Note: Descriptions are shown in the official language in which they were submitted.
CA 02308151 2000-04-17
WO 99123073 PCT/EP98106880
-1
BIPHENYL DERIVATIVES AS PHfARMACEUTICALS
The present invention relates to novel biphenyl derivatives, their
preparation, their use as -
pharmaceuticals and pharmaceutical compositions containing them.
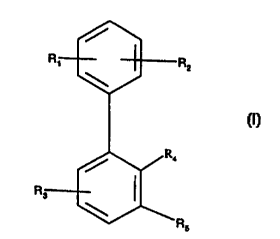
More particularly the invention provides a compound of formula I
s
wherein
R~ and R~, independently, are hydrogen, (C~.a)alkyl, (Cm)alkox~-,
(C~.4)alkylthio, halogen,
trifluoromethyl, trifluoromethoxy~, cyano or (C2_s)alkanoyl,
R3 is hydrogen, hydroxy, (Cm)alkyl, (Cm)alkoxy, (C~s)cycloalkylo~-, halogen,
cyano, (C2.-s)alkanoyl, carbamoyl, (C»)alkylsulfonyloxy or
trifluoromethylsulfonyloxy,
R,, is hydrogen, hydroxy or (C»)alkoxy, and
R5 is a group of formula (a), (b), (c) or (d)
N_R6
N-Ra N-R6
(a) (b) (c)
CA 02308151 2000-04-17
WO 99/23073 PCT/EP98/06880
-2-
i~
-X- Nw
(d)
wherein
R6 is (Cm)alkyl,
X is a straight or branched alkylene chain with 1 to 4 carbon atoms, and
R7 and R8, independently, are hydrogen, (Cl.a)alkyl, hydroxy(C2.a)alkyl or
phenyl(G.a)alkyl, or form together with the nitrogen atom to which they are
attached a pyrrolidinyl, piperidino, piperazinyl or morpholino group, or a
group of formula (e)
R~3
(e)
Rya
."
Rya
wherein Z is O, CH2 or CH2-CH2 and R9, Rio, Rn, Rl~, R3 and RI4,
independently,are H, halogen, (Cm)alkyl or (Cm)alkoxy,
in free base or acid addition salt form.
On account of the asymmetrical carbon atoms which rnay be present in the
compounds of
formula I and their salts, the compounds may exist in optically active form or
in form of
mixtures of optical isomers, e.g. in form of racemic mixtures. All optical
isomers and their
mixtures including the racemic mixtures are part of the present invention.
Halogen is fluorine, chlorine, bromine or iodine, preferably fluorine or
chlorine.
Any alkyl, alkoxy and alkylthio radicals preferably are straight chain
radicals. They
preferably have 1 to 3 carbon atoms, -more preferably they are methyl, methoxy
and
methylthio groups.
CA 02308151 2000-04-17
WO 99IZ3073 PC'flEP98/06880
-3
The following significances and their combinat'~ons are preferred:
R~ and R2, independently, are hydrogen, (Ca.a)alkyl, (Cm)alkoxy, halogen or
trifluoromethyl,
R3 is hydrogen or in para to the phenyl substituent is hydroxy, (C»)alkoxy,
(C~)
cycloalkyloxy, cyano or carbamoyl,
RS is a group of formula (a) or (d).
When R; is a group of formula (a), R6 is preferably methyl or propyl.
When Rs is a group of formula (d), R7 and Rs are preferably hydrogen,
(Cm)alkyl or,
together with the nitrogen atom to which they are attached, a group of formula
(e).
When R7 and Rs, together with the nitrogen atom to which they are attached,
form a
group of formula (e), Z is preferably O and R9 to Rya, independently, are
preferably
hydrogen or methyl.
In one group of compounds of formula I, Rl, R2, R3 and Ra are as defined above
and RS is
a group of formula (a), (b) or (c).
In another group of compounds of formula I, R~, R2, R3 and Ra are as defined
above and
RS is a group of formula (d).
In a further aspect, the invention provides a process for the production of
the compounds
of formula I and their salts, whereby a compound of formula II
_ Y
R~
I I
CA 02308151 2000-04-17
WO 99/23073 PCT/EP98/06880
-4
wherein R3, R4, and Rs are as defined above and Y is halogen or
trifluoromethyl sulfonate,
is reacted with a compound of formula III
Ill
R,
wherein RI and R2 are as defined above, and the resulting compound is
recovered in free
base form or in acid addition salt form.
The reaction may~ be effected in known manner, preferably by transition metal-
catalysed
aryl-an-1 coupling, e.g. as described in Example 1. Hal is preferably- bromine
or iodine,
particularly' iodine.
Alternatively the compounds may be obtained by other well established metal-
catalysed
aryl-aryl coupling procedures, using for example aryl-stannyl, -zinc, -halide
or Grignard
precursors.
For the preparation of compounds of formula I wherein Rs is a group of formula
(c), the
nitrogen in the group of formula (c) may be protected e.g. by an
alkoxycarbonyl group,
which can be removed after the reaction with the compound of formula III
according to
well=known procedures. See for example Example 11.
For the preparation of compounds of formula I wherein Rs is a group of formula
(b), a
compound having the formula I but wherein Rs is a 3-pyridyl group, may first
be prepared
as described above for the compounds of formula I, and the pyridyl group
subsequently
converted into the desired tetrahydropyridyl over a corresponding pyridinium
salt,
according to well-known procedures, for example over the pyridinium iodide as
described
in Example 1.
CA 02308151 2000-04-17
WO 99123073 PCT/EP98/OG880
-S
For the preparation of compounds of formula I wherein RS is a group of formula
(a), the
corresponding compounds wherein RS is of- formula (b) may be prepared first,
and
subsequently hydrogenated according to well-known procedures, e.g. as
described in
Example S.
Compounds of formula I in optically pure form can be obtained from the
corresponding
racernates according to well-known procedures, e.g. as described in Examples 9
and 10.
Alternatively, optically pure starting materials can be used as described in
Examples 28 and
29.
Acid addition salts may- be produced in known manner from the free base forms
and vice-
versa. Suitable pharmaceutically acceptable acid addition salts for use in
accordance with
the present invention include for example the hydrochloride, the hydrogen
maleate, the
hydrogen fumarate and the hydrogen malonate.
The starting materials of formula II are known or may be produced by
halogenating
compounds of formula IV
R,
R'
IV
R~
wherein R3, R4 and RS are as defined above, in accordance with known
procedures.
The starting materials of formulae III and N are known or may be produced in
analogous
manner to known procedures, e.g. as described in the Examples.
The compounds of formula I and their pharmaceutically acceptable acid addition
salts,
hereinafter referred-to collectively as "agents of the invention", exhibit
pharmacological
activity and are, therefore, useful as pharmaceuticals.
The agents of the invention provide long-lasting protection against maximal
electroshock-
induced convulsions in mice at doses of about 1 to 100 mg/kg p.o. and about
0.32 to 32
CA 02308151 2000-04-17
WO 99/23073 PCTIEP98/06880
-6
mg/kg i.p. [cf. E.A. Swinyard, J. Am.Pharrn. Assoc. Scient. Ed. 38, 201 (1949)
and
J.Pharmacol. Exptl. Therap. ltd, 319 (1952)}.=
The agents of the invention are therefore useful in the treatment of epilepsy
and other
convulsive states such as high pressure neurological syndrome.
Furthermore, the agents of the invention reduce ischaemia-induced neuronal
damage and
ensuing symptoms in the middle cerebral artery (MCA) occlusion model in rats
at a dosage
of 1-SO mg/kg i.p, i.v. and p.o. [cf. A. Tamura et al., J. Cereb. Blood Flow
Metabol. 1, S3-
60 (1981), A. Sauter, M. Rudin, Stroke I7, 1228-1234 (1986)].
The agents of the invention are therefore useful in the treatment of any
clinical condition
involving a component of cerebral anoxia, hypoxia and/or ischaernia, e.g.
ischemic damage
to grey and white matter, stroke, reperfusion injury, subarachnoid
haemorrhage, brain and
spinal cord injury/trauma, high intracranial pressure, mufti-infarct dementia
or vascular
dementia, and any surgical procedure potentially associated with cerebral
anoxia, hypoxia
andlor ischemia (e.g. cardiac bypass, operations on extracerebral vessels).
The agents of the invention display binding to the veratridine-sensitive
sodium channel
with IC;os of from about 0.1 to about 100 p.M. For the binding procedure see
for example
J.B Brown, Journal of Neuroscience ~, 2064-2070 (1986). They block veratridine-
induced
glutamate release in rat hippocampal slice preparations at concentrations of
about 0.1-1
mM. The experiment is performed according to a modification of M. J. Leach et
al. in
Epilepsia 27, 490-497 ( 1986) and Stroke 24, 1063-1067 ( 1993), using
exogenous
glutamate.
The agents of the. invention are accordingly indicated for use in the
treatment of any
pathology, disorder or clinical condition involving glutamate release in their
etiology,
including psychiatric disorders (such as schizophrenia, depression, anxiety,
panic attacks,
attention deficit and cognitive disorders, social withdrawal), hormonal
conditions (excess
GH [e.g. in the treatment of diabetes mellitus, angiopathy and acromegaly] or
LH [prostate
CA 02308151 2000-04-17
WO 99123073 PCT/EP98/06880
hypertrophy, menopausal syndrome] secretion, corticosterone secretion in
stress), metabolic
induced brain damage (hypoglycaemia,- =non-ketotic hyperglycinaemia [glycine
encephalopathy], sulphite oxidase deficiency, hepatic encephaiopathy
associated with liver
failure), ernesis, spasticity, tinnitus, pain (e.g. cancer pain, arthritis)
and drug (ethanol,
opiates [including synthetics with opiate-like effects, e.g. pethidine,
methadone etc.],
cocaine, amphetamine, barbiturates and other sedatives, benzodiazepines) abuse
and
withdrawal.
Moreover the agents of the invention are indicated for use in the treatment of
any
pathology involving neuronal damage, for example neurodegenerative disorders
such as
Alzheimer's, Huntington's or Parkinson's diseases, virus (including HN)-
induced
neurodegeneration, Amyotrophic lateral sclerosis (ALS), supra-nuclear palsy,
olivoponto-
cerebellar atrophy {OPCA), and the actions of environmental, exogenous
neurotoxins.
For the above-mentioned indications, the appropriate dosage will of course
vary- depending
upon, for example, the compound employed, the host, the mode of administration
and the
nature and severity of the condition being treated. However, in general,
satisfactory results
in animals are indicated to be obtained at a daily dosage of from about 0.1 to
about 100,
preferable from about 0.5 to about 100 mg/kg animal body weight. In larger
mammals,
for example humans, an indicated daily dosage is in the range from about 1 to
about 500,
preferably from about 1 to about 300 mg of an agent of the invention,
conveniently
administered, for example, in divided doses up to four times a day or in
sustained release
form.
The agent of the invention may be administered by any conventional route, in
particular
enterally, preferably orally, for example in the form of tablets or capsules,
or parenterally,
for example in the form of injectable solutions or suspensions.
In accordance with the foregoing, the present invention also provides an agent
of the
invention, for use as a pharmaceutical, e.g. for the treatment of epilepsy,
stroke and brain
or spinal trauma.
CA 02308151 2000-04-17
WO 99/23073 PCT/EP98106880
-g-
The present invention furthermore provides a pharmaceutical composition
comprising an
agent of the invention in association with at least one pharmaceutical carrier
or diluent.
Such compositions may be manufactured in conventional manner. Unit dosage
forms
contain, for example, from about 0.25 to about 150, preferably from 0.25 to
about 2S mg
of a compound according to the invention.
Moreover the present invention provides the use of an agent of the invention,
for the
manufacture of a medicament for the treatment of any condition mentioned
above, e.g.
epilepsy, stroke and brain or spinal trauma.
In still a further aspect the present invention provides a method for the
treatment of any
condition mentioned above, e.g. epilepsy, stroke and brain or spinal trauma,
in a subject in
need of such treatment, which comprises administering to such subject a
therapeutically
effective amount of an agent of the invention.
The following examples illustrate the invention. The temperatures are given in
degrees
Celsius and are uncorrected.
Exam_"ple 1~ 3-(4-methoxy-4'-trifluoromethyl-biphenyl-3~11-1-methvl-12.5.6-
tetrahvdro-
A. 3-( S-bromo-2-methoxy-phenyl)-pyridine
A solution of bromine (12.2 g, 3.9 ml, 0.076 mol) in glacial acetic acid (40
ml) is added
dropwise over 1S minutes to a solution of 3-(2-methoxyphenyl)-pyridine (14.0
g, 0.076
mol) and anhydrous sodium acetate (6.8 g, 0.083 rnol) in glacial acetic acid
(140 ml), held
at 1S-20°. A precipitate is observed, which dissolves gradually upon
stirring of the
suspension. This suspension is left to stir for 18 hours at room temperature,
after which a
clear orange solution is obtained. The acetic acid is removed under reduced
pressure, and
the residue taken up in EtOAc (2S0 rnl). The extract is washed with water (1S0
ml),
saturated agueous NaHC03 (100 ml) and brine (7S ml), dried (MgSOa), and
evaporated to
CA 02308151 2000-04-17
WO 99123073 PCTIEP98/06880
_g_
give the product as an orange oil. TLC (silica gel, toluene-EtOH-NHaOH
85:15:1) Rf =
0.5.
B. 3-(4-methoxy-4'-trifluorornethyl-biphenyl-3-yl)-pyridine
A mixture of 3-(S-bromo-2-methoxy-phenyl)-pyridine (19.0 g, 0.072 mol), 4-
trifluoromethylphenylboronic acid (14.3 g, 0.076 mol), palladiurn(II)acetate
(520 mg,
0.0023 mol), tri-o-toluylphosphine (2.1 g, 0.0069 mol), 2M aqueous Na2C03 (39
ml,
0.077 mol), MeOH (80 ml) and toluene (350 ml) is heated to reflux under an
argon
atmosphere during 18 hours. The mixture is allowed to cool, filtered through
Hyflo,
diluted with water ( 100 ml), and the phases separated. The aqueous phase is
extracted
with toluene (150 rnl) and the combined organic phases are washed with water
(100 ml)
and brine (100 ml), dried (MgS04), treated with activated charcoal (1 g),
filtered through
Hyflo and evaporated to give a yellow oil. This oil is dissolved in EtOH (50
ml) and
treated with 3N ethanolic HCl (25 ml). The hydrochloride salt precipitates
upon addition
of ether (20 g, 76%), m.p. 229-231°. TLC (silica gel, toluene-EtOH-
NH40H 85:15:1) Rf
= 0.55.
C. 3-(4-methoxy-4'-triffuorornethyl-biphenyl-3-yl)-1-methyl-pyridi~niurn
iodide
A solution of methyl iodide (9.4 g, 66 mmol) in acetone (25 ml) is added
dropwise over 15
minutes to a cool (15°) solution of 3-(4-methoxy-4'-trifluoromethyl-
biphenyl-3-yl)-
pyridine (10.9 g, 33 mmol) in acetone (100 ml). The reaction mixture is then
heated to
reflux for 2 hours, after which a second portion of methyl iodide ( 1 ml, 2.2
g, 0.015 mmol)
is added, and the mixture is allowed to reflux for a further 1.5 hours. After
evaporation of
the solvent, the title compound is obtained as a yellow solid, which is used
directly in the
following reaction. TLC (silica gel, toluene-EtOH-NH40H 85:15:1 ) Rf = 0 -
0.02.
D. 3-~4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-1-methyl-1,2,5,6-
tetrahydro-pyridine
A solution of sodium hydroxide ( 1.5 g; '~6 mmol) in water ( 100 rnl) is added
to a solution
of 3-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-1-methyl-pyridinium iodide
in MeOH
( 150 ml). To the resulting mixture, sodium borohydride (2.5 g, 66 mrnol) are
added in
CA 02308151 2000-04-17
WO 99/23073 PCT/EP98106880
-10-
portions over 30 minutes, and the mixture is lefr to stir for 60 hours at room
temperature.
The mixture is filtered through Hyflo and the filtrate concentrated to ca. 100
ml, upon
which a yellow oil separates. This mixture is extracted with EtOAc (3 x 125
ml). The
combined organic extracts are washed with brine (75 ml), dried (MgS04) and
evaporated
to yield the title compound as a yellowish-brown oil. TLC (silica gel, toluene-
EtOH-
NHaOH 85:15:1) Rf = 0.4. The hydrogen maleinate has a m.p. of 123-125°
(EtOH/Et20).
The following compounds of formula I are prepared in analogous manner to
Example 1:
Examule 2: 3-i4-methoxy-biphenyl-3~r1~ 1-methyl-12.5.6-tetrahs dr~o-pyridine
The hydrochloride has a m.p. of 187-194°.
Example 3: 3-(2'-chloro-4-methoxy-biphenyl-3-yl)-1-meth 1-X1.2_,5,6-tetrahydro-
p
The by drogen oxalate has a m.p. of 137-142°.
Example 4: 3-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-1-propyl-1.2,5.6-
tetrahydro-
P
The compound is obtained as a yellow oil. TT,C (silica gel, toluene-EtOH-NH40H
85:15:1)Rf=0.25.
Examy,ple 5: 3-~(4-methoxy-4'-trifluoromethyl-biphenyl-3-~ -1-meth,~piperidine
Palladium on charcoal ( 10%, 700 mg) is added to a degassed solution of 3-(4-
methoxy-4'-
trifluoromethyl-biphenyl-3-yl)-1-methyl-1,2,5,6-tetrahydro-pyridine (obtained
according to
Example 1 ) (5.3 g, 15.9 mmol) in glacial acetic acid (75 ml) and the mixture
hydrogenated
on a Parr Hydrogenator for 18 hours at room temperature and 5 atm. hydrogen
pressure,
during which time 70 ml of hydrogen are taken up. The catalyst is then
filtered off, and
fresh catalyst added (0.800 g), and the mixture further hydrogenated for 18
hours at 45°
CA 02308151 2000-04-17
WO 99/23073 PCT/EP98/06880
-11
and 5 atm, hydrogen, after which time 400 ml of hydrogen are taken up. The
suspension
is then filtered, the solid washed with AcOH-~d the filtrates evaporated. The
residue is
treated with saturated aqueous K2C03 until a basic solution ensues, which is
extracted
with EtOAc. The organic extracts are washed with brine (30 mI), dried (MgS04)
and
evaporated to give the product as a light brown oil. The hydrogen maleinate
has a m.p. of
93-96° (EtOH/Et~O, dec.).
The following compounds of formula I are prepared in analogous manner to
Example 5.
Example 6: 3-(4-methoxy-biphenyl-3-yl)-1-methyl-piperidine
The h~fdrochloride has a m.p. of 254-262°.
Example 7: 3-!2'-chloro-4-methox~-biphen~3-yl1-1-meth~rl-piperidine
The by drochloride has a m.p. of 237-247°.
Example 8: 3-(4-methoxy-4'-trifluoromethgirl-biphenyl-3-vl)-1-prowl-
pipericline
The hydrogen fumarate of the racemate has a m.p. of 178-180°
(EtOH/Et20, dec.).
The racernate is resolved into its enantiomers by HPLC on Chiralcel OJ, column
25 x 0.46
cm, Mobile phase: Hexane-EtOH 9:1 with 0.1% TFA. Flow rate: 1mL/min. The first
enantiomer elutes with retention time 8.35 min and the second with 10.25 min.
The
enantiomers are crystallised as their corresponding fumarate salts, the first
has (a)DZO
+24.4 (c = 1.0, MeOH), and the second (aJD2o _ _24.3 (c = 1.0, MeOH).
Examyle 9: (-)-3-(4-methoxy-4'-trifluoromethyl-biphenyl-3=y11-1-meth ~I-
piperidine
A solution of 3-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-1-methyl-
piperidine
(obtained according to Example 5) (7.7 g, 22 rnmol) and (-)-2,3-di-o-
toluyltartaric acid
CA 02308151 2000-04-17
WO 99123073 PCTIEP98/06880
-12-
hydrate (8.9 g, 22 mmol) in warm (70°) EtOH (I00 ml) is allowed to cool
to room
temperature and left to stand for 18 hours. - the crystals so formed are
filtered off and
recrystallised from 100 ml EtOH, to give crystals with a rn.p. of 155-
156°; [a)D2o - _77.8
(c = 1.0 MeOH). From the crystals thus obtained the free base is formed (oil;
[a)D2o - -9,3
(c = 0.95, MeOH). These crystals are recrystallised again from EtOH (80 ml),
to give
crystals with a mp of 159-160°, [a)D2o - _g3.0 (c = 1.0, MeOH); free
base (oil): [a)D''° -
12.5 (c = 0.9, MeOH}. The hydrogen maleate has a mp of 124-126°
(EtOHIEt20); [a)D2o
- -6.2 (c = 1.0, MeOH).
Example 10: t+)-3-(4-methoxy-4'-triffuoromethvl-biphenyl-3-vll-1-meth l~l-
piperidine
The mother liquors from Example 9 are reserved, and free base is prepared from
each
by treating with sat. aqueous K2C03 and extraction with EtOAc. The free base
obtained from the first mother liquors (2,4 g, 6.88 mmol) is treated with (+)-
2,3-di-o-
toluyltartaric acid hydrate (2.6 g, 6.88 mmol) in boiling EtOH (40 ml), which
upon
cooling yields crystals which are recrystallised from EtOH (25 ml) to give
colourless
crystals with a mp of 164-165°, [a)D2° _ + 81.1 (c = 1.1, MeOH)
from which the free
base has a [a)D2° of + 11.3 (c = 1.0, MeOH). The free base from the
combined second and
third mother liquors (3.6 g, 10.3 mmol) are combined with (+)-2,3-di-o-
toluyitartaric acid
hydrate (3.9 g, 10.3 mmol) in boiling EtOH (50 ml). After complete
crystallisation , the
crystals are recrystallised from EtOH (45 ml) to give a crop of crystals
having a [a]n2° of +
85.2 (c = 1.0, MeOH) from which the free base has a [a)D2o of + 9.8 (c = 1.4,
MeOH), and
are further recrystallised from EtOH (30 ml) to give crystals with a m.p. of
164-165°;
[a)DZO = +87,3 (c = i.0, MeOH) from which the free base has a [a)D2o of + 11.3
(c = 1.0,
MeOH). The hydrogen maleate has a m.p. of 125-127° (EtOH/Et20); [a)DZO
= + 5.4 (c =
1.1, MeOH) -
Example 11: 3-(4-methoxy-4'-trifluorom~yl-biphenyl-3-vl~l-methyl-pyrrolidine
A. 1-ethoxycarbonyl-3-(2-methoxyphenyl)-pyrrolidine
CA 02308151 2000-04-17
WO 99/23073 PCTIEP98/06880
-13-
A solution of ethylchloroformate (0.61 ml, 673 mg, 6.21 mmol) in CH2Cl2 (3
rnl) is added
dropwise over 10 minutes to a cold (~5°, ice-bath) solution of N-ethyl-
N,N-
diisopropylamine (1.3 ml, 911 mg, 7.01 mmoi) and 3-(2-methoxyphenyi)-
pyrrolidine (1.00
g, 5.65 mmol) in CH~C12 ( 15 rnl). The yellow reaction mixture is left to stir
for 3.5 hours
at room temperature, after which it is washed with 1N HCl ( 15 ml), saturated
aqueous
NaHC03 ( 15 ml) and brine ( 10 rnl), dried (MgS04) and the solvent evaporated
to .give the
product as a yellow oil. TLC (silica gel, toluene-EtOH-NH40H 85:15:1 ) Rf =
0.6.
B. 1-ethoxycarbonyl-3-(5-bromo-2-methoxyphenyl)-pyrrolidine
A solution of bromine (835 mg, 5.22 matg) in glacial acetic acid (3 ml) is
added dropwise
to a solution of 1-ethoxycarbonyl-3-(2-methoxyphenyl)-pyrrolidine (1.300 g,
5.22 mmol)
and sodium acetate (470 mg, 5.70 mmol) in glacial acetic acid (15 ml), and the
mixture is
left to stir for 18 hours at room temperature. The mixture is filtered through
Hyflo and
the solvent evaporated. The residue is taken up in ethyl acetate (30 ml) and
the solution
washed with water (20 ml), saturated aqueous Na2C03 (20 ml) and brine (15
rnl). The
aqueous phases are extracted with EtOAc (25 ml) and the combined organic
extracts dried
(MgSOa) and evaporated to give the product as a yellow-brown oil [TLC (silica
gel,
toluene-EtOH-NH40H 85:15:1) Rf = 0.6] which is used directly in the following
reaction.
C. 1-ethoxycarbonyl-3-(4-methoxy-4'-tritluoromethlyl-biphenyl-3-yl)-
pyrroliaine
Obtained from 1-ethoxycarbonyl-3-(5-bromo-2-methoxyphenyl)-pyrrolidine and 4-
trifluoromethylphenyl-boronic acid by palladium-catalysed coupling in
analogous manner
to Example 1B. TLC (silica gel, toluene-EtOH-NH40H 85:15:1) Rf = 0.78. The
crude
product is used directly in the following reaction.
D. 3-_(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-1-methyl-pyrrolidine
The crude product obtained under C (2.00 g) is dissolved in THF (15 ml) and
added
dropwise over 15 minutes to a cold (0-5°) suspension of lithium
aluminium hydride
(350 mg, 9.2 mmol) in THF (25 ml). The reaction mixture is allowed to stir at
room
CA 02308151 2000-04-17
WO 99/23073 PCT/EP98/06880
-14-
temperature for 1 hour and heated to reflux for 18 hours, after which it is
allowed to
cool to room temperature, and treated by t~ careful sequential addition of
saturated
aqueous NazSOa (2m1), 2N NaOH (2 ml), and Et20 (SO ml). The mixture is stirred
vigorously 1 hour at room temperature, and the precipitate filtered off. The
precipitate
is further washed with Et20-THF (1:1 30 ml), and the combined filtrates dried
(MgS04) and evaporated to give a reddish oil. This crude product is dissolved
in Et20
(SO ml) and extracted with 2N HCl (2x 1S ml). The combined acid phases are
further
extracted with Et20 (20 ml), cooled (ice-bath), made alkaline with sat. aq.
K2C03 and
extracted with Et20 (SO ml). The ether extracts are washed with brine (30 ml),
dried
(MgSOa) and evaporated to give the product as a reddish oil. TLC (silica gel,
toluene-
EtOH-NHaOH 8S:IS:1) Rf = 0.1. The hydrogen fumarate has a m.p. of I76-
180°
(dec.).
Example 12 2 (4' trifluorometh~phenyl-3-vl)ethvlamine
A mixture of 3-(bromophenyl)-ethylamine (1.400 g, 7.0 mmol) , 4-
trifluoromethyl-
phenylboronic acid (1.33 g, 7.0 mmol), tris-(ortho-toluyl)phosphine (212 mg,
0.70
mmol), palladium (II) acetate (160 mg, 0.7 rnmol), aqueous sodium carbonate
solution
(2M, 3.S ml), MeOH (2 ml) and toluene (2S ml) is heated to reflux under argon
for 18
hours. The mixture is then allowed to cool to room temperature, and the phases
are
separated. The aqueous phase is extracted with toluene (2S ml). The combined
organic
phases are washed with water (2S rnl) and brine (30 ml), dried (MgS04) and
evaporated. The residue is purified by chromatography (silica gel, toluene-
ethanol-
NH40H 85:15:1) to yield the product as a light yellow oil. TLC (silica gel,
toluene-
ethanol-NHaOH 8S:1S:1)Rf = 0.30.
Example I3 N N dimethvl 12 (4'-trifluoromethvl-biphenyl-3-vllethvllamine
Obtained analogously to Example 12. The hydrogen maleate has a m.p. of 150-
153°
(EtOH/Et~O).
CA 02308151 2000-04-17
WO 99/23073 PCT/EP98106880
-15-
The compound can also be obtained by dimethylation of the compound of Example
12
according to known procedures, e.g. by Eschwe~ler-Clarke methylation.
Example 14: 2-(6-methoxy-4'-tritluoromethLl-biphen~~yl)ethylamine
Obtained analogously to Example 12 as a yellow oil. TLC (silica gel, toluene-
EtOH-
NH40H 85:15:1 ) Rf = 0.30.
Example 1S: N.N-dimethyl-f2-(6-methoxy-4'-trifluoromethyl-biphenyl-3-
yl)ethyllarnine
Obtained analogously to Example 12. The hydrochloride has a m.p, of 210-
212°
(EtOH/Et20)..
The compound can also be obtained by Eschweiler-Clarke methylation of the
compound
of Example 14.
Example 16: 2-(4-methoxy-4'-tiifluoromethyl-biphenyl-3-yl)~thvlamine
Obtained analogously to Example I2. The hydrogen rnaleate has a m.p. of 157-
160°
(EtOH).
Example 17: N.N-dimeth~rl-2-f(4-methoxy-4'-trifluoromethyl-biphenyl-3-vl)ethyl-
]amine
Obtained analogously to Example 12. The hydrogen maleate has a m.p. of 136-
137°
(EtOH).
The compound can also be obtained by Eschweiler-Clarke methylation of the
compound
of Example 16. -
Example 18: N-propyl-2-[4-methoxy-4'-trifluorometh~phenyl-3-yl?Leth~lamine
CA 02308151 2000-04-17
WO 99123073 PCTIEP98/06880
-16-
Obtained analogously to Example 12. The hydrogen maleate has a m.p. of 178-
180°
(EtOH/Et~O). -
Example 19: 1-[2-f4-methoxv-4'-trifluommeth~phenyl-3-yllethyl]py~olidine
Obtained analogously to example 12. The hydrogen maleate has a m.p. of 131-
133°
(EtOHlEt20).
The compound can also be obtained as follows:
A. 1-[2-(4-methoxy-4'-tritluoromethyl-biphenyl-3-yl)-ethyl-pyrroIidine-
2,5-dione
A solution of 2-(4-methoxy-4'-trifluoromethylbiphenyl-3-yi)-ethylarnine (1.4
g, 4.74
mmol) and succinic anhydride (475 mg, 4.74 mmol) in THF (60 ml) is heated to
reflux for
I8 hours. The solution is then evaporated to dryness, and the residue heated
to 190° to
give an oil that crystallises on standing, and is recrystallised (Et20) to
yield the product,
m.p. 116-120°.
B. 1-[2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-ethylJpyrrolidine
A solution of 1-(2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-ethyl-
pyrrolidine-2,5-
dione (1.3 g, 3.45 mmol) in THF (10 ml) is added dropwise over 10 minutes to a
suspension of lithium aluminium hydride (262 mg, 6.9 mmol) in THF (15 ml) held
at 0-
10°. When the addition is complete, the mixture is allowed to warm up
to room
temperature, and stirred for 1 hour, after which the mixture is heated to
reflux for i8
hours. After cooling, the reaction mixture is treated sequentially with
saturated aqueous
Na2SOa (2 ml) and aqueous NaOH (2N, 1 ml). After addition of Et20 (25 rnl) the
resulting mixture is stirred for 1 hour, and then filtered. The precipitate is
washed with
Et20, and the washings combined with the filtrate. The Et20 solution is dried
(MgS04)
and evaporated to give a yellow oil which is purified by crystallisation as
its hydrogen
maleate.
Example 20: (1S''.2S~',6R"'.7R~)- 4-[2-(4-methoxy-4'-trifluoromethyl-biphenyl-
3-yl)-
ethyll-10-oxa-4-aza-tricyclo [S .2.1.0 (,2 L6~] decane
CA 02308151 2000-04-17
WO 99/23073 PCTIEP98/06880
-17-
Obtained analogously to Example 12. The-hydrogen male ate has a rn.p. of 163-
164°
(EtOH/Et20). The compound can also be obtained as follows:
A. (2S ;2R*,6S*',7R~')-4-[2-(4-methoxy- 4'-trifluoromethyl-biphenyl-3-yl)-
ethyl]-10-oxa-4-aza-tricyclo[5.2.1.0(2,6)]decane-3,5-dione
A solution of 2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)ethylamine (1.4 g,
4.74
mmol) and (IS*,2R *,6S*,7R *)-4,10-dioxa-tricyclo[5.2.1.0(2,6)]decane-3,5-
dione (850
mg, 5.1 mmol) in THF (60m1) is heated to reflux for 18 hours, in similar
fashion to
example 8A, to give the product, mp. 166-168°.
B.(1S*',2S ;6R ;7R*')-4-[2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-
ethylJ-10-oxa-4-aza-tricyclo[5.2.1.0(2,6)]decane
The product from Example 20A is reduced with lithium aiuminium hydride in THF
to
give the product as a brown oil which is crystallised as its hydrogen maleate
salt.
Example 21: f 1S'~.2S*.6R*.7R*1- 4-[2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-
eth~l~,-10-oxa-4-aza-2.6-dimeth I-~yclo[5.2.1.0(2.6)]decane
Obtained analogously to Example 12. The hydrochloride has a m.p. of 229-
231°. The
compound can also be obtained as follows:
A. (IS~',2R ;6S*,7R*')-4-[2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-
ethyl]-10-oxa-4-aza-2,6-dimethyl-tricyclo [5 .2.1.0(2,6 ) J decane-3,5-dione
Obtained in a similar fashion to example 20A from of 2-(4-methoxy-4'-
trifluoromethyl-biphenyl-3-yl)-ethylamine and (1 R *,2S*,6R *,7S*)-2,6-
dimethyl-4,10-
dioxa-tricyclo [5.2:1.0(2,6)]decane-3,S-dione. Mp. 115-117°
(Et20/hexane)
B. (ZS*,2Sx,6R*',7R*)-4-[2~4-methoxy-4'-trifluorornethyl-biphenyl-3-yl)-
ethyl]- I 0-oxa-4-aza-2,6-dimethyl-tricyclo [S .2.1.0 ( 2,6 ) ] decane
CA 02308151 2000-04-17
WO 99123073 PCT1EP98106880
-18
Obtained in similar fashion to Example 20B by reduction of (I R *,2S *,6R *,7S
*)-4-[2-
(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl~~:ethyl]-10-oxa-4-aza-2,6-dimethyl-
tricyclo[5.2.1.0(2,6)]decane-3,5-dione.
Example 22 2-~,4'-isopropyl-4-methoxy-biphenyl-3-v11-ethvlamine
Obtained analogously to Example 12. TLC (silica gel, EtOAc-MeOH-NH40H 0:20:2)
Rf = 0.22.
Examvle 23~ N11T dimethYl_-2-(4'-isovropyl-4-methoxy-biphenyl-3-vl)-ethvlamine
Obtained analogously to Example 12. The hydrogen maleate has a m.p. of 123-
124°
(EtOH/Et~O).
The compound can also be obtained by Eschweiler-Clarke methylation of the
compound of Example 22.
Examule 24- 2-(2'-chloro-4-methoxy-biphen~yll-ethvlamine
Obtained analogously to Example 12. The hydrochloride has a m.p. of 191-
205°.
ExamQ,le 25' N N-dimeth~l-2-!2'-chloro-4-methoxY biphenyl-3-vl)-ethvlamine
Obtained analogously to Example 12. The hydrochloride has a m.p. of 151-
159°.
The compound can also be obtained by Eschweiler-Clarke methylation of the
compound of Example 24.
Exam,~le 26~ (2'-ehloro-4-methoxy-biphenyl-3-yl-methyl)-N,N-dimethvlamine
Obtained analogously to Example 12. The hydrogen oxalate has a m.p. of 145-
159°.
Example 27~ N.N dimethyl_-2-(2'-chloro-4-methoxy-biphen~-3-yl)-1-methyl-
ethvlamine
CA 02308151 2000-04-17
WO 99123073 PCT/EP98/06880
-19
Obtained analogously to Example 12. The hydrochloride has a rn.p. of 141-
145°.
Example 28~ (+) f2 f4 Methoxy-4'-trifluorometh~phenyl-3-ill-1-methyl-ethvll-
N,N-dimethy_I-amine
A. Z-1-Methoxy-2-(2-nitropropenyl)-benzene
A solution of 10 g of 2-methoxybenzaldehyde, 6.07 g of nitroethane, 0.8 ml of
1-
butylamine and 30 ml of ethanol is refluxed for 3 days, the ethanol evaporated
and the
remaining mixture dissolved in ethyl acetate. After extraction with water and
brine the
solvent is evaporated and the residue bulb to bulb distilled. The fraction
distilling at
120 - 170° I 0.01 mbar is purified on silicagel by elution with
methylenechloride I
cyclohexane 1:1 giving the title compound in form of yellow plates, m.p. 39 -
42°.
B. rac-2-(2-Methoxy-phenyl)-1-methyl-ethylamine
A solution of 45.01 g (233 mmol) of Z-1-methoxy-2-(2-nitropropenyl)-benzene in
250
ml of ether is slowly dropped into a flask equipped with a mechanical stirrer,
thermometer and reflux condenser containing 40.84 g of LiAlH4 in 600 ml of
diethyl
ether. The exothermic reaction is cooled with ice and the temperature kept
between 5 -
10°. After stirring overnight at room temperature 330 ml of 2M Na2C03
are added,
the resulting suspension filtered and the ether phase extracted with 2M HCI.
The acidic
aqueous phase is made alkaline with 1.2 equivalents of conc. ammonia and
extracted
with diethyl ether. The ether phase is washed with brine, dried over Na2S04
and
concentrated to afford the title compound as a yellow oil.'H-NMR (360 MHz,
CDCl3):7.2t,1H;7.1d,1H;7.0-6.8m,2H;3.9s,3H;3.2m,1H;2.8m,1H;2.6
m, 1H; 1.1 d, 3H.
Further purification is achieved by transformation of the compound into the
naphthalene-1,5-disulfonate salt.
C.1 (-)-2-(2-Methoxy-phenyl)-1-methyl-ethylamine
CA 02308151 2000-04-17
WO 99123073 PCT/EP98/06880
-20-
To 19.16 g of rac-2-{2-methoxy-phenyl)-1-methyl-ethylamine dissolved in 300 ml
of
methanol a solution of 17.49 g of D-(-)-tarta~c acid in 334 ml of methanol is
added
and the mixture kept at 4° for 3 hours. The solid is filtered, the
mother liquor put aside
and the filter cake washed with ice-cold methanol and recristallised twice
from
methanol until the optical rotation remained constant. One obtains 11.11 g of
(-)-2-(2-
methoxy-phenyl)-1-methyl-ethylamine D-tartaric acid salt as fine white plates,
m.p.
144 - 149°. The free base show a specific rotation of [a]ss9= -
35.4° (c=1, MeOH).
Analytical chiral capillary electrophoresis of the free base reveals an
optical purity of
>98%.
C.2 (+)-2-(2-Methoxy-phenyl)-1-methyl-ethylamine
The first mother liquor obtained in the preparation of the (-)-enantiomer of
C.1 is
concentrated and the residue liberated from the tartrate by treatment with
conc.
ammonia and ethyl acetate. After evaporation of the organic layer, 11.14 g of
a yellow
oil are obtained and combined with 10.12 g of L-(+)-tartaric acid in a total
volume of
140 ml of methanol. After 3 h at 4°, the formed solid is collected,
washed with ice-cold
methanol and recristallised from methanol until the specific rotation remains
constant.
One obtains 11.92 g of (+)-2-(2-methoxy-phenyl)-1-methyl-ethylamine-L-tartaric
acid
salt as white plates. The free base show a specific rotation of [a]ss9 =
+37.7° (c=1,
MeOH). Analytical chiral capillary electrophoresis reveals an optical purity
of >98%.
D. (-)-[2-(2-Methoxy-phenyl)-1-methyl-ethyl]-N,N-dimethyl-amine
11.8 g of the tartaric acid salt of (+)-2-(2-methoxy-phenyl)-1-methyl-
ethylamine are
liberated with conc. ammonia in ethyl acetate and the obtained oil is
dissolved in 56
ml of methanol. To this solution 22.3 ml of 36.5% aqueous formaldehyde
solution are
added, the mixture cooled to 3° and treated in small portions with
10.66 g of
NaCNBH3. After 22 h of stirring at room temperature, the solvent is evaporated
and
the residue distributed between ethyl acetate and water. The organic phase is
washed
with water and brine, dried over Na2SC~, concentrated and the residue purified
on
silicagel by elution with toluene l ethyl acetate I methanol l conc. ammonia
60:30:10:1
yielding 5.02 g of the title compound as a yellow oil with [a]ss9 = -
19.9° (c = 1,
CA 02308151 2000-04-17
WO 99/23073 PCT/EP98106880
-2I-
MeOH).'H-NMR (360 MHz, CDC13): 7.2 dxt, 1H; 7.1 dxd, 1H; 7.0 - 6.8 m, 2H;
3.85s,3H;3.1-3.Om,1H;2.95-2.85m,k~i;2.4brs,7H;0.95d,3H.
E. (+)-[2-(S-bromo-2-methoxy-phenyl)-1-methyl-ethyl]-N,N-dimethyl-
amine
To a mixture of 4.47 g of (-)-[2-(2-methoxy-phenyl)-1-methyl-ethyl]-dimethyl-
amine,
2.09 g of sodium acetate and 40 ml of glacial acetic acid in a mechanically
stirred
flask, 1.19 mI of bromine in 8.5 ml of glacial acetic acid are added dropwise
at 20 -
30°. The reaction mixture is stirred for 16 h, neutralised with conc.
ammonia and
extracted with ethyl acetate. Drying of the organic layer with brine and
NazS04,
evaporation of the solvent and purification of the obtained residue on
silicagel with
toluene I ethyl acetate I methanol I conc. ammonia 60:30:10:1 give 4.32 g of
the title
compound as a brown oil, [a)589 = +1.5° (c = 1.01, MeOH). 1H-NMR (360
MHz,
CDC13):7.2-7.Om,2H;6.60d, 1H;3.70s,3H;2.95-2.75m,2H;2.3-2.2m+s,
7H; 0.85 d, 3H.
F. (+)-[2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-1-methyl-ethyl]-
N,N-dimethyl-amine
A 500 rnl-flask is charged with 140 ml of toluene and 28 ml of 2M Na2C03 and
gased
with argon for 1 h. Then 3.94 g of (+)-[2-(5-bromo-2-methoxy-phenyl)-1-methyl-
ethyl]-dimethyl-amine, 4.95 g of 4-trifluoromethyl-phenylboronic acid and 374
mg of
tetrakis(triphenylphosphine)palladium are added and the mixture is refluxed
for 12 h.
The aqueous layer is extracted with ethyl acetate and the combined organic
phases are
washed with brine, dried over NazS04 and concentrated to afford a brown oil.
Formation of the naphthalene-1,5-disulfonate salt of this oil and liberation
of the free
base allow to cristallise the product as hydrochloride salt from HCl in
diethyl ether.
Recristallisation ffom ethanol I ether afford the hydrochloride of the title
compound as
fine white plates, rn.p. 155 - 163°.
The free base solidifies with a m.p. of 36~- 37° and shows a specific
rotation of [a]s89 =
+13.0° (c = 0.995, MeOH). Analytical chiral HPLC (Chiracel OJ) reveals
an optical
purity of >99%.'H-NMR (360 MHz, CDC13): 7.70 s, 4H; 7.45 dxd, 1H; 7.40 d, 1H;
CA 02308151 2000-04-17
WO 99/23073 PCT/EP98/06880
-22-
6.95d,1H;3.90s,3H;3.25-3.20m,1H;3.0-2.9m,1H;2.55-2.45m,1H;2.40s,
bH; 1.00 d, 1H.
Example 29: (-1-'[2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-1-methyl-
ethyl~-
N.N-dirnethyl-amine
A. (+)-[2-(2-methoxy-phenyl)-1-methyl-ethyl]-N,N-dimethyl-amine
The product prepared as described in example 28C.1 is reduced with NaCNBH3 and
formaldehyde according to the description in example 28D to afford the title
compound as a yellow oil, [a]ss9 = +21.6° (c = 1.03, MeOH).
B. (-)-[2-(5-bromo-2-methoxy phenyl)-1-methyl-ethyl]-N,N-dimethyl-
amine
The product prepared under A is brominated as described in example 28E to
afford the
title compound as a yellow oil, [a]ss9 = -1.0 (c = 1.05, MeOH).
C. (-)-[2-(4-methoxy-4'-triffuoromethyl-biphenyl-3-yl)-1-methyl-ethyl]-
N,N-dimethyl-amine
The product prepared under B is arylated with 4-trifluoromethyl-phenylboronic
acid
according to example 28F to afford the hydrochloride of the title compound as
white
plates, m.p. 148 - 163°.
The free base solidifies with a m.p. of 31° and shows a specific
rotation of [a]ss9 =
-12.8° (c = 1.0, MeOH). Analytical chiral HPLC (Chiracel OJ) reveals an
optical purity
of >99%.
Example 30: ~1-(4-methoxy-4'-triffuorometh~phenyl-3-ylmethyll-propyll-N.N-
dimethyl-amine
A. 1-methoxy-2-(2-vitro-but-1-enyl)-benzene
A solution of 2.72 g of 2-methoxybenzaIdehyde, 1.96 g of 1-nitropropane, 0.4
rnl of 1-
butylamine and 10 ml of toluene is refluxed for 16 h. The toluene is
evaporated and
the residue dissolved in ethyl acetate and extracted with water and brine.
CA 02308151 2000-04-17
WO 99/Z3073 PCT/EP98/06880
- 23
Concentration of the organic phase affords the title compound as yellow oil,
sufficiently pure for the next step.'H-NMR (;,~60 MHz, CDCl3): 8.6 s (olefinic
H of E-
isorner); 8.2 s (olefinic H of Z-isomer).
B. 1-( 2-methoxybenzyl)-propylamine
To a magnetically stirred mixture of 2.28 g of LiAIHa and 3S ml of diethyl
ether a
solution of 3.79 g of 1-methoxy-2-(2-nitro-but-1-enyl)-benzene in 1S ml of
diethyl
ether is dropped at 0 - S°. After stirring overnight at room
temperature 20 ml of 2M
NazC03 are added, the resulting suspension is filtered and the organic phase
extracted
with 2M HCI. The acidic aqueous phase is made alkaline with 2M NaOH and
extracted with methyl-t-butyl ether. The organic phase is washed with brine,
dried over
Na2S04 and concentrated to give the title compound as a yellow oil. 'H-NMR
(360
MHz,CDCl3):7.2-7.Om,2H;6.9-6.7m,2H;3.8s,3H;2.9m,1H;2.8dxd,lH;
2.4dxd,1H;l.Sm,lH;l.3m,1H;0.9t,3H.
C. 1-(S-bromo-2-methoxy-benzyl)-propylamine
To a mixture of 2.25 g of 1-(2-methoxybenzyl)-propylamine, 1.13 g of sodium
acetate
and S7 ml of glacial acetic acid in a magnetically stirred flask, 0.65 ml of
bromine in 3
ml of glacial acetic acid are added dropwise at 20 - 30°. The reaction
mixture is stirred
for 4 h, concentrated and the obtained residue distributed between water and
ethyl
acetate. The organic phase is extracted with 2M acetic acid and the combined
acidic
aqueous phases are made alkaline with conc. ammonia. Re-extraction with ethyl
acetate, drying of the organic phase over Na2SOa and concentration give the
title
compound as colorless powder.'H-NMR (360 MHz, CDC13): 7.2 d, 1H; 7.1 s, 1H;
6.7 d, 1H; 3.8 s, 3H; 2.9 m, 1H; 2.7 m, 1H; 2.S m, 1H; 1.6 - 1.3 m, 2H; 1.0 t,
3H.
D. 1-(4-methoxy-4'-trifluoromethyl-biphenyl-3-ylmethyl )-propyl]-amine
A mixture of 1.91 g of 1-(S-bromo-2-methoxy-benzyl)-propylamine, 0.3 g of
tetrakis(triphenylphosphine)palladium, 2.61 g of 4-trifluoromethyl-
phenylboronic
acid, 24 ml of 2M Na2C03 and 2S ml of toluene is refluxed under argon for 7 h.
After
extraction of the water phase with diethyl ether, the organic phases are
combined,
CA 02308151 2000-04-17
WO 99/23073 PCT/EP98/06880
-24
dried over Na2S04 and concentrated to afford the title compound as a brown oil
which
is purified on silicagel by elution with toluepe I ethanol I conc. ammonia
8S:1S:1.'H-
NMR (360 MHz, CDC13): 7.65 s, 4H; 7.S dxd, 1H; 7.4 d, 1H; 7.0 d, 1H; 3.9 s,
3H;
3.1m,1H;2.9dxd,1H;2.6dxd,1H;1.6m,1H;1.4m,1H,1.1t,3H.
E. [1-(4-methoxy-4'-trifluoromethyl-biphenyl-3-ylmethyl)-propyl]-N,N-
dimethyl-amine hydrochloride
To a mixture of 1.25 g of 1-(4-methoxy-4'-trifluoromethyl-biphenyl-3-ylmethyl)-
propyl)-amine, 2.S ml of 36.5% formaline solution and 10 rnl of methanol under
argon 1.66 g of NaCNBH3 are added in several portions at 3°. After
stirring overnight
at room temperature the solvent is evaporated and the residue distributed
between
ethyl acetate and water. The organic phase is washed with water and brine and
concentrated to afford the crude product which is treated with HCl in diethyl
ether.
The formed solid is filtered and recristallised from aceton / diethyl ether to
give the title
compound as white needles, m.p. 1SS - 172°.
Examine 31 1 f2 (4 methoxy 4' trifluoromethvl-biphenyl-3-~~)-1-methyl-eth
~ioeridine_
A. 2-(S-Bromo-2-methoxy-phenyl)-1-methyl-ethylamine
A mixture of 6.54 g of 2-(2-methoxy-phenyl)-1-methyl-ethylamine (free base,
prepared
as described in example 28B), 3.56 g of sodium acetate and 180 ml of glacial
acetic
acid is treated with 6.31 g of bromine as described in example 30C, affording
the title
compound as a yellow oil.'H-NMR (360 MHz, CDCl3): 7.4 - 7.2 m, 2H; 6.7 d, 1H;
3.8s,3H;3.2hex,1H;2.8-2.Sm,2H;1.1d,3H.
B. 2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-1-methyl-ethylamine
hydrochloride
A mixture of 7.62 g of 2-(S-bromo-2-methoxy-phenyl)-1-methyl-ethylamine, 0.71
g of
tetrakis(triphenylphosphine)palladium, Z.98 g of 4-trifluoromethyl-
phenylboronic
acid, 24 ml of 2M Na2C03, 40 ml of toluene and 10 ml of ethanol is refluxed
for 9 h
under argon. After workup similar to Example 30D, the crude oil is purified by
CA 02308151 2000-04-17
WO 99123073 PCTIEP98/06880
- 25
transformation into the hydrochloride salt which is obtained in form of white
plates.
'H-NMR (360 MHz, DMSO-d6): 7.90 - 7.7.~ dxd, 4H; 7.65 dxd, 1H; 7.60 d, 1H;
7.15d,1H;3.85s,3H;3.Sm,1H;3.1-2.8rn,2H;1.15d,3H.
C. 4-[2-(4-methoxy-4'-triffuoromethyl-biphenyl-3-yl)-1-methyl-ethyl-
carbamoyl]butyric acid
The base of 1.1 g of 2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-1-methyl-
ethylamine hydrochloride is liberated and reacted with 0.37 g of glutaric
anhydride in
THF under reflux for 20 h. The solvent is evaporated and the residue dissolved
in ethyl
acetate. After extraction of the organic phase with 1M HCI, water and brine,
drying
over Na~SOa and evaporation the title compound is obtained as a white powder,
m.p.
88° (dec.).
D. 1-(2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-1-methyl-ethyl]-
piperidine-2,6-dione
A mixture of 1.02 g of the carboxylic acid obtained under C and 4.3 g of
acetyl
chloride in 20 ml of chloroform was refluxed for I7 h and, after cooling,
extracted
with water and 2M Na2C03. After drying of the organic phase over Na2SOa the
solvent is evaporated and the residue purified on silicagel by elution with
ethyl acetate I
cyclohexane 1:2, yielding the title compound as a bright yellow oil,
solidifying in the
refrigerator. 'H-NMR (360 MHz, CDCl3): 7.6 - 7.5 m, 4H; 7.4 dxd, 1H; 7.2 d,
1H;
6.8d,1H;5.2m,1H;3.8s,3H;3.2-3.Om,2H;2.4-2.3m,4H;1.5brm,ca.2H
(+HOD); 1.35 d, 3H.
E. 1-[2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-1-methyl-ethyl]-
piperidine hydrochloride
To a suspension of 0.054 g of LiAlH4 in 6 ml of diethyl ether under argon a
solution of
0.3 g of 1-[2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-yl)-1-methyl-ethyl]-
piperidine-
2,6-dione in 2 ml of diethyl ether is drc.~gped under slight cooling. After
stirring for 30
min 0.5 ml of 2M Na~COa are added, the resulting mixture is filtered and the
filtrate
acidified and extracted with 1M HCI. The aqueous phase is made alkaline with
conc.
CA 02308151 2000-04-17
WO 99/23073
-26-
ammonia and extracted with diethyl ether. The organic phase, after drying over
Na2S04, is concentrated and the residue treated with a solution of HCl in
diethyl ether
thereby affording the title compound as colorless plates, m.p.185°
(dec.).
Example 32~ N N-diethyl-j2-(4-methoxv-4'-trifluoromethyl-biphenyl-3-yl)-1-
methvl-
ethyl],=amine
Obtained by reductive aIkylation of 2-(4-methoxy-4'-trifluoromethyl-biphenyl-3-
yl)-1-
rnethyl-ethylamine (obtained as described in Example 31B) with acetaldehyde
over
10% Pd/C in ethylacetate. The hydrochloride has a m.p. of 105-107°.