the
primers to form primer extension products of the first and second primers.
where at least one of
the two primers is a cleavable primer.
CA 02301875 2000-02-25
WO 98112355 PCTIUS97/17101
19
- BRIEF DESCRIPTION OF THE DRAWINGS
FIG. IA is a resolved spectrum of nucleic acid fragments (DNA) in the 20,000
to 25,000
Da range using MALDI-TOF mass spectrometry. This positive ion time of flight
mass spectrum
was obtained from 200 fmoles of DNA in 3-HPA the summation of 100 laser pulses
at 266 nm.
The spectrum is of a single-stranded 72-mer which also shows a 71-mer. The
FWHM resolution
is 240 clearly resolving matrix adducts (labeled M}.
FIG. IB displays the positive ion TOF mass spectrum of a 88-mer parent peak
and has a
resolution of 330. This MALDI-TOF spectrum is a sum of 100 laser pulses at 266
nm was
obtained from 200 fmoles of DNA in 3-HPA.
FIG. 2A shows the mass spectrum of a heterozygous mix of wild type and mutant
DNA
fragments where an A has mutated to a T giving spectral peaks separated by 9
mass units.
FIG.2B illustrates the effect on mass resolution of a mass-substituted base.
The
spectrum in FIG. 2B consists of a mass spectrum of a heterozygous mix of wild
type and mutant
DNA fragments where A has mutated to T and the T has been replaced by
heptynyldeoxyuridine
during amplification of the mutant region (R = heptynye). The spectral peaks
are now separated
by 65 mass units as compared to only 9 mass units in FIG. 2A.
FIG. 3 is a diagram illustrating the effect of analyzing full-length double-
stranded
amplified target nucleic acid, where the blunt-ended double-strands result in
unresolved peaks in
the mass spectrum of the unresolved double-stranded fragments. In this
instance, the source
nucleic acid may be amplified, for example, by PCR, and then mass analyzed as
the full-length,
double-stranded product. The amplified target nucleic acid should typically be
no greater than
about 100 base pairs in length.
FIG. 4 is a diagram illustrating the effect of analyzing reduced-length double-
stranded
amplified target nucleic acid, where one of the strands has a 4 nucleotide
overhang which results
in fully resolved peaks of the double-stranded fragments in the mass spectrum.
In this case, the
source nucleic acid is amplified (e.R. PCR) in step (a), then reduced in
length by restriction
digestion of the double-stranded product in step (b) to yield an uneven ended
product. The
reduced-length, double-stranded product is then mass analyzed to yield to
fully resolved peaks.
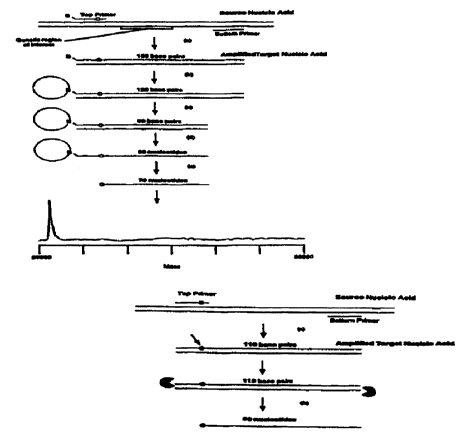
FIG. 5 illustrates that analyzing only a single-stranded amplified target
nucleic acid
reduces the number of strands and simplifies the mass spectrum. The source
nucleic acid is
amplified (e.g. PCR) in step (a) and then captured to a solid phase
streptavidin bead in step (b).
The solid phase is then rigorously washed to remove salts and unwanted
biomolecules including
CA 02301875 2000-02-25
WO 98/12355 PCT/US97I17101
the bottom complementary strand in step (c). The isolated full-length, single-
stranded amplified
target strand is finally released into solution to be mass analyzed in step
(d). Although this
diagram as well as the diagrams in FIG. 6, FIG. 7, FIG. 8, FIG. 9, FIG. 10,
FIG. 11 and FIG. 12
depicts the use of a biotinylate top primer (B) and a streptavidin solid
support, these schemes are
5 also generally applicable to other methods of selectively binding one strand
of an amplified
target nucleic acid to a solid support to facilitate the washing away of
unwanted biomolecules,
salts and the other complementary strains.
FIG. 6 employs a cleavable primer to reduce in length an amplified target
nucleic acid
that is larger than 100 base pairs to less than 100 nucleotides in length. The
cleaving at the
10 cleavable primer site also releases the single-stranded amplified target
nucleic acid from the solid
support. In this instance, the top primer is biotinylated and cleavable. In
step (a) the source
nucleic acid is amplified (e.g. PCR) and captured to solid phase streptavidin
bead in step (b).
The solid phase is then rigorously washed to remove salts and unwanted
biomolecules including
the bottom complementary strand in step (c). The isolated, reduced-length,
single-stranded
1 S amplified target is released into solution by cleaving the primer at the
cleavable site in step (d)
for mass analysis.
FIG. 7 is a diagram illustrating the isolation of a single-stranded amplified
target nucleic
acid that has been reduced in length by cleaving off at least a portion of
both flanking regions.
The first flanking region contains a cleavable site in the cleavable primer,
located outside of the
20 region of interest. The second flanking region is on the opposite end of
the amplified target
nucleic acid and the portion of that second flanking region is cleaved off by
digestion with a
restriction endonuclease. The top primer is biotinylated and cleavable. The
source nucleic acid
is first amplified (e.g. PCR) (step (a)) and captured to the solid phase (e.g.
streptavidin bead)
(step (b)). The double-stranded target is then selectively restricted outside
the genetic region of
interest (step (c)). The order of steps (b) and (c) may be reversed. The solid
phase is rigorously
washed to remove salts and unwanted biomolecules including the bottom
complementary strand
(step (d)). Finally, the isolated, reduced-length, single-stranded amplified
target strand is
released into solution to be mass analyzed by cleaving the cleavable primer
(step (e)).
FIG. 8 shows the isolation of a single-stranded amplified target nucleic acid,
where the
length of the amplified target nucleic acid is reduced by cleaving off a
portion of both flanking
regions by ( 1 ) using a first or top primer having a chemically cleavable
site incorporated during
amplification; and (2) using a bottom primer having a Type IIS restriction
endonuclease
CA 02301875 2000-02-25
WO 98112355 PCTII1S97117101
21
recognition site at the 5' end during amplification. The target nucleic acid
is first amplified (e.g.,
PCR) (step (a)). The use of the bottom primer with a type IIS restriction
enzyme recognition site
on the 5' end allows for the incorporation of this site on the end of the
target nucleic acid. The
top primer is then captured to a soiid phase (e.g., a streptavidin bead) in
step (b). One flanking
region is then cleaved off by digesting with a Type IIS restriction
endonuclease in step (c). After
rigorously washing to remove salts and unwanted biomolecules, including the
unwanted
(unbound) complementary strand in step (d), the single-stranded amplified
target nucleic acid is
released from the solid support by cleaving at the cleavable site within the
cleavable primer in
step (e) for mass spectral analysis. The order of steps (b) and (c) are
reversible.
FIG. 9 depicts another embodiment of the invention, wherein one primer has an
exonucIease blocker (~). After amplification of the target nucleic acid. step
(a), the amplified
target nucleic acid contains an exonuclease blocking group. The amplified
target nucleic acid is
then treated with a 5' to 3' exonuclease, step (b), which degrades the strand
containing the
exonuclease blocking group only up to the blocking group. The 5' to 3'
exonuclease completely
I 5 degrades the other complementary strand of the amplified target nucleic
acid as the other strand
does not have an exonuclease blocking group. The treatment with the 5' to 3'
exonuclease, thus,
leaves a single stranded amplified target nucleic acid for mass spectrometric
analysis.
FIG. 10 is a diagram illustrating yet another embodiment, in which one primer
contains a
Type IIS restriction recognition site and a binding moiety, e.g., biotin {B),
wherein the Type IIS
restriction cleavage site is located between the Type IIS restriction
recognition site and the
binding moiety. The source nucleic acid is first amplified using this primer
and another primer
complementary to the other strand, step (a). The amplified target nucleic acid
is then restricted
with the Type IIS restriction endonuclease corresponding to the Type IIS
restriction recognition
and cleavable sites in the primer (step (b)), leaving a reduced-length
amplified target nucleic acid
comprising a binding moiety, e.g. biotin, which can then be captured to a
solid phase
(streptavidin bead) (step (c)). The reduced-length amplified target nucleic
acid is then rigorously
washed to remove salts and the unbound complementary strand. step ( d ). Then
the reduced
length, single-stranded amplified target nucleic acid is released from the
solid support for mass
spectrometric analysis by denaturing the biotin streptavidin bond, e.g,. by
boiling under low salt
conditions. step (e).
FIG. 11 is a diagram illustrating a variation of the embodiment illustrated in
FIG. 10,
wherein instead of isolating the bound reduced length, single~>tranded
amplified target nucleic
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
22
acid, the complementary (unbound) strand is released from the bound strand and
isolated for
mass spectrometric analysis. Thus, the source nucleic acid is amplified (step
(a)). The amplified
target nucleic acid is then cleaved with a type IIS restriction endonuclease
(step (b)) and captured
to a solid phase (e.g., by biotin (b) interacting with a streptavidin bead)
(step (c)). Unwanted
S salts and biomolecules are removed by washing (step (d)). This time, in
contrast to the schemes
depiction FIG. 5, FIG. 6, FIG. 7, FIG. 8 and FIG. 10. The initial washing is
not so rigorous as to
disrupt the interaction between the bound strand and its complement. In step
(e) the
complementary strand is released for mass analysis.
FIG. 12 is another embodiment wherein the double stranded DNA is mass
analyzed. The
source nucleic acid is amplified (step (a)). In this embodiment the top primer
is biotinylated and
contains a type IIS restriction site such that the cleavage site is between
the region of interest and
the biotin moiety. The amplified target nucleic acid is captured to the solid
phase (step (b)) to
facilitate washing away unwanted salts and biomolecules (step (c)). A
restriction endonuclease
is then used to release the double stranded fragment for mass spectral
analysis (step (d)).
1 S FIG. 13 is a mass spectrum of single-stranded amplified short tandem
repeats from the
tyrosine hydroxylase gene THO1 locus.
FIG. 14 is a mass spectrum of an allelic set (ladder) of single-stranded
amplified target
nucleic acids from the THOI gene locus, wherein the single-stranded amplified
target nucleic
acids ranged in length from 71 to 95 nucleotides in length. The method used to
produce this
spectrum is the one depicted in FIG. 6.
FIG. 15 is a mass spectrum of a set of single-stranded amplified target
nucleic acids
wherein the single-stranded amplified target nucleic acids were the same as
those depicted in
FIG. 14 except that the lengths of the amplified target nucleic acids had been
reduced by 31 base
pairs by endonuclease cleavage. The method employed to prduce this spectrum is
the one
illustrated in FIG. 7.
FIG. 16A shows the chemical formula for 2'-deoxythymidine-3'-(S)-
phosphorothioate
FIG. 16B shows the chemical formula for 2'-deoxythymidine-S'-(S)-
phosphorothioate.
DESCRIPTION OF SPECIFIC EMBODIMENTS
The present invention, directed to methods of and (kits for preparing target
nucleic acids
for mass spectrometric analysis and for detecting poiymorphisms. provides
advantages of
technical ease, speed, and high sensitivity. Additionally, only minute samples
of femtomole
CA 02301875 2000-02-25
WO 98/12355 PCTNS97/17101
23
amounts are required. The methods and kits described herein yield a minimal
set of products
with improved mass resolution and accuracy and detailed information about the
nature of the
polymorphisms detected in the target nucleic acids screened.
One embodiment of the present invention involves methods of detecting
polymorphisms
in one or more target nucleic acids comprising (a) amplifying at least one of
said target nucleic
acids, wherein each of said target nucleic acids comprises a region of
interest and optionally one
or more flanking regions, (b) isolating either a positive or a negative strand
of interest of each of
said target nucleic acids in the form of one or more single-stranded amplified
target nucleic
acids, wherein said isolating preferably comprises binding said strand of
interest of each of said
amplified target nucleic acids to a solid support, and (c) determining the
masses of each of said
single-stranded amplified target nucleic acids using a mass spectrometer,
wherein said
determining preferably does not involve sequencing of said amplified single-
stranded target
nucleic acids. The amplifying step may include the use of a specialized primer
that can be used
in the isolating step to bind the amplified target nucleic acids to a solid
support. The primer may
also have attached a cleavable or reversible linker, or the primer itself may
contain a cleavable
site. If a cleavable site is introduced into one of the amplified target
nucleic acids by using a
cleavable or reversible linker during said amplifying step, the determining
does not involve
sequencing of the amplified target nucleic acids. The primer may also be
biotinylated or
modified in other ways such as to effect binding of the amplified target
nucleic acids to a solid
support. The primer may also optionally be bound or attached to the solid
support prior to being
amplified. One of ordinary skill in the art will appreciate the multiplicity
of methods to effect
such attachment.
The isolating may further comprise denaturing and washing to remove the
complementary strand from the strand of interest which is bound to a solid
support, followed by
release of the bound single-stranded target nucleic acids from the solid
support. Alternatively,
the unbound complementary strand may be released and isolated for mass
spectrometric analysis.
After amplifying and either before or after the amplified target nucleic acids
have been
bound to a solid support, the amplified target nucleic acid may be reduced in
length by a number
of different techniques. For example. one or more flanking regions may be
cleaved using one or
more restriction endonucleases. such as Type II or Type I1S restriction
endonucleases or
combinations thereof. The amplified target nucleic acid may also be reduced in
length by using a
cleavable primer. Another method of reducing length comprises using an
exonuclease blocking
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
24
moiety in one of the two primers for amplification, and digesting said
amplified target nucleic
acid with a 5' to 3' exonuclease.
The target nucleic acid may be single-stranded or double-stranded DNA, RNA or
hybrids
thereof, from any source. The target nucleic acid is generally a nucleic acid
which must be
screened to determine whether it contains a polymorphism. The corresponding
target nucleic
acid derived from a wild type source is referred to as a wild type target
nucleic acid. The
amplified target nucleic acids can be obtained from a source sample containing
nucleic acids and
can be produced from the nucleic acid by PCRTM amplification or other
amplification techniques.
Although human sources are preferred, any source which one is interested in
screening for
polymorphisms may be used in the methods described herein. When the target
nucleic acid is
RNA, the RNA strand is the + strand. If desired, the target nucleic acid may
be an. RNA/DNA
hybrid, wherein either strand can be designated the + strand and the other,
the - strand.
In cases where the amplified target nucleic acid contains RNA, the methods
using
restriction endonucleases described herein cannot be used to directly reduce
the length of the
final product. A restriction endonuclease may be used to reduce the length of
the doubie-
stranded DNA intermediates prior to the RNA transcription step.
The amplified target nucleic acids are typically less than 100 bases in length
because
current mass spectrometric methods do not have the mass accuracy and
resolution necessary to
identify a single base change in polynucleotides larger than 100 base pairs.
However, as mass
spectrometric techniques for analyzing nucleic acids improve. the single-
stranded or double-
stranded amplified target nucleic acids of this invention may be larger than
100 bases in length.
Due to the simpler mass spectrum that results from mass analysis of single-
stranded
amplified target nucleic acids, it is preferred to determine the masses of
sets of single-stranded
amplified target nucleic acids. The amplified target nucleic acids may also
contain mass-
modified nucleotides, which can enhance ease of analysis, especially when a
point
polymorphism has resulted in a very small mass change (on the order of 9 Da)
in a target nucleic
acid as compared to the corresponding wild type target nucleic acid. The
methods described
herein use mass spectrometry to determine the masses of a single-stranded
amplified target
nucleic acid or set of single-stranded amplified target nucleic acids to
detect polymorphisms in at
least one target nucleic acid.
The amplified target nucleic acids comprise a region of interest and
optionally, one or
more flanking regions. A region of interest contains or is suspected of
containing a
CA 02301875 2000-02-25
WO 98/12355 PCT/US97l17101
polymorphism, whereas a flanking region is generally believed not to contain a
polymorphism or
a polymorphism in that region is considered unimportant. The region of
interest may be as small
as a single nucleotide. A flanking region may contain a cleavable site or
cleavable moiety that
can be selectively cleaved to release single-stranded nucleic acids from a
solid support prior to
5 mass spectrometric analysis. An amplified target nucleic acid may also
optionally comprise
another flanking region on the end of the target nucleic acid opposite from
the cleavable site used
for release from the solid support. This second flanking region may contain
one or more
restriction cleavable sites that do not occur in the region of interest.
The methods described herein may be performed on a single amplified target
nucleic acid
10 or on a set of different amplified target nucleic acids, each containing a
different region of
interest. The various steps of reducing length, binding to a solid support,
releasing from the solid
support may differ with respect to each different target nucleic acid in a
set, or may be the same,
and the resulting set of single-stranded or double-stranded amplified target
nucleic acids can be
mass analyzed simultaneously. Accordingly, another advantage of the methods
described herein
15 is that they can be used to prepare a set or collection of two or more
different target nucleic acids
in a single reaction or a single container, possibly using at least one common
reagent, which
results in increased efficiency and more informative data from a single mass
spectrum of the
prepared target nucleic acids.
Wild type refers to a standard or reference nucleotide sequence, or number of
repeat di-,
20 tri-. or tetra-nucleotides, to which variations are compared. As defined,
any variation from wild
type is considered a polymorphism, including naturally occurring sequence
polymorphisms, and
mutations which are pathogenic.
Two nucleic acids are considered "complementary" if they are capable of
specifically
hybridizing to one another (i) under typical hybridization and wash conditions
(see, eg., Maniatis
25 et al.. 1982) or (ii) using reduced stringency wash conditions that allow
at most about 25-30%
base pair mismatches, for example, 2 x SSC, 0.1 % SDS, room temperature twice,
30 minutes
each: then 2 x SSC, 0.1% SDS, 37°C once. 30 minutes; then 2 x SSC room
temperature twice, 10
minutes each.
The types of mass spectrometry used in the invention include ESI or MALDI,
wherein
these methods may optionally include time-of flight. The significant multiple
charging of
molecules in ESI and the fact that complex mixture analysis is often required
mean that the ESI
mass spectra will consist of a great many spectral peaks, possibly overlapping
and causing
CA 02301875 2000-02-25
wo 9a~mss rc~r~s97nmoi
26
confusion. Because the MALDI MS approach produces mass spectra with fewer
major peaks,
this method is preferred. Thus, a MALDI MS time-of flight instrument is
prefen:ed for the mass
analysis of the invention.
The methods described herein do not require sequencing of one or more target
nucleic
acids (using the sequencing methods that require four different base-specific
chain termination
reactions or chemical cleavages to determine the complete nucleotide sequence
of a nucleic acid)
in order to determine the nature and presence of a polymorphism within any of
said target nucleic
acids. However, the methods described herein, separately or in combination,
may be used in the
sequencing of one or more amplified target nucleic acids using mass
spectrometric techniques.
For an initial polymorphism screen, a useful range of amplified target nucleic
acid sizes
that will allow detection of a point polymorphism is around 10 to 100 bases.
This size range is
where mass spectrometry presently has the necessary level of mass resolution
and accuracy.
Thus, the methods used in this invention are designed to produce amplified
target nucleic acids
ranging up to about 100 bases in size, but can also be used to produce larger
amplified target
I S nucleic acids.
Existing mass spectrometric instrumentation in the case of MALDI-TOF MS
optimally
has a mass accuracy of about 1 part in 10,000 (0.01%), four times what is
necessary for detecting
a single base change in a 50-base long single-stranded DNA fragment.
Utilization of mass-
modified nucleotides (described herein) and nearby masses as internal
calibrants, provides
optimal resolution and mass accuracy of larger nucleic acids, and can extend
the usable point
polymorphism detection range up to 100 bases, if not higher. Continued
advances in mass
spectrometric instrumentation will also push this range higher.
Examples of the resolving capabilities of MALDI-TOF MS are displayed in FIG.
lA and
FIG. 1 B which show the positive ion TOF mass spectra obtained from 200 fmoles
of DNA in the
matrix 3-HPA. FIG. IA shows two single-stranded PCR products of lengths 71 and
72 (mass
difference = 305 Da = Adenosine) as well as the 72mer and 72mer + a single
matrix adduct (M)
(mass difference = 139 Da) to be well resolved (FWHM resolution = 240). FIG.
IB shows an 88
base length single-stranded product having a resolution of 330. Both spectra
display high
enough accuracy and resolution to detect a point polymorphism if one were
present.
CA 02301875 2000-02-25
WO 98/IZ355 PCT/US97117101
27
BEiVEFITS OF ANALYZING SINGLE-STRANDED NUCLEIC ACIDS
One object of this invention is the accurate mass determination of a single-
stranded
amplified target nucleic acid or a set of single-stranded amplified target
nucleic acids to
determine presence and character of any polymorphisms. The embodiments of this
invention
S include mass spectrometric mass determination of the single-stranded
amplified target nucleic
acid or set of different single-stranded amplified target nucleic acids, as
well as mass
determination of a mass-modified, single-stranded amplified target nucleic
acid or set thereof. A
preferred embodiment is to detect polymorphisms in an amplified target nucleic
acid in single-
stranded form, wherein the single-stranded amplified target nucleic acids) are
derived from one
of either the positive or the negative strand of the genome. The examples of
single-stranded
methods described herein focus on single-stranded amplified target nucleic
acids derived from
the positive strand, although the methods disclosed in the present invention
are equally
applicable to target nucleic acid as derived from the negative strand as well.
FIG.3 illustrates that a double-stranded target nucleic acid comprising two
complementary strands, produces two difficult to resolve peaks in the mass
spectrum
corresponding to the denatured single strands. The additional peaks from
double-stranded
amplified target nucleic acids as compared to single-stranded amplified target
nucleic acids add
to the congestion of mass peaks in the mass spectra, as well as introducing
the possibility that it
may be extremely difficult to resolve the complementary fragments if they have
nearly or exactly
identical base compositions. Furthermore, some portion of the double-stranded
amplif ed target
nucleic acids do not fully denature. and mass peaks corresponding to the
double-stranded
products increase the spectral congestion.
Spectra using both strands may also contain a two-fold redundancy in data,
since any
polymorphism in one strand may be present within its complement. One strand my
be removed
prior to mass spectrometric analysis while still producing all data necessary
for a complete
polymorphism analysis. Therefore, it is often preferable to analyze a set of
single strands, using
only one of~the two complementary sets of amplified target nucleic acids
representing the full set
' of target nucleic acids.
FIG. 4 shows the expected spectrum if only the positive strand of a target
nucleic acid
from FIG. 3 is analyzed by mass spectrometry as analysis of one of the two
complementary
Strands UI' the double-stranded amplified target nucleic acids halves the
number of expected
peaks within the mass spectra, allowing for better resolution. Thus. removal
of one of the two
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
28
strands from each amplified target nucleic acids eliminates the greatest
source of complication
for each spectra. A number of methods for isolating and preparing single-
stranded amplified
target nucleic acids for mass spectrometry are described herein.
S PURIFICATION METHODS
For mass spectrometry analyses, the target nucleic acids should be within the
resolvable
range and high mass accuracy range of the mass spectrometer. Additionally,
nucleic acid
fragments that do not contribute to the analysis and may unnecessarily
convolute the mass
spectra should be eliminated, if feasible.
With analysis methods such as gel electrophoresis, a mixture of specifically
labeled
nucleic acid fragments (radioactively or fluorescently tagged) can be
visualized in the presence
of other unlabeled nucleic acid fragments that comigrate but are invisible and
therefore do not
convolute analysis of the gel data. The mass spectrometric methods described
herein do not use
any form of labeling that could render certain fragments invisible, e.g. the
complementary strand
1 S in a double-stranded product, and it is therefore preferable to remove
such fragments prior to
analysis.
The samples should preferably be of relatively high purity prior to
introduction to the
mass spectrometer. The presence of impurities, especially salts, may greatly
affect the
resolution, accuracy and intensity of the mass spectrometric signal.
Contaminating primers,
residual sample genomic DNA, and proteins, all can affect the quality of the
mass spectra.
The purification methods of the present invention are well-suited to mass
spectrometric
analysis of nucleic acids. For example, the methods herein physically isolate
selected sets of
single-stranded or double-stranded amplified target nucleic acids from a
multiplicity of
impurities including undesirable nucleic acid fragments (including the
complementary strand and
2S flanking regions), proteins and salts, that would result in a poor quality
mass spectrum. These
isolation methods offer significant advantages due to the physical separation
of a desired set of
single-stranded or double-stranded amplified target nucleic acids from other
impurities in
preparation.
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
29
APPROACHES TO ISOLATING SINGLE-STRANDED OR DOUBLE-STRANDED AMPLIFIED TARGET
NUCLEIC ACIDS
. As described earlier, analysis of single-stranded amplified target nucleic
acids is
generally preferable since it provides a complete set of data with the minimal
number of
fragments and therefore simplif es the spectra and facilitates an increase in
the total number of
target nucleic acids that can be analyzed in a single assay. A number of
approaches can be taken
toward the production of single-stranded amplified target nucleic acids and
their purification
which includes the elimination of undesired oligonucleotides. In some cases,
it may be
preferable to use a method of amplification that yields primarily single-
stranded amplified target
nucleic acids, such as asymmetric PCR or transcription-mediated amplification.
To isolate the single-stranded amplified target nucleic acids, the amplified
target nucleic
acids may be designed to be attached or bound to a solid support. Several
means are available to
effect this attachment to a solid support, including: (a) hybridization to a
complementary, solid-
phase bound nucleic acid capture probe (which can be an oligonucleotide or one
strand of the
amplified target nucleic acid) comprising a first binding moiety that
specifically binds to a
second binding moiety attached to a solid phase; (b) direct binding of the
amplified target nucleic
acid strands of interest, each comprising a polynucleotide region of interest
and a first binding
moiety, to a second binding moiety attached to a solid phase (e.g.
biotin/streptavidin or avidin or
antigen/antibody pairs); or (c) direct covalent attachment of the strands of
interest to a solid
support.
A capture probe is an oligonucleotide that comprises a portion capable of
hybridizing to a
nucleic acid, such as an amplified target nucleic acid, and a binding moiety
that binds the capture
probe to a solid phase, either through covalent binding or affinity binding.
or a mixture thereof.
A capture probe can itself bind to a solid support via binding moieties
(direct capture) or can
bind to a solid support via another capture probe that binds to a solid
support (indirect capture).
A preferred embodiment is the use of a biotinylated amplified target nucleic
acid coupled
to streptavidin or avidin attached to a solid support where the strand of
interest is itself bound.
Biotin coupling to streptavidin (or avidin) requires that any amplified target
nucleic acid or acids
contain a biotin. The biotin is part of the linker molecule. It is
straightforward to capture the
amplified target nucleic acid because biotinylated primers can be used in the
PCR amplification.
If only one of the two strands of an amplified target nucleic acid is to be
analyzed by mass
spectrometry, only one of the two PCR primers for each different target
nucleic acid should be
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
bibtinylated. For each target nucleic acid, the PCR primer to be biotinylated
should be the
primer that is extended to form the single-stranded amplified target nucleic
acid of interest.
The amplified target nucleic acid or set of amplified target nucleic acids can
be covalently
attached to a solid support using any of the number of methods commonly
employed in the art to
5 immobilize an oligonucleotide or polynucleotide on a solid support. The
amplified target nucleic
acid or set of amplified target nucleic acids covalently attached to the solid
support should also
be stable and accessible for base hybridization.
Covalent attachment of the amplified target nucleic acid or set of amplified
target nucleic
acids to the solid support may occur by reaction between a reactive site or a
binding moiety on
10 the solid support and a reactive site or another binding moiety attached to
the target or via
intervening linkers or spacer molecules, where the two binding moieties can
react to form a
covalent bond. Coupling of an amplified target nucleic acid or set of
amplified target nucleic
acids to a solid support may be carried out through a variety of covalent
attachment functional
groups. Any suitable functional group may be used to attach the amplified
target nucleic acid or
15 set of amplified target nucleic acids to the solid support, including
disulfide, carbamate,
hydrazone. ester, N-functionalized thiourea, functionalized maleimide,
mercuric-sulfide, gold-
sulf de, amide, thiolester, azo, ether and amino.
The solid support may be made from a wide variety of materials, such as
cellulose,
nitrocellulose, nylon membranes, controlled-pore glass beads, acrylamide gels,
polystyrene,
20 activated de~ctran, agarose, polyethylene, functionalized plastics, glass,
silicon, aluminum, steel,
iron. copper. nickel and gold. Some solid support materials may require
functionalization prior
to attachment of an oligonucleotide or capture probe. Solid supports that may
require such
surface modification include aluminum, steel, iron, copper, nickel, gold,
silicon, and
nonfunctionalized polymers. Solid support materials for use in coupling to a
capture probe
25 include functionalized supports such as the l , l'-carbonyldiimidazole
activated supports available
from Pierce ( Rockford, IL) or functionalized supports such as those
commercially available from
Chiron Corp. (Emeryville, CA). Binding of an amplified target to a solid
support can be carried
out by reacting a free amino group of an amino-modified target with the
reactive imidazole
carbamate of the solid support. Displacement of the imidazole group results in
formation of a
30 stable N-alkyl carbamate linkage between the amplified target and the
support.
The amplified target nucleic acid or set of amplified target nucleic acids may
also be
bound to a solid support comprising a gold surface. The amplified target
nucleic acid or set of
CA 02301875 2000-02-25
WO 98112355 PCT/US9'7/17101
31
amplified target nucleic acids can be modified at their 5'-end with a linker
arm terminating in a
thiol group, and the modified amplified target nucleic acid or set of modified
amplified target
nucleic acids can be chemisorbed with high affinity onto gold surfaces
(Hegner, et al., 1993b).
In methods in which a solid-phase approach is used, preferably the double-
stranded
amplified target nucleic acid or set of amplified target nucleic acids may be
washed to remove
deleterious contaminants. However, when the amplified target nucleic acid
strands of interest are
directly bound, either covalently or via biotin/streptavidin biotin/avidin
interactions, it is
preferable to rigorously wash the sample to yield the highest purity. Such a
rigorous wash
typically removes the complementary strand, if present, isolating the single-
stranded amplified
target nucleic acid. Following washing, it is necessary to release single-
stranded amplified target
nucleic acids from the solid support for mass spectrometric analysis. The
isolation of a set of
single-stranded amplified target nucleic acids may be performed on the same
plate that is used
within the mass spectrometer or on a separate surface such as beads or a
filter. Both the capture
probe hybridization and biotin/streptavidin or biotin/avidin approaches can
use a number of
means of denaturation to disrupt the noncovalent interactions and afford
release of the set of
single-stranded amplified target nucleic acids bound to the solid support.
Alternatively, a cleavable linkage may be incorporated between the first
binding moiety
and the amplified target nucleic acids. Any covalent coupling chemistry may be
reversible or it
may be necessary to include a separate chemically cleavable linkage somewhere
within the
bound product. It may also be useful to use a chemically cleavable linkage
approach with the
biotinlstreptavidin (or avidin) strategies so that release of the double-
stranded target nucleic acids
can be performed under relatively mild conditions. In all cases the cleavable
linkage can be
located within the linker molecule connecting the biotin and the base (e.g. a
disulfide bond in the
linker), within the base itself (e.g. a more labile glycosidic linkage), or
within the phosphate
backbone linkage (e.g. replacement of phosphate with a phosphoramidate).
Another way to isolate single-stranded amplified target nucleic acids is to
use a primer
comprising an exonuclease blocking moiety and to treat with a 5' to 3'
exonuclease, which
digests the strand lacking an exonuclease blocking moiety and the portion of
the other strand up
to the exonuclease blocking moiety. leaving just the portion of the strand
containing the
exonuclease blocking moiety and the portion of that strand 3' to the
exonuclease blocking
moiety. This method is described in the methods of reducing length section
herein. Single-
CA 02301875 2000-02-25
WO 98/12355 PCT/US97l17101
32
stranded amplified target nucleic acids can also be isolated by using a DNA-
specific or RNA-
specific nuclease to digest an RNA/DNA hybrid.
The use of two primers each comprising an exonuclease blocking moiety, wherein
each
primer binds to a different complementary strand, is another way to isolate a
double-stranded
amplif ed target nucleic acid.
METHODS OF REDUCING LENGTH OF AMPLIFIED TARGET NUCLEIC ACIDS
After the amplification of target nucleic acids, the amplified target nucleic
acids, which
are in double-stranded form, can be cleaved with restriction endanucleases to
remove flanking
regions that are not within the region of interest, wherein said region of
interest is suspected of
containing a polymorphism.
If DNA restriction endonucleases are used to remove one or more flanking
regions from
an amplified target nucleic acid prior to isolating the single-stranded or
double-stranded
amplified target nucleic acid(s), it may be necessary for the amplified target
nucleic. acid to have
a double-stranded form prior to restriction, or more specifically, that the
restriction endonuclease
recognition sites and cleaving sites be located in double-stranded DNA regions
flanking or
outside the region of interest. The alternative to having fully double-
stranded DNA prior to
restriction is to hybridize restriction site oligonucleotide probes to single-
stranded DNA, wherein
the restriction site oligonucleotide probes are complementary to the
restriction sites for selected
restriction endonucleases.
The basic known methods for DNA isolation - precipitation, dialysis,
filtration and
chromatography do not isolate single-stranded from double-stranded DNA. If
these purification
methods are employed, and it may be desired to produce a single-stranded
product, it is
necessary to add a separate step where single-strand isolation is performed.
If restriction endonucleases are used to cleave off one or more regions from
an amplified
target nucleic acid. a preferred method for isolating single-stranded
amplified target nucleic acids
from these products is to use at least one biotinylated primer located at one
end of an amplified
target nucleic acid.
The production of reduced length amplified target nucleic acids can provide
benefits of
increased accuracy and resolution in the mass spectrometric analysis of even
double-stranded
amplified target nucleic acids. Double-stranded amplified target nucleic acids
can have their
length reduced in a similar manner to that used for processing single-stranded
amplified target
CA 02301875 2000-02-25
WO 98/12355 PCTIUS97/17101
33
nucleic acids. Either endogenous restriction recognition sites outside the
region of interest or
primer-incorporated restriction recognition sites, as described below, or
combinations thereof can
be used. Endogenous restriction recognition sites are those that are found
naturally within one or
more flanking regions.
In cases where one or more endogenous restriction recognition sites cannot be
found
outside the region of interest, an alternative method is necessary for
reducing the length of the
amplified target nucleic acid. Use of a modified primer during the
amplification process can
mediate the incorporation of a Type II or Type IIS restriction endonuclease
recognition site
within a primer region of the amplified target nucleic acid. Type IIS
restriction endonucleases
recognize a particular double-stranded sequence region and selectively cleave
the double strand a
defined distance away from the recognition site. As an example, the
restriction enzymes Bpml
and Bsgl cleave the double strands 14 nucleotides (top strand) and 16
nucleotides (bottom strand)
away from the recognition sites. Other representative Type IIS restriction
enzymes include
BseRl, BsmRl and Fokl. See New England Biolabs 1996 Product Catalog. Use of
Type IIS
restriction for the reduction of amplified target nucleic acids is illustrated
in FIG. 7.
The restriction method for reducing the length of an amplified target nucleic
acid affords
significant advantages where double-stranded amplified target nucleic acids
are to be analyzed
by mass spectrometry. For instance, the smaller molecules are easier to
resolve. Moreover, a
second beneficial effect of using restriction endonucleases to reduce length,
specifically one that
does not produce blunt ends, is the production of two strands of different
lengths and hence
different masses. The creation of two complementary strands of different
lengths, e.g. 4 to 6
nucleotides difference in size, yields dramatically improved separation and
resolution of two
complementary strands during mass spectrometric analysis as shown in FIG. 4.
In many cases,
reduction of length by restriction endonuclease digestion can eliminate the
need for single-strand
isolation.
In one .embodiment, the restriction endonuclease recognition site can be the
same as the
site of cleavage, located in the flanking region opposite from the end of the
amplified target
nucleic acid that is bound to the solid support. In another embodiment, the
restriction
endonuclease recognition site is different from the site of cleavage, as in
the case of Type IIS
restriction endonucleases, which cleave at a defined distance (20-40 bases)
from one side of their
recognition sequence. When a Type IIS restriction endonuclease is used to
reduce the length of
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
34
an-amplified target nucleic acid, both the recognition site and the site of
cleavage are commonly
located outside of the region of interest and in the flanking region.
One of ordinary skill will readily appreciate that many combinations arid
variations of
these methods for reducing length are possible. For example, endogenous Type
II restriction
S recognition sites for one or more Type II restriction endonuclease can be
used to reduce length
on one or both regions flanking a region of interest. Alternatively,
endogenous Type II and
endogenous Type IIS restriction recognition and cleavage sites can be used to
reduce length by
cleaving in one or more flanking regions. Also, one or more restriction
recognition sites can be
introduced using a mismatch primer or an overhang primer containing one or
more new
restriction recognition sites. A mismatch primer is one which contains at
least a single base
mismatch with the target nucleic acid to be amplified and can include primers
that have an
overhang region that does not hybridize to the target nucleic acid. Mismatch
primers are a type
of cleavable primer. Alternatively, endogenous restriction recognition sites
and primer-
introduced restriction recognition sites can be combined. CIeavable sites can
also occur outside
I S the primer region, ranging from 20-50 nucleotides away from the end of the
primer region. All
of these types of primers are cleavable primers because they contain a site,
moiety or group, that
can be used to reduce the length of the target nucleic acid. For example,
cleavable primers may
contain a recognition site, a cleavable site. or an exonuclease blocking
moiety.
Another method of reducing length involves the use of a primer comprising an
exonuclease blocking moiety, wherein said exonuclease blocking moiety prevents
a 5' to 3'
exonuciease from digesting a region of interest that is 3' to said exonuclease
blocking moiety.
The exonuclease blocking moiety can include modified nucleotides that prevent
5' to 3'
exonuclease activity from continuing, o.g. phosphorothioates, methyl
phosphonates, borano
phosphates, and peptide nucleic acids (PNA). FIG. 9 depicts the use of an
exonuclease blocking
moiety and an exonuclease to reduce the length of a target nucleic acid. The
nucleotides that are
degraded by the exonuclease are a multiplicity of cleavable sites. including
many that are
adjacent to one another.
Peptide nucleic acids are modified DNA mimics in which the sugar-phosphate
backbone
has been replaced with a backbone based on amino acids. Peptide nucleic acids
exhibit
sequence-specific binding to DNA and RNA and are resistant to nuclease and
protease attack.
See Buchardt et al., 1993. A preferred 5' to 3' exonuclease is Exonuclease
1II. FIG. 9 illustrates
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/1'7101
hover the use of this method results in isolation of a reduced-length single-
stranded amplified
target nucleic acid.
The exonuclease approach to reducing length can be also used in combination
with one or
more of the restriction endonuclease cleavage techniques described above to
reduce length. In
S such a combined approach, the restriction cleavage should occur before the
exonuclease
digestion.
One of ordinary skill in the art will appreciate that the above methods of
reducing length
may also be used as means of isolating a reduced-length amplified target
nucleic acid or a
reduced-length single-stranded amplif ed target nucleic acid. For example, a
chemically
10 cleavable site can be incorporated in a flanking region and cleavage at
that site can accomplish
both length reduction and release from a solid support at the same time. Thus,
for example, after
the amplified target nucleic acid is reduced in length, either the bound
strand or the unbound
strand or both strands of said amplified target nucleic acid can be isolated
for mass spectrometric
analysis.
15 In cases where the single-stranded amplified target nucleic acid strand to
be analyzed is
directly bound to the solid phase, it can be rigorously washed to remove
unbound components,
including any number of deleterious contaminants, including the unwanted
complementary
strand, nucleic acid fragments containing at least a portion of a flanking
region, salts, enzymes,
and other reagents. These unbound components can be removed from any nucleic
acid bound to
20 a solid support in any of the embodiments described herein and combinations
thereof. If the
strand to be analyzed (the strand of interest) is not bound directly but
rather via hybridization to a
complementary nucleic acid, it cannot be as rigorously washed and thereby
cannot be purified to
as great an extent. Direct binding of the amplified target nucleic acid strand
to be analyzed
ultimately produces a higher quality signal, e.g. less salt adducts, durino
mass spectrometric
25 analysis. thus improving mass resolution and accuracy.
Following the necessary wash steps, the single-stranded amplified target
nucleic acids are
released from the solid support and analyzed by mass spectrometry. Note that
regions that are
cleaved off by one or more restriction endonucleases are released into
solution and washed away,
and are therefore not analyzed. Loss of these flanking regions can enhance the
ability for mass
30 spectrometry to quickly identify the existence of a polymorphism. The
isolation of the single-
stranded amplified target nucleic acids occurs prior to the mixing of the
single-stranded
amplified target nucleic acids with the matrix material for mass spectrometric
analysis.
CA 02301875 2000-02-25
WO 98/12355 PGT/US97/17101
36
To release the reduced-length single-stranded amplified target nucleic acid
from the solid
support, several methods can be used, depending on the methods used to bind
the amplified
target nucleic acid to the solid support. Both the hybridization and
biotin/streptavidin (or avidin)
methods can use a number of means of denaturation to disrupt noncovalent
interactions and
S cause the release of the bound single-stranded amplified target nucleic
acids. Any covalent
chemistry used must be either reversible or include a separate chemically
cleavable site
somewhere within the bound product. It may be preferred to use a chemically
cleavable site with
the biotin/streptavidin (or avidin) method so that release of the target
nucleic acids can be
performed under relatively mild conditions. In all cases, the cleavable site
can be located within
a linker molecule connecting the biotin and the base (e.g. a disulfide bond in
the linker), within
the base itself (e.g. a more labile glycosidic linkage), or within the
phosphate backbone linkage
(e.g. replacement of phosphate with a phosphoramidate).
In a preferred embodiment, the cleavable site is located near the 3' end of
the primer used
to bind the amplified target nucleic acid to the solid support. By locating
the cleavable site near
the 3' end, it is possible to further reduce the length of the amplified
target nucleic acid,
eliminating a flanking region from the polynucleotide region of interest.
Cleavable primers are
described in PCT/US96/06116, filed April 26. 1996 (incorporated herein by
reference).
IMPROVING MASS ACCURACY BY INTERNAL CALIBRATION AND INTERNAL SELF-CALIBRATION
Mass spectrometers are typically calibrated using analyses of known mass. A
mass
spectrometer can then analyze an analyte of unknown mass with an associated
mass accuracy and
precision. However. the calibration, and associated mass accuracy and
precision, for a given
mass spectrometry system (including MALDI-TOF MS) can be significantly
improved if
analyses of known mass are contained within the sample containing the
analyte(s) of unknown
mass(es). The inclusion of these known mass analyses within the sample is
referred to as use of
internal calibrants. External calibrants, i.e. analyses of known mass that are
not mixed in with
the set of target nucleic acids of unknown mass and simultaneously analyzed in
a mass
spectrometer. are analyzed separately. External calibrants can also be used to
improve mass
accuracy. but because they are not analyzed simultaneously with the set of
target nucleic acids of
unknown mass. they will not increase mass accuracy as much as internal
calibrants do. Another
disadvantage of using external calibrants is that it requires an extra sample
to be analyzed by the
mass spectrometer. For MALDI-TOF MS. generally only two calibrant molecules
are needed for
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
37
complete calibration, although sometimes three or more calibrants are used.
All of the
embodiments of the invention described herein can be performed with the use of
internal
caIibrants to provide improved mass accuracy.
Using the methods described herein, one can obtain a mass spectrum with
numerous mass
peaks corresponding to the set of single-stranded amplified target nucleic
acids under study. If
no polymorphism is present in any of said target nucleic acids, all of the
mass peaks
corresponding to the amplified target nucleic acids will be at mass-to-charge
ratios associated
with the set of amplified target nucleic acids from the wild type target
nucleic acids. However, if
a target nucleic acid contains a polymorphism, usually no more than one or two
of the mass
peaks will be shifted in mass, leaving the majority of mass peaks at unaltered
locations. In a
preferred embodiment of the invention. a self calibration algorithm uses these
nonpolymorphic
or unmutated target nucleic acids for internal calibration to optimize the
mass accuracy for
analysis of the single-stranded amplified target nucleic acids containing a
polymorphism, thus
requiring no added calibrant(s), simplifying the calibration, and avoiding
potential spectral
overlaps. In a given sample, however. it will not be known a priori which mass
peaks, if any,
are altered or shifted from their expected masses for the wild type target
nucleic acids.
The self calibration algorithm begins by dividing up the observed mass peaks
into
subsets. each subset consisting of all but one or two of the observed mass
peaks. Each data
subset has a different one or two mass peaks deleted from consideration. For
each subset, the
algorithm divides the subset further into a first group of two or three masses
which are then used
to generate a new set of calibration constants, and a second group which will
serve as an internal
consistency check on those new constants. The internal consistency check
begins by calculating
the mass difference between the m/z values calculated for the second group of
mass peaks and
the values corresponding to reasonable choices for the associated wild-type
target nucleic acids.
The internal consistency check can thus take the form of a chi-square
minimization where the
key parameter is this mass difference. The algorithm finds which data subset
has the lowest sum
of the squares of these mass differences resulting in a choice of optimized
calibration constants
associated with group one of this data subset.
After new self optimized calibration constants are obtained, the mass-to-
charge ratios are
determined for the mass peaks omitted from the data subset; these are the
amplified target
nucleic acids suspected to contain a polymorphism. The differences from the
observed mass
peaks for the wild type amplified target nucleic acids are then used to
determine whether a
CA 02301875 2000-02-25
WO 98/12355 3 g PGT/I1S97/17101
polymorphism is present, and if so, what the nature of this polymorphism is
(e.g. the exact type
of deletion, insertion, or point polymorphism). This self calibration
procedure should yield a
mass accuracy of approximately 1 part in 10,000.
The methods described herein permit MALDI-TOF MS analysis of single-stranded
S amplified target nucleic acids which has a mass accuracy of approximately 1
part in 10,000. The
use of internal self caiibrants makes it possible to extend this level of
accuracy up to and
potentially beyond 30,000 Da or 100 bases. This mass accuracy enables exact
sizing of one or
more target nucleic acids and the determination of the presence and nature of
any polymorphism,
including point polymorphisms, insertions and deletions. Further described
herein are methods
for improving the resolution of individual target nucleic acids by means
including elimination of
equal-length complementary pairs through the use single-strand-targeted
isolation procedures,
and the incorporation of mass-modified nucleotides to enhance the mass
difference between
similar sized amplified target nucleic acids and/or wild type amplified target
nucleic acids. In
addition, these methods provide for the removal of salts and other deleterious
materials as well as
a means for the removal of unwanted nucleic acid fragments prior to mass
spectroscopic
analysis.
MASS RESOLUTION, MASS ACCURACY, AND THE USE OF MASS-MODIFIED NUCLEOTIDES
Any of the embodiments of the invention described herein optionally include
amplified
target nucleic acids having one or more nucleotides replaced with mass-
modified nucleotides,
wherein said mass-modified nucleotides comprise nucleotides or nucleotide
analogs having
modifications that change their mass relative to the nucleotides that they
replace. The mass-
modified nucleotides incorporated into the target nucleic acids of the
invention must be amenable
to the enzymatic and nonenzymatic processes used for the amplification of
target nucleic acids.
For example, the mass-modified nucleotides must be able to be incorporated by
DNA or RNA
polymerase during amplification of the target nucleic acid. Moreover. the mass-
modified
nucleotides must not inhibit the processes used to process the target nucleic
acids, including,
inter alia, specific cleavage by restriction endonucleases, whenever such
steps are used. Mass-
modifications can also be incorporated in the target nucleic acids of the
invention after the
enzymatic steps have been concluded. For example, a number of small chemicals
can react to
modify specific bases, such as kethoxal or formaldehyde.
CA 02301875 2000-02-25
WO 98112355 39 PCTIUS97117101
- - Any or all of the nucleotides in the target nucleic acids can be mass-
modified, if
necessary, to increase the spread between their masses. It has been shown that
modifications at
the CS position in pyrimidines or the N7 position in purines do not prevent
their incorporation
into growing nucleic acid chains by DNA or RNA polymerase (Lee et al., 1992).
For example,
an octynyl moiety can be used in place of methyl on thymidine to alter the
mass by 94 Da.
Mass-modifying groups can be, for example, halogen, alkyl, ester or polyester,
ether or
polyether, or of the general type XR, wherein X is a linking group and R is a
mass-modifying
group. The mass-modifying group can be used to introduce defined mass
increments into the
target nucleic acids. One of skill in the art will recognize that there are
numerous possibilities
for mass-modifications useful in modifying nucleic acid fragments or
oligonucleotides, including
those described in Oligonucleotides and Analogues: A Practical Approach,
Eckstein ed. (Oxford
1991 ) and in PCT/US94/00193, which are both incorporated herein by reference.
At larger mass ranges (30,000-90,000 Da), the mass resolution and mass
accuracy of
current MALDI-TOF mass spectrometers will not typically be sufficient to
identify a single base
1 S change. For this reason, it may be preferable to increase the useful mass
range artificially by
substituting standard nucleotides within a target nucleic acid with mass-
modified nucleotides
having significantly larger mass differentials. Use of mass-modified
nucleotides applies as well
to the mass range below 30,000 Da. Mass modification can generally increase
the quality of the
mass spectra by enlarging the mass differences between different amplified
target nucleic acids
of similar size and composition. For example, mass-modified nucleotides can
increase the
minimum mass difference between two amplified target nucleic acids that happen
to be identical
in base composition except for a single base which is an A in one and is a T
in the other.
Normally, these two target nucleic acids will differ in mass by only 9 Da. By
replacing one of
- the bases with a mass-modified version during amplification, the mass
difference can be > 20 Da.
The illustrations of spectra in FIG. 2A and FIG. 2B depict the influence mass-
modified
nucleotides can have on target nucleic acid resolution. One example of the
many possible mass
modifications useful in this invention is the use of 5-(2-heptynyl)-
deoxyuridine in place of
thymidine. The replacement of a methyl group by heptynyl changes the mass of
this particular
nucleotide by 65 Da. An A to T transversion in a nucleic acid in which all
thvmidine bases have
been replaced with 5-(2-heptynyl)-deoxyuridine would produce a peak shift of
~6 Da as opposed
to 9 Da for the same nucleic acid fragments without the mass-modified
nucleotides. The use of
mass-modified nucleotides is especially important in the analysis of single-
stranded target
CA 02301875 2000-02-25
WO 98/12355 40 PCT/US97/17101
nucleic acids derived from RNA. Normally, the masses of C and U vary by only 1
Da, making it
practically impossible to detect C to U or U to C point polymorphisms within a
given target
nucleic acid.
Each of the techniques described herein can be used in combination with any of
the
isolation methods also described herein. Moreover the techniques can be used
in combination
with each other, as one of ordinary skill in the art using the techniques
described herein how to
combine the different aspects of the invention. All of these methods and
combinations thereof
can optionally include the use of mass-modified nucleotides and internal
calibrants.
KtTs
The present invention also includes kits for preparing nucleic acids for mass
spectrometric analysis. In one embodiment, a kit comprises: a first primer
capable of binding a
first strand of one of said target nucleic acids at a region 5' to a region of
interest of said target
nucleic acid; a second primer capable of binding a second strand complementary
to said first
strand at a region 5' to said region of interest of said target nucleic acid;
a DNA polymerase
capable of extending said primers to form primer extension products of said
first and second
primers; and a restriction endonuclease capable of reducing length of
amplified target nucleic
acids. The restriction endonuclease can be a type II restriction endonuclease
or a Type IIS. The
first primer may be biotinylated, i.e. comprise at least one biotin. The kit
may also comprise a
solid support capable of selectively binding either a positive strand or a
negative strand
comprising the region of interest of an amplified target nucleic acids, and a
matrix solution.
In another embodiment, a kit comprises a first primer capable of binding a
first strand of
one of said target nucleic acids at a region 5' to a region of interest of
said target nucleic acid; a
second primer capable of binding a second strand complementary to said first
strand at a region
5' to said region of interest of said target nucleic acid; a DNA polymerase
capable of extending
said primers to form primer extension products of said first and second
primers: wherein the first
primer is cleavable by chemical or enzymatic treatment. The cleavable primers
of the invention
include those comprising a chemically cleavable site, an exonuclease blocking
moiety, a Type
IIS restriction endonuclease recognition site, or at least one biotin, but
does not include a Type II
restriction endonuclease recognition site where one of the complementary
strands cannot be
cleaved by said Type II restriction endonuclease. This kit may optionally
comprise a solid
support and a matrix solution.
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
41
- - The following examples are provided to illustrate embodiments of the
invention, but do
not limit the scope of the invention.
EXAMPLES
Example 1. PCR Amplification of a Single Target Nucleic Acid.
An example PCR protocol that may be employed in this invention is as follows.
A sample containing 10-10,000 copies of a source DNA may be mixed with two
antiparallel
DNA primers that surround a target nucleic acid, e.g. the coding region for a
gene involved in
carcinogenesis. The target nucleic acid may be any sequence that is known or
suspected to be
polymorphic, including STRs, SSLP, and genetic deletions, insertions, or point
polymorphisms.
The PCR mix may typically be composed of: 8 ~l 2.5 mM deoxynucleoside
triphosphates, 10 ~1
1 OX PCR buffer, 10 ~ 1 25 mM MgCl2, 3 ~ 1 10 pM forward primer, 3 p l 10 ~M
reverse primer,
0.3 ~1 thermostable Taq DNA polymerase, 64.? pl HzO, and 1 pl source DNA. The
sample tube
may then be sealed and placed into a thermal cycling device. A typical cycling
protocol is as
follows:
Step 95C 2 min.
1
Step 95C 15 sec.
2
Step 55C 15 sec.
3
Step 72C 1 min.
4
Step repeat Steps 2-4 35
5 times
Step 72C 5 min.
6
Step stop
7
Example 2. Production of Single-Stranded Nucleic Acids by Asymmetric PCR.
The basic PCR procedure of Example 1 can be modified in order to produce
predominantly one of the two strands. These asymmetric procedures involve
modifying the
ratios of the two primers, a typical ratio is 10:I. These procedures are
described in Molecular
Cloning: A Laboratory Manual, Sambrook et al., 1989 (incorporated by reference
herein).
CA 02301875 2000-02-25
WO 98/12355 42 PCT/US97/17101
Example 3. Production of Single-Stranded DNA via Biotinylated PCR Products.
For the preparation and capturing of amplified target nucleic acids to a solid
support, one
of the two primers used in PCR amplification may be synthesized with a biotin
moiety internally
or at the 5' end of the oligonucleotide. Following a standard PCR, the double-
stranded product
S can be bound to a solid-phase surface coated with streptavidin. For example,
10 pmol of double-
stranded PCR product is mixed with 5 p, l of 10 mg/ml paramagnetic
streptavidin-coated beads in
a binding/washing buffer of 2.0 M NaCI, 10 mM TrisCl, 1 mM EDTA, pH 8Ø The
solution is
incubated for 15 min. at room temperature with mixing. Following incubation
the tube is placed
next to a high field, rare earth magnet and the paramagnetic beads with the
bound biotinylated
PCR product are precipitated to the wall of the tube. The supernatant is
removed, and the
particles, outside the influence of the magnetic field, are resuspended into
binding/washing
buffer. The beads and wash solution are mixed and then subjected once again to
the magnetic
field to precipitate the magnetic particles. The supernatant is once again
removed and either the
wash step is repeated or the alkaline denaturation step commences. To release
the unbiotinylated
strand from the double-stranded product, the beads are mixed with an alkaline
denaturation
solution such as, 0.1 M NaOH. The beads are incubated at room temperature for
10 min. which
denatures the PCR product and releases the unbiotinylated product into
solution. The
biotinylated strand, bound to the magnetic heads is precipitated from the
solution under the
magnetic field and unbiotinylated strand, now single-stranded, can optionally
be transferred to a
new tube with the supernatant and readied for mass spectrometric analysis.
The bound single-stranded amplified target nucleic acid can be released from
the
streptavidin-coated beads using one of a number of different procedures. These
procedures
include denaturation of biotin/streptavidin bond by heat denaturation (95
°C for ~ min.), or the
use of a denaturant, such as NaOH ( 1 mM NaOH for 15 min. at 65°C), and
use of a secondary
cleavable site, such as a disulfide linkage ( 100 mM DTT (dithiothreitol) for
1 ~ min. at room
temperature) or a ~' thiolated nucleotide (0.1 mM AgN03 for I S min. at room
temperature)
present within the primer.
Example 4. Mass Modification of Target Nucleic Acids.
Mass modification of the target nucleic acid is typically performed during the
amplification step. One or more standard deoxynucleoside triphosphates are
replaced with
modified deoxynucleoside triphosphates. As an example thymidine may be
replaced with a 5-
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
43
alkynyl-substituted-2'-deoxyuridine triphosphate. Because the modified
nucleotides may not be
efficient substrates for DNA polymerase it may be necessary to increase the
concentration of the
corresponding triphosphate by a factor of 2 to 100 over normal levels.
Example 5. Analysis of Single-Stranded Amplified Tetranucleotide Repeat Region
of
THO1 Gene.
A sample of human genomic DNA is subjected to PCR amplification with the
primer
pair: (SEQ ID NO: 2) 5'-Biotin-GTGATTCCCATTGGCCTGT(sT)CCTC-3' and (SEQ ID
N0:2)
5'- AGTGCAGGTCACAGGGAACACAGA-3', which selectively amplify the tetranucleotide
repeat region of the tyrosine hydroxylase ("THOI ") gene to give 90-114 by PCR
products where
sT is 5'-thiolated thymidine, also known as 2'-deoxythymidine-5'-(S)-
phosphorothioate (see FIG.
16). These PCR amplified target nucleic acids are about a factor of 2 smaller
products than
current commercially available primers provide. The PCR reaction is performed
on a 50 p.L
scale using 17.5 pmol of each primer and 50-100 ng of template, with 30 3-step
thermal cycles,
following an initial step at 94°C for 2 min, of 94°C for 45 s
followed by 55°C for 30 s and then
72°C for 30 s. A final step at 72°C for S min is added to
complete the reaction. The biotinylated
product is bound to streptavidin-coated magnetic beads MPG (CPG Inc., Great
Neck, NY) and
then subjected to denaturation conditions of 0.1 M NaOH for 10 minutes. The
solid-support-
bound single-stranded target nucleic acid is then subjected to extensive
washing with 10 mM
ammonium acetate and deionized water. The single-stranded amplified target
nucleic acid is
then released from the beads by cleavage of the P-S bond in the cleavable
primer with 0.1 mM
AgN03. After 2 pL of I00 mM dithiothreitol is added to sequester the Ag+ ion,
the sample is
evaporated to dryness in a Speed-vac concentrator. For analysis. the sample is
redissolved in
1 pL deionized H20 and is mixed with 1 pL of matrix solution consisting of 3-
HPA
(3-hydroxypicolinic acid) in acetonitrile:H20 1:1. The sample is deposited
onto a silicon stage,
dried under a gentle flow of nitrogen and is placed into the mass
spectrometer.
The experimental apparatus used for analyzing the sample amplified target
nucleic acids
is composed of an excitation source, a sample manipulator and a TOF mass
spectrometer. The
excitation source used for desorption is a Nd:YAG laser. The laser is operated
at a 10 Hz
repetition rate, with a 5 nanosecond pulse width. The desorption laser beam
maintained at an
incident angle of 45° is focused onto the sample with a 250 mm focal
length AL-2 coated
spherical mirror to an elliptical spot size of approximately 100 by 150 ym. n
Clan-laser
CA 02301875 2000-02-25
WO 98/12355 44 PCT/US97/17101
polarizer (Newport Corporation, Fountain Valley, CA) is placed in a rotation
stage in the beam
path for continuously variable attenuation, allowing adjustment of the
polarized Nd:YAG laser
energy density from below 1 mJ/cm2 to 100 mJ/cm2. The optimum energy density
for desorption
is in the range of 10 to 20 mJ/cm2.
Mass spectra are recorded in positive-ion mode at room temperature. The sample
region
is evacuated by a 300 liter per second turbomolecular pump. The drift and
detection regions are
evacuated using a cryopump with nominal 1500 liter per second pumping speed.
The base
pressure of the chamber is 3 x 10-9 Torr, and the normal working pressure,
within about five
minutes of sample introduction, is S x 10-8 Torr. A total of 100 laser shots
are summed to obtain
a spectrum. The spectrum shown in FIG. 13 reveals two clear peaks
corresponding to an 84-mer
and a 91-mer, which are the expected product sizes corresponding to 8 and 9.3
repeats with one
extra adenine base added to the 3'-end of each, due to the well-known property
of Tag DNA
polymerise to yield one-base over-extensions. The two peaks arise because the
final sample,
though single-stranded, derives from amplification of a heterozygous allele.
Example 6. Comparison of mass spectra for a single-stranded amplified THOl
ladder of
nucleic acids and a single-stranded amplified THOI ladder that have had their
lengths
reduced by endonuclease cleavage.
A I pL sample of TH01 ladder (Promega Inc., Madison, WI) which contains PCR
products ranging in size from 179-203 by is reamplified in a 100 pL reaction
volume according
to the same amplification protocol as described above for genomic DNA to yield
a ladder of
products in the size range of 90-I 14 bp. One half of the product mixture
(amplified target
nucleic acids) is bound to streptavidin-coated magnetic beads, denatured,
washed and cleaved
from the beads as described in the previous example. The other half of the
product mixture is
then bound to the streptavidin-coated magnetic beads and washed. Then, 2 ~.L
of lOX NEB
Buffer 4 is added to the second half of the amplified target nucleic acids,
followed by the
addition of I6 pL of H20 and 20 units of Nco I restriction endonuclease (New
England Biolabs,
Beverly, MA) recognizing CCATGG. This restriction endonuclease was chosen to
reduce the
length of the amplified target nucleic acid because the amplifed target
nucleic acid contained a
Nco I recognition site (in the wild type or consensus sequence) which was
located in a flanking
region and not in the region of interest. The mixture is incubated at
37°C for I hour after which
the enzyme is washed away, the amplified target nucleic acids are bound
through biotinylated
CA 02301875 2000-02-25
WO 98/12355 PCTIUS97/17101
primers, denatured, washed and single-stranded amplified target nucleic acids
are then cleaved
from the beads with AgN03. The restriction enzyme-digested ladder has a size
range of 40 to 64
bases as a result of the enzyme cutting 3 by past the end of the final CATT
repeat. Samples of
both undigested/full-length and digested/reduced-length products are prepared
for mass
. 5 spectrometry analysis by reducing the volume by evaporation and adding I
p,L of 3-
hydroxypicoIinic acid matrix solution and allowing to dry on the sample plate.
The resulting
positive-ion mass spectra of the undigested and digested ladders are shown in
FIG. 14 and
FIG. 15, respectively. The mass resolution of the peaks for the digested
ladder (FIG. 15) is much
greater than that for the undigested ladder (FIG. 14). This is due to the use
of Nco I to reduce the
10 length of the single-stranded amplified target nucleic acids that were
subjected to mass
spectrometric analysis.
Example 7. Mass Spectrometry Analysis.
The single-stranded amplified target nucleic acid sample to be analyzed is
typically
15 mixed with an equal volume of matrix solution consisting of 0.5 M 3-
hydroxypicolinic acid (3-
HPA) and SO mM diammonium hydrogen citrate. Typically, a 1 ~L portion of the
sample is
applied to the mass spectrometer sample stage and allowed to dry under a
gentle stream of
nitrogen gas at room temperature. When the sample has completely dried to form
crystals
(typically 5 min.) the sample is inserted into the mass spectrometer for
analysis. The usual
20 analysis conditions employ the use of a Nd:YAG laser operating at 266 nm
with an average pulse
energy of 15 mJ/cm'. An average of 100 laser shots is typically used to obtain
a spectrum.
All publications and patent applications mentioned in this specification are
herein
incorporated by reference to the same extent as if each individual publication
or patent
application was specifically and individually indicated to be incorporated by
reference.
25 The invention now being fully described, it will be apparent to one of
ordinary skill in the
art that many changes and modifications can be made thereto without departing
from the spirit or
scope of the invention and the appended claims.
CA 02301875 2000-02-25
WO 98/12355 PCT/US97/17101
46
- REFERENCES
The following references, to the extent that they provide exemplary procedural
or other
details supplementary to those set forth herein, are specifically incorporated
herein by reference.
Ahrams et al., "Comprehensive Detection of Single Base Changes in Human
Genomic DNA
Using Denaturing Gradient Gel Electrophoresis and a GC Clamp," Genomics,
7:463,
1990.
Buchardt et al., Trends Biotechn., 11(9):384-386, 1993.
Eckstein ed., A Practical Approach, Oxford, 1991.
l 0 Fenn et al., Science, 246:64-71, 1989.
Hegner et al., Surface Sci., 291:39-46, 1993b.
Lee et al., "DNA Sequencing with Dye-Labeled Terminators and T7 DNA
Polymerase: Effect of
Dyes and dNTPs on Incorporation of Dye-Terminators and Probability Analysis of
Termination Fragments." Nuc. Acids. Res., 20:2471, 1992.
Nelson et al., "Detection of Human IgM at m/z ~ I MDa, "Rapid Commun. Mass
,Spectrum.,
9:625, 1995.
Orita et al., "Detection of Polymorphisms of Human DNA by Gel Electrophoresis
as Single-
Stranded Conformation Polymorphisms," Proc. Natl. Acad Sci. USA, 86:2766,
1989.
PCT/US94/00193
Saleeba and Cotton. "Chemical Cleavage of Mismatch to Detect Mutations,"
Methods in
Enzymology 217:286, I 993.
Sambrook, Fritsch and Maniatis, eds., Molecular Cloning: A Laboratory Mantrcrl
2d ed.,
14.28-14.29, 1989.
Spengler et al., "Laser Mass Analysis in Biology," Ber. Bunsenges. Phys.
Chem., 93:396-402,
1989.
Tanaka et al., "Protein and Polymer Analyses up to m/z I 00,000 by Laser
Ionization Time-of
flight Mass Spectrometry," Rapid Commun. el~lass ,Spectrum., 2:151-153, 1988.
Tang et al., Rapid Commun. in Mass Spectrum., 8:183-I 86. 1994.
Wu, et al., Rapid C'ommuns. in Nlass Spectrometry, 7:142-146,1993.
Youil et al., "Screening ti>r Mutations by Enzyme Mismatch Cleavage with T4
Endonuclease
VII," Proc. Natl. ~1 ccrct. Sci. USA, 92:87, 1995.
CA 02301875 2000-02-25
WO 98112355 4,~ PCT/US97/17101
- SE(~UENCE LISTING
(1) GENERAL INFORMATION:
(i) APPLICANT:
(A) NAME: GENETRACE SYSTEMS, INC.
(B) STREET: 333 Ravenswood Avenue, PN 083
(C) CITY: Menlo Park
(D) STATE: CA
(E) COUNTRY: US
(F) POSTAL CODE (ZIP): 94025
(G} TELEPHONE: (512) 418-3000
(H) TELEFAX: (713) 789-2679
(ii) TITLE OF INVENTION: METHODS OF PREPARING NUCLEIC ACIDS FOR MASS
SPECTROMETRIC ANALYSIS
(iii) NUMBER OF SEQUENCES: 2
(iv) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
(C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: PatentIn Release #1.0, Version #1.30 (EPO)
(vi) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: US 08/759,993
(B) FILING DATE: 02-DEC-1996
(vi) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: US 60/032,369
(B) FILING DATE: 02-DEC-1996
(vi) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER: US 08/715,582
(B) FILING DATE: 19-SEP-1996
(2) INFORMATION FOR SEQ ID NO: 1:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 24 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ix) FEATURE:
(A) NAME/KEY: misc_feature
(B) LOCATION:1
(D) OTHER INFORMATION:/note= "5' end is biotinylated"
(ix) FEATURE:
(A) NAME/KEY: modified_base
(B) LOCATION:20
(D) OTHER INFORMATION:/note= "5'-thio-thymidine"
CA 02301875 2000-02-25
WO 98/12355 PCT/US97I17101
48
- -(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 1:
GTGATTCCCA TTGGCCTGTT CCTC 24
(2) INFORMATION FOR SEQ ID NO: 2:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 24 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(xi) SEQUENCE DESCRIPTION: SEQ ID NO: 2:
AGTGCAGGTC ACAGGGAACA CAGA 24