Note: Descriptions are shown in the official language in which they were submitted.
CA 02309609 2000-OS-04
WO 99/24421 PCT/US98/23224
IMIDAZOYLA,~KYL SUBSTITUTED WITH A FIVE SIX OR
SEVEN MEMBERED HETEROCYCLIC RING CONTAINING ONE
NITROGEN ATOM
BACKGROUND
H3 receptor sites are known and are of current interest to those
skilled in the art--for example, see: West, Jr. et al., "Biexponential
Kinetics
of (R)-a-[3H]Methylhistamine Binding to the Rat Brain H3 Histamine
Receptor", Journal of Neurochemistry, Vol. 55, No. 5, pp. 1612-1616,
1990; West, Jr. et al., "Identification of Two H3-Histamine Receptor
Subtypes", Molecular Pharmacology, 38:610-613; and Korte et ai.,
"Characterization and Tissue Distribution of H~ Histamine Receptors in
Guinea Pigs by Na-Methylhistamine", Biochemical and Biophysical
Research Communications, Vol. 168, No. 3, pp. 979-986.
Arrang et al. in U.S. 4,767, 778 (Issued August 30, 1988) disclose a
pharmaceutical composition containing a histamine derivative of the
formula:
Ra R3
~C-C-NH2
HN~N R2 R~
wherein each of R~, R2, and R4, represents a hydrogen or a methyl, or R1
and R2 taken together represent a methylene, and Rg is a hydrogen, a
methyl or a carboxy, with the proviso that R1, R2, R3, and R4 are not
simultaneously methyl groups. It is disclosed that the derivatives behave
as complete agonists of the H3 receptors in rat brain and produce a
maximal inhibition of release identical to that induced by histamine
CA 02309609 2000-OS-04
WO 99/24421 PCT/US98/23224
-2-
(approximately 60%). It is also disclosed that the histamine derivatives
powerfully inhibit the release and synthesis of histamine by very
selectively stimulating the H3 receptors. Consequently, according to
Arrang et al., the derivatives are likely to decrease histaminergic
transmission in the digestive tract and in the nervous, cardiovascular and
immune systems. Arrang et al. disclose that the derivatives can be used in
therapy as a drug having sedative effects, as a sleep regulator,
anticonvulsant, regulator of hypothalmic-hypophyseal secretion,
antidepressant, and modulator of cerebral circulation. According to
Arrang et al., inhibition of the release of inflammation messengers in
various allergic conditions (e.g., asthma) is expected to result from
stimulation of the H3 receptors of the lung. It is further disclosed that the
inhibition of release of gastric histamine is likely to exert antisecretory
and
anti ulcerative effects. According to Arrang et al., modification of release
of
the messengers of immune responses is likely to modulate the latter
responses.
Derwent abstract 86-273706/42 for EP 0 197 840 discloses
imidazole derivatives of the formula:
R1
R_N \ a
~N 3 ~N_R2
wherein R~ is H, methyl ar ethyl; R is H or R2; and R2 is 1-6C alkyl,
piperonyl, 3-(benzimidazolon-1-yl)propyl, -CZ-NHRS or a group (i):
-(CH2)~ X ~ '~R3
wherein n is 0-3; X is a bond, O, S, NH, CO, CH=CH or a group (ii):
Rs
CH '
R3 is H, methyl, halo, CN, CF3 or COR4; R4 is 1-6C alkyl, 3-6C cycloalkyl
or phenyl (optionally substituted by methyl or F); Z is O, S, NH, N-methyl or
N-CN; and R5 is 1-8C alkyl, 3-6C cycloalkyl (optionally substituted by
*rB
CA 02309609 2000-OS-04
WO 99/24421 PCT/US98/23224
-3-
phenyl), 3-6C cycioalkyl(1-3C)aikyl, phenyl (optionally substituted by
methyl, halo or CF3), phenyl(1-3C)alkyl, naphthyl, adamantyi or
p-toluenesulphonyl. It is disclosed that these compounds are
psychotropic agents. It is also disclosed that these compounds
antagonize the histamine H3 receptors and increase the speed of cerebral
histamine renewal.
Derwent abstract 90-184730!24 for U.S. 4,925,851 discloses 2- or
4-(2-(1 H-imidazol-1-yl)ethyl) piperidine compounds useful as antitumour
agents for inhibiting lymphoma, sarcoma, myeloma and leukemia. The
compounds have the formula:
2
CH2CH2 N
N
R- N 4
R~
wherein R is -CH2(CH2)m-Me, -CO-(CH2)m-Me or -CO-CMe2-R2; m is 2-
18; R2 is H or Me; Ri is -(CH2)~-R3; n is 0-13; R3 is H, i-Pr or t-Bu; and the
floating group is at the 2- or 4- position; with the proviso that (1 ) the sum
of
C atoms in R~ does not exceed 13; and (2) the sum of C atoms in R and R~
does not exceed 25.
WO 93!12107 published June 24, 1993 discloses a compound of
the formula:
R2 (C) Ra
___ T N-R5
)m"'~ X (1.0)
HN~N (C)P R4
or a pharmaceutically acceptable salt or solvate thereof, wherein:
(A) m is an integer selected from the group consisting
of: 1 and 2;
(B) n and p are integers and are each independently selected
from the group consisting of: 0, 1, 2, 3, and 4 such that the sum of n and p
is 4 and T is a 6-membered ring;
C) R3 and R4 are each independently bound to the same or
different carbon atom of ring T such that there is only one R3 group and
CA 02309609 2000-OS-04
WO 99/24421 PCT/US98/23224
-4-
one R4 group in ring T, and each R~, R2, R3, and R4 is independently
selected from the group consisting of:
(1 ) H;
(2) C1 to C6 alkyl; and
(3) -(CH2)q-R6 wherein q is an integer of: 1 to 7, and R6 is
selected from the group consisting of: phenyl,
substituted phenyl, -ORS, -C(O)ORS, -C(O)RD,
-OC(O)R~, -C(O)NR~RB, CN and -SRS wherein R~ and
R8 are as defined below, and wherein the substituents
on said substituted phenyl are each independently
selected from the group consisting of: -OH, -O-(C~ to
C6)alkyl, halogen, C1 to C6 alkyl, -CF3, -CN, and -
N02, and wherein said substituted phenyl contains
from 1 to 3 substituents;
(D) R5 is selected from the group consisting of:
(1) H;
(2) Ci to C2o alkyl;
(3) C3 to C6 cycioalkyl;
(4) -C(O)OR~~; wherein R~~ is the same as R~ defined
below except that R~~ is not H;
(5) -C(O)RD;
(6) -C(O)NR~RB;
(7) allyl;
(8) propargyl; and
(9) -(CHz)q-R6, wherein q and R6 are as
defined above,
and when q is equal to 1, then R6 is not OH or SH;
(E) R~ and R8 are each independently selected from the group
consisting of: H, C1 to C6 alkyl, and C3 to C6 cycloalkyl;
(F) the dotted line (------) represents a double bond that is
optionally present when m is 1, and n is not 0, and p is not
0, and when said double bond is present then R2 is absent;
and
CA 02309609 2000-OS-04
WO 99/24421 PCT/US98/23224
-5-
(G) when m is 2, each R~ is the same or different substituent for
each m, and each RZ is the same or different substituent for each m,
and at feast two of the substituents R1 and/or R2 are H.
These two latter documents claim the use of the compounds for
treatment of allergy and other disorders.
EP 0 428 434 A2 as well as WO 96/29315 and WO 95/06037
describe a wide range of compounds and claim their use as H3 receptor
(ant)agonist. The above documents also include a comprehensive
summary of the art dealing with this chemical field.
US Application Serial. No. 08/689951 filed August 16, 1996 and
U.S. Application Serial No. 081909319 filed August 14, 1997 disclose
compositions for the treatment of the symptoms of allergic rhinitis using a
combination of at feast one histamine H1 receptor antagonist and at least
one histamine H3 receptor antagonist.
In view of the art's interest in compounds which affect the H3
receptors, novel compounds having antagonist activity on H3 receptors
would be a welcome contribution to the art. This invention provides just
such a contribution by providing novel compounds having H3 antagonist
activity.
SUMMARY OF THE INVENTION
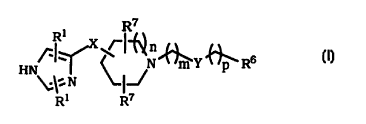
This invention relates to compounds of formula I
Ri ~~~'~n
HN~~~ ~~N~Y~R6 ~
~:~:N ~~J
Ri R~
or pharmaceutically acceptable salts or solvates thereof, wherein:
X is a straight chain alkyl group having 1 to 7 carbon atoms or an
alkene or alkyne group with 2 to 4 carbon atoms; wherein said alkyl or
alkene groups are optionally substituted with up to two (i.e., 1 or 2) R'
groups;
n is 0, 1 or 2 ,
CA 02309609 2000-OS-04
WO 99/24421 PCT/US98/23224
-6-
mandpare0to4;
when m is 0 to 4, Y represents -S02-; -CS-; -CO-; -CONR5 -;
-CO(CH2 )WO- (with w being 1 to 4); -COO-; -CON(OR5)-; -C(NR~)NR5-;
-S02NR~ - or -CSNRS-;
when m is 2 to 4, Y represents all the groups above when m is 0 to
4 and, in addition, Y represents -CHORS -; -O-; -NRSCONRS ; -NR5C0-;
-NR5 -; -OCONR~ -; -NRSC(NR5)NRSV -NRSCSNR5~ -NRSCS- or -NR5S02-;
-NR5C(O)O-; or -CSNRS-;
each R5 independently represents hydrogen, alkyl or benzyi;
R6 represents aryl, heteroaryl, or a 3- to 7- membered heterocyclic
group having one to three heteroatoms in the ring, wherein the
heteroatoms are selected from N, S and O, and wherein said R6 group is
optionally substituted by one to three substituents as defined below;
when Y is -S02 -, then R6, in addition to the above groups, also
represents alkyl having 1 to 7 carbon atoms or a group -NR1~R1 ~ wherein
R~ ~ and R1 ~ are independently selected from H, alkyl or trihalomethyl;
each R~ is independently hydrogen, alkyl or trihalomethyl;
each R~ is independently selected from hydrogen, alkyl,
trihalomethyl, phenyl or benzyl, , wherein said phenyl and benzyl are
optionally substituted by one to three substituents independently selected
from of alkyl, halogen, trihalomethyl, CN, N02, OR» or NR»R> >, wherein
R» and R» wherein R» and R» as above defined.
This invention also provides pharmaceutical compositions
comprising a pharmaceutically acceptable carrier and an effective amount
of a compound (or a salt or solvate thereof) of Formula I.
This invention further provides a method of treating allergy, (for
example asthma), inflammation, cardiovascular disease, hypotension,
raised intraocular pressure (such as glaucoma)--i.e., a method of lowering
intraocular pressure, sleeping disorders (e.g., hypersomnia, somnolence,
narcolepsy and sleeplessness, such as insomnia), diseases of the GI tract,
states of hyper and hypo motility and acidic secretion of the gastro-
intestinal tract, disturbances of the central nervous system, hypo and
CA 02309609 2000-OS-04
WO 99/24421
PCT/US98/23224
_7_
hyperactivity of the central nervous system (for example, agitation and
depression) and other CNS disorders (such as Alzheimer's,
schizophrenia, obesity and migraine) comprising administering an
effective amount of a compound, or a salt or solvate thereof, of Formula I
to a patient in need of such treatment.
This invention further provides a method for treating upper airway
allergic responses by comprising administering an effective amount of a
compound, or a salt or solvate thereof, of Formula I in combination or
admixture with a suitable H~ receptor antagonist.
DETAILED DESCRIPTION OF THE INVENTION
As used herein the following terms have the following meanings
unless indicated otherwise:
alkyl - represents a straight or branched, saturated hydrocarbon
chain having from 1 to 6 carbon atoms;
lower alkyl (including the alkyl portions of lower alkoxy) -
represents a straight or branched, saturated hydrocarbon chain having
from 1 to 6 carbon atoms, preferably from 1 to 4;
cycloalkyl - represents a saturated carbocyclic ring having from
3 to 6 carbon atoms, optionally substituted by 1 to 3 groups independently
selected from the group consisting of lower alkyl, trihalomethyl and
NR»R~ ~ , wherein R» and R1 ~ as defined above;
halogen (halo) - represents fluoro, chloro, bromo or iodo;
aryl - represents a carbocyclic group having from 6 to 14 carbon
atoms and having at least one benzenoid ring, with all available
substitutable aromatic carbon atoms of the carbocyclic group being
intended as possible points of attachment, said carbocyclic group being
optionally substituted with 1 to 3 groups, each independently selected
from halo, alkyl, hydroxy, phenoxy, amino, loweralkylamino,
diloweralkylamino, (e.g., NRI~Ri ~ wherein R~o and R> > are independently
selected from hydrogen, lower alkyl or trihalomethyl), loweralkoxy,
polyhaloloweralkoxy, (e.g., OR'° wherein R'° is as above
defined)
polyhaloloweralkyl (e.g., trihalomethyl), CN, or N02; preferred aryl groups
CA 02309609 2000-OS-04
WO 99/24421
PCT/US98/23224
-g-
include 1-naphthyl, 2-naphthyl and indanyl, and especially phenyl and
substituted phenyl;
heterocycfic - represents saturated and unsaturated non-
aromatic cyclic organic groups having at least one O, S and/or N atom
interrupting a carbocyclic ring structure that consists of one ring or two
fused rings, wherein each ring is 3 to 7 membered (e.g., 5-, 6- or 7-
membered), which ring structure has from 2 to 8, preferably from 3 to 6
carbon atoms; e.g., 2- or 3-pyrrolidinyl, 2-, 3- or 4-piperidinyl, 2- or 3-
piperazinyl, 2- or 3-morpholinyl, or 2- or 3-thiomorpholinyl; said
heterocycfic group being optionally substituted by 1 to 3 groups
independently selected from alkyl, trihalomethyl and NR1~R~1, wherein
R» and R~ 1 are independently selected from hydrogen, alkyl or
trihalomethyl, said substituents being bound to carbon atoms
(substitutable carbon atoms) in the ring such that the total number of
substituents in the ring is 1 to 3; and wherein said heterocyclic ring
contains nitrogen atoms, said nitrogen atoms (i.e., the substitutable
nitrogen atoms) being optionally substituted with lower alkyl (e.g., methyl),
e.g., N-methylpyrrolidinyl;
heteroaryl - represents a cyclic organic group having at least
one O, S and/or N atom interrupting a carbocyclic ring structure and
having a sufficient number of defocalized pi electrons to provide aromatic
character, with the aromatic heterocyclic group having from 2 to 14,
preferably 4 or 5 carbon atoms, e.g., 2-, 3- or 4-pyridyl, 2- or 3-furyl, 2-
or 3-
thienyl, 2-, 4- or 5-thiazolyl, 2- or 4-imidazolyl, 2-, 4- or 5-pyrimidinyl, 2-
pyrazinyl, or 3- or 4-pyridazinyl, etc.; preferred heteroaryl groups are 2-, S-
and 4-pyridyl; said heteroaryl groups being optionally substituted with 1 to
3 groups, each optional substituent being independently selected from
alkyl, halogen, trihalomethyl, CN, N02, OR~c or NR~cRI~, wherein R» and
R> > are independently selected from hydrogen, alkyl or trihalomethyl, said
substituents being bound to carbon atoms (substitutable carbon atoms) in
the ring such that the total number of substituents in the ring is 1 to 3;
DMF - stands for N, N,-dimethylformamide;
SEM - stands for 2-(trimethylsilyl)ethoxymethyl;
CA 02309609 2000-OS-04
WO 99/24421 PCT/US98/23224
_g-
THF - stands for tetrahydrofuran;
DMAP - stands for dimethylaminopyridine;
DIPA - stands for diisopropylamine;
DMSO - stands for dimethyl sulfoxide;
DBU - stands for diazabicycloundecene;
DBN - stands for diazabicyclononane;
LAH - stands for lithium aluminum hydride;
FAB - stands for fast atom bombardment;
CI - stands for chemical ionization;
EI - stands for electron impact;
HOBT - stands for 1-hydroxybenzotriazole; .
EDCI - stands for 1-(3-dimethylaminopropyl)-3-ethylcarbo-
diimide hydrochloride;
LC/MS - stands for liquid chromatography/mass spectrometry;
TFA - stands for trifluoroacetic acid;
Tr - stands for trityl; and
LRMS - stands for low resolution mass spectrometry.
Also, unless stated otherwise, the substituents for the various
embodiments described below are as defined for Formula I.
Preferred compounds are represented by of formula II
R~ R~
R1
q ~N~Y~Rs III)
m P
R~
HN~\ 1 R~
iN
R
wherein q is 1 to 7, m is 0 to 4, n is 0 or 1, p is 0 to 4, Y is selected from
-S02 -, -S02NH-, -CONH-, -CO-, -C(NH)NH-, or -CO(CH2)W O-, or, when m
is 2 to 4, Y, in addition to the groups above, also represents -NHCONH-,
-O- or -NHC(NH)NH-; and w, R~, R6 , and R~ are as defined above.
Preferably R6 is phenyl or substituted phenyl.
Most preferred are compounds of formula II wherein (1) q is 1 to 4; (2) n is
0 or 1; (3) m is 0 to 4 (more preferably, 0 to 3, and even more preferably 0
to 2); (4) p is 0 to 2; (5) Y is -CONH-, -CO-, -S02-, -CO(CH2)20- or -O-
(when m is greater than or equal to 2, i.e., Y can also be -O- when m is 2-
CA 02309609 2000-OS-04
WO 99/24421 PCT/US98/23224
-10-
4); (6) R6 is phenyl, wherein said phenyl is optionally substituted by one,
two or three substituents independently selected from halogen, preferably
fluorine or chlorine, CF3, C1 to C4 alkoxy, OCF3 , N02, or NR»R» with
R1~ and R~1 being as defined above.
For the compounds of formula II, R~ and R~ are preferably
hydrogen.
For the compounds of formula II, preferably when Rs is mano-
substituted phenyl said substitutent is in the 3- or 4-position and said
substituent is selected from fluorine, chlorine, methoxy or trifluoromethoxy,
, and when R6 is disubstituted phenyl said substitutents are in the 3,5-
positions and said substituents are the same and are selected from
fluorine, chlorine, methoxy or trifluoromethoxy.
Compounds of this invention include, but are not limited to
N '-"'(O .N '"'1
HN~ N N \ / Cl H ~ N O \ /
' H
O
'N~O C' ~ 'N~
HNV N N \ / HN~ N O \ /
H
Cl
.N 'J'(O . N '-'(O
F-INV N H \ / HN~ N H \ /
C1
N ~O , I C1
HNV N Y N
' H
H
N
G i _F
N H
N
100 H
N O
CA 02309609 2000-OS-04
WO 99/24421
PCT/US98/23224
-11 -
H
N\~O
HN ~ 101 '' H~'N
~N
and
Compounds of this invention also include, but are not limited to
H l, \ HN~\
~N
N~O
\ ~ / \ I
HN, \
L''lNN1
HN
O I \ I
NH
0 , cl
HN \
L H~\
N
N N
N
O I \ ~ \ F
O
cl and ''
Certain compounds of the invention may exist in different isomeric
(e.g., enantiomers and diastereoisomers) forms. The invention
contemplates all such isomers both in pure form and in admixture,
including racemic mixtures. Enol forms are also included.
The compounds of Formula I can exist in unsolvated as well as
solvated forms, including hydrated forms, e.g., hemi-hydrate. In general,
the solvated forms, with pharmaceutically acceptable solvents such as
water, ethanol and the like are equivalent to the unsolvated forms for
purposes of the invention.
Certain basic compounds of the invention also form
pharmaceutically acceptable salts, e.g., acid addition salts. For example,
the nitrogen atoms may form salts with acids. Examples of suitable acids
CA 02309609 2000-OS-04
WO 99/24421
-12-
PCT/US98/23224
for salt formation are hydrochloric, sulfuric, phosphoric, acetic, citric,
oxalic, malonic, salicylic, malic, fumaric, succinic, ascorbic, malefic,
methanesulfonic and other mineral and carboxylic acids well known to
those in the art. The salts are prepared by contacting the free base form
with a sufficient amount of the desired acid to produce a salt in the
conventional manner. The free base forms may be regenerated by
treating the salt with a suitable dilute aqueous base solution such as dilute
aqueous sodium hydroxide, potassium carbonate, ammonia and sodium
bicarbonate.
The free base forms differ from their respective salt forms somewhat
in certain physical properties, such as solubility in polar solvents, but the
acid and base salts are otherwise equivalent to their respective free base
forms for purposes of the invention.
All such acid and base salts are intended to be pharmaceutically
acceptable salts within the scope of the invention and all acid and base
salts are considered equivalent to the free forms of the corresponding
compounds for purposes of the invention.
Numerous chemical substances are known to have histamine H~
receptor antagonist activity. Many useful compounds can be classified as
ethanolamines, ethylenediamines, alkylamines, phenothiazines or
piperidines. Representative H1 receptor antagonists include, without
limitation: astemizole, azatadine, azefastine, acrivastine, bromphenir-
amine, cetirizine, chlorpheniramine, clemastine, cyclizine, carebastine,
cyproheptadine, carbinoxamine, descarboethoxyloratadine (also known
as SCH-34117), diphenhydramine, doxylamine, dimethindene, ebastine,
epinastine, efletirizine, fexofenadine, hydroxyzine, ketotifen, loratadine,
levocabastine, mizolastine, mequitazine, mianserin, noberastine,
meclizine, norastemizole, picumast, pyrilamine, promethazine,
terfenadine, tripelennamine, temelastine, trimeprazine and triprolidine.
Other compounds can readily be evaluated to determine activity at H1
receptors by known methods, including specific blockade of the contractile
response to histamine of isolated guinea pig ileum. See for example,
W098/06394 published February 19, 1998.
CA 02309609 2000-OS-04
WO 99/24421
-13-
PCT/US98/23224
For example, the H3 antagonists of this invention can be combined
with an H~ antagonist selected from astemizole, azatadine, azelastine,
brompheniramine, cetirizine, chlorpheniramine, clemastine, carebastine,
descarboethoxyloratadine (also known as SCH-34117), diphen-
hydramine, doxylamine, ebastine, fexofenadine, ioratadine,
levocabastine, mizolastine, norastemizole, or terfenadine.
Also, for example, the H3 antagonists of this invention can be
combined with an H~ antagonist selected from, azatadine, bromphen-
iramine, cetirizine, chlorpheniramine, carebastine, descarboethoxy-
loratadine (also known as SCH-34117), diphenhydramine, ebastine,
fexofenadine, loratadine, or norastemizoie.
Representative combinations include: the H3 antagonists of this
invention with loratadine, H3 antagonists of this invention with
descarboethoxyloratadine, H3 antagonists of this invention with
fexofenadine, and H3 antagonists of this invention with cetirizine.
Those skilled in the art will know that the term "upper airway"
means the upper respiratory system--i.e., the nose, throat, and associated
structures.
The compounds of this invention may be prepared according to
suitable processes known in the art for making similar compounds, e.g.
processes described in the literature referred to above.
The following processes may be employed to produce compounds
of Formula I. Unless stated otherwise, reactions are conducted at an
appropriate temperature which allows the reaction to proceed at a
reasonable rate to completion.
GENE AL PREPARATIfJN ,SCHEMES.
In general the compounds of this invention are prepared by first
providing starting compounds of the general formula
R~
R~
-~ x~r~~ n
Z NI'~ ~~ H
,N
~R~ R~
CA 02309609 2000-OS-04
WO 99/24421
-14-
PCT/US98/23224
which then in a further step are reacted with a compound of the general
formula
~-(Cf"~2 )m -Y-(CH2 )p -R6
followed by elimination of the protecting group Z to yield a compound of
formula I.
In the above formulae R~ , R6 , R~ , X, Y, m, n and p are as defined
for formula I above. L represents a leaving group such as CI, Br, I, and
activated versions of OH such as OS02CF3 generated independently or in
situ.
The following reaction schemes illustrate the various steps of the
processes used.
PREPARATION OF P1PERIDINES In = 1 )
j~,~action Scheme 1 - Compounds wherein X is -(CH~~~_~
Ste~_1
Z Rl Rl R~
/N f 1) EtMgBr, CH2C12 /:~=N ~~N
\N~~ ~ R~ Z N~M
R D 2) N~ ~ , R~
~'-J~' MCHO OH 3
R~
0°C-RT
Compound 1, wherein (1 ) D is halogen, preferably iodide, (2) Z
represents a protecting group such as triphenylmethyl, 2-(trimethylsilyl)-
ethoxymethyl and the like, and (3) R~ can be either hydrogen, alkyl or
trihalomethyl, is dissolved in a suitable solvent, such as methylene
chloride, and treated with a Grignard reagent, such as ethylmagnesium
bromide. Subsequent addition of an appropriate aldehyde 2 (M = (CH2)p_
6) produces compound 3.
Rl R~ Rl R~
/'-~:N ~~N ACpO, Et3N ~ /'-I=N r~'~N
Z N\:~~M '-~\J DMAP, CH2CI2 Z N\:~~M
Rl OH 3 R~ Rl OAc 4 R~
CA 02309609 2000-OS-04
WO 99/24421 PCT/US98/23224
-15-
In step 2, compound 3 is dissolved in an organic solvent, such as
methylene chloride, and treated with a tertiary amine base, such as
triethylamine, and an acylation catalyst, such as dimethylaminopyridine.
Subsequent treatment with acetic anhydride provides the compound of
formula 4.
R1 R~ R1 R~
~=~=N r~=~N Pt02, HOAC ~:,=N ~'~ NH
Z N~~~ 1 M l' , IJ H2, 60 psi zlN~%~~~M
\R OAc 4 R~ 2 days R 5 R~
In step 3, compound 4 is dissolved in a suitable organic acid, such
as acetic acid, and hydrogenated under pressure (16-60 psi) in the
presence of an appropriate catalyst, such as platinum oxide, to provide
compound 5.
Reaction Scheme 2 - Compounds wherein X is -~(CH~ ).2=
Ste~1
z
I Ri
~i_'. R~ C%
N. Rl R2-N ~ C=CH N ~~
Rl C\~ R~
W
Ny 7
1 R1 D (Ph3P)2PdCl2, CuI
THF, DIPA ~~N~R2
R~
In Step 1, compound 1, wherein (1) D = halogen, preferably iodide,
(2) Z represents a protecting group such as triphenylmethyl, 2-(trimethyl-
silyl)ethoxymethyl and the like, and (3) R2 represents benzyl or substituted
benzyl, is dissolved in a suitable solvent or a mixture of solvents selected
from ethereal and dialkylamine solvents. A tetrahydrofuran/diisopropyl-
amine mixture is preferred. Addition of a compound of structure 6
followed by addition of a suitable catalyst, such as bistriphenylphosphine-
palladium dichloride and copper iodide, and stirring at temperatures from
21-60 °C provides compound 7.
CA 02309609 2000-OS-04
WO 99/24421
-16-
PCT/US98/23224
Z R1 Z
NY I R1
H2 (60 psi) N~
i J
R C~ C R~ Pd/C of Pd(OH)2 N' R'
i
1 MeOH, HOAc, CH2CI2 R
I~N~R2 8 ~~N~R2
R~ R~
In Step 2, compound 7 is dissolved in a suitable organic solvent or
mixtures thereof (examples of solvents include methylene chloride,
methanol, and acetic acid) and hydrogenated with a catalyst, such as
palladium or palladium hydroxide, at pressures ranging from 16-60 psi to
provide compound 8.
~te~3,
Z Z
I Ri I Ri
N/ N./
J R' H2 (1 atm), Pd(OH)2
R
MeOH, HCI
~1 R
~N~ ~~NH
8 ~ R2 5p
R~ R~
In Step 3, compound 8 is dissolved in a suitable alcohol, such as
methanol, and treated with a few drops of hydrochloric acid (1 M) and
hydrogenated with a suitable catalyst, such palladium or palladium
hydroxide, at pressures ranging from 16-60 psi to provide compound 5A.
Reaction Scheme 3 - Preparation of Compound 10
R1 R~ Ri R~
_ r(-N r~~ NH ~~-N \~rJiR
Z Ny~M ~~ ~ Z N~~~M ~ J
'Rl 5 R~ \Rl 9 ~R~
i
HCI, MeOH ~.'=N R~.~N ~R
80 °C HN~;~~M ~ J (HCI)r
i \
R 10 ~R~
CA 02309609 2000-OS-04
WO 99/24421
-17-
PCT/US98123224
(wherein R is the group -(CH2 )m -Y-(CH2 )p -Rs ).
Those skilled in the art will appreciate that the number of HCI
molecules (r) is based on the number of basic groups present in
compound 10.
Compound 5 is reacted with L-(CHZ )m-Y-(CH2 )p-R6 to produce
compound 9. L is a leaving group, such as CI, Br, I and activated
versions of OH, such as OS02CF3 generated independently or in situ.
When Y is -C(O)NH-, -OCO and -S02-, and m is 2, then compound 5 is
reacted with reactants such as (CH2=CH)C(O)O(CH2)PRs,
(CH2=CH)C(O)NR5(CHZ)pRs, and (CH2=CH)S02(CH~pRs.
The reactions are conducted in suitable solvents, such as ether,
tetrahydrofuran, dioxane, dimethylsulfoxide, dimethylformamide, water,
methylene chloride, and toluene, with or without the presence of a suitable
bases, such as triethylamine, lithium diisopropylamide or sodium hydride,
at temperatures ranging from -78° to 200°C.
When Z is triphenylmethyl, compound 9 is deprotected by treatment
with dilute aqueous acid, such as HCI or HBr, at a temperature of about
25° to 100°C to produce compound 10. Other protecting groups are
removed by methods well known in the art.
PREPARATION OF COMPOUNDS HAVING A 7-MEMBERED HETERO-
(CYCLIC RING
Reaction Scheme 4 - Compounds wherein X is -CH2_
Ho2c i1 ' Ho2c ~1'
NH ~ N-Z
'R \R
11 12
In Step 1, compound 11 (prepared in an analogous manner to the
procedures outlined in European J. Med Chem. 1979,14, 157-164 and
Tetrahedron Letts. 1990, 31, 933-936) is reacted with a compound ZCI in
a suitable organic solvent at a temperature of from 0°to about
50°C in the
presence of an organic base to produce compound 12. Z represents a
protecting group, preferably carbobenzyloxy. Suitable solvents include
*rB
CA 02309609 2000-OS-04
WO 99/24421 PCTlUS98/23224
_18_
THF, ether, dioxane or the like. Suitable bases include triethylamine and
the like.
H02C /1 ~ OHC
N-Z -'~" N-Z
~/ ~/
R' R~
12 13
In Step 2, compound 12 is reduced to the aldehyde 13 using a
suitable reducing agent, such as BH3~SMe2 or the like, in a suitable
organic solvent, such as THF, ether, dioxane or the like, at a temperature
of 0°to 100°C.
Step 3
OH
OHC /1 ~ ~ \Rl ~ R~
/N-Z ~ TrN' N
N-Z
~R~ Ri
13 14 R
In Step 3, compound 13 is reacted with the Grignard reagent
formed from iodoimidazole in the same manner as described for Step 1 of
Reaction Scheme 1 to give the alcohol 14.
Step 4
OH
~~R1 ~R7 -f-TrN~/R
TrN\ ..N 'N -Z ~ ~ i N ~\ JNH
R1 ~R R 15 R~
14
In Step 4, compound 14 is reduced to compound 15 in a suitable
polar organic solvent using H2 in the presence of a metal catalyst and a
trace of acid at a temperature of from 25°to 75°C. Suitable
solvents
include MeOH, EtOH and i-PrOH, with EtOH being preferred, and catalysts
can include Pd/C or Pt02 or the like.
CA 02309609 2000-OS-04
WO 99/24421 PCT/US98/23224
-19-
St~~ 5
1
R ~1 LR ~iR ~1
~'i, -~.~.
TrN~N NH TrN\ 'N N-R
R1 \ ~ Ri \J
15 R lb R
In Step 5, compound 15 is reacted with LR in a suitable solvent
such as THF, ether, or the like in the presence of a suitable tertiary amine
base such as triethylamine at a temperature from 0°to 100°C,
preferably
25°C, to produce compound 16. R is -(CH2 )m -Y-(CH2 )p -Rs and L is a
leaving group as defined in Reaction Scheme 3 above.
~te~6 .
R1 R~ R~ R~
TrN~/ ' N-R - > HN~/ ' N-R
~''-r1 ~ ~ ',~N
Rl 16 \R' Rl 17 \R~
Step 6 is performed in a similar manner to the deprotection step in
Reaction Scheme 3 above to give compound 17.
Reaction Scheme 5 - X is -(CH2.~2-
t 1
Me0
OHC R~ I R~
'1 '1
N -Z '-'~' N -Z
MeOCH2PPh3 \R~
13 19
In Step 1, aldehyde 13 is reacted with the Wittig reagent in a
suitable ethereal solvent in the presence of a strong base at a temperature
from -25° to 80°C to give compound 19. Suitable solvents include
THF,
ether, dioxane or the like. Strong bases can include lithium or potassium
diisopropylamide, and lithium, sodium or potassium bis(trimethylsilyl)-
amide or the like. Other suitable bases can include NaH or KH in a
suitable polar aprotic solvent, such as DMSO.
CA 02309609 2000-OS-04
WO 99/24421
-20-
PCT/US98/23224
Me0
CHO
R~ R~
1 ~1
N-Z "'~ ,N-Z
~R~ ~R~
19 20
In Step 2, the enol ether 19 is hydrolyzed to the aldehyde 20 by
treatment with a dilute mineral acid, such as HCI or HBr, at a temperature
from 0° to about 80°C. Aldehyde 20 can then be converted to the
desired
targets in a manner similar to that described in Reaction Scheme 4, Steps
3 to 6.
F3,eaction Scheme 6 - X is ~CH2~-_to -(CH2),~-
CHO
OHC
R~ '
~1
/N_z _z
\R~ R~
21 22
Aldehyde 20 can be converted to aldehyde 21 in a similar manner
to that described in Reaction Scheme 5. Compound 21 can then be
converted to the desired targets in a manner similar to that described in
Reaction Scheme 4, Steps 3 to 6. A similar sequence can be applied to
compound 22 and to higher homologs.
PREPARATION OF PYRROL.IDINES (n = 0)
Reaction Scheme 7
Step 1
Tr Ri Tr
I I R1
N/
~I
N \ CHO N ~ ~ CO2R14
23 Rl s 24 Rl s
wherein s is 0 to 5, and R'4 represents lower alkyl (e.g., methyl or ethyl).
In Step 1, a suitable Homer-Emmons reagent such as trimethyl- or
triethyl phosphonoacetate is treated with a strong base, such as NaH, KH,
CA 02309609 2000-OS-04
WO 99/24421
-21 -
PCT/US9$/23224
lithium diisopropylamide or the like, in a suitable ethereal solvent such as
THF, ether, dioxane or the like. The phosphonate carbanion is then
reacted with the aldehyde 23 for 30 min. to 24 h at a temperature suitable
to complete the reaction and give ester 24.
Steep 2
Tr
N/R1 Nr Ri
I
N, Ri ~ CO2R~4 ~ N~' 1 C02Rla
S R /g
24 25 ~ No2
R~
In Step 2, ester 24 is reacted with a substituted or unsubstituted
nitroalkane, such as nitromethane or nitroethane, in a polar aprotic
solvent, such as acetonitrile, THF or the like, preferably acetonitrile, in
the
presence of an amine base, such as DBU, DBN, triethylamine or the like,
preferably DBU, at a temperature from 0°to 80°C, preferably
25°C, for 24h
to yield nitro ester 25.
Step 3
Tr Tr
I R1 I R1
C\ I <N~
N ~ 1 S C02R 14 --~. N ~R1
!s
25 ,~ ,~ No2 26 ~ ,~N
R R \
H
In Step 3, the vitro group of vitro ester 25 is reduced to the amine
using hydrogen and a suitable metal catalyst, such as Pd/C, Ra-Ni or the
like, in a suitable erotic solvent, such as methanol, ethanol or the like, at
a
temperature of from 25° to 80°C. The resulting amino ester is
cyclized to
the iactam by heating in a suitable erotic solvent, such as methanol or
ethanol, at a temperature of up to 80°C in the presence of a small
amount
of a base such as potassium carbonate or the like to give compound 2fi.
CA 02309609 2000-OS-04
WO 99/24421
-22-
Step 4
PCTNS98/23224
Tr Rl it
i
N / N /R
_.
\_
Rl JS ~~ R1 JS
26
R7 \H R~ \H
In Step 4, compound 26 is reacted with a suitable reducing agent,
such as LAH, BH3, or the like, preferably LAH, in a suitable solvent, such
as THF, ether, dioxane or the like, at a temperature ranging from 0°
to
80°C, preferably 60°C, for a time ranging from 30 min. to 24h,
preferably
3h to give compound 27.
Compound 27 is then reacted with a compound of the formula L-
(CH2 )m -Y-(CH2 )p -R6 followed by deprotection in a manner similar to the
procedure outlined for Reaction 3 above.
The starting compounds of formula 23 are either known
compounds or may be obtained according to procedures well known in
the art, for example by following the preparations in the steps outlined for
compounds 13, 20, and 22 above.
A person skilled in the art will easily see that several variations of
the above processes are possible. For example, the substituents R~ and
R~ may be present in the starting materials or may be introduced at any
convenient stage of the process.
The following examples are intended to illustrate, but not to limit,
the present invention.
*rB
CA 02309609 2000-OS-04
WO 99/24421
- 23 -
EXAMPLE 1
PCT/US98/23224
Tr n.
1
N N
--'' y
N OH N
CHO
Tr 29
I
N
-" ~~ I
N
C02Me
To a flask containing oxalyl chloride (13.8 g, 9.5 ml, 109 mmols) in
5 methylene chloride (300 ml) at -78°C was added DMSO (19.9 g, 255
mmols) dropwise. When gas evolution stopped, the mixture was stirred for
8 min., and a solution of the alcohol 28 (10.0 g, 27.2 mmols) in methylene
chloride (50 ml) was added. The reaction was maintained at -78°C for 50
min., triethylamine (45 ml, 255 mmol) was added, and the reaction
10 allowed to warm to room temperature aver 45 min. The contents were
diluted with NH4C1 solution and extracted with methylene chloride. The
combined organics were washed with brine, dried over MgS04, filtered
and concentrated. The crude product was chromatographed over silica
gel (10 to 30% acetone in methylene chloride ) to give the product 29 as a
15 faint yellow oil (7.7 g, 77%): LRMS (CI, M+H) = 367.
To a flask containing NaH (95%, 2.0 g, 79 mmol), was added dry
THF (600 ml) under a nitrogen atmosphere. To this mixture was added
trimethylphosphonoacetate (14.0 g, 77.5 mmols) dropwise via syringe.
Gas evolution was observed and a viscous white mixture resulted. The
20 mixture was warmed to 35°C for 30 min. and then allowed to cool back
to
room temperature. The aldehyde 29 (14.5 g, 39.6 mmols) in dry THF (200
ml) was added via syringe to the reaction mixture. TLC (40% EtOAc-Hex)
indicated the reaction to be complete after stirring for 45 min. at r.t. The
contents were diluted with water, and the aqueous portion was extracted
25 with EtOAc. The combined organics were washed with brine, dried over
MgS04, filtered and concentrated to give a solid which was recrystallized
from Et20-hexane (1:2 v/v) to give 5.9 g of pure material. The mother
CA 02309609 2000-OS-04
WO 99/24421
PCTNS98/23224
-24-
liquors were chromatographed on silica gel (40% EtOAc-hexane ---> 60%
EtOAc) to afford another 7.7 g of material, 81% combined yield. LRMS (CI,
M+H) = 423. Analytical CHN for (C28H26N202): C, 79.25; H, 6.22; N,
6.60: Found C, 79.11; H, 6.39; N, 6.66. mp = 129-130.5°C
Tr Tr
I I
N N
y I -~ y
N COZMe N ~ C02Me
30 31
N02
To a CH3CN solution (300 ml) of 30 (11.0 g, 26.1 rr~mots) was
added CH3N02 (29.3 g, 26 ml, 480 mmols) followed by DBU (5.1 g, 5.0
m(, 33.4 mmols). The reaction mixture was stirred under a nitrogen
atmosphere for 18 h, at which time no starting material was observed by
TLC (40% EtOAc-Hex). The solvents were evaporated under reduced
pressure, and the residue was chromatographed directly on silica gel
(50% EtOAc-Hex ---> 70% EtOAc) affording 13.1 g (>100% crude yield) of
the product as a colorless oil. LRMS (Ci, M+H) = 484. Analytical CHN for
(C2gH2gN3O4): C, 72.03; H, 6.04; N, 8.69: Found C, 72.06; H, 6.34; N,
8.66. mp = 97.5-99.5°C.
Ste_~C
Tr Tr
I I
N ~ N
y I
N ~ C02Me N
31 ~ 32
N02 N
H
Compound 31 (2 x 5 g, 20.7 mmols) was dissolved in a solution of
absolute EtOH-THF (60-20, v/v). Ra-Ni (-2 x 5 g) was added, and the Parr
vessel was pressurized to 50 psi with hydrogen. After shaking for 4-6 h,
TLC indicated the reduction to the amino-ester was complete (10%
MeOH-EtOAc). The catalyst was removed by filtering through celite.
Evaporation under reduced pressure afforded the amino-ester
intermediate which was subsequently cyclized to the lactam by refluxing in
CA 02309609 2000-OS-04
WO 99/24421
- 25 -
PCT/US98/23224
MeOH with a small amount of K2C03 for 3 h. Removal of the K2C03 by
filtration and evaporation of the solvent afforded an oil which was
chromatographed on silica gel (10% MeOH-CH2C12 --->10% MeOH + 2%
NH40H) to give the product as an off white amorphous solid, 8.1 g (92%).
LRMS (CI, M+H) = 422. Analytical CHN for (C28H2~N30 x 1.5 mol H20):
C, 74.91; H, 6.57; N, 9.36: Found C, 74.76; H, 6.17; N, 9.14. MP = 171-
173.5 ~C.
Step ~
Tr
I I
I .-.-~ ~\ I .
N N
32 ~ 33
N N
\ \
H H
To a flask containing a THF (12 ml) solution of LAH (180 mg, 4.80
mmols, 10 equiv.) was added a THF solution of 32 at room temperature.
The mixture was heated to 60°C for 3 h, then allowed to cool to
r.t. The
reaction was quenched by the addition of solid Na2S04 x 10H20. After 20
min. 5% NaOH (~ 1 ml) was added causing the viscous gray mixture to
become colorless and homogeneous. After another 20 min. the mixture
was filtered through celite and the filter cake was washed well with THF
and MeOH. The effluent was concentrated under reduced pressure and
then chromatographed on silica gel (10% MeOH-CH2C12 --->10% MeOH
+ 2% NH40H) to afford 168 mg (73%) of the product 33 as hygroscopic
foam. LRMS (Cl, M+H) = 408.
Ste,~E
Tr Tr
I I
I
N N
33 g4
N N
\ \
S \ / C1
02
CA 02309609 2000-OS-04
WO 99/24421
-26-
PCT/US98/23224
To a CH2C12 solution (6 ml) of (+/-)33 (315 mg, 0.774 mmols) was
added Et3N (2 ml, 14.4 mmols) followed by p-chlorosulfonyl chloride (215
mg, 1.09 mmols} at r.t. The mixture was stirred under a nitrogen
atmosphere for 21 h, then evaporated to 1/2 volume and
chromatographed on silica gel (1% MeOH-CH2C12 ---> 3 % MeOH) which
afforded an amorphous white solid. Trituration with hexane-acetone
followed by evaporation yielded a fluffy white foam. LRMS (CI, M+H) _
582.
to
Tr H
I I
N N
~\ ~ "' <\
N ~,/ N
34 1~ 35
\ S CI 'S CI
02 ~ ~ O
Standard HCI deprotection of 34 provides the hydrochloride salt of
35 as a light tan solid. LRMS (CI, M+H) = 340.
Example 1 A: Chiral Synthesis
t A
H H
.,~~~~CHZOH
_~~~~~CH2-OH
1. H2 /Pd
2. (BOC)20 N
'( 36 BOC 37
Ph
A solution of 36 (2.Og) in ethanol (20 ml) and 10% palladium-on-
carbon (0.3g) is hydrogenated in a Parr shaker at 60 psi for 24 hr. The
catalyst is then filtered and the filtrate is evaporated under reduced
pressure. The residual oil is dissolved in dichloromethane (20 ml}. Di-Pert
butyldicarbonate (2g) is added to the solution followed by 4-dimethyl-
aminopyridine (0.05g). The reaction mixture is stirred at 70°C for 1
hour
and is then evaporated under reduced pressure. The product is then flash
chromatographed on silica gel (50 ml). Elution with 8% methanol-
CA 02309609 2000-OS-04
WO 99/24421 PCT/US98/23224
-27-
dichtoromethane afforded after evaporation under reduced pressure the
title compound 37 as a colorless oil (1.1 g), MS (CI) m/e=146 (M-56).
Steg B
H H
.~a~CH2-OHM .~~~~CH2-OS02CH3 ~ .~~ACH2-I
BoC N 37 BOC N 37a BOC N 38
A solution of 37 {1.1 g) and triethylamine (0.84 ml) in
dichloromethane is cooled in an ice-bath and stirred while adding
dropwise a solution of mesyl chloride (0.47 ml) in dichloromethane (5 ml).
The reaction mixture is stirred for 1 hour and is then washed with water,
dried over sodium sulfate and filtered through a silica-gel plug. The filtrate
is evaporated to afford the mesylate 37a which is then dissolved in
acetone (30 ml) containing sodium iodide (1.6 g). The reaction mixture is
heated with stirring in an oil-bath (70° C) for 24 hours and then
cooled.
The insolubte salts are removed by filtration and the filtrate is evaporated
under reduced pressure. The residual product is dissolved in
dichtoromethane and washed with water, dried over sodium sulfate and
filtered through a silica-gel plug. The filtrate is evaporated under reduced
pressure to afford the title compound 38 as an oil (1.53 g), MS (FAB) m/e
280 (MH)+
Step C
H H
.,v~H2'I PPh .,.ACHa-P+tPh)3 j_
'-
BoC 38 BoC~ 39
A solution of 38 (1.53 g) and triphenylphosphine (1.9 g) in
dimethylformamide (10 ml) is heated in an oil-bath (90°C) for24 hr. The
reaction mixture is then evaporated under reduced pressure and the
residual product is flash chromatographed on silica-gel (50 ml). Elution
with 10% methanol-dichloromethane afforded after evaporation under
reduced pressure the title compound 39 as a white powder (1.56g), MS
(FAB) m/e=446 (M)+
CA 02309609 2000-OS-04
WO 99/24421
- 28 _
PCT/US98/23Z24
N CHO
H \N ~ N,BOC
.avCH2'P+IPhj3I' Boc N
N ~39 N H
BOC H
2.5 M butyllithium solution in hexanes (0.9 ml) is added to a
solution of 39 (1.0 g) in tetrahydrofuran (10 ml) at -78°. The solution
is
5 then stirred at room temperature for 30 minutes and the resulting solution
is re-cooled to -78° followed by the addition of a solution of the
aldehyde
{0.38 g) in tetrahydrofuran ( 5 ml). The reaction mixture is the filtered and
the filtrate is evaporated under reduced pressure. The resulting crude
product is flash chromatographed on silica-gel (50 ml). Elution with 5%
10 methanol-dichloromethane afforded after evaporation under reduced
pressure the title compound 40 as a white powder (0.34 g), MS (FAB)
m/e=264 (MH)+
Ste'P E
Boc Boc
N N
N ~ H, / Pt0 N
\N , 40 \N ~ 41
H H H H
15 A solution of 40 ( 0.32 g) in ethanol (5 ml) containing Pt0 (0.085g)
is hydrogenated at atmospheric pressure for 24 hours. The catalyst is
then filtered and the filtrate is evaporated under reduced pressure. The
resulting crude product is flash chromatographed on silica-gel (30 ml).
Elution with 10% methanol-dichloromethane afforded after evaporation
20 under reduced pressure the title compound 41 as a resinous gum (0.23
g), MS (FAB) m/e=266 (MH)+ .
Steti FF
BOC H
N N
N N
/ /
~N ~ 41 ~N ~ ~ S~2
H H H ~ ~ H
CA 02309609 2000-OS-04
W O 99/24421
PCT/US98/23224
-29-
Compound 41 (0.1 g) is stirred with 4M HCI in dioxane (2 ml) for 30
minutes and the reaction mixture is then evaporated under reduced
pressure. The residual product is dissolved in methanol (2 ml) and the
solution is stirred while adding Biorad AG 1-X8 (OH' form) ion-exchange
resin until the pH of the solution is above 8. The resin is removed by
filtration and the filtrate is then evaporated to afford the title compound 42
as a resinous gum ( 0.061 g), MS (CI) m/e=165 (MH)''.
The R-enantiomer can be obtained in a similar manner.
SAMPLE 2
Tr Tr
I I
N N
y I
N v~~~ N
33 N 43 N~
H
C02Me
To a MeOH solution (15 ml) of 33 obtained in Step D of Example 1
(600 mg, 1.48 mmols) at r.t. was added methyl acrylate {0.300 ml, 3.33
mmols). The reaction mixture was stirred for 2h at r.t., then heated to
60°C
overnight. The solvents were evaporated and the residue was
chromatographed directly on silica gel (5% MeOH-CH2C12 ---> 10%
MeOH) providing 574 mg (78%) of 43 as an off-white solid. LRMS (CI,
M+H) = 494.
Tr Tr
I I
N N
~ I ~~ I
N ''w/ N ~/
+ - '~ 43 44
N N
H
N
COZMe (+/-)
CI
rB
CA 02309609 2000-OS-04
WO 99/24421
-30-
PCT/US98/23224
To a toluene (5 ml) solution of p-chloroaniline (0.160 g, 1.25
mmols) was added trimethylaluminum (0.700 ml, 2M in toluene) at 0°C.
The mixture was stirred at 0°C for 15 min., and at r.t. for 40 min.
Then a
toluene-CH2C12 solution of the compound 43 (10 ml, 1:1, v/v) was added
at 0°C to the aniline complex. After 30 min., the mixture was heated
to
80°C for 3h, and left at r.t. overnight. The reaction was quenched by
the
addition of solid Na2S04x10 H20, followed by the addition of MeOH.
After stirring for 20 min., the mixture was filtered through celite, and
concentrated under reduced pressure. Chromatography on silica gel
i 0 (10% MeOH-EtOAc ---> 15% MeOH with 1 % NH40H) gives 624 mg (97%)
of 44 as a white foam. Irms (CI, M+H) = 589.
Ste~~
Tr H
I 1
N N
~ 1 .-~ ~~ I
N N
rj H N 45
H
N "'~. N
Cl
C1
To a dioxane solution (10 ml) of the compound 44 from the
previous step was added a solution of 4M HCI-dioxane (2 x 2 ml) and the
mixture was heated to 80°C for 6h. The mixture was cooled to r.t., and
evaporated under reduced pressure affording a gummy foam. The
residue was rinsed with Et20 (3 x 10 ml) and the supernatant was
decanted. The product was stored under high vacuum affording 45 as a
tan solid (400 mg of dihydrochloride salt). MS(CI) 347 (M+1 ).
krB
CA 02309609 2000-OS-04
WO 99/24421
-31 -
EXAMPLE 3
PCTNS98/23224
Tr
1 ) EtMgl3r, CH2Ci2 /=N ~ ~N
Tr -N
~l
N 2) N_ ',--CHO OH 47
46 II
0°C-RT
Ethylmagnesium bromide (23 mL, 69.1 mmol, 3M in ether) was
dropwise added to a 0°C solution of 4-iodo-triphenylmethylimidazole
(25.1 g, 57.6 mmol) in methylene chloride (280 mL). The mixture was
stirred at 0°C for 30 min., the cooling bath was removed and the
resulting
yellow solution was stirred at room temperature for 60 min. 4-pyridine-
carboxaldehyde (6.1 ml, 63.4 mmol) was added dropwise. The reaction
becomes very thick. A small aliquot of the reaction mixture is partitioned
between ethyl acetate and saturated ammonium chloride. TLC (5%
methanol/methylene chloride) indicated consumption of starting material.
The reaction was quenched with saturated ammonium chloride. The
resulting mixture was dissolved in methylene chloride (required ~1.5 L),
transferred to a separatory funnel, and extracted with methylene chloride.
The extracts were combined, washed with brine, dried over anhydrous
sodium sulfate, filtered and concentrated onto enough silica gel such that
a free flowing powder was obtained. The powder was loaded onto a
chromatography column prepacked with 10% methanol/methylene
chloride. Elution with the same solvent provided 22.5 g (93%) of 47 as a
white solid. NMR ~ H (400 MHz, CDCi3): 8.56( 2H, d, J=6.0 Hz), 7.47(1 H,
d, J=1.4 Hz), 7.36{11 H, m), 7.13(6H, s), 6.63(1 H, s), 5.79(1 H, s), 4.43(1
H,
s). MS (CI): 418 (M+1, 26}, 243(100), 167(45).
S,~g~B_
/=-N ~ ~N AC2O, Et3N /'=N ~ ~N
Tr-N / ~ ~ Tr-N
DMAP, CH2CI2
o ff 47 oAc 48
Acetic anhydride (9.7 mL, 51.4 mmol) was added to a room
temperature suspension of 47 (21.4 g, 51.1 mmol), triethylamine (35.6
ML, 255.7 mmol) and dimethylaminopyridine (0.13 g, 1.0 mmol) in
CA 02309609 2000-OS-04
WO 99/24421
-32-
PCT/US98/23224
methylene chloride (800 mL). The suspension was allowed to stir
overnight. All of the solid eventually dissolves. TLC (10%
methanol/methylene chloride) indicated consumption of starting material.
The mixture was transferred to a separatory funnel, diluted with methylene
chloride, washed with saturated ammonium chloride and brine, dried over
anhydrous sodium sulfate and filtered. The filtrate was concentrated and
the resulting residue was azeotroped (3X) with toluene (to remove
residual acetic acid and acetic anhydride) to give 22.8 g (97%) of 48 as a
white solid. NMR 1 H (400 MHz, CDCI3): 8.61 ( 2H, d, J=6.1 Hz), 7.46(1 H,
d, J=1.4 Hz), 7.38(11 H, m), 7.15(6H, s), 6.83(1 H, s), 6.80(1 H, s), 2.20(3H,
s).
SteI~C
r-N ~ ~N PtO2, HOAC ~=N ~NH
Tr-N ~ ~ ~ Tr-N
H2, 60 psi
oAc ag 2 days 49
48 was dissolved in acetic acid (100 mL) with warming, transferred
to a Parr hydrogenation flask and purged with nitrogen. Platinum oxide
(1.13 g, 4.96 mmol) was added. The resulting mixture was hydrogenated
on a Parr apparatus at 60 psi overnight. A small aliquot was quenched
into 1 N NaOH and ethyl acetate. TLC (10% MeOH/methylene chloride)
indicated consumption of starting material and the formation of lower Rf
products. The mixture was resubmitted to hydrogenation for an additional
day. TLC indicated consumption of starting material. The mixture was
filtered through celite and concentrated. The residue was partitioned
between 1 N sodium hydroxide and methylene chloride. Solid sodium
chloride was added to increase separation and the mixture was extracted
with methylene chloride. The extracts were combined, washed with brine,
dried over anhydrous sodium sulfate and concentrated onto enough silica
gel such that a free flowing powder results. This powder was loaded onto
a chromatography column prepacked with silica and 10%
methanol/methyiene chloride. Elution with 5% NH40H(conc.)/10%
methanol/85% dichloromethane to give 15.9 g (79%) of 49 as a white
glass. NMR ~ H (400 MHz, CDCI3): 7.33(10H, m), 7.14(6H, m), 6.51 (1 H, s),
*rB
CA 02309609 2000-OS-04
WO 99/24421
PCT/US98/23224
-33-
3.04(2H, m), 2.57(2H, dd, J=2.4, 12.1 Hz), 2.44(2H, d, J=7.0 Hz), 1.76(1 H,
m), 1.66(2H, d, J =12.5 Hz), 1.10(2H, dd, J=3.7, 12.4 Hz). MS (LC/MS):
408 (M+)
-N NH RSO2CI ~ ~N N. S02R
Tr-N / Et3N, CH2CI2 Tr-N / 50
5 49 (R is 4-chlorophenyl)
4-Chlorobenzenesulfonyl chloride (0.12 g, 0.56 mmol) was added
to a room temperature solution of 49 (0.21 g, 0.51 mmol) and
triethylamine (0.11 ml, 0.76 mmol) in methylene chloride (3' ml). The
resulting mixture was stirred overnight. TLC (10 % methanol/methylene
10 chloride) indicated consumption of starting material. The solution was
transferred to a separatory funnel, diluted with methylene chloride,
washed with water and brine, dried over anhydrous sodium sulfate,
filtered and concentrated onto enough silica gel such that a free flowing
powder was obtained. The resulting powder was loaded onto a
15 chromatography column prepacked with silica and methylene chloride.
Elution with methylene chloride followed by 10% methanol/methylene
chloride gave 0.26 g of 50 as a white solid. NMR ~ H (400 MHz, CDCI3):
7.68(2H, d, J=8.6 Hz), 7.49(2H, d, J=8.5 Hz), 7.32(10H, m), 7.12(6H, m),
6.49(1 H, s), 3.74(2H, d, J=11.5 Hz), 2.41 (2H, d, J=7.0 Hz), 2.24(2H, dd,
20 J=2.36, 11.8 Hz), 1.69(2H, d, J=13.0 Hz), 1.61 (1 H, m), 1.28(2H, dd,
J=4.2,
12.8 Hz). MS (LC/MS): 582 (M+).
Step E_,
~N N.SOZR HCI ~" ,SOZR
__ N
~N ~ MeOH H-N / ~ HCI
50 51
A mixture of 50 (0.299 g, 0.56 mmol) in methanol (6 ml) and 1 N HCI
25 (3 ml) was warmed to 80°C. After 3h a small aliquot was quenched
into
1 N sodium hydroxide and ethyl acetate. TLC (10% methanol/methylene
chloride) indicated consumption of starting material. The mixture was
cooled to room temperature and concentrated. The residue was dissolved
in water and ether and transferred to a separatory funnel. The water layer
CA 02309609 2000-OS-04
WO 99/24421
- 34 -
PCT/(TS98/23224
was washed with ether. The aqueous layer was concentrated to give
0.154 g (75%) of 51 as a glass. NMR ~ H {400 CD30D): 8.80{1 H, d, J=1.4
Hz), 7.75(2H, d, J=8.8 Hz), 7.62(2H, d, J=8.8 Hz), 7.32(1 H, s), 3.77( d,
J=11.8 Hz), 2.66(2H, d, J=7.2 Hz), 2.29(2H, DT, J=2.5,12.1 Hz), 1.73(2H,
d, J=11.7 Hz), 1.60(1 H, m), 1.32{2H, m). MS (CI): 340 (M+1 )
EXAMPLE 4
R
/'= HOBT ' EDCI, ~N N~O
N -NH
TrN / 49 NMM, CH2C12 TrN / 52
R is 4-chlorophenyl. 1-3-(Dimethylaminopropyl)-3-ethylcarbo-
diimide hydrochloride (0.20 g, 0.68 mmol) was added to a room
temperature solution of 49 (0.21 g, 0.52 mmol), 4-chlorobenzoic acid
(0.07 g, 0.57 mmol), N-methylmorpholine (0.17 ml, 1.56 mmol) and
hydroxybenzotriazole (0.08 g, 0.62 mmol) in dimethylformamide {2 ml)
and methylene chloride (2 ml). The resulting mixture was stirred
overnight. TLC (10% methanol/methylene chloride) indicated
consumption of starting material. The mixture transferred to a separatory
funnel, diluted with methylene chloride, washed with water and brine,
dried over anhydrous sodium sulfate, filtered and concentrated onto
enough silica gel such that a free flowing powder was obtained. The
resulting powder was loaded onto a chromatography column prepacked
with silica and 10% methanol/methyiene chloride. Elution with the same
solvent gave 0.26 g of a clear oil. NMR shows the product was
contaminated with dimethylformamide. The product was dissolved in ethyl
acetate, washed with water, dried over anhydrous sodium sulfate, filtered
and concentrated to give 0.237 g (83%) of 52. NMR 1 H (400 MHz,
CDC13): 7.34(14H, m), 7.13(6H), 6.52(1 H, s), 4.65(1 H, m), 3.68(1 H, m),
2.98(1 H, m), 2.74(1 H, m), 2.47(2H, d, J=7 Hz), 1.96 (1 H, m), 1.70(2H, m),
1.16(2H, m). MS (LC/MS): 546 (M+).
CA 02309609 2000-OS-04
WO 99/24421
-35-
EXAMPLE 5
PCT/US98/23224
O
~ o
1 2
NH ~ N--~ ~ 1 2
jN PhCH3 ~ _N N ~ N NR R
49 53
R' is H and R2 is 4- chlorophenyl. A mixture of 49 (2.0 g, 4.9 mmol)
and N-(4-chlorophenyl)acrylamide (0.98 g, 5.4 mmol) in toluene (50 ml)
was heated to reflux overnight. TLC (10% methanol/methylene chloride)
indicated consumption of starting material. The mixture was cooled to
room temperature and concentrated onto enough silica gel~such that a
free flowing powder was obtained. The resulting powder was loaded onto
a chromatography column prepacked with silica and 10%
methanol/methylene chloride. Elution with 10% methanol/methylene
chloride followed by 5% ammonia (conc)/10% methanol/85% methylene
chloride gave 1.17 g of the title compound with a trace of impurity and 1.50
g of pure 53 as oils. Combined yield 2.67 g (92%). NMR 1 H (400 MHz,
CDCl3): 7.46(2H, d, J=11.8 Hz), 7.34(1H, s), 7.32(10H, m), 7.23(2H, d,
J=11.8 Hz), 7.14(6H, m), 6.55(1H, s) 3.04(2H, d, J=15.3) 2.68(2H, m),
2.5i (4H, d, J=8.3 Hz), 2.07{2H, t, J=14.7 Hz), 1.80(3H, m), 1.28 {2H, m).
MS (LC/MS): 589 (M+).
EXAMPLE 6
0
~N NH RNCO ~-N N- ' NHR
Tr-N / CH2CI2, RT ~ Tr-N /
49 54
R is 3,5-dichlorophenyl. 3,5-dichlorophenylisocyanate (0.21 g, 1.1
mmol) was added to a room temperature solution of 49 (0.3 g, 0.74 mmol)
in methylene chloride (5 ml). The resulting mixture was stirred overnight.
TLC (5% ammonia (conc)/10% methanol/85% methyiene chloride)
indicated consumption of starting material. The mixture was concentrated
onto enough silica gel such that a free flowing powder was obtained. The
resulting powder was loaded onto a chromatography column prepacked
CA 02309609 2000-OS-04
WO 99/24421
PCT/US98/23224
-36-
with silica and 20% acetone/methylene chloride. Elution with 20%
acetone/methylene chloride follow~ad by 5% methanol/methylene chloride
gave 0.37 g (83%) of 54 as a whites solid. NMR ~ H (400 MHz, CDCI3):
7.53(1 H, m), 7.36(10H, m), 7.12(61-1, m), 6.97(1 H, m), 6.71 (1 H, m),
6.56(1 H, s), 4.04(2H, d, J=17.3 Hz), 2.86{2H, m), 2.52(2H, d, J=9.1 Hz),
1.95(1 H, m), 1.72{2H, d, J=17.1 Hz:), 1.16(2H, m). MS (LC/MS): 596 (M+).
F~~MP~.E 7
Ste .~A
Cl ~ ~ NHZ
~ N N ~ C02Me
N ~ Me3Al, PhCH3 Cl
/=N N~N
10 Tr N / 56 H
Trimethylaluminum (1.2 ml, 2.4 mmol, 2M in toluene) was added to
a 0°C solution of 3-chloroaniline (0.10g, 0.8 mmol) in toluene (7.5
ml).
After 5 minutes the cooling bath w,as removed and the mixture was stirred
at room temperature for 30 minutes. 55 (0.48 g, 0.1 mmol) in toluene (10
15 ml) was added via cannula. The mixture was refluxed overnight. TLC
(10% methanol/85% methyiene chloride) indicated consumption of
starting material. The mixture was. cooled to room temperature, diluted
with ethyl acetate and quenched with a saturated solution of sodium
sulfate. The resulting mixture was stirred overnight. The mixture was
20 made basic with 1 N NaOH (3 ml). The resulting mixture was transferred to
a separatory funnel and extracted with ethyl acetate. The extracts were
combined, washed with water andl brine, dried over anhydrous sodium
sulfate and concentrated onto enough silica gel such that a free flowing
powder was obtained. The resulting powder was loaded onto a
25 chromatography column prepacke~d with silica and 3%
methanol/methylene chloride. Elution with 3-10% methanol/methylene
chloride gave 0.31 g (66%) of 56 as a white Foam. NMR 1 H (400 MHz,
CDC13): 7.71 (1 H, m) 7.29(12H, m), 7.14(6H, m), 7.05(2H, m), 6.55(1 H, s),
CA 02309609 2000-OS-04
WO 99/24421 PCT/US98/23224
-37-
3.04(2H, m), 2.68(2H, m), 2.51 (3H, m), 2.09(2H, m), 1.81 (2H, m), 1.58(2H,
m), 1.3(2H, m). MS (LC/MS): 589 (~JI+).
teg, B
o ~ c1
r-N N'~'~ N w. -,..
Tr-N / H p / Cl
56 ~ ~
r=N N~N
H-N'/ 5~ H
Compound 56 (0.6 g, 1.0 mrnol) in methanol (18 ml) and 1 N HCI (6
ml) was warmed to 60°C. Progress of the reaction was monitored by
quenching a small aliquot of the reaction with 1 N sodium hydroxide and
ethyl acetate. TLC (5% ammonia(c:onc)/10% methanoU85% methylene
chloride) indicated consumption of ;starting material. The mixture was
cooled to room temperature and concentrated. The residue was not totally
soluble in ether/water. The residue was made basic with 1 N NaOH,
diluted with methylene chloride, transferred to a separatory funnel and
extracted with methylene chloride. The extracts were combined, washed
with brine, dried over anhydrous sodium sulfate, filtered and concentrated
onto enough silica gel such that a free flowing powder was obtained. The
powder was loaded onto a chromatography column prepaclced with silica
and 10% methanol/methylene chloride. Elution with 10%
methanol/methylene chloride followed by 5%
ammonia(concentrated)/10% meth;~nol/ chloride) gave the title compound
as a clear oil. The oil was redissoh~ed in methylene chloride and treated
with an excess of HCI (4M in dioxane) and concentrated in vacuum to give
0.205 g (44%) of 57 as a clear glass. NMR 1 H (400 CD30D): 8.85(1 H, s),
7.50(2H, d, J=11.4 Hz), 7.40(1 H, s), 7.05(2H, d, J=11.4 Hz), 4.251 (2H, s),
4.i5(2H, t, J=7.5 Hz), 3.46(2H, d, J:=16.2 Hz), 3.35(3H, m), 3.02(2H, t,
J=16.2 Hz), 2.95(6H, s), 2.74(2H, d, J=9.0 Hz), 2.25(2H, m), 1.93(2H, d,),
1.60(2H, m). MS (FAB): 357 (M+1 ).
CA 02309609 2000-OS-04
WO 99/24421
.. 3g _
.EyM L
Ste~A_
PCT/US98/23224
1 ) LDA
Ph '--N\~O '; Ph '-N ~ OSO CF
2 3
5g 2) PhN(S03CF3)2 59
n-butyl lithium (30.4 ml, 48.E~ mmol, 1.6 M in hexane) was added to
a -78°C solution of diisopropyl amune (6.63 ml, 50.6 mmol) in
tetrahydrofuran (75 ml). After 30 minutes 58 (7.5 ml, 40.5 mmol) in
tetrahydrofuran (30 ml) was added slowly via cannula. The reaction was
stirred at -78°C for 1.5 hours, then N-
phenyltrifluoromethanesulfonamide
(15.3 g, 44.5 mmol) in tetrahydrofuran (50 ml) was added via cannula.
The mixture was allowed to warm to room temperature overnight. TLC
(20% ethyl acetate/hexanes) indicated consumption of starting material.
Triethylamine (added to prevent acid hydrolysis of the triflate on silica gel)
was added and the resulting mixture was concentrated onto enough silica
gel such that a free flowing powder was obtained. The powder was
loaded onto a chromatography column prepacked with silica and 20%
ethyl acetate/hexanes. Elution with the same solvent provided 10.8 g
(83%) of 59 as a yellow oii. NMR '~ H (400 MHz, CDCI3): 7.30(5H, m),
5.73(1 H, m), 3.63(2H, s), 3.13(2H, dd, J=3.0, 6.4 Hz), 2.72(2H, t, J =5.7
Hz), 2.45(2H, m).
a B
Ph ~N~ OS02CFg '-"' -TMS ~ Ph ~ N ~ - TMS
~/~~59 (PhsP)~'PdCh, Cul
THF, DIPA
Trimethylsilylacetylene (5.9 ml, 42.1 mmol) was added to a room
temperature solution of 59 (10.8 g, 33.7 mmol) in a 3:1 mixture of
25 tetrahydrofuran and diisopropylamine (50 ml). Dichlorobis(triphenyl-
phosphine)palladium (II) (1.42 g, 2.0 mmol) and copper (I) iodide (1.1 g,
5.7 mmol) were added. The color of the reaction progressed from red to
brown to black. After 1 hour, TLC (5% ethyl acetatelhexanes) indicated
consumption of starting material. '('he reaction was diluted with ethyl
30 ether, transferred to a separatory funnel, washed with water, 3/1 saturated
CA 02309609 2000-OS-04
WO 99/24421
-39-
PCT/US98/23224
ammonium chloride/ammonia (conc) and brine, dried over anhydrous
sodium sulfate, filtered and concentrated onto enough silica gel such that
a free flowing powder was obtained. The powder was loaded onto a
chromatography column prepacked with silica and 10% ethyl
acetate/hexanes. Elution with the same solvent provided 6.1 g (67%) of
60 as a yellow solid. NMR ~H (400 MHz, CDCI3): 7.35(5H, m), 6.14(1 H,
m), 3.63(2H, s), 3.08(2H, m), 2.63(~!H, t, J =5.7 Hz), 2.33(2H, m), 0.23(9H,
s).
Step C
Ph~-N ~ ._. TMS . TBAF ~ Phi
N\~ CH
THF
6~ 61
Tetrabutylammonium fluoride (27 ml, 27.0 mmol, 1 M in
tetrahydrofuran) was added to a room temperature solution of 60 (6.1 g,
22.5 mmol) in tetrahydrofuran (100 ml). After --2 hours, TLC (20% ethyl
acetate/hexanes) indicated consumption of starting material. The reaction
mixture was diluted with ethyl acetate, transferred to a separatory funnel,
washed with water and brine, dried over anhydrous sodium sulfate,
filtered and concentrated onto enough silica gei such that a free flowing
powder was obtained. The powder was loaded onto a chromatography
column prepacked with silica and '10% ethyl acetate/hexanes. Elution
with the same solvent provided 3.4 g (76%) of 61 as a yellow solid. NMR
~ H (400 MHz, CDC13): 7.36(5H, m), 6.17(1 H, m), 3.65(2H, s), 3.11 {2H, m),
2.91 (1 H, s), 2.64(2H, t, J =5.6 Hz), 2.35(2H, m).
Step D
,Tr
,Tr
Ph~N\~CH ~Ph3P)2PdC~h,~Cul- Ph,~ ~ - ~ N
N
61 T~F/DIF~A N
62
67 (3.42 g, 17.3 mmol) and 1-triphenylmethyl-4-iodoimidazole (6.3
g, 14.4 mmol) were dissolved in tetrahydrofuran (100 ml) and diisopropyl-
amine (40 ml). Dichlorobis(triphenylphosphine)palladium (II) {1.22 g, 1.7
mmol) and copper (I) iodide (0.4 g, 1.7 mmol) were added. The reaction
CA 02309609 2000-OS-04
WO 99/24421
PCT/US98/23224
- 40 -
mixture was allowed to stir at room temperature overnight. TLC (5%
rnethanol/methylene chloride) indicated consumption of starting material.
The reaction was diluted with methylene chloride, transferred to a
separatory funnel, washed with water, 3/1 saturated ammonium
chloride/ammonia (conc) and brine, dried over anhydrous sodium sulfate,
filtered and concentrated. The residue was recrystallized fom~ ethyl
acetate to give 7.02 g (9$%) of 62 as a slightly yellow solid. NMR 1 H
(400 MHz, CDCI3): 7.44(1 H, d, J=1.1 Hz), 7,40(14H, m), 7.1$(6H, m),
7.06(1 H, d, J=1.5 Hz), 6.12(1 H, m), 3.64(2H, s), 3.12(2H, m), 2.64(2H, t, J
=5.7 Hz), 2.39(2H, m). MS(FAB): 505 (M+).
,Tr
Ph ~ ~ ~ N~~ HZ X60 Psi) Ph ~
N _ ~ ~ N N
N 10% Pd/C
62 63
62 (7.0 g, 14.1 mmol) was dissolved in a mixture of tetrahydrofuran
(250 ml), methanol (200 ml) and methylene chloride (100 ml) and purged
with nitrogen. 10% palladium on carbon (1.0 g) was added and the
resulting suspension was hydrogenated on a Parr apparatus overnight at
60 psi. TLC (5% methanol/methylene chloride) indicated a considerable
amount of remaining starting material. The mixture was filtered through
celite, fresh 10% palladium on carbon was added and the mixture was
again hydrogenated on a Parr apparatus at 60 psi for two days TLC (5%
methanol/methylene chloride) indicated a considerable amount of
remaining starting material. 20% palladium hydroxide on carbon (1.0 g)
and acetic acid (60 ml) were added and the mixture was again
hydrogenated on a Parr apparatus at 60 psi overnight. The mixture was
filtered and concentrated. TLC (5% ammonia (conc)/10%
methanol/methylene chloride) indicated a number of new spots. The
residue was redissolved in acetic acid (75 ml) and 20% palladium
hydroxide on carbon (1.0 g) was added and the mixture was
hydrogenated at 50 psi for two days. The reaction was filtered through
celite, the filter cake was well washed with methanol. The filtrate was
concentrated and the residue was azeotroped with toluene (3X) to remove
CA 02309609 2000-OS-04
WO 99/24421
-41
PCT/US98/23224
residual acetic acid. The residue was dissolved with 1 N NaOH and
methylene chloride, transferred to a separatory funnel and extracted with
methylene chloride. The extracts were combined, washed with water and
brine, dried over anhydrous sodium sulfate and concentrated to provide
6.8 g of an amber oil. Chromatography on silica eluting with 4%
ammonia(conc)/10% methanol/86% methylene chloride provided 0.7 g
(10%) of 63. NMR ~ H (400 MHz, CD3C13): 7.29(15H, m), 7.13(6H, m),
6.49(1 H, s), 3.58(2H, m), 2.94(2H, m), 2.54(2H, t, J =8.0 Hz), 1.98(2H),
1.66(2H, m), 1.54(3H, m), 1.29(2H).
_teQF
,Tr ,Tr
N H2 (1 atm) N
Ph~N /N ~ 20% Pd(OH)p/C HN /N
MeOH, HCI
63 64
63 (0.019 g, 0.054 mmol) was dissolved in methanol (1 ml), 1 M HCI
(2 drops) was added. The resulting solution was purged with nitrogen.
10% palladium on carbon (0.005 g) was added and the mixture was
stirred under a balloon of hydrogen gas overnight. The mixture was
filtered through celite, the filter cake was well washed with methanol and
concentrated to give 0.0128 g of a clear oil. ~ H NMR analysis indicated no
reaction had occurred. The oil was redissolved in methanol (1 ml), HCI (1
drop) was added. The resulting solution was purged with nitrogen. 20%
palladium hydroxide on carbon (0.01 g) was added and the mixture was
stirred under a balloon of hydrogen gas overnight. The mixture was
filtered through celite, the filter cake was well washed with methanol and
concentrated to give 0.0085 g of 64 as a clear oil. NMR ~H (400 MHz,
CD30D): 8.87(1 H, s), 7.41 (1 H, s), 3.45(2H, m), 3.04(2H, rri), 2.85(2H, m),
2.05(2H, m), 1.75(3H, m), 1.53(3H, m).
Compound (64) was then used to produce compounds of
formula I, e.g. by following the procedures of the examples above.
CA 02309609 2000-OS-04
WO 99/24421
- 42 -
AMPLE 9
PCT/US98/23224
N-H (t-Boc)20, . t-Boc
Et0 Dioxane/Wate~EtO N LAH~ N-t-Boc
HO~~~
O 65 O 66 67
1. LDA, 300mo1%
2. CH3 I (1000mo1%) or CICF3, DAST
N. t-Boc
6$ Et0
O R R= CHg
N , t-Boc Iz~ PPh~ N, t-Boc P~ . t-Boc
HO~~,~ I~y~ I- PhgP+~\~,~,
67 69 70
N . t-Boc 1. t-BuOK, THF, /'N I
I - Ph3P+~V~~ -'~' \N N
70 O ~.i,' ' t-Boc
2' ?2
N H
N 71
Tr
,N I 1. H2, 10% Pd-C
''N ~N 2. TFA, RT, 1 h
t ' t-Boc
Tr 72 Tr
Commercially available ethyl isonipecotate was protected with di-
tert-butyl Bicarbonate, the ethyl ester reduced with lithium aluminum
hydride and the intermediate alcohol was transformed into the desired
iodide 69 with iodine according to the procedure described by A.
Villalobos in the Journal of Medicinal Chemistry 1994, 37, 2721-2734.
A 500 mL round bottomed flask was charged with iodide 69 (lO.Og,
30.75 mmol), triphenylphosphine (16.9g, 64.6 mmol) and 150 mL
acetonitrile.
The solution was heated at refiux for 16h, cooled to room
'N
N
73
N
15 temperature and then concentrated in vacuo to a yellow oil. The crude
product was further purified by chromatography on silica using a gradient
from 4:1 hexane: ethyl acetate to 100% ethyl acetate and final elution with
CA 02309609 2000-OS-04
WO 99/24421
-43-
PCT/US98/23224
95:5 methylene chloride: methanol to afford phosphonium salt 70 (7.13 g)
in 40% yield.
A 500 mL round bottomed flask was charged with phosphonium
salt 70 (7.13g, 12.14 mmol), n-trityl imidazole-4-carboxaldehyde (4.5g,
13.14 mmol) and 250 mL dry tetrahydrofuran an the reaction mixture was
cooled to 4°C. Potassium t-Butoxide (14 ml of a 1 M in Dioxane, 14
mmol)
was added dropwise and the solution was allowed to warm slowly to room
temperature and the disappearance of aldehyde was monitored by TLC.
Additional potassium t-butoxide was added at 4 h (2.4 mL, 2.4 mmol) and
the reaction was allowed to stir at room temperature. After a total of 16 h
the reaction was filtered and the filtrate was concentrated to an oil. Elution
on silica gel column with hexanes: ethyl acetate afforded pure alkene 72
(3.2g) in 51 % yield as a mixture of E/Z isomers.
A 500 mL round bottomed flask was charged with alkene 72 (3.2
g), Pt02 (0.75g), and 150 mL methanol and affixed with a three-way
stopper with a hydrogen bladder. The heterogeneous reaction was stirred
under hydrogen for 2 h. The catalyst was filtered and the filtrate was
concentrated to an oil (3.2g). The crude intermediate was redissolved in
180 mL dioxane and treated at room temperature with 1 M TFA in dioxane
(20 mL, 20 mmol) for 24 h. The pH of the reaction mixture was adjusted to
greater than 8 with sodium hydroxide (1 M), ethyl acetate was added and
the layers were separated. The organic layer was washed with brine,
dried over magnesium sulfate and concentrated to a semi-solid. The
crude product was purified by chromatography (methylene chloride:
methanol eluent) to afford pure 73 (1.8 g, 69% yield).
EXAMPLE 10
Tr
Tr
IPhgP H ' N ~ N I H
CHO
Via..
N
N~ t-BuOK, THF or ~4 N>
39 NaHMDS, THF or
Boc BuLi, THF
CA 02309609 2000-OS-04
WO 99/24421
- 44 -
PCT/US98/23224
Tr
N Pt02, MeOH,
y I H
N /n.,, H2 Balloon ~~ ~ H
i,,..
<3h
74 75
N N
Boc ~Boc
HCl Dioxane
N room temp N
v I H --~--.a. y I H
N ~~~.. N
75 N HCl 76. R = Trityl N~
~Boc dioxane
H
reflux 77, R = H
To a flask containing the phosphonium salt 39 (3.5 g, 6.11 mmol)
was added dry THF (30 mL) under a nitrogen atmosphere. The mixture
was cooled to 0°C and t-BuOK was added (1.0 M solution, 8 g, 8 mmols)
dropwise via syringe. The resulting yellow mixture was stirred for 20 min,
then the 3-carbon aldehyde (2.4 g, 6.55 mmol) was added in 8 mL THF
via syringe. The reaction mixture was stirred for 24 h at 25°C then
quenched by the addition of NH4Cl solution. The aqueous portion was
extracted with EtOAc. The combined organics were washed with brine,
dried over MgS04, filtered and concentrated. Chromatogrphy on silica
gel (40% EtOAc-hexane ---> 60% EtOAc) afforded 74 2.6 g (71 %) of
material. MS (electrosrpay, M+H) = 534.
To Compound 74 (2.3g, 4.3 mmol) dissolved in MeOH was added
Pt02 (0.4 g). A hydrogen balloon was placed over the reaction mixture,
and stirring was continued for 2-3 h at 25°C. The reaction mixture was
then chromatographed on Si02 (100% hexane increasing to 75% EtOAc-
hexane) to remove the catalyst and obtain the pure product 75, 2.24 g
(97%). MS (electrosrpay, M+H) = 536.
To a dioxane solution of compound 75 (2.0 g, 3.7 mmol) was
added a 4M HCI-dioxane solution (10 mL) at 25°C. The mixture was
stirred for about 6 h, then cooled to 0°C, and 5% NaOH was added to
bring the pH to 7. The mixture was extracted with EtOAc, and the
combined organics were washed with brine, dried over MgS04, filtered
and concentrated, to give 1.i4 g (100%) of compound 76. MS
(electrosrpay, M+H) = 436.
CA 02309609 2000-OS-04
WO 99/24421
PCT/US98/23224
- 45 -
Treatment of 76 ( 200 mg, 0.46 mmoi) again with 4M HCI-dioxane
(5 mL) at 80°C for 4 h affords 740 mg of compound n. MS (Ci, M+H)
194.
The compounds below were prepared following procedures similar
to those described above.
H~~
H ~ ~ '\N
~N N
C1 79 ~o
78 N'S \ / ~ \
02 , i
HN
HN ~ ~ N
'N N H
C1 $j N
\ C1
80 \
0
CA 02309609 2000-OS-04
WO 99/24421
-46-
C1
H ~~ i \
~N
N
82 ~ NH
O
H~~
~.~. N
N H
83 ~ N
o I \ ci
ci
PCT/US98/23224
HN ~ HN
'N 'N
' ' H
H ~
84 N N 85 N I r N ~ C1
\ o l
o I
c~ . ci
HN ~ HN
~N ~N
N N
86 ~-0 87 ~l~--o
o I \ o I \
ci
HN ~ HN
'N 'N
N N H
88 ~ c g9 ~ N
I \ ~ I \
Cl,
CA 02309609 2000-OS-04
WO 99/24421
- 47 -
PCT/US98/23224
H~~
~N
HN ~ ~ H
~N
91 ~N \
N ,, ~ C1 p /
90 ~N \ / ''
H . C1
HN
~~N > HN
N H ...
N N
92 ~ / ~ N w- Cl
93
ci _ o
H~~
'~~N
N H
94 ~ N
o / ~ F
HN ~ HN
~N 'N
N H N
95 ~ N 96
o p / \ Ct o / \
OCF3 CI
HN \ N~ ~~ HN \ N,
~N S ~N S
97 02 _ 98 oa
CA 02309609 2000-OS-04
WO 99/24421
-48-
H I,\ H
'N ~N~N \
99 p ~
F
N
I F
N H
N
100 H
N p
H
~. N~~O
H ~ ~ 101 HN
N \
F
H ~\
.N I H
_ ~N~N \
102 p
F
H F
HN~N N~N / I
103 p
,.
PC'T/US98/23224
H F
HN \ N
~N ~ N
104 '' ~~p /
CA 02309609 2000-OS-04
WO 99/24421
- 49 -
0
N
C1
105 O
HN~ N w C1
NCO I \
'N l 106 Cl / Cl
N
H
,
O / I O. CF3
N~N \
N H
vlo~
N
H
/ C1
I
N N ~,
~~ l ~'~o
N 108 O C1
H
,
/ C1
I
N N~~ \
~~ l o
N 109 CI
H
N l N ~ C1
N 110 O I
H C1
PCT/US98/23224
CA 02309609 2000-OS-04
WO 99/24421
-50-
I H
N N N ~ C1
113 ~ ~ /
H
I H
N N N ~ CI
112
H
HN ~ N CH3
r
O
F F
I F
NON
N H
~N ~ y 114
H
O / C1
N ~ C1
N
115
N
H
PCT/US98/23224
CA 02309609 2000-OS-04
WO 99/24421
-51 -
c1
'N N
N H
116
N
H
F F
f 'F
N N
N H
117
N
H
O
N ~ C1
N H
s
N
H
N
HN ~ 119 O
~N
'N O C1
HN 120 N
iN H
C1
PCT/US9$/23224
CA 02309609 2000-OS-04
WO 99/24421
-52-
0
N
HN~ 121 O
~N
PCT/US98/23224
.N __~O
HN 122 N ~
~N H
C1, and
'N ~C / Cl
\'
HN ~ 123 N
~i N H
MASS SPErTROMETRY DATA
COMPOUND MASS SPEC COMPOUND MASS SPEC
NO. NO.
78 (CI) 340 (M+1 79 (FAB) 300 (M+1
) )
80 (CI) 304 (M+1 81 (EI) 346 (M+)
)
82 (EI) 374 {M+) 83 (EI) 380 (M+)
84 (CI) 347 {M+1 85 (CI) 353 (M+1
) )
86 (EI) 313 (M+) 87 (EI) 347 (M+)
88 (EI) 333 (M+) 89 (CI) 439 (M+1
)
90 (FAB) 333 (IVI+191 (FAB) 319 (M+1
) )
92 (FAB) 319 (M+193 (FAB) 332 (M+1
) )
94 (FAB) 331 (NI+195 (FAB) 361 (M+1
) )
96 (FAB) 312 (M+197 (CI) 390 (M+1
) )
98 (CI) 340 (M+1 99 (C1) 331 (M+1
) )
100 (CI) 359 (M+1 101 Elect~osp~ay
) 359 (M+1 )
CA 02309609 2000-OS-04
WO 99/24421
-53-
PCT/US98/23224
102 Eiectrospray 103 FAB 303 (M+1
345 (M+1 ) )
104 (CI) 329 (M+1105 High Resolution
)
Calc. 410.1402
Found 410.1410
106 High Resolution-
Calc.396.1609------ __---
Found 396.1618
The compounds of this invention are either agonists or antagonists
of the histamine H3 receptor. The binding affinity of the compounds of the
invention to the H3 receptor may be demonstrated by the procedure
described below:
H_3 Recc~~tor Bindin ss
The source of the H3 receptors in this experiment was guinea pig
brain. The animals used weighed 400-600 g. The tissue was
homogenized using a Polytron in a solution of 50 mM Tris, pH 7.5. The
final concentration of tissue in the homogenization buffer was 10% w/v.
The homogenates were centrifuged at 1000 x g for 10 min. in order to
remove clumps of tissue and debris. The resulting supernatants were
then centrifuged at 50,000 x g for 20 min. in order to sediment the
membranes, which were next washed 3 times in homogenization buffer
(50,000 x g for 20 min. each). The membranes were frozen and stored at -
70°C until needed.
All compounds to be tested were dissolved in DMSO and
then diluted into the binding buffer (50 mM Tris, pH 7.5) such that the final
concentration was 2 ~,g/ml with 0.1 % DMSO. Membranes were then
added (400 ~g of protein) to the reaction tubes. The reaction was started
by the addition of 3 nM [3HJR-a-methylhistamine (8.8 Ci/mmol) or [3HJ-N-
methylhistamine (80 Ci/mmol) and incubated at 30° for 30 min. Bound
ligand was separated from unbound ligand by filtration, and the amount of
radioactive ligand bound to the membranes was quantitated by liquid
scintillation spectrometry. All incubations were performed in duplicate and
CA 02309609 2000-OS-04
WO 99/24421
PCT/US98/23224
-54-
the standard error was less than 10% in all instances. Compounds that
inhibited greater than 70% of the specific binding of radioactive ligand to
the receptor were serially diluted to determine a K; (nM).
Compounds 45, 78, 79, 81-97, and 113-118 had a K; in the range of
0.1 to 220 nM. Compounds 45, 79, 81, 82, 83, 84, 86, 87, 88, 89, 91, 94,
96, and 116 had a K; in the range of 0.1 to 20 nM.
Compounds described by this invention, inert, pharmaceutically
acceptable carriers can be either solid or liquid. Solid form preparations
include powders, tablets, dispersible granules, capsules, cachets and
suppositories. The powders and tablets may be comprised of from about
5 to about 70 percent active ingredient. Suitable solid carriers are known
in the art, e.g. magnesium carbonate, magnesium stearate, talc, sugar,
lactose. Tablets, powders, cachets and capsules can be used as solid
dosage forms suitable for oral administration.
For preparing suppositories, a low melting wax such as a mixture of
fatty acid glycerides or cocoa butter is first melted, and the active
ingredient is dispersed homogeneously therein as by stirring. The molten
homogeneous mixture is then poured into convenient sized molds,
allowed to cool and thereby solidify.
, Liquid form preparations include solutions, suspensions and
emulsions. As an example may be mentioned water or water-propylene
glycol solutions for parenteral injection.
Liquid farm preparations may also include solutions for intranasal
administration,
Aerosol preparations suitable for inhalation may include solutions
and solids in powder form, which may be in combination with a
pharmaceutically acceptable carrier, such as an inert compressed gas.
Also included are solid form preparations which are intended to be
converted, shortly before use, to liquid form preparations for either oral or
parenteral administration. Such liquid forms include solutions,
suspensions and emulsions.
The compounds of the invention may also be deliverable
transdermally. The transderma! compositions can take the form of creams, ,
lotions, aerosols and/or emulsions and can be included in a transdermal
CA 02309609 2000-OS-04
WO 99/24421
PCT/US98/23224
- 55 -
patch of the matrix or reservoir type as are conventional in the art for this
purpose.
Preferably the compound is administered orally.
Preferably, the phamlaceuticaf preparation is in unit dosage form.
5 In such form, the preparation is subdivided into unit doses containing
appropriate quantities of the active component, e.g., an effective amount to
achieve the desired purpose.
The quantity of active compound in a unit dose of preparation may
be varied or adjusted from about O.i mg to 1000 mg, more preferably from
. about 1 mg to 500 mg, according to the particular application.
The actual dosage employed may be varied depending upon the
requirements of the patient and the severity of the condition being treated.
Determination of the proper dosage for a particular situation is within the
skill of the art. Generally, treatment is initiated with smaller dosages which
15 are less than the optimum dose of the compound. Thereafter, the dosage
is increased by small increments until the optimum effect under the
circumstances is reached. For convenience, the total daily dosage may
be divided and administered in portions during the day if desired.
The amount and frequency of administration of the compounds of
20 the invention and the pharmaceutically acceptable salts thereof will be
regulated according to the judgment of the attending clinician considering
such factors as age, condition and size of the patient as well as severity of
the symptoms being treated. A typical recommended dosage regimen is
oral administration of from 1 mg to 2000 mg/day preferably 10 to 1000
25 mg/day, in one to four divided doses to achieve relief of the symptoms.
The compounds are non-toxic when administered within this dosage
range.
The following are examples of pharmaceutical dosage forms which
contain a compound of the invention. As used therein, the term "active
30 compound" is used to designate one of the compounds of the formula I or
salt thereof. The scope of the invention in its pharmaceutical composition
aspect is not to be limited by the examples provided.
CA 02309609 2000-OS-04
WO 99/24421
-56-
Pharmaceutical,~t osage Form Examolp,~
EXAMPLE ~
T et
PCT/US98/23224
N~c Ingredients - m /tablet malts
. lI~ t
1. Active compound 100 500
2. Lactose USP 122 113
3. Corn Starch, Food Grade, 30 40
as a 10% paste in
Purified Water
4. Com Starch, Food Grade 45 40
5. Magnesium Stearate
Total 300 700
mecnoa of nnanutacture
Mix Item Nos. 1 and 2 in a suitable mixer for 10-15 minutes. Granulate
the mixture with Item No. 3. Mill the damp granules through a coarse
screen (e.g., 1/4", 0.63 cm) if necessary. Dry the damp granules. Screen
the dried granules if necessary and mix with Item No. 4 and mix for 10-15
minutes. Add Item No. 5 and mix for 1-3 minutes. Compress the mixture
to appropriate size and weigh on a suitable tablet machine.
EXAMPLE B
sule
Ingredient m~/ca~pSUlemq/cal sule
1. Active compound 100 500
2. Lactose USP 106 123
3. Corn Starch, Food Grade 40 70
4. Magnesium Stearate NF 4 7
Total 250 700
CA 02309609 2000-OS-04
WO 99/24421
-57-
PCTNS98lZ3224
Method of Manufacture
Mix Item Nos. 1, 2 and 3 in a suitable blender for 10-15
minutes. Add Item No. 4 and mix for 1-3 minutes. Fill the mixture into
suitable two-piece hard gelatin capsules on a suitable encapsulating
machine.
While the present invention has been described in
conjunction with the specific embodiments set forth above, many
alternatives, modifications and variations thereof will be apparent to those
of ordinary skill in the art. All such alternatives, modifications and
variations are intended to fall within the spirit and scope of.the present
invention.