Note: Descriptions are shown in the official language in which they were submitted.
i i
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
ANTIVIRAL AGENTS
The present invention relates to the use of purine acyclonucleosides as agents
in the
' treatment and/or prophylaxis of hepatitis B, pharmaceutical compositions for
use in such
therapy and novel purine acyclonucleosides.
Infection with human hepatitis B virus is a major public health problem
because of the
ability of the virus to cause acute and chronic infections. Chronic hepatitis
B virus
infection (hereinafter referred to as HBV ) causes serious liver disease in
humans and
frequently results in cirrhosis and hepatocellular carcinoma. Currently there
is no
effective therapy for the successful management of chronic HBV infections. The
> 250
million chronic HBV carriers throughout the world are unable to benefit from
the
commercial vaccine now available.
Currently available therapies for HBV provide inadequate levels of efficacy or
are
I 5 accompanied by deleterious side effects. Accordingly, a need exists for
effective
treatments for HBV.
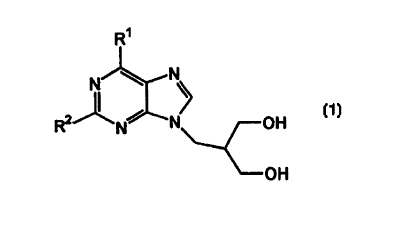
It has now been discovered that compounds of formula (1) are active agents
against
hepatitis B virus.
Accordingly, in one aspect of the present invention there is provided the use
of a
compound of formula ( 1 )
R'
N \ N
R2 N N OH
OH
(I)
CA 02302630 2000-03-06
WO 99!12927 PCT/AU98/f:0748
-2-
where:
R' is hydrogen, halogen, hydroxy, azido, alkoxy, aryloxy, thio, alkylthio,
amino,
alkylamino, hydrazino, hydroxylamino, benzyloxy, NRR' or NRCOR';
RZ is hydrogen, halogen, hydroxy, azido, alkoxy, aryloxy, thin, alkylthio,
amino,
alkylamino, hydrazino, hydroxytamino, benzyloxy, NRR' or NRCOR'; and
R and R' are independently selected from hydrogen, alkyl and aryl;
or salts or pharmaceutically acceptable derivatives thereof;
in the treatment and/or prophylaxis of hepatitis B viral infection.
These compounds have been found to exhibit surprisingly good activity in an
anti-
hepatitis B assay .
Preferably R' is hydroxy or a group capable of being converted in vivo to
hydroxy.
Preferably R'- is amino; or a group which is capable of being converted in
vivo to
amino.
The invention further provides a method for the treatment or prophylaxis of
hepatitis B viral infection which method includes administering to a patient
in need thereof
an effective amount of a compound of formula (1), its salts, and
pharmaceutically
acceptable derivatives.
The present invention also provides a compound of formula (1), its salts, and
pharmaceutically acceptable derivatives for use in treatment or prophylaxis of
HBV.
The compounds of the invention may further be used in the manufacture of a
medicament for the treatment or prophylaxis of HBV. Accordingly, the present
invention
provides pharmaceutical compositions for said treatment or prophylaxis which
include a
compound of formula (1), its salts or pharmaceutically acceptable derivatives
in
association with a pharmaceutically acceptable carrier or diluent.
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-3-
The present invention also provides the use of a compound of formula (1), its
salts
or pharmaceutically acceptable salts thereof in the manufacture of a
medicament for use in
the treatment or prophylaxis of HBV.
The salts of the compounds of formula ( 1 ) are preferably pharmaceutically
acceptable, but it will be appreciated that non-pharmaceutically acceptable
salts also fall
within the scope of the present invention, since these are useful as
intermediates in the
preparation of pharmaceutically acceptable salts. The pharmaceutically
acceptable salts
may include conventional non-toxic salts or quaternary ammonium salts of these
compounds, which may be formed for example from organic or inorganic acids or
bases.
Examples of such acid addition salts include, but are not limited to, those
formed with
pharmaceutically acceptable acids such as acetic, propionic, citric, lactic,
methanesulphonie, toluenesulphonic, benzenesulphonic, salicylic, ascorbic,
hydrochloric,
orthophosphoric, sulphuric and hydrobromic acids. Base salts includes, but is
not limited
to, those formed with pharmaceutically acceptable cations, such as sodium,
potassium,
lithium, calcium magnesium, ammonium and alkylammonium. They may be formed by
treating a compound of formula (1) with an appropriate metal hydroxide. Also,
basic
nitrogen-containing groups may be quaternised with such agents as lower alkyl
halides,
such as methyl, ethyl, propyl, and butyl chlorides, bromides and iodides;
dialkyl sulfates
like dimethyl and diethyl sulfate; and others.
The compounds of the invention may be in crystalline form or as solvates (e.g.
hydrates) and it is intended that both forms are within the scope of the
present invention.
Methods of solvation are generally known within the art.
Pharmaceutically acceptable derivatives may include any pharmaceutically
acceptable salt, hydrate, prodrug, or any other compound which, upon
administration to a
subject, is capable of providing (directly or indirectly) a compound of
formula (1) or an
antivirally active metabolite or residue thereof. For example, compounds where
a
hydroxy group on the acyclic sidechain has been replaced with a phosphate
ester are
within the scope of pharmaceutically acceptable derivatives.
The term "prodrug" is used in its broadest sense and encompasses those
derivatives
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-4-
that are converted in vivo to the compounds of the invention. Such derivatives
would
readily occur to those skilled in the art, and include, for example, compounds
where R' is
a group which is capable of being converted in vivo to hydroxy; or RZ is a
group which is
capable of being converted in vivo to amino; or compounds where a free hydroxy
group
S on the acyclic sidechain is converted into a group, for example an ester, a
carbonate or a
carbamate, which is capable of being converted in vivo back to a hydroxy
group. A
prodrug may include modifications to one or more of the functional groups of a
compound
of the invention.
Throughout this specification the phrase "a group which is capable of being
converted in vivo" used in relation to another functional group includes all
those functional
groups or derivatives of such groups which upon administration into a mammal
may be
converted into the stated functional group. Those skilled in the art may
readily determine
whether a group may be capable of being converted in vivo into the stated
functional group
using routine enzymatic or animal studies.
It will be appreciated that some derivatives of compounds of formula (1) may
have
an asymmetric centre, and therefore are capable of existing in more than one
stereoisomeric form. The invention extends to each of these forms individually
and to
mixtures thereof, including racemates. The isomers may be separated
conventionally by
chromatographic methods or using a resolving agent. Alternatively, the
individual
isomers may be prepared by asymmetric synthesis using chiral intermediates, or
enzymes.
Preferably R' is hydroxy or a group which is capable of being converted in
vivo to
hydroxy.
Preferably R' is amino, or a group which is capable of being converted in vivo
to
ammo.
Some of the compounds of formula ( 1 ) are novel and accordingly the invention
also provides compounds of formula (la):
CA 02302630 2000-03-06
WO ~~9/12927 PCT/AU98/00748
-5-
R~
N \ N
R2/ \ ~ N OH
N
OH
(la)
where:
R' is hydrogen, halogen, hydroxy, azido, alkoxy, aryloxy, thio, alkylthio,
amino,
alkylamino, hydrazino, hydroxylamino, benzyloxy, NRR' or NRCOR';
Rz is hydrogen, halogen, hydroxy, azido, alkoxy, aryloxy, thio, alkylthio,
amino,
alkylamino, hydrazino, hydroxylamino, benzyloxy, NRR' or NRCOR'; and
R and R' are independently selected from hydrogen, alkyl and aryl;
and salts and pharmaceutically acceptable derivatives thereof;
provided that the following compounds are excluded;
9-[3-Hydroxy-2-hydroxymethylprop-1-yl]-guanine, 9-[3-Hydroxy-2-
hydroxymethylprop-1-yl]-adenine, 9-[(2-isopropyl-1,3-dioxan-5-yl)methyl]-
guanine, and
9-[(2-isopropyl-1,3-dioxan-5-yl)methyl]-adenine.
The compound of formula (1) where R' is OH and RZ is NH2 has been reported by
Martin et al. (1986) as being inactive against herpes simplex virus type 1 and
as a poor
substrate for the thymidine kinase of that virus.
Throughout this specification the term alkyl , used either alone or in
compound
words such as haloalkyl or alkyl acids is denoted, unless otherwise defined,
to mean
both the straight chain C,_3"alkyl or branched chain C3_3~alkyl and the
branched or
unbranched C3_3~,cycloalkyl. Unless otherwise defined, such groups may be
saturated or
unsaturated.
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-6-
The term"aryl" as used herein refers to any compound which includes ~r
consists
of one or more aromatic rings. The aromatic rings may be carbocyclic,
heterocyclic or
pseudoaromatic, and may be mono or polycyclic ring systems and preferably have
2 to 20
carbon atoms. The aromatic rings may also have one or more heteroatoms
selected from
N, S, O and P. Examples of suitable rings include but are not limited to
benzene,
biphenyl, terphenyl, quaterphenyl, naphthalene, tetradyronaphthalene, 1-
benzylnaphthalene, anthracene, dihydroanthracene, benzanthracene,
dibenzanthracene,
phenanthracene, peryiene, pyridine, 4-phenylpyridine, 3-phenylpyridine,
thiophene,
benzothiophene, naphthothiophene, thianthrene, furan, pyrene, isobenzofuram,
chromene, xanthene, phenoxathiin, pyrrole, imidazole, pyrazole, pyrazine,
pyrimidine,
pyridazine, indole, indolizine, isoindole, purine, quinoline, isoquinoline,
phthalazine,
quinoxaline, quinazoline, pteridine, carbazole, carboline, phenanthridine,
acridine,
phenanthroline, phenazine, isothiazole, isooxazole, phenoxazine and the like,
each of
which may be optionally substituted. The term "pseudoaromatic" refers to a
ring system
which is not strictly aromatic, but which is stabilized by means of
delocalization of
electrons and behaves in a similar manner to aromatic rings. Examples of
pseudoaromatic
rings include but are not limited to furan, thiophene, pyrrole and the like.
Throughout this specification the term alkoxy , used either alone or in
compound
words such as haloalkoxy is denoted, unless otherwise defined, to mean both
the straight
chain C,_3~alkoxy or branched chain Cj_3~alkoxy and the branched or unbranched
C3_
3~eycloalkoxy. Unless otherwise defined, such groups may be saturated or
unsaturated.
Unless otherwise stated the term "ester" is denoted to mean those compounds or
derivatives which correspond to the ester formed by reaction of an alcohol
with an organic
acid, preferably a carboxylic acid. Particularly preferred carboxylic acids
from which
esters may be formed include amino acids and alkyl acids. Preferred amino
acids are
aliphatic amino acids such as valine and isoleucine, preferably in the L-form.
Preferred
alkyl acids include C,-C4 alkyl acids, and fatty acids from C"-Cz2 such as
lauryl,
myristoyl, palmitoyl, stearoyl, eicasanoyl, behenoyl, myristoleic,
myristelaidic,
palmitoleic, palmitelaidie, n6-octadecenoic, oleic, elaidic, erucic or
brassidic acids.
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
A preferred group of compounds of formula (1) and formula (la) and
pharmaceutically acceptable derivatives of the compounds of formula (1) and
formula (la)
are those compounds of formula (4)
X
N
N
NH ~ N ORS
2 N
O R6
(4)
where:
X is hydrogen or hydroxy;
RS and R6 are the same or different and together with the oxygen atom to which
they are attached form a hydroxy group, an ester, a carbonate, a carbamate, or
a
thiocarbonate; preferably a hydroxy or an ester;
and salts thereof.
Examples of some compounds of formula (4) are shown in Table 1.
Table 1
X RS R6
H H H
OH H H
H acet 1 ace 1
OH ace 1 acetyl
H acet 1 H
OH acetyl H
H val 1 val 1
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
_g_
OH val 1 vale
H val 1 H
OH v al 1 H
H val I ace I
S OH val 1 acct I
H isoleucyl isoleuc 1
OH isoleuc 1 isoleuc 1
H isoleuc 1 ace I
OH isoleuc 1 ace 1
H valyl stea 1
OH valyl stea 1
H valI octadecen 1
OH valyl octadecen 1
H isoleuc 1 stea 1
OH isoleucyl stea I
H isoleuc 1 octadecen 1
OH isoleuc 1 octadecen 1
H val 1 almit 1
OH val 1 almi l
H val 1 elaid 1
OH val 1 elaid 1
H isoleuc 1 almi I
OH isoleuc 1 almi I
H isoleuc~ I elaid 1
OH isoleuc 1 elaid I
H H stea l
OH H ste 1
H H octadecen 1
OH H octadecen 1
3 H H almi 1
0
OH H almi 1
H H elaid 1
OH H elaid 1
H chol 1
3 OH H chol 1
5
H val 1 chol 1
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-9-
OH val 1 chol l
H ro ion 1 ro ion 1
OH ro ion 1 ro ion I
H iso ro ionvl iso ro ion 1
5OH iso ro ion 1 iso ro ion 1
H H bu l
OH H bu 1
H bu I bu 1
OH bu 1 bu 1
In yet another aspect of the invention there is provided a method for the
manufacture of
the compounds of formula (1), their salts and pharmaceutically acceptable
derivatives.
The compounds may be prepared by reacting a purine derivative (2) with a
compound of
formula (3). The group R' of purine derivative (2) may be any group listed
under R' for
I 5 the compound of formula ( 1 ) or any group that may be converted by
methods known in
the art to such groups, such groups include chlorine, bromine or iodine; and
RZ may be
any group listed under RZ for the compound of formula (1) or any group that
may be
converted by methods known in the art to such groups. In compound (3) Z is any
suitable
leaving group, such as methane sulfonate or bromine, and R3 and R4 are hydroxy
or a
group that may be converted into hydroxy, such groups include ethers and
esters.
Methods for such conversion are known to those skilled in the art. R3 and R4
may be
joined together to form an optionally substituted 5 or 6 membered ring system.
R~
R3
N ~ N
~~ / ~ R4 Z
R2~ N
N H
(2) (3)
Compounds of formula (2) are commercially available or may be prepared by
literature
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
- 10-
procedures. Compounds of formula (3) may be prepared by literature procedures
or along
the lines of the procedures described in Steps A to C of Example 1, Steps A to
E of the
alternative route to Example 1, see Schemes l and 2 respectively. Those in the
art will
appreciate that a number of variations may be made to the methodology actually
exemplified.
The procedure for manufacture of the compound of formula (3) outlined in
Example 1 Scheme 1 is particularly useful and may be more broadly applicable
to the
synthesis of acyclic nucleoside analogues than is outlined in the specific
example. The
general procedure of Step B forms a further aspect of the present invention.
Accordingly,
a symmetrical triol (5), preferably where n is 1 or 2, may be converted into a
diester (6)
by treatment with about one equivalent of a trialkylorthoester, preferably
triethylorthoacetate, under appropriate conditions, followed by treatment with
about one
equivalent of water under appropriate conditions, followed by treatment with
water under
appropriate conditions. The diester may then be isolated by conventional
procedures,
conveniently this is done via a careful neutralisation of the mixture with a
mild base, for
example sodium bicarbonate, followed by extraction into an organic solvent.
Those
skilled in the art may readily determine which conditions are appropriate in
view of the
particular triol and trialkylorthoester selected.
(~~~ ~ (~~(-oH
HO-(CI~~ (C~'~n'a'~ RQ(CI-L~n (C~"~1f1-OR
-OR = ~ ester
(5) (6)
The remaining hydroxyl group on the diester (6) may be converted into a
leaving
group, Z, to give diester compounds, which where n is 1 are of general formula
(3).
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
Compounds of formula (6), such as 2-hydroxymethyl-1,3-propanediol diacetate,
and diester compounds including those of general formula (3), such as 3-
acetoxy-2-
acetoxymethylprop-1-yl methanesulfonate, are novel and form a further aspect
of the
invention. Preferably n is 1 in compound (5) and (6).
In accordance with conventional processes known in the art, the acyclic
hydroxyl
groups of compounds of formula (1) may be readily converted into esters,
ethers or
phosphate groups or a mixture of these groups on the acyclic chain. In some
instances it
may be useful to utilise protected intermediates of the compound of formula
(I) in order to
prepare the desired tinal derivative. Such intermediates may be prepared in
accordance
with standard procedures and when no longer required the protecting groups
removed
using standard procedures, such as those described by Greene. Examples of
suitable
protecting groups are trimethylsilyl and monomethoxytrityl groups.
Acylation and alkylation may be carried out using any conventional procedure
such
as those generally known in the art or described or referenced in the Third
Edition of
I S March's Advanced Organic Chemistry published by Wiley-Interscience.
Examples of
acylating agents suitable for the process of acylating the compounds of
formula (1) are
carboxylic acids, acid halides and acid anhydrides. The reaction may be
carried out in a
conventional manner, for example in a solvent such as pyridine,
dimethylformamide, etc. ,
optionally in the presence of a coupling agent such as N,N'-
dicyclohexylcarbodiimide, and
optionally in the presence of a catalytic base such as 4-
dimethylaminopyridine. The
product of the reaction may be isolated in a conventional manner. Examples of
alkylating
agents suitable for the process of alkylating compounds of formula (1) are
alkyl halides,
such as methyl, ethyl, propyl, and benzyl chlorides, bromides and iodides; and
dialkyl
sulfates like dimethyl and diethyl sulfate.
In the case of amino acids or their functional equivalents, for example acid
halides,
it may be advantageous, in order to avoid side reactions, to use amino
protected
derivatives of the amino acid or amino acid equivalent, for example
benzyloxycarbonyl
derivatives. Such derivatives are commercially available. The protecting
groups may be
removed utilising standard procedures.
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
- 12-
The acylation or alkylation reactions may produce a single derivative of
compound
(1), incorporating one or more acyl or alkyl groups, or may produce a mixture
of
compounds incorporating acyl or alkyl groups. The outcome depends on a number
of
factors, such as the relative amounts and chemical nature of the reactants,
the physical
conditions of the reaction, and the solvent system. Any mixture produced in
this way may
be separated using standard techniques, preferably chromatography.
It will be appreciated by one skilled in the art that it is possible to
produce
derivatives of compounds of formula (1) that may have a mixture of different
acyl and/or
alkyl groups. Such derivatives are within the scope of the present invention.
Protected intermediates of the compounds of formula (1) may also be used to
prepare derivatives of compound (1) incorporating phosphate esters.
The compounds of this invention may also be useful in combination with known
antiviral or antiretroviral agents or other pharmaceuticals used in the
treatment of viral
infections. Representative examples of these additional pharmaceuticals
include
immunomodulators, immunostimulants, and antibiotics. Exemplative anti-viral
agents
include AZT, 3TC, acyclovir, famciclovir, ddI, ddC, ganciclovir, saquanivir,
loviride,
other non-nucleotide reverse transcriptase (RT) inhibitors and protease
inhibitors.
Exemplative immunomodulators and immunostimulants include various
interleukins,
cytokines, antibody preparations, blood transfusions and cell transfusions.
Exemplative
antibiotics includes antifungal agents, antibacterial agents and anti-
Pneumocystis carinii
agents.
If formulated as a fixed dose, such combination products employ the compounds
of
this invention in the dosage ranges described below and the other
pharmaceutically active
agent within its approved dosage range. The compounds of the invention may be
used
sequentially with known anti-viral, anti-retroviral or pharmaceutical agents
when a
combination formulation is inappropriate.
By an effective amount is meant a quantity of active compound which will upon
single or multiple dose administration to the patient be effective in
controlling the viral
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98100748
-13-
infections, such as HBV, or in achieving a blood or tissue level in the
patient that
corresponds to a concentration of the active compound that has been shown to
inhibit a
virus, such as HBV, in an assay known to predict for clinical anti-viral
activity of
chemical compounds. For example the assay described by Korba and Gerin.
As used herein the term controlling the viral infections refers to slowing,
interrupting, arresting or stopping its growth or replication and does not
necessarily
indicate a total elimination of the virus.
Controlling the viral infections will be useful in the treatment and/or
prophylaxis
of such viral infections.
When a compound of the invention is administered to a human subject the daily
dosage can normally be determined by the attending physician with the dosage
generally
varying according to the age, weight, and response of the individual patient,
as well as the
severity of the patient's symptoms. In general a suitable dose of the compound
of the
invention will be in the range of 0.1 to 50 mg per kilogram body weight of the
recipient
I 5 per day, preferably in the range of 0.5 to 10 mg per kilogram body weight
per day. The
desired dose is preferably presented as two, three, four, five, six or more
sub-doses
administered at appropriate intervals throughout the day. These sub-doses may
be
administered in unit dosage forms, for example, containing 1 to 1000 mg,
preferably 10 to
500 mg of active ingredient per unit dosage form.
The compounds according to the invention, also referred to herein as the
active
ingredient, may be administered for therapy by any suitable route, including
oral, rectal,
nasal, topical (including buccal and sublingual), vaginal and parenteral
(including
subcutaneous, intramuscular, intravenous and intradermal). Preferably,
administration
will be by the oral route, however it will be appreciated that the preferred
route will vary
with the condition and age of the recipient, the nature of the invention and
the chosen
active ingredient. When administered by the oral route a prodrug of the active
compound
which is more efficiently absorbed than the unmodified compound is generally
preferred.
The compositions of the present invention comprise the compound of formula ( 1
),
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-14-
optionally as a salt or other pharmaceuticall;~ acceptable derivative,
together with one or
more pharmaceutically acceptable carriers, diluents or excipients therefor,
and optionally
other therapeutic agents. Each carrier, diluent or excipient must be
pharmaceutically
"acceptable" in the sense of being compatible with the other ingredients of
the composition
and not injurious to the patient. Compositions include those suitable for
oral, rectal,
nasal, topical (including buccal and sublingual), vaginal or parenteral
(including
subcutaneous, intramuscular, intravenous and intradermal) administration. The
compositions may conveniently be presented in unit dosage form and may be
prepared by
methods well known in the art of pharmacy. Such methods include the step of
bringing
into association the active ingredient with the carrier, diluent or excipient
which includes
one or more accessory ingredients. In general, the compositions are prepared
by
uniformly and intimately bringing into association the active ingredient with
liquid carriers
or finely divided solid carriers or both, and then if necessary shaping the
product.
Compositions of the present invention suitable for oral administration may be
presented as discrete units such as capsules, sachets or tablets each
containing a
predetermined amount of the active ingredient; as a powder or granules; as a
solution or a
suspension in an aqueous or non-aqueous liquid; or as an oil-in-water liquid
emulsion or a
water-in-oil liquid emulsion. The active ingredient may also be presented as a
bolus,
electuary or paste.
Tablets may be made by compression or moulding, optionally with one or more
accessory ingredients. Compressed tablets may be prepared by compressing in a
suitable
machine the active ingredient in a free-flowing form such as a powder or
granules,
optionally mixed with a binder (e. g. inert diluent, preservative disintegrant
(e. g. sodium
starch glycollate, cross-linked povidone, cross-linked sodium carboxymethyl
cellulose)
surface-active or dispersing agent. Moulded tablets may be made by moulding in
a
suitable machine a mixture of the powdered compound moistened with an inert
liquid
diluent. The tablets may optionally be coated or scored and may be formulated
so as to
provide slow or controlled release of the active ingredient therein using, for
example,
hydroxypropylmethyl cellulose in varying proportions to provide the desired
release
profile. Tablets may optionally be provided with an enteric coating, to
provide release in
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98~ J0748
-15-
parts of the gut other than the stomach.
Compositions suitable for topical administration in the mouth include lozenges
comprising the active ingredient in a flavoured base, usually sucrose and
acacia or
tragacanth gum; pastilles comprising the active ingredient in an inert base
such as gelatin
and glycerin, or sucrose and acacia gum; and mouthwashes comprising the active
ingredient in a suitable liquid carrier.
Compositions for rectal administration may be presented as a suppository with
a
suitable base comprising, for example, cocoa butter.
Compositions suitable for vaginal administration may be presented as
pessaries,
tampons, creams, gels, pastes, foams or spray formulations containing in
addition to the
active ingredient such carriers as are known in the art to be appropriate.
Compositions suitable for parenteral administration include aqueous and non-
aqueous isotonic sterile injection solutions which may contain anti-oxidants,
buffers,
bacteriostats ~.nd solutes which render the composition isotonic with the
blood of the
1 S intended recipient; and aqueous and non-aqueous sterile suspensions which
may include
suspending agents and thickening agents. The compositions may be presented in
unit-dose
or mufti-dose sealed containers, for example, ampoules and vials, and may be
stored in a
freeze-dried (lyophilised) condition requiring only the addition of the
sterile liquid carrier,
for example water for injections, immediately prior to use. Extemporaneous
injection
solutions and suspensions may be prepared from sterile powders, granules and
tablets of
the kind previously described.
Preferred unit dosage compositions are those containing a daily dose or unit,
daily
sub-dose, as herein above recited, or an appropriate fraction thereof, of an
active
ingredient.
The compounds according to the invention may also be presented for use in the
form of veterinary compositions, which may be prepared, for example, by
methods that
are conventional in the art. Examples of such veterinary compositions include
those
adapted for:
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-16-
(a) oral administration, external application, for example drenches (e.g.,
aqueous or non-aqueous solutions or suspensions); tablets or boluses;
powders, granules or pellets for admixture with feed stuffs; pastes for
application to the tongue;
(b) parenteral administration for example by subcutaneous, intramuscular
or intravenous injection, e.g. as a sterile solution or suspension;
(c) topical application, e.g. as a cream, ointment or spray applied to the
skin; or
(d) intravaginally, e. g. as a pessary, cream or foam.
It should be understood that in addition to the ingredients particularly
mentioned
above, the compositions of this invention may include other agents
conventional in
the art having regard to the type of composition in question, for example,
those
suitable for oral administration may include such further agents as
sweeteners,
thickeners and flavouring agents.
IS
EXAMPLES
Examples are provided to assist in the further understanding of the invention.
Particular materials, and conditions employed are intended to be illustrative
of the
invention and not limitative of the reasonable scope thereof.
2-Amino-6-chloropurine was obtained commercially in 98 % purity from Chugai
Boyeki Co. , Ltd. 2-Amino-6-chloropurine was converted to 2-amino-6-iodopurine
according to the method of Bisacchi et al. Unreferenced reagents were obtained
commercially and used as supplied, unless otherwise specified.
All temperatures are given in degrees Celsius.
" CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
- 17-
Example 1
chem 1
Reaction sequence described in Example 1
O OEt OH
O O Step A
i
OEt OEt OH OH
Step B
OMs OH
~ Step C
OAc OAc OAc OAc
Step D
t OH
Steps
H2N N N OAc H2N N N OH
OAc OH
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
_18_
S tep A
2-Hydroxymethyl-1, 3-propanediol
A modified procedure of Harnden et al was used. To a solution of borane-
methylsulfide complex ( lOM) { 14 ml, 0.14 mol) in toluene (65 ml) under a
nitrogen
atmosphere, at gentle reflux, was added dropwise triethylmethane
tricarboxylate
(9.68 g, 0.0417 mol). The reaction mixture was retluxed for 7.5 hr with
distillation
of dimethyl sulfide. The reaction mixture was cooled to room temperature and
methanol (40 ml) was added dropwise. The reaction mixture was stirred at room
temperature overnight, then the solvents were removed and the residue co-
evaporated with methanol repetitively. The residue was chromatographed on
silica
with 25% methanol in dichloromethane to afford 2-hydroxymethyl-I,3-propanediol
as a pa:c lemon oil (3.18 g, 72%). 'H n.m.r. (DMSO-db) 1.60, septet, J =
5. SHz, 1 H; 3.40, t, J = 5. SHz, 2H; 4. 31, t, J = S. SHz, 3H.
Step B
2-Hydroxymethyl-1,3-propanediol diacetate
To a solution of 2-hydroxymethyl-1,3-propanediol (3.18 g, 0.03 mol),
trifluoroacetic acid (1.54 ml, 0.02 mol) in N,N-dimethylformamide (35 ml)
under a
nitrogen atmosphere, was added triethylorthoacetate (6.09 ml, 0.033 mol). The
reaction mixture was stirred for 1.5 hr then water (0.63 ml, 0.035 mol) was
added.
The reaction mixture was stirred for a further 1.5 hr then
triethylorthoacetate (6.26
ml, 0.034 mol) was added. The reaction mixture was stirred for a further 1.5
hr
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
- 19-
then water (1.74 ml, 0.097 mol) was added. After 1 hr sodium bicarbonate was
carefully added until the reaction mixture was neutralized. Water was added
and the
solution was extracted with diehloromethane (3x). The combined extracts were
washed with water, dried over magnesium sulfate and concentrated to afford 2-
hydroxymethyl-1,3-propanediol diacetate as a colourless oil (4.50 g, 79%). 'H
n.m.r. (CDCl3) 2.06, s, 6H; 2.18, septet, J = 6Hz, 1H, 2.54, br s, 1H, 3.62,
d,
J = 6Hz, 2H; 4.15, d, J = 6Hz, 4H.
Step C
3-Acetoxy-2-acetoxymethylprop-1-yl methanesulfonate
A solution of 2-hydroxymethyl-1,3-propanediol diacetate (4.49 g, ~'.~J236
mol) in dichloromethane (40 ml) under a nitrogen atmosphere, was cooled to -5
° .
Triethylamine (4.93 ml, 0.0354 mol) was added, followed by dropwise addition
of
a solution of methanesulfonyl chloride (2.19 ml, 0.0283 mol) in
dichloromethane
(20 ml). The reaction was stirred a further 2 hr at 0° then warmed to
room
temperature. The reaction mixture was washed with 1.5M hydrochloric acid ( 3 x
30 ml), saturated sodium bicarbonate solution ( 30 ml) and brine ( 30 ml),
dried
over magnesium sulfate and concentrated to afford 3-acetoxy-2-
acetoxymethylprop-
1-yl methanesulfonate as a pale brown oil (5.02 g, 79% ). 'H n.m.r. (CDCl3)
2.0, s, 6H; 2.48, septet, 3 = 6Hz, 1H; 3.03, s, 3H; 4.07 - 4.24, m, 4H; 4.29,
d, J
= 6Hz, 2H.
Step D
9-[3-Acetoxy-2-acetoxymethylprop-1-yl]-2-amino-6-iodopurine
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-20-
A mixture of 3-acetoxy-2-acetoxymethylprop-1-yl methanesulfonate (5.02 g,
0.0187 mol), 2-amino-6-iodopurine (4.66 g, 0.0178 mol) and potassium carbonate
(7.38 g, 0.0534 mol) in N,N-dimethylformamide (250 ml) under a nitrogen
atmosphere, was stirred at 60° for 2 days. The N,N-dimthylformamide was
removed in vacuo, water was added and the residue was extracted with ethyl
acetate
(3 x 100 ml). The extracts were washed with water and brine, dried over
magnesium sulfate, then filtered through a plug of silica, washing thoroughly
with
ethyl acetate. The filtrate was concentrated to give the crude product as a
yellow
solid (5.96 g). This was recrystallised from methanol and diethyl ether to
afford 9-
I0 [3-acetoxy-2-acetoxymethylprop-1-yl]-2-amino-6-iodopurine as pale lemon
crystals
(4.43 g, 55 % ). 'H n. m. r. (DMSO-db) 1. 95, s, 6H; 2.56 - 2.78, m, 1 H; 3.
90 -
4.10, m, 4H; 4.12, d, J = 7Hz, 2H; 6.83, s, 2H; 8.09, s, 1H.
Step E
9-[3-Hydroxy-2-hydroxymethylprop-1-yl]-guanine
9-[3-Acetoxy-2-acetoxymethylprop-1-yl]-2-amino-6-iodopurine (2.5 g,
0.00576 mol ) in 1.5M hydrochloric acid was heated at retlux for 3 hr. The
reaction
mixture was cooled to room temperature and the pH adjusted to 14 with sodium
hydroxide. The reaction mixture was stirred for a further 1.5 hr, then
neutralized
with concentrated hydrochloric acid. The resulting precipitate was filtered
off and
recrystallised from water to afford 9-[3-hydroxy-2-hydroxymethylprop-1-yl]-
guanine (1.267 g, 92%) as colourless fluffy crystals. M.p. 294 - 296°.
'H n.m.r.
(DMSO-d~,) 1.9 - 2.05, m, 1H; 3.3, t, J = 5.5 Hz, 4H; 3.95, d, J = 7.0 Hz,
2H; 4.6, t, J = 5.5 Hz, 2H; 6.5, s, 2H; 7.6, s, 1H; 10.1, s, 1H.
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-21 -
Example 1 (Alternate Routed
eme 2
Diagramatic representation of the alternate reaction sequence.
HOH2C~CO2Et H3C\- Step H3C~ CO2Et
CHO ~
C~
HOH
C CO
Et H
2 H3C
2 CO2Et
3 O
Step B
H3C~--~ CO H teP C H3C~--~ O C02H
HaC ~ H3C~ CO H
O ~ 2
O
Step D
H3C~---~ CH20H Step E~ H3C
H H
C ~ C ~OSO
CH
a 3
O 2
O a
C~
N ~ N
Step F
NH2"N H
OH CI
\ N~ E-eP G ~ \ N
H2N N~ N OH NH2 N N O CH3
~CH3
OH O
s
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-22-
Step A
5, 5-Dicarboxyethyl-2-isopropyl-1, 3-dioxane
S
5,5-Dicarboxyethyl-2-isopropyl-I,3-dioxane was prepared according to a
modified method of Eliel et al. A solution of diethyl
bis(hydroxymethyl)malonate (25.0
g, 0.113 mol), isobutylaldehyde (0.226 moI, 20.6 ml) and p-toluene sulfonic
acid (0.300
g) in petroleum spirit (50 ml) was heated at ret7ux and water collected in a
Dean-Stark
I O apparatus. The petroleum spirit was removed and the product distilled to
afford 5,5-
dicarboxyethyl-2-isopropyl-1,3-dioxane (22.5 g, 73~) as a colourless oil. B.p.
103 /0.15
mm Hg. 'H n.m.r. (CDCI3) 0.9, d, J = 6.9 Hz, 6H; 1.2, t, J = 6.9 Hz, 3H; 1.3,
t, J
= 6.9 Hz, 3H; 1.7 - I.85, m, 1H; 3.9, d, JAB = 11.5 Hz, ZH; 4.15, q, J = 6.9
Hz,
2H; 4.22, d, J = 4.8 Hz, 'H; 4.3, q, J = 6.9 Hz, 2H; 4.7, d, JAB = 11.5 Hz,
2H.
Step B
5,5-Dicarboxylic-2-isopropyl-I ,3-dioxane
5,5-Dicarboxylic-2-isopropyl-1,3-dioxane was synthesised from 5,5-
dicarboxyethyl-2-isopropyl-1,3-dioxane (20.0 g, 72.9 mmol) according to the
method of
Eliel et al. The crude product was re-crystallised from ethyl acetate and
petroleum spirit
to give 5,5-dicarboxylic-2-isopropyl-1,3-dioxane (14.1 g, 80%) as a colourless
solid.
M.p. 143.5 - 145°. 'H n.m.r. (DMSO-db) 0.8, d, J = 6.9 Hz, 6H; I.55 -
1.75, m,
1H; 3.8, d, JAB = 11.3 Hz, 2H; 4.3, d, J = 4.8 Hz, 1H; 4.5, d, JAB = 11.3 Hz,
2H;
12.8, br s, 2H.
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
- 23 -
Step C
5-Carboxy-2-i sopropyl-1, 3-dioxane
5-Carboxy-2-isopropyl-1,3-dioxane was synthesised from 5,5-dicarboxylic-2-
isopropyl-1,3-dioxane (15.7 g, 64.8 mmoI) according to the method of Eliel et
al. The
crude product was re-crystallised from ethyl acetate and petroleum spirit to
give 5-
carboxy-2-isopropyl-1,3-dioxane (9.75 g, 76%) as a colourless solid. M.p. 131-
134°. 'H
n.m.r. (DMSO-d~) 0.8, d, J = 6.9 Hz, 6H; 1.6 - 1.8, m, 1H; 2.7 - 2.85, m, 1H;
3.65, t, JAB = 11.5 Hz, 2H; 4.15, t, JAB = 10.1 Hz, 2H; 4.2, t, J = 4.8 Hz, 1
H.
Step D
5-Hydroxymethyl-2-isopropyl-1,3-dioxane
To a solution of 5-carboxy-2-isopropyl-1,3-dioxane (8.75 g, 44.1 mmol) in
anhydrous diethyl ether (65 ml) under a nitrogen atmosphere, was added borane
methylsulfide complex (9.0 ml, IOM, 90 mmol). The reaction mixture was heated
to
reflux for 1 hr, cooled to room temperature and quenched with water (20 ml)
and
methanol (30 ml). The methanol was removed and the aqueous phase extracted
several
times with diethyl ether. The combined diethyl ether extracts were washed with
water and
brine, dried over magnesium sulfate and the solvent removed to give 5-
hydroxymethyl-2-
isopropyl-1,3-dioxane (6.50 g, 80%) as a colourless oil. 'H n.m.r. (DMSO-db)
0.85,
d,J = 7 Hz, 6H; 1.6 - 1.8, m, 1H; 1.85 - 2.05, m, 1H; 3.2, t, JAB = 11.3 Hz;
2H; 3.3
- 3.45, m, 4H; 4.15, d, J = 4.8 Hz, 1H.
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98I00748
-24-
S tep E
2-Isopropyl-5-(methanesulfoxy)methyl-1, 3-dioxane
2-Isopropyl-5-(methanesulfoxy)methyl-1,3-dioxane was synthesised from 5-
carboxy-2-isopropyl-1,3-dioxane (7.30 g, 39.6 mmol) according to the method
used for
3-acetoxy-2-acetoxymethylprop-1-yl methanesulfonate to give the product as a
colourless
oil (10.1 g, 97%). 'H n.m.r. (DMSO-d~) 0.85, d, J = 6.9 Hz, 6H; I.6 - 1.8, m,
1H;
2.15 - 2.35, m, 1H; 3.2, s, 3H; 3.5, t, JAB = 11.3 Hz, 2H; 4.0 - 4.15, m, 4H;
4.2, d, J
= 4.8 Hz, 1H.
S tep F
2-Amino-6-chloro-9-[(2-i sopropyl-1, 3-dioxan-5-y 1)methyl]purine
2-Amino-6-chloro-9-[(2-isopropyl-1,3-dioxan-5-yl)methyl]purine was synthesised
from 2-isopropyl-5-(methanesulfoxy)methyl-1,3-dioxane (10.1 g, 38.5 mmol)
according
to method used for 9-[3-acetoxy-2-acetoxymethylprop-1-yl]-2-amino-6-iodopurine
to give
the product as a colourless solid (4.80 g, 40%). M.p. 204 - 205°. 'H
n.m.r. (DMSO-d~)
0.85, d, J = 7 Hz, 6H; 1.6 - 1.75, m, 1H; 2.4 - 2.55, m, 1H; 3.45, t, J = 11.3
Hz,
2H; 3.85 - 4.0, m, 4H; 4.2, d, J = 5 Hz, 1H; 6.9, s, 2H; 8.1, s, 1H. '3C
n.m.r.
(DMSO-db) 16.8, 31.9, 34.3, 41.4, 68.6, 104.5, 123.3, 143.1, 149.4, 154.2,
159.7.
Mass spectrum: m/z 312 ((M+1)+, 100%), 340 ((M+29)+, 15), 314 ((M+3)+, 30),
313 ((M+2)+, 18), 276 (20). Accurate mass: found 312.1209 (M+1)+,
C,3H,~NSO~C1, required 312.1227.
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
- 25 -
Step G
9-[3-Hydroxy-2-hydroxymethylprop-1-yl]-guanine
9-[ 1-Hydroxy-2-hydroxymethylpropyl]guanine was synthesised from 2-amino-6-
chloro-9-[(2-isopropyl-1,3-dioxan-5-yl)methyl]purine (1.28 g, 4.11 mmol)
according to
the method used for 9-[3-hydroxy-2-hydroxymethylprop-1-yl]-guanine (step E
above) to
afford the product as a colourless solid (0.60 g, 61 %). M.p. 285° dec.
'H n.m.r.
(DMSO-d6) 1.9 - 2.05, m, 1H; 3.3, t, J = 5.5 Hz, 4H; 3.95, d, JJ = 7.0 Hz, 2H;
4.6, t, J = 5.5 Hz, 2H; 6.5, s, 2H; 7.6, s, 1H; 10.1, s, 1H. '3C n. m. r.(DMSO-
d6)
41.5, 43.6, 58.9, 116.3, 138.1, 151.3, 153.4, 156.8. Mass spectrum: m/z 340
((M+1)+, 100%), 368 ((M+29)+, 20), 341 ((M+2)+, 12).
Example 2
IS
9-[3-Hydroxy-2-hydroxymethylprop-1-yl]-2-amino-6-methoxypurine
A mixture of 9-[3-acetoxy-2-acetoxymethylprop-1-yl]-2-amino-6-iodopurine (307
mg, 0.708 mmol ) , sodium hydroxide (7.75 g, 194 mmol), methanol (30 mI) and
water
(3 ml) was stirred at room temperature for 18 hr. The reaction mixture was
neutralised with aqueous HCl and the solvent was removed by rotary
evaporation. Hot
methanol was added to the residue and the solution decanted off and filtered
through a
short silica column. The filtrate was concentrated and recrystallised from
water to afford
9-[3-hydroxy-2-hydroxymethylprop-1-yl]-2-amino-6-methoxypurine as colourless
crystals
( 179 mg, quantitative). 'H nmr (DMSO-db) 1.93 - 2.16, m, 1H; 3.22 - 3.40, m,
4H;
3.95, s, 3H; 4.19, d, J = 7Hz, 2H; 4.69, t, J = SHz, 2H; 6.45, s, 2H; 7.79, s,
1H.
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-26-
Example 3
9-[3-Acetoxy-2-acetoxymethy lprop-1-yl J-2-amino-6-hydrazi no-purine
S
A mixture of 9-[3-acetoxy-2-acetoxymethylprop-1-yl1-2-amino-6-iodopurine (300
mg, 0.69 mmol), hydrazine hydrate (85 % , 2101, 6.68 mmol) and ethanol (35 ml)
under
a nitrogen atmosphere, was stirred at retlux for 3 hr and then at room
temperature for 16
hr. The resulting colourless solid was filtered oft, washing well with ethanol
and dried in
vacuo to afford 9-[3-acetoxy-2-acetoxymethylprop-1-yi]-2-amino-6-hydrazino-
purine (196
mg, 84 % ). 'H n. m. r. (DMSO-db) 1. 98, s, 6H; 2.54 - 2.77, m, 1 H; 3. 87 -
4.08, m,
4H; 4.05, d, J = 7Hz, 2H; 4.40, br s, 2H; 5.89, br s, 2H; 7.67, s, 1H; 8.40,
s, 1H.
Example 4
IS
2-Amino-6-hydrazino-9-[3-hydroxy-2-hydroxymethylprop-1-yl]purine
A mixture of 9-[3-acetoxy-2-acetoxymethylprop-1-yl]-2-amino-6-iodopurine (300
mg, 0.69 mmol), hydrazine hydrate (85%, 7001, 22.27 mmol) and ethanol (35 ml)
under a nitrogen atmosphere, was stirred at retlux for 6 hr and then at room
temperature
for 16 hr. The reaction mixture was concentrated to dryness and the residue
recrystallised
from water to afford 2-amino-6-hydrazino-9-[3-hydroxy-2-hydroxymethylprop-1-
yl]purine {86 mg, 49%o) as cream crystals. 'H n.m.r. (DMSO-db) 1.90 - 2.12, m,
1H;
3.20 - 3.40, m, 4H; 3.98, d, J = 7Hz, 2H; 4.42, br s, 2H; 4.74, t, J = SHz,
2H; 5.98,
s, 2H; 7.62, s, 1H; 8.45, s, 1H.
. r
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-27-
Exam
2-Amino-9-[3-hydroxy-2-hydroxymethylprop-1-yl]-6-iodopurine
A mixture of 9-[3-acetoxy-2-acetoxymethylprop-1-yl]-2-amino-6-iodopurine (304
mg, 0.70 mmol) and methanolic ammonia (10 ml) were stirred for 2 hr in a
stoppered
t7ask. The supper was removed and the reaction mixture left to stand for 16
hr. The
reaction mixture was filtered giving Z-amino-9-[3-hydroxy-2-hydroxymethylprop-
1-yl]-6-
iodopurine (201 mg, 82%) as colourless needles. 'H n.m.r. (DMSO-d6) 1.96 -
2.19,
m, 1H; 3.25 - 3.43, m, 4H; 4.01, d, J = 7Hz, 2H; 4.61, t, J = SHz, 2H; 6.84,
s, 2H;
8. 02, s, 1 H.
Example 6
2,6-Diamino-9-[3-hydroxy-2-hydroxymethylprop-1-yl]purine
A mixture of 9-[3-acetoxy-2-acetoxymethylprop-1-yl]-2-amino-6-iodopurine (299
mg, 0.69 mmol) and methanolic ammonia ( 10 ml) were stirred for 18 hr in a
bomb at
100°. The reaction mixture was cooled to room temperature and after 1
hr a precipitate
formed. This was filtered off and dried in vacuo to afford 2,6-diamino-9-[3-
hydroxy-2-
hydroxymethylprop-1-yl]purine (128 mg, 78%) as colourless crystals. 'H n.m.r.
(DMSO-d~) 1.90 - 2.10, m, 1H; 3.20 - 3.40, m, 4H; 3.96, d, J = 6.SHz, 4.75, t,
J =
CA 02302630 2000-03-06
WO 99/12927 PCT/~.U98/00748
-2s-
SHz, 2H; 5.84, s, 2H; 6.70, s, 2H; 7.78, s, 1H.
Ex a 7
2-(Dimethylaminomethylene)amino-9-[3-hydroxy-2-hydroxymethylprop-1-yl]guanine
A mixture of 9-[3-hydroxy-2-hydroxymethylprop-1-yl]-guanine (200 mg, 0.84
mmol), N,N-dimethylformalnide dimethyl acetal (1.5 ml, 11.3 mmol) and N,N-
dimethylformamide (20 ml) under a nitrogen atmosphere was stirred at room
temperature
for 2 days. Solvents were removed in vacuo at 60° and the residue was
recrystallised
from ethanol to afford 2-(dimethylaminomethylene)amino-9-[3-hydroxy-2-
hydroxymethylprop-1-yl]guanine ( 180 mg, 73 % ) as colourless crystals. 'H n.
rn. r.
(DMSO-d~) 1.95 - 2.17, m, 1H; 3.03, s, 3H; 3.15, s, 3H; 3.21 - 3.47, m, 4H;
4.03,
d, J = 7Hz, 2H; 4.62, t, J = SHz, 2H; 7.74, s, 1H; 8.53, s, 1H; 11.26, br s,
1H.
Exam le
2-Amino-9-[(2-isopropyl-1,3-dioxan-5-yl)methyl]purine
A mixture of 2-amino-6-chloro-9-[(2-isopropyl-1,3-dioxan-5-yl)methyl]purine
(350 mg, 1.12 mmol), triethylamine (172 ~cl, 1.23 mmol), 10% palladium on
carbon (35
mg) and ethanol (5 ml) was stirred under an atmosphere of hydrogen for 3 days.
The
reaction mixture was then filtered through GFA paper washing copiously with
dichloromethane. The filtrate was concentrated to dryness, dissolved in
dichloromethane,
washed with water (2 x) and brine, dried over sodium sulfate and concentrated
to afford
r
CA 02302630 2000-03-06
, WO 99/12927 PCT/AU98/00748
-29-
2-amino-9-[(2-isopropyl-1,3-dioxan-5-yl)methyl]purine (260 mg, 84%) as a
colourless
solid. 'H n.m. r. (DMSO-d~,) 0. 83, d, J = 7Hz, 6H; 2.35 - 2.63, m, 1 H; 3.45,
t, J =
llHz, 2H; 3.87, d, J = 7Hz, 2H; 3.82 - 3.95, m, 2H; 4.17, d, J = SHz, 1H;
6.53, s,
2H; 8.02, s, 1H; 8.57, s, IH.
S
Example 9
2-Amino-9-[3-hydroxy-2-hydroxymethy (prop-1-yl]purine
2-Amino-9-[(2-isopropyl-1,3-dioxan-5-yl)methyl]purine (160 mg, 0.58 mmol) was
stirred in trifluoroacetic acid (5 ml) for 2 hr. After this time a few drops
of water was
added and the reaction mixture was stirred for a further 3 hr. Solvent was
removed in
vacuo and the residue was neutralized with saturated sodium bicarbonate. The
solution
was concentrated to dryness and hot methanol was added. The solution was
decanted and
passed through a short silica column. The crude material was then hplc
chromatographed
with 2% acetonitrile in water as the eluting solvent . The fractions
containing the desired
product were combined and freeze dried to afford 2-amino-9-[3-hydroxy-2-
hydroxymethylprop-1-yl]purine (70 mg, 55%) as a fluffy colourless solid. 'H
n.m.r.
(DMSO-db) 2.00 - 2.20, m, 1 H, 3.20 -3.40, m, 4H; 4.06, d, J = 7Hz, 2H; 4.68,
br s,
2H; 6.53, s, 2H; 8.00, s, IH; 8.57, s, IH.
CA 02302630 2000-03-06
WO 99/12927 PCTJAU98/00748
-30-
Examl I~ a 10
9-[3-Hydroxy-2-hydroxymethylprop-1-ylJ-guanine sodium salt
9-[3-Hydroxy-2-hydroxymethylprop-1-yl]-guanine (147.5 mg, 0.616 mmol) was
dissolved in aqueous sodium hydroxide (1.0 M, 616 ~cl, 0.616 mmol). The
solution was
filtered, washing with a small amount of water, then freeze dried to afford 9-
[3-hydroxy-
2-hydroxymethylprop-I-yl]-guanine sodium salt (160.7 mg, quantitative) as a
fluffy
colourless solid. 'H n.m.r. (DMSO-db) 1.78 - 2.02, m, 1H; 3.06 - 3.29, m, 4H;
3.91,
d, J = 6Hz, 2H; 5.16, br s, 2H; 5.42, br s, 2H; 7.32, s, 1H.
Example 11
9-[ 3-Acetoxy-2-hydroxymethylprop-1-yIJ-guanine
To a solution of 9-[3-hydroxy-2-hydroxymethylprop-1-yl]-guanine (1.0 g, 4.18
mmol) in N,N-dimethylformamide (5 ml) under a nitrogen atmosphere, was added
trit7uoroacetic acid(510 ~l, 6.63 mmol) and triethylorthoacetate (790 ~cl,
4.31 rnmol)
resulting in a cloudy mixture. The reaction was monitored by hplc (5 %
acetonitrile in
water) and more triethylorthoacetate (720 ~cl, 3.92 mmol) was added
portionwise until the
reaction was complete. Water (243 ~1, 13,5 mmol) was added and the reaction
stirred for
a further 1.5 hr. The reaction mixture was neutralised with sodium bicarbonate
and
solvent was removed at room temperature. The crude product was recrystallised
from
water to give a colourless product (901 mg). This was preadsorbed and flash
chromatographed on silica eluting with 10%, 15% and 17 % methanol in
dichloromethane. The fractions containing pure product were combined and
concentrated
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-31-
to afford 9-[3-acetoxy-2-hydroxymethylprcp-I-yl]-guanine (520 mg, 44 %) as a
colourless solid. 'H n.m.r. {DMSO-d~) 1.94, s, 3H; 2.20 - 2.40, m, 1H; 3.26 -
3.43,
m, 2H; 3.84 - 4.03, m, 4H; 4.82, t, J = SHz, 1H; 6.46, s, 2H; 7.64, s, IH;
10.58, s,
1H.
Example 12
9-[3-Hydroxy-2-hydroxymethylprop-1-yl]-guanine triphosphate tetraammonium salt
The procedure used for the preparation followed that of Ludwig et al. A
solution of 2-
chloro-4H-1,3,2-benzodioxaphosphorin-4-one (150 mg, 0.74 mmol) in dry dioxane
(2
ml) was added dropwise to the stirred 9-[3-acetoxy-2-hydroxymethylprop-1-yl]-
guanine
(187.8 rng, 0.668 mmol, dried under high vac. at 85°C for ca. 7-8 h) in
dry N,N-
dimethylformamide ( 10 ml) and dry pyridine (2 ml) over approximately 5 min.
Stirring
was continued for 0.75 hr. Bis[(tri-rt-butyl)ammonium]pyrophosphate hemi DMF
(590
mg; 1.0 mmol) dissolved in dry N,N-dimethylformamide (2.5 ml) containing dry n-
BujN(0.75 ml) was then added dropwise to the stirred solution.
After ca. 2.5 h stirring at room temperature, the yellow reaction solution was
treated
dropwise with iodine solution (9.5 ml) made by dissolving iodine (3.56 g) in
pyridine
(200 ml ) and water (5 ml). The slight excess of iodine added was destroyed on
addition
of a few drops of 5 % NaHS03 solution.
After stirring for ca. 1.5 hr at room temperature, the solvent was removed
below 30° to
give an orange-yellowish oil which was treated with 25 ml water at room
temperature for
1 h with strong stirring. Concentrated NH40H (50 ml) was added and the
reaction
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-32-
mixture was stirred for 3 hr at room temperature before removing the NH~OH
solution at
25°C. The semi-solid product was treated twice with acetone to give a
pale yellow solid
(0.56 g).
The product (0.5 g) was dissolved in a little water, centrifuged and made up
to 5 ml in
volume and HPLC chromatographed in lots of 0.5 ml eluting with water (flow
rate of 12
ml/min) The fractions between 13-14 mins and 18-19 mins were collected and
were
freeze dried to give a pale yellow solid (320 mg).
The material was dissolved in water (4 ml) and re-chromatographed in 0.5 ml
lots. For
each fraction the leading and trailing section of the peak containing the
triphosphate was
cut. This was repeated a further 4 times. Finally the product was dialysed
(100 MWCO
tubing, 6.5 h, H20) before again purifying by HPLC. This treatment removed a
small
amount of phosphorus impurity (small 3'P nmr peak at 8 +0.93), due,
presumably, to
some inorganic phosphate.
'H nmr (DZO); 8 2.41, s, 1H; 3.60, d, J=5.38 Hz, 2H; overlapping doublets
4.00, d,
J=4.98 Hz and 4.17, d, J=5.76 Hz, 4H; 7.87, s, 0.8H.
3'P nmr (D20): In the nmr of the crude triphosphate the phosphate peaks were
the best
detined; viz. Pp (t, S-21.16, J,>y=19.66 Hz); P" (doublet of triplets at ~-
9.87 (JPP 19.40
Hz; JPH 5.39 Hz); Pa (d, 8-5.96; J~P 20.01 HZ). The 3'P nmr peaks broadened
and
moved on HPLC purification so that the purified sample had two broad 3'P peaks
at 8-
9.96 (Pa+Pa) and 8-22.07 (P~3) of intensity ratio 2:1.
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-33-
Example 13
2-Amino-6-cyclopropylamino-9-[3-hydroxy-2-hydroxymethylprop-1-yl]purine
S A mixture of 9-[3-acetyloxy-2-acetyloxymethylprop-I-yl]-2-amino-6-iodopurine
(557 mg,
1.24 mmol) and cyclopropylamine ( 1.0 ml, 14.4 mmol)) were stirred for 18 hr
in a bomb at
80". The reaction mixture was cooled to room temperature and concentrated to
give a crude
orange oil (989 mg). A portion of this was then hplc chromatographed with 3%
acetonitrile
in water as the eluting solvent . The fractions containing the desired product
were combined
and freeze dried to af~'ord 2-amino-6-cyclopropylamino-9-[3-hydroxy-2-
hydroxymethylprop-
I-yl]purine as yellow oil which solidified on standing. 'H n.m.r. (DMSO-db)
0.55 - 0.76.
m, 4H; 1.9I - 2.12, m, 1H; 2.95 - 3.13, m, 1H; 3.21 - 3.45, m, 4H; 3.97, d, .l
= 7 Hz, 2H;
4.75, t, J = S Hz, 2H; 5.91, br s, 2H; 7.09, d, J = 5 Hz, 1H; 7.62, s, 1H.
Example 14
9-[3-Acetyloxy-2-acetyloxymethylprop-1-yl]-2-aminopurine
9-[3-Acetyloxy-2-acetyloxymethylprop-I-yl]-2-aminopurine was prepared
according to the
method of Example 8 from 9-[3-acetyloxy-2-acetyloxymethylprop-1-yl]-2-amino-6-
iodopurine (5.0 g, 1 I.5 mmol), triethylamine (1.76 ml, 12.65 mmol) and 10%
palladium on
carbon (500 mg) in ethanol (200 ml). Yield: 2.33 g (66%). 'H n.m.r. (DMSO-d~)
1.93, s,
6H; 2.59 - 2.80, m, 1H; 3.96, dd, .7 = 5.6 & I 1.4 Hz, 2H; 4.03, dd, J = 5.6 &
11.4 Hz, 2H;
4.15, d, .I = 7 Hz, 2H; 6.49, br s, 2H; 8.04, s, 1H; 8.57, s, 1H.
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-34-
Examl 1~ a 15
9-[3-Acetyloxy-2-acetyloxymethylprop- I -yl]-guanine
A mixture of 9-[3-hydroxy-2-hydroxymethylprop-1-yl]-guanine (560 mg, 2.34
mmol) and 4-
dimethylaminopyridine (50 mg) in acetic anhydride ( I 5 ml) was stirred at
room temperature
for 16 hr. The reaction mixture was concentrated to dryness and the residue
was partitioned
between water and chloroform. The resulting solid was filtered off and
recrystallised from
methanol to afford 9-[3-acetyloxy-2-acetyloxymethylprop-I-yl]-guanine as
colourless
crystals (593 mg, 78%). 'H n.m.r. (DMSO-d~) 1.97, s, 6H; 2.53 - 2.70, m, 1H;
3.87 -
4.09, m, 6H; 6.42, br s, 2H; 7.67, s, IH; 10.56, br s, 1H.
Example 16 & 17
IS 9-[2-L-Valyloxymethyl-3-L-valyloxyprop-1-yl]-guanine bishydrochloride salt
and
9-[3-Hydroxy-2-L-valyloxymethylprop-I-yl]-guanine hydrochloride salt
A mixture of 9-[3-hydroxy-2-hydroxymethylprop-1-yl]-guanine (3.02 g, 12.6
mmol), N-
benzyloxycarbonyl-L-valyl-N-carboxy anhydride (Z-L-valyl-NCA) (purchased from
Isochem
or SNPE North American Inc.) (3.69 g, 13.32 mmol) and 4-dimethylaminopyridine
(75 mg)
in N,N-dimethylformamide (75 ml), under a nitrogen atmosphere, was stirred at
room
temperature for I 6 hr. HPLC analysis indicated incomplete reaction. A further
portion of Z-
L-valyl-NCA (1.84 g, 6.67 mmol) was added and the reaction mixture was stirred
a further
24 hr. Concentrated to dryness and ethyl acetate was added to precipitate out
unreacted 9-
[3-hydroxy-2-hydroxymethylprop-I-yl]-guanine. The filtrate was concentrated,
preadsorbed
onto silica and flash chromatographed on silica with 5%, 7%, 10% & 15%
methanol in
dichloromethane as the eluting solvents. Fractions containing a single spot on
tlc were
combined to give 9-[2-(N-benzyloxycarbonyl-L-valyloxymethyl)-3-(N-
benzyloxycarbonyl-L-
valyloxyprop-1-yl] guanine as a colourless solid (2.54 g). 'H n.m.r. (DMSO-d~)
0.86, d,
CA 02302630 2000-03-06
WO 99/12927 PCTlAU98/00748
-35-
12H; 1.85 - 2.15, m, 2H; 2.51 - 2.70, m, 1H; 3.80 - 4.18, m, 8H; 5.04, s, 4H;
6.41, br s, 2H;
7. 31, br s, 1 OH; 7. S 9, s, 1 H; 7. 75, d, J = 8 Hz, 2H; 10. 62, s, 1 H.
The remaining fractions were combined and rechromatographed with 5%, 7%, 10% &
15%
methanol in dichloromethane as the eluting solvents to give a further 229 mg
of the divalyl
compound and a colourless solid (880 mg). This was dissolved in
dichloromethane then
washed with saturated sodium bicarbonate {2x) and brine, then dried over
magnesium sulfate
to give 9-[2-(N-benzyloxycarbonyl-L-valyloxy methyl)-3-hydroxyprop-I-yl]
guanine as a
colourless foamy solid (266 mg) 'H n.m.r. (DMSO-d~) 0.86, d, .I = 6.8 Hz, 6H;
1.88 -
2.11, m, 1H; 2.20 - 2.39, m, 1H; 3.21 - 3.43, m, 2H; 3.78 - 4.10, m, SH; 4.82,
t, .I = 5 Hz,
1H; 5.04, s, 2H; 6.46, br s, 2H; 7.34, br s, SH; 7.6I, d, J = 3 Hz, 1H; 7.65 -
7.75, m, IH;
10.59, br s, 1H.
9-[2-L-Valyloxymethyl-3-L-valyloxyprop-1-yl] guanine bishydrochloride salt
A mixture of 9-[2-(N-benzyloxycarbonyl-L-valyloxymethyl)-3-(N-
benzyloxycarbonyl-L-
valyloxyprop-1-yl]guanine (858 mg, 1.22 mmol), 1M aqueous hydrochloric acid
(2.44 ml,
2.43 mmol) and 10% palladium on carbon (215 mg) in ethanol (SO ml) was stirred
in an
atmosphere of hydrogen for 3 hr. The reaction mixture was filtered through GFA
paper
washing copiously with ethanol. The filtrate was concentrated to dryness at
40°. The residue
was taken up in water and filtered through a short column packed with GFA
paper, Celite
577 and more GFA paper. The filtrate was freeze dried to give 9-[2-L-
valyloxymethyl-3-L-
valyloxyprop-1-yl] guanine bishydrochloride salt as a cream solid (517 mg,
83%). 'H n.m.r.
(DMSO-d~) 0.87 - 1.02, m, 12H; 2.05 - 2.30, m, 2H; 2.60 - 2.80, m, 1H; 3.85,
dd, J = 4.6
& 1 I.1 Hz, 2H; 4.00 - 4.32, m, 6H; 6.59, s, 2H; 7.78. s, 1H; 8.63, br s, 6H;
10.78, br s, IH.
CA 02302630 2000-03-06
WO 99112927 PCT/AU98/00748
-36-
9-[3-Hydroxy-2-L-valyloxymethylprop-1-yl]guanine hydrochlo:ide salt
9-[3-Hydroxy-2-L-valyloxymethylprop-1-yl] guanine hydrochloride salt was
prepared
according to the method of Example 16 from 9-[2-(N-benzyloxycarbonyl-L-
valyloxy
S methyl)-3-hydroxyprop-1-yl]-guanine (212 mg, 0.45 mmol), IM aqueous
hydrochloric acid
(0.45 ml, 0.45 mmol) and 10% palladium on carbon (S6 mg) in ethanol (1S ml) to
give the
product as a colourless fluffy solid (I 62 mg, 96%). 'H n.m.r. (DMSO-d~) 0.96,
dd, J =
7.2 & I 1.9 Hz, 6H; 2.00 - 2.25, m, I H; 2.25 - 2.47, m, I H; 3.31 - 3.51, m,
2H; 3 .82, dd, J =
4, 6 & 13 . 5 Hz, 1 H; 3 . 96 - 4.14, m, 4H; 4. 9 I , br s, 1 H; 6. S 9, br s,
2 H; 7. 69, s, 1 H; 8. S 2, br s,
3H; 10.72, br s, 1H.
Example 18
9-[3-Acetoxy-2-L-valyloxymethyl)prop-l-yl]-guanine hydrochloride salt
1S
A mixture of 9-[3-acetloxy-2-hydroxymethylprop-I-yl] guanine (330 mg, 1.17
mmol), Z-L-
valyl-NCA (3S8 mg, 1.29 mmol) and 4-dimethylaminopyridine (30 mg) in N,N-
dimethylformamide (20 ml), under a nitrogen atmosphere, was stirred at room
temperature
for 2 days. The reaction mixture was concentratred to dryness and the residue
preadsorbed
onto silica and flash chromatographed with 10% methanol in dichloromethane.
Fractions
containing the desired product were combined and concentrated to give 9-[3-
acetoxy-2-(N-
benzyloxycarbonyl-L-valyloxy methyl)prop-I-yl] guanine as a colourless solid
(313 mg,
S2%). 'H n.m.r. (DMSO-d~) 0.87, d, J = 7 Hz, 6H; 1.97, s, 3H; 1.88 - 2.13, m,
1H; 2.53
- 2.71, m, 1H; 3.82 - 4.15, m, 7H; 5.04, s, 2H; 6.43, br s, 2H; 7.35, br s,
SH; 7.63, d, J = 3.6
2S Hz, 1H; 7.75, d, .I = 8.1 Hz, 1H.
9-[3-Acetoxy-2-L-valyloxymethyl)prop-1-yl]-guanine hydrochloride salt was
prepared
according to the method of Example 16 from 9-[3-acetoxy-2-(N-benzyloxycarbonyl-
L-
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-37-
vaIyloxymethyl)prop-1-yl]-guanine (201 mg, 0.39 mmol), 1M aqueous hydrochloric
acid
(0.39 ml, 0.39 mmol) and 10% palladium on carbon (62 mg) in ethanol (10 ml) to
give the
product as a colourless fluffy solid (162 mg, quantitative). 'H n.m.r. (DMSO-
d~) 0.95, dd,
J = 3.8 & 6.8 Hz, 6H; 1.99, s, 3H; 2.03 - 2.25, m, IH; 2.57 - 2.78, m, IH;
3.84, dd, J = 4.7
& 10.2 Hz, IH; 3.93 - 4.23, m, 6H; 6.54, br s, 2H; 7.71, s, 1H; 8.33, br s,
3H; 10.71, br s,
I H.
Example 19
9-[3-Hydroxy-2-palmityloxymethylprop-I-yI] guanine
Palmitoyl chloride (2.07 g, 7.53 mmol) was dissolved in dry dichloromethane
and made up
to a volume of 10 ml and used as a stock solution.
To a suspension of 9-[3-hydroxy-2-hydroxymethylprop-I-yl] guanine (I.0 g, 4.18
mmol) in
I S pyridine (20 ml) and N,N-dimethylformamide (1 Oml), under a nitrogen
atmosphere, was
added the stock solution of palmitoyl chloride (3.5 ml). The reaction mixture
was stirred for
16 hr at room temperature and a further aliquot (3.5 ml) of stock solution was
added. The
reaction mixture was stirred for 24 hr abd the final aliquot of stock solution
was added. The
reaction mixture was stirred for 2 more days, the solvents were removed in
vacuo at 60°.
The crude solid (3.7 g) was preadsorbed and flash chromatographed on silica
eluting with
5% to 23% methanol in dichloromethane. The fractions containing pure product
were
combined to give 9-[3-hydroxy-2-palmityloxymethylprop-1-yl] guanine as a
colourless solid
(595 mg). 'H n.m.r. (DMSO-d~) 0.85, t, J = 6.8 Hz, 3H; 1.12 - 1.38, m, 24H;
1.33 -
1.60, m, 2H; 2.19, q, J = 7.2 Hz, 2H; 3.32 - 3.40, m, 2H; 3.95, t, .I = 6 Hz,
2H; 4.07 - 4.20,
m, 2H; 4.87, br s, 1H; 6.65, br s, 2H; 7.63, s, 1H; 10.76, br s, IH.
Example 20
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-38-
9-[3-Palmityloxy-2-L-valyloxymethylprop-1-yl] guanine hydrochloride salt
A mixture of 9-[3-hydroxy-2-palmityloxymethylprop-1-yl] guanine {534 mg, 1.12
mmol), Z-
L-valyl-NCA (620 mg, 2.24 mmol) and 4-dimethylaminopyridine (25 mg) in N,N-
S dimethylformamide (25 ml), under a nitrogen atmosphere, was stirred at room
temperature
for 16 hr. Solvent was removed in vacrso at 60° and the residue
partitioned between
dichloromethane and water. The layers were separated and the aqueous was
extracted twice
more with dichloromethane. The combined organic layers were washed with
saturated
sodium bicarbonate solution (2x) and brine, dried over magnesium sulfate and
concentrated
to give the crude product as a colourless oil (509 mg). This was flash
chromatographed on
silica eluting with 5% methanol in dichloromethane. The fractions containing
the pure
product were combined to give 9-[3-palmityloxy-2-(N-benzyloxycarbonyl-L-
valyloxymethyl)prop-I-yl] guanine as a colourless foamy solid (290 mg). 'H
n.m.r.
(DMSO-db) 0.75-0.95, m, 9H; 1.22, br s, 24H; 1.33 - 1.60, m, 2H; 1.90 - 2.15,
m, IH;
IS 2.23, t, J = 7.2 Hz, 2H; 2.50 - 2.75, ra, 1H; 3.85 - 4.14, m, 7H; 5.04, s,
2H; 6.45, br s, SH;
7.62, d, ,l = 3.8 Hz, 1 H; 7.74, d, J = 8.2 Hz, 1 H; 10.66, br s, 1 H.
9-[3-Palmityioxy-2-L-valyloxymethylprop-1-yl] guanine hydrochloride salt was
prepared
according to the method of Example I 6 from 9-[3-palmityloxy-2-(N-
benzyloxycarbonyl-L-
valyloxymethyl)prop-1-yl] guanine (174 mg, 0.24 mmol), IM aqueous hydrochloric
acid
(0.24 ml, 0.24 mmol) and 10% palladium on carbon (50 mg) in ethanol (10 ml) to
give the
product as a colourless fluffy solid (124 mg, 84%). 'H n.m.r. (DMSO-d~) 0.78 -
0.98, m,
9H; 1.22, br s, 24H; I .37 - 1.58, m, 2H; 1.99 - 2.22, m, IH; 2.25, t, J = 7.2
Hz, 2H; 2.55 -
2.76, m, I H; 3 .74, dd, J = 4. 8 & I 0.0 Hz, 1 H; 3 . 90 - 4.20, m, 6H; 6.
51, br s, 2H; 7.3 0 -
8.10, br s, 3H; 7.69, s, 1H; 10.88, br s, 1H.
CA 02302630 2000-03-06
WO 99112927 PCT/AU98/00748
-39-
Example 2.1
9-[3-Hydroxy-2-cholyloxymethylprop-I-yl]-guanine
To a solution of cholic acid (1.71 g, 4.18 mmol) and diisopropylethylamine
(567 mg, 4.39
mmol) in N,N-dimethylformamide (25 ml), under a nitrogen atmosphere, at
10°, was added
ethyl chloroformate (4001, 4.18 mmol} This was stirred for 30 minutes, then a
mixture of 9-
[3-hydroxy-2-hydroxymethylprop-1-yl] guanine (1.0 g, 4.18 mmol) in N,N-
dimethylformamide ( 100 ml) was added and the reaction mixture was stirred for
a further 2
days. HPLC analysis (70% methanol in water) indicated ~ 20 - 30% product had
formed.
The reaction mixture was concentrated to dryness and methanol was added to
precipitate out
unreacted 9-[3-hydroxy-2-hydroxymethylprop-I-yl] guanine. The filtrate was
concentrated
to dryness to give a yellow oil (2.0 g), which was purified by semi-
preparative HPLC eluting
with 70% methanol in water. 9-[3-Hydroxy-2-cholyloxymethylprop-I-yl] guanine
was
obtained as a colourless glassy solid (124 mg). 'H n.m.r. (DMSO-d~) 0.57, s,
3H; 0.80, s,
3H; 0.70 - 2.40, m, 27H; 3.08 - 3.28, m, 2H; 3.37, d, J = 5.2 Hz, 2H; 3.61, s,
1H; 3.78, s,
1H; 3.87 - 4.05, m, 5H; 4.12, d, J = 3.4 Hz, 1H; 4.33, d, J = 4.3 Hz, 1H;
4.82, t, J = 4.5
Hz, I H; 6. 52, s, 2H; 7.63, s, 1 H; 10. 74, br s, 1 H.
Example 22
9-[3-Cholyloxy-2-L-valyloxy-methylprop-I-yl] guanine hydrochloride salt
9-[2-(N-Benzyloxycarbonyl-L-valyloxy-3-cholyloxymethylprop-I-yl]-guanine was
prepared
according to the method of Example 20 from crude 9-[3-hydroxy-2-
cholyloxymethylprop-1-
yl]-guanine (1.21 g, 1.92 mmol), Z-L-valyl-NCA (0.99 g, 3.57 mmol) and 4-
dimethylaminopyridine (20 mg) in N,N-dimethylformamide (10 ml). The crude
material was
flash chromatographed on silica eluting with 10% methanol in dichloromethane.
The
fractions containing the pure product were combined to give 9-[2-(N-
benzyloxycarbonyl-L-
CA 02302630 2000-03-06
WO 99 12927 PCT/AU98/00748
-40-
valyloxy-3-cholyloxymethylprop-1-yl] guanine as a colourless foamy solid (258
mg). 'H
n.m.r. (DMSO-d~) 0.57, s, 3H; 0.80, s, 3H; 0.74 - 2.40, m, 34 H; 2.50 - 2.78,
m, IH; 3.05
- 3.30, m, 2H;~3.60, s, IH; 3.77, s, 1H; 3.83 - 4.20, m, 8H; 4.33, d, .l = 4.3
Hz, 1H; 5.04, s,
2H; 6.44, br s, 2H; 7.34, br s, 5H; 7. 62, d, J = 3 .7 Hz, 1 H; 7. 74, d, J =
8.2 Hz, 1 H; 10.61,
S br s, 1 H.
9-[3-Cholyloxy-2-L-valyloxy-methylprop-I-yl] guanine hydrochloride salt was
prepared
according to the method of Example 16 from 9-[2-(N-benzyloxycarbonyl-L-
valyloxy-3-
cholyloxymethylprop-1-yl] guanine (248 mg, 0.29 mmol), IM aqueous hydrochloric
acid
1 (1 (0.29 mI, 0.29 mmol) and 10% palladium on carbon (30 mg) in ethanol ( 10
ml) to give the
product as a cream solid
(185 mg, 84%). 'H n.m.r. (DMSO-d~) 0.57, s, 3H; 0.80, s, 3H; 0.74 - 2.40, m,
34H; 2.50
- 2.78, m, 1H; 3.05 - 3.30, m, 2H; 3.60, s, 1H; 3.77, s, IH; 3.85, dd, .I =
4.6 & 10.5 Hz, 1H;
3.93 - 4.26, m, 7H; 4.34, d, .l = 4.0 Hz, IH; 6.56, br s, 2H; 7.71, s, 1H;
8.25 - 8.80, br s,
15 3H; 10.72, br s, IH.
Example 23
9-[2-Elaidyloxy-3-hydroxy-methylprop-1-yl] guanine
9-[2-Elaidyloxy-3-hydroxy-methylprop-1-yl] guanine was prepared according to
the method
of Example 19 from 9-[3-hydroxy-2-hydroxymethylprop-1-yl] guanine (1.3 g, 5.43
mmol),
elaidoyl chloride (3.1 g, 9.70 mmol) dissolved in dry dichloromethane {7 ml),
in pyridine (25
ml) and N,N-dimethylformamide (12 ml) After work-up, some elaidic acid was
removed by
distillation, to give a crude yellow solid (2.14 g). This was chromatographed
on silica eluting
with 7.5% methanol in dichloromethane. The fractions containing pure product
were
combined to give 9-[2-elaidyloxy-3-hydroxy-methylprop-1-yl] guanine (640 mg).
'H n.m.r.
(DMSO-dG) 0.84, t, .I = 6.7 Hz, 3H; 1.23, br s, 20H; 1.30 - 1.59, m, 2H; 1.85 -
2.00, m,
CA 02302630 2000-03-06
WO 99/12927 PCT AU98/00748
-41 -
4H; 2.20, t, .I = 7.3 Hz, 2H; 2.22 - 2.41, m, 1H; 3.36, d, J = 5.4 Hz, 2H;
3.85 - 4.07, m, 4H;
4.82, t, J = 5 Hz, 1H; 5.23 - 5.48, m, 2H; 6.44, br s, 2H; 7.63, s, 1H; 10.58,
br s, IH.
Example 24
9-[2-Stearoyloxy-3-valyloxymethyl)prop-I-yl] guanine hydrochloride salt
A mixture of 9-[-2-elaidyloxy-3-hydroxymethylprop-1-yl] guanine (200 mg, 0.40
mmol), Z-
L-valyl-NCA (121 mg, 0.44 mmol) and 4-dimethylaminopyridine (5 mg) in
dichloromethane
(20 ml), under a nitrogen atmosphere, was stirred at room temperature for 3
days. A further
portion of Z-L-valyl-NCA (40 mg, 0.14 mmol) was added and the reaction was
stirred at 40°
for 6 hr and then at room temperature for 16 hr. Solvent was removed in vacuo
at 60°, the
residue preadsorbed and flash chromatographed on silica eluting with 6%
methanol in
dichloromethane. The fractions containing the pure product were combined to
give 9-[2-(N-
benzyloxycarbonyl-L-valyloxy-3-elaidyloxy-methyl)prop-1-yl]-guanine as a
colourless foamy
solid (174 mg, 60%). 'H n.m.r. (DMSO-d~) 0.78 - 0.93, m, 9H; 1.22, br s, 20H;
1.30 -
1.59, m, 2H; 1.89 - 2.13, m, SH; 2.23, t, .I = 7.2, Hz, 2H; 2.50 - 2.72, m,
1H; 3.85 - 4.14, m,
6H; 5.04, s, 2H; 5.24 - 5.45, m, 2H;6.43, s, 2H; 7.34, br s, SH; 7.61, d, J =
3.9 Hz, IH;
7. 74, d, J = 8.1 Hz, 1 H; 10.64, br s, 1 H.
9-[2-Stearoyloxy-3-valyloxymethyl)prop-1-yl] guanine hydrochloride salt was
prepared
according to the method of Example 16 from 9-[2-(N-benzyloxycarbonyl-L-
valyloxy-3-
elaidyloxy-methyl)prop-I-yl] guanine (162 mg, 0.22 mmol), IM aqueous
hydrochloric acid
(0.22 mI, 0.22 mmol) and 10% palladium on carbon (60 mg) in ethanol (10 ml) to
give the
product as a colourless fluffy solid (108 mg, 77%). 'H n.m.r. (DMSO-d~) 0.84,
t, J = 6.3
Hz, 3H; 0.93, dd, J = 3.2 & 6.8 Hz, 6H; I .22 br s, 28H; I .36 - 1.57, m, 2H;
1.98 - 2.23, m,
1H; 2.25, t, J = 7.3 Hz, 2H; 2.55 - 2.75, m, 1H; 3.77, dd, J = 4.6 & 10,0 Hz,
1H; 3.90 -
4.22, m, 6 H; 6.51, br s, 2H; 7.69, s, IH; ?.65 - 8.15, br s, 3H; 10.48 -
10.82, br s, 1H.
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-42-
Example 25
2-Amino-9-[2-L-valyloxy-3-L-valyloxymethylprop-I-yl]purine bishydrochloride
salt
A mixture of 2-amino-9-[3-hydroxy-2-hydroxymethylprop-I-yl]purine (9)(500 mg,
2.24
mmol), Z-L-valyl-NCA (1.30 g, 4.70 mmol) and 4-dimethylaminopyridine (5 mg) in
N,N-
dimethylformamide ( 10 ml), under a nitrogen atmosphere, was stirred at room
temperature
for 18 hr. The reaction mixture was concentrated to dryness and the residue
chromatographed on silica with ethyl acetate as the eluting solvent. The
fractions containing
pure product were combined and concentrated to give 2-amino-9-[2-(N-
benzyloxycarbonyl-
L-valyloxymethyl)-3-(N-benzyloxycarbonyl-L-valyloxyprop-1-yl]purine as a honey
coloured
glassy solid ( 1.12 g, 73%). 'H n.m.r. (DMSO-d~) 0.86, d, .l = 6.7 Hz, 12H;
1.85 - 2.1 S,
m, 2H; 2.57 - 2.78, m, 1H; 3.83 - 4.19, m, 8H; 5.04, s, 4H; 6.49, s, 2H; 7.34,
br s, 10H;
7. 76, d, J = 8.2 Hz, 2H; 7.96, s, 1 H; 8. 5 9, s, 1 H.
2-Amino-9-[2-L-valyloxy-3-L-valyloxymethylprop-I-yl]purine bishydrochloride
salt was
prepared according to the method of Example 16 from 2-amino-9-[2-(N-
benzyloxycarbonyl-
L-valyloxymethyl)-3-(N-benzyloxycarbonyl-L-valyloxyprop-1-yl]purine (500 mg,
0.73
mmol), 1M aqueous hydrochloric acid (1.45 ml, 1.45 mmol) and 10% palladium on
carbon
(90 mg) in ethanol (25 ml) to give the product as a colourless fluffy solid
(319 mg, 89%).
'H n.m.r. (DMSO-d~) 0.87 - 1.03, m, 12H; 2.03 - 2.27, m, 2H; 2.70 - 2.80, m,
IH; 3.83,
dd, .l = 4.8 & 11.2 Hz, 2H; 4.07 - 4.42, m, 6H; 6.50, s, 2H; 8.15 - 8.88, br
s, 3H; 8.20, s,
1 H; 8 . 60, s, 1 H.
Example 26
9-[3-Acetoxy-2-acetoxymethylprop-1-yl]-6-thioguanine
A mixture of 9-[3-acetoxy-2-acetoxymethylprop-I-yl]-2-amino-6-iodopurine (576
mg, 1.33
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
- 43 -
mmol) and thiourea (100 mg, 1.33 mmol) in ethanol (7 ml) under a nitrogen
atmosphere,
was heated at reflux for I hr. The reaction mixture was chilled and the
product filtered ofl',
washing with ethanol. The pale lemon solid was dried in vacuo at 80° to
give 9-(3-acetoxy-2-
acetoxymethylprop-1-yIJ-6-thioguanine (288 mg, 64%). 'H n.m.r. (DMSO-db) 1.98,
s,
6H; 2.52 - 2.74, m, 1H; 3.87 - 4.13, m, 6H; 6.77, s, 2H; 7.88, s, 1H; 11.89,
s, 1H.
Example 27 Antiviral Activity
Tests of antiviral activity in human cells infected with hepatitis B were
performed
according to the method of Korba and Gerin. The effective concentration for
50% and
90% inhibition of the replication of the virus was determined from dose
response curves.
Results for some compounds of the invention are shown in Table 2.
Table 2
Test Compound EC50 p,M EC9o p.M
Exam le 1 0.16 1.9
Exam le 13 4.4 13
Example 28
Bioavailability Testing
The oral bioavailability of various compounds of the invention was compared in
rats.
Briefly, the compounds were administered by oral gavage at 0.2mmol/kg of body
weight.
The compounds were suspended in 1 mL of an aqueous vehicle containing 1 %
carboxymethylcellulose and 0.05 % Tween 80. Plasma was sampled over an 8 hour
period and the concentration of the parent compound, in this case the compound
of
CA 02302630 2000-03-06
WO 99/12927 PCT/AU98/00748
-44-
example 1, was determined by hplc.
Aliquots of rat plasma ( I SO ~cL) were acidified with 3 0% trichloroacetic
acid (37.5 uL) and
centrifuged at 3000 rpm for 10 min. The supernatant was filtered through 0.22
~m
cellulose acetate centrifuge filters. Samples (100 ~cL) were then injected
onto the C18
Waters Symmetry HPLC column (3.9 and 150 mm, 5 ~cm) equilibrated at
40°C. The two
component mobile phase (A - 0.05% trifluoroacetic acid and 20 mM heptane
sulfonic acid in
distilled, deionised water; B - 0.05% trifluoroacetic acid, 20 mM heptane
sulfonic acid and
70% acetonitrile in distilled, deionised water) was pumped at 0.5 mL/min with
the
percentage of mobile phase component B increasing linearly from 0 to 25% over
25 min.
Analysis was by ultraviolet detection at 254 nm. The parent compound eluted at
17 min.
The concentration of drug versus time profile was plotted and the area under
the curve
determined. This was compared with the area under the curve provided by
intravenous
administration of the sodium salt of the parent compound to provided a measure
of total
bioavailabilty as a percentage. Results are shown in Table 3.
Test Compound % Bioavailabilty
Exam le 1 2.8
Exam le 9 53.7
Example 14 66.2
Exam le 16 16.3
Exam le 17 33.1
Exam le 18 6.5
Exam le 20 21
It will be appreciated by persons skilled in the art that numerous variations
and/or
modifications may be made to the invention as described without departing from
the spirit
or the scope of the invention. The present examples and specific details are,
therefore, to
CA 02302630 2000-03-06
WO 99/i2927 PCT/AU98/00748
- 45 -
be considered in all respects as illustrative of the invention and not
restrictive.
The steps, features, compositions and compounds disclosed herein or referred
to or
indicated in the specification and/or claims of this application,
individually, collectively, and
any and all combinations of any two or more of said steps or features.
Throughout this specification and the claims which follow, unless the context
requires otherwise, the word "comprise", and variations such as "comprises"
and
"comprising", will be understood to imply the inclusion of a stated integer or
step or
group of integers or steps but not the exclusion of any other integer or step
or group of
integers or steps.
lU
References:
Cullen et al., Antimicrob. Agents Chemother., 41 (10), 2076-82 (1997)
Martin et al., J. Med. Chem. 29, 1384-1389 (1986).
Greene T. W., Protective Groups in Organic Synthesis, John Wiley and Sons,
Inc. (1991)
Bisacchi et al., J. Org. Chem. 60, 2902 - 2905 (1995).
Harnden et al., J. Med. Chem. 33, 187-196 ( 1990)
Korba and Gerin, Antiviral Research, 19, 55-70 (1992)
Eliel et al., J. Am. Chem. Soc. 94(1), 171-176 (1972)
Dunn et al., J. Org. Chem. 55, 6368-6373 (1990)
Ludwig et al., J. Org. Chem. ,54, 631-635 (1989)