Note: Descriptions are shown in the official language in which they were submitted.
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
1
NUCLEIC ACID TRANSFER VECTOR FOR THE INTRODUCTION OF
NUCLEIC ACID INTO THE DNA OF A CELL
Field of the Invention
This invention relates to methods for functional genomics including
identifying expression control sequences, coding sequences and the function of
coding sequences in the genomic DNA of a cell. The invention also relates to
transposons and transposases.
Background of the Invention
Transposons
Transposons or transposable elements include a short piece of nucleic
acid bounded by inverted repeat sequences. Active transposons encode enzymes
that facilitate the insertion of the nucleic acid into DNA sequences.
In vertebrates, the discovery of DNA-transposons, mobile elements that
move via a DNA intermediate, is relatively recent (Radice, A.D., et al., 1994.
Mol. Gen. Genet. 244, 606-612). Before then, only inactive, highly mutated
members of the Tc 1 /mariner as well as the hAT (hobolAclTam) superfamilies of
eukaryotic transposons had been isolated from different fish species, Xenopus
and human genomes (Oosumi et al., 1995. Nature 378, 873; Ivics et al. 1995.
Mol. Gen. Genet. 247, 312-322; Koga et al., 1996. Nature 383, 30; Lam et al.,
1996. J. Mol. Biol. 257, 359-366 and Lam, W. L., et al. Proc. Natl. Acad. Sci.
USA 93, 10870-10875).
DNA transposable elements transpose through a cut-and-paste
mechanism; the element-encoded transposase catalyzes the excision of the
transposon from its original location and promotes its reintegration elsewhere
in
the genome (Plasterk, 1996 Curr. Top. Microbiol. Immunol. 204, 125-143).
Autonomous members of a transposon family can express an active transposase,
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
2
the traps-acting factor for transposition, and thus are capable of transposing
on
their own. Nonautonomous elements have mutated transposase genes but may
retain cis-acting DNA sequences. These cis-acting DNA sequences are also
referred to as inverted terminal repeats. Some inverted repeat sequences
include
one or more direct repeat sequences. These sequences usually are embedded in
the terminal inverted repeats (IRs) of the elements, which are required for
mobilization in the presence of a complementary transposase from another
element or from itself.
Not a single autonomous transposable element has been isolated from
vertebrates; all transposon-like sequences isolated to date are defective,
apparently as a result of a process called "vertical inactivation" (Lobe et
al.,
1995 Mol. Biol. Evol. 12, 62-72). According to one phylogenetic model (Hartl
et
al., 1997 Trends Genet. 13, 197-201 ), the ratio of nonautonomous to
autonomous elements in eukaryotic genomes increases as a result of the trans-
complementary nature of transposition. This process leads to a state where the
ultimate disappearance of active, transposase-producing copies in a genome can
be inevitable. Consequently, DNA-transposons can be viewed as transitory
components of genomes which, in order to avoid extinction, must find ways to
establish themselves in a new host. Indeed, horizontal gene transmission
between species is thought to be one of the important processes in the
evolution
of transposons (Lohe et al., 1995 Mol. Biol. Evol. 12, 62-72 and Kidwell,
1992.
Curr. Opin. Genet. Dev. 2, 868-873).
The natural process of horizontal gene transfer can be mimicked under
laboratory conditions. In plants, transposable elements of the AclDs and Spm
families have been routinely introduced into heterologous species (Osborne and
Baker, 1995 Curr. Opin. Cell Biol. 7, 406-413). In animals, however, a major
obstacle to the transfer of an active transposon system from one species to
another has been that of apparent species-specificity of transposition due to
the
requirement for factors produced by the natural host. For this reason,
attempts
have been unsuccessful to use the P element transposon of Drosophila
melanogaster for genetic transformation of non-drosophilid insects, zebrafish
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
3
and mammalian cells (Gibbs et al., 1994 Mol. Mar. Biol. Biotech. 3, 317-326;
Handler et al., 1993. Arch. Insect Biochem. Physiol. 22, 373-384; and Rio et
al.,
1988 J. Mol. Biol. 200, 411-415). In contrast to P elements, members of the
Tcllmariner superfamily of transposable elements may not be as demanding for
species-specific factors for their transposition. These elements are
widespread
in nature, ranging from single-cellular organisms to humans (Plasterk, 1996
Curr. Top. Microbiol. Immunol. 204, 125-143). In addition, recombinant Tcl
and mariner transposases expressed in E coli are sufficient to catalyze
transposition in vitro (Vos et al, 1996 Genes. Dev. 10, 755-761 and Lampe et
al.,
1996. EMBO J. 15, 5470-5479 and PCT International Publication No. WO
97/29202 to Plasterk et al.). Furthermore, gene vectors based on Minos, a Tcl-
like element (TcE) endogenous to Drosophila hydei, were successfully used for
germline transformation of the fly Ceratitis capitata (Loukeris et al., 1995
Science 270, 2002-2005).
Molecular phylogenetic analyses have shown that the majority of the fish
TcEs can be classified into three major types: zebrafish-, salmonid- and
Xenopus
TXr-type elements, of which the salmonid subfamily is probably the youngest
and thus most recently active (Ivics et al., 1996, Proc. Natl. Acad. Sci. USA
93,
5008-5013). In addition, examination of the phylogeny of salmonid TcEs and
that of their host species provides important clues about the ability of this
particular subfamily of elements to invade and establish permanent residences
in
naive genomes through horizontal transfer, even over relatively large
evolutionary distances.
TcEs from teleost fish (Goodier and Davidson, 1994 J. Mol. Biol. 241,
26-34), including Tdrl in zebrafish (Izsvak et al., 1995 Mol. Gen. Genet. 247,
312-322) and other closely related TcEs from nine additional fish species
(Ivics
et al., 1996. Proc. Natl. Acad. Sci. USA 93, 5008-5013) are by far the best
characterized of all the DNA-transposons known in vertebrates. Fish elements,
and other TcEs in general, are typified by a single defective gene encoding a
transposase enzyme flanked by inverted repeat sequences. Unfortunately, all
the
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98124348
4
fish elements isolated so far are inactive due to one or more mutations in the
transposase genes.
Functional Genomics
There are estimated to be between 50,000 and 100,000 genes in the
genome of vertebrates. The expression of these genes is carefully orchestrated
such that most genes are not expressed most of the time in most tissues. The
roles of most genes in vertebrate genomes are unknown. Yet, most diseases
have a genetic basis. Accordingly, finding the sites and roles of expression
of the
genes in a vertebrate, especially human, genome is an important task. The task
is
exceedingly difficult.
Most studies to date in the field of genomics have concentrated on
identifying in cells of various types the sequences of expressed mRNAs encoded
by the coding sequence of a gene. However, this procedure does not often
provide insights into the functions of the genes, nor their importance.
An alternative method of finding genes and their functions is to interrupt
(mutate) genes with a molecular tag. Then, the interrupted genetic locus can
be
isolated based on the inserted genetic tag and the gene can be correlated with
a
phenotype, i.e., a physical result due to the loss of function of the
interrupted
gene. Genetic tags called gene-traps have been devised wherein a marker gene
is
inserted randomly into a genome (reviewed in Mountford, P. S., et al. Trends
Genet., I 1, 179-84 (1995)). When a critical gene is interrupted, and the
marker
gene is inserted in just the right way (in the correct direction, in-frame,
and in an
exon of the interrupted gene), the marker gene is expressed in the tissue in
which the interrupted gene normally is expressed.
A variation of the gene trap is to employ a splice acceptor site followed
by an internal ribosome entry site (IRES) placed in front of a marker gene.
Splice acceptor sites provide signals to target the sequences following the
splice
acceptor site to be expressed as mRNA provided there is an intron upstream of
the splice acceptor site (Padgett, T., et al., Ann. Rev. Biochem. J., 55, 1119-
1150
(1988)). An IRES allows ribosomal access to mRNA without a requirement for
cap recognition and subsequent scanning to the initiator AUG (Pelletier, J.A.,
et
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
al., Nature, 334, 320-325 (1988)). This expands the probability that the
marker
gene will be expressed when inserted into a gene. With a construct containing
a
splice acceptor site followed by an IRES is placed in front of a marker gene,
it is
possible to get expression of the marker gene even if the construct integrates
in
5 an intron or if it integrates out of frame with respect to the interrupted
gene. The
splice acceptor increases the likelihood that the inserted sequences will be
present in the resulting mRNA, and the IRES increases the likelihood of
translation of the inserted sequences. This approach, known to the art as a
"gene-trap," requires that the molecular tag insert within the coding sequence
where it will be expressed at approximately the same levels as the gene that
is
disrupted. However, the level of expression of the disrupted gene may be low
and the "target-size" (the length of the coding sequence in base-pairs) may be
small.
The encephalomycarditis virus (EMCV) IRES has been used for gene-
trapping (von Melchner et aL, J. Yirol, 63, 3227-3233 (1989)), is well
characterized (Jung, S. K., et al., Genes Dev 4, 1560-1572 (1990); Kaminski,
A.,
et al., EMBO J 13, 1673-1681 (1994}; Hellen, C. U., et al., Curr. Top.
Microbiol. Immunol. 203, 31-63 (1995)) and has been shown to function
efficiently in mammalian (Borman, A. M., et al., Nucleic Acids Res. 25, 925-32
(1997), Borman, A. M., et al., Nucleic Acids Res. 23, 3656-63 (1995)) and
chicken cells (Ghattas, I. R., et al., Mol. Cell. Biol. l l, 5848-59 (1991)).
The
use of an 1RES between the splice acceptor and reporter molecule has been
shown to lead to as much as 10-fold greater numbers of 6418-resistant colonies
in mouse embryonic stem cells than a non-IRES vector (see Mountford P. S., et
al. Trends Genet., 11, 179-84 (1995)). But this rate is still unacceptably
low,
which is why it is not used for mass screening of genes.
IRESs have been adapted into dicistronic vectors for the expression of
two open reading frames. For instance, using an IRES in a dicistronic vector
can result in more than 90% of transfected cells producing both the biological
gene of interest and the selectable marker (Ghattas et al. Mol. Cell. Biol.,
11,
5848-59 (1991)).
CA 02309000 2000-OS-04
WO 99JZ5817 PCT/US98/24348
6
Another strategy results in the "trapping" of sequences 3' of the inserted
marker gene. This entails the use of a retrovirus to deliver a marker gene
that is
placed between a promoter and a splice donor site (Zambrowicz, B.P., et al.,
Nature, 392, 608-611 (1998)). Splice donor sites provide signals to target the
RNA sequences encoding the marker gene to be spliced to the next downstream
splice acceptor site. When the marker gene is expressed, and there is a
downstream splice acceptor site, the mRNA may contain a poly(A) tail and
therefore be more stable and more efficiently translated. This expands the
probability that the marker gene will be expressed only when inserted into a
gene.
An alternative strategy is to use an enhancer-trap (Weber, F., et al.,
Cell, 36, 983-992 (1984)). In this strategy, the marker gene is placed behind
a
weak promoter to give a minimal promoter-marker gene construct. The minimal
promoter by itself does not have the ability to direct high expression of the
marker gene. However, when the minimal promoter is located in the vicinity of
certain regulatory sequences called enhancers, it can direct the expression of
the
marker gene at levels and in tissues in which the enhancers are active. Thus,
the
enhancer-trap tag does not have to insert only within a coding sequence; it
can
be activated by insertion outside of the transcription unit. An enhancer-trap
may
direct higher levels of expression than a gene-trap vector, which may increase
the ability of a researcher to detect the insertion of the molecular tag.
Many methods for introducing DNA into a cell in order to perform
various types of mutational analysis such as described above are known. These
include, but are not limited to, DNA condensing reagents such as calcium
phosphate, polyethylene glycol, and the like, lipid-containing reagents, such
as
liposomes, mufti-lamellar vesicles, and the like, virus-mediated strategies,
ballistic methods and microinjection and the like. These methods all have
their
limitations. For example, there are size constraints associated with DNA
condensing reagents and virus-mediated strategies. Further, the amount of
nucleic acid that can be introduced into a cell is limited in virus
strategies. Not
all methods facilitate integration of the delivered nucleic acid into cellular
nucleic acid and while DNA condensing methods and lipid-containing reagents
*rB
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98I24348
7
are relatively easy to prepare, the incorporation of nucleic acid into viral
vectors
can be labor intensive. Moreover, virus-mediated strategies can be cell-type
or
tissue-type specific and the use of virus-mediated strategies can create
immunologic problems when used in vivo. Most non-viral mediated methods
often result in concatamerization of input DNA as well as random break points
within the delivered DNA. Consequently, currently available vectors are
limited
in the ability to insert either gene-traps or enhancer-traps into genomes at
high
rates for high throughput screening for mutations and associated
identification of
tissues in which the marker gene is expressed. Thus, there remains a need for
new methods for introducing into a cell constructs that contain molecular tags
that can provide information regarding sites and roles of expression of genes.
Summary of the Invention
The present invention is directed to novel transposon-derived vectors and
methods of using them for insertional mutagenesis. A nucleic acid fragment is
provided that includes a nucleic acid positioned between at least two inverted
repeats wherein the inverted repeats can bind to a transposase, preferably an
SB
protein. The nucleic acid sequence includes a coding sequence. In some
embodiments of the invention the coding sequence is a detectable marker coding
sequence that encodes a detectable marker or a selectable marker, such as
green
fluorescent protein, luciferase or neomycin. The nucleic acid sequence
optionally includes at least one of (i) a weak promoter, for instance a carp
13-
actin promoter, (ii) a splice acceptor site and (iii) an internal ribosome
entry site,
each of which is operably linked to the detectable marker coding sequence.
Alternatively, the nucleic acid sequence can include an analyte coding
sequence
located 5' of the detectable marker coding sequence and an internal ribosome
entry site located therebetween, the internal ribosome entry site being
operably
linked to the detectable marker coding sequence. In some embodiments the
analyte coding sequence is operably linked to a promoter.
The present invention further provides a method for identifying an
expression control region, such as an enhancer, in a cell. A nucleic acid
fragment of the invention containing a nucleic acid sequence that includes a
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
8
detectable marker coding sequence is introduced into a cell, together with a
source of transposase. The detectable marker coding sequence is operably
linked to a weak promoter, and the nucleic acid sequence is positioned between
at least two inverted repeats, wherein the inverted repeats can bind to
transposase. The detectable marker or the selectable marker is then detected
in
the cell or its progeny containing the nucleic acid fragment, wherein the
expression of the detectable marker or the selectable marker indicates that
the
nucleic acid fragment has integrated into the DNA of the cell or its progeny
within a domain that contains an enhancer. The transformed cell or its progeny
can be evaluated for any changes in phenotype resulting from the insertion. In
order to determine the location in the cell DNA into which the nucleic acid
fragment has inserted, the DNA of the cell can be cleaved with a restriction
endonuclease to yield one or more restriction fragments that contain at least
a
portion of the inverted repeat and genomic DNA of the cell that is adjacent to
1 S the inverted repeat. The restriction fragment can be sequenced to
determine the
nucleotide sequence of the adjacent genomic DNA, and this sequence can then
be compared with sequence information in a computer database.
Also provided by the invention is a method for identifying a genomic
coding sequence in a cell. A nucleic acid fragment of the invention containing
a
detectable marker coding sequence, a splice acceptor site and an internal
ribosome entry site is introduced into along with a source of transposase. The
splice acceptor site and internal ribosome entry site are each operably linked
to
the detectable marker coding sequence, and the nucleic acid sequence is
positioned between at least two inverted repeats wherein the inverted repeats
can
bind to the transposase. The detectable marker or the selectable marker is
detected in the cell or its progeny containing the nucleic acid fragment,
wherein
expression of the detectable marker or the selectable marker indicates that
the
nucleic acid fragment has integrated within a genomic coding sequence of the
cell or its progeny. The detectable marker or the selectable marker can be
expressed spatially and temporally in the same way as the genomic coding
sequence is expressed when not interrupted. The cell or its progeny can be
evaluated for any change in phenotype resulting from the insertion. The DNA of
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
9
the cell can be cleaved with a restriction endonuclease and the resulting
restriction fragments sequenced in order to determine the location in the cell
DNA into which the nucleic acid fragment has inserted.
Another aspect of the invention provides a method for identifying the
S function of an analyte coding sequence. A nucleic acid fragment containing a
detectable marker coding sequence, an analyte coding sequence located S' of
the
detectable marker coding sequence, and an internal ribosome entry site located
therebetween is introduced into a cell along with a source of transposase. The
internal ribosome entry site is operably linked to the detectable marker
coding
sequence, and the nucleic acid fragment is positioned between at least two
inverted repeats that can bind to a transposase. The detectable marker or the
selectable marker is detected in the cell or its progeny containing the
nucleic
acid fragment, wherein the expression of the detectable marker or the
selectable
marker indicates that the nucleic acid fragment has integrated into the DNA of
the cell and that the analyte coding sequence is expressed. The cell or its
progeny can be evaluated for any change in phenotype resulting from the
insertion, wherein an altered phenotype indicates that the analyte coding
sequence plays a function in the phenotype. The DNA of the cell can be cleaved
with a restriction endonuclease and the resulting restriction fragments
sequenced
in order to determine the location in the cell DNA into which the nucleic acid
fragment has inserted
The invention also provides a gene transfer system to introduce a nucleic
acid sequence into the DNA of a cell. The system includes a nucleic acid
fragment and a source of transposase, wherein the nucleic acid fragment
includes a nucleic acid sequence that contains a coding sequence and is
positioned between at least two inverted repeats that can bind the
transposase.
In some embodiments of the invention the coding sequence is a detectable
marker coding sequence that encodes a detectable marker or a selectable
marker,
including green fluorescent protein, luciferase or neomycin. The nucleic acid
sequence of the gene transfer system can include one or more of {i) a weak
promoter, for instance a carp D-actin promoter, (ii) a splice acceptor site
and (iii)
an internal ribosome entry site, each being operably linked to the detectable
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
marker coding sequence. Alternatively, the nucleic acid sequence of the gene
transfer system can include an analyte coding sequence located 5' of the
detectable marker coding sequence and an internal ribosome entry site located
therebetween, the internal ribosome entry site being operably linked to the
5 detectable marker coding sequence. In some embodiments the analyte coding
sequence is operably linked to a promoter. The nucleic acid fragment of the
gene transfer system can by part of a plasmid or a recombinant viral vector.
The invention provides a method for producing a transgenic animal
including introducing a nucleic acid fragment and a transposase source into a
10 cell wherein the nucleic acid fragment includes a nucleic acid sequence
that
contains a heterologous coding sequence. The nucleic acid sequence is
positioned between at least two inverted repeats wherein the inverted repeats
can
bind to the transposase to yield a transgenic cell. The cell is grown into a
transgenic animal, and progeny can be derived from the transgenic animal.
Further provided by the present invention is a gene transfer system to
introduce a nucleic acid sequence into the DNA of a fish, preferably a
zebrafish,
which includes a nucleic acid fragment containing a nucleic acid sequence that
includes an internal ribosome entry site, wherein the nucleic acid fragment is
capable of integrating into the genomic DNA of a fish. The nucleic acid
sequence of the gene transfer system can further include a first coding
sequence
located 3' to and operably linked to the internal ribosome entry site and a
second
coding sequence located 5' to both the first coding sequence and the internal
ribosome entry site.
Also provided by the present invention is a transgenic fish or fish cell,
preferably a zebrafish or zebrafish cell, that comprises a heterologous
internal
ribosome entry site.
Abbreviations
EMCV encephalomycarditis virus
GFP green fluorescent protein
IRES internal ribosome entry site
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
11
Brief Description of the Figures
Fig. 1 illustrates the molecular reconstruction of a salmonid Tc 1-like
transposase gene. Fig. l(A) is a schematic map of a salmonid TcE. The TcE
includes inverted repeat/direct repeat (IR/DR) flanking sequences. Depicted on
the nucleotide sequence between the inverted repeat/direct repeat sequences is
the location of conserved domains in the transposase encoded by the nucleotide
sequence. The numbers 1 and 340 refer to the amino acids of the transposase
encoded by the nucleotide sequence. Abbreviations: DNA-recognition, a DNA-
recognition/binding domain; NLS, a bipartite nuclear localization signal; the
boxes marked D and E comprising the DDE domain (Doak, et al., Proc. Natl.
Acad, Sci., USA, 91, 942-946 (1994)) that catalyzes transposition; G-rich,
glycine-rich box; Fig. l(B) provides an exemplary strategy for constructing an
open reading frame for a salmonid transposase (SB1-SB3) and then
systematically introducing amino acid replacements into this gene (SB4-SB10).
Amino acid residues are shown using single letter code, typed black when
different from the consensus. Positions within the transposase polypeptide
that
were modified by site-specific mutagenesis are indicated with arrows.
Translational termination codons appear as asterisks, frameshift mutations are
shown as #. Residues changed to the consensus are check-marked and typed in
white italics. In the right margin, the results of various functional tests
that were
done at various stages of the reconstruction are indicated.
Fig. 2(A) is a double-stranded nucleic acid sequence encoding the SB
protein (SEQ ID N0:3). Fig. 2(B) is the amino acid sequence (SEQ ID NO:1) of
an SB transposase. The major functional domains are highlighted; see the
legend
to Fig. 1 A for abbreviations.
Fig. 3 illustrates the DNA-binding activities of an N-terminal derivative
(N123) of the SB transposase. Fig. 3(A) provides the SDS-PAGE analysis
illustrating the steps in the expression and purification of N123. Lanes: 1)
extract of cells containing expression vector pET21 a; 2) extract of cells
containing expression vector pET21a/N123 before induction with IPTG; 3)
extract of cells containing expression vector pET21 a/N 123 after 2.5 hours of
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
12
induction with IPTG; 4) partially purified N123 using Ni2+-NTA resin.
Molecular weights in kDa are indicated on the right. Fig. 3(B) illustrates the
results of mobility-shift analysis studies to determine whether N123 bound to
the inverted repeats of fish transposons. Lanes: 1 ) probe (a radiolabeled 300
by
DNA fragment comprising the left IR of the Tdrl transposon (T)) only without
any protein; 2) extract of cells containing expression vector pET2la; 3)
10,000-
fold dilution of the N123 preparation shown in lane 4 of Panel A; 4) same as
lane 3 plus a 1000-fold molar excess of unlabelled probe as competitor DNA; 5)
same as lane 3 plus a 1000-fold molar excess of an inverted repeat fragment of
a
zebrafish Tdrl element (z-IR) as competitor DNA; 6-13) 200,000-, 100,000-,
50,000-, 20,000-, 10,000-, 5,000-, 2,500-, and 1,000-fold dilutions of the
N123
preparation shown in lane 4 of Panel A.
Fig. 4 provides the DNase I footprinting of deoxyribonucleoprotein
complexes formed by N123. Fig. 4(A) is a photograph of a DNase I footprinting
gel containing a 500-fold dilution of the N 123 preparation shown in lane 4 of
Fig. 3A using the same transposon inverted repeat DNA probe as in Fig. 3B.
Reactions were run in the absence (lane 3) or presence (lane 2) of N123.
Maxam-Gilbert sequencing of purine bases in the same DNA was used as a
marker (lane 1 ). Fig 4(B) provides a sequence comparison of the salmonid
transposase-binding sites illustrated in Panel A with the corresponding
sequences in the zebrafish Tdrl elements. Fig. 4(C) is a sequence comparison
between the outer and internal transposase-binding sites in the SB
transposons.
Fig. 5 illustrates the integration activity of SB in human HeLa cells. Fig.
5(A) is a schematic illustrating the genetic assay strategy for SB-mediated
transgene integration in cultured cells. Fig. 5(B) demonstrates HeLa cell
integration using Petri dishes of HeLa cells with stained colonies of G418-
resistant HeLa cells that were transfected with different combinations of
donor
and helper plasmids. Plate: 1 ) pT/neo plus pSB 10-AS; 2) pT/neo plus pSB 10;
3)
pT/neo plus pSB 10-ODDE; 4) pT/neo plus pSB6; S) pT/neo-DIR plus pSB 10.
Fig. 6 summarizes the results of transgene integration in human HeLa
cells. Integration was dependent on the presence of an active SB transposase
and a transgene flanked by transposon inverted repeats. Different combinations
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
13
of the indicated donor and helper plasmids were cotransfected into cultured
HeLa cells and one tenth of the cells, as compared to the experiments shown in
Fig. 5, were plated under selection to count transformants. The efficiency of
transgene integration was scored as the number of transformants surviving
antibiotic selection. Numbers of transformants at right represent the numbers
of
6418-resistant cell colonies per dish. Each column represents the average
obtained from three transfection experiments.
Fig. 7 illustrates the integration of neomycin resistance-marked
transposons into the chromosomes of HeLa cells. Fig. 7(A) illustrates the
results of a southern hybridization of HeLa cell genomic DNA with neomycin-
specific radiolabeled probe from 8 individual HeLa cell clones that had been
cotransfected with pT/neo and pSB 10 and survived 6418 selection. Genomic
DNA was digested with the restriction enzymes NheI, XhoI, BgIII, SpeI and
XbaI, enzymes that do not cut within the neo-marked transposon, prior to
agarose gel electrophoresis and blotting. Fig. 7(B) is a diagram of the
junction
sequences of T/neo transposons integrated into human genomic DNA. The
donor site is illustrated on top with plasmid vector sequences that originally
flanked the transposon (black arrows) in pT/neo. Human genomic DNA serving
as target for transposon insertion is illustrated as a white box containing
the base
pairs TA, i.e., the site of DNA integration mediated by the SB transposase. IR
sequences and the flanking TA base pairs are uppercase, and the flanking
genomic sequences are in lowercase.
Fig. 8 is a schematic demonstrating an interplasmid assay for excision
and integration of a transposon. The assay was used to evaluate transposase
activity in zebrafish embryos. Two plasmids plus an RNA encoding an SB
transposase protein were coinjected into the one-cell zebrafish embryo. One of
the plasmids had an ampicillin resistance gene (Ap) flanked by IR/DR sequences
(black arrows) recognizable by the SB transposase. Five hours after
fertilization
and injection, low molecular weight DNA was isolated from the embryos and
used to transform E. coli. The bacteria were grown on media containing
ampicillin and kanamycin (Km) to select for bacteria harboring single plasmids
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
14
containing both the Km and Ap antibiotic-resistance markers. The plasmids
from doubly resistant cells were examined to confirm that the Ap-transposon
was excised and reintegrated into the Km target plasmid. Ap-transposons that
moved into either another indicator Ap-plasmid or into the zebrafish genome
were not scored. Because the amount of DNA in injected plasmid was almost
equal to that of the genome, the number of integrations of Ap-transposons into
target plasmids should approximate the number of integrations into the genome.
Fig. 9 illustrates two preferred methods for using the gene transfer
system of this invention. Depending on the integration site of the nucleic
acid
fragment of this invention the effect can be either a loss-of function or a
gain-of
function mutation. Integrations, as depicted with functional coding sequences
in
a transposon, typically result in gain-of function gene transfer. A subset are
also
a loss-of function or gene inactivation event. Both types of activity can be
exploited, for example, for gene discovery and/or functional genomics or gene
delivery, i.e., human gene therapy.
Fig.10 illustrates a preferred screening strategy using IRS-PCR
(interspersed repetitive sequence polymerase chain reaction). Fig.10(A)
illustrates a chromosomal region in the zebrafish genome containing the
retroposon DANA (D), Tdrl transposons (T, and TZ), and the highly reiterated
miniature inverted-repeat transposable element Angel (A). The arrows below
the elements represent specific PCR primers.
The X superimposed on the central DANA element to represents a
missing element or a mutated primer binding site in the genome of another
zebrafish strain. The various amplified sequence tagged sites (STSs) are
identified by lowercase letter (a through g), beginning with the longest
detectable PCR product. The products marked with an X are not produced in
the PCR reaction if genomes with defective "X-DNA" are amplified. Elements
separated by more than about 2000 base pairs (bp) and elements having the
wrong orientation relative to each other are not amplified efficiently.
Fig.10(B)
is a schematic of the two sets of DNA amplification products from both
genomes with (lane 1 ) and without (lane 2) the DANA element marked with an
X. Note that bands "a" and "d" are missing when the marked DANA sequence
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
is not present.
Fig. l l illustrates a preferred method for using an expression control
sequence-trap transposon vector. Abbreviations: I, intron; E, exon.
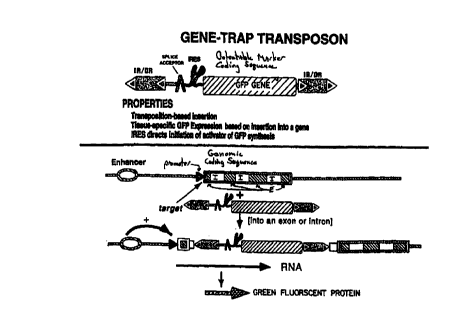
Fig.12 illustrates a preferred method for using a gene-trap transposon
5 vector. Fig.12(A) is a gene-trap that contains a GFP operably linked to a
splice
acceptor site and an IRES. Fig.12(B) is a gene trap similar to Fig. 12(A), but
encodes an activator which activates expression of a GFP coding sequence,
elsewhere in the genome, thereby amplifying the level of GFP expression over
what it would be were the GFP coding sequence in the gene trap vector.
10 Abbreviations: I, intron; E, exon.
Fig.13 illustrates the dicistronic vectors pBeL, phBeL, and pBL. The
promoters are indicated by the large arrows on the left; the smaller raised
arrows indicate the transcriptional initiation sites for the dicisctronic
mRNAs.
The IRES is depicted by a set of stem-loops. Changes in the control vectors
15 phBeL and pBL are circled. CMV/T7, CMV/T7 promoters;13-gal,13-
galactosidase coding sequence; hp, hairpin structure; Luc, luciferase coding
sequence; HGH(A), human growth hormone poly(A) signal.
Fig.14 The expression levels of 13-galactosidase and luciferase are
shown for embryos at 6 hours after injection with either pBeL, phBeL, and pBL
mRNA. The error bars indicate 95% confidence intervals. Abbreviation: RLU,
relative light units.
Fig.15 illustrates a strategy for using dicistronic coding sequence
expression transposon vectors.
Fig.16 illustrates an inverse PCR strategy to identify genomic DNA
adjacent to an inserted nucleic acid fragment.
Detailed Description
The present invention relates to novel transposases and the transposons
that are used to introduce nucleic acid sequences into the DNA of a cell. A
transposase is an enzyme that is capable of binding to DNA at regions of DNA
termed inverted repeats. Preferably a transposon contains two inverted repeats
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
16
that flank an intervening nucleic acid sequence, i.e., there is an inverted
repeat 5'
to and 3' to the intervening nucleic acid sequence. Inverted repeats of an SB
transposon can include two direct repeats and preferably include at least one
direct repeat. The transposase binds to recognition sites in the inverted
repeats
and catalyzes the incorporation of the transposon into DNA.
Transposons are mobile, in that they can move from one position on
DNA to a second position on DNA in the presence of a transposase. There are
two fundamental components of any mobile cut-and-paste type transposon
system, a source of an active transposase and the DNA sequences that are
recognized and mobilized by the transposase. Mobilization of the DNA
sequences permits the intervening nucleic acid between the recognized DNA
sequences to also be mobilized.
DNA-transposons, including members of the Tcllmariner superfamily,
are ancient residents of vertebrate genomes (Radice et al., 1994 Mol. Gen.
Genet., 244, 606-612; Smit and Riggs, 1996 Proc. Natl. Acad. Sci. USA 93,
1443-1448). However, neither antonomous copies of this class of transposon nor
a single case of a spontaneous mutation caused by a TcE insertion have been
proven in vertebrate animals. While evidence has been presented suggesting
that the zebrafish genome contains active transposons, (Lam et al W.L., et
al.,
Proc. Natl. Acad. Sci., USA, 93, 10870-10875 (1996)), neither autonomous
copies of this class of transposon nor a single case of a spontaneous mutation
caused by a TcE insertion have been rigorously proven in vertebrate animals.
This is in contrast to retroposons whose phylogenetic histories of mutating
genes
in vertebrates is documented (Izsvak et al., 1997). Failure to isolate active
DNA-transposons from vertebrates has greatly hindered ambitions to develop
these elements as vectors for germline transformation and insertional
mutagenesis. However, the apparent capability of salmonid TcEs for horizontal
transmission between two teleost orders (Ivics et al., 1996, supra) suggested
that
this particular subfamily of fish transposons might be transferred through
even
larger evolutionary distances.
Reconstructions of ancestral archetypal genes using parsimony analysis
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
17
have been reported (Jermann et al., 1995. Nature 374, S7-S9; Unnikrishnan et
al., 1996, Stewart, 1995 Nature 374, 12-13). However, such a strategy requires
vertical transmission of a gene through evolution for phylogenetically
backtracking to the root sequence. Because parsimony analysis could not
resolve
S the phylogenetic relationships between salmonid TcEs, the present invention
utilizes the approach of reconstructing a consensus sequence from inactive
elements belonging to the same subfamily of transposons. The resurrection of a
functional promoter of the L1 retrotransposon in mouse (Adey et al., 1994
Proc.
Natl. Acad. Sci. USA 91, 1 S69-1 S73) has previously been reported.
A strategy for obtaining an active gene is not without risks. The
consensus sequence of transposase pseudogenes from a single organism may
simply reflect the mutations that had occurred during vertical inactivation
that
have subsequently been fixed in the genome as a result of amplification of the
mutated element. For instance, most Tdrl elements isolated from zebrafish
1 S contain a conserved, 3 SO-by deletion in the transposase gene (Izsvak et
al., 1995,
supra). Therefore, their consensus is expected to encode an inactive element.
In
the present invention, because independent fixation of the same mutation in
different species is unlikely, a consensus from inactive elements of the same
subfamily of transposons from several organisms is derived to provide a
sequence for an active transposon.
Both the transposase coding regions and the inverted repeats (IRs) of
sahnonid-type TcEs accumulated several mutations, including point mutations,
deletions and insertions, and show about S% average pairwise divergence (Ivics
et al., 1996, supra). Example 1 describes the methods that were used to
2S reconstruct a transposase gene of the salinonid subfamily of fish elements
using
the accumulated phylogenetic data. This analysis is provided in the EMBL
database as DS30090 from FTP.EBLAC.AK in
directory/pub/databases/embl/align and the product of this analysis was a
consensus sequence for an inactive SB protein. All the elements that were
examined were inactive due to deletions and other mutations. A salmonid
transposase gene of the SB transposase family was created using PCR-
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
18
mutagenesis through the creation of 10 constructs as provided in Fig. 1 and
described in Example 1.
This sequence can then be modified further, as described here, to
produce active members of the SB protein family.
The SB protein typically recognizes nucleotide sequences located within
inverted repeats on a nucleic acid fragment and each inverted repeat includes
at
least one direct repeat. The gene transfer system of this aspect of the
invention,
therefore, comprises two components: a transposase and a cloned,
nonautonomous (i.e., non-self inserting) salmonid-type element or transposon
(referred to herein as a nucleic acid fragment having at least two inverted
repeats) that carries the inverted repeats of the transposon substrate DNA.
When
put together these two components provide active transposon activity. In use,
the transposase binds to the direct repeats in the inverted repeats and
promotes
integration of the intervening nucleic acid sequence into DNA of a cell
including
chromosomes and extra chromosomal DNA of fish as well as mammalian cells.
This transposon does not appear to exist in nature.
The transposase that was reconstructed using the methods of Example 1
represents one member of a family of proteins that can bind to the inverted
repeat region of a transposon to effect integration of the intervening nucleic
acid
sequence into DNA, preferably DNA in a cell. One example of the family of
proteins of this invention is provided as SEQ ID NO:1 (see Fig. 2B). This
family of proteins is referred to herein as SB proteins. The proteins of this
invention are provided as a schematic in Fig. lA. The proteins include, from
the
amino-terminus moving to the carboxy-terminus, a paired-like domain with
leucine zipper, one or more nuclear localizing domains (NLS) domains and a
catalytic domain including a DD(34)E box (i.e., a catalytic domain containing
two invariable aspartic acid residues, D(153} and D(244), and a glutamic acid
residue, E(279), the latter two separated by 43 amino acids) and a glycine-
rich
box as detailed in an example in Fig. 2. The SB family of proteins includes
the
protein having the amino acid sequence of SEQ ID NO: 1. Preferably, a
member of the SB family of proteins also includes proteins with an amino acid
sequence that shares at least an 80% amino acid identity to SEQ ID NO:1.
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
19
Amino acid identity is defined in the context of a homology comparison
between the member of the SB family of proteins and SEQ ID NO:1. The two
amino acid sequences are aligned in a way that maximizes the number of amino
acids that they have in common along the lengths of their sequences; gaps in
S either or both sequences are permitted in making the alignment in order to
maximize the number of shared amino acids, although the amino acids in each
sequence must nonetheless remain in their proper order. The percentage amino
acid identity is the higher of the following two numbers: (a) the number of
amino acids that the two polypeptides have in common within the alignment,
divided by the number of amino acids in the member of the SB family of
proteins, multiplied by 100; or (b) the number of amino acids that the two
polypeptides have in common within the alignment, divided by the number of
amino acids in the reference SB protein, i.e., SEQ ID NO:1, multiplied by 100.
Proteins of the SB family are transposases, that is, they are able to
catalyze the integration of nucleic acid into DNA of a cell. In addition, the
proteins of this invention are able to bind to the inverted repeat sequences
of
SEQ ID NOs:4-5 and direct repeat sequences (SEQ ID NOs:6-9) from a
transposon as well as a consensus direct repeat sequence (SEQ ID NO:10). The
SB proteins preferably have a molecular weight range of about 35 kD to about
40 kD on about a 10% SDS-polyacrylamide gel.
To create an active SB protein, suitable for further modification, a number of
chromosomal fiagments were sequenced and identified by their homology to the
zebrafish tlansposon-like sequence Tdrl, from eleven species of fish (Ivics et
al.,
1996, supra). Next these and other homologous sequences were compiled and
aligned. The sequences were identified in either GenBank or the EMBL database.
Others have suggested using parsimony analysis to arrive at a consensus
sequence
but in this case parsimony analysis could not resolve the phylogenetic
relationships
among the salmonid-type TcEs that had been compiled. A consensus transposon
was then engineered by changing selected nucleotides in codons to restore the
amino acids that were likely to be in that position. This strategy assumes
that the
most common amino acid in a given position is probably the original (active)
amino
acid for that locus. The consensus sequence was examined far sites at which it
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
appeared that C->T mutations had been fixed where deamination of SmC residues
may have occurred (which leads to C being converted to T which in tum can lead
to
the "repair" of the mismatched G residue to an A). In these instances, the
"majority-
rule" consensus sequence was not always used. Next various expected activities
of
5 the resurrected transposase were tested to ensure the accuracy of the
engineering.
The amino acid residues described herein employ either the single letter
amino acid designator or the three-letter abbreviation. Abbreviations used
herein are in keeping with the standard polypeptide nomenclature. All amino
acid residue sequences are represented herein by formulae with left-to-right
10 orientation in the conventional direction of amino-terminus to carboxy-
terminus.
Although particular amino acid sequences encoding the transposases of
this invention have been described, there are a variety of conservative
changes
that can be made to the amino acid sequence of the SB protein without altering
SB activity. These changes are termed conservative mutations, that is, an
amino
15 acid belonging to a grouping of amino acids having a particular size or
characteristic can be substituted for another amino acid, particularly in
regions
of the protein that are not associated with catalytic activity or DNA binding
activity, for example. Other amino acid sequences of the SB protein include
amino acid sequences containing conservative changes that do not significantly
20 alter the activity or binding characteristics of the resulting protein.
Substitutes
for an amino acid sequence may be selected from other members of the class to
which the amino acid belongs. For example, the nonpolar (hydrophobic) amino
acids include alanine, leucine, isoleucine, valine, proline, phenylalanine,
tryptophan, and tyrosine. The polar neutral amino acids include glycine,
serine,
threonine, cysteine, tyrosine, asparagine and glutamine. The positively
charged
(basic) amino acids include arginine, lysine and histidine. The negatively
charged (acidic) amino acids include aspartic acid and glutamic acid. Such
alterations are not expected to substantially affect apparent molecular weight
as
determined by polyacrylamide gel electrophoresis or isoelectric point.
Particularly preferred conservative substitutions include, but are not limited
to,
Lys for Arg and vice versa to maintain a positive charge; Glu for Asp and vice
versa to maintain a negative charge; Ser for Thr so that a free -OH is
maintained; and Gln for Asn to maintain a free NH2.
The SB protein has catalytic activity to mediate the transposition of a
nucleic acid fragment containing recognition sites that are recognized by the
SB
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98124348
21
protein. The source of the SB protein can be the protein introduced into a
cell,
or a nucleic acid introduced into the cell. The SB protein can be introduced
into
the cell as ribonucleic acid, including mRNA; as DNA present in the cell as
extrachromosomal DNA including, but not limited to, episomal DNA, as
S plasmid DNA, or as viral nucleic acid. In addition to a ribonucleotide
sequence
that is translated to yield a sequence of amino acids, an mRNA typically
includes a guanine added to the 5' end of the mRNA to form a 5' cap. The 5'
cap
region can be methylated at several locations as described by Lewin, B., Genes
VI, Oxford University Press, pp. 171-172 (1997). An mRNA also typically
includes a sequence of polyadenylic acid (i.e., a poly(A) tail) at the 3' end
of the
mRNA.
Further, DNA encoding the SB protein can be stably integrated into the
genome of the cell for constitutive or inducible expression. Where the SB
protein is introduced into the cell as nucleic acid, the SB encoding sequence
is
preferably operably linked to a promoter. There are a variety of promoters
that
could be used including, but not limited to, constitutive promoters, tissue-
specific promoters, inducible promoters, and the like. Promoters are
regulatory
signals that bind RNA polymerase in a cell to initiate transcription of a
downstream (3' direction) coding sequence. A DNA sequence is operably
linked to an expression control sequence, such as a promoter when the
expression control sequence controls and regulates the transcription and
translation of that DNA sequence. The term "operably linked" includes having
an appropriate start signal (e.g., ATG) in front of the DNA sequence to be
expressed and maintaining the correct reading frame to permit expression of
the
DNA sequence under the control of the expression control sequence to yield
production of the desired protein product.
One nucleic acid sequence encoding the SB protein is provided as SEQ
ID N0:3. In addition to the conservative changes discussed above that would
necessarily alter the SB-encoding nucleic acid sequence, there are other DNA
or
RNA sequences encoding an SB protein having the same amino acid sequence
as an SB protein such as SEQ ID N0:3, but which take advantage of the
degeneracy of the three letter codons used to specify a particular amino acid.
For example, it is well known in the art that the following RNA codons (and
therefore, the corresponding DNA codons, with a T substituted for a U) can be
used interchangeably to code for each specific amino acid:
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
22
Phenylalanine (Phe or F) UUU or UUC
Leucine (Leu or L) UUA, UUG, CUU, CUC, CUA or CUG
Isoleucine (Ile or I) AUU, AUC or AUA
Methionine (Met or M) AUG
Valine (Val or V) GUU, GUC, GUA, GUG
Serine (Ser or S) UCU, UCC, UCA, UCG, AGU,
AGC
Proline (Pro or P) CCU, CCC, CCA, CCG
Threonine (Thr or T) ACU, ACC, ACA, ACG
Alanine (Ala or A) GCU, GCG, GCA, GCC
Tyrosine (Tyr or Y) UAU or UAC
Histidine (His or H) CAU or CAC
Glutamine (Gln or Q) CAA or CAG
Asparagine (Asn or N) AAU or AAC
Lysine (Lys or K) AAA or AAG
Aspartic Acid (Asp or D) GAU or GAC
Glutamic Acid (Glu or E) GAA or GAG
Cysteine (Cys or C) UGU or UGC
Arginine (Arg or R) CGU, CGC, CGA, CGG, AGA,
AGC
Glycine {Gly or G) GGU or GGC or GGA or GGG
Termination codon UAA, UAG or UGA
Further, a particular DNA sequence can be modified to employ the
codons preferred for a particular cell type. For example, the preferred codon
usage for E. coli is known, as are preferred codon usages for animals and
humans. These changes are known to those of ordinary skill in the art and are
therefore considered part of this invention.
Also contemplated in this invention are antibodies directed to an SB
protein of this invention. An "antibody" for purposes of this invention is any
immunoglobulin, including antibodies and fragments thereof that specifically
binds to an SB protein. The antibodies can be polyclonal, monoclonal and
chimeric antibodies. Various methods are known in the art that can be used for
the production of polyclonal or monoclonal antibodies to SB protein. See, for
example, Antibodies: A Laboratory Manual, Harlow and Lane, eds., Cold
Spring Harbor Laboratory Press: Cold Spring Harbor, New York (1988).
Nucleic acid encoding the SB protein can be introduced into a cell as a
nucleic acid vector such as a plasmid, or as a gene expression vector,
including a
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
23
viral vector. The nucleic acid can be circular or linear. Methods for
manipulating DNA and protein are known in the art and are explained in detail
in the literature such as Sambrook et al, (1989) Molecular Cloning: A
Laboratory Manual., Cold Spring Harbor Laboratory Press or Ausubel, R.M.,
ed. (1994). Current Protocols in Molecular Biology. A vector, as used herein,
refers to a plasmid, a viral vector or a cosmid that can incorporate nucleic
acid
encoding the SB protein or the nucleic acid fragment of this invention. The
term
"coding sequence" or "open reading frame" refers to a region of nucleic acid
that
can be transcribed and/or translated into a polypeptide in vivo when placed
under the control of the appropriate regulatory sequences.
Another aspect of this invention relates to a nucleic acid fragment,
sometimes referred to as a transposon or transposon element, that includes a
nucleic acid sequence positioned between at least two inverted repeats. Each
inverted repeat preferably includes at least two direct repeats (hence, the
name
IR/DR). A direct repeat is typically between about 25 and about 35 base pairs
in
length, preferably about 29-31 base pairs in length. Notwithstanding the
above,
however, an inverted repeat can contain only one direct "repeat," in which
event
it is not actually a "repeat" but is nonetheless a nucleotide seqeunce having
at
least about 80% identity to a consensus direct repeat sequence as described
more
fully below. The transposon element is a linear nucleic acid fragment
(extending from the 5' end to the 3' end, by convention) that can be used as a
linear fragment or circularized, for example in a plasmid.
In a preferred embodiment of the transposon element, there are two
direct repeats in each inverted repeat sequence. The direct repeats (which
number, in this embodiment, four) have similar nucleotide sequences, as
described in more detail below. An inverted repeat on the 5' or "left" side of
a
nucleic acid fragment of this embodiment typically comprises a direct repeat
(i.e., a left outer repeat), an intervening region, and a second direct repeat
(i.e., a
left inner repeat). An inverted repeat on the 3' or "right" side of a nucleic
acid
fragment of this embodiment comprises a direct repeat (i.e., a right inner
repeat),
an intervening region, and a second direct repeat (i.e., a right outer
repeat).
Because they are inverted with respect to each other on the nucleic acid
fragment, the direct repeats in the 5' inverted repeat of the nucleic acid
fragment
are in a reverse orientation compared to the direct repeats in the 3' inverted
repeat of the nucleic acid fragment. The intervening region within an inverted
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
24
repeat is generally at least about 150 base pairs in length, preferably at
least
about 160 base pairs in length. The intervening region is preferably no
greater
than about 200 base pairs in length, more preferably no greater than about 180
base pairs in length. The nucleotide sequence of the intervening region of one
inverted repeat may or may not be similar to the nucleotide sequence of an
intervening region in another inverted repeat.
Most transposons have perfect inverted repeats, whereas the inverted
repeats that bind SB protein generally have at least about 80% to identity to
a
consensus direct repeat, preferably about 90% identity to a consensus direct
repeat. A preferred consensus direct repeat is 5'-
CAKTGRGTCRGAAGTTTACATACACTTAAG-3' (SEQ ID NO:10) where K
is G or T, and R is G or A. The presumed core binding site of SB protein is
nucleotides 4 through 22 of SEQ ID NO:10. Nucleotide identity is defined in
the context of a homology comparison between a direct repeat and SEQ ID
NO:10. The two nucleotide sequences are aligned in a way that maximizes the
number of nucleotides that they have in common along the lengths of their
sequences; gaps in either or both sequences are permitted in making the
alignment in order to maximize the number of shared nucleotides, although the
nucleotides in each sequence must nonetheless remain in their proper order.
The
percentage nucleotide identity is the higher of the following two numbers: (a)
the number of nucleotides that the two sequences have in common within the
alignment, divided by the number of nucleotides in the direct repeat,
multiplied
by 100; or (b) the number of nucleotides that the two sequences have in common
within the alignment, divided by the number of nucleotides in the reference
direct repeat, i.e., SEQ ID NO:10, multiplied by 100. Examples of direct
repeat
sequences that bind to SB protein include: a left outer repeat 5'-
GTTGAAGTCGGAAGTTTACATACACTTAG-3' (SEQ ID N0:6); a left inner
repeat 5'-CAGTGGGTCAGAAGTTTACATACACTAAGG-3' (SEQ ID
N0:7); a right inner repeat 5'-
TTAACTCACATACAATTGAAGACTGGGTGAC-3' (SEQ ID N0:8); and a
right outer repeat S'-GATTCCACATACATTTGAAGGCTAAGTTGA-3' (SEQ
ID N0:9). As written, the right side direct repeats (SEQ ID NOs:8 and 9) are
depicted as they would appear on the transposon, i.e., the nucleotides are in
a
reverse complement order when compared for homology to the nucleotide
sequence of the left side repeats (SEQ ID NOs:S and 6).
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
In one embodiment the direct repeat sequence includes at least the
following sequence: ACATACAC (SEQ ID NO:11 ).
One preferred inverted repeat sequence of this invention is SEQ ID
N0:4
5 5'-AGTTGAAGTC GGAAGTTTAC ATACACTTAA GTTGGAGTCA TTAAAACTCG
TTTTTCAACT ACACCACAAA TTTCTTGTTA ACAAACAATA GTTTTGGCAA
GTCAGTTAGG ACATCTACTT TGTGCATGAC ACAAGTCATT TTTCCAACAA
TTGTTTACAG ACAGATTATT TCACTTATAA TTCACTGTAT CACAATTCCA
GTGGGTCAGA AGTTTACATA CACTAA-3'
and another preferred inverted repeat sequence of this invention is SEQ ID
NO:S
5'-TTGAGTGTAT GTTAACTTCT GACCCACTGG GAATGTGATG AAAGAAATAA
AAGCTGAAAT GAATCATTCT CTCTACTATT ATTCTGATAT TTCACATTCT
TAAAATAAAG TGGTGATCCT AACTGACCTT AAGACAGGGA ATCTTTACTC
GGATTAAATG TCAGGAATTG TGAAAAAGTG AGTTTAAATG TATTTGGCTA
AGGTGTATGT AAACTTCCGA CTTCAACTG-3'.
The inverted repeat (SEQ ID NO:S) contains the poly{A) signal AATAAA at
nucleotides 104-109. This poly(A) signal can be utilized by a coding sequence
present in the nucleic acid fragment to result in addition of a poly(A) tail
to an
mRNA. The addition of a poly(A) tail to an mRNA typically results in increased
stability of that mRNA relative to the same mRNA without the poly(A) tail.
Preferably, the inverted repeat (SEQ ID NO:S) is present on the 3' or "right
side"
of a nucleic acid fragment that comprises two direct repeats in each inverted
repeat sequence.
The direct repeats are preferably the portion of the inverted repeat that
bind to the SB protein to permit insertion and integration of the nucleic acid
fragment into the cell. The site of DNA integration for the SB proteins occurs
at
TA base pairs (see Figure 7B).
The inverted repeats flank a nucleic acid sequence which is inserted into
the DNA in a cell. The nucleic acid sequence can include all or part of an
open
reading frame of a gene (i.e., that part of a gene encoding protein), one or
more
expression control sequences (i.e., regulatory regions in nucleic acid) alone
or
together with all or part of an open reading frame. Preferred expression
control
sequences include, but are not limited to promoters, enhancers, border control
elements, locus-control regions or silencers. In a preferred embodiment, the
nucleic acid sequence comprises a promoter operably linked to at least a
portion
CA 02309000 2000-OS-04
WO 99/25817 PCT/t3S98/24348
26
of an open reading frame.
As illustrated in the examples, the combination of the nucleic acid
fragment of this invention comprising a nucleic acid sequence positioned
between at least two inverted repeats wherein the inverted repeats can bind to
an
SB protein and wherein the nucleic acid fragment is capable of integrating
into
DNA in a cell, in combination with an SB protein (or nucleic acid encoding the
SB protein to deliver SB protein to a cell) results in the integration of the
nucleic
acid sequence into the cell. Alternatively, it is possible for the nucleic
acid
fragment of this invention to be incorporated into DNA in a cell through non-
homologous recombination through a variety of as yet undefined, but
reproducible mechanisms. In either event the nucleic acid fragment can be used
for gene transfer.
As described in the examples, the SB family of proteins mediates
integration in a variety of cell types and a variety of species. The SB
protein
facilitates integration of the nucleic acid fragment of this invention with
inverted
repeats into both pluripotent (i.e., a cell whose descendants can
differentiate into
several restricted cell types, such as hematopoietic stem cells or other stem
cells)
and totipotent cells (i.e., a cell whose descendants can become any cell type
in an
organism, e.g., embryonic stem cells). It is likely that the gene transfer
system of
this invention can be used in a variety of cells including animal cells,
bacteria,
fungi (e.g., yeast) or plants. Animal cells can be vertebrate or invertebrate.
Cells
such as oocytes, eggs, and one or more cells of an embryo are also considered
in
this invention. Mature cells from a variety of organs or tissues can receive
the
nucleic acid fragment of this invention separately, alone, or together with
the SB
protein or nucleic acid encoding the SB protein. Cells receiving the nucleic
acid
fragment or the SB protein and capable of receiving the nucleic acid fragment
into the DNA of that cell include, but are not limited to, lymphocytes,
hepatocytes, neural cells, muscle cells, a variety of blood cells, and a
variety of
cells of an organism. Example 4 provides methods for determining whether a
particular cell is amenable to gene transfer using this invention. The cells
can be
obtained from vertebrates or invertebrates. Preferred invertebrates include
crustaceans or mollusks including, but not limited to shrimp, scallops,
lobster,
clams, or oysters.
Vertebrate cells also incorporate the nucleic acid fragment of this
invention in the presence of the SB protein. Cells from fish, birds and other
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
27
animals can be used, as can cells from mammals including, but not limited to,
rodents, such as rats or mice, ungulates, such as cows or goats, sheep, swine
or
cells from a human.
The DNA of a cell that acts as a recipient of the nucleic acid fragment of
this invention includes any DNA in contact with the nucleic acid fragment of
this
invention in the presence of an SB protein. For example, the DNA can be part
of
the cell genome or it can be extrachromosomal, such as an episome, a plasmid,
a
circular or linear DNA fragment. Targets for integration are double-stranded
DNA.
The combination of the nucleic acid fragment of this invention including
a nucleic acid sequence positioned between at least two inverted repeats
wherein
the inverted repeats can bind to an SB protein and wherein the nucleic acid
fragment is capable of integrating into DNA of a cell and a transposase or
nucleic acid encoding a transposase, wherein the transposase is an SB protein,
including SB proteins that include an amino acid sequence that is at least
about
80% identical to SEQ ID NO:1 is useful as a gene transfer system to introduce
nucleic acid sequence into the DNA of a cell. In a preferred embodiment, the
SB
protein comprises the amino acid sequence of SEQ ID NO:1 and in another
preferred embodiment the DNA encoding the transposase can hybridize to the
DNA of SEQ ID N0:3 under the following hybridization conditions: in 30%
(v/v) forniamide in O.Sx SSC, 0.1% (w/v) SDS at 42°C for 7 hours.
Gene transfer vectors for gene therapy can be broadly classified as viral
vectors or non-viral vectors. The use of the nucleic acid fi~agment of this
invention as a transposon in combination with an SB protein represents a
tremendous advancement in the field of non-viral DNA-mediated gene transfer.
Up to the present time, viral vectors have been found to be more efficient at
introducing and expressing genes in cells. There are several reasons why non-
viral gene transfer is superior to virus-mediated gene transfer for the
development of new gene therapies. For example, adapting viruses as agents for
gene therapy restricts genetic design to the constraints of that virus genome
in
terms of size, structure and regulation of expression. Non-viral vectors are
generated largely from synthetic starting materials and are therefore more
easily
manufactured than viral vectors. Non-viral reagents are less likely to be
immunogenic than viral agents making repeat administration possible. Non-viral
vectors are more stable than viral vectors and therefore better suited for
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98124348
28
pharmaceutical formulation and application than are viral vectors.
Current non-viral gene transfer systems are not equipped to promote
integration of nucleic acid into the DNA of a cell, including host
chromosomes.
As a result, stable gene transfer frequencies using non-viral systems have
been
very low; 0.1% at best in tissue culture cells and much less in primary cells
and
tissues. The present system is a non-viral gene transfer system that
facilitates
integration and markedly improves the frequency of stable gene transfer.
In the gene transfer system of this invention the SB protein can be
introduced into the cell as a protein or as nucleic acid encoding the protein.
In
one embodiment the nucleic acid encoding the protein is RNA and in another,
the nucleic acid is DNA. Further, nucleic acid encoding the SB protein can be
incorporated into a cell through a viral vector, anionic or cationic lipid, or
other
standard transfection mechanisms including electroporation, particle
bombardment or microinjection used for eukaryotic cells. Following
introduction of nucleic acid encoding SB, the nucleic acid fragment of this
invention can be introduced into the same cell.
Similarly, the nucleic acid fragment can be introduced into the cell as a
linear fragment or as a circularized fragment, preferably as a plasmid or as
recombinant viral DNA. Preferably the nucleic acid sequence comprises at least
a portion of an open reading frame to produce an amino-acid containing
product.
In a preferred embodiment the nucleic acid sequence encodes at least one
protein
and includes at least one promoter selected to direct expression of the open
reading frame or coding region of the nucleic acid sequence. The protein
encoded by the nucleic acid sequence can be any of a variety of recombinant
proteins new or known in the art. In one embodiment the protein encoded by the
nucleic acid sequence is a marker protein such as GFP, chloramphenicol
acetyltransferase (CAT), f3-galactosidase (lack and luciferase (LUC). In
another embodiment, the protein encoded by the nucleic acid is a growth
hormone, for example to promote growth in a transgenic animal, or insulin-like
growth factors (IGFs).
In one embodiment of a transgenic animal, the protein encoded by the
nucleic acid fragment is a product for isolation from a cell. Transgenic
animals
as bioreactors are known. Protein can be produced in quantity in milk, urine,
blood or eggs. Promoters are known that promote expression in milk, urine,
blood or eggs and these include, but are not limited to, casein promoter, the
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
29
mouse urinary protein promoter, (3-globin promoter and the ovalbumin promoter
respectively. Recombinant growth hormone, recombinant insulin, and a variety
of other recombinant proteins have been produced using other methods for
producing protein in a cell. Nucleic acid encoding these or other proteins can
be
incorporated into the nucleic acid fragment of this invention and introduced
into
a cell. Efficient incorporation of the nucleic acid fragment into the DNA of a
cell occurs when an SB protein is present. Where the cell is part of a tissue
or
part of a transgenic animal, large amounts of recombinant protein can be
obtained. There are a variety of methods for producing transgenic animals for
research or for protein production including, but not limited to those
described
by Hackett et al. (The molecular biol~of transgenic fish. In Biochemistry and
Molecular Biology of Fishes (Hochachka & Mommsen, eds) Vol.2, pp., 207-240
(1993)). Other methods for producing transgenic animals include the teachings
of M. Markkula et al., Rev. Reprod., l, 97-106 (1996); R. T. Wall et al., J.
Dairy
1 S Sci., 80, 2213-2224 ( 1997); J. C. Dalton, et al., Adv. Exp. Med. Biol. ,
411, 419-
428 (1997); and H. Lubon et al., Transfus. Med. Rev.,10, 131-143 (1996).
Transgenic zebrafish were made, as described in Example 6. The system has
also been tested through the introduction of the nucleic acid with a marker
protein into mouse embryonic stem cells (ES) and it is known that these cells
can
be used to produce transgenic mice (A. Bradley et al., Nature, 309, 255-256
( 1984)).
In general, there are two methods to achieve improved stocks of
commercially important animals. The first is classical breeding, which has
worked
well for land animals, but it takes decades to make major changes. Controlled
breeding, growth rates in coho salmon (Oncorhynchus Icisutch) increased 60%
over
four generations and body weights of two strains of channel catfish (Ictalurus
punctatus) were increased 21 to 29% over three generations. The second method
is
genetic engineering, a selective process by which genes are introduced into
the
chromosomes of animals or plants to give these organisms a new trait or
characteristic, like improved growth or greater resistance to disease. The
results of
genetic engineering have exceeded those of breeding in some cases. In a single
generation, increases in body weight of 58% in common carp (C~primss carpio)
with
extra rainbow trout growth hormone I genes, more than 1000% in salmon with
extra
salmon growth hormone genes, and less in trout were obtained. The advantage of
CA 02309000 2000-OS-04
WO 99/Z5817 PCT/US98/24348
genetic engineering in fish, for example, is that an organism can be altered
directly in
a very short periods of time if the appropriate gene has been identified. The
disadvantage of genetic engineering in fish is that few of the many genes that
are
involved in growth and development have been identified and the interactions
of
5 their protein products is poorly understood. Procedures for genetic
manipulation are
lacking in many economically important animals. The present invention provides
an
efficient system for performing insertional mutagenesis (gene tagging) and
efficient
procedures for producing transgenic animals. Prior to this invention,
transgenic DNA
is not efficiently incorporated into chromosomes. Only about one in a million
of the
10 foreign DNA molecules integrates into the cellular genome, generally
several
cleavage cycles into development. Consequently, most transgenic animals are
mosaic. As a result, animals raised from embryos into which transgenic DNA has
been delivered must be cultured until gametes can be assayed for the presence
of
integrated foreign DNA. Many transgenic animals fail to express the transgene
due
15 to position effects. A simple, reliable procedure that directs early
integration of
exogenous DNA into the chromosomes of animals at the one-cell stage is needed.
The present system helps to fill this need, as described in more detail below.
The transposon system of this invention has applications to many areas of
biotechnology. Development of transposable elements for vectors in animals
20 permits the following: 1 ) efficient insertion of genetic material into
animal
chromosomes using the methods given in this application. 2) identification,
isolation,
and characterization of genes involved with growth and development through the
use of transposons as insertional mutagens (e.g., see Kaiser et al., 1995,
"Eukaryotic
transposable elements as tools to study gene structure and fi~nction." In
Mobile
25 Genetic Elements, IRL Press, pp. 69-100). 3) identification, isolation and
characterization of transcriptional regulatory sequences controlling growth
and
development. 4) use of marker constructs for quantitative trait loci (QTL)
analysis.
5) identification of genetic loci of economically important traits, besides
those for
growth and development, i.e., disease resistance (e.g., Anderson et al., 1996,
Mol.
30 Mar. Biol. Biotech., S, 105-113). In one example, the system of this
invention can be
used to produce sterile transgenic fish. Broodstock with inactivated genes
could be
mated to produce sterile offspring for either biological containment or for
maximizing growth rates in aquacultured fish.
In yet another use of the gene transfer system of this invention, the
nucleic acid fragment is modified to incorporate a gene to provide a gene
therapy
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
31
to a cell. The gene is placed under the control of a tissue specific promoter
or of
a ubiquitous promoter or one or more other expression control sequences for
the
expression of a gene in a cell in need of that gene. A variety of genes are
being
tested for a variety of gene therapies including, but not limited to, the CFTR
gene for cystic fibrosis, adenosine deaminase (ADA) for immune system
disorders,
factor IX globins and interleukin-2 (IZ;-2) genes for blood cell diseases,
alpha-1-
antitrypsin for lung disease, and tumor necrosis factors (TNFs),
phenylalanine/hydroxylase for PKU (phenylketouria), and multiple drug
resistance
(MDR) proteins for cancer therapies.
These and a variety of human or animal specific gene sequences
including gene sequences to encode marker proteins and a variety of
recombinant proteins are available in the known gene databases such as
GenBank, and the like.
Further, the gene transfer system of this invention can be used as part of a
process for working with or for screening a library of recombinant sequences,
for
example, to assess the function of the sequences or to screen for protein
expression, or to assess the effect of a particular protein or a particular
expression control sequence on a particular cell type. In this example, a
library
of recombinant sequences, such as the product of a combinatorial library or
the
product of gene shuffling, both techniques now known in the art and not the
focus of this invention, can be incorporated into the nucleic acid fragment of
this
invention to produce a library of nucleic acid fragments with varying nucleic
acid sequences positioned between constant inverted repeat sequences. The
library is then introduced into cells together with the SB protein as
discussed
above.
An advantage of this system is that it is not limited to a great extent by
the size of the intervening nucleic acid sequence positioned between the
inverted
repeats. The SB protein has been used to incorporate transposons ranging from
1.3 kilobases (kb) to about 5.0 kb and the mariner transposase has mobilized
transposons up to about 13 kb. There is no known limit on the size of the
nucleic acid sequence that can be incorporated into DNA of a cell using the SB
protein.
Rather, what is limiting can be the method by which the gene transfer
system of this invention is introduced into cells. For example, where
microinjection is used, there is very little restraint on the size of the
intervening
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
32
sequence of the nucleic acid fragment of this invention. Similarly, lipid-
mediated strategies do not have substantial size limitations. However, other
strategies for introducing the gene transfer system into a cell, such as viral-
mediated strategies could limit the length of the nucleic acid sequence
positioned
between their terminal repeats, according to this invention.
The two-part SB transposon system can be delivered to cells via viruses,
including retrovimses (including lentiviruses), adenoviruses, adeno-associated
viruses,
herpesviruses, and others. There are several potential combinations of
delivery
mechanisms for the transposon portion containing the transgene of interest
flanked by
the inverted terminal repeats (IRs) and the gene encoding the transposase. For
example,
both the transposon and the transposase gene can be contained together on the
same
recombinant viral genome; a single infection delivers both parts of the SB
system such
that expression of the transposase then directs cleavage of the transposon
from the
recombinant viral genome for subsequent integration into a cellular
chromosome. In
another example, the transposase and the transposon can be delivered
separately by a
combination of viruses and/or non-viral systems such as lipid-containing
reagents. In
these cases either the transposon and/or the transposase gene can be delivered
by a
recombinant virus. In every case, the expressed transposase gene directs
liberation of
the tiansposon from its carrier DNA (viral genome) for integration into
chromosomal
DNA.
This invention also relates to methods for using the gene transfer system
of this invention. In one method, the invention relates to the introduction of
a
nucleic acid fragment comprising a nucleic acid sequence positioned between at
least two inverted repeats into a cell. In a preferred embodiment, efficient
incorporation of the nucleic acid fragment into the DNA of a cell occurs when
the cell also contains an SB protein. As discussed above, the SB protein can
be
provided to the cell as SB protein or as nucleic acid encoding the SB protein.
Nucleic acid encoding the SB protein can take the form of RNA or DNA. The
protein can be introduced into the cell alone or in a vector, such as a
plasmid or a
viral vector. Further, the nucleic acid encoding the SB protein can be stabiy
or
transiently incorporated into the genome of the cell to facilitate temporary
or
prolonged expression of the SB protein in the cell. Further, promoters or
other
expression control sequences can be operably linked with the nucleic acid
encoding the SB protein to regulate expression of the protein in a
quantitative or
in a tissue-specific manner. As discussed above, the SB protein is a member of
a
*rB
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
33
family of SB proteins preferably having at least an 80% amino acid sequence
identity to SEQ ID NO:1 and more preferably at least a 90% amino acid
sequence identity to SEQ ID NO:1. Further, the SB protein contains a DNA-
binding domain, a catalytic domain (having transposase activity) and an NLS
signal.
The nucleic acid fragment of this invention is introduced into one or
more cells using any of a variety of techniques known in the art such as, but
not
limited to, microinjection, combining the nucleic acid fragment with lipid
vesicles, such as anionic or cationic lipid vesicles, particle bombardment,
electroporation, microinjection, DNA condensing reagents (e.g., calcium
phosphate, polylysine or polyethyleneimine) or incorporating the nucleic acid
fragment into a viral vector and contacting the viral vector with the cell.
Where
a viral vector is used, the viral vector can include any of a variety of viral
vectors
known in the art including viral vectors selected from the group consisting of
a
1 S retroviral vector, an adenovirus vector or an adeno-associated viral
vector.
The gene transfer system of this invention can readily be used to produce
transgenic animals that carry a particular marker or express a particular
protein
in one or more cells of the animal. Methods for producing transgenic animals
are known in the art and the incorporation of the gene transfer system of this
invention into these techniques does not require undue experimentation. The
examples provided below teach methods for creating transgenic fish by
microinjecting the gene transfer system into a cell of an embryo of the fish.
Further, the examples also describe a method for introducing the gene transfer
system into mouse embryonic stem cells. Methods for producing transgenic
mice from embryonic stem cells are well known in the art. Further a review of
the production of biopharmaceutical proteins in the milk of transgenic dairy
animals (see Young et al., BIO PHARM (1997), 10, 34-38) and the references
provided therein detail methods and strategies for producing recombinant
proteins in milk. The methods and the gene transfer system of this invention
can
be readily incorporated into these transgenic techniques without undue
experimentation in view of what is known in the art and particularly in view
of
this disclosure.
The nucleic acid fragments of this invention in combination with the SB
protein or nucleic acid encoding the SB protein is a powerful tool for
germline
transformation, for the production of transgenic animals, as methods for
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
34
introducing nucleic acid into DNA in a cell, for insertional mutagenesis, and
for
gene-tagging in a variety of species. Two strategies are diagramed in Figure
9.
Due to their inherent ability to move from one chromosomal location to
another within and between genomes, transposable elements have been exploited
as genetic vectors for genetic manipulations in several organisms. Transposon-
tagging is a technique in which transposons are mobilized to "hop" into genes,
thereby inactivating them by insertional mutagenesis. These methods are
discussed by Evans et al., (TIG, 13, 370-374 (1997)). In the process, the
inactivated genes are "tagged" by the transposable element which then can be
used to recover the mutated allele. The ability of the human and other genome
projects to acquire gene sequence data has outpaced the ability of scientists
to
ascribe biological function to the new genes. Therefore, the present invention
provides an efficient method for introducing a tag into the genome of a cell.
Where the tag is inserted into a location in the cell that disrupts expression
of a
protein that is associated with a particular phenotype, expression of an
altered
phenotype in a cell containing the nucleic acid of this invention permits the
association of a particular phenotype with a particular gene that has been
disrupted by the nucleic acid fragment of this invention. Here the nucleic
acid
fragment functions as a tag. Primers designed to sequence the genomic DNA
flanking the nucleic acid fragment of this invention can be used to obtain
sequence information about the disrupted gene.
In another application of this invention, the invention provides a method
for mobilizing a nucleic acid sequence in a cell. In this method the nucleic
acid
fragment of this invention is incorporated into DNA in a cell, as provided in
the
discussion above. Additional SB protein or nucleic acid encoding the SB
protein
is introduced into the cell and the protein is able to mobilize (i.e. move)
the
nucleic acid fragment from a first position within the DNA of the cell to a
second position within the DNA of the cell. The DNA of the cell can be
chromosomal DNA or extrachromosomal DNA. The term "genomic DNA" is
used herein to include both chromosomal DNA and extrachromosomal DNA.
The method permits the movement of the nucleic acid fragment from one
location in the genome to another location in the genome, or for example, from
a
plasmid in a cell to the genome of that cell.
Additional modifications of the transposable elements disclosed herein
can further increase the e~ciency of insertion of genetic material into animal
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
chromosomes so as to allow the identification, isolation, and characterization
of genes involved with growth, development and disease, and the
identification, isolation and characterization of transcriptional regulatory
sequences controlling growth, development and disease. Examples of the types
5 of modifications that can be made to the transposable elements disclosed
herein
include the construction of transposable elements taking the form of
expression
con~ol sequence-trap transposon vectors, gene-trap transposon vectors, and
dicistronic gene expression transposon vectors.
In a preferred embodiment of the nucleic acid fragment of the invention,
10 the nucleic acid sequence that is flanked by the inverted repeats
(sometimes
referred to herein as the "intervening nucleic acid sequence") comprises at
least
one coding sequence. In an embodiment that is particularly suited for use in
functional genomic analysis as well as gene discovery, the coding sequence
encodes a detectable and/or selectable marker. For ease of reference, a coding
15 sequence that encodes a detectable and/or selectable marker will be
referred to as
a "detectable marker coding sequence," however it is to be understood that
this
coding sequence can encode any type of detectable or selectable marker, or a
protein that activates a detectable or selectable marker supplied in traps or
in cis.
An example of a selectable marker is neomycin. Preferred detectable markers
20 include luciferase,13-galactosidase, fluorescent proteins, chloramphenicol
acetyl
transferase (CAT) and other exogenous proteins detectable by their
fluorescence,
enzymatic activity or immunological properties. Non-limiting examples of
fluorescent proteins include GFP, Yellow Fluorescent Protein and Blue
Fluorescent Protein. Typically, a detectable marker coding sequence is
operably
25 linked to a poly(A) signal that is present 3' to the detectable marker
coding
sequence. Useful activators of detectable markers supplied in traps or in cis
(see, e.g., Fig. 12(B)) include those that can bind to specific promoters and
cause
the transcription of a coding sequence operably linked to the promoter. In
embodiments of the method of the invention that utilize activatable detectable
30 markers, the cells into which the nucleic acid fragment is introduced
preferably
also contain a detectable marker coding sequence operably linked to a promoter
that can be activated by an activator protein. An example of a protein encoded
by a detectable marker coding sequence of an expression control sequence-trap
vector or a gene-trap vector is the traps-acting activator protein tTA
{tetracycline
35 controlled transactivator) (Clontech, Palo Alto, CA), which interacts with
a
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
36
tetracycline response element to which a detectable marker coding sequence is
operably linked.
Optionally, the intervening nucleic acid sequence of the nucleic acid
fragment of the invention further comprises at least one expression control
sequence that is operably linked to the detectable marker coding sequence. In
one preferred embodiment, the expression control sequence comprises a
promoter, more preferably a weak promoter. As used herein, the terms "weak
promoter" or "minimal promoter" refer to a promoter that by itself does not
have the ability to direct high expression of the coding sequence to which it
is
operably linked. However, when the nucleic acid fragment inserts into a cell's
genomic DNA so that the weak promoter is operably linked to at least one
expression control sequence already present in the cell's DNA, preferably at
least one of which is an enhancer (see, for instance, Fig. 11), the weak
promoter can direct the expression of the detectable marker coding sequence in
1 S tissues in which the enhancer is active and at levels higher than the weak
promoter would direct expression when not operably linked to the enhancer. An
enhancer is a cis-acting nucleotide sequence that generally increases the
activity
of promoters and typically can function in either orientation and either
upstream
or downstream of a promoter. Examples of suitable weak promoters useful in
vertebrate cells are the promoter for the carp 13-actin coding sequence (Liu
et al.,
BioTechnol., 8_, 1268-1272 (1990); (Caldovic, L., et al., Mol Mar Biol
Biotechnol., 4, 51-61 (1995)), and the Herpes Simplex Virus thymidine kinase
promoter.
The invention includes a method for using the nucleic acid fragment of
the invention to identify or "trap" expression control sequences present in
genomic DNA. Preferably, the coding sequence of the nucleic acid fragment
encodes a detectable marker and is operably linked to at least one expression
control sequence present in the nucleic acid sequence of the nucleic acid
fragment. The detectable marker is preferably a fluorescent protein or a
selectable marker. In a nucleic fragment especially well-suited for this use,
the
intervening nucleic acid sequence comprises a detectable marker coding
sequence operably linked to a promoter, preferably a weak promoter. In the
method of the invention, an expression control sequence-trap transposon vector
comprising the nucleic acid fragment is introduced into a cell, preferably
along
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
37
with a source of transposase, such that the nucleic acid fragment inserts into
the
DNA of the cell. The transposase source can be a nucleic acid and/or a protein
as described in detail hereinbelow. For instance, a vector containing the
nucleic
acid fragment can contain a second coding sequence encoding a transposase. In
another aspect, the cell can contain a coding sequence that encodes an SB
transposase. Alternatively, an mRNA encoding an SB transposase or an SB
transposase itself can be introduced into the cell.
The nucleic acid fragment can insert within a coding sequence present in
a cell's DNA that can result in the insertional inactivation of that coding
sequence, or the nucleic acid fragment can insert into DNA outside of a coding
sequence. Either type of insertion can result in expression of the detectable
marker provided the nucleic acid fragment inserts near an appropriate
expression control sequence. Preferably, the nucleic acid fragment integrates
into the DNA of the cell or its progeny within a domain that contains an
expression control sequence, more preferably an enhancer. It is possible that
the
nucleic acid fragment of this embodiment will insert in-frame into a coding
sequence in a cell's DNA and is expressed by virtue of the endogenous promoter
and not the weak promoter. When this happens, the nucleic acid fragment will
be operating as a gene trap.
The nucleic acid fragment comprising a detectable marker operably
linked to a weak promoter can be used to detect the presence of an expression
control sequence that regulates the expression of the promoter. Preferably,
enhancers are detected. As enhancers activate promoters located within the
same
domain defined by border elements as the enhancer, the expression of the
detectable marker generally indicates that the nucleic acid fragment has
inserted
within the same domain as an enhancer.
Expression control sequences can be detected in accordance with the
invention in any type of cell, without limitation. Preferred cells are
pluripotent
or totipotent cells, including an oocyte, a cell of an embryo, an egg and a
stem
cell. However, cells can be derived from any type of tissue, differentiated or
undifferentiated. Cells from fish, birds and other animals can be used, as can
cells from mammals including, but not limited to, rodents, such as rats or
mice,
ungulates, such as cows or goats, sheep, swine or cells from a human.
It is possible for enhancers to be active only at specific times or specific
tissues within an animal. Thus, evaluation of expression of the detectable
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
38
marker encoded by an inserted nucleic acid fragment in an animal can result in
identification of enhancers that have distinct spatial and/or temporal
expression.
For instance, detection of the detectable marker only at specific times during
the
cell cycle or during development of the animal indicates that the enhancer is
active only at specific times (i.e., developmental stage-specific expression).
Detection of the detectable marker only in specific tissues of the whole
animal
indicates that the trapped enhancer is a tissue-specific enhancer.
Preferably the cells are grown into an animal and the cells assayed for
expression of the detectable marker are present in an animal. Thus, cells that
can
be detected include progeny of a cell that contain the nucleic acid fragment
comprising the detectable marker coding sequence. The animal can be an
embryo, an adult, or at a developmental phase between embryo and adult.
Preferably the animal is an embryo. Expression of the detectable marker in the
animal can be assayed by methods known to the art. For instance, assay of 13-
I S galactosidase expression or immunological detection of a foreign protein
like
CAT can be used. Another example of evaluating expression of a detectable
marker in an embryo is the expression of fluorescent proteins in the optically
clear zebrafish embryo.
Optionally the expression control sequence detection method includes
observing at least one phenotype of a cell that contains the integrated
nucleic
acid fragment, and comparing it to a cell that does not contain the nucleic
acid
fragment to determine whether the phenotype of the first cell is altered. An
altered phenotype can be detected by methods known to the art. Alternatively,
the cell that contains the integrated nucleic acid fragment can be grown into
an
animal, and animal phenotypes similarly compared.
The method can be used to make a transgenic animal having tissue-
specific expression of a preselected coding sequence. For instance, a first
transgenic animal can be produced that contains an expression control sequence-
trap that is expressed in a particular tissue, and the detectable marker
coding
sequence encodes a trans-acting activator. A second and independent transgenic
animal can be produced that contains a preselected coding sequence that is
operably linked to a promoter that is activated by the activator encoded by
the
expression control sequence-trap that is present in the first transgenic
animal.
Crossing the two transgenic animals can result in transgenic progeny that
contain
i) the expression control sequence-trap that is expressed in a particular
tissue and
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
39
ii) the preselected coding sequence operably linked to a promoter that is
activated by the activator encoded by the expression control sequence-trap.
Tissue-specific expression of the activator protein will cause tissue-specific
expression of the preselected coding sequence. This aspect of the invention is
particularly useful in those animals where tissue-specific promoters have not
yet
been identified.
To obtain information about the location in the cell genome into which
the nucleic acid fragment has inserted, the method optionally includes
cleaving
the DNA of the cell with a restriction endonuclease capable of cleaving at a
restriction site within the intervening nucleic acid sequence of the nucleic
acid
fragment to yield at least one restriction fragment containing at least a
portion of
the integrated nucleic acid fragment, which portion comprises at least a
portion
of an inverted repeat sequence along with an amount of genomic DNA of the
cell, which genomic DNA is adjacent to the inverted repeat sequence. The
1 S specificities of numerous endonucleases are well known and can be found in
a
variety of publications, e.g. Sambrook et al.; Molecular Cloning: A Laboratory
Manual; Cold Spring Harbor Laboratory: New York (1989). The intervening
nucleic acid sequence thus preferably includes a restriction endonuclease
recognition site, preferably a 6-base recognition sequence. Following
integration
of the nucleic acid fragment into the cell DNA, the cell DNA is isolated and
digested with the restriction endonuclease. Where a restriction endonuclease
is
used that employs a 6-base recognition sequence, the cell DNA is cut into
about
4000- base pair restriction fragments on average. Since the site of DNA
integration mediated by the SB proteins generally occurs at TA base pairs and
the TA base pairs are typically duplicated such that an integrated nucleic
acid
fragment is flanked by TA base pairs, TA base pairs will be immediately
adjacent to an integrated nucleic acid fragment. The genomic DNA of the
genomic fragment is typically immediately adjacent to the TA base pairs on
either side of the integrated nucleic acid fragment.
After the DNA of the cell is digested, the genomic fragments can be
cloned in a vector using methods well known to the art allowing individual
clones containing genomic fragments comprising at least a portion of the
integrated nucleic acid fragment and genomic DNA of the cell adjacent to the
inserted nucleic acid fragment to be identified. A non-limiting example of
identifying the desired genomic fragments include hybridization with a probe
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
complementary to the sequence of the inverted repeats. Alternatively, linkers
can be added to the ends of the digested fragments to provide complementary
sequence for PCR primers. Where linkers are added, PCR reactions are used to
amplify fragments using primers from the linkers and primers binding to a
nucleotide sequence within the inverted repeats.
Nucleotide sequences of the genomic DNA on either or both sides of the
inserted nucleic acid fragment (i.e., flanking the inverse repeats) can be
determined by nucleotide sequencing using methods well known to the art. The
resulting nucleotide sequences are then used to search computer databases such
10 as GenBank or EMBL for related sequences; if the nucleotide sequences
encode
a putative protein, the encoded amino acid sequences can also be used to
search
protein data bases such as SwissProt for related or homologous polypeptide
sequences.
Alternatively, the restriction endonuclease used to cleave the cell DNA is
15 one that is incapable of cleaving the nucleic acid sequence of the nucleic
acid
fragment. Non-limiting examples of characterizing the resulting restriction
fragments include adding linkers to the ends of the digested fragments to
provide
complementary sequences for PCR primers described above or for inverse PCR.
For instance, to identify fragments that contain nucleotides on either or both
20 sides of the inserted nucleic acid fragment using inverse PCR, genomic DNA
is isolated from cells that express a detectable marker such as GFP or show a
consequential phenotypic response after mutagenesis with a transposon of the
present invention (see, e.g., Fig. 16). The DNA is then cleaved with one or
more
restriction endonucleases that cut outside of the transposon and the resulting
25 fragments of DNA are circularized using DNA ligase. About one in a million
genomic fragments may contain the transposon. The genomic sequence can then
be PCR amplified in two steps. The first PCR amplification uses the P2
external
primers IR/DR(L)-p2 CCACAGGTACACCTCCAATTGACTC (SEQ ID
N0:72) and IR/DR(R)-P2 GTGGTGATCCTAACTGACCTTAAGAC (SEQ ID
30 N0:73). Following 10-15 cycles of amplification, the products of round 1 of
amplification are reamplified using internal P 1 primers that further augment
the
number of copies of the interrupted genetic sequence. The internal primers are
IR/DR(L)-pl GTGTCATGCACAAAGTAGATGTCC (SEQ ID N0:74) and
IR/DR(R)-P1 CTCGGATTAAATGTCAGGAATTGTG (SEQ ID N0:75).
35 Primers P1 and P2 are complementary to sequences within the DR elements of
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
41
the SB transposon. The amplified DNA sequences are isolated for sequencing
and/or other analysis, and nucleotide sequences of the genomic DNA on either
or
both sides of the inserted nucleic acid fragment can thus be determined.
In another preferred embodiment of the nucleic acid fragment of the
invention comprising a coding sequence operably linked to at least one
expression control sequence, the intervening nucleic acid sequence includes a
splice acceptor site and/or an internal ribosomal entry site (IRES), each of
these
expression control sequences being operably linked to the coding sequence,
preferably a detectable marker coding sequence. Preferably the intervening
nucleic acid sequence comprises both a splice acceptor site and an IRES, and
the
IRES is positioned between the splice acceptor site and the detectable marker
coding sequence so as to ultimately permit ribosome binding to the detectable
marker mRNA and thereby initiate translation of the detectable marker
nucleotide sequence (see, for instance, Fig. 12(A), 12(B)). In this regard,
the
1 S splice acceptor site and/or an IRES are considered operably linked to a
coding
sequence when the splice acceptor site and/or the IRES is located 5' of the
detectable marker coding sequence and is present in an mRNA containing the
detectable marker coding sequence prior to processing of the mRNA.
Preferably, the splice acceptor site is located 5' to the IRES, and the IRES
is
located S' to the coding sequence to which a splice acceptor site and an IRES
are
operably linked.
The splice acceptor site acts to provide signals to target the sequences 3'
to, i.e., following, the splice acceptor site, including the detectable marker
coding sequence, to be present in the mRNA containing the detectable marker
coding sequence provided there is an intron upstream of the splice acceptor
site
(Padgett, T., et al., Am. Rev. Biochem. J., 55, 1119-1150 (1988)). Typically,
a
splice acceptor site includes a branch site and a 3' splice site. The
consensus
sequence of a branch site is typically a nucleotide sequence S'-PyBO N PY~
PyB, Pyg~
P"~s A P~,s, where PY is T or C, P" is A or G, and the subscripted number is
the
approximate percent occurrence of the appropriate nucleotide (see, for
instance,
Lewin, B., Genes VI, Oxford University Press, pp. 891-893 (1997)). The branch
site is typically located 10 to 60 nucleotides 5' to the splice site,
preferably 15 to
50 nucleotides 5' to the splice site. The 3' splice site is typically the
nucleotide
sequence CssAG, where the subscripted number is the percent occurrence of the
C, and the intron is cleaved after the G. Preferably the splice acceptor site
is
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
42
derived from the 3' end (i.e., the splice acceptor end) of the first intron
(intron A)
of the Li-actin coding sequence of carp (Liu, Z., et al., DNA Sequence -J. DNA
Sequencing and Mapping, Vol. 1, pp. 125-136 (1990)). Preferably nucleotides
1335-1571 of the nucleotides sequence available at GenBank Accession No.
M24113, more preferably nucleotides 1485-1571, are particularly suitable for
use in the present invention.
The maximum distance between splice acceptor site and IRES is
unknown. However, the overall size of the nucleic acid fragment can have an
effect on the efficiency of transposition of the nucleic acid fragment. For
instance, the SB protein has been used to incorporate transposons ranging from
1.3 kilobases (kb) to about 6.0 kb and the mariner transposase has mobilized
transposons up to about 13 kb. There is no known limit on the size of the
nucleic acid sequence that can be incorporated into DNA of a cell using the SB
protein. The IRES is typically positioned within about 0 to 7 bases of the
translation initiation codon, e.g., ATG, of the coding sequence to which the
IRES is operably linked. Typically, an IRES contains at least two translation
initiation codons. Preferably, the IRES includes at least one translation
initiation
codon, and the IRES is ligated to the translation coding region such that an
IRES
translation initiation codon replaces the translation initiation codon of the
coding
sequence. An IRES allows ribosomal access to mRNA without a requirement
for cap recognition and subsequent scanning to the initiator AUG (Pelletier,
J.A.,
et al., Nature, 334, 320-325 (1988)). An IRES that can be used in the
invention
typically includes a viral IRES, preferably a picornavirus IRES, poliovirus
IRES,
mengovirus IRES, or EMCV IRES, more preferably a poliovirus IRES,
mengovirus IRES, or EMCV IRES, and most preferably an EMCV IRES. An
example of an EMCV IRES that can be used in the invention is nucleotides 234-
848 of the nucleotide sequence available at GenBank Accession No. M81861. In
some embodiments nucleotides 827-831 (GATA) are replaced with TGCT. This
615 base pair nucleotide sequence contains ATG codons at nucleotides 834-836
and 846-848. Typically, the ATG codon at nucleotides 834-836 is used as the
translation initiation codon by the ribosome and a coding sequence (for
instance
a GFP coding sequence) can be fused to this ATG codon. However, in some
embodiments it is preferable to fuse a coding sequence (for instance a GFP
coding sequence) to the EMCV IRES so that the first codon of the coding
sequence, i.e., the ATG codon,replaces the ATG codon at nucleotides 846-848 of
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
43
the EMCV IRES. This typically results in the fused coding sequence beginning
with the amino acid sequence MATT (SEQ ID N0:70), which is the sequence
encoded by nucleotides 834-845 of the EMCV 1RES.
Although the coding sequence included in the intervening nucleic acid
sequence of the nucleic acid fragment of the invention typically contains a
polyadenylation signal, it need not. In embodiments of the nucleic acid
fragment containing a coding sequence that does not include a polyadenylation
signal, the detectable marker coding sequence is preferably operably linked to
a promoter located S' to the coding sequence and a splice donor site located
3'
of the coding sequence. When the nucleic acid fragment inserts into a region
of genomic DNA that is not an exon of a genomic coding sequence, the
resulting mRNA will generally be unstable due to the lack of a poly(A) tail.
However, when the nucleic acid fragment of this aspect of the invention
inserts
into an exon and the exon is part of a coding sequence is then expressed, the
mRNA containing the detectable marker may be stabilized when the splice
donor splices with a downstream exon that encodes a poly(A) tail. This is
known as a poly(A) trap.
The invention includes a method for using the nucleic acid fragment of
the invention to identify or "trap" coding sequences present in genomic DNA,
i.e. a "gene trap" transposon method that allows for gene discovery and
functional analysis. Insertion of a transposon into genomic DNA can interrupt
or mutate a genomic coding sequence. When a genomic coding sequence
present in a cell is interrupted, and the detectable marker coding sequence is
inserted in just the right way (in the correct direction, in-frame, and in an
exon
of the interrupted coding sequence), typically the detectable marker coding
sequence is expressed spatially and temporally in the same way as the
interrupted genomic coding sequence is expressed when not interrupted. This
aspect of the invention can be used, for example, in gene discovery by
providing
for a method to insert a nucleic acid fragment into genomic DNA so that a
genomic coding sequence no longer expresses a functional product, i.e., the
insertion results in a loss-of function mutation. Successful utilization of
the
transposon-derived vectors in the gene-trap and enhancer-trap methods of the
invention without further modification was surprising in view of the
possibility
that the IR/DR sequences might contain cryptic promoter or splicing signals
that
would have interfered with the use of these vectors.
*rB
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
44
A genomic coding sequence in a cell's DNA can be identified according
to the present invention by introducing a nucleic acid fragment comprising a
coding sequence, preferably a detectable marker coding sequence, into a cell,
preferably along with a source of transposase as described above, then
detecting
the detectable marker in the cell or its progeny. Preferably, the intervening
nucleic acid sequence of the nucleic acid fragment includes a splice acceptor
site
and/or an IRES, each of which is operably linked to the coding sequence. As
previously noted, the IRES is preferably located between the splice acceptor
site
and the detectable marker coding sequence. Additionally, the detectable
marker coding sequence is preferably not operably linked to a promoter. The
use of a splice acceptor site and an internal ribosome binding site operably
linked to the detectable marker coding sequence expands the probability that
the detectable marker coding sequence will be expressed when inserted into a
genomic coding sequence: it is possible to get expression of the detectable
marker coding sequence even if the transposon integrates in an intron or if it
integrates out of frame with respect to the interrupted genomic coding
sequence. Detection of the detectable marker in the cell or in progeny of the
cell containing the nucleic acid fragment is indicative that the nucleic acid
fragment has integrated within a genomic coding sequence of the cell.
Genomic coding sequences can be detected in any type of cell as generally
described above, including but not limited to an oocyte, a cell of an embryo,
an
egg cell or a stem cell, and in any type of tissue, differentiated or
undifferentiated. Preferably, the detectable marker is expressed spatially and
temporally in the same way as the genomic coding sequence is expressed when
not interrupted.
Optionally the genomic coding sequence detection method includes
observing at least one phenotype of a cell that contains the integrated
nucleic
acid fragment, and comparing it to a cell that does not contain the nucleic
acid
fragment to determine whether the phenotype of the first cell is altered.
Alternatively, the cell that contains the integrated nucleic acid fragment can
be
grown into an animal, and animal phenotypes similarly compared. Additionally,
the method optionally comprises cleaving the DNA of the cell with a
restriction
endonuclease to yield at least one restriction fragment containing at least a
portion of the integrated nucleic acid fragment, which portion comprises an
inverted repeat sequence along with an amount of genomic DNA of the cell,
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
which genomic DNA is adjacent to the inverted repeat sequence. The
intervening nucleic acid sequence thus preferably includes a restriction
endonuclease recognition site, as described above in connection with the
expression control region detection method. Restriction fragments containing
5 portions of the inverted repeats and genomic DNA are sequenced, and the DNA
flanking the inverted repeats and/or the amino acid sequences encoded thereby
are used to search computer databases such as GenBank or SwissProt.
In yet another preferred embodiment of the nucleic acid fragment of the
invention, the intervening nucleic acid sequence comprises a coding sequence,
10 preferably a detectable marker coding sequence, and a second coding
sequence
located 5', i.e., upstream, of the detectable marker coding sequence. The
detectable marker coding sequence typically is not operably linked to a
promoter.
Preferably the intervening nucleic acid sequence further comprises an IRES
located between the detectable marker coding sequence and the second coding
1 S sequence, wherein the IRES is operably linked to the detectable marker
coding
sequence (see, for instance, Fig. 1 S). Optionally, the second coding sequence
is
operably linked to at least one expression control sequence. The expression
control sequence to which the second coding sequence is optionally operably
linked can include a splice acceptor site, an IRES or a promoter, preferably a
20 promoter.
For reference, this second coding sequence is referred to as an "analyte
coding sequence." The analyte coding sequence can include any coding
sequence of interest including, for instance, a randomly inserted coding
sequence
from a library of DNA fragments or a preselected coding sequence. The nucleic
25 acid sequence comprising the analyte coding sequence preferably includes at
least one expression control sequence, including but not limited to expression
control sequences that are associated with the analyte coding sequence in its
wild
type or native state, i.e., those expression control sequences operably linked
to
the coding sequence as it naturally exists in a cell. Preferably, at least one
of the
30 expression control sequences is a promoter. Useful promoters include
constitutive and inducible promoters. Alternatively, the promoter can be the
native promoter, i.e., the promoter that is normally operably linked to the
analyte
coding region. The detectable marker coding sequence can be operably linked
to a splice acceptor site and/or an IRES. Preferably, in this aspect of the
35 invention the detectable marker coding sequence is operably linked to an
IRES
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
46
{see, e.g., Fig. 15).
The analyte coding sequence can encode a protein that is biologically
active, thereby allowing, for example, the evaluation andlor verification of
the
function of coding sequences and/or their protein products, as well as mutant
rescue and transgenic analysis. Generally, insertion of a dicistronic vector
that
has an analyte coding sequence that encodes a biologically active protein can
cause a gain-of function mutation.
Alternatively, the analyte coding sequence can encode a protein that is
incapable of performing the function of the wild-type, i.e., native, protein.
This
type of protein is typically inactive by virtue of an amino acid sequence
altered
relative to the native protein and can be used for the functional analysis of
proteins using, for example, dominant-negative mutant analysis.
The nucleic acid sequence of this aspect of the invention can encode two
mRNAs, one encoded by the detectable marker coding sequence and a second
mRNA encoded by the analyte coding sequence. Preferably, the nucleic acid
sequence of this aspect of the invention encodes one mRNA that includes two
coding sequences, i.e., a dicistronic mRNA. While not intended to be limiting,
a
dicistronic vector of this aspect of the invention generally provides for the
expression of a detectable marker coding sequence primarily when the analyte
coding sequence is expressed.
The invention includes a method for identifying or analyzing the function
of an analyte coding sequence that involves introducing into a host cell a
dicistronic nucleic acid fragment of the invention that includes the analyte
coding sequence and a detectable marker coding sequence, preferably together
with a source of transposase, followed by detection of the detectable marker.
The development of transposable elements for vectors in animals according to
the present invention thus makes possible the identification, isolation, and
characterization of coding sequences involved with growth, development and
disease, and also the transcriptional regulatory sequences that control
growth,
development and disease. Preferably, the nucleic acid fragment used in this
method of the invention, when read from left to right, contains at least the
following elements in the following order: inverted repeats, the analyte
coding
sequence, the detectable marker coding sequence, and inverted repeats. In
other words, the analyte coding sequence is located 5' of the detectable
marker
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
47
coding sequence, and the analyte coding sequence is transcribed first,
followed
by the detectable marker coding sequence. In this embodiment transcription of
the two coding sequences in the nucleic acid fragment can result in a
dicistronic
mRNA. Preferably, the analyte coding sequence is not operably linked to either
a splice acceptor site or an IRES, although it can be. While it is anticipated
that
insertion of the nucleic acid fragment into genomic DNA can result in the
interruption of a genomic coding sequence, identification of an analyte coding
sequence does not require the interruption of a genomic coding sequence.
Preferably, the ana.lyte coding sequence is operably linked to a promoter, as
described above. The detectable marker coding sequence can be operably
linked to a splice acceptor site and/or an IRES. Preferably, in this aspect of
the
method of the invention the detectable marker coding sequence is operably
linked to an IRES (see, e.g., Fig. 15).
Thus, when the dicistronic transposon vector of this aspect of the
invention inserts into DNA of a cell, the two coding sequences present in the
transposon are transcribed, and a dicistronic mRNA is typically produced.
Generally, the analyte coding sequence of the nucleic acid fragment will be
translated by virtue of ribosome initiation via scanning from the 5' end of
the
mRNA. Typically the detectable marker coding sequence of the nucleic acid
fragment will be translated by virtue of internal initiation mediated by the
IRES element. Thus, the translation of the detectable marker, i.e., the second
coding sequence of the dicistronic mRNA, provides a method to detect
expression of the analyte coding sequence of the nucleic acid fragment. This
is a significant advantage, as the expression of some biological coding
sequences of interest can be difficult to monitor directly. The dicistronic
gene
expression transposon vectors of the invention will generally allow the
expression of a biological coding sequence of interest to be detected.
Notably, there have been no previous reports of an IRES that functions in
zebrafish. The EMCV IRES and others are derived from mammalian sources,
and it is surprising that these sequences are able to direct internal ribosome
entry
in zebrafish. Millions of years of evolutionary divergence could easily have
altered the domains of the translation factors) and/or other proteins that
direct
the interaction.
A use of a dicistronic transposon vector of this aspect of the invention is
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
48
depicted schematically in Fig. 1 S. The dicistronic transposon vector and mRNA
encoding SB transposase can be microinjected into zebrafish embryos which are
allowed to mature. Expression of GFP marks cells in which "Gene X" is also
expressed. This allows analysis of the effects of "Gene X" on specific
tissues; a
form of mosaic analysis. Gene X may encode a protein or a portion of a
protein,
and the encoded protein can be beneficial or deleterious to the cells. It
should be
understood that the function of analyte coding sequences can be analyzed in
any
type of cell, including but not limited to an oocyte, a cell of an embryo, an
egg
cell or a stem cell, and in any type of tissue, differentiated or
undifferentiated.
An alternative use of the dicistronic vector is to inject dicistronic mRNA
encoded by a vector containing a nucleic acid fragment comprising the analyte
coding sequence and the detectable marker coding sequence. An example of this
embodiment is described in Example 9.
Optionally the method for identifying or analyzing the function of an
analyte coding sequence includes observing the phenotype of a cell that
contains
the integrated nucleic acid fragment, and comparing it to a cell that does not
contain the nucleic acid fragment to determine whether the phenotype of the
first
cell is altered, wherein an altered phenotype is indicative that the analyte
coding
sequence plays a function in the identified phenotype. Alternatively, the cell
that
contains the integrated nucleic acid fragment can be grown into an animal, and
animal phenotypes similarly compared.
It can be seen that the nucleic acid fragments of the invention have
applications to many areas of biotechnology and functional genomics. The
invention allows e~cient insertion of genetic material into the genomic DNA
of a cell of animals, preferably vertebrate animals, for the mutation,
evaluation
of function, and subsequent cloning of a genomic coding sequence and/or
genomic expression control sequences. The invention has the property of
allowing identification of organisms in which the detectable marker that is
encoded by the inserted nucleic acid fragment is expressed in specific tissues
or at specific times in development: Another property of the invention is the
ability to insert a biological coding sequence of interest into a cell's
genomic
DNA and evaluate the location and time of expression of the biological coding
sequence of interest by assaying for the co-expressed downstream detectable
marker coding sequence.
In a preferred embodiment of the gene transfer system of the invention,
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98I24348
49
the system has two components: a nucleic acid fragment that comprises a
nucleic
acid sequence comprising a coding sequence, wherein the nucleic acid sequence
is positioned between at least two inverted repeats that can bind to an SB
protein,
and a source of transposase. It is to be understood that the intervening
nucleic
acid sequence of the nucleic acid fragment can include any variation or
feature
herein disclosed, without (imitation, and the nucleic acid fragment is one
that is
capable of integrating into DNA of a cell, as described more fully
hereinabove.
The nucleic acid fragment is preferably part of a plasmid or a recombinant
viral
vector. As already noted, the transposase source can be either a nucleic acid
encoding the transposase or the transposase protein itself, and the
transposase is
preferably an SB protein.
Another embodiment of the gene transfer system is directed to the
introduction of a nucleic acid fragment into the DNA of a human or a fish.
This
embodiment of the gene transfer system includes a nucleic acid fragment
comprising a nucleic acid sequence that comprises an IRES, and the nucleic
acid
fragment is capable of integrating into the fish or human DNA. Preferably, the
nucleic acid sequence of this embodiment further comprises a coding sequence
located 3' to and operably linked to the 1RES. Optionally, the nucleic acid
sequence of this embodiment comprises a first coding sequence located 3' to
and
operably linked to the IRES, and a second coding sequence located 5' to both
the
first coding sequence and the IRES. It should be noted that in this particular
embodiment of the gene transfer system, the nucleic acid sequence of the
nucleic
acid fragment need not be flanked by inverted repeats that bind an SB protein,
nor is a source of transposase necessary, although these features are
optionally
included. The invention is further directed to a transgenic human or fish,
preferably zebrafish, whose cells contain a nucleic acid fragment comprising
an
IRES as described, and its progeny. In a preferred embodiment, the invention
is
directed to a transgenic fish or fish cell comprising a IRES that is
heterologous
with respect to the fish genome, for example a viral IRES.
The invention also includes a method for producing a transgenic animal.
A nucleic acid fragment of the invention, including any variation or feature
herein disclosed, without limitation, and a source of transposase as described
above are introduced into a cell. The nucleic acid fragment preferably
contains a
coding sequence that is heterologous with respect to the animal, i.e., it is
not
found in the animal's genome. However, the coding sequence can also be one
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
that is endogenous to the animal. The cell or cells containing the nucleic
acid
fragment are then grown into an animal. The resulting animal can be
transgenic,
including a mosaic. Preferably, the nucleic fragment is integrated into both
somatic and germline cells of the transgenic animal, and the transgenic animal
is
5 capable of transmitting the nucleic acid fragment to its progeny. The
invention
is further directed to a transgenic animal whose cells contain a nucleic acid
fragment of the invention, and its progeny.
Examples
The following examples, while exemplary of the present
invention, are not to be construed as specifically limiting the invention.
Accordingly, variations and equivalents, now known or later developed, that
would be within the purview of one skilled in the art are to considered to
fall
1 S within the scope of this invention.
Example 1
Reconstruction of an SB transposase
Recombinant DNA
Gene reconstruction-Phase 1: Reconstruction of a transposase open reading
frame. The Tss 1.1 element from Atlantic salmon (GenBank accession number
L12206) was PCR-amplified using a primer pair flanking the defective
transposase gene, FTC-Start and FTC-Stop to yield product SB 1. Next, a
segment of the defective transposase gene of the Tssl.2 element (L12207) was
PCR-amplified using PCR primers FTC-3 and FTC-4, then further amplified
with FTC-3 and FTC-5. The PCR product was digested with restriction enzymes
NcoI and BIpI, underlined in the primer sequences, and cloned to replace the
corresponding fragment in SB 1 to yield SB2. Then, an approximately 250 by
HindIII fragment of the defective transposase gene of the Tsgl element from
rainbow trout (L12209) was isolated and cloned into the respective sites in
SB2
to result in SB3. The Tssl and Tsgl elements were described in (Radice et al.,
1994) and were kind gifts from S.W. Emmons.
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
51
FTC-Start: 5'-CCTCTAGGATCCGACATCATG (SEQ ID N0:17)
FTC-Stop: 5'-TCTAGAATTCTAGTATTTGGTAGCATTG (SEQ ID
N0:18)
FTC-3: 5'-AACACCATGGGACCACGCAGCCGTCA (SEQ ID N0:19)
FTC-4: 5'-CAGGTTATGTCGATATAGGACTCGTTTTAC (SEQ ID
N0:20)
FTC-5: 5'-CCTTGCTGAGCGGCCTTTCAGGTTATGTCG (SEQ ID
N0:21 )
Gene reconstruction-Phase 2: Site-specific PCR mutagenesis of the SB3
open reading frame to introduce consensus amino acids. For PCR
mutagenesis, two methods have been used: megaprimer PCR (Sarkar and
Sommer, 1990 BioTechniques 8, 404-407) from SB4 through SB6, and Ligase
Chain Reaction {Michael, 1994 BioTechniques 16, 410-412) for steps SB7 to
SB10.
Oligonucleotide primers for product SB9 were the following:
FTC-7: 5'-TTGCACTTTTCGCACCAA for Gln->Arg(74) and Asn->Lys(75)
(SEQ ID N0:22);
FTC-13: 5'-GTACCTGTTTCCTCCAGCATC for Ala->Glu(93) (SEQ ID
N0:23);
FTC-8: 5'-GAGCAGTGGCTTCTTCCT for Leu->Pro( 121 ) (SEQ ID N0:24);
FTC-9: 5'-CCACAACATGATGCTGCC for Leu->Met(193) (SEQ ID
N0:25);
FTC-10: 5'-TGGCCACTCCAATACCTTGAC for Ala->Val(2b5) and Cys-
>Trp(268) (SEQ ID N0:26);
FTC-11: 5'-ACACTCTAGACTAGTATTTGGTAGCATTGCC for Ser-
>Ala(337) and Asn->Lys(339) (SEQ ID N0:27).
Oligonucleotide primers for product SBS:
BS-PTV: 5'-GTGCTTCACGGTTGGGATGGTG (SEQ ID N0:28) for Leu-
>Pro(183), Asn->Thr(184) and Met->Val(185) (SEQ ID N0:28).
Oligonucleotide primers for product SB6:
FTC-DDE: 5'-ATTTTCTATAGGATTGAGGTCAGGGC for Asp->Glu(279)
CA 02309000 2000-OS-04
WO 99/25817 PCT1US98/24348
52
(SEQ ID N0:29).
Oligonucleotide primers for products SB7 and SBB, in two steps:
PR-GAIS: 5'-GTCTGGTTCATCCTTGGGAGCAATTTCCAAACGCC for
Asn->Ile(28), His->Arg(31) and Phe->Ser(21) (SEQ ID N0:30).
Oligonucleotide primers for product SB9:
KARL: 5'-CAAAACCGACATAAGAAAGCCAGACTACGG for Pro-
>Arg( 126) (SEQ ID N0:31 );
RA: 5'-
ACCATCGTTATGTTTGGAGGAAGAAGGGGGAGGCTTGCAAGCCG for
Cys->Arg{166) and Thr->Ala(175) (SEQ ID N0:32);
EY: 5'-GGCATCATGAGGAAGGAAAATTATGTGGATATATTG for Lys-
>Glu(216) and Asp->Tyr(218) (SEQ ID N0:33);
KRV: 5'-CTGAAAAAGCGTGTGCGAGCAAGGAGGCC for Cys->Arg(288)
(SEQ ID N0:34);
VEGYP: S'-GTGGAAGGCTACCCGAAACGTTTGACC for Leu->Pro(324)
(SEQ ID N0:35).
Oligonucleotide primers for product SBIO:
FATAH: 5'-GACAAAGATCGTACTTTTTGGAGAAATGTC for Cys-
>Arg{143) (SEQ ID N0:36).
Plasmids. For pSB 10, the SB 10 transposase gene was cut with EcoRI and
BamHI, whose recognition sequences are incorporated and underlined above in
the primers FTC-Start and FTC-Stop, filled in with Klenow and cloned into the
Klenow-filled NotI sites of CMV-~igal {Clonetech), replacing the IacZ gene
originally present in this plasmid. Because of the blunt-end cloning, both
orientations of the gene insert were possible to obtain and the antisense
direction
was used as a control for transposase. For pSB 10-tIDDE, plasmid pSB 10 was
cut with MscI, which removes 322 by of the transposase-coding region, and
recircularized. Removal of the MscI fragment from the transposase gene deleted
much of the catalytic DDE domain and disrupted the reading frame by
introducing a premature translational termination codon.
Sequence alignment of 12 partial salmonid-type TcE sequences found in
8 fish species (available under DS30090 from FTP.EBLAC.AK in
directory/pub/databases/embl/align from the EMBL database) allowed us to
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98124348
53
derive.a majority-rule, salmonid-type consensus sequence, and identify
conserved protein and DNA sequence motifs that likely have functional
importance (Fig. 1 A).
Conceptual translation of the mutated transposase open reading frames
and comparison with functional motifs in other proteins allowed us to identify
five regions that are highly conserved in the SB transposase family (Fig. lA):
I) a
paired box/leucine zipper motif at the N-terminus; ii) a DNA-binding domain;
iii} a bipartite nuclear localization signal (NLS); iv) a glycine-rich motif
close to
the center of the transposase without any known function at present; and v) a
catalytic domain consisting of three segments in the C-terminal half
comprising
the DDE domain that catalyzes the transposition. DDE domains were identified
by Doak et al. in Tc 1 mariner sequences (Doak et al., 1994 Proc. Natl. Acad.
Sci. USA 91, 942-946). Multiple sequence alignment also revealed a fairly
random distribution of mutations in transposase coding sequences; 72% had
occurred at non-synonymous positions in codons. The highest mutation
frequencies were observed at CpG dinucleotide sites which are highly mutable
(Adey et al., 1994, supra). Although amino acid substitutions were distributed
throughout the transposases, fewer mutations were detected at the conserved
motifs (0.07 non-synonymous mutations per codon), as compared to protein
regions between the conserved domains (0.1 non-synonymous mutations per
codon}. This observation indicated that some selection mechanism had
maintained the functional domains before inactivation of transposons took
place
in host genomes. The identification of these putative functional domains was
of
key importance during the reactivation procedure.
The first step of reactivating the transposase gene, was to restore an open
reading frame (SB 1 through SB3 in Fig. 1 B) from bits and pieces of two
inactive
TcEs from Atlantic salmon (Salmo salary and a single element from rainbow
trout (Oncorhynchus mykiss) (Radice et al., 1994, supra). SB3, which has a
complete open reading frame after removal of stop codons and frameshifts, was
tested in an excision assay similar to that described by Handler et al. (1993)
but
no detectable activity was observed. Due to non-synonymous nucleotide
substitutions, the SB3 polypeptide differs from the consensus transposase
sequence in 24 positions (Fig. 1B) which can be sorted into two groups; nine
residues that are probably essential for transposase activity because they are
in
the presumed functional domains and/or conserved in the entire Tcl family, and
CA 02309000 2000-OS-04
WO 99125817 PCT1US98/24348
54
another f fteen residues whose relative importance could not be predicted.
Consequently, a dual gene reconstruction strategy was undertaken. First, the
putative functional protein domains of the transposase were systematically
rebuilt one at a time by correcting the former group of mutations. Each domain
for a biochemical activity was tested independently when possible. Second, in
parallel with the first approach, a full-length, putative transposase gene was
synthesized by extending the reconstruction procedure to all of the 24 mutant
amino acids in the putative transposase.
Accordingly, a series of constructs was made to bring the coding
sequence closer, step-by-step, to the consensus using PCR mutagenesis (SB4
through SB 10 in Fig. 1 B). As a general approach the sequence information
predicted by the majority-rule consensus was followed. However, at some
codons deamination of SmC residues of CpG sites occurred, and C ->T
mutations had been fixed in many elements. At R(288), where TpG's and CpG's
1 S were represented in equal numbers in the alignment, the CpG sequence was
chosen because the CpG ->TpG transition is more common in vertebrates than
the TpG -> CpG. The result of this extensive genetic engineering is a
synthetic
transposase gene encoding 340 amino acids (SB10 in Figs. 1B and 2).
The reconstituted functional transposase domains were tested for activity.
First, a short segment of the SB4 transposase gene (Fig. 1 B) encoding an NLS-
like protein motif was fused to the IacZ gene. The transposase NLS was able to
mediate the transfer of the cytoplasmic marker-protein,13-galactosidase, into
the
nuclei of cultured mouse cells (Ivics et al., 1996, supra), supporting
predictions
that a bipartite NLS was a functional motif in SB and that our approach to
resurrect a full-length, multifunctional enzyme was viable.
Ezample 2
Preparation of a nucleic acid fragment with inverted repeat sequences.
In contrast to the prototypic Tcl transposon from Caenorhabditis elegans
which has short, 54-by indirect repeat sequences (IRs) flanking its
transposase
gene, most TcEs in fish belong to the IR/DR subgroup of TcEs (Ivics et al.,
1996; Izsvak et al., 1995, both supra) which have long, 210-250 by 1Rs at
their
termini and directly repeated DNA sequence motifs (DRs) at the ends of each IR
(Fig, lA). However, the consensus IR sequences are not perfect repeats (i.e.,
similar, but not identical) indicating that, in contrast to most TcEs, these
fish
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
SS
elements naturally possess imperfect inverted repeats. The match is less than
80% at the center of the IRs, but is perfect at the DRs, suggesting that this
nonrandom distribution of dissimilarity could be the result of positive
selection
that has maintained functionally important sequence motifs in the IRs (Fig.
3).
Therefore, DNA sequences at and around the DRs might carry cis-acting
information for transposition and mutations within the IRs, but outside the
DRs,
would probably not impair the ability of the element to transpose. As a model
substrate, a single salmonid-type TcE substrate sequence from Tanichthys
albonubes (hereafter referred to as T), which has intact DR motifs whose
sequences are only 3.8% divergent from the salmonid consensus, was chosen.
The variation in the DNase-protected regions of the four DR sequences varied
from about 83% to about 95 %, see SEQ ID NOS:6-9.
A TcE from Tanichthys albonubes (L48685) was cloned into the SmaI
site of pUC 19 to result in pT. The donor construct for the integration
assays,
pT/neo, was made by cloning, after Klenow fill-in, an EcoRIlBamHI fragment of
the plasmid pRc-CMV (Invitrogen, San Diego, CA) containing the SV40
promoter/enhancer, the neomycin resistance gene and an SV40 poly(A) signal
into the StuIlMscI sites of pT. The StuIlMscI double digest of pT leaves 352
by
on the left side and 372 by on the right side of the transposon and thus
contains
the terminal inverted repeats. An EcoRI digest of pT/neo removed a 350 by
fragment including the left inverted repeat of the transposon, and this
plasmid,
designated pT/neo-DIR, was used as a control for the substrate-dependence of
transposase-mediated transgene integration {see Example 4)
Example 3
DNA specificity of an SB transposase
There are at least two distinct subfamilies of TcEs in the genomes of
Atlantic salmon and zebrafish, Tssl/Tdrl and Tss2/Tdr2, respectively. Elements
from the same subfamily are more alike, having about 70% nucleic acid
identity,
even when they are from two different species (e.g., Tssl and Tdrl) than
members of two different subfamilies in the same species. For example, Tdrl
and Tdr2 are characteristically different in their encoded transposases and
their
inverted repeat sequences, and share only about 30% nucleic acid identity. It
may
be that certain subfamilies of transposons must be significantly different
from
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98I24348
56
each other in order to avoid cross-mobilization. A major question is whether
substrate recognition of transposases is sufficiently specific to prevent
activation
of transposons of closely related subfamilies.
The 12-by DRs of salmonid-type elements, identical to the DRs of
zebrafish-type TcEs, are part of the binding sites for SB. However, these
binding-sites are 30 by long. Thus, specific DNA-binding also involves DNA
sequences around the DRs that are variable between TcE subfamilies in fish.
Such a difference in the sequences of transposase binding sites might explain
the
inability of N123 to bind efficiently to zebrafish Tdrl IRs, and may enable
the
transposase to distinguish even between closely related TcE subfamilies.
Indeed,
mutations of four base pairs in the 20-by Tc 1 binding site can abolish
binding of
transposase (Vos and Plasterk, 1994 EMBO J. 13, 6125-6132). The DR core
motifs are likely involved primarily in transposase-binding while sequences
around the DR motifs likely provide the specificity for this binding.
SB has four binding-sites in its transposon substrate DNA that are located
at the ends of the IRs. These sites share about a 83% to about a 95% identity
(by
comparison of SEQ ID NOS:6-9). However, a zebrafish Tdrl element lacking
an internal transposase-binding site was apparently able to transpose. This
observation agrees with the finding that removal of internal transposase-
binding
sites from engineered Tc3 elements did not lessen their ability to transpose
(Colloms et al., 1994 Nucl. Acids Res. 22, 5548-5554), suggesting that the
presence of internal transposase-binding sites is not essential for
transposition.
Multiple binding-sites for proteins, including transposases, are frequently
associated with regulatory functions (Gierl et al., 1988 EMBO J. 7, 4045-
4053).
Consequently, the internal binding-sites for transposases in the IR/DR group
of
TcEs serve one or more regulatory purposes affecting transposition and/or gene
expression.
Once in the nucleus, a transposase must bind specifically to its
recognition sequences in the transposon. The specific DNA-binding domains of
both the Tcl and Tc3 transposases have been mapped to their N-terminal regions
(Colloms et al., 1994, supra; Vos and Plasterk, 1994, supra). However, there
is
very little sequence conservation between the N-terminal regions of TcE
transposases, suggesting that these sequences are likely to encode specific
DNA-
binding functions in these proteins. On the other hand, the N-terminal region
of
SB has significant structural and sequence similarities to the paired DNA-
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
57
binding domain, found in the Pax family of transcription factors, in a novel
combination with a leucine zipper-like motif (Ivics et al., 1996, supra). A
gene
segment encoding the first 123 amino acids of SB (N123), which presumably
contains all the necessary information for specific DNA-binding and includes
the
NLS, was reconstructed (SB8 in Fig. 1B), and expressed in E. coli. N123 was
purified via a C-terminal histidine tag as a 16 KDa polypeptide (Fig. 3A).
Induction of N 123 was in E. col i strain BL21 (DE3 ) (Novagen) by the
addition of 0.4 mM IPTG at 0.5 O.D. at 600 nm and continued for 2.5 h at 30oC.
Cells were sonicated in 25 mM HEPES, pH 7.5, 1 M NaCI, 15% glycerol, 0.25%
Tween 20, 2 mM [i-mercaptoethanol, 1 mM PMSF) and 10 mM imidazole (pH
8.0) was added to the soluble fraction before it was mixed with Ni2+-NTA resin
(Qiagen) according to the recommendations of the manufacturer. The resin was
washed with 25 mM HEPES (pH 7.5), 1 M NaCI, 30% glycerol, 0.25% Tween
20, 2 mM [i-mercaptoethanol, 1 mM PMSF and 50 mM imidazole (pH 8.0) and
bound proteins were eluted with sonication buffer containing 300 mM
imidazole, and dialyzed overnight at 4oC against sonication buffer without
imidazole.
In addition to the NLS function, N 123 also contains the specific DNA-
binding domain of SB, as tested in a mobility-shift assay (Fig. 3B). A 300 by
EcoRIlHindIII fragment of pT comprising the left inverted repeat of the
element
was end-labeled using [a32P]dCTP and Klenow. Nucleoprotein complexes were
formed in 20 mM HEPES {pH 7.5), 0.1 mM EDTA, 0.1 mg/ml BSA, 150 mM
NaCI, 1 mM DTT in a total volume of 10 ~tl. Reactions contained 100 pg labeled
probe, 2 ~g poly[dI][dC] and 1.5 pl N123. After 15 min incubation on ice, 5
p,l
of loading dye containing 50% glycerol and bromophenol blue was added and
the samples loaded onto a 5% polyacrylamide gel (Ausubel). DNaseI
footprinting was done using a kit from BRL according to the recommendations
of the manufacturer. Upon incubation of a radiolabeled 300-by DNA fragment
comprising the left IR of T, deoxyribonucleoprotein complexes were observed
(Fig. 3B, left panel- lane 3), as compared to samples containing extracts of
bacteria transformed with the expression vector only (lane 2) or probe without
any protein (lane 1 ). Unlabelled IR sequences of T, added in excess to the
reaction as competitor DNA, inhibited binding of the probe (lane 4), whereas
the
analogous region of a cloned Tdrl element from zebrafish did not appreciably
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
58
compete with binding {lane S). Thus, N123 is able to distinguish between
salmonid-type and zebrafish-type TcE substrates.
The number of the deoxyribonucleoprotein complexes detected by the
mobility-shift assay at increasingly higher N123 concentrations indicated two
protein molecules bound per IR (Fig. 3B, right panel), consistent with either
two
binding sites for transposase within the 1R or a transposase dimer bound to a
single site. Transposase-binding sites were further analyzed and mapped in a
DNaseI footprinting experiment. Using the same fragment of T as above, two
protected regions close to the ends of the IR probe were observed (Fig. 4).
The
two 30-by footprints cover the subterminal DR motifs within the IRs. Thus, the
DRs are the core sequences for DNA-binding by N 123. The DR motifs are
almost identical between salmonid- and zebrafish-type TcEs (Ivics et al.,
1997).
However, the 30-by transposase binding-sites are longer than the DR motifs and
contain 8 base pairs and 7 base pairs in the outer and internal binding sites,
respectively, that are different between the zebrafish- and the salmonid-type
IRs
(Fig. 4B).
Although there are two binding-sites for transposase near the ends of
each IR, apparently only the outer sites are utilized for DNA cleavage and
thus
excision of the transposon. Sequence comparison shows that there is a 3-by
difference in composition and a 2-by difference in length between the outer
and
internal transposase-binding sites (Fig. 4C). In summary, our synthetic
transposase protein has DNA-binding activity and this binding appears to be
specific for salmonid-type IR/DR sequences.
For the expression of an N-terminal derivative of SB transposase, a gene
segment of SB8 was PCR-amplified using primers FTC-Start and FTC-8, 5'-
phosphorylated with T4 polynucleotide kinase, digested with BamHI, filled in
with Klenow, and cloned into the NdeIlEcoRI digested expression vector
pET2la (Novagen) after Klenow fill-in. This plasmid, pET2lalN123 expresses
the first 123 amino acids of the transposase (NI23) with a C-terminal
histidine
tag.
Example 4
Transposition of DNA by an SB transposase
The following experiments demonstrate that the synthetic, salmonid-type
SB transposase performed all of the complex steps of transposition, i.e.,
*rB
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
59
recognized a DNA molecule, excised the substrate DNA and inserted it into the
DNA of a cell, such as a cell chromosome. This is in contrast to control
samples
that did not include the SB transposase and therefore measured integration
through non-homologous recombination.
Upon cotransfection of the two-component SB transposon system into
cultured vertebrate cells, transposase activity manifested as enhanced
integration
of the transgene serving as the DNA substrate for transposase. The binding of
transposase to a donor construct and subsequent active transport of these
nucleoprotein complexes into the nuclei of transfected cells could have
resulted
in elevated integration rates, as observed for transgenic zebrafish embryos
using
an SV40 NLS peptide (Collas et al., 1996 Transgenic Res. 5, 451-458).
However, DNA-binding and nuclear targeting activities alone did not increase
transformation frequency, which occurred only in the presence of full-length
transposase. Although not sufficient, these functions are probably necessary
for
transposase activity. Indeed, a single amino acid replacement in the NLS of
mariner is detrimental to overall transposase function (Lohe et al., 1997
Proc.
Natl. Acad. Sci. USA 94, 1293-1297). The inability of SB6, a mutated version
of
the transposase gene, to catalyze transposition demonstrates the importance of
the sequences of the conserved motifs. Notably, three of the 11 amino acid
substitutions that SB6 contains, F(21), N(28) and H(31) are within the
specific
DNA-binding domain (Figs. 1 and 2). Sequence analysis of the paired like
DNA-binding domain of fish TcE transposases indicates that an isoleucine at
position 28 is conserved between the transposases and the corresponding
positions in the Pax proteins (Ivics et al., 1996, supra). Thus, this motif is
probably crucial for DNA-binding activity. SB exhibits substrate-dependence
for
specific recognition and integration; only those engineered transposons that
have
both of the terminal inverted repeats can be transposed by SB. Similarly, in P
element transformation in Drosophila, the transposase-producing helper
construct is often a "wings-clipped" transposase gene which lacks one of the
inverted repeats of P which prevents the element from jumping (Cooley et al.,
1988 Science 239, 1121-1128). In our transient assay, transposition can only
occur if both components of the SB system are present in the same cell. Once
that happens, multiple integrations can take place as demonstrated by the
finding
of up to 11 integrated transgenes in neomycin-resistant cell clones (Fig. 7A).
In
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
contrast to spontaneous integration of plasmid DNA in cultured mammalian cells
that often occurs in the form of concatemeric multimers into a single genomic
site (Perucho et al., 1980 Cell 22, 309-317), these multiple insertions appear
to
have occurred in distinct chromosomal locations.
Integration of the synthetic, salmonid transposons was observed in fish as
well as in mouse and human cells. In addition, recombination of genetic
markers
in a plasmid-to-plasmid transposition assay (Lampe et al., 1996, supra) was
significantly enhanced in microinjected zebrafish embryos in the presence of
transposase. Consequently, SB apparently does not need any obvious, species-
10 specific factor that would restrict its activity to its original host.
Importantly, the
most significant enhancement, about 20-fold, of transgene integration was
observed in human cells as well as fish embryonic cells.
Integration activity of SB
In addition to the abilities to enter nuclei and specifically bind to its
sites
15 of action within the inverted repeats, a fully active transposase is
expected to
excise and integrate transposons. In the C-terminal half of the SB
transposase,
three protein motifs make up the DD(34)E catalytic domain; the two invariable
aspartic acid residues, D(153) and D(244), and a glutamic acid residue,
E(279),
the latter two being separated by 34 amino acids (Fig. 2). An intact DD(34)E
box
20 is essential for catalytic functions of Tcl and Tc3 transposases (van
Luenen et
al., 1994 Ce1179, 293-301; Vos and Plasterk, 1994, supra).
Two different integration assays were used. A first assay was designed to
detect chromosomal integration events into the chromosomes of cultured cells.
The assay is based on traps-complementation of two nonautonomous
25 transposable elements, one containing a selectable marker gene (donor) and
another that expresses the transposase (helper) (Fig. SA). The donor, pT/neo,
is
an engineered, T based element which contains an SV40 promoter-driven neo
gene flanked by the terminal IRs of the transposon containing binding sites
for
the transposase. The helper construct expresses the full-length SB10
transposase
30 gene driven by a human cytomegalovirus (CMV) enhancer/promoter. In the
assay, the donor plasmid is cotransfected with the helper or control
constructs
into cultured vertebrate cells, and the number of cell clones that are
resistant to
the neomycin analog drug 6418 due to chromosomal integration and expression
of the neo transgene serves as an indicator of the efficiency of gene
transfer. If
35 SB is not strictly host-specific, transposition should also occur in
CA 02309000 2000-OS-04
WO 99/25817 PCf/US98/24348
61
phylogenetically distant vertebrate species. Using the assay system shown in
Fig.
SA, enhanced levels of transgene integration were observed in the presence of
the helper plasmid; more than S-fold in mouse LMTK cells and more than 20-
fold in human HeLa cells (Figs. SB and 6). Consequently, SB appears to be able
to increase the efficiency of transgene integration, and this activity is not
restricted to fish cells.
To analyze the requirements for enhanced transgene integration, further
experiments were conducted. Fig. SB shows five plates of transfected HeLa
cells
that were placed under 6418 selection, and were stained with methylene blue
two weeks post-transfection. The staining patterns clearly demonstrate a
significant increase in integration of neo-marked transposons into the
chromosomes of HeLa cells when the SB transposase-expressing helper
construct was cotransfected (plate 2), as compared to a control cotransfection
of
the donor plasmid plus the SB transposase gene cloned in an antisense
orientation (pSBlO-AS; plate 1). This result indicates that the production of
transposase protein was essential for enhanced chromosomal integration of the
transgene and demonstrates that the transposase is precise even in human
cells.
In a second assay, an indicator plasmid containing the transposase
recognition sequence and a marker gene (ampicillin resistance) was co-injected
with a target plasmid containing a kanamycin gene and SB transposase.
Resulting plasmids were isolated and used to transform E. coli. Colonies were
selected for ampicillin and kanamycin resistance (see Figure 8). While SB
transposase was co-microinjected in these assays, mRNA encoding the SB
transposase could also be co-microinjected in place of or in addition to, the
SB
transposase protein.
Cell transfections
Cells were cultured in DMEM supplemented with 10% fetal bovine
serum, seeded onto 6 cm plates one day prior to transfection and transfected
with
5 pg Elutip (Schleicher and Schuell)-purified plasmid DNA using Lipofectin
from BRL. After 5 hrs of incubation with the DNA-lipid complexes, the cells
were "glycerol-shocked" for 30 sec with 15% glycerol in phosphate buffered
saline (PBS), washed once with PBS and then refed with serum-containing
medium. Two days post-transfection, the transfected cells were trypsinized,
resuspended in 2 m1 of serum-containing DMEM and either 1 ml or 0.1 ml
aliquots of this cell suspension were seeded onto several 10 cm plates in
medium
CA 02309000 2000-OS-04
WO 99/25817 PCTlUS98/24348
62
containing 600 pg/ml 6418 (BRL). After two weeks of selection, cell clones
were either picked and expanded into individual cultures or fixed with 10%
formaldehyde in PBS for 15 min, stained with methylene blue in PBS for 30
min, washed extensively with deionized water, air dried and photographed.
These assays can also be used to map transposase domains necessary for
chromosomal integration. For this assay, a frameshift mutation was introduced
into the SB transposase gene which put a translational stop codon behind
G( 161 ). This construct, pSB 10-ODDE, expresses a truncated transposase
polypeptide that contains specific DNA-binding and NLS domains, but lacks the
catalytic domain. The transformation rates obtained using this construct
(plate 3
in Fig. SB) were similar to those obtained with the antisense control (Fig.
6).
This result suggests that the presence of a full-length transposase protein is
necessary and that DNA-binding and nuclear transport activities themselves are
not sufficient for the observed enhancement of transgene integration.
As a further control of transposase requirement, the integration activity of
an earlier version of the SB transposase gene was tested, SB6 which differs
from
SB 10 at 11 residues, Fig. 1 B), using the same assay. The number of
transformants observed using SB6 (plate 4 in Fig. SB) was about the same as
with the antisense control experiment (Fig. 6), indicating that the amino acid
replacements that we introduced into the transposase gene were critical for
transposase function. In summary, the three controls shown in plates 1, 3, and
4
of Fig. SB establish the traps-requirements of enhanced, SB-mediated transgene
integration.
True transposition requires a transposon with intact IR sequences. One
of the IRs of the neo-marked transposon substrate was removed, and the
performance of this construct, pT/neo-SIR, was tested for integration. The
transformation rates observed with this plasmid (plate 5 in Fig. SB) were more
than 7-fold lower than those with the full-length donor (Fig. 6). These
results
indicated that both of the IRs flanking the transposon are required for
efficient
transposition and thereby establish some of the cis-requirements of the two-
component SB transposon system.
To examine the structures of integrated transgenes, eleven colonies of
cells growing under 6418 selection from an experiment similar to that shown in
plate 2 in Fig. SB were picked and their DNAs analyzed using Southern
hybridization. Genomic DNA samples of the cell clones were digested with a
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
63
combination of five restriction enzymes that do not cut within the 2233 by
T/neo
marker transposon, and hybridized with a neo-specific probe (Fig. 7). The
hybridization patterns indicated that all of the analyzed clones contained
integrated transgenes in the range of 1 (lane 4) to 11 (lane 2) copies per
transformant. Moreover, many of the multiple insertions appear to have
occurred
in different locations in the human genome.
The presence of duplicated TA sequences flanking an integrated transposon
is a hallinark of TcE transposition. To reveal such sequences, junction
fragments of
integrated transposons and human genomic DNA were isolated using a ligation-
mediated PCR assay (Devon et al., Nucl. Acids. Res., 23,1644-1645 (1995),
Izsvak,
et al., BioTechniques,15, 814-816 (1993)). Junction fragments of five
integrated
transposons were cloned and sequenced. All of them showed the predicted
sequences of the llZs which continue with TA dinucleotides and sequences that
are
different in all of the junctions and different from the plasmid vector
sequences
originally flanking the transposon in pT/neo (Fig. 7B). The same results were
obtained from nine additional junctions containing either the left or the
right IR of
the transposon (data not shown). These results indicated that the marker
transposons
had been precisely excised from the donor plasmids and subsequently spliced
into
various locations in human chromosomes. Next, the junction sequences were
compared to the corresponding "empty" chromosomal regions clod from wild-
type HeLa DNA. As shown in Fig. 7B, all of these insertions had occurred into
TA target sites, which were subsequently duplicated to result in TA's flanking
the
integrated transposons. These data demonstrate that SB uses the same, cut-and-
paste-type mechanism of transposition as other members of the Tcllmariner
superfamily and that fidelity of the reaction is maintained in heterologous
cells.
These data also suggest that the frequency of SB-mediated transposition is at
least
15-fold higher than random recombination. Since none of the sequenced
recombination events were mediated by SB-transposase, the real rate of
transposition over random recombination could be many fold higher. If the
integration is the result of random integration that was not mediated by the
SB
protein, the ends of the inserted neo construct would not correspond to the
e~s of
the plasmids; there would have been either missing IR sequences and/or
additional
plasmid sequences that flank the transposon. Moreover, there would not have
been
duplicated TA base pairs at the sites of integration.
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
64
Taken together, the dependence of excision and integration, from
extrachromosomal plasmids to the chromosomes of vertebrate cells, of a
complete transposon with inverted repeats at both ends by a full-length
transposase enzyme demonstrates that the gene transfer system is fully
functional.
Example 5
Transposition of DNA in cells from different species
Host-requirements of transposase activity were assessed using five
different vertebrate cells, NIH 3T3, LMTK and embryonic stem cells from
mouse, HeLa cells from human and embryonic cells from the zebarafish.
An assay was designed to demonstrate that the transposase worked in a
functioning set of cells (i.e., embryonic cells that were differentiating and
growing in a natural environment). The assay involved inter-plasmid transfer
where the transposon in one plasmid is removed and inserted into a target
plasmid and the transposase construct was injected into 1-cell stage zebrafish
embryos. In these experiments the Indicator (donor) plasmids for monitoring
transposon excision and/or integration included: 1 ) a marker gene that when
recovered in E. coli or in fish cells, could be screened by virtue of either
the loss
or the gain of a function, and 2) transposase-recognition sequences in the IRs
flanking the marker gene. The total size of the marked transposons was kept to
about 1.6 kb, the natural size of the TcEs found in teleost genomes. The
transposition activity of Tsl transposase was evaluated by co-microinjecting
200
ng/~.1 of Tsl mRNA, made in vitro by T7 RNA polymerase from a Bluescript
expression vector, plus about 250 ng/~,1 each of target and donor plasmids
into 1-
cell stage zebrafish embryos. Low molecular weight DNA was prepared from
the embryos at about S hrs post-injection, transformed into E.coli cells, and
colonies selected by replica plating on agar containing 50 ~,g/ml kanamycin
and/or ampicillin. In these studies there was a transposition frequency into
the
target plasmid was about 0.041 % in experimental cells as compared to 0.002%
in control cells. This level did not include transpositions that occurred in
the
zebrafish genome. In these experiments we found that about 40% to 50% of the
embryos did not survive beyond 4 days. Insertional mutagenesis studies in the
mouse have suggested that the rate of recessive lethality is about 0.05 (i.e.,
an
average of about 20 insertions will be lethal). Assuming that this rate is
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
applicable to zebrafish, the approximate level of mortality suggests that with
the
microinjection conditions used in these experiments, about 20 insertions per
genome, the mortality can be accounted for.
Example 6
Stable gene expression from SB transposons
A transposon system will be functional for gene transfer, for such purposes
as gene therapy and gene delivery to animal chromosomes for bioreactor
systems,
only if the delivered genes are reliably expressed. To determine the fidelity
of gene
10 expression following Sleeping Beauty transposase-mediated delivery, we co-
microinjected a transposon containing the GFP (GFP) gene under the direction
of an
Xenopus eF 1 a promoter plus in vitro-synthesized mRNA encoding Sleeping
Beauty
transposase into 1-cell zebrafish embryos. 34 of the injected embryos, that
showed
some expression of GFP during embryogenesis, were allowed to grow to maturity
15 and were mated with wild-type zebrafish. From these coatings we found that
4 of
the 34 fish could transfer a GFP gene to their progeny (Table 1). The
expression of
GFP in the offspring of these four FO fish, identified as A, B, C, and D, was
evaluated and the fish were grown up. From the original four founders, the
rate of
transmission of the GFP gene ranged from about 2% to 12% {Table 1 ), with an
20 average of about 7%. The expression of GFP in these fish was nearly the
same in all
individuals in the same tissue types, suggesting that expression of the GFP
gene
could be revived following transmission through eggs and spernl. These data
suggest that the germ-lines were mosaic for expressing GFP genes and that the
expression of the genes was stable. The F1 offspring of Fish D were mated with
25 each other. In this case we would expect about 75% transmission and we
found that
indeed 69/90 (77%) F2 fish expressed the GFP protein at comparable levels in
the
same tissues; further testimony of the ability of the SB transposon system to
deliver
genes that can be reliably expressed through at least two generations of
animals.
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
66
10
Table 1
Stability of gene expression in zebrafish following injection of a SB
transposon containing the GFP gene.
Expression of GFP
Transeenic Line FO F1 F2
34 founders 34 (of which 4 progeny, A-D, passed on the transgene)
A 25/200 (12%)
B 76/863 (9%)
C 12/701 (2%)
D 86/946 ( 10%) 69/90 (77%)
The numbers in the columns for fish A-D show the numbers of GFP expressing
fish followed by the total number of offspring examined. The percentages of
GFP-expressing offspring are given in parentheses.
Example 7
SB Transposons for Insertional Mutagenesis and Gene Discovery
Due to their inherent ability to move from one chromosomal location to
another within and between genomes, transposable elements have revolutionized
genetic manipulation of certain organisms including bacteria (Gonzales et al.,
1996 Vet. Microbiol. 48, 283-291; Lee and Henk, 1996. Vet. Microbiol. 50, 143-
148), Drosophila (Ballinger and Benzer, 1989 Proc. Natl. Acad. Sci. USA 86,
9402-9406; Bellen et al., 1989 Genes Dev. 3, 1288-1300; Spradling et al., 1995
Proc. Natl. Acad. Sci. USA 92, 10824-10830), C. elegans (Plasterk, 1995. Meth.
Cell. Biol., Academic Press, Inc. pp. 59-80) and a variety of plant species
(Osborne and Baker, Curr. Opin. Cell Biol, 7, 406-413 (1995)). Transposons
have been harnessed as useful vectors for transposon-tagging, enhancer
trapping
and transgenesis. However, the majority, if not all, animals of economic
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
67
importance lack such a tool. For its simplicity and apparent ability to
function in
diverse organisms, SB should prove useful as an efficient vector for species
in
which DNA transposon technology is currently not available.
An SB-type transposable element can integrate into either of two types of
chromatin, fimctional DNA sequences where it may have a deleterious effect due
to
insertional mutagenesis or non-fiuictional chromatin where it may not have
much of
a consequence (Fig. 9). This power of "transposon tagging" has been exploited
in
simpler model systems for nearly two decades (Bingham et al., Cell, 25, 693-
704
( 1981 ); Bellen et al. , 1989, supra). Transposon tagging is an old technique
in which
transgenic DNA is delivered to cells so that it will integrate into genes,
thereby
inactivating them by insertional mutagenesis. In the process, the inactivated
genes
are tagged by the transposable element which then can be used to recover the
mutated allele. Insertion of a transposable element may disrupt the fimction
of a gene
which can lead to a characteristic phenotype. As illustrated in Fig. 9,
because
insertion is approximately random, the same procedures that generate
inserkional,
loss-of fimction mutants can often be used to deliver genes that will confer
new
phenotypes to cells. Gain-of function mutants can be used to understand the
roles
that gene products play in growth and development as well as the importance of
their
regulation.
There are several ways of isolating the tagged gene. In all cases genomic DNA
is isolated finm cells finm one or more tissues of the mutated animal by
conventional techniques (which vary for different tissues and animals). The
DNA is
cleaved by a restriction endonuclease that may or may not cut in the
transposon tag
(more often than not it does cleave at a known site). The resulting fiagments
can
then either be directly cloned into plasmids or phage vectors for
identification using
probes to the transposon DNA (see Kim et al.,1995 for references in Mobile
Genetic
Elements, IRL Press, D. L. Sheratt eds.). Alternatively, the DNA can be PCR
amplified in any of many ways; including the LM-PCR procedure of Izsvak and
Ivics (1993, supra) and a modification by Devon et al. (1995, supra) and
identified
by its hybridization to the transposon probe. An alternative method is inverse-
PCR
(e.g., Allende et al., Genes Dev.,10, 3141-3155 (1996)). Regardless of method
for
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
68
cloning, the identified clone is then sequenced. The sequences that flank the
transposon (or other inserted DNA) can be identified by their non-identity to
the
insertional element. The sequences can be combined and then used to search the
nucleic acid databases for either homology with other previously characterized
gene(s), or partial homology to a gene or sequence motif that encodes some
function. In some cases the gene has no homology to any known protein. It
becomes
a new sequence to which others will be compared. The encoded protein will be
the
center of further investigation of its role in causing the phenotype that
induced its
recovery. For gene traps and poly(A) traps, mRNA can be used to detemline the
nucleotide sequence of the genomic DNA flanking the inserted nucleic acid
fiagment. For instance, the use of sequence-specific primers that hybridize to
nucleotide sequences of the inserted nucleic acid fragment that would be
present in a
resulting mRNA, subsequent reverse transcription and 5' or 3' RACE (rapi
Example 8
SB transposons as markers for gene mapping
Repetitive elements for mapping transgenes and other genetic loci have
also been identified. DANA is a retroposon with an unusual substructure of
distinct cassettes that appears to have been assembled by insertions of short
sequences into a progenitor SINE element. DANA has been amplified in the
Danio lineage to about 4 x 105 copies/genome. Angel elements, which are nearly
as abundant as DANA, are inverted-repeat sequences that are found in the
vicinity of fish genes. Both DANA and Angel elements appear to be randomly
distributed in the genome, and segregate in a Medelian fashion. PCR
amplifications using primers specific to DANA and Angel elements can be used
as genetic markers for screening polymorphisms between fish stocks and
localization of transgenic sequences. Interspersed repetitive sequence-PCR
(IRS-PCR) can be used to detect polymorphic DNA. IRS-PCR amplifies
genomic DNA flanked by repetitive elements, using repeat-specific primers to
produce polymorphic fragments that are inherited in a Medelian fashion (Fig.
l0A). Primers that can be used in IRS-PCR to detect polymorphic DNA include
S'-GGCGACRCAGTGGCGCAGTRGG (SEQ ID N0:13) where R is G or A
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
69
and 5'-GAAYRTGCAAACTCCACACAGA (SEQ ID N0:14) where Y is T or C
and R is G or A, each of which anneal to nucleotides present in the retroposon
DANA (D); 5'-TCCATCAGACCACAGGACAT (SEQ 1D NO:15) and 5'-
TGTCAGGAGGAATGGGCCAAAATTC (SEQ m N0:16), each of which anneal
to nucleotides present in Tdrl transposons; and 5'-
TTTCAGTTTTGGGTGAACTATCC (SEQ ID N0:12), which anneals to
nucleotides present in Angel (A) (a highly reiterated miniature inverted-
repeat
transposable element). Polymorphic DNA fragments can be generated by
DANA or Angel specific primers in IRS-PCR and the number of detectable
polymorphic bands can be significantly increased by the combination of various
primers to repetitive sequences in the zebrafish genome, including SB-like
transposons.
Polymorphic fragments can be recovered from gels and cloned to provide
sequence tagged sites (STSs) for mapping mutations. Fig. l OB illustrates the
general principles and constraints for using IRS-PCR to generate STSs. It is
estimated that about 0.1% of the zebrafish genome can be directly analyzed by
IRS-PCR using only 4 primers. The four conserved (C1-4) regions of DANA
seem to have different degrees of conservation and representation in the
zebrafish genome and this is taken into account when designing PCR primers.
The same method has a potential application in fingerprinting fish stocks
and other animal populations. The method can facilitate obtaining subclones of
large DNAs cloned in yeast, bacterial and bacteriophage P1-derived artificial
chromosomes (YACs, BACs and PACs respectively) and can be used for the
detection of integrated transgenic sequences.
Example 9
SB Transposon for Insertional Mutagenesis and Functional
Analysis of Genes
I. Dicistronic vector construction
Dicistronic vectors in zebrafish would allow researchers to track the
expression of a biological gene of interest in living embryos simply by using
a
CA 02309000 2000-OS-04
WO 99/Z5817 PCT/US98/24348
reporter molecule, like GFP. Knowing where and when an introduced DNA or
mRNA construct is expressed could greatly facilitate interpretation of over-
expression and mutant-rescue experiments. In order for a dicistronic vector to
be
useful for all of these purposes, it must determined in which cells and
tissues,
5 and at what developmental stages, detectable expression of a second cistron
encoding a marker gene occurs.
Accordingly, several dicistronic vectors using the EMCV IRES to
determine the parameters under which a mammalian IRES could be used for
dicistronic technology in zebrafish have been constructed. The EMCV IRES can
10 function in developing zebrafish from early cleavage stages to larval
stages. The
products of both genes in mRNAs co-localize within the embryo, indicating that
both products are made in many cell types within the embryo.
a. Methods
15 phBeL: phBeL was constructed from component fragments of
pRC/CMV (Invitrogen), pCMVl3 (Clontech), pGem/eLuc, and CMV4
{Andersson et al. 1989). The vector backbone consists of the 3.1 S kilobase
(kb)
fragment obtained after digestion with the restriction endonucleases XhoI and
NotI. The XhoI to NotI fragment contains the CoIE 1 origin of replication,
20 ampicillin resistance gene (amp), and CMV promoter found in the complete
pRC/CMV vector. Fused to the NotI site of the pRC/CMV XhoI/NotI fragment
is the 3.74-kb fragment obtained after digestion of pCMVl3 with the
restriction
endonuclease NotI. This NotI fragment contains the complete 13-galactosidase
(Bgal) coding region found in pCMV(3. At the NotI site after the 13ga1 coding
25 sequence was fused the 2.34-kb fragment obtained after digestion of
pGem/eLuc
with the restriction endonucleases NotI and StuI. The NotI/StuI fragment
contains the EMCV IRES and luciferase coding regions. A 1.11-kb fragment of
CMV4 was obtained after digestion with the restriction endonucleases SmaI and
SaII. This SmaI/SaII fragment contains the human growth hormone poly(A)
30 signal , the SV40 origin of replication, and the SV40 early
enhancer/promoter
region. The SmaI/SaII CMV4 fragment completes the vector since StuI and
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
71
SmaI create blunt cuts while XhoI and SaII have compatible single-stranded
overhangs. The hairpin structure found in the mRNA of this vector is due to
the
large number of restriction sites upstream of the 13-galactosidase coding
region
due to the incorporation of partial multiple cloning sites from both pRC/CMV
and pCMVLi.
pGem/eLuc: pGem/eLuc was created from component fragments of
pGem Luc (Promega) and SK/EMCV IRES. The vector pGem Luc was digested
with the BamHI; the single-stranded overhang left by digestion with BamHI was
removed by treatment with S 1 nuclease. The linearized vector was then cut by
NotI which cuts within 20 base pairs of the BamHI site. SK/EMCV was digested
first with XhoI and the single-stranded overhang left after digestion was
removed
by treatment with S 1 nuclease. The linearized SK/EMCV was then digested by
NotI. The 0.64-kb NotI/XhoI (S 1 nuclease treated) fragment was then cloned
into pGem Luc modified as above.
SK/EMCV IRES: SK/EMCV IRES was created from component
fragments of pBluescriptSK- (Stratagene) and pED4 {R. J. Kaufrnan et al.,
Nucleic Acids Res.,19(16), 4485-90 (1991)). pBluescriptSK- was digested by
the restriction endonucleases EcoRI and XhoI, which both cut within the
multiple
cloning site of pBluescriptSK-. pED4 was digested with EcoRI and Xhol to
obtain the 0.60-kb fragment corresponding to the EMCV IRES. The
EcoRI/XhoI fragment was then ligated into pBluescriptSK- modified as above.
pBL: pBL was created from phBeL. phBeL was digested by the
restriction endonucleases KpnI and NotI. The single stranded overhangs left by
these restriction enzymes was then removed by treatment with S1 nuclease. The
two large fragments, the 6.02-kb KpnI/KpnI fragment and the 3.47-kb Not1/NotI
fragment, were Iigated together. This resulted in a loss of a 70-base pair
fragment within the multiple cloning site that disrupted the hairpin structure
found in phBeL, and a loss of a 0.51-kb fragment corresponding to the all but
100 base pairs of the EMCV IRES.
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
72
pBeL: pBeL was made from phBeL and pBL. Both vectors were cut
with the restriction endonucleases ScaI and BssHII. The ScaI recognition site
is
within the amp resistance gene and the BssHII recognition site is within the
13-
galactosidase coding region. The 7.03-kb fragment of phBeL was combined
with the 2.97-kb fragment of pBL. This resulted in a loss of the hairpin
structure
of phBeL while maintaining the complete EMCV IRES.
pnBeG: pnBeG was constructed of component fragments of
SK/nBeG(afinx) and pBL. Both vectors were digested with the restriction
endonucleases SacI and XmaI. SacI cuts within the amp resistance gene and
XmaI cuts just upstream of the B-galactosidase gene in either vector. The 6.67-
kb XmaI/SacI fragment of SK/nBeG(afinx) was ligated to the 1.35-kb SacI/XmaI
fragment of pBL. This regenerated the amp resistance gene and replaced the T7
promoter region of SK/nBeG(afinx) with the CMV/T7 promoters located within
pBL. pnBeG was further optimized by PCR mutagenesis of the IRES-GFP
junction to GAAAAACACGATTGCTATATGGCCACA ACCATGGCTAGC
(SEQ ID N0:64). This sequence restored wild-type EMCV IRES spacing from
the polypyrimidine tract to the ATG start codon (double underline), as well as
restoring the wild-type sequence around the start codon. Incorporation of an
Nhel restriction endonuclease site (italics) allowed a fusion with the unique
NheI
restriction site in the Affymax GFP (Affymax, Santa Clara, CA). One fusion
site
is four amino acids downstream of the EMCV IRES initiation codon. The fission
also recreated the NcoI restriction endonuclease site (underlined), which is
found
in some strains of EMCV. The Affymax GFP is a GFP that has been modified to
fluoresce more than GFP. Also, the 0.56-kb MscI/EcoRI fragment of pXex-GM2
(Obtained from Shao Lin, Dept. of Biochemistry and Molecular Biology,
Medical College of Georgia, Augusta, GA) was used to replace the 0.55-kb
MscIlEcoRI fragment of pnBeG. This moved the chromophore and C-terminus
of an enhanced GFP (GM2) (B.P. Cormack et al., Gene,173(1 Spec No), 33-8
(1996)) into pnBeG. GM2 is a GFP that has been modified to fluoresce more
than Affymax GFP. Teh construct with the optimal spacing between the EMCV
IRES and the GM2 was named pnBeG*.
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
73
SK/nBeG(afmx): SK/nBeG(afmx) was constructed from component
fragments of SK/eG(afmx) and KS/NCOnlsBgal. SK/eG(afinx) was digested
with the restriction endonuclease EcoRV . This linearized the vector upstream
of the EMCV IRES and Affymax GFP. KS/NCOnlsBgal was digested with the
restriction endonucleases DraI and SpeI. Following this digestion, the single-
stranded overhangs created by these enzymes were completely filled by using
T4 polymerise. The 3.28-kb SpeI/DraI fragment, which contained a nuclear
localized variant of B-galactosidase, was ligated into the EcoRV digested
SK/eG(afmx). Recombinants with the B-galactosidase coding region on the
same coding strand as the GFP were selected.
SK/eG(afmx): SK/eG(afmx) was created with component fragments of
SK/B-globin 3'UTR 2a, SK/EMCV IRES, and pBAD-GFP (A. Crameri et al.,
Nat. Biotechnol., 14(3), 315-9 (1996), available from Affymax). SK/B-globin
3'UTR 2a was digested with EcoRI. This linearized SK/B-globin 3'UTR 2a 5'
of the Xenopus B-globin 3'UTR. SK/EMCV IRES was digested first with
XhoI. The single-stranded overhang created by the XhoI enzyme was restored
to double-stranded DNA by filling in nucleotides with Klenow polymerise.
SK/EMCV IRES was then digested with EcoRI. The resultant 0.60-kb
EcoRI/XhoI (filled) fragment contained the EMCV IRES. pBAD-GFP was
first digested with XbaI. The single-stranded overhang created by XbaI
digestion was completely filled using Klenow polymerise. The pBAD-GFP
was then digested with EcoRI. The 0.73-kb XbaI/EcoRI fragment contained
the complete coding region for the Affymax GFP. Successful recombinants of
these three fragments have the EMCV IRES fused to the Affymax GFP
upstream of the Xenopus B-globin 3' UTR.
SK/B-globin 3'UTR 2a: SK/B-globin 3' UTR 2a was created from
component fragments of pBluescriptSK- (Stratagene) and XenB3UTR (a gift of
H. Joseph Yost, Huntman Cancer Center, Univeristy of Utah, Salt Lake City,
UT). The XenB3UTR was digested with EcoRI and XbaI. The single-
stranded overhang resulting from digestion with these enzymes was completely
filled using Klenow polymerise. This fragment containing the Xenopus B-
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
74
globin 3' UTR cDNA in the orientation from EcoRI to XbaI was cloned into
the SmaI site of pBluescriptSK- (Stratagene). The recombinants with the
EcoRI/XbaI fragment of the XenB3UTR in the orientation from the T7 to the
T3 primer binding sites of pBluescriptSK- were SK/B-globin 3' UTR 2a.
KS/NCOnlsBgal: KS/NCOnls13ga1 was constructed from component
fragments of pBluescript KS-, pPD1.27 (A. Fire et al., Gene, 93(2), 189-98
(1990)), and a short adapter (AGCCATGGCT) (SEQ ID N0:65). pBluescript
KS- was cut with XbaI and NotI. Both of these enzymes cut within the
multiple cloning site of pBluescript KS- and therefore the digest results in a
linearization of the pBluescript KS-. pPD1.27 was also cut with XbaI and
NotI. From this digest a 3.61-kb fragment, that contained the complete coding
sequence of the nuclear localized B-galactosidase and the SV40 poly{A) signal,
was ligated to linearized pBluescript KS-. The resultant plasmid KS/nlsBgal
was digested with XbaI. The single-stranded DNA resulting from digestion
with XbaI was completely filled in using Klenow polymerise. This linearized
fragment of KS/nlsBgal was then religated with the addition of an adapter
(AGCCATGGCT) (SEQ ID N0:65) that contained an NcoI restriction site.
This also insured a good Kozak context for the initiation codon.
Microinjection of Zehrafish. Embryos from wild-type zebrafish were
obtained and maintained as described (M. Westerfield, The Zebrafish Book,
University of Oregon Press, Eugene, OR (1995)). For dicistronic mRNA
injections, capped synthetic mRNA was prepared using Ambion's mMessage
machine and diluted to 200 pg/ml with DEPC-treated H20 prior to injection.
Purified supercoiled DNA was diluted to 50 pg/ml with H20 prior to injection.
One nanoliter of capped mRNA or DNA was injected into or just under the
cytoplasm of single-cell embryos. Post-injection embryos were incubated at
28.5°C.
13-galactosidase and Luciferase Expression Levels. Embryos injected
with pBeL, pBL, or phBeL mRNAs were collected in groups of five embryos at
0.5, 2, 4, 6, 8, 10, and 12 hours postinjection. The embryos were lysed with
50
~.1 of lx reporter lysis buffer (Promega) and a micropestal. Embryonic lysates
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
?5
were stored at -80°C prior to further analysis. Frozen lysates were
thawed by
hand, and microfuged at 8,000 x g at 4°C for S minutes. Lysates were
kept on
ice at all times during preparation. One embryonic equivalent (10 ~1 of
lysate)
was tested for 13-galactosidase and luciferase activity. 13-galactosidase and
luciferase activity were measured using a Berthold Lumat LB9501 luminometer
with Galacto-Light (Tropix, Bedford, MA) and luciferase (Promega) assay
systems, respectively.
Immunohistochemistry. Embryos at various stages of development
were manually dechorinated and fixed overnight at 4°C with 4%
paraformaldehyde in PBST [200mM phosphate, 0.8% NaCI, 0.02% KCI, and
0.1% Tween-20, pH ?.3]. Batches of no more than 100 embryos were washed 8
times with immunowash solution [1% BSA, 1% DMSO in PBST] for 15
minutes at room temperature. Following washing, the embryos were incubated
in immunoblock solution [5% goat serum in immunowash solution] for 3 hours.
They were then incubated overnight at 4°C in 100 ul of immunoblock
solution
containing a 1:1500 dilution of mouse monoclonal antibody against !3-
galactosidase (Boehringer-Mannheim, Indianapolis, IN) and a 1:40 dilution of
rabbit polyclonal antibody to luciferase (Cortex Biochem, San Leandro, CA).
The embryos were then washed and blocked as above and incubated overnight at
4°C in 100 ul of immunoblock containing a 1:1500 dilution of FITC-
conjugated
goat monoclonal antibody to mouse IgG and a 1:1000 dilution of rhodamine-
conjugated goat monoclonal antibody to rabbit IgG. The embryos were washed
as above and mounted in 50% glycerol in PBST. Imaging was done on a BioRad
MRC-1000/1024 laser scanning confocal microscope.
GFP Detection. GFP expression was visualized in manually
dechorinated living embryos anesthetized with tricaine as described
(Westerfield, M. The Zebrafish Book. University of Oregon Press. (1995)).
Imaging was done on a BioRad MRC-1000/1024 laser scanning confocal
microscope.
b. Results
Dicistronic Vectors and mRNAs. pBeL (Figure 13) encodes 13-
galactosidase in the first cistron and luciferase in the second cistron. The
two
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
76
cistrons are separated by the EMCV IRES. 13-galactosidase was expected to be
translated by standard cap-dependent scanning whereas luciferase is expected
to
be translated only if the EMCV IRES directs internal initiation in a
developing
zebrafish. Alternatively, luciferase activity detected from pBeL could be due
to
leaky scanning through the 13-galactosidase initiation codon or reinitiation
of
ribosomes at the luciferase initiation codon. To prevent these
misinterpretations
two dicistronic control vectors, phBeL and pBL (Fig. 13), were constructed. In
phBeL, an additional sequence in the 5' UTR forms a stable hairpin structure
in
the mRNA that should prevent ribosomal scanning to the 13-galactosidase open
reading frame. If the luciferase activity observed in the test vector, pBeL,
is due
to leaky scanning, the luciferase activity observed in phBeL should be reduced
to
the same extent as the 13-galactosidase expression. However, if the EMCV IRES
promotes internal initiation, luciferase expression levels should be
unaffected by
the incorporation of a hairpin structure in the S' UTR of phBeL. In pBL, the
majority of the EMCV IRES was removed. If the luciferase activity observed in
the test vector, pBeL, is from ribosomes that have translated the 13-
galactosidase
open reading frame followed by reinitiation at the luciferase initiation
codon,
luciferase levels from pBL should be comparable to those in pBeL. However, if
the expression of luciferase in pBeL, is due to internal initiation directed
by the
EMCV IRES, there should be little to no luciferase activity in pBL-injected
embryos.
Expression from dicistronic mRNAs in zebrafish. mRNA was
transcribed in vitro using the T7 promoter present in pBeL, pBL, and phBeL
(Fig. 13). Shown in Fig. 14 are the >3-galactosidase and luciferase activities
of
pBeL, phBeL, or pBL mRNA-injected embryos at 6 hours postinjection. pBeL-
injected embryos expressed significant amounts of both 13-galactosidase and
luciferase. This was the first indication that a dicistronic message could
produce
protein from both of its open reading frames in developing zebrafish embryos.
In phBeL-injected embryos, a hairpin structure in front of the first open
reading
frame,13-galactosidase, blocked its production but did not affect production
of
luciferase in the second cistron. Deletion of the EMCV IRES blocked the
production of luciferase from the second cistron in pBL-injected embryos, but
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
77
did not affect 13-galactosidase production. Thus, the EMCV IRES is required
for
translation of luciferase from the dicistronic message pBeL in zebrafish
embryos,
and translation of the second cistron is not occurring by a leaky scanning or
reinitiation mechanism.
Immunolocalization of Dicistronic Reporters. To determine whether
or not translation of both cistrons could occur equally well in the various
tissues
of a developing zebrafish,13-galactosidase and luciferase were localized by
immunohistochemistry. pBeL plasmid DNA was injected into or just under the
cytoplasm of single cell embryos. The embryos were then fixed and
immunostained. The embryos displayed highly mosaic expression patterns
characteristic of DNA injections. Cells positive for 13-galactosidase also
stained
for luciferase. Occasionally, weakly expressing cells were observed to express
only one of the two reporters. Presumably this is because the luciferase
expression in this cell is below the detection limits of our
immunohistochemical
assay since other myotomes with higher 13-galactosidase expression stain quite
well for luciferase. Approximately 200 fish embryos of greater than 10,000
cells
each have been observed. There has been no observation of a brightly
expressing cell for one reporter which did not express the other reporter.
Alternative reporters expressed from the EMCV IRES. In order to
increase the functionality of dicistronic vector usage in zebrafish, the
ability of
the EMCV IRES to express detectable quantities of GFP was examined. GFP is
a powerful reporter in the optically clear embryos of the zebrafish because it
allows non-invasive analysis of expression in living embryos. Embryos injected
with pnBeG DNA were examined for GFP expression at 24 hours postinjection.
Although the observed GFP expression was only 5-15% of what is seen when
standard monocistronic GFP expression cassettes are injected into zebrafish,
its
expression was readily detectable. GFP was expressed in a wide variety of
cells
derived from ectoderm, mesoderm, and endoderm. Expression of GFP was seen
in several myotomes and cells in the blood island, which are derived from
mesoderm and endoderm respectively. Several cells in the head region of a 24-
hour embryo that express GFP were observed, including several ectoderm-
derived cells along the dorsal edge of the hindbrain.
*rB
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
78
II. Gene-trap vector construction
A gene-trap transposon vector has been constructed and injected into
zebrafish embryos (see, e.g., Fig. 12(A)). At least one specific cell in
several
embryos at approximately 28 hours post-injection tested positive for the
detectable marker encoded by the gene-trap, indicating that the gene-trap had
transposed into a coding sequence present in the zebrafish genome.
a. Methods
pFGT/eGFP-b: pFGT/eGFP-b was formed from component fragments
of pT/HB and pFV/e(nls)G. The parental vector, pTIHB, was cut with the
restriction endonucleases BgIII and EagI. Prior to the cloning the BgIII and
EagI
recessed ends were completely filled in using Klenow polymerase. pFV/e(nls)G
was cut with the restriction endonuclease NaeI and the fragment containing
approximately the last 200 nucleotides of the carp 13-actin intron 1, the EMCV
IRES, GFP, and the Chinook salmon growth hormone (CSGH) poly(A) signal
(otained from Dr. Choy Hew, Department of Biochemistry, Hospital for Sick
Children, Toronto, Canada) was cloned into the pT/HB vector modified as
above. The orientation of pFGT/eGFP-b has IR/DR(R) of the sleeping beauty
transposon followed by the remnant BgIII site, the 3' end of carp I3-actin
intron I,
the EMCV IRES, GFP, the CSGH poly(A) signal, the remnant EagI site, and the
IR/DR(L) of the sleeping beauty transposon.
pT/HB: pT/HB was constructed from components of pBluescript KS-
(Stratagene) and pT/SVNeo (Z. Ivics et al. Cell, 91(4), 501-10 (1997}).
pBluescript KS- was digested with the restriction endonucleases SacI and AccI;
this digest removes most of the multiple cloning site found within pBluescript
KS-. pT/SVNeo was also cut with SacI and AccI. This digest gave two products
one of them being the SVNeo sleeping beauty transposon complete with both
IR/DRs. The transposon piece was then cloned into pBluescript KS-. This
vector, pT/HindIII-precursor, was then digested with the restriction
endonuclease
HindIII. This digest removed the internal portion of the transposon containing
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/Z4348
79
the SV40 promoter, neomycin resistance gene, and SV40 poly(A) signal. The
remaining vector piece was ligated to create the plasmid pT/HindIII, a vector
containing a single HindIII site between the IR/DRs of the sleeping beauty
transposon system. pT/HindIII was then cut with XbaI. XbaI cut pT/HindIII
once, and the recessed ends of this digestion were completely filled in using
Klenow polymerase. The resultant fragment was then ligated to form pTlMCS-
precursor. pT/MCS-precursor was then cut with HindIII. Into this vector a
short
double-stranded oligo was ligated to produce a multiple cloning region
containing restriction endonuclease sites for HindIII, EcoRV, EcoRI, Spel,
EagI,
NotI, XbaI, and BgIII. pT/HB has the multiple cloning oligo inserted so that
the
sites go from HindIII to BgIII with respect to the orientation of IR/DR(L) to
IR/DR(R).
pFV/e(nls)G: pFV/e(nls)G was formed from components of pFV3
(Caldovic L., et al., Mol. Mar. Biol. Biotechnol., 4, 51-61 (1995)) and
pnBeG*.
1 S pFV3 was first digested by EcoRI. This linearized pFV3 just 3' of the CSGH
poly(A) signal. After digestion with EcoRI, the recessed ends of pFV3 were
completely filled in using Klenow polymerase. The resultant fragment was self
ligated to form pFV30Ri. A double-stranded oligo, FV7-MCS
(CGGGGTACCGAATTCCCGGGTACCCCG) (SEQ ID N0:66) containing an
EcoRI and SmaI sites within KpnI sites, was digested with KpnI. This oligo was
then cloned into pFV3ARI cut with KpnI, which cuts once just after the carp b-
actin intron 1. There were two products of this ligation, pFV7a and pFV7b.
pFV7a has the SmaI site preceding the EcoRI site in relationship to the carp
13-
actin promoter, carp b-actin exon 1, carp 13-actin intron 1, and the CSGH
poly(A)
signal. pnBeG* was then cut with EcoRI. One of the resulting fragments of this
digest contained only the EMCV IRES and GFP. This fragment was then cloned
into pFV7a digested with EcoRI. The product that contained the EMCV IRES
and GFP in the proper orientation with respect to the fish elements (i.e.
promoter, exon, intron, poly(A) signal) was named pFV/eG. pFV/eG was then
digested with the restriction endonuclease NheI that cuts just after the
initiation
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
codon of GFP. Into this site a short double-stranded oligo, NLS2
(TACTCCACCAAAGAAGAGAAAGGT GGAGGACG (SEQ ID N0:67) with
CTAG 5' end overhangs), was ligated. One of the resulting products of this
ligation, pFV/e(nls)G, has an additional 12 amino acids (TPPKKRKVE DAS)
5 (SEQ ID N0:68) encoding the SV40 nuclear localization signal.
pFGT/etTA: pFGT/etTA was formed from component fragments of
pFGT/eGFP-b and pTet-Off (Clontech). The parental vector, pFGT/eGFP-b was
cut with NcoI and SpeI. This digest removed the GFP and CSGH poly(A) signal
from the remaining pFGT/eGFP-b vector. The tetracycline responsive
10 transcriptional activator (tTA) of pTet-Off was PCR mutagenized to create
an
NcoI site at the initiator AUG using the sense primer KJC-008
(CATCCATGGCTAGATTAGATAAAAGTAAAG TAAAG) (SEQ ID N0:69).
This allowed in-frame fusion of the tTA behind the first four amino acids
(MATT) (SEQ ID N0:70) of the EMCV polypeptide, insuring efficient
15 translation from the IRES. The antisense primer KJC-009
(GCTCTAGACTAGTGATTTTTTTCTCCATTTTAGC) (SEQ ID N0:71)
incorporated a SpeI recognition site just after the SV40 poly(A) signal in the
pTet-Off vector. The NcoI-tTA PCR product was cut with the restriction
endonucleases NcoI and SpeI and cloned into the pFGT/eGFP-b vector modified
20 as above.
pSBRNAX: The pSBRNAX vector was made with component
fragments from SK/11-globin 3'UTR 2a and SB10 transposase (Z. Ivics et al.
Cell, 91 (4), 501-10 (1997)). SK/b-globin 3'UTR 2a was digested with the
restriction endonuclease EcoRV. The SB10 transposase was ampified by
25 polymerase chain reaction that incorporated an BamHI restriction site
upstream
of the SB 10-coding sequence and an EcoRI restriction site downstream of the
SB10-coding sequence as described by Z. Ivics et al. Cell, 91(4), SO1-10
(1997).
This fragment was digested with BamHI and EcoRI, and the resulting single-
strand DNA overhangs were completely filled in using Klenow polymerase. The
30 resulting 1.03-kb fragment was then ligated into the linearized SK/13-
globin3' UTR-2a.
CA 02309000 2000-OS-04
WO 99/Z5817 PCTNS98/24348
81
Microinjection of Zebrafish. Embryos from wild-type zebrafish were
used for all experiments as described (M. Westerfield, The Zebrafish Book,
University of Oregon Press, Eugene, OR {1995)). For injections of gene trap
vectors, 3 pl of 50 ~g/ml of pFGT/eGFP-b DNA was mixed with 1 pl of 100
~g/ml Sleeping Beauty mRNA. pFGT/eGFP-b was injected as a supercoiled
plasmid or as linear DNA. The linear form of pFGT/eGFP-b was obtained by
digestion with the restriction endonuclease BspHI, which has two recognition
sites within the vector backbone. The two resultant fragments were separated
by
gel electrophoresis, and the transposon containing fragment was purified using
Qiagen's gel extraction kit. The Sleeping Beauty mRNA was produced using
Ambion's mMessage Machine with pSBRNAX digested with NotI as template.
One nanoliter of solution [37.5 ,uglml pFGT/eGFP-b and 25 ~g/ml sleeping
beauty mRNA] was injected into the cytoplasm or within the yolk just below the
cytoplasm of 1-cell zebrafish embryos.
b. Results
Embryos were injected with linear pFGT/eGFP-b DNA and Sleeping
Beauty mRNA, grown to about the 28-hour stage and illuminated with blue
light. Expression of GFP in selective cells (for instance, muscle pioneer
cells
and myotomes) emitted a green fluorescence, indicating that the transposon had
integrated into a gene that was expressed in these cells.
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
82
Sequence Listing Free Text
SEQ ID NO:1; An SB transposase.
SEQ ID N0:2; Junction sequence of T/neo transposon integrated into
human genomic DNA.
SEQ ID N0:3; Nucleic acid sequence encoding an SB protein.
SEQ ID N0:4-5; An inverted repeat sequence.
SEQ ID N0:6; 5' outer direct repeat.
SEQ iD N0:7; 5' inner direct repeat.
SEQ ID NO:B; 3' inner direct repeat.
SEQ ID N0:9; 3' outer direct repeat.
SEQ ID NO:10; A consensus direct repeat.
SEQ ID NO:11; A portion of a direct repeat sequence.
SEQ ID N0:12-36; Oligonucleotide primer.
SEQ ID N0:37; Salmonid transposase-binding sites.
SEQ ID N0:38; Zebrafish Tdrl transposase-binding sites.
SEQ ID N0:39: Salmonid transposase-binding sites.
SEQ ID N0:40; Zebrafish Tdrl transposase-binding sites.
SEQ ID N0:41; Outer transposase-binding site in SB transposon
SEQ ID N0:42; Internal transposase-binding site in SB transposon.
SEQ ID N0:43-44; Junction sequence of T/neo transposon integrated
into pUC 19 DNA.
SEQ ID N0:45-63; Junction sequence of T/neo transposon integrated
into human genomic DNA.
SEQ ID N0:64; IRES-GFP junction in pnBeG.
SEQ ID N0:65; An adaptor.
SEQ ID N0:66-67; A double stranded oligonucleotide.
SEQ ID N0:68; SV40 nuclear localization signal.
SEQ ID N0:69; Oligonucleotide primer.
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
83
SEQ ID N0:70; Amino acids of EMCV polypeptide.
SEQ ID N0:71-75; Oligonucleotide primer.
CA 02309000 2000-OS-04
WO 99125817 PCTNS98/24348
1
SEQUENCE LISTING
<110> Regents of the University of Minnesota
<120> DNA-BASED TRANSPOSON SYSTEM FOR THE INTRODUCTION OF
NUCLEIC ACID INTO DNA OF A CELL
<130> 110.00870201
<140> Not Assigned
<141> 1998-11-13
<150> 60/065,303
<151> 1997-11-13
<160> 75
<170> PatentIn Ver. 2.0
<210> 1
<211> 340
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: An SB
transposase
<400> 1
Met Gly Lys Ser Lys Glu Ile Ser Gln Asp Leu Arg Lys Lys Ile Val
1 5 10 15
Asp Leu His Lys Ser Gly Ser Ser Leu Gly Ala Ile Ser Lys Arg Leu
20 25 30
Lys Val Pro Arg Ser Ser Val Gln Thr Ile Val Arg Lys Tyr Lys His
35 40 45
His Gly Thr Thr Gln Pro Ser Tyr Arg Ser Gly Arg Arg Arg Val Leu
50 55 60
Ser Pro Arg Asp Glu Arg Thr Leu Val Arg Lys Val Gln Ile Asn Pro
65 70 75 80
Arg Thr Thr Ala Lys Asp Leu Val Lys Met Leu Glu Glu Thr Gly Thr
85 90 95
Lys Val Ser Ile Ser Thr Val Lys Arg Val Leu Tyr Arg His Asn Leu
100 105 110
Lys Gly Arg Ser Ala Arg Lys Lys Pro Leu Leu Gln Asn Arg His Lys
115 120 125
Lys Ala Arg Leu Arg Phe Ala Thr Ala His Gly Asp Lys Asp Arg Thr
130 135 140
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98I24348
2
Phe Trp Arg Asn Val Leu Trp Ser Asp Glu Thr Lys Ile Glu Leu Phe
145 150 155 160
Gly His Asn Asp His Arg Tyr Val Trp Arg Lys Lys Gly Glu Ala Cys
165 170 175
Lys Pro Lys Asn Thr Ile Pro Thr Val Lys His Gly Gly Gly Ser Ile
180 185 190
Met Leu Trp Gly Cys Phe Ala Ala Gly Gly Thr Gly Ala Leu His Lys
195 200 205
Ile Asp Gly Ile Met Arg Lys Glu Asn Tyr Val Asp Ile Leu Lys Gln
210 215 220
His Leu Lys Thr Ser Val Arg Lys Leu Lys Leu Gly Arg Lys Trp Val
225 230 235 240
Phe Gln Met Asp Asn Asp Pro Lys His Thr Ser Lys Val Val Ala Lys
245 250 255
Trp Leu Lys Asp Asn Lys Val Lys Val Leu Glu Trp Pro Ser Gln Ser
260 265 270
Pro Asp Leu Asn Pro Ile Glu Asn Leu Trp Ala Glu Leu Lys Lys Arg
275 280 285
Val Arg Ala Arg Arg Pro Thr Asn Leu Thr Gln Leu His Gln Leu Cys
290 295 300
Gln Glu Glu Trp Ala Lys Ile His Pro Thr Tyr Cys Gly Lys Leu Val
305 310 315 320
Glu Gly Tyr Pro Lys Arg Leu Thr Gln Val Lys Gln Phe Lys Gly Asn
325 330 335
Ala Thr Lys Tyr
340
<210> 2
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 2
tgtttattgc ggcactattc 20
<210> 3
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
3
<211> 1023
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Nucleic acid
sequence encoding an SB protein
<400> 3
atgggaaaat caaaagaaat cagccaagac ctcagaaaaa aaattgtaga cctccacaag 60
tctggttcat ccttgggagc aatttccaaa cgcctgaaag taccacgttc atctgtacaa 120
acaatagtac gcaagtataa acaccatggg accacgcagc cgtcataccg ctcaggaagg 180
agacgcgttc tgtctcctag agatgaacgt actttggtgc gaaaagtgca aatcaatccc 240
agaacaacag caaaggacct tgtgaagatg ctggaggaaa caggtacaaa agtatctata 300
tccacagtaa aacgagtcct atatcgacat aacctgaaag gccgctcagc aaggaagaag 360
ccactgctcc aaaaccgaca taagaaagcc agactacggt ttgcaactgc acatggggac 420
aaagatcgta ctttttggag aaatgtcctc tggtctgatg aaacaaaaat agaactgttt 480
ggccataatg accatcgtta tgtttggagg aagaaggggg aggcttgcaa gccgaagaac 540
accatcccaa ccgtgaagca cgggggtggc agcatcatgt tgtgggggtg ctttgctgca 600
ggagggactg gtgcacttca caaaatagat ggcatcatga ggaaggaaaa ttatgtggat 660
atattgaagc aacatctcaa gacatcagtc aggaagttaa agcttggtcg caaatgggtc 720
ttccaaatgg acaatgaccc caagcatact tccaaagttg tggcaaaatg gcttaaggac 780
aacaaagtca aggtattgga gtggccatca caaagccctg acctcaatcc tatagaaaat 840
ttgtgggcag aactgaaaaa gcgtgtgcga gcaaggaggc ctacaaacct gactcagtta 900
caccagctct gtcaggagga atgggccaaa attcacccaa cttattgtgg gaagcttgtg 960
gaaggctacc cgaaacgttt gacccaagtt aaacaattta aaggcaatgc taccaaatac
1020
tag
1023
<210> 4
<211> 226
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: An inverted
repeat sequence
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
4
<400> 4
agttgaagtc ggaagtttac atacacttaa gttggagtca ttaaaactcg tttttcaact 60
acaccacaaa tttcttgtta acaaacaata gttttggcaa gtcagttagg acatctactt 120
tgtgcatgac acaagtcatt tttccaacaa ttgtttacag acagattatt tcacttataa 180
ttcactgtat cacaattcca gtgggtcaga agtttacata cactaa 226
<210> 5
<211> 229
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: An inverted
repeat sequence
<400> 5
ttgagtgtat gttaacttct gacccactgg gaatgtgatg aaagaaataa aagctgaaat 60
gaatcattct ctctactatt attctgatat ttcacattct taaaataaag tggtgatcct 120
aactgacctt aagacaggga atctttactc ggattaaatg tcaggaattg tgaaaaagtg 180
agtttaaatg tatttggcta aggtgtatgt aaacttccga cttcaactg 229
<210> 6
<211> 29
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: 5' outer
direct repeat
<400> 6
gttgaagtcg gaagtttaca tacacttag 29
<210> 7
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: 5' inner
direct repeat
<400> 7
cagtgggtca gaagtttaca tacactaagg 30
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
<210> 8
<211> 31
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: 3~ inner
direct repeat
<400> 8
cagtgggtca gaagttaaca tacactcaat t 31
<210> 9
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: 3~ outer
direct repeat
<400> 9
agttgaatcg gaagtttaca tacaccttag 30
<210> 10
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: A consensus
direct repeat
<400> 10
caktgrgtcr gaagtttaca tacacttaag 30
<210> 11
<211> 8
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: A portion of a
direct repeat sequence
<400> 11
acatacac 8
<210> 12
<211> 23
<212> DNA
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
6
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 12
tttcagtttt gggtgaacta tcc 23
<210> 13
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 13
ggcgacrcag tggcgcagtr gg 22
<210> 14
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 14
gaayrtgcaa actccacaca ga 22
<210> 15
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 15
tccatcagac cacaggacat 20
<210> 16
<211> 25
<212> DNA
<213> Artificial Sequence
<220>
CA 02309000 2000-OS-04
WO 99/Z5817 PCT/US98/24348
7
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 16
tgtcaggagg aatgggccaa aattc 25
<210> 17
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 17
cctctaggat ccgacatcat g 21
<210> 1B
<211> 28
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 18
tctagaattc tagtatttgg tagcattg 28
<210> 19
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 19
aacaccatgg gaccacgcag ccgtca 26
<210> 20
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
CA 02309000 2000-OS-04
WO 99/Z5817 PCT/US98I24348
B
<400> 20
caggttatgt cgatatagga ctcgttttac 30
<210> 21
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 21
ccttgctgag cggcctttca ggttatgtcg 30
<210> 22
<211> 18
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 22
ttgcactttt cgcaccaa 1g
<210> 23
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 23
gtacctgttt cctccagcat c 21
<210> 24
<211> 18
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 24
gagcagtggc ttcttcct 18
CA 02309000 2000-OS-04
PCT/US98/24348
WO 99/25817
9
<210> 25
<211> 18
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 25
ccacaacatg atgctgcc 18
<210> 26
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 26
tggccactcc aataccttga c 21
<210> 27
<211> 31
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 27
acactctaga ctagtatttg gtagcattgc c 31
<210> 2$
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 2B
gtgcttcacg gttgggatgg tg 22
<210> 29
<211> 26
<212> DNA
CA 02309000 2000-OS-04
WO 99/25817
PCT/US98/24348
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 29
attttctata ggattgaggt cagggc 26
<210> 30
<211> 35
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 30
gtctggttca tccttgggag caatttccaa acgcc 35
<210> 31
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 31
caaaaccgac ataagaaagc cagactacgg 30
<210> 32
<211> 44
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 32
accatcgtta tgtttggagg aagaaggggg aggcttgcaa gccg 44
<210> 33
<211> 36
<212> DNA
<213> Artificial Sequence
<220>
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
11
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 33
ggcatcatga ggaaggaaaa ttatgtggat atattg 36
<210> 34
<211> 29
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 34
ctgaaaaagc gtgtgcgagc aaggaggcc 29
<210> 35
<211> 27
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 35
gtggaaggct acccgaaacg tttgacc 27
<210> 36
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 36
gacaaagatc gtactttttg gagaaatgtc 30
<210> 37
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Salmonid
transposase-binding sites
CA 02309000 2000-OS-04
WO 99/25817 PCTNS98/24348
12
<400> 37
gttgaagtcg gaagtttaca tacacttagg 30
<210> 38
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Zebrafish Tdri
transposase-binding sites
<400> 38
gtttaaacca gaagtttaca cacactgtat 30
<210> 39
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Salmonid
transposase-binding sites
<400> 39
ccagtgggtc agaagtttac atacactaag 30
<210> 40
<211> 28
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Zebrafish Tdr1
transposase-binding sites
<400> 40
cttgaaagtc aagtttacat acaataag 28
<210> 41
<211> 34
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Outer
transposase-binding site in SB transposon
<400> 41
tacagttgaa gtcggaagtt tacatacact tagg 34
CA 02309000 2000-OS-04
WO 99/Z5817 PCT/US98/24348
13
<210> 42
<211> 32
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Internal
transposase-binding site in SB transposon
<400> 42
tccagtgggt cagaagttta catacactaa gt 32
<210> 43
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into pUCl9
DNA
<400> 43
tgaattcgag ctcggtaccc tacagt 26
<210> 44
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into pUCl9
DNA
<400> 44
actgtagggg atcctctaga gtcgac 26
<210> 45
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 45
aaatttattt aatgtgtaca tacagt 26
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
14
<210> 46
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 46
actgtataag aacctttaga acgaag 26
<210> 47
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 47
aaatttattt aatgtgtaca to 22
<210> 48
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 48
taagaacctt tagaacgaag 20
<210> 49
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 49
gaataaacag tagttcaact tacagt 26
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/2434$
<210> 50
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 50
actgtatatg ttttcatgga aaatag 26
<210> 51
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 51
gaataaacag tagttcaact to 22
<210> 52
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 52
tatgttttca tggaaaatag 20
<210> 53
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 53
tcactgactc attcaacatc tacagt 26
CA 02309000 2000-OS-04
WO 99/25817 PCT/US98/24348
16
<210> 54
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 54
actgtattta ttgaatgcct gctgaa 26
<210> 55
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 55
tcactgactc attcaacatc to 22
<210> 56
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 56
tttattgaat gcctgctgaa 20
<210> 57
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 57
acttacataa ttataagttt tacagt 26
CA 02309000 2000-OS-04
WO 99/25817
17
PCT/US98/24348
<210> 58
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 58
actgtatata atgatgacat ctatta 26
<210> 59
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 59
acttacataa ttataagttt to 22
<210> 60
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 60
tataatgatg acatctatta 20
<210> 61
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 61
tataaagaca cattcacatg tacagt 26
CA 02309000 2000-OS-04
WO 99/25817
1s
PCTNS98/24348
<210> 62
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 62
actgtatgtt tactgcggca ctattc 26
<210> 63
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Junction
sequence of T/neo transposon integrated into human
genomic DNA
<400> 63
tataaagaca catgcacacg to 22
<210> 64
<211> 39
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: IRES-GFP
junction in pnBeG
<400> 64
gaaaaacacg attgctatat ggccacaacc atggctagc 39
<210> 65
<211> 10
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: An adaptor
<400> 65
agccatggct 10
<210> 66
<211> 27
CA 02309000 2000-OS-04
WO 99/25817
19
PCT/US98/24348
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: A double
standed oligonucleotide
<400> 66
27
cggggtaccg aattcccggg taccccg
<210> 67
<211> 36
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: A double
standed oligonucleotide
<400> 67
ctagtactcc accaaagaag agaaaggtgg aggacg 36
<210> 68
<211> 12
<212> PRT
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: SV40 nuclear
localization signal
<400> 68
Thr Pro Pro Lys Lys Arg Lys Val Glu Asp Ala Ser
1 5 10
<210> 69
<211> 35
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 69
catccatggc tagattagat aaaagtaaag taaag 35
<210> 70
<211> 4
<212> PRT
<213> Artificial Sequence
CA 02309000 2000-OS-04
WO 99/25817
PCTNS98/24348
<220>
<223> Description of Artificial Sequence: Amino Acids
of EMCV polypeptide
<400> 70
Met Ala Thr Thr
1
<210> 71
<211> 34
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 71
gctctagact agtgattttt ttctccattt tagc 34
<210> 72
<211> 25
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 72
ccacaggtac acctccaatt gactc 25
<210> 73
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide primer
<400> 73
gtggtgatcc taactgacct taagac 26
<210> 74
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
CA 02309000 2000-OS-04
WO 99125817
21
PCT/US98/24348
Oligonucleotide primer
<400> 74
24
gtgtcatgca caaagtagat gtcc
<210> 75
<211> 25
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oiigonucleotide primer
<400> 75
ctcggattaa atgtcaggaa ttgtg 25