Note: Descriptions are shown in the official language in which they were submitted.
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
METHODS OF SYNTHESIZING POLYNUCLEOTIDES BY LIGATION
OF MULTIPLE OLIGOMERS
FIELD OF THE IN'VENTION
The present invention relates generally to methods of
synthesizing polynucleotides. More specifically, the
invention relates to methods of synthesizing
polynucleotides by ligation of a plurality of oligomeric
units onto a template-bound primer. The plurality of
oligomers can be preselected to contain oligomers which are
complementary to the template strand or the oligomers can
be supplied as a library and allowed to self select. The
synthesis by ligation can proceed unidirectionally or
bidirectionally from the primer and can be used to
synthesize both strands simultaneously by the use of two
primers. Amplification can be performed linearly or
exponentially and can be used to copy DNA and RNA. The
methods of the invention are useful in a variety of
applications, including cloning, preparing labeled
polynucleotides for diagnostic use, mutation analysis and
screening, gene expression monitoring and sequence
analysis.
BACKGROUND OF THE INVENTION
The enzymatic ligation of pairs of oligonucleotides
bound to a target nucleic acid is widely known. It is
generally thought that the oligomers must each be of a
minimum length to be ligated efficiently. Recent work has
shown this minimum length to be about 6-8 bases (C.E.
1
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
Pritchard and E.M. Southern, Nucl. Acids Res., 25, 3403-
3407 (1997)). It is generally thought that ligation of
oligonucleotides shorter than about 6 bases is not
possible.
Under certain conditions, primer independent ligation
can be accomplished using oligomers of at least six bases
long. In this manner, PCR primers were prepared in situ
from concatenated groups of a small number hexamers,
heptamers or octamers (T. Kaczorowski and W. Szybalski,
Gene, 179, 189-193 (1996); L.E. Kotler, D. Zevin-Sonkin,
I.A. Sobolev, A.D. Beskin and L.E. Ulanovsky, Proc. Natl.
Acad. Sci. USA, 90, 4241-4245 (1993)). Such ligation in the
absence of a primer is undesirable in the present methods
and must be avoided. The success in replicating a
polynucleotide sequence in a controlled and defined manner
rests in knowing the point of origination of the newly
synthesized strand.
Nucleic acids can be synthesized from a template,
primer and nucleotide triphosphates (NTPs) by the action of
a polymerase action. Labels can be incorporated by
substituting a percentage of labeled NTPs. The ability to
achieve a high degree of label incorporation is limited and
the precise spacing of labels is not controllable.
The polymerase chain reaction (PCR) is a method of
amplifying the amount of a polynucleotide by the use of a
primer complementary to each strand which span the region
to be replicated. Nucleic acid synthesis proceeds by
extension of each primer with a polymerase and the four
dNTPs. Thermal cycling allows multiple copies of the
template to be synthesized, approximately doubling the
2
CA 02304258 2000-03-17
WO 00/05412 PCTIUS99/14160
quantity of amplicon in each cycle. A variant termed Ligase
Chain Reaction (LCR) involves the ligation of two pairs of
oligonucleotides with a ligase enzyme to replicate the
sequence of interest (D.Y. Wu and R.B.Wallace, Genomics, 4,
560-569 (1989)). The two oligonucleotides to be ligated
constitute the entire length of the strand. Ligation of a
large number of small oligomers to a primer to replicate a
nucleic acid has not been achieved to the best of
Applicant's knowledge.
Methods of providing sequence information using
oligonucleotide ligation are disclosed in US 5,750,341 and
US 5,770,367 and a publication (S. Dubiley, E. Kirilov, Y.
Lysov and A Mirzabekov, Nucl. Acids Res., 25, 2259-2265
(1997)). The reported methods differ fundamentally from
those of the present invention in requiring that oligomers
be ligated one at a time and the sequence be analyzed after
each step. These methods are therefore far more laborious
than those of the present invention.
Methods of Labeling Nucleic Acids - Present methods of
labeling nucleic acids or oligonucleotides include the
tailing method, random primed labeling, nick translation,
the labeled branched DNA and end labeling using a labeled
primer. Each method suffers disadvantages in certain
applications. Use of an end labeled primer extended by PCR
with unlabeled bases leads to only one or a few labels per
product nucleic acid.
The tailing method incorporates an indeterminate and
uncontrolled number of labels by appending a tail of
noncomplementary bases onto the nucleic acid of interest.
This adds many additional bases, which not only adds
3
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
expense, but may interfere with hybridization and lead to
nonspecific binding. In addition it is not readily
applicable to the synthesis,of short nucleic acids or
oligonucleotides since the length of the tail could exceed
the length of the sequence of interest.
The random prime method, applicable to the labeling of
long nucleic acids, uses a mixture of primers which are
extended by a polymerase with a mixture of labeled and
unlabeled bases. The number of bases which can be
incorporated is variable and arbitrary in number. A mixture
of numerous nucleic acid fragments of varying lengths are
produced from both strands. Similarly, nick translation
produces a mixture of numerous nucleic acid fragments of
varying lengths from both strands. Breaks in both strands
of DNA are created and new nucleic acid strands are
synthesized from the position of the nick using a mix of
labeled and unlabeled bases. Since the position of nicking
is arbitrary, label incorporation is not controlled either.
The branched DNA technology has been used in diagnostic
tests as a means to attach several labels to a target DNA.
The methodology relies on the creation of several branches
of synthetic nucleic acid each bound to a probe, followed
by hybridization of multiple labeled oligonucleotides to
each of the branched amplification multimers. The method
requires the costly preparation of many probes and branched
DNA and is not generally applicable, especially for the
generation of short pieces of highly labeled DNA.
SUMMARY OF THE INVENTION
It is an object of the present invention to provide
4
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
methods of synthesizing single or double stranded
polynucleotides. It is another object of the present
invention to provide methods of synthesizing
polynucleotides by ligating a plurality of oligonucleotide
5'-phosphates to a primer hybridized to a template
polynucleotide. it is another object of the present
invention to provide methods of synthesizing
polynucleotides using a library of oligonucleotide 5'-
phosphates. It is another object of the present invention
to provide methods of synthesizing labeled polynucleotides
with a specified position and degree of label
incorporation. Another object of the present invention is
to provide methods for amplifying the amount of a nucleic
acid by primer-directed ligation. Another object of the
present invention is to provide methods for the detection
of genes and the analysis of gene expression. Yet another
object of the present invention is to provide methods for
the detection of genetic mutations. Still another object of
the present invention is to provide methods for the
analysis of the base sequence of a nucleic acid.
GENERAL DESCRIPTION
It has been discovered that a series of short
oligonucleotide-5'-phosphates can be simultaneously ligated
onto a template-bound primer in a contiguous manner to
produce the complementary strand of a template
polynucleotide or nucleic acid. The nucleic acid produced
can be either labeled or unlabeled by using either labeled
or unlabeled short oligomers. The oligomers in the set each
preferably contain the same number of bases. When a
5
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
sequence to be synthesized is known exactly, a set
containing the minimum number of oligomers can be used. The
oligomers are ligated in the correct order starting from
the primer, to produce the correct sequence. Primer-
independent ligation does not occur when using
oligonucleotides of length <_ 5 bases. When the sequence to
be synthesized is not known, a library of a large number of
the total possible pool of oligomers is used. The latter
situation occurs in sequence analysis and mutation
screening. Ligations are preferably conducted by means of a
ligase enzyme. Known chemical agents for ligating
nucleotides and oligonucleotides can be employed as well.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 schematically depicts the ligation of two
oligomers P1 and P2 onto a template-bound primer in the
presence of a competing nonligatable complementary oligomer
P3.
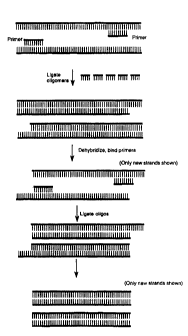
Figure 2 schematically depicts a method for amplifying
the amount of a nucleic acid using template-bound primer
directed ligation of multiple oligomers. Synthesis is shown
occurring in one direction in each strand, but can also be
accomplished bidirectionally as described below.
Figure 3 schematically depicts a method for detecting a
point mutation in a gene by the ligation of detectably
labeled mutation specific oligomers onto a template-bound
primer.
Figure 4 schematically depicts a method for detecting
two different genotypes of a mutation in a gene by the
ligation of different sets of detectably labeled mutation
6
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
specific or wild-type specific oligomers onto a template-
bound primer. The mutation specific oligomers bear a first
label while the wild-type specific oligomers bear a second
label.
Figure 5 depicts an example of branched DNA or
amplification multimers.
Figure 6 depicts an adaptation of branched DNA in which
at least the first branch is prepared by ligation of
labeled oligomers for providing branch points at regularly
spaced intervals.
Figure 7 schematically depicts a method for determining
the sequence of a nucleic acid by the ligation of unique
labeled oligomers onto a template-bound primer, cleaving
the labels and analysis of the mass of each unique label.
DETAILED DESCRIPTION OF THE INVENTION
Definitions
Oligomer, oligonucleotide - as used herein will refer
to a compound containing a phosphodiester internucleotide
linkage and a 5'-terminal monophosphate group. The
nucleotides can be the normally occurring ribonucleotides
A, C, G, and U or deoxyribonucleotides, dA, dC, dG and dT.
Primer or probe/primer - refers to an oligonucleotide
used to direct the site of ligation and is required to
initiate the ligation process. Primers are of a length
sufficient to hybridize stably to the template and
represent a unique sequence in the template. Primers will
usually be about 15-30 bases in length although longer
7
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
primers can be used. Labeled primers containing detectable
labels or labels which allow solid phase capture are within
the scope of the term as used herein. Primer also
contemplates contiguously stacked oligomers of at least six
bases as is known in the art (T. Kaczorowski and W.
Szybalski, Gene, 179, 189-193 (1996)).
Template, test polynucleotide, target are used
interchangeably and refer to the nucleic acid whose length
is to be replicated.
Sample - A fluid containing or suspected of containing
one or more analytes to be assayed. Typical samples which
are analyzed by the chemiluminescent reaction method are
biological samples including body fluids such as blood,
plasma, serum, urine, semen, saliva, cell lysates, tissue
extracts and the like. Other types of samples include food
samples and environmental samples such as soil or water.
Short oligonucleotide - As used herein, a
oligonucleotide 5'-phosphate of at lest two and up to about
10 base length. The bases can be ribonucleotides or
deoxyribonucleotides or analogs thereof. The length of a
short oligonucleotide useful in a given context can vary
within this range and may be less than the whole range. The
preferred length varies depending on the particular
application.
Specific binding pair - Two substances which exhibit a
mutual binding affinity. Examples include antigen-antibody,
hapten-antibody or antibody-antibody pairs, complementary
oligonucleotides or polynucleotides, avidin-biotin,
streptavidin-biotin, hormone-receptor, lectin-carbohydrate,
IgG-protein A, nucleic acid-nucleic acid binding protein
8
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
and nucleic acid-anti-nucleic acid antibody and metal
complex-ligand.
One object of the invention therefore is method for
synthesizing a strand of a nucleic acid complementary to at
least a portion of a target single stranded nucleic acid
template comprising:
a) providing a primer which is complementary to a
portion of the target single stranded nucleic acid
template;
b) hybridizing the primer with the template to form a
primer-template hybrid having a single stranded region and
a double stranded region;
c) contacting the primer-template hybrid with a
plurality of oligonucleotide 5'-monophosphates;
d) ligating to the primer-template hybrid in sequence
at least some of the plurality of oligonucleotide 5'-
monophosphates to extend the double stranded region and
thereby synthesize a nucleic acid strand which is
complementary to the portion of the template.
A preferred method of ligation uses a ligase such as a
DNA ligase. Representative ligases include T4 ligase, T7
ligase, Tth ligase, Taq ligase and E. coli DNA ligase. The
ligase can be a thermostable ligase, in which case thermal
cycling techniques as discussed below are possible. Thermal
cycling with a thermostable ligase is useful in methods of
amplifying nucleic acids in a manner analogous to the
polymerase chain reaction, but using oligomers and a ligase
in place of dNTPs and a polymerase. Methods of performing
enzymatic ligation reactions are generally described in
e.g., Sambrook, et al., Molecular Cloning: A Laboratory
9
*rB
CA 02304258 2008-07-09
Manual, 2nd Ed., Cold Spring Harbor Laboratory, New York,
1989.
Enzymatic ligation reactions are generally performed in
a buffer solution, optionally in the presence of additives
to promote hybridization. The buffer has a pH typically in
the range of 6 9, more usually 7 - 8.5 and preferably in
the range 7.5 - 8. Buffers capable of maintaining a pH in
this range are suitable. The reaction can be performed over
a range of temperatures in the range of 0 to about 50 C.
Optimal-temperatures will vary over the range depending on
the nature and size of oligonucleotide phosphates to be
ligated, the enzyme, presence and amount of additive and
can be optimized empirically with reference to the general
literature on ligases and by reference to the specific
examples below. The length of time for performing the
ligation can be as short as a few minutes up to several
hours, although it is desirable to conduct the reaction as
rapidly as possible. Single stranded DNA binding proteins
can be added to oligonucleotide ligation reactions to
improve their efficiency. Their effect is due to their
relaxation of any secondary structure that is in the
template strand thus allowing the complementary
oligonucleotides to bind and ligate. E. coli single
stranded binding protein (PromegaTM, Madison, WI or
AmershamT"/USB) and T4 Gene 32 protein (Boehringer Mannheim,
Indianapolis, IN) can be used. The use of volume excluding
agents such as polyethylene glycols (PEG) may be
advantageous in promoting ligations. Inclusion of up to 200
mM NaCl may also be useful for promoting ligations. The use
of other additives in enzymatic ligations is contemplated
CA 02304258 2000-03-17
WO 00/05412 PCTIUS99/14160
and is within the scope of the present methods. Additives
include phosphate transfer agents such as ATP, sulfhydryl
reagents, including DTT and 2-mercaptoethanol, and divalent
cations such as Mg+2 salts.
Ligation of oligomer 5'-phosphates also comprehends
nonenzymatic methods of ligation as well. Chemical reagents
which effect the formation of the phosphodiester
internucleotide bond are known (CNBr: K.D. James, A.D.
Ellington, Chemistry & Biology, 4,595,605, (1997); N-
cyanoimidazole: T. Li, K.C. Nicalaou, Nature, 369, 218-221
(1994); EDAC: D. Sievers, G. Von Kiedrowski,Nature, 369,
221-224 (1994)). Chemical ligation methods have not been
applied to methods of sequence analysis.
Incorporation of mismatched oligomers can occur as in
other techniques, especially when the sequence has a high
G-C content. The occurrence of mismatches is controllable
as is the case with other hybridization methods.
Temperature, salt concentration, and additives can all be
employed in art-recognized manners to control the
stringency of the hybridization process. Since the effect
of a mismatch on a small oligomer should be proportionately
greater than on a larger one, discrimination of improper
sequences may show improvement over other ligation
techniques.
Another embodiment uses a library of possible sequences
to achieve the ligation of a series of short oligomers of
length n bases to synthesize a complementary nucleic acid.
The library contains many more possible combinations of the
n bases (n-mers) than are required to form the product
nucleic acid. When n=5, for example, there are 45 or 1024
11
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
possible 5-mers which contain the four naturally occurring
bases A, C, G, T and U. The library can contain all 4n
possible oligomers or less than the full set, but should
contain at least a substantial proportion (> 50%, and
preferably > 75%, most preferably >90%) of the possible
oligomers.
Known methods of synthesizing polynucleotides, by
polymerase extensions with dntps or ligation of preformed
oligonucleotides, function by providing only a small number
of different reactants for incorporation into the product
molecule. Known ligation-based methods usually preselect
the one oligonucleotide with the correct sequence.
Polymerase extension methods supply the four individual
bases for incorporation. The present methods differ
fundamentally in providing a large number of potential
reactants into the reaction mixture. Moreover, a
significant number of the short oligonucleotides have a
sequence appropriate for hybridization to the target but,
if hybridized, would block or prematurely terminate the
ligation process.
As seen in Figure 1, oligomer P3 is complementary to a
portion of the target sequence, but, if hybridized, would
block ligation of P1 and P2 to the primer. Surprisingly,
the presence of complementary oligomers which can not be
ligated onto the primer does not interfere with or prevent
the successful ligation of the desired oligomers to the
template-bound primer.
The library also will contain a majority of
oligonucleotide sequences which are not complementary to
the target or only partially complementary. This excess of
12
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
oligonucleotides, in effect, competes with the correct
sequences for recognition and ligation. Nevertheless,
ligation of short oligonucleotides in the correct order
does occur effectively in spite of the statistical
unlikelihood. The ability to faithfully replicate a nucleic
acid by successive ligation of many short oligonucleotides
in one step is unexpected and greatly simplifies the
process compared to others known in the art.
The length of oligonucleotides to use in the present
methods is governed by the interplay of several competing
factors. Larger oligomers will hybridize more strongly
under a given set of conditions (salt concentration,
temperature) and can therefore hybridize at a higher
temperature. As the length of the oligonucleotide
increases, the number of discrete compounds required to
assemble the complete library of all possible n-mers
increases by a factor of 4 for each unit increase of n.
Lenath of Oligomer Total # of Seauences
1 4
2 16
3 64
4 256
5 1024
6 4096
7 16,384
8 65,536
9 262,144
10 1,048,576
13
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
Shorter oligomers require less compounds to construct the
entire library, but become more difficult, e.g. lower
temperature, to hybridize and ligate as their length
decreases. This, in turn, translates to greater stringency
at a given temperature. Still another factor is the ability
of the oligonucleotide to hybridize and initiate extension
at a site not associated with the primer. Primer-
independent hybridization has been demonstrated to occur,
under the right conditions, with oligonucleotides as small
as 6 bases. Ligation of 2 or more contiguous hexamers to
produce e.g., a dodecamer or octadecamer, then effectively
produces a new primer. If this happens, the ability to
control the starting point for polynucleotide synthesis is
compromised. On the other hand, the probability of finding
multiple occurrences of a given sequence in a nucleic acid
of hundreds of bases increases substantially as shorter
oligonucleotides are used. In applications involving
sequence determination, it is desirable to avoid or
minimize the occurrence of duplicate sequence elements. The
selection of the optimum length oligonucleotide to use is a
compromise among these conflicting effects. The optimum
length will be different in different end uses.
In practice it may not be necessary to use the full
library of oligonucleotides of length n. When the number of
oligonucleotides required to produce the given sequence is
small compared to the total number of oligonucleotides in
the library, partial libraries can be used and still
maintain a high probability that all of the required
oligonucleotides will be present. In some instances it may
be desirable to exclude certain sequence oligonucleotides
14
CA 02304258 2000-03-17
WO 00/05412 PCTNS99/14160
which hybridize too weakly or strongly.
It is not necessary in the present methods, except as
explicitly noted below, that each component of the set of
oligonucleotide 5'-phosphates used in a given method be of
the same number of bases. It can be advantageous in some
embodiments to use a combination of oligomers of two or
more different lengths, such as pentamers and hexamers, in
order to avoid the occurrence of duplicate oligomers.
In another embodiment,template-directed ligation of a
plurality of a set of short oligonucleotides of the same
length onto a primer can be performed in a manner which
controls the endpoint of the ligation by the use of
nonextendable oligomers. A nonextendable oligomer can
contain the same or a different number of bases as the
other oligomers in the set. The nonextendable oligomer
contains a 5'-phosphate so that it can be ligated, but
lacks the 3'-OH group. It could, for example, have a
dideoxy base at the 3'-end of the oligomer so that there is
no 3'-OH for ligation. Another type of nonextendable
oligomer contains a blocked 3'-OH group for example where
the hydroxyl group is blocked with a methyl group or a
phosphate group, to prevent subsequent ligation.
Modifications to the terminal base which prevent ligation
are another possible type of nonextendable oligomer. The
nonextendable oligomer can be labeled or unlabeled,
depending on the need. A preferred embodiment is to use
oligomers containing a dideoxy base at the 3'-terminus.
Another aspect of the present invention is a method for
synthesizing a nucleic acid by ligation of a plurality of
oligonucleotide 5'-phosphates onto a template-bound primer
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
in both the 5'-43' and 3'--->5' direction at the same time.
This process can be performed, for example, by providing a
5'-phosphate group on the primer. Ligation can occur
simultaneously from both termini of the primer as long as
the appropriate ligatable oligomers are provided. The point
of termination of synthesis in either or both directions
can be controlled by the use of nonextendable oligomers or
by excluding selected oligomers. A nonextendable oligomer
for terminating synthesis in the 3'-45' direction would not
have the 5'-phosphate group.
The template-directed ligation of a plurality of a set
of short oligonucleotides onto a primer can be used in a
method of amplifying the quantity of a target DNA.
Accordingly, another aspect of the invention comprises a
method of amplifying a target nucleic acid using a ligase,
two primers and a set of short oligonucleotides where the
probes are complementary to regions on opposing strands
spanning the region of the target to be amplified. At a
minimum, the oligomer set supplied for reaction must
contain those oligomers required to extend both primers on
their respective strands as far as the position
corresponding to the 5' end of the other primer. Additional
oligomers can be included, for example as would occur when
using the entire library of oligomers instead of
preselecting the set of oligomers. The process, shown
schematically in Figure 2, is distinct from the polymerase
chain reaction, PCR, but using a library of oligomers and a
ligase instead of the four deoxyribonucleotides and a
polymerase. Each cycle of annealing, ligase extension and
dehybridization results in a two-fold amplification of the
16
*rB
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
target sequence. Since heating is generally required for
separating the newly synthesized duplex nucleic acid,
additional ligase may need to be added in subsequent rounds
of ligation-extension. Alternatively, the process can be
performed with a thermostable ligase. Thermal cycling can
then be performed without replacing the ligase every cycle.
In accordance with the above description there is
provided a method for amplifying the amount of a portion of
a double stranded nucleic acid having a first strand and a
second strand comprising:
a) providing a first primer which is complementary to a
region of the first strand and a second primer which is
complementary to a region of the second strand wherein the
first and second regions define the portion of the double
stranded nucleic acid to be amplified;
b) providing a plurality of oligonucleotide 5'-
monophosphates;
c) separating the first and second strands of the
double stranded nucleic acid;
d) hybridizing the first and second primers with the
separated strands;
e) ligating onto the hybridized first and second
primers in sequence at least some of the plurality of
oligonucleotide 5'-monophosphates to extend the double
stranded region and thereby synthesize a nucleic acid
strand which is complementary to the portion of the
template; and
f) repeating steps c-e as many times as desired to
increase the amount of the amplified portion of double
stranded nucleic acid.
17
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
In a preferred embodiment of an amplification process,
the set of oligomers is preselected to contain only those
oligomers necessary to replicate the two strands, i.e.
those oligomers occurring on the two strands in the region
spanned by the two primers. In another preferred
embodiment, nonextendable oligomers are used for the
terminal positions of each strand. These two terminating
oligomers, by definition, have a base sequence
complementary to the first group of bases of the length of
the oligomer at the 5' end of each primer.
Amplification methods in accordance with the present
invention can be achieved by synthesis of each strand in
both the 5'--->3' and 3'--~5' direction at the same time. This
bidirectional amplification process can be performed, for
example, by providing a 5'-phosphate group on the primer.
Ligation can occur simultaneously from both termini of each
primer as long as the appropriate ligatable oligomers are
provided. The point of termination of synthesis in either
or both directions can be controlled by the use of
nonextendable oligomers or by excluding selected oligomers
as described above.
As is the case with other uses of the present oligomer
ligation method of synthesizing nucleic acid, either
labeled or unlabeled oligomers can be used. The set of
oligomers used can be the entire library, a substantial
portion of the library or a preselected subset if the
sequence to be amplified is known in advance.
In another aspect, the method of synthesizing specific
nucleic acid sequences by ligating oligomers onto target
bound primers can be used in diagnostic applications.
18
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
Specific sequences characteristic of the target of interest
can be detected using labeled oligomers in the method of
synthesizing the new strand. When the base sequence of the
target nucleic acid region is known, the corresponding
oligomers needed to complete this sequence are used, at
least some of which should carry a detectable label. Such
methods have use in many areas of nucleic acid diagnostics,
including detection of infectious agents such as C.
trachomatis and N. gonorrhoeae, P. carinii, M.
tuberculosis, detection of food borne pathogens such as
Salmonella and E. coli, methods of detecting the expression
of genes in high throughput screening assays, methods of
detecting genetic abnormalities, forensic testing of DNA
samples from suspected criminals, identity matching of
human remains and paternity testing. a specific sequence
and differentiating it from other related sequences.
In the area of genetic abnormality testing, one
application is a method for the detection of genetic
mutations. The mutations can be a point mutation (a and 9-
Thalassemia), a single base substitution (Sickle Cell
Anemia), a deletion (Cystic Fibrosis OF508, Tay-Sachs), an
insertion, a duplication, a transposition of bases or a
combination of the above. Labeled oligomers are selected
for ligation to a probe/primer such that the resulting
extended primer is a labeled mutation-specific
polynucleotide.
The methods of the present invention can be used to
provide a method for the differentiation of heterozygotes
from homozygotes for such a genetic condition. Since two
copies of a chromosome containing a DNA sequence of
19
*rB
CA 02304258 2000-03-17
WO 00/05412 PCTIUS99/14160
interest are present in a sample, the method of
synthesizing labeled complementary DNA provides a means for
distinguishing heterozygotes from either homozygote. A set
of oligomers carrying a first label which, when ligated,
produce a portion of a strand complementary to the normal
sequence is provided for ligation. Another set of oligomers
carrying a second label produces a portion of a strand
complementary to the mutant sequence upon ligation (Figure
4). Ligating the sets of oligomers to a probe hybridized to
target DNA in the sample creates a polynucleotide
complementary to the sample genotype. The three genotypes
are resolved by the determining which labels are present in
the newly synthesized DNA. Homozygous DNA will contain one
label or the other; heterozygous DNA will contain both.
When the sequence to be synthesized is not known, a
library of a large number of the total possible pool of
oligomers is used. The latter situation occurs in sequence
analysis and mutation screening. When used in conjunction
with the methods of ascertaining the base sequence of a
newly synthesized polynucleotide described in detail below,
numerous mutations of a particular gene can be analyzed and
identified simultaneously. The ability to test for multiple
mutations in a gene would enable screening for genetic
diseases such as cystic fibrosis for which more than 500
mutations have been identified.
In yet another aspect, there is provided a method of
synthesizing an immobilized single stranded nucleic acid
having a region whose base sequence is complementary to a
portion of the base sequence of a test nucleic acid
comprising:
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
a) providing a capture probe/primer which is
complementary to a portion of the test single stranded
nucleic acid;
b) contacting the capture probe/primer with the test
single stranded nucleic acid under hybridizing conditions
to capture the test single stranded nucleic acid and form a
captured probe-test nucleic acid hybrid having a single
stranded region and a double stranded region;
c) contacting the captured hybrid with a plurality of
oligonucleotide 5'-monophosphates;
d) ligating at least some of the plurality of
oligonucleotide 5'-monophosphates to the capture
probe/primer to extend the double stranded region;
e) removing the oligonucleotide 5'-monophosphates which
are not ligated; and
f) denaturing the captured probe-test nucleic acid
hybrid having an extended double stranded region to remove
the test nucleic acid from the solid support and produce
the immobilized single stranded nucleic acid.
The method controls the point of origin and is not
limited by the size of oligomers to be ligated. When the
sequence to be transcribed is exactly known, the oligomers
can be pre-selected to reduce cost and complexity.
Another aspect of the invention is a method of
synthesizing multiply labeled nucleic acid where the extent
of label incorporation is controlled and provides a high
density of labeling. When using an immobilized primer to
serve the dual purpose of capture probe and primer the
process comprises: a method of synthesizing an immobilized
multiply labeled single stranded nucleic acid comprising:
21
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
a) providing a capture probe/primer which is
complementary to a portion of the test single stranded
nucleic acid;
b) contacting the capture probe/primer with the test
single stranded nucleic acid under hybridizing conditions
to capture the test single stranded nucleic acid and form a
captured probe-test nucleic acid hybrid having a single
stranded region and a double stranded region;
c) contacting the captured hybrid with a plurality of
labeled oligonucleotide 5'-monophosphates;
d) ligating at least some of the plurality of labeled
oligonucleotide 5'-monophosphates to the capture
probe/primer to form a captured probe-test nucleic acid
hybrid having an extended double stranded region;
e) removing the labeled oligonucleotide 5'-
monophosphates which are not ligated; and
f) denaturing the captured probe-test nucleic acid
hybrid having an extended double stranded region to remove
the test nucleic acid from the solid support and produce an
immobilized labeled single stranded nucleic acid containing
a plurality of labels.
The primer can alternatively be a nonimmobilized primer
for the purposes of synthesizing a multiply labeled nucleic
acid. This embodiment comprises:
a) providing a primer which is complementary to a
portion of a test single stranded nucleic acid;
b) contacting the primer with the test single stranded
nucleic acid under hybridizing conditions to form a primer-
test nucleic acid hybrid having a single stranded region
and a double stranded region;
22
*rB
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
c) contacting the hybrid with a plurality of labeled
oligonucleotide 5'-monophosphates;
d) ligating at least some of the plurality of labeled
oligonucleotide 5'-monophosphates to the primer to form an
extended primer-test nucleic acid hybrid having an extended
double stranded region; and
e) removing the labeled oligonucleotide 5'-
monophosphates which are not ligated.
The method can further comprise the step of separating
the extended primer strand from the template nucleic acid
strand if desired. The label borne on each oligonucleotide
5'-monophosphate can be different or all can be the same
label. Alternatively, a limited number of different labels,
e.g. 2-5 labels, can be employed. The choice of labels used
will be governed by the final application.
The present methods, in contrast to other methods of
labeling nucleic acids described in the Background section,
can prepare virtually any length nucleic acid, but would
probably be most useful for products of at least about 50
bases. Shorter products would have less labels attached.
One of the main advantages is that the degree and position
of label attachment is precisely controlled. For example,
pentamers bearing one label each lead to product in which
every fifth base is labeled, providing a label density of
20 %. Still higher densities can be achieved with shorter
oligomers or with pentamers bearing two or more labels
each.
The ability to controllably label at these high
densities will be particularly advantageous in diagnostic
23
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
tests where detection sensitivity is paramount. Higher
label densities should translate to improved limits of
detection. Controlled labeling will contribute to improved
assay precision. The labels can be virtually detectable
species, including radioisotopes, chemiluminescent labels
and fluorescent labels, colorimetric labels detected on the
basis of absorption of light, specific binding molecules
including antigens and antibodies, binding proteins such as
streptavidin and haptens such as biotin and digoxigenin. In
addition, when the label is a small hapten, the detectable
label can be a species such as an enzyme which is bound to
the nucleic acid via an enzyme-anti-hapten conjugate. In
the latter regard, the use of pentamer ligation to produce
labeled nucleic acid provides still another advantage. The
bulky enzyme labels would be attached at every fifth base,
which places them at nearly 180 angles along the double
helix from the nearest neighboring label. Consideration of
the internucleotide separation and molecular diameters of
enzymes, reveals that even relatively large globular
proteins can be accommodated at this labeling density
without severe steric congestion.
Still higher label densities can be achieved by
adopting the branching label principle in conjunction with
the incorporation of regular labeled oligomers as depicted
in Figures 5 and 6. In practice, some or all of the
oligomers would constitute a "handle" such as a hapten or
short recognition sequence which is used to bind to a
branched amplification multimer.
Alternately, the arms of the branches could be prepared
by the ligation of labeled short oligomers, so that each of
24
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
the multiple arms carries detectable labels. Synthesis of
densely labeled nucleic acids by ligation of labeled
oligomers can be adapted to other types of branching DNA
technology such as the DNA dendrimers (Polyprobe,
Philadelphia).
The oligonucleotide 5'-phosphates used in the above-
disclosed methods of synthesis, amplification, preparing
labeled polynucleotides or immobilized polynucleotides are
preferably relatively short. In these applications, it is
not necessary to use a substantial fraction of the total
library of oligomers of a given length in order to be able
to synthesize the desired nucleic acid of known sequence.
The size of the oligomers can take any convenient value,
typically from 2 to about 20 bases. When high density
labeling is desired, it is preferred that the oligomers
contain less than about 10 bases and preferably from about
4 to about 8 bases.
In another aspect of the invention, methods are
provided for determining the sequence (sequencing) of an
unknown single stranded nucleic acid. The method can be
applied to RNA, ssDNA and denatured dsDNA sequences of
suitable lengths provided that at least a portion of the
sequence is known. The latter restriction is necessary in
order that a capture probe/primer may be designed.
The capture probe/primer is immobilized or capable of
being immobilized onto a solid support such as a bead,
tube, filter, membrane microtiter plate or chip. The
capture probe should be of sufficient base length to.
guarantee efficient hybridization and represent a unique
partial sequence on the test nucleic acid. These conditions
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
will generally be satisfied with a length of at least 10
bases and preferably at least 15 bases. The capture probe
can be immobilized onto the solid support in any art-
recognized way. A commonly used means is to provide a
biotin label for binding to a streptavidin-coated support.
Streptavidin-coated beads and microtiter plates are
commercially available.
The oligonucleotide 5'-phosphates used in sequence
analysis determinations performed in accordance with the
methods disclosed herein are preferably relatively short.
It is necessary to use a substantial fraction of all
possible oligomers of a given length in order to be able to
synthesize long stretches of nucleic acid of unknown
sequence. In order to keep the total library size
manageable, it is desirable to limit the size of the
oligomer to less than about 8 bases. It is more preferred
that the oligomers contain 5 or 6 bases. A further
requirement in embodiments involving sequence analysis is
that all oligonucleotide 5'-phosphates be of the same
number of bases.
In this aspect of the invention, a method for
determining the sequence of a portion of a single stranded
nucleic acid comprises the steps of:
a) providing a capture probe/primer which is
complementary to a portion of the single stranded nucleic
acid;
b) hybridizing the capture probe/primer with the single
stranded nucleic acid to capture the single stranded
nucleic acid and form a captured probe-nucleic acid hybrid
having a single stranded region and a double stranded
26
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
region;
c) contacting the captured hybrid with a plurality of
labeled oligonucleotide 5'-monophosphates of the same
number of bases each oligonucleotide 5'-monophosphate
having a unique label;
d) ligating at least some of the plurality of labeled
oligonucleotide 5'-monophosphates to the capture
probe/primer to form a captured probe-nucleic acid hybrid
having an extended double stranded region;
e) removing the labeled oligonucleotide 5'-
monophosphates which are not ligated;
f) denaturing the captured probe-nucleic acid hybrid
having an extended double stranded region to remove the
nucleic acid from the solid support and produce an
immobilized complementary single stranded nucleic acid
containing a plurality of labels and a region whose
sequence is complementary to a region of the nucleic acid;
g) detecting the plurality of labels;
h) relating the plurality of detected labels to the
identity of their corresponding oligonucleotide 5'-
monophosphates; and
i) determining the base sequence of the portion of the
nucleic acid from the identity of the plurality of
oligonucleotide 5'-monophosphates.
The process of converting the collection of partial
base sequences derived from the plurality of detected
labels involves performing a set of analyses to relate the
collection of partial base sequences to their correct
relative order or position in the total sequence to be
determined. A subset of partial base sequences is
27
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
identified in the initial ligation experiment. The order of
occurrence of each partial sequence in the full sequence is
then deduced from a set of experiments in which one
oligomer representing one partial sequence is excluded from
the set of all identified partial sequences. For a nucleic
acid sequence of N bases, the number of identified partial
sequences of n bases would be N/n, assuming no duplicates.
The number of such sets, each containing (N/n)-l oligomers
and lacking a different oligomer, equals the number of
partial sequences, N/n. The ligation reaction of these sets
to the hybridized primer produces a collection of extended
primers of different lengths, ranging from 0 to N/n
additional sets of n bases plus the length of the primer,
i.e. primer+n, +2n, +3n, etc. Since the identity (sequence)
of the excluded oligomer is known for each experiment, its
relative position in the total sequence is given by the
formula: 1 + number of additional n-base units which were
incorporated in that experiment.
There are several ways in which the reaction product of
each of the aforementioned N/n experiments can be
identified. Each method constitutes a different embodiment
of the invention. In one embodiment, unique cleavable
labels are provided on each oligomer. The plurality of
unique labels is cleaved from the extended probe/primer to
produce a set of label fragments, each having a unique
molecular mass. This method of sequence analysis is
depicted schematically in Figure 7. The set of label
fragments is analyzed by introduction into a mass
spectrometer. In a preferred mode, the mass analysis is
performed under conditions where the parent ion of each
28
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
label fragment can be detected. The experimental output of
each experiment consists of a set of molecular masses, each
set containing a different number of values. The collection
of sets of molecular masses is compared to determine the
relative position of each unique label and its associated
partial base sequence in the total sequence being
determined. This analysis is most conveniently done by a
computer algorithm.
The cleavable labels can be any molecular fragment
capable of being controllably released from the extended
probe/primer. Preferred labels are small organic molecules
of molecular mass less than about 50,000 amu. It is
desirable that the labels all be of one structural type,
having a common functional group so that all are cleavable
by a common means. One means for effecting cleavage is by
thermolysis of a thermally labile group. A preferred
thermally labile group for use in cleavable labels is a
1,2-dioxetane. It is well known that 1,2-dioxetanes undergo
a thermal fragmentation of the dioxetane ring to produce
two carbonyl fragments. Dioxetane-labeled oligomers can be
prepared which release a carbonyl compound when heated by
tethering a dioxetane group to a ribonucleotide or
deoxyribonucleotide.
O-O O O
A_~0 /k +
Base
Base
A library of oligomers would comprise the set of all
possible sequences of n bases, each covalently attached to
a unique dioxetane moiety. For convenience of synthesis,
29
CA 02304258 2000-03-17
WO 00/05412 PCTIUS99/14160
the linking functionality connecting the dioxetane ring
group to the oligomer should be common to all members of
the family of labeled oligomers. Substituents on the
dioxetane ring at the carbon which is cleaved will vary
among the members of the set of compounds.
Other thermally cleavable functional groups such as
noncyclic peroxides are known and can be used. The
temperature required for thermolysis must be low enough so
that oligonucleotide fragmentation does not occur.
The means of cleaving the cleavable label is not
limited to thermal cleavage. Any means of controllably
releasing the label from the extended probe/primer can be
employed. Other means include, without limitation,
enzymatic reactions, chemical reactions including
nucleophilic displacements such as fluoride-induced silyl
ether cleavage, basic or acidic hydrolytic fragmentations
such as ester hydrolysis or vinyl ether hydrolysis,
photochemical fragmentations, reductive cleavage such as
metal-induced reductive cleavage of a disulfide or
peroxide, oxidative cleavage of alkenes or diols.
An exemplary enzymatic reaction for label cleavage
utilizes enzymatically triggerable dioxetanes as labels.
Enzymatic deprotection of a protected phenolic substituent
triggers cleavage of the dioxetane ring into two carbonyl
compounds as depicted above. The reaction can be performed
at room temperature and the rate of cleavage controlled by
the amount and nature of the triggering enzyme and the
characteristics of the reaction solution, e.g. pH. Numerous
triggerable dioxetane structures are well known in the art
and have been the subject of numerous patents. The
CA 02304258 2000-03-17
WO 00/05412 PCTIUS99/14160
spiroadamantyl-stabilized dioxetanes disclosed in U.S.
5,707,559 are one example, others containing alkyl or
cycloalkyl substituents as disclosed in U.S. 5,578,253
would also be suitable. A linking substituent from the
aforementioned spiroadamantyl, alkyl or cycloalkyl groups
would be required to attach the dioxetane label to the
oligomer. Linkable dioxetanes are disclosed in U.S.
5,770,743.
O-OOR
f~go COOR
o Cleave O-X
to Trigger
t+~
OX
O
Chemical methods of cleaving triggerable dioxetanes are
also well known and would be similarly useful in the
methods of the invention. In the example above, X can be a
trialkylsilyl group and the triggering agent fluoride.
Other triggering agent/cleavable group pairs are described
in, for example, the aforementioned 5,707,559, 5,578,253
and 5,770,743 patents.
The foregoing method comprised the steps of:
1) performing an initial ligation experiment with
labeled oligomers,
2) releasing the labels, 3) detecting the labels, 4)
determining the set of partial base sequences associated
with the labels, 5) performing a set of ligation reactions
with a subset of oligomers identified in the preliminary
analysis to relate the collection of partial base sequences
to their correct relative order or position in the total
sequence. Alternatively, the preliminary ligation and
31
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
analysis can be omitted and the sequence can be determined
by performing sets of ligation reactions, excluding one
oligomer in each set. In this mode, the sets would need to
comprise the whole library of partial sequences less the
excluded one. Detection and/or quantitation of the labels
is then performed in the same manner as described above.
In the foregoing methods where arrays are prepared
lacking one oligomer from the set of oligomers, an
alternative approach would be to incorporate nonextendable
oligomers for the particular oligomer which would otherwise
be excluded. The nonextendable oligomer can be labeled or
unlabeled, depending on the need. Such nonextendable
oligomers could have a dideoxy base at the 3'-end of the
oligomer so that there is no 3'-OH for ligation. The 3'-OH
could be blocked, for example with a methyl group or a
phosphate group, to prevent subsequent ligation.
Modifications to the terminal base which prevent ligation
are another possibility.
In another aspect, the method of ligating oligomers
onto a template-bound primer for the purpose of sequence
analysis can be performed using a single label with
quantitative analysis. The process of determining the
sequence can be achieved by performing a set of ligation
reactions each reaction containing the full library of
oligomers of n bases less one as described generally above.
Each oligomer carries the same detectable label. Each
ligation reaction produces an extended primer of a length,
ranging from 0 to N/n additional sets of n bases plus the
length of the primer, i.e. primer+n, +2n, +3n, etc. The
quantity of detectable label in each reaction is
32
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
proportional to the number of ligated oligomers.
Collectively the set of extended primers produces all
values from 0 to N/n with the maximum value resulting in
all reactions in which the excluded oligomer is not present
in the template sequence. The value of 0 occurs when the
excluded oligomer represents the first five bases in the
template sequence. Since the identity (sequence) of the
excluded oligomer is known for each experiment, its
relative position in the total sequence is given by the
formula: 1 + number of additional n-base units which were
incorporated in that experiment. The template sequence is
then deduced from the order of occurrence of each partial
sequence (oligomer).
There are several ways in which the label can be
detected. Each method or type of label constitutes a
different embodiment of the invention. In one embodiment,
the label is a fluorescent molecule such as the fluorescers
FAM, JOE, ROX and TAMRA commonly used in automated dideoxy
sequencing. Numerous methods of labeling nucleotides and
oligonucleotides are known in the art and include direct
attachment of label (Haugland, Handbook of Fluorescent
Probes and Research Chemicals, (Molecular Probes, Eugene,
OR), 1992). Labeling can also be accomplished by indirect
means where, for example, where a universal linker such as
biotin is provided as the primary label and a fluorescer-
labeled binding partner for biotin provides the label.
In another embodiment, the label is a chemiluminescent
compound and the quantity of label is detected by the light
intensity produced by triggering the generation of
chemiluminescence from the label. Several types of
33
CA 02304258 2008-07-09
chzmiluminescent compounds are known and can be used as
labels. Representative examples include acridinium esters
and sulfonamides, luminol or isoluminol derivatives, and
dioxetanes (R. Handley, H. Akhavan-Tafti, A.P. Schaap, J.
Clin. Ligand Assay, 20(4) 302-312 (1997)). A preferred
chemiluminescent label is an acridan phosphate compound as
disclosed in Applicant's U.S. Patent No. 6,017,763.
The latter compounds are used advantageously because of
their stability, high chemiluminescence quantum efficiency,
ease of.conjugation and ability to be triggered under a
wide range of conditions, including in electrophoresis
gels. Bioluminescent and electrochemiluminescent compounds
are considered within the scope of detectable
chemiluminescent labels.
In another embodiment, the label is a chromogenic
compound and the quantity of label is detected by light
absorbance. Another label type is a radioisotope such as
32P and 35 S whose presence can be detected using
scintillation counting or x-ray imaging. The label can also
be an enzyme such as alkaline phosphatase, 9-galactosidase,
luciferase and horseradish peroxidase. The quantity of
enzyme is determined by measuring the action of the enzyme
on a fluorogenic, chromogenic or chemiluminogenic
substrate.
The quantitative detection techniques described above
rely on the ability to discriminate signal from 0 to N/n
with unit resolution where N is the total number of bases
to be sequenced and n is the number of bases in the
oligomer phosphates used. The resolution demand of the
detection process can be relaxed by performing m parallel
34
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
sets of reactions where only a predetermined fraction(1/m)
of the oligomers are labeled. If m sets of experiments are
then performed in which a different portion of the library
(N/m) is labeled in each set and the rest unlabeled,
ligation of the library and detection produces a set of
values in the range 0 to N/Sm in each set of experiments.
The sum of the information in the m sets combines to
produce the same information (total sequence). This reduces
the measurement precision requirement and provides m-fold
redundancy of results. As an example using pentameric
oligomers, pentamers arbitrarily designated 1-205 would be
labeled and the rest unlabeled in the first set. Numbers
206-410 would be labeled in a second set, numbers 411-615
in a third, 616-820 in a fourth and 821-1024 in a fifth
set. Each of the five sets of ligations will produce data
with numeric values from 0 to N/5m. The individual
reactions responsible for producing these values will
differ among the five sets.
In still a further embodiment, unlabeled oligomers can
be used in a method for sequencing by ligation when applied
to polynucleotides of up to a few hundred bases. Methods of
DNA sequence analysis using MALDI-TOF mass spectrometry
have been developed to accurately determine the molecular
mass of a series of polynucleotides differing in length by
one base generated by exonuclease digestion of a nucleic
acid. The technique is easily capable of discriminating
polynucleotides differing in length by 5 bases on the basis
of molecular mass. Current technology can accurately
identify polynucleotides up to about 80-100 bases with
adequate (single base) mass resolution. A series of ligated
*rB
CA 02304258 2008-07-09
polynucleotide products formed in accordance with the
methods of the present invention containing from 0 to about
100 ligated short oligonucleotides, such as pentamers,
would require no better instrumental resolution and would
extend the mass range which could be sequenced several
fold.
Another aspect of the invention comprises a method of
detecting a target nucleic acid by detecting a labeled
extended nucleic acid which is complementary to the target,
the method being a simpler alternative than traditional
Southern and northern blotting. Preparation of the labeled
extended complementary nucleic acid is performed by
ligation of a plurality of labeled short oligomers onto a
probe/primer which is hybridized to the target. Extension
is followed by denaturing electrophoretic separation and
detection of the labeled species. The presence of the
labeled extended primer is indicative of the presence of
the target since ligation only takes place when the primer
is hybridized to the target. It is preferred that the label
is detectable in the gel. Suitable labels include acridan
alkenes as described in U.S. Patent No. 6,017,769
issued January 1, 2000, which can be detected by
chemiluminescence, and fluorescers which are readily
detectable in gels. In this embodiment, no blotting is
performed. If the label is such that detection in the gel
is not feasible, then blotting onto membrane is performed
and then detection of the label is performed on the
membrane. In no case is hybridization on the membrane,
antibody binding, enzyme-conjugate binding, substrate
addition or other commonly used methods necessary.
36
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
In an alternate embodiment of this method, the labeled
extended complementary nucleic acid remains hybridized to
the target and after electrophoresis, the band detected at
the appropriate molecular weight in the manner described
above. This mode may be desirable when accurate molecular
weight information is needed. In this method it is more
convenient to provide substantially the full library of
possible oligomer 5'-phosphates of n bases when the target
is much longer than the probe/primer. In cases where the
target sequence is known and where its length makes it more
practical, it may be preferred to preselect the subset of
oligonucleotide 5'-phosphates.
Yet another embodiment comprises providing a suitable
fluorescent donor as a label on a probe/primer and a
suitable fluorescent acceptor as a label on the oligomers.
It is not necessary to label each oligomer. Ligation is
performed on hybridized primer to form an extended primer
bearing a fluorescent donor and one or more fluorescent
acceptor labels. Under suitable conditions, i.e. when the
donor and acceptor possess sufficient spectral overlap for
energy transfer to be feasible and the spatial separation
between donor and acceptors are within the Forster
distance, energy transfer between fluorescers can occur
within the extended primer. Irradiation of the extended
primer at a wavelength absorbed by the fluorescent donor on
the primer results in fluorescence from the acceptor on the
extended portion. This method can therefore serve as the
basis for a homogeneous assay for detecting a target
nucleic acid since the presence of target is required to
permit the ligation to occur and thereby bring the
37
CA 02304258 2000-03-17
WO 00/05412 PCTIUS99/14160
fluorophores within energy transfer distance.
Another method for detecting a target nucleic acid
based on the ligation of a plurality of labeled oligomers
comprises using a fluorescent intercalating dye as a label.
It is known that certain dyes become fluorescent when
intercalated within the double helix of double stranded
nucleic acids. An example is the widely used compound
ethidium bromide. Accordingly, a method for detecting a
target nucleic acid comprises:
a) providing plurality of oligonucleotide 5'-phosphates
wherein at least some contain a fluorescent intercalating
dye as a label;
b) providing an oligonucleotide primer which is
complementary to a portion of a target nucleic acid;
c) contacting the primer with the target nucleic acid
under hybridizing conditions to form a primer-target duplex
having a single stranded region and a double stranded
region;
c) contacting the duplex with the plurality of
oligonucleotide 5'-monophosphates;
d) ligating at least some of the plurality of
oligonucleotide 5'-monophosphates to the duplex to extend
the double stranded region;
e) detecting fluorescence from the intercalated bound
label.
As an optional step, agarose can be added to the
reaction to enhance fluorescence. In the absence of target,
ligation does not occur, so the detection of fluorescence
is evidence of the presence of the target and additionally
is evidence that the primer was sufficiently complementary
38
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
to the target to hybridize. The collection of oligomers can
be the full library of all possible sequences, or a subset
containing preselected members if the target sequence is
known.
The fraction of labeled oligomers to use can be
selected empirically with regard to the desired degree of
detection sensitivity by using a range of different label
densities. It may be desirable, depending on the size of
the oligonucleotide 5'-phosphates, to limit the fraction of
labeled oligomers to avoid self quenching of fluorescence.
Another aspect of the present invention comprises a
library of short oligonucleotide 5'-phosphates. It is
preferred that the oligonucleotides consist of 5 bases or
less, with pentamers being more preferred. The number of
pentamers required for the full library is 45 or 1024
individual compounds.
In practice it may not be necessary to include all
oligonucleotide 5-phosphates of length n in forming a
library. In applications where the number of
oligonucleotides required to produce the given sequence of
N nucleotides is small compared to the total number of
oligonucleotides in the library (N/n << 4n), partial
libraries will often suffice to maintain a high probability
of providing all of the required oligonucleotides. As an
illustration of this point, a polynucleotide of 500 bases
consists of 100 pentameric units. The full library of all
pentamers contains 1024 compounds, a more than 10-fold
excess.
It may be desirable to use a partial library which
excludes selected sequence oligonucleotides which hybridize
39
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
too weakly or strongly. In several methods described above
for sequence analysis of a test nucleic acid, the library
will be predetermined to consist of a selected number, x,
of oligomers determined in a proceeding step from the
identification of x+1 labels. In the sequencing method, a
collection of partial libraries of x bases each will be
used. Each partial library will lack a different one of the
x+l oligomers identified on the basis of the preceding
step.
The partial libraries can be preformed by preparing all
of the possible combinations of x oligomers beforehand.
Alternatively, the partial libraries can be assembled as
needed from the individual oligomers. The assembly of such
partial libraries can be accomplished by robotic
workstations with automatic fluid handling capabilities.
In general, the library of oligonucleotide 5'-
monophosphates will contain labeled oligonucleotide 5'-
monophosphates, in particular those bearing detectable
labels. In some uses, all of the members of the library
will bear a detectable label. In other applications, a
preselected fraction of the members will be labeled. An
example is the method of quantitative analysis disclosed
above where, for example, five libraries of all possible
oligomers are formed, each library having a different one-
fifth fraction of the members being labeled.
In another embodiment of a library, at least one of the
constituents of a library is a nonextendable oligomer. The
nonextendable oligomer can be labeled or unlabeled. Such
nonextendable oligomers could have a dideoxy base at the 3-
end of the oligomer so that there is no 3'-OH for ligation.
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
The 3'-OH could be blocked, for example with a methyl group
or a phosphate group, to prevent subsequent ligation.
Modifications to the terminal base which prevent ligation
are another possibility.
41
CA 02304258 2008-07-09
Synthesis of oligomers - Oligonucleotides are readily
synthesized using standard methods of synthesis well known
to those of skill in the art including, e.g:,
phosphoramidate chemistry. Phosphorylation of
oligonucleotides is performed using a polynucleotide kinase
and ATP or by chemical methods of phosphorylation as
described in (L.A. Slotin, Synthesis, 737-752 (1977); T.
Horn, M. Urdea, Tetrahedon Lett., 2,7, 4705-4708 (1986)). A
kit is commercially available for carrying out 5'-
phosphorylation (Phosphate-ONT", Clontech, Palo Alto, CA).
Methods for the automated synthesis of oligonucleotides
are well known in the art and in common commercial use. A
common method uses a solid support of immobilization and
automated reagent handling to add nucleotides sequentially.
All addition, blocking and deblocking steps are under
computer control. Such instruments are available from
several commercial suppliers such as Applied BiosystemsTm, CA
(Model 392 and 394). Automated instruments for transfer of
liquid reagents and samples can be performed under computer
control using laboratory robots such as are commercially
available (Perkin-ElmerTM, model 800 Catalyst, BeckmanTM
Instruments BiomekT"'). Newer techniques for the high speed
synthesis or synthesis of large numbers of oligonucleotides
utilize photolithographic techniques or ink jet technology
for the rapid and precise delivery of reagents and
reactants.
Still a further aspect of the invention comprises
annealing a primer oligonucleotide 5'-phosphate to a
single-stranded template and, in the manner disclosed
above, ligating a library of oligomers to extend the primer
42
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
from both ends to duplicate the template strand. The method
is useful in a method to render a single stranded template
double stranded in order to clone it. This would find
utility in methods for isolating related genes or gene
families.
In an exemplary method, a primer oligonucleotide is
hybridized to a template strand in the presence of a
library of all possible combinations of pentamers, a DNA
ligase, and an appropriate reaction buffer. Pentamers that
are complementary to the template strand, and in exact
register with the 5' and 3' ends of the primer
oligonucleotide, anneal and are sequentially ligated by the
action of the DNA ligase. The template strand thereby
becomes substantially copied or rendered double-stranded.
This procedure can be used to detect target templates in a
mixture of nucleic acid strands and to prepare double-
stranded nucleic acids for cloning using cloning vectors
and techniques known in the art.
In order to more fully describe various aspects of the
present invention, the following examples are presented
which do not limit the scope of the invention in any way.
43
CA 02304258 2008-07-09
EXAMPLES
Example 1. General Procedure
The template used in this experiment was a PCR
amplified product (200 bp) of exon 10 region of the cystic
fibrosis transmembrane regulator (CFTR) gene. The PCR-
amplified DNA of the CFTR gene was purified either by
running it through a column (Qiaquick PCR purification kit,
QiagenT"', Santa Clarita, CA) or by ethanol precipitation. The
DNA was resuspended in distilled water at a concentration
of approximately 0.5 g/ L. Pentamers bearing a 5'-
phosphate group and primers were obtained commercially
(Oligos Etc., Wilsonville, OR).
The primer and pentamers were designed to be
complimentary to either the sense or the antisense strand
of the template used. The length of the primer used in
these experiments ranged from 21 to 26 nucleotides. The
pentamers were designed in such a way that the first
pentamer anneals to the template immediately adjacent to
the 3' end of the primer. The subsequent primers line up
contiguously starting at the 3' end of the first pentamer.
Hybridization of the primer and pentamers to the template
followed by ligation by T4 DNA ligase results in back to
back ligation at the 5'-3' junctions. To enable the
detection of the ligated primer-pentamer products, biotin-
dUTP labeled pentamers (at the internal dTTP position) were
used.
Hybridization of the primer and pentamers to the
template and their ligation to each other was accomplished
in a 3-step process. First, the template-primer-pentamer
mix was heated to 94 C and kept for 5 min to allow the
44
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
denaturation of the double stranded template. The mix was
cooled to 60 C or 65 C,depending on the size and base
composition of the primer, to anneal the primer to the
teinplate for 2 min. Finally, the reaction tubes were cooled
to 16 C. After about 2 min at 16 C, ligation buffer (66
mM Tris HC1, pH 7.6, 6.6 mM MgC121 10mM DTT, 66 M ATP,
Amersham) and T4 DNA ligase, 1 U (Amersham, 1:10 dilution)
were added and ligated at 16 C for 2 hours. The ligation
reaction was stopped by adding 1/10th volume of loading dye
(0.01% xylene cyanol and 0.01% bromophenol blue, and 0.01 M
EDTA in deionized formamide).
The ligation reactions were electrophoresed on a
denaturing polyacrylamide gel along with biotin-labeled
oligonucleotide size markers. The DNA was capillary
transferred to a nylon membrane, bound with anti-biotin
antibody-HRP conjugate, and detected by reacting with
Lumigen PS-3 (a chemiluminescent HRP substrate) and
exposing to an x-ray film. The size of the ligated product
varies depending on the number of pentamers ligated to the
primer.
Example 2. Determining Optimal Concentrations of Template,
Primer and Pentamers.
Template: A 200 bp PCR product of CFTR exon 10 (See the
attachment for the template DNA sequence) was obtained by
PCR amplification using a set of sense (5'
ACTTCACTTCTAATGATGATTATG 3') (Seq. ID#1) and an antisense
(5' CTCTTCTAGTTGGCATGCTTTGAT 3') (Seq. ID#2) primers.
A 26 base oligonucleotide complementary to the sense
strand of the template DNA was designed as a primer (5'
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
AGTGGAAGAATTTCATTCTGTTCTCA 3') (Seq. ID#3).
Pentamers: Six 5-base long oligonucleotide 5'-
phosphates complementary to the sense strand immediately
adjacent the 3' end of the primer were prepared. The 5' end
of the first pentamer aligns immediately next to the 3' end
of the primer, the 5' end of the second pentamer aligns
immediately next the 3' end of the first pentamer and so
on. To facilitate ligation, the 5' end of each pentamer was
phosphorylated. To enable the detection of the ligation
products, pentamers 1 and 3 were labeled with biotin-dUTP
at the central dTTP position and the last pentamer was
labeled with biotin at the 3' end. The pentamers were as
follows:
Pentamer 1: 5' P04-GTTTT 3'
Pentamer 2: 5' P04-CCU*GG 3' U* = U-Biotin
Pentamer 3: 5' P04-ATTAT 3'
Pentamer 4: 5' P04-GCCU*G 3'
Pentamer 5: 5' P04-GCACC 3'
Pentamer 6: 5' P04-ATTAA 3'-Biotin.
Ligations were performed using T4 DNA ligase and
ligation buffer (Amersham), according to the ligation
conditions described in Example 1. The ligations were
performed in a volume of 20 L. The amount of template was
kept constant at about 1 g per reaction. The amount of
primer was varied from 100 ng to 1 pg between reactions.
The amount of each pentamer was varied from 2 ng to 0.2 pg
in each reaction. The reaction with 1 g of template, 100
ng of primer, and 2 ng of each pentamer contained
46
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
approximately equimolar concentrations of the template,
primer and pentamers. The primer and pentamers were varied
systematically to determine the lowest amount of detectable
ligation product.
The ligation reactions were electrophoresed, capillary
transferred to a nylon membrane, bound with anti-biotin
antibody-HRP conjugate, and detected with Lumigen PS-3 as
described in Example 1. A full-length primer-pentamer
ligation product of expected size (56 bp) was detected in
the ligation reaction containing 100 ng of primer and 20 ng
of each pentamer (0.6 gM). Lower concentrations of the
primer and pentamers in the ligation reaction yielded very
low amount or no detectable ligation product under these
conditions.
Example 3. Ligations with varying number of pentamers.
To show that the pentamers are sequentially ligated to
the primer starting from the first pentamer (immediately
downstream from the primer), ligations were performed using
the template, primer and pentamers of Example 2 by
incrementing the number of pentamers in each reaction. All
the reactions contained equimolar concentrations (0.6 M)
of the template, primer and each pentamer. The ligations
were performed as described above and the products detected
by binding with anti-biotin-HRP antibody and reacting with
Lumigen PS-3 substrate.
As expected, the size of the ligation product increased
incrementally by five bases with the addition of each
pentamer starting from the first pentamer and so on. There
was no ligation product in the absence of the first
47
*rB
-------- --- ---
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
pentamer and with the rest of the pentamers in the reaction
demonstrating the requirement of the primer and specificity
of the pentamers for the ligation to occur. In one ligation
reaction containing the first four pentamers, there were
two bands of the ligated product one of which was the
expected size and the other was the size expected when all
five pentamers are present in the reaction. Comparing the
sequences of the pentamers revealed a single base
difference between the third and fifth pentamers. The third
pentamer appears to hybridize at the fifth pentamer
position when the fourth pentamer is present in the
reaction.
Reaction Pentamers Used Product Lenath
1 2, 3, 4, 5, 6 26 (primer)
2 1, 2 36
3 1, 2, 3 41
4 1, 2, 3, 4 46
5 1, 2, 3, 4, 5 51, 56
6 1, 2, 3, 4, 5, 6 56
In reaction 1, the product, which consisted of the primer
alone, was detected by virtue of a label on the primer.
Example 4. Competition from "Out of Register" Pentamer Set.
This example demonstrates that the ligation of a set of
pentamers to a primer in a contiguous chain starting from
the 3' end of the primer is not affected by the presence of
a second set of pentamers which are also complementary to
the template and also align contiguously, but begin at a
position `one-base-out' from the 3' end of the primer. Both
the correct pentamer sets (1-8) and one-base-out pentamer
48
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
sets (la-5a) were included in the reaction along with the
template and primer during the denaturing, annealing at 60
C and ligation steps. The ligation reactions contained
equimolar (0.6 M) concen-trations of template, primer and
each of the pentamers.
Primer (5' ATTAAGCACAGTGGAAGAATTTCAT 3') (Seq. ID#4).
Pentamer 1: 5' P04-TCU*GT 3' Pentamer la: 5' P04-CTGTT 3'
Pentamer 2: 5' P04-TCTCA 3' Pentamer 2a: 5' P04-CTCAG 3'
Pentamer 3: 5' P04-GTTTU* 3' Pentamer 3a: 5' P04-TTTTC 3'
Pentamer 4: 5' P04-CCU*GG 3' Pentamer 4a: 5' P04-CTGGA 3'
Pentamer 5: 5' P04-ATTAT 3' Pentamer 5a: 5' P04-TTATG 3'
Pentamer 6: 5' P04-GCCU*G 3' Pentamer 6a: 5' P04-CCTGG 3'
Pentamer 7: 5' P04-GCACC 3'
Pentamer 8: 5' P04-ATTAA 3'-biotin
The target region of the template comprises the sequence:
3' TAA TTC GTG TCA CCT TCT TAA AGT AAG ACA AGA GTC AAA AGG
ACC TAA TAC GGA CCG TGG TAA TT 5' (Seq. ID#5).
Reaction Pentamers Used Product Lenath
1 1, la-6a 31 (primer + 5)
2 1, 2, la-6a 36
3 1-3, la-6a 41
4 1-4, la-6a 46
5 1-5, la-6a 51
6 1-6, la-6a 56
7 1-7, la-6a 61
8 1-8, la-6a 66
9 2-8, la 26 (primer)
10 1-8, la-6a(no primer) none
49
CA 02304258 2000-03-17
WO 00/05412 PCTIUS99/14160
Even in the presence of a set of six competing pentamers,
the ligation products formed were the result of ligation of
the "correct" pentamers being ligated to the primer.
Reaction 10 confirmed that the presence of primer is
required for ligation to occur. The one-base-out pentamers
did not appear to interfere with the ligation of the
correct pentamers.
Example 5. Competition from Labeled "Out of Register"
Pentamer.
The experiment as Example 4 but pentamer la was
biotinylated. This afforded the opportunity to directly
observe the formation of any ligation products from the set
of pentamers la-6a. No ligation products were observed from
the set of one-base-out pentamers.
Example 6. Ligation Experiments Using JH Downstream
Template.
Primer-directed pentamer ligation products were also
obtained using as the template a 700 bp DNA downstream of
immunoglobulin heavy chain joining region (JH) cloned into
a plasmid vector. The JH downstream region was amplified by
PCR, cloned into a plasmid vector which was then digested
with Eco RI to obtain a sufficient amount of the template
DNA. The restriction digest was separated on an agarose
gel, and the DNA band of interest extracted using a gel
extraction kit (Qiagen). The DNA was resuspended in
distilled water at a concentration of approximately 0.5
kg; l .
The primer and pentamers used with this template are
CA 02304258 2000-03-17
WO 00/05412 PCTIUS99/14160
shown below:
21mer Primer: 5' GAAACCAGCTTCAAGGCACTG 3' (Seq. ID#6)
Pentamer 1: 5' Phosphate AGGU*C
Pentamer 2: 5' Phosphate CU*GGA 3'
Pentamer 3: 5' Phosphate GCCU*C 3'
Pentamer 4: 5' Phosphate CCU*AA 3'
Pentamer 5: 5' Phosphate GCCCC 3'-Biotin
Ligations were performed with 500 ng of template, 100
ng of primer, and 20 ng of each pentamer in each 20 L
ligation reaction. The number of pentamers was
incrementally increased in each successive ligation
reaction to show that the size of the ligation product grew
in 5 base increments with each addition of a pentamer.
After performing the ligation reactions according to
the general method of Example 1, there was a 5-base
incremental increase in the size of the ligation product
with each addition of successive pentamers. There were two
bands in the ligation reaction containing the first four
pentamers, the upper band being more intense than the lower
band. The size of the upper band was the same as when all
five pentamers were used for the ligation. This is probably
because there is sequence similarity between the third and
fifth pentamers, so the third pentamer was ligated also at
the fifth pentamer position.
Example 7. Ligation at Various Temperatures.
Ligations using the template, primer and the first four
pentamers of Example 6 were performed at 30 C, 37 C, 40 C
and 45 C to examine the effect of ligation temperature on
mismatch discrimination. The template, primer and pentamer
51
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
concentrations were the same as in the previous experiment.
Parallel reactions were perfoz-med and the relative amount
of the four pentamer and five pentamer extension products
assessed. At 30 C ligation temperature, the correct size
and the non-specific ligation products were of the same
intensity. More of the correct size ligation product was
detected at ligation temperatures of 37 C and 40 C. At 45
C, the amount of correct size ligation product detected
was diminished.
Example 8. Ligation Using Octamers.
Ligations of octamers with 5' phosphate and an internal
biotin label to a 23mer primer specific to pUC18 plasmid
template were performed using T4 DNA ligase (Amersham), Taq
DNA ligase (New England Biolabs), and Ampligase-
(Epicenter Technologies, Madison, WI), an NAD-dependent
thermostable ligase, each using their respective ligation
buffers. In these experiments, 100 to 800 ng of EcoRI
linearized plasmid template was used. The concentrations of
the primer and the octamers were 12 pmols per reaction.
In experiments with T4 DNA ligase and Taq DNA ligase,
first the template, primer and pentamer mix was heated at
94 C for 5 min, cooled to 60 C for 2 min for annealing
the primer to the template, and further lowered to 16 C
(for T4 DNA ligase) or 45 C (for Taq DNA ligase) for 2
min. Then the respective ligation buffers and enzymes (1 U
/reaction) were added and ligated for 2 h.
For ligation with Ampligase, the ligation mix
containing the template, primer and octamers (same as
above) was heated at 94 C for 5 min, then cycling
52
CA 02304258 2008-07-09
temperatures of 94 C for 5 min and 45 C for 5 min (35
cycles) were used for ligation. In another experiment using
the Ampligase, a short PCR product of pUC18 (100 bp) was
used as template and the conditions of ligation were 94 C
for 1 min, 55 C for 1 min, and 15 C for 5 min for a total
of 20 cycles.
The ligation products were electrophoresed on an 8%
PAGE-Urea gel, semi-dry blotted (HoeferT" TE90 blotter) onto
a positively charged nylon membrane, UV cross-linked,
incubated with anti-biotin antibody conjugated to HRP and
detected with Lumigen PS-3 substrate.
With T4 DNA ligase and Ampligase using the linearized
pUC18 plasmid, a ligation product of the expected size was
detected. However, under the conditions used, Ampligase
appeared to be much less efficient than T4 DNA ligase as
was evident from the amounts of ligation product detected.
Taq DNA ligase reaction did not yield a detectable ligation
product. Using the short pCR product of pUC18, Ampligase
ligation produced more ligation product than using the
full-length pUC18.
Example 9. Bidirectional Ligation
In this experiment, a 5'-phosphorylated primer was
ligated at both ends to a set of pentamers with 5'-
phosphate using T4 DNA ligase. A CFTR PCR product template,
primer and pentamer mix was heated at 94 C for 5 min, the
primer annealed at 65 C for 5 min, and the pentamers
ligated at 16 C for 2 h. The ligation buffer and the
enzyme were added after the reaction mix was at 16 C for 2
min. The reaction mixture was electrophoresed, blotted and
53
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
detected as in Example 8. The pentamers were ligated to the
primer in both the 5' and 3' directions from the primer.
Product bands of the size observed could only be produced
by ligation of each of the pentamers in the set. The
signals of ligation products resulting from the ligation of
pentamers at the 5' end of the primer were less intense
than those ligated at the 3' end of the primer. This could
be either due to differences in the ligation efficiency in
two directions, or in the labelling with biotin of the two
pentamer sets.
Example 10. Isothermal Ligation at Various Temperatures.
Ligation of a set of 5-phosphorylated octamers onto a
template-bound primer was performed at a constant
temperature without prior heating and reannealing of the
primer to the template. All the components of the ligation
reaction - the template, primer, octamers, ligation buffer
and ligase were added at room temperature and ligated at
either 25 C, 30 C, 37 C or 45 C for 2 h. Linearized
pUC18 (EcoRI) at concentrations ranging from 10 ng to 800
ng was used as template for ligations using T4 DNA ligase.
The primer and octamer concentrations used were 12 pmols in
each reaction.
Isothermal ligations (37 C, 45 C, 55 C, 65 C) were
also performed with Taq DNA ligase, Ampligase and Pfu DNA
ligase using their respective ligation buffers. With T4 DNA
ligase, ligation product was detectable at all the
isothermal conditions used. At 37 C, as low as 10 ng of
the template produced a detectable ligation product. Larger
amounts of template yielded ligation products with intense
54
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
signal.
None of the other ligases produced any detectable
ligation product under isothermal conditions.
The foregoing description and examples are illustrative
only and not to be considered as restrictive. It is
recognized that modifications of the specific compounds and
methods not specifically disclosed can be made without
departing from the spirit and scope of the present
invention. The scope of the invention is limited only by
the appended claims.
CA 02304258 2000-03-17
WO 00/05412 SEQlJENCE LISTING PCT/US99/14160
<110> Akhavan-Tafti, Hashem
<120> METHODS OF SYNTHESIZING POLYNUCLEOTIDES BY LIGATION OF
MULTIPLE OLIGOMERS
<130> LUM-4.1-53
<140> 09/121,887
<141> 1998-07-24
<160> 7
<170> Patentln Ver. 2.0
<210> 1
<211> 24
<212> DNA
<213> primer
<400> 1
acttcacttc taatgatgat tatg 24
<210> 2
<211> 24
<212> DNA
<213> primer
<400> 2
ctcttctagt tggcatgctt tgat 24
<210> 3
<211> 26
<212> DNA
<213> primer
<400> 3
agtgqaagaa tttcattctg ttctca 26
<210> 4
<211> 25
<212> DNA
<213> primer
<400> 4
attaagcaca gtggaagaat ttcat 25
<210> 5
<211> 65
<212> DNA
<213> DNA template
<400> 5
ttaatggtgc caggcataat ccaggaaaac tgagaacaga atgaaattct tccactgtgc 60
ttaat 65
<210> 6
<211> 21
<212> DNA
<213> primer
1/2
CA 02304258 2000-03-17
WO 00/05412 PCT/US99/14160
<400> 6
gaaaccagct tcaaggcact g 21
<210> 7
<211> 21
<212> DNA
<213> primer
<400> 7
attcagtgcc atgggacata g 21
2 / 2